(green chemistry and sustainable technology) an hui lu, sheng dai (eds.)-porous materials for carbon...
TRANSCRIPT

Green Chemistry and Sustainable Technology
An-Hui LuSheng Dai Editors
Porous Materials for Carbon Dioxide Capture

Green Chemistry and Sustainable Technology
For further volumes: http://www.springer.com/series/11661
Series editors
Prof. Liang-Nian He State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin, China
Prof. Robin D. Rogers Department of Chemistry, Center for Green Manufacturing, The University of Alabama, Tuscaloosa, USA
Prof. Dangsheng SuShenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, Shenyang, ChinaandDepartment of Inorganic Chemistry, Fritz Haber Institute of the Max Planck Society, Berlin, Germany
Prof. Pietro TundoDepartment of Environmental Sciences, Informatics and Statistics, Ca’ Foscari University of Venice, Venice, Italy
Prof. Z. Conrad ZhangDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China

Aims and Scope
The series Green Chemistry and Sustainable Technology aims to present cutting-edge research and important advances in green chemistry, green chemical engineering and sustainable industrial technology. The scope of coverage includes (but is not limited to):
– Environmentally benign chemical synthesis and processes (green catalysis, green solvents and reagents, atom-economy synthetic methods etc.)
– Green chemicals and energy produced from renewable resources (biomass, carbon dioxide etc.)
– Novel materials and technologies for energy production and storage (biofuels and bioenergies, hydrogen, fuel cells, solar cells, lithium-ion batteries etc.)
– Green chemical engineering processes (process integration, materials diversity, energy saving, waste minimization, efficient separation processes etc.)
– Green technologies for environmental sustainability (carbon dioxide capture, waste and harmful chemicals treatment, pollution prevention, environmental redemption etc.)
The series Green Chemistry and Sustainable Technology is intended to provide an accessible reference resource for postgraduate students, academic researchers and industrial professionals who are interested in green chemistry and technologies for sustainable development.
Green Chemistry and Sustainable Technology

An-Hui Lu · Sheng Dai Editors
1 3
Porous Materials for Carbon Dioxide Capture

EditorsAn-Hui LuState Key Laboratory of Fine ChemicalsSchool of Chemical Engineering Dalian University of Technology DalianChina
Library of Congress Control Number: 2014936432
© Springer-Verlag Berlin Heidelberg 2014This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. Exempted from this legal reservation are brief excerpts in connection with reviews or scholarly analysis or material supplied specifically for the purpose of being entered and executed on a computer system, for exclusive use by the purchaser of the work. Duplication of this publication or parts thereof is permitted only under the provisions of the Copyright Law of the Publisher’s location, in its current version, and permission for use must always be obtained from Springer. Permissions for use may be obtained through RightsLink at the Copyright Clearance Center. Violations are liable to prosecution under the respective Copyright Law.The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use.While the advice and information in this book are believed to be true and accurate at the date of publication, neither the authors nor the editors nor the publisher can accept any legal responsibility for any errors or omissions that may be made. The publisher makes no warranty, express or implied, with respect to the material contained herein.
Printed on acid-free paper
Springer is part of Springer Science+Business Media (www.springer.com)
ISSN 2196-6982 ISSN 2196-6990 (electronic)ISBN 978-3-642-54645-7 ISBN 978-3-642-54646-4 (eBook)DOI 10.1007/978-3-642-54646-4Springer Heidelberg New York Dordrecht London
Sheng DaiOak Ridge National Laboratory Chemical Sciences Division Oak Ridge, TN USA

v
Carbon capture and storage (CCS) and potentially carbon capture and utilization (CCU) have received increasing attention from both the scientific community and industry during the past several decades, because day-to-day carbon dioxide (CO2) emissions arising from fossil fuel combustion may cause detrimental changes to the earth’s environment. To reach the CCS and CCU goals, the primary step is CO2 capture, through which CO2 is separated from gas mixtures. CO2 also represents a ubiquitous, renewable carbon source that enables the production of methanol and dimethyl ether and efficient alternative transportation fuels, as well as their various derived products. Furthermore, sequestration of low-partial-pressure CO2 from an enclosed space is of importance in life-support systems for submarines and space vehicles. Hence, the selective capture and separation of CO2 in an economical, energy-efficient fashion is of positive significance not only in terms of academic interest but also to social and economic progress. Compared with liquid phase ammonia scrubbing, adsorption processes based on porous solids are considered to be a promising alternative separation technique because of their low energy con-sumption, ease of regeneration, and superior cycling capability. The critical factor in these processes is the design and synthesis of high-performance sorbents. With rapid developments in novel sorbent materials, CO2 capture-based sorption, sepa-ration, and purification have become more and more dominant for carbon capture. In view of their past, current, and potential future importance, it is time to assem-ble key achievements in relevant aspects of CO2 capture materials and methods that underpin progress in this field.
The book Porous Materials for Carbon Dioxide Capture is aimed at providing researchers with the most pertinent and up-to-date advances related to the fields of porous materials design and fabrication and subsequent evaluation in innova-tive cyclic CO2 adsorption processes, with special emphasis on uncovering the relationships between structural characteristics and CO2 capture performance. The book is divided into seven chapters that provide a resume of the current state of knowledge of porous CO2 capture materials, which include ionic liquid-derived carbonaceous adsorbents, porous carbons, metal-organic frameworks, porous aro-matic frameworks, microporous organic polymers, sorption techniques such as cyclic calcination and carbonation reactions, and membrane separations.
The main benefit of the book is that it highlights the synthesis principles, advanced characterization methods, and structural merits of most of the advanced
Preface

Prefacevi
CO2 capture solids and presents some of the most important CO2 separation methods and related computational simulations. It may serve as a self-contained major reference that appeals to scientists and researchers. The book can be used in the classroom for graduate students who focus on CO2 separation processes. The material in this book will also benefit engineers active in the research and development of CO2 capture technologies.
January 2014 An-Hui LuSheng Dai

vii
Contents
1 Ionic Liquid-Derived Carbonaceous Adsorbents for CO2 Capture . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1Xiang Zhu, Chi-Linh Do-Thanh and Sheng Dai
2 Porous Carbons for Carbon Dioxide Capture . . . . . . . . . . . . . . . . . . . . 15An-Hui Lu, Guang-Ping Hao and Xiang-Qian Zhang
3 Metal-Organic Frameworks (MOFs) for CO2 Capture . . . . . . . . . . . . . 79Hui Yang and Jian-Rong Li
4 Carbon Dioxide Capture in Porous Aromatic Frameworks . . . . . . . . . 115Teng Ben and Shilun Qiu
5 Microporous Organic Polymers for Carbon Dioxide Capture . . . . . . . 143Yali Luo and Bien Tan
6 CO2 Capture via Cyclic Calcination and Carbonation Reactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 181Marcin Broda, Roberta Pacciani and Christoph R. Müller
7 Functionalized Inorganic Membranes for High-Temperature CO2/N2 Separation . . . . . . . . . . . . . . . . . . . . . . . . . . 223Mayur Ostwal and J. Douglas Way

ix
Teng Ben Department of Chemistry, Jilin University, Changchun, People’s Republic of China
Marcin Broda Laboratory of Energy Science and Engineering, ETH Zurich, Zürich, Switzerland
Sheng Dai Oak Ridge National Laboratory, Chemical Sciences Division, Oak Ridge, TN, USA
Chi-Linh Do-Thanh Oak Ridge National Laboratory, Chemical Sciences Divi-sion, Oak Ridge, TN, USA
Guang-Ping Hao State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian, People’s Republic of China
Jian-Rong Li Department of Chemistry and Chemical Engineering, College of Environmental and Energy Engineering, Beijing University of Technology, Beijing, People’s Republic of China
An-Hui Lu State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian, People’s Republic of China
Yali Luo Department of Chemistry, Huazhong University of Science and Tech-nology, Wuhan, People’s Republic of China
Christoph R. Müller Laboratory of Energy Science and Engineering, ETH Zurich, Zürich , Switzerland
Mayur Ostwal Department of Mechanical Engineering, University of Colorado, Boulder, USA
Roberta Pacciani Air Products and Chemicals, Campus de la UAB, Bellaterra, Barcelona, Spain
Shilun Qiu State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University, Changchun, People’s Republic of China
Bien Tan Department of Chemistry, Huazhong University of Science and Technology, Wuhan, People’s Republic of China
Contributors

Contributorsx
J. Douglas Way Chemical and Biological Engineering Department, Colorado School of Mines, Golden, CO, USA
Hui Yang Department of Chemistry and Chemical Engineering, College of Environmental and Energy Engineering, Beijing University of Technology, Beijing, People’s Republic of China
Xiang-Qian Zhang State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian, People’s Republic of China
Xiang Zhu Oak Ridge National Laboratory, Chemical Sciences Division, Oak Ridge, TN, USA

1
Abstract Removal of CO2 from major emission sources, such as power plants and industrial facilities for environmental remediation has attracted significant interest. Among currently accessible CO2 capture technologies, the use of porous solids is considered to be one of the most promising approaches. The use of ionic liquids (ILs) composed of an organic cation and an inorganic anion as precursors for the synthesis of carbonaceous materials has been an emerging field. Porous carbons with a high specific surface area can be facilely made by directly annealing ILs or using appropriate porous templates. By choosing different ILs, materials with various het-eroatoms doping and good pore properties can be produced. The attractive features of IL-derived materials such as facile synthesis, high specific surface area, and nitro-gen content make them promising candidates for CO2 capture. In this chapter, we review the recent research progress on IL-derived carbonaceous materials and their potential CO2 separation application.
1.1 Introduction
As global environmental standards are becoming evermore stringent regarding the emission of designated greenhouse gases, large fixed carbon dioxide (CO2) sources, such as power plants, are in pursuit of novel methods for the sequestration of CO2 [1]. Substantial scale carbon capture and sequestration (CCS) is considered as one of the most promising strategies to mediate the atmospheric CO2 concentra-tion for environmental remediation [2]. Conventional processes widely employed in industry for CO2 capture involve chemical absorption of CO2 with ethanola-mine solutions. Though this method is well-established and offers a high CO2 absorption capacity, it suffers from several serious drawbacks, including solvent
Chapter 1Ionic Liquid-Derived Carbonaceous Adsorbents for CO2 Capture
Xiang Zhu, Chi-Linh Do-Thanh and Sheng Dai
A.-H. Lu and S. Dai (eds.), Porous Materials for Carbon Dioxide Capture, Green Chemistry and Sustainable Technology, DOI: 10.1007/978-3-642-54646-4_1, © Springer-Verlag Berlin Heidelberg 2014
X. Zhu · C.-L. Do-Thanh · S. Dai (*) Chemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USAe-mail: [email protected]

2 X. Zhu et al.
loss, a high parasitic energy cost for the regeneration and equipment corrosion [3]. In this regard, alternative processes such as physical adsorption separation by porous solid adsorbents have been proposed.
Thus far, significant research efforts have been devoted to exploring porous mate-rials with high specific surface area and excellent thermal stability toward revers-ible CO2 adsorption. These materials include hybrid microporous and mesoporous materials such as metal-organic frameworks (MOFs) [4, 5], zeolitic imida-zolate frameworks (ZIFs) [6], microporous organic polymers (MOPs) [2, 7–16], and amine-modified silicas [17–20] (e.g., “molecular basket” sorbents, hyper-branched aminosilica). In comparison with the traditional CCS technologies, these porous solids with high CO2 uptake capacities and lower energy for regeneration have been proven to be a more attractive solution for CO2 separation. However, in spite of these beneficial properties, multi-step synthesis processes of such solid adsorbents, commonly involving surface modification steps with CO2-philic moie-ties, may limit the scale-up preparation for CO2 capture. Facile and cost-effective preparation processes combined with excellent gas adsorption properties are keys to make porous solid adsorbents as promising candidates for practical applications in CO2 separation from flue gas. Therefore, sorbents based on porous carbons are considered to be promising candidates for CO2 capture [21–27]. Several potential strategies like introducing N-doped CO2-philic moieties (as shown in Fig. 1.1) into the porous carbonaceous networks have been developed to increase CO2 loading capacity and the adsorption selectivity for CO2 over N2, which is another crucial parameter for CO2 capture materials. The large availability of carbon precursors and synthetic routes to design sorbents with tailored pores, large specific surface areas, and surface groups make carbons even more attractive for the development of future CCS technologies.
Recently, the use of ionic liquids (ILs) composed of an organic cation and an inorganic anion as precursors for the synthesis of carbonaceous materials has been an emerging field [28–31]. Porous carbons with high specific surface areas can
Fig. 1.1 N-doped sites within carbonaceous framework (Reproduced with permission Ref. [31] Copyright 2010, The Royal Society of Chemistry). 1 Amine, 2 pyrollic, 3 nitro, 4 pyridinic, 5 quaternary graphitic

31 Ionic Liquid-Derived Carbonaceous Adsorbents for CO2 Capture
be facilely made by directly annealing ILs or using appropriate porous templates. By choosing different ILs, materials with various heteroatoms doping and good pore properties can be produced [29]. The attractive features of IL-derived materi-als such as facile synthesis, high specific surface area and nitrogen content make them promising candidates for CO2 capture. Exceptional CO2 separation perfor-mance can be achieved by these facilely made carbonaceous adsorbents. Thus, in this chapter, a summary of recent research progress on IL-derived carbonaceous materials and their potential CO2 separation application is provided.
1.2 Nanoporous Carbons Derived from Task-Specific Ionic Liquids
Traditional carbonaceous adsorbents synthesis involves the carbonization of low-vapor pressure polymeric precursors derived from either synthetic (e.g., polyacrylonitrile (PAN), phenolic resins) or natural sources such as pitch and shell nuts. These poly-meric species possess low vapor pressures so that cross-linking reactions can proceed with concomitant char formation and without vaporization of the corresponding pre-cursor units. Nonpolymeric carbon sources are rarely used to form carbon because of their uncontrolled vaporization during high-temperature pyrolysis [32]. Recently, ILs with cross-linkable functional groups, namely task-specific ionic liquids (TSILs), have been considered as highly promising precursors for the synthesis of functional carbo-naceous materials due to their negligible volatility and molecular tunability (Fig. 1.2). The intrinsic nonvolatility suggests favorable conditions for an intriguing carboniza-tion process based on well-behaved cross-linking reactions of monomeric TSIL pre-cursor units with minimal loss of reactant. The key structural prerequisite of TSIL precursors is the presence of certain functional groups that can undergo cross-linking reactions under pyrolysis conditions. Given the tunability of TSILs, either cations or anions can be functionalized with cross-linking groups. To date, nitrile groups, the key factors in determining the high carbon yields of PAN under charring conditions, have been mostly appended onto the structure of ILs because of their cyclotrimeriza-tion of triazine rings at high temperatures [32, 33]. In addition, TSILs further allow for the preparation of graphitizable carbons with heteroatom doping (such as nitrogen and boron with their ratios in the carbon materials controlled by their amounts initially present in the cross-linkable ions).
1.2.1 Nitrile-Functionalized Cations
The first success of TSILs-derived carbonaceous materials was demonstrated by appending nitrile functionality onto imidazolium backbones. N-doped porous car-bons with high specific surface area can be prepared through a simple, convenient, and catalyst-free process. The structural morphology (porosity and surface area) of
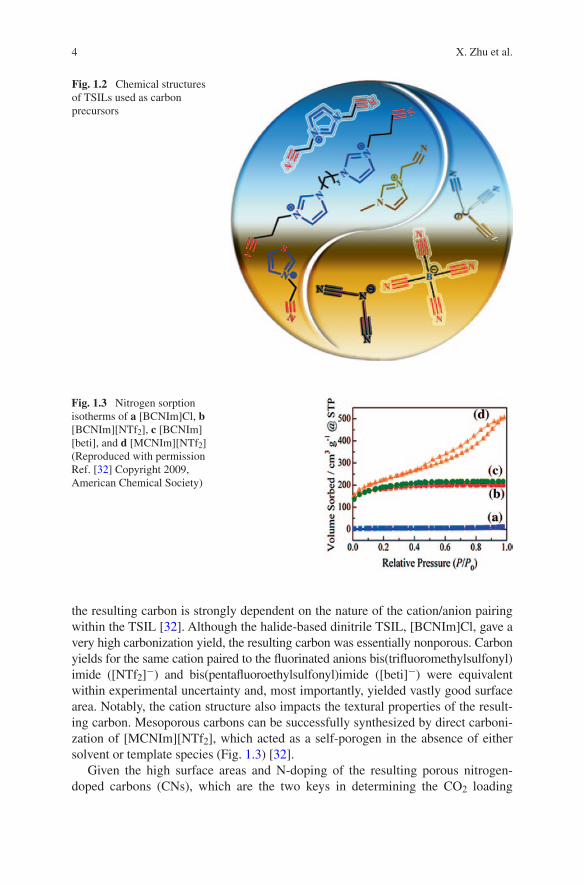
4 X. Zhu et al.
the resulting carbon is strongly dependent on the nature of the cation/anion pairing within the TSIL [32]. Although the halide-based dinitrile TSIL, [BCNIm]Cl, gave a very high carbonization yield, the resulting carbon was essentially nonporous. Carbon yields for the same cation paired to the fluorinated anions bis(trifluoromethylsulfonyl)imide ([NTf2]−) and bis(pentafluoroethylsulfonyl)imide ([beti]−) were equivalent within experimental uncertainty and, most importantly, yielded vastly good surface area. Notably, the cation structure also impacts the textural properties of the result-ing carbon. Mesoporous carbons can be successfully synthesized by direct carboni-zation of [MCNIm][NTf2], which acted as a self-porogen in the absence of either solvent or template species (Fig. 1.3) [32].
Given the high surface areas and N-doping of the resulting porous nitrogen-doped carbons (CNs), which are the two keys in determining the CO2 loading
Fig. 1.2 Chemical structures of TSILs used as carbon precursors
Fig. 1.3 Nitrogen sorption isotherms of a [BCNIm]Cl, b [BCNIm][NTf2], c [BCNIm][beti], and d [MCNIm][NTf2] (Reproduced with permission Ref. [32] Copyright 2009, American Chemical Society)

51 Ionic Liquid-Derived Carbonaceous Adsorbents for CO2 Capture
capacities of carbonaceous adsorbents, the CO2 capture performance of these new materials was investigated. As the carbonization temperatures change, specific sur-face areas and nitrogen contents of the resulting material have also undergone a significant change. In addition, though no chemical/physical activation processes were involved in their preparation, the obtained porous CNs displayed excep-tional CO2 adsorption capacity of 4.39 mmol g−1 at 0 °C and 1 bar (Fig. 1.4). This results from the strong interactions between CO2 molecules and abundant num-bers of nitrogen-containing groups in these frameworks, which provided an isos-teric heat of adsorption value of 32.1 kJ mol−1. In this regard, the use of TSILs as precursors for the preparation of tailor-made porous adsorbents opens interesting avenues in the area of carbon capture because of the simplicity of this method to prepare nitrogen-doped carbons [25].
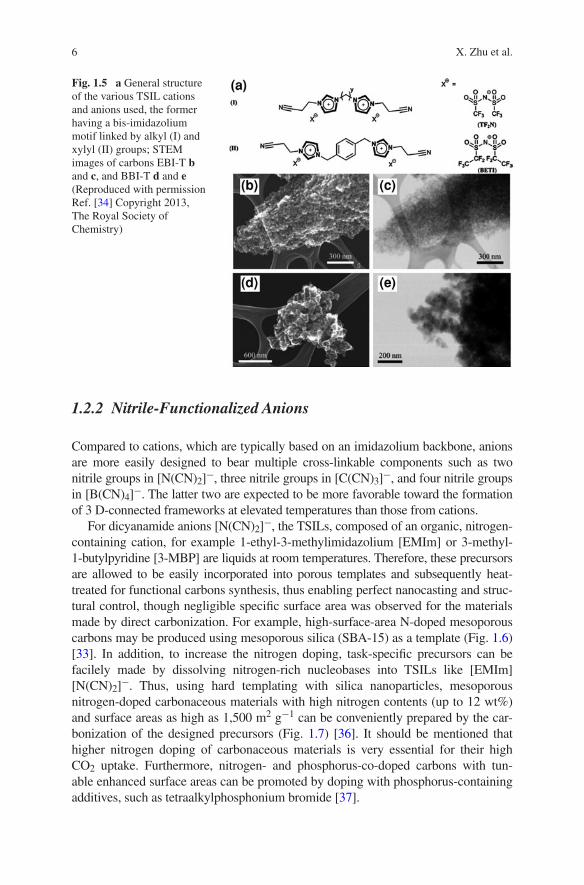
Very recently, TSILs having a bis-imidazolium motif linked with various organic groups may allow for the synthesis of hierarchical nanoporous nitrogen-doped carbons (Fig. 1.5). Microporous–mesoporous carbon with the specific surface area up to 1,300 m2 g−1 was facilely obtained through the ionothermal synthesis process as well [34]. Besides the potential good CO2 uptake abilities of these N-doped mesoporous carbons, they may also be considered as the “bas-kets” for polyamines-derived CO2 adsorbents based on their good mesoporous architectures.
Moreover, carbonaceous materials that are intrinsically co-doped with nitrogen and sulfur heteroatoms can be synthesized by facile annealing of nitrile-function-alized thiazolium salts. The obtained materials exhibit an aromatic graphite-like carbon backbone with remarkably high degrees of heteroatom doping of about 6–8 wt% of both S and N, even at the highest temperature (1,000 °C) [35]. It is worth mentioning that multiple doping is a promising way for functionalizing car-bon materials. Given the high degree of S/N co-doping, the resulting porous solids may be promising candidates for carbon dioxide capture.
Fig. 1.4 CO2 and N2 uptake of TSILs-derived CNs at 273 and 298 K, respectively
6 X. Zhu et al.
1.2.2 Nitrile-Functionalized Anions
Compared to cations, which are typically based on an imidazolium backbone, anions are more easily designed to bear multiple cross-linkable components such as two nitrile groups in [N(CN)2]−, three nitrile groups in [C(CN)3]−, and four nitrile groups in [B(CN)4]−. The latter two are expected to be more favorable toward the formation of 3 D-connected frameworks at elevated temperatures than those from cations.
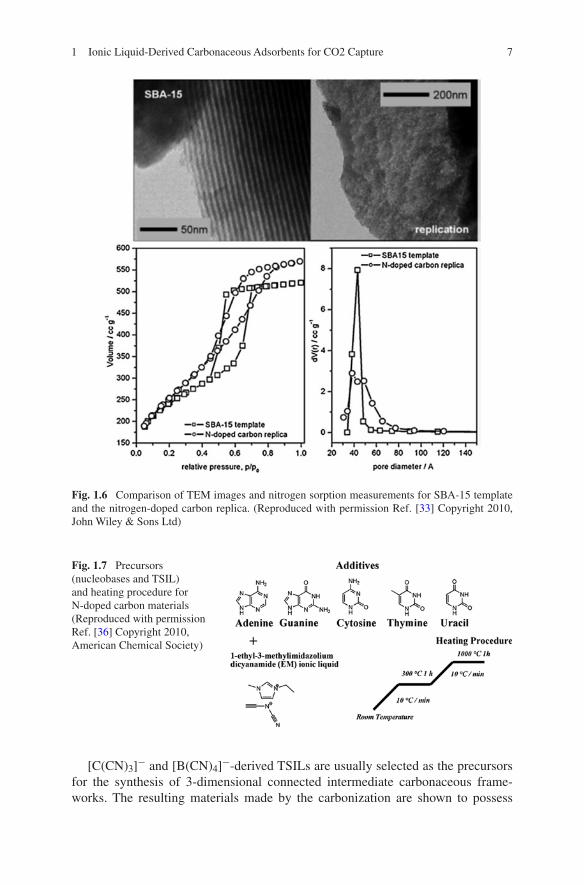
For dicyanamide anions [N(CN)2]−, the TSILs, composed of an organic, nitrogen-containing cation, for example 1-ethyl-3-methylimidazolium [EMIm] or 3-methyl-1-butylpyridine [3-MBP] are liquids at room temperatures. Therefore, these precursors are allowed to be easily incorporated into porous templates and subsequently heat-treated for functional carbons synthesis, thus enabling perfect nanocasting and struc-tural control, though negligible specific surface area was observed for the materials made by direct carbonization. For example, high-surface-area N-doped mesoporous carbons may be produced using mesoporous silica (SBA-15) as a template (Fig. 1.6) [33]. In addition, to increase the nitrogen doping, task-specific precursors can be facilely made by dissolving nitrogen-rich nucleobases into TSILs like [EMIm] [N(CN)2]−. Thus, using hard templating with silica nanoparticles, mesoporous nitrogen-doped carbonaceous materials with high nitrogen contents (up to 12 wt%) and surface areas as high as 1,500 m2 g−1 can be conveniently prepared by the car-bonization of the designed precursors (Fig. 1.7) [36]. It should be mentioned that higher nitrogen doping of carbonaceous materials is very essential for their high CO2 uptake. Furthermore, nitrogen- and phosphorus-co-doped carbons with tun-able enhanced surface areas can be promoted by doping with phosphorus-containing additives, such as tetraalkylphosphonium bromide [37].
Fig. 1.5 a General structure of the various TSIL cations and anions used, the former having a bis-imidazolium motif linked by alkyl (I) and xylyl (II) groups; STEM images of carbons EBI-T b and c, and BBI-T d and e (Reproduced with permission Ref. [34] Copyright 2013, The Royal Society of Chemistry)
71 Ionic Liquid-Derived Carbonaceous Adsorbents for CO2 Capture
[C(CN)3]− and [B(CN)4]−-derived TSILs are usually selected as the precursors for the synthesis of 3-dimensional connected intermediate carbonaceous frame-works. The resulting materials made by the carbonization are shown to possess
Fig. 1.6 Comparison of TEM images and nitrogen sorption measurements for SBA-15 template and the nitrogen-doped carbon replica. (Reproduced with permission Ref. [33] Copyright 2010, John Wiley & Sons Ltd)
Fig. 1.7 Precursors (nucleobases and TSIL) and heating procedure for N-doped carbon materials (Reproduced with permission Ref. [36] Copyright 2010, American Chemical Society)

8 X. Zhu et al.
either small or negligible surface areas, which have been observed for [N(CN)2]− anion-derived carbons [38, 39]. However, interestingly, by mixing these two dif-ferent anion-based TSILs and changing the ratios, nonporous samples and carbons with some accessible micropores and broad distributions of slit-like mesopores were obtained with higher surface areas exceeding 500 m2 g−1 (Fig. 1.8) [39]. Large boron (B) and nitrogen (N) contents were found in these materials even after carbonizations at 800 °C. Consequently, such nitrogen-rich carbons exhibited high adsorption capacity for CO2 adsorption and selectivity for CO2/N2 separa-tion [39]. Compared with the [C(CN)3]− anion, [B(CN)4]−-based TSILs are more favorable for carbon-nitride synthesis because the N and B atoms are capable of adding favorable properties to carbonaceous networks when structurally incorpo-rated. “Salt templating,” a new technique for the preparation of functional carbons, was shown to allow the preparation of highly porous nitrogen- or nitrogen/boron-doped carbons derived from three eutectic mixtures and different [B(CN)4]−-based TSILs (Fig. 1.9) [40]. This offers the opportunity of tuning the morphologies of the materials from micro- to mesoporous with apparent specific surface areas up to 2,000 m2 g−1. Since a high specific surface area is very essential for CO2 capture, the obtained N/B co-doped porous solids may play well in CO2 uptake processes.
1.2.3 Nitrile-Containing Polymeric Ionic Liquids
Besides the monomer-based TSILs, nitrile-containing polymeric ionic liquids (PILs) have also been synthesized for functional carbons synthesis. Different from
Fig. 1.8 Proposed reaction scheme of the trimerization of a nitrile-containing anion, leading to the formation of a 3-dimensional extended framework (Reproduced with permission Ref. [39] Copyright 2011, The Royal Society of Chemistry)

91 Ionic Liquid-Derived Carbonaceous Adsorbents for CO2 Capture
TSILs, which will thermally polymerize into triazine-based polymers in the early stages of the low-temperature reaction, the synthesis approach starting from an already prepolymerized monomer may allow for typical polymer operations, such as molding, extrusion, coating, or casting under preservation of a given shape [41]. For example, porous nitrogen-doped carbon nanotubes loaded with Fe2O3 nano-particles and porous carbon films with tunable thickness can be made. The nitrile-containing PIL backbone, poly(3-cyanomethyl-1-vinylimidazolium) (PCMVIm), acts as both a carbon precursor and a nitrogen source, while the anion [NTf2]− plays a role like the template (Fig. 1.10) [42]. The good nitrogen doping and high carbon yields of the resulting carbonaceous materials may allow the preparation of good CO2 adsorbents with high specific surface area and good CO2 uptake by
Fig. 1.9 Description of product and surface area formation using the salt templating approach (Reproduced with permission Ref. [40] Copyright 2013, John Wiley & Sons Ltd)
Fig. 1.10 Synthetic routes and the chemical structure of ionic liquid monomer CMVImTf2N and PIL polymer PCMVImTf2N (Reproduced with permission Ref. [42] Copyright 2013, The Royal Society of Chemistry)

10 X. Zhu et al.
either physical activation or chemical activation of these solids. It is worth men-tioning that recently, a mesoporous PIL with low-specific surface area ranging typ-ically between 150 and 220 m2 g−1 has been synthesized using the silica template and screened for its potential in CO2 separation/utilization [43]. In addition, PILs-derived nitrogen-doped carbonaceous membranes can also be utilized in the mem-brane-based CO2/N2 separation process built on the enhanced interaction between CO2 molecules and nitrogen-containing sites within the membrane framework.
Fig. 1.11 SEM micrographs of DES-derived CO2 adsorbents. Insets show pictures of the monoliths obtained after thermal treatment at 800 °C (Reproduced with permission Ref. [44] Copyright 2011, The Royal Society of Chemistry)

111 Ionic Liquid-Derived Carbonaceous Adsorbents for CO2 Capture
1.3 Nanoporous Carbons Derived from Deep Eutectic Solvents
Recently, deep eutectic solvents (DESs), a new class of IL obtained by the complexion of quaternary ammonium salts with hydrogen bond donors such as acids, amines, and alcohols among others, have also been used as both precursors and structure-directing agents in the synthesis of nitrogen-doped carbonaceous materials. The use of DESs is attractive because, when compared with nitrile-containing TSILs, they are less expen-sive and easy to be prepared owing to a wide range of compounds such as regular car-bonaceous precursors (e.g., resorcinol). The application of resorcinol is by no means trivial because it provides high carbonization yields (up to ca. 85 wt%). This feature, besides the capability of recovering the second component of DES that is not involved in carbon formation (e.g., choline chloride), makes the synthetic processes based on DESs especially attractive in terms of efficiency and sustainability [44, 45]. These advantages may allow for efficient synthesis of CO2 adsorbents with good CO2 sepa-ration performance. Typically, the combination of good specific surface area and high nitrogen content provided an extraordinary CO2 capture capacity (up to 3.3 mmol g−1 at 25 °C and 1 bar) of the DES-derived adsorbents (Fig. 1.11) [44], which was made by the carbonization of the designed DESs composed of resorcinol, 3-hydroxypyri-dine, and choline chloride. Moreover, by changing the composition of DESs, the pore architectures of the synthesized carbon monoliths can also be facilely modified. The use of DESs-containing resorcinol, 4-hexylresorcinol, and tetraethylammonium bro-mide may allow for the synthesis of CO2 adsorbents with outstanding CO2/CH4 selec-tivity (especially at low pressures) [45]. Overall, features such as facile and low cost synthesis of carbonaceous materials from DESs open interesting perspectives for the application of the carbons in CO2 separation technologies for low-pressure post-com-bustion processes and natural gas upgrading.
1.4 Conclusions
The application of ILs as carbon precursors is an attractive field within the research area of carbon chemistry. Nitrile-containing task-specific ILs and DESs are a promising class of monomers for the synthesis of carbonaceous materials. The architecture of TSILs allows for a wide combination of nitrile-modified cati-ons and anions, tailoring the properties of the resulting porous solids. The attrac-tive properties of IL-derived nitrogen-doped carbons make them a promising material for potential application in CO2 separation from flue gas.
Acknowledgments The research was supported financially by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, US Department of Energy.

12 X. Zhu et al.
References
1. D’Alessandro DM, Smit B, Long JR (2010) Carbon dioxide capture: prospects for new mate-rials. Angew Chem Int Ed 49:6058–6082
2. Lu W, Sculley JP, Yuan D, Krishna R, Wei Z, Zhou H-C (2012) Polyamine-tethered porous pol-ymer networks for carbon dioxide capture from flue gas. Angew Chem Int Ed 51:7480–7484
3. Lu W, Yuan D, Sculley J, Zhao D, Krishna R, Zhou H-C (2011) Sulfonate-grafted porous polymer networks for preferential CO2 adsorption at low pressure. J Am Chem Soc 133:18126–18129
4. Sumida K, Rogow DL, Mason JA, McDonald TM, Bloch ED, Herm ZR, Bae T-H, Long JR (2011) Carbon dioxide capture in metal-organic frameworks. Chem Rev 112:724–781
5. Li J-R, Sculley J, Zhou H-C (2011) Metal-organic frameworks for separations. Chem Rev 112:869–932
6. Phan A, Doonan CJ, Uribe-Romo FJ, Knobler CB, O’Keeffe M, Yaghi OM (2009) Synthesis, structure, and carbon dioxide capture properties of zeolitic imidazolate frameworks. Acc Chem Res 43:58–67
7. Dawson R, Cooper AI, Adams DJ (2013) Chemical functionalization strategies for carbon dioxide capture in microporous organic polymers. Polym Int 62:345–352
8. Furukawa H, Yaghi OM (2009) Storage of hydrogen, methane, and carbon dioxide in highly porous covalent organic frameworks for clean energy applications. J Am Chem Soc 131:8875–8883
9. Ben T, Li Y, Zhu L, Zhang D, Cao D, Xiang Z, Yao X, Qiu S (2012) Selective adsorption of car-bon dioxide by carbonized porous aromatic framework (PAF). Energy Environ Sci 5:8370–8376
10. Chen Q, Luo M, Hammershøj P, Zhou D, Han Y, Laursen BW, Yan C-G, Han B-H (2012) Microporous polycarbazole with high specific surface area for gas storage and separation. J Am Chem Soc 134:6084–6087
11. Du N, Park HB, Robertson GP, Dal-Cin MM, Visser T, Scoles L, Guiver MD (2011) Polymer nanosieve membranes for CO2-capture applications. Nat Mater 10:372–375
12. Dawson R, Stevens LA, Drage TC, Snape CE, Smith MW, Adams DJ, Cooper AI (2012) Impact of water coadsorption for carbon dioxide capture in microporous polymer sorbents. J Am Chem Soc 134:10741–10744
13. Dawson R, Stockel E, Holst JR, Adams DJ, Cooper AI (2011) Microporous organic polymers for carbon dioxide capture. Energy Environ Sci 4:4239–4245
14. Zhu X, Tian C, Mahurin SM, Chai S-H, Wang C, Brown S, Veith GM, Luo H, Liu H, Dai S (2012) A superacid-catalyzed synthesis of porous membranes based on triazine frameworks for CO2 separation. J Am Chem Soc 134:10478–10484
15. Zhu X, Do-Thanh C-L, Murdock CR, Nelson KM, Tian C, Brown S, Mahurin SM, Jenkins DM, Hu J, Zhao B, Liu H, Dai S (2013) Efficient CO2 capture by a 3D porous polymer derived from Tröger’s base. ACS Macro Letters 2:660–663
16. Lu W, Verdegaal WM, Yu J, Balbuena PB, Jeong H-K, Zhou H-C (2013) Building multiple adsorption sites in porous polymer networks for carbon capture applications. Energy Environ Sci 6:3559–3564
17. Hicks JC, Drese JH, Fauth DJ, Gray ML, Qi G, Jones CW (2008) Designing adsorbents for CO2 capture from flue gas-hyperbranched aminosilicas capable of capturing CO2 reversibly. J Am Chem Soc 130:2902–2903
18. Ma X, Wang X, Song C (2009) “Molecular basket” sorbents for separation of CO2 and H2S from various gas streams. J Am Chem Soc 131:5777–5783
19. Goeppert A, Czaun M, May RB, Prakash GKS, Olah GA, Narayanan SR (2011) Carbon dioxide capture from the air using a polyamine based regenerable solid adsorbent. J Am Chem Soc 133:20164–20167
20. Kuwahara Y, Kang D-Y, Copeland JR, Brunelli NA, Didas SA, Bollini P, Sievers C, Kamegawa T, Yamashita H, Jones CW (2012) Dramatic enhancement of CO2 uptake by poly(ethyleneimine) using zirconosilicate supports. J Am Chem Soc 134:10757–10760

131 Ionic Liquid-Derived Carbonaceous Adsorbents for CO2 Capture
21. Hao G-P, Li W-C, Qian D, Lu A-H (2010) Rapid synthesis of nitrogen-doped porous carbon monolith for CO2 capture. Adv Mater 22:853–857
22. Hao G-P, Li W-C, Qian D, Wang G-H, Zhang W-P, Zhang T, Wang A-Q, Schüth F, Bongard H-J, Lu A-H (2011) Structurally designed synthesis of mechanically stable poly(benzoxazine-co-resol)-based porous carbon monoliths and their application as high-performance CO2 cap-ture sorbents. J Am Chem Soc 133:11378–11388
23. Sevilla M, Valle-Vigón P, Fuertes AB (2011) N-doped polypyrrole-based porous carbons for CO2 capture. Adv Funct Mater 21:2781–2787
24. Xing W, Liu C, Zhou Z, Zhang L, Zhou J, Zhuo S, Yan Z, Gao H, Wang G, Qiao SZ (2012) Superior CO2 uptake of N-doped activated carbon through hydrogen-bonding interaction. Energy Environ Sci 5:7323–7327
25. Zhu X, Hillesheim PC, Mahurin SM, Wang C, Tian C, Brown S, Luo H, Veith GM, Han KS, Hagaman EW, Liu H, Dai S (2012) Efficient CO2 capture by porous, nitrogen-doped carbo-naceous adsorbents derived from task-specific ionic liquids. ChemSusChem 5:1912–1917
26. Zhu X, Chai S, Tian C, Fulvio PF, Han KS, Hagaman EW, Veith GM, Mahurin SM, Brown S, Liu H, Dai S (2013) Synthesis of porous, nitrogen-doped adsorption/diffusion carbonaceous membranes for efficient CO2 separation. Macromol Rapid Commun 34:452–459
27. Zhu X, Tian C, Chai S, Nelson K, Han KS, Hagaman EW, Veith GM, Mahurin SM, Liu H, Dai S (2013) New tricks for old molecules: development and application of porous N-doped, carbonaceous membranes for CO2 separation. Adv Mater 25:4152–4158
28. Ma Z, Yu J, Dai S (2010) Preparation of inorganic materials using ionic liquids. Adv Mater 22:261–285
29. Paraknowitsch JP, Thomas A (2012) Functional carbon materials from ionic liquid precur-sors. Macromol Chem Phys 213:1132–1145
30. Zhai Y, Dou Y, Zhao D, Fulvio PF, Mayes RT, Dai S (2011) Carbon materials for chemical capacitive energy storage. Adv Mater 23:4828–4850
31. Paraknowitsch JP, Thomas A, Antonietti M (2010) A detailed view on the polycondensation of ionic liquid monomers towards nitrogen doped carbon materials. J Mater Chem 20:6746–6758
32. Lee JS, Wang X, Luo H, Baker GA, Dai S (2009) Facile ionothermal synthesis of microporous and mesoporous carbons from task specific ionic liquids. J Am Chem Soc 131:4596–4597
33. Paraknowitsch JP, Zhang J, Su D, Thomas A, Antonietti M (2010) Ionic liquids as precursors for nitrogen-doped graphitic carbon. Adv Mater 22:87–92
34. Fulvio PF, Hillesheim PC, Oyola Y, Mahurin SM, Veith GM, Dai S (2013) A new family of fluidic precursors for the self-templated synthesis of hierarchical nanoporous carbons. Chem Commun 49:7289–7291
35. Paraknowitsch JP, Wienert B, Zhang Y, Thomas A (2012) Intrinsically sulfur- and nitrogen-co-doped carbons from thiazolium salts. Chem Eur J 18:15416–15423
36. Yang W, Fellinger T-P, Antonietti M (2010) Efficient metal-free oxygen reduction in alkaline medium on high-surface-area mesoporous nitrogen-doped carbons made from ionic liquids and nucleobases. J Am Chem Soc 133:206–209
37. Paraknowitsch JP, Zhang Y, Wienert B, Thomas A (2013) Nitrogen- and phosphorus-co-doped carbons with tunable enhanced surface areas promoted by the doping additives. Chem Commun 49:1208–1210
38. Lee JS, Wang X, Luo H, Dai S (2010) Fluidic carbon precursors for formation of functional carbon under ambient pressure based on ionic liquids. Adv Mater 22:1004–1007
39. Fulvio PF, Lee JS, Mayes RT, Wang X, Mahurin SM, Dai S (2011) Boron and nitrogen-rich carbons from ionic liquid precursors with tailorable surface properties. Phys Chem Chem Phys 13:13486–13491
40. Fechler N, Fellinger T-P, Antonietti M (2013) “Salt templating”: a simple and sustainable pathway toward highly porous functional carbons from ionic liquids. Adv Mater 25:75–79
41. Yuan J, Giordano C, Antonietti M (2010) Ionic liquid monomers and polymers as precursors of highly conductive, mesoporous, graphitic carbon nanostructures. Chem Mater 22:5003–5012

14 X. Zhu et al.
42. Zhao Q, Fellinger T-P, Antonietti M, Yuan J (2013) A novel polymeric precursor for micro/mesoporous nitrogen-doped carbons. J Mater Chem A 1:5113–5120
43. Wilke A, Yuan J, Antonietti M, Weber J (2012) Enhanced carbon dioxide adsorption by a mesoporous poly(ionic liquid). ACS Macro Lett 1:1028–1031
44. Gutierrez MC, Carriazo D, Ania CO, Parra JB, Ferrer ML, del Monte F (2011) Deep eutectic solvents as both precursors and structure directing agents in the synthesis of nitrogen doped hierarchical carbons highly suitable for CO2 capture. Energy Environ Sci 4:3535–3544
45. Patino J, Gutierrez MC, Carriazo D, Ania CO, Parra JB, Ferrer ML, Monte Fd (2012) Deep eutectic assisted synthesis of carbon adsorbents highly suitable for low-pressure separation of CO2–CH4 gas mixtures. Energy Environ Sci 5:8699–8707

15
Abstract Porous carbons play an important role in CO2 adsorption and separation due to their developed porosity, excellent stability, wide availability, and tunable surface chemistry. In this chapter, the synthesis strategies of porous carbon materi-als and evaluation of their performance in CO2 capture are reviewed. For clarity, porous carbons are mainly classified into the following categories: conventional activated carbons (ACs), renewable-resources-derived porous carbons, synthetic polymer-based porous carbons, graphitic porous carbons, etc. In each category, macroscopic and microscopic morphologies, synthesis principles, pore structures, composition and surface chemistry features as well as their CO2 capture behavior are included. Among them, porous carbons with targeted functionalization and a vast range of nanostructured carbons (carbon nanofibers, CNTs, graphene, etc.) for CO2 capture are being created at an increasing rate and are highlighted. After that, the main influence factors determining CO2 capture performance including the pore features and heteroatom decoration are particularly discussed. In the end, we briefly summarize and discuss the future prospectives of porous carbons for CO2 capture.
2.1 Introduction
The term “Carbon Filter Process (CFP)” has been proposed and accepted widely. Selected carbonaceous materials, e.g., activated carbons (ACs), carbon aerogels, and carbon fibers, act as filter materials, which deliver a high affinity (and, hence, high capacity) to CO2 but not to its balance gas. This, in turn, leads to a high selectivity of CO2/balance gas (in most case, N2). Most importantly, along with the improvement in science and technology, it has been possible to synthesize carbon materials with
Chapter 2Porous Carbons for Carbon Dioxide Capture
An-Hui Lu, Guang-Ping Hao and Xiang-Qian Zhang
A.-H. Lu and S. Dai (eds.), Porous Materials for Carbon Dioxide Capture, Green Chemistry and Sustainable Technology, DOI: 10.1007/978-3-642-54646-4_2, © Springer-Verlag Berlin Heidelberg 2014
A.-H. Lu (*) · G.-P. Hao · X.-Q. Zhang State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, People’s Republic of Chinae-mail: [email protected]

16 A.-H. Lu et al.
defined nanostructure and morphology, tunable surface area, and pore size. Because of the advanced porous carbon materials, current CFP can recover more than 90 % of flue gas CO2 (with purity of higher than 90 %) at a fraction of the cost normally associated with the conventional amine absorption process.
2.2 Conventional Activated Carbons (ACs) for CO2 Capture
ACs are the most commonly used form of porous carbons for a long time. Typically, they refer to coal and petroleum pitch as well as coconut shells-based AC. In most cases, ACs are processed to be filled with rich micropores that increase the surface area available for gas sorption and separation. For this category, to get a definite clas-sification on the basis of pore structure is difficult due to their countless products as well as their complex pore features. Based on the physical characteristics, they can be widely classified into the following types: powdered, granular, extruded, bead ACs, etc. For the pore structure of ACs, actually, all the three types of pores (micropore, mesopore, and macropore) are included in one product (Fig. 2.1), with a wide pore size distribution [1, 2]. Up to now, many kinds of ACs have been well commercial-ized in gas sorption/separation including CO2 capture. For example, the BPL type with specific area of 1,141 m2 g−1 is able to adsorb 7 mmol g−1 CO2 under the con-ditions of 25 °C and 35 bar, while under the same conditions MAXSORB-activated carbon with specific area of 3,250 m2 g−1 can capture up to 25 mmol g−1 [3].
2.3 Renewable-Resources-Derived Porous Carbons for CO2 Capture
New types of porous carbons for CO2 capture have been created through carboniza-tion and activation of renewable biomass precursors, such as coconut husk, bamboo, wood, peat, cellulose, and lignite due to their wide availability and renewable features (Fig. 2.2). Interestingly, for this category, as new precursors are discovered, new types of ACs can be created through carbonization and activation. For example, the precur-sors can be extended to microorganism, celtuce leaves, fungi, algae, bean dreg, and so on [4–6]. And thus such carbon “family” is enriched and will be further expanded due to the widely available carbon precursors and their high effectiveness in CO2 capture.
2.3.1 Direct Pyrolysis Method
Direct pyrolysis combined with activation of renewable biomass precursors has been widely studied and employed for fabricating the porous carbon materials. For example, waste celtuce leaves were used to prepare porous carbons by air-drying,

172 Porous Carbons for Carbon Dioxide Capture
pyrolysis at 600 °C in argon, followed by KOH activation. The as-prepared porous carbons show a very high specific surface area of 3,404 m2 g−1 and a large pore volume of 1.88 cm3 g−1. They show an excellent CO2 adsorption capacity at 1 bar, which is up to 6.04 and 4.36 mmol g−1 at 0 and 25 °C, respectively. Wang et al. [7] reported a series of porous carbons with adjustable surface areas and narrow micropore size distribution by KOH activation of fungi-based carbon sources. The high CO2 uptake of 5.5 mmol g−1 and CO2/N2 selectivity of 27.3 at 1 bar, 0 °C of such fungi-based carbons made them promising for CO2 capture and separation. Similarly, Shen et al. [8] found that yeast is a promising carbon precursor for the synthesis of hierarchical microporous carbons, which show a high CO2 adsorption capacity (4.77 mmol g−1) and fast adsorption rate (equilibrium within 10 min) at 25 °C. This may stem from their large surface area and hierarchical pore systems as well as the surface-rich basic sites.
Fig. 2.1 Schematic representation three-dimensional (a) and two-dimensional (b) structures of the ACs. Reprinted from Ref. [1], Copyright 1998, with permission from Elsevier
Fig. 2.2 Biomass feed stocks as carbon precursors: a coconut shell; b bamboo; c yeast; d fungi; e celtuce leaves; and f algae

18 A.-H. Lu et al.
Different from microstructure tuning, their macrostructures (form, density, etc.) modification is also crucial for reducing pressure drop, mitigating adsorption heat as well as enhancing volumetric capture capacity. Linares-Solano et al. [9] sys-tematically investigated this issue by using carefully selected carbon monoliths (A series, M3M and K1M). The properties of the three types are characterized and compared in their work. As seen, A series monoliths show high-density values but moderate porosities, while M3M and K1M represent the other type with moderate densities and high porosity developments (Table 2.1).
From the systematical CO2 adsorption investigation, the authors found that (1) the gravimetric storage capacities of the adsorbents depend on their textural prop-erties, while the volumetric adsorption capacity is directly related to their textural properties and densities. It is worth to note that the density shows the most impor-tant impact on gas storage capacity. (2) due to their singular high density, the A series carbon monoliths, as well as its CO2 activated carbon monoliths, present exceptionally high volumetric storage capacity for CO2 at room temperature.
In parallel with the structure tuning, the carbon surface properties can also be modified by selecting the N-containing precursors [6, 10]. For example, Xing et al. [6] prepared a series of N-doped ACs from bean dreg by KOH activation and investigated their CO2 capture properties. The resulting materials possess a very high CO2 uptake of up to 4.24 mmol g−1 at 25 °C under 1 atm. They demonstrated that the CO2 uptake is independent of the specific surface area and micropore vol-ume, but closely related to the N content of the ACs. In their opinion, the intro-duction of N into a carbon surface facilitates the hydrogen-bonding interactions (Fig. 2.3) between the carbon surface and CO2 molecules rather than the acid–base interactions between N-containing basic functional groups and acidic CO2 mole-cules, which accounts for the superior CO2 uptake of the N-doped porous carbons.
Similarly, Sevilla et al. [11] reported a chemical activated synthesis (KOH as activating agent) of highly porous N-doped carbons for CO2 capture. In their syn-thesis method, polypyrrole (PPy) was selected as carbon precursor. The activation process was carried out under severe (KOH/PPy = 4) or mild (KOH/PPy = 2)
Table 2.1 Porosity characterization of the carbon monoliths selected in the present study, deduced from N2 (–196 °C) and CO2 (0 °C) adsorption isotherms and densities of the carbon monoliths
Reproduced from Ref. [9] by permission of The Royal Society of Chemistry
Monolith SBET (m2 g−1) VDR (N2) (cm3 g−1) VDR (CO2) (cm3 g−1) Density (g cm−3)
A1 928 0.43 0.44 1.00A3 941 0.43 0.45 1.07A3–12 988 0.56 0.50 0.99A3–24 1,145 0.66 0.57 0.93A3–36 1,367 0.71 0.50 0.87A3–48 1,586 0.77 0.50 0.80M3 3,180 1.31 0.70 –K1 3,120 1.25 0.72 –M3M 2,610 0.93 0.60 0.42K1M 2,320 0.91 0.59 0.50
192 Porous Carbons for Carbon Dioxide Capture
activation conditions at different temperatures in the 600–800 °C range. Mildly activated carbons have two important characteristics: (1) they contain a large num-ber of nitrogen functional groups (up to 10.1 wt% N) identified as pyridonic-N with a small proportion of pyridinic-N groups, and (2) they exhibit, in relation to the carbons prepared with KOH/PPy = 4, narrower micropore size. The above two properties ensure the mildly activated carbons a large CO2 adsorption capaci-ties. Furthermore, the capture of CO2 over this type of carbon takes place at high adsorption rates, more than 95 % of the CO2 being adsorbed in ca. 2 min. In con-trast, N2 adsorption occurs at slower rates; approximately 50 min are necessary to attain maximum adsorption uptake (0.77 mmol N2 g−1).
2.3.2 Sol-gel Process and Hydrothermal Carbonization Method
Another new but rapidly expanding research area is the production of porous carbons from renewable resources (e.g., collagen, cellulose, and starch) based on a sol-gel process (Fig. 2.4) [12, 13]. One of the successful examples is polysaccharide-derived “Starbons®” carbon, which exhibits outstanding mesoporous textural properties. More importantly, their pore volumes and sizes are comparable to materials prepared via the hard template routes or soft template methods based on the self-assembly and polymerization of aromatic precursors (e.g., phenols). In this technology, three main stages are involved. Selected precursors are first gelatinized by heating in water. Then, the water inside of the gel is exchanged with the lower surface tension solvent (e.g., ethanol). After drying, the porous gel is doped with a catalytic amount of acid and pyrolyzed under vacuum, ending up in highly porous carbons.
Sol-gel method is indeed a simple and direct approach for the synthesis of bulky carbons and is already widely used in both laboratory and industry. However, the major disadvantage is the long synthesis period and the rigorous drying process of
Fig. 2.3 Theoretical models for a N-doped carbon surface and b pure carbon surface (red ball oxygen atom; blue ball nitrogen atom; gray ball carbon atom; small gray ball hydrogen atom). c Hydrogen bond energies at different adsorption sites. Reproduced from Ref. [6] by permission of The Royal Society of Chemistry

20 A.-H. Lu et al.
the wet gel (i.e., solvent exchange or supercritical drying), in which slight variations may cause drastic variations in the structural features, and hence properties [14]. In addition, pore blocking and sometimes uncontrolled dispersion of active sites both on the surface and in carbon pore walls remain to be solved.
Concurrently with the “Starbons ®” technology, Titirici et al. [15, 16] have been particularly active in the development and production of useful carbonaceous mate-rials from sugar-based biomass via a hydrothermal carbonization (HTC) approach. HTC is a spontaneous, exothermic process, producing materials where the major-ity of the original carbons are incorporated into the final structure. The initial prod-ucts of the sugar dehydration (e.g., furfuryl derivatives) are thought to polymerize to form condensed spherical functional carbons after autoclave processing at 180 °C for 20–24 h (Fig. 2.5) [17]. Manipulation of particle size was possible via the utilization of different sugar-based carbon sources, while the surface and bulk chemical struc-ture of the material may be directed by the utilization of hexose- or pentose-based biomass, as demonstrated by 13CP MAS NMR investigations [16]. HTC is relatively straightforward, affording small colloidal carbon spheres (CS), the surface texture and chemistry of which can be controllable via the introduction of co-monomers, and selection of biomass precursor. However, HTC materials demonstrate low or negligi-ble surface areas, very small particle size, and little developed or structured porosity.
Fig. 2.4 Examples of organized hierarchical structures found in biological systems. Reproduced from Ref. [12] by permission of The Royal Society of Chemistry

212 Porous Carbons for Carbon Dioxide Capture
The CO2 capture behavior over HTC-based porous carbons has recently been investigated. For example, Sevilla and Fuertes [18] reported a series of sustainable porous carbon capture materials, which are produced from the chemical activation of hydrothermally treated precursors (polysaccharides and biomass) using KOH as an activating agent. The CO2 adsorption properties, kinetics, and regeneration of these materials were investigated. Compared with the raw HTC materials, the chemical activated counterparts show a significant increase of micropores, deliv-ering a high surface area of 1,260 and 2,850 m2 g−1 depending on the activation conditions. The CO2 capture properties at 0, 25, and 50 °C and 1 bar are studied. As shown in Fig. 2.6, these HTC-based porous carbons show a high capacity even up to 4.8 mmol g−1 at 25 °C and 1 atm. They found that the remarkable CO2 cap-ture capacity arises from the presence of rich and narrow micropores (<1 nm), and the surface area plays a less important role. More interestingly, they found that this type of porous carbons showed very fast adsorption kinetics. Around 95 % CO2 uptake can be achieved in 2 min. Under the same conditions, the N2 adsorption uptake is 1/9 of that of CO2, indicating a CO2/N2 selectivity of ca. 9.
Subsequently, Sevilla et al. [19] prepared the highly porous N-doped carbons through chemical activation of hydrothermal carbons derived from mixtures of algae and glucose. They demonstrate that the control of the activation conditions (temperature and amount of KOH) allows the synthesis of exclusively micropo-rous biomass-based materials. These materials possess surface areas in the range of 1,300–2,400 m2 g−1 and pore volumes up to 1.2 cm3 g−1. They additionally exhibit
Fig. 2.5 Conversion of cellulose into HTC: A via HMF resulting in a furan-rich aromatic network and B direct aromatization. Reproduced from Ref. [17] by permission of The Royal Society of Chemistry

22 A.-H. Lu et al.
the N contents in the range of 1.1–4.7 wt%, and these heteroatoms being mainly present as pyridone-type structures. When tested as CO2 sorbents at subatmos-pheric conditions, they show a large CO2 capture capacity of up to 7.4 mmol g−1 at 0 °C and 1 bar, which is among the highest values for porous materials. However, the results indicate that the large CO2 capture capacity is exclusively due to their high volume of narrow micropores and not to the high surface areas or pore vol-umes, neither to the presence of heteroatoms.
2.4 Synthetic Polymer-Based Porous Carbons for CO2 Capture
Compared with conventional ACs and biomass-derived carbons, the use of synthetic polymers as porous carbons precursors enables better chemical composition con-trol, easy-to-achieve precise morphology, tunable pore system, and targeted surface chemistry. Thus, synthetic polymers-based porous carbons are extensively investi-gated nowadays. The strategies toward advanced porous carbons mainly rely on pro-tocols such as precursor-controlled pyrolysis, rational synthesis by chemical vapor deposition (CVD), templating and surface-mediated synthesis, self-assembly, surface-grafting and modification, and others. The CO2 capture performances are strongly depending on these microstructure features. This can be achieved by a designed synthesis methodology. Thus, a precise controlled synthesis on carbon structure will provide a promising opportunity to authentically understand the physical and chemi-cal properties of carbon materials from molecular level and thereby efficiently guide practical applications. For clarity, in this part, the porous carbon materials are classi-fied into the following groups according to synthesis methods: template-free synthesis, self-assembly strategy, and hard template method. In each group, several subgroups
Fig. 2.6 a CO2 adsorption isotherms at 0, 25, and 50 °C and b adsorption kinetics of CO2 and N2 at 25 °C for the HTC-based porous carbons AS-2-600 sample. Reproduced from Ref. [18] by permission of The Royal Society of Chemistry

232 Porous Carbons for Carbon Dioxide Capture
are further included according to their morphologies: 3D monolithic structure, 2D films, membranes, and spheres. To note, ionic liquids or related derivates derived porous carbons have been discussed in Chap. 1 and thus are not within the scope here.
2.4.1 Template-Free Synthesis
2.4.1.1 Monoliths
Porous carbons are versatile materials that possess a wide range of morpholo-gies not only on the microscopic level but also on the macroscopic level. Macroscopically, a monolith generally shows wide flexibility of operation in contrast to its powder counterparts [20]. Microscopically, monolithic structure is characterized by its 3D bicontinuous hierarchical porosity, which usually leads to several distinct advantages such as low pressure drop, fast heat and mass trans-fer, high contacting efficiency, and easy to deal with [21–24]. Thus, the monolithic carbons well apply to gas sorption and separation, including CO2 capture.
The synthesis of monolithic carbons generally relies on the means including sol-gel method and self-assembly approach [25, 26]. In recent years, much efforts have been devoted to create new types of carbon monoliths with enhanced func-tions, which are developing new polymerization systems (solvents and/or precur-sors), precise pore engineering toward multimodal porosities, and targeted surface/bulk functionalization for a high performance in CO2 capture [27–30].
The sol-gel method is one of the most conventional methods to prepare bulk carbon materials with fully interconnected pores. Carbon aerogels are the repre-sentative monolithic materials, whose synthesis generally involves the transforma-tion of molecular precursors into highly cross-linked organic gels based on sol-gel chemistry [31]. Since the pioneering work of Pekala [32], the polymer-based mon-olithic carbons have scored remarkable achievements in the new polymerization system and further surface/bulk functionalization. Fairén-Jiménez et al. [33] syn-thesized carbon aerogels with monolith density ranging from 0.37 to 0.87 g cm−3 by carbonization of organic aerogels deriving from resorcinol–formaldehyde (RF) polymer prepared in various solvents such as water, methanol, ethanol, tetrahydro-furan, or acetone solution. They found that the samples with a density higher than 0.61 g cm−3 had micropores and mesopores but no macropores.
Using deep eutectic salts either as solvents, or as carbonaceous precursors and structure-directing agents, Monte’s group prepared carbon monoliths with high yield (80 %) and tailored mesopore diameters [34, 35]. Sotiriou-Leventis, and coworkers, in recent years, have developed several new polymerization systems such as isocyanate-cross-linked RF gels, polyurea (PUA) gels, and polyimide gels, which offer a high degree of flexibility in producing the monolithic carbons [36–38]. The carbon products show interconnected hierarchical pore networks and 3D bicontinuous morphology, high surface area, and large pore volume. For example, PUA gels, which eventually convert to highly porous (up to 98.6 % v/v) aerogels

24 A.-H. Lu et al.
over a very wide density range, can be prepared by carefully controlling of the relative Desmodur RE (isocyanate)/water/triethylamine (catalyst) ratios in acetone (Fig. 2.7). It is worthy of exploration of their applications as CO2 capture materi-als in the forthgoing research.
Alternatively, the copolymerization and/or cooperative assembly between car-bon precursors, and one or more additional modifiers (i.e., heteroatom-containing components), can be used to directly synthesize functional carbons with enhanced CO2 adsorption capability [39]. Sepehri et al. [40] synthesized a series of nitro-gen–boron codoped carbon cryogels by homogenous dispersion of ammonia borane in RF hydrogel during solvent exchange and followed by freeze-drying and pyrolysis. The nitrogen–boron codoping results in a big improvement in porous structure and thus accelerates molecule/ions transport properties as compared to the non-modified carbons. Recently, Lu’s group reported a time-saving synthesis toward to a new type of nitrogen-doped carbon monolith through a sol-gel copoly-merization of resorcinol, formaldehyde, and l-lysine [41]. Based on N2 sorption, TEM and SEM results (Fig. 2.8), it is clear that this carbon monolith possesses a hierarchical porous structure, i.e., contains both macropores and micropores. This should be advantageous for a CO2 sorption process, since the macropores provide low-resistant pathways for the diffusion of CO2 molecules, while the micropores are most suitable for trapping of CO2.
As expected, such a monolithic carbon performs very well in CO2 capture with the capacity of 3.13 mmol g−1 at 25 °C. With an increase in adsorption tempera-ture, the adsorption capacities decrease from 3.13 to 1.64, 1.22 to 0.62 mmol g−1, at the corresponding temperatures of 60, 80, and 120 °C, but are still at a high level
Fig. 2.7 SEM of carbon aerogels derived from polyurea aerogels made of Desmodur RE triiso-cyanate. Densities (inset) are those of the parent polyurea aerogels. Scale bar: 5 μm. Densities of the actual C samples (from left to right): top row, not measured (sample broke to pieces); 0.29 ± 0.06 g cm−3; 0.40 ± 0.02 g cm−3; lower row, 0.62 ± 0.08 g cm−3; 0.72 ± 0.03 g cm−3; 0.78 ± 0.01 g cm−3). Reprinted with the permission from Ref. [36]. Copyright 2010 American Chemical Society

252 Porous Carbons for Carbon Dioxide Capture
as compared to its non-doped analogous carbon monoliths (Fig. 2.9). Also to note, the decrease in the CO2 adsorption capacity at high temperature is the common effect of porous carbons. The high uptake at the initial stage of the isotherm may be attributed to the affinity between basic nitrogen groups and CO2 via acid–base interaction. Interestingly, they undergo an easy regeneration process, i.e., by argon purge at room temperature.
2.4.1.2 Spheres
Dimensionally, porous carbons can also be processed into size-shortened spheres. Commonly, carbon-based spheres are prepared by carbonization of polymer ana-logs. In this case, polymer precursors are required to be thermally stable and are able to form carbon residue after a high-temperature pyrolysis. Phenolic resins derived from the polymerization of phenols (e.g., phenol, resorcinol, phloroglucinol) with aldehyde (e.g., formaldehyde, furfuraldehyde, hexamethylenetetramine) are attrac-tive due to their excellent performance characteristics such as high-temperature
Fig. 2.8 a Photograph of as-made polymer monolith and its carbonized product. b N2-sorption isotherms of the obtained carbon monolithic pyrolyzed at different temperatures (P/P0 is the rela-tive pressure). c, d SEM and TEM images of sample carbonized at 500 °C (the inset in c) show an overview of the macroscopic structure. Reproduced from Ref. [41] by permission of John Wiley & Sons Ltd

26 A.-H. Lu et al.
resistance, thermal abrasiveness, and high yield of carbon conversion. As a result, varieties of chemical syntheses have been reported for the preparation and CO2 adsorption performance of CS [42–46].
Yuan et al. [47] reported a new type of spherical nitrogen-containing polymer and microporous carbon materials for CO2 adsorption analysis. In their synthesis, a nitrogen-containing compound, hexamethylenetetramine, was selected as both a nitrogen source and one of the carbon precursors under solvothermal conditions, without using any surfactant or toxic reagent such as formaldehyde. Thus, the syn-thesis strategy is user-friendly, cost-effective and can be easily scaled up for pro-duction. The microporous CS exhibit high surface areas of 528–936 m2 g−1 with a micropore size of 0.6–1.3 nm. The synthesized microporous carbons show a good CO2 capture capacity, which is mainly due to the presence of nitrogen-containing groups and a large amount of narrow micropores (<1.0 nm). At 1 atm, the equilib-rium CO2 capture capacities of the obtained microporous carbons are in the range of 3.9–5.6 mmol g−1 at 0 °C and 2.7–4.0 mmol g−1 at 25 °C.
Further, they normalized the CO2 capture capacities in accordance with narrow micropore volume and nitrogen content, with the aim to determine the influence of both textural and surface chemistry properties on their capture performance. The nor-malization of the CO2 capture capacities by the narrow micropore volume shows the effect of surface chemistry properties of the samples on the CO2 uptake, while the nor-malization by nitrogen content exhibits the contribution of textural properties. From the results, we can assume that samples prepared from low treatment temperature (i.e., 600 °C) exhibit the greatest capacity per narrow micropore volume, while high-tem-perature pyrolyzed samples show increased contribution by the micropores.
Fig. 2.9 a CO2 multicircle sorption isotherms and N2 sorption isotherm for sample at 25 °C, b temperature-dependent CO2 adsorption isotherms at 25, 60, 80, and 120 °C. Reproduced from Ref. [41] by permission of John Wiley & Sons Ltd

272 Porous Carbons for Carbon Dioxide Capture
Gu et al. [48] have developed a template-free synthesis for new types of porous CS, which show a good performance in CO2 capture. In their synthesis, the azide–alkyne 1,3-dipolar Huisgen cycloaddition reaction was employed for the condensa-tion of 1,4-bis(azidomethyl)benzene and 1,3,5-ethynylbenzene (Fig. 2.10). Because the resulting solid product contains periodically arranged aromatic 1,2,3-triazole rings in the polymer backbone, such carbon precursors contain a large percentage of nitrogen atom sources for the preparation of N-doped carbon materials. More importantly, it may be possible to control the N-doping level of products by sim-ply changing the degree of polymerization for the carbon precursors. As expected, the N contents and surface area can be tuned to 4.30 wt% and 423 m2 g−1 after a pyrolysis under 800 °C. Based on the developed pores and high N content, the sam-ple can adsorb 126.8 cm3 g−1 (5.66 mmol g−1), 69.6 cm3 g−1 (3.10 mmol g−1), and 53.9 cm3 g-1 (2.41 mmol g-1) of CO2 at 196, 273, and 298 K, respectively.
2.4.2 Self-assembly Method
2.4.2.1 Monolith
Imparting mesoscale pores is an effective way to enhance sorption kinetics, which can go beyond that of microporous ACs. Particularly, mesoporous car-bons with defined pore size and symmetry provide a possibility for the fabrica-tion of model carbons, which are highly valuable for fundamental investigation of the adsorption processes. Thus, besides the porous carbon monoliths show-ing irregular mesopores, great progress has also been achieved in the synthesis of monolithic carbons with ordered mesoporosity by self-assembly of copolymer molecular template and carbon precursors. However, it remains a great challenge to achieve highly developed mesoporosity while to remain good monolith mor-phology due to the following requirements. Firstly, a perfect matching interac-tion between the carbon-yielding precursors and the pore-forming component is required, which allows self-assembling of a stable micelle nanostructure; secondly,
Fig. 2.10 Synthetic scheme for the N-doped porous carbon materials. Several key nitrogen spe-cies are indicated in NPC materials. Reprinted from Ref. [48]. Copyright 2013, with permission from Elsevier

28 A.-H. Lu et al.
the micelle structures should be stable during sustaining the temperature required for curing a carbon-yielding component, but can be readily decomposed during carbonization; thirdly, the carbon-yielding component should be able to form a highly cross-linked polymeric materials that can retain their nanostructure during the decomposition or the extraction of the pore-forming component. In order to achieve a monolithic carbon with well-developed mesoporosity, not a single one of these conditions can be dispensed with.
Dai’s group first synthesized highly ordered mesoporous carbon through a sol-vent annealing accelerated self-assembly method using polystyrene-block-poly(4-vinylpyridine) (PS-P4VP) as soft templates and N,N-dimethylformamide (DMF) as solvent [49]. However, the samples are in film form. Since then, using self-assembly method to prepare porous carbons has been extensively investigated. At present, the products are mostly in a form of powder or film. For example, Valkama et al. [50] reported a soft template method to achieve carbon products in any desired shape, and the porosity can be tuned from mesoporous to hierarchi-cally micro- and mesoporous simply by varying pyrolysis conditions for the cured block copolymer–phenolic resin complexes.
Recently, based on the soft-templating principle, Dai’s group reported a versatile synthesis of porous carbons (monolith, film, fiber, and particle) by using phenol-, resorcinol-/phloroglucinol-based phenolic resins as carbon precursors and tri-block copolymer (F127) as the soft template. They found that due to the enhanced hydrogen-bonding interaction with triblock copolymers, phloroglucinol with three hydroxyl groups is an excellent precursor for the synthesis of mesoporous carbons with well-organized mesostructure [51]. This type of mesoporous carbon monolith shows good performance in gas capture [52]. At 800 Torr and 298 K, the adsorption equilibrium capacity of the ordered mesoporous carbon for CO2 is 1.49 mmol g−1. Significantly higher adsorption uptake was observed for CO2 to be 3.26 mmol g−1 at 100 bar and 298 K. More interestingly, the diffusion time constant of CO2 decreased with adsorbate pressures due to the obvious mesoscale pore system.
Later, they prepared carbons with ordered mesopores based on self-assembly approach of RF polymer and block copolymers under strong acidic conditions and by subsequent centrifugation and shaping techniques. The I+X−S+ mecha-nism and hydrogen bonding are believed to be the driving force for self-assem-bly between the RF resol and F127 template [53]. The polymerization-induced spinodal decomposition in glycolic solutions of phloroglucinol/formaldehyde polymers and block copolymers also leads to successful formation of the bimodal meso-/macroporous carbon monoliths [54].
Alternatively, Zhao’s group developed a hydrothermal synthesis by using F127 and P123 as double soft templates and phenol/formaldehyde as carbon precursor (molar ratio between phenol and surfactant about 46:1), followed by hydrothermal aging at 100 °C for 10 h [55]. Recently, the same group reported a controllable one-pot method to synthesize N-doped ordered mesoporous carbon with a high N content by using dicyandiamide as a nitrogen source via an evaporation-induced self-assembly process [56]. In this synthesis, resol molecules can bridge the Pluronic F127 template and dicyandiamide via hydrogen-bonding and electrostatic

292 Porous Carbons for Carbon Dioxide Capture
interactions. During thermosetting at 100 °C for formation of rigid phenolic resin and subsequent pyrolysis at 600 °C for carbonization, dicyandiamide pro-vides closed N species, while resol can form a stable framework, thus ensur-ing the successful synthesis of ordered N-doped mesoporous carbon (Fig. 2.11). Such N-doped ordered mesoporous carbons possess tunable mesostructures (p6m and Im3m symmetry) and pore size (3.1–17.6 nm), high surface area (494–586 m2 g−1), and high N content (up to 13.1 wt%). Ascribed to the unique feature of large surface area and high N contents, the materials show high CO2 capture of 2.8–3.2 mmol g−1 at 25 °C and 1.0 bar (Fig. 2.11).
Similarly, Xiao’s group also reported a hydrothermal synthesis at even higher temperature and longer time (i.e., 260 °C for more than 17 h) to prepare car-bon monoliths with well-ordered hexagonal or cubic mesopore systems [57]. Meanwhile, Gutiérrez et al. [58] synthesized a low-density monolithic carbon exhibiting a 3D continuous micro- and macroporous structure, which derived from a PPO15-PEO22-PPO15 block copolymer-assisted RF polymerization. Zhang’s group reported an organic–organic aqueous self-assembly approach to prepare B-/P-doped ordered mesoporous carbons using boric acid and/or phosphoric acid as B- or P-heteroatom source, RF resin as the carbon precursor and triblock copolymer Pluronic F127 as the mesoporous structure template [59]. Lu’s group established a rapid and scalable synthesis of crack-free and nitrogen-doped car-bon monolith with fully interconnected macropores and an ordered mesostructure through the soft template method. The monolith is achieved by using organic base lysine as a polymerization agent and mesostructure assembly promotor, through
Fig. 2.11 a The formation process of ordered N-doped mesoporous carbon from a one-pot assembly method using dicyandiamide (DCDA) as a nitrogen source. b CO2 and N2 adsorption isotherms at 25 °C for the N-doped mesoporous carbon H-NMC-2.5. c CO2 adsorption isotherms at different temperature for A-NMC after activation by KOH. Reproduced from Ref. [56] by permission of John Wiley & Sons Ltd

30 A.-H. Lu et al.
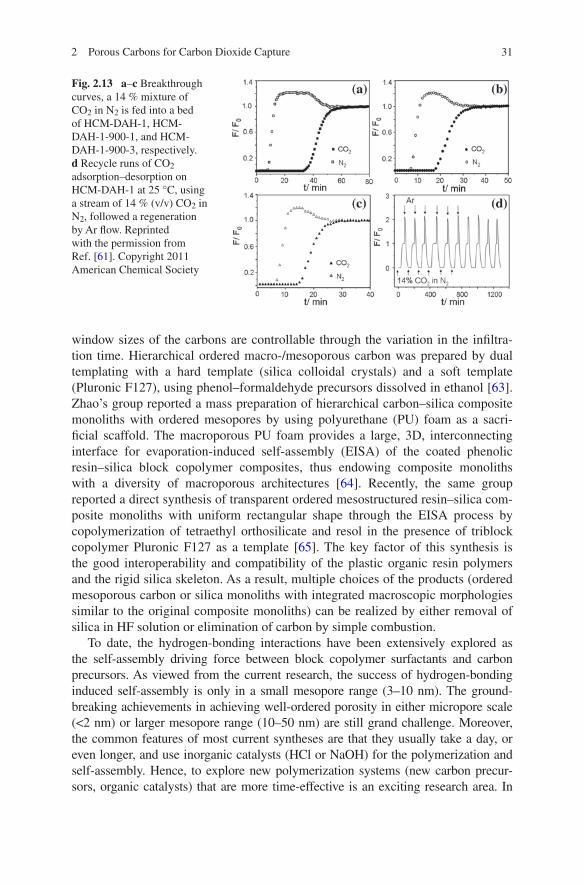
rapid sol-gel process at 90 °C [60]. Later, the same group reported a new type of porous carbon monolith, which was synthesized through a self-assembly approach based on benzoxazine chemistry [61]. The obtained carbon monoliths show crack-free macromorphology, well-defined multilength-scale pore structures, a nitrogen-containing framework, and high mechanical strength (Fig. 2.12).
With such designed structure, the carbon monoliths show outstanding CO2 cap-ture and separation capacities, high selectivity, and facile regeneration at room temperature. At ~1 bar, the equilibrium capacities of the monoliths are in the range of 3.3–4.9 mmol g−1 at 0 °C and of 2.6–3.3 mmol g−1 at 25 °C, while the dynamic capacities are in the range of 2.7–4.1 wt% at 25 °C using 14 % (v/v) CO2 in N2 (Fig. 2.13). The carbon monoliths exhibit high selectivity for the capture of CO2 over N2 from a CO2/N2 mixture, with a separation factor ranging from 13 to 28. Meanwhile, they undergo a facile CO2 release in argon stream at 25 °C, indi-cating a good regeneration capacity.
Due to the high precision in pore engineering by nanocasting pathway and the great variety of micelle nanostructures deriving from soft templating, many researchers try to combine both techniques into an interdependent and interactive module with the aim of achieving porous carbons with controlled pore structure in a cost-effective manner. Wang et al. [62] prepared 3D ordered macro-/mesoporous porous carbons by using colloidal crystals and surfactants as dual templates through a gas-phase process. In a vapor-phase infiltration, the wall thickness and
Fig. 2.12 Photograph of the synthesized polymer and carbon monolith (a); SEM images of carbon monolith HCM-DAH-1 (b); TEM images (c, d, and e: images viewed in the [100, 110, 111] direction; the insets are the corresponding fast Fourier transform (FFT) diffractograms) and HR-SEM images (f, g) of the carbon monolith HCM-DAH-1. Reprinted with the permission from Ref. [61]. Copyright 2011 American Chemical Society
312 Porous Carbons for Carbon Dioxide Capture
window sizes of the carbons are controllable through the variation in the infiltra-tion time. Hierarchical ordered macro-/mesoporous carbon was prepared by dual templating with a hard template (silica colloidal crystals) and a soft template (Pluronic F127), using phenol–formaldehyde precursors dissolved in ethanol [63]. Zhao’s group reported a mass preparation of hierarchical carbon–silica composite monoliths with ordered mesopores by using polyurethane (PU) foam as a sacri-ficial scaffold. The macroporous PU foam provides a large, 3D, interconnecting interface for evaporation-induced self-assembly (EISA) of the coated phenolic resin–silica block copolymer composites, thus endowing composite monoliths with a diversity of macroporous architectures [64]. Recently, the same group reported a direct synthesis of transparent ordered mesostructured resin–silica com-posite monoliths with uniform rectangular shape through the EISA process by copolymerization of tetraethyl orthosilicate and resol in the presence of triblock copolymer Pluronic F127 as a template [65]. The key factor of this synthesis is the good interoperability and compatibility of the plastic organic resin polymers and the rigid silica skeleton. As a result, multiple choices of the products (ordered mesoporous carbon or silica monoliths with integrated macroscopic morphologies similar to the original composite monoliths) can be realized by either removal of silica in HF solution or elimination of carbon by simple combustion.
To date, the hydrogen-bonding interactions have been extensively explored as the self-assembly driving force between block copolymer surfactants and carbon precursors. As viewed from the current research, the success of hydrogen-bonding induced self-assembly is only in a small mesopore range (3–10 nm). The ground-breaking achievements in achieving well-ordered porosity in either micropore scale (<2 nm) or larger mesopore range (10–50 nm) are still grand challenge. Moreover, the common features of most current syntheses are that they usually take a day, or even longer, and use inorganic catalysts (HCl or NaOH) for the polymerization and self-assembly. Hence, to explore new polymerization systems (new carbon precur-sors, organic catalysts) that are more time-effective is an exciting research area. In
Fig. 2.13 a–c Breakthrough curves, a 14 % mixture of CO2 in N2 is fed into a bed of HCM-DAH-1, HCM-DAH-1-900-1, and HCM-DAH-1-900-3, respectively. d Recycle runs of CO2 adsorption–desorption on HCM-DAH-1 at 25 °C, using a stream of 14 % (v/v) CO2 in N2, followed a regeneration by Ar flow. Reprinted with the permission from Ref. [61]. Copyright 2011 American Chemical Society

32 A.-H. Lu et al.
addition, hierarchical structured monolithic carbons with multimodal porosity would be more suitable for application in gas capture and separation. More desirably, the effluences of multilength-scale pores on the sorption kinetics and storage capability should be figured out by experimental and computational works.
2.4.2.2 Films
Besides the investigation of monolithic carbons with well-ordered mesoporosity as CO2 sorbents, the synthesis of ordered mesoporous carbon films for CO2 capture (membrane gas separation) also attracts much interest. Noticeably, highly ordered mesoporous carbon with cubic Im3m symmetry has been synthesized successfully via a direct carbonization of self-assembled F108 (EO132PO50EO132) and RF com-posites obtained in a basic medium of non-aqueous solution [66]. Dai et al. [49] demonstrated a stepwise self-assembly approach to the preparation of large-scale, highly ordered nanoporous carbon films (Fig. 2.14).
The synthesis of well-defined porous carbon films involves four steps: (1) mono-mer–block copolymer film casting, (2) structure refining through solvent annealing, (3) polymerization of the carbon precursor, and (4) carbonization. Zhao et al. [67] reported the fabrication of free-standing mesoporous carbon thin films with highly ordered pore architecture via a simple coating-etching method. The mesoporous carbon films were first synthesized by coating a resol precursors/Pluronic copoly-mer solution on a pre-oxidized silicon wafer, forming highly ordered polymeric mesostructures based on organic–organic self-assembly, followed by carbonizing at 600 °C and finally etching of the native oxide layer between the carbon film and the silicon substrate. Mild reacting conditions and wide composition ranges are the obvi-ous advantages of the method over the techniques previously reported [51, 68, 69].
Based on mesoporous RF-derived carbon films, Yoshimune et al. [70] inves-tigated the permeation properties of different gases, including H2, He, N2, CF4, and condensable gases of CO2 and CH4. As expected, these membranes exhib-ited relatively high permeances due to their well-developed mesoporous structure. By comparing permeances of the six gases on the RF carbon films at 303 K, the authors found that the gas permeances exhibited a sublinear dependence on the inverse square of the molecular weights, which are predominantly governed by the Knudsen mechanism. Interestingly, the permeances of condensable gases of CO2 and CH4 were accelerated. This is because the RF carbon films have both mesopores and a small number of micropores, and the enhanced permeance is attributed to the strong adsorption of condensable gases in the narrower micropo-res due to the surface diffusion mechanism. However, the effect of molecular siev-ing mechanism, which allows smaller gas molecules such as those of He to diffuse faster than larger gas molecules such as those of CF4, is negligible since the gas permeances of the RF carbon membranes are independent of the kinetic diame-ter of the permeating molecules. Consequently, the transport mechanism of these membranes is mainly governed by Knudsen diffusion, and the existence of a small number of micropores might have induced the surface diffusion.

