heterogeneous activation of the fanconi anemia pathway by patient
TRANSCRIPT

Heterogeneous activation of the Fanconi anemiapathway by patient-derived FANCA mutants
Daiki Adachi1,2, Tsukasa Oda1, Hiroshi Yagasaki1,2, Keiko Nakasato1, Toshiyasu Taniguchi3,
Alan D. D’Andrea3, Shigetaka Asano2 and Takayuki Yamashita1,*
1Division of Genetic Diagnosis, The Institute of Medical Science, The University of Tokyo, Tokyo, Japan,2Department of Molecular Therapy, Advanced Clinical Research Center, The Institute of Medical Science,
The University of Tokyo, Tokyo, Japan and 3Department of Pediatric Oncology, Dana-Farber Cancer Institute,
Harvard Medical School, Boston, MA, USA
Received July 22, 2002; Revised September 26, 2002 and Accepted October 4, 2002
Fanconi anemia (FA) is an autosomal recessive disorder of hematopoiesis characterized by hypersensitivityto DNA crosslinkers such as mitomycin C (MMC). There is growing evidence for a model of the FA pathway,wherein a nuclear multiprotein complex of five FA proteins (FANCA, C, E, F and G) regulates activation ofFANCD2 into a monoubiquitinated form, which, collaborating with the BRCA1 machinery, affects cellularresponse to DNA damage. However, the role of the FA pathway in defective DNA damage response caused byvarious mutant forms of FA proteins has not been fully assessed. In the present study, 21 patient-derivedFANCA mutants with a missense or a small in-frame deletion were expressed in FANCA-deficient fibroblastsand examined for complementation of MMC sensitivity and for reconstitution of the FA pathway: FANCAphosphorylation, interaction with FANCC, FANCF and FANCG and nuclear localization and FANCD2monoubiquitination. The altered FANCA proteins complemented MMC sensitivity at different grades: fiveproteins (group I) behaved like wild-type FANCA, whereas the other proteins were either mildly (group II,n¼ 4) or severely (group III, n¼ 12) impaired. Group I proteins showed an apparently normal reconstitution ofthe FA pathway, thus they may be pathogenic by reducing endogenous expression or possibly benignpolymorphisms. Reconstitution of the FA pathway by group II and III mutants closely correlated with cellularsensitivity to MMC. The different activation of the FA pathway may partly account for the phenotypic variationseen in FA patients.
INTRODUCTION
Fanconi anemia (FA) is an autosomal recessive disease withcongenital anomalies, progressive bone marrow failure andleukemia susceptibility (for reviews, see 1–3). A characteristiccellular phenotype of FA is chromosomal instability andhypersensitivity to DNA crosslinkers such as mitomycin C(MMC) (1–3). FA has at least eight different complementationgroups (FA–A to FA–G) (4–8). Growing evidence indicatesthat protein products of six FA genes (FANCA, C, D2, E, F andG) which have been cloned to date (8–14) function in acommon pathway, termed the FA pathway (15–27).
In a current model of the FA pathway, FANCA, C, E, F and Gassemble into a nuclear multiprotein complex, which convertsFANCD2 into a monoubiquitinated form (15–27). This activeisoform is likely to affect DNA damage response in collabora-tion with the BRCA1 machinery, through homology-directedrepair (26). Furthermore, a recent study showed that biallelic
inactivating mutations of BRCA2, a component involved inhomologous recombination, cause a clinical phenotype of FA(28). The model of the FA pathway has been largely based onstudies using FA protein-deficient cells. However, it has notbeen verified that various mutant forms of FA proteins similarlyaffect the FA pathway. It is also conceivable that some mutantscause aberrant DNA damage response or hematopoiesisthrough impairment of distinct pathways. This notion isconsistent with recent findings that anti-apoptotic functions ofFANCC and the ATM-dependent cell cycle checkpoint functionof FANCD2, the defects of which may partly account for FAphenotypes, are separated from activation of the FA pathway bymutational analyses (29,30).
FANCA is a relatively large protein with 1455 amino acids(Mr¼ 163 kDa) and contains a bipartite nuclear localizationsignal (NLS) at its N-terminus and a leucine zipper-like motifbetween amino acids 1069 and 1090 (10,11). Subsequentstudies revealed several features of this protein: (i) FANCA is
*To whom correspondence should be addressed at: The Institute of Medical Science, The University of Tokyo, Division of Genetic Diagnosis,4-6-1 Shirokanedai, Minato-ku, Tokyo 108-8639, Japan. Tel: þ81 354495765; Fax: þ81 354495764; Email: [email protected]
# 2002 Oxford University Press Human Molecular Genetics, 2002, Vol. 11, No. 25 3125–3134
Downloaded from https://academic.oup.com/hmg/article-abstract/11/25/3125/578773by gueston 26 March 2018

predominantly localized in the nuclei (15–23,32); (ii) FANCAindirectly interacts with FANCC and FANCF (15–19,23–25)and directly interacts with FANCG and possibly FANCE(19–22,24,27); (iii) a serine kinase binds and phosphorylatesFANCA (17,31). However, little is known of functionaldomains of FANCA, except that its N-terminal portion,including NLS, is required for nuclear localization and fordirect interaction with FANCG (16,19–22,32). The FANCAgene abnormalities are seen in the majority (60–70%) of FApatients and more than 100 types of mutations throughout thegene have been found to date (10,11,33–38). While most ofthese mutations are either small insertion/deletion mutationsresulting in premature termination or intragenic large deletionsand presumably lack protein expression (null-mutations), 30 ormore mutations are predicted to produce altered proteins with asingle amino acid substitution or a small in-frame deletion(33–38). Although a few mutants, such as H1110P, R1117Gand delF1263, were shown to be defective in complementationof MMC sensitivity and reconstitution of the FA pathway(17–20), other mutants have not been thoroughly characterized.
