gmp trends: contamination control according to … · gmp trends: contamination control according...
TRANSCRIPT

ETIF 2008Costa Salguero
Centre -
Buenos Aires -
Argentina
24 October 2008
Giovanni Bini
“Revision of Annex 1, 14/02/2008”
GMP Trends:
Contamination control according to Annex I effective from 2009

2
Contents
Revision History
Introduction
Cleanrooms Classification
Cleanrooms Routine Monitoring
Blow/fill/seal
Media Fill
Bioburden
Capping of vials
Conclusions

3
EU GMP –
Annex 1 –
Manufacture of Sterile Medicinal Products
Revision September 2003
EU GMP –
Annex 1 –
Manufacture of Sterile Medicinal Products (1997)
Revision Annex 1Revisions History
EU GMP –
Annex 1 –
Manufacture of Sterile Medicinal Products
Revision 14 February 2008Revision 14 February 2008
Aim of the revision: •
Keep pace with technological change;
•
Solve some interpretation problems
•
Harmonize with the ISO guidelines and FDA regulations.

4
Revision Annex 1Introduction
•
Classification of Cleanrooms;
•
Media Fill execution method;
•
Advices concerning bioburden for sterilization loads;
•
Advices concerning capping of vials.
The Annex 1 of EC Guide to GMP, “Manufacture of Sterile Medicinal Products, has been recently reviewed (February 2008) concerning on the following topics:
Annex 1 becomes effective on March 1st, 2009, except for capping, in order to give enough time to industry to set on the new requirements; this part will be active starting from March 1st, 2010.

5
•
“Production of sterile products is subject to special requirements in order to minimize the risk of microbial and pirogen
contamination.
•
Sterile areas should have an appropriate cleaning standard with air passing through efficient filters.”
Revision Annex 1Introduction
Two different methods of production
Aseptic production
Production with final sterilization
Volume 4, EU GMP, Annex 1“Manufacture of Sterile Medicinal Products”
February 2008
“Where possible, heat sterilization is the method of choice”

6
•
Grade A: The local zone for high risk operations. Laminar air flow systems should provide a homogeneous airspeed in a range of 0.36 –
0.54 m/s
(guidance value) at the working position in open cleanroom applications;
•
Grade B: For aseptic preparation and filling, this is the background environment for the grade A zone;
•
Grade C e D: Clean areas for carrying out less critical stages in the manufacture of sterile products.
Revision Annex 1Introduction
For the manufacture of sterile medicinal products 4 cleanroom cleanliness grades can be distinguished:
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008

7
Revision Annex 1Introduction
Examples of operations to be carried out in the various grades:
Grade Examples of operations for terminally sterilised products.
A Filling of products, when unusually at risk
C Preparation of solutions, when unusually at risk. Filling of products
D Preparation of solution and components for subsequent filling
Grade Examples of operations for aseptic preparations.
A Aseptic preparation and filling
C Preparation of solution to be filtered
D Handling of components after washing
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008

8
The requirements for classification are clearly defined
Routinary
monitoringInitial Classification
The following is applied to
Clean Air Devices
(hoods, LAF, isolator, RABS,…)
Cleanrooms
Revision Annex 1Cleanrooms Classification