332 Porous Carbons for Carbon Dioxide Capture
Very recently, Hao et al. [71] report a wet-chemistry synthesis of a new type of porous carbon nanosheets whose thickness can be precisely controlled over the nanometer length scale. This feature is distinct from conventional porous carbons that are composed of micron-sized or larger skeletons, and whose structure is less controlled. Smartly, the synthesis uses graphene oxides (GO) as the shape-direct-ing agent and asparagine as bridging molecules that connect the GO and in situ grown polymers by electrostatic interaction between the molecules. The assembly of the nanosheets can produce macroscopic structures, i.e., hierarchical porous carbon monoliths which have a mechanical strength up to 28.9 MPa, the highest reported for the analogs. The synthesis provides precise control of porous car-bons over both microscopic and macroscopic structures at the same time. In all syntheses, the graphene content used was in the range 0.5~2.6 wt%, which is sig-nificantly lower than that of common surfactants used in the synthesis of porous materials. This indicates the strong shape-directing function of GO. In addition, the overall thickness of the nanosheets can be tuned from 20 to 200 nm (Fig. 2.15) according to a fitted linear correlation between the carbon precursor/GO mass ratio and the coating thickness.
The porous carbon nanosheets show impressive CO2 adsorption capacity under equilibrium, good separation ability of CO2 from N2 under dynamic conditions, and easy regeneration. The highest CO2 adsorption capacities can reach 5.67 and
Fig. 2.14 Electron microscopy images of the carbon film. a Z-contrast image of the large-scale homogeneous carbon film in a 4 × 3 mm area. The scale bar is 1 mm. b Z-contrast image show-ing details of the highly ordered carbon structure. The scale bar is 300 nm. c HR-SEM image of the surface of the carbon film with uniform hexagonal pore array. The pore size is 33.7 ± 2.5 nm, and the wall thickness is 9.0 ± 1.1 nm. The scale bar is 100 nm. d SEM image of the film cross section, which exhibits all parallel straight channels perpendicular to the film surface. The scale bar is 100 nm. Reproduced from Ref. [49] by permission of John Wiley & Sons Ltd

34 A.-H. Lu et al.
3.54 CO2 molecules per nm3 pore volume and per nm2 surface area at 25 °C and ~1 bar (Fig. 2.16). The probable reason is that the interaction of PCNs samples with CO2 molecules is strong due to (a) the large amount of microporosity with pore size of ca. 8 Å and (b) the polar surface caused by the residual heteroatom-containing (e.g., O, N) species. These dynamic data provide clear evidence that PCN-17 is extremely selective for adsorbing CO2 over N2, which represents a sig-nificant step forward in rationally designing a material for dilute CO2 separation in humid conditions. These values are ideally consistent with the pure CO2 adsorp-tion data at partial pressures of 0.14, 0.09 and 0.04 bar, indicating its extraordinary moisture resistance. The cycling experiments using CO2/H2O/N2 of 4/3/93 v% also verified the selective and reversible CO2 adsorption capacity of PCN-17 (Fig. 2.16e). A sample saturated with CO2 was subjected to an Ar purge flow of 15 mL min−1 at 50 °C. After approximately 30 min, no CO2 was detected in the effluent. Successive regenerations reveal that the sample retains more than 97 % of its intrinsic capacity after such mild regeneration (Fig. 2.16f). The 3 % loss of CO2 capacity may be due to the strongly adsorbed CO2 on highly active sites. This can be explained by the high Qst at the ultra-low CO2 uptake. As the regeneration temperature increases to 100 °C, the residual CO2 (ca. 3 %) can also be recovered.
Fig. 2.15 FE-SEM images with low and high magnification of the obtained porous carbon nanosheets with different thickness: a, b PCN-17, c, d PCN-71, e, f PCN-82. The marked coating thickness on the top of b, d, f is the average thickness based on the measurements. Reproduced from Ref. [71] by permission of The Royal Society of Chemistry

352 Porous Carbons for Carbon Dioxide Capture
2.4.2.3 Spheres
The synthesis of porous carbon nanospheres for CO2 capture is another important topic due to their combined features such as shortened dimensions, increased sur-face area, easy surface or bulk functionalization. Generally, CS include solid spheres and hollow spheres, which can be prepared through either self-assembly method or templating way [72, 73]. For example, Liu et al. [74] developed a methodology to synthesize monodisperse RF resin polymer colloidal spheres and their carbonaceous analogs through a modified Stöber method. The key to the successful synthesis is using ammonia in the reaction system; its role, they consider, lies in not only accel-erating the polymerization of RF, but also supplying the positive charges that adhere to the outer surface of spheres to prevent particle aggregation. The particle size of the RF resin colloidal spheres obtained can be finely tuned by changing the ratio of alcohol/water, the amounts of ammonia and RF precursor, using alcohols with short alkyl chains, or introducing the triblock copolymer surfactant.
Following the same synthesis principle, Jaroniec et al. reported the prepara-tion of a series of CS by carbonization of phenolic resin spheres via the modified Stöber method. As shown in Fig. 2.17, all samples show spherical morphology with the average diameter of 570, 420, 370, and 200 nm for as-synthesized CS-6, CS-6-CD-4, CS-6-CD-8, CS-6-CD-12 carbon spheres, respectively. The particle
Fig. 2.16 CO2 adsorption evaluation of the PCNs. a, b CO2 adsorption isotherms for high and low CO2 partial pressures at 25 °C, where the solid line represents a Toth model fit to the CO2 isotherms. c The number of CO2 molecules adsorbed per nm3 pore volume and d per nm2 surface area for PCNs with different thickness. e, f CO2 separation evaluation of PCN-17 in dynamic breakthrough tests. e Breakthrough curves and f cycling of CO2 separation from a stream of CO2/H2O/N2 of 4/3/93 v% at 25 °C, following a regeneration by an Ar purge at 50 °C. Reproduced from Ref. [71] by permission of The Royal Society of Chemistry
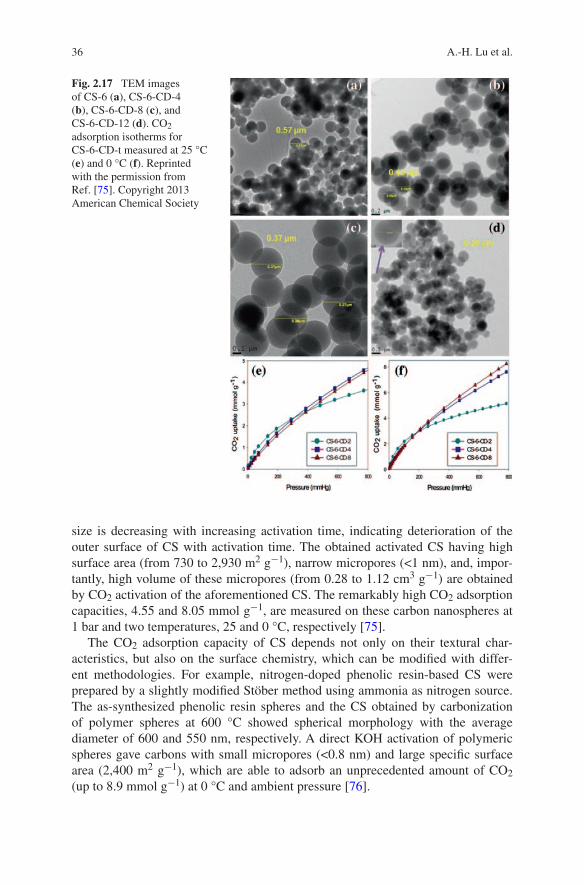
36 A.-H. Lu et al.
size is decreasing with increasing activation time, indicating deterioration of the outer surface of CS with activation time. The obtained activated CS having high surface area (from 730 to 2,930 m2 g−1), narrow micropores (<1 nm), and, impor-tantly, high volume of these micropores (from 0.28 to 1.12 cm3 g−1) are obtained by CO2 activation of the aforementioned CS. The remarkably high CO2 adsorption capacities, 4.55 and 8.05 mmol g−1, are measured on these carbon nanospheres at 1 bar and two temperatures, 25 and 0 °C, respectively [75].
The CO2 adsorption capacity of CS depends not only on their textural char-acteristics, but also on the surface chemistry, which can be modified with differ-ent methodologies. For example, nitrogen-doped phenolic resin-based CS were prepared by a slightly modified Stöber method using ammonia as nitrogen source. The as-synthesized phenolic resin spheres and the CS obtained by carbonization of polymer spheres at 600 °C showed spherical morphology with the average diameter of 600 and 550 nm, respectively. A direct KOH activation of polymeric spheres gave carbons with small micropores (<0.8 nm) and large specific surface area (2,400 m2 g−1), which are able to adsorb an unprecedented amount of CO2 (up to 8.9 mmol g−1) at 0 °C and ambient pressure [76].
Fig. 2.17 TEM images of CS-6 (a), CS-6-CD-4 (b), CS-6-CD-8 (c), and CS-6-CD-12 (d). CO2 adsorption isotherms for CS-6-CD-t measured at 25 °C (e) and 0 °C (f). Reprinted with the permission from Ref. [75]. Copyright 2013 American Chemical Society

372 Porous Carbons for Carbon Dioxide Capture
Later on, the same group also reported a series of cysteine-stabilized phenolic resin-based polymer and CS prepared by the modified Stöber method [77]. They believe cysteine plays a very important role in the formation of such nanospheres, acting as a particle stabilizer and a source of heteroatoms (nitrogen and sulfur) that can be intro-duced into these spheres. The diameter of these spheres can be tuned in the range of 70–610 nm by adjusting the cysteine amount and reaction temperature. Since polymer spheres obtained in the presence of cysteine contain sulfur and nitrogen heteroatoms, they are promising in CO2 capture.
For the research regarding CS, another important trend is to prepare monodis-perse, uniform, and colloidal spheres, which can serve as model carbons for fun-damental analysis in many applications including CO2 capture. For example, Xu’s group has created a novel and general method to prepare monodisperse carbon nano-spheres with a regular round ball-like shape [73, 78]. In their synthesis, three steps are involved: (1) synthesize monodisperse polystyrene spheres by soap-free emulsion polymerization; (2) increase the surface cross-linking degree of polystyrene spheres via Friedel Crafts alkylation as a post-cross-linking reaction; (3) carbonize the reac-tion product. By adjusting the post-cross-linking reaction time, the size of hollow core can be finely tuned. This is a novel method in the field of fabrication of CS.
Furthermore, it has been known that monodisperse colloidal spheres have the ability to self-assemble into three-dimensional periodic colloidal crystals, only when their size distributions are less than 5 % [79]. That is a particular challenge to establish a new and facile synthesis strategy toward truly monodispersed carbon nanospheres [80]. Wang et al. [81] have established a new strategy of synthesis of highly uniform carbon nanospheres with precisely tailored sizes and high mono-dispersity on the basis of the benzoxazine chemistry.
2.4.3 Hard-Templated Porous Carbons
Nanocasting is a process in which a mold (may be called as hard template, scaf-fold) over nanometer scale is filled with a precursor, and after processing, the initial mold is afterward removed [82–87]. In this way, the space once occupied by the host mold is thus transferred into pores of the final carbon products, and the carbon in the original template pores is released as a continuous carbon framework. Nanocasting usually involves the following steps: (1) preparation of a porous template with con-trolled porosity; (2) introduction of a suitable carbon precursor into the template pores through techniques such as wet impregnation, CVD, or their combination; (3) polymerization and carbonization of the carbon precursor to generate an organic–inorganic composite; and (4) removal of the inorganic template. In the past decades, nanocasting pathway has been demonstrated as a controllable method in preparing carbon monoliths with tailorable pore size over several length scales. The keys rely on preparing a template with accessible porosity and a thermal stable carbon precur-sor such as phenolic resin, sucrose, furfuryl alcohol (FA), acrylonitrile, acetonitrile, and mesophase pitch. In the following part, we discuss the detailed synthesis princi-ple based on several representative examples.

38 A.-H. Lu et al.
2.4.3.1 Porous Carbons Replicated from Porous Silica
Porous carbons replicated from porous silica are extensively investigated. For exam-ple, Lindén and coworkers prepared hierarchical porous monolithic carbon contain-ing wormholelike mesopores and macropores [88–90]. In a similar way, Shi et al. [91] prepared a novel porous carbon with co-continuous structure and trimodal pores using a hierarchical silica monolith as the template. Hu et al. [92] synthesized hierar-chically porous carbons with a relatively higher graphite-like ordered carbon struc-ture by using meso-/macroporous silica as a template and using mesophase pitch as a precursor. Yin’ group also reported a preparation of mesoporous nitrogen-doped carbon (N-MC) with highly ordered two-dimensional hexagonal structures using diaminobenzene (DAB) as carbon and nitrogen sources, ammonium peroxydisulfate (APDS) as an oxidant, and SBA-15 as a hard template [93]. By adjusting the synthe-sis temperatures in a range of 70–100 °C, the pore diameter of the as-made materials can be tuned from 3.4 to 4.2 nm, while the specific surface area of the N-MC with a nitrogen content of 26.5 wt% can be tuned from 281.8 to 535.2 m2 g−1. The C/N molar ratio of the samples can be tuned in a range of 3.25–3.65 by adjusting the mole ratio of DAB/APDS precursors at a synthesis temperature of 80 °C, while the pore diameter of the N-MC can be tuned in a range of 4.1–3.7 nm.
The above nanocasting method was far more successful, but the multisteps and long synthesis period it involved are impressive. To simplify the tedious proce-dures, researchers made massive efforts. Han and coworkers developed a one-step nanocasting technique to synthesize micro-/mesoporous carbon monoliths with very high BET surface area (ca. 1,970 m2 g−1) and ca. 2 nm mesopores by the cocondensation of β-cyclodextrin with tetramethylorthosilicate [94].
Carbon nitride (CN) is a well-known and fascinating material that has attracted worldwide attention because the incorporation of nitrogen atoms in the carbon nanostructure can improve the mechanical properties and surface chemistry. As a result, increasing efforts have been recently devoted to the synthesis of CN. Vinu et al. prepared two-dimensional mesoporous carbon nitride (MCN) with tunable pore diameters using SBA-15 materials with different pore diameters as templates through a simple polymerization reaction between ethylenediamine (EDA) and carbon tetrachloride (CTC) by a nano hard-templating approach [95]. HRTEM was used to further examine the structural order and morphology of the mesoporous CN materials with different pore diameters. The pore diameter of the MCN materials can be easily tuned from 4.2 to 6.4 nm without affecting their structural order. The carbon-to-nitrogen ratio of the MCN decreases from 4.3 to 3.3 with increasing weight ratio of EDA to CTC from 0.3 to 0.9. The optimum EDA-to-CTC weight ratio required for fabricating the well-ordered MCN materi-als with excellent textural parameters and high nitrogen content is around 0.45. The specific surface area and the specific pore volume of MCN materials can be adjusted ranging between 505 and 830 m2 g−1 and 0.55–1.25 cm3 g−1.
In a similar way, Zhao’s group reported the preparation of porous CN spheres with partially crystalline frameworks via a nanocasting approach by using spheri-cal mesoporous cellular silica foams (MCFs) as a hard template, and EDA and

392 Porous Carbons for Carbon Dioxide Capture
CTC as precursors. The elemental analyses show that the material has high nitro-gen content (17.8 wt%) with nitrogen-containing groups and abundant basic sites. The obtained CN spheres have mesostructure with small and large mesopores with pore diameters centered at ca. 4.0 and 43 nm, respectively, a relatively high BET surface area of ~550 m2 g−1, and a pore volume of 0.90 cm3 g−1. The adsorption isotherms of the mesoporous CN spheres (Fig. 2.18a) show that after adsorption for 150 min, CO2 uptake reaches 2.90 mmol g−1 at 25 °C. When the temperature was increased to 75 °C, the uptake greatly decreased to 0.97 mmol g−1. The CO2 uptake (2.50 mmol g−1) of the pristine carbon material (Fig. 2.18b) was similar to that of the mesoporous CN sample at 25 °C. However, when the temperature was increased to 75 °C, its CO2 uptake decreased dramatically to 0.30 mmol g−1, which is much lower than the corresponding value of the mesoporous CN spheres, suggesting that there is a weak interaction between the carbon pore walls and CO2 molecules [96].
2.4.3.2 Porous Carbons Replicated from Crystalline Microporous Materials
As shown in Fig. 2.19, the nanocasting pathway is also powerful to produce nano-porous carbons with remarkably high surface areas (up to 4,000 m2 g−1) and pre-cisely controlled microporous structures (0.5–1.5 nm) that are well suitable for CO2 capture [97]. The efforts to construct such molecular-sieve-type porous car-bon using well-crystalline zeolite or MOFs-related materials (ZIFs, MOCPs) as sacrificial templates still continue.
Banerjee et al. [98] reported a synthesis and gas adsorption properties of porous carbons synthesized by a nanocasting method at 1,000 °C, in which isoreticular zeolitic imidazolate frameworks (IRZIFs) acts as the template and FA as carbon source. Similarly, Deng et al. [99] synthesized a series of porous carbons from non-porous metal–organic coordination polymers (MOCPs), using in situ polym-erized phenol resin as the carbon precursor. The textural properties of highly
Fig. 2.18 CO2 capture capacities of a the mesoporous CN materials and b the pristine CS at 25 and 75 °C. Reprinted from Ref. [96], with kind permission from Springer Science+Business Media

40 A.-H. Lu et al.
microporous ZIF-templated carbons derived from commercially available ZIFs (Basolite Z1200) can be further improved via mild chemical activation, as demon-strated by Almasoudi and Mokaya [100].
Moving one-step forward, the direct transformation of MOFs-related materi-als (PCPs, ZIFs, PAFs, etc.) to porous carbons with nanopores of a precise and uni-form size has been achieved through simple carbonization at elaborate temperature. This synthesis strategy for nanoporous carbons is a burgeoning new area of research, which will further broaden the library of MSCs. Yamauchi et al. [101] have pioneered the synthesis of a novel nanoporous carbon with highly developed porosity (surface area of 5,500 m2 g−1 and pore volume of 4.3 cm3 g−1) by a direct carbonization of Al-PCPs. Porous carbons with hierarchical pore structures could also be achieved by direct carbonization of the selected IRMOFs (IRMOF-1, IRMOF-3, and IRMOF-8), indicating the tunable pore characteristics of MOFs-derived nanoporous carbons [102]. Ariga et al. [103] reported a nanoporous carbon with high surface areas (up to 1,110 m2 g−1), and narrow pore size distributions that are close to its parent ZIF-8.
A family of nanoporous carbons has also been prepared by thermal decomposi-tion of guest-free MOFs (even non-porous MOFs) by Kim and colleagues [104]. They found that the porosity of the carbon materials depends linearly on the Zn/C ratio of MOFs precursors, which allow a precise control of the porosity of the car-bon materials in a predictable manner.
Through a simple pyrolysis of crystalline polymer (PAF-1), Qiu et al. [105] have prepared a series of nanoporous carbons having high surface areas and narrow micropore size distributions. The carbonized (at 450 °C) sample PAF-1-450 showed very excellent adsorption capacities (4.5 mmol g−1) for CO2 than that of original PAF-1 at ambient conditions (Fig. 2.20). These aforementioned works exemplify a
Fig. 2.19 Synthesis of molecular sieve carbon (MSC) through nanocasting pathway: The sacri-ficial templates include the crystalline zeolites and MOFs. Reproduced from Ref. [97] by permis-sion of John Wiley & Sons Ltd

412 Porous Carbons for Carbon Dioxide Capture
striking indication that the facile and one-step pathway replicated from crystalline microporous materials is highly efficient toward highly nanoporous carbons.
2.4.3.3 Porous Carbons Replicated from Colloidal Crystals
Colloidal crystals are the self-assembly periodic structures consisting of close-packed uniform particles. In most cases, replication of the colloidal crystals (colloidal sil-ica/polymer spheres) will lead to a high degree of periodicity in three dimensions. Subsequent removal of the crystal templates results in a replica with 3D-ordered macroporous (3DOM) structures. The groups of Stein, Velev, and Lenhoff have inde-pendently achieved many great results in the field of colloidal crystal and their related areas. Here, we only discuss a small aspect where the templating of colloidal crystal is used as an effective path to get porous carbons with highly ordered macroporosity. For example, Lee et al. [106] synthesized 3DOM carbons via a RF sol-gel process using poly(methyl methacrylate) colloidal crystal templates. Similarly, Adelhelm et al. [107] also synthesized a hierarchical meso- and macroporous carbon using mesophase pitch as precursors and PS or PMMA as templates through spinodal decomposition.
By carbonization of a thin layer of phenolic resin on the suitable templates, Gierszal et al. [108] reported the synthesis of one kind of uniform carbon film with large pore volumes (6 cm3 g−1 for 24 nm silica colloids), uniform pore sizes, and controlled thickness. This synthetic route involves the formation of a uniform poly-meric film on the silica pore walls of silica colloidal crystals or colloidal aggregates and its carbonization and template removal. After proper pre-treatment of the silica
Fig. 2.20 a CO2 adsorption (solid symbols) and desorption (open symbols) isotherms of PAF-1 and carbonized samples at 273 K; b Qst CO2 of PAF-1 and carbonized samples as a function of the amount of CO2 adsorbed. Reproduced from Ref. [105] by permission of The Royal Society of Chemistry

42 A.-H. Lu et al.
template and under controlled experimental conditions, the mixture of resorcinol and crotonaldehyde copolymerizes on the silica surface and form a uniform film. Recently, Zhang and coworkers presented a one-pot method to synthesize hierar-chically bimodal-ordered porous carbons with interconnected macropores and mesopores, via in situ self-assembly of colloidal polymer (280, 370, and 475 nm) and silica spheres (50 nm) using sucrose as the carbon source. Compared with the classical nanocasting procedure, this approach is veritably simple; neither pre-syn-thesis of crystal templates nor additional infiltration is needed, and the self-assem-bly of polymer spheres into the crystal template and the infiltration are finished simultaneously in the same system [109]. Similarly, a hierarchically porous carbon with multimodal (macropore and mesopore) porosity have also been prepared by using dual-template (PS/colloidal silica and PMMA/colloidal silica), where PS (or PMMA) is used for creating 3D-ordered macropores, colloidal silica is responsible for creating spherical mesopores [110, 111]. The unique hierarchical structures of 3DOM carbons, i.e., the open larger mesopores located in the ordered macropores may enhance the kinetics greatly when used as CO2 sorbents.
2.4.3.4 Porous Carbons Replicated from MgO nanoparticles
Park et al. [112] reported a series of porous carbons with well-developed pore structures, which were directly prepared from a weak acid cation exchange resin (CER) by the carbonization of a mixture with Mg acetate in different ratios (Fig. 2.21). By dissolving the MgO template, the porous carbons exhibited high specific surface areas (326–1,276 m2 g−1) and high pore volumes (0.258–0.687 cm3 g−1). The CO2 adsorption capacities of the porous carbons were enhanced to 164.4 mg g−1 at 1 bar and 1,045 mg g−1 at 30 bar by increasing the Mg-acetate-to-CER ratio. This result indicates that CER is one of the desirable carbon precursors for producing the porous structure, as well as improving the CO2 adsorption capacities of the carbon species.
Recently, Jang’s group reported a time-saving synthesis toward ordered mesoporous carbon supported MgO (Mg-OMC) materials, which were fabricated by the carbonization of sulfuric acid-treated silica/triblock copolymer/sucrose/Mg(NO3)2 composites. In the current approach, triblock copolymer P123 and sucrose were employed as both structure-directing agents for the self-assembly of rice husk ash silica solution and carbon precursor. Sulfuric acid was used to cross-link P123 and sucrose in the as-synthesized composites in order to improve the carbon yield. The CO2 adsorption capacity of Mg-OMC-1 was observed to be 92 mg g−1, which is comparable with that of the well-established CO2 sorbents [113].
Przepiórski et al. reported the competitive uptake of SO2 and CO2 on the porous carbon materials containing CaO and MgO, prepared by carbonization of poly(ethylene terephthalate) mixed with a natural dolomite. The as-prepared porous carbon was examined as a sorbent for simultaneous removal of CO2 and SO2 from air in dry conditions and in a presence of humidity, at temperatures ranging from 20 to 70 °C. The attained results clearly confirmed the crucial effect of water on the amounts of gases removed from air streams and the removal mechanisms. The

432 Porous Carbons for Carbon Dioxide Capture
breakthrough curves registered clearly revealed the overshoot in concentration of the gas during the first minutes of removal tests (Fig. 2.22) [114].
Recently, Park et al. reported the preparation of nanoporous (styrene-divinylbenzene)-based ion exchange resin-based carbons (MPCs) by MgO-templating synthesis and KOH activation. MPCs were prepared from a (styrene-divinylbenzene)-based ion exchange resin by the carbonization of a mixture with Mg gluconate at 900 °C. And then, the prepared MPCs were treated with KOH at KOH/MPCs ratios ranging from 0.5 to 4 at 800 °C. Low KOH/MPCs ratios (KOH/MPCs ratio = 1, MCK-1) tended to favor the formation of micropores, whereas higher KOH/MPCs (KOH/MPCs ratio = 4, MCK-4) led to the formation of mesopores. The treated MPCs with a KOH/MPCs ratio = 1 exhibited the best CO2 adsorption value of 266 mg g−1 at 1 bar. However, the treated MPCs with a KOH/MPCs ratio of 3 (MCK-3) exhibited the best CO2 adsorption value of 1,385 mg g−1 at 30 bar. This result indicated that the CO2 adsorption capacity of nanoporous carbons is attributed to the mesopore volume frac-tion at higher pressure [115].
2.4.4 Porous Carbon-Based Composites
Post-modification is a versatile method for the preparation of advanced carbons with powerful functions through processes such as CVD [116, 117], impregnation [118–120], and metal transfer reactions [121].
Fig. 2.21 Schematic representation of the preparation of MgO-templated nanoporous carbons. Reprinted from Ref. [112]. Copyright 2012, with permission from Elsevier

44 A.-H. Lu et al.
García-Martínez et al. [122] reported a solvent-free, liquid-phase synthesis of self-assembled carbon foams, which can be prepared in variable shapes and mor-phologies without the need of any binders. Long et al. [123] prepared carbon aerogels by sol-gel polymerization of phenol, melamine and formaldehyde, fol-lowed by subsequent carbonization process. The as-prepared samples showed 3D mesoporosity and large pore volume, which allow easy diffusion of reactants and products, and served as the reservoir for the guest molecules. Using similar pro-cedure, Nielsen et al. [124] prepared 2LiBH4-MgH2/carbon aerogel systems as hydrogen storage materials through the nanoconfined chemistry. In this designed composite, LiBH4 and MgH2 nanoparticles were embedded in a nanoporous car-bon aerogel scaffold with pore size (Dmax) of 21 nm and reacted to form MgB2 during release of hydrogen.
Besides post-modification, a direct copolymerization is suitable not only for introduction of molecular functional groups to the carbon products, but also allows well dispersion of nanoparticles throughout the carbon framework. Researchers from
Fig. 2.22 Breakthrough curves for MgO-/CaO-loaded carbon sorbent exposed to air contami-nated with CO2 or SO2 at different experimental temperatures: 20, 40, 70 °C (a and b) and at 20 °C after pre-humidification conducted for different times (c and d). Reprinted from Ref. [114]. Copyright 2013, with permission from Elsevier

452 Porous Carbons for Carbon Dioxide Capture
Lawrence Livermore National Laboratory have made major advances in the synthe-sis and functionalization of monolithic carbon aerogels [125, 126]. Recently, they reported TiO2/C, TiCN/C, ZnO/C composite aerogels by carbothermal reduction in the titania (or ZnO)-coated carbon aerogels. The resulting monoliths consisting of nitrogen-rich titanium carbonitride (TiC1−xNx, x = 0.90) nanocrystals or well-crystallized ZnO nanoparticles exhibited surface areas of 1,838 and 1,500 m2 g−1, respectively. Also, they successfully integrated CNTs or graphene sheets into the sol-gel reaction, leading to the formation of the advanced monolithic carbons with significantly improved mechanical properties [127–129]. This strategy has used the organic RF binder that is reducible concurrently with the GO or CNTs to thus pro-duce carbon cross-links in the graphene or CNTs network which are virtually indis-tinguishable from those in the graphene sheets or CNTs networks (Fig. 2.23).
Recently, Jin et al. [130] reported a new type of CNT-modified carbon monoliths that were prepared from a commercial phenolic resin mixed with just 1 wt% of CNTs followed by carbonization and physical activation with CO2. The products possess a hierarchical macro-/microporous structure and superior CO2 adsorption properties. In particular, they show the top-ranked CO2 capacity (52 mg CO2 per g adsorbent at 25 °C and 114 mmHg) under low CO2 partial pressure, which is of more relevance for flue gas applications. This study demonstrates an effective way to create narrow micropores through structural modification of carbon composites by CNTs.
From an application point of view, the volumetric capacities are even more important than that on a gravimetric basis, due to the limited volume of the gas storage tank. Under this consideration, Qian et al. [131] optimized the structural features of hierarchical porous carbon monolith by incorporating the advantages of MOFs (Cu3(BTC)2) to maximize the volumetric based CO2 capture capabil-ity (CO2 capacity in cm3 per cm3 adsorbent). The mesoscopic structure of the HCM-Cu3(BTC)2 composites and the parent materials (HCM and Cu3(BTC)2) were characterized by SEM. The SEM micrograph (Fig. 2.24) clearly displays that Cu3(BTC)2 crystallites are born within the macropores of the HCM matrix. The sponge-like skeleton of HCM before and after the MOF growth remains
Fig. 2.23 Synthesis procedure for the GO-RF aerogel and graphene aerogel. Reprinted with the permission from Ref. [128]. Copyright 2010 American Chemical Society

46 A.-H. Lu et al.
unchanged. The octahedral Cu3(BTC)2 crystallites are well dispersed within the HCM matrix. The equilibrium CO2 adsorption measurements were carried out at 25 °C, and the results (Fig. 2.24) reveal that HCM-Cu3(BTC)2 composites exhibit an obvious increment in CO2 adsorption capacity on a volumetric basis com-pared with the original HCM. The HCM-Cu3(BTC)2-3 composite with the highest Cu3(BTC)2 loading can achieve maximum CO2 uptake of 22.7 cm3 STP per cm3 at ~1 bar, which is almost as twice as the uptake of HCM (12.9 cm3 STP per cm3) under the same conditions. This result encourages a new principle on a rational design of CO2 capture material by maximizing the capacity on a volumetric basis.
2.5 Graphitic Porous Carbons for CO2 Capture
2.5.1 Bulk Crystalline Porous Carbons
Porous carbons obtained through the above-mentioned methods, in most cases, have the amorphous carbon walls, which contain either adventitious micropores or templated open mesopores. Due to the long range randomly arrangement of the primary carbon fragments, the amorphous carbon possesses abundant active sites and displays various kinds of porosity, thus leading to a high surface area. These properties endowed by the amorphous feature combined with the ease of handling render the amorphous carbons to be widely used in many fields such as catalysis, adsorption/separation, hydrogen storage, and desalination.
Besides amorphous carbons, graphitic porous carbons are also widely inves-tigated and may be valuable for figuring out the CO2 capture behavior due to its
Fig. 2.24 SEM micrographs of HCM (a), Cu3(BTC)2 (b), and HCM-Cu3(BTC)2-3 (c–e). (f) XRD patterns of HCM, Cu3(BTC)2, and HCM-Cu3(BTC)2−3. (g) CO2 adsorption isotherms on a volu-metric basis. Reprinted with the permission from Ref. [131]. Copyright 2012 American Chemical Society

472 Porous Carbons for Carbon Dioxide Capture
ordering at the atomic scale. Though high-temperature thermal treatments above 2,000 °C can facilitate the transition to graphitization phase, unfortunately, it often results in a partial or total collapse of the pore structures and reduces the accessi-ble surface areas. By employing graphitization catalysts (i.e., Fe or Co salt) [132], one can obtain graphitic porous carbons, in which an additional leaching process is required to remove the final metal oxide derived from the catalyst precursors. Liang et al. [133] reported the synthesis of graphitic carbon column with bimodal pores, which was prepared by pyrolyzing a rod made of a copolymer of a resorcinol/iron(III) complex and formaldehyde in the presence of silica beads through a nano-casting process. Very recently, Dai’s group pioneered a “brick-and-mortar” self-assembly approach toward ordered graphitic mesoporous carbon nanocomposites with tunable mesopore sizes below 850 °C without using graphitization catalysts or high-temperature thermal treatments [134]. In this strategy, phenolic resin-based mesoporous carbons act as mortar, while the highly conductive carbon blacks or carbon onions were introduced as bricks that are responsible for the graphitic domains of the pore walls. This breakthrough provides a new approach to the syn-thesis of porous carbons with high level of graphitization under a facile condition.
2.5.2 2D Nanosheets/Films
The emergence of graphene nanosheet has opened up an exciting new field in the science and technology of two-dimensional nanomaterials [135–137]. Graphite oxide (GO) is a derivative of graphene and consists of oxygen functional groups on their basal planes and edges, so surface modification of GO with amines or amine-containing molecules takes place easily through the corresponding nucle-ophilic substitution reactions. If polyamines covalently attach to their layers, the residual unreacted amine groups can react with CO2 and have potential for the removal of CO2. Under this consideration, Zhao et al. prepared GO–amine com-posites based on the intercalation reaction of GO with amines, including EDA, diethylenetriamine (DETA), and triethylene tetramine (TETA). Dynamic CO2 breakthrough test revealed that the aminated GO was an efficient adsorbent for CO2 capture. For example, the typical sample of GO/EDA showed an adsorption capacity of 53.62 mg g−1 sample [138].
Srinivas et al. [136] reported a method to obtain a wide range of highly porous carbon adsorbents through chemical activation of exfoliated graphene oxide pre-cursors with KOH. Through tuning the synthesis, they successfully prepared a series of GO-derived carbons (GODCs) that showed different porous structures (Table 2.2). By comparing porous properties with respect to the gas adsorption capacity of this new class GODCs with a range of other porous solids including ACs and metal–organic frameworks, they believe that the GODCs have a great potential in gas adsorption applications.
Very interestingly, Koenig et al. [139] pioneered one type of interesting graphene membrane-based molecular sieve. In their synthesis method, ultraviolet-induced

48 A.-H. Lu et al.
Tabl
e 2.
2 G
OD
Cs
with
syn
thes
is c
ondi
tions
(K
OH
/exf
-GO
con
cent
ratio
n an
d ac
tivat
ion
tem
pera
ture
), y
ield
by
wei
ght
of p
recu
rsor
exf
-GO
, th
e po
rosi
ty
para
met
ers:
BE
T s
urfa
ce a
rea,
ave
rage
por
e si
ze, p
oros
ity b
ased
on
a sk
elet
on d
ensi
ty o
f 2
g cm
−3 ,
and
hig
h-pr
essu
re C
O2
and
met
hane
ads
orpt
ion
capa
citie
s at
20
and
35 b
ar, r
espe
ctiv
ely,
at 3
00 K
Rep
rodu
ced
from
Ref
. [13
6] b
y pe
rmis
sion
of
The
Roy
al S
ocie
ty o
f C
hem
istr
y
Sam
ple
Yie
ld b
y
wei
ght (
%)
BE
T s
urfa
ce
area
/m2
g−1
Tota
l por
e
volu
me/
cm3
g−1
Ave
rage
por
e
size
/nm
Poro
sity
(%
)C
O2
adso
rptio
n/
mg
mg−
1M
etha
ne a
dsor
ptio
n/
mg
mg−
1
GO
DC
4-60
054
916
0.48
1.3
490.
390
0.09
6G
OD
C9-
600
3661
90.
361.
5442
0.36
00.
084
GO
DC
4-70
053
1,09
60.
561.
5453
0.48
00.
119
GO
DC
9-70
028
1,32
61.
101.
8569
0.55
50.
137
GO
DC
4-80
038
1,27
60.
721.
6059
0.49
70.
126
GO
DC
9-80
026
1,70
41.
652.
677
0.65
20.
163
GO
DC
sol-
800
331,
894
1.60
2.4
760.
721
0.17
5G
OD
Cso
l-90
010
1,27
20.
991.
8666
0.52
50.
137

492 Porous Carbons for Carbon Dioxide Capture
oxidative etching can create pores in micrometer-sized graphene sheets (Fig. 2.25). A pressurized blister test and mechanical resonance are used to measure the transport of a range of gases (H2, CO2, Ar, N2, CH4, and SF6) through the pores. The molecular selectivity of the fabricated porous graphene membrane was demonstrated by meas-uring the rate of change of δ with time (−dδ/dt) for the same membrane pressurized with a number of different gases. Figure 2.25a shows δ versus t for H2, CO2, Ar, and CH4 before and after etching, and N2 after etching. At short times, −dδ/dt is approxi-mately linear. This rate was plotted versus kinetic diameter for all the gases, using
Fig. 2.25 Measuring leak rates in porous graphene membranes. a Schematic of a microscopic graphene membrane on a silicon oxide substrate. b After removing the graphene membrane from the pressure chamber, the membrane bulges upwards. c Following etching pore(s) in the gra-phene membrane bigger than H2, the H2 is able to rapidly leak out of the microchamber through the membrane pore(s). d Once all the H2 molecules have leaked out of the microchamber, the membrane will deflect downwards. Comparison of leak rates of pristine and porous graphene membranes. e Maximum deflection δ versus t before and after etching. f Average −dδ/dt versus molecular size found from the slopes of membrane deflection versus t (in e) before and after introducing pores into the same graphene membrane. The connecting lines show the measure-ments before (black) and after (red) etching. Reprinted by permission from Macmillan Publishers Ltd.: Ref. [139], Copyright 2012

50 A.-H. Lu et al.
the same membrane/microcavity, before and after etching (Fig. 2.25b). After etching, there is an increase in −dδ/dt by two orders of magnitude for the H2 and CO2 leak rates, whereas those for Ar and CH4 remain relatively unchanged. This suggests that the etched pores change the transport mechanism for H2 and CO2, but leave the trans-port of Ar and CH4 nearly unchanged. The experimental results reveal the realization of graphene gas separation membranes by molecular sieving and represent an impor-tant step toward the realization of size-selective porous graphene membranes.
Besides graphene sheets tested as CO2 capture materials, the introduction of other active sites onto graphene sheets for high-performance CO2 sorbents was also extensively investigated recently [140–142]. For example, PPy function-alized graphene sheets are employed to fabricate N-doped porous carbons via chemical activation [143]. This type of material shows selective adsorption of CO2 (4.3 mmol g−1) over N2 (0.27 mmol g−1) at 25 °C. Similarly, the highest CO2 uptake (75 mmol g−1) at 11 bar and 25 °C over polyaniline–graphene nanocom-posites were reported by Mishra and Ramaprabhu [144].
2.5.3 1D Carbon Nanotubes/Fibers/Ribbons/Scrolls
Another familiar carbon nanostructure is carbon nanotube. Since the first synthesis of carbon nanotubes (CNTs) via arcing between graphite-like electrodes by Iijima in 1991 [145], 1D carbon materials have been extensively researched, due to their outstanding properties such as excellent chemical and thermal stability, high sur-face area, and potential applications in electronics, adsorption, and catalysis. So far, CVD [146, 147] and the electrospinning technique [148] have been widely used in the production of 1D carbon materials. One of their potential applications is as sorbents for CO2 capture due to their shortened size, easy functionalization, and/or integration with foreign active species for selective CO2 recognition [149–153].
For example, Dillon et al. [154] reported a covalent attachment of branched polyethyleneimine (PEI) to the sidewalls of SWNTs through the use of fluorinated single-wall CNTs as precursors. The structural integrity of the original purified SWNT is maintained upon covalent functionalization with PEI. Solid-state 13C NMR shows the presence of carboxylate substituents due to carbamate formation as a consequence of the reversible CO2 absorption to the primary amine substit-uents of the PEI. Desorption of CO2 is accomplished by heating under argon at 75 °C, while the dependence of the quantity of CO2 absorbed on temperature and the molecular weight of the PEI is also observed.
Besides the experimental investigation, the theoretical researches on the CO2 capture over CNT were also reported. For example, Liu et al. [155] have shown, from molecular dynamics simulations, that the windowed CNTs are able to sepa-rate CO2 from the CO2/CH4 mixture with a CO2 permeance several orders of mag-nitude higher than the conventional analogs (Fig. 2.26).
The aforementioned examples open the door for the design and preparation of highly effective carbonaceous CO2 adsorbents with controlled pore features and

512 Porous Carbons for Carbon Dioxide Capture
tailored surface chemistry. These porous carbons would combine the merits of designed synthesis (controlled pore structure and task-specific surface chemistry) and intrinsic properties (excellent chemical and thermal stability, developed poros-ity) of carbon materials and meet the complex requirements of efficient adsorbents for CO2 capture.
2.5.3.1 Carbon Nanofibers
Besides CNTs, another 1D carbon nanostructure is carbon nanofibers. For exam-ple, Shen et al. [156] prepared a series of hierarchical porous carbon fibers with a BET surface area of 2,231 m2 g−1 and a pore volume of 1.16 cm3 g−1. In this syn-thesis method, the polyacrylonitrile (PAN) nanofibers (prepared by dry–wet spin-ning) were selected as precursors, and pre-oxidation and chemical activation were involved to get the developed porosities. This type of material contained a large amount of nitrogen-containing groups (N content > 8.1 wt%) and consequently basic sites, resulting in a faster adsorption rate and a higher adsorption capacity for CO2 than the commercial zeolite 13X that is conventionally used to capture CO2, in the presence of H2O (Fig. 2.27).
2.5.3.2 Carbon Nanoribbons and Nanoscrolls
Asai et al. [157] reported a new CO2 sorption behavior over graphitic nanorib-bons, which was distinctly different from the behavior of nanoporous carbon and carbon blacks. They found a remarkable irreversibility in adsorption of CO2 and H2O on such kind of graphitic nanoribbons at ambient temperature (Fig. 2.28).
Fig. 2.26 a 4N4H windows or pores on the wall of the inner tube. b Initial setup of the simula-tion where CO2/CH4 gas mixture is inside the windowed inner tube; on the outside is a pristine tube. The following color code is adopted throughout: carbon (cyan), oxygen (red), hydrogen (white), and nitrogen (blue). Reprinted with the permission from Ref. [155]. Copyright 2012 American Chemical Society

52 A.-H. Lu et al.
The irreversible adsorptions of both CO2 and H2O are due to the large number of sp3-hybridized carbon atoms located at the edges. The authors believe that the observed irreversible adsorption capacity of the edge surfaces of graphitic nanorib-bons for CO2 and H2O indicates a high potential in the fabrication of novel types of catalysts and highly selective gas sensors.
Through molecular dynamics simulations, Mantzalis and Asproulis [158] investigated the layering behavior of carbon dioxide transported through carbon nanoscrolls. The layering arrangements were investigated for carbon nanoscrolls with interlayer distances spanning from 4.2 to 8.3 Ǻ at temperature of 300 K and pressures ranging from 5 to 20 bars. It was shown that the number of layers, their
Fig. 2.27 Effect of H2O on the adsorption of CO2 by PAN-PK (a) and zeolite 13X (b) at 25 °C. Reproduced from Ref. [156] by permission of The Royal Society of Chemistry
Fig. 2.28 Adsorption isotherms of CO2 on GNRs and well-crystalline CBs at 303 K. Reprinted with the permission from Ref. [157]. Copyright 2011 American Chemical Society

532 Porous Carbons for Carbon Dioxide Capture
relative strength, and the starting point of bifurcation phenomena vary as a func-tion of the nanoscrolls’ interlayer distance, scroll’s core radius, CO2 density, and gas structure interactions. It is also shown that the number of carbon dioxide mol-ecules adsorbed per scroll’s carbon particles is a function of the scroll’s surface-to-volume ratio and is maximized under certain structural configurations.
2.6 Critical Material Aspects of Porous Carbon Design on CO2 Capture
2.6.1 Micropore Size and Volume Influences
It is highly necessary to discuss the key parameters of materials for CO2 capture. To do this, the first thing is the selection of model carbon. Carbide-derived carbon (CDC) is such kind of carbon, which can be fabricated through hydrothermal decom-position of carbide precursors on various substrates and then selectively etching metals from metal carbides using chlorine at elevated temperature. The most attrac-tive aspects of these materials lie in their precisely controlled micropore size (with a sub-angstrom accuracy) and tunable specific surface area (up to 3,200 m2 g−1) [159–161]. This characteristics allow them to serve as carbon models for fundamen-tal investigating the influences of micropore sizes on CO2 adsorption. In research-ing hydrogen and methane adsorption over CDC, it has been shown that the sorption capability does not necessarily scale with the surface area or the pore size, but it depends on the volume of pores smaller than 1 nm. Similarly, Presser et al. [161] systematically investigate CO2 adsorption at atmospheric and subatmospheric pres-sures at near-ambient temperature (0 °C) on the basis of a series of CDC with well-controlled PSD and surface areas (Table 2.3) synthesized from TiC powders.
They found that the average pore size and the total pore volume are not ade-quate measures to predict the CO2 uptake of microporous carbon sorbents, the pore volume of micropores strongly governs the amount of adsorbed CO2 [161]. Neither high surface area CDC after chemical activation (surface area 3,101 m2 g−1) nor high pore volume nano-TiC-CDC (Vtotal 1.61 cm3 g−1) cor-respond with the highest CO2 adsorption capacity. At ambient pressure, the CO2 uptake closely follows a linear correlation with the volume of pores smaller or equal to a diameter of 1.5 nm. Pores smaller than 0.5 nm contribute to the amount of adsorbed CO2, but the best correlation is found for pore volume smaller than 0.8 nm (Fig. 2.29). The correlation between the amount of adsorbed CO2 at low partial pressures and volume of smaller pores is the basis for the well-known application of CO2 sorption as a method to calculate the pore characteristics of microporous materials. Subatmospheric pressures are of particular interest for industrial applications, where partial pressure of CO2 is below 1 bar, and here, the best prediction of the CO2 uptake capacity at 0.1 bar would be based on the vol-ume of pores smaller or equal to a diameter of 0.5 nm (Fig. 2.29). This correlation can be used to design better CO2 sorbents and CCS devices.

54 A.-H. Lu et al.
Tabl
e 2.
3 P
ore
char
acte
rist
ics
and
CO
2 up
take
at 0
.1 a
nd 1
.0 b
ar f
or n
ano-
TiC
-CD
C, m
icro
-TiC
-CD
C a
nd a
ctiv
ated
mic
ro-T
iC-C
DC
*Dat
a de
rive
d fr
om N
2 so
rptio
n**
Dat
a de
rive
d fr
om C
O2
sorp
tion
Rep
rodu
ced
from
Ref
. [16
1] b
y pe
rmis
sion
of
The
Roy
al S
ocie
ty o
f C
hem
istr
y
Sam
ple
Chl
orin
atio
n
tem
pera
ture
(°C
)A
nnea
ling
atm
osph
ere
te
mpe
ratu
re (
°C)
Mea
n po
re*
size
(nm
)D
FT S
SA*
(m2 /
g)B
ET
SSA
*
(m2 /
g)To
tal p
ore
vo
lum
e* (
cm3 /
g)C
O2
upta
ke
@0.
1 ba
r**
(m
ol/k
g)
CO
2 up
take
@
1 ba
r**
(m
ol/k
g)
Nan
o-T
ic-C
DC
200
H2@
200
0.56
336
324
0.26
0.86
2.15
200
H2@
600
0.58
444
434
0.36
1.10
2.74
400
H2@
400
0.62
780
740
0.54
1.13
3.26
400
H2@
600
0.69
1,03
81,
002
0.73
0.90
3.47
600
H2@
600
0.85
843
952
0.81
0.54
2.54
800
H2@
600
0.86
1,62
41,
855
1.22
0.75
4.23
1,00
0H
2@60
00.
961,
674
1,92
01.
610.
623.
701,
200
H2@
600
1.15
563
619
0.88
0.10
1.40
Mic
ro-T
ic20
0H
2@20
00.
5258
249
70.
230.
892.
8240
0H
2@60
00.
5396
891
00.
301.
153.
9060
0H
2@60
00.
601,
294
1,38
30.
561.
626.
2370
0H
2@60
00.
662,
015
1,83
20.
841.
517.
0980
0H
2@60
00.
801,
687
1,77
20.
721.
566.
791,
000
H2@
600
0.78
1,66
21,
669
0.75
1.18
5.45
1,20
0H
2@60
00.
941,
319
1,54
00.
860.
683.
34A
ctiv
ated
mic
ro-
TIC
-CD
C60
0va
cuum
@1,
500
0.85
1,58
91,
776
0.87
1.06
4.77
800
NH
3@60
00.
761,
840
2,09
40.
781.
155.
3340
0K
OH
@60
00.
722,
628
2,91
10.
921.
065.
9150
0K
OH
@60
00.
962,
778
3,10
10.
971.
186.
3180
0K
OH
@60
00.
792,
300
2,56
51.
051.
316.
9260
0C
O2@
875
0.69
2,03
71,
810
0.70
1.54
6.79
600
CO
2@92
50.
822,
167
2,46
80.
991.
195.
9760
0C
O2@
950
0.83
2,14
22,
489
1.00
1.03
5.42
600
CO
2@96
00.
832,
045
2,22
90.
881.
276.
18
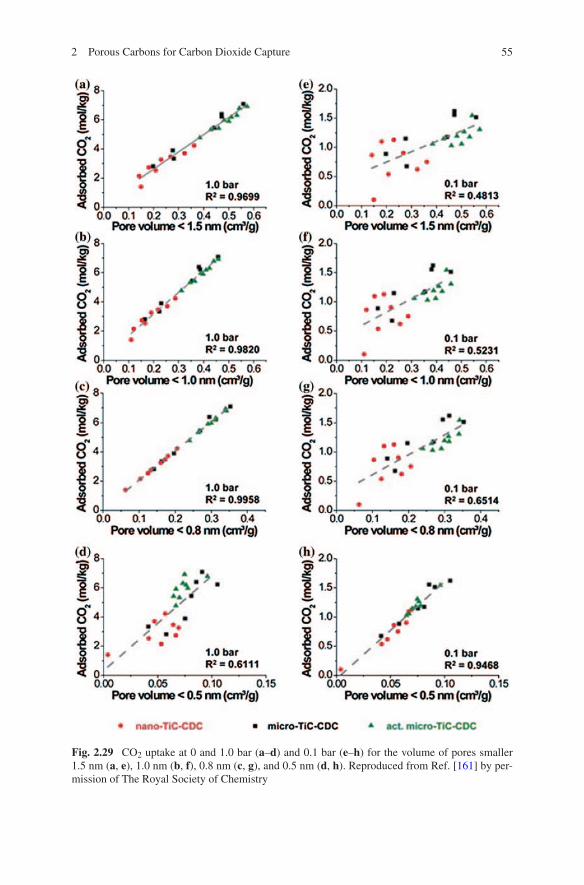
552 Porous Carbons for Carbon Dioxide Capture
Fig. 2.29 CO2 uptake at 0 and 1.0 bar (a–d) and 0.1 bar (e–h) for the volume of pores smaller 1.5 nm (a, e), 1.0 nm (b, f), 0.8 nm (c, g), and 0.5 nm (d, h). Reproduced from Ref. [161] by per-mission of The Royal Society of Chemistry

56 A.-H. Lu et al.
The important role of micropores on CO2 capture has been widely accepted. Therefore, besides the above-mentioned ways, i.e., hard template methods (e.g., zeo-lites as templates), CDC pathways, and so on, new strategies to fabricate such pore systems are highly expected. For instance, Qian et al. [162] report a novel synthesis approach for the fabrication of microporous carbon materials (HCMs) by using discrete chelating zinc species as dynamic molecular porogens to create extra micropores that enhance their CO2 adsorption capacity and selectivity. During carbonization process, the evaporation of the in situ formed Zn species would create additional nanopaths that contribute to the additional micropore volume for CO2 adsorption (Fig. 2.30).
The resulted HCMs show increased amount of micropores with sizes in the range of 0.7–1.0 nm, and a high CO2 adsorption capacity of 5.4 mmol g−1 (23.8 wt%) at 273 K and 3.8 mmol g−1 (16.7 wt%) at 298 K and 1 atm, which are superior to most carbon-based adsorbents with N-doping or high specific surface area. As shown in Fig. 2.31, the dynamic gas separation measurement, using 16 % (v/v) CO2 in N2 as feedstock, demonstrates that CO2 can be effectively separated from N2 under ambi-ent conditions and shows a high separation factor (SCO2/N2 = 110) for CO2 over N2, reflecting a strongly competitive CO2 adsorption capacity. When the feedstock con-tains water vapor, the dynamic capacity of CO2 is almost identical as that measured under dried conditions, indicating the carbon material has an excellent tolerance to humidity. An easy CO2 release can be realized by purging an argon flow through the fixed-bed adsorber at 298 K, indicating the good regeneration ability.
2.6.2 Heteroatoms Doping Influences
Besides micropores, the heteroatom doping is another possible factor that may influence the CO2 adsorption behavior. Among them, the influence of N-doping is reported most frequently. A wide range of N-doped carbons with diverse morphol-ogies have been developed and tested for CO2 capture. The N-doping ways can be broadly classified into three categories: (1) using nitrogen-containing precursors and pyrolysis; (2) high-temperature reaction and transformation based on pre-made car-bons, i.e., NH3 activation; (3) direct loading of liquid amines into the pores of pre-made carbons. For the first two approaches, the introduction of nitrogen-containing
Fig. 2.30 Synthesis principle of microporous carbon using zinc species as dynamic molecular porogens for the creation of abundant microporosity. Reproduced from Ref. [162] by permission of John Wiley & Sons Ltd

572 Porous Carbons for Carbon Dioxide Capture
functional groups has little effect on pore structures, and these functional groups are highly dispersed either in the surface or in the carbon matrix. For the third way, a high concentration of functional groups can be obtained; however, a large part of pores will be blocked by filling of liquid amines. As regards to the role of heteroatom-involving sites, particularly N-containing groups in CO2 capture, it still remains controversial. At present, there are three viewpoints regarding this issue: acid–base interactions, hydrogen-bonding interaction, and electrostatic interactions. The role of heteroatoms is discussed along with the review of each type of porous carbons.
2.6.2.1 Using Nitrogen-Containing Precursors
The rationally selected polymeric carbon precursors mainly involve p-diaminoben-zene [163], polyacrylonitrile [164], melamine [165], and so on. Zhao et al. [163, 166] have prepared a series of nitrogen-doped carbon materials with a very high nitrogen doping concentration (ca. 13 wt%) and rich micropores (<1 nm), which lend them the highly desired characteristics for selective CO2 capture. For exam-ple, they reported a nitrogen-doped mesoporous carbon materials (Fig. 2.32) that can be nanocasted from tri-continuous mesoporous silica (IBN-9) by using a mixed
Fig. 2.31 a Breakthrough curve of HCM-ZC-1 using a stream of 16 % (v/v) CO2 in N2 at 25 °C; b breakthrough curve of CO2 under a moisture condition; c recycle runs of CO2 adsorption–des-orption on HCM-ZC-1 at 25 °C, using a stream of 16 % (v/v) CO2 in N2, followed by a regen-eration under argon flow; d CO2 uptake of each adsorption run. Reproduced from Ref [162] by permission of John Wiley & Sons Ltd

58 A.-H. Lu et al.
carbon precursor comprised of nitrogen-containing p-diaminobenzene and nitrogen-free furfural alcohol. After a chemical activation process, a high content of nitrogen and a large proportion of micropores in one material were integrated. The optimized material exhibited excellent adsorption properties in terms of both CO2 uptake and CO2/N2 selectivity (Fig. 2.32). Particularly, its CO2 adsorption uptake in the typi-cal condition of flue gas (e.g., CO2 partial pressure of 0.2 bar, 25 °C) is as high as 1.75 mmol g−1, which is far superior to most reported carbon materials. These new
Fig. 2.32 a, d TEM images and SAED patterns (insets) of IBN9-NC (a) and IBN9-NC1 (d) taken along the [100] direction. b, c High-resolution TEM images of IBN9-NC taken along the [100] (b) and [110] (c) directions. e, f Energy-filtered TEM images of the particle in (d), showing carbon mapping (e) and nitrogen mapping (f). Images d–f have the same magnification as image (a); image (b) has the same magnification as image (c). CO2 adsorption isotherms at 0, 25, and 55 °C and N2 adsorption isotherm at 25 °C of IBN9-NC1 (g) and IBN-9-NC1-A (h). Reproduced from Ref. [163] by permission of The Royal Society of Chemistry

592 Porous Carbons for Carbon Dioxide Capture
materials showed high CO2 adsorption heats (ca. 40 kJ mol−1 at initial adsorption stages), suggesting an enhanced physical adsorption effect by nitrogen doping [163].
Later, they designed and prepared a series of porous carbons, including micropo-rous carbon, mesoporous carbon, which are used for selective CO2 capture. The authors found that the combination of a high N-doping concentration (>10 wt%) and extra-framework cations endowed N-doped microporous carbons with exceptional CO2 adsorption capabilities, especially at low pressures (CO2 uptake of 1.62 mmol g−1 at 25 °C and 0.1 bar) [166]. Single component adsorption isotherms indicated that its CO2/N2 selectivity was 48, which also significantly surpasses the selectivity of conven-tional carbon materials. Furthermore, the dynamical breakthrough experiments using CO2/N2 (10:90 v/v) mixtures reveal that the CO2/N2 selectivity was as high as 44, comparable to that predicted from equilibrium adsorption data. More interestingly, they conducted theoretical calculations that correlate the polarizing capabilities of various functional groups (K+, Cl− ions as well as N-containing sites, see Fig. 2.33) with their enhancement effects on CO2 adsorption and demonstrated that such effects are essen-tially based on electrostatic interactions. This represents a new perspective to explain the positive role of heteroatoms in contribution to a high CO2 uptake.
Kowalewski et al. [167] reported another type of nitrogen-enriched porous carbon nanostructure as CO2 capture materials, which has been prepared via the carboni-zation of polyacrylonitrile containing block copolymer. The typical sample exhib-ited good selectivity for CO2 manifested by sevenfold to tenfold larger amount of adsorbed CO2 over N2 (Fig. 2.34). The analysis of isosteric heats of CO2 adsorption
Fig. 2.33 Optimized configurations of CO2 adsorption on carbon clusters with different polar groups (cyan C; white H; blue N; red O; light green K) and the corresponding contour plots of the differential charge density. The contour value is ±0.001. The purple and lime regions repre-sent the charge accumulation and charge depletion regions, respectively. Reprinted with the per-mission from Ref. [166]. Copyright 2012 American Chemical Society
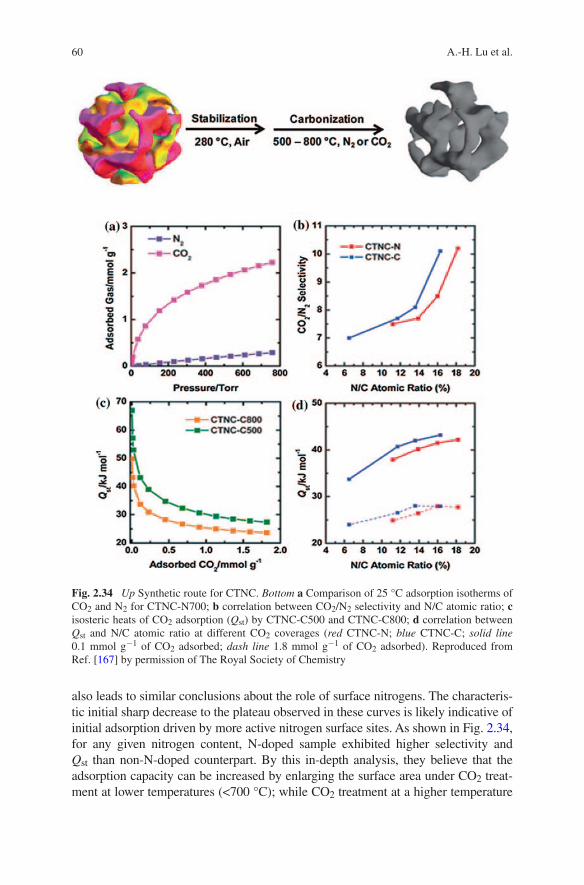
60 A.-H. Lu et al.
also leads to similar conclusions about the role of surface nitrogens. The characteris-tic initial sharp decrease to the plateau observed in these curves is likely indicative of initial adsorption driven by more active nitrogen surface sites. As shown in Fig. 2.34, for any given nitrogen content, N-doped sample exhibited higher selectivity and Qst than non-N-doped counterpart. By this in-depth analysis, they believe that the adsorption capacity can be increased by enlarging the surface area under CO2 treat-ment at lower temperatures (<700 °C); while CO2 treatment at a higher temperature
Fig. 2.34 Up Synthetic route for CTNC. Bottom a Comparison of 25 °C adsorption isotherms of CO2 and N2 for CTNC-N700; b correlation between CO2/N2 selectivity and N/C atomic ratio; c isosteric heats of CO2 adsorption (Qst) by CTNC-C500 and CTNC-C800; d correlation between Qst and N/C atomic ratio at different CO2 coverages (red CTNC-N; blue CTNC-C; solid line 0.1 mmol g−1 of CO2 adsorbed; dash line 1.8 mmol g−1 of CO2 adsorbed). Reproduced from Ref. [167] by permission of The Royal Society of Chemistry

612 Porous Carbons for Carbon Dioxide Capture
(800 °C) produced even more significant increase in surface area and CO2 capacity but reduced selectivity, due to the loss of nitrogen.
Recently, Nandi and coworkers have fabricated a series of highly porous N-doped porous carbon monoliths as CO2 capture materials. This series of N-doped carbons were obtained from the mesoporous PAN monolith via thermal treatment in two steps (Fig. 2.35) [164]. The monoliths were pre-treated in air at 503 K for activation, which led to cyclization inside the polymer framework, generating a ladder polymer. Then, the ladder polymer gradually underwent aromatization, generating an aromatic lad-der. In the next heating step, the aromatized polymer was converted to its carbon with a lamellar phase by carbonization in Ar or an Ar-CO2 mixture. CO2 adsorption iso-therms (Fig. 2.35) show reversible adsorption characteristics indicating weak interac-tion of CO2 molecules with the pore walls. More impressively, these carbon monoliths show unprecedentedly high CO2 uptake of 5.14 mmol g−1 at ambient pressure and temperature and 11.51 mmol g−1 at ambient pressure and 0 °C. As shown in Fig. 2.35, the typical sample shows high initial Qst values of up to 65.2 kJ mol−1. High initial isosteric heats of adsorption (Qst) values indicate strong adsorbent–adsorbate interac-tion between the N-containing carbon framework and CO2 molecules.
2.6.2.2 High-Temperature Reaction and Transformation
Treating as-made porous carbons with gaseous ammonia under a high temperature (e.g., 900 °C) is another popular way for preparation of N-doped carbon. In prin-ciple, the reaction with ammonia is expected to take place at carboxylic acid sites formed by the oxidation of side groups and the ring system. At a high tempera-ture, ammonia decomposes with the formation of radicals, such as NH2, NH, and H [168, 169]. These radicals may react with the carbon surface to form functional groups, such as –NH2, –CN, pyridinic, pyrrolic, and quaternary nitrogen.
Many researchers believe that the introduction of nitrogen will increase basic-ity of carbons and thus will facilitate the removal of trace amounts of acidic gases including CO2. For example, Przepiórski et al. [170] found that high-temperature ammonia treatment of activated carbon clearly enhance the CO2 adsorption. In their work, the ammonia treatment was performed for 2 h at elevated temperatures rang-ing from 200 to 1,000 °C. The CO2 capture tests confirm that the adsorption of CO2 was enhanced by ammonia treatment. The enhancement was attributed to the pres-ence of C–N and C=N groups. And further, the largest CO2 uptake was found to be at 400 °C of the ammonia treatment temperature. From their opinion, the higher tem-perature treatment may cause the close of micropores or changes in the size of pores.
Pevida et al. [171] demonstrated that ammonia treatment at temperatures higher than 600 °C incorporated nitrogen mainly into aromatic rings, while at lower tem-peratures nitrogen was introduced into more labile functionalities, such as amide-like functionalities. The CO2 capture capacities at 25 °C of the treated carbons increased with respect to the parent carbons. In particular, after ammonia modifica-tion at 800 °C, the CO2 capture capacities of wood-derived carbons rose from 7.0 to 8.4 wt%. It is worth pointing out that it is the specific nitrogen functionalities rather

62 A.-H. Lu et al.
than the total nitrogen content that are responsible for increasing the CO2-adsorbent affinity. The same group also investigated the ammonia treatment of pristine carbons in the presence of air (ammoxidation) [172]. They found that, during ammoxidation (amination in the presence of air), the formation of nitriles and amide-like function-alities were favoured and a great amount of nitrogen was incorporated onto the car-bon surface. Further, nitrogen uptake by ammoxidation at 300 °C was found to be
Fig. 2.35 a Carbonization of PAN monolith. b CO2 sorption up to 1 bar, at 273 K and 298 K: adsorp-tion (filled symbols) and desorption (empty symbols). c Isosteric heat of CO2 adsorption (Qst) as a func-tion of CO2 adsorbed. Reproduced from Ref. [164] by permission of The Royal Society of Chemistry

632 Porous Carbons for Carbon Dioxide Capture
proportional to the oxygen content of the starting carbon. Ammonia seems to react preferentially with the CO2-evolving groups of the starting carbon, such as carbox-yls, while the remaining oxygen mainly forms part of CO-evolving groups, such as amides or lactams. CO2 capture result shows that CO2 capture capacity is related to the narrow micropore volume of the samples, and this relationship is approximately linear at room temperature (Fig. 2.36). However, above room temperature, the trend deviates from linearity due to the possible influence of surface basicity.
Fig. 2.36 Relationship between the CO2 capture capacity and: a the narrow micropore volume, W0, and b the point of zero charge, pHPZC. Reprinted from Ref. [172]. Copyright 2010, with per-mission from Elsevier

64 A.-H. Lu et al.
2.6.2.3 Impregnation with Liquid Amines
Inspired with silica-based hybrid sorbents (molecular basket) with grafted or impregnated amine groups on porous silica substrates [173, 174], Zhao et al. reported an aminated adsorbents generated from sustainable biomass (glucose) [175]. Two steps are involved in this synthesis: (1) HTC of glucose; (2) transfor-mation into porous carbon–amine composite by a post-synthetic modification with a branched tetramine. The authors first prepared the substrate carbons with novel raspberry morphology, which may be beneficial for the loading of liquid amines. Interestingly, the raspberry morphology is maintained after modification of the car-bons with grafted polyamines, as illustrated in the SEM images in Fig. 2.37.
CO2 capture results show a very high CO2 uptake (up to 4.3 mmol g−1) at −20 °C (Fig. 2.37). More importantly, this type of composites delivered a very high CO2 selectivity at low (–20 °C, CO2/N2 of 65–85) and high (70 °C, CO2/N2 of 90–110) temperatures. These high capacities and selectivities are consistent
Fig. 2.37 SEM images of amine-rich carbonaceous materials: a HC-2AA-NH2, and b HC-10AA-NH2. The numbers 2 and 10 refer to the amounts of acrylic acid (AA) relative to the total amount of glucose (in wt%). Temperature- and pressure-dependent CO2 uptake of c HC-2AA-NH2 and d HC-10AA-NH2. The lines are numerical estimates using Freundlich models (one- or two-site). 1 torr = 1.333 × 102 Pa. Reproduced from Ref. [175] by permission of John Wiley & Sons Ltd

652 Porous Carbons for Carbon Dioxide Capture
with the high amine loadings. The high capacity is remarkable given that the pre-pared absorbents have only a moderately large specific surface area. Clearly, some of the CO2 absorbs within the amine-rich, liquid-grafted surface layer. Thus, a more ideal CO2 capture material can be imagined with a high active amine content combined with a more optimized channel.
It should be noted that the introduction of liquid amines through impregnation might result in some other negative effects, such as blockage of pores, the unstable basic sites on the surface in long time cycling. To address this issue, Tour’s group developed a route to synthesize polymer–mesocarbon composites that would lead to higher degrees of CO2 adsorption by the in situ polymerization of amine species to produce polyethylenimine (PEI) and polyvinylamine (PVA) inside the meso-carbon CMK-3 (Fig. 2.38) [176]. This structured composites exhibit high stabil-ity due to the formation of interpenetrating composite frameworks between the entrapped polymers and mesocarbon CMK-3. CO2 uptake measurements showed that the 39 % PEI-CMK-3 composite had ca. 12 wt% CO2 uptake capacity and the 37 % PVA-CMK-3 composite had ca. 13 wt% CO2 uptake capacity at 30 °C and 1 atm. More importantly, the composite can easily be regenerated at 75 °C and cycles stably (even up to 500 min, see Fig. 2.38).
Besides the most commonly investigated N-containing functional groups, the oxygen-containing or sulfur-containing functional groups were also investigated
Fig. 2.38 a Synthesis processes to produce mesoporous polymer–carbon composites PEI-CMK-3 and PVA-CMK-3. b Sorption cycles of CO2 studied by TGA at 30 °C on the 37 % PVA-CMK-3 sorbent. Reprinted with the permission from Ref. [176]. Copyright 2011 American Chemical Society

66 A.-H. Lu et al.
to enhance CO2 adsorption in microporous carbon materials, particularly in the absence of water vapor, and the hydrated graphite was found to hinder CO2 adsorp-tion [177]. Liu and Wilcox [178] theoretically analyzed the role of oxygen-con-taining groups in CO2 capture based on an assumption, in which complex pore structures for natural organic materials (e.g., coal and gas shale) and carbon-based porous materials are modeled as a collection of independent, non-interconnected, functionalized graphitic slit pores with surface heterogeneities. Electronic structure calculations coupled with van der Waals-inclusive corrections have been performed to investigate the electronic properties of functionalized graphitic surfaces. With Bader charge analysis, electronic structure calculations can provide the initial frame-work comprising both the geometry and corresponding charge information required to carry out statistical modeling. Grand canonical Monte Carlo simulations were car-ried out to determine the adsorption isotherms for a given adsorbent–adsorbate inter-action at temperature/pressure conditions relevant to carbon capture applications to focus on the effect of the surface functionalities.
With the above-mentioned simulation techniques, the authors summarized the results in Fig. 2.39. The main points include: (1) the CO2 molecules are more organized and aligned when they are adsorbed in the functionalized slit pores, and thus, the adsorption capacity is enhanced by the higher efficient side-by-side pack-ing; (2) in the ultramicropores (less than 7 Å), due to the overlapping potentials from the strong pore wall−wall interactions and the strong CO2−wall interaction, the condensed adsorbed-CO2 density is even higher than that of the larger pores.
In general, as the pore width decreases, the surface functionalities dictate the adsorption, and thus, the surface functionalities play a more important role in increasing the CO2 adsorption capacity. The surface heterogeneity changes the adsorbates’ accumulation configuration by changing the geometry of the pore surface and the charge distribution of the surface, which is consistent with the Bader charge results of DFT study. Also, other molecular-level simulations of CO2 adsorption behavior in micropores of porous carbons show that heteroatom doping greatly enhances CO2 uptake and selectivity at low coverage [179]; while the CO2 capture performance of porous carbons at high pressure is largely dependent on their structure parameters.
2.7 Summary and Outlook
In summary, porous carbon-based materials for CO2 capture have experienced rapid development in the last several decades and will continue to blossom. The requirements of CO2 captures vary a lot depending on different processes, namely post-combustion (low pressure, predominantly CO2/N2 separation), pre-combus-tion (high pressure, predominantly CO2/H2 separation) capture and natural gas sweetening (predominantly CO2/CH4 separation). Thus, various kinds of new car-bon materials with defined textural properties as well as tailored surface chemistry have been synthesized for a specific CO2 capture process. Another advantage lies

672 Porous Carbons for Carbon Dioxide Capture
Fig. 2.39 Comparisons of CO2 adsorbed in functionalized micropores with that in the perfect graphite slit pore: left side views of adsorbed CO2 in various functionalized graphite slit pores with pore width of 9.2 Å; right side views of adsorbed CO2 in various functionalized graphite slit pores with pore width of 20 Å. Reprinted with the permission from Ref. [178]. Copyright 2012 American Chemical Society

68 A.-H. Lu et al.
in the relatively hydrophobic properties, which ensure a high capacity even under hydrated condition. Although much progress has been made, many challenges still exist from both scientific and technological points of view.
Firstly, there are completely irregular features over nanometer length scale. Researchers should pay more attention to the in-depth understanding of the phys-ics and chemistry of carbon, which is highly important for the design and syn-thesis of tailored porous carbons as next-generation CO2 capture materials and model carbons for fundamental investigations. For example, the important scien-tific issues may include the contribution of pores in different length scale (micro-, meso-, and macropores) to CO2 capture capability, the CO2 molecules trans-port behavior in pore systems, and so on. These depend on the development of advanced characterization methods, which can support, guide and provide further refinement to the most promising structures. For instance, the solid-state NMR techniques can provide reliable information regarding CO2 diffusion at molecu-lar scale. From this measurement, researchers can correlate microscopic absorbate dynamics with the structural information on the basis of one kind of carbon model after CO2 loading. By comparing the behaviors of the microscopic mobility and the macroscopic diffusion, one can get an insight into the mechanism of selective transport through these materials.
Secondly, the mechanism as well as the contribution of surface heterogeneity (e.g., N-, S-doped sites) for CO2 binding is controversial. Up to now, the reported carbonaceous CO2 capture materials show great complexity in pore structures and, in particular, in composition and surface chemistry (functional groups binding and dispersion in accessible surface). Thus, this condition put a big obstacle to achieve a reliable understanding of the specific contribution of each parameter. Therefore, along with developed characterizations, computational predication based on an ideal structure may be also needed.
Thirdly, the evaluation for CO2 capture should be conducted under a simulated or even an actual flue gas condition, rather than the most often used equilibrium CO2 sorption for the current studies. In our opinion, the international facilities in this field should create the benchmarking materials and further the prototypical materials column, which can be used for the evaluation of the CO2 capture perfor-mance of newly emerging capture materials on the same standards. Beyond these considerations, the engineering economics of the new materials must be evaluated upon the scale-up of the materials for industrial applications, and economic mod-els must be established to cover lifecycle CO2 separation, capture, and sequestra-tion costs for various technologies.
Acknowledgments This work was financially supported by the National Natural Science Foundation of China (No. 21225312) and the National Basic Research Program of China (No. 2013CB934104).

