gtp-binding proteins of the rho/rac family: regulation, effectors and functions in vivo
TRANSCRIPT

GTP-binding proteins of theRho/Rac family: regulation,effectors and functions in vivoXose R. Bustelo,* Vincent Sauzeau, and Inmaculada M. Berenjeno
SummaryRho/Rac proteins constitute a subgroup of the Rassuperfamily of GTP hydrolases. Although originallyimplicated in the control of cytoskeletal events, it iscurrently known that these GTPases coordinate diversecellular functions, including cell polarity, vesiculartrafficking, the cell cycle and transcriptomal dynamics.In this review, we will provide an overview on therecent advances in this field regarding the mechanismof regulation and signaling, and the roles in vivo ofthis important GTPase family. BioEssays 29:356–370,2007. � 2007 Wiley Periodicals, Inc.
Introduction
The isolation of rhoA,(1) the first member of the Rho/Rac family
ever identified, was achieved by Richard Axel’s group in
1985 during the search for ras-related genes in Aplysia.(1) The
subsequent use of conventional cloning techniques and the
more-recent characterization of genomes revealed that
the original gene is not alone, having numerous family
counterparts in other species including, among many others,
S. cerevisiae (7 genes), A. taliana (11 genes), C. elegans
(9 genes), D. melanogaster (9 genes) and H. sapiens
(23 genes). In humans, these twenty-three different loci can
generate at least twenty-six different proteins due to alter-
native splicing events. In accordance with their homology at
the amino acid sequence level, these proteins are classified
into six subfamilies: Rho, Rac, Cdc42, Rnd, RhoBTB and
RhoT/Miro (Fig. 1). RhoBTB and RhoT proteins are also
referred to as ‘‘atypical’’ Rho/Rac GTPases because they are
very different from the other GTPase subfamilies according to
structural, regulatory and functional criteria.
Like the majority of Ras superfamily proteins, most Rho/
Rac GTPases behave as ‘‘molecular switches’’ that fluctuate
between inactive and active states, two conformations that
depend on the binding of either GDP or GTP to the GTPases,
respectively (Fig. 2). Two types of regulatory proteins control
this cycling: GEFs and GAPs (Fig. 2). GEFs promote the
exchange of GDP for GTP molecules, thereby producing the
activation of these proteins during signal transduction. GAPs
promote the hydrolysis of the bound GTP molecules, thus
allowing the transfer of the GTPase back to the inactive state at
the end of the stimulation cycle. In the GTP-bound state, these
GTPases bind to a large collection of effector molecules that,
Centro de Investigacion del Cancer and Instituto de Biologıa Molecular
y Celular del Cancer (IBMCC), CSIC-University of Salamanca,
Salamanca, Spain.
Funding agencies: XRB’s work is supported by grants from the US
National Cancer Institute (5R01-CA73735-10), the Spanish Ministry of
Education and Science (SAF2003-00028), the Castilla-Leon Autono-
mous Government (SA053A05), and the Red Tematica de Investiga-
cion Cooperativa en Cancer (RD06/0020/0001, Spanish Ministry of
Health). VS was supported by an EMBO long term postdoctoral
fellowship. IMB was partially supported by a Spanish Ministry of
Education and Science FPU fellowship (FP2000-6489). The Centro de
Investigacion del Cancer is supported by endowments from the CSIC,
University of Salamanca, Castilla-Leon Autonomous Government, the
Red Tematica de Investigacion Cooperativa de Centros de Cancer
(C03/10, Spanish Ministry of Health), and the Foundation for Cancer
Research of Salamanca.
*Correspondence to: Xose R. Bustelo, Centro de Investigacion del
Cancer and Instituto de Biologıa Molecular y Celular del Cancer
(IBMCC). CSIC-University of Salamanca, Campus Unamuno, E-37007
Salamanca, Spain. E-mail: [email protected]
DOI 10.1002/bies.20558
Published online in Wiley InterScience (www.interscience.wiley.com).
356 BioEssays 29.4 BioEssays 29:356–370, � 2007 Wiley Periodicals, Inc.
Abbreviations: Akt, v-akt murine thymoma viral oncogene homolog;
BAFF, B-cell activating factor; Baiap, brain-specific angiogenesis
inhibitor 1-associated protein; Bcl, B-cell chronic lymphocytic leuke-
mia/lymphoma 2 (Bcl2) like; Bcl2L, Bcl2 like; Borg, binder of rho
GTPases; Cdc42Ep, Cdc42 effector protein; C3orf10, chromosome 3
open reading frame 10; Cyfip, cytoplasmic fragile X mental retardation 1
(FMR1) interacting protein; Erk, extracellular-regulated MAP kinase;-
GAP, GTPase activating protein; GEF, Guanosine nucleotide exchange
factor; GTPase, GTP hydrolase; HSC, Hematopoietic stem cell; Hspc,
Hematopoietic stem progenitor cell; Ictm, Isoprenylcysteine carboxyl
methyltransferase; Ig, immunoglobulin; IQGAP, IQ motif containing
GTPase activating protein; Limk, LIM (Lin-11, Isl-1 and Mec-3) domain
kinase 2; NADPH, nicotinamide adenine dinucleotide phosphate; Nap,
non-catalytic region of tyrosine kinase (Nck)-associated protein; Nckap,
Nck-activated protein; Pak, p21-activated kinase; Pir, p53-inducible
mRNA; Pix, Pak-interacting exchange factor; Pkn, protein kinase N;
Rce, Ras and a factor converting enzyme; RhoGDI, Rho GDP
dissociation inhibitor; Rhpn, Rhophilin; Rock, Rho-associated, coiled-
coil containing protein kinase; Smurf, Smad ubiquitination regulatory
factor; Was, Wiskott-Aldrich syndrome protein; Wasf, Was protein
family; Wave, Was protein family verprolin-homologous protein; Wrch,
Wint-1 responsive Cdc42 homolog.
Review articles

in turn, lead to the stimulation of signaling cascades that
promote general cellular responses such as cytoskeletal
change, microtubule dynamics, vesicle trafficking, cell
polarity and cell cycle progression.(2) The plasticity of Rho/
Rac proteins both in terms of subcellular localization, regula-
tion, binding to effectors and crosstalk with other cellular
pathways has put them in a central regulatory point for a quite
large number of cellular processes. Unfortunately, the toll that
we have to pay for this is the development of diseases
when these routes become dysfunctional.(3,4) This crucial role
has led to a comprehensive study on their mechanism of
regulation, to the identification of additional elements of their
signal transduction pathways, and to the characterization
of their roles in vivo. In the present work, we will give an
overall view of the recent developments in those areas,
placing special emphasis on their regulatory and biological
properties in vivo. Given that Rho, Rac and Cdc42 are the
best-characterized Rho/Rac subfamilies, we will limit our
review to these molecules. Readers can find additional
information on other aspects of Rho/Rac biology in recent
publications.(2,5)
Regulation of Rho/Rac protein activity
In order to ensure proper signaling responses to extracellular
stimuli, cells control the activity of Rho/Rac proteins through a
number of regulatory steps. These include: (1) the control of
nucleotide binding and hydrolysis by GEFs and GAPs, a
process that has been already the object of recent reviews,(6,7)
(2) the regulation of their subcellular localization, (3) the
modulation of their protein expression levels, and (4) other
regulatory events. We summarize below the advances in the
understanding of these additional regulatory layers.
Regulation of Rho/Rac proteins by changesin the subcellular localizationIn addition to GDP/GTP exchange, most Rho/Rac proteins
require the docking onto cell membranes in order to perform
their biological functions. However, unlike other Ras super-
family proteins, this anchoring step is not achieved by default
during their biosynthesis and requires, instead, a combination
of intrinsic tethering signals and cooperative signaling
events.(8) The first and most crucial of the intrinsic tethering
signals is the progressive post-translational modification of the
Figure 1. Dendrogram showing the classification of Rho/Rac subfamily members according to structural similarity criteria. Members of
each subfamily are highlighted using the same color code and grouped by shaded areas. The first symbol used for each GTPase
corresponds to that approved by the Human Genome Organization Gene Nomenclature Committee. When appropriate, other commonly
used names are also included. The same criterium has been followed in the rest of this review article.
Review articles
BioEssays 29.4 357

Figure 2. Schematic representation of the biosynthesis (top), sequestration (middle) and regulatory (bottom) cycles of Rho/Rac proteins.
In the latter case, we have included the prototypical GDP/GTP cycle as well as other regulatory steps mediated by the action of either
effectors or other biological pathways (ubiquitination, protease cleavage, internalization). The main steps in each cycle are highlighted
using dark-gray arrows. Other less common regulatory interactions are indicated in light-gray arrows (when resulting in an activation signal)
or blunted lanes (when resulting in a downmodulation signal). The enzymes catalyzing those steps are shown in green. For the sake of
simplicity, we have not included here other post-translational events of Rho/Rac proteins that have been described in the main text such as
palmitoylation. It is also still unclear whether the insertion of the GTPase into the docking membrane is achieved when in the GDP or GTP-
bound state. The latter case is not contemplated in the scheme and would require the activation of the GTPase by GEFs, the re-association
of the GTP-bound GTPase with either RhoGDI or other carrier proteins, and the subsequent delivery of the GTPase to the target membrane.
Abbreviations used are: CAAX, an acronym derived from the combination of C¼ cysteine, A¼aliphatic amino acids and X¼Met, Ser, Ala
or Gln; Cyt, cytosol; EM, endomembranes; ER, endoplasmic reticulum; FT, farnesyl transferase; GGT, geranyl-geranyl transferase;
PM, plasma membrane; PRR BP, proline rich region binding protein. Consult main text for further details.