9
Clean rooms and clean air devices should be classified in accordance with EN ISO 14644-1; the table for particles limits has (almost) been
harmonized with itMaximum permitted number of particles per m3 equal to or greater than
the tabulated size
At rest In operation
Grade 0.5µm 5.0 µm 0.5µm 5.0 µm
A 3520 20 3520 20
B 3520 29 352000 2900
C 352000 2900 3520000 29000
D 3520000 29000 Not detect Not detect
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008
For classification purposes EN/ISO 14644-1 methodology defines both the minimum number of sample locations and the sample size based on the classlimit of the largest considered particle size and the method of evaluation of
the data collected
Revision Annex 1Cleanrooms Classification
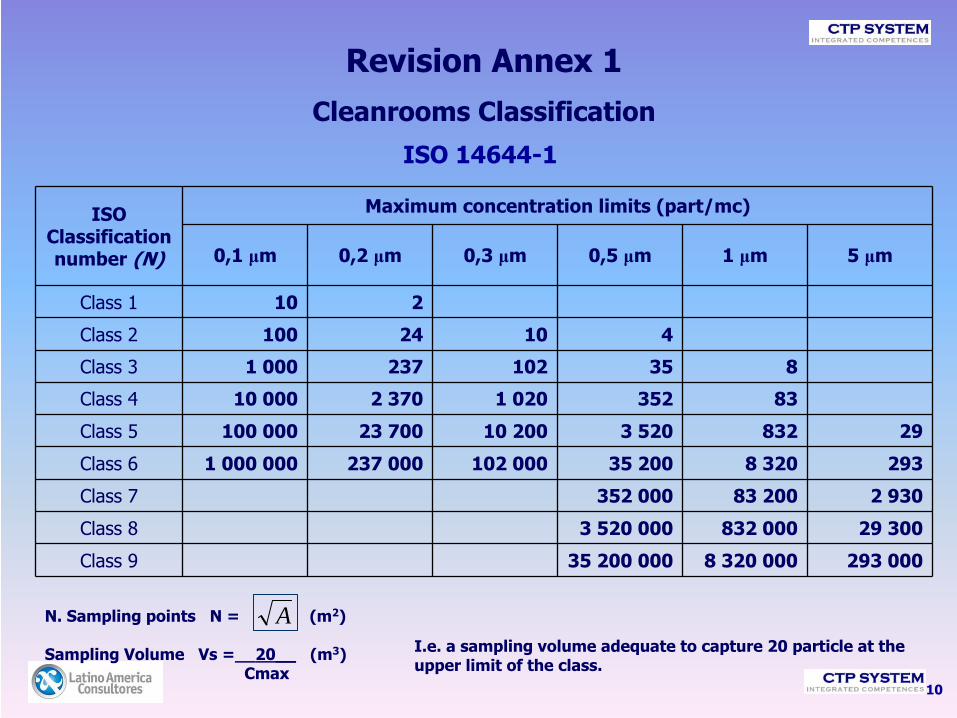
10
ISO 14644-1
N. Sampling points N = (m2)
Sampling Volume Vs =__20__
(m3)Cmax
ISO Classification number (N)
Maximum concentration limits (part/mc)
0,1 μm 0,2 μm 0,3 μm 0,5 μm 1 μm 5 μm
Class 1 10 2
Class 2 100 24 10 4
Class 3 1 000 237 102 35 8
Class 4 10 000 2 370 1 020 352 83
Class 5 100 000 23 700 10 200 3 520 832 29
Class 6 1 000 000 237 000 102 000 35 200 8 320 293
Class 7 352 000 83 200 2 930
Class 8 3 520 000 832 000 29 300
Class 9 35 200 000 8 320 000 293 000
AI.e. a sampling volume adequate to capture 20 particle at the upper limit of the class.
Revision Annex 1Cleanrooms Classification

11
An important topic for revision is the requirement on particle size ≥
5.0 µm.
FDA does not plan to monitor them, while for EMEA it is a valid "diagnostic tool" to track the loss of benefits of the cleanroom and to report the incidence of abnormal situations not easily detectable by other parameters.
The Annex 1 has retained the obligation to control the particle sized ≥
5.0 μm, establishing a limit of 29 part/m3 for grade B at rest.
Despite that, the limit for such particles for grade A, formerly
of ≤
1 part/m3
was changed to ≤
20 part/m3:
“The occasional indication of ≥5.0 μm particle counts may be false counts due to electronic noise, stray light, coincidence, etc. However consecutive or regular counting of low levels is an indicator of a possible contamination event and should be investigated. Such events may indicate early failure of the HVAC system, filling equipment failure or may also be diagnostic of poor practices during machine set-up and routine operation.”
Particle size ≥
5.0 µm
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008
Revision Annex 1Cleanrooms Classification

12
Grade“at rest” “in operation”
Particles ≥
0.5µm
Particles≥
5µm
Particles ≥
0.5µm
Particles ≥
5µm
A 3.520 (3.500)
20 (1)
3.520 (3.500)
20 (1)
B 3.520 (3.500)
29 (1)
352.000 (350.000)
2.900 (2.000)
C 352.000 (350.000)
2.900 (2.000)3.520.000
(3.500.000)29.000
(20.000)
D 3.520.000 (3.500.000)29.000
(20.000)No defined No defined
(limits set in 2003 revision of Annex 1 are between brackets)
Comparison between particles limits (n. part./m3)
Annex 1 (2003) vs. Annex 1 (2008)
Revision Annex 1Cleanrooms Classification