692 Porous Carbons for Carbon Dioxide Capture
References
1. Rodríguez-Reinoso F (1998) The role of carbon materials in heterogeneous catalysis. Carbon 36:159–175
2. Kyotani T (2006) Synthesis of various types of nano carbons using the template technique. Bull Chem Soc Jpn 79:1322–1337
3. Himeno S, Komatsu T, Fujita S (2005) High-pressure adsorption equilibria of methane and carbon dioxide on several activated carbons. J Chem Eng Data 50:369–376
4. Olivares-Marín M, Maroto-Valer M (2012) Development of adsorbents for CO2 capture from waste materials: a review. Greenhouse Gas Sci Technol 2:20–35
5. Wang R, Wang P, Yan X, Lang J, Peng C, Xue Q (2012) Promising porous carbon derived from celtuce leaves with outstanding supercapacitance and CO2 capture performance. ACS Appl Mater Interfaces 4:5800–5806
6. Xing W, Liu C, Zhou Z, Zhang L, Zhou J, Zhuo S, Yan Z, Gao H, Wang G, Qiao SZ (2012) Superior CO2 uptake of N-doped activated carbon through hydrogen-bonding interaction. Energy Environ Sci 5:7323–7327
7. Wang J, Heerwig A, Lohe MR, Oschatz M, Borchardt L, Kaskel S (2012) Fungi-based porous carbons for CO2 adsorption and separation. J Mater Chem 22:13911–13913
8. Shen W, He Y, Zhang S, Li J, Fan W (2012) Yeast-based microporous carbon materials for carbon dioxide capture. ChemSusChem 5:1274–1279
9. Marco-Lozar JP, Kunowsky M, Suárez-García F, Carruthers JD, Linares-Solano A (2012) Activated carbon monoliths for gas storage at room temperature. Energy Environ Sci 5:9833–9842
10. Thote JA, Iyer KS, Chatti R, Labhsetwar NK, Biniwale RB, Rayalu SS (2010) In situ nitro-gen enriched carbon for carbon dioxide capture. Carbon 48:396–402
11. Sevilla M, Valle-Vigón P, Fuerte AB (2011) N-doped polypyrrole-based porous carbons for CO2 capture. Adv Funct Mater 21:2781–2787
12. White RJ, Budarin V, Luque R, Clark JH, Macquarrie DJ (2009) Tuneable porous carbona-ceous materials from renewable resources. Chem Soc Rev 38:3401–3418
13. Grzyb B, Hildenbrand C, Berthon-Fabry S, Bégin D, Job N, Rigacci A, Achard P (2010) Functionalisation and chemical characterisation of cellulose-derived carbon aerogels. Carbon 48:2297–2307
14. ElKhatat AM, Al-Muhtaseb SA (2011) Advances in tailoring resorcinol-formaldehyde organic and carbon gels. Adv Mater 23:2887–2903
15. Titirici MM, Thomas A, Antonietti M (2007) Back in the black: hydrothermal carbonization of plant material as an efficient chemical process to treat the CO2 problem? New J Chem 31:787–789
16. Titirici MM, Antonietti M, Baccile N (2008) Hydrothermal carbon from biomass: a com-parison of the local structure from poly-to monosaccharides and pentoses/hexoses. Green Chem 10:1204–1212
17. Titirici MM, White RJ, Falco C, Sevilla M (2012) Black perspectives for a green future: hydrothermal carbons for environment protection and energy storage. Energy Environ Sci 5:6796–6822
18. Sevilla M, Fuertes AB (2011) Sustainable porous carbons with a superior performance for CO2 capture. Energy Environ Sci 4:1765–1771
19. Sevilla M, Falco C, Titirici M-M, Fuertes AB (2012) High-performance CO2 sorbents from algae. RSC Adv 2:12792–12797
20. Gaweł B, Gaweł K, Øye G (2010) Sol–gel synthesis of non-silica monolithic materials. Materials 3:2815–2833
21. Kadib AE, Chimenton R, Sachse A, Fajula F, Galarneau A, Coq B (2009) Functionalized inorganic monolithic microreactors for high productivity in fine chemicals catalytic synthe-sis. Angew Chem Int Ed 48:4969–4972
22. Davis ME (2002) Ordered porous materials for emerging applications. Nature 417:813–821

70 A.-H. Lu et al.
23. Yuan Z-Y, Su B-L (2006) Insights into hierarchically meso–macroporous structured materi-als. J Mater Chem 16:663–667
24. Lu AH, Hao GP (2013) Porous materials for carbon dioxide capture. Annu Rep Sect A: Inorg Chem 109:484–503
25. Lu A-H, Schüth F (2006) Nanocasting: a versatile strategy for creating nanostructured porous materials. Adv Mater 18:1793–1805
26. Lee J, Kim J, Hyeon T (2006) Recent progress in the synthesis of porous carbon materials. Adv Mater 18:2073–2094
27. Hoheisel TN, Schrettl S, Szilluweit R, Frauenrath H (2010) Nanostructured carbonaceous materials from molecular precursors. Angew Chem Int Ed 49:6496–6515
28. Tao Y, Endo M, Kaneko K (2009) Hydrophilicity-controlled carbon aerogels with high mes-oporosity. J Am Chem Soc 131:904–905
29. Silva AMT, Machado BF, Figueiredo JL, Faria JL (2009) Controlling the surface chemistry of carbon xerogels using HNO3 hydrothermal activation. Carbon 47:1670–1679
30. Stein A, Wang Z, Fierke MA (2009) Functionalization of porous carbon materials with designed pore architecture. Adv Mater 21:265–293
31. Biener J, Stadermann M, Suss M, Worsley MA, Biener MM, Rose KA, Baumann TF (2011) Advanced carbon aerogels for energy applications. Energy Environ Sci 4:656–667
32. Pekala RW (1989) Organic aerogels from the polycondensation of resorcinol with formalde-hyde. J Mater Sci 24:3221–3227
33. Fairén-Jiménez D, Carrasco-Marín F, Moreno-Castilla C (2008) Inter- and intra-primary-particle structure of monolithic carbon aerogels obtained with varying solvents. Langmuir 24:2820–2825
34. Gutiérrez MC, Rubio F, del Monte F (2010) Resorcinol-formaldehyde polycondensation in deep eutectic solvents for the preparation of carbons and carbon−carbon nanotube compos-ites. Chem Mater 22:2711–2719
35. Carriazo D, Gutiérrez MC, Ferrer ML, del Monte F (2010) Resorcinol-based deep eutectic solvents as both carbonaceous precursors and templating agents in the synthesis of hierar-chical porous carbon monolith. Schem Mater 22:6146–6152
36. Mulik S, Sotiriou-Leventis C, Leventis N (2008) Macroporous electrically conducting car-bon networks by pyrolysis of isocyanate-cross-linked resorcinol-formaldehyde aerogels. Chem Mater 20:6985–6997
37. Leventis N, Sotiriou-Leventis C, Chandrasekaran N, Mulik S, Larimore ZJ, Lu H, Churu G, Mang JT (2010) Multifunctional polyurea aerogels from isocyanates and water. A structure-property case study. Chem. Mater. 22:6692–6710
38. Chidambareswarapattar C, Larimore Z, Sotiriou-Leventis C, Mang JT, Leventis N (2010) One-step room-temperature synthesis of fibrous polyimide aerogels from anhydrides and isocyanates and conversion to isomorphic carbons. J Mater Chem 20:6978–9666
39. Wan Y, Qian X, Jia N, Wang Z, Li H, Zhao D (2008) Direct triblock-copolymer-templating synthesis of highly ordered fluorinated mesoporous carbon. Chem Mater 20:1012–1018
40. Sepehri S, García BB, Zhang Q, Cao G (2009) Enhanced electrochemical and structural properties of carbon cryogels by surface chemistry alteration with boron and nitrogen. Carbon 47:1436–1443
41. Hao G-P, Li W-C, Qian D, Lu A-H (2010) Rapid synthesis of nitrogen-doped porous carbon monolith for CO2 capture. Adv Mater 22:853–857
42. Fang Y, Gu D, Zou Y, Wu Z, Li F, Che R, Deng Y, Tu B, Zhao D (2010) A low-concentration hydrothermal synthesis of biocompatible ordered mesoporous carbon nanospheres with tun-able and uniform size. Angew Chem Int Ed 49:7987–7991
43. Liu J, Qiao SZ, Liu H, Chen J, Orpe A, Zhao D, Lu GQ (2011) Extension of the Stöber method to the preparation of monodisperse resorcinol-formaldehyde resin polymer and car-bon spheres. Angew Chem Int Ed 50:5947–5951
44. Tien BM, Xu MW, Liu JF (2010) Synthesis and electrochemical characterization of carbon spheres as anode material for lithium-ion battery. Mater Lett 64:1465–1467
45. Horikawa T, Hayashi J, Muroyama K (2004) Size control and characterization of spherical carbon aerogel particles from resorcinol–formaldehyde resin. Carbon 42:169–175

712 Porous Carbons for Carbon Dioxide Capture
46. Fujikawa D, Uota M, Sakai G, Kijima T (2007) Shape-controlled synthesis of nanocarbons from resorcinol–formaldehyde nanopolymers using surfactant-templated vesicular assem-blies. Carbon 45:1289–1295 (Original Research Article)
47. Liu L, Deng QF, Hou XX, Yuan ZY (2012) User-friendly synthesis of nitrogen-contain-ing polymer and microporous carbon spheres for efficient CO2 capture. J Mater Chem 22:15540–15548
48. Gu JM, Kim WS, Hwang YK, Huh S (2013) Template-free synthesis of N-doped porous carbons and their gas sorption properties. Carbon 56:208–217
49. Liang CD, Hong KL, Guiochon GA, Mays JW, Dai S (2004) Synthesis of a large-scale highly ordered porous carbon film by self-assembly of block copolymers. Angew Chem Int Ed 43:5785–5789
50. Valkama S, Nykänen A, Kosonen H, Ramani R, Tuomisto F, Engelhardt P, Brinke G, Ikkala O, Ruokolainen J (2007) Hierarchical porosity in self-assembled polymers: post-modifica-tion of block copolymer-phenolic resin complexes by pyrolysis allows the control of micro- and mesoporosity. Adv Funct Mater 17:183–190
51. Liang CD, Dai S (2006) Synthesis of mesoporous carbon materials via enhanced hydrogen-bonding interaction. J Am Chem Soc 128:5316–5317
52. Saha D, Deng S (2010) Adsorption equilibrium and kinetics of CO2, CH4, N2O, and NH3 on ordered mesoporous carbon. J Colloid Interface Sci 345:402–409
53. Wang X, Liang C, Dai S (2008) Facile synthesis of ordered mesoporous carbons with high thermal stability by self-assembly of resorcinol-formaldehyde and block copolymers under highly acidic conditions. Langmuir 24:7500–7505
54. Liang C, Dai S (2009) Dual phase separation for synthesis of bimodal meso-/macroporous carbon monoliths. Chem Mater 21:2115–2124
55. Huang Y, Cai H, Feng D, Gu D, Deng Y, Tu B, Wang H, Webley PA, Zhao D (2008) One-step hydrothermal synthesis of ordered mesostructured carbonaceous monoliths with hierar-chical porosities. Chem Commun 23:2641–2643
56. Wei J, Zhou D, Sun Z, Deng Y, Xia Y, Zhao D (2013) A controllable synthesis of rich nitro-gen-doped ordered mesoporous carbon for CO2 capture and supercapacitors. Adv Funct Mater 23:2322–2328
57. Liu L, Wang F-Y, Shao G-S, Yuan Z-Y (2010) A low-temperature autoclaving route to syn-thesize monolithic carbon materials with an ordered mesostructure. Carbon 48:2089–2099
58. Gutiérrez MC, Picó F, Rubio F, Amarilla JM, Palomares FJ, Ferrer ML, Monte F, Rojo JM (2009) PPO15-PEO22-PPO15 block copolymer assisted synthesis of monolithic macro- and microporous carbon aerogels exhibiting high conductivity and remarkable capacitance. J Mater Chem 19:1236–1240
59. Zhao X, Wang A, Yan J, Sun G, Sun L, Zhang T (2010) Synthesis and electrochemical perfor-mance of heteroatom-incorporated ordered mesoporous carbons. Chem Mater 22:5463–5473
60. Hao G-P, Li W-C, Wang S, Wang G-H, Qi L, Lu A-H (2011) Lysine-assisted rapid synthesis of crack-free hierarchical carbon monoliths with a hexagonal array of mesopores. Carbon 49:3762–3772
61. Hao G-P, Li W-C, Qian D, Wang G-H, Zhang W-P, Zhang T, Wang A-Q, Schüth F, Bongard H-J, Lu A-H (2011) Structurally designed synthesis of mechanically stable poly(benzoxazine-co-resol)-based porous carbon monoliths and their application as high-performance CO2 capture sorbents. J Am Chem Soc 133:11378–11388
62. Wang Z, Li F, Ergang NS, Stein A (2006) Effects of hierarchical architecture on electronic and mechanical properties of nanocast monolithic porous carbons and carbon−carbon nano-composites. Chem Mater 18:5543–5553
63. Deng Y, Liu C, Yu T, Liu F, Zhang F, Wan Y, Zhang L, Wang C, Tu B, Webley PA, Wang H, Zhao D (2007) Facile synthesis of hierarchically porous carbons from dual colloidal crystal/block copolymer template approach. Chem Mater 19:3271–3277
64. Xue C, Tu B, Zhao D (2008) Evaporation-induced coating and self-assembly of ordered mesoporous carbon-silica composite monoliths with macroporous architecture on polyure-thane foams. Adv Funct Mater 18:3914–3921

72 A.-H. Lu et al.
65. Wei H, Lv Y, Han L, Tu B, Zhao D (2011) Facile synthesis of transparent mesostructured composites and corresponding crack-free mesoporous carbon/silica monoliths. Chem Mater 23:2353–2360
66. Liu CY, Li LX, Song HH, Chen XH (2007) Facile synthesis of ordered mesoporous carbons from F108/resorcinol–formaldehyde composites obtained in basic media. Chem Commun 7:757–759
67. Feng D, Lv YY, Wu ZX, Dou YQ, Han L, Sun ZK, Xia YY, Zheng GF, Zhao DY (2011) Free-standing mesoporous carbon thin films with highly ordered pore architectures for nan-odevices. J Am Chem Soc 133:15148–15150
68. Rodriguez AT, Li XF, Wang J, Steen WA, Fan HY (2007) Facile synthesis of nanostructured carbon through self-assembly between block copolymers and carbohydrates. Adv Funct Mater 17:2710–2716
69. Meng Y, Gu D, Zhang FQ, Shi YF, Yang HF, Li Z, Yu CZ, Tu B, Zhao DY (2005) Ordered mesoporous polymers and homologous carbon frameworks: amphiphilic surfactant templat-ing and direct transformation. Angew Chem Int Ed 44:7053–7059
70. Yoshimune M, Yamamoto T, Nakaiwa M, Haraya K (2008) Preparation of highly mesoporous carbon membranes via a sol–gel process using resorcinol and formaldehyde. Carbon 46:1031–1036
71. Hao G-P, Jin Z-Y, Sun Q, Zhang X-Q, Zhang J-T, Lu A-H (2013) Porous carbon nanosheets with precisely tunable thickness and selective CO2 adsorption properties. Energy Environ Sci 2013(6):3740–3747
72. Sun X, Li Y (2005) Hollow carbonaceous capsules from glucose solution. J Colloid Interface Sci 291:7–12
73. Li Y, Chen J, Xu Q, He L, Chen Z (2009) Controllable route to solid and hollow monodis-perse carbon nanospheres. J Phys Chem C 113:10085–10089
74. Liu J, Qiao SZ, Liu H, Chen J, Orpe A, Zhao D, Lu GQ (2011) Extension of the Stöber method to the preparation of monodisperse resorcinol-formaldehyde resin polymer and car-bon spheres. Angew Chem Int Ed 50:5947–5951
75. Wickramaratne NP, Jaroniec M (2013) Activated carbon spheres for CO2 adsorption. ACS Appl Mater Interfaces 5:1849–1855
76. Wickramaratne NP, Jaroniec M (2013) Importance of small micropores in CO2 capture by phenolic resin-based activated carbon spheres. J Mater Chem A 1:112–116
77. Wickramaratne NP, Perera VS, Ralph JM, Huang SD, Jaroniec M (2013) Cysteine-assisted tailoring of adsorption properties and particle size of polymer and carbon spheres. Langmuir 29(12):4032–4038
78. Zeng Q, Wu D, Zou C, Xu F, Fu R, Li Z, Liang Y, Su D (2010) Template-free fabrication of hierarchical porous carbon based on intra-/inter-sphere crosslinking of monodisperse sty-rene–divinylbenzene copolymer nanospheres. Chem Commun 46:5927–5929
79. Jiang P, Bertone JF, Colvin VL (2001) A lost-wax approach to monodisperse colloids and their crystals. Science 291:453–457
80. Lu A-H, Hao G-P, Sun Q (2011) Can carbon spheres be created through the Stöber method? Angew Chem Int Ed 50:9023–9025
81. Wang S, Li W-C, Hao G-P, Hao Y, Sun Q, Zhang X-Q, Lu A-H (2011) Temperature-programmed precise control over the sizes of carbon nanospheres based on benzoxazine chemistry. J Am Chem Soc 133:15304
82. Nakanishi K, Tanaka N (2007) Sol–gel with phase separation. Hierarchically porous mate-rials optimized for high-performance liquid chromatography separations. Acc Chem Res 40:863
83. Brun N, Prabaharan SRS, Morcrette M, Sanchez C, Pécastaings G, Derré A, Soum A, Deleuze H, Birot M, Backov R (2009) Hard macrocellular silica Si(HIPE) foams templating micro/macroporous carbonaceous monoliths: applications as lithium ion battery negative electrodes and electrochemical capacitors. Adv Funct Mater 19:3136
84. Alvarez S, Esquena J, Solans C, Fuertes AB (2004) Meso/macroporous carbon monoliths from polymeric foams. Adv Eng Mater 6:897

732 Porous Carbons for Carbon Dioxide Capture
85. Yang H, Shi Q, Liu X, Xie S, Jiang D, Zhang F, Yu C, Tu B, Zhao D (2002) Synthesis of ordered mesoporous carbon monoliths with bicontinuous cubic pore structure of Ia3d sym-metry. Chem Commun 23:2842
86. Wang X, Bozhilov KN, Feng P (2006) Facile preparation of hierarchically porous carbon monoliths with well-ordered mesostructures. Chem Mater 18:6373–6381
87. Xia Y, Mokaya R (2007) Ordered mesoporous carbon monoliths: CVD nanocasting and hydrogen storage properties. J Phys Chem C 111:10035–10039
88. Taguchi A, Smått J-H, Lindén M (2003) Carbon monoliths possessing a hierarchical, fully interconnected porosity. Adv Mater 15:1209–1211
89. Lu A-H, Smått J-H, Lindén M (2005) Combined surface and volume templating of highly porous nanocast carbon monoliths. Adv Func Mater 15:865–871
90. Lu A-H, Smått J-H, Backlund S, Lindén M (2004) Easy and flexible preparation of nano-casted carbon monoliths exhibiting a multimodal hierarchical porosity. Microporous Mesoporous Mater 72:59–65
91. Shi Z-G, Feng Y-Q, Xu L, Da S-L, Zhang M (2003) Synthesis of a carbon monolith with trimodal pores. Carbon 41:2677–2679
92. Hu Y-S, Adelhelm P, Smarsly BM, Hore S, Antonietti M, Maier J (2007) Synthesis of hier-archically porous carbon monoliths with highly ordered microstructure and their application in rechargeable lithium batteries with high-rate capability. Adv Funct Mater 17:1873–1878
93. Liu N, Yin L, Wang C, Zhang L, Lun N, Xiang D, Qi Y, Gao R (2010) Adjusting the tex-ture and nitrogen content of ordered mesoporous nitrogen-doped carbon materials prepared using SBA-15 silica as a template. Carbon 48:3579–3591
94. Han B-H, Zhou W, Sayari A (2003) Direct preparation of nanoporous carbon by nanocast-ing. J Am Chem Soc 125:3444–3445
95. Vinu A (2008) Two-dimensional hexagonally-ordered mesoporous carbon nitrides with tun-able pore diameter, surface area and nitrogen content. Adv Funct Mater 18:816–827
96. Li Q, Yang J, Feng D, Wu Z, Wu Q, Park SS, Ha CS, Zhao D (2010) Facile synthesis of porous carbon nitride spheres with hierarchical three-dimensional mesostructures for CO2 capture. Nano Res 3:632–642
97. Nishihara H, Kyotani T (2012) Templated nanocarbons for energy storage. Adv Mater 24:4473–4498
98. Pachfule P, Biswal BP, Banerjee R (2012) Control of porosity by using isoreticular zeolitic imidazolate frameworks (IRZIFs) as a template for porous carbon synthesis. Chem Eur J 18:11399–11408
99. Deng H, Jin S, Zhan L, Wang Y, Lu B, Qiao W, Ling L (2012) Synthesis of porous carbons derived from metal-organic coordination polymers and their adsorption performance for car-bon dioxide. New Carbon Mater 27:194–199
100. Almasoudi A, Mokaya R (2012) Preparation and hydrogen storage capacity of templated and activated carbons nanocast from commercially available zeolitic imidazolate frame-work. J Mater Chem 22:146–152
101. Hu M, Reboul J, Furukawa S, Torad NL, Ji Q, Srinivasu P, Ariga K, Kitagawa S, Yamauchi Y (2012) Direct carbonization of Al-based porous coordination polymer for synthesis of nanoporous carbon. J Am Chem Soc 134:2864–2867
102. Yang SJ, Kim T, Im JH, Kim YS, Lee K, Jung H, Park CR (2012) MOF-derived hierarchically porous carbon with exceptional porosity and hydrogen storage capacity. Chem Mater 24:464–470
103. Chaikittisilp W, Hu M, Wang H, Huang H-S, Fujita T, Wu KC-W, Chen L-C, Yamauchi Y, Ariga K (2012) Nanoporous carbons through direct carbonization of a zeolitic imidazolate framework for supercapacitor electrodes. Chem Commun 48:7259–7261
104. Lim S, Suh K, Kim Y, Yoon M, Park H, Dybtsev DN, Kim K (2012) Porous carbon mate-rials with a controllable surface area synthesized from metal–organic frameworks. Chem Commun 48:7447–7449
105. Ben T, Li Y, Zhu L, Zhang D, Cao D, Xiang Z, Yao X, Qiu S (2012) Selective adsorption of carbon dioxide by carbonized porous aromatic framework (PAF). Energy Environ Sci 5:8370–8376

74 A.-H. Lu et al.
106. Lee KT, Lytle JC, Ergang NS, Oh SM, Stein A (2005) Synthesis and rate performance of monolithic macroporous carbon electrodes for lithium-ion secondary batteries. Adv Funct Mater 15:547–556
107. Adelhelm P, Hu Y-S, Chuenchom L, Antonietti M, Smarsly BM, Maier J (2007) Generation of hierarchical meso- and macroporous carbon from mesophase pitch by spinodal decompo-sition using polymer templates. Adv Mater 19:4012–4017
108. Gierszal KP, Jaroniec M (2006) Carbons with extremely large volume of uniform mesopores synthesized by carbonization of phenolic resin film formed on colloidal silica template. J Am Chem Soc 128:10026–10027
109. Zhang S, Chen L, Zhou S, Zhao D, Wu L (2010) Facile synthesis of hierarchically ordered porous carbon via in situ self-assembly of colloidal polymer and silica spheres and its use as a catalyst support. Chem Mater 22:3433–3440
110. Fang B, Kim M-S, Kim JH, Lim S, Yu J-S (2010) Ordered multimodal porous carbon with hierarchical nanostructure for high Li storage capacity and good cycling performance. J Mater Chem 20:10253–10259
111. Liang Y, Liang F, Wu D, Li Z, Xu F, Fu R (2011) Construction of a hierarchical architec-ture in a wormhole-like mesostructure for enhanced mass transport. Phys Chem Chem Phys 13:8852–8856
112. Meng LY, Park SJ (2012) Influence of MgO template on carbon dioxide adsorption of cation exchange resin-based nanoporous carbon. J Colloid Interface Sci 366:125–129
113. Bhagiyalakshmi M, Hemalatha P, Ganesh M, Mei PM, Jang HT (2011) A direct synthesis of mesoporous carbon supported MgO sorbent for CO2 capture. Fuel 90:1662–1667
114. Czyèwski A, Kapica J, Moszynski D, Pietrzak R, Przepiórski J (2013) On competitive uptake of SO2 and CO2 from air by porous carbon containing CaO and MgO. Chem Eng J 226:348–356
115. Meng L-Y, Park S-J (2012) MgO-templated porous carbons-based CO2 adsorbents produced by KOH activation. Mater Chem Phys 137:91–96
116. Su F, Zhao XS, Wang Y, Lee JY (2007) Bridging mesoporous carbon particles with carbon nanotubes. Microporous Mesoporous Mater 98:323–329
117. Wang X, Bozhilov KN, Feng P (2006) Facile preparation of hierarchically porous carbon monoliths with well-ordered mesostructures. Chem Mater 18:6373–6381
118. Huwe H, Froeba M (2007) Synthesis and characterization of transition metal and metal oxide nanoparticles inside mesoporous carbon CMK-3. Carbon 45:304–314
119. Wikander K, Hungria AB, Midgley PA, Palmqvist AEC, Holmberg K, Thomas JM (2007) Incorporation of platinum nanoparticles in ordered mesoporous carbon. J Colloid Interface Sci 305:204–208
120. Jang JH, Han S, Hyeon T, Oh SM (2003) Electrochemical capacitor performance of hydrous ruthenium oxide/mesoporous carbon composite electrodes. J Power Sources 123:79–85
121. Kim H, Kim P, Joo JB, Kim W, Song IK, Yi J (2006) Fabrication of a mesoporous Pt-carbon catalyst by the direct templating of mesoporous Pt-alumina for the methanol electro-oxida-tion. J Power Sources 157:196–200
122. García-Martínez J, Lancaster TM, Ying JY (2008) Synthesis and catalytic applications of self-assembled carbon nanofoams. Adv Mater 20:288–292
123. Long D, Chen Q, Qiao W, Zhan L, Liang X, Ling L (2009) Three-dimensional mesoporous carbon aerogels: ideal catalyst supports for enhanced H2S oxidation. Chem Commun 26:3898–3900
124. Nielsen TK, Bösenberg U, Gosalawit R, Dornheim M, Cerenius Y, Besenbacher F, Jensen TR (2010) A reversible nanoconfined chemical reaction. ACS Nano 4:3903–3908
125. Worsley MA, Kuntz JD, Cervantes O, Han TY-J, Gash AE, Satcher JH, Baumann TF (2009) Route to high surface area TiO2/C and TiCN/C composites. J Mater Chem 19:7146–7150
126. Han TY-J, Worsley MA, Baumann TF, Satcher JH (2011) Synthesis of ZnO coated activated carbon aerogel by simple sol–gel route. J Mater Chem 21:330–333
127. Worsley MA, Kucheyev SO, Satcher JH, Hamza AV, Baumann TF (2009) Mechanically robust and electrically conductive carbon nanotube foams. Appl Phys Lett 94:073115

752 Porous Carbons for Carbon Dioxide Capture
128. Worsley MA, Pauzauskie PJ, Olson TY, Biener J, Satcher JH, Baumann TF (2010) Synthesis of graphene aerogel with high electrical conductivity. J Am Chem Soc 132:14067–14069
129. Worsley MA, Olson TY, Lee JRI, Willey TM, Nielsen MH, Roberts SK, Pauzauskie PJ, Biener J, Satcher J, Baumann TF (2011) High surface area, sp2-cross-linked three-dimen-sional graphene monoliths. J Phys Chem Lett 2:921–925
130. Jin Y, Hawkins SC, Huynh CP, Su S (2013) Carbon nanotube modified carbon composite monoliths as superior adsorbents for carbon dioxide capture. Energy Environ Sci 6:2591–2596
131. Qian D, Lei C, Hao G-P, Li W-C, Lu A-H (2012) Synthesis of hierarchical porous carbon monoliths with incorporated metal-organic frameworks for enhancing volumetric based CO2 capture capability. ACS Appl Mater Interfaces 4:6125
132. Lu A-H, Li W-C, Salabas E-L, Spliethoff B, Schüth F (2006) Low temperature catalytic pyrolysis for the synthesis of high surface area. Nanostruct Graphitic Carbon Chem Mater 18:2086–2094
133. Liang C, Dai S, Guiochon G (2003) A graphitized-carbon monolithic column. Anal Chem 75:4904–4912
134. Fulvio PF, Mayes RT, Wang X, Mahurin SM, Bauer JC, Presser V, McDonough J, Gogotsi Y, Dai S (2011) “Brick-and-Mortar” self-assembly approach to graphitic mesoporous car-bon nanocomposites. Adv Funct Mater 21:2208–2215
135. Ghosh A, Subrahmanyam KS, Krishna KS, Datta S, Govindaraj A, Pati SK, Rao CNR (2008) Uptake of H2 and CO2 by graphene. J Phys Chem C 112:15704–15707
136. Srinivas G, Burress J, Yildirim T (2012) Graphene oxide derived carbons (GODCs): synthe-sis and gas adsorption properties. Energy Environ Sci 5:6453–6459
137. Shan M, Xue Q, Jing N, Ling C, Zhang T, Yan Z, Zheng J (2012) Influence of chemical functionalization on the CO2/N2 separation performance of porous graphene membranes. Nanoscale 4:5477–5482
138. Zhao Y, Ding H, Zhong Q (2012) Preparation and characterization of aminated graphite oxide for CO2 capture. Appl Surf Sci 258:4301–4307
139. Koenig SP, Wang L, Pellegrino J, Bunch JS (2012) Selective molecular sieving through porous graphene. Nature Nanotechnol 7:728–732
140. Carrillo I, Rangel E, Magana LF (2009) Photochemical deposition of Ag nanoparticles on multiwalled carbon nanotubes. Carbon 47:2752–2760
141. Garcia-Gallastegui A, Iruretagoyena D, Gouvea V, Mokhtar M, Asiri AM, Basahel SN, Al-Thabaiti SA, Alyoubi AO, Chadwick D, Shaffer MSP (2012) Graphene oxide as support for layered double hydroxides: enhancing the CO2 adsorption capacity. Chem Mater 24:4531–4539
142. Zhou D, Liu Q, Cheng Q, Zhao Y, Cui Y, Wang T, Han B (2012) Graphene-manganese oxide hybrid porous material and its application in carbon dioxide adsorption. Chin Sci Bull 57:3059–3064
143. Chandra V, Yu SU, Kim SH, Yoon YS, Kim DY, Kwon AH, Meyyappan M, Kim KS (2012) Highly selective CO2 capture on N-doped carbon produced by chemical activation of polypyrrole functionalized graphene sheets. Chem Commun 48:735–737
144. Mishra AK, Ramaprabhu S (2012) Nanostructured polyaniline decorated graphene sheets for reversible CO2 capture. J Mater Chem 22:3708–3712
145. Iijima S (1991) Helical microtubules of graphitic carbon. Nature 354:56–58 146. Nitze F, Hamad EA, Wågberg T (2011) Well-dispersed Pd3Pt1 alloy nanoparticles in large
pore sized mesocellular carbon foam for improved methanol-tolerant oxygen reduction reac-tion. Carbon 49:1101–1107
147. Hata K, Futaba DN, Mizuno K, Namai T, Yumura M, Iijima S (2004) Water-assisted highly efficient synthesis of impurity-free single-walled carbon nanotubes. Science 306:1362–1364
148. Yang KS, Edie DD, Lim DY, Kim YM, Choi YO (2003) Preparation of carbon fiber web from electrostatic spinning of PMDA-ODA poly(amic acid) solution. Carbon 41:2039–2046
149. Kowalczyk P, Furmaniak S, Gauden PA, Terzyk AP (2010) Optimal single-walled carbon nanotube vessels for short-term reversible storage of carbon dioxide at ambient tempera-tures. J Phys Chem C 114:21465–21473
150. Mishra AK, Ramaprabhu S (2012) Polyaniline/multiwalled carbon nanotubes nanocompos-ite—an excellent reversible CO2 capture candidate. RSC Adv 2:1746–1750

76 A.-H. Lu et al.
151. Ye Q, Jiang J, Wang C, Liu Y, Pan H, Shi Y (2012) Adsorption of low-concentration carbon diox-ide on amine-modified carbon nanotubes at ambient temperature. Energy Fuels 26:2497–2504
152. Su F, Lu C, Chen H-S (2011) Adsorption, desorption, and thermodynamic studies of CO2 with high-amine-loaded multiwalled carbon nanotubes. Langmuir 27:8090–8098
153. Lu C, Bai H, Wu B, Su F, Hwang JF (2008) Comparative study of CO2 capture by carbon nanotubes, activated carbons, and zeolites. Energy Fuels 22:3050–3056
154. Dillon EP, Crouse CA, Barron AR (2008) Synthesis, characterization, and carbon dioxide adsorption of covalently attached polyethyleneimine-functionalized single-wall carbon nanotubes. ACS Nano 2:156–164
155. Liu H, Cooper VR, Dai S, Jiang D (2012) Windowed carbon nanotubes for efficient CO2 removal from natural gas. J Phys Chem Lett 3:3343–3347
156. Shen W, Zhang S, He Y, Li J, Fan W (2011) Hierarchical porous polyacrylonitrile-based activated carbon fibers for CO2 capture. J Mater Chem 21:14036–14040
157. Asai M, Ohba T, Iwanaga T, Kanoh H, Endo M, Campos-Delgado J, Terrones M, Nakai K, Kaneko K (2011) Marked adsorption irreversibility of graphitic nanoribbons for CO2 and H2O. J Am Chem Soc 133:14880–14883
158. Mantzalis D, Asproulis N (2011) Enhanced carbon dioxide adsorption through carbon nano-scrolls. Phys Rev E84:066304
159. Chmiola J, Largeot C, Taberna PL, Simon P, Gogotsi Y (2010) Monolithic carbide-derived carbon films for micro-supercapacitors. Science 328:480
160. Rose M, Korenblit Y, Kockrick E, Borchardt L, Oschatz M, Kaskel S, Yushin G (2011) Hierarchical micro- and mesoporous carbide-derived carbon as a high-performance elec-trode material in supercapacitors. Small 7:1108–1117
161. Presser V, McDonough J, Yeon S-H, Gogotsi Y (2011) Effect of pore size on carbon dioxide sorption by carbide derived carbon. Energy Environ Sci 4:3059–3066
162. Qian D, Lei C, Wang E-M, Li W-C, Lu A-H (2013) A method for creating microporous carbons with excellent CO2 adsorption capacity and selectivity. ChemSusChem. doi: 10.1002/cssc.201300585
163. Zhao Y, Zhao L, Yao KX, Yang Y, Zhang Q, Han Y (2012) Novel porous carbon materials with ultrahigh nitrogen contents for selective CO2 capture. J Mater Chem 22:19726–19731
164. Nandi M, Okada K, Dutta A, Bhaumik A, Maruyama J, Derks D, Uyama H (2012) Unprecedented CO2 uptake over highly porous N-doped activated carbon monoliths pre-pared by physical activation. Chem Commun 48:10283–10285
165. Chen C, Kim J, Ahn W-S (2012) Efficient carbon dioxide capture over a nitrogen-rich car-bon having a hierarchical micro-mesopore structure. Fuel 95:360–364
166. Zhao Y, Liu X, Yao KX, Zhao L, Han Y (2012) Superior capture of CO2 achieved by introduc-ing extra-framework cations into N-doped microporous carbon. Chem Mater 24:4725–4734
167. Zhong M, Natesakhawat S, Baltrus JP, Luebke D, Nulwala H, Matyjaszewski K, Kowalewski T (2012) Copolymer-templated nitrogen-enriched porous nanocarbons for CO2 capture. Chem Commun 48:11516–11518
168. Stohr B, Boehm HP, Schlogl R (1991) Enhancement of the catalytic activity of activated carbons in oxidation reactions by thermal treatment with ammonia or hydrogen cyanide and observation of a superoxide species as a possible intermediate. Carbon 29:707–720
169. Boehm HP, Mair G, Stoehr T, De Rincon AR, Tereczki B (1984) Carbon as a catalyst in oxi-dation reactions and hydrogen halide elimination reactions. Fuel 63:1061–1063
170. Przepiórski J, Skrodzewicz M, Morawski AW (2004) High temperature ammonia treatment of activated carbon for enhancement of CO2 adsorption. Appl Surf Sci 225:235–242
171. Pevida C, Plaza MG, Arias B, Fermoso J, Rubiera F, Pis JJ (2008) Surface modification of activated carbons for CO2 capture. Appl Surf Sci 254:7165–7172
172. Plaza MG, Rubiera F, Pis JJ, Pevida C (2010) Ammoxidation of carbon materials for CO2 capture. Appl Surf Sci 256:6843–6849
173. Angeletti E, Canepa C, Martinetti G, Venturello P (1989) Amino groups immobilized on silica gel: an efficient and reusable heterogeneous catalyst for the Knoevenagel condensa-tion. J Chem Soc 1:105–107

772 Porous Carbons for Carbon Dioxide Capture
174. Yue MB, Chun Y, Cao Y, Dong X, Zhu JH (2006) CO2 capture by as-prepared SBA-15 with an occluded organic template. Adv Funct Mater 16:1717–1722
175. Zhao L, Bacsik Z, Hedin N, Wei W, Sun Y, Antonietti M, Titirici MM (2010) Carbon dioxide capture on amine-rich carbonaceous materials derived from glucose. ChemSusChem 3:840–845
176. Hwang CC, Jin Z, Lu W, Sun Z, Alemany LB, Lomeda JR, Tour JM (2011) In situ synthesis of polymer-modified mesoporous carbon CMK-3 composites for CO2 sequestration. ACS Appl Mater Interfaces 3:4782–4786
177. Xia Y, Zhu Y, Tang Y (2012) Preparation of sulfur-doped microporous carbons for the stor-age of hydrogen and carbon dioxide. Carbon 50:5543–5553
178. Liu Y, Wilcox J (2012) Effects of surface heterogeneity on the adsorption of CO2 in microporous carbons. Environ Sci Technol 46:1940–1947
179. Babarao R, Dai S, Jiang D (2012) Nitrogen-doped mesoporous carbon for carbon capture—a molecular simulation study. J Phys Chem C 116:7106–7110

79
Abstract Metal-organic frameworks (MOFs) composed of metal nodes linked by organic linkers are a class of newly developed crystalline hybrid porous solids. In the past few years, MOFs have seen a very rapid development both in terms of synthesis of novel structures and their potential applications in a wide variety of fields. Nearly all metals and a large diversity of organic species can be used to construct MOFs, so that a huge variety of materials of MOFs with different structures and properties are accessible. Due to their uniform yet tunable pore sizes, high-surface areas, and easy pore functionalization, MOFs have emerged as superior porous materials for adsorption and membrane-based applications. Particularly, recent studies have demonstrated that MOFs are perfect and quite promising in CO2 capture. This chapter starts with an introduction of MOFs, including their design and synthesis, structural features, properties, and potential applications. Then, their implementation and performance in CO2 capture-related aspects including selective CO2 adsorption in MOFs, CO2 separation in MOFs, MOF-based membrane for CO2 separation, the design of MOFs for CO2 capture, and computational simulation in MOFs for CO2 capture are discussed.
3.1 Introduction
Economical and efficient CO2 capture and storage (CCS) from existing emission sources has been attracting tremendous attention due to the culprit causing disadvan-tageous environmental issues, such as the greenhouse effect [1, 2]. Anthropogenic emission resulting in the unparallel increase in the CO2 concentration in the
Chapter 3Metal-Organic Frameworks (MOFs) for CO2 Capture
Hui Yang and Jian-Rong Li
A.-H. Lu and S. Dai (eds.), Porous Materials for Carbon Dioxide Capture, Green Chemistry and Sustainable Technology, DOI: 10.1007/978-3-642-54646-4_3, © Springer-Verlag Berlin Heidelberg 2014
H. Yang · J.-R. Li (*) Department of Chemistry and Chemical Engineering, College of Environmental and Energy Engineering, Beijing University of Technology, Beijing 100124, People’s Republic of Chinae-mail: [email protected]

80 H. Yang and J.-R. Li
atmosphere comes mainly from the burning of fossil fuels, which continues to play an important role as primary energy source in the foreseeable future [1]. Based on the generation of CO2, several options and technologies have been considered for the CO2 capture and separation from fossil fuel-burned streams, particularly at station-ary point sources such as coal-fired power plants, including pre-combustion capture, oxy-fuel combustion, and post-combustion capture wherein [1, 3].
Pre-combustion capture involves the separation of H2 from CO2, which is pro-duced from the fuel burning in O2 or air. The produced gas mixture is mainly com-posed of CO and H2, which is known as synthesis gas (syn gas) [4]. The CO formed is further reacted with steam to give CO2 and more H2. The separation of CO2 and H2 is thus the center of this option, with great challenge. The major product of oxy-fuel combustion is CO2, which has the stringent requirement of nearly pure O2. High purity O2 can be obtained by the separation of air or by other novel techniques, which is involved in the oxy-fuel combustion capture. Post-combustion capture requires to remove CO2 from flue gas, comprised mainly of N2 and CO2, before its emission from power plants into the atmosphere. Post-combustion capture is actu-ally the most feasible option in a short time because many of the proposed technolo-gies can be retrofitted to existing fossil fuel consuming power plants.
Purification of natural gas (mainly CH4) is another advantage of developing CO2 separation techniques, which is typically soured with over 40 % CO2 and N2, and is only useable at low concentrations of CO2. A huge challenge in this separa-tion is that the special technology and material are needed to endure high pres-sures presented during the mining of natural gas [5, 6].
Among separation methods, adsorptive and membrane separations are favora-ble in CO2 separation mentioned above due to their high efficiency, easy imple-ment, and low energy import, to some extent. Adsorption methods are divided into physical and chemical adsorption [7, 8]. The former is primarily dependent on the temperature and pressure, while the latter for CO2 usually depends on the acid–base neutralization reaction. Right now, chemical absorption technologies have been utilized in the post-combustion capture of CO2 from flue gas, where aque-ous alkanolamine solutions are used as sorbent. However, high energy combus-tion (about 30 % of the output of the power plant) involved in the regeneration of the sorbent, as well as the corrosion of equipments and environmental pollu-tion from used sorbents limit greatly the development and pervasive implement of this technology. Alternately, physical adsorption and membrane separation are relatively low in energy cost and environmentally benign, therefore attracting intense attention. The development of adsorptive and membrane separation of CO2 mainly depends on the selection of a sound adsorbent medium. In particular, for the separation of CO2/N2, CO2/H2, and CO2/CH4, partially because of their simi-lar kinetic diameters (3.3, 3.64, 2.8, and 3.76 Å, respectively), it would require materials to possess a suitable pore size or specific pore surface property capa-ble of highly selective adsorption of CO2 molecule [9]. Consequentially, conven-tional solid adsorbents including activated carbons, zeolites, meso-porous silicates, activated alumina, porous metal oxides, and others, as well as newly developed metal-organic frameworks (MOFs), have been checked for CO2 adsorption

813 Metal-Organic Frameworks (MOFs) for CO2 Capture
and separation [10]. Particularly, MOFs with high-surface areas and tunable structures and properties have attracted considerable interest in CCS, because they can store greater amounts of CO2 than most other porous materials and be deco-rated with functional chemical moieties suitable for the molecular recognition of CO2 [11–15].
Clearly, ether chemical absorption, physical adsorption or membrane separa-tion, and the progress and achievement of associated technologies rely heavily on the development of materials. Therefore, looking for or evaluating economical and effective material is the key to solve the problem of CO2 capture.
3.2 Metal-Organic Frameworks (MOFs)
In the past two decades, a new class of crystalline porous solid materials, MOFs, have emerged and attracted great attention owing to their potential applications in gas storage [16, 17], separation [18], sensing [19], drug delivery [20], heterogene-ous catalysis [21], and so on. These materials are inorganic–organic hybrid materi-als comprised of single metal ions (or their clusters) and organic ligands, which assembled together to form extended frameworks principally through coordination bonds. MOFs have crystallographically well-defined framework structures and in most cases, these structures are robust enough to allow the removal of the included guest species resulting in permanent porosity. Particularly, the structure and asso-ciated properties of MOFs can be easily tuned and modified through the rational design and/or control of ligands, metal-based nodes, and synthetic conditions to meet a specific requirement [22, 23]. This is unique for MOFs and not easy acces-sible in most of other traditional porous solids. In addition, the inherent crystallin-ity of MOFs also allows precise structural characterization by X-ray diffraction, thus facilitating their rational design and formulation of structure–function rela-tionships. In addition, it should be pointed out that MOFs can be divided into rigid and flexible/dynamic classes [12]. The former have comparatively robust frame-work structure with permanent porosity, similar to zeolites, whereas the latter pos-sess dynamic or “soft” frameworks that respond to external stimulations, such as pressure, temperature, and guest molecules, being inaccessible from traditional inorganic porous materials, in most cases.
3.2.1 Design, Synthesis, and Structure of MOFs
The structural architectures of MOFs are mainly dependent on the assembly of metal-containing centers and organic ligands linkers (Fig. 3.1) [24–27]. In spite of wide utilization of single metal ions, it has been recognized that the second-ary building units (SBUs) approach offers a prospective avenue toward the rational design and construction of novel MOFs [22]. As a result, the structures

82 H. Yang and J.-R. Li
and properties of some MOFs can be designed and systematically tuned by the judicious choice of SBUs. On the other hand, particular importance is the design and synthesis of organic ligands, because they can add flexibility and diversity to the structures and functions of these materials [23]. Synthesis of MOFs is a self-assembly process, which is affected by many factors, including the nature and coordination mode of ligand and metal-containing center, cationic, anionic, tem-plate molecules, solvent, temperature, pH value, the ratio between the reactants, and others [28]. There have also been a lot of synthetic methods developed during these years. Structurally, a complex MOF structure is usually simplified by net and topology, which facilitate the structural analysis and further design of new MOF materials [29]. Particularly, Yaghi et al. define reticular chemistry, which concerns with the linking of molecular building blocks into predetermined structures by the geometrical and synthesis design. Where, a MOF structure is only considered as joints and links, which correspond to vertices and edges of a net. Besides the pre-design in synthesis, post-synthetic modifications have also been successfully used in modifying the pore properties of MOFs for target applications [30].
Organic ligands’ design and selection are among most important in the syn-thesis of desired MOFs (Fig. 3.2). The geometric predictability of organic ligands makes them excellent controllable building blocks for the MOF construction. Ligands design can thus be used not only in enriching the diversity of MOFs, con-trolling MOF structures, studying supramolecular isomerism, but also in modify-ing the functionalities of MOFs for specific applications. As a typical example in the structural control of MOFs through the ligand design, HKUST-1 [Cu3(btc)2
(H2O)3]n (btc = benzene-1,3,5-tricarboxylate), one of most popular MOFs, was firstly reported by Williams’s group in 1999, which has a framework structure
Fig. 3.1 Schematic representation of the construction of MOFs. Reproduced from Ref. [27] by permission of The Royal Society of Chemistry
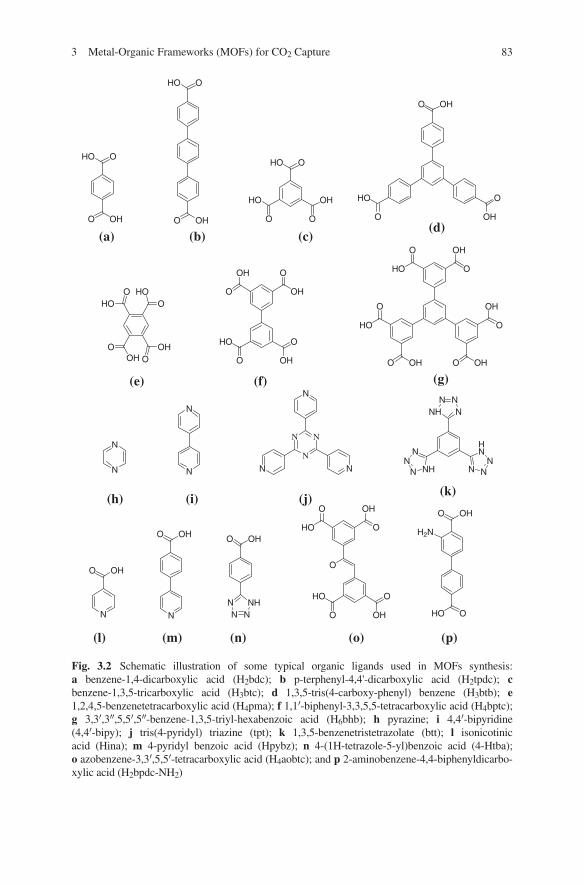
833 Metal-Organic Frameworks (MOFs) for CO2 Capture
(a)
(e) (f) (g)
(k)(j)(i)(h)
(l) (m) (n) (o) (p)
(b) (c)(d)
HO
OH
O
O
HO
OH
O
O
O
OO
OHHO
HO
O
OH
OH
OHO
O
OHO
HOO
OHOOOH
OHO
OOH
OH
O
O
HO
O
OH
HO
O
OH OHO
O
OH
O
HO
O
N
N
N
N
N
N
N
N
N
N
NNN
NH
N NN
HNN
NN NH
N
O OH
N
O OH
NN N
NH
O OH
O
OH
O
O
HO
O
OH
HO
O
O OH
OHO
H2N
Fig. 3.2 Schematic illustration of some typical organic ligands used in MOFs synthesis: a benzene-1,4-dicarboxylic acid (H2bdc); b p-terphenyl-4,4'-dicarboxylic acid (H2tpdc); c benzene-1,3,5-tricarboxylic acid (H3btc); d 1,3,5-tris(4-carboxy-phenyl) benzene (H3btb); e 1,2,4,5-benzenetetracarboxylic acid (H4pma); f 1,1′-biphenyl-3,3,5,5-tetracarboxylic acid (H4bptc); g 3,3′,3″,5,5′,5″-benzene-1,3,5-triyl-hexabenzoic acid (H6bhb); h pyrazine; i 4,4′-bipyridine (4,4′-bipy); j tris(4-pyridyl) triazine (tpt); k 1,3,5-benzenetristetrazolate (btt); l isonicotinic acid (Hina); m 4-pyridyl benzoic acid (Hpybz); n 4-(1H-tetrazole-5-yl)benzoic acid (4-Htba); o azobenzene-3,3′,5,5′-tetracarboxylic acid (H4aobtc); and p 2-aminobenzene-4,4-biphenyldicarbo-xylic acid (H2bpdc-NH2)

84 H. Yang and J.-R. Li
with both cages and channels [31]. Subsequently, an analogous ligand, H3tatb (H3tatb = 4,4′,4″-s-triazine-2,4,6-triyl-tribenzoic acid) with the same geometry but only different length was designed and used to construct MOFs by Zhou’s group [32]. A MOF, [Cu3(tatb)2(H2O)3]n (PCN-6′), has been obtained, which has the same structural topology with HKUST-1 but enhanced porosity due to the elongated ligand. On the other hand, ligands with different geometry, length, and/or containing different functional groups always have the opportunity to sys-tematically study the relationship between the ligand and resulting MOFs or their supramolecular building units such as metal-organic polyhedra (MOPs), in both structures and properties. For instance, a series of angular carboxylic acid ligands were designed and synthesized by Li et al. and successfully used in the construc-tion of MOPs with controllable molecular shape, size, and properties [33, 34]. Through the ligand design, these MOPs have been further constructed into MOFs. For example, several (3, 24)-connected isoreticular MOFs were constructed by using similar hexacarboxylate ligands containing three isophthalate groups linked by trigonal linkers with different length. The structures of all these MOFs contain cuboctahedral MOPs as supramolecular building units, which can be assembled by only isophthalate ligands [35]. These MOFs exhibited not only graceful architec-tures, but also excellent gas adsorption properties.
The metal-containing centers in MOFs can be either a mononuclear metal ion or a cluster unit, which are assembled by two or more metal ions. The metal clus-ter center commonly refers to as SBUs, which offer a prospective avenue toward the design and construction of MOFs [22]. The formation of the SUBs can be con-trolled through the selection of suitable synthetic conditions, to some extent. Until now, more than hundred kinds of geometries of SBUs have been reported, among them, most are transition-metal carboxylate assemblies produced during the for-mation of MOFs under given conditions (Fig. 3.3).
SBUs generated in situ usually have predefined and rigid linkage geom-etries, which provide a versatile tool of connectors for the rational construc-tion of targeted MOFs. Typical SBUs can be four-, and six-points of extension in the assembly of MOFs network. Many well-known MOFs, such as MOF-5 (or IRMOF-1, Zn4O(bdc)3) [24], MIL-101 (Cr3F(H2O)2O(bdc)3) [26], and HKUST-1 [31] are constructed by several common SBUs as shown in Fig. 3.3. MOF-5 has a tetrameric [Zn4O(CO2)6] SBU, in which a cationic oxide-cen-tered Zn4O tetrahedron is edge-bridged by six carboxylates to give the neutral octahedral node. This inorganic node reticulates through the linkage of ben-zene groups into a three-dimensional (3D) cubic network. MIL-101 is based on a trimeric [Cr3O(CO2)6] SBU, which consists of three metal octahedra, each coordinated by four bridging carboxylate groups, sharing a μ3-O common ver-tex. Combination of these SBUs with a two connected ligand bdc formed the 3D structure of the MOF. HKUST-1 is composed of binuclear paddle-wheel units [Cu2(CO2)4] linked by btc ligands, resulting in a (3, 4)-connected net-work. It should be pointed out that since many SBUs are generated in situ during the synthesis of MOFs, synthetic experimental conditions (such as reac-tant ratio, temperature, pH, and solvent used) always play crucial roles in the

853 Metal-Organic Frameworks (MOFs) for CO2 Capture
formation of a SBU with a given shape and geometry. Under different synthetic conditions, for example, the assembly between a transition-metal ion and car-boxylate group may lead to a variety of SBUs, thereby different MOFs with the same ligand.
In the synthesis of MOFs, various methods/strategies have been employed, including solvent evaporation or diffusion, hydrothermal, solvothermal, using ionic liquids, electrochemistry, mechanochemistry, and ultrasonic methods [28]. And new ones are being explored with the development of MOFs field. Particularly, high-throughput screen has also been used in finding new MOFs materials [36, 37].
A straightforward and simple method for the synthesis of MOFs is the evapo-ration of solvent, by which the reaction solution is concentrated to precipitate product. Experimentally, a reaction solution formed by mixing metal ions and organic ligands dissolved in suitable solvents are allowed to evaporate the sol-vents; after a given time, resulting products are harvested. Solvent diffusion includes several cases; typically one type of solvent having bad solubility toward product diffuses slowly into the reaction solution of metal ions and ligands, to participate the products. In another case, slow diffusion of two solutions of the metal ion and the ligand allows the process of the reaction very slowly, to form MOF product, particularly as single crystals. For example, a MOF Ag-tpha (tpha = tris(4-(1-(2-pyridin-2-ylhydrazono)ethyl)-phenyl)) was synthesized by layering acetonitrile solution of AgBF4 onto a solution of tpha in CH3OH/CHCl3 (v: v = 1:4) [38]. Hydrothermal and solvothermal methods have been widely used in the synthesis of MOFs. Many well-known MOFs, such as MOF-5,
Fig. 3.3 Single metal nodes and selected SBUs used in the construction of MOFs: a tetrahe-dral metal ion; b octahedral metal ion; c [M2(COO)4] paddle-wheel SBU; d [MO(COO)6] trimer SBU; e [M4O(COO)6] quad-core SBU; f [M4Cl(COO)8] quad-core SBU; g [M4Cl(trz)8] quad-core SBU; and h [Fe10O20(COO)10] pentagonal antiprism SBU

86 H. Yang and J.-R. Li
MIL-101, and HKUST-1, were synthesized by this method. Ion thermal synthesis refers to the use of ionic liquids as reaction solvents to synthesize MOFs. Several MOFs have been synthesized by using ionic liquids as solvents. For example, Lin et al. [39] reported a chiral MOF, which was synthesized by employing the chiral ionic liquid.
Increasing attention of MOFs in practical applications has promoted the devel-opment of fast, large-scale, and low-cost synthetic methods for MOFs, such as microwave and mechanochemistry syntheses. In 2005, the microwave synthesis method was first applied in the synthesis of Cr-MIL-100 [40]. HKUST-1 also has been synthesized by the microwave method later [41]. In the progress of the mech-anochemistry synthesis, the mechanical breakage of intramolecular bonds followed by a chemical transformation takes place. This method was used in the MOF syn-thesis began in 2006. Pichon et al. [42] got [Cu(INA)2] (INA= isonicotinic acid) by grinding a mixture of Cu(OAc)2·3H2O and INA in ten minutes. In addition, high-throughput methods are a powerful tool to accelerate the discovery of new compounds and to optimize syntheses procedures. For example, a series of new MOFs including 25 zeolitic imidazolate frameworks (ZIFs) were synthesized by this method typically [36].
With the continuous improvement in the understanding of structural chemistry, the net or topology was widely used in the description of different chemical con-texts [29]. It was realized that the edges and vertices of a net could be polyatomic linkers and clusters, such as complicated organic ligands and SBUs in MOFs, respectively. The development of net topology thus provides a straightforward way to simplify and understand the structure of MOFs and is very helpful in the design of new MOFs.
A MOF structure usually can be simplified into a simple topological net. For example, the structure of MOF-5 has a pcu-type topology (Fig. 3.4a), when consid-ering [Zn4O(COO)6] SBUs as six-connected nodes and benzene group as linkers to form a net with primitive cubic lattice. As shown in Fig. 3.4b, similarly, HKUST-1 is formed by binuclear paddle-wheel units [Cu2(CO2)4] linked by four btc ligands, connected into a (3, 4)-connected tbo-type topology (Fig. 3.4b). Furthermore, 4-connected dia network is also very common (Fig. 3.4c). Not only SBUs, but also organic ligands can be simplified as a node, with different connectivity based on different construction consideration in some cases. Such as for Cu2(bptc)(H2O)2
Fig. 3.4 Three typical topological nets in MOFs: a pcu, b tbo, c dia

873 Metal-Organic Frameworks (MOFs) for CO2 Capture
(MOF-505) [43], if the paddle-wheel SBU is simplified as a 4-connected vertex and the carboxylic ligand is considered as a 4-connected branching point (vertex), the underlying net would be a cubic 4-connected net with nbo-type topology. If the organic linker is considered as two 3-connected vertices, the overall structure can be simplified as a fof-type topology based on a (3, 4)-connected net.
Finally, it should be mentioned that different from zeolite and activated car-bon materials, which usually perform rigid pore frameworks, MOFs could afford not only rigid, but also flexible and dynamic features. The framework structure of flexible MOFs can respond to external stimulus, such as light, electric field, and guest molecules, and change their pore properties reversibly. After removal of guest molecules, the pores of a flexible MOF may become close. So flexible MOFs usu-ally give a stepwise adsorption and/or show hysteretic desorption isotherms for gases, such as CO2 typically. Flexible MOFs are also commonly used in the selec-tive adsorption and separation of small molecules. An example is the MIL-53 sys-tem developed by Férey’s group, whose frameworks show an abnormal “breathing effect” in the adsorption of CO2 and CH4 [44–46]. The structure of MIL-53 is com-posed of infinite chains of corner sharing MO4(OH)2 (M = Al3+, Cr3+ or In3+) enti-ties linked by bdc ligands, creating one-dimensional (1D) rhombic tunnels. It was found that the “breathing effect” of the MIL-53 framework is mostly related to the flexible linkage between the metal centers and the carboxylate groups of ligands.
3.2.2 Properties and Potential Applications of MOFs
As pointed out above MOFs are unique in their degree of structural diversity and tunability, thereby their range of chemical and physical properties. MOFs have thus great potential in various applications, such as luminescent, magnetic, sensor, drug delivery, biomedicine, gas storage, selective adsorption and separations, and heterogeneous catalysis.
A lot of research work with MOFs as sensor based on their fluorescent prop-erties was reported [47]. These studies include the detection of anions and cati-ons, small organic molecules, explosives, gas and steam. For examples, Chen’s group reported a luminescent MOF, Eu(3,5-pdc)1.5(dmf) (3,5-pdc = pyridine-3,5- dicarboxylate), which exhibited a sensing function toward metal ions: alkali and alkaline earth metal ions have a negligible effect on the luminescent intensity of the MOF, but Cu2+ can reduce the luminescent intensity significantly. Li’s group reported two fluorescent Zn-based MOFs capable of sensing nitro-containing mol-ecules relevant to the detection of explosives [48, 49].
Another very important application of MOFs is H2 storage [17]. For instance, in 2010, Furukawa et al. [50] reported the highest total H2 storage capacity of MOFs: about 176 mg/g (excess 86 mg/g) in MOF-210 (Zn4O(bte)4/3(bpdc), bte = 4,4′,4″-(benzene-1,3,5-triyltris(ethyne-2,1-diyl))tribenzoate, bpdc = 4,4'- biphenyldicarboxylate) at 77 K and 80 bar. In addition to H2 storage, MOFs are also explored in CH4 storage. It should be pointed out that unlike H2 storage where

88 H. Yang and J.-R. Li
the low heat of adsorption is the bottleneck of most of MOFs, the heat of adsorption for CH4 is already within the ideal scope to be used under ambient conditions [51]. Several MOFs have showed very high CH4 storage ability. For example, Zhou’s group showed that CH4 uptake capacity of Cu2(sbtc) (PCN-11, sbtc = trans-stilbene-3,3′,5,5′-tetracarboxylate) is 171 v/v at 298 K and 35 bar, which is very close to the DOE target (180 v/v, at ambient conditions within 35 bar) [52]. Following that, through the MOF design, the real breakthrough for MOF-based CH4 storage was achieved in Cu2(adip) (PCN-14, adip = 5,5′-(9,10-Anthracenediyl)di-isophthalic acid) by the same group [53]. At 290 K and 35 bar, the absolute CH4 adsorption capacity in PCN-14 is 230 v/v, which is 28 % higher than the DOE target at that time.
MOFs have also been widely explored in catalysis, starting from the earlier state of MOF development [21]. For example, in 2000, Seo et al. [54] reported homo-chiral MOF, POST-1 (Zn3(μ3-O)(L31-H)6), which exhibited an interesting catalytic activity for an asymmetric chemical reaction based on the special organic groups on the pore surface of the MOF. Following that, the asymmetric catalysis promoted by metal ions on a MOF framework was reported in 2001 by Lin’s group [55].
In addition, bio-MOFs have emerged and been used in biomedical applications, such as drug delivery, which requires a biologically friendly composition of the material [20]. For example, Férey’s group reported the capacity for drug hosting and controlled delivery by MIL-100 (Cr) and MIL-101 (Cr). Their results showed that per gram MIL-100 (Cr) adsorbed around 0.35 g of ibuprofen. On the other hand, MIL-101 (Cr) indicated a surprising result, which adsorbed 1.4 g ibuprofen per gram. In the second step, adsorbed ibuprofen in the materials can be released by simulated body fluid at 310 K. Further investigation indicated that ibuprofen delivery for MIL-100 (Cr) can be completely released after 3 hours. However, achieving the same release level in MIL-101 (Cr) needs 6 h [56].
Of course, as a new class of porous materials, MOFs possess great potential in separation, including adsorptive separation and membrane separation [18]. The investigation of MOFs for selective adsorption and separation not only can greatly extend the scope of MOFs applications, solve requirements of some practi-cal applications, but could lead to answers of some crucial problems in adsorp-tion/separation science and related technologies. Among them, CO2 separation using MOFs as supporting materials is quite attractive and has been studied for several years. Some MOFs have represented excellent performance in the selective adsorption and separation of CO2 over CH4, N2, and H2. The details of these appli-cations will be discussed as below.
3.3 Metal-Organic Frameworks (MOFs) for CO2 Capture
As a newly developed class of porous materials, MOFs have presented great potential in CO2 capture, which were explored and demonstrated from both exper-imental exploration and computational simulation [16, 57, 58]. In this section, sev-eral important aspects related to this topic are introduced based on current research

893 Metal-Organic Frameworks (MOFs) for CO2 Capture
progress, including: (1) selective CO2 adsorption in MOFs, which covers tuned CO2 adsorption, the enthalpy of adsorption, CO2 adsorption site, size/shape exclu-sion, interaction between CO2 and pore surface, and the selective adsorption in flexible MOFs; (2) CO2 separations using MOFs, such as in post-combustion cap-ture, pre-combustion capture, and oxy-fuel combustion capture; (3) MOF-based membrane for CO2 separation, including those in both MOF thin film and MOF-based mixed matrix membrane (MMM); (4) the design of MOFs for CO2 capture, which was realized through pore size control, pore surface functionalization, using single-molecule trap (SMT) or based on the flexibility of MOFs; and (5) computa-tional simulation in MOFs for CO2 capture.
3.3.1 Selective CO2 Adsorption in MOFs
For CO2 capture, CO2 adsorption capacity and selectivity of a MOF are the central issues. The critical parameters of adsorption capacity are pore size/volume and surface features of an adsorbent. Compared to other porous materials, such as zeolites and activated carbon, MOFs continue to attract much attention because of their higher pore volume and surface area, as well as tunable pore surface func-tionalization/modification. Some well-known MOFs (e.g., HKUST-1, MOF-5, MOF-74 (M2(dhtp), dhtp = 2,5-dihydroxyterephthalate, M = Mg, Co, Zn, Ni, Mn), MOF-177, MOF-200 (Zn4O(bbc)2, bbc = 4,4′,4,4″ -[benzene-1,3,5-triyl-tris(benzene-4,1-diyl)]tribenzoate), MOF-210, MIL-53, MIL-101) have showed a high CO2 adsorption capacity [18, 57]. For example, at 298 K and 1 bar, the volumetric CO2 adsorption capacity of MOF-74-Mg can achieve 27.5 wt% [59]. It should be pointed out that the high-pressure CO2 adsorption capacity at (or near) room temperature is directly related to the ultimate storage ability of a MOF, which is another significant issue in CCS. Indeed, for CO2 storage, the great-est uptake capacities at high pressures are related to the surface area of sorbent material. Furthermore, excellent CO2 adsorption properties have also been dem-onstrated in a number of MOFs with modest surface areas that, however, have a significant density of high-affinity adsorption sites, such as exposed metal sites. This will be discussed in detail later.
A brief qualitative evaluation of CO2 adsorption selectivity in MOFs is the direct observation on differences in uptakes between separated gases under given condi-tions, usually based on the single-component adsorption isotherms. These isotherms can also be used to quantitatively estimate the adsorption selectivity. If adsorp-tion species are presented at low loadings, namely within the Henry’s regime, the adsorption selectivity for an equimolar mixture is close to the ratio of the Henry’s constants for each species. At non-dilute loadings, however, more information is required to estimate multi-component adsorption. One common approach is to use ideal adsorbed solution theory (IAST) to predict multi-component adsorption iso-therms and selectivity based on the single-component adsorption isotherms [60]. This approximate theory is known to work accurately in many porous materials,

90 H. Yang and J.-R. Li
including MOFs. For example, the adsorption equilibrium selectivity of CO2 over N2 in MOF-5 and MOF-177 is 17.48 and 17.73 respectively, which were calculated from the ratio of Henry’s constants of single-component adsorptions by Deng’s group [61]. For IAST application in this context, for example, the selectivity of CO2 over N2 was evaluated to be 33 and 22 at 298 K and 20 bar for Cu3(tpbtm) and Cu3(btei) (PCN-61, tpbtm= N,N',N''-tris(isophthalyl)-1,3,5-benzene tricarbox amide btei = 5,5′,5″-benzene-1,3,5-triyltris(1-ethynyl-2-isophthalate), respectively, reported by Zheng et al. [62]. It should be pointed out that most of previous reports have approximated the adsorption selectivity factors by comparing the adsorption capacity of single-component adsorption isotherms. For instance, Demessence et al. [63] briefly evaluated the adsorption selectivity of CO2 over N2 in Fe-btt (btt = 1,3,5-benzenetristetrazolate), resulting in a separation factor of approxi-mately 5.5:1 at room temperature. Of course, breakthrough experiments of a gas mixture containing CO2 are the direct demonstration of its adsorption selectivity in given adsorbent materials. This will be discussed in the following section.
3.3.1.1 Tuned CO2 Adsorption in MOFs
A lot of strategies in tuning CO2 adsorption of MOFs have been proposed, such as, introducing functional groups, ion exchange, dropping strategy, post-synthesis, (to modify their porous properties) [57]. For example, Bae et al. [64] reported a framework Zn2(bttb)(CF3-py)2, bttb = 4,4′,4″,4″′-benzene-1,2,4,5-tetrayltetrabenzo-late), which contained highly polar py-CF3 groups in its pore surface, leading to an enhancement of CO2 uptake compared with N2 at low pressure. Furthermore, Kondo et al. [65] demonstrated that changing metal ions can tune CO2 adsorp-tion in two isostructural MOFs, M(4,4′-bipy)2(otf)2 (M = Cu and Co, otf = tri-fluoromethanesulfonate). Although the difference of metal ions in the two MOFs resulted in only slightly different structures, it was found that the CO2 adsorp-tion properties were largely dependent on the metal ions. Furthermore, An and Rosi [66] demonstrated that a post-synthetic exchange of extra-framework cations within an anionic MOF, [Zn8(ad)4(bpdc)6O](cation) (bio-MOF-1, ad = adeninate, bpdc = 4,4-biphenyldicarboxylate), can be used to systematically modify its pore sizes, thereby, tuning the CO2 adsorption capacity. They also concluded that smaller pores in MOFs (with large cations in this case) may be ideal for condensing CO2 at temperatures relevant to practical application. In addition, the cation exchange to tune CO2 adsorption in a flexible MOF, [Ni(bpe)2(N(CN)2)](cation) (bpe = 1,2-bis (4-pyridyl)ethane) has also been demonstrated by Kitagawa’s group. As an alterna-tive strategy, a Co(II) doping strategy was applied to tune the CO2 adsorption of MOF-5, reported by the same group [67]. In addition, the presence of water mol-ecules coordinated to metal sites in the framework capable of enhancing the CO2 uptake in HKUST-1 has also been demonstrated by Snurr’s group [68]. Their results indicated that enhanced CO2 adsorption can be attributed to the strong interaction between the quadrupole moment of the CO2 molecule and the electric field created from water molecules. Similarly, MIL-53(Cr) also showed such a phenomenon for CO2 adsorption relative to CH4. [69].

913 Metal-Organic Frameworks (MOFs) for CO2 Capture
3.3.1.2 The Enthalpy of Adsorption of CO2 in MOFs
The enthalpy (heat) of adsorption (Qst) of CO2 is a critical parameter that has a significant influence on the performance of a given material for CO2 capture appli-cation. Keskin et al. [70] gave a detailed analysis of the CO2 adsorption heat in the examined MOFs in their review. For most MOFs, the Qst is ranging from 20 to 50 kJ mol−1, and it decreases with increased CO2 loading in most cases whereas Demessence et al. reported an ethylenediamine-functionalized MOF, in which the Qst of CO2 can reach 90 kJ mol−1. The high Qst can be attributed to the ethylene-diamine groups in the MOF, which form very strong interactions (partially chemi-cal bond) with CO2 molecules [63]. In general, the higher of the adsorption heat, the stronger of the interaction between the CO2 molecules with the pore chan-nels, so it is advantageous to the capture of CO2 in MOFs with high Qst of CO2. However, it should be pointed out that a very high heat of adsorption is not neces-sarily good in terms of the CO2 separation application, because of the large energy requirement associated with the regeneration (i.e., desorption) of the materials. For gas adsorption, Qst is usually calculated from the adsorption isotherms at differ-ent temperatures. Complementary to this typical method, measurements of the Qst by pulse-response experiments in a ultrahigh-vacuum reactor, referred to as a TAP reactor (temporal analysis of products), have been performed for the adsorption of CO2 in three MOFs: IRMOF-1, Zn4O(NH2-bdc)3(IRMOF-3), and HKUST-1 [71].
3.3.1.3 CO2 Adsorption Site in MOFs
Identifying adsorption site of a porous material is quite important in exploring its adsorption property. In MOFs, unsaturated metal sites and special chemical groups such as -NH2, -OH, and rich N donors are preferable as special adsorp-tion sites in the design of MOFs for their modified high CO2-selective adsorption. As a breakthrough achievement, the direct observation of CO2 adsorption behav-ior from the X-ray crystallographic resolution has been realized in MOF, Zn2(atz)(ox) (ox = oxalate) by Vaidhyanathan et al. [72]. As shown in Fig. 3.5, two inde-pendent CO2 binding sites were located in the pores of this MOF: one is near the amine group and another is close to the oxalate group. The O(CO2)···H-N(NH2) hydrogen bond and interactions between the O of oxalate group and O of CO2 and between the N lone pair of the amine group and C of CO2 have been con-firmed. Furthermore, the CO2···CO2 cooperative interaction was also observed. Accompanied with computational simulation, authors concluded that the com-bination of appropriate pore size, strong interaction between CO2 and functional groups, and cooperative binding of CO2 molecules is responsible for the observed binding of CO2 in this MOF. Furthermore, unsaturated metal sites in MOFs also play an important role in their CO2 adsorption. For examples, Bordiga et al. [73] demonstrated the influence of unsaturated Cu(II) centers in HKUST-1 by IR spec-troscopy. Similarly, Dietzel et al. [74] showed that CO2 molecule adopts the “end-on” coordination mode when interacting with the coordinatively unsaturated Ni(II) sites in Ni2(dhtp) (H4dhtp = 2,5-dihydroxyterephthalic acid), which gave rise to

92 H. Yang and J.-R. Li
high CO2 adsorption capacity. The CO2 adsorption mode in MIL-53(Cr) was also studied by Férey’s group through IR spectra [75]. The observed red shift of the ν3 band and splitting of the ν2 mode of CO2, as well as the shifts of the ν(OH) and the δ(OH) bands of the MIL-53(Cr) hydroxyl groups confirmed that CO2 inter-acted with the O atoms of framework -OH groups.
3.3.1.4 Selective Adsorption Based on Size/Shape Exclusion
Selective adsorption of CO2 over N2 or CH4 based mainly on the molecular siev-ing effect has been confirmed in some MOFs. This effect is determined by the sizes and shapes of both the gas adsorbates and the pores in a given MOF. For exam-ple, an experimental selective adsorption of CO2 and O2 over N2 in MOF, Zn(dtp) (H2dtp = 2,3-di-1H-tetrazol-5-ylpyrazine) has been early reported by Li et al. [76] (Fig. 3.6). This highly selective adsorption of CO2 over N2 was attributed to the small aperture of the channels in this MOF. Another similar selective adsorp-tion of CO2 over N2 was observed in Mn(HCO2)2 by Kim’s group. This MOF also exhibited a highly selective adsorption for H2/N2 and CO2/CH4 at low tem-perature, being attributed to its small window size [77]. In addition, some MOFs were found to selectively adsorb CO2 over CH4 based on size/shape exclusion, such as MIL-96 (Al12O(OH)18(H2O)3(Al2(OH)4)[btc]6) and Zn2(cnc)2(dpt) (cnc = 4-carboxycinnamic, dpt = 3,6-di-4-pyridyl-1,2,4,5-tetrazine) [78, 79]. In addition, Ma et al. also reported the PCN-17, which is a coordinatively linked interpenetrated framework. The pore (window) size of PCN-17 is about 3.5 Å, which leads to the observed selective adsorption of H2 and O2 over N2 and CO [80].
Fig. 3.5 Structure (a) and gas adsorption properties (b) of Zn2(atz)(ox). (c) The CO2 binding (directly determined by X-ray structure refinement at 173 K) within the pores of Zn2(atz)(ox) (c). Reprinted from Ref. [57] Copyright 2011, with permission from Elsevier

933 Metal-Organic Frameworks (MOFs) for CO2 Capture
3.3.1.5 Selective Adsorption Based on Interaction Between CO2 and Pore Surface
Besides the size/shape exclusion, the nature of the interaction between gas molecule and pore surface is also important in determining the adsorption selectivity of a MOF toward a given gas. In most of the rigid MOFs, observed adsorption selectivity of CO2 over other gases can be attributed to the thermodynamic equilibrium effect or the kinetic effect in a given equilibrium time, namely the preferential adsorp-tion (based on the interaction) but not the molecular sieving effect. In this case, the selectivity is related to adsorbate properties such as polarity, quadruple moment, and H-bonding, as well as to the surface properties of the MOF pores.
A large number checked MOFs indeed showed the selective adsorption of CO2 over N2 and CH4, particularly at room or higher temperature. The primary reason is that CO2 has a larger quadrupole moment than other gases. For example, a pillared-layer MOF Zn2(2,6-ndc)2(dpni) (2,6-H2ndc = 2,6-naphthalic acid; dpni = N,N′-di-(4-pyridyl)-1,4,5,8-naphthalenetetracarboxydiimide) was reported by Bae et al. [81], which has a large pore, but CO2 was more preferentially adsorbed than CH4 in it. Similarly, the same group also reported a carborane-based MOF with coordi-natively unsaturated metal sites, which showed a highly selective adsorption of CO2 over CH4 [82]. This result has also shown that open metal sites in a MOF can aid in the adsorptive separation of (quadru)polar/non-polar gas pairs, such as CO2/CH4. Selective adsorptions of CO2 over other gases were also observed in several ZIFs. The frameworks of these ZIFs contain large cages interconnected by small aper-tures. For instance, ZIF-95 and ZIF-100 have unique combinations of large cavities and highly constricted apertures of 3.65 and 3.35 Å, respectively [83]. At 298 K, both ZIFs showed high affinity and storage capacity for CO2 over CH4, CO, and N2. These observed high selectivities were attributed to the combined effects of the slit width of the pore apertures in the MOFs being similar in size to CO2, and the strong quadrupolar interactions of carbon with nitrogen atoms present in their links.
0.0 0.2 0.4 0.6 0.8 1.0
0
20
40
60
80
100
Vad
s(cm
3 /g)
P/P0
N2 at 77 K
O2 at 77 K
CO2 at 195 K
Fig. 3.6 Crystal structure of [Zn(dtp)] (left) and its gas sorption isotherms (right). Reprinted from Ref. [57] Copyright 2011, with permission from Elsevier

94 H. Yang and J.-R. Li
3.3.1.6 Selective Adsorption in Flexible MOFs
A lot of flexible MOFs have been synthesized and characterized by gas adsorption, and some of them showed unique selective adsorptions of CO2 over other gases. The mechanisms of selective adsorption in flexible MOFs are more complicated than that in rigid MOFs. In some cases, the gas sorption isotherms show hyster-etic behaviors due to framework rearrangements during adsorption/desorption pro-cesses. Besides size/shape exclusion and adsorbate–surface interactions, structural rearrangement must thus also be taken into account in these cases.
Based on size/shape exclusion effect, for example, the selective adsorption of CO2 over N2 and CH4 at low temperature was observed in a pillared-layer flex-ible MOF Cu(fma)(bpee)0.5 (fma = fumarate; bpee = 1,2-bis(4-pyridyl)ethane) [84]. Similar to rigid MOFs, the surface property of pores also plays an important role in determining gas adsorption selectivity of flexible MOFs. A typical exam-ple is MIL-53, which has a flexible 3D framework structure with a 1D channel of 8.5 Å in dimension (evaluated in its hydrated form) (Fig. 3.7a) [85–87]. This MOF exhibited a “breathing effect” upon hydration–dehydration, and showed different adsorption behaviors toward CH4 and CO2 (Fig. 3.7b). The adsorption isotherm of CH4 showed a typical type I behavior, while the CO2 isotherm exhibited two steps; above the first step at low pressure, the CO2 uptake greatly exceeded that of CH4. The difference between the CH4 and CO2 adsorptions was attributed to the quadru-pole moment of the CO2 molecules. Another typical example is Ni(bpee)[Ni(CN)4] (bpee = trans-1,2-bis(4-pyridyl)ethylene) [88]. Although the pore size of this MOF is large enough, CO2 molecule can diffuse into its micropores, whereas O2 and N2 cannot. This phenomenon was attributed to the strong interactions of O2 or N2 with the pore windows, which blocked other molecules from passing into the pore. Whereas, the phenomenon that CO2 can diffuse into pores was suggested to be due to its much stronger interaction with pore windows, which led to the opening of pores of this MOF. It was also proposed that the NiII atoms, the func-tional groups, and the p-electron clouds of the bpee ligands in this MOF gave rise to an electric field, which was effective in the adsorption of CO2 due to its high quadrupole moment.
It should be mentioned that some flexible MOFs have small or even no pores to allow guest molecule to enter. But the pores expand, being dependent on the adsorbate–surface interactions when exposed to certain gas adsorbates, a gate-opening process thus works in the gas adsorption. A typical example is ZIF-20 (Zn(pur)2, pur = purinate) with a 3D porous structure containing large cages con-nected by small windows, in which the CO2 uptake is five times higher than that of CH4 at 1 bar and 273 K [89]. According to the crystal structure determination, the maximum window aperture of this MOF is only about 2.8 Å, which is smaller than the kinetic diameter of both CO2 and CH4. Therefore, it was believed that the large cage space in the structure becomes accessible through a dynamic window-opening process induced by the strong interaction between the pore window surface and the CO2 molecules. Another interesting case is such a type of flexible MOFs, in which each gas has its own gate-opening pressure. Below a certain pressure,

953 Metal-Organic Frameworks (MOFs) for CO2 Capture
the gas molecules cannot enter into the pores. Several types of gases may thus be separated at different pressures by just one adsorbent. One example is Cu(dhbc)2(4,4′-bipy) (dhbc = 2,5-dihydroxybenzolate, 4,4′-bipy = 4,4′-bipyridine), which has different “gate”-opening pressures toward CO2, N2, and CH4 [90]. At dif-ferent pressure ranges, these gases can thus be selectively adsorbed by this MOF.
3.3.2 CO2 Separation in MOFs
Until now, the reports of adsorptive separation of CO2 and other gases in a mixture by using MOFs, conducted by experimental separation process, are limited. Among various characterization methods in separation, the breakthrough experiment and gas chromatographic separation are simple and straightforward in the evaluation of the separation performance of a MOF toward a gas mixture. On the other hand, as previ-ously mentioned, reported CO2 separation in MOFs mainly includes CO2/N2 sepa-ration for post-combustion capture, CO2/H2 from synthesis gas for pre-combustion capture, and O2/N2 and CO2/CO separation for oxy-combustion capture, which will be detailed as following.
3.3.2.1 Characterization of CO2 Separation
Breakthrough experiments have been used in CO2 separation by MOFs, which can give qualitative evaluations in most cases. As a typical example, several ZIFs have been checked to separate CO2 from other gases through breakthrough experi-ments performed by Yaghi’s group [91]. They selected ZIF-68 (Zn(bIm)(nIm), bIm = benzimidazolate, nIm = 2-nitroimidazolate), ZIF-69 (Zn(cbIm)(nIm)), and ZIF-70 (Zn(Im)1.13(nIm)0.87, Im = imidazolate) to separate mixture of CO2/CO at
Fig. 3.7 a The structure of MIL-53. b Gas adsorption isotherms of MIL-53(Cr) at 304 K (inset, schematic illustration of the “breathing effect” in MIL-53(Cr)). Reprinted from Ref. [57] Copyright 2011, with permission from Elsevier

96 H. Yang and J.-R. Li
room temperature, ZIF-78 (Zn(nbIm)(nIm), nbIm = 5-nitrobenzimidazolate) and ZIF-82 (Zn(cnIm)(nIm), cnIm = 4-cyanoimidazolate) to separate CO2/CH4, and ZIF-95 and ZIF-100 to separate CO2/CH4, CO2/CO or CO2/N2. Their results indi-cated that these materials are feasible in the separation of CO2 from other gases and have a longer retention time for CO2 than BPL carbon under the same condi-tions. Another example is using Mg-MOF-74 in the separation of CO2 and CH4. A 20 % mixture of CO2 in CH4 was tested in this study by the same group [92]. The results showed that the adsorption for CO2 in this MOF is highly preferred over CH4 with a dynamic capacity of 8.9 wt% CO2 uptake, being higher than that of zeolite NaX under similar conditions (Fig. 3.8). Subsequently, they studied the effect of metal ion of this series of MOFs on CO2 adsorptive separation by break-through in isostructural Zn-MOF-74, which took up just 0.35 wt% CO2, a reduc-tion in 96 % compared with Mg-MOF-74.
Breakthrough experiments in the CO2 separation have also been performed on flexible MOFs. A typical example is MIL-53 series materials, which have bistable structures, as described above, with narrow pores at low CO2 pressure and larger ones at higher [85, 93]. The separation of CO2/CH4 mixtures using a fixed bed packed with MIL-53(Al) was performed by Finsy et al. [94]. The results indicated that the adsorption of CO2 in MIL-53(Al) at higher pressures leads to an expan-sion of the framework and an increase in the adsorbed amount, but the CO2 selec-tivity over CH4 decreased from ca. 7 to ca. 4 at pressures above 5 bar. In addition, the separation and removal of CO2 from binary CO2/N2 and CO2/CH4 and ternary CO2/CH4/N2 mixtures using MOF-508b checked by breakthrough experiments were explored by Chen’s group [95, 96]. They indicated that the sorption selectivi-ties of CO2 over CH4 or N2 in this MOF are moderate (3–6) at 303 K, which were lower than that of activated carbon, and decrease with increasing temperature.
Gas chromatography is another important method used for the evaluation of gas separations in an adsorbent. For example, Yoon et al. [97] studied the gas chroma-tographic separation of a gas mixture composed of H2/O2/N2/CH4/CO2 (0.6: 2: 28: 10: 27, mol%) by passing it through a column packed with Co3(2,4-pdc)2(μ3-OH)2
Fig. 3.8 Breakthrough experiment of Mg-MOF-74 using a 20 % mixture of CO2 in CH4 (inset highlights the structure of the MOF). Reprinted from Ref. [57] Copyright 2011, with permission from Elsevier

973 Metal-Organic Frameworks (MOFs) for CO2 Capture
(CUK-1, 2,4-pdc = pyridine-2,4-dicarboxylate). The results indicated that these gases can be easily separated by this material. Quartapelle Procopio et al. [98] also explored the gas chromatographic separation of a C2H2/N2/CH4/CO2 mixture using an ionic MOF, A[Cu3(μ3-OH)(μ3−4-carboxypyrazolato)3] (A = NH4+ or Et3NH+) (Fig. 3.9). They found that CO2 and C2H2 have a strong interaction with the mate-rial, while the interaction with N2 and CH4 is negligible, thus revealed the effec-tiveness of ionic MOFs as a viable option for the CO2 separation.
3.3.2.2 Separation in Post-Combustion Capture
After the combustion of fossil fuels, the main product is flue gas with CO2, N2, and other minor components. In adsorption-based CCS, an adsorbent thus is required to separate/capture CO2 from the post-combustion flue gas. An ideal adsorbent for the post-combustion capture of CO2 should exhibit (1) a high selectivity for CO2 over the other flue gas components; (2) a high CO2 adsorp-tion capacity; (3) a rapid diffusion of the gas through the adsorbent; (4) a minimal energy penalty for its regeneration; and (5) a long-term stability under the operat-ing conditions. Actually, some MOFs nearly meet all of these requirements, being promising in the post-combustion CO2 capture.
To the best of our knowledge, up to now there is no experimentally reported CO2/N2 binary adsorption isotherms yet reported. So the calculated selectivity factors are useful for preliminary evaluations of MOFs in post-combustion CO2 capture. It was believed that for a high adsorption selectivity, the CO2 adsorption should be maximized at pressures near 0.15 bar. Theoretically, as the adsorbent for the post-combustion CO2 capture, it is expected that a MOF has such pore surface properties that can increase the adsorption selectivity and capacity toward CO2 yet
Fig. 3.9 Structure of A[Cu3(μ3-OH)(μ3−4-carboxypyrazolato)3] (A = NH4+ or Et3NH+) and the results of variable-temperature pulse gas chromatography experiments based on an equimo-lecular C2H2/N2/CH4/CO2 gas mixture passing through a chromatographic column packed with this material. Reprinted from Ref. [57] Copyright 2011, with permission from Elsevier

98 H. Yang and J.-R. Li
minimize the regeneration energy. A MOF can indeed enhance its CO2 adsorption and selectivity by the modification of functional groups, such as -NH2 and -OH, as well as the location of special adsorption sites (such as unsaturated metal cent-ers). For instance, Yaghi’s group reported the IRMOF series, which provide a basis for elucidating the effects of aromatic amines within MOFs. IRMOF-1 (MOF-5) can be adsorbed about 4.6 wt% CO2 at 298 K and 1.1 bar [99]. Under the same conditions, amine-functionalized variant IRMOF-3 adsorbs 5.0 wt% CO2, while the BET surface area decreases from 2,833 to 2,160 m2/g. Another much more typical example is an alkylamine functional MOF, H3[(Cu4Cl)3(BTTri)8(mmen)12] (mmen-Cu-BTTri; H3BTTri = 1,3,5-tri(1H-1,2,3-triazol-4-yl)benzene) reported by Long’s group, which was prepared by the incorporation of the N,N′-dimethylethylenediamine (mmen) into the Cu-BTTri. The mmen-Cu-BTTri drasti-cally enhanced the CO2 adsorption capacity, reaching about 9.5 wt% CO2 at 298 K and 0.15 bar [63]. The evaluated IAST selectivity of CO2/N2 in mmen-Cu-BTTri was 372 at 298 K, and the isosteric heat of CO2 adsorption was -96 kJ mol−1 at zero coverage. Similarly, mmen-Mg2(dobpdc) (dopbdc = 4,4′-dioxido-3,3′-biphenyldicarboxylate) also displayed an exceptional capacity for CO2 adsorp-tion at low pressures, taking up 8.1 wt% at 0.39 mbar and 298 K, reported by the same group [100]. In addition, a majority of the amine-functionalized MOFs were reported, such as Ni2(NH2-bdc)2(dabco) (dabco = 1,4-diazabicyclo[2.2.2]octane), Al(OH)(NH2-bdc) (NH2-MIL-53(Al)), In(OH) (NH2-bdc), which all showed a high CO2 separation ability from N2 [101, 102]. Furthermore, other organic linkers (other than amines) with heteroatom functional groups have also been investigated for their effects on the CO2 adsorption and selectivity over N2, such as functioned ZIFs (69, 70, 78, 81, 82), MTV-MOF-5-EHI [36, 103].
3.3.2.3 Separation in Pre-combustion Capture
At high temperature and pressure, a mixture of mostly H2, CO, CO2, and H2O is named synthesis gas (syn gas), which is produced by the steam reforming and partial oxidation of hydrocarbons or a combination of both processes (tandem reforming) in chemical industry [104]. The syn gas will produce H2 and CO2 through the water gas shift reaction at high pressure and slightly elevated temperature (5-40 bar and 313 K). In order to obtain pure H2 for further utilizations, pre-combustion CO2 cap-ture, which refers to the separation of CO2 from H2 within the gas mixture, is required.
A variety of materials are currently under consideration as potential candidates for the pre-combustion CO2 capture, such as porous solid adsorbents and liquid absorbers. Few MOFs have also been investigated as adsorbents for the pressure-swing adsorption-based separation of CO2 from H2. For example, Long’s group reported the separation of CO2 and H2 by Mg-, Ni-MOF-74, and MOF-177 [16, 105, 106]. The results indicated that MOF-74 (Mg- and Ni-) not only have the highest CO2 saturation capacity, but also have a relatively steep CO2 adsorption isotherm. The balance between selectivity and working capacity rendered them extremely promising for the pre-combustion CO2 capture. In contrast, MOF-177 has a high CO2 capacity and a shallow initial rise in its CO2 adsorption isotherm.