The goal of the present study was to assess the role of the FApathway in defective DNA damage response caused by variousmutants of FANCA proteins. For this, 19 uncharacterized orpoorly characterized mutants were expressed in FANCA-deficient cells and systematically analysed for complementationof MMC sensitivity and for reconstitution of the FA pathway, incomparison with the well-characterized mutants H1110P anddelF1263 (17–20). We found that various mutants showdifferent complementation of MMC sensitivities. FANCAinteractions with FANCC and FANCF, FANCA phosphoryla-tion and FANCD2 monoubiquitination were closely associatedwith MMC sensitivity, whereas the nuclear localization ofFANCA was dissociated from these events. Our presentfindings provide fundamental information which will serve tobetter understand mechanisms and the pathophysiological roleof the FA pathway.
RESULTS
Table 1 summarizes data on the 21 patient-derived FANCAmutations examined in the present study. Sixteen mutationswere predicted to result in substitution of a single amino acid,and five were presumed to result in in-frame small deletions.Many of the mutations were detected in a single patient,i.e. ‘private mutation’ and reported without identification of amutation in a second allele. Figure 1 shows a schematicpresentation of the FANCA protein structure and locations ofthe patient-derived mutations.
To characterize these mutants, we introduced into GM6914FANCA-null fibroblasts wild-type (wt) and mutant FANCAproteins carrying a Flag epitope at their N-termini. Theseproteins were stably expressed at similar levels (Fig. 2A). MMChypersensitivity of GM6914 cells was corrected by introductionof Flag-wt-FANCA (Fig. 2B). Different mutants corrected MMCsensitivities of GM6914 cells, to various degrees (Fig. 2B andC). Five mutants (D598N, Q1128E, T1131A, F1262L andH1417D: group I) behaved like wt-FANCA, whereas 12 mutants(R435C, H492R, L845P, FQ868–869del, R1055L, R1055W,H1110P, F1135del, W1174del, 1239–43del, F1263del andW1302R: group III) failed to correct MMC sensitivities. Fourmutants (L817P, P1324L, D1359Y and M1360I: group II)exhibited partial correction.
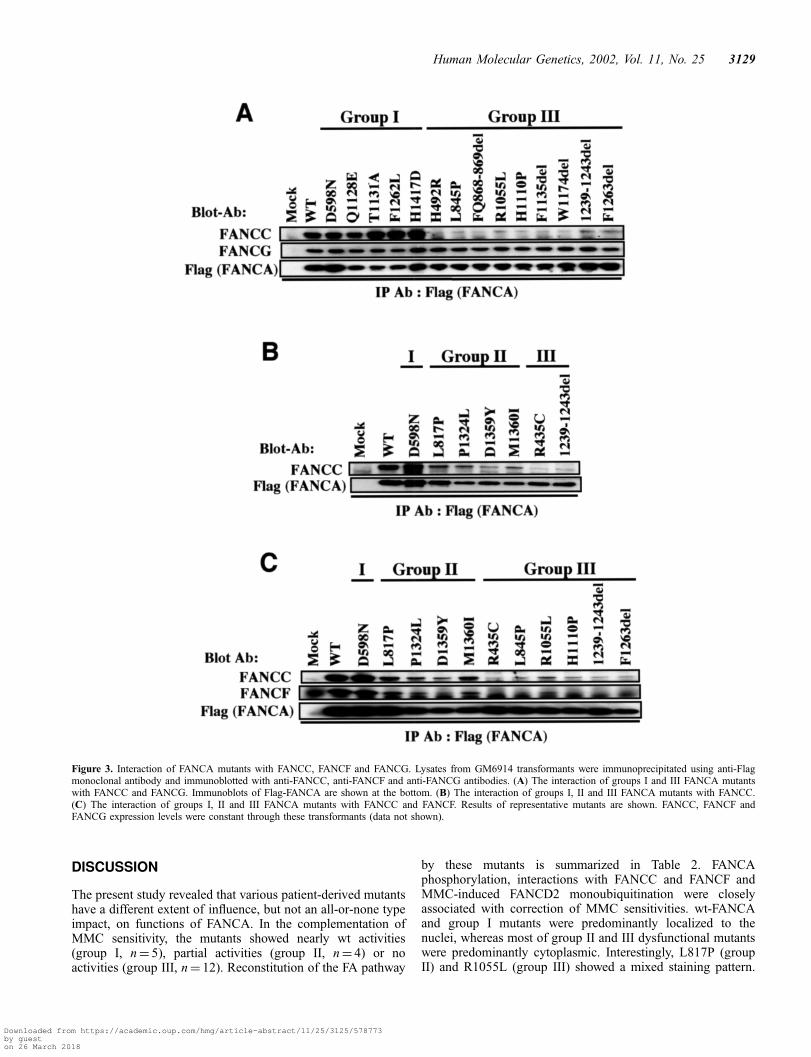
In a subsequent series of experiments, we assessed effects ofmutations on reconstitution of the FA pathway. For this, we firstexamined the interaction of the FANCA mutants with FANCC,FANCF and FANCG. Flag-FANCA immunoprecipitates fromGM6914 cell lysates were analysed by immunoblotting withanti-FANCC (17), anti-FANCF (25) and anti-FANCG (19)antibodies. Group I mutants interacted with FANCC as stronglyas did wt-FANCA, whereas group III mutants interacted to amuch lesser extent with FANCC (Fig. 3A). Unlike FANCC,FANCG constantly interacted with all group I and III mutants(Fig. 3A). The interaction of group II mutants with FANCC
Table 1. Mutations in the FANCA gene
Nucleotide change Effect No. of alleles No. of patients Second Mutation (No.) Reference
1303C>T R435C 3 2 33,371475A>G H492R 1 1 331792G>A D598N 2 2 1627–1900del (1) 36,382450T>C L817P 1 1 332534T>C L845P 1 1 33IVS27–1G>A FQ868–869del 1 1 2546delC (1) 373163A>T R1055W 1 1 2546delC (1) 353164G>T R1055L 1 1 333329A>C H1110P 2 1 18,383382C>G Q1128E 1 1 333391A>G T1131A 2 2 333403–3405del F1135del 1 1 383520–3522del W1174del 4 2 333715–3729del 1239–1243del 2 1 333786C>G F1262L 1 1 4080G>C (1) 383788–3790del F1263del 42 37 1360–1826del (1), 50UTR–3066del (1), 1471–1626del (1) 33,36,383904T>C W1302R 1 1 333971C>T P1324L 1 1 1191–1194del (1) 384075G>T D1359Y 1 1 344080G>C M1360I 1 1 3786C>G (1) 384249C>G H1417D 2 1 33
3126 Human Molecular Genetics, 2002, Vol. 11, No. 25
