hepatitis b virus large surface antigen promotes liver ... · hepatitis b virus large surface...
TRANSCRIPT

Molecular and Cellular Pathobiology
Hepatitis B Virus Large Surface Antigen Promotes LiverCarcinogenesis by Activating the Src/PI3K/Akt Pathway
Haiou Liu1,3, Jiejie Xu1,2, Lei Zhou1, Xiaojing Yun1, Lin Chen1, Shanshan Wang1, Linlin Sun1, Yumei Wen2, andJianxin Gu1
AbstractOf the three envelope glycoproteins encoded by hepatitis B virus (HBV) that are collectively referred to as HBV
surface antigen (HBsAg), the large HBsAg (LHBs) glycoprotein is expressed preferentially in HBV-associatedhepatocellular carcinoma. LHBs can act as an oncogene in transgenicmice, but how it contributes functionally tohepatocarcinogenesis remains unclear. In this study, we determined the molecular and functional roles of LHBsduring HBV-associated hepatocarcinogenesis. LHBs increased tumor formation of hepatoma cells. Moreover,expression of LHBs but not other HBV envelope glycoproteins specifically promoted proliferation of hepatomaand hepatic cells in vitro. Mechanistic investigations revealed that these effects were caused by activation of theSrc/PI3K/Akt pathway through proximal stimulation of PKCa/Raf1 signaling by LHBs. Proliferation induced bystable LHBs expression was associated with increased G1–S cell-cycle progression and apoptosis resistancemediated by Src kinase activation, as established in hepatocellular carcinoma clinical specimens. Importantly,LHBs-induced cellular proliferation and tumor formation were reversed by administration of the Src inhibitorsaracatinib. Together, our findings suggest that LHBs promotes tumorigenesis of hepatoma cells by triggering aPKCa/Raf1 to Src/PI3K/Akt signaling pathway, revealing novel insights into the underlying mechanisms of HBV-associated hepatocarcinogenesis. Cancer Res; 71(24); 7547–57. �2011 AACR.
Introduction
More than 350 million people worldwide are chronicallyinfected with hepatitis B virus (HBV), which is a leading causefor chronic hepatitis, cirrhosis, and hepatocellular carcinoma(HCC; refs. 1, 2). Although chronic HBV infection has beennoted to be associatedwith the development of HCC soon afterthe discovery of HBV surface antigen (HBsAg), the oncogenicfunctions of HBV are not completely known, partly because ofits noncytopathic characteristic (3). Progression from chronichepatitis B toHCC involves direct effects of the virus protein onthe cellular function of infected hepatocytes, as well as indirecteffects through the process of inflammation, regeneration, and
cirrhosis because ofHBV infection (4, 5). Regarding the effect ofviral proteins expression on hepatocellular malignant trans-formation, several HBV proteins have been found in infectedtissues more frequently than others, including large HBsAg(LHBs), C-terminally truncated middle HBsAg (MHBs), HBV Xprotein (HBx), and a novel spliced transcript of HBV, referredto as hepatitis B–spliced protein (HBSP; ref. 6).
The viral genome of HBV encodes for 4 overlapping openreading frames, which include preS1/preS2/S, preC/C, X, and P(2). The preS1/preS2/S region of the virus genome encodes the3 viral surface antigens by differential initiation of translationat each of 3 in-frame initiation codons (7). Initiation at thenearest start codon (S) produces the small HBsAg (SHBs),which is the most abundant protein in hepatitis B patients.Initiation at the more upstream start codon (preS2) generatesthe MHBs, the function of which is still unknown. Initiation atthe most upstream start codon (preS1) yields the LHBs, whichis thought to play pivotal roles in binding of the virus to hostcell receptors and in the assembly of the virion and its releasefrom the cell (7, 8).
Previous study has shown that HBV sequences introducedinto transgenic mice are able to confer only a tissue-specificexpression of HBsAg rather than any other HBV proteins (9).Chisari and colleagues have shown that HBV transgenic micethat overproduce LHBs and accumulate toxic quantities ofSHBs within the hepatocytes develop severe, prolonged hepa-tocellular injury that initiates a programmed responsewith theliver, characterized by inflammation, regenerative hyperplasia,transcriptional deregulation, aneuploidy, and eventually pro-gresses to neoplasia (10). Another study has also proved that
Authors' Affiliations: 1Department of Biochemistry andMolecular Biology,2Key Laboratory of Medical Molecular Virology, Ministry of Education andHealth, Shanghai Medical College, Fudan University, Shanghai; and 3Insti-tute of Nautical Medicine, Nantong University, Nantong, China
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
H. Liu and J. Xu contributed equally to this work.
Corresponding Author: Jianxin Gu, Department of Biochemistry andMolecular Biology, Shanghai Medical College, Fudan University, Shanghai200032, China. Phone: 86-21-54237704; Fax: 86-21-64437703; E-mail:[email protected] and Yumei Wen, Key Laboratory of Medical MolecularVirology, Ministry of Education and Health, Shanghai Medical College,Fudan University, Shanghai 200032, China. Fax: 86-21-54237603; E-mail:[email protected]
doi: 10.1158/0008-5472.CAN-11-2260
�2011 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 7547
on January 28, 2020. © 2011 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2011; DOI: 10.1158/0008-5472.CAN-11-2260

HBsAg-positive ground-glass hepatocytes emerged through-out the liver parenchyma in nearly all HBV transgenicmice, butthe extensive expression of HBsAg is gradually downregulatedduring neoplastic transformation, just as the morphologic andbiochemical phenotypes of foci of altered hepatocytes, hepaticadenoma, and HCC in transgenic mice, resembling thosedescribed in chemical hepatocarcinogenesis (11). Althoughthe critical indirect roles of LHBs during malignant transfor-mation in transgenic mice have been observed previously, thedirect effects of LHBs on hepatocellular function remain poorlyunderstood.
Extensive studies over past years have identified aberrantactivation of major signaling cascades such as PI3K/Akt/mTOR pathway, Raf/MEK/ERK pathway, WNT/b-cateninpathway, and HGF/c-MET pathway involved in pathogenesisof HCC (12–15). Abrupt alterations that occur in liver tissueswithHBV infection cause significant changes in several cellularsignaling including WNT/b-catenin, p53, pRb, mitogen—acti-vated protein kinase (MAPK), NF-kB pathways and alter geneexpression resulting in hepatoma formation because ofincreased proliferation, cell-cycle progression, and apoptosisresistance (5). As a dominant nonreceptor tyrosine kinaseactivated in HCC carcinogenesis, abnormal Src signaling acti-vation conferred by HBx expression has been reported toparticipate in HBV-associated hepatocarcinogenesis (16–18).However, the interaction between Src signal activation withLHBs expression during HBV infection has not beencharacterized.
In this study, we sought to determine the direct oncogenicfunction of LHBs expression in HBV-associated hepatocarci-nogenesis. Our present investigation reveals that LHBs expres-sion promotes tumorigenesis of hepatoma cells dependent onPKCa/Raf1/Src/PI3K/Akt signal activation in vitro and in vivo,which may shed a new light into the molecular mechanismsunderlying HBV-associated hepatocarcinogenesis and providea promising therapeutic target for patients with HCC withchronic HBV infection.
Materials and Methods
Cell culture and human HCC samplesOne immortalized hepatic cell line (L02) and 2 hepatoma cell
lines (Huh7 and SK-Hep1), in addition to an African greenmonkey kidney epithelial cell line (Vero), were obtained direct-ly from Shanghai Cell Bank of Chinese Academy of Sciences(Shanghai, China) and cultured in Dulbecco's Modified Eagle'sMedium supplemented with 10% FBS at 37�C in a humidified5%CO2 incubator. The cell lines have been characterized at thebank by DNA fingerprinting analysis using STR (short tandemrepeat)markers. All cell lineswere placed under cryostage afterthey were obtained from the bank and used within 6months ofthawing fresh vials. Thirty-seven pairs of frozen fresh tumorliver tissues and their peripheral nontumor tissues after sur-gical resection were collected frompatients withHCCwho hadreceived neither chemotherapy nor radiotherapy before sur-gical resection in Nantong Tumor Hospital (Jiangsu) withinformed consent and Institutional Review Board approvalbetween 2004 and 2008.