993 Metal-Organic Frameworks (MOFs) for CO2 Capture
Because its internal surface imparts little CO2/H2 selectivity, MOF-177 is thus a bad pre-combustion CO2 capture material. These two examples also indicated that the MOFs with high-surface area and CO2 capacity, however, may not be suitable for the pre-combustion CO2 capture.
3.3.2.4 Separation in Oxy-Fuel Combustion Capture
Fossil fuels, such as carbon burning in a nearly pure O2 will produce a relatively small number of CO2, so capture CO2 from this system was defined as oxy-fuel combustion capture. So the gas separation in the oxy-fuel combustion capture is essentially of an O2/N2 separation to get pure O2.
Some MOFs have been checked in the selective adsorption of N2 over O2. For instance, MOF-177 and Zn4O(bdc)(btb)4/3 (UMCM-1) are both high-surface area frameworks with BET surface areas of greater than 4,000 m2/g, in which O2 and N2 adsorption isotherms at 298 K indicate that the selectivity of N2 over O2 is less than 1 (calculated as the number of moles of O2 adsorbed at 0.21 bar divided by the number of moles of N2 adsorbed at 0.79 bar) [107–110]. It is interesting that a MOF, Cr3(btc)2 bearing exposed Cr2+ adsorption sites, showed a highly selec-tive binding of O2 over N2, reported by Long’s group [111]. This MOF adsorbed 11 wt% O2 at 298 K and a pressure of just 2 mbar, while the quantity of N2 adsorp-tion was just 0.58 wt% at 1 bar. The evaluated selectivity of O2 over N2 was 19.3 at room temperature. This observed high adsorption selectivity is mainly due to that the Cr2+ sites prefer to bind O2 molecules. The steep initial rise in the O2 adsorp-tion isotherm is indicative of a strong (chemical) interaction between the Cr2+ sites and O2 molecules. Another example is Fe2(dobdc) reported by the same group, which contains porous channels lined with five-coordinated Fe2+ centers in their surface [112]. The O2 uptake value in this MOF was 10.4 wt% at 298 K and 1 bar, while the N2 uptake was lower, only reaching 1.3 wt% under these conditions. Thus this material exhibited a selectivity factor of 6.6; but the adsorption of O2 at 298 K was irreversible. The isosteric heat of adsorption of N2 and O2 in Fe2(dobdc) was 35 and 41 kJ mol−1, respectively, at zero surface coverage, which showed that this higher propensity for O2 was mainly due to the acceptance of charge from Fe2+. Accordingly, Fe2(dobdc) exhibited a high O2/N2 selectivity ranging from 4.4 to 11 at the temperature range of 201–226 K. Using IAST to simulate breakthrough curves indicated that this MOF was a promising material for the separation of O2 from air at temperatures above those currently used for cryogenic distillations.
3.3.3 MOF-Based Membrane for CO2 Separation
Membrane-based gas separations have attracted intense interest because of their far less energy consumptive and continuous simple process. MOFs, as newly developed porous materials, are attractive in membrane separation; however, there are only a few reports that include gas permeation data available to date,

100 H. Yang and J.-R. Li
and very limited discussions on CO2 separation. Challenges associated with MOF membranes fabrication such as poor substrate-MOF interaction, moisture instability, and easy microscopic and macroscopic crack indeed slowed down the investiga-tion progress of these materials as membranes for the gas separation.
3.3.3.1 MOF Thin Films for CO2 Separation
Pure MOF membranes were fabricated derived from 2009. Resulted MOF-5 thin film was well-intergrown and randomly oriented polycrystalline, with which gas permeation results indicated that small gas molecules exhibited Knudsen diffu-sion due to the large pore size of this MOF [113, 114]. Later on, Keskin and Sholl [115] demonstrated through computational simulation, that the MOF-5 membrane can be used to assess the separation of CO2/CH4 mixtures. The single-component permeation results predicted that MOF-5 membrane showed a strong selectiv-ity for CH4 in CO2/CH4 mixtures whereas the predictions for mixture permea-tion suggested that this membrane offered only weak selectivity for CO2. Another example is HKUST-1 thin film fabricated by Guo et al. [116], which was used to separate H2/N2, H2/CO2, and H2/CH4. The results showed that this MOF film had a high H2 permeation flux (0.107 mol m−2 s−1, better than a lot of zeolite thin films), good separation selectivity of H2 from CO2 with a separation factor of 6.84, but may face a long-term mechanical stability problem due to its freestand-ing nature. Liu et al. [117] also reported the c-oriented ZIF-69 film fabricated on a porous α-alumina. This membrane exhibited a high CO2 permeance better than that expected for Knudsen diffusion despite the large pore size of ZIF-69 (about 7.8 Å), and the CO2/CO selectivity reached 3.5 at room temperature. It was notice-able that the permeability of CO2 was equivalent to a zeolite membrane (zeolite Y) with similar pore size when measured as a single component. However, when CO2 permeability was measured in binary mixture, it was about 5 times higher for the ZIF-69 membrane, which indicated that this MOF membrane has a pref-erential adsorption of CO2 and may be useful in CO2 capture. Particularly, this membrane maintained their crystal structure in boiling benzene, boiling methanol, boiling water, and supercritical CO2, indicating their potential suitability in real environments.
3.3.3.2 MOF-Based Mixed Matrix Membrane for CO2 Separation
Hybrid membrane or MMM using MOFs as the filler material is another option for the application of MOFs in membrane separation. Adams et al. [118] reported an MMM comprised of poly (vinyl acetate) (PVAc) and a MOF composed of cop-per and bdc ligand (Cu-bdc), which exhibited an increased selectivity for many gases, including CO2 upon inclusion of the MOF compared with the pure PVAc membrane. Ordonez et al. reported the ZIF-based polymer MMM using ZIF-8 as the filler phase and Matrimid® as the polymer phase, respectively as shown in

1013 Metal-Organic Frameworks (MOFs) for CO2 Capture
Fig. 3.10. Pure Matrimid® exhibited an ideal selectivity for CO2/CH4 about 43, while at 50 % loading of the ZIF-8 crystals, the ideal selectivity of the MMM increased to 124 [119]. Pure Matrimid® exhibited an adsorption selectivity of 42 for CO2 over CH4 (gas mixture CO2/CH4= 10:9). At 50 % loading, the selectivity of the MMM increased to 89.
3.3.4 Design of MOFs for CO2 Capture
Since the MOFs materials have the characteristics of the structural diversity and tunability, the pore size and property can thus be adjusted according to require-ments. Several strategies have been deduced or proposed to enhance the CO2-selective adsorption and separation ability of MOFs. On the other hand, the design strategies of MOFs for separation are also related to the methods and processes, which imply that adsorptive separation and membranes-based separation have dif-ferent emphases in used materials. For example, for adsorptive separation, high CO2 uptake capacity and selectivity from other gases are equally important for
Fig. 3.10 a Top–down and b cross section FE-SEM images of ZIF-8 membrane with larger, well-intergrown crystals grown from organic-linker-modified supports. c Top-down and d cross-sectional FE-SEM images of a ZIF-8 film with poorly intergrown crystals also grown on modi-fied supports. Reprinted from Ref. [57] Copyright 2011, with permission from Elsevier

102 H. Yang and J.-R. Li
an adsorbent material, while the high penetrability and selectivity are primary for membranes-based separation. In accordance with the requirements of the practi-cal applications in CO2 capture, it is also noticeable that first, the materials should have a good thermal and chemical stability in the real environment; second, effica-cious separation can be done under given practical conditions; last, it must be cost-effective in the material preparation and regeneration.
3.3.4.1 Pore Size Control
Pore size is the most important factor of porous materials used for separations. MOFs have great potential applications in gas separation, due to their adjustable pore size and shape.
For example, Banerjee et al. [91] reported a series of ZIF materials with differ-ent pore sizes for CO2 separation. The pore diameters of this series of ZIFs were varied incrementally from 7.1 to 15.9 Å. The results showed that these ZIFs have a pore-size-dependent selectivity for CO2 capture from its binary mixtures of CH4 and N2, respectively. Furthermore, the cation exchange can also adjust the pore size of MOFs. An et al. reported the cation exchange in modifying the pore size of bio-MOF-1. Their results showed that such modifications can be used to system-atically tune the CO2 adsorption capacity of the material [66]. In addition, control-ling the structural interpenetration of the MOF framework also can adjust the pore size, thereby CO2-selective adsorption properties. For example, Cu(fma)(bpee)0.5 was rationally designed from a primitive cubic net and its pores were tuned by double framework interpenetration [84]. The pore cavities changed from 3.6 to 2.0 × 3.6 Å, leading to MOF presented a highly selective adsorption behavior of CO2 over N2.
3.3.4.2 Pore Surface Functionalization
Compared with non-polar or weakly polar N2, CH4, and H2, the CO2 is highly quadrupolar. This means that there are profound differences in the interaction between these gas molecules and the pore surface of a porous material. This can be taken advantage of through modifying the surface properties of MOFs to enhance their adsorption and separation ability toward different gases. For MOFs, the sur-face properties can be tuned not only by the pre-design of ligands, metal-containing nodes, and MOF construction, but also by the post-modification of existing MOFs. Open active metal sites located on the pore walls of a MOF provide an approach for the enhanced separation of quadrupolar/non-polar gas pairs, such as CO2/CH4. These active adsorption centers are usually created by a post-treatment of MOFs. For example, there are two unsaturated coordination sites in paddle-wheel Cu2(COO)4 units after removing the solvent molecules in the axial positions, which are extensively studied in many MOFs. Typically, activated HKUST-1 has demon-strated a preferential adsorption of CO2 over CH4 and N2 [73, 120].

1033 Metal-Organic Frameworks (MOFs) for CO2 Capture
As mentioned above, the introduction of functional groups with a high affinity for CO2 into pores of MOFs can be employed as another strategy to enhance the adsorption capacity and selectivity of CO2. For example, an amine-functionalized MIL-53 presented an enhanced CO2 uptake relative to CH4 compared with its par-ent MIL-53(Al) was reported by Couck et al. [121]. Furthermore, filling the metal salts inside the pore has also been demonstrated to be an effectively approach in enhancing CO2 binding. Bloch et al. [122] showed that when Al(OH)(bpydc) (MOF-253, bpydc = 2,2′-bipyridine-5,5′-dicarboxylate) was modified to Al(OH)(bpydc)·0.97Cu(BF4)2 by post-synthetic method, the selectivity factor for CO2 over N2 under typical flue gas conditions increased from 2.8 to 12 in the latter. Indeed, the heat of adsorption for CO2 increased from 23 to 30 kJ mol−1 upon the insertion of Cu(BF4)2 into MOF-253, which led to the enhancement of adsorption selectivity.
3.3.4.3 Single-Molecule Trap
Although many MOFs have been prepared for CO2-selective adsorption and sepa-ration, a precise construction implemented at the true molecular level is challenged. Recently, Li et al. [123] demonstrated how it is possible to design a cavity specifi-cally for the capture of a single CO2 molecule, based on both pore size and sur-face functionalization considerations. As shown in Fig. 3.11a, the first step is the design and synthesis of a molecular cavity based on the size of CO2 molecule, which is called “single-molecule trap” (SMT). As molecular building blocks, these SMTs can be linked to construct a 3D MOF to ensure efficient transport of CO2 in and out of the cavities. An alternative route is the impregnation of an existing porous material such as mesoporous silica with SMTs directly. In both cases, the selectivity of the SMT for CO2 would be preserved. Combining experimental and calculated results, they indicated that such a SMT can strengthen CO2-host inter-actions without evoking chemical bonding, thus showing great potential applica-tion in CO2 capture. The results also demonstrated the efficiency of this molecular SMT in the preferential adsorption of CO2 over N2 and CH4, which are important for CO2 capture from flue gas and CH4 upgrading. Furthermore, it was confirmed that the synthesized MOF (PCN-88, [Cu(L3)1/2]n, H4L3 = 5,5′-(naphthalene-2,7-diyl)diisophthalic acid) with the SMTs acting as molecular building units pre-serves the SMT properties in capturing CO2 molecules, therefore showing good performance in the selective adsorption of CO2 over N2 and CH4 (Fig. 3.11b and c).
3.3.4.4 Flexible MOFs
As mentioned above, flexible MOFs have shown additional advantages for the selective adsorption and separation of CO2. External stimuli such as pressure and temperature can be used to induce gating effects, which are desired for the selec-tive adsorption and separation in some cases. This phenomenon has been observed in several flexible MOFs and used in CO2-selective adsorption and separation.
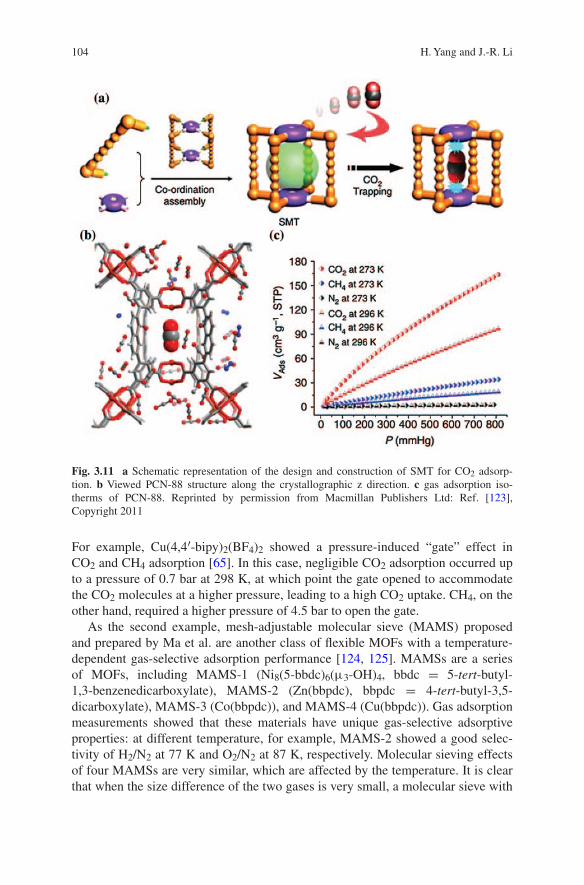
104 H. Yang and J.-R. Li
For example, Cu(4,4′-bipy)2(BF4)2 showed a pressure-induced “gate” effect in CO2 and CH4 adsorption [65]. In this case, negligible CO2 adsorption occurred up to a pressure of 0.7 bar at 298 K, at which point the gate opened to accommodate the CO2 molecules at a higher pressure, leading to a high CO2 uptake. CH4, on the other hand, required a higher pressure of 4.5 bar to open the gate.
As the second example, mesh-adjustable molecular sieve (MAMS) proposed and prepared by Ma et al. are another class of flexible MOFs with a temperature-dependent gas-selective adsorption performance [124, 125]. MAMSs are a series of MOFs, including MAMS-1 (Ni8(5-bbdc)6(μ3-OH)4, bbdc = 5-tert-butyl-1,3-benzenedicarboxylate), MAMS-2 (Zn(bbpdc), bbpdc = 4-tert-butyl-3,5- dicarboxylate), MAMS-3 (Co(bbpdc)), and MAMS-4 (Cu(bbpdc)). Gas adsorption measurements showed that these materials have unique gas-selective adsorptive properties: at different temperature, for example, MAMS-2 showed a good selec-tivity of H2/N2 at 77 K and O2/N2 at 87 K, respectively. Molecular sieving effects of four MAMSs are very similar, which are affected by the temperature. It is clear that when the size difference of the two gases is very small, a molecular sieve with
Fig. 3.11 a Schematic representation of the design and construction of SMT for CO2 adsorp-tion. b Viewed PCN-88 structure along the crystallographic z direction. c gas adsorption iso-therms of PCN-88. Reprinted by permission from Macmillan Publishers Ltd: Ref. [123], Copyright 2011

1053 Metal-Organic Frameworks (MOFs) for CO2 Capture
the precise mesh size is not always readily available. So MAMSs can always meet the separation needs, being highly desirable. Structural tailoring from modifiable ligands is expected to get this type of MOFs with highly selective adsorption abili-ties toward CO2 at near room temperature.
3.3.5 Computational Simulation in MOFs for CO2 Capture
Computational simulation plays an important role in the exploration of CO2 adsorption and separation in porous materials. For MOFs system, a comprehensive review contributed by Zhong’s group can be documented [126]. Compared with experiments, molecular simulations are much easier to be used to study in detail the MOF systems for their optimized performances, such as in CO2 uptake, diffusion, selective adsorption, and separation.
For examples, equilibrium molecular dynamics (EMD) was used to study CO2 diffusion in MOF-5 by Skoulidas and Sholl [127]. The results indicated that self-diffusivity and transport diffusivity had an order of H2 > N2 ≈ CH4 ≈ Ar > CO2 at room temperature and the computed transport diffusivity of CO2 was a non-monotonic function of pore loading. Zhong’s group also studied the separa-tion behaviors of gas mixtures including CO2, CH4, and C2H6 in HKUST-1 by Grand Canonical Monte Carlo (GCMC) simulations [128]. They showed that dif-ferent adsorption properties of the microdomains in this MOF structure led to a significant enhancement of gas separation ability. Furthermore, the adsorption of CO2/CH4/H2 mixture was also studied by computational method [129]. The results showed that gas adsorptive separation could be influenced by the geometry and pore size of the MOF structure. GCMC simulation was also used in the explora-tion of the adsorption sites of light gases in MOF-5 [130]. The results showed that the positions and occupations of the binding sites can be correctly predicted when an appropriate force field was applied. For CO2, the primary adsorption site is located near the Zn4O cluster with its planar orientation pointing toward the zinc atom. In addition, Babarao et al. [131] investigated the adsorptive separation of CO2/CH4 in seven different MOFs, including IRMOF-5, metal-exposed HKUST-1, PCN-6′ and PCN-6, catenated IRMOF-13 and non-catenated IRMOF-14, and charged soc-MOF through molecular simulations. Their results indicated that the adsorption behaviors changed with different pressures and catenated MOFs showed a higher CO2/CH4 selectivity than their non-catenated counterparts.
Heats of adsorption play an important role in gas adsorption and separation, which can be evaluated through computational simulation. For example, the heats of adsorption of seven gases including Kr, Xe, N2, CO2, CH4, n-C4H10, and i-C4H10 in three MOFs of MOF-5, IRMOF-3, and HKUST-1 were studied by Snurr’s group through the molecular simulation [71]. It was found that in general, the experimental and simulated heats of adsorption matched very well each other for IRMOF-1 and IRMOF-3. However, for HKUST-1, the simulation gave a larger adsorption energies (21.8 kJ mol−1) than that evaluated experimentally.

106 H. Yang and J.-R. Li
Besides real MOFs, conceptually modified or hypothetical MOFs can also be explored for CO2 adsorption and separation by the molecular simulation. For example, Zhong’s group systematically investigated nine typical MOFs (IRMOF-1, IRMOF-8, IRMOF-10, IRMOF-11, IRMOF-14, and IRMOF-16, Mn-MOF, MOF-177, and HKUST-1) with various pore sizes, topologies, organic linkers, and electrostatic properties using molecular simulation method [132]. Their results showed that a suitable pore size of a MOF with high CO2 adsorption capacity is around 10–20 Å. And, it is an integrative result of several influencing factors, including the CO2-MOF interactions, surface area, free volume, and electrostatic interactions, leading to a high CO2 uptake in MOFs. However, at low pressures, CO2 uptake is mainly governed by the strength of the CO2-MOF interactions.
CO2 separation in MOF-based membranes was also explored by computa-tional simulations by Sholl’s group [115]. Single-component penetration results predicted a high selectivity for CH4 in the mixtures, while the mixture adsorption and diffusion showed that MOF-5 membrane only had a weak selectivity for CO2. Finally, their results revealed that the performance of MOF membranes cannot be correctly predicted only using the properties of single-component gases, whereas the mixture effect was crucial in determining the membrane separation properties.
On the other hand, new computational methodologies or modified simulation (theory) methods have also been developed toward MOFs for their application in gas adsorption and separation [126]. For instance, density functional theory (DFT) can be used in the accurate description of gas adsorption in MOFs, but is very time-consuming. On the bases of the DFT, the “connectivity-based atom contribution” (CBAC) theory was developed by Zhong’s group, which is built on the assump-tion that the atomic charge of an atom in a MOF is fully determined by its bond-ing environment [133]. Dubbeldam et al. [134] also developed a new computational method to efficiently calculate the unit-cell shape and size of flexible MOF struc-tures loaded with adsorbates. This method is based on the model following tech-nique for rigid bodies, being suitable to treat rigid molecules in a flexible system.
3.4 Conclusion and Outlook
Along with social development, the growing demand for fossil fuels and resulting environmental pollution problems from burning these fuels are the major issues facing humanity in this and even coming centuries. Solving the related energy and environmental problems is the strategic objective to achieve sustainable develop-ment. In this context, more and more anthropogenic CO2 emission is contributing the increasing atmospheric pollutions. Therefore, limiting the CO2 emission and lowering its concentration in air are extremely urgent, where CCS could play an important role. In CCS, adsorption and separation are two of central issues, in which the exploitation of sound adsorbents is critical yet difficult because of the many requirements posed by industry and environmental protection. MOFs rep-resent a new class of crystalline porous materials with advantages, such as easy

1073 Metal-Organic Frameworks (MOFs) for CO2 Capture
structure design and synthesis, high porosity, tailored pore properties, and large numbers of selections. They are thus promising candidates as adsorption and sepa-ration materials for CO2 capture.
In the past 20 years, thousands of MOFs have been synthesized and charac-terized structurally; however, only a small part was checked for CO2 adsorption, storage, and separation. Because it was unreasonable to measure all of these new materials, a further development of scanning techniques was recommended. In exploring CO2 separation and capture in MOFs, there are still some issues wait-ing to be elucidated. Such as, the mixed gas treated in practical CO2 separations always contain water vapor, there have, however, only been a very limited num-ber of evaluations of the effect of water on the separation performances of MOFs. Furthermore, a large number of MOFs were checked by only single-component CO2 adsorption, while about the co-adsorption of CO2 and other gases have been rarely reported. There is no doubt that the measurement of adsorption of a multi-component system is much more challenging than a single-component one. However, it is critical in actual applications. Experimental data with mixed gas systems would also aid in the evaluation of MOFs for their applications not only in adsorptive separation but also in membrane separation of CO2.
Separation using MOF-based membranes is an effective method for the CO2 capture; however, the related investigations are very limited. The development of MOF-based membranes meets with difficulties due to the bad stability of some MOFs, challenging fabrication methods, and expensive organic ligands in some cases. In the practical applications, low cost and high stability of MOFs are neces-sary to realize their potential as membrane for the CO2 capture. In addition, new fabrication methods for MOF-based membranes should be developed to overcome some of the unique challenges this type of new materials faced, such as ease of forming crack during cooling and drying, and poor adherence with support.
Beside the experimental explorations, molecular simulations must be further developed. It is clear that classical and quantum-mechanical-based molecular simu-lations are playing a significant role in the elucidation of adsorption and separa-tion mechanisms, the evaluation of existent materials, and the design of promising new materials. Molecular simulations can not only be used to identify some spe-cial structural MOFs as promising materials for CO2 capture, but also helped to elucidate other factors, such as the structure of geometry, pore size, pore volume, molecular interactions on preferential adsorption, impurities in the mixed gases and structural changes caused by breathing effects. In particular, molecular simulations play important roles in detailing selectivity properties, which help to understand the process of adsorption and desorption and the affecting factors like pressure, loading, and temperature. In addition, molecular simulations are extremely use-ful to evaluate the adsorption performance of hypothetical materials based on the atomistic model of their structures. So it is very helpful in synthesizing new MOFs. Finally, based on these advantages, molecular simulations become more and more important in the study of gas adsorption and separation, such as in MOFs.
On the other hand, it must be pointed out that the thermal/water stability and cyclic application of MOFs during adsorption/desorption are also very important

108 H. Yang and J.-R. Li
when connected to practical applications. In the separation of CO2 from other gases, such as in a post-combustion capture, MOFs are needed in a long-term exposure into gas streams containing water and other components. Some MOFs even have good thermal stability, but bad water stability in most cases, such as MOF-5 decomposed upon contacting with humid air [135]. The synthesis of stable MOFs, especially those being water stable, is thus urgently required for CO2 cap-ture, but still one of challenges in this field.
Clearly, a huge progress has already been made in exploring and improving the properties of MOFs for their application in CO2 adsorption and separation, which confirmed that these classes of newly developed porous materials are quite prom-ising for the real application in CO2 capture. We remain optimistic that MOFs capable of serving as new generation CO2 capture adsorbent will be discovered and utilized in practice, of course through our continued efforts in this field.
References
1. IPCC (2005) IPCC special report on carbon dioxide capture and storage. Cambridge University Press, Cambridge
2. Rackley SA (2010) Carbon capture and storage. Elsevier, Amsterdam 3. Wilson EJ, Gerard D (2007) Carbon capture and sequestration: integrating technology, mon-
itoring, regulation. Wiley, New York 4. Bolhàr-Nordenkampf J (2009) Chemical looping for syngas and power generation with CO2 cap-
ture: pilot plant study and process modeling. Suedwest-deutscher Verlag fuer Hochschulschriften 5. Mokhatab S, Poe WA, Speight JG (2006) Handbook of natural gas transmission and pro-
cessing. Gulf Professional Publishing, Houston 6. Tagliabue M, Farrusseng D, Valencia S et al (2009) Natural gas treating by selective adsorp-
tion: material science and chemical engineering interplay. Chem Eng J 155:553–566 7. Hasib-ur-Rahman M, Siaj M, Larachi F (2010) Ionic liquids for CO2 capture—development
and progress. Chem Eng Process 49:313–322 8. Wappel D, Gronald G, Kalb R et al (2010) Ionic liquids for post-combustion CO2 absorp-
tion. Int J Greenhouse Gas Control 4:486–494 9. Beck DW (1974) Zeolite molecular sieves. Wiley, New York 10. Choi S, Drese JH, Jones CW (2009) Adsorbent materials for carbon dioxide capture from
large anthropogenic point sources. ChemSusChem 2:796–854 11. Yaghi OM, O’Keeffe M, Ockwig NW et al (2003) Reticular synthesis and the design of new
materials. Nature 423:705–714 12. Kitagawa S, Kitaura R, Noro S (2004) Functional porous coordination polymers. Angew
Chem Int Ed 43:2334–2375 13. Ferey G (2008) Hybrid porous solids: past, present, future. Chem Soc Rev 37:191–214 14. Batten SR, Neville SM, Turner DR (2009) Coordination polymers: design, analysis and
application. RSC Publishing, Cambridge 15. MacGillivray L (2010) Metal-organic frameworks: design and application. Wiley, New York 16. Sumida K, Rogow DL, Mason JA et al (2012) Carbon dioxide capture in metal-organic
frameworks. Chem Rev 112:724–781 17. Suh MP, Park HJ, Prasad TK et al (2012) Hydrogen storage in metal-organic frameworks.
Chem Rev 112:782–835 18. Li JR, Sculley J, Zhou HC (2012) Metal-organic frameworks for separations. Chem Rev
112:869–932 19. Kreno LE, Leong K, Farha OK et al (2012) Metal-organic framework materials as chemical
sensors. Chem Rev 112:1105–1125

1093 Metal-Organic Frameworks (MOFs) for CO2 Capture
20. Horcajada P, Gref R, Baati T et al (2012) Metal-organic frameworks in biomedicine. Chem Rev 112:1232–1268
21. Yoon M, Srirambalaji R, Kim K (2012) Homochiral metal-organic frameworks for asym-metric heterogeneous catalysis. Chem Rev 112:1196–1231
22. Tranchemontagne DJ, Mendoza-Cortés JL, O’Keeffe M et al (2009) Secondary build-ing units, nets and bonding in the chemistry of metal–organic frameworks. Chem Soc Rev 38:1257–1283
23. Zhao D, Timmons DJ, Yuan DQ et al (2011) Tuning the topology and functionality of metal-organic frameworks by ligand design. Acc Chem Res 44:123–133
24. Li H, Eddaoudi M, O’Keeffe M et al (1999) Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 402:276–279
25. Kitaura R, Kitagawa S, Kubota Y et al (2002) Formation of a one-dimensional array of oxy-gen in a microporous metal-organic solid. Science 298:2358–2361
26. Férey G, Mellot-Draznieks C, Serre C et al (2005) A chromium terephthalate-based solid with unusually large pore volumes and surface area. Science 309:2040–2042
27. Li JR, Kuppler RJ, Zhou HC (2009) Selective gas adsorption and separation in metal–organic frameworks. Chem Soc Rev 38:1477–1504
28. Stock N, Biswas S (2012) Synthesis of metal-organic frameworks (MOFs): routes to various MOF topologies, morphologies, and composites. Chem Rev 112:933–969
29. O’ Keeffe M, Yaghi OM (2012) Deconstructing the crystal structures of metal-organic frameworks and related materials into their underlying nets. Chem Rev 112:675–702
30. Cohen SM (2012) Postsynthetic methods for the functionalization of metal-organic frame-works. Chem Rev 112:970–1000
31. Chui SSY, Lo SMF, Charmant JPH et al (1999) A chemically functionalizable nanoporous material [Cu3(TMA)2(H2O)3]n. Science 283:1148–1150
32. Ma SQ, Sun DF, Ambrogio M et al (2007) Framework-catenation isomerism in metal−organic frameworks and its impact on hydrogen uptake. J Am Chem Soc 129:1858–1859
33. Li JR, Zhou HC (2010) Bridging-ligand-substitution strategy for the preparation of metal–organic polyhedra. Nat Chem 2:893–898
34. Li JR, Yakovenko AA, Lu W et al (2010) Ligand bridging-angle-driven assembly of molecular architectures based on quadruply bonded Mo–Mo dimers. J Am Chem Soc 132:17599–17610
35. Yuan DQ, Zhao D, Sun DF et al (2010) An isoreticular series of metal-organic frameworks with dendritic hexacarboxylate ligands and exceptionally high gas-uptake capacity. Angew Chem Int Ed 49:5357–5361
36. Banerjee R, Phan A, Wang B et al (2008) High-throughput synthesis of zeolitic imidazolate frameworks and application to CO2 capture.Science 319:939–943
37. Stock N (2010) High-throughput investigations employing solvothermal syntheses. Microporous Mesoporous Mater 129:287–295
38. Jing X, He C, Dong D et al (2012) Homochiral crystallization of metal-organic silver frameworks: asymmetric [3 + 2] cycloaddition of an azomethine ylide. Angew Chem Int Ed 40:10127–10131
39. Lin ZJ, Slawin AMZ, Morris RE (2007) Chiral induction in the ionothermal synthesis of a 3D coordination polymer. J Am Chem Soc 129:4880–4881
40. Jhung SH, Lee JH, Chang JS (2005) Microwave synthesis of a nanoporous hybrid material, chromium trimesate. Bull Korean Chem Soc 26:880–881
41. Schlesinger M, Schulze S, Hietschold M et al (2010) Evaluation of synthetic methods for microporous metal–organic frameworks exemplified by the competitive formation of [Cu2(btc)3(H2O)3] and [Cu2(btc)(OH)(H2O)]. Microporous Mesoporous Mater 132:121–127
42. Pichon A, Lazuen-Garaya A, James SL (2006) Solvent-free synthesis of a microporous metal–organic framework. CrystEngComm 8:211–214
43. Chen BL, Ockwig NW, Millward AR et al (2005) High H2 adsorption in a microporous metal-organic framework with open metal sites. Angew Chem Int Ed 44:4745–4749
44. Millange F, Serre C, Férey G (2002) Synthesis, structure determination and properties of MIL-53 as and MIL-53ht: the first CrIII hybrid inorganic–organic microporous solids: CrIII(OH) · {O2C–C6H4–CO2} · {HO2C–C6H4–CO2H}x. Chem Commun 38:822–823

110 H. Yang and J.-R. Li
45. Serre C, Millange F, Thouvenot C et al (2002) Very large breathing effect in the first nanopo-rous chromium(III)-based solids: MIL-53 or CrIII(OH) · {O2C–C6H4–CO2} · {HO2C–C6H4–CO2H}x · H2Oy. J Am Chem Soc 124:13519–13526
46. Loiseau T, Serre C, Huguenard C et al (2004) A rationale for the large breathing of the porous aluminum terephthalate (MIL-53) upon hydration. Chem Eur J 10:1373–1382
47. Cui YJ, Yue YF, Qian GD et al (2012) Luminescent functional metal-organic frameworks. Chem Rev 112:1126–1162
48. Lan An J, Li KH, Wu HH et al (2009) A luminescent microporous metal-organic framework for the fast and reversible detection of high explosives. Angew Chem Int Ed 48:2334–2338
49. Pramanik S, Zheng C, Zhang X et al (2011) New microporous metal–organic framework demonstrating unique selectivity for detection of high explosives and aromatic compounds. J Am Chem Soc 133:4153–4155
50. Furukawa H, Ko N, Go YB et al (2010) Ultrahigh porosity in metal-organic frameworks. Science 329:424–428
51. Makal TA, Li JR, Lu W et al (2012) Methane storage in advanced porous materials. Chem Soc Rev 41:7761–7779
52. Wang XS, Ma SQ, Rauch K et al (2008) Metal–organic frameworks based on double-bond-coupled di-isophthalate linkers with high hydrogen and methane uptakes. Chem Mater 20:3145–3152
53. Ma SQ, Sun DF, Simmons JM et al (2008) Metal-organic framework from an anthracene derivative containing nanoscopic cages exhibiting high methane uptake. J Am Chem Soc 130:1012–1016
54. Seo JS, Whang D, Lee H et al (2000) A homochiral metal–organic porous material for enan-tioselective separation and catalysis. Nature 404:982–986
55. Evans OR, Ngo HL, Lin W (2001) Chiral porous solids based on lamellar lanthanide phos-phonates. J Am Chem Soc 123:10395–10396
56. Horcajada P, Serre C, Vallet-Regí M et al (2006) Metal-organic frameworks as efficient materials for drug delivery. Angew Chem Int Ed 45:5974–5978
57. Li JR, Ma YG, McCarthy MC et al (2011) Carbon dioxide capture-related gas adsorption and separation in metal-organic frameworks. Coord Chem Rev 255:1791–1823
58. Mason JA, Sumida K, Herm ZR et al (2011) Evaluating metal-organic frameworks for post-combustion carbon dioxide capture via temperature swing adsorption. Energy Environ Sci 4: 3030-3040
59. Bao Z, Yu L, Ren Q et al (2011) Adsorption of CO2 and CH4 on a magnesium-based metal organic framework. J Colloid Interface Sci 353:549–556
60. Myers AL, Prausnitz JM (1965) Thermodynamics of mixed-gas adsorption. AIChE J 11:121–127
61. Saha D, Bao ZB, Jia F et al (2010) Adsorption of CO2, CH4, N2O, and N2 on MOF-5, MOF-177, and zeolite 5A. Environ Sci Technol 44:1820–1826
62. Zheng B, Bai J, Duan J et al (2011) Enhanced CO2 binding affinity of a high-uptake rht-type metal–organic framework decorated with acylamide groups. J Am Chem Soc 133:748–751
63. Demessence A, D’Alessandro DM, Foo ML et al (2009) Strong CO2 binding in a water-stable, triazolate-bridged metal–organic framework functionalized with ethylenediamine. J Am Chem Soc 131:8784–8786
64. Bae YS, Farha OK, Hupp JT et al (2009) Enhancement of CO2/N2 selectivity in a metal-organic framework by cavity modification. J Mater Chem 19:2131–2134
65. Kondo A, Chinen A, Kajiro H et al (2009) Metal-ion-dependent gas sorptivity of elastic layer-structured MOFs. Chem Eur J 15:7549–7553
66. An J, Rosi NL (2010) Tuning MOF CO2 adsorption properties via cation exchange. J Am Chem Soc 132:5578–5579
67. Maji TK, Matsuda R, Kitagawa S (2007) A flexible interpenetrating coordination framework with a bimodal porous functionality. Nat Mater 6:142–148
68. Yazaydin AÖ, Benin AI, Faheem SA et al (2009) Enhanced CO2 adsorption in metal-organic frameworks via occupation of open-metal sites by coordinated water molecules. Chem Mater 21:1425–1430

1113 Metal-Organic Frameworks (MOFs) for CO2 Capture
69. Llewellyn PL, Bourrrelly S, Serre C et al (2006) How hydration drastically improves adsorption selectivity for CO2 over CH4 in the flexible chromium terephthalate MIL-53. Angew Chem Int Ed 4:7751–7754
70. Keskin S, van Heest TM, Sholl DS (2010) Can metal-organic framework materials play a useful role in large-scale carbon dioxide separations? ChemSusChem 3:879–891
71. Farrusseng D, Daniel C, Gaudillere C et al (2009) Heats of adsorption for seven gases in three metal-organic frameworks: systematic comparison of experiment and simulation. Langmuir 25:7383–7388
72. Vaidhyanathan R, Iremonger SS, Shimizu GKH et al (2010) Direct observation and quan-tification of CO2 binding within an amine-functionalized nanoporous solid. Science 330:650–653
73. Bordiga S, Regli L, Bonino F et al (2007) Adsorption properties of HKUST-1 toward hydro-gen and other small molecules monitored by IR. Phys Chem Chem Phys 9:2676–2685
74. Dietzel PDC, Johnsen RE, Fjellvag H et al (2008) Adsorption properties and structure of CO2 adsorbed on open coordination sites of metal–organic framework Ni2(dhtp) from gas adsorption, IR spectroscopy and X-ray diffraction. Chem Commun 41:5125–5127
75. Vimont A, Travert A, Bazin P et al (2007) Evidence of CO2 molecule acting as an elec-tron acceptor on a nanoporous metal–organic-framework MIL-53 or Cr3+(OH)(O2C–C6H4–CO2). Chem Commun 31:3291–3293
76. Li JR, Tao Y, Yu Q et al (2008) Selective gas adsorption and unique structural topology of a highly stable guest-free zeolite-type MOF material with N-rich chiral open channels. Chem Eur J 14:2771–2776
77. Dybtsev DN, Chun H, Yoon SH et al (2004) Microporous manganese formate: a simple metal–organic porous material with high framework stability and highly selective gas sorp-tion properties. J Am Chem Soc 126:32–33
78. Loiseau T, Lecroq L, Volkringer C et al (2006) MIL-96, a porous aluminum trimesate 3D structure constructed from a hexagonal network of 18-membered rings and μ3-oxo-centered trinuclear units. J Am Chem Soc 128:10223–10230
79. Xue M, Ma SQ, Jin Z et al (2008) Robust metal–organic framework enforced by triple-framework interpenetration exhibiting high H2 storage density. Inorg Chem 47:6825–6828
80. Ma SQ, Wang XS, Yuan DQ et al (2008) A coordinatively linked Yb metal-organic frame-work demonstrates high thermal stability and uncommon gas-adsorption selectivity. Angew Chem Int Ed 47:4130–4133
81. Bae YS, Mulfort KL, Frost H et al (2008) Separation of CO2 from CH4 using mixed-ligand metal–organic frameworks. Langmuir 24:8592–8598
82. Bae YS, Farha OK, Spokoyny AM (2008) Carborane-based metal–organic frameworks as highly selective sorbents for CO2 over methane. Chem Commun 35:4135–4137
83. Wang B, Côte AP, Furukawa H et al (2008) Colossal cages in zeolitic imidazolate frame-works as selective carbon dioxide reservoirs. Nature 453:207–211
84. Chen BL, Ma SQ, Zapata F et al (2007) Rationally designed micropores within a metal–organic framework for selective sorption of gas molecules. Inorg Chem 46:1233–1236
85. Bourrelly S, Llewellyn PL, Serre C et al (2005) Different adsorption behaviors of methane and carbon dioxide in the isotypic nanoporous metal terephthalates MIL-53 and MIL-47. J Am Chem Soc 127:13519–13521
86. Hamon L, Llewellyn PL, Devic T, Ghoufi A et al (2009) Co-adsorption and separa-tion of CO2–CH4 mixtures in the highly flexible MIL-53(Cr) MOF. J Am Chem Soc 131:17490–17499
87. Férey G, Serre C (2009) Large breathing effects in three-dimensional porous hybrid matter: facts, analyses, rules and consequences. Chem Soc Rev 38:1380–1399
88. Culp JT, Smith MR, Bittner E et al (2008) Hysteresis in the physisorption of CO2 and N2 in a flexible pillared layer nickel cyanide. J Am Chem Soc 130:12427–12434
89. Hayashi H, Côte AP, Furukawa H et al (2007) Zeolite A imidazolate frameworks. Nat Mater 6:501–506
90. Kitaura R, Seki K, Akiyama G et al (2003) Porous coordination-polymer crystals with gated channels specific for supercritical gases. Angew Chem Int Ed 42:428–431

112 H. Yang and J.-R. Li
91. Banerjee R, Furukawa H, Britt D et al (2009) Control of pore size and functionality in iso-reticular zeolitic imidazolate frameworks and their carbon dioxide selective capture proper-ties. J Am Chem Soc 131:3875–3877
92. Britt D, Furukawa H, Wang B et al (2009) Highly efficient separation of carbon dioxide by a metal–organic framework replete with open metal sites. Proc Natl Acad Sci USA 106:20637–20640
93. Ramsahye NA, Maurin G, Bourrelly S (2007) On the breathing effect of a metal–organic framework upon CO2 adsorption: monte carlo compared to microcalorimetry experiments. Chem Commun 31:3261–3263
94. Finsy V, Ma L, Alaerts L et al (2009) Separation of CO2/CH4 mixtures with the MIL-53(Al) metal–organic framework. Micropor Mesopor Mater 120:221–227
95. Bárcia PS, Bastin L, Hurtado EJ et al (2008) Single and multicomponent sorption of CO2, CH4 and N2 in a microporous metal organic framework. Sep Sci Technol 43:3494–3521
96. Bastin L, Bárcia PS, Hurtado EJ et al (2008) A microporous metal−organic frame-work for separation of CO2/N2 and CO2/CH4 by fixed-bed adsorption. J Phys Chem C 112:1575–1581
97. Yoon JW, Jhung SH, Hwang YK et al (2007) Gas-sorption selectivity of CUK-1: a porous coordination solid made of cobalt(II) and pyridine-2,4- dicarboxylic acid. Adv Mater 19:1830–1834
98. Quartapelle Procopio E, Linares F, Montoro C et al (2010) Cation-exchange porosity tuning in anionic metal-organic frameworks for the selective separation of gases and vapors and for catalysis. Angew Chem Int Ed 49:7308–7311
99. Millward AR, Yaghi OM (2005) Metal–organic frameworks with exceptionally high capac-ity for storage of carbon dioxide at room temperature. J Am Chem Soc 127:17998–17999
100. McDonald TM, Lee WR, Mason JA et al (2012) Capture of carbon dioxide from air and flue gas in the alkylamine-appended metal-organic framework mmen-Mg2(dobpdc). J Am Chem Soc 134:7056–7065
101. Arstad B, Fjellvåg H, Kongshaug KO et al (2008) Amine functionalised metal organic frameworks (MOFs) as adsorbents for carbon dioxide. Adsorption 14:755–762
102. Stavitski E, Pidko EA, Couck S et al (2011) Complexity behind CO2 capture on NH2-MIL-53(Al). Langmuir 27:3970–3976
103. Deng HX, Doonan CJ, Furukawa H et al (2010) Multiple functional groups of varying ratios in metal-organic frameworks. Science 327:846–850
104. Higman C, Burgt V (2003) Gasification. Elsevier, Amsterdam 105. Dietzel PDC, Besikiotis V, Blom R (2009) Application of metal–organic frameworks with
coordinatively unsaturated metal sites in storage and separation of methane and carbon dioxide. J Mater Chem 19:7362–7370
106. Herm ZR, Swisher JA, Smit B et al (2011) Metal–organic frameworks as adsorbents for hydro-gen purification and precombustion carbon dioxide capture. J Am Chem Soc 133:5664–5667
107. Mu B, Schoenecker PM, Walton KS (2010) Gas adsorption study on mesoporous metal–organic framework UMCM-1. J Phys Chem C 114:6464–6471
108. García-Ricard OJ, Hernández-Maldonado A (2010) Cu2(pyrazine-2,3-dicarboxylate)2(4,4′-bipyridine) porous coordination sorbents: activation temperature, textural properties, and CO2 adsorption at low pressure range. J Phys Chem C 114:1827–1834
109. Li Y, Yang RT (2007) Gas adsorption and storage in metal–organic framework MOF-177. Langmuir 23:12937–12944
110. Wang QM, Shen D, Bülow M et al (2002) Metallo-organic molecular sieve for gas separa-tion and purification. Microporous Mesoporous Mater 55:217–230
111. Murray LJ, Dincă M, Yano J et al (2010) Highly-selective and reversible O2 binding in Cr3(1,3,5-benzenetricarboxylate)2. J Am Chem Soc 132:7856–7857
112. Bloch ED, Murray LJ, Queen WL et al (2011) Selective binding of O2 over N2 in a redox-active metal–organic framework with open iron (II) coordination sites. J Am Chem Soc 133:14814–14822
113. Liu YY, Ng ZF, Khan EA et al (2009) Synthesis of continuous MOF-5 membranes on porous α-alumina substrates. Micropor Mesopor Mater 118:296–301

1133 Metal-Organic Frameworks (MOFs) for CO2 Capture
114. Yoo Y, Lai ZP, Jeong HK (2009) Fabrication of MOF-5 membranes using microwave-induced rapid seeding and solvothermal secondary growth Micropor Mesopor Mater 123:100–106
115. Keskin S, Sholl D (2007) Screening meta–organic framework materials for membrane-based methane/carbon dioxide separations. J Phys Chem C 111:14055–14059
116. Guo H, Zhu G, Hewitt IJ et al (2009) “Twin copper source” growth of metal–organic frame-work membrane: Cu3(BTC)2 with high permeability and selectivity for recycling H2. J Am Chem Soc 131:1646–1647
117. Liu Y, Hu E, Khan EA et al (2010) Synthesis and characterization of ZIF-69 membranes and separation for CO2/CO mixture. J Membr Sci 353:36–40
118. Adams R, Carson C, Ward J et al (2010) Metal organic framework mixed matrix membranes for gas separations. Micropor Mesopor Mater 131:13–20
119. Ordoñez MJC, Balkus KJ Jr, Ferraris JP et al (2010) Molecular sieving realized with ZIF-8/Matrimid® mixed-matrix membranes. J Membr Sci 361:28–37
120. Liang ZJ, Marshall M, Chaffee AL (2009) CO2 adsorption-based separation by metal organic framework (Cu-BTC) versus zeolite (13X). Energy Fuels 23:2785–2789
121. Couck S, Denayer JFM, Baron GV et al (2009) An amine-functionalized MIL-53 metal–organic framework with large separation power for CO2 and CH4. J Am Chem Soc 131:6326–6327
122. Bloch ED, Britt D, Lee C et al (2010) Metal insertion in a microporous metal–organic framework lined with 2,2′-bipyridine. J Am Chem Soc 132:14382–14384
123. Li JR, Yu JM, Lu WG et al (2013) Porous materials with pre-designed single-molecule traps for CO2 selective adsorption. Nat Comm 4:1538–1545
124. Ma SQ, Sun DF, Wang XS et al (2007) A mesh-adjustable molecular sieve for general use in gas separation. Angew Chem Int Ed 46:2458–2462
125. Ma SQ, Sun DF, Yuan DQ et al (2009) Preparation and gas adsorption studies of three mesh-adjustable molecular sieves with a common structure. J Am Chem Soc 131:6445–6451
126. Yang QY, Liu DH, Zhong C et al (2013) Development of computational methodolo-gies for metal-organic frameworks and their application in gas separations. Chem Rev 113:8261–8323
127. Skoulidas AI, Sholl DS (2005) Self-diffusion and transport diffusion of light gases in metal-organic framework materials assessed using molecular dynamics simulations. J Phys Chem B 109:15760–15768
128. Yang Q, Zhong C (2006) Electrostatic-field-induced enhancement of gas mixture separation in metal-organic frameworks: a computational study. ChemPhysChem 7:1417–1421
129. Yang Q, Zhong C (2006) Molecular simulation of carbon dioxide/methane/hydrogen mix-ture adsorption in metal–organic frameworks. J Phys Chem B 110:17776–17783
130. Dubbeldam D, Frost H, Walton KS et al (2007) Molecular simulation of adsorption sites of light gases in the metal-organic framework IRMOF-1. Fluid Phase Equilib 261:152–161
131. Babarao R, Jiang J, Sandler SI (2009) Molecular simulations for adsorptive separation of CO2/CH4 mixture in metal-exposed, catenated, and charged metal–organic frameworks. Langmuir 25:6590–6590
132. Yang QY, Zhong CL, Chen JF (2008) Computational study of CO2 storage in metal–organic frameworks. J Phys Chem C 112:1562–1569
133. Xu Q, Zhong C (2010) A general approach for estimating framework charges in metal–organic frameworks. J Phys Chem C 114:5035–5042
134. Dubbeldam D, Krishna R, Snurr RQ (2009) Method for analyzing structural changes of flexible metal–organic frameworks induced by adsorbates. J Phys Chem C 113:19317–19327
135. Lu CM, Liu J, Xiao KF et al (2010) Microwave enhanced synthesis of MOF-5 and its CO2 capture ability at moderate temperatures across multiple capture and release cycles. Chem Eng J 156:465–470

115
Abstract Porous solids have been proved to be good candidates as the carbon dioxide recycling sorbents. In the last decades, many efforts were devoted to improving the surface area and heat of adsorption of artificial porous materials. Among those synthesized porous solids with ultrahigh surface area, porous aro-matic frameworks (PAFs) possess ultrahigh Brunauer–Emmett–Teller (BET) sur-face area and excellent physicochemical stability, which can meet the criteria of carbon dioxide storage and separation. PAFs are the new generation of a whole new class of organic networks with an intrinsic nanoporosity. They are character-ized by a rigid aromatic open-framework structure constructed by covalent bonds that remain accessible to small molecules. In this chapter, the design, synthesis, and carbon dioxide adsorption properties of PAFs are discussed.
4.1 Introduction of Porous Aromatic Frameworks
The pore is the central structural motif in host–guest functional materials, such as zeolites, and is also the ubiquitous object in nature from microscopic to macro-scopic points of view. Most importantly, the porous structure provides inherently accessible space, for guest, where chemical reaction or physical sorption could take place; therefore, they are always addressed as open frameworks. Accordingly, if one of the open frameworks could be selectively synthesized, assembled, or con-structed for molecules, supramolecules, or polymers, they should form continuously
Chapter 4Carbon Dioxide Capture in Porous Aromatic Frameworks
Teng Ben and Shilun Qiu
A.-H. Lu and S. Dai (eds.), Porous Materials for Carbon Dioxide Capture, Green Chemistry and Sustainable Technology, DOI: 10.1007/978-3-642-54646-4_4, © Springer-Verlag Berlin Heidelberg 2014
T. Ben Department of Chemistry, Jilin University, Changchun, Jilin, People’s Republic of China
S. Qiu (*) State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University, Changchun, Jilin, People’s Republic of Chinae-mail: [email protected]

116 T. Ben and S. Qiu
communicating pore passages. Inspired by the success of sophisticated natural and artificial zeolites that are key for their elaborate functional in modern chemical industry involving molecular storage, recognition, separation, and catalytic activity [1, 2], chemists have been challenged to develop porous organic–inorganic hybrid and organic frameworks, supramolecules, and oligomers with a controlled func-tional pore, not only to mimic zeolite structures and functions but also for their potential applications in material science, such as molecular separation [3], clean energy storage [4, 5], photoelectric materials [6, 7], molecular motor [8], and cataly-sis [9]. Based on such ideas, in the last decades, porous organic frameworks (POFs) such as metal-organic frameworks (MOFs) [10–12], covalent organic frameworks (COFs) [13–15], polymers of intrinsic microporosity (PIMs) [16, 17], conjugated microporous polymers (CMPs) [18–23], hyper-cross-linked polymers (HCPs) [24, 25], and porous aromatic frameworks (PAFs) [26–33] have been prepared and the functions of nanopores are also explored. The first example of introducing organic framework into pore materials was reported by Kitagawa et al. [34]. With that, Yaghi et al. and Williams et al. established the gas sorption method to detect the pore structure of POFs by Langmuir and Brunauer–Emmett–Teller (BET) theories, respectively, in 1998 and 1999, which was proved to be a standard of characteriza-tion of porosity of microporous organic frameworks [35, 36]. Since then, gas sorp-tion experiment was also used to explore the surface area and pore size of organic porous polymer. In 2002, Budd and Mckeown described the first PIM material with surface area about 950 m2 g−1 [37]. After 3 years, in 2005, Yaghi and coworkers synthesized first crystalline covalent linked organic framework [13]. The surface area of COFs could achieve 4,200 m2 g−1, which surpasses all the porous polymer networks at that time [14]. Sherrington et al. showed the “Davankov-type” hyper-cross-linked polymer with the surface area up to 2,000 m2 g−1 in 2006 [38, 39]. One year later, a first CMP was discovered by Cooper et al. which expressed multiple functions with high surface area [18].
In 2009, Qiu et al. have developed a method to synthesize the first long-range-ordered PAF with dia topology (PAF-1) [26], which shows a record surface area (SBET = 5,640 m2 g−1) at that time and exceptional physicochemical stability via a nickel(0)-catalyzed Yamamoto-type Ullmann cross-coupling [40, 41]. Besides, PAF-1 also shows very high uptakes of carbon dioxide (1.3 g g−1 at 40 bar, 298 K) to make it a good candidate for carbon dioxide storage. This new type of three-dimen-sional homogeneous, rigid, and open-network structure provides the PAF of “new generation” with several unusual, even peculiar properties. This chapter describes the significant progress in the development of PAFs, with a particular focus on the rela-tionship between structure design, synthesis method, and carbon dioxide properties.
4.2 Carbon Dioxide Capture by Microporous Materials
World climate and environment are now strongly suffered from greenhouse gas which is over emitted associated with human activities. Though there are many efforts to develop new techniques for alternated energy such as wind, solar, and

1174 Carbon Dioxide Capture in Porous Aromatic Frameworks
tidal to alternate the traditional energy sources, there are about 86 % energy originating from fossil fuels, coal, petroleum, and natural gas which have signifi-cantly increased the CO2 concentration in atmosphere from 280 to 385 ppm and methane concentration has almost doubled [42]. The Intergovernmental Panel on Climate Change (IPCC) pointed out that carbon capture and storage (CCS) [43] was an optional approach to reduce CO2 concentration and stop continuous global warming. Another approach is seeking and developing renewable and clean energy techniques and sources such as hydrogen and methane to alternate the dependence of traditional carbon energy.
CCS [43], also referred to as carbon capture and sequestration, is recently a hot topic and important technology to reduce the release of greenhouse, e.g., carbon dioxide, into the atmosphere from the use of fossil fuel, biomass consuming power generation plant and other industries [44, 45]. Pre-combustion, post-combustion capture, and oxyfuel combustion are three main types of CCS technologies which require different conditions and evaluation criterion for carbon capture. In case of oxyfuel combustion, the final outlet is carbon dioxide and water which is readily be separated. In pre-combustion, which is widely applied in fertilizer, chemi-cal, gaseous fuel, and power production, CO2 acts as final outcome and impuri-ties which need to be removed from hydrogen gas flue. In the case of large point sources such as power stations, post-combustion capture is often applied, in which the CO2 needs to be removed after combustion of the fossil fuel and biomass. During this procedure, the flue gas at power plants mainly consists of carbon diox-ide (15–16 %), nitrogen (70–75 %), water vapor (5–7 %) at ~1 bar. Besides, with the same goal as CCS, applying clean energy such as methane is also critical to reduce the CO2 emission level. However, as a main source of methane, natural gas always contains a certain percent of CO2 impurities. Therefore, separation or cap-ture of CO2 from the natural gas is very significant in the industrial application. Another very important application of carbon dioxide capture is in enclosed space such as aircraft, spacecraft, or submarine. In this case, carbon dioxide is highly diluted. The technique of how to extract carbon dioxide from air is also desired.
All the above-mentioned cases express the urgent needs of CO2 separation from gas mixtures. Conventional method to remove CO2 employed in power station is “wet scrubbing” by amine solutions such as monoethanolamine (MEA). But regeneration of MEA solvents needs more energy which equate about 25–40 % of the power plant’s energy output. Moreover, when consider the prerequisite of cor-rosive protection of MEA solvents, such technique increases the costs for electric-ity generation and reduces efficiencies. This encourages the interest of pursuing advanced CO2 capture materials with high selectivity for CO2 (none CO2 gases).
Microporous materials have been proved to have the potential applica-tion in gas storage and separation by physical adsorption during last decades [46]. Such application is usually based on the presence of a large permanent surface area, suitable pore size distribution, and suitable enthalpy of adsorp-tion between micropore and gas molecules of porous sorbents. Recent research revealed that microporous materials are considered as potential carbon capture media due to their relatively weak physical interaction with carbon dioxide, which renders the recovery of the gas and recycling of the sorbent less energy

118 T. Ben and S. Qiu
consuming than processes where carbon dioxide is bound chemically. Pressure swing adsorption (PSA), vacuum swing adsorption (VSA), and temperature swing adsorption (TSA) are three main processes which have been applied for carbon dioxide adsorption and recovery by microporous materials. PSA always applied in the case that adsorption occurred at pressure higher than 1 bar, which large amounts of carbon dioxide could be stored. VSA is similar to PSA, but uses different sorption pressure lower than 1 bar. TSA relies on the difference in carbon dioxide uptakes at different temperatures. It is advantageous for PSA, VSA, and TSA processes to develop microporous materials that can provide effective working capacity over a limited energy punishment to adsorb and recover the carbon dioxide.
4.3 Design of Porous Aromatic Framework for Carbon Dioxide Capture
4.3.1 High Surface Area
Before the appearance of PAFs, there are plenty of research on the synthesis and applications of crystalline zeolite analogs such as MOFs and COFs. A practi-cal drawback of MOFs and COFs is that most of them possess limited physico-chemical stability. By contrast, there also has a growing range of porous polymer networks comprising stable covalent C–C, C–H, and C–N bonds with high phys-icochemical stability. Unfortunately, the highest surface area of covalent linked porous polymer networks are among 3,000 m2 g−1 at that time. The emerging of PAFs first provides an ideal microporous material combining ultrahigh surface area and high physicochemical stability.
The original idea of synthesis of PAF-1 comes from the fantastic structure and properties of diamond, in which each carbon atom is tetrahedrally connected to four neighboring atoms by covalent bonds, when breaking the C–C covalent bond of diamond and inserting rigid phenyl rings should allow sufficient exposure of the faces and edges of phenyl rings with the expectation of increasing the inter-nal surface areas share dia topology. Before starting synthesis, we systemati-cally calculated the surface area and density of PAF-301, PAF-1, and PAF-303 (Fig. 4.1) by employing a multiscale simulation method combining first-principles calculations and grand canonical Monte Carlo (GCMC) simulation. Results indi-cate that PAF-301 share a Langmuir surface area of 2,350 m2 g−1 (BET surface area, 1,880 m2 g−1) with a density of 0.8364 g cm−3 and PAF-1 show a Langmuir surface area of 7,000 m2 g−1 (BET surface area, 5,640 m2 g−1) with a density of 0.315 g cm−3. The simulation result of PAF-303 indicated that its pore sizes were so large and were broken into the mesoscale range. Based on the simulation results, PAF-1 shows a record surface area which has a broad potential application in hydrogen storage and carbon capture.