Downloaded from https://academic.oup.com/hmg/article-abstract/11/25/3125/578773by gueston 26 March 2018

was intermediate between group I and group III mutants(Fig. 3B and C). The interaction of group II and III mutantswith FANCF was impaired in parallel with their interactionwith FANCC (Fig. 3C). D1359Y interacted with FANCC andFANCF more weakly than other group II mutants, whereasR1055L interacted with FANCC and FANCF more stronglythan other group III mutants. The deviation of these twomutants was observed in repeated experiments. The FANCA/FANCG interaction was not affected by group II mutations.Some data are not shown (see Table 2).
Group I mutants were phosphorylated much like wt-FANCA,whereas phosphorylation of group III mutants was markedlyreduced (Fig. 4A). Again, group II mutants were phosphory-
lated intermediately between group I and group III mutants(Fig. 4B).
To analyse the subcellular distribution of the mutants, we didimmunofluorescence studies, using an anti-FANCA antibody.Group I mutants, like wt-FANCA, were predominant in thenuclei in more than 90% of cells, whereas most group II andgroup III mutants were primarily cytoplasmic in more than95% of cells (Fig. 5). However, there were two exceptionalmutants, L817P (group II) and R1055L (group III), whichshowed homogenous staining mixed with nuclear and cyto-plasmic predominance: homogenous, nuclear and cytoplasmicstaining patterns were observed in 65, 20 and 15% of L817Ptransformants and 63, 2 and 35% of R1055L transformants,
Table 2. Functions of FANCA mutants
Group Mutant Localization Phosphorylation FANCCInteraction
FANCFinteraction
FANCGinteraction
FANCD2**ubiquitination
Mock — — — — — negativeWT N>C strong strong strong strong strong
I (n¼ 5) D598N N>C strong strong strong strong strongQ1128E N>C* strong strong strong* strong strongT1131A N>C strong strong strong* strong strongF1262L N>C* strong strong strong* strong strongH1417D N>C strong strong strong* strong strong
II (n¼ 4) L817P mixed (C¼¼N, N>C, C>N) moderate moderate moderate strong* mildP1324L C>N moderate moderate moderate strong* mildD1359Y C>N moderate* mild mild strong* mildM1360I C>N moderate moderate moderate strong* mild
III (n¼ 12) R435C C>N weak weak negative strong* weakH492R C>N* weak negative negative* strong weakL845P C>N* weak weak weak strong weakFQ868–869del C>N* weak weak negative* strong weakR1055L mixed (C¼¼N, C>N, N>C) weak mild mild strong weakR1055W C>N NT NT NT NT weakH1110P C>N* weak weak weak strong weakF1135del C>N* weak weak negative* strong* weak*W1174del C>N* weak weak negative* strong weak1239–1243del C>N* weak weak negative strong weakF1263del C>N* weak weak negative strong weakW1302R C>N weak weak* weak* strong* weak
Localization: N>C, Nucleus dominant; C>N, Cytoplasm dominant; C¼¼N, Homogenous; Other assays: strong (wt-like activity)>moderate>mild>weak>negative; NT, not tested; *Data not shown; **FANCD2 ubiquitination in MMC-treated cells.
Figure 1. Schematic representation of wt and patient-derived mutant forms of FANCA. The wt FANCA protein, containing 1455 amino acids, has a nuclearlocalization signal (NLS) at its N-terminus and a leucine zipper-like motif (Leu Zip-like) between amino acids 1069 and 1090.
Human Molecular Genetics, 2002, Vol. 11, No. 25 3127
Downloaded from https://academic.oup.com/hmg/article-abstract/11/25/3125/578773by gueston 26 March 2018

respectively (Fig. 5). Cytoplasmic and nuclear levels ofwt-FANCA and representative mutants of groups I, II and IIIwere determined using immunoblotting (Fig. 6). Group II andIII proteins in the nuclear fraction were significantly reduced incomparison with wt and group I proteins, whereas nodifference was noted between group II and group III mutants.