Construction of plasmidsThe plasmids containing LHBs (pcDNA3-LHBs-flag), MHBs
(pcDNA3-MHBs-flag), SHBs (pcDNA3-SHBs-flag), Akt(pcDNA3.1-Akt), and Raf1 (pcDNA3.1-Raf1) were generated aspreviously described (19–22). The dominant-negative mutantRaf1-S621A containing a serine-to-alanine mutation at aminoacid 621 was constructed on the basis of aforementioned Raf1expression plasmid. The plasmid containing Src (pcDNA3.1-Src) was a generous gift from Dr. Jianguo Gu (Tohoku Phar-maceutical University, Miyagi, Japan). The kinase deadmutantSrc-K295M plasmid and dominant-negative mutant Akt-K179M plasmid were kindly provided by Dr. Mien-Chie Hung(MD Anderson Cancer Center, Houston, TX). All plasmidconstructs were confirmed by DNA sequencing.
Plasmids transfection and RNA interferenceTransient and stable transfections with various plasmids
were carried out as previously described (23). Two siRNAsagainst SRC gene Src siRNA (h), 2 siRNAs against AKT1/2 geneAkt1/2 siRNA (h), 2 siRNAs against PKCa gene PKCa siRNA (h),2 siRNAs against RAF1 gene Raf1 siRNA (h), and correspondingcontrol siRNA-A (Santa Cruz Biotechnology) were transfectedinto Huh7 and SK-Hep1 cells in 6-well plates using theX-tremeGENE siRNA Transfection Reagent (Roche AppliedScience) according to the manufacturer's instructions. Genesilencing effect was confirmed by Western blot analysis andRT-PCR at 72 hours posttransfection.
Western blottingProtein extraction from cultured cells or tumor tissues and
Western blot analysis were carried out as previously described(23). Primary antibodies used included those against LHBs,GAPDH (Santa Cruz Biotechnology), Akt, pAkt(S473), Src, pSrc(Y416), Raf1, pRaf1(S338), PKCa, cyclin D1, cyclin D3, CDK4,CDK6, cleaved caspase-3 (Cell Signaling Technology), andproliferating cell nuclear antigen (PCNA; BD Biosciences).
Tumor xenograft experiments and saracatinib(AZD0530) treatment
Tumor xenograft experiments in nudemicewere carried outas previously described (23). The specific Src inhibitor sara-catinib (Selleck) was dissolvedwith dimethyl sulfoxide (DMSO)for 5 mg/mL additive stock solution. Saracatinib gavage solu-tion or vehicle control gavage solution was prepared freshly bycombination saracatinib additive stock solution or DMSOwithcorn oil at a ratio of 95% corn oil: 5% DMSO. Subcutaneoustumor xenografted nudemice were fedwith saracatinib gavagesolution or vehicle control gavage solution daily at a dose of25 mg/kg body weight in vehicle corn oil via oral gavage for4 weeks as hepatoma cells subcutaneous injection.
Histology, immunohistochemical analysis, andevaluation
Tumor sections from subcutaneous tumor xenograftednude mice and patients with HCC were hematoxylin and eosin(H&E) stained and immunohistochemically analyzed asdescribed previously (24, 25). Primary antibodies used includedthose against LHBs, Ki67 (Millipore), pAkt(S473), and pSrc
Liu et al.
Cancer Res; 71(24) December 15, 2011 Cancer Research7548
on January 28, 2020. © 2011 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2011; DOI: 10.1158/0008-5472.CAN-11-2260

(Y416). The intensity of immunohistochemical staining in thetumor cells was scored independently by 2 pathologists usingthe semiquantitative IRS (immunoreactive score) scale accord-ing to Remmele and Stegner (26).
Colony formation assay, cell proliferation assay,bromodeoxyuridine incorporation assay, cell-cycleanalysis, and Annexin V/PI stainingColony formation assay, cell proliferation assay, bromodeox-
yuridine (BrdUrd) incorporation assay, and cell-cycle analysiswere carried out as previously described (23). AnnexinV/propidium iodide (PI) staining was carried out by using theAnnexin V–FITC Apoptosis Detection Kit (BD Biosciences)according to the manufacturer's instructions.
Statistical analysisExperimental data were presented as mean� SD or SEM of
at least 3 independent replicates through analyzing withGraphPad Prism 5 (GraphPad Software) and assessing com-parisons between different groups by the Student t test, one-way ANOVA. The association between LHBs and clinicopath-ologic characteristics was assessed using the Fisher exact test.The correlation between LHBswith Ki67, pSrc(Y416), and pAkt(S473) staining obtained by immunohistochemistry was deter-mined using the Spearman correlation test. Differences wereconsidered significant at values of P < 0.05.
Results
LHBs expression promotes tumor formation ofhepatoma cells in vivo and in vitro
To elucidate the effect of LHBs expression on tumorformation in vivo, tumor xenograft experiments were carriedout in nude mice with Huh7 cells stably transfected withLHBs. As shown in Fig. 1A, stable LHBs expression in clones1 and 2 compared with control Huh7 cells was confirmed byWestern blot analysis, and Huh7-LHBs clone 2 was selectedfor further investigation because of higher stable LHBsexpression. Tumor xenograft experiments showed that LHBsexpression significantly accelerated overall tumor growthcompared with the control group as assessed by the tumorvolume (Fig. 1B). Four weeks after tumor xenograft, nudemice were sacrificed and subcutaneous tumor tissues wereexamined. Notably, LHBs stably expressing Huh7 cellsshowed increased tumor growth compared with controlcells as determined through tumor photography and weightmeasurement (Fig. 1C and D). H&E staining showed moreaggressive tumor growth gained by LHBs stably transfectedHuh7 cells than control cells in nude mice (Fig. 1E). Becausethe initiating ATG codons for the MHBs and SHBs have beenmutated to ACG in the plasmid containing LHBs, aforemen-tioned provocative tumor formation should be attributed toLHBs expression but not MHBs or SHBs expression. To
Figure 1. LHBs expressionpromotes tumor formation ofhepatoma cells in nude mice. A,Western blot analysis was carriedout to confirm stable LHBsexpression in Huh7 cells (clones 1and 2). B, in vivo subcutaneoustumor growth curves of Huh7-LHBs(clone 2) and Huh7-Control cells(n ¼ 6). �, P < 0.05 compared withHuh7-Control. C, images of allharvested 6 subcutaneous tumorsfrom 3 nude mice of each group areshown. D, total tumor weight fromeach group of mice was calculatedand shown. �, P ¼ 0.008. E, H&Estaining images of representativesubcutaneous tumors. Tu, tumortissue; black scale bar, 50 mm.GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
A C
B
E
D
Clone
LHBs
GAPDH
Huh7-Control
Huh7-Control
*(P = 0.008)
1.5
1.0
0.5
0.0
0.5
0.4
0.3
0.2
0.1
0.0
Huh7-LHBs
Huh7-Control Huh7-LHBs
Huh7-Control Huh7-LHBs
7 14 21
Days after tumor xenograft
Tum
or
volu
me
(cm
3 )
Tum
or
wei
gh
t (g
)
28
*
*
*
Huh7-LHBs
Con 1 2
LHBs Promotes Hepatocarcinogenesis via Src/PI3K/Akt Signal
www.aacrjournals.org Cancer Res; 71(24) December 15, 2011 7549
on January 28, 2020. © 2011 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2011; DOI: 10.1158/0008-5472.CAN-11-2260

further assess the oncogenic effect of LHBs, MHBs, and SHBsexpression on hepatocarcinogenesis, colony formation assayand cell proliferation assay were carried out in Huh7, SK-Hep1, and L02 cells after transiently transfection with LHBs,MHBs, and SHBs. LHBs expression, but not MHBs or SHBsexpression were found to significantly promote both colony
formation (Fig. 2A–C) and cellular proliferation (Fig. 2D) ofHuh7, SK-Hep1, and L02 cells. In contrast, Vero cells under-going identical experiments did not show increased colonyformation despite LHBs expression (data not shown). Takentogether, these data show that LHBs expression could pro-mote tumor formation of hepatoma cells in vivo and in vitro.