13
5. “For classification purposes in Grade A zones, a minimum sample volume of 1m3
should be taken per sample location.”Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008
Sampling Volume
•
In the 1997 edition of annex 1 the limit for particles ≥5.0 μm
was “0”
and
there were not any indication about the sampling volume, so the ISO 14644-1 formula was applied to 0,5 μm particles (0,1 ft3
could be enough, typically 1 ft3
was used).•
In the 2003 revision the limit was established as “1”
(specifying that 0 have not a statistical significance) and the correspondent test about the sampling volume was:“For routine test the total sample volume should not less than 1 m3 for grade A and B areas and preferably also in grade C areas”.This request was often applied to the whole area instead of each
sampling point.
•
The new 2008 revision:–
moves the 1 m3
sampling volume application from the routine monitoring to the “classification purpose”
sampling;–
limits the request to the grade A areas, with just an option for
grade B areas;–
specifies that 1 m3
sampling volume should be “taken for sample location”.
Revision Annex 1Cleanrooms Classification

14
“For Grade A the airborne particle classification is ISO 4.8 dictated by the limit for particles ≥5.0 μm.
For Grade B (at rest) the airborne particle classification is ISO 5 for both considered particle sizes.
For Grade C (at rest & in operation) the airborne particle classification is ISO 7 and ISO 8 respectively.
For Grade D (at rest) the airborne particle classification is ISO 8.
For classification purposes EN/ISO 14644-1 methodology defines both the minimum number of sample locations and the sample size based on the class limit of the largest considered particle size and the method of evaluation of the data collected.”
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008
Correspondence with ISO 14644-1
Revision Annex 1Cleanrooms Classification

15
Revision Annex 1Cleanrooms Classification
In practice we can interpret it…
•
The Grade A represents the ISO 5 class for particles ≥
0.5 μm
and a “ISO 4.8”
class for ≥
5 μm particles
•
Grade B at rest
corresponds to ISO 5, in operation to ISO 7. There are some
interpretative doubts on the limit for Grade B at rest, which would lead to a theoretical sampling volume of about 0.7 m3; it seems logical to interpret this requirement so as to use a sample volume of 1 m3
also able to B.
•
For Grade C (corresponding to ISO 7 at rest
and ISO 8 in operation) and D
(corresponding to ISO 8 at rest), however, due to the clear reference to ISO 14644-1, nothing prevents you from taking a “typical”
sample volume of 1 ft3
for "sampling location".
Correspondence with ISO 14644-1

16
The 2008 EU GMP Annex 1:
•
defines how to count the particles in operation
(or during the routine production or during a Media Fill simulation); also refers to the ISO 14644-2 regarding the frequency of repetition of tests for grading;
•
it requires that points to be monitored in operation are determined on the basis of risk analysis and the results of the classification;
Revision Annex 1Cleanrooms Classification

17
Routine Cleanrooms monitoring:
•
the sampling volume is accepted to be different from that used for
classification, encouraging to use a sampling rate appropriate to report transient spikes of contamination (and therefore should be possible not to sample 1 m3
in class A and B, but use repeated smaller volume samples;
•
Class A is supposed to have a continuous sampling
at all critical stages of production;
•
Class B admits a less frequent control, based on the effectiveness of segregation of the zone A compared to zone B;
•
Annex 1 implicitly does not recommend the use of particulate samplers like "manifold“, because an excessive length and too many curves of the sampling tube cause a decrease in collection efficiency of larger particles
Revision Annex 1Cleanrooms Classification

18
Routine Cleanrooms monitoring:
•
Monitoring in grade A is tolerated to be interrupted in case of process phases in which hazardous contaminants are present (e.g. radiopharmaceuticals, living organisms, viruses);
•
it admits that sampling near the point of filling bottles can produce off-specification values for ≥
5.0μm particles, although the trend of
the inspectors is to suggest that there are engineering solutions to avoid this problem, and therefore we assume that they are available in industry;
•
Finally, the need to define a recovery time of class after the
completion of the transactions is required; this would allow to return to the particles conditions set out for "at-rest" after 15-20 minutes.
Revision Annex 1Cleanrooms Routine Monitoring

19
9. “For Grade A zones, particle monitoring should be undertaken for the full duration of critical processing, including equipment assembly, except where justified by contaminants in the process that would damage the particle counter or present a hazard, e.g. live organisms and radiological hazards. In such cases monitoring during routine equipment set up operations should be undertaken prior to exposure to the risk.
Monitoring during simulated operations should also be performed.
The Grade A zone should be monitored at such a frequency and with suitable sample
size that all interventions, transient events and any system deterioration would be captured and alarms triggered if alert limits are exceeded. It is accepted that it may not always be possible to demonstrate low levels of ≥5.0 μm
particles at the point of fill when filling is in progress, due to the generation of particles or droplets from the product itself.”
10. “It is recommended that a similar system be used for Grade B zones although the sample frequency may be decreased. The importance of the particle monitoring system should be determined by the effectiveness of the segregation between the adjacent Grade A and B zones. The Grade B zone should be monitored at such a frequency and with suitable sample size that changes in levels of contamination and any system deterioration would be captured and alarms triggered if alert limits are exceeded.”
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008
Revision Annex 1Cleanrooms Routine Monitoring