Review articles
358 BioEssays 29.4

so-called GTPase ‘‘CAAX box’’ (Fig. 2). The first stage of this
modification is the incorporation of either a geranyl-geranyl or,
less frequently, a farnesyl group to the cysteine residue of the
CAAX box, a process catalyzed in the cytoplasm by either
type I geranyl-geranyl or farnesyl transferases, respectively
(Fig. 2).(8,9) The attachment of the isoprenoid group to the
CAAX box promotes the translocation of the GTPases to the
endoplasmic reticulum,(8,10) where the proteolytic cleavage of
the AAX tripeptide tail ensues via the isoprenyl, CAAX-specific
protease Rce1 (Fig. 2).(8,11) After this reaction, the newly
exposed a-carboxyl group of the C-terminal cysteine residue
becomes methylesterified by the carboxyl methyltransferase
Icmt (Fig. 2).(8,12) In some cases (i.e. RhoB), Rho/Rac
proteins are further modified in the endoplasmic reticulum by
the attachment of palmitate groups on additional cysteine
residues present nearby the CAAX motif.(8) The enzyme
responsible for this step is still uncharacterized in mammals.
Recent results have shown that the incorporation of farnesyl or
geranyl-geranyl groups is a conditio sine qua non for proper
membrane anchoring and biological activity of the majority of
Ras superfamily members. Instead, the endoproteolytic and
methylation steps are only essential for the subcellular
localization and biological responses of farnesylated
GTPases.(13)
The final destination of the post-translationally-modified
GTPases depends on the computation by cells of other
ancillary signals present in the GTPase C terminus (Fig. 2). In
the case of palmitoylated GTPases, one of these additional
signals is the nature of the isoprenyl group attached to the
CAAX box. Perhaps the best example for this type of regulation
is RhoB, since this protein is localized preferentially in
endomembraneswhen geranyl-geranylated and at the plasma
membrane when farnesylated.(14) In other cases, the signal
mediating proper membrane localization is a polybasic amino
acic sequence located just upstream of the CAAX box. This is
the case of Rac subfamily proteins, where small differences in
those regions are responsible for the differential localization
of Rac1, Rac2 and RhoG in lipid rafts, endosomes, and
caveolar vesicles, respectively.(15,16) Finally, in the case of
Rac1, a proline-rich domain located near the CAAX box
contributes to the translocation of this GTPase to focal
adhesion complexes via its interaction with SH3 domain
proteins such as b-Pix, a Rac1-specific GEF that is con-
stitutively located in those subcellular regions.(17)
In addition to the presence of the above structural cues,
Rho/Rac proteins need additional upstream signals in order
to move from the cytosol to target membranes and, sub-
sequently, to remain stably anchored in those structures.
RhoGDIs play important roles in this regulatory context,
because they hide the isoprenyl groups of the GTPases, an
action that favors the sequestration of the inactive GTPases in
the cytosol or organelles (Fig. 2). This property is also
important for the removal of the GTPase from the plasma
membrane at the end of the signaling process (Fig. 2). Due to
the interaction of RhoGDIs with the GTPase switch regions,
they also impede the release of GDP from the GTPase and,
consequently, contribute to the maintenance of the GTPases
in an inactive state in non-stimulated cells (Fig. 2).(18)
The dissociation of the RhoGDI from the GTPase, an
essential requisite for the activation of GTPases by GEFs and
for their subsequent association with membranes, is regulated
at different levels during signal transduction. These regulatory
steps have been mapped out extensively in the case of Rac1.
Thus, it has been shown that integrins play an important role in
this process, because they increase the affinity of Rac for lipid
rafts, a process that in turn favors the displacement of the
geranyl-geranyl motif of the GTPase from the hydrophobic
pocket of the RhoGDI and its insertion into the phospholipid
bilayer of the target membrane (Fig. 2).(19,20) Other factors
cooperating in this dissociation step include RhoGDIs dis-
placement factors (i.e. the cytoplasmic tail of the low-affinity
nerve growth factor receptor)(21) and the decrease of the
RhoGDI affinity towards Rac1 upon phosphorylation of
RhoGDI molecules by protein kinase C(22) and Pak1,(23) a
Rac1 downstream element(24) (Fig. 2). Some Rho/Rac
GTPases also require the cooperation of additional pathways
to remain anchored to membranes once they have been
liberated from the RhoGDIs. In the case of Rac1, its residence
at the plasma membrane requires in some instances integrin-
dependent signals that block the co-internalization of the
GTPase with lipid rafts(25) (Fig. 2). Taken together, these
observations indicate that the localization of Rho/Rac
GTPases is tightly modulated in time and space by a complex
system of cell type-dependent regulatory pathways.
Transcriptional regulation and/ordifferential degradationMany Rho/Rac GTPases show cell-type-specific and/or
stimulus-dependent expression. For instance, Rac2 is mostly
restricted to hematopoietic cells.(26) Rac3 is preferentially
expressed in neurons of ganglia and the central nervous
system.(27) Moreover, RhoG and RhoB proteins have been
shown to have fluctuations during the cell cycle.(28,29) RhoB
expression undergoes further regulation by extracellular
stimuli such as UV irradiation, growth factors, cytokines and
oncogenes,(30–34) a control that is facilitated by the relatively
instability of its mRNA (t1/2¼ 20 min).(32) Finally, rhoU (also
known as wrch1) is a Wnt-regulated gene.(35) Some Rho/Rac
proteins are also controlled through degradation at specific
sites in the cell. Thus, RhoA can be degraded by the ubiquitin
ligase Smurf1 in a Rac1- and Cdc42-dependent manner
(Fig. 2). This regulation contributes to inhibit the inappropriate
formation of stress fibers in certain areas of the leading edge of
the cell during the process of cell migration.(36) Partial
proteolytic cleavage also plays regulatory roles on Rho/Rac
proteins. This is the case for Cdc42, whose proteolytic
Review articles
BioEssays 29.4 359

degradation by caspases following the activation of the Fas
death receptor contributes to the activation of Fas-dependent
apoptotic events(37) (Fig. 2).
Other regulatory eventsRho/Rac GTPases can be also regulated by additional
signaling mechanisms. Thus, RhoU has a putative autoinhi-
bitory domain at its N-terminus that can be released by the
binding of Grb2, an SH3–SH2 adaptor protein. This interaction
is mediated by the recognition of an N-terminal proline-rich
region of RhoU by one of the Grb2 SH3 domains. This
interaction does not alter GDP/GTP exchange in RhoU but,
instead, promotes a more-efficient binding of this GTPase to
the downstream serine/threonine kinase Pak1.(38) Rho/Rac
proteins can also undergo phosphorylation in specific resi-
dues, a post-translational event that may influence their
interaction with RhoGDIs,(39) stability in the membrane(39,40)
and effector functions(40) (Fig. 2).
The understanding of all these regulatory steps has allowed
the development, for the first time, of drugs that can control
the signaling output of Rho family GTPases in specific
pathological states by interfering with their GDP/GTP
exchange,(41) post-translational modification,(8) and sub-
cellular localization.(42) Given the important contribution of
Rho/Rac GTPases to the progression of some human
diseases, it is likely that these current efforts will eventually
crystallize into new therapeutic agents.
Effector molecules of Rho/Rac proteins
Once activated and translocated to their specific subcellular
locations, Rho/Rac proteins interact with downstream effector
molecules to engage specific signaling cascades.(5,24) To date,
more than 70 potential effectors have been identified for
members of the Rho/Rac family (Table 1). From a structural
point of view, it is known that these effectors use distinct
residues within the switch I and switch II regions as the major
docking/recognition sites.(5,24) This structural property has
made it possible to generate GTPase point mutants that can
bind only to a subset of effectors and engage only a limited
number of downstream effects (Fig. 3A). In some instances,
the stable association of effectors requires the participation of
additional structural cues located in the polybasic C-terminal
region, the b2 sheet and/or the helices a3, a30 and a5 of the
upstream GTPases(24) (Fig. 3A,B). In other cases, it requires
the localization of the upstream GTPase in specific sites of the
cell. For instance, the functional specificity found for Rac1 and
Rac2 in neutrophils is mainly due to their differential
subcellular localization within these hematopoietic cells.(15)
Despite the large structural diversity of Rho/Rac effectors,
we have learned a number of common regulatory themes that
take place during the activation of the downstream effectors by
Rho/Rac GTPases. Thus, the tethering of effector molecules
to membranes is part of the mechanism by which they become
activated (Fig. 4). Indeed, translocation of Pak, Pkn, citron,
Rock and other effector proteins to signaling hot spots of cells
has been shown recently.(43–47) Moreover, it has been shown
that the activation of Pak1 only occurs when active Rac1 is at
the plasma membrane but not when free in the cytosol.(19)
Other results indicate that the interaction of effectors with Rho/
Rac GTPases provokes conformational changes that
shift them from autoinhibitory conformations to fully active
structures (Fig. 4). Such regulatory mechanism has been
observed for a wide collection of both catalytic (i.e. Pak,
Rock, Pkn) and non-catalytic, adaptor-like (i.e. Diaphanous,
Was, and Baiap2) effectors.(24) These changes could be self-
sufficient for activation or, alternatively, may cooperate with
other signals to promote optimal effector activation (Fig. 4A).