A
D
E F
B CHuh7Control LHBs
MHBs
NSNS
NS
NS
NS
NS
* * *
**
*
*
*
**
**
**
**
SHBs
Control
0 12 24 48
Num
ber
of soft a
gar
colo
nie
s
Num
ber
of soft a
gar
colo
nie
s
Num
ber
of soft a
gar
colo
nie
s
40
30
20
10
0
Absorb
ance a
t 450 n
mR
atio o
f absorb
ance a
t 450 n
m
rela
tive
to u
ntr
eate
d c
ontr
ol
Ratio o
f absorb
ance a
t 450 n
m
rela
tive
to u
ntr
eate
d c
ontr
ol
3
2
1
0
2.0
1.5
1.0
0.5
0.0
2.0
1.5
1.0
0.5
0.0
Absorb
ance a
t 450 n
m 2.5
2.0
1.5
1.0
0.5
0.0 Absorb
ance a
t 450 n
m 2.5
2.0
1.5
1.0
0.5
0.0
30
20
10
0
15
10
5
0LHBs MHBs SHBs
ControlLHBsMHBsSHBs
LHBs LHBs
ControlLHBsMHBsSHBs
ControlLHBsMHBsSHBs
Control LHBs MHBs SHBs Control LHBs MHBs SHBs
SK-Hep1Control LHBs
MHBs SHBs
L02
Huh7
Time (h)0 12 24 48
Time (h)0 12 24 48
Time (h)
SK-Hep1
Huh7 SK-Hep1
L02
Control LHBs
MHBs SHBs
Con
trol
DM
SO
LY29
4002
Wor
tman
nin
PD
-980
59
U01
26
SB
2035
80
PD
TC
PP
2
Rap
amyc
in
BA
Y11
7082
Con
trol
DM
SO
LY29
4002
Wor
tman
nin
PD
-980
59
U01
26
SB
2035
80
PD
TC
PP
2
Rap
amyc
in
BA
Y11
7082
Figure 2. LHBs expression rather thanMHBs or SHBs expression exerts oncogenic promotion in hepatic cells. A–C, top, colony formation assay for Huh7 cells(A), SK-Hep1 cells (B), and L02 cells (C) transiently transfected with empty vector (control), LHBs plasmid (LHBs), MHBs plasmid (MHBs), and SHBs plasmid(SHBs), respectively. Bottom, quantification of colonies. �, P < 0.05 compared with control. NS, not significant. D, cell proliferation assay for Huh7 cells (left),SK-Hep1 cells (middle), and L02 cells (right) transiently transfected with empty vector (control), LHBs plasmid (LHBs), MHBs plasmid (MHBs), and SHBsplasmid (SHBs), respectively. �,P < 0.05 comparedwith control. E and F, cell proliferation assay for Huh7 (E) and SK-Hep1 cells (F) transiently transfectedwithempty vector (control) and LHBs plasmid (LHBs), respectively, after treatment for 24 hours with DMSO, LY294002 (50 mmol/L), Wortmannin (10 nmol/L),PD098059 (50 mmol/L), U0126 (20 mmol/L), SB203580 (10 mmol/L), PDTC (10 mmol/L), BAY117082 (10 mmol/L), PP2 (10 mmol/L), or rapamycin (10 nmol/L).�, P < 0.05.
Liu et al.
Cancer Res; 71(24) December 15, 2011 Cancer Research7550
on January 28, 2020. © 2011 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2011; DOI: 10.1158/0008-5472.CAN-11-2260

LHBs expression activates PKCa/Raf1/Src/PI3K/Aktsignal pathway in hepatoma cellsTo characterize the mechanistic signaling pathway under-
lying instigated tumor formation conferred by LHBs expres-sion in hepatoma cells, cell proliferation assay was carried outin LHBs expressing Huh7 and SK-Hep1 cells after treatmentwith small molecular inhibitors against phosphoinositide 3-kinase (PI3K; LY294002, Wortmannin), extracellular signal–regulated kinase (ERK; PD98059), MAP/ERK kinase (MEK;U0126), p38/MAPK (SB203580), NF-kB (PDTC, BAY117082),Src (PP2), and mTOR (rapamycin). As shown in Fig. 2E and F,no significant change of the proliferation ratio was observedafter PD98059, U0126, SB203580, PDTC, and BAY117082 treat-ment compared with DMSO treatment in LHBs-Huh7 andLHBs-SK-Hep1 cells. However, a considerable inhibition of theproliferation ratio was noted after LY294002, Wortmannin,rapamycin, and PP2 treatment compared with DMSO treat-ment in LHBs-Huh7 and LHBs-SK-Hep1 cells (Fig. 2E and F).Moreover, Western blot analysis showed that the phosphory-
lation level of Src(Y416) and Akt(S473) were much higher inLHBs-Huh7 cells with respect to the control Huh7 cells(Fig. 3A), indicating LHBs expression could activate Src andAkt signals in hepatoma cells. Consistent with aforementionedphenomenon in vitro, Western blot analysis of subcutaneoustumor tissues also showed increased pSrc(Y416) and pAkt(S473) levels in 3 intersected tumor tissues generated byHuh7-LHBs cells compared with control Huh7 cells fromxenografted nude mice (Supplementary Fig. S1). All these dataindicate that bothPI3K/Akt/mTORand Src signaling pathwaysmight involve in instigated tumor formation conferred by LHBsexpression.
To further illuminate the regulatory relationship betweenSrc and PI3K/Akt signals activation induced by LHBs expres-sion, Western blot analysis was carried out in Huh7 cells afterSrc and PI3K/Akt signal inhibition. To assess RNA interferenceknockdown effect on Src and Akt signals activation, 2 siRNAsagainst Src and 2 siRNAs against Akt1/2 were prepared andtransfected into Huh7 cells to evaluate their knockdown
A B C
D E F
Control
pSrc(Y416)
pAkt(S473)
Src
Akt
LHBs
GAPDH
pSrc(Y416)
pAkt(S473)
Src
Akt
LHBs
GAPDH
pSrc(Y416)
pAkt(S473)
Src
Akt
LHBs
GAPDH
LHBsDMSO
PP2Fold difference
Fold difference Fold difference
+
* *
* *
–––
–+––
–++–
–+–+
1.16±0.10
1.16±0.05 1.14±0.10 0.94±10.05 * * * *1.25±0.11 1.28±0.13 1.09±0.08
0.9±0.11.22±0.13
Control
pSrc(Y416)
pAkt(S473)
Src
Akt
LHBs
GAPDH
pSrc(Y416)
pAkt(S473)
Src
Akt
LHBs
GAPDH
pSrc(Y416)
pAkt(S473)
Src
Akt
LHBs
GAPDH
LHBsDMSO
LY294002Fold difference
ControlLHBs
DMSOAkt K179M
Fold difference
ControlLHBs
NS siRNAAkt siRNA #1Akt siRNA #2
Fold difference
Fold difference Fold difference
+
* * * * * * * * * *
* * * * **
–––
–+––
–++–
–+–+
+–––
+–
–
––
–+
–
––
–+
+
––
–+
–
+–
–+
–
–+
–+––
–++–
–+–+
1.24±0.13 1.37±0.21 1.49±0.25 1.42±0.2 1.32±0.13 1.46±0.071.83±0.15 1.8±0.06
1.37±0.2 1.32±0.221.03±10.08 1.26±0.18 1.47±0.28 1.02±0.1 2.05±0.08 2.32±0.03 1.02±0.04 0.92±0.06
1.11±0.051.18±0.12
ControlLHBs
Src WTSrc K295M
Fold difference
ControlLHBs
NS siRNASrc siRNA #1Src siRNA #2
Fold difference
+
* * **
–––
+––––
–+–––
–++––
–+–+–
–+––+
–+––
–++–
–+–+
1.17±0.09 1.66±0.32
2.08±0.14 1.8±0.09 1.12±0.04 1.17±0.08
1.67±0.260.96±0.02 0.82±0.030.92±0.081.28±0.12