Finally, we examined effects of FANCA mutations onFANCD2 monoubiquitination. Group I transformants expressa monoubiquitinated form of FANCD2 (D2-Ub) to someextent, like GM6914/wt-FANCA, whereas D2-Ub was not
detected in group III transformants (Fig. 7A, top). Thedifference between these two groups was much morepronounced when these cells were treated with MMC(Fig. 7A, middle). The same blot shows that D2-Ub wasslightly evident in group III cells, but not in mock cells(Fig. 7A, bottom). Although the difference in D2-Ublevels between group II and group III cells was not clearunder basal conditions, MMC treatment revealed a higherinduction of D2-Ub in group II cells than in group III cells(Fig. 7B).
Figure 2. Complementation of MMC sensitivity by various FANCA mutants. (A) Expression of Flag-tagged wt and mutant FANCA proteins in GM6914 cells.Whole cell lysates of transformants were analysed using immunoblotting with anti-Flag (M2) antibody. Results of representative mutants are shown. (B) Cellsurvival assay of GM6914 transformants. Results of representative cells are shown. (C) MMC IC50 values for each transformant. Values represent means�SEof triplicate assays. FANCA mutants are categorized into three groups (groups I, II and III) based on the cellular sensitivities to MMC. MMC IC50 values forgroups I, II and III are: >30, 10–30 and <10 nM, respectively.
3128 Human Molecular Genetics, 2002, Vol. 11, No. 25
Downloaded from https://academic.oup.com/hmg/article-abstract/11/25/3125/578773by gueston 26 March 2018
DISCUSSION
The present study revealed that various patient-derived mutantshave a different extent of influence, but not an all-or-none typeimpact, on functions of FANCA. In the complementation ofMMC sensitivity, the mutants showed nearly wt activities(group I, n¼ 5), partial activities (group II, n¼ 4) or noactivities (group III, n¼ 12). Reconstitution of the FA pathway
by these mutants is summarized in Table 2. FANCAphosphorylation, interactions with FANCC and FANCF andMMC-induced FANCD2 monoubiquitination were closelyassociated with correction of MMC sensitivities. wt-FANCAand group I mutants were predominantly localized to thenuclei, whereas most of group II and III dysfunctional mutantswere predominantly cytoplasmic. Interestingly, L817P (groupII) and R1055L (group III) showed a mixed staining pattern.
Figure 3. Interaction of FANCA mutants with FANCC, FANCF and FANCG. Lysates from GM6914 transformants were immunoprecipitated using anti-Flagmonoclonal antibody and immunoblotted with anti-FANCC, anti-FANCF and anti-FANCG antibodies. (A) The interaction of groups I and III FANCA mutantswith FANCC and FANCG. Immunoblots of Flag-FANCA are shown at the bottom. (B) The interaction of groups I, II and III FANCA mutants with FANCC.(C) The interaction of groups I, II and III FANCA mutants with FANCC and FANCF. Results of representative mutants are shown. FANCC, FANCF andFANCG expression levels were constant through these transformants (data not shown).
Human Molecular Genetics, 2002, Vol. 11, No. 25 3129
Downloaded from https://academic.oup.com/hmg/article-abstract/11/25/3125/578773by gueston 26 March 2018

Thus, various mutant forms of FANCA affect DNA damageresponse through different activation of the FA pathway.
We analysed 16 missense variants and five mutants withsmall in-frame deletions (Table 1, Fig. 1). All of the deletionmutants were classified into group III. Eleven (seven of groupIII and four of group II proteins) of 16 missense variants provedto be disease-associated, whereas five missense variants ofgroup I behaved like wt-FANCA. There are possible inter-pretations for wt activities of group I proteins. Firstly, theseproteins may be pathogenic by increased degradation of pro-teins resulting in reduced endogenous expression. Althoughgroup I proteins did not show significantly reduced levels orincreased degradation rates (not shown) in the present system,we cannot exclude the possibility that enforced overexpressionmasked the increased degradation. A similar problem is true forother mutants. Although group II mutants showed partialactivities in the present system, these mutations may result inmore severe effects at endogenous expression levels. Secondly,when only genomic DNA is sequenced, the possibility shouldbe considered that a nucleotide change in coding regions,predicted to be a missense or nonsense mutation, may result indefective pre-mRNA splicing (39). Finally, group I variantsmay be benign polymorphisms, although they have not beendetected in the normal population. To distinguish thesepossibilities, analyses of mRNAs and proteins and comple-mentation tests, using patient cells, will be required.
FANCA interacts with other FA proteins (C, E, F and G) anda protein kinase phosphorylating FANCA in a multiproteincomplex (15–25,27,31), but little is known regarding thestructural basis of these protein interactions, except for thedirect and constitutive interaction between FANCA andFANCG (19–22,24). Previous analyses using several mutantsled to the notion that specific regions of FANCA may beinvolved in interactions with FANCC and FANCA-proteinkinase (17–19,31). However, the present results showed thatvarious point-mutations, regardless of their locations, similarlyimpaired FANCA phosphorylation and association withFANCC and FANCF in a correlative manner. Thus, it is likelythat these mutations prevent FANCA from proper interactionwith other proteins by affecting tertiary, but not local, structureof the molecule.
Monoubiquitination of FANCD2 was shown to be essentialfor normal cellular response to MMC (26). However, it has notbeen determined if FANCD2 is the only downstream transducerof the FA pathway signaling. It can be hypothesized that theFA–protein complex activates distinct signaling pathways,required for normal DNA damage response. However,dysfunction of this hypothetical pathway is, if present, unlikelyto play a major role in MMC hypersensitivity in FA–A cells,since effects of diverse FANCA mutations on MMC-triggeredFANCD2 monoubiquitination were tightly linked with thosefor MMC-induced cell death.