For example, Pkn needs RhoA binding, lipid association and
autophosphorylation events to became fully active.(24,48)
Some downstream signaling elements are also activated by
the release of trans-inhibitory factors upon the binding of the
GTPase (Fig. 4B). This is at least the case of Wasf/Wave/Scar
proteins, which get released from an inhibitory complex
formed with Nckap1–Nap125, Cyfip2–Pir121 and C3orf10–
Hspc300 upon the binding of GTP-bound Rac1 to Nckap1 and
Cyfip2.(49) It should be noted, however, that the binding of the
activated GTPase results in the inhibition, not the stimulation,
of the bound effector (Fig. 4C). This is the case, for instance, of
the interaction of Cdc42 with Cdc42Eps (also referred to as
Borgs).(50)
The final result of the modulation of the activity of these
effectors is the generation of multibranched signals that
promote, among other responses, cytoskeletal change,
vesicle trafficking and cell cycle entry (Table 1). All these
pathways have been extensively reviewed before and will not
be re-mentioned here.(2,5) However, it should be noted that the
activation of effectors might fire back on the GTPases
themselves, thus contributing to the generation of balanced
and time-restricted signals by Rho/Rac proteins. Pak family
proteins are very active in this regulatory context, since they
can modify the activity of both RhoGDIs and Rho/Rac
GEFs(23,51) (Fig. 2). These results underscore the high level
of plasticity and large number of feed-back loops taking place
in the signal transduction pathways of these GTPases.
As in the case of the regulatory elements that mediate Rho/
Rac activation, the understanding of the mode of action of the
downstream molecules has allowed the development of
inhibitory molecules for Pak and Rock family proteins.(52–54)
One of the Rock inhibitors, fasudil (also known as HA-1077
and AT877), is already being used for the treatment of patients
with cardiovascular disorders.(55)
Genetic analysis of Rho/Rac GTPase
functions in vivo
Most of the functional observations obtained with Rho/Rac
proteins have been derived from cultured cells. While this
Review articles
360 BioEssays 29.4

Table
1.
Alis
tof
sele
cte
deff
ecto
rpro
tein
sfo
rR
hoA
,R
ac1
and
Cdc42
Effecto
raTypeofPro
tein
bUpstream
GTPasec
Main
biologicalAction
Cnksr1
Scaff
old
pro
tein
(Ksr)
RhoA
Inte
racts
with
RhoA
(Rhophili
n)
and
Ras
eff
ecto
rs(R
alG
DS
)
Rtk
n1,2
Scaff
old
pro
tein
RhoA
Inte
raction
with
PD
Zpro
tein
s,
NFkB
activation
Rhpn1,2
Scaff
old
,P
DZ
conta
inin
gpro
tein
RhoA
Cyto
ske
leta
lre
gula
tion?
Ktn
1S
caff
old
pro
tein
RhoA
,R
ac1,
Cdc42
Kin
esin
bin
din
g,
vesic
ula
rtr
affi
ckin
gth
rough
mic
rotu
bule
s
Dia
ph1,2
Scaff
old
pro
tein
(Dia
1,2
)R
hoA
,R
ac1
Cyto
ske
leta
lchange
via
pro
filin
and
Baia
p
Arfi
p2
Scaff
old
pro
tein
(Por1
)R
ac1
Cyto
ske
leta
lre
gula
tion
Pard
6A
,GS
caff
old
pro
tein
(Par6a,g)
Rac1,
Cdc42
Cell
pola
rity
.Lin
ks
GT
Pases
and
atipic
alP
KC
s
Baia
p2
Scaff
old
pro
tein
(p53
IRS)
Rac1,
Cdc42
Cyto
ske
leta
lorg
aniz
ation
via
regula
tio
nof
Wasf/
Wave
pro
tein
s
IQG
AP
1,2
RhoG
AP
and
scaff
old
pro
tein
Rac1,
Cdc42
Regula
tor
of
the
cyto
ske
leto
n,
cell-
cell
conta
cts
,and
pro
lifera
tion
Was
Scaff
old
pro
tein
(Wasp)
Rac1
Cyto
ske
leta
lre
gula
tion
via
the
Arp
2/3
com
ple
x
Nck1
Scaff
old
pro
tein
with
SH
2/S
H3
dom
ain
sR
ac1
Com
ple
xfo
rmation
with
Wasp.
Sig
naltr
ansduction
Nckap1
Scaff
old
pro
tein
(Nap125,
Nap1)
Rac1
Regula
tio
nof
the
cyto
ske
leto
nvia
Wasf
pro
tein
s
Cyfip2
Scaff
old
pro
tein
(Pir121)
Rac1
Regula
tio
nof
the
cyto
ske
leto
nvia
Wasf
pro
tein
s
Cdc42S
E1,2
Scaff
old
pro
tein
(Spec1,2
)R
ac1,
Cdc42
Modula
tion
of
GT
Pase
sig
nalin
goutp
uts
IL1R
ap1
Scaff
old
pro
tein
Rac1
Inte
rleukin
sig
nalli
ng
Hspc121
Scaff
old
pro
tein
Rac1,
RhoA
,C
dc42
Regula
tio
nof
kin
ase
cascades
and
gene
expre
ssio
n
WasL
Scaff
old
pro
tein
(N-W
asp)
Cdc42
Cyto
ske
leta
lre
gula
tion
via
de
Arp
2/3
com
ple
x
Trip10
Scaff
old
pro
tein
Cdc42
Bin
din
gof
Was
tom
icro
tubule
s
Cdc42E
P1,3
,5S
caff
old
pro
tein
(Borg
s1,3
,5)
Cdc42
Regula
tio
nof
septins
Mig
-6S
caff
old
pro
tein
Cdc42
Activ
ation
of
the
Jnk
route
Wasf1
,2S
caff
old
pro
tein
(Wave
1,2
;S
car1
,2)
Cdc42,
Rac1
Cyto
ske
leta
lre
gula
tion
via
the
Arp
2/3
com
ple
x
CopG
2C
oato
mer
pro
tein
(g2-C
op)
Cdc42
Vesic
letr
affi
ckin
g(c
lath
rin
route
)
Itpr1
Inositol1,4
,5-t
riphospha
tere
cepto
rR
hoA
Calc
ium
entr
yin
endoth
elia
lcells
Plc
G1
Phospholip
ase,
Cty
pe
(PLC
-g1)
RhoA
Pro
duction
of
second
messengers
DgkQ
Dia
cylg
lycero
lkin
asey
RhoA
Dia
cylg
lycero
ldeple
tion
PI-
5-p
5K
Lip
idkin
ase
RhoA
Modula
tion
of
phospha
tidylin
ositolbip
hospha
tele
vels
SynJ2
Poly
phosphoin
ositid
ephospha
tase
Rac1
Inhib
itio
nof
recepto
rendocyto
sis
via
the
cla
thrin
route
PIK
3R
1R
egula
tory
p85
subunit
of
PIK
3C
Rac1,
Cdc42
Regula
tio
nof
PIK
3C
activi
ty,
sig
naltr
ansduction
Pld
1P
hospholip
ase,
Dty
pe
RhoA
,R
ac,
Cdc42
Pro
duction
of
phospha
tidic
acid
and
cholin
e(s
econd
messengers
)
Plc
B2
Phospholip
ase,
Cty
pe
(PLC
-b2)
Cdc42,
Rac1
Pro
duction
of
second
messengers
Cit
Serine/t
hre
onin
ekin
ase
(Citro
n)
RhoA
Citokin
esis
Pkn1,2
Serine/t
hre
onin
ekin
ase
(Prk
)R
hoA
Vesic
lere
cyc
ling,
cell
cyc
lere
gula
tion,
Pld
1activa
tion
Rock1,2
Serine/t
hre
onin
ekin
ase
(Roka,b)
RhoA
Cyto
ske
leto
n,
cyto
kin
esis
,blo
ckage
of
cell
conta
ct
inhib
itio
n
Pak1-7
Serine/t
hre
onin
ekin
ases
Rac1,
Cdc42
Cyto
ske
leta
lorg
aniz
ation,
activation
of
kin
ase
cascades
Map3K
11
Serine/t
hre
onin
ekin
ase
(Mlk
3)
Rac1,
Cdc42
Activ
ation
of
kin
ase
cascades
Prk
cA
Serine/t
hre
onin
ekin
ase
(PK
Ca)
RhoA
,R
ac1,
Cdc42
Sig
naltr
ansduction
Cdc42bpgA
,BS
erine/t
hre
onin
ekin
ase
(MR
CKa,b)
Rac1,
Cdc42
Cyto
ske
leta
lre
gula
tion
Rps6kB
1S
erine/t
hre
onin
ekin
ase
(p70
S6K,
S6K
1)
Cdc42
Regula
tio
nof
transla
tion,
cell
cyc
le
Map3K
10
Serine/t
hre
onin
ekin
ase
(Mlk
2)
Cdc42
Activ
ation
of
kin
ase
cascades
Map3K
4S
erine/t
hre
onin
ekin
ase
(Mekk4)
Cdc42
Activ
ation
of
the
Jnk
route
Tnk2
Tyro
sin
ekin
ase
(Ack)
Cdc42
Sig
naltr
ansduction,
activa
tion
of
GE
Fs
Ncf1
,2N
AD
PH
oxid
ase
com
ple
xsubunit
Rac1,
Cdc42
Supero
xid
epro
duction
CybA
NA
DP
Hoxid
ase
com
ple
xsubunit
Rac1
Supero
xid
epro
duction
Ppp1r1
2A
Regula
tory
subunit
of
phospha
tase1
RhoA
Myo
sin
light
chain
inactivation,
cyto
ske
leta
lre
gula
tio
n
(Continued)
Review articles
BioEssays 29.4 361

approach has allowed the discovery of important aspects of
their regulation and function, it has obvious limitations. For
instance, it is rather difficult to verify whether the observations
obtained using in vitro conditions can be extrapolated to more
complex situations in which primary, non-immortalized cells
are exposed to limited amounts of stimuli or to different tissue-
and cell-type crosstalk. These approaches do not provide
faithful information regarding the level of functional overlaps
among closely related GTPases, either to verify the role of
specific GTPases in the tissues where they are actually
expressed, or to study their participation in complex physio-
logical responses. To surmount some of these problems,
different research groups have generated animal models in
which specific members of the Rho/Rac subfamily and
effectors have been disrupted using homologous recombina-
tion techniques. Below, we summarize the most-recent results
achieved in this area. As a note of caution, please be adviced
that the strict comparison of the phenotypes displayed
by different genetically modified mouse strains cannot be
interpreted in absolute terms, because the lack of effect of a
null gene may not necessarily indicate less-important roles of
its encoded protein but, rather, functional complementation
events by related proteins or parallel signal transduction
pathways. The reader can find additional information about
phenotypes of transgenic mice expressing Rho/Rac proteins
and knockout animals lacking genes for Rho/Rac GEFs in
recent review articles.(6,56,57)
Genetic analysis of members of the Rac subgroupThe inactivation of the rac1, rac2, rac3 and rhoG loci has
been already achieved in mice using standard homologous
recombination techniques or, in some cases, via the use of
either tissue-specific or inducible knockout strategies. In
addition, some Rac1 effectors belonging to the Pak and Wasf
families have also been knocked out to address their role
in vivo.