20
Recommended limits for microbial contamination (a)
Grade Air sample cfu/m3
Settle plates (diameter 90 mm)
cfu/4 hours (b)
Contact plates (diameter 55
mm) cfu/plate
Glove print 5 fingers
cfu/glove
A <1 <1 <1 <1
B 10 5 5 5
C 100 50 25 -
D 200 100 50 -
No significant news emerge relative to microbial contamination, the limits have remained unchanged, and no explanation has been given to the fact that they are referred to mean values, important point of difference from the U.S. legislation
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008
Revision Annex 1Cleanrooms Routine Monitoring

21
Revision Annex 1Blow/fill/seal
Blow/fill/seal machine should be installed in class C areas. Manufacturing machine for final sterilized products should be installed in class D areas.
27. “Because of this special technology particular attention should be paid to, at least the following:
•
equipment design and qualification;•
validation and reproducibility of cleaning-in-place and sterilisation-in-
place;•
background clean room environment in which the equipment is
located;•
operator training and clothing;
•
interventions in the critical zone of the equipment including any aseptic assembly prior to the commencement of filling”.
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008
Blow/fill/seal units are purpose built machines in which, in one
continuous operation, containers are formed from a thermoplastic granulate, filled and then sealed, all by the one automatic machine.

22
Revision Annex 1Media Fill
Paragraphs on Media Fill have been expanded:
66. “Validation of aseptic processing should include a process simulation test using a nutrient medium (media fill). Selection of the nutrient medium should be made based on dosage form of the product and selectivity, clarity, concentration and suitability for sterilisation of the nutrient medium”.
67. “The process simulation test should imitate as closely as possible the routine aseptic manufacturing process and include all the critical subsequent manufacturing steps. It should also take into account various interventions known to occur during normal production as well as worst-case situations”.
68. “Process simulation tests should be performed as initial validation with three consecutive satisfactory simulation tests per shift and repeated at defined intervals and after any significant modification to the HVAC-system, equipment, process and number of shifts. Normally process simulation tests should be repeated twice a year per shift and process”.
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008

23
Revision Annex 1Media Fill
Limits of number of contaminating units depending on total filled units, are defined:
69. “The number of containers used for media fills should be sufficient to enable a valid evaluation. For small batches, the number of containers for media fills should at least equal the size of the product batch. The target should be zero growth and the following should apply:
•
When filling fewer than 5000 units, no contaminated units should
be detected.
•
When filling 5,000 to 10,000 units:a)One (1) contaminated unit should result in an investigation,
including consideration of a repeat media fill;b)Two (2) contaminated units are considered cause for
revalidation, following investigation.
•
When filling more than 10,000 units:a)One (1) contaminated unit should result in an investigation;b)Two (2) contaminated units are considered cause for
revalidation, following investigation”.
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008

24
Revision Annex 1Media Fill
An investigation of microbiological contamination events is required for any size Media Fill executed; even if the contamination detected is low, but not zero, the cause should be clarified. If instead the Media Fill highlights problems of contamination (e.g. Beyond the limits specified), an analysis
of their impact
on sterility of batches of product processed starting from the last Media Fill succeeded until then, is required.
70. “For any run size, intermittent incidents of microbial contamination may be indicative of low-level contamination that should be investigated. Investigation of gross failures should include the potential impact on the sterility assurance of batches manufactured since the last successful
media fill”.
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008
Requirements concerning Media Fill
have been “harmonized”
with FDA.