Figure 4. In vivo phosphorylation of FANCA mutants in GM6914 cells. (A) Comparison between group I and group III mutants. Immunoblots of Flag-FANCA areshown at the bottom. (B) Intermediate phosphorylation of group II mutants in comparison with group I and group III mutants. Immunoblots of Flag-FANCA areshown at the bottom. Results of representative mutants are shown.
3130 Human Molecular Genetics, 2002, Vol. 11, No. 25
Downloaded from https://academic.oup.com/hmg/article-abstract/11/25/3125/578773by gueston 26 March 2018

Previous studies showed that not only an N-terminal regionincluding NLS but also a C-terminal region plays a pivotal rolein the nuclear localization of FANCA (16,19,32), yet little isknown of regulatory mechanisms of its subcellular localization.Diverse mutations had similar effects on nuclear localization of
FANCA, thereby suggesting that alteration of its tertiarystructure, rather than local structures, prevents FANCA fromproper interaction with nuclear import machinery (40). Earlierobservations led to the notion that interaction with other FAproteins and phosphorylation may regulate the nuclear import
Figure 5. Immunofluorescence localization of FANCA mutants in GM6914 cells. Cells expressing indicated proteins were stained using an anti-FANCAantibody. Cell nuclei were counterstained with DAPI. Results of representative mutants are shown.
Human Molecular Genetics, 2002, Vol. 11, No. 25 3131
Downloaded from https://academic.oup.com/hmg/article-abstract/11/25/3125/578773by gueston 26 March 2018

of FANCA (15–19,22). While this notion is consistentwith results of most mutants examined in the present study,the results of L817P (group II) and R1055L (group III)showeda dissociation between nuclear localization andphosphorylation or interactions with other FA proteins.Unlike R1055L, R1055W (group III) was predominantly
cytoplasmic, which suggests that a side chain of an aminoacid at this position specifically affects nuclear import ofFANCA.
FA is characterized by variability of clinical phenotypes(1,2,41), which are likely to be affected not only by genotypesbut also by ethnic and individual genetic backgrounds (modi-fier genes) and environmental factors (42–45). A recentstudy revealed a significant genotype–phenotype correlationin FA–A: patients homozygous for FANCA null-mutationshave an earlier onset of anemia and a higher incidence ofleukemia than did those with mutations to produce an alteredprotein (46). The present study indicates that altered FANCAproteins can activate the FA pathway to various degrees, inresponse to DNA damage, as assessed by FANCD2 mono-ubiquitination, while no activation of this pathway was detectedin FANCA-null cells. The different activation of the FApathway may, in part, account for the genotype–phenotypecorrelation in FA–A patients. Measurement of FANCD2monoubiquitination levels in cells treated with DNA cross-linkers provides a sensitive and specific assay for functionof the FA pathway. Determination of the relationshipbetween function of the FA pathway in patient cells andclinical phenotypes will aid in a better understanding of themolecular basis of the genotype–phenotype correlation con-cerning FA.
Figure 6. Subcellular fractionation of FANCA mutants. GM6914 cells expres-sing indicated proteins were fractionated, then the nuclear fraction (N) and thecytoplasmic fraction (C) were analysed using immunoblotting with an anti-Flag(M2) antibody. The same samples were analysed using immunoblottingwith anti-c-Abl and b-tubulin antibodies, nuclear and cytoplasmic markers,respectively.
Figure 7. Effects of FANCA mutants on FANCD2 monoubiquitination in GM6914 cells. (A) Comparison between group I and group III mutants. Whole celllysates from GM6914 transformants cultured with 40 ng/ml MMC [MMC (þ)] or without MMC [MMC (�)] for 24 h were analysed. For analyses of MMC-treatedcells, chemiluminescence signals were obtained by exposure of X-ray films to a blotted membrane for 5 min (Short Exp) and 10 min (Long Exp). Protein bandscorresponding to FANCD2 (D2) and a monoubiquitinated form of FANCD2 (D2-Ub) are indicated by arrows. (B) Intermediate effects of group II mutants onmonoubiquitination of FANCD2 in comparison with group I and group III mutants. Results of representative mutants are shown.
3132 Human Molecular Genetics, 2002, Vol. 11, No. 25
Downloaded from https://academic.oup.com/hmg/article-abstract/11/25/3125/578773by gueston 26 March 2018

MATERIALS AND METHODS
Cell culture
SV40-immortalized fibroblasts GM6914 were maintained inDMEM containing 10% fetal calf serum (FCS), as describedpreviously (16,19). 293T cells were maintained in DMEMcontaining 10% FCS.
Generation of FANCA mutant constructs and retroviralinfection of GM6914 cells
cDNAs of FANCA mutants with a Flag epitope at itsN-terminus were generated using a QuikChange Site-DirectedMutagenesis Kit (Stratagene, CA) and synthetic oligonucleo-tide primers containing patient-derived mutations. cDNAinserts of wt and mutant FANCA, verified by DNA sequencing,were subcloned into a retroviral expression vector, pMMP, asdescribed previously (16). The indicated pMMP constructswere co-transfected by lipofection using Fugene (RocheDiagnostics, IN, USA) into 293T producer cells with cDNAsof VSV-G envelope protein. Retroviral supernatants werecollected on day 2 following the lipofection. Infection ofGM6914 cells was carried out as described previously (16).After 2 days, selection with 0.5 mg/ml of puromycin was begun.
Cell survival assay
Cell survival assay was carried out as described previously (16).
Immunoprecipitation and immunoblotting
Cell extracts of GM6914 cells (�107 cells/sample) wereprepared in lysis buffer supplemented with protease inhibitorsand subjected to immunoprecipitation using an anti-Flagmonoclonal antibody (M2, Sigma), as described previously(19). Immunoprecipitates or lysates were separated on SDS–polyacrylamide gels, transferred to PVDF membranes andimmunoblotted with the indicated antibodies. Protein bandswere visualized using enhanced chemiluminescence detectionreagents (DuPont).