The deletion of the rac1 gene provokes embryonic lethality
caused by both gastrulation defects and apoptosis of
mesodermal cells.(58) In contrast, the inactivation of the other
Rac subfamily members gives rise to more limited defects,
which are usually found in the tissues where these GTPases
are preferentially expressed. Thus, rac2�/� mice show
hematopoietic defects (see below), rac3�/� animals present
slight motor coordination problems and enhanced learning
abilities,(59) and rhoG�/� mice have some hyperactivation of
T-cell responses to antigens and minor defects in the super-
oxide pathway of neutrophils,(60,61) the cells responsible for
inflammatory responses.
The generation of inducible, cell-type-specific knockouts
for the rac1 gene and their side-by-side comparison with both
rac2�/� and double rac1�/�; rac2�/� mutant animals has
permited a comprehensive understanding of the physiological
roles of these two GTPases and their level of signaling overlap/
Table
1.
(Continued)
Effecto
raTypeofPro
tein
bUpstream
GTPasec
Main
biologicalAction
Fm
l1Form
in-lik
em
ole
cule
Rac1
Cyto
ske
leta
lorg
aniz
ation,
cell
pola
rity
and
cyto
kin
esis
Fhod1
Form
in-
like
Rac1
Cyto
ske
leta
land
transcriptionalre
gula
tio
n
Cyfip1
Actin
bin
din
gpro
tein
Rac1
Cyto
ske
leta
lorg
aniz
ation
Fln
AA
ctin
bin
din
gpro
tein
RhoA
,R
ac1,
Cdc42
Cyto
ske
leta
lre
gula
tio
n,
actin
fila
ment
cro
sslin
kin
g
Tbc1d3
Unknow
n,
Tbc
oncopro
tein
fam
ilyC
dc42
Cyto
ske
leta
lre
gula
tio
n
TubA
1Tubulin
Rac1
Inte
gra
lcom
ponent
of
mic
rotu
bule
s
KcnA
2P
ota
siu
mC
hannelsubunit
RhoA
Pota
siu
mentr
y
Sta
t3Tra
nscriptionalfa
cto
rR
ac1,
Cdc42
Tra
nscription
Nos2A
Nitric
oxid
esynth
ase
Rac1
Nitric
oxid
epro
duction
Sm
urf
2E
3ubiq
uitin
pro
tein
ligase
2R
ac1
Sm
ad
and
RhoA
ubiq
uitin
ation,
TG
Fb
recepto
rsig
nalli
ng
aE
ffecto
rnam
es
are
giv
en
inth
econsensus
nom
encla
ture
esta
blis
hed
by
the
Hum
an
Genom
eO
rganiz
ation
and
have
been
gro
uped
accord
ing
tofu
nctions.
bIn
som
ein
sta
nces,altern
ative
nam
es
used
for
the
eff
ecto
rare
giv
en
inpare
nth
esis
.cD
ata
are
derive
dfr
om
invitro
,in
viv
o,pro
teom
icand/o
rcello
mic
techniq
ues.
An
usefu
llin
kfo
rexplo
ring
furt
her
these
inte
ractions
and
the
eff
ecto
rsofoth
er
Rho/R
ac
subfa
mily
pro
tein
sis
the
Bio
GR
ID
data
base
availa
ble
onlin
e(w
ww
.thebio
gri
d.o
rg).
Review articles
362 BioEssays 29.4
specificity (Fig. 5). In the case of HSCs, the role of Rac1 and
Rac2 has been addressed using reconstitution experiments in
sublethaly irradiated immunocompromised mice and inducible
approaches of gene inactivation. These studies revealed
important functional differences between these two family
members. Rac1 has been shown to be important for the
optimal reconstitution of the hematopoietic system, having
roles both in the engraftment and retention of HSCs in the bone
marrow. Instead, rac2�/� HSCs show normal behaviour in all
these reponses. Rac1 and Rac2 also differ in the type of
intracellular responses that they regulate in HSCs. Thus, Rac1
is essential for the entry of HSCs into the cell cycle upon
extracellular stimulation as well as for their progression
through S and G2/M phases whereas Rac2 is important for
cytoskeletal responses, adhesion, spreading and Akt-depen-
dent HSC survival(62,63) (Fig. 5).
In the case of T-cells, rac2�/� mice show no apparent
problems in the differentiation of those cells in the thymus. In
contrast, mature rac2�/� T-cells display defects in T-cell
receptor clustering, actin polymerization, generation of Ca2þ
fluxes and Erk activation upon engagement of the T-cell
receptor(64) (Fig. 5). These defects are rather marginal,
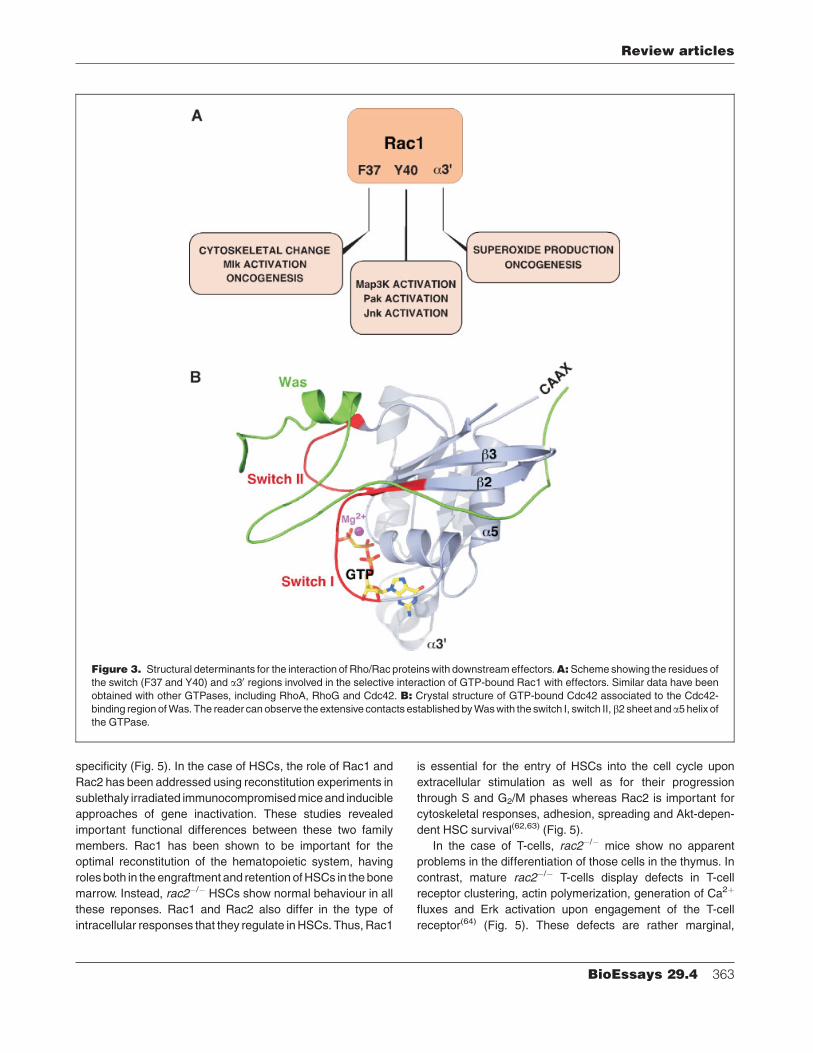
Figure 3. Structural determinants for the interaction of Rho/Rac proteins with downstream effectors.A:Scheme showing the residues of
the switch (F37 and Y40) and a30 regions involved in the selective interaction of GTP-bound Rac1 with effectors. Similar data have been
obtained with other GTPases, including RhoA, RhoG and Cdc42. B: Crystal structure of GTP-bound Cdc42 associated to the Cdc42-
binding region of Was. The reader can observe the extensive contacts established by Was with the switch I, switch II,b2 sheet anda5 helix of
the GTPase.
Review articles
BioEssays 29.4 363
probably due to the compensation exerted by the endogenous
Rac1 protein present at high levels in those cells. Rac2 is also
important for the differentiation of helper T-cells to the TH1
subtype because it regulates the p38- and NFkB-dependent
induction of interferon-g, an important mediator of this
maturation step(65) (Fig. 5). The effects of the Rac1 deficiency
in T-cells have not been addressed as yet. However, as inferred
from the results obtained by Rac1 GEFs,(57) we can predict
that this GTPase will have roles in T-cell differentiation, positive
and negative selection, stimulation of phosphatidylinositol-3
kinase/Akt and the Ras/Erk routes, and overall responses to
antigens.