1194 Carbon Dioxide Capture in Porous Aromatic Frameworks
After carefully design of the second building unit (SBU), a proper growing method should be chosen for link all SBUs into an open framework. Based on our targeted network, reactions of formation of C–C bond are always taken into account. Particularly for synthesis of PAF-1, optimized nickel(0)-catalyzed Yamamoto-type Ullmann cross-coupling was used. The Yamamoto coupling in general is an aryl–aryl coupling of aryl-halogenide compounds mediated mostly by stoichiometric amounts of bis(1,5-cyclooctadiene)nickel(0) (Ni(COD)2 for short). The polymerization is advantageous as self-polycondensation with just a single, halogen-functionalized SBU can be used to form organic framework. The mechanism of Yamamoto coupling is shown in Scheme 4.1 [40]. Firstly, the oxi-dative addition is triggered between Ni(0)Lm and halogen-functionalized mono-mer. After the disproportionation of two complex (I) and complex (II) of nickel, reductive elimination of complex (III) leads to regeneration of Ni(0)Lm and addi-tion product. During the recycling period of the coupling reaction, stoichiomet-ric Ni(0)Lm is consumed when aryl–aryl bond is formed. That is, the Ni(COD)2 has to be used in excess (1.2–2.8 equiv of per aryl–aryl bond formed) to allow high yield of the desired product. Compared with other C–C coupling reactions such as Sonogashira-Hagihara routes [47–56] and Suzuki cross-coupling [57–66], Yamamoto coupling shows unexpected halogen elimination ability of ending group.
Fig. 4.1 Structure model of synthesized and simulated PAFs (C purple; N blue; Si yellow, O green; Ge brown)

120 T. Ben and S. Qiu
This makes it unique to prepare ultrahigh porosity solid because heavy ending halogen atoms evidently decrease the surface area. By using Yamamoto-type Ullmann cross-coupling, PAF-1 was synthesized successfully, and for the first time, a covalent linked porous organic framework showed unexpected high surface area of 5,640 m2 g−1 by BET theory. Furthermore, we continuously synthesized PAFs using the same method containing quadricovalent Si (PAF-3) and Ge (PAF-4) [29]. These PAFs were thermally stable up to 465 °C for PAF-3 and 443 °C for PAF-4. Like PAF-1, PAF-3 also shows high surface area (up to 2,932 m2 g−1) and excellent adsorption ability to carbon dioxide, hydrogen, and methane. The synthesis of PAF-3 via Yamamoto coupling was first reported by Cooper et al. in 2010 (SBET = 1,102 m2 g−1) [67]. Recently, this structure was synthesized by optimized Yamamoto coupling again, known as PPN-4 (Fig. 4.2), and a world record BET surface area as 6,461 m2 g−1 was revealed [68]. Combined with such
Fig. 4.2 a Synthetic route for PPN-3 (X: Adamantane), PPN-4 (X: Si), PPN-5 (X: Ge), and PAF-1 (X: C). b The default non-interpenetrated diamondoid network of PPN-4 (black C; pale gray H; gray Si). Reproduced from Ref. [68] with permission from Wiley
Scheme 4.1 Mechanism of Yamamoto of coupling

1214 Carbon Dioxide Capture in Porous Aromatic Frameworks
an impressive surface area, PPN-4 can adsorb 2,121 mg g−1 CO2 (212 wt%) at 50 bar/295 K, 158 mg g−1 hydrogen at 77 K/80 bar, and 389 mg g−1 methane at 298 K/55 bar. This result proved again that Yamamoto coupling is an efficient method for the synthesis of ultrahigh surface area microporous networks.
The microporous organic framework sharing dia topology structure was further explored via Suzuki cross-coupling reaction by Yuan et al. The targeted PAF-11 [32] was linked by quaterphenyl group. That is, the pore size of PAF-11 should reach mesoporous theoretically. Results show that the yield is lower than that of PAF-1, and the CP/MAS NMR and FTIR revealed bromo-capping species were found. As an inevitable result, the apparent surface area derived from the BET model is 704 m2 g−1, which is much lower than PAF-1. Compared with very nar-row pore size distribution observed in PAF-1, a very widespread pore size distribu-tion of PAF-11 between 0.5 and 5 nm was found. Though there is no proof of how much possibility and degree of interpenetration of PAF-11 may take to reduce the surface area, a doubtless fact should be noted that Yamamoto-type Ullmann cross-coupling has more efficiency and specificity for aryl–aryl coupling.
Compared with other ultrahigh surface area solid such as porous carbons, porous silicas, zeolites, MOFs, and microporous polymers, PAFs show very high surface area and excellent physicochemical stability. PAF-1 is the first example of PAFs with a recorded surface area of 5,640 m2 g−1 at that time. Based on its powder X-ray diffraction (PXRD) patterns, PAF-1 shows long-range order and a certain extent amorphous nature as well as the TEM results reveal all the pores are wormlike but with uniform pore size. PPN-4 share ultrahigh surface area and predominantly amorphous. These results broke a misleading notion that exception-ally high surface areas are the preserve of highly ordered molecular networks. We believe that rigid biphenyl framework, three-dimensional dia topology and elimi-nation of ending bromo group (heavy atoms) are keys to prepare high surface area materials. Because there are so many porous polymer networks that are reported with lower surface area even some of those also share dia topology, they are worth to be explored again by more efficiently synthesis route.
4.3.2 Effective Binding Site
Combined with high surface area, PAFs also appear high physicochemical stabil-ity which makes it unique in those ultrahigh surface area materials. Most of PAFs we have reported do not dissolve in organic solvents and can sustain in boiling water and cold acid or base solution. This is due to its covalent bonding nature and cross-linked rigid phenyl framework. High-pressure hydrogen storage of PAF-1, PAF-3, and PAF-4 was determined at 77 K, and the excess hydrogen uptake capacity is 7.0 wt% at 48 bar, 5.5 wt% at 60 bar, and 4.2 wt% at 60 bar for PAF-1, PAF-3, and PAF-4, respectively [26, 29]. The heat of adsorption related to hydrogen– adsorbents (QstH2
for short) interactions were calculated from the hydrogen adsorption isotherms recorded at 77 and 87 K. Calculated by Clausius–Clapeyron

122 T. Ben and S. Qiu
equation, it appears to be 5.4, 6.6, and 6.3 kJ mol−1 for PAF-1, PAF-3, and PAF-4, respectively. Accordingly, the low-pressure uptakes of hydrogen are 186, 232, and 169 cm3 g−1 for PAF-1, PAF-3, and PAF-4, respectively. The results coming from the high-pressure and low-pressure hydrogen storage and QstH2
tell a fact that for high-pressure gas storage, Qst is not so important to determine how many gas mol-ecules could be stored. The energy for high pressure could remedy the weak inter-action between gases and porous frameworks. In the case of high-pressure gases storage, surface area is more important because it reveals how much porosity a framework is. More surface area of a material means more gases could be stored. But in the case of low-pressure gases storage especially when ambient condition gas storage (1 bar, room temperature) was applied, both surface area and Qst should be considered. Usually, the Qst is more important and results often show higher gas uptakes material express higher Qst at ambient condition.
PAFs show not only high surface area and excellent physicochemical stability, but also exceptional selectivity to green house gases. At 273 K and 1 bar, PAF-1, PAF-3, and PAF-4 exhibit very high selectivity to adsorb carbon dioxide and methane over hydrogen, nitrogen, oxygen, and argon. In particular, PAF-3 shows extraordinarily selectivity for the adsorption of CO2 over nitrogen with the value of 87/1 as well as of methane, which is about 30 times higher than that of nitrogen. That is, PAFs could efficiently recognize and enrich greenhouse gases from dry air which have potential applications in CCS. To prove this, a CO2 reversible adsorp-tion (25 °C) and regeneration (80 °C) experiment was designed and performed. As shown in Fig. 4.3, PAF-3 is the best one which shows a very high average weight change of 4.40 wt% during the cycles. When considering the high stability of PAF-3, this carbon dioxide adsorption procedure exhibits more energy savings and more cost-effectiveness.
The former results reveal the fantastic gas storage and carbon capture properties of PAFs. Among those PAFs reported, PAF-1 has the best physicochemical stabil-ity and ultrahigh surface area. But one of a practical drawback of such a material is the Qsts between PAF-1 and gases are not high enough to meet the request of adsorption at ambient condition. Very recently, Zhou et al. present a post-synthetic way to improve the heat of adsorption of PAF-1 by introducing of sulfone groups
Fig. 4.3 CO2 cyclic adsorption (25 °C) and regeneration (85 °C) of PAF-1 (black), PAF-3 (blue), and PAF-4 (green) (red is temperature). Reproduced from Ref. [29] with permission from the Royal Society of Chemistry

1234 Carbon Dioxide Capture in Porous Aromatic Frameworks
onto the biphenyl frameworks [69]. By reaction with chlorosulfonic acid, PAF-1 was modified to give sulfonate-grafted acid (PPN-6-SO3H) and lithium salt (PPN-6-SO3Li) species. After sulfonation, the surface area of PPN-6-SO3H and PPN-6-SO3Li reduced as low as 1,254 and 1,186 m2 g−1, but the Qst increased as high as 30.4 and 35.7 kJ mol−1. Strong interaction between sulfonate-grafted samples and carbon dioxide leads to very high uptakes capacity, with values of 13.1 and 13.5 wt% (equivalent to 3.6 and 3.7 mmol g−1) for PPN-6-SO3H and PPN-6-SO3Li, respec-tively. Calculated by the ideal adsorption solution theory (IAST) of Myers and Prausnitz [70–77], at post-combustion carbon capture procedure condition (typi-cally 15 % CO2 and 85 % N2), sulfonate-grafted samples show unexpected high adsorption selectivity for carbon dioxide and nitrogen which surpass the selectiv-ity of NaX zeolite [78]. Thus, these materials hold considerable promise for post-combustion carbon capture application.
4.4 Modification of Porous Aromatic Frameworks
4.4.1 Sulfonation
In 2011, Zhou’s group reported that the porous polymer networks grafted with sul-fonate (Scheme 4.2) exhibited preferential CO2 adsorption at low-pressure range [69].
For sufficient surface area being retained after the functionality, PPN-6 (also known as PAF-1 [26]) with ultrahigh surface area and physicochemical stability was chosen as the starting material. The uniformly large pores would shuttle the reac-tants and improve the diffusion rates. In addition, the robust all-carbon scaffold of PPN-6 makes it an ideal candidate for attachment of polar functionalities on biphe-nyl species under harsh reaction condition. In detail, the sulfonate-grafted PAFs (PPN-6-SO3H) was synthesized by reaction with chlorosulfonic acid, and its lithium salt (PPN-6-SO3Li) was obtained by further neutralized with LiOH solution [69].
N2 sorption measurements were used to detect the porosity of the materials. It shows an ideal I-type isotherm in PPN-6-SO3H and the large desorption hys-teresis disappeared after sulfonate grafted on PPN-6 (Fig. 4.4). Furthermore, besides the decrease in BET surface areas from 4,023 m2 g−1 for PPN-6 to 1,254 and 1,186 m2 g−1 for PPN-6-SO3H and PPN-6-SO3Li, respectively, the pore size of the grafted PPN-6 fall into 5.0–10.0 Å range. It seems that the pore sizes are more suitable with the kinetic diameter of a CO2 molecule, which will enhance the interaction between CO2 and the network. In addition, the relatively small pore size can separate CO2 from other gases with relatively larger kinetic diameters, such as N2 and CH4.
In low-pressure range, the interaction between CO2 molecules and the frame-works plays the dominant role in CO2 capture. In the sulfonation case, three fac-tors enhancing the CO2-philic effect of the materials should be noticed. Firstly, functionalization of all-carbon-scaffold frameworks was expected to create elec-tric fields on the surface that impart to the networks a strong affinity toward CO2

124 T. Ben and S. Qiu
molecules through its high quadrupole moment. It can be observed in the CO2 sorption at 295 K and 1 bar. The gravimetric CO2 uptake of PPN-6 was 5.1 wt%, and it rose up to 13.1 and 13.5 wt% for PPN-6-SO3H and PPN-6-SO3Li, respec-tively, under the same condition (Fig. 4.5). Notably, the CO2 uptake value of PPN-6-SO3Li was the highest one among all microporous organic polymers at the time it was published. Secondly, small pore size and polar functionalities will increase the heat of adsorption, and it has been proved by sulfonate-grafted PAFs here. For example, the zero-coverage CO2 isosteric enthalpy of PPN-6-SO3H and PPN-6-SO3Li are 30.4 and 35.7 kJ mol−1, which is extremely higher than that of PPN-6 (17 kJ mol−1). Thirdly, the Li+ cation which acted as an open coordination sites after full activation will lead to stronger electrostatic interactions between CO2 and the Li+ cation and will also raise the heat of sorption [69].
Absolute uptakes of CO2 are not the only important factor in choosing a suc-cessful CO2 sorbents, selectivity against other competing sorbates is also a key. The adsorption selectivity of the sulfonate-grafted PPN-6 estimated from the
Fig. 4.4 a N2 adsorption (closed)/desorption (open) isotherms at 77 K; b pore size distribution curves. PPN-6 (red), PPN-6-SO3H (green), and PPN-6-SO3Li (blue)
Scheme 4.2 Synthesis and grafting of PPN-6

1254 Carbon Dioxide Capture in Porous Aromatic Frameworks
experimental single-component isotherms by the IAST of Myers and Prausnitz [70] exhibits exceptionally high value (Fig. 4.6). The selectivity for CO2 over N2 is 150 for PPN-6-SO3H and 414 for PPN-6-SO3Li, which demonstrated potential application in the post-combustion CO2 capture. Based on the constituent of CO2 in flue gases, i.e., 15 %, the mass of CO2 uptake at 0.15 bar divided by the mass of N2 uptake at 0.75 bar often used as another method to evaluate the CO2/N2 selec-tivity. In this case, the value will jump from 3.0 for PPN-6 to 15 for PPN-6-SO3H to 17 for PPN-6-SO3Li.
After being boiled in water for 6 h, the surface areas and CO2 uptake capacities of PPN-6-SO3H and PPN-6-SO3Li showed no obvious loss, which indicated their
Fig. 4.5 Gravimetric CO2 and N2 adsorption (solid circle)/desorption (open circle) isotherms at 295 K
Fig. 4.6 IAST-predicted adsorption selectivities for PPN-6 (red), PPN-6-SO3H (green), PPN-6-SO3Li (blue), and NaX zeolite (black)

126 T. Ben and S. Qiu
ultrahigh hydrothermal stability. Accompany with the reversibility, sulfonate-modified PAFs will save the regeneration cost, which is also an important consideration in prac-tical carbon capture and sequestration application.
4.4.2 Lithiation
In 2012, Konstas et al. [79] reported the exceptional gas storage properties of lithiated PAFs (Scheme 4.3). Based on the previous lithiation method applied on CMPs [80], they prepared Li@PAF-1 with lithium naphthalenide and tuned the Li loading as desired. For removal of naphthalene and activation of lithium ions, higher reaction temperatures were used to reduce framework. The identity of the Li@PAF-1 confirmed by 1H, 13C, 6Li magic-angle spinning nuclear magnetic res-onance (MAS NMR). As shown in 1H NMR, the resonances of the aromatic pro-tons in 5 %_Li@PAF-1 shift to upfield as a result of the increasing local electron density of the reduced framework. In addition, in both the 1H and 13C NMR, naph-thalene cannot be observed, which proved the completed activated framework.
As expected, lithiation leads to shrinkage in pores and decrease in surface areas. The pore size reduced from 14 Å in PAF-1 to 11 Å in 5 %_Li@PAF-1 (Fig. 4.7). These pores filling coincide well with the data calculated from posi-tron annihilation lifetime spectroscopy (PALS) and atomistic simulation predic-tions. At the same time, the BET surface area dropped from 3,639 m2 g−1 for PAF-1 to 1,358 m2 g−1 and 479 m2 g−1 for 1 %_Li@PAF-1 and 5 %_Li@PAF-1, respectively.
Low-pressure CO2 sorption measurements at 273 and 298 K performed on Li@PAF-1, 1 %_Li@PAF-1, 2 %_Li@PAF-1, 5 %_Li@PAF-1, and 10 %_Li@PAF-1 to detect the CO2 storage properties (Fig. 4.8). The CO2 uptakes at 273 K reveal a remarkable enhancement in CO2 capture capacities upon reduc-tion by lithiation. 5 %_Li@PAF-1 (8.99 mmol g−1) shows the highest CO2 uptake at 273 K and 1.22 bar. It should be noticed that CO2 uptake exhibits a sharp rise
Scheme 4.3 Synthetic route to Li-PAF-1. Lithiation of PAF-1 (blue structure) reduces the framework at the π groups, activating it for gas storage

1274 Carbon Dioxide Capture in Porous Aromatic Frameworks
at low pressure, which possible dominates the exceptional capacity. However, the pseudo-chemisorptive behavior between exposed Li+ sites and CO2 cannot be observed in the higher Li+-loaded sample. It seems that the agglomeration of lith-ium ions will degrade the sample.
4.4.3 Amine Introduction
Amination is another useful strategy to introduce CO2-philic moieties on the PAFs. The polarizability and the large quadrupole moment of CO2 will create strong interaction between the networks and CO2 molecules. Due to the stabil-ity of the entirely covalently bonded network PPN-6 (also known as PAF-1) and high porosity (both surface area and pore volume), many different amines can be
Fig. 4.7 Pore size distribution of PAF-1 (blue), 1 %_Li@PAF-1 (black), and 5 %_Li@PAF-1 (red) using results obtained from DFT fits to Ar adsorption isotherm data at 87 K
Fig. 4.8 Experimental CO2 adsorption (solid square) and desorption (open square) isotherms of PAF-1 (blue) and the lithiated PAFs (1 %_Li@PAF-1, black; 2 %_Li@PAF-1, green; 5 %_Li@PAF-1, red) at 273 K

128 T. Ben and S. Qiu
modified and amine loadings could easily be tuned [81]. In the polyamine-tethered PAFs case, Zhou’s group synthesized PPN-6-CH2Cl first and then introduced sev-eral kinds of polyamine groups on it (Scheme 4.4). The efficiency of amine substi-tution was confirmed by elemental analysis. For example, the nitrogen content of PPN-6-CH2DETA was 11.95 %, which corresponds to 0.3 functional groups load-ing on one phenyl ring.
Nitrogen sorption measurements were used to detect the porosity of these pol-yamine-tethered PAFs (Fig. 4.9a). After modification, the BET surface areas decreased obviously, from 4,023 m2 g−1 for PPN-6 to 1,740, 1,014, 663, 634, 555 for PPN-6-CH2Cl, PPN-6-CH2EDA, PPN-6-CH2TAEA, PPN-6-CH2TETA, PPN-6-CH2DETA, respectively. It should be noticed that the huge hysteresis loop in PPN-6-CH2Cl disappeared into a nearly type I isotherm for the polyamine-modified PAFs.
The CO2 storage capacities were detected at 295 K in low-pressure range (Fig. 4.9b). Interestingly, the highest CO2 uptake at 295 K and 1 bar belonged to PPN-6-CH2DETA as 4.3 mmol g−1 (15.8 wt%), which has the smallest surface area. The recorded CO2 uptake value demonstrated that strong interaction between CO2 molecules and the polarity species modified PAFs played a more significant role than the surface area. As expected, the isosteric heats of adsorption (Qst) cal-culated from dual-site Langmuir fits of the polyamine-tethered PAFs are higher
Scheme 4.4 Synthetic route to polyamine-tethered PPNs. a CH3COOH/HCl/H3PO4/HCHO, 90 °C, 3 days; b amine, 90 °C, 3 days

1294 Carbon Dioxide Capture in Porous Aromatic Frameworks
than that of PPN-CH2Cl and PPN-6 (Fig. 4.9d). Since the CO2 constituent in flue gases is 15 %, the CO2 uptake at 0.15 bar attracts much attention. At 295 K and 0.15 bar, PPN-6-CH2Cl can storage 0.25 mmol g−1 (1.1 wt%), while PPN-6-CH2DETA takes up 3.0 mmol g−1 (11.8 wt%). Furthermore, the CO2 uptake value only slightly dropped to 10.0 wt% when the temperature rises to 313 K. The high CO2 uptake capacities maintained at elevated temperatures make the materials more useful under realistic flue gas condition.
Ideal adsorbed solution theory (IAST) model of Myers and Prausnitz [70] is used to evaluate the CO2/N2 selectivities (Fig. 4.9c). The poor N2 uptake due to the low porosity of PPN-6-CH2DETA leads to high CO2/N2 selectivity. The value is 442 and 115 for PPN-6-CH2DETA and PPN-6-CH2EDA, respectively, which is higher than that of PPN-6-CH2Cl (S = 13).
Fig. 4.9 a N2 adsorption (solid circle) and desorption (open circle) isotherms at 77 K. b CO2 adsorption (solid circle) and desorption (open circle) isotherms, as well as PPN-6-CH2DETA N2 adsorption, at 295 K. c The component loadings of N2 and CO2 calculated by IAST with bulk gas-phase partial pressures of 85 and 15 kPa for N2 and CO2, respectively, with PPN-6-CH2DETA, PPN-6-CH2EDA, PPN-6-CH2Cl, PPN-6-SO3Li, NaX zeolite, Mg-MOF-74, mmen-CuBTTri. Loadings for calculated selectivities of 200 and 400 are shown as a guide. d Isosteric heats of adsorption Qst for the adsorption of CO2, calculated using the dual-site Langmuir iso-therm fits

130 T. Ben and S. Qiu
Besides the CO2/N2 selectivity and the CO2 uptake capacity, cyclability is another important factor in post-combustion CO2 capture. TSA and VSA are com-bined to test the cyclability of the polyamine-tethered PAFs. After 20 cycles, no capacity loss can be observed (Fig. 4.10). Although regeneration energy for PPN-6-CH2DETA with more primary amines species in the network are higher than those for mmen-CuBTTri [82] with only secondary amines, it is still lower than that for amine solution.
Capture of CO2 from the atmosphere (400 ppm CO2, 78.96 % N2, 21 % O2) with solid adsorbents is another important strategy in CCS. This will address the two-thirds of total CO2 emissions that are generated from the transportation and other industries, such as concrete, paper, and chemical production [83]. However, direct air capture (DAC) under such dilute CO2 concentrations condition is a great challenge for technology and sorbents. One well-studied DAC technology was to choose sodium hydroxide solutions as sorbents. But the sorbent should be heated up to 900 °C to regenerate, and this makes the cost of CO2 as high as $600 per ton [84]. Another state-of-the-art technology relies on amine solutions. However, the large energy requirement, highly corrosive solutions, and solvent boil-off still embarrass the wide application of this DAC strategy [85]. For effective CO2 capture under these ultradilute conditions, three factors should be considered: (1) large working capacities, (2) low regeneration energies, and (3) stability toward contaminants.
By IAST, CO2 selectivity of PPN-6-CH2DETA was 3.6 × 1010 under “air” con-dition (400 ppm CO2 78.96 % N2, 21 % O2, total pressure was 100 kPa), and the purity of CO2 after separation can reach 99.999993 % (Fig. 4.11) [86]. It indicates that this material is useful in a true mixed component system since the resulting
Fig. 4.10 Twenty cycles of CO2 uptake at 273 K. After saturation, the sample was regenerated with a temperature swing to 80 °C and then under vacuum (0.1 mmHg) for 100 min. Data were collected with an ASAP2020 analyzer

1314 Carbon Dioxide Capture in Porous Aromatic Frameworks
CO2 has a high enough purity to be compressed and stored or used as a UHP grade chemical commodity. Besides, the loading capacity of PPN-6-CH2DETA calcu-lated by IAST was 1.04 mol kg−1 under the same “air” condition.
Breakthrough calculation which uses mixed gases in air composition can better reflect the true CO2 capture under practical condition [87]. As shown in the break-through calculation, PPN-6-CH2DETA exhibits the longest breakthrough time, which reflects the highest CO2 uptake capacities (Fig. 4.12).
Working capacity was calculated from mixed component (IAST) loadings at 295 K/400 ppm CO2 and desorption at 1 bar over a range of temperatures (Fig. 4.13). Regeneration of PPN-6-CH2DETA can be operated at 98 °C and the CO2 can be almost totally desorbed at 170 °C. The result, low regeneration energy penalty, coincides with the available adsorption enthalpies of PPN-6-CH2DETA (Qst are around 60 kJ mol−1).
4.4.4 Carbonization
In 2012, Qiu et al. developed the carbonized PAF-1 [donated as PAF-1-x, where x is the carbonization temperature (in °C)] in nitrogen with trace of oxygen [88]. The resulting carbonized PAF-1 derivatives showed significant increase in CO2 uptake capacities and exceptionally high CO2/H2, CO2/N2 and CO2/CH4 adsorp-tion selectivity under ambient conditions.
The carbonization of PAF-1 leads to a continuously shrinking of pore size and surface area (Fig. 4.14). The apparent surface areas calculated from BET models for relative pressure between 0.01 and 0.1 were 4,033, 2,881, 2,292, and 1,191 m2 g−1 for PAF-1-350, PAF-1-380, PAF-1-400, and PAF-1-450, respec-tively. The total pore volumes of those samples calculated by using the den-sity functional theory (DFT) method drop from 2.43 cm3 g−1 of PAF-1 to
Fig. 4.11 IAST calculations of component loadings of CO2 and (N2 + O2) in the adsorbed phase with selected materials. The total bulk gas pressure is 100 kPa and contains 400 ppm CO2, 78.96 % N2, and 21 % O2. Loadings for calculated selectivities are shown as a guide
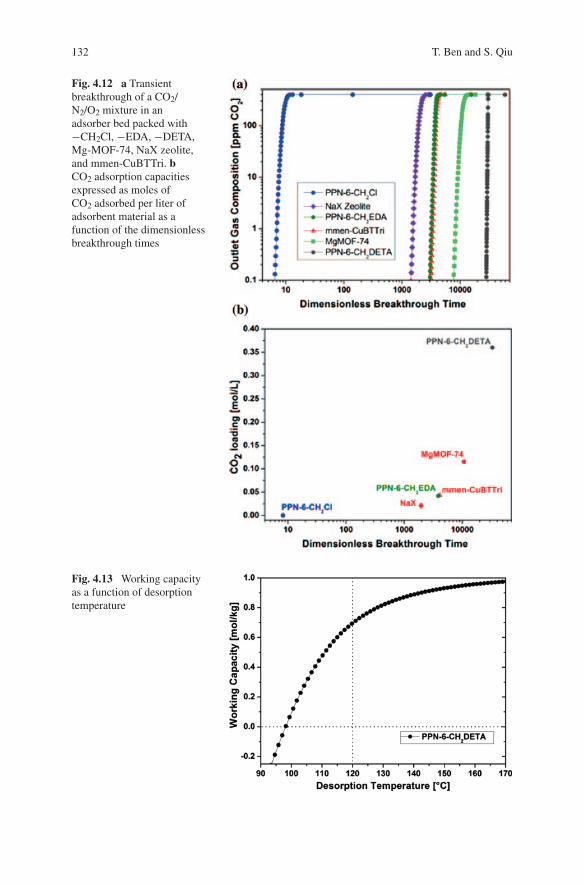
132 T. Ben and S. Qiu
Fig. 4.12 a Transient breakthrough of a CO2/N2/O2 mixture in an adsorber bed packed with −CH2Cl, −EDA, −DETA, Mg-MOF-74, NaX zeolite, and mmen-CuBTTri. b CO2 adsorption capacities expressed as moles of CO2 adsorbed per liter of adsorbent material as a function of the dimensionless breakthrough times
Fig. 4.13 Working capacity as a function of desorption temperature
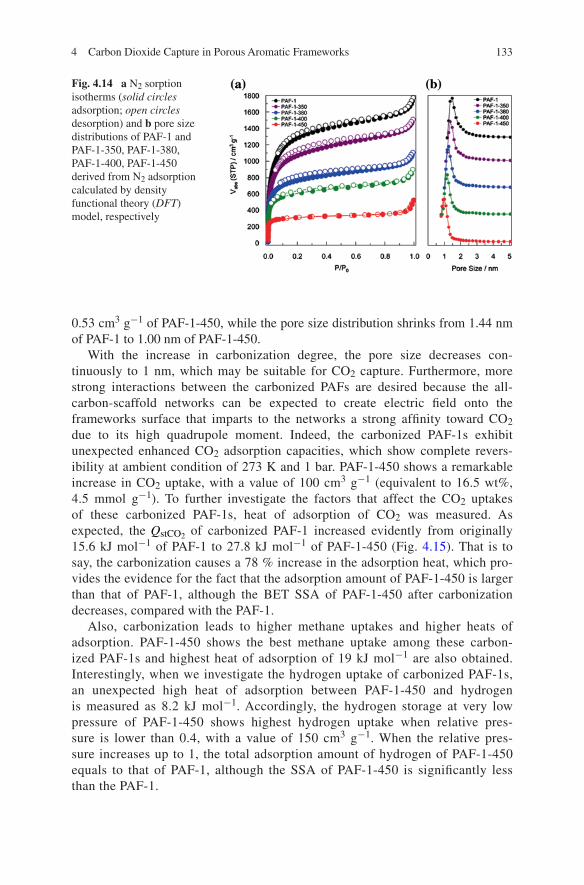
1334 Carbon Dioxide Capture in Porous Aromatic Frameworks
0.53 cm3 g−1 of PAF-1-450, while the pore size distribution shrinks from 1.44 nm of PAF-1 to 1.00 nm of PAF-1-450.
With the increase in carbonization degree, the pore size decreases con-tinuously to 1 nm, which may be suitable for CO2 capture. Furthermore, more strong interactions between the carbonized PAFs are desired because the all-carbon-scaffold networks can be expected to create electric field onto the frameworks surface that imparts to the networks a strong affinity toward CO2 due to its high quadrupole moment. Indeed, the carbonized PAF-1s exhibit unexpected enhanced CO2 adsorption capacities, which show complete revers-ibility at ambient condition of 273 K and 1 bar. PAF-1-450 shows a remarkable increase in CO2 uptake, with a value of 100 cm3 g−1 (equivalent to 16.5 wt%, 4.5 mmol g−1). To further investigate the factors that affect the CO2 uptakes of these carbonized PAF-1s, heat of adsorption of CO2 was measured. As expected, the QstCO2
of carbonized PAF-1 increased evidently from originally 15.6 kJ mol−1 of PAF-1 to 27.8 kJ mol−1 of PAF-1-450 (Fig. 4.15). That is to say, the carbonization causes a 78 % increase in the adsorption heat, which pro-vides the evidence for the fact that the adsorption amount of PAF-1-450 is larger than that of PAF-1, although the BET SSA of PAF-1-450 after carbonization decreases, compared with the PAF-1.
Also, carbonization leads to higher methane uptakes and higher heats of adsorption. PAF-1-450 shows the best methane uptake among these carbon-ized PAF-1s and highest heat of adsorption of 19 kJ mol−1 are also obtained. Interestingly, when we investigate the hydrogen uptake of carbonized PAF-1s, an unexpected high heat of adsorption between PAF-1-450 and hydrogen is measured as 8.2 kJ mol−1. Accordingly, the hydrogen storage at very low pressure of PAF-1-450 shows highest hydrogen uptake when relative pres-sure is lower than 0.4, with a value of 150 cm3 g−1. When the relative pres-sure increases up to 1, the total adsorption amount of hydrogen of PAF-1-450 equals to that of PAF-1, although the SSA of PAF-1-450 is significantly less than the PAF-1.
Fig. 4.14 a N2 sorption isotherms (solid circles adsorption; open circles desorption) and b pore size distributions of PAF-1 and PAF-1-350, PAF-1-380, PAF-1-400, PAF-1-450 derived from N2 adsorption calculated by density functional theory (DFT) model, respectively
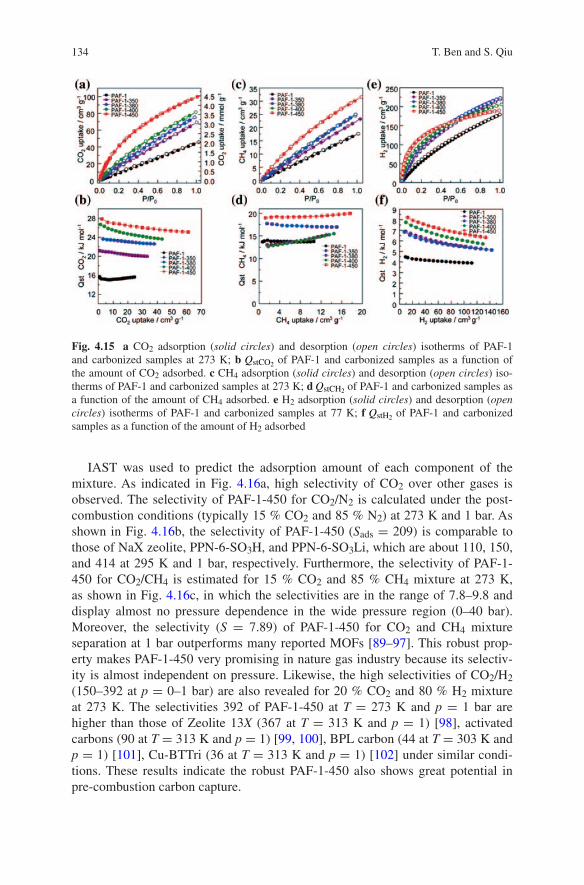
134 T. Ben and S. Qiu
IAST was used to predict the adsorption amount of each component of the mixture. As indicated in Fig. 4.16a, high selectivity of CO2 over other gases is observed. The selectivity of PAF-1-450 for CO2/N2 is calculated under the post-combustion conditions (typically 15 % CO2 and 85 % N2) at 273 K and 1 bar. As shown in Fig. 4.16b, the selectivity of PAF-1-450 (Sads = 209) is comparable to those of NaX zeolite, PPN-6-SO3H, and PPN-6-SO3Li, which are about 110, 150, and 414 at 295 K and 1 bar, respectively. Furthermore, the selectivity of PAF-1-450 for CO2/CH4 is estimated for 15 % CO2 and 85 % CH4 mixture at 273 K, as shown in Fig. 4.16c, in which the selectivities are in the range of 7.8–9.8 and display almost no pressure dependence in the wide pressure region (0–40 bar). Moreover, the selectivity (S = 7.89) of PAF-1-450 for CO2 and CH4 mixture separation at 1 bar outperforms many reported MOFs [89–97]. This robust prop-erty makes PAF-1-450 very promising in nature gas industry because its selectiv-ity is almost independent on pressure. Likewise, the high selectivities of CO2/H2 (150–392 at p = 0–1 bar) are also revealed for 20 % CO2 and 80 % H2 mixture at 273 K. The selectivities 392 of PAF-1-450 at T = 273 K and p = 1 bar are higher than those of Zeolite 13X (367 at T = 313 K and p = 1) [98], activated carbons (90 at T = 313 K and p = 1) [99, 100], BPL carbon (44 at T = 303 K and p = 1) [101], Cu-BTTri (36 at T = 313 K and p = 1) [102] under similar condi-tions. These results indicate the robust PAF-1-450 also shows great potential in pre-combustion carbon capture.
Fig. 4.15 a CO2 adsorption (solid circles) and desorption (open circles) isotherms of PAF-1 and carbonized samples at 273 K; b QstCO2
of PAF-1 and carbonized samples as a function of the amount of CO2 adsorbed. c CH4 adsorption (solid circles) and desorption (open circles) iso-therms of PAF-1 and carbonized samples at 273 K; d QstCH2
of PAF-1 and carbonized samples as a function of the amount of CH4 adsorbed. e H2 adsorption (solid circles) and desorption (open circles) isotherms of PAF-1 and carbonized samples at 77 K; f QstH2
of PAF-1 and carbonized samples as a function of the amount of H2 adsorbed

1354 Carbon Dioxide Capture in Porous Aromatic Frameworks
4.5 Outlook and Conclusion
In the last ten years, there has been great progress in the molecular engineering of ultrahigh surface area organic frameworks. The records of surface area, heat of adsorption, and selectivity of molecular recognition are broken continually. With the development of molecular simulation method and computer technique, more and more structures which have high possibility for application in carbon capture, gas storage, molecular recognition, and separation were revealed. It seems PAFs have great potential to be a more fantastic host material when functional groups are introduced onto the aromatic frameworks. Cao et al. and we evaluate the hydrogen storage performance of designed porous materials PAF-30X (X = 1–4)
Fig. 4.16 a CO2, CH4, N2, and H2 sorption of PAF-1-450 at 273 K. b IAST-predicted adsorption selectivity of CO2/N2 of PAF-1-450 using 15/85 CO2/N2 ratio. c IAST-predicted adsorption selec-tivity of CO2/CH4 of PAF-1-450 using 15/85 CO2/CH4 ratio. d IAST-predicted adsorption selectiv-ity of CO2/H2 of PAF-1-450 using 20/80 CO2/H2 ratio

136 T. Ben and S. Qiu
(Fig. 4.17) with dia topology structure by multiscale simulation method [103]. Results show that hydrogen uptakes of PAFs mainly depend on their densities and free volumes. Results show that PAF-304 and PAF-303 possesses higher gravimet-ric hydrogen uptakes than PAF-1. In particular, the gravimetric hydrogen uptake of PAF-304 reaches 6.53 wt% at room temperature and 100 bars, which is the highest among all of the present porous materials. The structure of PAF-304 is also known as PAF-11, which was synthesized by Suzuki cross-coupling with a relative low surface area of 704 m2 g−1. The contradiction between PAF-11 and theoretical cal-culation comes from the inefficiency synthesis method and possibility of interpen-etration of aromatic frameworks. It is worthy to explore again by more carefully design of SBU and synthesis method.
Fig. 4.17 Unit cells of PAFs, a PAF-301, b PAF-302, c PAF-303, and d PAF-304, derived from topology design and geometry optimization with the force field method. Here, gray and pink spheres represent carbon and hydrogen atoms, respectively, while the blue polyhedron represents the tetrahedrally bonded carbon atoms. In addition, the yellow sphere denotes the pores in 3D PAFs. Reproduced from Ref. [103] with permission from the American Chemical Society

1374 Carbon Dioxide Capture in Porous Aromatic Frameworks
Jiang et al. designed new PAFs by introducing polar organic groups to the biphenyl unit (Fig. 4.18) and investigated their separating power toward carbon dioxide by using GCMC simulations [104]. Results indicated that PAF-1 functionalized with tetrahydrofuran-like ether groups shows high adsorption capacity for carbon dioxide (10 mmol g−1, 1 bar, r. t.) and high selectivity for CO2/H2, CO2/CH4, and CO2/N2 mixtures at ambient conditions. Whether the above-mentioned novel PAFs could be effectively synthesized, the theoretical simulation and calculation provide an effec-tive method to guide the developing direction of more attractive PAFs.
At last, it should be noted that PAFs, especially synthesized by Yamamoto-type Ullmann cross-coupling, are costly to deploy on a large scale as a carbon capture adsorbent. There is still a great challenge to find a more convenient and low-cost synthesis method to prepare PAFs. Besides, the incorporation of useful and novel functions within robust PAFs with ultrahigh surface area remains a formidable challenge and an exciting opportunity for material chemists.
References
1. Schuth F, Sing KSW, Weitkamp J (2002) Handbook of porous solids. Wiley-VCH, New York 2. Valtchev V, Mintova S, Tsapatsis M (2009) Ordered porous solids: recent advances and
prospects. Elsevier B. V, Oxford 3. Li J, Sculley J, Zhou H (2012) Metal-organic frameworks for separations. Chem Rev
112:869–932 4. Suh MP, Park HJ, Prasad TK, Lim D (2012) Hydrogen storage metal-organic frameworks.
Chem Rev 112:782–835
Fig. 4.18 The unit cell of the proposed porous aromatic framework containing tetrahydrofuran-like moieties and the simulation pattern of CO2/N2 selectivity and CO2 capacity. Reproduced from Ref. [104] with permission from the American Chemical Society

138 T. Ben and S. Qiu
5. Makowski P, Thomas A, Kuhn P, Goettmann F (2009) Organic materials for hydrogen stor-age applications: from physisorption on organic solids to chemisorption organic molecules. Energy Environ Sci 2:480–490
6. Ding X, Guo J, Feng X, Honsho Y, Guo J, Seki S, Maitarad P, Saeki A, Nagase S, Jiang D (2011) Bowl-shaped fragments of C70 or higher fullerenes: synthesis, structural analysis, and inversion dynamics. Angew Chem Int Ed 50:1289–1293
7. Wan S, Guo J, Kim J, Ihee H, Jiang D (2009) A photoconductive covalent organic frame-work: self-condensed arene cubes composed of eclipsed 2D Polypyrene sheets for photocur-rent generation. Angew Chem Int Ed 48:5439–5442
8. Comotti A, Bracco S, Valsesia P, Beretta M, Sozzani P (2010) Fast molecular rotor dynam-ics modulated by guest inclusion in a highly organized nanoporous organosilica. Angew Chem Int Ed 49:1760–1764
9. Yoon M, Srirambalaji R, Kim K (2012) Homochiral metal-organic frameworks for asym-metric heterogeneous catalysis. Chem Rev 112:1196–1231
10. Yaghi OM, O’Keeffe M, Ockwig NW, Chae HK, Eddaoudi M, Kim J (2003) Reticular syn-thesis and the design of new materials. Nature 423:705–715
11. Mulfort KL, Farha OK, Malliakas CD, Kanatzidis MG, Hupp JT (2010) An interpenetrated framework material with hysteretic CO2 uptake. Chem Eur J 16:276–281
12. Horcajada P, Chalati T, Serre C, Gillet B, Sebrie C, Baati T, Eubank JF, Heurtaux D, Clayette P, Kreuz C, Chang J, Hwang YK, Marsaud V, Bories P, Cynober L, Gil S, Férey G, Couvreur P, Gref R (2010) Porous metal-organic-framework nanoscale carriers as a poten-tial platform for drug delivery and imaging. Nat Mater 9:172–178
13. Côté AP, Benin AI, Ockwig NW, O’Keeffe M, Matzger AJ, Yaghi OM (2005) Porous, crys-talline, covalent organic frameworks. Science 310:1166–1170
14. El-Kaderi HM, Hunt JR, Medoza-Cortés JL, Côté AP, Taylor RE, O’Keeffe M, Yaghi OM (2007) Designed synthesis of 3D covalent organic frameworks. Science 316:268–272
15. Wan S, Guo J, Kim J, Ihee H, Jiang D (2008) A Belt-shaped, blue luminescent, and semi-conducting covalent organic framework. Angew Chem Int Ed 47:8826–8830
16. McKeown NB, Budd PM, Msayib KJ, Ghanem BS, Kingston HJ, Tattershall CE, Makhseed S, Reynolds KJ, Fritsch D (2005) Polymers of intrinsic microporosity (PIMs): bridging the void between microporous and polymeric materials. Chem Eur J 11:2610–2620
17. McKeown NB, Budd PM (2006) Polymers of intrinsic microporosity (PIMs): organic mate-rials for membrane separations, heterogeneous catalysis and hydrogen storage. Chem Soc Rev 35:675–683
18. Jiang J, Su F, Trewin A, Wood CD, Campbell NL, Niu H, Dickinson C, Ganin AY, Rosseinsky MJ, Khimyak YZ, Cooper AI (2007) Conjugated microporous poly (aryleneethynylene) networks. Angew Chem Int Ed 46:8574–8578
19. S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang (2008) A photoconductive covalent organic framework: self-condensed arene cubes composed of eclipsed 2d polypyrene sheets for pho-tocurrent generation. Angew Chem Int Ed 47:8826–8830
20. Chen L, Honsho Y, Seki S, Jiang D (2010) Conjugated microporous polymer. J Am Chem Soc 132:6742–6748
21. Cooper AI (2009) Conjugated microporous polymers. Adv Mater 21:1291–1295 22. Jiang JX, Su F, Trewin A, Wood CD, Niu H, Jones TA, Khimyak YZ, Cooper AI (2008)
Synthetic control of the pore dimension and surface area in conjugated microporous poly-mer and copolymer networks. J Am Chem Soc 130:7710–7720
23. Dawson R, Laybourn A, Clowes R, Khimyak YZ, Adams DJ, Cooper AI (2009) Functionalized conjugated microporous polymers. Macromolecules 42:8809–8816
24. Wood CD, Tan B, Trewin A, Niu H, Bradshaw D, Rosseinsky MJ, Khimyak YZ, Campbell NL, Kirk R, Stöckel E, Cooper AI (2007) Hydrogen storage in microporous hypercrosslinked organic polymer networks. Chem Mater 19:2034–2048
25. Tsyurupa MP, Davankov VA (2002) Hypercrosslinked polymers: basic principle of prepar-ing the new class of polymeric materials. React Funct Polym 53:193–203

1394 Carbon Dioxide Capture in Porous Aromatic Frameworks
26. Ben T, Ren H, Ma S, Cao D, Lan J, Jing X, Wang W, Xu J, Deng F, Simmons JM, Qiu S, Zhu G (2009) Targeted synthesis of a porous aromatic framework with high stability and exceptionally high surface area. Angew Chem Int Ed 48:9457–9461
27. Ren H, Ben T, Wang E, Jing X, Xue M, Liu B, Cui Y, Qiu S, Zhu G (2010) Targeted synthe-sis of a 3D porous aromatic framework for selective sorption of benzene. Chem Commun 46:291–293
28. Peng Y, Ben T, Xu J, Xue M, Jing X, Deng F, Qiu S, Zhu G (2011) A covalently-linked microporous organic-inorganic hybrid framework containing polyhedral oligomeric silses-quioxane moieties. Dalton Trans 40:2720–2724
29. Ben T, Pei C, Zhang D, Xu J, Deng F, Jing X, Qiu S (2011) Gas storage in porous aromatic frameworks (PAFs). Energy Environ Sci 4:991–3999
30. Ben T, Shi K, Cui Y, Pei C, Zuo Y, Guo H, Zhang D, Xu J, Deng F, Tian Z, Qiu S (2011) Targeted synthesis of an electroactive organic framework. J Mater Chem 21:18208–18214
31. Ren H, Ben T, Sun F, Guo M, Jing X, Ma H, Cai K, Qiu S, Zhu G (2011) Synthesis of a porous aromatic framework for adsorbing organic pollutants application. J Mater Chem 21:10348–10353
32. Yuan Y, Sun F, Ren H, Jing X, Wang W, Ma H, Zhao H, Zhu G (2011) Targeted synthe-sis of a porous aromatic framework with a high adsorption capacity for organic molecules. J Mater Chem 21:13498–13502
33. Zhao H, Jin Z, Su H, Jing X, Sun F, Zhu G (2011) Targeted synthesis of a 2D ordered porous organic framework for drug release. Chem Commun 47:6389–6391
34. Kondo M, Yoshitomi T, Seki K, Matsuzaka H, Kitagawa S (1997) Three-dimensional framework with channeling cavities for small molecules: {[M2(4, 4′-bpy)3(NO3)4]·xH2O}n (M = Co, Ni, Zn). Angew Chem Int Ed 36:1725–1727
35. Li H, Eddaoudi M, Groy TL, Yaghi OM (1998) Establishing microporosity in open metal-organic frameworks: gas sorption isotherms for Zn(BDC) (BDC = 1,4-Benzenedicarboxylate). J Am Chem Soc 120:8571–8572
36. Chui SS, Lo SM, Charmant JP, Orpen HAG, Williams ID (1999) A chemically functionaliz-able nanoporous material [Cu3(TMA)2(H2O)3]n. Science 283:1148–1150
37. McKeown NB, Makhseed S, Budd PM (2002) Phthalocyanine-based nanoporous network polymers. Chem Commun 23:2780–2781
38. Ahn JH, Jang JE, Oh CG, Ihm SK, Cortez J, Sherrington DC (2006) Rapid generation and control of microporosity, bimodal pore size distribution, and surface area in Davankov-type hyper-cross-linked resins. Macromolecules 39:627–632
39. Davankov VA, Tsyurupa MP (1990) Structure and properties of hypercrosslinked polystyrene—the first representative of a new class of polymer networks. React Polym 13:27–42
40. Yamamoto T (1999) π-conjugated polymers bearing electronic and optical functionalities. Preparation by organometallic polycondensations, properties, and their applications. Bull Chem Soc Jpn 72:621–638
41. Zhou G, Baumgarten M, Müllen K (2007) Arylamine-substituted oligo(ladder-type penta-phenylene)s: electronic communication between bridged redox centers. J Am Chem Soc 129:12211–12221
42. IPCC Climate Change (2007) Synthesis report. Contribution of working groups I, II and III to the fourth assessment report of the intergovernmental panel on climate change [Core Writing Team ed. Pachauri RK, Reisinger A] Intergovernmental panel on climate change, Geneva, Switzerland, 2007
43. IPCC (Intergovernmental Panel on Climate Change) (2005) CO2 capture and storage, a spe-cial report of the intergovernmental panel on climate change. Cambridge University Press, Cambridge
44. Sumida K, Rogow DL, Mason JA, McDonald TM, Bloch ED, Herm ZR, Bae T-H, Long JR (2012) Carbon dioxide capture in metal-organic frameworks. Chem Rev 112:724–781
45. Li J, Sculley J, Zhou H-C (2012) Metal-organic frameworks for separations. Chem Rev 112:869–932

140 T. Ben and S. Qiu
46. Wright PA (2008) Microporous framework solids. Royal Society of Chemistry, Cambridge 47. Bunz UHF(2000) Poly(aryleneethynylene)s: syntheses, properties, structures, and applica-
tions. Chem Rev 100:1605–1644 48. Sonogashira K, Tohda Y, Hagihara N (1975) A convenient synthesis of acetylenes: catalytic
substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett 16:4467–4470
49. Trumbo DL, Marvel CS (1986) Polymerization using palladium (II) salts: Homopolymers and copolymers from phenylethynyl compounds and aromatic bromides. J Polym Sci Part A: Polym Chem 24:2311–2326
50. Hittinger E, Kokil A, Weder C (2004) Synthesis and characterization of cross-linked conju-gated polymer milli-, micro-, and nanoparticles. Angew Chem Int Ed 43:1808–1811
51. Mendez JD, Schroeter M, Weder C (2007) Hyperbranched Poly(p-phenylene ethynylene)s. Macromol Chem Phys 208:1625–1636
52. Donhauser ZJ, Mantooth BA, Kelly KF, Bumm LA, Monnell JD, Stapleton JJ, Price DW, Rawlett AM, Allara DL, Tour JM, Weiss PS (2001) Conductance switching in single mol-ecules through conformational changes. Science 292:2303–2307
53. Zhou Q, Swager TM (1995) Fluorescent chemosensors based on energy migration in con-jugated polymers: the molecular wire approach to increased sensitivity. J Am Chem Soc 117:12593–12602
54. Venkataraman D, Lee S, Zhang JS, Moore JS (1994) An organic solid with wide channels based on hydrogen bonding between macrocycles. Nature 371:591–593
55. Zhang W, Moore JS (2006) Shape-persistent macrocycles: structures and syn-thetic approaches from arylene and ethynylene building blocks. Angew Chem Int Ed 45:4416–4439
56. Kiang YH, Gardner GB, Lee S, Xu ZT, Lobkovsky EB (1999) Variable pore size, variable chemical functionality, and an example of reactivity within porous phenylacetylene silver salts. J Am Chem Soc 121:8204–8215
57. Miyaura N, Yamada K, Suzuki A (1979) A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett 36:343
58. Miyaura N, Yanagi T, Suzuki A (1981) The palladium-catalyzed cross-coupling reaction of phenylboronic acid with haloarenes in the presence of bases. Synth Commun 11:513–519
59. Zhang H, Xue F, Mak TCW, Chan KS (1996) Enantioselectivity increases with reactivity: electronically controlled asymmetric addition of diethylzinc to aromatic aldehydes catalyzed by a chiral pyridylphenol. J Org Chem 1996(61):8002–8003
60. Zhang H, Chan KS (1996) Enantioselectivity increases with reactivity: electronically con-trolled asymmetric addition of diethylzinc to aromatic aldehydes catalyzed by a chiral pyri-dylphenol. Tetrahedron Lett 37:1043–1044
61. Badone D, Baroni M, Cardamone R, Ielmini A, Guzzi U (1997) Highly efficient palladium-catalyzed boronic acid coupling reactions in water: scope and limitations. J Org Chem 62:7170–7173
62. Casalnuovo AL, Calabrese JC (1999) Palladium-catalyzed alkylations in aqueous media. J Am Chem Soc 112:4324–4330
63. Yamada YMA, Takeda K, Takahashi H, Ikegami S (2002) An assembled complex of palla-dium and non-cross-linked amphiphilic polymer: a highly active and recyclable catalyst for the Suzuki–Miyaura Reaction. Org Lett 4:3371–3374
64. Frenette R, Friesen RW (1994) Biaryl synthesis via Suzuki coupling on a solid support. Tetrahedron Lett 35:9177–9180
65. Ishiyama T, Murata M, Miyaura N (1995) Palladium (0)-catalyzed cross-coupling reaction of alkoxydiboron with haloarenes—A direct procedure for arylboronic esters. J Org Chem 60:7508–7510
66. Ishiyama T, Itoh Y, Kitano T, Miyaura N (1997) Synthesis of arylboronates via the Palladium(0)-catalyzed cross-coupling reaction of tetra(alkoxo)diborons with aryl triflates. Tetrahedron Lett 38:3447–3450

1414 Carbon Dioxide Capture in Porous Aromatic Frameworks
67. Holst JR, Stöckel E, Adams DJ, Cooper AI (2010) Chemical tuning of CO2 sorption in robust nanoporous organic polymers. Macromolecules 43:8531–8538
68. Yuan D, Lu W, Zhao D, Zhou H (2011) Highly stable porous polymer networks with excep-tionally high gas-uptake capacities. Adv Mater 23:3723–3725
69. Lu W, Yuan D, Sculley J, Zhao D, Krishna R, Zhou H (2011) Sulfonate-grafted polymer net-works for preferential CO2 adsorption at low pressure. J Am Chem Soc 133:18126–18129
70. Myers AL, Prausnitz JM (1965) Thermodynamics of mixed-gas adsorption. AIChE J 11:121–127
71. Bae YS, Farha OK, Hupp JT, Snurr RQ (2009) Enhancement of CO2/N2 selectivity in a metal-organic framework by cavity modification. J Mater Chem 19:2131–2134
72. Yazaydin AO, Benin AI, Faheem SA, Jakubczak P, Low JJ, Willis RR, Snurr RQ (2009) Enhanced CO2 adsorption in metal-organic frameworks via occupation of open-metal sites by coordinated water molecules. Chem Mater 21:1425–1430
73. Zheng BS, Bai JF, Duan JG, Wojtas L, Zaworotko MJ (2011) Enhanced CO2 binding affin-ity of a high-uptake rht-type metal-organic framework decorated with acrylamide groups. J Am Chem Soc 133:748–751
74. Simmons JM, Wu H, Zhou W, Yildirim T (2011) Carbon capture in metal-organic frame-works: a comparative study. Energy Environ Sci 4:2177–2185
75. Krishna R, van Baten JM (2011) In silico screening of metal-organic frameworks in separa-tion applications. Phys Chem Chem Phys 13:10593–10616
76. Belmabkhout Y, Sayari A (2009) Effect of pore expansion and amine functionaliza-tion of mesoporous silica on CO2 adsorption over a wide range of conditions. Adsorption 15:318–328
77. Goj A, Sholl DS, Akten ED, Kohen DI (2002) Atomistic simulations of CO2 and N2 adsorp-tion in silica zeolites: the impact of pore size and shape. J Phys Chem B 106:8367–8375
78. Belmabkhout Y, Pirngruber G, Jolimaitre E, Methivier A (2007) A complete experimental approach for synthesis gas separation studies using static gravimetric and column break-through experiments. Adsorption 13:341–349
79. Konstas K, Taylor JW, Thornton AW, Doherty CM, Lim WX, Bastow TJ, Kennedy DF, Wood CD, Cox BJ, Hill JM, Hill AJ, Hill MR (2012) Lithiated porous aromatic frameworks with exceptional gas storage capacity. Angew Chem Int Ed 51:6639–6642
80. Li A, Lu R-F, Wang Y, Wang X, Han K-L, Deng WQ (2010) Lithium-doped conjugated microporous polymers for reversible hydrogen storage. Angew Chem Int Ed 49:3330–3333
81. Lu W, Sculley JP, Yuan D, Krishna R, Wei Z, Zhou H-C (2012) Polyamine-tethered porous polymer networks for carbon dioxide capture from flue gas. Angew Chem Int Ed 2012(51):7480–7484
82. McDonald TM, D’Alessandro DM, Krishna R, Long JR (2011) Enhanced carbon dioxide capture upon incorporation of N, N′-Dimethylethylenediamine in the metal-organic frame-work CuBTTri. Chem Sci 2:2022–2028
83. MacDowell N, Florin N, Buchard A, Hallett J, Galindo A, Jackson G, Adjiman CS, Williams CK, Shah N, Fennell P (2010) An overview of CO2 capture technologies. Energy Environ Sci 2:1645–1669
84. Socolow RH et al (2011) American Physical Society, College Park, MD, 1 June 2011 85. Sumida K, Rogow DL, Mason JA, McDonald TM, Bloch ED, Herm ZR, Bae TH, Long JR
(2012) Carbon dioxide capture in metal-organic frameworks. Chem Rev 112:724–781 86. Lu W, Sculley JP, Yuan D, Krishna R, Zhou H (2013) Carbon dioxide capture from air using
amine-grafted porous polymer networks. J Phys Chem C 117:4057–4061 87. Krishna R, Long JR (2011) Screening metal-organic frameworks by analysis of transient
breakthrough of gas mixtures in a fixed bed adsorber. J Phys Chem C 115:12941–12950 88. Ben T, Li Y, Zhu L, Zhang D, Cao D, Xiang Z, Yao X, Qiu S (2012) Selective adsorption
of carbon dioxide by carbonized porous aromatic framework (PAF). Energy Environ Sci 5:8370–8376
89. Xiang Z, Peng X, Cheng X, Li X, Cao D (2011) Gas separation by adsorption on MIL-53(Al): natural gas and biogas application. J Phys Chem C 115:19864–19871

142 T. Ben and S. Qiu
90. Burd SD, Ma S, Perman JA, Sikora BJ, Snurr RQ, Thallapally PK, Tian J, Wojtas L, Zaworotko MJ (2012) Highly selective carbon dioxide uptake by [Cu(bpy-n)2(SiF6)] (bpy-1 = 4,4′-Bipyridine; bpy-2 = 1,2-Bis(4-pyridyl)ethene). J Am Chem Soc 134:3663–3666
91. Simmons JM, Wu H, Zhou W, Yildirim T (2011) Remote toehold: a mechanism for flexible control of DNA hybridization kinetics. Energy Environ Sci 4:2177–2182
92. Li JR, Kuppler RJ, Zhou H-C (2009) Selective gas adsorption and separation in metal-organic frameworks. Chem Soc Rev 38:1477–1504
93. Bae YS, Farha OK, Hupp JT, Snurr RQ (2009) Enhancement of CO2/N2 selectivity in a metal-organic framework by cavity modification. J Mater Chem 19:2131–2134
94. Nicholson D, Gubbins KEJ (1996) A microporous metal-organic frameworks for separation of CO2/N2 and CO2/CH4 by 51-2. J Chem Phys 104:8126–8134
95. Heuchel M, Davies GM, Buss E, Seaton NA (1999) Adsorption of carbon dioxide and methane and their mixtures on an activated carbon: simulation and experiment. Langmuir 15:8695–8705
96. Babarao R, Hu ZQ, Jiang JW, Chempath S, Sandler SI (2007) A comparative study from Monte Carlo simulation. Langmuir 23:659–666
97. Shao XH, Feng ZH, Xue RS, Ma CC, Wang WC, Peng X, Cao DP (2011) Adsorption of CO2, CH4, CO2/N2 and CO2/CH4 in novel activated carbon beads: preparation measure-ments and simulation. AIChE J 57:3042–3051
98. Belmabkhout Y, Pirngruber G, Jolimaitre E, Methivier A (2007) A complete experimental approach for synthesis gas separation studies using static gravimetric and column break-through experiments. Adsorption 13:341–349
99. Wu J, Zhou L, Sun Y, Su W, Zhou Y (2007) Measurement and prediction of adsorption equi-librium for a H2/N2/CH4/CO2 mixture. AIChE J 53:1178–1191
100. Cao D, Wu J (2005) Modeling the selectivity of activated carbon for efficient separation of hydrogen and carbon dioxide. Carbon 43:1364–1370
101. Sircar S, Golden TC, Rao MB (1996) Activated carbon for gas separation and storage. Carbon 1:1–12
102. Herm ZR, Swisher JA, Smit B, Krishna R, Long JR (2011) Metal-organic frameworks as adsorbents for hydrogen purification and precombustion carbon dioxide capture. J Am Chem Soc 133:5664–5667
103. Lan J, Cao D, Wang W, Ben T, Zhu G (2010) High-capacity hydrogen storage in porous aro-matic frameworks with diamond-like structure. J Phys Chem Lett 1:978–981
104. Babarao R, Dai S, Jiang D (2011) Functionalizing porous-aromatic frameworks with polar organic groups for high-capacity and selective CO2 separation: a molecular simulation study. Langmuir 27:3451–3460

143
Abstract Microporous organic polymers (MOPs) are a unique class of porous materials consisting solely of the light elements (C, H, O, N, etc.). A series of vivid characteristics of MOPs, such as high-specific surface area, good physicochemical stability, diverse pore dimensions, topologies, and chemical functionalities, make them suitable adsorbents for CO2 capture. In this chapter, MOPs are categorized into four classes according to the types of organic reactions and the chemical structures of the resulting materials: hypercrosslinked polymers (HCPs), covalent organic frame-works (COFs), polymers of intrinsic microporosity (PIMs), and conjugated micropo-rous polymers (CMPs). For each type of the polymer network, the state-of-the-art development in the design, synthesis, characterization, and the CO2 sorption perfor-mance is reviewed. Strategies for controlling CO2 uptake capacity and adsorption enthalpy via manipulation of surface area, pore size, and functionality are discussed in detail. These studies would open up many new possibilities for the development of the novel solid sorbents targeting the CO2 capture process. It is expected that this chapter will not only summarize the main research activities in this field, but also find possible links between basic studies and practical applications.
Abbreviations
ACMP Acetylene gas mediated conjugated microporous polymersBA Benzyl alcoholBC Benzyl chlorideBCMA 9,10-Bis(chloromethyl)anthracene
Chapter 5Microporous Organic Polymers for Carbon Dioxide Capture
Yali Luo and Bien Tan
A.-H. Lu and S. Dai (eds.), Porous Materials for Carbon Dioxide Capture, Green Chemistry and Sustainable Technology, DOI: 10.1007/978-3-642-54646-4_5, © Springer-Verlag Berlin Heidelberg 2014
Y. Luo · B. Tan (*) Department of Chemistry, Huazhong University of Science and Technology, Wuhan, People’s Republic of Chinae-mail: [email protected]

144 Y. Luo and B. Tan
BCMBP 4,4-Bis(chloromethyl)-1,1-biphenylBDM 1,4-BenzenedimethanolBET Brunauer–Emmett–TellerBILPs Porous benzimidazole-linked polymers(±)BINAM (±)-2,2′-Diamino-1,10-binaphthaleneBINOL BinaphtholCCS Carbon capture and storageCMPs Conjugated microporous polymersCOFs Covalent organic frameworksCPOP Carbazole-based porous organic polymerDCX DichloroxyleneDVB DivinylbenzeneFDA Formaldehyde dimethyl acetalGCMC Grand canonical Monte CarloHATP 2,3,6,7,10,11-HexaaminotriphenyleneHCPs Hypercrosslinked polymersMOFs Metal organic frameworksMOPs Microporous organic polymersPAE Poly(aryleneethynylene)PAFs Porous aromatic frameworksPBI Poly(benzimidazole)PIMs Polymers of intrinsic microporosityPMDA Pyromellitic acid dianhydridePPNs Porous polymer networksPy PyrroleQst Isosteric heat of CO2 adsorptionSt StyreneTFPM Tetrakis(4-formylphenyl)methaneTPI Triazine-based porous polyimideTpPa-1 1,3,5-Triformylphloroglucinol with p-phenylenediamineTpPa-2 1,3,5-Triformylphloroglucinol with 2,5-dimethyl-p-phenylenediamineTZPIMs MOPs functionalized with CO2-philic pendant tetrazole groupsVBC Vinyl benzyl chlorideZIFs Zeolitic imidazole frameworks
5.1 Introduction
The world climate and environment problems such as global warming, sea level rise, and an irreversible increase in the acidity levels of the oceans, caused by exces-sive greenhouse gases emissions have attracted widespread public concern in recent years. Carbon dioxide (CO2) is the major greenhouse gas that is responsible for 63 % of the warming attributable to all greenhouse gases. As in November 2011, CO2 in the earth’s atmosphere is at a concentration of approximately 390 ppm by

1455 Microporous Organic Polymers for Carbon Dioxide Capture
volume, as opposed to the pre-industrial level of ca. 280 ppm, about 40 % increase occurring within the last 30 years. This drastic rise has been attributed to increas-ing dependence on the combustion of fossil fuels (coal, petroleum, and natural gas), which account for 44 % of anthropogenic CO2 emissions. In the long term, the best strategy to lower CO2 emissions is to adopt energy solutions which do not produce CO2 by a by-product. But unfortunately, energy requirements for modern society are still highly dependent on fossil fuels in the twenty-first century. Since an immediate CO2 emission halt is impossible, in recent years, worldwide efforts have been devoted to developing new technologies/processes for reducing atmos-pheric CO2 levels and preventing continuous global warming. Among them, carbon capture and storage (CCS), also referred to as carbon capture and sequestration, has been proposed as a feasible way to mediate the atmospheric CO2 concentra-tion. Conventional CO2 capture processes involving the chemisorption of CO2 by amine-based web-scrubbing systems present several disadvantages, including low energy efficiency, high cost, and the toxicity and corrosion problems [1]. Therefore, alternative concepts based on physical adsorption of CO2 in porous solids were pro-posed [2]. The key features of suitable sorbents for CO2 capture are high selective uptake of CO2, facile regeneration at low energy penalty, low cost, chemical stabil-ity, as well as good thermal stability. In recent years, a variety of sorbent materials have been widely studied in CCS schemes. Traditional porous inorganic materials, such as zeolites, activated carbons, and porous silicas, are widely used in industry and daily life. Despite their excellent adsorption capacities, these adsorbents are not selective enough for CO2 separation from flue gases because they also adsorb considerable amounts of N2. Recently, organic–inorganic hybrids materials such as metal-organic frameworks (MOFs), absorbents made of metal ions and organic linkers with high surface areas and tunable pore size, have emerged to show high CO2 adsorption capacity and selectivity [3]. However, under practical application conditions, MOFs usually suffer from low thermal and hydrothermal instability due to the nature of a coordination bond [4]. By replacing coordination bonds with stronger covalent bonds, microporous organic polymers (MOPs) are constructed and they are much more stable under ambient conditions. These materials can be considered as organic analogs of MOFs and zeolites in terms of pore properties. The combination of large specific surface areas, synthetic diversification, and good physicochemical stability makes MOPs excellent candidates for CO2 capture. In the past few years, a significant research effort has been directed at synthesizing such materials and impressive progress has been made, yielding a wide variety of microporous materials including polymers of intrinsic microporosity (PIMs) [5], conjugated microporous polymers (CMPs) [6, 7], covalent organic frameworks (COFs) [8, 9], hypercrosslinked polymers (HCPs) [10–12], crystalline triazine-based frameworks (CTFs) [13], and porous aromatic frameworks (PAFs) [14]. The chemical reactions involved include metal-catalyzed couplings, click reactions, oxi-dative polymerizations, trimerizations, and various condensation polymerizations (Fig. 5.1). In this chapter, we will summarize the latest progress of the MOPs along with their potential application in CO2 capture and separation.

146 Y. Luo and B. Tan
5.2 Hypercrosslinked Organic Polymers
HCPs [12, 15] represent a family of robust microporous organic materials which are low cost and mainly prepared by the Friedel–Crafts alkylation reaction. The per-manent porosity in HCPs is a result of extensive crosslinking reactions which pre-vent the polymer chains from collapsing into a dense, non-porous state. Such highly
Fig. 5.1 Schematic representation of the dynamic chemical reactions for the preparation of MOPs

1475 Microporous Organic Polymers for Carbon Dioxide Capture
crosslinked nature of the materials confers them high thermal stability that is not commonly expected for organic polymers. The most well-studied hypercrosslinked materials are “Davankov-type” resins [16], prepared by post-crosslinking of polysty-renic networks (Fig. 5.2). These materials can exhibit apparent Brunauer–Emmett–Teller (BET) surface areas as high as 2,090 m2 g−1 [17]. Typically, most pores in such a material are about 1.8 nm in size. The high surface areas and small pore sizes make these materials excellent gas adsorbent candidates for clean energy and envi-ronmental applications. In 2006, Germain et al. [18] and Lee et al. [10] simultane-ously reported the use of hypercrosslinked polystyrenes for hydrogen storage. It is found that the total hydrogen storage capacity is of 5.4 wt% at 8.0 MPa, which can approach the capacities required by the DOE for hydrogen storage. Inspired by these results, our group [19] studied the CO2 uptake capacity of the hypercrosslinked materials. By using poly(divinylbenzene-co-vinylbenzyl chloride) (DVB-VBC) with different DVB content as the precursors, a series of hypercrosslinked materials HCP-DVB-VBC were synthesized. With the DVB content varying from 0 to 10 %, the pore size of HCP-DVB-VBC decreases from macropore to micropore, the pore size distribution becomes narrower, and the micropore volume content increases from 6.82 to 61.90 %. The CO2 sorption experiments indicated that the polymers with smaller pore size and larger microporous volume show higher CO2 uptake capacities (Table 5.1). For example, HCP-DVB-VBC with 10 % DVB shows pure microporous structure and highest micropore volume, which can adsorb the most CO2 gas (2.82 mmol g−1, 12.41 wt%) at 1.00 bar/273.15 K. The CO2 separation performance of the microporous polystyrene was evaluated by Kaliva et al. [20]. The polystyrene particles comprising 35 mol% styrene and 65 mol% divinylbenzene exhibited an excellent separation performance of CO2 over CH4, with separation factors in the range of ~7–13 (Fig. 5.3) at 268 K for a CO2/CH4 = 5/95 mixture. This CO2/CH4 selectivity is comparable or exceeds those of other high-performance polymeric membranes and inorganic materials including zeolitic imidazolate frame-works (5–10) [21], MOF-5 (2–3) [22, 23], Cu-BTC (6–10) [23, 24], MCM-41 (4–5) [25], and zeolite 13X (2–14) [26]. They ascribed the CO2/CH4 mixed-gas selec-tivity to the different adsorptive interactions between the probe molecules and the porous surface of the polystyrene framework. The high π-electron density of the
Fig. 5.2 Reaction scheme for the synthesis of a hypercrosslinked polymer prepared from gel poly(divinylbenzene-co-vinylbenzyl chloride) (Reprinted with permission from Ref. [17]. Copyright 2006, American Chemical Society)

148 Y. Luo and B. Tan
phenyl groups facilitates strong dispersion or induced-dipole interactions with quad-rupolar CO2, but not with the essentially non-polar CH4 molecules (small octupole moment). These adsorptive processes promote the discrimination of CO2 over CH4 and thus the effective separation of the CO2/CH4 mixture.
Besides the materials synthesized by crosslinking the polyprecursors like swol-len polystyrene and poly(chloromethylstyrene), HCPs can also be produced by the direct polycondensation of small molecule monomers such as dichloroxylene (DCX), bis(chloromethyl)biphenyl (BCMBP), and bis(chloromethyl) anthra-cene (BCMA) (Fig. 5.4) [27]. These materials are predominantly microporous and exhibit apparent BET surface areas of up to 1,904 m2 g−1 as measured by
Table 5.1 Composition, porosity, and gas adsorption properties of samples
a Surface area calculated from nitrogen adsorption isotherms at 77.3 K using BET equationb Pore volume calculated from nitrogen isotherm at P/P0 = 0.995c t-Plot micropore volumed CO2 uptake determined volumetrically using a Micromeritics ASAP 2020 M analyzer at 1.00 bar and 273.15 K
No. DVB (mol %) SBET (m2 g−1)a PV (cm3 g−1)b MPV (cm3 g−1)cCO2 uptake (mmol g−1)d
1 0 1,420 1.29 0.088 2.262 0.5 1,790 1.86 0.0045 2.273 1 1,860 1.26 0.078 2.274 1.5 1,890 1.20 0.098 2.285 2 2,060 1.23 0.086 2.296 3 1,920 1.10 0.19 2.527 4 1,840 1.04 0.26 2.578 5 1,760 0.99 0.27 2.579 6 1,520 0.82 0.39 2.6110 7 1,430 0.77 0.37 2.7011 8 1,370 0.70 0.39 2.7612 10 1,260 0.63 0.39 2.82
Fig. 5.3 The structure of the highly cross-linked polystyrene particle and its CO2/CH4 selectiv-ity predicted by IAST for CO2/CH4 = 5/95 mixtures at 268 K (Reprinted with permission from Ref. [20]. Copyright 2012, American Chemical Society)

1495 Microporous Organic Polymers for Carbon Dioxide Capture
nitrogen adsorption at 77.3 K. Martin et al. [11] studied the CO2 adsorption capac-ity of these polymers using a thermogravimetric analyzer (atmospheric pressure tests) and a high-pressure magnetic suspension balance (high-pressure tests). It was found that the CO2 capture capacities were related to the textural properties of the HCPs. The performance of these materials to adsorb CO2 at atmospheric pressure was characterized by maximum CO2 uptakes of 1.7 mmol g−1 (7.4 wt%) at 298 K. At higher pressures (30 bar), the polymers show CO2 uptake of up to 13.4 mmol g−1 (59 wt%), superior to zeolite-based materials (zeolite 13X [26], zeolite NaX [28]) and commercial activated carbons (BPL [9], Norit R [9, 28]). In addition, these polymers showed low isosteric heats of CO2 adsorption and good selectivity toward CO2. These results confirmed that the HCPs have potential to be applied as CO2 adsorbents in pre-combustion capture processes where high CO2 partial pressures are involved. Based on the similar synthetic strategy, many HCPs have been reported by Friedel–Crafts self-condensation of polyfunctional benzyl chloride monomers such as benzyl chloride-terminated double-four-ring (D4R) cubic siloxane cages [29], carborane monomer mCB-3 [30], or co-condensation of BCMBP with some non-functionalized fluorene-based monomers (Fig. 5.4) [31]. These networks exhibit large BET surface areas and high gas uptake capacities for hydrogen or methane. Regrettably, however, no data exit about the CO2 uptake ability of these materials. Similar to the self-condensation of DCX, bishydroxym-ethyl monomer, 1,4-benzenedimethanol (BDM), also can self-condensate to give the hypercrosslinked polymer network (HCP-BDM) [32]. N2 sorption isotherm for
Fig. 5.4 Monomers used for the synthesis of the HCPs networks

150 Y. Luo and B. Tan
the polymer showed type I isotherm, and BET surface area was calculated to be 847 m2 g−1, which is comparable to the DCX networks (815–1,431 m2 g−1). At 1 bar/273 K, HCP-BDM can store a significant amount of CO2 (12.6 wt%) despite its relatively modest surface area. This excellent performance was attributed to the high micropore volume of the polymer and the residual hydroxyl groups in the network. In addition, our group [32] also found that monofunctional compounds such as benzyl alcohol (BA) and benzyl chloride (BC) can be used for construct-ing HCPs by self-condensation with the BET surface areas of ~740 m2 g−1 (Fig. 5.5). This is different from the previous reports where multifunctional mon-omers or cross-linkers are crucial for constructing the porous polymer networks. Based on these results, a possible mechanism is proposed to explain the forma-tion of the pores in the as-synthesized polymer. The primary step in the poly-condensation reaction seems to be the generation of benzyl carbocation, which attacks another monomer molecule to form a dimer. It should be noted that, in the dimer, one phenyl ring contains two predominantly ortho–para orienting groups which significantly enhance the susceptibility of the ring to be attacked by cations. Therefore, the dimer can grow by attacking other molecules with its hydroxym-ethyl group (benzyl carbonium ion) and by being attacked by BA. Such growth by the addition of benzyl groups to the dimer will result in highly substituted rings and many short branches. The porosity arises from the inefficient packing of the highly branched polymer chains. The gas uptake capacity shows that HCP-BA (or HCP-BC) can adsorb 8.46 wt% CO2 at 1 bar/273 K and the isosteric heat of adsorption for CO2 was estimated to be about 27.4–24.1 kJ mol−1. Both values are slightly higher than the copolymers of DCX/BCMBP (7.0–7.4 wt%) reported by Martin et al. [11]. This study opens up the possibility of synthesizing porous mate-rials using monofunctional monomers.
Fig. 5.5 Synthesis of HCP-BA by Friedel–Crafts catalyzed self-condensation (Reprinted with permission from Ref. [32]. Copyright 2013, Royal Society of Chemistry)
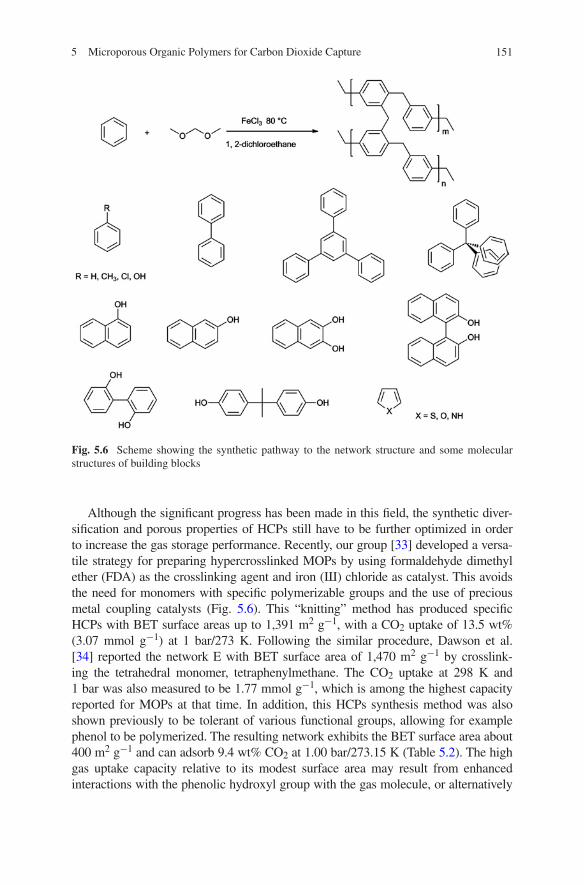
1515 Microporous Organic Polymers for Carbon Dioxide Capture
Although the significant progress has been made in this field, the synthetic diver-sification and porous properties of HCPs still have to be further optimized in order to increase the gas storage performance. Recently, our group [33] developed a versa-tile strategy for preparing hypercrosslinked MOPs by using formaldehyde dimethyl ether (FDA) as the crosslinking agent and iron (Ш) chloride as catalyst. This avoids the need for monomers with specific polymerizable groups and the use of precious metal coupling catalysts (Fig. 5.6). This “knitting” method has produced specific HCPs with BET surface areas up to 1,391 m2 g−1, with a CO2 uptake of 13.5 wt% (3.07 mmol g−1) at 1 bar/273 K. Following the similar procedure, Dawson et al. [34] reported the network E with BET surface area of 1,470 m2 g−1 by crosslink-ing the tetrahedral monomer, tetraphenylmethane. The CO2 uptake at 298 K and 1 bar was also measured to be 1.77 mmol g−1, which is among the highest capacity reported for MOPs at that time. In addition, this HCPs synthesis method was also shown previously to be tolerant of various functional groups, allowing for example phenol to be polymerized. The resulting network exhibits the BET surface area about 400 m2 g−1 and can adsorb 9.4 wt% CO2 at 1.00 bar/273.15 K (Table 5.2). The high gas uptake capacity relative to its modest surface area may result from enhanced interactions with the phenolic hydroxyl group with the gas molecule, or alternatively
Fig. 5.6 Scheme showing the synthetic pathway to the network structure and some molecular structures of building blocks
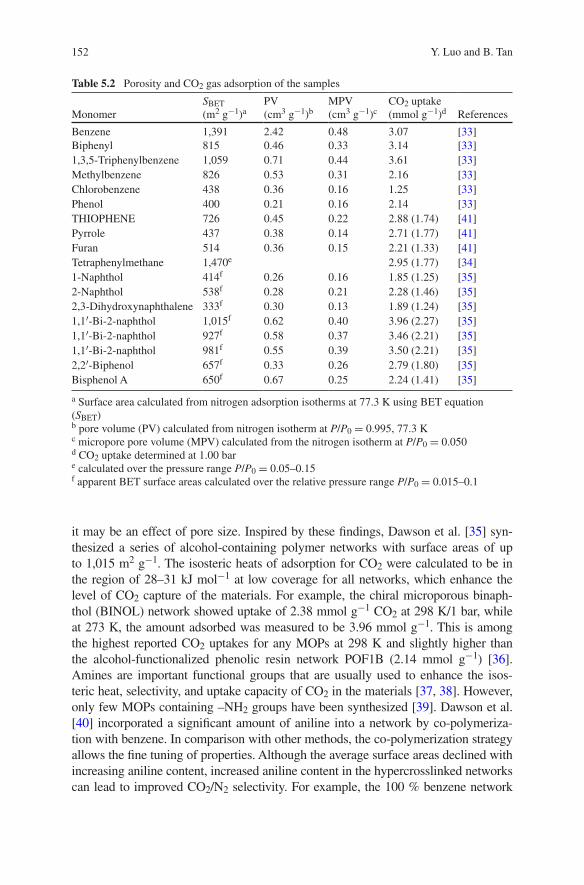
152 Y. Luo and B. Tan
it may be an effect of pore size. Inspired by these findings, Dawson et al. [35] syn-thesized a series of alcohol-containing polymer networks with surface areas of up to 1,015 m2 g−1. The isosteric heats of adsorption for CO2 were calculated to be in the region of 28–31 kJ mol−1 at low coverage for all networks, which enhance the level of CO2 capture of the materials. For example, the chiral microporous binaph-thol (BINOL) network showed uptake of 2.38 mmol g−1 CO2 at 298 K/1 bar, while at 273 K, the amount adsorbed was measured to be 3.96 mmol g−1. This is among the highest reported CO2 uptakes for any MOPs at 298 K and slightly higher than the alcohol-functionalized phenolic resin network POF1B (2.14 mmol g−1) [36]. Amines are important functional groups that are usually used to enhance the isos-teric heat, selectivity, and uptake capacity of CO2 in the materials [37, 38]. However, only few MOPs containing –NH2 groups have been synthesized [39]. Dawson et al. [40] incorporated a significant amount of aniline into a network by co-polymeriza-tion with benzene. In comparison with other methods, the co-polymerization strategy allows the fine tuning of properties. Although the average surface areas declined with increasing aniline content, increased aniline content in the hypercrosslinked networks can lead to improved CO2/N2 selectivity. For example, the 100 % benzene network
Table 5.2 Porosity and CO2 gas adsorption of the samples
a Surface area calculated from nitrogen adsorption isotherms at 77.3 K using BET equation (SBET)b pore volume (PV) calculated from nitrogen isotherm at P/P0 = 0.995, 77.3 Kc micropore pore volume (MPV) calculated from the nitrogen isotherm at P/P0 = 0.050d CO2 uptake determined at 1.00 bare calculated over the pressure range P/P0 = 0.05–0.15f apparent BET surface areas calculated over the relative pressure range P/P0 = 0.015–0.1
MonomerSBET (m2 g−1)a
PV (cm3 g−1)b
MPV (cm3 g−1)c
CO2 uptake (mmol g−1)d References
Benzene 1,391 2.42 0.48 3.07 [33]Biphenyl 815 0.46 0.33 3.14 [33]1,3,5-Triphenylbenzene 1,059 0.71 0.44 3.61 [33]Methylbenzene 826 0.53 0.31 2.16 [33]Chlorobenzene 438 0.36 0.16 1.25 [33]Phenol 400 0.21 0.16 2.14 [33]THIOPHENE 726 0.45 0.22 2.88 (1.74) [41]Pyrrole 437 0.38 0.14 2.71 (1.77) [41]Furan 514 0.36 0.15 2.21 (1.33) [41]Tetraphenylmethane 1,470e 2.95 (1.77) [34]1-Naphthol 414f 0.26 0.16 1.85 (1.25) [35]2-Naphthol 538f 0.28 0.21 2.28 (1.46) [35]2,3-Dihydroxynaphthalene 333f 0.30 0.13 1.89 (1.24) [35]1,1′-Bi-2-naphthol 1,015f 0.62 0.40 3.96 (2.27) [35]1,1′-Bi-2-naphthol 927f 0.58 0.37 3.46 (2.21) [35]1,1′-Bi-2-naphthol 981f 0.55 0.39 3.50 (2.21) [35]2,2′-Biphenol 657f 0.33 0.26 2.79 (1.80) [35]Bisphenol A 650f 0.67 0.25 2.24 (1.41) [35]

1535 Microporous Organic Polymers for Carbon Dioxide Capture
(SBET = 1,289 ± 156 m2 g−1) showed a CO2/N2 selectivity of 15.9: 1, while the 100 % aniline network (SBET = 7 ± 6 m2 g−1) has a selectivity of 49.2:1. This can be explained by the CO2 uptakes in the copolymers drop less sharply than the surface area, which led to materials with improved selectivity for CO2 over nitrogen.
Following the previous study, our group further widens the monomers used in HCPs to the aromatic heterocycle molecules and a series of microporous heterocy-clic polymers were obtained (Fig. 5.6) [41]. The BET surface area of these polymers is up to 726 m2 g−1. Owing to the narrow pore system and the heteroatoms-riched pore surface, the microporous aromatic heterocyclic polymers exhibit high adsorp-tion capacity for CO2 (12.7 wt%, 273 K, and 1 bar) (Table 5.2). These CO2 adsorp-tion capacities are comparable or even higher than those of amine- or carboxylic acid-functionalized materials. Specifically, Py-1 shows an extraordinarily high selec-tive adsorption of CO2 over N2 (about 117 at 273 K). To our knowledge, this selec-tive CO2 sorption is the highest among all the microporous materials reported to date. Collectively, these results emphasize the importance of utilizing heterocyclic molecules as building blocks to produce MOPs for CO2 capture application.
Recently, Lim et al. [42] reported the synthesis of a new class of 1,3,5-triazine-based microporous polymers by Friedel–Crafts reaction of 2,4,6-trichloro-1,3,5-tri-azine with aromatic compounds (Fig. 5.7). The surface areas of the polymers are in the range of 558–1,266 m2 g−1, depending on the aromatic linker length. At ambient pressure and temperature, the polymers exhibit high CO2 uptakes of 38–51 cm3 g−1. The isosteric heats determined at low loading were 36 kJ mol−1 for polymers 2 and 3 and 33 kJ mol−1 for polymer 4, which were comparable to those of functional-ized MOPs [39]. Polymers 2 and 3 showed a higher heat of adsorption than polymer
Fig. 5.7 Preparation of microporous polymers by Friedel–Crafts reaction of 2,4,6-trichloro-1,3,5-triazine (a) and 1-bromoadamantane (b) with an aromatic compound

154 Y. Luo and B. Tan
4 probably because they had more small pores. In addition, it is worth mentioning that the polymer prepared in this study has conjugated structures, which is differ-ent from the HCPs reported previous by Friedel–Crafts alkylation reaction. By replacing 2,4,6-trichloro-1,3,5-triazine with 1-bromoadamantane, adamantine-based microporous polymers also were prepared following the similar reaction [43]. The post-modification of the polymers were carried out by Friedel–Crafts reaction with 4-nitrobenzoyl chloride or BC. At 195 K, the CO2 adsorption was mainly dependent on the surface area of the materials. When the temperature was increased to 298 K, however, CO2 uptake capacity of the modified polymers was increased by 18–25 % owing to the interaction between the nitro group and CO2.
5.3 Covalent Organic Frameworks
COFs are a class of crystalline porous materials with pure organic groups con-nected via robust covalent bonds [44]. Since the seminal work of Yaghi and co-workers in 2005 [45], the rapid development in this research area has attracted worldwide attention and numerous chemical architectures with discrete (zero-dimensional, 0-D) to extended (1-D, 2-D, and 3-D) structures were constructed by assembling the diverse building units in different way [46]. In comparison with other crystalline porous solids (inorganic zeolites and hybrid MOFs), the COF materials possess the advantages of low density, large surface areas, tunable pore size and structure, facilely tailored functionality, and versatile covalent combina-tion of building units. All of these advantages make COFs as new candidates for the separation and adsorption of CO2. In 2009, Yaghi’s group [9] first experimen-tally reported the CO2 uptake capacity of both 2-D and 3-D COFs. It is found that with regard to the adsorbed amount of CO2, the surface excess masses increase in the order of group 1 < group 2 < group 3 COFs at high pressure (Fig. 5.8a). In particular, COF-102 with a BET surface area of 3,620 m2 g−1 exhibits a satu-rated uptake of 27 mmol g−1 (1,200 mg g−1) at 35 bar/298 K, which is higher than the uptake of both MOF-5 (22 mmol g−1) [47] and zeolite (5–8 mmol g−1) [26]. However, COF-6, possessing a smaller pore diameter, outperforms other COFs at 1 bar/273 K (85 cm3 g−1) (Fig. 5.8b).
To further understand the CO2 uptake of COFs, the interaction between CO2 molecules and COFs has been investigated using atomistic simulations [48–50]. According to Babarao’s simulations [49], the amount of excess CO2 uptake is directly related to the total pore volume. So the CO2 capacities in 2-D COFs at high pressures are lower compared to 3-D COFs because of their smaller avail-able free volumes. Choi et al. [50] also confirmed the opinion using grad canoni-cal ensemble Monte Carlo Calculations. Moreover, the binding sites and energies of CO2 on COFs were identified using ab initio calculations. It is found that CO2 acting both as Lewis acid and also Lewis base can bind to oxygen atoms and phe-nyl rings of linker units in COFs. In 2-D layered COFs, the CO2 bound is located between two layers with the binding energy of about 9.2 kcal mol−1. Besides the

1555 Microporous Organic Polymers for Carbon Dioxide Capture
oxygen atom, neighboring boron atoms in COFs also can enhance the binding of CO2 to COFs. However, 2-D COFs usually exhibit favorable π–π stacking inter-actions that can hinder the accessibility of boron sites. With this consideration in mind, Kahveci et al. [51] introduced the triptycene into the backbone of COFs to obtain the triptycene-derived covalent organic framework (TDCOF-5). Because the triptycene core in TDCOF-5 can reduce π–π interactions between the building units and thereby leads to more accessible boron sites for interaction with gas mol-ecules, TDCOF-5 can store up to 9.2 wt% (2.1 mmol g−1) of CO2 with Qst val-ues of 21.8 kJ mol−1. This value is higher than those of reported COFs and only exceeded by the uptake of COF-6 which has much narrower channels (9 Å). Other boron-containing porous materials should include the borazine-linked polymers (BLPs), which is prepared by the thermal decomposition of arylamine–borane or arylamine–boron trihalide adducts in aprotic solvents [52–55]. The borazine ring in these materials is structurally analogous to the boroxine building units found in COFs. Among them, BLP-12(H) exhibits high surface area of 2,244 m2 g−1 and can store significant amount of CO2 (12.8 wt%) at 273 K/1 bar [52]. However, any potential materials for CCS require not only high uptake of CO2 but also excellent selectivity for CO2. Both tailoring pore size and introducing functionalized groups are important strategies in constructing a porous material for gas separation. Nagai and co-workers [56] have established a simple and universal strategy for the pore surface engineering of COFs to allow the incorporation of variety of organic func-tionalities into the channels. This functionalization is made possible by the use of azide-appended N3-COF-5 (or N3-NiPc-COF) as the parent materials, which can undergo a quantitative click reaction with various alkynes to produce pore sur-faces with desired groups (such as alkyl chains, acetyl, aromatic units, ester, and chromophoric moieties) and preferred densities (Fig. 5.9). In addition, the pore size also can be finely tuned from 1.2 to 3.0 nm for the hexagonal COF-5 family and 0.75–2.2 nm for the tetragonal NiPc-COF. The gas adsorption studies revealed that the CO2/N2 selectivity of the surface-engineered COF-5 depended signifi-cantly on the surface groups. For example, the X%PyTrz-COF-5 exhibited an
Fig. 5.8 CO2 isotherms for COFs measured at 298 K (a) and 273 K (b) (Reprinted with permis-sion from Ref. [9]. Copyright 2009, American Chemical Society)

156 Y. Luo and B. Tan
increased tendency for selectivity with increasing pyrene content, and 25 %PyTrz-COF-5 exhibited a fivefold increased selectivity in comparison with COF-5. The 50 %BuTrz-COF-5, 50 %EsTrz-COF-5, and 50 %PhTrz-COF-5 displayed selec-tivity of 2.3, 1.1, and 1.6, respectively. Most significantly, 100 %AcTrz-COF-5 showed a 16-fold increase in selectivity relative to that of COF-5. All of these results indicate that the pore surface is an important factor that affects the sorption properties of the COFs. Recently, Lan et al. [57] used the multiscale simulation approach to comprehensively study the effects of the metal dopants on CO2 cap-ture. It is found that Li is the best modifier of COFs for CO2 capture among all the studied metals such as alkali, alkaline-earth, and transition metals. For example, the excess CO2 uptakes of the Li-doped COF-102 and COF-105 at 298 K/1 bar reach to 409 and 344 mg g−1, which are about eight and four times higher than those non-doped ones, respectively. Till date, however, metal complexes of those porous organic polymers are either created by the introduction of metal ions via post-functionalization [58], or by the use of predefined metal phthalocyanine [59] or porphyrin [60] building blocks as reactants. In 2012, Modak et al. [61] reported a series of iron-containing porous organic polymers (Fe-POPs) through a simple one-pot bottom-up approach involving extended aromatic substitution on pyrrole with aromatic dialdehydes. The Fe-POPs possess high BET surface area (875 m2 g−1) and large micropores and showed excellent CO2 capture (~19 wt%) at 273 K/1 bar. Salphens and salens are versatile tetradentate ligands, which have been used as linkers for construction of zinc-MOFs [62]. Mastalerz et al. [63] reported a one-pot three-component reaction of metal salphen containing porous
Fig. 5.9 Schematic representation of the surface engineering of COFs of a hexagonal COF-5, and b tetragonal NiPc-COF

1575 Microporous Organic Polymers for Carbon Dioxide Capture
organic polymers (MaSOFs). Due to the high BET surface areas (647 m2 g−1) and narrow pore size distribution, MaSOF-1 and MaSOF-2 can adsorb 8.1 and 9.8 wt% CO2 at 273 K/1 bar, respectively.
Besides the boron-containing COFs synthesized through the formation of boro-nate ester or boronate anhydride, imine-based COFs also have attracted considerable interest of the scientists. The imine-based COFs synthesized so far can be divided into two categories according to the covalent formation of distinct –C=N– bonds [44]. One is the “Schiff base” type formed through the condensation of aldehydes and amines [64]; another is hydrazine-linked COFs, which are synthesized by co-condensation reaction of aldehydes and hydrazides [65]. In general, porous net-works are synthesized by reactions between Ax + By monomers (where x and y > 2) [60, 66–69]. In 2012, Laybourn et al. [70] reported the MOP networks via one-pot polycondensation reaction between A2 aldehyde tereophthaldehyde and B2 amine 2,6-diaminopyridine (PI-1) or 2,4-diaminotoluene (PI-2). Due to introducing branching (and thus porosity) into linear networks, the resulting networks exhibit BET surface areas in the range 500–600 m2 g−1. At 298 K/1 bar, PI-1 showed a CO2 uptake of 1.41 mmol g−1 and CO2/N2 of 27:1, substantially higher than that of PI-2 (1.00 mmol g−1 and 12:1). These values are comparable to many other types of polymer network [71] as well as porous organic cages [72]. Furan-based porous organic frameworks (FOFs) also were prepared by condensation of biomass-derived 2,5-diformylfuran (DFF) and diamines via the formation of imine linkages. It is found that the materials obtained by DFF being condensed with nonlinear diamines showed higher specific surface areas (830 and 571 m2 g−1) than that of with linear diamines (96 and 307 m2 g−1). The accessible nitrogen sites and ether O atoms in the pore wall of the FOFs make them show higher adsorption capacity for CO2. For example, the FOF-1 can adsorb 77.0 mg g−1 CO2 at 273 K/1 bar, which is com-petitive with that of the reported microporous MOF SNU-150 (70.0 mg g−1) [73], and zeolitic imidazolate framework (ZIF-100, 74.8 mg g−1) [74]. However, since reversible reactions have been used for the synthesis of crystalline COFs, reversible back-reactions can occur after the synthesis and COFs in general completely decom-pose even in the presence of ambient humidity [75, 76]. Recently, Kandambeth et al. [77] synthesized two highly acid- and base-stable crystalline COFs, TpPa-1 and TpPa-2, by a combination of reversible Schiff base reaction and irreversible enol-to-keto tautomerization (Fig. 5.10). At 273 K/1 bar, the CO2 uptake of TpPa-1 (SBET = 535 m2 g−1) was measured as 78 cm3 g−1, which is comparable to the performance of COF-6 (85 cm3 g−1) [9]. TpPa-2 (SBET = 339 m2 g−1) showed a moderate CO2 uptake of 64 cm3 g−1 at the same temperature. In 2013, Xu et al. [78] prepared a series of imine-linked MOPs by condensation of 1,3,5-tris(4-ami-nophenyl)benzene and a number of dialdehyde monomers. Synthesis conditions were optimized to enhance CO2 uptake by the MOPs. It is found that the MOP A-B2 synthesized in an open system (condensation Ш) had the highest CO2 uptake (60.7 cm3 g−1, at 273 K/1 bar) among the synthesized MOPs with a heat of adsorp-tion of 33.4 kJ mol−1. The selectivity of CO2 over N2 at 273 K and 1 bar was 56, which requires either an influence of chemisorption on CO2 or a molecular sieving (or kinetic selection) of CO2 over N2.