In vivo phosphorylation of FANCA
In vivo phosphorylation of FANCA was examined as describedpreviously (17). Briefly, cells (�107 cells/sample) wereincubated in medium containing [32P]orthphosphate (1 mCi/ml) for 2 h and lysed in 1 ml of lysis buffer. Immunoprecipitateswith affinity-purified anti-FANCA antibody (2 mg) raisedagainst the N terminus (3) were separated on SDS–polyacry-lamide gels and blotted onto a PVDF membrane.Phosphorylation of proteins on the membrane was visualizedautoradiographically, and the same membrane was probedusing the indicated antibodies, as described previously (19).
Immunofluorescence microscopy
Cells were fixed with 2% paraformaldehyde in PBS (pH, 7.4)for 20 min followed by permeabilization with 0.3%Triton X-100 in PBS for 10 min. Next, the cells were incubated
for 1 h in blocking buffer [PBS containing 10% normal goatserum and 0.1% Nonidet P40 (NP40)], followed by incubationin blocking buffer containing anti-FANCA antibody (3) for 2 hat room temperature. Cells were washed with washing buffer(PBS containing 0.1% NP40) and incubated for 1 h in blockingbuffer containing goat anti-rabbit secondary antibody con-jugated to fluorescein isothiocyanate. Cells were again washedwith washing buffer. Cell nuclei were stained with a mountingmedium containing 40,6-diamidino-2-phenylindole (DAPI)(Vector Laboratories, CA). Fluorescence microscopy wascarried out as described previously (47). To determinepercentages of cells with different staining patterns, 100 cellswere scored in two independent experiments.
Mono-ubiquitination of FANCD2
A monoubiquitinated form of FANCD2 was detected as aprotein band with a slower mobility on immunoblotting usingan anti-FANCD2 monoclonal antibody (26).
Cell fractionation
Fractionation of GM6914 cells was carried out as describedpreviously (19). Each fraction was analysed on SDS–polyacrylamide gels. Proteins were analysed by immunoblot-ting with the indicated antibodies.
ACKNOWLEDGEMENTS
We thank T. Nakahata for support and M. Hoatlin forantibodies. This work was supported by a Grant-in-Aid forScientific Research from the Ministry of Education, Science,Technology, Sports and Culture of Japan and grants from theMinistry of Health, Labor and Welfare of Japan. The Divisionof Genetic Diagnosis is supported in part by OtsukaPharmaceutical Co. Ltd.
REFERENCES
1. Joenje, H. and Patel, K.J. (2001) The emerging genetic and molecularbasis of Fanconi anaemia. Nat. Rev. Genet., 2, 446–457.
2. Yamashita, T. and Nakahata, T. (2001) Current knowledge on thepathophysiology of Fanconi anemia: from genes to phenotypes.Int. J. Hematol., 74, 33–41.
3. Grompe, M. and D’Andrea, A.D. (2001) Fanconi anemia and DNA repair.Hum. Mol. Genet., 10, 2253–2259.
4. Strathdee, C.A., Duncan, A.M. and Buchwald, M. (1992) Evidence for atleast four Fanconi anemia genes including FACC on chromosome 9.Nat. Genet., 1, 196–198.
5. Joenje, H., Lo Ten Foe, J.R., Oostra, A.B., van Berkel, C.G.,Rooimans, M.A., Schroeder-Kurth, T., Wegner, R.D., Gille, J.J.,Buchwald, M. and Arwert, F. (1995) Classification of Fanconi anemiapatients by complementation analysis: evidence for a fifth genetic subtype.Blood, 86, 2156–2160.
6. Joenje, H., Oostra, A.B., Wijker, M., di Summa, F.M., van Berkel, C.G.,Rooimans, M.A., Ebell, W., van Weel, M., Pronk, J.C., Buchwald, M. andArwert, F. (1997) Evidence for at least eight Fanconi anemia genes.Am. J. Hum. Genet., 61, 940–944.
7. Joenje, H., Levitus, M., Waisfisz, Q., D’Andrea, A.D., Garcia-Higuera, I.,Pearson, T., van Berkel, C.G., Rooimans, M.A., Morgan, N., Mathew, C.G.and Arwert, F. (2000) Complementation analysis in Fanconi anemia:assignment of the reference FA–H patient to group A. Am. J. Hum. Genet.,67, 759–762.
Human Molecular Genetics, 2002, Vol. 11, No. 25 3133
Downloaded from https://academic.oup.com/hmg/article-abstract/11/25/3125/578773by gueston 26 March 2018

8. Timmers, C., Taniguchi, T., Hejna, J., Reifsteck, C., Lucas, L., Bruun, D.,Thayer, M., Cox, B., Olson, S., D’Andrea, A.D. et al. (2001) Positionalcloning of a novel Fanconi anemia gene, FANCD2. Mol. Cell., 7, 241–248.
9. Strathdee, C.A., Gavish, H., Shannon, W.R. and Buchwald, M. (1992)Cloning of cDNAs for Fanconi’s anaemia by functional complementation.Nature, 356, 763–767.
10. Lo Ten Foe, J.R., Rooimans, M.A., Bosnoyan-Collins, L., Alon, N.,Wijker, M., Parker, L., Lightfoot, J., Carreau, M., Callen, D.F., Savoia, A.et al. (1996) Expression cloning of a cDNA for the major Fanconi anaemiagene, FAA. Nat. Genet., 14, 320–323.