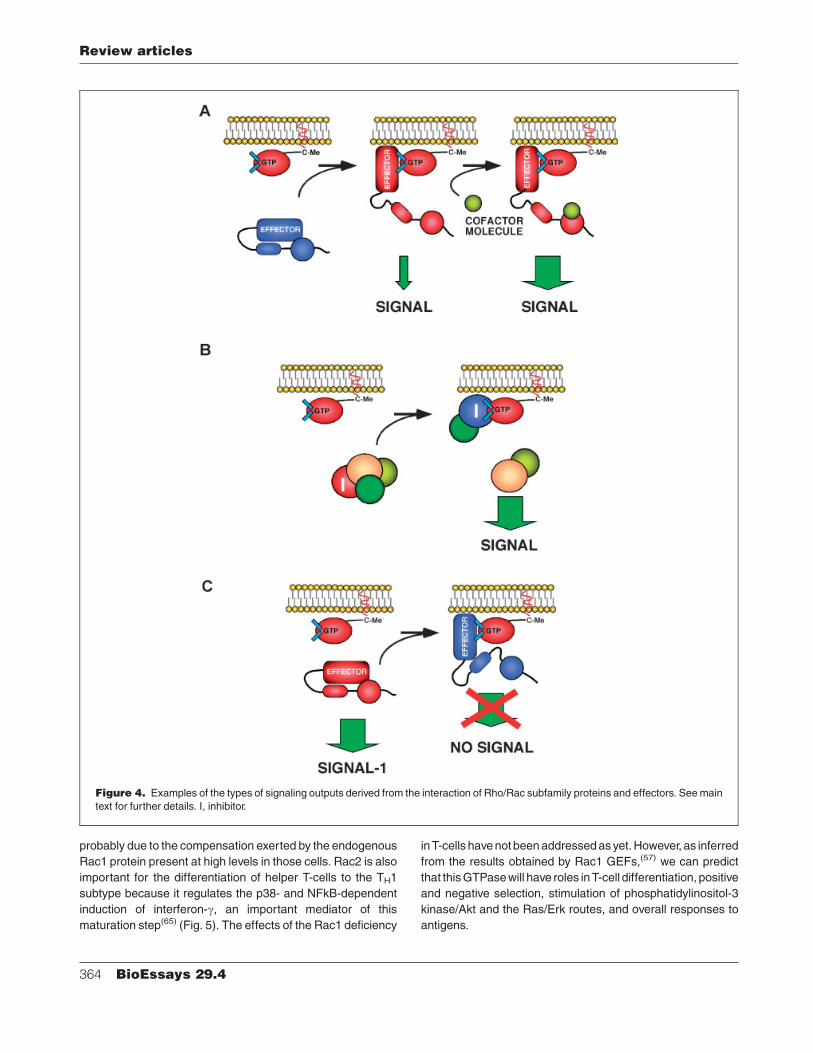
Figure 4. Examples of the types of signaling outputs derived from the interaction of Rho/Rac subfamily proteins and effectors. See main
text for further details. I, inhibitor.
Review articles
364 BioEssays 29.4

In the case of B-cells, rac2-deficient animals show defects
in the B-cell compartment, displaying reduced numbers of
peripheral B-cells, peritoneal B1 cells and IgM-secreting
plasma cells. Mature rac2�/� B-cells respond poorly to
stimulation of the B-cell receptor, showing reduced levels of
Ca2þ fluxes and of cell proliferation.(66) Rac1 seems to have
only a marginal and overlapping role with Rac2 in these
cells.(67) In agreement to this, the simultaneous elimination of
rac1 and rac2 genes induces an aggravation of the rac2�/�
phenotype, leading to a developmental block of B-cell
development at very immature stages.(67) This is caused by
low survival rates derived from the improper activation of the
Akt route and the inefficient expression of two anti-apoptotic
molecules, Bcl2L1 (most commonly known as Bcl-xL) and the
BAFF receptor.(67) Instead, the single rac1 gene knockout
has no detectable effects per se in this lymphoid lineage(67)
(Fig. 5).
Unlike the case of B-cells, Rac1 and Rac2 exert non-
overlapping functions in the neutrophil lineage. rac2 null
neurophils show a severe impairment of motility, adhesion,
chemotaxis and phagocytosis as well as a drastic reduction
(&60%) in the activity of the NADPH oxidase, the enzyme
complex responsible for the generation of anti-bacterial
superoxide molecules.(68,69) Recent results have shown that
the residual level of NADPH oxidase activity found in these
animals is due to the action of RhoG and, to a minor extent, of
Rac1(61,70) (Fig. 5). rac1�/� neutrophils have milder problems,
with defects detectable only in chemokine-dependent re-
sponses.(70,71) Unlike rac2�/� neutrophils, these cells do not
show significant problems in the cytoskeleton in the absence of
chemokines with the exception of defects in the RhoA-
dependent retraction of the uropod during stochastic migra-
tion(70,72) (Fig. 5).
The role of rac genes in macrophages has only begun to be
elucidated. Available reports indicate that Rac2 is important
for superoxide production and phagocytosis to some (i.e. FcgR
stimulation, IgG-sensitized sheep red blood cells) but not
all (i.e. serum-opsonized zymosan) stimuli.(73) In addition, it
is important for the migration of these cells, as evidenced by
the lack of the accumulation of exudate macrophages during
Figure 5. Representation of the main developmental routes for hematopoietic cells and the steps that are dependent on either Rho/Rac
subfamily proteins or Rho/Rac effectors. The GTPases and/or effectors involved in those steps are highlighted in blue. The processes
impaired by the gene inactivations in each hematopoietic lineage are summarized into light-brown boxes.
Review articles
BioEssays 29.4 365

the peritoneal inflammation of rac2�/� mice(73) (Fig. 5).
Contrary to Rac2, Rac1 seems to be important for regulating
macrophage cell morphology and proper lamellipodia forma-
tion(74) (Fig. 5). However, these defects do not induce any
significant defect on the migration and chemotactic responses
of this cell type.(74)
In agreement with the high levels of expression in platelets,
Rac1 seems to be the major player of the Rac subfamily in this
cell type. Its functions include the generation of lamellipodia
upon the stimulation of platelets with ADP, the induction of
proper spreading and aggregation of platelets, and the
formation of thrombi in vivo (Fig. 5). These defects are not
very severe, because Rac1-deficient animals do not experi-
ence hemorrhages.(75)
More recently, other tissue-specific rac1 gene knockouts
have begun to shed light on its function in non-hematopoietic
tissues. Thus, it has been shown that Rac1 is important for the
formation of myelin sheaths in the central nervous system.(76)
In the case of the skin, Rac1 has been shown to be important
for the integrity of hair follicles and, as consequence, mice with
a keratinocyte-specific inactivation of the rac1 locus develop a
hairless phenotype.(77) Finally, it has been shown that Rac1
and Rac2 proteins play important roles in the dendritic cells
that present antigens to T-cells.(78) Due to this, dendritic
cells lacking expression of both Rac1 and Rac2 show defective
cytoskeletal change, migration and antigen presentation
that, as a result, preclude adequate cell contacts with T-cells.
The development of this defect requires the simultaneous
deletion of both rac1 and rac2 genes, indicating that these
two proteins exert similar and additive roles in dendritic
cells.(78)
Consistent with the important role of Pak and Wasf
family proteins in Rac1 signaling, the deletion of some of
those cytoskeletal regulators has dire consequences during
embryonic development. Thus, the elimination of the pak4
gene leads to embryonic lethality due to heart development
problems. This protein is also important for the migration,
differentiation and axogenesis of spinal cord neurons (both
motorneurons and interneurons) and for the proper folding of
the caudal region of the neural tube.(79) The disruption of the
wasf2 gene also leads to embryonic lethality at later stages
(E12.5). These embryos show growth retardation, brain
ventricle malformations and vascularization deficiencies when
compared to wild-type embryos.(80,81) As expected from the
previous functional characterization of Wasf proteins, the
analysis of wasf2�/� mouse embryonic fibroblasts (MEFs)
indicates that this cytoskeletal regulator is important for the
generation of lamellipodia, Rac1-dependent actin polymeriza-
tion, and cell migration events.(81) Despite these examples, the
disruption of other Rac effectors in mice induces milder
phenotypes. Thus, wasf1�/� adult mice develop normaly but
have reduced size and experience anxiety, sensorimotor
retardation and deficits in hippocampal-dependent learning
and memory.(82) pak5�/� mice are fully viable and display no
obvious abnormalities.(83)
Genetic analysis of members of the Rho subgroupThe phenotypes of mice lacking functional rhoB and rhoC
genes have been recently described. These two mouse strains
are fully viable and fertile. When MEFs from these animals
were studied in vitro, it was found that RhoB is important for
proper cell motility but not for adhesion or spreading.(84)
However, these latter functions became diminished when
MEFs were transformed by both E1A and ras oncogenes,
suggesting that RhoB function is probably required for
oncogenic-dependent cytoskeletal responses.(84) rhoC�/�
MEFs show only cytoskeletal defects under serum-starved
conditions.(84) In contrast to these apparently mild pheno-
types, it has been observed that rhoB and rhoC have
important, although antagonistic, roles in tumor progression.