Figure 3. LHBs expression activates Src/PI3K/Akt signal pathway in hepatoma cells. A, Western blot analysis for Huh7 cells after transient LHBs expressionunder treatment for 24 hours with PP2 (10 mmol/L). B, Western blot analysis for Huh7 cells transiently transfected with empty vector, LHBs plasmid, orcotransfected LHBs plasmid with Src wild-type (WT) plasmid or Src K295M plasmid together. C, Western blot analysis for Huh7 cells transiently transfectedwith empty vector, LHBs plasmid, or cotransfected LHBs plasmid with nonsilencing (NS) siRNA, Src siRNA#1, or Src siRNA#2 together. D, Western blotanalysis for Huh7 cells after transient LHBs expression under treatment for 24 hours with LY294002 (50 mmol/L). E, Western blot analysis for Huh7cells transiently transfected with empty vector, LHBs plasmid, or cotransfected LHBs plasmid with Akt WT plasmid or Akt K179M plasmid together.F,Western blot analysis for Huh7 cells transiently transfectedwith empty vector, LHBs plasmid, or cotransfected LHBs plasmid with NS siRNA, Akt siRNA#1,or Akt siRNA#2 together. �, P < 0.05 compared with the left lane. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
LHBs Promotes Hepatocarcinogenesis via Src/PI3K/Akt Signal
www.aacrjournals.org Cancer Res; 71(24) December 15, 2011 7551
on January 28, 2020. © 2011 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2011; DOI: 10.1158/0008-5472.CAN-11-2260

efficiency by using RT-PCR and Western blot analysis (Sup-plementary Fig. S2A and S2B). Interestingly, specific Src inhib-itor PP2 treatment or Src siRNA cotransfection reversedincreased phosphorylation level of Akt(S473) by LHBstransfection in Huh7 cells, suggesting LHBs might promotePI3K/Akt activation through Src signal pathway (Fig. 3A andC). To clarify the functional role of Src tyrosine kinase activa-tion in LHBs-induced Akt activation, kinase dead mutant SrcK295M plasmid cotransfection was used to competing endog-enous Src tyrosine kinase activity. Inhibition of Src tyrosinekinase activationmediated by Src K295M plasmid transfectioncould dramatically reverse upregulated phosphorylation levelof Akt(S473) by LHBs expression in Huh7 cells (Fig. 3B).However, blockade PI3K activity with its specific inhibitorLY294002, which could significantly downregulate theincreased Akt(S473) phosphorylation level, had no effect onenhanced Src(Y416) phosphorylation level induced by LHBs
expression (Fig. 3D). Consistent with the aforementionedphenomenon, both inhibition of endogenic Akt expressionwith Akt siRNA transfection and prohibition endogenic Aktkinase activity with dominant-negative mutant Akt K179Mplasmid transfection could decrease Akt(S473) phosphoryla-tion level without effect on Src(Y416) phosphorylation level inLHBs expressing Huh7 cells (Fig. 3E and F). These results showthat LHBs expression could induce PI3K/Akt activation viainstigating Src tyrosine kinase activity in hepatoma cells.
Because previous study has proved that PreS2 activatorsincluding LHBs and C-terminally truncated MHBs triggeredPKCa/b-dependent activation of Raf1/Erk2 signaling, result-ing in an increased hepatocyte proliferation rate in transgenicmice (27), we hypothesized that PKC-dependent Raf1 activa-tion might establish a mechanistic link between LHBs expres-sion with Src kinase activation. As shown in Fig. 4A, significantdecreased Src(Y416) and Raf1 (S338) phosphorylation levels
A
D E
B CLHBs LHBs
LHBsVehicle
PKC pseudo substrate
NS siRNA
LHBsRaf1 WT
Raf1 S621A
Fold difference
LHBs
PKCα siRNA #1
PKCα siRNA #2
Go6976
7.03±0.52 0.5±0.11 0.73±0.14 6.25±0.23 7.76±0.34 1.01±0.14 2.52±0.3
1.42±0.14 1.29±0.12 0.79±0.28 0.94±0.13
2.91±0.24 2.93±0.11 1.05±0.03 2.2±0.17
1.75±0.23 1.39±0.15 1.86±0.21
2.4±0.2 1.02±0.11
Safingol
CGP53353LY333531
GW5074Fold difference Fold difference
NS siRNAPKCα siRNA #1PKCα siRNA #2
Fold difference
Fold difference
Huh7
pSrc(Y416)
Src
pRaf1(S338)
Raf1
LHBs
GAPDH
pSrc(Y416)
Src
pRaf1(S338)
Raf1
LHBs
GAPDH
pSrc(Y416)
Src
pRaf1(S338)
Raf1
LHBs
GAPDH
pSrc(Y416)
Src
pRaf1(S338)
Raf1
LHBs
GAPDH
pSrc(Y416)
Src
pRaf1(S338)
Raf1
LHBs
GAPDH
pSrc(Y416)
Src
PKCα
LHBs
GAPDH
pSrc(Y416)
Src
LHBs
GAPDH
pSrc(Y416)
Src
LHBs
GAPDH
pSrc(Y416)
Src
PKCα
LHBs
GAPDH
pSrc(Y416)
Src
pRaf1(S338)
Raf1
LHBs
GAPDH
Fold difference
SK-Hep1
Huh7
Fold difference
SK-Hep1
Huh7
Fold difference
SK-Hep1
Huh7
Fold difference
SK-Hep1
Huh7
Fold difference
SK-Hep1
––
–
–––
+––
++–
+–
––––
+–––
++––
+–+–
+––++
–––
+––
++–
+–+
–
–
–
+
–
–
–
+
+
–
–
+
–
+
–
+
–
–
+
––––
+
*
1.95±0.21
1.65±0.24 1.49±0.17 0.94±0.23
1.11±0.18 1.45±0.25 1.64±0.11 2.21±0.37 0.65±0.21*
*
1.16±0.16 1.34±0.13 0.95±0.11* 2.01±0.52 1.81±0.36 0.97±0.14 0.46±0.23*
1.84±0.12 1.56±0.25 0.81±0.15 0.95±0.12* *
**
*
* *
* * * * *
*
* *
*
*
–––––
++––––
+–+–––
+––+––
+–––+–
+––––+
Figure 4. LHBs expression induces Src activation dependent on PKCa/Raf1 signal pathway. A, Western blot analysis for Huh7 and SK-Hep1 cells aftertransient LHBs expression under treatment for 24 hours with Go6976 (100 nmol/L), safingol (10 mmol/L), LY333531 (10 nmol/L), CGP53353 (2 mmol/L), orGW5047 (1 mmol/L). B, Western blot analysis for Huh7 and SK-Hep1 cells transiently transfected with empty vector, LHBs plasmid, or cotransfectedLHBs plasmid with nonsilencing (NS) siRNA, PKCa siRNA#1, or PKCa siRNA#2 together. C,Western blot analysis for Huh7 and SK-Hep1 cells after transientLHBs expression under treatment for 24 hours with PKC pseudosubstrate (50 mmol/L) or its vehicle. D, Western blot analysis for Huh7 and SK-Hep1 cellstransiently transfected with empty vector, LHBs plasmid, or cotransfected LHBs plasmid with Raf1 WT or Raf1 S621A together. E, Western blot analysisfor Huh7 and SK-Hep1 cells transiently transfected with empty vector, LHBs plasmid, or cotransfected LHBs plasmid with NS siRNA, Raf1 siRNA#1,or Raf1 siRNA#2 together. �, P < 0.05 compared with the left lane. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Liu et al.