11. The Fanconi Anaemia/Breast Cancer Consortium (1996) Positional cloningof the Fanconi anaemia group A gene. Nat. Genet., 14, 324–328.
12. de Winter, J.P., Waisfisz, Q., Rooimans, M.A., van Berkel, C.G.,Bosnoyan-Collins, L., Alon, N., Carreau, M., Bender, O., Demuth, I.,Schindler, D. et al. (1998) The Fanconi anaemia group G gene FANCG isidentical with XRCC9. Nat. Genet., 20, 281–283.
13. de Winter, J.P., Rooimans, M.A., van Der Weel, L., van Berkel, C.G.,Alon, N., Bosnoyan-Collins, L., de Groot, J., Zhi, Y., Waisfisz, Q.,Pronk, J.C. et al. (2000) The Fanconi anaemia gene FANCF encodesa novel protein with homology of ROM. Nat. Genet., 24, 15–16.
14. de Winter, J.P., Leveille, F., van Berkel, C.G., Rooimans, M.A., van Der Weel,L., Steltenpool, J., Demuth, I., Morgan, N.V., Alon, N., Bosnoyan-Collins, L.et al. (2000) Isolation of a cDNA representing the Fanconi anemiacomplementation group E gene. Am. J. Hum. Genet., 67, 1306–1308.
15. Kupfer, G.M., Naf, D., Suliman, A., Pulsipher, M. and D’Andrea, A.D.(1997) The Fanconi anaemia proteins, FAA and FAC interact to form anuclear complex. Nat. Genet., 17, 487–490.
16. Naf, D., Kupfer, G.M., Suliman, A., Lambert, K. and D’Andrea, A.D.(1998) Functional activity of the Fanconi anemia protein FAA requires FACbinding and nuclear localization. Mol. Cell. Biol., 18, 5952–5960.
17. Yamashita, T., Kupfer, G.M., Naf, D., Suliman, A., Joenje, H., Asano, S.and D’Andrea, A.D. (1998) The Fanconi anemia pathway requires FAAphosphorylation and FAA/FAC nuclear accumulation. Proc. Natl Acad. Sci.USA, 95, 13085–13090.
18. Kupfer, G.M., Naf, D., Garcia-Higuera, I., Wasik, J., Cheng, A.,Yamashita, T., Tipping, A., Morgan, N., Mathew, C.G. and D’Andrea, A.D.(1999) A patient-derived mutant form of the Fanconi anemia protein,FANCA, is defective in nuclear accumulation. Exp. Hematol., 27, 587–593.
19. Garcia-Higuera, I., Kuang, Y., Naf, D., Wasik, J. and D’Andrea, A.D.(1999) Fanconi anemia proteins FANCA, FANCC, and FANCG/XRCC9interact in a functional nuclear complex. Mol. Cell. Biol., 19, 4866–4873.
20. Waisfisz, Q., de Winter, J.P., Kruyt, F.A., de Groot, J., van der Weel, L.,Dijkmans, L.M., Zhi, Y., Arwert, F., Scheper, R.J., Youssoufian, H. et al.(1999) A physical complex of the Fanconi anemia proteins FANCG/XRCC9 and FANCA. Proc. Natl Acad. Sci. USA, 96, 10320–10325.
21. Kruyt, F.A., Abou-Zahr, F., Mok, H. and Youssoufian, H. (1999) Resistanceto mitomycin C requires direct interaction between the Fanconi anemiaproteins FANCA and FANCG in the nucleus through an arginine-richdomain. J. Biol. Chem., 274, 34212–34218.
22. Garcia-Higuera, I., Kuang, Y., Denham, J. and D’Andrea, A.D. (2000) TheFanconi anemia proteins FANCA and FANCG stabilize each other andpromote the nuclear accumulation of the Fanconi anemia complex. Blood,96, 3224–3230.
23. de Winter, J.P., van Der Weel, L., de Groot, J., Stone, S., Waisfisz, Q.,Arwert, F., Scheper, R.J., Kruyt, F.A., Hoatlin, M.E. and Joenje, H. (2000)The Fanconi anemia protein FANCF forms a nuclear complex withFANCA, FANCC and FANCG. Hum. Mol. Genet., 9, 2665–2674.
24. Medhurst, A.L., Huber, P.A.J., Waisfisz, Q., de Winter, J.P. andMathew, C.G. (2001) Direct interactions of the five known Fanconianaemia proteins suggest a common functional pathway. Hum. Mol. Genet.,10, 423–429.
25. Siddique, M.A., Nakanishi, K., Taniguchi, T., Grompe, M. andD’Andrea, A.D. (2001) Function of the Fanconi anemia pathway inFanconi anemia complementation group F and D1 cells. Exp. Hematol., 29,1448–1455.
26. Garcia-Higuera, I., Taniguchi, T., Ganesan, S., Meyn, M.S., Timmers, C.,Hejna, J., Grompe, M. and D’Andrea, A.D. (2001) Interaction of theFanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell., 7,249–262.
27. Pace, P., Johnson, M., Tan, W.M., Mosedale, G., Sng, C., Hoatlin, M., deWinter, J.P., Joenje, H., Gergely, F. and Patel, K.J. (2002) FANCE: the linkbetween Fanconi anaemia complex assembly and activity. EMBO J., 21,3414–3423.
28. Howlett, N.G., Taniguchi, T., Olson, S., Cox, B., Waisfisz, Q.,de Die-Smulders, C., Persky, N., Grompe, M., Joenje, H., Pals, G.et al. (2002) Biallelic inactivation of BRCA2 in Fanconi anemia. Science,297, 606–609.