RhoB-deficient animals are more susceptible to developing
tumors when tested in skin carcinogenesis assays, indicating
that this GTPase may have tumor-suppressor properties, at
least in the case of skin.(84) Using crosses with transgenic mice
expressing the oncogenic polyomavirus middle T-antigen, it
was observed that the absence of RhoC is not important for
tumor development.(85) Despite this, rhoC�/� tumor cells are
less metastatic than the wild-type counterparts, a phenotype
attributed to the reduced migration, lower invasiveness and
poor survival of RhoC-deficient cells.(85) Despite these
advances, more work will be required to assess the relative
contributions of RhoB and RhoC to the life and pathogenesis
of animals. An important step in that direction will be the side-
by-side comparison of these two strains using identical genetic
backgrounds and conditions. In addition, it will be interesting to
generate the double RhoB/RhoC knockout to corroborate that
their functions are not overlapping in vivo.
Although the rhoA locus has not been targeted as yet,
several of the main RhoA effectors have been inactivated by
homologous recombination. These studies have revealed that
Rock1 and Rock2 are important for eyelid closure and fusion of
the ventral body wall, because the disruption of any of those
two genes give rise to neonates with omphalocele and open
eyes.(86,87) In agreement with the described routes modulated
by Rocks, keratinocytes derived from these tissues show
defective stress fiber formation and low myosin light chain
phosphorylation upon EGF stimulation.(87) This mild pheno-
type is highly dependent of the genetic background, because
the inactivation of the rock2 gene in another mouse strain
leads to placental defects and embryonic death.(88) cit�/�
animals also develop normally but they succumb to lethal
epileptic seizures during the first postnatal month.(89) This is
due to a marked reduction in the number of GABAergic
interneurons and of both dentate gyrus and cerebellar
neurons, a phenotype caused by cytokinesis defects in
neuroblast subsets.(89) More recently, it has been shown that
Review articles
366 BioEssays 29.4

cit�/� also have defects in both the survival and cytokinesis of
spermatogenic precursors, leading to a severe impairment of
testicular function.(90) The knockout of limk2, a locus encoding
a serine/threonine kinase that is activated by Rock,(5,24) also
induces defects in spermatogenesis, although they develop
normally and show no major disturbances in the adult
period.(91) Finally, rhpn2�/� animals show no detectable
defects.(92) The relatively mild phenotype of Rock-, Cit- and
Limk2-deficient animals is somewhat surprising, given the
crucial role attributed to these three kinases in general
cytoskeletal and cytokinesis events. At least in the case of
Limk2, this mild phenotype cannot be attributed to compensa-
tion effects by other Rho/Rac effectors, because Limk2-
deficient cells show a total impairment in the phosphorylation
of the main substrate of this kinase family, the cytoskeletal
regulator cofilin.(91) An intriguing possibility derived from these
results is that, at least during embryonic development, the
migration and adhesion of cells may follow different pathways
to those described in immortalized cultured cells.
Genetic analysis of members of the Cdc42 subgroupThe knockout of the cdc42 locus leads to embryonic lethality
prior to the E6.5 stage.(93) The isolation of embryonic stem
cells from E3.5 cdc42�/� blastocysts has allowed a glimpse
of the functional relevance of Cdc42 inside cells. Under
these conditions, it has been shown that Cdc42 is essential for
the phosphatidylinositol bisphosphate-mediated polymeriza-
tion of actin and, due to this, its deletion induces a highly
disorganized cytoskeleton, round-up morphologies, and
smaller cell sizes.(94) In contrast to these results, the cell-
specific inactivation of the cdc42 locus in fibroblasts does not
induce any impairment on cytoskeletal structures or cell
migration.(94) It has been argued that this result is due to
functional redundancy with other Rho/Rac proteins, because
the expression of a dominant negative mutant of Cdc42 in
the cdc42�/� fibroblasts significantly impairs most of those
biological processes.(95) cdc42�/� cells do show defects in
polarity, including minor disturbances in establishment of the
proper directionality and relocation of the Golgi apparatus in
migrating fibroblasts.(95) These results seem to be however
highly dependent on the fibroblast type, because a more
recent study has shown that primary fibroblasts do show
problems in filopodia formation, migration and proliferation in
the absence of Cdc42 expression.(94) More recently, the
specific inactivation of the cdc42gene in oligodendrocytes and
neuronal precursors has revealed a role for Cdc42 in the
central nervous system.(76) In the case of oligodendrocytes,
Cdc42 is important for the correct formation of myelin
sheaths.(76) In the case of neuronal precursors, Cdc42
plays crucial roles in the establishment of Par6-dependent
apico-basal polarity processes of stem cells.(96) In contrast, it
does not seem important for the adhesion, cell-cycle regula-
tion or cytokinesis of this stem cell population.(96)
Several Cdc42 effectors have been also targeted by
homologous recombination. Was-deficient mice show re-
duced numbers of thymocytes, mature lymphocytes and
platelets. The reduced production of thymocytes is due to
impaired progression from the CD44�/CD25þ to the CD44�/
CD25� stage of differentiation. was�/� thymocytes and
mature T cells show impaired T-cell receptor capping and
endocytosis, generation of Ca2þ fluxes and actin polymeriza-
tion. As a consequence, they proliferate poorly upon engage-
ment of the T-cell receptor(97) (Fig. 5). Probably due to all these
immunological disturbances, was�/� mice develop colitis as
they age.(97) These animals have also neurophils with reduced
phagocytic activity and osteoclasts with severe cytoskeletal
defects that generate abnormal patterns of bone resorp-
tion.(98,99) Iqgap1�/� animals show no detectable phenotypic
defects with the exception of the development of gastric
hyperplasia,(100) a result that suggests that this Cdc42 effector
may exert inhibitory properties for the proliferation of intestinal
epithelial cells.
Taken together, these studies confirm the important role of
specific Rho/Rac family members in the biological pathways
related to cytoskeletal dynamics, polarity, cell survival/
apoptosis, cell proliferation, immune system responses and
oncogenesis. In addition, they show that despite the high
structural homology, these proteins exert related, but not
identical, functions in vivo at least in certain cell types.
Conclusion
Since the isolation of the first Rho/Rac family more than
20 years ago, substantial information has been gained
regarding the number of family members, the type of effectors
they engage, the main regulatory layers controling their
activities and the biological processes that they are implicated
on. Despite these advances, more information remains to be
gathered in the near future. For instance, we have to delineate
the dynamics and kinetics of engagement of the different
interactive Rho/Rac-dependent networks during cell signaling.
Likewise, we need to get additional information regarding the
type of signaling networks engaged and signaling outputs
generated in function of the type, concentration, and/or
combination of the extracellular stimuli received by cells.
Given the complex array of signaling molecules involved and,
in some instances, the multifunctional nature of them, the
execution of this aim will not be an easy task. Fortunately,
the high-throughput techniques that are being developed in the
cellomics field to monitor the behavior of molecules in real time
will probably help tackling these issues. Likewise, proteomic
and genomic techniques will be also useful for isolating all the
signaling complexes and regulatory molecules involved in
these pathways. Given that most of the studies done up to now
have focused on few GTPases, more work remains be done to
elucidate the functions of the less-studied family counterparts.
In this context, the generation of new animal models will help
Review articles
BioEssays 29.4 367

assigning specific functional tasks to these GTPases and, in
addition, provide information about the level of signaling
overlap and/or cooperativity existing among them. Given
the important roles that these GTPases play in different
pathologies, it is likely that the progress in these areas of
research will contribute to a better understanding and
treatment of human disease.
Acknowledgments
The authors wish to apologize to the scientists not cited in this
work due to space constrains. We also like to thank M. Dosil
and P. Crespo for helpful comments on the manuscript.
References1. Madaule P, Axel R. 1985. A novel ras-related gene family. Cell 41:31–
40.
2. Etienne-Manneville S, Hall A. 2002. Rho GTPases in cell biology. Nature
420:629–635.
3. Boettner B, Van Aelst L. 2002. The role of Rho GTPases in disease
development. Gene 286:155–174.
4. Sahai E, Marshall CJ. 2002. RHO-GTPases and cancer. Nat Rev Cancer
2:133–142.
5. Jaffe AB, Hall A. 2005. Rho GTPases: biochemistry and biology. Annu
Rev Cell Dev Biol 21:247–269.
6. Rossman KL, Der CJ, Sondek J. 2005. GEF means go: turning on RHO
GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell
Biol 6:167–180.
7. Peck J, Douglas Gt, Wu CH, Burbelo PD. 2002. Human RhoGAP
domain-containing proteins: structure, function and evolutionary rela-
tionships. FEBS Lett 528:27–34.
8. Winter-Vann AM, Casey PJ. 2005. Post-prenylation-processing en-
zymes as new targets in oncogenesis. Nat Rev Cancer 5:405–412.
9. Casey PJ, Seabra MC. 1996. Protein prenyltransferases. J Biol Chem
271:5289–5292.
10. Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, et al. 1999.
Endomembrane trafficking of ras: the CAAX motif targets proteins to the
ER and Golgi. Cell 98:69–80.
11. Boyartchuk VL, Ashby MN, Rine J. 1997. Modulation of Ras and a-factor
function by carboxyl-terminal proteolysis. Science 275:1796–1800.
12. Dai Q, Choy E, Chiu V, Romano J, Slivka SR, et al. 1998. Mammalian
prenylcysteine carboxyl methyltransferase is in the endoplasmic
reticulum. J Biol Chem 273:15030–15034.
13. Michaelson D, Ali W, Chiu VK, Bergo M, Silletti J, et al. 2005.
Postprenylation CAAX processing is required for proper localization of
Ras but not Rho GTPases. Mol Biol Cell 16:1606–1616.
14. Lebowitz PF, Davide JP, Prendergast GC. 1995. Evidence that
farnesyltransferase inhibitors suppress Ras transformation by interfer-
ing with Rho activity. Mol Cell Biol 15:6613–6622.