Cancer Res; 71(24) December 15, 2011 Cancer Research7552
on January 28, 2020. © 2011 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2011; DOI: 10.1158/0008-5472.CAN-11-2260
were observed after Go6976 (PKCa/b inhibitor), safingol(PKCa inhibitor), and GW5047 (Raf1 inhibitor) treatment,despite no significant change of Src(Y416) phosphorylationlevels after LY333531 (PKCb inhibitor) and CGP53353 (PKCbIIinhibitor) treatment, compared with DMSO treatmentin LHBs-expressing Huh7 and SK-Hep1 cells, suggestingPKCa/Raf1 signal activity is required for LHBs-induced Srckinase activation. To verify the crucial role of PKCa/Raf1 signalin LHBs-induced Src kinase activation in hepatoma cells, Src(Y416) phosphorylation levels were assessed after endogenousPKCa or Raf1 activity inhibitions in LHBs-expressingHuh7 andSK-Hep1 cells. Consistent with the above mentioned result,both inhibition of endogenous PKCa activity with PKCa siRNAcotransfection (Fig. 4B) or competitive PKC pseudosubstratetreatment (Fig. 4C), and prohibition of endogenous Raf1 activ-ity with dominant-negative Raf1 S621A mutant cotransfection(Fig. 4D) or Raf1 siRNA cotransfection (Fig. 4E) decreased Src(Y416) phosphorylation levels in LHBs-transfected Huh7 andSK-Hep1 cells. Taken together, these data reveal that LHBs
expression induces Src/PI3K/Akt signal activation dependenton PKCa/Raf1 pathway in hepatoma cells.
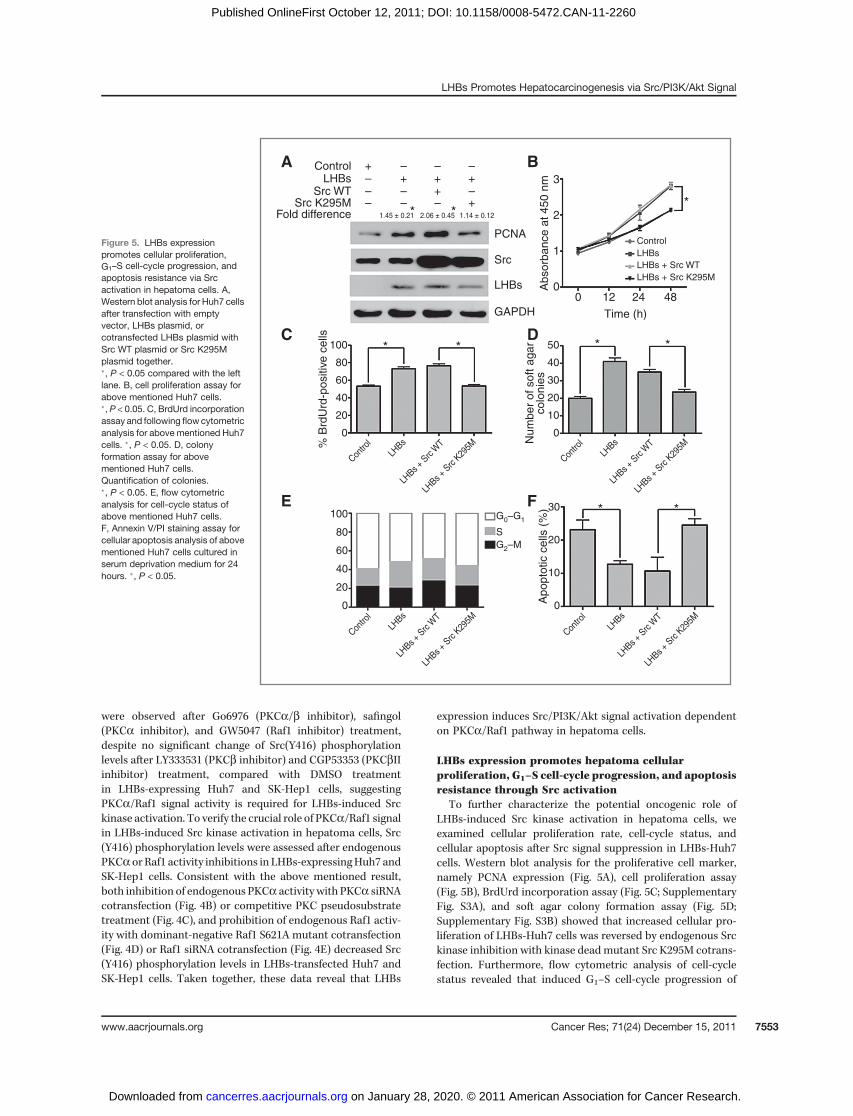
LHBs expression promotes hepatoma cellularproliferation, G1–S cell-cycle progression, and apoptosisresistance through Src activation
To further characterize the potential oncogenic role ofLHBs-induced Src kinase activation in hepatoma cells, weexamined cellular proliferation rate, cell-cycle status, andcellular apoptosis after Src signal suppression in LHBs-Huh7cells. Western blot analysis for the proliferative cell marker,namely PCNA expression (Fig. 5A), cell proliferation assay(Fig. 5B), BrdUrd incorporation assay (Fig. 5C; SupplementaryFig. S3A), and soft agar colony formation assay (Fig. 5D;Supplementary Fig. S3B) showed that increased cellular pro-liferation of LHBs-Huh7 cells was reversed by endogenous Srckinase inhibition with kinase deadmutant Src K295M cotrans-fection. Furthermore, flow cytometric analysis of cell-cyclestatus revealed that induced G1–S cell-cycle progression of
Figure 5. LHBs expressionpromotes cellular proliferation,G1–S cell-cycle progression, andapoptosis resistance via Srcactivation in hepatoma cells. A,Western blot analysis for Huh7 cellsafter transfection with emptyvector, LHBs plasmid, orcotransfected LHBs plasmid withSrc WT plasmid or Src K295Mplasmid together.�, P < 0.05 compared with the leftlane. B, cell proliferation assay forabove mentioned Huh7 cells.�, P < 0.05. C, BrdUrd incorporationassay and following flowcytometricanalysis for above mentioned Huh7cells. �, P < 0.05. D, colonyformation assay for abovementioned Huh7 cells.Quantification of colonies.�, P < 0.05. E, flow cytometricanalysis for cell-cycle status ofabove mentioned Huh7 cells.F, Annexin V/PI staining assay forcellular apoptosis analysis of abovementioned Huh7 cells cultured inserum deprivation medium for 24hours. �, P < 0.05.
A ControlLHBs
LHBs
LHBs + Src WT
LHBs + Src K295M
Src WTSrc K295M
PCNA
1.45 ± 0.21 2.06 ± 0.45 1.14 ± 0.12
Src
LHBs
GAPDH Time (h)
Ab
so
rba
nce
at
45
0 n
m
% B
rdU
rd-p
ositiv
e c
ells
Nu
mb
er
of
so
ft a
ga
rco
lon
ies
Ap
op
totic c
ells
(%
)
0 12
3
2
1
0
100
80
60
40
20
0
100
80
60
40
20
0
30
20
10
0
50
40
30
20
10
0
24 48
Control
LHBs
LHBs
+ Src
WT
LHBs
+ Src
K29
5M
Contro
l
LHBs
LHBs
+ Src
WT
LHBs
+ Src
K29
5M
Contro
l
LHBs
LHBs
+ Src
WT
LHBs
+ Src
K29
5M
Contro
l
LHBs
LHBs
+ Src
WT
LHBs
+ Src
K29
5M
Contro
l
* *
*
*
*
* *
**
Fold difference
+–––
–+––
–++–
–+–+
B
C D
E FG0–G1
G2–M
S
LHBs Promotes Hepatocarcinogenesis via Src/PI3K/Akt Signal
www.aacrjournals.org Cancer Res; 71(24) December 15, 2011 7553
on January 28, 2020. © 2011 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2011; DOI: 10.1158/0008-5472.CAN-11-2260

LHBs-Huh7 cells was also reversed by Src kinase inhibitionwith Src K295M cotransfection (Fig. 5E; Supplementary TableS1).Western blot analysis for G1–S cell-cycle regulators such ascyclin D1, cyclin D3, CDK4, and CDK6 showed that despite noalteration with cyclin D3 and CDK6 protein levels after LHBsexpression, increased cyclin D1 and CDK4 protein levels wasreversed by cotransfection with Src K295M in LHBs-Huh7 cells(Supplementary Fig. S3C). Moreover, Annexin V/PI stainingassay (Fig. 5F; Supplementary Fig. S3D) and Western blotanalysis for cleaved caspase-3 (Supplementary Fig. S3E) inLHBs-Huh7 cells under serum starvation indicated that apo-ptosis resistance conferred by LHBs expression was reversedby Src kinase inhibition with Src K295M cotransfection. Takentogether, these results indicate that LHBs expression couldpromote cellular proliferation, G1–S cell-cycle progression, andapoptosis resistance in hepatoma cells.