158 Y. Luo and B. Tan
CO2 uptakes for the porous benzimidazole-linked polymers (BILPs) have also been reported. Rabbani and El-Kaderi [79] synthesized BILP-1 (Fig. 5.11a) with a BET surface area of 1,172 m2 g−1, whose CO2 uptake reaches 188 mg g−1 (4.3 mmol g−1) at 273 K/1 bar. The Qst for CO2 is 26.5 kJ mol−1 at zero coverage, and the adsorption selectivity for CO2 over N2 is 70. The CO2 uptake and Qst are higher than the values reported for COFs, and comparable to CO2 selective MOFs [80, 81] or ZIF [82] which generally feature –NH2 or –OH functionalized pores. The notable enhanced CO2 capture capacity and selectivity were attributed to its subnano pore dimensions and imidazole-functionalized pore walls. Since trip-tycene-based porous materials have been reported to possess enhanced adsorptive gas uptake capacities [83–86], they also synthesized the triptycen-derived BILPs, BILP-3 (HATT+TFPM) and BILP-6 (HATT+TPAL). These polymers can store 5.12 mmol g−1 of CO2 at 273 K/1 bar and exhibit high CO2/N2 (63) and CO2/CH4 (8.4) selectivity [87]. The BILPs family was then expended by varying the amine- and aldehyde-containing building blocks (Fig. 5.11b) [88]. The resulting BILPs exhibit moderate surface area (SBET = 599–1,306 m2 g−1), high chemical and ther-mal stability, and remarkable gas uptake and selectivity. The highest selectivity based on initial slope calculations at 273 K was observed for BILP-2 (HATP+TPAL): CO2/N2 (113) and CO2/CH4 (17), while the highest CO2 storage capacity was recorded for BILP-4 (BTA+TFPM) (24 wt% at 273 K/1 bar). These selectivities and gas uptakes are among the highest values produced by porous organic poly-mers known to date. Although the above approaches are successful for constructing the poly(benzimidazole) networks, the synthetic strategy still has to be further opti-mized because the imine linkage was inevitably formed during the formation of imidazole ring. Additionally, it is not easy to control the bubbling of oxygen into the reaction system, which is necessary for the formation of imidazole ring. With these consideration in mind, Zhao et al. [89] reported the synthesis of microporous
Fig. 5.10 Schematic representation of the synthesis of TpPa-1 and TpPa-2 by the combined reversible and irreversible reaction of 1,3,5-triformylphloroglucinol with p-phenylenediamine and 2,5-dimethyl-p-phenylenediamine, respectively (Reprinted with permission from Ref. [77]. Copyright 2012, American Chemical Society)

1595 Microporous Organic Polymers for Carbon Dioxide Capture
poly(benzimidazole) networks through condensation reaction between dione, dial-dehyde, and ammonium acetate. This approach is a template-free and catalyst-free condensation and produces no by-products. The materials, with BET-specific surface area over 600 m2 g−1, possess a high CO2 uptake (up to 14.0 wt% at 273 K/1 bar). However, all the values are lower than those of BILP-1 network reported by Rabbani and El-Kaderi [79]. Recently, Yu et al. [90] also synthesized porous poly(benzimidazole) networks PBI-1 and PBI-2 through one-step polycondensation of 3,3′-diaminobenzidine with tris(4-carboxyphenyl)amine (TCA, Fig. 5.11b) and tetrakis(4-carboxyphenyl)silane (TCS, Fig. 5.11b). Compared with other synthetic methods [79, 87–89], the significant advantage of this strategy is that the reaction needs not to be tube-sealed and the reaction time can be reduced from 3 days to sev-eral hours. The analyses of CO2 adsorption isotherms reveal that PBI-1 and PBI-2 networks have ultramicroporous structures with surface areas of 694 and 241 m2 g−1, respectively. In addition, at 273 K/1 bar, PBI-1 displays a high CO2 uptake capacity of 13.2 wt% and CO2/N2 selectivity of 12.3. Similar to the synthetic route of BILPs reported by Rabbani and El-Kaderi [79], polycondensation reactions between hexa-chlorocyclotriphosphazene and diaminobenzidine also can give the porous networks PECONFs [91]. The CO2 uptake of these materials are about 1.81–3.49 mmol g−1 at 273 K/1 atm, and CO2/N2 selectivity are 74–109 according to the ratios of the Henry law constants. Such high selectivity for CO2 over N2 can be explained by Lewis acid–base interactions between the electron-poor CO2 and the electron-rich sorbent.
Fig. 5.11 The synthesis of BILP-1 (a) and the monomers used for constructing BILPs (b)

160 Y. Luo and B. Tan
Another type of COFs is the covalent triazine frameworks (CTFs) [13, 92–96], which were developed by Thomas and co-workers via the cyclical trimerization of cyano reaction groups under ionothermal conditions (Fig. 5.12a). In comparison with those COFs obtained via the solvothermal method, the CTFs are often of lower crystallinity because the reversible cyclotrimerization reaction takes place under harsh reaction conditions. To date, only two building units (1,4-dicyanobenzene and 2,6-naphthalenedicarbonitrile) have been successfully utilized to synthesize the crys-talline CTFs [13, 94]. The high amount of nitrogen atoms may render CTFs with potential application for CO2 capture. For example, Wang et al. [97] reported that PAF-16 which is synthesized by trimerization reaction of tetrakis(4-cyanophenyl)silica (TCPSi) can adsorb 8.3 wt% of CO2 at 273 K/1 atm. The initial adsorption heat is 30.32 kJ mol−1. However, while the ionothermal method is efficient for tria-zine ring formation in these CTFs, the high temperature (≥400 °C) and long reac-tion time required might limit practical applications. In 2012, Ren et al. [98] and Zhu et al. [99] reported almost simultaneously the synthesis of CTFs using trifluo-romethanesulfonic acid (CF3SO3H) as the catalyst. This synthesis strategy makes the reaction being carried out at a much lower temperature (<100 °C) for a shorter time. Most importantly, porous membranes also can be obtained (Fig. 5.12b). At 273 K/1 bar, the membrane TFM-1, with a high BET surface area of 738 m2 g−1, exhibited a good CO2 uptake of 1.73 mmol g−1. It also exhibits increased selectivity for the membrane separation of CO2 over N2. An ideal CO2/N2 permselectivity of 29 ± 2 was achieved, with a CO2 permeability of 518 ± 25 barrer [99].
Fig. 5.12 a Synthesis of CTF-1 and the extended crystalline structures. b Trimerization reaction of 4,4′-biphenyldicarbonitrile in CF3SO3H at 100 °C and the photograph of a directly synthe-sized sample of the transparent and flexible triazine-framework-based membrane TFM-1

1615 Microporous Organic Polymers for Carbon Dioxide Capture
5.4 Polymers of Intrinsic Microporosity
PIMs introduced by Budd and Mckeown, are amorphous organic microporous mate-rials in which the pores are derived from the space-inefficient packing of highly rigid and contorted non-network polymer chains [5, 100, 101]. Usually, there are two key points for design and synthesis of PIMs [102]: (1) Polymer chains are generally aro-matic heterocyclic ladder polymers with a non-network structure, and (2) polymer chains should be rigid and contorted enough to prevent rotational freedom along the polymer backbone, otherwise the conformation of the polymer chains can rearrange to collapse the porous structure. The first examples consisted of phthalocyanines [103] or porphyrins [104] were linked by rigid spirobisindane groups. These materi-als exhibited microporosity, with the BET surface areas of 895 and 910 m2 g−1 for phthalocyanine- and porphyrin-based networks, respectively. Since then, a variety of PIMs have been prepared using different rigid molecular components and nonlinear linking groups, in which the main synthetic strategy is the dioxane-forming reaction (Fig. 5.13). All of these polymers exhibit surface areas ranging from approximately 500 to over 100 m2 g−1. More importantly, when bifunctional monomers are used, the resulting polymers can be soluble in organic solvents and are frequently cast as films, which allows for membrane gas separation applications [5, 105].
Among all the PIMs synthesized, PIM-1 is one of the most well-known types for membrane gas separation [106, 107]. The pristine PIM-1 membrane has dem-onstrated outstanding permeability with moderate separation factor for several important gas pairs (e.g., O2/N2, CO2/N2, and CO2/CH4), which overcome the upper bound trade-off proposed by Robeson [108, 109]. In order to further enhance the gas-pair selectivity, several works have attempted to modify the PIM-1 by incor-porating some function groups in the polymer chains. For example, Du et al. [110] synthesized the disulfonyl-based PIMs by co-condensation of A1 (Fig. 5.13), B4 (Fig. 5.13), and disulfone-based monomers. Compared with the PIM-1, the sulfone-based copolymers have higher gas selectivities for CO2/N2. However, there is some reduction in pure-gas permeabilities. In addition, the gas separation properties also can be improved through post-modification of nitrile-based PIM membranes. In the work of Du et al. [111], the nitrile groups of PIM-1 were converted to carboxyl groups via a simple hydrolysis reaction (Fig. 5.14a, Reaction A). The degree of con-version could be controlled by the hydrolysis time and temperature. Similar to the previous report, the hydrolyzed PIM-1 film (i.e., carboxylated-PIM film) showed an obvious increase in gas-pair selectivity but a decrease in gas permeability. By using ab initio methods, it is found there are two different interactions between CO2 and N-containing aromatic molecules (such as pyridine, imidazole, and tetrazole), which are Lewis acid–Lewis base interactions and hydrogen bonding [112]. Thus, incorpo-ration of such heterocyclics into the frameworks will improve the CO2-philic nature of the polymers. With this consideration in the mind, Du et al. [113] incorporated tetrazole groups into PIM-1 by the [2+3] cycloaddition post-polymerization reac-tion between nitriles and azides (Fig. 5.14a, Reaction B). The resulting membranes show both high CO2 permeability and high CO2/light gas selectivity (Fig. 5.14b, c). This strategy provides a direction in the design of porous membrane materials for

162 Y. Luo and B. Tan
economic CO2 capture processes. Amidoximes are known to be CO2-philic [114], and functionalization of PIM-1 by amidoxime will create sites that have strong affinity toward CO2 through its high quadruple moment. In 2012, Patel and Yavuz [115] synthesized the amidoxime-PIM-1 successfully through a conventional and non-invasive synthetic route (Fig. 5.14a, Reaction C). CO2 uptake of amidoxime-PIM-1 (72.4 mg g−1) was doubled to 17 % when compared to PIM-1 (62 mg g−1) at 298 K/1 bar. In order to enhance the gas-pair selectivity and at the same time main-taining the gas permeability of PIM-1 membranes, Li et al. [116] prepared the ther-mally self-cross-linked PIM-1 membranes via the inherent cross-linking reaction of aromatic nitrile groups to form triazine rings. With the nature of the contorted struc-ture of PIM-1 backbone, the cross-linking reaction would lead to the pronounced inefficient chain packing, thus an increase in free volume and gas permeability with the increase in thermal soaking time. The results showed that PIM-1 thermally treated at 300 °C for 2 days has the CO2 permeability of 4,000 barrer with CO2/CH4 and CO2/N2 ideal selectivity of 54.8 and 41.7, respectively. The ideal CO2/CH4 selectivity is almost four times higher than the host PIM-1 membrane.
In addition to the dibenzodioxin reaction, the more classical polymerization reac-tion of imide formation has been used to form PIMs. Weber and co-workers [117, 118] firstly reported the synthesis of soluble poly(amide) and poly(imide) using monomers derived from 9,9′-spirobifluorene. Such a spirobifluorene generates a 90° kink per repeating unit and thus prevents space-efficient chain packing, result-ing in microporous materials with high surface areas. For example, by reacting 2,2′-diamino-9,9′-spirobifluorene (Fig. 5.15, A1) with pyromellitic dianhydride (PMDA) (Fig. 5.15, B1), a soluble polymer with a BET surface area of 551 m2 g−1 was obtained [118]. Binaphthalene-based polyimide was synthesized by Ritter et al.
Fig. 5.13 Some monomers used in the synthesis of PIMs
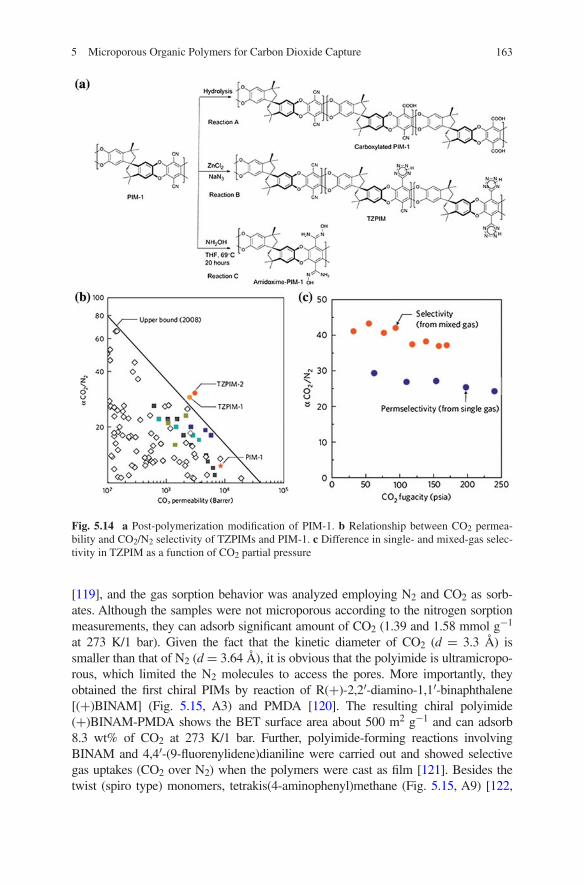
1635 Microporous Organic Polymers for Carbon Dioxide Capture
[119], and the gas sorption behavior was analyzed employing N2 and CO2 as sorb-ates. Although the samples were not microporous according to the nitrogen sorption measurements, they can adsorb significant amount of CO2 (1.39 and 1.58 mmol g−1 at 273 K/1 bar). Given the fact that the kinetic diameter of CO2 (d = 3.3 Å) is smaller than that of N2 (d = 3.64 Å), it is obvious that the polyimide is ultramicropo-rous, which limited the N2 molecules to access the pores. More importantly, they obtained the first chiral PIMs by reaction of R(+)-2,2′-diamino-1,1′-binaphthalene [(+)BINAM] (Fig. 5.15, A3) and PMDA [120]. The resulting chiral polyimide (+)BINAM-PMDA shows the BET surface area about 500 m2 g−1 and can adsorb 8.3 wt% of CO2 at 273 K/1 bar. Further, polyimide-forming reactions involving BINAM and 4,4′-(9-fluorenylidene)dianiline were carried out and showed selective gas uptakes (CO2 over N2) when the polymers were cast as film [121]. Besides the twist (spiro type) monomers, tetrakis(4-aminophenyl)methane (Fig. 5.15, A9) [122,
Fig. 5.14 a Post-polymerization modification of PIM-1. b Relationship between CO2 permea-bility and CO2/N2 selectivity of TZPIMs and PIM-1. c Difference in single- and mixed-gas selec-tivity in TZPIM as a function of CO2 partial pressure
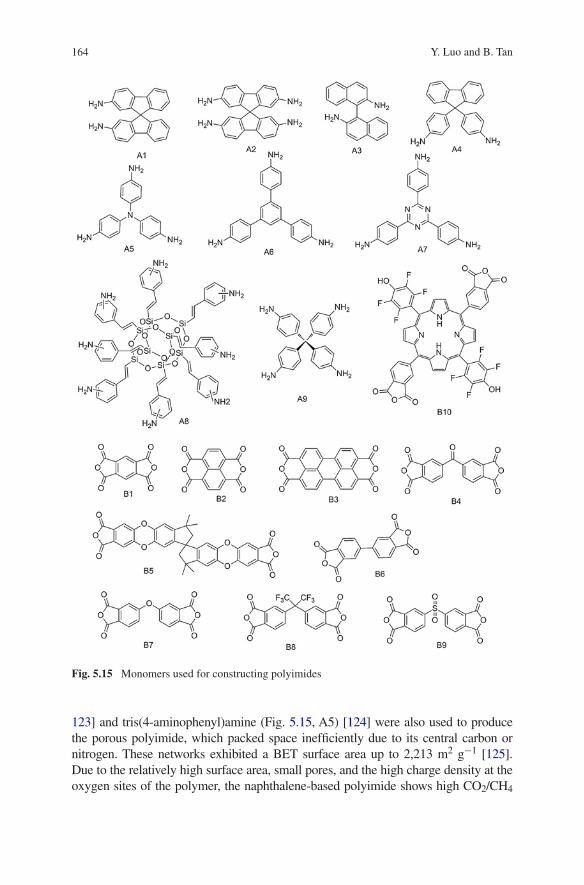
164 Y. Luo and B. Tan
123] and tris(4-aminophenyl)amine (Fig. 5.15, A5) [124] were also used to produce the porous polyimide, which packed space inefficiently due to its central carbon or nitrogen. These networks exhibited a BET surface area up to 2,213 m2 g−1 [125]. Due to the relatively high surface area, small pores, and the high charge density at the oxygen sites of the polymer, the naphthalene-based polyimide shows high CO2/CH4
Fig. 5.15 Monomers used for constructing polyimides

1655 Microporous Organic Polymers for Carbon Dioxide Capture
selectivity of 12–28 in the typical condition of natural gas purification (yCH4= 0.95
and 2 bar), which are similar to that of zeolites 13X (2–24) in the same condition [123]. Then, Farha et al. [126] prepared the Li-doped polyimide by chemical reduc-tion in the naphthalene diimide material using lithium metal as reductant. The reduced material (i.e., Li-doped polyimide) remains porosity and demonstrates a drastic increase in selectivity (38) for CO2 over CH4. Recently, polyimide-containing triazine rings have been reported by using cheap, commercially available melamine and anhydrides as the monomers [127]. The use of PMDA with melamine produced the material with BET surface area of 660 m2 g−1. Due to the high percentages of nitrogen contained, it can adsorb 7.3 wt% of CO2 at 273 K/1.13 bar in spite of its moderate surface area. Subsequently, a series of triazine-based porous poyimide (TPI) polymer networks were synthesized by Liebl et al. [128]. The high degree of func-tionalization led to comparatively high CO2 adsorption heats for TPI polymer net-works between 29 and 34 kJ mol−1. TPI-1, with the BET surface area of 809 m2 g−1, can adsorb 2.45 mmol g−1 of CO2 at 273 K/1 bar, while TPI-7 gave the highest bind-ing selectivity (56) for CO2 over N2. Although Trewin et al. [129] predicted the crys-tal structures and pore structures of some polyimides through atomistic simulations, all the porous polyimide mentioned above are amorphous. In 2010, Zeng et al. [130] synthesized the crystalline polyimide spheres with the BET surface area up to 350 m2 g−1. It is worth noting that the high surface area of this porous material is mainly stemmed from the mesopores formed by the oriented packing of nanowhisker.
5.5 Conjugated Microporous Polymers
CMPs [6], as the word implies, are the microporous polymers with conjugated frameworks. They can be considered a subclass of HCPs. CMPs differ significantly, however, from other HCPs in that they consist of multiple C–C bonds and/or aro-matic rings that form an extended conjugated network [131]. The first CMPs were porous poly(arylene ethynylene)s (PAEs), which were reported in 2007 by Cooper et al. and prepared by Pd-catalyzed Sonogashira–Hagihara cross-coupling between ary-lethynylenes and aryl halides (Fig. 5.16a). Although the monomers are planar, they can form 3-D networks due to the easy rotation about the alkyne bonds. In addition, the porous structure (BET surface area, pore volume, average pore size) could be finely tuned by changing the strut length of the monomers (Fig. 5.16b). Since then, various synthetic strategies have been used to synthesize CMPs, such as Yamamoto [132–136], Ullmann [137], Suzuki [138–145], Sonogashira–Hagihara coupling [146–152], oxidative coupling [145, 153], and ethynyl cyclotrimerization [154–156]. As a result, the CMPs family was expanded quickly in the last several years.
In 2009, Ben et al. [14] synthesized a porous aromatic framework, PAF-1, via Yamamoto homocoupling of tetrahedral tetrakis(4-bromophenyl)methane. This material had a record BET surface area of 5,640 m2 g−1 at that time and remark-able thermal/solvent stability. Besides, PAF-1 also showed very high uptake of CO2 (1,300 mg g−1 at 298 K/40 bar) to make it a good candidate for CO2 storage.

166 Y. Luo and B. Tan
Furthermore, by replacing tetrakis(4-bromophenyl)methane with 1,3,5,7-tetrakis(4-bromophenyl)adamantine (TBPA), Cooper group [157] and Zhou group [158] synthesized a microporous polymer with the BET surface area of 3,180 m2 g−1 (network 3) and 2,840 m2 g−1 (PPN-3), respectively. At 298 K/1.13 bar, network 3 exhibits the CO2 uptake of 7.59 wt%, which is higher than that reported for PAF-1(4.8 wt%, 298 K/1 atm) [159], despite having a lower surface area. The synthesis of PAF containing quadricovalent Si (network 2) was first reported by Cooper group in 2010, with the BET surface area of 1,102 m2 g−1 [157]. Subsequently, this struc-ture was synthesized by Qiu group (PAF-3) [159] and Zhou group (PPN-4) [160] using an optimized Yamamoto coupling again. For PPN-4, the BET surface area is about 6,461 m2 g−1, which is the highest BET surface area presently reported for any porous material [160]. Combined with such an impressive surface area, PPN-4 can adsorb 212 wt% CO2 at 295 K/50 bar. The CO2 selectivity of the materials at low pressure also was measured. PAF-3, with the BET surface area of 2,932 m2 g−1, exhibits extraordinarily promising selectivity (87/1) for the adsorption of CO2 over N2 under ambient conditions (760 mmHg, 273 K) as well as of CH4, which is 30 times higher than that of N2 [159]. Though PAFs are good practical candidates for CO2 capture and separation, the isosteric heat of CO2 adsorption (Qst) is relatively low at 15–19 kJ mol−1, which limited the application of gas storage at higher tem-perature. Babarao et al. [161] designed new PAFs by introducing polar organic groups to the biphenyl unit and then investigate their separating power toward CO2 by using grand canonical Monte Carlo (GCMC) simulations. The isosteric heat of CO2 adsorption for the functionalized PAF-1 is about 21.05 (NH2_PAF-1),
Fig. 5.16 a Chemical structure for series of five PAE CMP networks with varying “strut” lengths. b Nitrogen sorption analysis for series of materials CMP-0–CMP-5; the apparent BET surface area varies from 512 to 1,018 m2 g−1 as the strut length is reduced (Reprinted with per-mission from Ref. [6]. Copyright 2009, Wiley-VCH)

1675 Microporous Organic Polymers for Carbon Dioxide Capture
33.68 (MO_PAF-1), and 30.19 kJ mol−1 (DHF_PAF-1). All these values are far higher than that of the pristine PAF-1 (12.76 kJ mol−1). Inspired by these simula-tion results, Lu et al. [162] synthesized the sulfonate-grafted PPN-6 (also known as PAF-1)—PPN-6-SO3H (grafted with sulfonic acid) and PPN-6-SO3Li (grafted with lithium sulfonate) (Fig. 5.17A). Although the surface area of PPN-6-SO3H and PPN-6-SO3Li reduced to 1,254 and 1,186 m2 g−1, the isosteric heat of CO2 adsorption increased to 30.4 and 35.7 kJ mol−1. Strong interactions between the sul-fonate-grafted PPN-6 samples and CO2 make them display significantly enhanced CO2-uptake capacities. For example, non-grafted PPN-6 (SBET = 4,023 m2 g−1) has a gravimetric CO2 uptake of 5.1 wt% at 295 K/1 bar, whereas PPN-6-SO3H and PPN-6-SO3Li can adsorb 13.1 and 13.5 wt% CO2 at the same condition, respec-tively. IAST calculations using single-component-isotherm data and a 15/85 CO2/N2 ratio at 295 K/1 bar revealed that the sulfonate-grafted PPN-6 networks show exceptionally high CO2/N2 selectivity, with the values of 155 and 414 for PPN-6-SO3H and PPN-6-SO3Li, respectively. The high CO2 adsorption capacity and selectivity over N2 of the PPN-6-SO3Li can be largely attributed to stronger electro-static interactions between CO2 and Li+ cations [162]. Subsequently, Konstas et al. [163] prepared the lithiation of PAF-1 (Li@PAF-1) by directly incorporating lith-ium ions within the pores of PAF-1. 5 %_Li@PAF-1, with the BET surface area of 473 m2 g−1, has a CO2 storage capacity of 8.99 mmol g−1 at 273 K/1.22 bar, which is among the highest reported for any material and increase of 320 % compared to the native PAF-1. Xiang et al. [164] and Ma et al. [165] also synthesized the lith-ium-loaded porous aromatic frameworks Li@COP-1 and PAF-18-OLi, respectively. Similarly, the enhancement effects of lithium modification on CO2 uptake and selec-tivity have also been observed, and the corresponding data are shown in Table 5.3. Besides the Li+, the present of other alkali metal ions (such as Na+, K+, and Cs+) in
Fig. 5.17 Idealized structure of PPN-6 and its derivatives

168 Y. Luo and B. Tan
Table 5.3 Summary of CO2 uptakes in CMPs
Network SBETa
(m2 g−1)CO2 uptakeb (mmol g−1)
CO2/N2 selectivity Qst (kJ mol−1) References
IAST Henry’s law
PAF-1 5,600 2.05c 15.6 [159]PAF-3 2,932 3.48c 19.2 [159]PAF-4 2,246 2.41c 16.2 [159]PPN-4 6,461 48.2d [160]PPN-6 (PAF-1) 4,023 1.16e 17 [162]PPN-6–SO3H 1,254 3.6e 150 30.4 [162]PPN-6–SO3Li 1,186 3.7e 414 35.7 [162]PPN-6–
CH2DETA555 4.3e 329 56 [168]
COP-1 827 4.41f 30.71 [164]Li@COP-1 573 5f 33.03 [164]PAF-18–OH 1,121 2.5 34 28 [165]PAF-18–OLi 981 3.27 129 29.5 [165]A (PAF-1) 4,077 2.65 8.1k 8.7k 23.7 [71]C (click
network)1,237 3.86 19.8k 14.2k 33.7 [71]
D (tetrahedral- CMP)
1,213 2.42 14.9k 12.2k 26.1 [71]
F (CMP- NH2CH3)
653 1.8 18k 12.2k 26.7 [71]
G (CMP carbazole)
1,056 2.15 25.2k 15.1k 26.6 [71]
CMP-1 837 2.05 26.8 [39]CMP-1–(OH)2 1,043 1.8 27.6 [39]CMP-1–(CH3)2 899 1.64 26.9 [39]CMP-1–NH2 710 1.64 29.5 [39]CMP-1–
COOH522 1.6 32.6 [39]
Tet1 3,160 2.55 [157]Tet2 1,102 1.96 [157]Tet3 3,180 2.66 [157]Tet4 1,917 3.03 [157]Tet5 1,470 1.26g [157]PON-1 1,422 2.48g [177]COP-1 168 1.36g [174]COP-2 158 0.93g [174]ACMP-C 629 1.56h [7]ACMP-C6 380 0.78h [7]ACMP-N 46 1.15h [7]LMOP-1 411 1.11i [170]LMOP-2 391 1.52i [170]LMOP-3 791 2.34i [170]TNCMP-2 995 2.62 [171]
(continued)

1695 Microporous Organic Polymers for Carbon Dioxide Capture
the porous frameworks also benefit the CO2 uptake of the materials. In 2012, Kiskan et al. [166] synthesized the phenolphthalein-based network N1. Treatment of N1 with alkali bases (LiOH, NaOH, KOH, and CsOH) can easily get alkali metal ion doped networks Li–N1, Na–N1, K–N1, and Cs–N1. It is found that the Qst followed the order Li–N1 (23 kJ mol−1) < N1 (26.8 kJ mol−1) < Cs–N1 (30 kJ mol−1). This result is in agreement with reports on cation-modified zeolites, where higher interac-tion strength was also found for Cs+-modified materials compared to Li+ and partly related to a coupling to the framework. In addition, alkylamine groups are the much stronger CO2-philic moieties and incorporate such functional group into the porous frameworks would be more effective in increasing the heat of CO2 adsorption [167]. Take this condition in mind, Lu et al. [168] prepared the polyamine-tethered PPNs by post-modification (Fig. 5.17B). As expected, the amine-modified PPN-6 exhib-its higher Qst (40–63 kJ mol−1). Although PPN-6–CH2DETA has the lowest surface area (555 m2 g−1), it exhibits an exceptionally high binding affinity and the largest selectivity for CO2 of any porous material reported at that time. The cyclability of PPN-6–CH2DETA was also measured. After 20 cycles, there was no apparent loss in capacity, thus indicating complete desorption during each regeneration cycle.
Although the enhancement gravimetric CO2 uptake has been made by post-syn-thetically modified PAF-1, the extent of modification greatly depends on the ability of the reagents to penetrate its whole morphological structure. As a result, inhomogeneity may very well exist in a PAF particle that the outer “shell” would have more func-tional groups than the inner “core” [169]. With these consideration in mind, Garibay et al. [169] described a de novo approach to synthesize the functionalized PAF-1 by using the corresponding functionalized monomers as the building blocks. It is found that the introduction of aminomethyl groups into PAF-1 resulted in outstanding CO2 adsorption: The loading capacity of PAF-1–CH2NH2 was almost double that of PAF-1 (98 vs. 55 cm3 g−1) at 273 K/1 bar. The Qst increases from the original 15.6 kJ mol−1
Network SBETa
(m2 g−1)CO2 uptakeb (mmol g−1)
CO2/N2 selectivity Qst (kJ mol−1) References
IAST Henry’s law
NCMP-2 900 2.10 [171]Ni-Por-1 1711 3.13j 14.4 [172]PPBI-1 385 1.8 [175]PPBI-2 158 1.39 [175]
a BET surface areab conditions: 273 K/1.0 barc 273 K/1 atmd 295 K/50 bare 295 K/1 barf 298 K/18 barg 298 K/1 barh 273 K/1.06 bari 273 K/722 mmHgj 298 K/1.08 bark 298 K
Table 5.3 (continued)

170 Y. Luo and B. Tan
value for PAF-1 to 57.6 kJ mol−1 for PAF-1–CH2NH2. The Qst of the PAF-1–CH2NH2 also is slight higher than that obtained for the PPN-6–CH2DETA (~55 kJ mol−1) [168]. In fact, the de novo approach used for synthesized functionalized CMP materials have been reported by Dawson et al. [39]. Several function groups, such as carboxylic acids (–COOH), amines (–NH2), hydroxyl groups (–OH), and methyl groups (–CH3), have been incorporated in the CMP-1 by reaction of 1,3,5-triethynylb-enzene with the corresponding functionalized monomers, respectively (Fig. 5.18). The experimental isosteric heats showed the following order in terms of appended func-tional groups: –COOH > –(OH)2 > –NH2 > –H > –(CH3)2 (Fig. 5.18). This result suggested that polar groups were effective in increasing CO2 capture, while bulky non-polar groups had a negative impact. In addition, the carboxylic acid-functionalized net-work, rather than its amine analog, shows the highest isosteric heat of sorption for CO2 (32.6–26.1 kJ mol−1).
Fig. 5.18 Functionalities incorporated into a series of CMPs and their effects on the heat of adsorption for CO2. Color-coding is as follow: unsubstituted networks (black); –(CH3)2 (green); –(OH)2 (orange); –NH2 (blue); and –COOH (red) (Reprinted with permission from Ref. [39]. Copyright 2011, Royal Society of Chemistry)

1715 Microporous Organic Polymers for Carbon Dioxide Capture
Besides the post-synthetic modification and the de novo approach to introduce the CO2-philic groups into the CMPs, the heteroatoms in the porous frameworks also can increase the CO2 adsorption property of the materials. Three building units with/without nitrogen atom, 1,3,5-tri(4-iodophenyl)benzene, tri(4-iodophe-nyl)amine, and tetrakis(4-bromophenyl)methane, have been selected to construct CMPs by Choi et al. [7] and Sun et al. [170] via the Sonogashira and Heck cou-pling reactions, respectively. They both discovered that the CMPs constructed from tri(4-iodophenyl)amine exhibit higher CO2 uptakes (ACMP-N, 51 mg g−1; LMOP-2, 67 mg g−1) than that of the networks constructed from 1,3,5-tri(4-iodophenyl)benzene (ACMP-C6, 34.7 mg g−1; LMOP-1, 49 mg g−1), despite the fact that the surface area of the former are much smaller than that of the latter (Table 5.3). The characteristics for adsorption of CO2 on ACMP-N or LMOP-2 are considered to be attributed to the lone pair electrons of centered nitrogen atoms, which can provide interaction sites through dipole–dipole interactions, thus enhancing the CO2 adsorption properties. Ren et al. [171] compared the CO2 adsorption properties of the 1,3,5-trizaine-based CMPs (TCMPs) with the analo-gous 1,3,5-connected benzene CMP systems. It was found that the TCMP net-works adsorbed more CO2 than CMP analogs with comparable BET surface areas (Table 5.3). Since then, various nitrogen-containing monomers such as porphyrin [172], piperazine [173], 4,4′-bipiperidine [174], and halo-benzimidazole [175] are used as the building blocks for constructing the electron-rich porous organic networks. The CO2 uptake properties of the resulting materials also are listed in Table 5.3. In particular, Chen et al. [176] prepared the microporous polycarbazole (CPOP-1) via carbazole-based oxidative coupling polymerization (Fig. 5.19). The BET surface area for obtained polymers is up to 2,220 m2 g−1. Gas adsorption isotherms showed that its CO2 uptake capacity can reach 21.2 wt% at 273 K/1 bar. Due to the cost-effective preparative strategy and the good performance for the gas storage and separation, this material exhibits potential use for clean energy appli-cations and environmental field.
5.6 Conclusions and Outlook
MOPs are one of the most exciting areas in current materials sciences, and the development of MOPs for CO2 capture and separation (CCS) is particularly important for energy and environmental applications. In general, an ideal CCS material will have good stability, a high surface area, and a CO2-philic surface in order to increase the amount of CO2 adsorbed. It seems likely that MOPs are well on the way to fulfilling most of these criteria. Synthetic micoporous polymers pos-sess some of the highest reported surface areas (SBET = 6,461 m2 g−1) to date and can exhibit good physicochemical stability. Moreover, the ability to incorporate various functional groups into MOPs can be advantageous for tuning the inter-action of their surface with different types of guest molecules. Thus, the porous materials with CO2-philic surface can be obtained easily by choosing proper
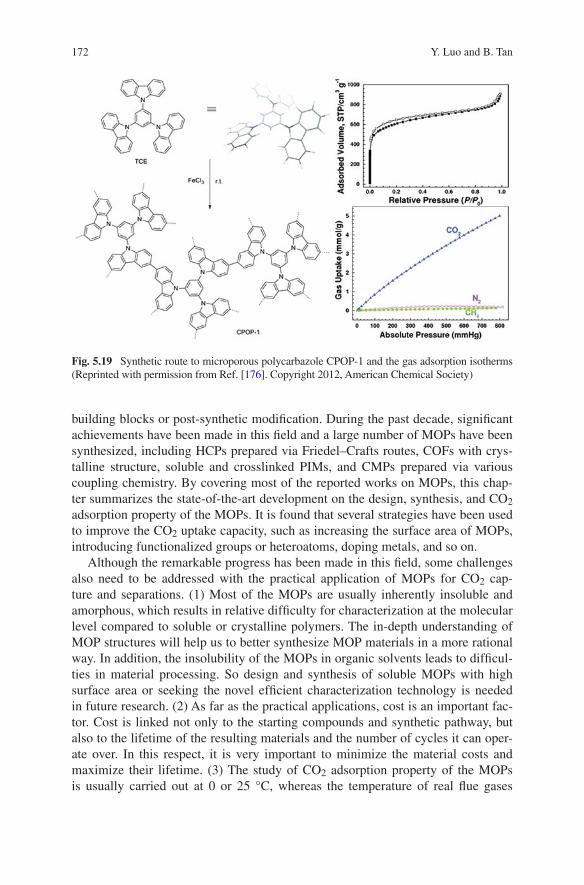
172 Y. Luo and B. Tan
building blocks or post-synthetic modification. During the past decade, significant achievements have been made in this field and a large number of MOPs have been synthesized, including HCPs prepared via Friedel–Crafts routes, COFs with crys-talline structure, soluble and crosslinked PIMs, and CMPs prepared via various coupling chemistry. By covering most of the reported works on MOPs, this chap-ter summarizes the state-of-the-art development on the design, synthesis, and CO2 adsorption property of the MOPs. It is found that several strategies have been used to improve the CO2 uptake capacity, such as increasing the surface area of MOPs, introducing functionalized groups or heteroatoms, doping metals, and so on.
Although the remarkable progress has been made in this field, some challenges also need to be addressed with the practical application of MOPs for CO2 cap-ture and separations. (1) Most of the MOPs are usually inherently insoluble and amorphous, which results in relative difficulty for characterization at the molecular level compared to soluble or crystalline polymers. The in-depth understanding of MOP structures will help us to better synthesize MOP materials in a more rational way. In addition, the insolubility of the MOPs in organic solvents leads to difficul-ties in material processing. So design and synthesis of soluble MOPs with high surface area or seeking the novel efficient characterization technology is needed in future research. (2) As far as the practical applications, cost is an important fac-tor. Cost is linked not only to the starting compounds and synthetic pathway, but also to the lifetime of the resulting materials and the number of cycles it can oper-ate over. In this respect, it is very important to minimize the material costs and maximize their lifetime. (3) The study of CO2 adsorption property of the MOPs is usually carried out at 0 or 25 °C, whereas the temperature of real flue gases
Fig. 5.19 Synthetic route to microporous polycarbazole CPOP-1 and the gas adsorption isotherms (Reprinted with permission from Ref. [176]. Copyright 2012, American Chemical Society)

1735 Microporous Organic Polymers for Carbon Dioxide Capture
normally is about 90 °C. The current choice is to first cool down the flue gas, but this will be energy consuming. Moreover, typical coal-fired flue gas usually con-tains a relatively low proportion of CO2 (1–14 vol%) in N2 (>70 vol%). So CO2 capture under real operating conditions still needs much more effort. (4) Another major challenge in this field is to develop the stimuli responsive porous materials and achieve the control release of the CO2 adsorbed in the MOPs.
References
1. Johnson J (2004) Putting a lid on carbon dioxide–carbon sequestration, clean-coal research mark government response to climate-change threat. Chem Eng News 82:36–42
2. Wang QA, Luo JZ, Zhong ZY et al (2011) CO2 capture by solid adsorbents and their appli-cations: current status and new trends. Energy Environ Sci 4:42–55
3. D’Alessandro DM, Smit B, Long JR (2010) Carbon dioxide capture: prospects for new materials. Angew Chem Int Edit 49:6058–6082
4. Czaja AU, Trukhan N, Muller U (2009) Industrial applications of metal-organic frame-works. Chem Soc Rev 38:1284–1293
5. McKeown NB, Budd PM (2006) Polymers of intrinsic microporosity (PIMs): organic mate-rials for membrane separations, heterogeneous catalysis and hydrogen storage. Chem Soc Rev 35:675–683
6. Cooper AI (2009) Conjugated microporous polymers. Adv Mater 21:1291–1295 7. Choi JH, Choi KM, Jeon HJ et al (2010) Acetylene gas mediated conjugated microporous polymers
(ACMPs): first use of acetylene gas as a building unit. Macromolecules 43:5508–5511 8. El-Kaderi HM, Hunt JR, Mendoza-Cortes JL et al (2007) Designed synthesis of 3D covalent
organic frameworks. Science 316:268–272 9. Furukawa H, Yaghi OM (2009) Storage of hydrogen, methane, and carbon dioxide in highly porous
covalent organic frameworks for clean energy applications. J Am Chem Soc 131:8875–8883 10. Lee JY, Wood CD, Bradshaw D et al (2006) Hydrogen adsorption in microporous hyper-
crosslinked polymers. Chem Commun 2670–2672 11. Martin CF, Stockel E, Clowes R et al (2011) Hypercrosslinked organic polymer networks as
potential adsorbents for pre-combustion CO2 capture. J Mater Chem 21:5475–5483 12. Tsyurupa MP, Davankov VA (2006) Porous structure of hypercrosslinked polystyrene: state-
of-the-art mini-review. React Funct Polym 66:768–779 13. Kuhn P, Antonietti M, Thomas A (2008) Porous, covalent triazine-based frameworks pre-
pared by ionothermal synthesis. Angew Chem Int Edit 47:3450–3453 14. Ben T, Ren H, Ma SQ et al (2009) Targeted synthesis of a porous aromatic framework with
high stability and exceptionally high surface area. Angew Chem Int Edit 48:9457–9460 15. Xu S, Luo Y, Tan B (2013) Recent development of hypercrosslinked microporous organic
polymers. Macromol Rapid Commun 34:471–484 16. Davankov VA, Tsyurupa MP (1990) Structure and properties of hypercrosslinked polysty-
rene—the first representative of a new class of polymer networks. React Polym 13:27–42 17. Ahn JH, Jang JE, Oh CG et al (2006) Rapid generation and control of microporosity,
bimodal pore size distribution, and surface area in davankov-type hyper-cross-linked resins. Macromolecules 39:627–632
18. Germain J, Hradil J, Frechet JMJ et al (2006) High surface area nanoporous polymers for reversible hydrogen storage. Chem Mater 18:4430–4435
19. Li B, Gong R, Luo Y et al (2011) Tailoring the pore size of hypercrosslinked polymers. Soft Matter 7:10910–10916
20. Kaliva M, Armatas GS, Vamvakaki M (2012) Microporous polystyrene particles for selec-tive carbon dioxide capture. Langmuir 28:2690–2695

174 Y. Luo and B. Tan
21. Banerjee R, Furukawa H, Britt D et al (2009) Control of pore size and functionality in iso-reticular zeolitic imidazolate frameworks and their carbon dioxide selective capture proper-ties. J Am Chem Soc 131:3875–3877
22. Babarao R, Hu Z, Jiang J et al (2007) Storage and separation of CO2 and CH4 in sili-calite, C168 schwarzite, and IRMOF-1: a comparative study from monte carlo simulation. Langmuir 23:659–666
23. Yang Q, Zhong C (2006) Molecular simulation of carbon dioxide/methane/hydrogen mix-ture adsorption in metal-organic frameworks. J Phys Chem B 110:17776–17783
24. Yang Q, Zhong C (2006) Electrostatic-field-induced enhancement of gas mixture separation in metal-organic frameworks: a computational study. ChemPhysChem 7:1417–1421
25. Belmabkhout Y, Sayari A (2009) Adsorption of CO2 from dry gases on MCM-41 silica at ambient temperature and high pressure. 2: Adsorption of CO2/N2, CO2/CH4 and CO2/H2 binary mixtures. Chem Eng Sci 64:3729–3735
26. Cavenati S, Grande CA, Rodrigues AE (2004) Adsorption equilibrium of methane, carbon dioxide, and nitrogen on zeolite 13X at high pressures. J Chem Eng Data 49:1095–1101
27. Wood CD, Tan B, Trewin A et al (2007) Hydrogen storage in microporous hypercrosslinked organic polymer networks. Chem Mater 19:2034–2048
28. Llewellyn PL, Bourrelly S, Serre C et al (2008) High uptakes of CO2 and CH4 in mesoporous metals-organic frameworks MIL-100 and MIL-101. Langmuir 24:7245–7250
29. Chaikittisilp W, Kubo M, Moteki T et al (2011) Porous siloxane-organic hybrid with ultra-high surface area through simultaneous polymerization-destruction of functionalized cubic siloxane cages. J Am Chem Soc 133:13832–13835
30. Yuan S, White D, Mason A et al (2011) Porous organic polymers containing carborane for hydrogen storage. Int J Energy Res. doi:10.1002/er.1886
31. Schwab MG, Lennert A, Pahnke J et al (2011) Nanoporous copolymer networks through multiple Friedel-Crafts-alkylation-studies on hydrogen and methane storage. J Mater Chem 21:2131–2135
32. Luo Y, Zhang S, Ma Y et al (2013) Microporous organic polymers synthesized by self-con-densation of aromatic hydroxymethyl monomers. Polym Chem 4:1126–1131
33. Li B, Gong R, Wang W et al (2011) A new strategy to microporous polymers: knitting rigid aromatic building blocks by external cross-linker. Macromolecules 44:2410–2414
34. Dawson R, Stockel E, Holst JR et al (2011) Microporous organic polymers for carbon diox-ide capture. Energy Environ Sci 4:4239–4245
35. Dawson R, Stevens L, Drage TC et al (2012) Impact of water co-adsorption for carbon diox-ide capture in microporous polymer sorbents. J Am Chem Soc 134:10741–10744
36. Katsoulidis AP, Kanatzidis MG (2011) Phloroglucinol based microporous polymeric organic frameworks with −OH functional groups and high CO2 capture capacity. Chem Mater 23:1818–1824
37. Couck S, Denayer JFM, Baron GV et al (2009) An amine-functionalized MIL-53 metal-organic framework with large separation power for CO2 and CH4. J Am Chem Soc 131:6326–6327
38. Chen S, Zhang J, Wu T et al (2009) Multiroute synthesis of porous anionic frameworks and size-tunable extraframework organic cation-controlled gas sorption properties. J Am Chem Soc 131:16027–16029
39. Dawson R, Adams DJ, Cooper AI (2011) Chemical tuning of CO2 sorption in robust nano-porous organic polymers. Chem Sci 2:1173–1177
40. Dawson R, Ratvijitvech T, Corker M et al (2012) Microporous copolymers for increased gas selectivity. Polym Chem 3:2034–2038
41. Luo Y, Li B, Wang W et al (2012) Hypercrosslinked aromatic heterocyclic microporous pol-ymers: a new class of highly selective CO2 capturing materials. Adv Mater 24:5703–5707
42. Lim H, Cha MC, Chang JY (2012) Preparation of microporous polymers based on 1,3,5-tri-azine units showing high CO2 adsorption capacity. Macromol Chem Phys 213:1385–1390
43. Lim H, Cha MC, Chang JY (2012) Synthesis of microporous polymers by Friedel-Crafts reaction of 1-bromoadamantane with aromatic compounds and their surface modification. Polym Chem 3:868–870

1755 Microporous Organic Polymers for Carbon Dioxide Capture
44. Ding S-Y, Wang W (2013) Covalent organic frameworks (COFs): from design to applica-tions. Chem Soc Rev 42:548–568
45. Côté AP, Benin AI, Ockwig NW et al (2005) Porous, crystalline, covalent organic frame-works. Science 310:1166–1170
46. Feng X, Ding X, Jiang D (2012) Covalent organic frameworks. Chem Soc Rev 41:6010–6022
47. Millward AR, Yaghi OM (2005) Metal-organic frameworks with exceptionally high capacity for storage of carbon dioxide at room temperature. J Am Chem Soc 127:17998–17999
48. Yang Q, Zhong C (2009) Molecular simulation study of the stepped behaviors of gas adsorption in two-dimensional covalent organic frameworks. Langmuir 25:2302–2308
49. Babarao R, Jiang J (2008) Exceptionally high CO2 storage in covalent-organic frameworks: atomistic simulation study. Energy Environ Sci 1:139–143
50. Choi YJ, Choi JH, Choi KM et al (2011) Covalent organic frameworks for extremely high reversible CO2 uptake capacity: a theoretical approach. J Mater Chem 21:1073–1078
51. Kahveci Z, Islamoglu T, Shar GA et al (2013) Targeted synthesis of a mesoporous trip-tycene-derived covalent organic framework. CrystEngComm 15:1524–1527
52. Jackson KT, Rabbani MG, Reich TE et al (2011) Synthesis of highly porous borazine-linked polymers and their application to H2, CO2, and CH4 storage. Polym Chem 2:2775–2777
53. Jackson KT, Reich TE, El-Kaderi HM (2012) Targeted synthesis of a porous borazine-linked covalent organic framework. Chem Commun 48:8823–8825
54. Reich TE, Behera S, Jackson KT et al (2012) Highly selective CO2/CH4 gas uptake by a halogen-decorated borazine-linked polymer. J Mater Chem 22:13524–13528
55. Reich TE, Jackson KT, Li S et al (2011) Synthesis and characterization of highly porous borazine-linked polymers and their performance in hydrogen storage application. J Mater Chem 21:10629–10632
56. Nagai A, Guo Z, Feng X et al (2011) Pore surface engineering in covalent organic frame-works. Nat Commun 2:536
57. Lan J, Cao D, Wang W et al (2010) Doping of alkali, alkaline-earth, and transition metals in covalent-organic frameworks for enhancing CO2 capture by first-principles calculations and molecular simulations. ACS Nano 4:4225–4237
58. Shultz AM, Farha OK, Hupp JT et al (2011) Synthesis of catalytically active porous organic polymers from metalloporphyrin building blocks. Chem Sci 2:686–689
59. Ding X, Chen L, Honsho Y et al (2011) An n-channel two-dimensional covalent organic framework. J Am Chem Soc 133:14510–14513
60. Chen X, Addicoat MA, Irle S et al (2012) Control crystallinity and porosity of covalent organic frameworks through managing interlayer interactions based on self-complementary π-electronic force. J Am Chem Soc 135:546–549
61. Modak A, Nandi M, Mondal J et al (2012) Porphyrin based porous organic polymers: novel syn-thetic strategy and exceptionally high CO2 adsorption capacity. Chem Commun 48:248–250
62. Wang C, Zheng M, Lin W (2011) Asymmetric catalysis with chiral porous metal-organic frameworks: critical issues. J Phys Chem Lett 2:1701–1709
63. Mastalerz M, Hauswald H-JS, Stoll R (2012) Metal-assisted salphen organic frameworks (MaSOFs) with high surface areas and narrow pore-size distribution. Chem Commun 48:130–132
64. Jin Y, Zhu Y, Zhang W (2013) Development of organic porous materials through schiff-base chemistry. CrystEngComm 15:1484–1499
65. Uribe-Romo FJ, Doonan CJ, Furukawa H et al (2011) Crystalline covalent organic frame-works with hydrazone linkages. J Am Chem Soc 133:11478–11481
66. Uribe-Romo FJ, Hunt JR, Furukawa H et al (2009) A crystalline imine-linked 3-D porous covalent organic framework. J Am Chem Soc 131:4570–4571
67. Wan S, Gándara F, Asano A et al (2011) Covalent organic frameworks with high charge car-rier mobility. Chem Mater 23:4094–4097
68. Ding S-Y, Gao J, Wang Q et al (2011) Construction of covalent organic framework for catalysis: Pd/COF-LZU1 in suzuki–miyaura coupling reaction. J Am Chem Soc 133:19816–19822

176 Y. Luo and B. Tan
69. Pandey P, Katsoulidis AP, Eryazici I et al (2010) Imine-linked microporous polymer organic frameworks. Chem Mater 22:4974–4979
70. Laybourn A, Dawson R, Clowes R et al (2012) Branching out with aminals: microporous organic polymers from difunctional monomers. Polym Chem 3:533–537
71. Dawson R, Stockel E, Holst JR et al (2011) Microporous organic polymers for carbon diox-ide capture. Energy Environ Sci 4:4239–4245
72. Jiang S, Bacsa J, Wu X et al (2011) Selective gas sorption in a [2+3] ‘propeller’ cage crys-tal. Chem Commun 47:8919–8921
73. Cheon YE, Suh MP (2009) Selective gas adsorption in a microporous metal-organic frame-work constructed of CoII
4 clusters. Chem Commun 0:2296–2298 74. Wang B, Cote AP, Furukawa H et al (2008) Colossal cages in zeolitic imidazolate frame-
works as selective carbon dioxide reservoirs. Nature 453:207–211 75. Spitler EL, Giovino MR, White SL et al (2011) A mechanistic study of lewis acid-catalyzed
covalent organic framework formation. Chem Sci 2:1588–1593 76. Du Y, Mao K, Kamakoti P et al (2012) Experimental and computational studies of pyridine-
assisted post-synthesis modified air stable covalent-organic frameworks. Chem Commun 48:4606–4608
77. Kandambeth S, Mallick A, Lukose B et al (2012) Construction of crystalline 2D covalent organic frameworks with remarkable chemical (acid/base) stability via a combined revers-ible and irreversible route. J Am Chem Soc 134:19524–19527
78. Xu C, Hedin N (2013) Synthesis of microporous organic polymers with high CO2-over-N2 selectivity and CO2 adsorption. J Mater Chem A 1:3406–3414
79. Rabbani MG, El-Kaderi HM (2011) Template-free synthesis of a highly porous benzimida-zole-linked polymer for CO2 capture and H2 storage. Chem Mater 23:1650–1653
80. An J, Geib SJ, Rosi NL (2010) High and selective CO2 uptake in a cobalt adeninate metal-organic framework exhibiting pyrimidine- and amino-decorated pores. J Am Chem Soc 132:38–39
81. Demessence A, D’Alessandro DM, Foo ML et al (2009) Strong CO2 binding in a water-stable, triazolate-bridged metal-organic framework functionalized with ethylenediamine. J Am Chem Soc 131:8784–8786
82. Panda T, Pachfule P, Chen Y et al (2011) Amino functionalized zeolitic tetrazolate framework (ZTF) with high capacity for storage of carbon dioxide. Chem Commun 47:2011–2013
83. Ghanem BS, Hashem M, Harris KDM et al (2010) Triptycene-based polymers of intrinsic microporosity: organic materials that can be tailored for gas adsorption. Macromolecules 43:5287–5294
84. Mastalerz M, Schneider MW, Oppel IM et al (2011) A salicylbisimine cage compound with high surface area and selective CO2/CH4 adsorption. Angew Chem Int Edit 50:1046–1051
85. Vagin SI, Ott AK, Hoffmann SD et al (2009) Synthesis and properties of (triptycenedicarbo-xylatio) zinc coordination networks. Chem Eur J 15:5845–5853
86. Chong JH, Ardakani SJ, Smith KJ et al (2009) Triptycene-based metal salphens—exploiting intrinsic molecular porosity for gas storage. Chem Eur J 15:11824–11828
87. Rabbani MG, Reich TE, Kassab RM et al (2012) High CO2 uptake and selectivity by trip-tycene-derived benzimidazole-linked polymers. Chem Commun 48:1141–1143
88. Rabbani MG, El-Kaderi HM (2012) Synthesis and characterization of porous benzimida-zole-linked polymers and their performance in small gas storage and selective uptake. Chem Mater 24:1511–1517
89. Zhao Y-C, Cheng Q-Y, Zhou D et al (2012) Preparation and characterization of triptycene-based microporous poly(benzimidazole) networks. J Mater Chem 22:11509–11514
90. Yu H, Tian M, Shen C et al (2013) Facile preparation of porous polybenzimidazole networks and adsorption behavior of CO2 gas, organic and water vapors. Polym Chem 4:961–968
91. Mohanty P, Kull LD, Landskron K (2011) Porous covalent electron-rich organonitridic frame-works as highly selective sorbents for methane and carbon dioxide. Nat Commun 2:401–406
92. Kuhn P, Forget A, Hartmann J et al (2009) Template-free tuning of nanopores in carbona-ceous polymers through ionothermal synthesis. Adv Mater 21:897–901

1775 Microporous Organic Polymers for Carbon Dioxide Capture
93. Kuhn P, Thomas A, Antonietti M (2009) Toward tailorable porous organic polymer net-works: a high-temperature dynamic polymerization scheme based on aromatic nitriles. Macromolecules 42:319–326
94. Bojdys MJ, Jeromenok J, Thomas A et al (2010) Rational extension of the family of layered, covalent, triazine-based frameworks with regular porosity. Adv Mater 22:2202–2205
95. Ren H, Ben T, Wang ES et al (2010) Targeted synthesis of a 3D porous aromatic framework for selective sorption of benzene. Chem Commun 46:291–293
96. Hug S, Tauchert ME, Li S et al (2012) A functional triazine framework based on n-heterocy-clic building blocks. J Mater Chem 22:13956–13964
97. Wang W, Ren H, Sun F et al (2012) Synthesis of porous aromatic framework with tuning porosity via ionothermal reaction. Dalton Trans 41:3933–3936
98. Ren S, Bojdys MJ, Dawson R et al (2012) Porous, fluorescent, covalent triazine-based frame-works via room-temperature and microwave-assisted synthesis. Adv Mater 24:2357–2361
99. Zhu X, Tian C, Mahurin SM et al (2012) A superacid-catalyzed synthesis of porous mem-branes based on triazine frameworks for CO2 separation. J Am Chem Soc 134:10478–10484
100. McKeown NB (2012) Polymers of intrinsic microporosity. ISRN Mater Sci. doi:10.5402/2012/513986
101. McKeown NB, Budd PM (2010) Exploitation of intrinsic microporosity in polymer-based materials. Macromolecules 43:5163–5176
102. Wu D, Xu F, Sun B et al (2012) Design and preparation of porous polymers. Chem Rev 2012:3959–4015
103. McKeown NB, Makhseed S, Budd PM (2002) Phthalocyanine-based nanoporous network polymers. Chem Commun 2780–2781
104. McKeown NB, Hanif S, Msayib K et al (2002) Porphyrin-based nanoporous network poly-mers. Chem Commun 2782–2783
105. Budd PM, Ghanem BS, Makhseed S et al (2004) Polymers of intrinsic microporosity (PIMs): robust, solution-processable, organic nanoporous materials. Chem Commun 230–231
106. Thomas S, Pinnau I, Du N et al (2009) Hydrocarbon/hydrogen mixed-gas permeation prop-erties of PIM-1, an amorphous microporous spirobisindane polymer. J Membr Sci 338:1–4
107. Ahn J, Chung W-J, Pinnau I et al (2010) Gas transport behavior of mixed-matrix mem-branes composed of silica nanoparticles in a polymer of intrinsic microporosity (PIM-1). J Membr Sci 346:280–287
108. Budd PM, McKeown NB, Ghanem BS et al (2008) Gas permeation parameters and other physicochemical properties of a polymer of intrinsic microporosity: polybenzodioxane PIM-1. J Membr Sci 325:851–860
109. Budd PM, Msayib KJ, Tattershall CE et al (2005) Gas separation membranes from polymers of intrinsic microporosity. J Membr Sci 251:263–269
110. Du N, Robertson GP, Pinnau I et al (2009) Polymers of intrinsic microporosity derived from novel disulfone-based monomers. Macromolecules 42:6023–6030
111. Du N, Robertson GP, Song J et al (2009) High-performance carboxylated polymers of intrinsic microporosity (PIMs) with tunable gas transport properties†. Macromolecules 42:6038–6043
112. Vogiatzis KD, Mavrandonakis A, Klopper W et al (2009) Ab initio study of the interactions between CO2 and N-containing organic heterocycles. ChemPhysChem 10:374–383
113. Du N, Park HB, Robertson GP et al (2011) Polymer nanosieve membranes for CO2-capture applications. Nat Mater 10:372–375
114. Zulfiqar S, Karadas F, Park J et al (2011) Amidoximes: promising candidates for CO2 cap-ture. Energy Environ Sci 4:4528–4531
115. Patel HA, Yavuz CT (2012) Noninvasive functionalization of polymers of intrinsic micropo-rosity for enhanced CO2 capture. Chem Commun 48:9989–9991
116. Li FY, Xiao Y, Chung T-S et al (2012) High-performance thermally self-cross-linked poly-mer of intrinsic microporosity (PIM-1) membranes for energy development. Macromolecules 45:1427–1437
117. Weber J, Antonietti M, Thomas A (2008) Microporous networks of high-performance poly-mers: elastic deformations and gas sorption properties. Macromolecules 41:2880–2885

178 Y. Luo and B. Tan
118. Weber J, Su Q, Antonietti M et al (2007) Exploring polymers of intrinsic microporosity—microporous, soluble polyamide and polyimide. Macromol Rapid Commun 28:1871–1876
119. Ritter N, Antonietti M, Thomas A et al (2009) Binaphthalene-based, soluble polyimides: the limits of intrinsic microporosity. Macromolecules 42:8017–8020
120. Ritter N, Senkovska I, Kaskel S et al (2011) Towards chiral microporous soluble poly-mers—binaphthalene-based polyimides. Macromol Rapid Commun 32:438–443
121. Ritter N, Senkovska I, Kaskel S et al (2011) Intrinsically microporous poly(imide)s: struc-ture−porosity relationship studied by gas sorption and X-ray scattering. Macromolecules 44:2025–2033
122. Wang Z, Zhang B, Yu H et al (2010) Microporous polyimide networks with large surface areas and their hydrogen storage properties. Chem Commun 46:7730–7732
123. Farha OK, Spokoyny AM, Hauser BG et al (2009) Synthesis, properties, and gas separation studies of a robust diimide-based microporous organic polymer. Chem Mater 21:3033–3035
124. Wang Z, Zhang B, Yu H et al (2011) Synthetic control of network topology and pore struc-ture in microporous polyimides based on triangular triphenylbenzene and triphenylamine units. Soft Matter 7:5723–5730
125. Rao KV, Haldar R, Kulkarni C et al (2012) Perylene based porous polyimides: tunable, high surface area with tetrahedral and pyramidal monomers. Chem Mater 24:969–971
126. Farha OK, Bae Y-S, Hauser BG et al (2010) Chemical reduction of a diimide based porous polymer for selective uptake of carbon dioxide versus methane. Chem Commun 46:1056–1058
127. Luo Y, Li B, Liang L et al (2011) Synthesis of cost-effective porous polyimides and their gas storage properties. Chem Commun 47:7704–7706
128. Liebl MR, Senker J (2013) Microporous functionalized triazine-based polyimides with high CO2 capture capacity. Chem Mater 25:970–980
129. Trewin A (2010) Predicting crystalline polyamic acids as precursors to porous polyimides. CrystEngComm 12:2315–2317
130. Zeng S, Guo L, Cui F et al (2010) Template-free synthesis of crystalline polyimide spheres with radiate branches. Mater Lett 64:625–627
131. Dawson R, Cooper AI, Adams DJ (2012) Nanoporous organic polymer networks. Prog Polym Sci 37:530–563
132. Xiang ZH, Cao DP (2012) Synthesis of luminescent covalent-organic polymers for detect-ing nitroaromatic explosives and small organic molecules. Macromol Rapid Commun 33:1184–1190
133. Chaikittisilp W, Sugawara A, Shimojima A et al (2010) Microporous hybrid polymer with a certain crystallinity built from functionalized cubic siloxane cages as a singular building unit. Chem Mater 22:4841–4843
134. Kassab R, Jackson K, El-Kadri O et al (2011) Nickel-catalyzed synthesis of nanoporous organic frameworks and their potential use in gas storage applications. Res Chem Intermed 37:747–757
135. Zhang Q, Zhang S, Li S (2012) Novel functional organic network containing quaternary phosphonium and tertiary phosphorus. Macromolecules 45:2981–2988
136. Schmidt J, Werner M, Thomas A (2009) Conjugated microporous polymer networks via yamamoto polymerization. Macromolecules 42:4426–4429
137. Ren H, Ben T, Sun F et al (2011) Synthesis of a porous aromatic framework for adsorbing organic pollutants application. J Mater Chem 21:10348–10353
138. Yuan Y, Sun F, Ren H et al (2011) Targeted synthesis of a porous aromatic framework with a high adsorption capacity for organic molecules. J Mater Chem 21:13498–13502
139. Jing X, Sun F, Ren H et al (2013) Targeted synthesis of micro-mesoporous hybrid mate-rial derived from octaphenylsilsesquioxane building units. Microporous Mesoporous Mater 165:92–98
140. Brandt J, Schmidt J, Thomas A et al (2011) Tunable absorption and emission wavelength in conjugated microporous polymers by copolymerization. Polym Chem 2:1950–1952
141. Xu Y, Nagai A, Jiang D (2013) Core-shell conjugated microporous polymers: a new strategy for exploring color-tunable and -controllable light emissions. Chem Commun 49:1591–1593

1795 Microporous Organic Polymers for Carbon Dioxide Capture
142. Chen L, Honsho Y, Seki S et al (2010) Light-harvesting conjugated microporous polymers: rapid and highly efficient flow of light energy with a porous polyphenylene framework as antenna. J Am Chem Soc 132:6742–6748
143. Xiao D, Li Y, Liu L et al (2012) Two-photon fluorescent microporous bithiophene polymer via suzuki cross-coupling. Chem Commun 48:9519–9521
144. Chen Q, Luo M, Wang T et al (2011) Porous organic polymers based on propeller-like hexa-phenylbenzene building units. Macromolecules 44:5573–5577
145. Chen Q, Wang J-X, Yang F et al (2011) Tetraphenylethylene-based fluorescent porous organic polymers: preparation, gas sorption properties and photoluminescence properties. J Mater Chem 21:13554–13560
146. Dawson R, Laybourn A, Clowes R et al (2009) Functionalized conjugated microporous pol-ymers. Macromolecules 42:8809–8816
147. Dawson R, Laybourn A, Khimyak YZ et al (2010) High surface area conjugated micropo-rous polymers: the importance of reaction solvent choice. Macromolecules 43:8524–8530
148. Jiang JX, Laybourn A, Clowes R et al (2010) High surface area contorted conjugated microporous polymers based on spiro-bipropylenedioxythiophene. Macromolecules 43:7577–7582
149. Jiang JX, Trewin A, Su FB et al (2009) Microporous poly(tri(4-ethynylphenyl)amine) net-works: Synthesis, properties, and atomistic simulation. Macromolecules 42:2658–2666
150. Jiang J-X, Su F, Trewin A et al (2008) Synthetic control of the pore dimension and sur-face area in conjugated microporous polymer and copolymer networks. J Am Chem Soc 130:7710–7720
151. Morisaki Y, Gon M, Tsuji Y et al (2011) Stacked 1,3,5-tris[(2,5-dimethylphenyl)-ethynyl]benzenes: dimer and conjugated microporous polymer. Tetrahedron Lett 52:5504–5507
152. Stockel E, Wu XF, Trewin A et al (2009) High surface area amorphous microporous poly(aryleneethynylene) networks using tetrahedral carbon- and silicon-centred monomers. Chem Commun 212–214
153. Xia J, Yuan S, Wang Z et al (2010) Nanoporous polyporphyrin as adsorbent for hydrogen storage. Macromolecules 43:3325–3330
154. Yuan S, Dorney B, White D et al (2010) Microporous polyphenylenes with tunable pore size for hydrogen storage. Chem Commun 46:4547–4549
155. Yuan S, Kirklin S, Dorney B et al (2009) Nanoporous polymers containing stereocontorted cores for hydrogen storage. Macromolecules 42:1554–1559
156. Yuan SW, White D, Mason A et al (2012) Improving hydrogen adsorption enthalpy through coordinatively unsaturated cobalt in porous polymers. Macromol Rapid Commun 33:407–413
157. Holst JR, Stöckel E, Adams DJ et al (2010) High surface area networks from tetrahedral monomers: metal-catalyzed coupling, thermal polymerization, and “click” chemistry. Macromolecules 43:8531–8538
158. Lu WG, Yuan DQ, Zhao D et al (2010) Porous polymer networks: synthesis, porosity, and applications in gas storage/separation. Chem Mater 22:5964–5972
159. Ben T, Pei C, Zhang D et al (2011) Gas storage in porous aromatic frameworks (PAFs). Energy Environ Sci 4:3991–3999
160. Yuan D, Lu W, Zhao D et al (2011) Highly stable porous polymer networks with exception-ally high gas-uptake capacities. Adv Mater 23:3723–3725
161. Babarao R, Dai S, Jiang D (2011) Functionalizing porous aromatic frameworks with polar organic groups for high-capacity and selective CO2 separation: a molecular simulation study. Langmuir 27:3451–3460
162. Lu W, Yuan D, Sculley J et al (2011) Sulfonate-grafted porous polymer networks for prefer-ential CO2 adsorption at low pressure. J Am Chem Soc 133:18126–18129
163. Konstas K, Taylor JW, Thornton AW et al (2012) Lithiated porous aromatic frameworks with exceptional gas storage capacity. Angew Chem Int Edit 51:6639–6642
164. Xiang ZH, Cao DP, Wang WC et al (2012) Postsynthetic lithium modification of covalent-organic polymers for enhancing hydrogen and carbon dioxide storage. J Phys Chem C 116:5974–5980

180 Y. Luo and B. Tan
165. Ma H, Ren H, Zou X et al (2013) Novel lithium-loaded porous aromatic framework for effi-cient CO2 and H2 uptake. J Mater Chem A 1:752–758
166. Kiskan B, Antonietti M, Weber J (2012) Teaching new tricks to an old indicator: pH-switch-able, photoactive microporous polymer networks from phenolphthalein with tunable CO2 adsorption power. Macromolecules 45:1356–1361
167. McDonald TM, D’Alessandro DM, Krishna R et al (2011) Enhanced carbon dioxide cap-ture upon incorporation of N,N′-dimethylethylenediamine in the metal-organic framework CuBTTri. Chem Sci 2:2022–2028
168. Lu W, Sculley JP, Yuan D et al (2012) Polyamine-tethered porous polymer networks for car-bon dioxide capture from flue gas. Angew Chem Int Edit 51:7480–7484
169. Garibay SJ, Weston MH, Mondloch JE et al (2013) Accessing functionalized porous aro-matic frameworks (PAFs) through a de novo approach. CrystEngComm 15:1515–1519
170. Sun L, Liang Z, Yu J et al (2013) Luminescent microporous organic polymers containing the 1,3,5-tri(4-ethenylphenyl)benzene unit constructed by heck coupling reaction. Polym Chem 4:1932–1938
171. Ren S, Dawson R, Laybourn A et al (2012) Functional conjugated microporous polymers: from 1,3,5-benzene to 1,3,5-triazine. Polym Chem 3:928–934
172. Wang Z, Yuan S, Mason A et al (2012) Nanoporous porphyrin polymers for gas storage and separation. Macromolecules 45:7413–7419
173. Pachfule P, Dhavale VM, Kandambeth S et al (2013) Porous-organic-framework-templated nitrogen-rich porous carbon as a more proficient electrocatalyst than Pt/C for the electro-chemical reduction of oxygen. Chem Eur J 19:974–980
174. Patel HA, Karadas F, Canlier A et al (2012) High capacity carbon dioxide adsorption by inexpensive covalent organic polymers. J Mater Chem 22:8431–8437
175. Zhang XJ, Bian N, Mao LJ et al (2012) Porous polybenzimidazoles via template-free suzuki coupling polymerization: preparation, porosity, and heterogeneous catalytic activity in kno-evenagel condensation reactions. Macromol Chem Phys 213:1575–1581
176. Chen Q, Luo M, Hammershøj P et al (2012) Microporous polycarbazole with high specific surface area for gas storage and separation. J Am Chem Soc 134:6084–6087
177. Jeon HJ, Choi JH, Lee Y et al (2012) Highly selective CO2-capturing polymeric organic net-work structures. Adv Energy Mater 2:225–228

181
Abstract The reversible carbonation and calcination reactions of, respectively, CaO and CaCO3 have very promising CO2 capture characteristics with regard to CO2 capture costs, theoretical CO2 uptake per gram of sorbent and mate-rial availability. However, CaO derived from naturally occurring Ca-based materials, predominantly limestone, shows a rapid decrease in its CO2 capture capacity with number of carbonation/calcination cycles. The loss of the CO2 capture capacity of unsupported CaO has been attributed to dramatic changes in the material’s morphology due to sintering and pore blockage. However, since the molar volume of CaCO3 is more than twice as large as that of CaO, accessible pore volume in pores of diameter <100 nm is critical to yield high CO2 uptakes. In this chapter, we review the fundamentals of the carbonation and calcination reaction, with a particular focus on the morphology of CaO and changes thereof. Furthermore, a detailed overview over kinetic models to describe the carbonation and calcination reaction is provided, followed by a critical review of the effect of typical flue gas impurities such as H2O and SO2 on the CO2 capture characteristics of CaO. We conclude the chapter with a presentation of recent advances in the development of synthetic CaO-based CO2 sorbents which substantially exceed the cyclic CO2 capture capacity of limestone.
Chapter 6CO2 Capture via Cyclic Calcination and Carbonation Reactions
Marcin Broda, Roberta Pacciani and Christoph R. Müller
A.-H. Lu and S. Dai (eds.), Porous Materials for Carbon Dioxide Capture, Green Chemistry and Sustainable Technology, DOI: 10.1007/978-3-642-54646-4_6, © Springer-Verlag Berlin Heidelberg 2014
M. Broda · C. R. Müller (*) Laboratory of Energy Science and Engineering, ETH Zurich, Leonhardstrasse 27, 8092 Zurich, Switzerlande-mail: [email protected]
R. Pacciani Air Products and Chemicals, Campus de la UAB, Bellaterra, Barcelona, Spain