15. Filippi MD, Harris CE, Meller J, Gu Y, Zheng Y, et al. 2004. Localization
of Rac2 via the C terminus and aspartic acid 150 specifies superoxide
generation, actin polarity and chemotaxis in neutrophils. Nat Immunol
5:744–751.
16. Prieto-Sanchez RM, Bustelo XR. 2003. Structural basis for the signaling
specificity of RhoG and Rac1 GTPases. J Biol Chem 278:37916–
37925.
17. ten Klooster JP, Jaffer ZM, Chernoff J, Hordijk PL. 2006. Targeting and
activation of Rac1 are mediated by the exchange factor beta-Pix. J Cell
Biol 172:759–769.
18. DerMardirossian C, Bokoch GM. 2005. GDIs: central regulatory
molecules in Rho GTPase activation. Trends Cell Biol 15:356–363.
19. del Pozo MA, Price LS, Alderson NB, Ren XD, Schwartz MA. 2000.
Adhesion to the extracellular matrix regulates the coupling of the small
GTPase Rac to its effector PAK. Embo J 19:2008–2014.
20. Del Pozo MA, Kiosses WB, Alderson NB, Meller N, Hahn KM, et al. 2002.
Integrins regulate GTP-Rac localized effector interactions through
dissociation of Rho-GDI. Nat Cell Biol 4:232–239.
21. Yamashita T, Tohyama M. 2003. The p75 receptor acts as a
displacement factor that releases Rho from Rho-GDI. Nat Neurosci
6:461–467.
22. Price LS, Langeslag M, ten Klooster JP, Hordijk PL, Jalink K, et al. 2003.
Calcium signaling regulates translocation and activation of Rac. J Biol
Chem 278:39413–39421.
23. DerMardirossian C, Schnelzer A, Bokoch GM. 2004. Phosphorylation of
RhoGDI by Pak1 mediates dissociation of Rac GTPase. Mol Cell
15:117–127.
24. Bishop AL, Hall A. 2000. Rho GTPases and their effector proteins.
Biochem J 348Pt 2:241–255.
25. del Pozo MA, Alderson NB, Kiosses WB, Chiang HH, Anderson RG,
et al. 2004. Integrins regulate Rac targeting by internalization of
membrane domains. Science 303:839–842.
26. Didsbury J, Weber RF, Bokoch GM, Evans T, Snyderman R. 1989. rac, a
novel ras-related family of proteins that are botulinum toxin substrates. J
Biol Chem 264:16378–16382.
27. Bolis A, Corbetta S, Cioce A, de Curtis I. 2003. Differential distribution of
Rac1 and Rac3 GTPases in the developing mouse brain: implications
for a role of Rac3 in Purkinje cell differentiation. Eur J Neurosci 18:2417–
2424.
28. Zalcman G, Closson V, Linares-Cruz G, Lerebours F, Honore N, et al.
1995. Regulation of Ras-related RhoB protein expression during the cell
cycle. Oncogene 10:1935–1945.
29. Vincent S, Jeanteur P, Fort P. 1992. Growth-regulated expression of
rhoG, a new member of the ras homolog gene family. Mol Cell Biol
12:3138–3148.
30. Jahner D, Hunter T. 1991. The ras-related gene rhoB is an immediate-
early gene inducible by v-Fps, epidermal growth factor, and platelet-
derived growth factor in rat fibroblasts. Mol Cell Biol 11:3682–
3690.
31. Fritz G, Kaina B. 1997. rhoB encoding a UV-inducible Ras-related small
GTP-binding protein is regulated by GTPases of the Rho family and
independent of JNK, ERK, and p38 MAP kinase. J Biol Chem
272:30637–30644.
32. Fritz G, Kaina B, Aktories K. 1995. The ras-related small GTP-binding
protein RhoB is immediate-early inducible by DNA damaging treat-
ments. J Biol Chem 270:25172–25177.
33. Fritz G, Kaina B. 2001. Transcriptional activation of the small GTPase
gene rhoB by genotoxic stress is regulated via a CCAAT element.
Nucleic Acids Res 29:792–798.
34. Jiang K, Sun J, Cheng J, Djeu JY, Wei S, et al. 2004. Akt mediates Ras
downregulation of RhoB, a suppressor of transformation, invasion, and
metastasis. Mol Cell Biol 24:5565–5576.
35. Tao W, Pennica D, Xu L, Kalejta RF, Levine AJ. 2001. Wrch-1, a novel
member of the Rho gene family that is regulated by Wnt-1. Genes Dev
15:1796–1807.
36. Wang HR, Zhang Y, Ozdamar B, Ogunjimi AA, Alexandrova E, et al.
2003. Regulation of cell polarity and protrusion formation by targeting
RhoA for degradation. Science 302:1775–1779.
37. Tu S, Cerione RA. 2001. Cdc42 is a substrate for caspases
and influences Fas-induced apoptosis. J Biol Chem 276:19656–
19663.
38. Shutes A, Berzat AC, Cox AD, Der CJ. 2004. Atypical mechanism of
regulation of the Wrch-1 Rho family small GTPase. Curr Biol 14:2052–
2056.
39. Forget MA, Desrosiers RR, Gingras D, Beliveau R. 2002. Phosphoryla-
tion states of Cdc42 and RhoA regulate their interactions with Rho GDP
dissociation inhibitor and their extraction from biological membranes.
Biochem J 361:243–254.
40. Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M,
et al. 1996. Protein kinase A phosphorylation of RhoA mediates the
morphological and functional effects of cyclic AMP in cytotoxic
lymphocytes. Embo J 15:510–519.
41. Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. 2004. Rational design
and characterization of a Rac GTPase-specific small molecule inhibitor.
Proc Natl Acad Sci USA 101:7618–7623.
42. Pelish HE, Peterson JR, Salvarezza SB, Rodriguez-Boulan E, Chen JL,
et al. 2006. Secramine inhibits Cdc42-dependent functions in cells and
Cdc42 activation in vitro. Nat Chem Biol 2:39–46.
Review articles
368 BioEssays 29.4

43. Phee H, Abraham RT, Weiss A. 2005. Dynamic recruitment of PAK1 to
the immunological synapse is mediated by PIX independently of SLP-76
and Vav1. Nat Immunol 6:608–617.
44. Brown MC, West KA, Turner CE. 2002. Paxillin-dependent paxillin
kinase linker and p21-activated kinase localization to focal adhesions
involves a multistep activation pathway. Mol Biol Cell 13:1550–1565.
45. Dharmawardhane S, Sanders LC, Martin SS, Daniels RH, Bokoch GM.
1997. Localization of p21-activated kinase 1 (PAK1) to pinocytic
vesicles and cortical actin structures in stimulated cells. J Cell Biol
138:1265–1278.
46. Mellor H, Flynn P, Nobes CD, Hall A, Parker PJ. 1998. PRK1 is targeted
to endosomes by the small GTPase, RhoB. J Biol Chem 273:4811–
4814.
47. Kosako H, Yoshida T, Matsumura F, Ishizaki T, Narumiya S, et al. 2000.
Rho-kinase/ROCK is involved in cytokinesis through the phosphoryla-
tion of myosin light chain and not ezrin/radixin/moesin proteins at the
cleavage furrow. Oncogene 19:6059–6064.
48. Mukai H, Kitagawa M, Shibata H, Takanaga H, Mori K, et al. 1994.
Activation of PKN, a novel 120-kDa protein kinase with leucine zipper-
like sequences, by unsaturated fatty acids and by limited proteolysis.
Biochem Biophys Res Commun 204:348–356.
49. Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. 2002.
Mechanism of regulation of WAVE1-induced actin nucleation by Rac1
and Nck. Nature 418:790–793.
50. Joberty G, Perlungher RR, Sheffield PJ, Kinoshita M, Noda M, et al.
2001. Borg proteins control septin organization and are negatively
regulated by Cdc42. Nat Cell Biol 3:861–866.
51. Callow MG, Zozulya S, Gishizky ML, Jallal B, Smeal T. 2005. PAK4
mediates morphological changes through the regulation of GEF- H1. J
Cell Sci 118:1861–1872.
52. Zhao L, Ma QL, Calon F, Harris-White ME, Yang F, et al. 2006. Role of
p21-activated kinase pathway defects in the cognitive deficits of
Alzheimer disease. Nat Neurosci 9:234–242.
53. Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, et al. 1997. Calcium
sensitization of smooth muscle mediated by a Rho-associated protein
kinase in hypertension. Nature 389:990–994.
54. Nagumo H, Sasaki Y, Ono Y, Okamoto H, Seto M, et al. 2000. Rho
kinase inhibitor HA-1077 prevents Rho-mediated myosin phosphatase
inhibition in smooth muscle cells. Am J Physiol Cell Physiol 278:C57–
C65.
55. Mueller BK, Mack H, Teusch N. 2005. Rho kinase, a promising drug
target for neurological disorders. Nat Rev Drug Discov 4:387–398.
56. Bustelo XR. 2002. Understanding Rho/Rac biology in T-cells using
animal models. Bioessays 24:602–612.
57. Turner M, Billadeau DD. 2002. VAV proteins as signal integrators for
multi-subunit immune-recognition receptors. Nat Rev Immunol 2:476–
486.
58. Sugihara K, Nakatsuji N, Nakamura K, Nakao K, Hashimoto R, et al.
1998. Rac1 is required for the formation of three germ layers during
gastrulation. Oncogene 17:3427–3433.
59. Corbetta S, Gualdoni S, Albertinazzi C, Paris S, Croci L, et al. 2005.
Generation and characterization of Rac3 knockout mice. Mol Cell Biol
25:5763–5776.