Saracatinib administration alleviates provocative tumorformation conferred by LHBs expression
To further determine whether Src signal activation con-ferred by LHBs expression could be used as a novel moleculartherapeutic target in HBV-associated hepatocarcinogenesis,
cellular proliferative rate, and tumor formation were assessedin LHBs stably expressing Huh7 and SK-Hep1 cells after aspecific Src inhibitor saracatinib treatment, which was used tosuppress Src kinase activity. Results of cell proliferation assayshowed that saracatinib treatment could significantly atten-uate promotive hepatoma cellular proliferation conferred bystable LHBs expression in Huh7 and SK-Hep1 cells (Fig. 6A andB). More importantly, tumor xenograft experiments in nudemice showed that orally saracatinib administration could alsoalleviate instigative tumor formation induced by stable LHBsexpression in Huh7 and SK-Hep1 cells (Fig. 6C–F). Theseresults show saracatinib administration could suppress cellu-lar proliferation in vitro and tumor formation in vivo throughintervening Src signal activation conferred by LHBs expression,whichmight be used as a potential therapeutic target for HBV-associated patients with HCC.
LHBs expression positively correlates with increasedKi67, pSrc(Y416), and pAkt(S473) staining in tumortissues from patients with HCC
To ascertain the correlation between LHBs expression withSrc/Akt signal activation and cellular proliferative status
A B
C E
D F
3
2
1
0
* *
**
***
*
*
* * * * * *
*
*
*
Absorb
ance a
t 450 n
m 3
2
1
0Absorb
ance a
t 450 n
m
Huh7 SK-Hep1
Huh7 SK-Hep1
Huh7 SK-Hep1
0 12 24 48
Control
LHBs
LHBs + DMSO
LHBs + saracatinib
Control + DMSO
Control + saracatinib
Con
trol
LHBs
LHBs
+ DM
SO
LHBs
+ sa
raca
tinib
Con
trol +
DM
SO
Con
trol +
sar
acat
inib
Con
trol
LHBs
LHBs
+ DM
SO
LHBs
+ sa
raca
tinib
Con
trol +
DM
SO
Con
trol +
sar
acat
inib
Control
LHBs
LHBs + DMSO
LHBs + saracatinib
Control + DMSO
Control + saracatinib
Control
LHBs
LHBs + DMSO
LHBs + saracatinib
Control + DMSO
Control + saracatinib
Control
LHBs
LHBs + DMSO
LHBs + saracatinib
Control + DMSO
Control + saracatinib
Time (h)
Tum
or
volu
me (
cm
3)
Tum
or
volu
me (
cm
3)
Days after tumor xenograft
7
1.5
1.0
0.5
0.0
Tum
or
weig
ht (g
)
0.6
0.4
0.2
0.0
Tum
or
weig
ht (g
)
0.6
0.4
0.2
0.0
1.0
0.8
0.6
0.4
0.2
0.014 21 28 7 14 21 28
Days after tumor xenograft
0 12 24 48
Time (h)
Figure 6. Saracatinib (AZD0530)administration alleviatesprovocative tumor formationconferred by LHBs expression. Aand B, cell proliferation assay forHuh7 cells (A) andSK-Hep1 cells (B)after stable LHBs expression undertreatment with saracatinib(1 mmol/L). �, P < 0.05. C and D,in vivo subcutaneous tumor growthcurves (C) and tumor weightquantification of intersectedsubcutaneous tumor tissues (D)of Huh7 cells after stable LHBsexpression under saracatinibtreatment (25 mg/kg body weightdaily for 4 weeks; n ¼ 18).�, P < 0.05. E and F, in vivosubcutaneous tumor growth curves(E) and tumor weight quantificationof intersected subcutaneous tumortissues (F) of SK-Hep1 cells afterstable LHBs expression undersaracatinib treatment (25 mg/kgbody weight daily for 4 weeks;n ¼ 18). �, P < 0.05.
Liu et al.
Cancer Res; 71(24) December 15, 2011 Cancer Research7554
on January 28, 2020. © 2011 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2011; DOI: 10.1158/0008-5472.CAN-11-2260

during HBV-associated hepatocarcinogenesis in vivo, immu-nohistochemistry was carried out to analyze the stainingpatterns of proliferative marker Ki67, pSrc(Y416), and pAktand Src in tumor tissues from 37 patients with HCC. On thebasis of the immunohistochemical results of LHBs staining, all37 patients with HCC were divided into 2 groups: LHBsnegative group (n ¼ 18) and LHBs positive group (n ¼ 19).LHBs expression in immunohistochemistry correlated posi-tively with the tumor grade (P ¼ 0.004) but did not showassociation with any other clinicopathologic parameters (Sup-plementary Table S2). Immunohistochemical analysis showedthat LHBs expression was positively correlated with Ki67 (n¼37, r ¼ 0.502, P ¼ 0.001), pSrc(Y416) (n ¼ 37, r ¼ 0.373, P ¼0.023), and pAkt(S473) (n¼ 37, r¼ 0.444, P¼ 0.005) staining inHCC tumor tissues (Fig. 7A–D; Supplementary Table S3). Theseresults suggest that Src/Akt signal activation and increasedcellular proliferation positively correlate with LHBs expressionduring the development of HBV-associated HCC.
Discussion
Although chronic HBV infection has been linked epidemi-ologically to the development of HCC for more than 30 years,the molecular mechanisms underlying viral-induced hepato-carcinogenesis remain largely controversial (3, 28). Whilesignificant advances have been noted in understanding ofindirect roles of chronic HBV infection proposed on themolecular basis of HBV-associated HCC, including virus per-sistence, genetic alterations conferred by HBV DNA integra-tion, and hepatic cell clone expansion because of chronicinflammation and fibrosis, increasing studies indicate thatexpression of HBV proteins such as HBx, PreS2 activators, andHBSP could modulate hepatic malignant transformation(5, 6, 29–32). In the absence of a dominant oncogene encodedbyHBV genome,molecular pathways engaged in growth signal
transduction being hijacking by viral proteins for malignanttransformation in hepatic cells served as underlying mechan-isms for HBV-associated tumorigenesis (5). Among all putativeoncopromotive proteins encoded by HBV genome, LHBsaddressed our attentions because of its potential of activatingtranscription factors such as activator protein-1 (AP-1) andNF-kB to trigger cellular proliferation, both ofwhich could playessential roles during inflammation-related tumorigenesis(27, 33, 34). Our current study shows the provocative effect ofLHBs on tumor formation of hepatoma cells in vivo and in vitro,which provides further evidence on the oncogenic function ofLHBs during HBV-related HCC development.
Because 2 signal transduction cascades including insulin/IGF/IRS-1/MAPK and WNT/Frizzled/b-catenin pathways areactivated early in over 90% of HCC tumors (35), we firstinvestigated the contribution of these 2 pathways to tumorformation instigation conferred by LHBs expression. MAPK/MEK/ERK signal inhibition with specific inhibitors PD98059,U0126, and SB203580 could not alleviate increased hepatomacellular proliferation induced by LHBs expression, and LHBsexpression could not alter protein expression or nuclear local-ization of b-catenin (data not shown), which excluded thepossibilities of these 2 pathways involved in this phenomenon.Perturbation of hepatic NF-kB signal activity provides amech-anistic link between inflammation and cancer in hepaticinflammation–fibrosis–cancer axis, and NF-kB is a majorfactor controlling both proneoplastic and malignant cells toresist apoptosis-based tumor surveillance mechanisms (36,37). ButNF-kBactivity inhibitionwith specific inhibitors PDTCand BAY117082 in our experiments could not attenuate pro-vocative hepatoma cellular proliferation conferred by LHBsexpression, which excluded the dedication of NF-kB activity onthis phenomenon and conflicted with previous finding thatLHBs could activate NF-kB to trigger cellular proliferation (27).However, our current investigation could not exclude the
Figure 7. LHBs expressionpositively correlates with increasedKi67, pSrc(Y416), and pAkt(S473)staining in tumor tissues frompatients with HCC. A,representative positive (case 1)and negative (case 2)immunohistochemical staining withLHBs, Ki67, pSrc(Y416), and pAkt(S473) in human hepatoma tissues(black scale bar, 50 mm). B–D,correlation analysis between LHBswith Ki67 (B), LHBs withpSrc(Y416) (C), and LHBs withpAkt(S473) (D) in human hepatomatissues.