182 M. Broda et al.
6.1 Introduction
The currently most advanced CO2 capture and storage (CCS) technology with regard to industrial implementation is scrubbing with amines. However, the high CO2 capture costs of ~55$ per ton of CO2 avoided [1] of this process, in com-bination with issues associated with amine degradation and loss, are directing substantial research efforts towards the development of solid CO2 sorbents such as hydrotalcites, zeolites, metal oxides, activated carbons or metal organic frame-works (MOFs) [2]. Indeed, an emerging, second-generation CO2 capture process is the use of alkaline earth-metal-based CO2 sorbents [3, 4]. Duan and Sorescu [4] analysed the thermodynamic constraints of CO2 capture using the oxides of Be, Mg, Ca, Sr and Ba. Theoretically, SrO and BaO would possess very prom-ising CO2 capture characteristics, but the carbonates formed would have to be regenerated at probably prohibitively high temperatures (1,200–1,400 °C). On the other hand, BeO has to be excluded from the list of viable candidates for CO2 capture due to health issues. Indeed, Duan and Sorescu [4] indicated that, based on thermodynamic arguments, MgO is the most promising candidate among the earth alkali metal oxides due to its attractive temperature range of operation (300–400 °C). However, for MgO, the slow carbonation kinetics at typical operat-ing temperatures is a serious drawback. Therefore, CaO is arguably the best com-promise between sufficiently high carbonation kinetics, CO2 uptake capacity and temperature range of operation and it is currently the most studied alkaline earth-metal-based CO2 sorbent [5]. In particular, CaO is very attractive due to the fol-lowing characteristics: (1) high theoretical CO2 capture capacity of 0.786 g CO2/g sorbent (17.8 mmol CO2/g sorbent) (2) low cost if provided via the decomposi-tion of naturally occurring limestone and (3) fast kinetics of the CO2 capture and release reaction. For CaO, the cyclic capture and release of CO2, i.e. the so-called calcium looping process, is described by the following reversible reaction:
Here, the forward and reverse reactions are typically referred to as the carbonation and calcination reaction, respectively. The “optimal” operation temperature for the reactors in which the carbonation and calcination reactions proceed can be easily determined by plotting the equilibrium partial pressure of CO2 as a function of tem-perature using, e.g. the correlation of Barin and Platzki [6], as shown in Fig. 6.1.
For example, tolerating a CO2 partial pressure of 0.0026 bar in the gas leaving the carbonation reactor would set the operating temperature to 600 °C. However, the operating temperature of the carbonation reactor is typically a compromise between an increased reaction rate at higher temperatures and a higher CO2 cap-ture efficiency at lower temperatures. Since the carbonation reaction is highly exothermic and proceeds at high temperatures, the heat released can be utilized to generate electricity, e.g. via a steam cycle, thus dramatically decreasing the CO2 capture costs. Subsequently, the carbonated CO2 sorbent is transferred to the cal-ciner, as shown in Fig. 6.2, in which CaCO3 is regenerated to CaO while releasing
(6.1)CaO(s) + CO2(g) ↔ CaCO3(s), �H025◦C = ±178 kJ/mol

1836 CO2 Capture via Cyclic Calcination and Carbonation Reactions
a pure stream of CO2. As shown in the equilibrium diagram in Fig. 6.1, a calcina-tion temperature of ~900 °C is required to obtain a CO2 partial pressure of 1 bar. However, since the calcination reaction is endothermic, heat must be provided to regenerate the sorbent. This can be accomplished by burning fuel in situ with oxy-gen provided by an air separation unit (ASU), thereby introducing an energy pen-alty to the overall efficiency of the system. This cycle is continuously repeated and the spent material replaced by fresh sorbent. In a practical arrangement, the cyclic carbonation and calcination reactions will be most likely conducted in two inter-connected bubbling fluidized beds or a circulating fluidized bed [7].
6.2 Fundamentals of the Calcination and Carbonation Reaction
The thermal decomposition of CaCO3 is probably the most intensively studied reaction of the type:
(6.2)Xsolid → Ysolid + Zgas
Fig. 6.1 Equilibrium partial pressure of CO2 for the reaction CaO + CO2 ↔ CaCO3 as a function of temperature using the correlation of Barin and Platzki [6]
Fig. 6.2 Schematic diagram of the calcium looping process

184 M. Broda et al.
For example, Maciejewski and Reller [8] reported that up to the year 1987, 168 different estimates for the activation energy of the calcination reaction could be found in literature, ranging from 46.9 to 3,831 kJ/mol, the most common value being close to 168 kJ/mol. Most limestones are composed of the CaCO3 poly-morph calcite, which do not shrink upon calcinations in the absence of significant impurities. A particle of limestone contains more than 90 % by weight of calcium carbonate with very low porosity, with values for its specific surface area ranging from 1 to 10 m2 g−1. The remaining 10 % by weight of limestone comprises impu-rities, which might crucially affect the rate of reaction. Depending on the type of impurities and the morphology of the CaCO3, calcination might not proceed in the same way, i.e. the reaction occurring either at a sharp interface between CaO and CaCO3, or uniformly inside the particle.
Although the calcination reaction has been extensively studied, there is still no consensus as to its rate-limiting step. As a typical non-catalytic, gas–solid reaction, one or more of the following steps may limit calcination [9]:
1. Heat transfer from the surrounding gas to the surface of a particle of CaCO3 and then through the product layer to the reaction zone.
2. The actual chemical reaction.3. The diffusion of CO2 produced through the outer layer of CaO to the particle’s
exterior.
The conflicting results reported in literature with regard to the rate-limiting step may be at least partially explained by the fact that a wide range of experimental equipment, particle size and operating temperatures were employed, making it dif-ficult to reconcile results from different studies.
Importantly, it is currently also not clear how the morphology of the solid, impurities and the “type” of the CaCO3-containing material, i.e. natural versus synthetic sorbent, affect the rate of reaction. These issues will be described in more detail in Sect. 6.3.
With regard to the carbonation reaction, i.e. the reaction of CaO with CO2 to form CaCO3, it can be thought to occur via the following steps:
1. Diffusion of CO2 from the bulk to the particle’s external surface2. Diffusion of CO2 from the external surface through the pores3. Diffusion of CO2 through the product layer of newly formed CaCO3
4. The intrinsic chemical reaction of unreacted CaO with CO2.
An important aspect of the carbonation reaction is the very large difference in the molar volume of CaO (16.9 cm3/g) and the product CaCO3 (36.9 cm3/g). Thus, when a particle of CaCO3 is calcined, the resulting particle of CaO, assuming that there is no change in the overall dimensions of the particle during the reaction, will have a porosity εo = 1 − 16.9/36.9 = 0.54. On the other hand, when a particle of CaO reacts with CO2 in the backward reaction, the resulting CaCO3 will form a product layer of high molar volume with all its consequences, e.g. blocking the pores.
Indeed, very early studies of the carbonation reaction of CaO, such as the one from Dedman and Owen [10], showed that the carbonation of CaO stopped already at CaO conversion levels markedly below unity, i.e. a sensible quantity of CaO, derived through

1856 CO2 Capture via Cyclic Calcination and Carbonation Reactions
the calcination of limestone, did not form CaCO3 [5, 11, 12, 13]. Based on these experi-mental findings, Barker [12] proposed that the carbonation of CaO occurred in two stages, i.e. a very rapid, kinetically controlled reaction stage, followed by a second, markedly slower reaction stage, in which diffusion of CO2 through the product layer of CaCO3 is the rate-limiting step. This two-stage mechanism is exemplified very well in Fig. 6.3a, which plots the CO2 uptake of limestone-derived CaO as a function of reac-tion time (as obtained from a thermo-gravimetric analyser, TGA).
Performing multi-cycle experiments, Barker [12] measured a CaO conversion of ~72 % at the end of the first reaction stage, and ~96 % after 24 h. Subsequently, the sample was re-calcined to completion in N2, and another 24-h carbonation step was performed. For the second cycle, Barker [12] reported a CaO conversion of 70 % after the first reaction stage and 95 % after 24 h. It should be noted that, even though the conversion attained after 24 h was high and close to 100 %, such long carbonation time would be impractical for large-scale operations. Moreover, Barker [12] showed that when the sample was re-calcined directly after the first reaction stage of the carbonation reaction, the extent of the conversion in the kinet-ically controlled reaction stage, marked in green in Fig. 6.3a, decreased more rap-idly with cycle number, as shown in Fig. 6.4.
Barker [12] compared the ratio of the reaction rate of CO2 with CaO in the first to the second reaction stage to the different diffusivities of CO2 in CaO and CaCO3. Indeed, both ratios had the same value of 102. This excellent agreement gave a strong indication that once the product layer of CaCO3 reached a critical thickness, the reaction entered a diffusion-limited reaction regime. For a parti-cle of CaO with a surface area of 25 m2/g obtained by decomposing a particle of CaCO3 with dp = 10 μm, assumed to be spherical, a critical thickness of 22 nm for the product layer of CaCO3 was calculated by Barker [12].
Fig. 6.3 a The two reaction stages of the carbonation reaction, and b a schematic diagram of the reaction regimes occurring during carbonation

186 M. Broda et al.
Similar to the work of Barker [12], Dennis and Pacciani [14] observed two distinctive regimes in the carbonation reaction. Dennis and Pacciani [14] argued that in the first, fast reaction stage, the volume available in pores of diameter dpore < 100 nm is filled. The accessible pore volume is crucial for the carbona-tion reaction to proceed rapidly since the molar volume of the product, CaCO3 (36.9 cm3/mol), is more than twice as large as that of CaO (16.9 cm3/mol). Once the pores have been filled, the second, significantly slower, reaction stage takes over, in which a layer of CaCO3 is deposited on the outer surface of the grains as shown in Fig. 6.3b. Based on these findings, Dennis and Pacciani [14] proposed a correlation between the volume V0 available in pores with a diameter dp < 100 nm and the overall CO2 capture capacity (expressed in g CO2/g of calcined sorbent):
Here, ρCaO = 3,340 kg/m3 is the density of CaO and Z = 2.17 is the ratio of the molar volumes of CaCO3 and CaO.
In an effort to measure the diffusion rate in the second, slow stage of the car-bonation reaction, Mess et al. [15] studied the carbonation of non-porous CaO crystals (equivalent diameter of 10.3 m) over a temperature range of 550–1,100 °C and a CO2 pressure range of 1–11.7 atm in a TGA. It was found that at tempera-tures higher than 600 °C, the carbonation rate decreases more rapidly with time than what would be expected from diffusion through a uniform CaCO3 layer [9]. Based on this observation, Mess et al. [15] postulated that the product layer con-sists of crystalline grains, which grow with time and coalesce until the grain diam-eter reaches the dimension of the particle. Accordingly, the rate of carbonation was described by a dual mechanism where an activated, CO2 pressure-independent process acts in parallel with bulk diffusion of CO2 through grain boundaries.
In a recent study, Alvarez and Abanades [16] determined the critical prod-uct layer thickness at which the transition from the fast to the diffusion-limited
(6.3)X ′=
V0ρCaO
Z − 1
MCO2
MCaO
Fig. 6.4 Weight of sample against reaction time: a 24-h cycle; b short carbonation time (<1 h). The reaction temperature was 866 °C. Adapted from Ref. [12], Copyright 1973, with permission from John Wiley & Sons

1876 CO2 Capture via Cyclic Calcination and Carbonation Reactions
reaction stage occurs by submitting limestone to several carbonation and calci-nation cycles in a fixed bed reactor. Mercury porosimetry curves of the calcined materials and their carbonated counterparts are obtained in Fig. 6.5, and their dif-ferences interpreted with a simple model, which correlates pore volume with the thickness of the product layer. Alvarez and Abanades [16] estimated the critical CaCO3 layer thickness to be approximately ~50 nm.
6.3 Cyclic CO2 Capture Performance
As shown in Fig. 6.6, CaO derived via the decomposition of naturally occurring limestone has the serious drawback of a rapidly decreasing CO2 capture capacity with a number of repeated carbonation and calcination cycles [13].
The dramatic reduction in the cyclic CO2 capture capacity has been attributed to thermal sintering owing to the low Tammann temperature (temperature at which sintering starts) of CaCO3 (533 °C) that is substantially lower than the operating temperature of the process, i.e. 650–950 °C [13, 17]. The sintering-induced change
Fig. 6.5 Pore size distributions of fresh and cycled (87 cycles) materials: (solid line) calcined stage (square) carbonated stage and (dashed line) model predictions for the carbonated material. Adapted with the permission from Ref. [16]. Copyright 2005, American Chemical Society
Fig. 6.6 Repeated carbonation/calcination cycles of naturally occurring limestone conducted in a TGA. The experiment was performed isothermally at 750 °C using a carbonation atmosphere containing 40 vol.% CO2

188 M. Broda et al.
in the pore structure of a limestone-derived CO2 sorbent is plotted in Fig. 6.7. Freshly calcined limestone shows a pore size distribution peaking at ~85 nm, whereas a basically non-porous material is obtained after the first carbonation step. These mercury porosimetry measurements are in agreement with SEM images showing a non-porous “stone-paved” -like appearance of the external surface of carbonated limestone (Fig. 6.8). Interestingly, after the second calcination step, a bimodal pore size distribution is observed, comprising small pores with a mean
Fig. 6.7 Pore size distribution of limestone in its a calcined and b carbonated form after being exposed to different numbers of carbonation/calcination cycles. Reprinted with the permission from Ref. [17]. Copyright 2005, American Chemical Society
Fig. 6.8 Scanning electron micrographs of limestone after the first carbonation step: a interior of a carbonated grain (the arrows indicate “saturated” regions where calcium carbonate cannot grow any further whereas b shows the typical “stone-paved” appearance of the external sur-face of re-carbonated limestone. Reprinted with the permission from Ref. [18]. Copyright 2003, American Chemical Society

1896 CO2 Capture via Cyclic Calcination and Carbonation Reactions
diameter of ~85 nm, attributed to the calcination of newly formed CaCO3, and larger pores which continuously increase in size with cycle number, as shown by the dashed lines in Fig. 6.7. The formation of larger pores was explained by inter-nal sintering of unreacted CaO during calcination.
As shown in Fig. 6.6, the largest reduction in the cyclic CO2 uptake capacity of limestone occurs over roughly the first 10 cycles, in particular for high calcination temperatures (T > 950 °C) and long calcination times [13]. After approximately 20 cycles, the CO2 uptake stabilizes around an asymptotic value of ~0.08 mol CO2/mol CaO that is maintained over the following 500 cycles [13]. To correlate the CO2 uptake as a function of cycle number, Grasa and Abanades [13] proposed the following equation:
where XN is the conversion in the Nth cycle, Xr is the residual conversion, and k is a deactivation constant. In an attempt to explain the residual conversion of CaO-based CO2 sorbents, Lysikov et al. [19] proposed a model in which the carbonation/calcination cycles result in the formation of an interconnected net-work of CaO that serves as an inner skeletal refractory support for an outer layer of reactive CaO. According to Lysikov et al. [19], the initial decomposition of CaCO3 produces highly dispersed CaO which, however, re-carbonates incom-pletely (Fig. 6.9). In subsequent cycles, CaO grains grow and agglomerate form-ing an interconnected CaO skeleton. However, due to diffusion limitations, only the outer layer of the CaO skeleton is able to re-carbonate. Nonetheless, carbona-tion of the outer layer of the stable CaO skeleton provides an asymptotic CO2
(6.4)XN =1
11−Xr
+ kN+ Xr
Fig. 6.9 Schematic sketch of the morphological transformation of CaO-based CO2 sorbents over multiple carbonation/calcination cycles. Adapted with the permission from Ref. [19]. Copyright 2007, American Chemical Society

190 M. Broda et al.
capture capacity of CaO. Indeed, the existence of a residual CO2 uptake capacity, which was initially not expected [20], is a further positive aspect of the calcium looping process.
Finally, a CO2 capture process at industrial scale requires an absorbent mate-rial that maintains a high CO2 capacity in the presence of the numerous impu-rities found in the flue gas of a coal-fired power plant, which has a typical composition of 10–15 % CO2, 3–4 % O2, 5–7 % H2O, 0.05–0.3 vol.% of SO2 and 0.015–0.05 vol.% of NOx, together with trace quantities of other compounds such as HCl, arsenic, mercury and selenium [21]. Understanding the effect that these impurities have on the rate and extent of the carbonation and calcination reaction is critical to most efficiently integrate a CO2 capture unit into the overall power plant. Depending on the interaction between the impurities and the capture mate-rial, a pre-treatment stage might be required, thereby potentially introducing addi-tional energy penalties to the system. The effect of the presence of SO2 and H2O on the rate and extent of the carbonation and calcination reactions is addressed in the following.
6.3.1 Effect of the Presence of SO2
Sulphur dioxide (SO2) is one of the main impurities in the flue gas from a coal-fired power plant. Indeed, calcium-based sorbents, such as limestone, are also commonly used to remove SO2 from flue gases of fluidized bed combustors [22–24]. At the typical combustion temperatures of 800–900 °C, CaCO3 decomposes to CaO and CO2. Subsequently, CaO reacts with SO2 and O2, or with SO3, to form CaSO4. Thus, the overall SO2 capture process proceeds according to the following two-step process:
According to Eqs. (6.5) and (6.6), sulphation theoretically requires 1 mol of Ca for each mol of S released during combustion. However, previous work using lime-stone and dolomite as CaO precursors [22, 24–26] showed that the overall conver-sion of calcined limestone is low, typically 20–30 % on a molar basis. Dennis and Hayhurst [27] found that the conversion of CaO is very sensitive to the pore size distribution developed during calcination. They modelled a particle of calcined limestone as an assembly of parallel, wedge-shaped pores each of finite length. During sulphation, CaSO4 builds up on the pore walls and eventually blocks them, as shown in Fig. 6.10.
Here, it is worth noticing that the molar volume of the product CaSO4 (~52.2 cm3/mol) is about three times that of CaO (~16.9 cm3/mol). Consequently, owing to diffusional mass transfer limitations of SO2 within a particle of calcined
(6.5)CaCO3(s) → CaO(s) + CO2(g), �Ho850 ◦C = +169 kJ mol−1
(6.6)CaO(s) + SO2(g) + 1/2O2(g) → CaSO4(s), �Ho850 ◦C = −481.4 kJ mol−1

1916 CO2 Capture via Cyclic Calcination and Carbonation Reactions
limestone, pores typically plug at the entrances with product. This common sul-phation characteristic of limestone-derived CaO has been imaged using electron microscopy and is shown in Fig. 6.11.
Therefore, the reaction ceases prematurely, explaining the low CaO conver-sion. Hence, a large proportion of the sorbent leaves a fluidized bed combustor unreacted. Increasing the conversion of a sorbent would, therefore, decrease the amount of raw materials required, and the costs of operation and disposal. Several methods have been proposed to achieve better conversion, e.g.:
1. Hydration of the spent sorbent [29–33]. During hydration, water penetrates to the core of the partly sulphated limestone particles and reacts with CaO to form Ca(OH)2, which possesses a larger molar volume than CaO. The formation of
Fig. 6.10 Idealized pore model to predict the SO2 uptake of CaO. ABFE and CDHG are the original pore walls before sulphation. The grey areas represent the build-up of CaSO4 on the walls. Adapted from Ref. [28], Copyright 1986, with permission from Elsevier
Fig. 6.11 SEM images of the surfaces of a calcined and b sulphated Luscar limestone. Reprinted from Ref. [29], Copyright 2000, with permission from Elsevier

192 M. Broda et al.
Ca(OH)2 leads to an expansion of the core and the fracture of the CaSO4 shell, thus increasing the diffusion rate of SO2. When the hydrated particles are sub-jected to re-sulphation, simultaneous dehydration and sulphation take place, the former enhancing the porosity of the sorbent and the latter leading to a re-plugging of the sorbent’s pores. This sulphation and hydration mechanism, pro-posed by Laursen et al. [29], is sketched schematically in Fig. 6.12.
2. Mixing with additives [34, 35]. For example, Jozewicz and Kirchgessner [34] treated particles of calcined limestone by hydrating them in the presence of a com-mercial additive named Lignosite™ (Georgia Pacific Corporation), i.e. a partially desugared variety of calcium lignosulfonate available in the form of a dry pow-der. The effect of this treatment was an increase in the final sulphation conversion, owing to the ability of the surfactant to enhance the porous structure of the parti-cle. Wei et al. [36] reported the development of mesoporous CaCO3 by bubbling CO2 through a supersaturated aqueous solution of Ca(OH)2 mixed with an anionic dispersant. This material achieved a remarkable 97 % conversion when sulphated at 900 °C in 3,000 ppm SO2 (balance N2). However, the particles used were very small, i.e. <10 μm dia., which would make their application in a fluidized bed challenging.
3. Synthetic sorbents [36–39]. In particular, Dennis and Hayhurst [27] devel-oped a low-cost, high-porosity sorbent from cement flue dust. The syn-thetic sorbent achieved CaO conversions well above 50 %, which compares very favourable to the 25–30 % conversion commonly reported for lime-stone. Chen et al. [39] synthesized a sorbent that consisted of pure mayenite (Ca12Al14O33). Interestingly, mayenite was found to react with SO2. However, the conversion obtained was much lower than that of limestone. Finally, Pacciani et al. [40] synthesized mayenite-stabilized CaO and subjected it to sul-phation over a range of SO2 concentrations. The sorbent was able to achieve conversions exceeding 50 % owing to the presence of pore volume in the
Fig. 6.12 Schematic sequence of calcination, sulphation and hydration reactions of a limestone particle. Reprinted from Ref. [29], Copyright 2000, with permission from Elsevier

1936 CO2 Capture via Cyclic Calcination and Carbonation Reactions
macroporous region; these bigger pores were not prematurely blocked by the newly formed CaSO4 and therefore allowed sulphation to proceed uniformly throughout the particle.
6.3.2 Effect of the Presence of Steam on the CO2 Capture Capacity of CaO
The presence of steam can have substantially different effects on the CO2 capture capacity of a CaO-based sorbent depending on whether it is present during (1) cal-cination (2) carbonation or (3) if spent CaO-based sorbents are exposed to a satu-rated atmosphere of steam at relatively low temperatures.
Calcining the carbonated CO2 sorbent in H2O could be a viable option to obtain a pure stream of CO2, since H2O and CO2 could be easily separated via condensa-tion. However, Borgwardt [41] observed that the presence of steam during calcina-tion enhances sintering resulting in a further decrease in the CO2 uptake capacity of CaO. Borgwardt [41] attributed the increased sintering rate in the presence of H2O to the surface interaction of H2O and CaO. He proposed that under these con-ditions, short-lived hydroxyl groups form at the surface, which in turn enhance the mobility of O2− along the surface [41]. However, the results of Borgwardt [41] also indicated that, in the presence of H2O in the calcining atmosphere, not only lattice diffusion, but additional sintering mechanisms are likely to occur, e.g. grain boundary and surface diffusion. In order to clarify the effect of the presence of H2O during the calcination reaction on the cyclic CO2 uptake capac-ity of limestone, Sun et al. [42] performed cyclic carbonation/calcination experi-ments in which calcination was performed in a mixture of 95 % H2O and 5 % N2. Surprisingly, Sun et al. [42] found no appreciable difference in the cyclic CO2 cap-ture capacity and pore structure of sorbents calcined in either N2 or H2O, suggest-ing that calcination of the sorbent in the presence of H2O could be a viable option.
On the other hand, it has also been observed that the presence of H2O dur-ing carbonation at temperatures above the thermodynamic stability of Ca(OH)2 allows higher CaO conversions. Manovic and Anthony [43] reported that higher CO2 uptakes were attained by freshly calcined limestones when H2O was pre-sent during carbonation, possibly owing to H2O enhancing the diffusion of CO2 through the layer of newly formed CaCO3 during the diffusion-controlled stage of the carbonation reaction. Furthermore, Donat et al. [44] reported higher CO2 uptakes for four natural limestones after 30 cycles of repeated carbonation and calcination reactions when both steps, i.e. the calcination and carbonation reac-tions, were performed in the presence of steam. The authors attributed this phe-nomenon on the one hand to a shift in the pore size distribution to larger pore diameters, which are less susceptible to pore blockage during carbonation, and on the other hand to the higher diffusion rate of CO2 through the product layer of CaCO3 during carbonation. However, it should be mentioned that other studies

194 M. Broda et al.
showed that the presence of H2O during carbonation has only a little or even neg-ligible effect on the rate and extent of the carbonation reaction, e.g. Sun et al. [42]. Finally, Arias et al. [45] performed a study aimed at understanding the influ-ence of H2O on the carbonation reaction by focusing only on its possible effect during the fast reaction stage of the carbonation reaction, i.e. when it is kinetically controlled. The experimental results obtained in a differential reactor were inter-preted by means of a simplified reaction model and showed that the presence of H2O during carbonation had little influence on both the rate and the extent of the carbonation reaction as shown in Fig. 6.13.
Arias et al. [45] attributed the surprising discrepancy between their results and the ones reported by Manovic and Anthony [43] to the different experimental set-up employed, i.e. Manovic and Anthony’s [43] results were obtained in a TGA, which does not operate under differential conditions. In such an experimental set-up, further factors, such as external diffusion owing to the large sample mass used (30 mg vs. <3 mg in the experiments by Arias et al. [45]) and internal diffusion in the large sorbent particles, may affect the results.
Finally, it has been reported recently that exposing spent CaO-based sorbents to an atmosphere saturated with H2O allows the CO2 sorbent to regain some of its lost CO2 capture capacity. For example, Manovic and Anthony [46] prepared both freshly calcined and heavily sintered samples of limestone and hydrated them at 200 °C for 30 min. After re-activation, the CO2 sorbent had a significantly higher activity reaching a CaO conversion of ~75 % in the first cycle, as shown in Fig. 6.14.
Furthermore, the re-activated sorbent achieved higher conversions in subse-quent cycles when compared to the untreated material. However, one drawback of this re-activation approach is the appearance of large cracks in the particles, resulting in poor mechanical strength and leading to attrition and disintegration of the material over repeated carbonation/calcination cycles. Manovic et al. [47] proposed recently a new method of hydration whereby spent or highly unreactive sorbents are pelletized by intermittently spraying water on the powder. The addi-tion of a binder further improved the mechanical strength of the sorbent. The SEM
Fig. 6.13 CaO conversion as a function of cycle number for two different limestones (a and b) demonstrating the negligible effect of steam on the cyclic CaO conversion. Reprinted with the permission from Ref. [45]. Copyright 2012, American Chemical Society
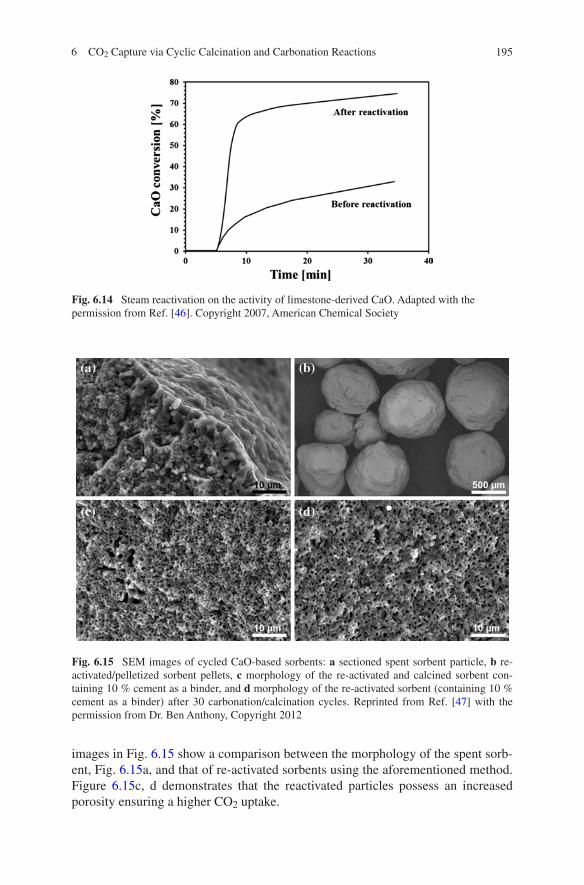
1956 CO2 Capture via Cyclic Calcination and Carbonation Reactions
images in Fig. 6.15 show a comparison between the morphology of the spent sorb-ent, Fig. 6.15a, and that of re-activated sorbents using the aforementioned method. Figure 6.15c, d demonstrates that the reactivated particles possess an increased porosity ensuring a higher CO2 uptake.
Fig. 6.14 Steam reactivation on the activity of limestone-derived CaO. Adapted with the permission from Ref. [46]. Copyright 2007, American Chemical Society
Fig. 6.15 SEM images of cycled CaO-based sorbents: a sectioned spent sorbent particle, b re-activated/pelletized sorbent pellets, c morphology of the re-activated and calcined sorbent con-taining 10 % cement as a binder, and d morphology of the re-activated sorbent (containing 10 % cement as a binder) after 30 carbonation/calcination cycles. Reprinted from Ref. [47] with the permission from Dr. Ben Anthony, Copyright 2012

196 M. Broda et al.
6.4 Kinetics of Calcination
6.4.1 Modelling Calcination
One of the most widely employed models to describe the mechanism of the calcina-tion reaction is the shrinking core model. Borgwardt [48] studied the calcination of limestone particles of diameters 1–90 μm in a differential reactor in the temperature range 670–1,075 °C. Borgwardt [48] found that calcination is kinetically controlled and occurs at a sharp interface, except for the final stage, when the diffusion of CO2 through the CaO product layer becomes rate-limiting. Interestingly, Beruto and Searcy [49] studied the thermal decomposition of crystals of calcite in vacuo. By interrupt-ing the reaction before completion, they were able to observe a 30-nm layer of poorly crystalline material between the un-decomposed limestone and the newly formed poly-crystalline CaO. Beruto and Searcy [49] could prove that this layer is composed of a metastable form of CaO, which transforms into its stable form during calcination. Turning now to an expression for the calcination reaction, Borgwardt [48] correlated the rate of reaction with the surface area of a particle of limestone according to:
where X is the conversion of CaCO3 to CaO at time t, So is the initial BET surface area of the limestone, and kc is the rate constant for the calcination reaction. Good agreement was found with the measured rate of calcination up to 1,000 °C, and an activation energy of 201 kJ/mol was calculated. Interestingly, Borgwardt [48] also concluded that the model was independent of the type of limestone used since it could be applied to both limestones studied. However, this statement should be further confirmed by applying it to the calcination of several other types of lime-stone. The shrinking core model with a sharp interface, originally observed by Borgwardt [48], was confirmed in subsequent studies. For example, Dennis and Hayhurst [50] carried out a thorough study of the kinetics of the calcination reac-tion using limestone particles (0.2–2 mm dia.) at conditions relevant for a CO2 capture system, i.e. both at atmospheric and high pressure (1–18 atm) using a flu-idized bed reactor. At the conditions employed, interphase and internal mass trans-fer as well as intraparticle heat transfer were negligible suggesting that calcination is kinetically controlled and follows a shrinking core mechanism. They also calcu-lated a value for the activation energy for the calcination reaction of 178 kJ/mol. The shrinking core mechanism for the calcination reaction was corroborated and extended by subsequent studies. Silcox et al. [51] described the decomposition of limestone as a shrinking core process for a spherical particle, proportional to the BET area of limestone as suggested by Borgwardt [48]. Using the experimental results of Borgwardt [48, 52], Silcox et al. [51] corrected the expression for the rate constant to take into account the effect of sintering by introducing a second exponential factor in the expression for the activation energy, viz:
(6.7)ln(1 − X) = Sokct
(6.8)k = A exp
(
−E
RT
)
exp
(
BPm
RT
)

1976 CO2 Capture via Cyclic Calcination and Carbonation Reactions
where A is the pre-exponential factor, E is the activation energy, P is the CO2 partial pressure (in atm), m is a dimensionless exponent, and B is a constant. The second exponential expression on the right in Eq. 6.8 effectively lowers the activa-tion energy for the sintering process (B < 0). Satterfield and Feakes [53] worked with pellets of diameter up to 20 mm at temperatures up to 900 °C and derived the rate of calcination per unit of superficial reaction area at three different thick-nesses of penetration of the reaction zone. Initially, it was assumed that the reac-tion followed a shrinking core mechanism. Using this assumption, an activation energy that was an order of magnitude greater than the enthalpy of reaction was determined. They attributed this rather unbelievable value to their apparently faulty initial assumption and concluded that the calcination reaction must occur homogeneously in porous agglomerates as a zone-type reaction, rather than at a sharply defined interface. Finally, Khinast et al. [54] used a TGA to study the cal-cination of limestone of different size (5–10 μm, 50–63 μm and 80–100 μm dia.) at 780 °C and a total pressure of P = 1 bar with various partial pressures of CO2. Khinast et al. [54] stated that the assumption whereby the reaction follows a shrinking core mechanism [50, 55], or the particles react uniformly [53, 56], is valid only when the internal surface area is not accessible, i.e. when a particle is initially non-porous, or when the reaction is relatively fast. To explain their exper-imental measurements, Khinast et al. [54] formulated a one-dimensional mathe-matical model based on a modified random pore model, similar to that proposed by Bhatia and Perlmutter [57, 58].
6.4.2 Effect of the Partial Pressure of CO2 on the Kinetics of the Calcination Reaction
A very important aspect to consider with regard to the calcination kinetics of CaCO3 is its dependence on the partial pressure of CO2 in the reacting gas. For a CO2 capture system employing CaO-based sorbents, the regeneration of the sorbent, i.e. its calcination, must be performed in atmospheres containing a high partial pressure of CO2. To elucidate the effect of the CO2 partial pressure on the kinetics of the calcination reaction, Garcia-Labiano et al. [56] studied CO2 partial pressures ranging from 0 to 80 % at temperatures ranging from 1,073 to 1,173 K. Their results indicate that calcination is significantly slowed down by the presence of CO2 in agreement with other authors. For example, Khinast et al. [54] showed that the reaction rate decreases exponentially with increasing par-tial pressure of CO2 up to PCO2
= 0.065 bar. They could not provide a conclu-sive explanation for this phenomenon, but suggested sorption effects at the CaO/CaCO3/CO2 interface as a possible reason for this exponential decay. To con-clude, the reaction mechanism and the influence of the CO2 partial pressure on the calcination reaction are far from understood. This is probably demonstrated best by the conclusion of Dennis and Hayhurst [50], stating that “very complex processes” must occur at the reaction interface, which include the breakdown

198 M. Broda et al.
of the rhombohedral CaCO3 lattice, followed by the desorption of CO2, and the nucleation and growth of cubic CaO.
6.4.3 Effect of Total Pressure
The last aspect of the calcination reaction to be considered here is the effect of the total pressure of the system on the rate of the calcination reaction. In fact, so far no conclusive explanation has been drawn on why the total pressure of the system affects the rate of reaction, even in the absence of CO2. For example, Dennis and Hayhurst [50] observed that increasing the pressure results in a decreasing reac-tion rate even in the absence of CO2; they suggested an empirical expression for Qc (rate of reaction of one particle) of the form:
where k is the rate constant for the carbonation (backward) reaction, yeCO2
is the equi-librium mole fraction of CO2 at temperature T, yr
CO2 is the mole fraction of CO2 at
the reaction front and YI is a spurious mole fraction of CO2. The authors found it difficult to explain the meaning of YI; nevertheless, YI proved to account satisfac-torily for the effect of pressure on the calcination reaction. Garcia-Labiano et al. [56] investigated the effect of pressure on the kinetics of the calcination reaction by performing experiments at 10 atm and different partial pressures of CO2. They con-firmed that the total pressure of the system affected the rate of reaction, even in the absence of CO2, as already observed by Dennis and Hayhurst [50]. Garcia-Labiano et al. [56] proposed a change in the molecular diffusivity as the most probable expla-nation for it; the molecular diffusivity of a gas is generally inversely proportional to the total pressure of the system, e.g. as in the correlation proposed by Fuller et al. [59]. However, Garcia-Labiano et al. [56] found that the effect of increasing pressure on the molecular diffusivity was too weak to adequately explain the dependence of the rate of calcination on the total pressure. Therefore, they proposed a modified ver-sion of Fuller’s [59] correlation, whereby the molecular diffusivity is proportional to 1/Pm, with m = 1.6 being a fitting parameter.
To summarize, despite the wealth of studies found in the literature, there is still little agreement on the mechanism that governs the rate of calcination of CaCO3. The major sources of discrepancy seem to be the size of the particles and the “type” of the CaCO3-containing material used. For particles smaller than, say, 60 μm, the rate of reaction was generally considered to be controlled by surface reaction. However, discrepancies were found in describing the mechanism of cal-cination for particles larger than, 60 μm. In this case, various authors identified the rate-limiting step of calcination as either heat transfer, diffusion in pores, true kinetics, or a combination of these. Furthermore, the morphology and the pres-ence of impurities seem to critically affect the reaction rate; however, it has not been possible to quantify these effects in a satisfactory manner. Also, the reaction
(6.9)Qc = kp
(
yeCO2
− yrCO2
)
+ Y1

1996 CO2 Capture via Cyclic Calcination and Carbonation Reactions
zone in different limestones might not develop in the same way; the reaction could occur either at a sharp interface between CaO and CaCO3, or uniformly inside the particle, depending on, e.g. whether the CaCO3 containing material is natural or synthetic, as well as on the type of impurities in natural limestone.
One last aspect to consider is the wide range of experimental equipment, parti-cle size and operating temperatures employed in the studies reviewed above. These experimental variations make it extremely difficult to reconcile results from dif-ferent studies. For example, Borgwardt [48] and Dennis and Hayhurst [50] used reactors, particle sizes and masses which ensured operation under differential con-ditions. However, it is rather doubtful whether experimental TGA studies can be performed under differential conditions.
Finally, when larger particle sizes were employed, results differed greatly, possibly owing to heat transfer effects. Satterfield and Feakes [53] worked with pellets of diameter up to 20 mm at temperatures up to 900 °C and con-cluded that heat transfer through the product layer to the reaction zone was a major resistance. Similarly, Hills [55] used spherical pellets, 10 mm in diame-ter, prepared by compacting powdered pure CaCO3 (5–8 μm dia.), which were then calcined in either air or mixtures of CO2 and air. He concluded that calci-nation occurred at a sharp spherical interface and that it was controlled by heat transfer to the reaction surface and mass transfer through the product layer of CaO [55].
6.5 Kinetics of the Carbonation Reaction of CO2
As mentioned in Sect. 6.2, the carbonation reaction can be split into two stages:
1. A fast stage in which CO2 reacts on the available surface, while at the same time, a layer of product CaCO3 builds up
2. A slow stage in which pores in the original calcium oxide have been filled or plugged by calcium carbonate. Thus, access to the unreacted calcium oxide requires diffusion through the product layer of carbonate.
The shift from the kinetically controlled to the diffusion-limited reaction regime of the carbonation reaction has been attributed to the build-up of a critical product layer thickness.
6.5.1 The First Stage of the Carbonation Reaction
Despite the importance of the carbonation reaction for the CO2 capture process, far fewer studies can be found in the literature on this matter than for the kinetics of the calcination reaction. Furthermore, there is still little consensus on the values of the kinetic constant, the reaction order and the activation energy.
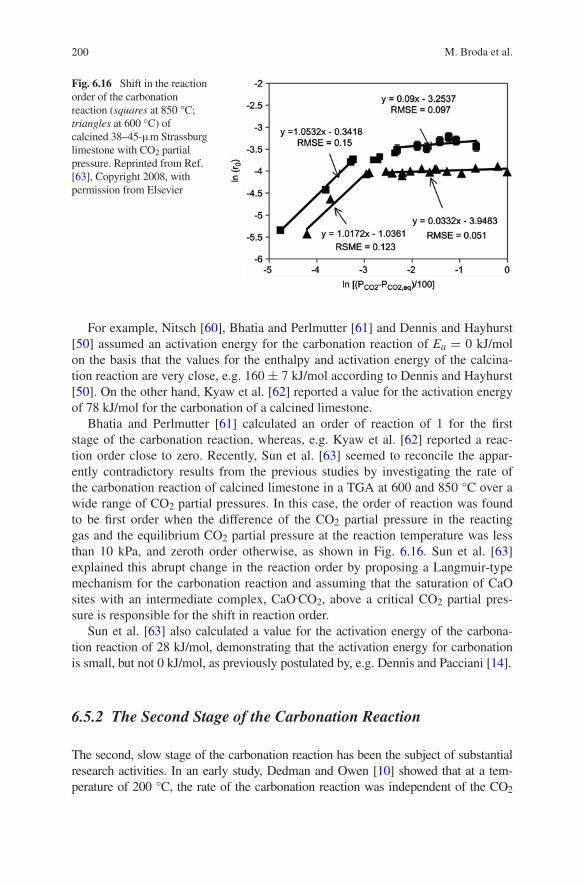
200 M. Broda et al.
For example, Nitsch [60], Bhatia and Perlmutter [61] and Dennis and Hayhurst [50] assumed an activation energy for the carbonation reaction of Ea = 0 kJ/mol on the basis that the values for the enthalpy and activation energy of the calcina-tion reaction are very close, e.g. 160 ± 7 kJ/mol according to Dennis and Hayhurst [50]. On the other hand, Kyaw et al. [62] reported a value for the activation energy of 78 kJ/mol for the carbonation of a calcined limestone.
Bhatia and Perlmutter [61] calculated an order of reaction of 1 for the first stage of the carbonation reaction, whereas, e.g. Kyaw et al. [62] reported a reac-tion order close to zero. Recently, Sun et al. [63] seemed to reconcile the appar-ently contradictory results from the previous studies by investigating the rate of the carbonation reaction of calcined limestone in a TGA at 600 and 850 °C over a wide range of CO2 partial pressures. In this case, the order of reaction was found to be first order when the difference of the CO2 partial pressure in the reacting gas and the equilibrium CO2 partial pressure at the reaction temperature was less than 10 kPa, and zeroth order otherwise, as shown in Fig. 6.16. Sun et al. [63] explained this abrupt change in the reaction order by proposing a Langmuir-type mechanism for the carbonation reaction and assuming that the saturation of CaO sites with an intermediate complex, CaO.CO2, above a critical CO2 partial pres-sure is responsible for the shift in reaction order.
Sun et al. [63] also calculated a value for the activation energy of the carbona-tion reaction of 28 kJ/mol, demonstrating that the activation energy for carbonation is small, but not 0 kJ/mol, as previously postulated by, e.g. Dennis and Pacciani [14].
6.5.2 The Second Stage of the Carbonation Reaction
The second, slow stage of the carbonation reaction has been the subject of substantial research activities. In an early study, Dedman and Owen [10] showed that at a tem-perature of 200 °C, the rate of the carbonation reaction was independent of the CO2
Fig. 6.16 Shift in the reaction order of the carbonation reaction (squares at 850 °C; triangles at 600 °C) of calcined 38–45-μm Strassburg limestone with CO2 partial pressure. Reprinted from Ref. [63], Copyright 2008, with permission from Elsevier

2016 CO2 Capture via Cyclic Calcination and Carbonation Reactions
partial pressure and therefore not limited by the diffusion of CO2 to the exterior of a particle of CaO. Indeed, it was proposed that the second stage of the carbonation reaction corresponded to the diffusion of CO2 along (carbonated) grain boundaries. Oakeson and Cutler [64] carbonated non-porous particles (40 μm dia.) in a TGA under CO2 pressures of 2.35–24.67 atm and in the temperature range of 853–1,044 °C. They found that carbonation was limited by diffusion through the CaCO3 product layer and that the CO2 partial pressure had a major effect on the rate of diffusion. Based on these observations, they postulated a Langmuir-type expression to explain their experimental observations, viz.
where D is the diffusion coefficient, PCO2 is the pressure of CO2 and b and K
are constants. Oakeson and Cutler [64] concluded that the most probable diffu-sion species was either CO2 or CO3
2− (with O2− diffusing countercurrently). Furthermore, by plotting values of ln(KD) versus 1/T, T being the reaction tem-perature, Oakeson and Cutler [64] calculated an activation energy for the second stage of the carbonation reaction of ~121 kJ/mol.
Bhatia and Perlmutter [61] investigated the kinetics of the carbonation reaction of CaO, focussing on how it is affected by the deposition of the layer of newly formed CaCO3 during the reaction. The incomplete conversion of CaO to CaCO3 was attributed to the uniform closure of narrow pores, after which only the larger pores kept filling up with product, albeit much more slowly. They suggested that below 515 °C, the counterdiffusion of CO3
2− and O2− ions in the solid state could be the rate-controlling mechanism. Furthermore, they calculated the activa-tion energy for the second stage of the carbonation reaction as 88.9 ± 3.7 kJ/mol for temperatures below 515 °C. Another interesting observation of Bhatia and Perlmutter [61] is the abrupt change in the activation energy to 179.2 ± 7.0 kJ/mol for temperatures higher than 515 °C. This transition suggests a change to a reaction mechanism, dominated by the sequential decompositions of carbonate ions in the calcium carbonate layer.
Mess et al. [15] studied the carbonation of non-porous crystals of CaO (15–20 μm dia.) at 550–1,100 °C and at pressures of CO2 of 1–11.7 atm using a TGA. They found that, for a given particle size, the reaction rate was initially rapid, but decreased with time more rapidly than predicted for the case that diffusion through a uniform product layer is the rate-controlling step. They explained this discrepancy by ascribing a dual mechanism to the carbonation reaction, i.e. a process that involves activated CO2 and in parallel bulk diffusion of CO2 through grain boundaries, independent of the pressure of CO2. The acti-vation energy calculated from their experimental data was 238 kJ/mol, i.e. some-what higher than the value determined by Bhatia and Perlmutter [61], but lower than the value reported for lattice diffusion in single calcite crystals, i.e. 352 kJ/mol. This finding suggests that defects in natural calcite may crucially affect the diffusivity of CO2.
(6.10)1
rate=
1
KD
(
1
bPCO2
+ 1
)

202 M. Broda et al.
Finally, Sun et al. [63] calculated the activation energy for the slow stage of the carbonation reaction by modelling the carbonation reaction with a random pore model. They reported a value of 215 kJ/mol for temperatures between 500 and 850 °C indicative of solid-state diffusion.
6.5.3 Modelling of the Carbonation Reaction
As mentioned before, the carbonation of CaO is a typical non-catalytic, gas–solid reaction. As such, it has been extensively modelled using either random pore or grain models. The random pore model was developed and first applied by Bhatia and Perlmutter [57] to model the sulphation of lime and subsequently extended by Sun et al. [65]. The pores were assumed as an assembly of randomly oriented cylinders of uniform diameter, which initially overlapped (Fig. 6.17). The initial increase in the reaction rate was attributed to the growth of the surface area of the CaO–CaCO3 interface, which is, however, overshadowed in later stages by the intersection of the growing surfaces, leading subsequently to a decrease in the reaction rate.
Other authors, such as Stendardo and Foscolo [66] and Khoshandam et al. [67] used a grain model approach to model the carbonation reaction. Here, the sorb-ent particle is considered to be composed of grains surrounded by pores through which CO2 can diffuse to reach the surface of the grains. A very novel approach to describe the mechanism of the carbonation of CaO was recently proposed by Li et al. [68, 69]. The model was developed from the observation that for the case that the temperature is increased during the carbonation of CaO, when the reaction is in the diffusion-limited stage, carbonation enters again the fast, kinetically con-trolled reaction stage Symonds et al. [70]. Using atomic force microscopy (AFM), Li et al. [69] showed that the product layer of CaCO3 grows with an island-like morphology on the CaO surface with most of the islands being formed at positions which contain terraces, steps and kinks (Fig. 6.18).
Fig. 6.17 Schematics of the simplified two-pore model developed by Sun et al. before (left) and after (right) formation of a CaCO3 product layer. Adapted from Ref. [65], Copyright 2008, with permission from Elsevier

2036 CO2 Capture via Cyclic Calcination and Carbonation Reactions
On the basis of the aforementioned observations, Li et al. [68] developed a model based on rate equation theory that comprises the following steps: (1) surface reac-tion (2) nucleation and growth and (3) grain boundary and lattice diffusion. Li et al. [68] demonstrated that the developed rate equation could explain successfully some macroscopic behaviour, such as the effect of temperature on the rate and mechanism of the reaction. Particularly, with the increase in the reaction temperature, the size of the CaCO3 islands increased while the island density decreased. This leads to an increase in CaO conversion in the fast reaction stage. When the carbonation reaction approaches equilibrium, the rates of nucleation and growth are fast, while the surface reaction is the limiting step, thus leading to the macroscopically observed slow rate of CaO conversion. The approach of Li et al. [68] is novel so far as it is a micro-scopic approach. In contrast with previous studies, e.g. Bhatia and Perlmutter [61] and Sun et al. [63], in which macroscopic approaches were employed, Li et al.’s [68] work can be used to link the macroscopic behaviour of a gas–solid reaction with the microscopic mechanism described at a molecular level.
Fig. 6.18 AFM images of CaCO3 forming on a single crystal of CaO as a function of reaction time. Reprinted with the permission from Ref. [69]. Copyright 2012, American Chemical Society

204 M. Broda et al.
6.5.4 Development of Sintering-Resistant CO2 Sorbents
To reduce the significant drop in the CO2 capture capacity of limestone, various attempts to develop more effective, CaO-based CO2 sorbents have been reported. The different approaches of these research efforts can be categorized in the fol-lowing two groups: (1) synthetic unsupported CaO with improved morphol-ogy and (2) stabilization of CaO on a high Tammann temperature support, e.g. oxides of Al, Zr or Mg.
6.5.4.1 Unsupported CaO
The CO2 capture performance of CaO is strongly affected by its morphological characteristics, e.g. its surface area, pore size distribution and particles size. It has been argued that if the CaO particles are sufficiently small, the carbonation reac-tion will occur exclusively in the kinetically controlled stage of the carbonation reaction, thus avoiding diffusion limitations. This method originates from an early finding of Barker [71] who used nanosized CaCO3 (<10 nm) as a CaO precur-sor and reported molar conversions of 93 % over 30 cycles without any noticeable decrease with cycle number. However, the duration of the carbonation reaction was 24 h, which is an impractically long time for industrial processes. Florin and Harris [72] repeated Barker’s experiment and noticed that by reducing the carbon-ation time to 20 min, the characteristic decay of the molar CaO conversion with cycle number was observed. This was attributed to thermal sintering resulting in the agglomeration of the CaCO3 nanoparticles, as shown in Fig. 6.19.
Lu et al. [73] assessed five different CaO precursors, i.e. Ca(OH)2, Ca(CH3-COO)2·H2O, CaO, CaCO3 and Ca(NO3)2·4H2O, with regard to their ability to produce CaO with a high surface area and a stable pore structure. After 27 carbonation/calcination cycles, the highest CO2 uptake (0.49 g CO2/g sorbent) was obtained for CaO derived from calcium acetate. On the other hand, already in the first cycle, Ca(NO3)2 exhibited a negligible CO2 uptake capacity. These observations were in agreement with N2 adsorption measurements, which deter-mined a high BET surface area of 20 m2/g (BJH pore volume of 0.23 cm3/g) for the sorbent synthesized using Ca(CH3COO)2 as the calcium precursor. In contrast, the BET surface area of CaO derived from Ca(NO3)2 was below the accuracy of the equipment, i.e. <1 m2/g. The decomposition characteristics of the various calcium oxide precursors tested give some explanations for the different CaO morphologies obtained. For example, calcium acetate decomposes first to CaCO3 (at ~400 °C) and subsequently to CaO (at ~600 °C). On the other hand, calcium nitrate decomposes in a single step to CaO in the temperature range 540–620 °C [74]. Thus, the significantly higher BET surface area and BJH pore volume as well as the bimodal pore size distribution of CaO derived from calcium acetate (Fig. 6.20a) are probably due to the two-step decomposition process. The extremely low surface area and pore volume of CaO derived from Ca(NO3)2 were
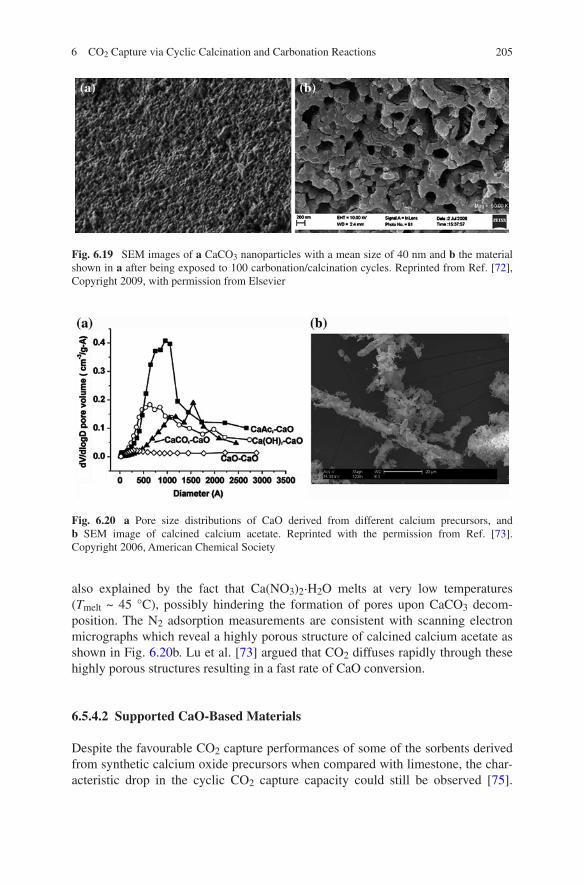
2056 CO2 Capture via Cyclic Calcination and Carbonation Reactions
also explained by the fact that Ca(NO3)2·H2O melts at very low temperatures (Tmelt ~ 45 °C), possibly hindering the formation of pores upon CaCO3 decom-position. The N2 adsorption measurements are consistent with scanning electron micrographs which reveal a highly porous structure of calcined calcium acetate as shown in Fig. 6.20b. Lu et al. [73] argued that CO2 diffuses rapidly through these highly porous structures resulting in a fast rate of CaO conversion.
6.5.4.2 Supported CaO-Based Materials
Despite the favourable CO2 capture performances of some of the sorbents derived from synthetic calcium oxide precursors when compared with limestone, the char-acteristic drop in the cyclic CO2 capture capacity could still be observed [75].
Fig. 6.19 SEM images of a CaCO3 nanoparticles with a mean size of 40 nm and b the material shown in a after being exposed to 100 carbonation/calcination cycles. Reprinted from Ref. [72], Copyright 2009, with permission from Elsevier
Fig. 6.20 a Pore size distributions of CaO derived from different calcium precursors, and b SEM image of calcined calcium acetate. Reprinted with the permission from Ref. [73]. Copyright 2006, American Chemical Society

206 M. Broda et al.
Therefore, to overcome the loss in the cyclic CO2 uptake capacity, the develop-ment of materials in which CaO is supported on a high Tammann temperature matrix, e.g. Al2O3, MgO or ZrO2, has been proposed. However, so far mostly “simple” techniques, such as co-precipitation, hydrolysis or mechanical mixing, have been applied to develop new materials. For example, Florin et al. [76] syn-thesized a CaO-based CO2 sorbents by bubbling CO2 through an aqueous solution containing Ca(OH)2 and Al(NO3)3. Upon calcination (at temperatures >800 °C), a reaction between CaO and Al2O3 was observed resulting in the formation of mayenite (Ca12Al14O33). Indeed, the formation of mayenite is thermodynamically favoured in the temperature range 800–900 °C [77, 78]. Mayenite is inert towards the cyclic calcination and carbonation reactions, i.e. it does not react with CO2 at the relevant process conditions [77, 79]. Florin et al. [76] reported that the sorb-ent synthesized with a weight ratio of CaO to Ca12Al14O33 of 80:20 possessed the highest initial CO2 uptake of ~0.43 g CO2/g sorbent. However, with cycle num-ber, the CO2 uptake decreased rapidly reaching only ~0.19 g CO2/g sorbent after 30 cycles (Fig. 6.21a). In order to obtain some insights into the morphological changes occurring over repeated carbonation and calcination cycles, Florin et al. [76] determined the BET surface area and BJH pore volume distribution of the fresh (calcined) and reacted material (Fig. 6.20b). Consistent with the decay in the cyclic CO2 capture capacity, a decrease in the BET surface area from 12 to 8 m2/g, associated with a reduction in the volume of pores with diameters ranging from 30 to 90 nm was observed.
Besides mayenite, encouraging results with respect to the manufacture of supported CaO-based sorbents via co-precipitation techniques have also been achieved for MgO as the inert support. For example, Filitz et al. [80] synthesized Ca-based materials by co-precipitating solutions of Ca(NO3)3 and Mg(NO3)2
Fig. 6.21 a CO2 uptake characteristics of a CO2 sorbent synthesized using a precipitation tech-nique. The weight ratio of CaO to Ca12Al14O33 was 80:20; b pore size distributions of the fresh (calcined) material and after being exposed to 10 carbonation/calcination cycles. Adapted with the permission from Ref. [76]. Copyright 2010, American Chemical Society

2076 CO2 Capture via Cyclic Calcination and Carbonation Reactions
using either Na2CO3 or a mixture of NH3aq and (NH4)2CO3. The best sorbent contained 71 wt.% CaO and possessed the CO2 uptake of 0.51 g CO2/g sorb-ent after 15 cycles, a value which was ~100 % higher than that of the reference limestone. Filitz et al. [80] emphasized the influence the co-precipitation param-eters, such as the precipitation agent, ageing time and the pH, on the morphol-ogy of the sorbents and, subsequently, their cyclic CO2 uptake capacities. It was found that sorbents, in which Ca and Mg were mixed on the crystal lattice, possessed a very high and stable cyclic CO2 capture capacity (Fig. 6.22a). On the other hand, the formation of separate crystals of CaCO3 and MgCO3 poly-morphs resulted in a rapid decay in the cyclic CO2 uptake, indicating that the presence of a support alone is not sufficient to stabilize the morphology of CaO (Fig. 6.22b).
In a further study, Li et al. [81] prepared a CO2 sorbent via hydrolysis. In a typical synthesis, Al(NO3)3 and powdered CaO were mixed into a solution of 2-propanol and distilled water. The weight ratio of CaO to Ca12Al14O33 (mayen-ite) was 75:25. The resulting slurry was dried, pelletized and calcined at 900 °C. Subsequently, the material was tested in a TGA to determine its cyclic CO2 capture capacity. Figure 6.23a, which plots the CO2 uptake as a function of the carbonation time, demonstrates clearly that the CO2 uptake of the material was largely occurring in the fast, kinetically controlled reaction stage (except for the first cycle). Interestingly, for this material, the CO2 uptake increased over the first five cycles. This intriguing observation was explained by a follow-up study of Pacciani et al. [79] who reported that in the first cycle, the volume inside pores of dp < 100 nm increased due to structural changes, resulting subsequently in an increased CO2 uptake. After ~5 cycles, the CO2 uptake was fairly stable and was equal to 0.45 g CO2/g sorbent in the 13th cycle. The favourable CO2 capture char-acteristics of the sorbent were attributed to the formation of ultrafine CaO particles stabilized by an inert Ca12Al14O33 matrix, as shown in Fig. 6.23b.
Fig. 6.22 SEM images of Ca-based, MgO-stabilized CO2 sorbents via co-precipitation tech-nique using a a mixture of NH3aq and (NH4)2CO3 or b Na2CO3. Reprinted with the permission from Ref. [80]. Copyright 2012, American Chemical Society

208 M. Broda et al.
So far, the most extensively used technique to stabilize CaO with an inert support is mechanical mixing. Depending on the precursors used, this method can be categorized into the following three groups: (1) mixing of soluble precursors (2) mixing of one soluble and one insoluble precursor and (3) mixing of two insol-uble precursors. For example, Liu et al. [82] prepared CaO-based CO2 sorbents by dissolving a wide range of calcium and magnesium salts in distilled water. The two best sorbents synthesized used calcium and magnesium gluconate as the calcium and magnesium precursor, respectively. The weight ratios of CaO to MgO of the two best sorbents were 85:15 and 75:25, and the materials possessed CO2 uptakes of, respectively, ~0.58 and 0.56 g CO2/g sorbent after 24 cycles. Interestingly, the CO2 uptakes determined are very close to their theoretical value (Fig. 6.24). Figure 6.24 also confirms that there is a maximum weight fraction of CaO in the sorbent, viz. ~85 wt.%, beyond which the material loses its CO2 capture stabil-ity. The favourable CO2 capture characteristics of the sorbents were attributed to a homogeneously dispersed, high Tammann temperature support (the Tammann temperature of MgO is 1,563 °C [83]), which effectively stabilized the nanostruc-tured morphology of CaO [82]. SEM images of the material synthesized, shown in Fig. 6.25a, confirm this hypothesis in that nanocrystalline MgO (~9 nm) adhered to the surface of CaO particles of size ~120 nm. Additionally, the BET surface area of the new material was 25 m2/g, a value that is significantly higher than that of com-mercial CaO (12 m2/g).
The simplest method to manufacture CO2 sorbents is the mixing of two insoluble precursors. This method would probably have the highest poten-tial to reduce the costs of sorbent preparation since inexpensive raw materials such as limestone or clay are often used. For example, Qin et al. [84] devel-oped CO2 sorbents using a suspension mixing method. Here, a calcium precur-sor, e.g. calcium hydroxide or calcium carbonate, was mixed in water with an
Fig. 6.23 a CO2 uptake as a function of carbonation time, and b SEM image of the calcined CO2 sorbent. Adapted with the permission from Ref. [81]. Copyright 2005, American Chemical Society

2096 CO2 Capture via Cyclic Calcination and Carbonation Reactions
insoluble support, e.g. calcium aluminate cement, clay or fly ash. Subsequently, the resulting slurry was dried and calcined at 900 °C. The cycling experiments, performed in a TGA as shown in Fig. 6.26, showed the highest CaO conversion of 0.47 mol CO2/mol CaO (after 18 cycles) for the sorbent synthesized using
Fig. 6.24 CO2 uptake of CaO-based materials synthesized using a wet mixing technique. The CO2 uptake is plotted as a function of the molar ratio of Ca2+ to Mg2+. Carbonation was con-ducted for 30 min at 650 °C in 15 vol.% CO2, whereas calcination was performed for 10 min at 900 °C in 100 vol.% N2. Adapted with the permission from Ref. [82]. Copyright 2005, American Chemical Society
Fig. 6.25 a Scanning transmission electron microscopy image of MgO-stabilized CO2 sorb-ents (Ca—red, Mg—cyan), and b pore size distribution. The CO2 sorbent contained a weight ratio of CaO to MgO of 75:25 %. Adapted with the permission from Ref. [82]. Copyright 2005, American Chemical Society

210 M. Broda et al.
calcium hydroxide and calcium aluminate cement (the weight ratio of CaO to calcium aluminate cement was 75:25). In contrast, clay and fly ash were found to be unsuitable support materials due to the reaction between CaO and silica. The formation of calcium silicates negatively affected the CO2 uptake character-istics of the materials.
Recently, wet-impregnation techniques in which CaO is deposited on to a sup-port material have attracted significant attention. For example, Huang et al. [85] developed CaO-based CO2 sorbents supported on mesoporous SBA-15. First, SBA-15 was synthesized according to the procedure reported by Vinu et al. [86]. Subsequently, 0.1 mol of calcium acetate was dissolved in 80 mL water and then 0.5 g of SBA-15 was added to the solution and soaked for 24 h. The excess solu-tion was removed by filtration or evaporation and the impregnated SBA-15 was subsequently calcined at 500 °C. Importantly, it was found that the procedure used to remove the excess solution significantly influenced the amount of calcium loaded on SBA-15, i.e. by using filtration and evaporation techniques, the weight ratio of Ca to SBA-15 was 5:94 and 49:51, respectively. In turn, the CaO-loading critically affected the surface area and pore volume of the CO2 sorbent as shown in Fig. 6.27a. Surface areas of 367 m2/g and 155 m2/g (0.67 cm3/g and 0.43 cm3/g) were determined for Ca-Si-filtr (filtration) and Ca-Si-evap (evaporation), respec-tively. The lower surface area and pore volume of CaO-impregnated SBA-15 were attributed to the blockage of the mesoporous channels of SBA-15 by CaO particles (Fig. 6.28). After 40 cycles (calcination was performed at 910 °C in 100 % N2),
Fig. 6.26 Performance of CO2 sorbents synthesized by mixing Ca(OH)2 with different support materials. The mass ratio of CaO to the support material was 75:25 %. The following supports were studied: (filled square) calcium aluminate cement, (filled circle) fly ash, (filled triangle) clay. Carbonation was performed for 30 min at 650 °C in 15 vol.% CO2, whereas calcination was conducted for 10 min at 900 °C in 100 vol.% N2. Adapted with the permission from Ref. [84]. Copyright 2012, American Chemical Society

2116 CO2 Capture via Cyclic Calcination and Carbonation Reactions
the best CO2 sorbent, that is Ca-Si-evap, showed a molar CaO conversion of 80 % (0.30 g CO2/g sorbent), Fig. 6.27b.
The potential of advanced synthesis techniques to develop CaO-based sorb-ents stabilized by a support material has been demonstrated recently by Broda et al. [74] who synthesized Al2O3-stabilized CO2 sorbents by employing a sol-gel technique. Here, aluminium iso-propoxide was added to reverse osmosis water (15 MΩ·cm) and allowed to hydrolyse for 30 min at 75 °C. Subsequently, a cal-cium precursor, i.e. calcium acetate hydrate (Ca(CH3OO)2·H2O), calcium nitrate tetrahydrate (Ca(NO3)2·4H2O) or calcium acetylacetonate (Ca(C5H7O2)), was dissolved in water and, together with an acid (either acetic or nitric acid), added
Fig. 6.27 a Pore size distribution of (filled square) SBA-15, (square) calcined Ca-Si-filtr, (cir-cle) calcined Ca-Si-evap, and b the CO2 capture characteristics of Ca-Si-evap. Adapted from Ref. [85], Copyright 2010, with permission from Elsevier
Fig. 6.28 TEM images of SBA-15. Reprinted from Ref. [85], Copyright 2010, with permission from Elsevier

212 M. Broda et al.
to the hydrolysed aluminium iso-propoxide. The condensation reaction was per-formed at 90 °C for times ranging from 6 to 24 h. The obtained gel was first dried overnight at 100 °C and then calcined for 2 h at 800 °C. It was found that the calcium precursor and the ratio of Ca2+ to Al3+ influenced the CO2 uptake characteristics of the synthesized material most. The use of weakly acidic cal-cium precursors, i.e. Ca(CH3OO)2 or Ca(C5H7O2)2, resulted in sorbents with a homogeneous, nanostructured morphology as well as a high pore volume as shown in Fig. 6.29a, b. In turn, these sorbents also possessed very favourable CO2 capture characteristics when compared with limestone. After 30 cycles of the repeated carbonation and calcination reactions, the CO2 uptake of the best sorb-ent was 0.51 g CO2/g sorbent which is about 150 % higher than that of the refer-ence limestone (Fig. 6.30). On the other hand, using a strongly acidic calcium precursor, i.e. Ca(NO3)2·4H2O, a CO2 sorbent with a coarse and heterogeneous morphology and a low pore volume was obtained (Fig. 6.29c). As expected, this material showed a very low CO2 uptake, significantly under-performing lime-stone. These observations with regard to Ca(NO3)2·4H2O are in agreement with the aforementioned study of Lu et al. [73] reporting that Ca(NO3)2·4H2O was not a suitable CaO precursor due to its very low melting point of ~45 °C. Broda et al. [74] also highlighted the influence of the molar ratio of Ca2+ to Al3+ on the morphology and the CO2 uptake capacity of the material. Decreasing the ratio of Ca2+ to Al3+ resulted in materials with a higher surface area and pore volume; however, reducing the quantity of active CaO also meant lower CO2 uptakes of the CO2 sorbents.
Very recently, Broda and Müller [87] extended the carbon sol-gel technique first reported by Pekala [88] to synthesize Al2O3-stabilized, CaO-based CO2 sor-bents with a hierarchical pore structure. It was argued that by nanostructuring the material, diffusive limitations of the carbonation reaction could be avoided. Here, a carbon gel acted as a template for pores in the small micrometre range. A sche-matic sketch of the different steps of the synthesis protocol is shown in Fig. 6.31. In the first step, formaldehyde was added to an aqueous solution of resorcinol to obtain a molar ratio of resorcinol to formaldehyde of 1:2. Subsequently, an
Fig. 6.29 High-resolution SEM images of solgel-derived CO2 sorbents synthesized using different calcium precursors: a calcium acetylacetonate (Ca(C5H7O2)2), b calcium acetate (Ca(CH3OO)2) and c calcium nitrate (Ca(NO3)2). Reproduced from Ref. [74] by permission of John Wiley & Sons Ltd

2136 CO2 Capture via Cyclic Calcination and Carbonation Reactions
aqueous solution of Ca2+ and Al3+ precursors (1.5 M solution) was added to the resorcinol/formaldehyde solution to obtain a molar ratio of C to (Ca2+ + Al3+) of 80:20. The second step, i.e. gelation of the mixed solution, was performed in a water bath at 80 °C for 3 days. At the end of the gelation step, carbonaceous spheres which were fully covered with an amorphous Ca-Al-based film were formed. In the third step (pyrolysis at 500 °C in N2), the film was crystallized.
Fig. 6.30 CO2 uptake of sol-gel-derived Ca-based CO2 sorbents over 30 cycles of the repeated carbonation and calcination reactions using different calcium precursors. The molar ratio of Ca2+ to Al3+ was 90:10, and the CO2 uptake of (dashed line) limestone is shown for reference: (filled circle) Ca(C5H7O2)2 (square) Ca(CH3COO)2·H2O (triangle) Ca(NO3)2·4H2O. The dashed line gives the theoretical CO2 uptake of pure CaO. Reproduced from Ref. [74] by permission of John Wiley & Sons Ltd
Fig. 6.31 Schematic of the synthesis steps employed to produce CaO-based, Al2O3-stabilized CO2 sorbent with a hierarchical pore structure. Reproduced from Ref. [87] by permission of John Wiley & Sons Ltd

214 M. Broda et al.
XRD analysis confirmed the presence of two CaCO3 polymorphs in the film, i.e. calcite and vaterite. Upon removal of the carbon gel template via calcination in air, hollow microspheres, comprised of a nanostructured shell, were obtained. The best sorbent possessed a weight ratio of CaO to Al2O3 of 91:9 and gave a CO2 uptake of 0.56 g CO2/g sorbent after 30 cycles, a value which is 180 % higher than that of limestone. The favourable CO2 capture characteristics of the material were attrib-uted to the following two features of the material. First, the nanostructured shell was comprised of grains of an average diameter of approximately 170 nm, thus minimizing the slow, diffusion-limited reaction stage of the carbonation reaction. Additionally, the synthesis technique ensured that Ca2+ and Al3+ were homog-enously mixed, partially via the formation of Ca12Al14O33 (mayenite) which effectively stabilized the nanostructured morphology over many cycles. Indeed, Fig. 6.32a demonstrates convincingly that the new material maintained its nano-structured morphology over repeated carbonation/calcination cycles. On the other hand, limestone subjected to the same experimental conditions lost its initial nano-structured morphology due to sintering, resulting in a comparatively non-porous material after 30 cycles (Fig. 6.32b, c).
As a summary for the interested reader, Table 6.1 lists the cyclic CO2 capture capacities of various CaO-based CO2 sorbents synthesized using a wide range of synthesis methods. Since the calcination and carbonation temperature and the partial pressure of CO2 are the parameters that influence the CO2 capture perfor-mance, most of this information is also provided in Table 6.1.
6.6 Summary and Outlook
In this chapter, we have provided an overview of the main aspects of using CaO for capturing CO2, with a particular focus on how the morphology of CaO is related to the rate and extent of the carbonation and calcination reactions. The effect of the presence of gas impurities on the performance of CaO-based sorbents was also addressed. In the second part of the chapter, we have reviewed recent
Fig. 6.32 SEM images of a the cycled newly developed CaO-based sorbent, b calcined limestone and c cycled limestone. Reproduced from Ref. [87] by permission of John Wiley & Sons Ltd

2156 CO2 Capture via Cyclic Calcination and Carbonation Reactions
Tabl
e 6.
1 S
umm
ary
of th
e C
O2
capt
ure
capa
citie
s an
d te
stin
g co
nditi
ons
of v
ario
us s
ynth
etic
CaO
-bas
ed C
O2
sorb
ents
Ref
eren
ceC
O2
sorb
ent
Rea
ctor
Car
bona
tion
cond
ition
sC
alci
natio
n co
nditi
ons
CO
2 up
take
(g
CO
2/g
sorb
ent)
(cy
cle
num
ber)
Lu
et a
l. [7
3]C
alci
um a
ceta
teT
GA
700
°C, 3
0 vo
l.% C
O2
in
He,
300
min
700
°C, 1
00 v
ol.%
He,
30
min
0.49
(27
)
Liu
et a
l. [8
9]C
alci
um g
luco
nate
TG
A65
0 °C
, 15
vol.%
CO
2 in
N
2, 3
0 m
in90
0 °C
, 100
vol
.% N
2,
10 m
in0.
66 (
9)C
omm
erci
al n
anos
ized
CaO
0.46
(9)
Cal
cium
citr
ate
0.45
(9)
Cal
cium
ace
tate
0.41
(9)
Gra
sa e
t al.
[75]
Cal
cium
ace
tate
TG
A65
0 °C
, 30
vol.%
CO
2 in
N
2, 5
min
950
°C, 1
00 v
ol.%
CO
2,0.
16 (
100)
Flor
in a
nd H
arri
s [7
2]C
omm
erci
al n
anos
ized
C
aCO
3
TG
A65
0, 1
5 vo
l.% C
O2
in N
2,
20 m
in85
0 °C
, 100
vol
.% N
2,
10 m
in0.
17 (
100)
Gup
ta a
nd F
an [
90]
CaC
O3
TG
A70
0 °C
, 100
vol
.% C
O2,
60
min
700
°C, 1
00 v
ol.%
, ~2
0 m
in>
0.71
(2)
Filit
z et
al.
[80]
CaO
:MgO
= 7
1:29
wt.%
TG
A75
0 °C
, 35
vol.%
CO
2 in
N
2, 2
0 m
in75
0 °C
, 100
vol
.% N
2,
10–2
0 m
in0.
51 (
15)
Kie
rzko
wsk
a et
al.
[91]
CaO
:Al 2
O3 =
81:
19 w
t.%T
GA
750
°C, 4
0 vo
l.% C
O2
in
N2,
20
min
750
°C, 1
00 v
ol.%
N2,
20
min
0.36
(30
)
Flor
in e
t al.
[76]
CaO
:Ca 1
2Al 1
4O33
= 8
5:15
wt.%
Flui
dize
d be
d65
0 °C
, 15
vol.%
CO
2 in
N
2, 1
0 m
in90
0 °C
, 15
vol.%
CO
2 in
N
2, 5
min
0.13
(15
)
TG
A65
0 °C
, 15
vol.%
CO
2 in
N
2 an
d H
e, 1
0 m
in90
0 °C
, 15
vol.%
CO
2 in
N
2 an
d H
e, 5
min
0.29
(15
)
Pacc
iani
et a
l. [7
9]C
aO:C
a 12A
l 14O
33 =
75:
25 w
t.%Fl
uidi
zed
bed
750
°C, 1
4 vo
l.% C
O2
in
N2,
500
s75
0 °C
, 100
vol
.% N
2,
500
s0.
26 (
20)
Liu
et a
l. [8
2]C
aO:M
gO =
75:
25 w
t.%T
GA
650
°C, 1
5 vo
l.% C
O2
in
N2,
30
min
900
°C, 1
00 v
ol.%
N2,
10
min
0.57
(24
)
Li e
t al.
[92]
CaO
:MgA
l 2O
4 =
68:
32
wt.%
TG
A65
0 °C
, 30
vol.%
CO
2,
10 v
ol.%
% H
2O,
50 v
ol.%
Ar,
10 v
ol.%
N
2, 6
0 m
in
850
°C, 2
.5 v
ol.%
H2O
, 2.
5 vo
l.% N
2, 9
5 vo
l.% A
r, 34
min
0.34
(65
)
(con
tinue
d)
216 M. Broda et al.
Tabl
e 6.
1 (
cont
inue
d)
Ref
eren
ceC
O2
sorb
ent
Rea
ctor
Car
bona
tion
cond
ition
sC
alci
natio
n co
nditi
ons
CO
2 up
take
(g
CO
2/g
sorb
ent)
(cy
cle
num
ber)
Hua
ng e
t al.
[85]
CaO
:SiO
2 =
66.
6:33
.3T
GA
700
°C, 1
00 v
ol.%
CO
2,
60 m
in91
0 °C
, 100
vol
.% N
2,
30 m
in0.
42 (
40)
Qin
et a
l. [8
4]C
aO: c
alci
um a
lum
inat
e ce
men
t = 7
5:25
TG
A65
0 °C
, 15
vol.%
CO
2 in
N
2, 3
0 m
in90
0 °C
, 100
vol
.% N
2,
10 m
in0.
36 (
70)
Mar
tava
ltzi a
nd
Lem
onid
ou [
93]
CaO
:Ca 1
2Al 1
4O33
= 7
5:25
w
t.%T
GA
690
°C, 1
5 vo
l.% C
O2
in
N2,
30
min
850
°C, 1
00 v
ol.%
N2,
10
min
0.27
(45
)
Man
ovic
and
Ant
hony
[9
4]C
aO: c
alci
um a
lum
inat
e ce
men
t = 9
0:10
wt.%
TG
A75
0 °C
, 15
vol.%
CO
2,
10 m
in,
750
°C, 1
00 %
N2,
10
min
,0.
37 (
30)
Lu
et a
l. [9
5]C
aO:Z
rO2 =
69:
31T
GA
700
°C, 3
0 vo
l.% C
O2
in
He,
300
min
700
°C, 1
00 v
ol.%
He,
30
min
0.24
(10
0)
Lu
et a
l. [9
6]N
anos
ized
CaC
O3
TG
A70
0 °C
, 30
vol.%
CO
2 in
H
e, 3
00 m
in70
0 °C
, 100
vol
.% H
e,
30 m
in0.
39 (
60)
Bro
da e
t al.
[74]
CaO
:Al 2
O3 =
90:
10 w
t.%T
GA
750
°C, 4
0 vo
l.% C
O2
in
N2,
20
min
750
°C, 1
00 v
ol.%
N2,
20
min
0.51
(30
)C
aO:A
l 2O
3 =
65:
35 w
t.%0.
47 (
10)
Bro
da e
t al.
[97]
CaO
:Al 2
O3 =
90:
10 w
t.%65
0 °C
, 20
vol.%
CO
2 in
N
2, 2
0 m
in90
0 °C
, 100
vol
.% C
O2,
10
min
0.28
(10
)
Flui
dize
d be
d75
0 °C
, 20
vol.%
CO
2,
6 m
in75
0 °C
, 100
vol
.% N
2,
~20
min
0.25
(30
)
Bro
da a
nd M
ülle
r [8
7]C
aO:A
l 2O
3 =
90:
10 w
t.%T
GA
750
°C, 4
0 vo
l.% C
O2
in
N2,
20
min
750
°C, 1
00 v
ol.%
N2,
20
min
0.56
(30
)
Luo
et a
l. [9
8]C
aO:A
l 2O
3 =
80:
20 w
t.%T
ube
furn
ace
650
°C, 1
5 vo
l.% C
O2
in
N2,
20
min
850
°C, 1
00 v
ol.%
N2,
10
min
0.43
(20
)
Sant
os e
t al.
[99]
CaO
TG
A70
0 °C
, 15
vol.%
CO
2 in
N
2, 5
0 m
in80
0 °C
, 100
vol
.% N
2,
13 m
in0.
24 (
70)