60. Vigorito E, Bell S, Hebeis BJ, Reynolds H, McAdam S, et al. 2004.
Immunological function in mice lacking the Rac-related GTPase RhoG.
Mol Cell Biol 24:719–729.
61. Condliffe AM, Webb LM, Ferguson GJ, Davidson K, Turner M, et al.
2006. RhoG regulates the neutrophil NADPH oxidase. J Immunol 176:
5314–5320.
62. Gu Y, Filippi MD, Cancelas JA, Siefring JE, Williams EP, et al. 2003.
Hematopoietic cell regulation by Rac1 and Rac2 guanosine tripho-
sphatases. Science 302:445–449.
63. Cancelas JA, Lee AW, Prabhakar R, Stringer KF, Zheng Y, et al. 2005.
Rac GTPases differentially integrate signals regulating hematopoietic
stem cell localization. Nat Med 11:886–891.
64. Yu H, Leitenberg D, Li B, Flavell RA. 2001. Deficiency of small GTPase
Rac2 affects T cell activation. J Exp Med 194:915–926.
65. Li B, Yu H, Zheng W, Voll R, Na S, et al. 2000. Role of the guanosine
triphosphatase Rac2 in T helper 1 cell differentiation. Science 288:
2219–2222.
66. Croker BA, Tarlinton DM, Cluse LA, Tuxen AJ, Light A, et al. 2002. The
Rac2 guanosine triphosphatase regulates B lymphocyte antigen
receptor responses and chemotaxis and is required for establishment
of B-1a and marginal zone B lymphocytes. J Immunol 168:3376–
3386.
67. Walmsley MJ, Ooi SK, Reynolds LF, Smith SH, Ruf S, et al. 2003. Critical
roles for Rac1 and Rac2 GTPases in B cell development and signaling.
Science 302:459–462.
68. Kim C, Dinauer MC. 2001. Rac2 is an essential regulator of neutrophil
nicotinamide adenine dinucleotide phosphate oxidase activation in
response to specific signaling pathways. J Immunol 166:1223–1232.
69. Li S, Yamauchi A, Marchal CC, Molitoris JK, Quilliam LA, et al. 2002.
Chemoattractant-stimulated Rac activation in wild-type and Rac2-
deficient murine neutrophils: preferential activation of Rac2 and Rac2
gene dosage effect on neutrophil functions. J Immunol 169:5043–
5051.
70. Glogauer M, Marchal CC, Zhu F, Worku A, Clausen BE, et al. 2003.
Rac1 deletion in mouse neutrophils has selective effects on neutrophil
functions. J Immunol 170:5652–5657.
71. Sun CX, Downey GP, Zhu F, Koh AL, Thang H, et al. 2004. Rac1 is the
small GTPase responsible for regulating the neutrophil chemotaxis
compass. Blood 104:3758–3765.
72. Pestonjamasp KN, Forster C, Sun C, Gardiner EM, Bohl B, et al. 2006.
Rac1 links leading edge and uropod events through Rho and myosin
activation during chemotaxis. Blood 108:2814–2820.
73. Yamauchi A, Kim C, Li S, Marchal CC, Towe J, et al. 2004. Rac2-
deficient murine macrophages have selective defects in superoxide
production and phagocytosis of opsonized particles. J Immunol
173:5971–5979.
74. Wells CM, Walmsley M, Ooi S, Tybulewicz V, Ridley AJ. 2004. Rac1-
deficient macrophages exhibit defects in cell spreading and membrane
ruffling but not migration. J Cell Sci 117:1259–1268.
75. McCarty OJ, Larson MK, Auger JM, Kalia N, Atkinson BT, et al. 2005.
Rac1 is essential for platelet lamellipodia formation and aggregate
stability under flow. J Biol Chem 280:39474–39484.
76. Thurnherr T, Benninger Y, Wu X, Chrostek A, Krause SM, et al. 2006.
Cdc42 and Rac1 signaling are both required for and act synergistically
in the correct formation of myelin sheaths in the CNS. J Neurosci
26:10110–10119.
77. Chrostek A, Wu X, Quondamatteo F, Hu R, Sanecka A, et al. 2006. Rac1
is crucial for hair follicle integrity but is not essential for maintenance of
the epidermis. Mol Cell Biol 26:6957–6970.
78. Benvenuti F, Hugues S, Walmsley M, Ruf S, Fetler L, et al. 2004.
Requirement of Rac1 and Rac2 expression by mature dendritic cells for
T cell priming. Science 305:1150–1153.
79. Qu J, Li X, Novitch BG, Zheng Y, Kohn M, et al. 2003. PAK4 kinase is
essential for embryonic viability and for proper neuronal development.
Mol Cell Biol 23:7122–7133.
80. Yamazaki D, Suetsugu S, Miki H, Kataoka Y, Nishikawa S, et al. 2003.
WAVE2 is required for directed cell migration and cardiovascular
development. Nature 424:452–456.
81. Yan C, Martinez-Quiles N, Eden S, Shibata T, Takeshima F, et al. 2003.
WAVE2 deficiency reveals distinct roles in embryogenesis and Rac-
mediated actin-based motility. Embo J 22:3602–3612.
82. Soderling SH, Langeberg LK, Soderling JA, Davee SM, Simerly R, et al.
2003. Loss of WAVE-1 causes sensorimotor retardation and reduced
learning and memory in mice. Proc Natl Acad Sci USA 100:1723–
1728.
83. Li X, Minden A. 2003. Targeted disruption of the gene for the PAK5
kinase in mice. Mol Cell Biol 23:7134–7142.
84. Liu AX, Rane N, Liu JP, Prendergast GC. 2001. RhoB is dispensable for
mouse development, but it modifies susceptibility to tumor formation as
well as cell adhesion and growth factor signaling in transformed cells.
Mol Cell Biol 21:6906–6912.
85. Hakem A, Sanchez-Sweatman O, You-Ten A, Duncan G, Wakeham A,
et al. 2005. RhoC is dispensable for embryogenesis and tumor initiation
but essential for metastasis. Genes Dev 19:1974–1979.
86. Thumkeo D, Shimizu Y, Sakamoto S, Yamada S, Narumiya S. 2005.
ROCK-I and ROCK-II cooperatively regulate closure of eyelid and
ventral body wall in mouse embryo. Genes Cells 10:825–834.
Review articles
BioEssays 29.4 369

87. Shimizu Y, Thumkeo D, Keel J, Ishizaki T, Oshima H, et al. 2005. ROCK-
I regulates closure of the eyelids and ventral body wall by
inducing assembly of actomyosin bundles. J Cell Biol 168:941–
953.
88. Thumkeo D, Keel J, Ishizaki T, Hirose M, Nonomura K, et al. 2003.
Targeted disruption of the mouse rho-associated kinase 2 gene results
in intrauterine growth retardation and fetal death. Mol Cell Biol 23:5043–
5055.
89. Di Cunto F, Imarisio S, Hirsch E, Broccoli V, Bulfone A, et al. 2000.
Defective neurogenesis in citron kinase knockout mice by altered
cytokinesis and massive apoptosis. Neuron 28:115–127.
90. Cunto FD, Imarisio S, Camera P, Boitani C, Altruda F, et al. 2002.
Essential role of citron kinase in cytokinesis of spermatogenic
precursors. J Cell Sci 115:4819–4826.
91. Takahashi H, Koshimizu U, Miyazaki J, Nakamura T. 2002. Impaired
spermatogenic ability of testicular germ cells in mice deficient in the
LIM-kinase 2 gene. Dev Biol 241:259–272.
92. Behrends J, Clement S, Pajak B, Pohl V, Maenhaut C, et al. 2005.
Normal thyroid structure and function in rhophilin 2-deficient mice. Mol
Cell Biol 25:2846–2852.
93. Chen F, Ma L, Parrini MC, Mao X, Lopez M, et al. 2000. Cdc42 is
required for PIP(2)-induced actin polymerization and early development
but not for cell viability. Curr Biol 10:758–765.
94. Yang L, Wang L, Zheng Y. 2006. Gene Targeting of Cdc42 and
Cdc42GAP Affirms the Critical Involvement of Cdc42 in Filopodia
Induction, Directed Migration, and Proliferation in Primary Mouse
Embryonic Fibroblasts. Mol Biol Cell 17:4675–4685.
95. Czuchra A, Wu X, Meyer H, van Hengel J, Schroeder T, et al. 2005.
Cdc42 is not essential for filopodium formation, directed migration, cell
polarization, and mitosis in fibroblastoid cells. Mol Biol Cell 16:4473–
4484.
96. Cappello S, Attardo A, Wu X, Iwasato T, Itohara S, et al. 2006. The Rho-
GTPase cdc42 regulates neural progenitor fate at the apical surface.
Nat Neurosci 9:1099–1107.
97. Snapper SB, Rosen FS, Mizoguchi E, Cohen P, Khan W, et al. 1998.
Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP
in T but not B cell activation. Immunity 9:81–91.
98. Zhang H, Schaff UY, Green CE, Chen H, Sarantos MR, et al. 2006.
Impaired integrin-dependent function in Wiskott-Aldrich syndrome
protein-deficient murine and human neutrophils. Immunity 25:285–295.
99. Calle Y, Jones GE, Jagger C, Fuller K, Blundell MP, et al. 2004. WASp
deficiency in mice results in failure to form osteoclast sealing zones and
defects in bone resorption. Blood 103:3552–3561.
100. Li S, Wang Q, Chakladar A, Bronson RT, Bernards A. 2000. Gastric
hyperplasia in mice lacking the putative Cdc42 effector IQG AP1. Mol
Cell Biol 20:697–701.
Review articles
370 BioEssays 29.4