ALHBS
Case 1
Case 2
LHBs score
n = 37r = 0.373P = 0.023
n = 37r = 0.444P = 0.005
n = 37
11
9
7
5
3
1
–1
11
9
7
5
3
1
–1
11
9
7
5
3
1
–1
r = 0.502P = 0.001
Ki6
7 sc
ore
pS
rc(Y
416)
scr
ore
pA
kt(S
473)
scr
ore
LHBs score LHBs score
Ki67 pSrc(Y416) pAkt(S473)
B C D
–1 1 3 5 7 9 11 –1 1 3 5 7 9 11 –1 1 3 5 7 9 11
LHBs Promotes Hepatocarcinogenesis via Src/PI3K/Akt Signal
www.aacrjournals.org Cancer Res; 71(24) December 15, 2011 7555
on January 28, 2020. © 2011 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2011; DOI: 10.1158/0008-5472.CAN-11-2260

potential NF-kB activation conferred by LHBs expression, andthe mechanistic link between NF-kB activity and hepaticcellular proliferation remains largely debated and awaits fur-ther investigation (38).
Genetic approaches and microarray technologies for ana-lyzing gene expression profiles have revealed strikingly dis-tinctive molecular mechanisms operate in HBV-related HCC,which includes a high copy number of HBV, mutations inPIK3CA and TP53, and specific activation of the PI3K/Akt/mTOR pathway (5, 39). Moreover, the PI3K/Akt/mTOR signal-ing pathway could be overactivated by enhanced stimulation ofreceptor tyrosine kinases, particularly the IGF receptor andEGFR (40). Expression of both IGF and IGF receptor is upre-gulated in HCC and human cirrhotic liver, resulting in stim-ulation of the PI3K/Akt/mTOR signaling pathway (5). Evidencealso suggested that anomalies in PTEN function may lead tooveractivation of the PI3K/AKT/mTOR pathway in HCC (41).Interestingly, our present study confirmed that elevated hep-atoma cellular proliferation ratio because of LHBs expressionwas dependent on PI3K/Akt/mTOR signal activation by usingPI3K/Akt/mTOR pathway specific inhibitors LY294002, Wort-mannin, and rapamycin treatment, which further showed thefundamental effect of PI3K/Akt/mTOR pathway activation onHBV-related hepatocarcinogenesis. Moreover, mounting evi-dences indicated the substantial role of Src signaling in theprocess of HCC development. Previous studies reported thatincreased Src tyrosine kinase activity was observed in HCCspecimens on the basis of in vitro Src kinase assays, in com-parison with liver tissue from normal subjects and chronichepatitis (16, 17). In another report, stimulation of hepatocyteswith stromal cell–derived factor-1 (SDF-1) led to the activationof the Src kinases, which in turn stimulates the Akt signal (42).Our results presented here also indicated the crucial role of Srctyrosine kinase activation in promotive hepatoma cellularproliferation conferred by LHBs expression by using specificSrc inhibitor PP2 treatment.More importantly, we also showedthat LHBs-induced Akt signal activation was dependent on Srctyrosine kinase activity by using specific inhibitor treatment,kinase dead mutant cotransfection, and specific siRNAcotransfection. Consistent with previous study indicatingPreS2 activators triggered PKCa/b-dependent Raf1 activation(27), our present investigation further revealed a mechanisticlink between Src/Akt signal activation and LHBs expressionthrough PKCa/Raf1 pathway, which could be potential molec-ular basis for HBV-related hepatocarcinogenesis.
In addition to cellular proliferative promotion, our currentstudy found that LHBs expression could also accelerate G1–Scell-cycle progression and endue with apoptosis resistancethrough Src activation in hepatoma cells, all of which con-stitute the oncogenic function of LHBs expression in
HBV-associated tumorigenesis. Moreover, correlative analysisamong Src/Akt signal activation, cellular proliferative status,and LHBs expression in our investigation here also revealedthat LHBs expression was positively correlated with Src/Aktactivation and cellular proliferation in HCC tumor tissues. Ourpresent demonstrations of the increased pSrc(Y416) staining inHCC tissues and the potent inhibitory effect of Src kinase deadmutant on oncogenic functions conferred by LHBs expressionin hepatoma cells in vitro provide a strong rationale thatspecific Src inhibitor might be developed as an anticanceragent in patients with HCC with chronic HBV infection. Ourcurrent study also elucidates the potential therapeutic effect ofan oral Src specific inhibitor saracatinib on hepatoma cellxenografted tumor in nude mice, which merits further clinicalinvestigation on patients with HBV-associated HCC to assessits feasible therapeutic efficiency.
On the basis of our current results, we propose a schematicmodel illustrating a possible molecular mechanism and func-tional basis for HBV-associated hepatocarcinogenesis conferredby LHBs expression (Supplementary Fig. S4). In conclusion, ourresults presented here reveal a novel association between LHBsexpression and PKCa/Raf1/Src/PI3K/Akt signal activation inthe development of HBV-associated HCC, thus revealing aputative molecular mechanism for the development and pro-gression of HBV-associated HCC. These results shed a new lightfor potential therapeutic intervention to prevent hepatocarci-nogenesis in the high-risk group of chronic hepatitis B patientswith PKCa/Raf1/Src/PI3K/Akt signal suppression treatment.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Zhangmei Ma and Tengfang Zhu (Fudan University,Shanghai, China) for experimental technical help; Prof. Aiguo Shen (NantongUniversity, Jiangsu, China) for HCC patient sample collection; and Drs. JianguoGu (Tohoku Pharmaceutical University) and Mien-Chie Hung (MD AndersonCancer Center) for the generous gifts of plasmids.
Grant Support
This study was supported by grants from the State Key Project Specialized forInfectious Diseases of China (2012ZX10002-008, 2012ZX10002-012), the NationalBasic Research Program of China 973 Program (2010CB912104, 2012CB8221004),and National Natural Science Fund (30900266, 30930025, 31000348, 31000600,31010103906, 31170766).
The costs of publication of this article were defrayed in part by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received July 4, 2011; revised September 27, 2011; accepted September 27, 2011;published OnlineFirst October 12, 2011.
References1. Lai CL, Ratziu V, Yuen MF, Poynard T. Viral hepatitis B. Lancet 2003;
362:2089–94.2. Lee WM. Hepatitis B virus infection. N Engl J Med 1997;337:1733–45.3. Di Bisceglie AM. Hepatitis B and hepatocellular carcinoma. Hepatol-
ogy 2009;49(5 Suppl):S56–60.
4. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogene-sis: from genes to environment. Nat Rev Cancer 2006;6:674–87.
5. Neuveut C, Wei Y, Buendia MA. Mechanisms of HBV-related hepa-tocarcinogenesis. J Hepatol 2010;52:594–604.
Liu et al.
Cancer Res; 71(24) December 15, 2011 Cancer Research7556
on January 28, 2020. © 2011 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2011; DOI: 10.1158/0008-5472.CAN-11-2260

6. Brechot C. Pathogenesis of hepatitis B virus-related hepatocellularcarcinoma: old and new paradigms. Gastroenterology 2004;127(5 Suppl 1):S56–61.