2176 CO2 Capture via Cyclic Calcination and Carbonation Reactions
advances in the development of novel CaO-based CO2 sorbents which substan-tially exceed the cyclic CO2 capture capacity of limestone.
Generally and despite the wealth of scientific studies concerning the application of CaO for capturing CO2, there is a striking lack of agreement on fundamental aspects of both the carbonation and calcination reaction. With regard to the calci-nation reaction, the major points of discrepancy are the effect of particle size and the “type” of the CaCO3-containing material on the rate of calcination. Various authors identified the rate-limiting step of the calcination reaction as either heat transfer, diffusion in pores, true kinetics or a combination of these. Furthermore, the morphology and the presence of impurities seem to affect critically the reac-tion rate; however, so far it has not been possible to quantify these effects in a sat-isfactory manner. Furthermore, the wide range of experimental equipment, particle size and operating temperatures employed make it extremely difficult to reconcile the results from different studies.
As for the carbonation reaction, there seems to be a general agreement among the scientific community on the reaction mechanism. The carbonation reaction is envisaged as proceeding through a two-step mechanism, whereby the reac-tion shifts from a fast, kinetically controlled stage to a slow stage where diffusion through the product layer becomes the rate-limiting step once the product layer reaches a certain thickness, i.e. ~50 nm. However, this theory has not yet been supported by direct measurements of the product layer formation in pores. In this respect, the recent study of Li et al. [69] provides new insights into the mecha-nism of the carbonation reaction by showing, for the first time, direct measure-ments of CaCO3 forming on the surface of a CaO single crystal using AFM. We believe that this approach could be successful in supporting the development of a theory that could reconcile the contradicting results found in the litera-ture. Additionally, there is substantial disagreement in very fundamental kinetic parameters, such as the activation energy and the order of reaction. For the latter, we refer the reader to the comprehensive study of Sun et al. [63], which provides reliable results for the order of the carbonation reaction over a wide range of par-tial pressures. Finally, the role that impurities such as SO2 and H2O play on the cyclic CO2 capture capacity of CaO must be taken into account carefully. Several studies agree that the presence of SO2 is detrimental for the CO2 capture capac-ity of CaO-based sorbent because it reacts irreversibly with CaO to CaSO4 at typical operating conditions and thereby depleting CaO available for carbonation. However, the effect of H2O on the CO2 capture capacity of CaO is still unclear with results ranging from having no effect, to either enhancing or decreasing the capture capacity. Thus, future efforts should be directed to reconcile these results, as they can affect substantially the design and integration of the CO2 capture unit in a power plant.
The main issue associated with the use of CaO, derived via the calcination of naturally occurring limestone, as a CO2 sorbent is its rapidly decreasing CO2 cap-ture capacity with cycle number. Thus, substantial efforts are currently directed to the development of novel, CaO-based materials with high and stable CO2 capture characteristics. This objective requires the materials to possess two main features:

218 M. Broda et al.
(1) a nanostructured morphology to avoid the slow, diffusion-limited reaction regime of the carbonation reaction, i.e. the build-up of a CaCO3 layer thicker than approximately 50 nm has to be avoided and (2) the presence of a high Tammann temperature supports to stabilize the morphology of the sorbents over repeated cycles. Indeed, freshly calcined limestone is a nanostructured, porous material comprised of roughly spherical grains of diameter ~100 nm. However, due to its intrinsic lack of a high Tammann temperature support, limestone losses rapidly its nanostructured morphology and in turn its CO2 uptake capacity.
So far many different techniques, such as sol-gel, mechanical mixing, wet-impregnation or co-precipitation have been applied to develop novel CaO-based CO2 sorbents. However, the new materials were often tested under very different carbonation and calcination conditions than those which would be employed in a practical CO2 capture system, thus making a proper comparison between the dif-ferent sorbents developed essentially impossible. Therefore, it is critical that in the future, newly developed CO2 sorbents are tested under identical, standardized con-ditions, i.e. calcination at 900 °C in a pure CO2 atmosphere.
Another aspect that deserves further attention is to understand better how a sup-port effectively stabilizes the CaO morphology and, thus, the CO2 uptake capacity over repeated cycles. As shown in the literature, the Tammann temperature of the support is not the only indicator of its ability to retard thermal sintering. Indeed, the CO2 sorbent that possesses the highest CO2 capture capacity reported so far was stabilized by the comparatively low Tammann temperature support mayenite. In addition, the very recent study of Filitz et al. [80] demonstrated that the level of mixing between CaO and the support is critical, with only mixing within the crystal lattice (through the formation of a Ca-rich dolomite) providing an excellent cyclic CO2 capture stability.
We would also like to stress the importance of additional requirements related to the physical properties such as the mechanical strength of potential CO2 sor-bents. The calcium looping process will most likely be operated in a circulating fluidized bed reactor set-up. This requires the development of attrition-resistant materials. However, the mechanical strength or attrition resistance of new devel-oped CO2 sorbents is rarely assessed and, thus, requires more attention if the materials are to be relevant for practical applications.
Acknowledgments We are grateful to the Swiss National Science Foundation (SNF) for partial financial support (Project: 200021_135457/1).
References
1. Tuinier MJ, Hamers HP, van Sint Annaland M (2011) Techno-economic evaluation of cryogenic CO2 capture: a comparison with absorption and membrane technology. Int J Greenhouse Gas Control 5:1559–1565
2. Aaron D, Tsouris C (2005) Separation of CO2 from flue gas: a review. Sep Sci Technol 40:321–348

2196 CO2 Capture via Cyclic Calcination and Carbonation Reactions
3. Müller CR, Pacciani R, Bohn CD, Scott SA, Dennis JS (2009) Investigation of the enhanced water gas shift reaction using natural and synthetic sorbents for the capture of CO2. Ind Eng Chem Res 48:10284–10291
4. Duan Y, Sorescu DC (2010) CO2 capture properties of alkaline earth metal oxides and hydroxides: a combined density functional theory and lattice phonon dynamics study. J Chem Phys 133:074508–074511
5. Kierzkowska AM, Pacciani R, Müller CR (2013) CaO-based CO2 sorbents: from fun-damentals to the development of new, highly effective materials. ChemSusChem 6:1130–1148
6. Barin I, Platzki G (1995) Thermochemical data of pure substances. VCH Verlagsgesellschaft, Weinheim, Germany
7. Shimizu T, Hirama T, Hosoda H, Kitano K, Inagaki M, Tejima K (1999) A twin fluid-bed reactor for removal of CO2 from combustion processes. Chem Eng Res Des 77:62–68
8. Maciejewski M, Reller A (1987) How unreliable are kinetic data of reversible solid-state decomposition processes? Thermochim Acta 110:145–152
9. Bhatia SK, Perlmutter DD (1983) Effect of the product layer on the kinetics of the CO2-lime reaction. AlChE J 29:79–86
10. Dedman AJ, Owen AJ (1962) Calcium cyanamide synthesis. Part 4—the reaction CaO + CO2 = CaCO3. Trans Faraday Soc 58:2027–2035
11. Choi S, Drese JH, Jones CW (2009) Adsorbent materials for carbon dioxide capture from large anthropogenic point sources. ChemSusChem 2:796–854
12. Barker R (1973) The reversibility of the reaction CaCO3 ⇄ CaO + CO2. J Appl Chem Biotechnol 23:733–742
13. Grasa GS, Abanades JC (2006) CO2 capture capacity of CaO in long series of carbonation/calcination cycles. Ind Eng Chem Res 45:8846–8851
14. Dennis JS, Pacciani R (2009) The rate and extent of uptake of CO2 by a synthetic, CaO-containing sorbent. Chem Eng Sci 64:2147–2157
15. Mess D, Sarofim AF, Longwell JP (1999) Product layer diffusion during the reaction of cal-cium oxide with carbon dioxide. Energy Fuels 13:999–1005
16. Alvarez D, Abanades JC (2005) Determination of the critical product layer thickness in the reaction of CaO with CO2. Ind Eng Chem Res 44:5608–5615
17. Alvarez D, Abanades JC (2005) Pore-size and shape effects on the recarbonation perfor-mance of calcium oxide submitted to repeated calcination/recarbonation cycles. Energy Fuels 19:270–278
18. Abanades JC, Alvarez D (2003) Conversion limits in the reaction of CO2 with lime. Energy Fuels 17:308–315
19. Lysikov AI, Salanov AN, Okunev AG (2007) Change of CO2 carrying capacity of CaO in isothermal recarbonation-decomposition cycles. Ind Eng Chem Res 46:4633–4638
20. Wang J, Anthony EJ (2005) On the decay behavior of the CO2 absorption capacity of CaO-based sorbents. Ind Eng Chem Res 44:627–629
21. Pacciani R, Torres J, Solsona P, Coe C, Quinn R, Hufton J, Golden T, Vega LF (2011) Influence of the concentration of CO2 and SO2 on the absorption of CO2 by a lithium ortho-silicate-based absorbent. Environ Sci Technol 45:7083–7088
22. Dennis JS, Hayhurst AN (1990) Mechanism of the sulphation of calcined limestone particles in combustion gases. Chem Eng Sci 45:1175–1187
23. Anthony EJ, Granatstein DL (2001) Sulfation phenomena in fluidized bed combustion sys-tems. Prog Energy Combust Sci 27:215–236
24. Anthony EJ, Bulewicz EM, Jia L (2007) Reactivation of limestone sorbents in FBC for SO2 capture. Prog Energy Combust Sci 33:171–210
25. Hartman M, Pata J, Coughlin RW (1978) Influence of porosity of calcium carbonates on their reactivity with sulfur dioxide. Ind Eng Chem Process Des Dev 17:411–419
26. Yrjas P, Iisa K, Hupa M (1995) Comparison of SO2 capture capacities of limestones and dolomites under pressure. Fuel 74:395–400

220 M. Broda et al.
27. Dennis JS, Hayhurst AN (1989) Alternative sorbents for flue-gas desulfurization, especially in fluidized-bed combustors. J Energy Inst 62:202–207
28. Dennis JS, Hayhurst AN (1986) A simplified analytical model for the rate of reaction of SO2 with limestone particles. Chem Eng Sci 41:25–36
29. Laursen K, Duo W, Grace JR, Lim J (2000) Sulfation and reactivation characteristics of nine limestones. Fuel 79:153–163
30. Wu Y, Sun P, Anthony EJ, Jia L, Grace J (2006) Reinvestigation of hydration/reactivation characteristics of two long-term sulphated limestones which previously showed uniformly sulphating behaviour. Fuel 85:2213–2219
31. Manovic V, Anthony EJ (2007) SO2 retention by reactivated CaO-based sorbent from multi-ple CO2 capture cycles. Environ Sci Technol 41:4435–4440
32. Montagnaro F, Nobili M, Salatino P, Telesca A, Valenti GL (2008) Hydration products of FBC wastes as SO2 sorbents: comparison between ettringite and calcium hydroxide. Fuel Process Technol 89:47–54
33. Manovic V, Anthony EJ, Lu DY (2008) Sulphation and carbonation properties of hydrated sorbents from a fluidized bed CO2 looping cycle reactor. Fuel 87:2923–2931
34. Jozewicz W, Kirchgessner DA (1989) Activation and reactivity of novel calcium-based sorb-ents for dry SO2 control in boilers. Powder Technol 58:221–229
35. Wang C, Shen X, Xu Y (2002) Investigation on sulfation of modified Ca-based sorbent. Fuel Process Technol 79:121–133
36. Wei S-H, Mahuli SK, Agnihotri R, Fan L-S (1997) High surface area calcium carbonate: pore structural properties and sulfation characteristics. Ind Eng Chem Res 36:2141–2148
37. Wolff EHP, Gerritsen AW, van Bleek CMD (1993) Multiple reactor testing of a synthetic sorbent for regenerative sulfur capture in fluidized bed combustion of coal. Can J Chem Eng 71:83–93
38. Gavaskar VS, Abbasian J (2006) Dry regenerable metal oxide sorbents for SO2 removal from flue gases. 1. Development and evaluation of copper oxide sorbents. Ind Eng Chem Res 45:5859–5869
39. Chen Q, Yoshida K, Yamamoto H, Uchida M, Sadakata M (2007) Investigation on the appli-cation of C12A7 in flue gas desulfurization at low–moderate temperature. Energy Fuels 21:3264–3269
40. Pacciani R, Muller CR, Davidson JF, Dennis JS, Hayhurst AN (2009) Performance of a novel synthetic Ca-based solid sorbent suitable for desulfurizing flue gases in a fluidized bed. Ind Eng Chem Res 48:7016–7024
41. Borgwardt RH (1989) Calcium oxide sintering in atmospheres containing water and carbon dioxide. Ind Eng Chem Res 28:493–500
42. Sun P, Grace JR, Lim CJ, Anthony EJ (2008) Investigation of attempts to improve cyclic CO2 capture by sorbent hydration and modification. Ind Eng Chem Res 47:2024–2032
43. Manovic V, Anthony EJ (2010) Carbonation of CaO-based sorbents enhanced by steam addi-tion. Ind Eng Chem Res 49:9105–9110
44. Donat F, Florin NH, Anthony EJ, Fennell PS (2012) Influence of high-temperature steam on the reactivity of CaO sorbent for CO2 capture. Environ Sci Technol 46:1262–1269
45. Arias B, Grasa G, Abanades JC, Manovic V, Anthony EJ (2012) The effect of steam on the fast carbonation reaction rates of CaO. Ind Eng Chem Res 51:2478–2482
46. Manovic V, Anthony EJ (2007) Steam reactivation of spent CaO-based sorbent for multiple CO2 capture cycles. Environ Sci Technol 41:1420–1425
47. Manovic V, Wu Y, He I, Anthony EJ (2012) Spray water reactivation/pelletization of spent CaO-based sorbent from calcium looping cycles. Environ Sci Technol 46:12720–12725
48. Borgwardt R (1985) Calcination kinetics and surface area of dispersed limestone particles. AIChE J 31:103–111
49. Beruto D, Searcy A (1976) Calcium oxides of high reactivity. Nature 263:221–222 50. Dennis JS, Hayhurst AN (1987) The effect of CO2 on the kinetics and extent of calcination of
limestone and dolomite particles in fluidised beds. Chem Eng Sci 42:2361–2372

2216 CO2 Capture via Cyclic Calcination and Carbonation Reactions
51. Silcox G, Kramlich J, Pershing D (1989) A mathematical model for the flash calcination of dispersed CaCO3 and Ca(OH)2 particles. Ind Eng Chem Res 28:155–160
52. Borgwardt RH (1989) Sintering of nascent calcium oxide. Chem Eng Sci 44:53–60 53. Satterfield CN, Feakes F (1959) Kinetics of the thermal decomposition of calcium carbonate.
AIChE J 5:115–122 54. Khinast J, Krammer G, Brunner C, Staudinger G (1996) Decomposition of limestone: the
influence of CO2 and particle size on the reaction rate. Chem Eng Sci 51:623–634 55. Hills AWD (1968) The mechanism of the thermal decomposition of calcium carbonate.
Chem Eng Sci 23:297–320 56. Garcia-Labiano F, Abad A, de Diego L, Gayan P (2002) Calcination of calcium-based sorb-
ents at pressure in a broad range of CO2 concentrations. Chem Eng Sci 57:2381–2393 57. Bhatia SK, Perlmutter DD (1980) A random pore model for fluid–solid reactions: I.
Isothermal, kinetic control. AIChE J 26:379–386 58. Bhatia SK, Perlmutter DD (1981) A random pore model for fluid–solid reactions: II.
Diffusion and transport effects. AIChE J 27:247–254 59. Fuller E, Schettler P, Giddings J (1966) New method for prediction of binary gas-phase diffu-
sion coefficients. Ind Eng Chem 58:19–27 60. Nitsch W (1962) Über die Druckabhängigkeit der CaCO3-bildung aus dem Oxyd. Z
Elektrochem 66:8–9 61. Bhatia SK, Perlmutter DD (1983) Effect of the product layer on the kinetics of the CO2-lime
reaction. AIChE J 29:79–86 62. Kyaw K, Matsuda H, Hasatani M (1996) Applicability of carbonation/decarbonation reactions
to high-temperature thermal energy and temperature upgrading. J Chem Eng Jpn 29:119–125 63. Sun P, Grace JR, Lim CJ, Anthony EJ (2008) Determination of intrinsic rate constants of the
CaO–CO2 reaction. Chem Eng Sci 63:47–56 64. Oakeson WG, Cutler IB (1979) Effect of CO2 pressure on the reaction with CaO. J Am
Ceram Soc 62:556–558 65. Sun P, Grace JR, Lim CJ, Anthony EJ (2008) A discrete-pore-size-distribution-based gas–
solid model and its application to the reaction. Chem Eng Sci 63:57–70 66. Stendardo S, Foscolo PU (2009) Carbon dioxide capture with dolomite: a model for gas–
solid reaction within the grains of a particulate sorbent. Chem Eng Sci 64:2343–2352 67. Khoshandam B, Kumar R, Allahgholi L (2010) Mathematical modeling of CO2 removal
using carbonation with CaO: the grain model. Korean J Chem Eng 27:766–776 68. Li Z, Sun H, Cai N (2012) Rate equation theory for the carbonation reaction of CaO with
CO2. Energy Fuels 26:4607–4616 69. Li Z-S, Fang F, Tang X-Y, Cai N-S (2012) Effect of temperature on the carbonation reaction
of CaO with CO2. Energy Fuels 26:2473–2482 70. Symonds RT, Lu DY, Macchi A, Hughes RW, Anthony EJ (2009) CO2 capture from syngas
via cyclic carbonation/calcination for a naturally occurring limestone: modelling and bench-scale testing. Chem Eng Sci 64:3536–3543
71. Barker R (1974) The reactivity of calcium oxide towards carbon dioxide and its use for energy storage. J Appl Chem Biotechnol 24:221–227
72. Florin NH, Harris AT (2009) Reactivity of CaO derived from nano-sized CaCO3 particles through multiple CO2 capture-and-release cycles. Chem Eng Sci 64:187–191
73. Lu H, Reddy EP, Smirniotis PG (2006) Calcium oxide based sorbents for capture of carbon dioxide at high temperatures. J Ind Eng Chem 45:3944–3949
74. Broda M, Kierzkowska AM, Müller CR (2012) Application of the sol–gel technique to develop synthetic calcium-based sorbents with excellent carbon dioxide capture characteris-tics. ChemSusChem 5:411–418
75. Grasa G, González B, Alonso M, Abanades JC (2007) Comparison of CaO-based synthetic CO2 sorbents under realistic calcination conditions. Energy Fuels 21:3560–3562
76. Florin NH, Blamey J, Fennell PS (2010) Synthetic CaO-based sorbent for CO2 capture from large-point sources. Energy Fuels 24:4598–4604

222 M. Broda et al.
77. Li ZS, Cai NS, Huang YY (2006) Effect of preparation temperature on cyclic CO2 capture and multiple carbonation-calcination cycles for a new Ca-based CO2 sorbent. Ind Eng Chem Res 45:1911–1917
78. Mercury JMR, Aza AHD, Turrillas X, Pena PJ (2004) The synthesis mechanism of Ca3Al2O6 from soft mechanochemically activated precursors studied by time-resolved neutron diffrac-tion up to 1000 °C. Solid State Chem 177:866–874
79. Pacciani R, Müller CR, Davidson JF, Dennis JS, Hayhurst AN (2008) Synthetic Ca-based solid sorbents suitable for capturing CO2 in a fluidized bed. Can J Chem Eng 86:356–366
80. Filitz R, Kierzkowska AM, Broda M, Müller CR (2012) Highly efficient CO2 sorbents: development of synthetic, calcium-rich dolomites. Environ Sci Technol 46:559–565
81. Li Z, Cai N, Huang Y, Han H (2005) Synthesis, experimental studies, and analysis of a new calcium-based carbon dioxide absorbent. Energy Fuels 19:1447–1452
82. Liu W, Feng B, Wu Y, Wang G, Barry J, Diniz da Costa JC (2010) Synthesis of sintering-resistant sorbents for CO2 capture. Environ Sci Technol 44:3093–3097
83. Carreon MA, Guliants VV (2005) Ordered meso- and macroporous binary and mixed metal oxides. Eur J Inorg Chem 1:27–43
84. Qin C, Liu W, An H, Yin J, Feng B (2012) Fabrication of CaO-based sorbents for CO2 cap-ture by a mixing method. Environ Sci Technol 46:1932–1939
85. Huang C-H, Chang K-P, Yu C-T, Chiang P-C, Wang C-F (2010) Development of high-tem-perature CO2 sorbents made of CaO-based mesoporous silica. Chem Eng J 161:129–135
86. Vinu A, Srinivasu P, Miyahara M, Ariga K (2006) Preparation and catalytic performances of ultralarge-pore TiSBA-15 mesoporous molecular sieves with very high Ti content. J Phys Chem B 110:801–806
87. Broda M, Müller CR (2012) Synthesis of highly efficient, Ca-based, Al2O3 stabilized, carbon gel templated CO2 sorbents. Adv Mater 24:3059–3064
88. Pekala RW (1989) Organic aerogels from the polycondensation of resorcinol with formalde-hyde. J Mater Sci 24:3221–3227
89. Liu W, Low NWL, Feng B, Wang G, Diniz da Costa C (2010) Calcium precursors for the production of CaO sorbents for multicycle CO2 capture. Environ Sci Technol 44:841–847
90. Gupta H, Fan L-S (2002) Carbonation-calcination cycle using high reactivity calcium oxide for carbon dioxide separation from flue gas. Ind Eng Chem Res 41:4035–4042
91. Kierzkowska AM, Poulikakos LV, Broda M, Müller CR (2013) Synthesis of calcium-based, Al2O3-stabilized sorbents for CO2 capture using a co-precipitation technique. Int J Greenhouse Gas Control 15:48–54
92. Li L, King DL, Nie Z, Li XS, Howard C (2010) MgAI2O4 Spinel-stabilized calcium oxide absorbents with improved durability for high-temperature CO2 capture. Energy Fuels 24:3698–3703
93. Martavaltzi CS, Lemonidou AA (2008) Development of new CaO based sorbent materials for CO2 removal at high temperature. Microporous Mesoporous Mater 110:119–127
94. Manovic V, Anthony EJ (2009) CaO-based pellets supported by calcium aluminate cements for high-temperature CO2 capture. Environ Sci Technol 43:7117–7122
95. Lu H, Khan A, Pratsinis SE, Smirniotis PG (2009) Flame-made durable doped-CaO nanosor-bents for CO2 capture. Energy Fuels 23:1093–1100
96. Hong Lu, Smirniotis PG, Ernst FO, Pratsinis SE (2009) Nanostructured Ca-based sorbents with high CO2 uptake efficiency. Chem Eng Sci 64:1936–1943
97. Broda M, Kierzkowska AM, Müller CR (2012) The influence of the calcination and carbona-tion conditions on the CO2 uptake of synthetic Ca-based CO2 sorbents. Environ Sci Technol 46:10849–10856
98. Luo C, Zheng Y, Ding N, Wu QL, Zheng CG (2011) SGCS-made ultrafine CaO/Al2O3 sorb-ent for cyclic CO2 capture. Chin Chem Lett 22:615–618
99. Santos ET, Alfonsín C, Chambel AJS, Fernandes A, Soares Dias AP, Pinheiro CIC, Ribeiro MF (2012) Investigation of a stable synthetic sol–gel CaO sorbent for CO2 capture. Fuel 94:624–628

223
Abstract Inorganic membranes play an important role in the development of eco-nomical processes for pre-combustion and/or post-combustion capture of carbon dioxide (CO2) at high temperatures. Mesoporous silica, due to its chemical and mechanical properties, is considered as a candidate for the capture of CO2 at high temperatures. Bare silica membranes exhibit Knudsen diffusion behavior for most gases but also exhibit the contribution of surface diffusion for heavier or inter-acting gases such as CO2 and CH4. The CO2/N2 selectivity of mesoporous silica membranes can be enhanced by surface modification using aminosilanes such as APTS (3-aminopropyl-triethoxy silane). The important aspect of such modified membranes is that they can be operated at high temperatures typically encountered in post-combustion gas streams (flue gas). For modified silica membranes, mixed gas separation factors as high as 10 for CO2 over N2 were observed. The transport mechanism in such membranes is the reaction of CO2 with the amine groups (in aminosilanes) to form a carbamate species and subsequent surface “hopping” of carbon dioxide. Under ambient conditions, CO2 is strongly bounded to the amine groups and thus greatly inhibits the surface diffusion of CO2; however, as the tem-perature increases, the CO2 permeance increases and selective transport of CO2 is observed. Thus, in surface-modified facilitated transport membranes, economical CO2 separation is achieved through the combination of the chemical reaction of CO2 associated with amine absorption along with the simplicity and low operating costs of membrane processes.
Chapter 7Functionalized Inorganic Membranes for High-Temperature CO2/N2 Separation
Mayur Ostwal and J. Douglas Way
A.-H. Lu and S. Dai (eds.), Porous Materials for Carbon Dioxide Capture, Green Chemistry and Sustainable Technology, DOI: 10.1007/978-3-642-54646-4_7, © Springer-Verlag Berlin Heidelberg 2014
M. Ostwal (*) Department of Mechanical Engineering, University of Colorado, Boulder, CO 80309, USAe-mail: [email protected]
J. Douglas Way Chemical and Biological Engineering Department, Colorado School of Mines, Golden, CO 80401, USA

224 M. Ostwal and J. Douglas Way
7.1 Introduction
Fossil fuel combustion produces carbon dioxide [CO2] and there is growing sci-entific evidence that rising atmospheric CO2 levels have and will continue to contribute unfavorably to climate change. Approximately 80 % of global energy demand comes from the combustion of fossil fuels and is expected to increase until 2020 [1]. Thus, a number of ideas and strategies are being explored to effectively capture, store, and sequester CO2. According to a report from McKinsey and company [2], membranes were identified as having the poten-tial for breakthrough technologies to reduce the cost of carbon capture as mem-brane technology is energy efficient, mechanically simple, easy to scale-up, and requires a smaller footprint.
CO2 capture from coal and natural gas fired power plants is important if the atmospheric CO2 levels are to be kept within accepted limits. Currently uti-lized commercial technology for CO2 separation from natural or synthesis gas is amine solvent-based absorption. But this process has its own drawbacks such as amine loss, handling of corrosive liquids, and removal of SO2 when applied to post-combustion CO2 capture [3]. Plus amine absorption process is not fiscally viable as the cost associated with it is around 80 % of the total CO2 seques-tration cost [4]. Thus, efforts are being made to develop materials and methods that can significantly reduce the capture and storage costs along with efficient performance.
Of the three important technologies currently being pursued for CO2 cap-ture, namely liquid absorption, solid adsorption [5], and membrane separations [6], membrane-based separation is promising since it is more energy efficient, mechanically simple and easy to scale-up with a smaller footprint. Polymeric, inorganic (silica, zeolite etc.), mixed matrix, and facilitated transport membranes are currently being investigated for this application [7].
Polymeric membranes have shown promise for CO2 capture but suffer from issues such as plasticization, thermal degradation or polymer decomposition, and loss of selectivity or permeability over time or lower selectivity at high tem-peratures [6].
Inorganic and facilitated transport membranes have recently attracted atten-tion [8–11] for CO2 separation as they have the ability to withstand high tem-peratures as well as corrosive environments encountered in post-combustion streams. Amine functionalized facilitated transport membranes [10, 12, 13] have shown better CO2 separation characteristics than the conventional solution-dif-fusion (polymeric) membranes. The separation characteristics along with their ability to perform under corrosive environment and high temperature make these materials promising for CO2 separation from flue gas or natural gas. Therefore, in these membranes, the chemical reaction of CO2 associated with amine absorption along with the simplicity and low operating costs of membrane pro-cesses, by the surface modification of porous inorganic membranes with organic amines is utilized.

225
This chapter describes the surface modification of silica membranes using aminosilanes in order to synthesize membranes for CO2 separation. Mesoporous silica has large (4–5 nm) and uniform pores (and high surface area) which can be surface modified to introduce a large number of adsorption sites [CO2-phillic sites] for CO2 and achieve higher separation of CO2 from gas mixtures by reactive or facilitated transport mechanism.
Luebke et al. [14] reported improved separation performance for CO2/He separation using modified γ-alumina supports with alkyltricholosilanes. Numerous other studies [11, 15–21] have also used surface modification with hydrocarbon silanes to enhance the gas separation properties of the substrate/support.
Amines have long been used as reactive liquid solvents in CO2 absorption columns [3, 22]. The goal of the study presented in this chapter is to synthesize surface-modified inorganic membranes using an amine compound, 3-aminopro-pyltriethoxysilane (APTS), for CO2/N2 separation.
The rest of this chapter is organized as follows. In Sect. 7.2, we describe the synthesis of surface-modified membranes. We then describe the characterization techniques used and the permeation experiments as well as the methodology of the ab initio quantum simulations using density functional theory (DFT). The char-acterization and permeation results are then presented and discussed. Finally, we present and discuss the results from DFT calculations.
7.2 Experimental Section
7.2.1 Membrane Preparation
Mesoporous symmetric Vycor tubes (Corning, Inc.) were used as supports for the synthesis of APTS-modified membranes. Specifically, Vycor tubes with pore size of 4.7 nm, internal diameter of 8.3 mm, and wall thickness of about 0.95 mm were used. The supports were cut into 1.7-cm-long sections before modification.
Following this, the supports were cleaned by boiling in a 30 % aqueous H2O2 solution for 30 min, followed by boiling in distilled water for 30 min. They were then dried overnight at room temperature and subsequently placed under low vac-uum for 1 h to remove any residual water from the surface.
The surface modification of the support by APTS was performed under Helium atmosphere so as to negate and avoid any reaction of the silane molecules with atmospheric water. The modification was achieved by filling the Vycor tube with 1 ml of APTS solution and then heating it to about 383 K in an oven for 2–3 h under He atmosphere or until all the silane was evaporated. The reaction between the silane vapors with the surface hydroxyl groups formed brush-like structures or strands on the support pore walls as shown in Fig. 7.1.
7 Functionalized Inorganic Membranes for High-Temperature CO2/N2 Separation

226 M. Ostwal and J. Douglas Way
Modified membranes were cleaned, by rinsing them with 1 liter of toluene to remove any unreacted silane. They were then soaked in toluene for 24 h and sub-sequently dried at room temperature under He atmosphere for 24 h.
7.2.2 Membrane Characterization
The synthesized membranes were characterized using nuclear magnetic resonance (NMR) spectroscopy and pore size analysis by gas adsorption.
To perform the NMR spectroscopy, a clean, unmodified Vycor tube and a modi-fied Vycor tube were crushed to a fine powder. Firstly, 29Si NMR was performed on unmodified and modified Vycor samples to ensure that the desired surface modification was achieved. 13C NMR was then carried out on the modified tube before and after exposure to CO2 to show the formation of the carbamate species, as this was the hypothesized mechanism for CO2 transport through these mem-branes as shown in the reaction mechanism below (Eq. 7.1). The procedure for the NMR experiments reported by Singh et al. [11] was used in the characterization. The chemical shift in the NMR signal for Si or C atoms depends on their envi-ronment, and thus, the peaks can distinguish various Si and C moieties present in the membrane material and hence provide useful information about the membrane microstructure.
Fig. 7.1 Schematic of the brush-like structure in the silica pore after APTS modification (not to scale). Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier

227
To ascertain the differences in physical properties such as pore sizes, pore vol-umes, and surface areas between unmodified and modified samples, pore size analysis and measurements were performed. The pore size analysis also serves to confirm the anchoring of the aminosilanes on the silica substrate. All the meas-urements were taken using N2 adsorption/desorption isotherms at 77 K using a Micromeritics ASAP 2020 surface area and porosity analyzer.
7.2.3 Transport Measurements
In order to perform the transport measurements and be able to mount the mem-brane in the test module, the ends of the modified support were sealed with dense quartz pieces using an epoxy adhesive as shown in Fig. 7.2a. Figure 7.2b shows the permeation module in which the membrane is mounted. The membrane along with the attached quartz pieces is mounted in the module using Ultra-torr fittings and Viton o-rings. The feed gas flows through the inside bore of the membrane while the permeate flow is perpendicular to the feed flow. Sweep gas [helium] was used in all the measurements. The flow rates of the retentate and permeate streams were measured using a bubble flow meter, and the composition was obtained through a gas chromatograph [SRI 8600C]. Based on the flow rate and composi-tion, the permeate flux and permeance was calculated. Details of the experimental procedure and transport measurement setup are given in detail elsewhere [23].
(7.1)CO2 + 2R − NH2 ⇋ R − NH+
3+ R − NHCOO
−
Fig. 7.2 a Membrane assembly. Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier, and b membrane module
7 Functionalized Inorganic Membranes for High-Temperature CO2/N2 Separation

228 M. Ostwal and J. Douglas Way
Transport properties (permeance and separation factor) of the modified inorganic membranes were measured through steady state pure and mixed gas experiments. In pure gas experiments, the feed gas was pressurized and permeate flow rate was measured using a bubble flow meter, which in turn was used to cal-culate the permeance using Eq. 7.2.
where Pi is the permeance of component i (in cm3/cm2·s·cm Hg), fluxi is the flux of component i (cm3/cm2·s), and Δpi is the differential pressure (cm Hg).
For mixed gas experiments, the flow rates and composition of the feed gas were controlled using electronic mass flow controllers. The total feed pressure was kept steady at 227.5 cm Hg (303 kPa) while the permeate pressure and temperature were maintained at ambient conditions (62 cm Hg and room temperature). Permeate and retentate streams, with helium as the sweep gas (on permeate side), were analyzed for gas compositions using a gas chromatograph (SRI Instruments, model # 8610 C). The total steady state permeate flow rate and the gas composition of the permeate stream were used to calculate the analyte gas flux. From the flux, and the pressure differential across the membrane, the gas permeances and separation factor (ratio of permeance) can be calculated.
Figure 7.3 and Eq. 7.1 show the proposed mechanism of CO2 transport using surface-tethered amines [24, 25]. In this mechanism (shown below), CO2 reacts with one amine group and forms a carbamate species (NHCOO−) [26, 27]. After the formation of carbamate species, the combination of thermally induced undu-lations of the amine strands along with the partial pressure driving force results in the CO2 being transferred from one amine strand to another. At the same time, reduction in pore size of the support due to the surface modification with APTS results in blocking of the flow of N2 due to its larger kinetic diameter. The hand-shaking diffusion of CO2 together with the steric blocking of N2, thus achieves a significant degree of CO2/N2 separation.
(7.2)Pi =fluxi
�pi
Fig. 7.3 Mechanism of carbamate formation [24]. Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier

2297 Functionalized Inorganic Membranes for High-Temperature CO2/N2 Separation
7.2.4 Density Function Theory (DFT) Calculations
DFT calculations were performed in order to:
1. Elucidate the proposed mechanism of CO2 transport in APTS-modified membranes as described above
2. To compute the CO2 diffusivity through the membrane3. To calculate CO2 binding energy on an amine strand.
Three most likely configurations of amine strands in the membrane were used, which will be described in detail in Sect. 7.3. Real space, numerical atomic orbital, DFT computer code DMOL [28] was used to quantitatively and qualitatively measure the above-mentioned properties. DMOL was used because of its computational efficiency and ability to provide accurate estimates of activation barriers to molecular hopping.
7.3 Results and Discussion
7.3.1 NMR Results
Amino-silane-modified porous silica has been extensively characterized using NMR spectroscopy [29–32]. Only qualitative results are presented here, as the aim of the characterization was to check whether the support surface has been modi-fied using amino-silanes as well as to prove the formation of carbamate species, and thus, no quantitative analysis was done on the spectroscopy results. Results obtained through 29Si NMR for unmodified and modified supports are shown in Fig. 7.4. Expected peak assignments and the structure of different silicon moieties present on the APTS-modified silica surface are shown in Fig. 7.5.
Fig. 7.4 29Si NMR spectra for silica support before and after modification with APTS (peak assignments are shown in Fig. 7.5). Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier
-200 -150 -100 -50 0 50 100
T3
Q4
Q3
Modified Support
ppm
SupportQ2
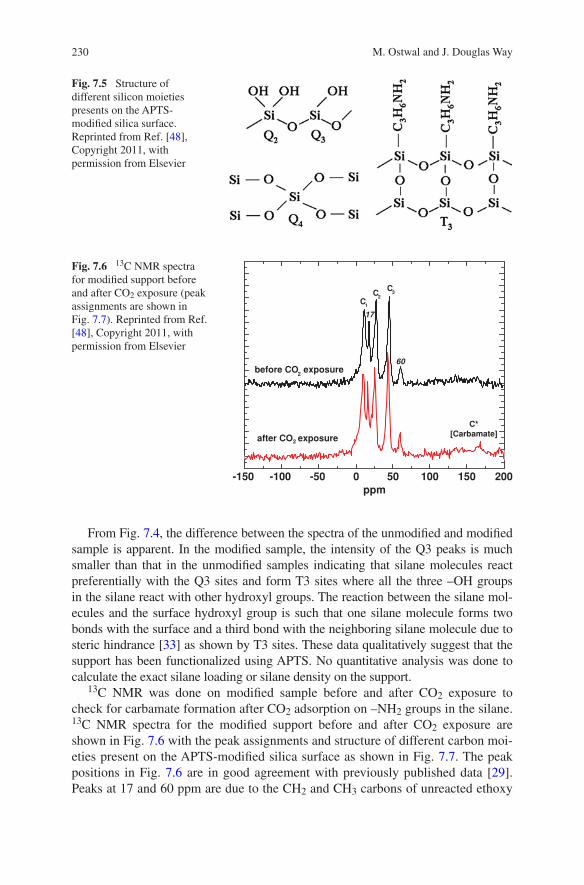
230 M. Ostwal and J. Douglas Way
From Fig. 7.4, the difference between the spectra of the unmodified and modified sample is apparent. In the modified sample, the intensity of the Q3 peaks is much smaller than that in the unmodified samples indicating that silane molecules react preferentially with the Q3 sites and form T3 sites where all the three –OH groups in the silane react with other hydroxyl groups. The reaction between the silane mol-ecules and the surface hydroxyl group is such that one silane molecule forms two bonds with the surface and a third bond with the neighboring silane molecule due to steric hindrance [33] as shown by T3 sites. These data qualitatively suggest that the support has been functionalized using APTS. No quantitative analysis was done to calculate the exact silane loading or silane density on the support.
13C NMR was done on modified sample before and after CO2 exposure to check for carbamate formation after CO2 adsorption on –NH2 groups in the silane. 13C NMR spectra for the modified support before and after CO2 exposure are shown in Fig. 7.6 with the peak assignments and structure of different carbon moi-eties present on the APTS-modified silica surface as shown in Fig. 7.7. The peak positions in Fig. 7.6 are in good agreement with previously published data [29]. Peaks at 17 and 60 ppm are due to the CH2 and CH3 carbons of unreacted ethoxy
Fig. 7.5 Structure of different silicon moieties presents on the APTS-modified silica surface. Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier
Fig. 7.6 13C NMR spectra for modified support before and after CO2 exposure (peak assignments are shown in Fig. 7.7). Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier
-150 -100 -50 0 50 100 150 200
17
C3C2
after CO exposure2
ppm
before CO exposure2
C*[Carbamate]
C1
60
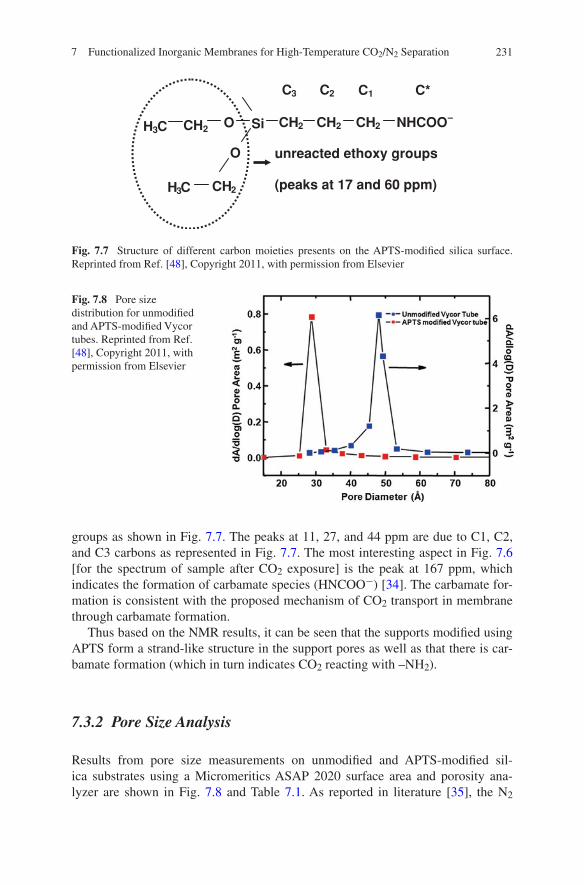
2317 Functionalized Inorganic Membranes for High-Temperature CO2/N2 Separation
groups as shown in Fig. 7.7. The peaks at 11, 27, and 44 ppm are due to C1, C2, and C3 carbons as represented in Fig. 7.7. The most interesting aspect in Fig. 7.6 [for the spectrum of sample after CO2 exposure] is the peak at 167 ppm, which indicates the formation of carbamate species (HNCOO−) [34]. The carbamate for-mation is consistent with the proposed mechanism of CO2 transport in membrane through carbamate formation.
Thus based on the NMR results, it can be seen that the supports modified using APTS form a strand-like structure in the support pores as well as that there is car-bamate formation (which in turn indicates CO2 reacting with –NH2).
7.3.2 Pore Size Analysis
Results from pore size measurements on unmodified and APTS-modified sil-ica substrates using a Micromeritics ASAP 2020 surface area and porosity ana-lyzer are shown in Fig. 7.8 and Table 7.1. As reported in literature [35], the N2
NHCOO–Si CH2 CH2 CH2O
O
CH2H3C
CH2H3C
unreacted ethoxy groups
(peaks at 17 and 60 ppm)
C*C3 C2 C1
Fig. 7.7 Structure of different carbon moieties presents on the APTS-modified silica surface. Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier
Fig. 7.8 Pore size distribution for unmodified and APTS-modified Vycor tubes. Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier

232 M. Ostwal and J. Douglas Way
adsorption isotherm for mesoporous materials follows the Type IV adsorption iso-therm with a H1 hysteresis loop. Pore size analysis results from both the modi-fied and unmodified samples follow this behavior (N2 adsorption isotherms are not shown here). Surface areas were calculated using the Brunauer–Emmett–Teller (BET) method while Barret–Joyner–Halenda (BJH) models were used to calcu-late the pore size distributions derived from the desorption branches of isotherms. From Table 7.1 and Fig. 7.8, it can be seen that the pore diameter of modified substrate is 29.7 Å, which is smaller than that of unmodified substrate, which is 47.1 Å. Thus, the size of the substrate is reduced by almost 17.4 Å after surface modification using APTS. Reduction in the surface area and pore volumes from unmodified to modified samples is also significant. Thus, the results from pore size analysis of the samples show that the aminosilanes are anchored to the pore walls of the substrate. These results are comparable to those reported in literature [24, 35, 36] for APTS-modified silica substrates.
7.3.3 Transport Measurement Results
A plot of pure gas permeances for a variety of penetrants such as N2, Ne, He, H2, CO2, CH4, C2H6 versus the reciprocal of the square root of their respective molecular weights for the unmodified Vycor tubes is shown in Fig. 7.9 [11, 37]. The relationship between permeance and inverse of the square root of molecu-lar weight is linear for non-interacting gases [N2, Ne, He, H2], indicating the gas permeation is governed by a Knudsen diffusion mechanism. For interacting gases [CO2, CH4, C2H6], the permeances are higher than the ideal values predicted by Knudsen diffusion theory, indicating an additional contribution due to surface dif-fusion arising from interaction of the gases with the silica membranes. For these gases, therefore, the permeance is governed by Knudsen diffusion along with con-tribution from surface flow [11, 37]. From Fig. 7.9, the ideal CO2/N2 separation factor for Vycor tubes is 1.12, which is slightly higher than the Knudsen ideal sep-aration factor of 0.79, due to the quadrupolar nature of CO2 and thus its interaction with the silica surface.
Thus, it can be concluded that, for interacting gases, the selectivity is achieved through a larger contribution from surface diffusion than from Knudsen diffusion. As an example, the ideal selectivity for butane over N2 of almost 10 was achieved for Vycor tubes (pore size ~5 nm) as reported by Singh et al. [11]. The high
Table 7.1 Pore structure parameters for modified mesoporous Vycor substrate with aminosilane
Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier
BET surface area Pore volume Pore diameter
m2/g cm3/g Å
Vycor tube 126 0.21 47.1APTS-modified Vycor tube 36.5 0.05 29.7
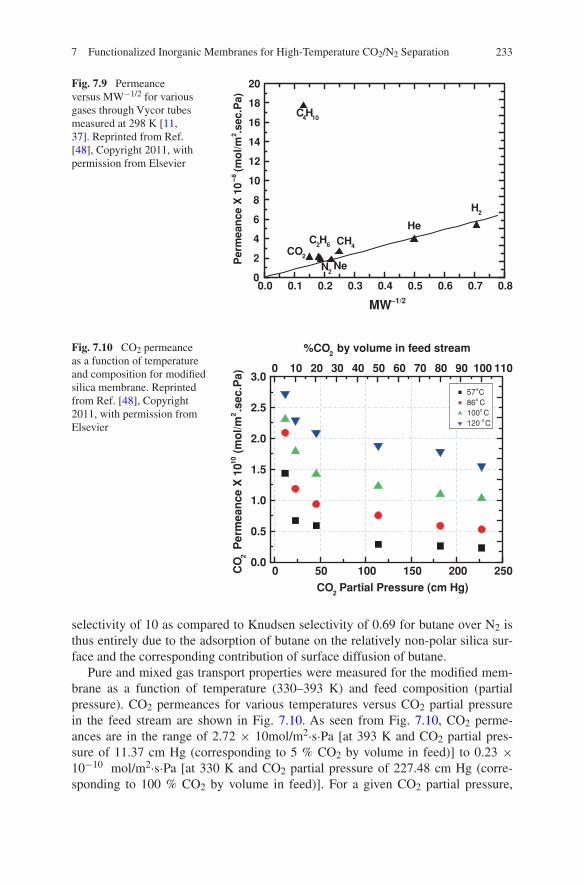
2337 Functionalized Inorganic Membranes for High-Temperature CO2/N2 Separation
selectivity of 10 as compared to Knudsen selectivity of 0.69 for butane over N2 is thus entirely due to the adsorption of butane on the relatively non-polar silica sur-face and the corresponding contribution of surface diffusion of butane.
Pure and mixed gas transport properties were measured for the modified mem-brane as a function of temperature (330–393 K) and feed composition (partial pressure). CO2 permeances for various temperatures versus CO2 partial pressure in the feed stream are shown in Fig. 7.10. As seen from Fig. 7.10, CO2 perme-ances are in the range of 2.72 × 10mol/m2·s·Pa [at 393 K and CO2 partial pres-sure of 11.37 cm Hg (corresponding to 5 % CO2 by volume in feed)] to 0.23 × 10−10 mol/m2·s·Pa [at 330 K and CO2 partial pressure of 227.48 cm Hg (corre-sponding to 100 % CO2 by volume in feed)]. For a given CO2 partial pressure,
Fig. 7.9 Permeance versus MW−1/2 for various gases through Vycor tubes measured at 298 K [11, 37]. Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.80
2
4
6
8
10
12
14
16
18
20
C4H10
CO2
C2H6
N2Ne
CH4
HeP
erm
ean
ce X
10
–8 (
mo
l/m2 .s
ec.P
a)
–1/2MW
H2
Fig. 7.10 CO2 permeance as a function of temperature and composition for modified silica membrane. Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier
0 50 100 150 200 2500.0
0.5
1.0
1.5
2.0
2.5
3.00 10 20 30 40 50 60 70 80 90 100 110
%CO2 by volume in feed stream
CO
2 P
erm
ean
ce X
1010
(m
ol/m
2.s
ec.P
a)
CO2 Partial Pressure (cm Hg)
57o C86o C100o C120 o C
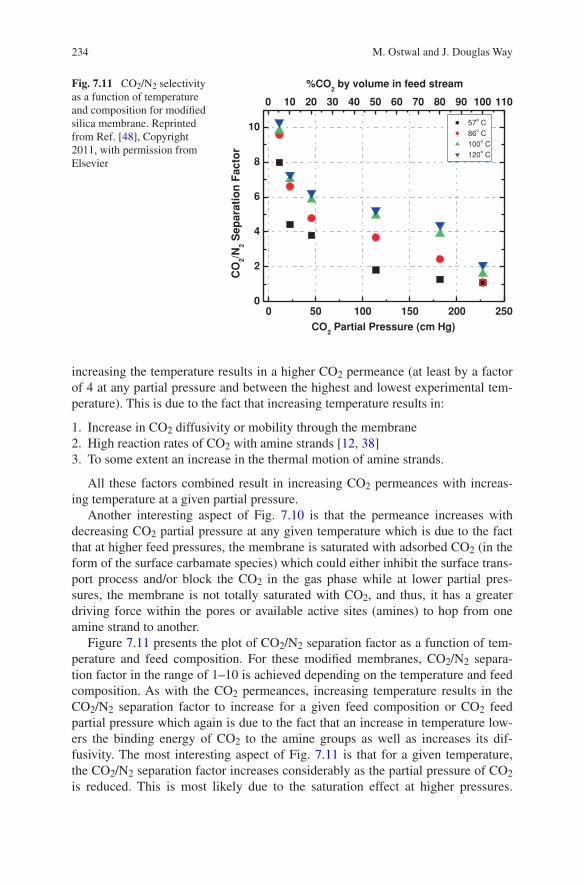
234 M. Ostwal and J. Douglas Way
increasing the temperature results in a higher CO2 permeance (at least by a factor of 4 at any partial pressure and between the highest and lowest experimental tem-perature). This is due to the fact that increasing temperature results in:
1. Increase in CO2 diffusivity or mobility through the membrane2. High reaction rates of CO2 with amine strands [12, 38]3. To some extent an increase in the thermal motion of amine strands.
All these factors combined result in increasing CO2 permeances with increas-ing temperature at a given partial pressure.
Another interesting aspect of Fig. 7.10 is that the permeance increases with decreasing CO2 partial pressure at any given temperature which is due to the fact that at higher feed pressures, the membrane is saturated with adsorbed CO2 (in the form of the surface carbamate species) which could either inhibit the surface trans-port process and/or block the CO2 in the gas phase while at lower partial pres-sures, the membrane is not totally saturated with CO2, and thus, it has a greater driving force within the pores or available active sites (amines) to hop from one amine strand to another.
Figure 7.11 presents the plot of CO2/N2 separation factor as a function of tem-perature and feed composition. For these modified membranes, CO2/N2 separa-tion factor in the range of 1–10 is achieved depending on the temperature and feed composition. As with the CO2 permeances, increasing temperature results in the CO2/N2 separation factor to increase for a given feed composition or CO2 feed partial pressure which again is due to the fact that an increase in temperature low-ers the binding energy of CO2 to the amine groups as well as increases its dif-fusivity. The most interesting aspect of Fig. 7.11 is that for a given temperature, the CO2/N2 separation factor increases considerably as the partial pressure of CO2 is reduced. This is most likely due to the saturation effect at higher pressures.
Fig. 7.11 CO2/N2 selectivity as a function of temperature and composition for modified silica membrane. Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier
0 50 100 150 200 2500
2
4
6
8
10
0 10 20 30 40 50 60 70 80 90 100 110
%CO2 by volume in feed stream
57o C 86o C 100o C 120o C
CO
2/N2 S
epar
atio
n F
acto
r
CO2 Partial Pressure (cm Hg)

2357 Functionalized Inorganic Membranes for High-Temperature CO2/N2 Separation
At higher CO2 pressures, the membrane material or the amine groups are saturated with adsorbed CO2 in the form of carbamate species, and thus, they hamper the movement (by essentially closing down the pores) for gas phase CO2 resulting in flow mostly due to Knudsen mechanism with little or no contribution from surface diffusion. On the other hand, at low partial pressures, however, CO2 has greater number of available sites to hop through as well as the driving force is high and hence higher CO2/N2 separation factors.
A recent study by Sakamoto et al. [10] on aminosilane [APTS] modified MCM-48 supports reported a CO2/N2 separation factor of ~800 at 373 K for a feed mixture of 20/80 CO2/N2. For the same conditions of 373 K and 20/80 CO2/N2, we observe a CO2/N2 selectivity of 5.85. This is much lower than the selectiv-ity reported by Sakamoto et al. [10]. One key difference between the two mem-branes was the starting pore size of the supports. Sakamoto et al. [10] prepared the APTS-modified membrane on a MCM-48 support whose starting pore size was ~2 nm, while our support had a pore size of ~4.7 nm. As seen from our pore size measurements, the pore size of support decreases from 4.7 to 2.9 nm after APTS modification, resulting in almost 1.7 nm change in the pore diameter. Thus, if the starting support pore size is 2 nm, APTS modification would effectively close down all pores. This closing down of pores in addition to the adsorbed CO2 would effectively block the N2 gas, resulting in higher CO2/N2 selectivity. The CO2 permeance for the APTS-modified Vycor tubes at 373 K and a feed mixture of 20/80 CO2/N2 is 1.42 × 10−10 mol/m2·s·Pa which is about seven times lower than that reported by Sakamoto et al. [10] (1.00 × 10−9 mol/m2.s.Pa), whereas the difference in the selectivity is more than two orders of magnitude. The dif-ference between the CO2/N2 selectivity can thus be attributed to the increased N2 permeance in APTS-modified Vycor tubes compared to that by Sakamoto et al. [10]. As explained above, the differences in the N2 flow could be due to the pore size differences in the mesoporous substrates. Another plausible explanation for the differences in selectivity could be due to the differences in the amine loading or density [39], but we do not have any quantitative data to support this argument. It may also be due to the differences in ambient humidity in Japan (humid) and Colorado, USA (dry). Sakamoto et al. conducted their work in Japan, where due to relatively high ambient humidity, water vapor may get adsorbed in the small pores which in turn would enhance the CO2 reaction with amines while at the same time contributing to N2 blocking. In Colorado, due to dry weather, this effect may not be as pronounced.
A CO2/N2 selectivity of around 1,800 for cross-linked polyvinyl alcohol poly-meric membranes containing amine functionality in the form of 2-aminoisobutyric acid–potassium salt (mobile amine carrier) and poly (allylamine) (fixed carrier), at 110 °C was reported by Zou et al. [13]. The main differences between this study and that reported by Zou et al. [13] are as follows: (1) they used two types of amine carriers while suggesting that the mobile carriers contributed more to the CO2 flux than the fixed carriers as opposed to APTS-modified membranes where the carriers were fixed [attached to the pore walls], (2) in their study, water was used both on the feed and permeate side, enhancing the reaction rate of CO2 with

236 M. Ostwal and J. Douglas Way
the amines as well as mobility of the carriers, (3) feed gas composition used in their study was 20/40/40 CO2/N2/H2. Combined effects of the above-mentioned key differences may have resulted in enhanced CO2/N2 selectivities.
Research by Bai et al. [12] reported CO2/N2 selectivity of 249 at the same experimental conditions and feed gas composition as Zou et al. [13], namely 100 °C and feed pressure of 30 psi for polymeric membranes using sulfonated polybenzimidazole (SPBI)–ethylenediamine (EDA) copolymer containing 30 % SPBI, 50 % polyethylenimine (PEI) (fixed amine carrier), and 20 % 2-aminoisobu-tyric acid–potassium salt (mobile amine carrier). In this case again, they had mobile and fixed amine carriers.
Kumar et al. [40] reported interesting data for membranes where MCM-48 supports (pore size ~2.4 nm) were modified with polyethyleneimine (PEI). They reported an N2/CO2 selectivity of 1.31 (@293 K) in the absence of water, 17.6 (@293 K) in the presence of water, and 1.35 (@363 K) in the presence of water for a feed mixture of 80/20 N2/CO2 and feed pressure of 20 psi (103.4 cm Hg). In the presence of water, the size of the diffusing unit (CO2) increased due to the clustering of water molecules, which in turn reduced the CO2 diffusivity at room temperature, and hence, the PEI-MCM 48 membranes were highly N2 selec-tive in the presence of water. This is opposite to what we and others [10, 12, 13] observe (CO2 selective membrane), and it may be due to the fact that in our case, the amine groups are readily accessible to the CO2 molecules (since they form a brush-like structure) for reactive separation whereas the PEI approach, in contrast, may be dominated by a solution-diffusion mechanism rather than reactive or facil-itated transport.
Reported data here as well as elsewhere [10, 12, 13] involving amine groups as carriers indicate that the membrane properties (i.e., selectivity) peak at 100–110 °C and hence can be suitable candidates for post-combustion CO2 separation and capture.
As mentioned previously, our hypothesis was that higher selectivities would be obtained if the pore size of the mesoporous support was significantly smaller prior to silane modification, similar to that reported by Sakamoto et al. [10]. To demonstrate the feasibility of this pore size hypothesis, Vycor tube substrates were modified, by depositing titanium oxide in the support pores, by plasma-assisted atomic layer deposition (ALD) [41]. The ALD of TiO2 was performed in a hot-wall tubular reactor using titanium tetraisopropoxide [TTIP or Ti(OCH(CH3)2)4] in conjunction with an O2–Ar plasma. The tube was mounted to a heated substrate stage maintained at 50 °C. TTIP was delivered from a bubbler using N2 as the car-rier gas. An O2–Ar plasma was generated by flowing a 1:1 O2–Ar mixture through inductively coupled plasma (ICP) source RF-powered at 100 W. The same gas mixture was used as purge between precursor exposures cycles since O2 does not react with the precursor at the temperature and pressure of the experiment. Ar was added to O2 to ensure that the plasma could be turned on and off reproducibly over several 100 s of times. The ALD cycles consisted of a 1-s TTIP exposure followed by a 120-s O2–Ar purge. The O2–Ar plasma was turned on for 15 s followed by a 60 s purge step. A total of 25 ALD cycles were carried out. As shown in Fig. 7.12,
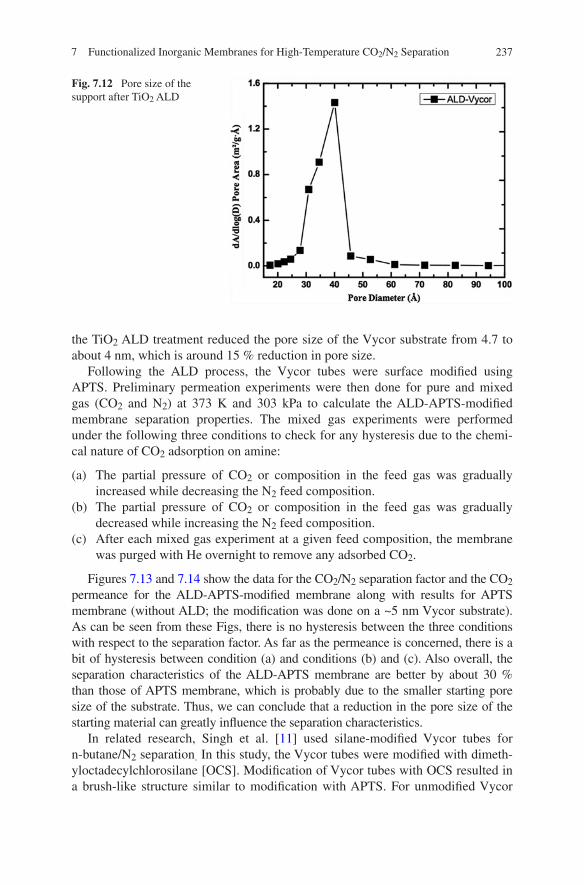
2377 Functionalized Inorganic Membranes for High-Temperature CO2/N2 Separation
the TiO2 ALD treatment reduced the pore size of the Vycor substrate from 4.7 to about 4 nm, which is around 15 % reduction in pore size.
Following the ALD process, the Vycor tubes were surface modified using APTS. Preliminary permeation experiments were then done for pure and mixed gas (CO2 and N2) at 373 K and 303 kPa to calculate the ALD-APTS-modified membrane separation properties. The mixed gas experiments were performed under the following three conditions to check for any hysteresis due to the chemi-cal nature of CO2 adsorption on amine:
(a) The partial pressure of CO2 or composition in the feed gas was gradually increased while decreasing the N2 feed composition.
(b) The partial pressure of CO2 or composition in the feed gas was gradually decreased while increasing the N2 feed composition.
(c) After each mixed gas experiment at a given feed composition, the membrane was purged with He overnight to remove any adsorbed CO2.
Figures 7.13 and 7.14 show the data for the CO2/N2 separation factor and the CO2 permeance for the ALD-APTS-modified membrane along with results for APTS membrane (without ALD; the modification was done on a ~5 nm Vycor substrate). As can be seen from these Figs, there is no hysteresis between the three conditions with respect to the separation factor. As far as the permeance is concerned, there is a bit of hysteresis between condition (a) and conditions (b) and (c). Also overall, the separation characteristics of the ALD-APTS membrane are better by about 30 % than those of APTS membrane, which is probably due to the smaller starting pore size of the substrate. Thus, we can conclude that a reduction in the pore size of the starting material can greatly influence the separation characteristics.
In related research, Singh et al. [11] used silane-modified Vycor tubes for n-butane/N2 separation. In this study, the Vycor tubes were modified with dimeth-yloctadecylchlorosilane [OCS]. Modification of Vycor tubes with OCS resulted in a brush-like structure similar to modification with APTS. For unmodified Vycor
Fig. 7.12 Pore size of the support after TiO2 ALD

238 M. Ostwal and J. Douglas Way
tubes, the ideal n-C4H10/N2 separation factor reported was 9.67 whereas that for OCS-modified membrane was 13.70. For a 50/50 mixture of n-C4H10 and N2, the mixed gas selectivity was reported to be 56.1, indicating blocking of N2 by adsorbed n-C4H10. This enhanced selectivity was attributed to the increased con-tribution of surface flow of n-C4H10 in addition to the blocking of N2. A similar observation with APTS-modified membranes was observed in the study presented here, where the CO2/N2 selectivity [pure gas] increases from 2.12 [@393 K] to 10.3 for mixed gas [5 % CO2 and 95 % N2], true for all temperature ranges.
Kajiwara et al. [42] deposited platinum on porous alumina tubes with pore diameter of 200 nm, using chemical vapor deposition to study H2 permeation. As reported, the deposition of platinum on alumina supports reduced the pore size of
Fig. 7.13 CO2 permeance as a function of composition for APTS-ALD-modified silica membrane
Fig. 7.14 CO2/N2 selectivity as a function of composition for APTS-ALD-modified silica membrane

2397 Functionalized Inorganic Membranes for High-Temperature CO2/N2 Separation
the alumina tubes to 1.6 nm. All the gases, He, N2, CH4, O2, and H2, they investi-gated for permeation showed Knudsen behavior except for H2 which had a much higher permeance than the Knudsen value. The H2 permeance increase was attrib-uted to the interaction of H2 with the platinum on the membrane surface, resulting in a large contribution of surface flow of atomic hydrogen in addition to Knudsen diffusion. We also see similar behavior for CO2 in APTS-modified membrane, where CO2 interacts with the amino groups resulting in greater contribution from surface flow in addition to some Knudsen diffusion.
7.3.4 DFT Results
Three probable and most likely model systems depicting APTS on silica surface were developed, and DFT calculations were performed on these systems to com-pute the CO2 diffusivity and binding energy. DFT was also used to understand the proposed hopping CO2 transfer mechanism in the membrane. A norm-conserving, spin-unrestricted, semi-core pseudopotential approach was employed with electron exchange and correlation accounted for through the Perdew-Wang generalized gradient approximation [43].
To start the DFT calculations, ground state configuration of each structure was obtained through geometrically optimizing it. To depict the nature of aminosi-lane anchoring on the support pore walls, the silicon atoms in all structures were anchored by fixing their atomic positions throughout the calculations and they were also saturated with hydrogen. The rest of the atoms were allowed to move.
Figure 7.15 shows the pathway toward arriving at a relaxed geometry for a given model system of APTS on anchored Si atoms along with a CO2 molecule. More particularly, the Fig shows a sequence of relaxation steps in a geometry opti-mization simulation that elucidates carbamate formation when CO2 interacts with an amine group. Initially, a hydrogen atom is placed on one amine strand, while a CO2 molecule is placed on the neighboring strand. As the structure relaxes, the proton [hydrogen atom] is pulled from its strand toward the strand with CO2 mol-ecule and bonds with the CO2 forming carbamate. All subsequent DFT computa-tions were therefore performed using this relaxed end state as a starting structure for subsequent CO2 hopping.
Figure 7.16 shows the reactant and product structures arrived at after geometry optimization for computing the binding energy associated with carbamate for-mation. The computed binding energy (reaction energy), which is the difference between the total energies of reactant and product states, for docking one CO2 molecule to an amine was calculated to be 15.5 kcal/mol. The computed binding energy of 15.5 kcal/mol is comparable with the reported experimental values for the heat of adsorption of CO2 on APTS-modified adsorbents (14.33–21.5 kcal/mol) [39]. Thus, based on the very good agreement between the computed and experimental values for binding energy, the proposed structural model used in DFT is reasonable.

240 M. Ostwal and J. Douglas Way
The second step in DFT computations was to compute the activation energy for a CO2 molecule to hop from one amine strand, in the form of a carbamate, to another. This was achieved and computed using transition state theory (TST). In particular, a linear synchronous transit/quadratic synchronous transit (LST/QST) algorithm was adopted [44]. LST/QST method uses the reactant and product con-figurations to generate an estimation for the reaction pathway with minimum sad-dle point energy and then iteratively refines this pathway until no further reduction in the saddle point energy is found.
Fig. 7.15 DFT geometry optimization sequence showing proton transfer between strands in association with carbamate formation. Reprinted from Ref. [48], Copyright 2011, with permis-sion from Elsevier
Fig. 7.16 Reactant and product structures for docking one CO2 molecule on an amine strand. Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier

2417 Functionalized Inorganic Membranes for High-Temperature CO2/N2 Separation
All three model configurations described previously were subjected to TST analysis, and Figs. 7.13, 7.14, 7.15 show the initial and final states used, to study the transfer of CO2 and compute activation energy. The distance between the N atoms that are involved in CO2 transfer is kept constant around 3–4 Å for all three configurations. Each model configuration reflects a possible means of CO2 hopping between strands. In each case, a strand with CO2 undulates, along with its neighbors, and a geometry is eventually achieved which favors CO2 trans-fer. Configuration # 1 (Fig. 7.17) shows the mechanism of CO2 transfer between strands associated with the same silicon anchor. Using this configuration, the computed barrier or activation energy was 17.7 kcal/mol, which could be due to the strand curvature that must be generated for transfer of CO2. The second con-figuration (Fig. 7.18) where the APTS strands are anchored on independent Si anchors gives an activation energy of 10.5 kcal/mol. This is much lower than the barrier obtained using configuration # 1 and is most likely due to the fact that in the second configuration, since the strands are on independent anchors, it mini-mizes the bending required due to their arrangement. In this case, CO2 hops from one strand to another in a zigzag fashion which minimizes the curvature required of each strand.
The hopping barrier is further reduced using configuration # 3 (Fig. 7.19). In this configuration, additional strands are introduced so as to reduce the depth of local minima in the potential energy. Thus, configuration # 3 [double inverted strands] is same as configuration # 2 but with additional strands. The associated hopping barrier is only 7.2 kcal/mol for this configuration, which is much lower than those computed using configuration # 1 and # 2. The dense packing and potential for inter-anchor hopping also make this the most physically reasonable configuration.
The activation energy calculated from the experimental permeances, based on pure CO2 experiments, and using Arrhenius equation, is 8.0 kcal/mol, very close to the one computed for configuration #3. The comparable values between the experimental and computational value for configuration # 3 demonstrate that con-figuration #3 is the most likely means by which CO2 is transferred.
CO2 diffusivity can be calculated from the estimated computational activation energy, using the following equation [45]:
D =nvl2
2αexp
[
−Ea
RT
]
,
Fig. 7.17 Configuration #1 for CO2 transfer between adjacent amine strands (sideways). Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier

242 M. Ostwal and J. Douglas Way
where D is the diffusivity (m2/s), n is the number of jump directions available, v is the vibrational frequency/mode (s−1) (computed by diagonalizing the associated is the dimensionality of the system (equal to 2 for surface Hessian matrix), diffusion), Ea is the activation energy (kcal/mol), T is temperature, and R is the gas constant. The CO2 diffusivity calculated from the above equation ranged from 4.92 × 10−11 m2/s (298 K) to 5.78 × 10−10 m2/s (373 K). For comparison, Xu et al. [46] reported CO2 diffusivity in aqueous 2-amino-2-methyl-1-propanol (AMP) solutions to be 0.78 × 10−9 m2/s (298 K) and 4.43 × 10−9 m2/s (348 K). Zhang et al. [47] reported it to be 1.04 × 10−9 m2/s (298 K) in diethanolamine (DEA) solutions. Our computed dif-fusivities, 4.92 × 10−11 m2/s (298 K) to 2.86 × 10−10 m2/s (348 K), are lower, by an order magnitude, than those reported by Xu et al. and Zhang et al. The discrepancy
Fig. 7.18 Configuration #2 for CO2 transfer between adjacent amine strands (cross fashion). Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier
Fig. 7.19 Configuration #3 for CO2 transfer between adjacent amine strands (cross fashion with two additional strands). Reprinted from Ref. [48], Copyright 2011, with permission from Elsevier

2437 Functionalized Inorganic Membranes for High-Temperature CO2/N2 Separation
could be due to the fact that their measurements were taken in aqueous solutions where water promotes and enhances CO2 reaction with the amine groups.
7.4 Conclusions
Mesoporous silica supports were functionalized using aminosilanes to fabricate fixed-carrier membranes for CO2 transport. Comprehensive experimental as well as theoretical analysis was done to elucidate the mechanism by which CO2 dif-fuses in these functionalized silica membranes. Vycor glass supports were mod-ified using APTS, in order to enhance its CO2/N2 selectivity through facilitated transport mechanism. In pure gas experiments, the maximum CO2/N2 selectivity achieved was 2 at 393 K. For mixed gas experiments, CO2/N2 selectivity as high as 10 was achieved at 393 K and 5/95 CO2/N2 composition. The membrane’s sep-aration characteristics were better with respect to selectivity at high temperatures and lower CO2 partial pressures, suggesting a facilitated or reactive separation mechanism. Anchoring of the aminosilane chains on the pore walls of the support was proved using pore size measurements. NMR spectroscopy data showed that the silica supports were modified and functionalized using APTS. It also proved the hypothesis of carbamate species formation when CO2 reacts with an amine group. When the pore size of the silica supports were reduced by 15 % using ALD process, the separation properties of the membrane went up by almost 30 %, showing that the pore size of the substrate plays an important role in these types of membranes. The binding energy of CO2, computed using DFT, was consistent with the range of experimental values in the literature. Based on the activation energy for CO2 hopping, it was shown that configuration #3 (Fig. 7.19) was the most likely mechanism by which CO2 transfers in these types of membranes. The computed diffusivities were within an order of magnitude with those reported in the literature for a similar setting but in aqueous solutions.
Acknowledgments The authors gratefully acknowledge financial support from the Department of Energy Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division under Grant DE-FG03-93ER14363. We also acknowledge the use computing resources provided through the Renewable Energy MRSEC program (NSF Grant No. DMR-0820518) and the Golden Energy Computing Organization (NSF Grant No. CNS-0722415) at the Colorado School of Mines.
The authors also like to acknowledge Prof. Sumit Agarwal, Dr. Vikrant Rai, Dr. Rajinder Singh, Dr. Steve Dec, and Dr. Mark Lusk for their valuable and important contributions in this research study.
References
1. Anonymous (2006) Key world energy statistics. International Energy Agency 2. Naucler T, Campbell W, Ruijs J (2008) Carbon capture and storage: assessing the economics.
McKinsey and Company, New York

244 M. Ostwal and J. Douglas Way
3. Rochelle GT (2009) Amine scrubbing for CO2 capture. Science 325(5948):1652–1654 4. IPCC (2005) Special report on carbon dioxide capture and storage. Cambridge University
Press, Cambridge 5. Choi S, Drese JH, Jones CW (2009) Adsorbent materials for carbon dioxide capture from
large anthropogenic point sources. Chemsuschem 2(9):796–854 6. Zhang Y et al (2013) Current status and development of membranes for CO2/CH4 separation:
a review. Int J Greenhouse Gas Control 12:84–107 7. Merkel TC et al (2010) Power plant post-combustion carbon dioxide capture: an opportunity
for membranes. J Membr Sci 359(1–2):126–139 8. Javaid A (2005) Membranes for solubility-based gas separation applications. Chem Eng J
112(1–3):219–226 9. Park DH et al (2003) Separation of organic/water mixtures with silylated MCM-48 silica
membranes. Microporous Mesoporous Mater 66(1):69–76 10. Sakamoto Y et al (2007) Preparation and CO2 separation properties of amine-modified
mesoporous silica membranes. Microporous Mesoporous Mater 101(1–2):303–311 11. Singh RP, Way JD, Dec SF (2005) Silane modified inorganic membranes: effects of silane
surface structure. J Membr Sci 259(1–2):34–46 12. Bai H, Ho WSW (2009) New carbon dioxide-selective membranes based on sulfonated
polybenzimidazole (SPBI) copolymer matrix for fuel cell applications. Ind Eng Chem Res 48(5):2344–2354
13. Zou J, Ho WSW (2006) CO2-selective polymeric membranes containing amines in crosslinked poly(vinyl alcohol). J Membr Sci 286(1–2):310–321
14. Luebke D, Myers C, Pennline H (2006) Hybrid membranes for selective carbon dioxide sep-aration from fuel gas. Energy Fuels 20(5):1906–1913
15. Javaid A, Ford DM (2003) Solubility-based gas separation with oligomer-modified inorganic membranes—part II. Mixed gas permeation of 5 nm alumina membranes modified with octa-decyltrichlorosilane. J Membr Sci 215(1–2):157–168
16. Javaid A et al (2001) Solubility-based gas separation with oligomer-modified inorganic mem-branes. J Membr Sci 187(1–2):141–150
17. Javaid A, Krapchetov DA, Ford DM (2005) Solubility-based gas separation with oligomer-modified inorganic membranes—part III. Effects of synthesis conditions. J Membr Sci 246(2):181–191
18. McCarley KC, Way JD (2001) Development of a model surface flow membrane by mod-ification of porous gamma-alumina with octadecyltrichlorosilane. Sep Purif Technol 25(1–3):195–210
19. Picard C et al (2001) Grafting of ceramic membranes by fluorinated silanes: hydrophobic features. Sep Purif Technol 25(1–3):65–69
20. Singh RP et al (2007) Dual-surface-modified reverse-selective membranes. Ind Eng Chem Res 46(22):7246–7252
21. Singh RP, Way JD, McCarley KC (2004) Development of a model surface flow membrane by modification of porous vycor glass with a fluorosilane. Ind Eng Chem Res 43(12):3033–3040
22. Kohl AL, Riesenfeld FC (1979) Gas purification. Gulf Publishing Company, Houston, Texas, pp 28–90
23. Singh RP (2005) Surface modified inorganic membranes: effects of silane function-ality and surface structure on gas and vapor separations. Colorado School of Mines, Golden
24. Hiyoshi N, Yogo K, Yashima T (2005) Adsorption characteristics of carbon dioxide on organ-ically functionalized SBA-15. Microporous Mesoporous Mater 84(1–3):357–365
25. Cussler EL, Aris R, Bhown A (1989) On the limits of facilitated diffusion. J Membr Sci 43(2–3):149–164
26. Leal O et al (1995) Reversible adsorption of carbon dioxide on amine surface-bonded silica gel. Inorg Chim Acta 240(1–2):183–189
27. Rocchia M et al (2003) Sensing CO2 in a chemically modified porous silicon film. Physica Status Solidi a-Appl Mater Sci 197(2):365–369

2457 Functionalized Inorganic Membranes for High-Temperature CO2/N2 Separation
28. Delley B (2000) From molecules to solids with the DMol(3) approach. J Chem Phys 113(18):7756–7764
29. Caravajal GS et al (1988) Structural characterization of (3-Aminopropyl)Triethoxysilane-modified silicas by Si-29 and C-13 nuclear magnetic-resonance. Anal Chem 60(17):1776–1786
30. Cheng JL, Fone M, Ellsworth MW (1996) Solid state NMR study on the conformation and mobility of n-octadecyl chains in a silane coupling agent attached to the surface of colloidal silica. Solid State Nucl Magn Reson 7(2):135–140
31. Sindorf DW, Maciel GE (1981) Si-29 Cp-Mas nmr-studies of methylchlorosilane reactions on silica-gel. J Am Chem Soc 103(14):4263–4265
32. Sindorf DW, Maciel GE (1983) Solid-state nmr-studies of the reactions of silica sur-faces with polyfunctional chloromethylsilanes and ethoxymethylsilanes. J Am Chem Soc 105(12):3767–3776
33. Fadeev AY, McCarthy TJ (2000) Self-assembly is not the only reaction possible between alkyltrichlorosilanes and surfaces: monomolecular and oligomeric covalently attached layers of dichloro- and trichloroalkylsilanes on silicon. Langmuir 16(18):7268–7274
34. Mani F, Peruzzini M, Stoppioni P (2006) CO2 absorption by aqueous NH3 solutions: spe-ciation of ammonium carbamate, bicarbonate and carbonate by a C-13 NMR. Green Chem 8(11):995–1000
35. Luan ZH et al (2005) Preparation and characterization of (3-aminopropyl)triethoxysilane-modified mesoporous SBA-15 silica molecular sieves. Microporous Mesoporous Mater 83(1–3):150–158
36. Qu RJ et al (2008) Chemical modification of silica-gel with hydroxyl- or amino-terminated polyamine for adsorption of Au(III). Appl Surf Sci 255(5):3361–3370
37. Lee D, Oyama ST (2002) Gas permeation characteristics of a hydrogen selective supported silica membrane. J Membr Sci 210(2):291–306
38. Tee YH, Zou J, Ho WSW (2006) CO2-selective membranes containing dimethylglycine mobile carriers and polyethylenimine fixed carrier. J Chin Inst Chem Eng, 37(1):37–47
39. Chaffee AL et al (2007) CO2 capture by adsorption: materials and process development. Int J Greenhouse Gas Control 1(1):11–18
40. Kumar P et al (2008) Polyethyleneimine-modified MCM-48 membranes: effect of water vapor and feed concentration on N2/CO2 selectivity. Ind Eng Chem Res 47(1):201–208
41. Rai VR, Agarwal S (2009) Surface reaction mechanisms during plasma-assisted atomic layer deposition of titanium dioxide. J Phys Chem C 113(30):12962–12965
42. Kajiwara M, Uemiya S, Kojima T (1999) Stability and hydrogen permeation behavior of supported platinum membranes in presence of hydrogen sulfide. Int J Hydrogen Energy 24(9):839–844
43. Perdew JP, Wang Y (1992) Accurate and simple analytic representation of the electron-gas correlation-energy. Phys Rev B 45(23):13244–13249
44. Halgren TA, Lipscomb WN (1977) The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem Phys Lett 49(2):225
45. Liu CL et al (1991) EAM study of surface self-diffusion of single adatoms of fcc metals Ni, Cu, Al, Ag, Au, Pd, and Pt. Surf Sci 253(1–3):334
46. Xu S, Otto FD, Mather AE (1991) Physical-properties of aqueous amp solutions. J Chem Eng Data 36(1):71–75
47. Zhang H-Y et al (2006) Modeling and experimental study of CO2 absorption in a hollow fiber membrane contactor. J Membr Sci 279(1–2):301
48. Ostwal M et al (2011) 3-Aminopropyltriethoxysilane functionalized inorganic membranes for high temperature CO2/N2 separation. J Membr Sci 369(1–2):139–147