25
Revision Annex 1Bioburden
80. The bioburden should be monitored before sterilisation. There should be working limits on contamination immediately before sterilisation, which are related to the efficiency of the method to be used.
Bioburden assay should be performed on each batch
for both aseptically filled product and terminally sterilised
products.
Where overkill sterilisation
parameters are set for terminally sterilised
products, bioburden might be monitored only at suitable scheduled intervals.
For parametric release systems, bioburden assay should be performed on each batch and considered as an in-process test.
Where appropriate the level of endotoxins should be monitored.
All solutions, in particular large volume infusion fluids, should be passed through a micro-organism-retaining filter, if possible sited immediately before filling.
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008

26
Revision Annex 1Capping of vials
The most relevant variation in the new Annex 1, considering impact on pharmaceutical products, regards the classification of the zones where capping of vials is performed.
These requirements are applied to each final form of sterile product:
•
Lyophilized
•
Liquids
•
Powders

27
Vials of products that are partially stoppered, should be kept in Grade A zone until the stopper is completely inserted; this obvious requirement was already effective in practice.
116. “Partially stoppered
freeze drying vials should be maintained under Grade A conditions at all times until the stopper is fully inserted”.
Revision Annex 1Capping of vials
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008

28
118. “The container closure system for aseptically filled vials is not
fully integral until the aluminium cap has been crimped into place on the stoppered
vial.
Crimping of the cap should therefore be performed as soon as possible after stopper insertion”.
120.“Vial capping can be undertaken as an aseptic process
using sterilised caps or as a clean process
outside the aseptic core. Where this latter
approach is adopted, vials should be protected by Grade A conditions up to the point of leaving the aseptic processing area, and thereafter
stoppered
vials should be protected with a Grade A air supply until the cap has been crimped.”
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008
Revision Annex 1Capping of vials
Until the previous revision, Annex 1 did not require to cap the vials in a specific grade zone; in the industrial practice, it was usually performed
in grade D or C,
with a local LAF protection on the machine and the conveyor belt.
The approach of the new Annex 1 have precise requirements

29
b.
Capping could be performed as a “clean”
instead of an aseptic process; in this case, vials should be protected during their transfer from the filling machine to the capping machine, until their complete closure.
The capping machine is allowed to be installed out of the sterile area, but the conveyor belt should stay inside the grade A zone until the exit
from the sterile
area; for the machine and the part of the belt outside the sterile area, a grade A air flow protection is required, without a specific requirement for a grade B background.
Paragraph n°122 suggests some options; among them, “RABS”
and isolators are mentioned as devices able to ensure the required conditions and minimize human intervention and, consequently, a possible contamination.
122. “Restricted access barriers and isolators may be beneficial in assuring the required conditions and minimising direct human interventions into the capping operation”.
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008
Revision Annex 1Capping of vials

30
In case the capping is performed as “aseptic process”, inside the sterile zone, it means…
• equipment suitable to be installed in a sterile area
(materials, finishing, maintenance mode, etc ...)
• personnel dressed with clothes for aseptic area
• microbial and particles monitoring (!!!)
• material’s paths suitable in the aseptic area
…
especially …
• Sterile caps and manipulated so as to maintain the sterility of the cap itself and the background environment (!!!)
Revision Annex 1Capping of vials

31
119. “As the equipment used to crimp vial caps can generate large quantities of non-viable particulates, the equipment should be located at a separate station equipped with adequate air extraction”.
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008
Note: capping machine should be separated from filling machine, in order to avoid contamination in the filling zone
Revision Annex 1Capping of vials

32
Another problem identified in the development of the new Annex 1
is the possibility of manual "not allowed" operations performed on partially closed bottles, for example, repositioning or replacement of poorly placed caps, which can lead to significant danger of contamination of the product .
Hence the request to provide identification systems for the capping machines, and the automatic discarding of bottles with missing or badly positioned caps before the corking, and to provide adequate systems to minimize the microbiological contamination in case of need for human intervention, such as precisely barrier or insulators systems.
In any case human intervention should be minimized.
121. “Vials with missing or displaced stoppers should be rejected prior to capping. Where human intervention is required at the capping station, appropriate technology should be used to prevent direct contact with the vials and to minimise
microbial contamination”.
Volume 4, EU GMP, Annex 1 -
“Manufacture of Sterile Medicinal Products”
February 2008
Revision Annex 1Capping of vials

33
Revision Annex 1Conclusions
•
“Risk Based”
approach for interpretation of the new requirements;
•
Harmonization with international standards;
•
A considerable change with high impact regarding the capping.
Doubts expressed by many European operators in the fields are shown (both manufacturers of finished drugs and manufacturers of packaging machinery) on the possibility to fully comply with the new requirements in less than two years, considering difficulties due to the need for considerable
investment and
technological difficulties.

Revisione Annex 1Risorse in rete
Annex 1, revision
2008
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/vol-4/pdfs-
en/2008_02_12_gmp_annex1.pdf
Comparison
between
old
and new
Annex 1
http://www.gmp-
navigator.com/elements/PDF/Gegenueberstellung_Annex_1_Feb_08.pdf