29. Pang, Q., Christianson, T.A., Keeble, W., Diaz, J., Faulkner, G.R.,Reifsteck, C., Olson, S. and Bagby, G.C. (2001) The Fanconi anemiacomplementation group C gene product: structural evidence ofmultifunctionality. Blood, 98, 1392–1401.
30. Taniguchi, T., Garcia-Higuera, I., Xu, B., Andreassen, P.R., Gregory, R.C.,Kim, S.T., Lane, W.S., Kastan, M.B. and D’Andrea, A.D. (2002)Convergence of the Fanconi anemia and ataxia telangiectasia signalingpathways. Cell, 109, 459–472.
31. Yagasaki, H., Adachi, D., Oda, T., Garcia-Higuera, I., Tetteh, N.,D’Andrea, A.D., Futaki, M., Asano, S. and Yamashita, T. (2001) Acytoplasmic serine protein kinase binds and may regulate the Fanconianemia protein FANCA. Blood, 98, 3650–3657.
32. Lightfoot, J., Alon, N., Bosnoyan-Collins, L. and Buchwald, M. (1999)Characterization of regions functional in the nuclear localization of theFanconi anemia group A protein. Hum. Mol. Genet., 8, 1007–1015.
33. Levran, O., Erlich, T., Magdolena, N., Gregory, J.J., Batish, S.D.,Verlander, P.C. and Auerbach, A.D. (1997) Sequence variation in theFanconi anemia gene FAA. Proc. Natl Acad. Sci. USA, 94, 13051–13056.
34. Savino, M., Ianzano, L., Strippoli, P., Ramenghi, U., Arslanian, A.,Bagnara, G.P., Joenje, H., Zelante, L. and Savoia, A. (1997) Mutations ofthe Fanconi anemia group A gene (FAA) in Italian patients. Am. J. Hum.Genet., 61, 1246–1253.
35. Nakamura, A., Matsuura, S., Tauchi, H., Hanada, R., Ohashi, H.,Hasegawa, T., Honda, K., Masuno, M., Imaizumi, K., Sugita, K. et al.(1999) Four novel mutations of the Fanconi anemia group A gene (FAA) inJapanese patients. J. Hum. Genet., 44, 48–51.
36. Wijker, M., Morgan, N.V., Herterich, S., van Berkel, C.G., Tipping, A.J.,Gross, H.J., Gille, J.J., Pals, G., Savino, M., Altay, C. et al. (1999)Heterogeneous spectrum of mutations in the Fanconi anemia group A gene.Eur. J. Hum. Genet., 7, 52–59.
37. Tachibana, A., Kato, T., Ejima, Y., Yamada, T., Shimizu, T., Yang, L.,Tsunematsu, Y. and Sasaki, M.S. (1999) The FANCA gene in JapaneseFanconi anemia: reports of eight novel mutations and analysis of sequencevariability. Hum. Mutat., 13, 237–244.
38. Morgan, N.V., Tipping, A.J., Joenje, H. and Mathew, C.G. (1999) Highfrequency of large intragenic deletions in the Fanconi anemia group A gene.Am. J. Hum. Genet., 65, 1330–1341.
39. Cartegni, L., Chew, S.L. and Krainer, A.R. (2002) Listening to silence andunderstanding nonsense: exonic mutations that affect splicing. Nat. Rev.Genet., 3, 285–298.
40. Jans, D.A., Xiao, C-Y. and Lam, H.C.M. (2000) Nuclear targeting signalrecognition: a key control point in nuclear transport? BioEssays, 22, 532–544.
41. Alter, B.P. (1993) Fanconi’s anaemia and its variability. Br. J. Haemat., 85,9–14.
42. Verlander, P.C., Lin, J.D., Udono, M.U., Zhang, Q., Gibson, R.A.,Mathew, C.G. and Auerbach, A.D. (1994) Mutation analysis of the Fanconianemia gene FACC. Am. J. Hum. Genet., 54, 595–601.
43. Yamashita, T., Wu, N., Kupfer, G., Corless, C., Joenje, H., Grompe, M. andD’Andrea, A.D. (1996) Clinical variability of Fanconi anemia (type C)results from expression of an amino terminal truncated Fanconi anemiacomplementation group C polypeptide with partial activity. Blood, 87,4424–4432.
44. Gillio, A.P., Verlander, P.C., Batish, S.D., Giampietro, P.F. andAuerbach, A.D. (1997) Phenotypic consequences of mutations in theFanconi anemia FAC gene: an international Fanconi anemia registry study.Blood, 90, 105–110.
45. Futaki, M., Yamashita, T., Yagasaki, H., Toda, T., Yabe, M., Kato, S.,Asano, S. and Nakahata, T. (2000) The IVS 4 þ 4 A to T mutation of theFanconi anemia gene FANCC is not associated with a severe phenotype inJapanese patients. Blood, 95, 1493–1498.
46. Faivre, L., Guardiola, P., Lewis, C., Dokal, I., Ebell, W., Zatterale, A.,Altay, C., Poole, J., Stones, D., Kwee, M.L. et al. (2000) Association ofcomplementation group and mutation type with clinical outcome in Fanconianemia. Blood, 96, 4064–4074.
47. Oda, T., Muramatsu, M., Isogai, T., Masuho, Y., Asano, S.and Yamashita, T. (2001) HSH2: a novel SH2 domain-containingadapter protein involved in tyrosine kinase signaling inhematopoietic cells. Biochem. Biophys. Res. Commun., 288,1078–1086.
3134 Human Molecular Genetics, 2002, Vol. 11, No. 25
Downloaded from https://academic.oup.com/hmg/article-abstract/11/25/3125/578773by gueston 26 March 2018