7. Ganem D, Prince AM. Hepatitis B virus infection—natural history andclinical consequences. N Engl J Med 2004;350:1118–29.
8. Seeger C,MasonWS. Hepatitis B virus biology.MicrobiolMol Biol Rev2000;64:51–68.
9. Babinet C, Farza H, Morello D, Hadchouel M, Pourcel C. Specificexpression of hepatitis B surface antigen (HBsAg) in transgenic mice.Science 1985;230:1160–3.
10. Chisari FV, KlopchinK,MoriyamaT, Pasquinelli C,DunsfordHA,Sell S,et al. Molecular pathogenesis of hepatocellular carcinoma in hepatitisB virus transgenic mice. Cell 1989;59:1145–56.
11. Toshkov I, Chisari FV, Bannasch P. Hepatic preneoplasia in hepatitis Bvirus transgenic mice. Hepatology 1994;20:1162–72.
12. Aravalli RN, Steer CJ, Cressman EN. Molecular mechanisms of hepa-tocellular carcinoma. Hepatology 2008;48:2047–63.
13. Llovet JM, Bruix J. Molecular targeted therapies in hepatocellularcarcinoma. Hepatology 2008;48:1312–27.
14. Zender L, Villanueva A, Tovar V, Sia D, Chiang DY, Llovet JM. Cancergene discovery in hepatocellular carcinoma. JHepatol 2010;52:921–9.
15. Whittaker S, Marais R, Zhu AX. The role of signaling pathways in thedevelopment and treatment of hepatocellular carcinoma. Oncogene2010;29:4989–5005.
16. Ito Y, Kawakatsu H, Takeda T, Sakon M, Nagano H, Sakai T, et al.Activation of c-Src gene product in hepatocellular carcinoma is highlycorrelated with the indices of early stage phenotype. J Hepatol2001;35:68–73.
17. Masaki T, Okada M, Shiratori Y, Rengifo W, Matsumoto K, Maeda S,et al. pp60c-src activation in hepatocellular carcinoma of humans andLEC rats. Hepatology 1998;27:1257–64.
18. Klein NP, Schneider RJ. Activation of Src family kinases by hepatitis Bvirus HBx protein and coupled signaling to Ras. Mol Cell Biol1997;17:6427–36.
19. Tian X, Zhao C, Zhu H, She W, Zhang J, Liu J, et al. Hepatitis B virus(HBV) surface antigen interacts with and promotes cyclophilin a secre-tion: possible link to pathogenesis of HBV infection. J Virol 2010;84:3373–81.
20. Wang YX, Xu X, Luo C, Ma ZM, Jiang HL, Ding JP, et al. A putative newdomain target for anti-hepatitis B virus: residues flanking hepatitis Bvirus reverse transcriptase residue 306 (rtP306). J Med Virol 2007;79:676–82.
21. Zhu X, Chen S, Yin X, Shen A, Ji S, Shen Z, et al. Constitutively activePKB/Akt inhibited apoptosis and down-regulated beta1,4-galactosyl-transferase 1 in hepatocarcinoma cells. Biochem Biophys Res Com-mun 2003;309:279–85.
22. Liu W, Shen X, Yang Y, Yin X, Xie J, Yan J, et al. Trihydrophobin 1 isa new negative regulator of A-Raf kinase. J Biol Chem 2004;279:10167–75.
23. Xu J, Yun X, Jiang J, Wei Y, Wu Y, Zhang W, et al. Hepatitis B virus Xprotein blunts senescence-like growth arrest of human hepatocellularcarcinoma by reducing Notch1 cleavage. Hepatology 2010;52:142–54.
24. Zhang W, Xu W, Xiong S. Blockade of Notch1 signaling alleviatesmurine lupus via blunting macrophage activation and M2b polariza-tion. J Immunol 2010;184:6465–78.
25. Zhang W, Xu W, Xiong S. Macrophage differentiation and polari-zation via phosphatidylinositol 3-kinase/Akt-ERK signaling pathwayconferred by serum amyloid P component. J Immunol 2011;187:1764–77.
26. RemmeleW, StegnerHE. Recommendation for uniformdefinition of animmunoreactive score (IRS) for immunohistochemical estrogen recep-tor detection (ER-ICA) in breast cancer tissue. Pathologe 1987;8:138–40.
27. Hildt E, Munz B, Saher G, Reifenberg K, Hofschneider PH. The PreS2activatorMHBs(t) of hepatitis B virus activates c-raf-1/Erk2 signaling intransgenic mice. EMBO J 2002;21:525–35.
28. Bruix J, Llovet JM. Hepatitis B virus and hepatocellular carcinoma.J Hepatol 2003;39 Suppl 1:S59–63.
29. Kremsdorf D, Soussan P, Paterlini-Brechot P, Brechot C. Hepatitis Bvirus-related hepatocellular carcinoma: paradigms for viral-relatedhuman carcinogenesis. Oncogene 2006;25:3823–33.
30. Lok AS. Prevention of hepatitis B virus-related hepatocellular carci-noma. Gastroenterology 2004;127(5 Suppl 1):S303–9.
31. Block TM,Mehta AS, Fimmel CJ, JordanR.Molecular viral oncology ofhepatocellular carcinoma. Oncogene 2003;22:5093–107.
32. Brechot C, Gozuacik D, Murakami Y, Paterlini-Brechot P. Molec-ular bases for the development of hepatitis B virus (HBV)-relatedhepatocellular carcinoma (HCC). Semin Cancer Biol 2000;10:211–31.
33. Hildt E, Saher G, Bruss V, Hofschneider PH. The hepatitis B virus largesurface protein (LHBs) is a transcriptional activator. Virology 1996;225:235–9.
34. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, andcancer. Cell 2010;140:883–99.
35. Branda M, Wands JR. Signal transduction cascades and hepatitis Band C related hepatocellular carcinoma. Hepatology 2006;43:891–902.
36. Elsharkawy AM, Mann DA. Nuclear factor-kappaB and the hepaticinflammation-fibrosis-cancer axis. Hepatology 2007;46:590–7.
37. Karin M. Nuclear factor-kappaB in cancer development and progres-sion. Nature 2006;441:431–6.
38. Karin M, Greten FR. NF-kappaB: linking inflammation and immunityto cancer development and progression. Nat Rev Immunol 2005;5:749–59.
39. Boyault S, Rickman DS, de Reynies A, Balabaud C, Rebouissou S,Jeannot E, et al. Transcriptome classification of HCC is related to genealterations and to new therapeutic targets. Hepatology 2007;45:42–52.
40. Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY,et al. Integrative transcriptome analysis reveals common molecularsubclasses of human hepatocellular carcinoma. Cancer Res 2009;69:7385–92.
41. Ito Y, Sasaki Y, Horimoto M, Wada S, Tanaka Y, Kasahara A, et al.Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology1998;27:951–8.
42. Liu HY, Wen GB, Han J, Hong T, Zhuo D, Liu Z, et al. Inhibition ofgluconeogenesis in primary hepatocytes by stromal cell-derived fac-tor-1 (SDF-1) through ac-Src/Akt-dependent signaling pathway. JBiolChem 2008;283:30642–9.
LHBs Promotes Hepatocarcinogenesis via Src/PI3K/Akt Signal
www.aacrjournals.org Cancer Res; 71(24) December 15, 2011 7557
on January 28, 2020. © 2011 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2011; DOI: 10.1158/0008-5472.CAN-11-2260

2011;71:7547-7557. Published OnlineFirst October 12, 2011.Cancer Res Haiou Liu, Jiejie Xu, Lei Zhou, et al. Carcinogenesis by Activating the Src/PI3K/Akt PathwayHepatitis B Virus Large Surface Antigen Promotes Liver
Updated version
10.1158/0008-5472.CAN-11-2260doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2011/12/13/0008-5472.CAN-11-2260.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/71/24/7547.full#ref-list-1
This article cites 42 articles, 10 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/71/24/7547.full#related-urls
This article has been cited by 4 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/71/24/7547To request permission to re-use all or part of this article, use this link
on January 28, 2020. © 2011 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2011; DOI: 10.1158/0008-5472.CAN-11-2260