hippocampal damage induced by ischemia and … · and glycine binding sites v. crepel,* a. represa,...
TRANSCRIPT

Neuroscience Vol. 31, No. 3. pp. 605-612, 1989 Printed in Great Britain
0306-4522/89 $3.00 + 0.00 Pergamon Press plc
0 1989 IBRO
HIPPOCAMPAL DAMAGE INDUCED BY ISCHEMIA AND INTRA-AMYGDALOID KAINATE INJECTION:
EFFECTS ON N-METHYL-D-ASPARTATE,
N-( l-[2-THIENYL]CYCLOHEXYL)PIP~RIDINE AND GLYCINE BINDING SITES
V. CREPEL,* A. REPRESA, M. BEAUDOIN and Y. BEN-ARI INSERM U 29, 123 Bd Port Royal, 75014 Paris, France
Abstract-The ~‘-methyl-D-aspartate receptor channel-complex is widely distributed in the hippocampus, particularly in the CA1 region, in the terminal field of CA3 pyramidal axons and in the fascia dentata, in the terminal field of the perforant pathway. In the present study, we have examined, in the rat, the effect of specific lesions of various neuronal populations of the hippocampus on the distribution of several markers of the N-methyl-D-aspartate receptor-channel complex. Anoxic-ischemic treatment produced a destruction of CA1 pyramidal cells (postsynaptic element); this was associated with a 50% loss of N-methyl-D-aspartate, glycine and N-(I-phenylcyclohexyl)pi~ridine binding sites. In contrast, the destruction of CA3 pyramidal cells and their axons ~resynaptic element) by kainate treatment did not induce significant changes in the density of binding sites.
The present results therefore strongly support an exclusively postsynaptic localization of the N-methyl- D-aspartate receptor-channel complex in CAI; the possibility of a localization of the remaining binding sites on glial cells or interneurons is discussed. In the molecular layer of the fascia dentata, the anoxic-ischemic treatment produced a partial destruction of the median perforant pathway (presynaptic element) associated with a decrease in the density of N-methyl-D-aspartate. N-(I-[2-thienylk~yclo- hexyl)piperidine and glycine binding sites; this suggests that, in contrast to CA], in the molecular layer of the fascia dentata, N-methyl-D-aspartate receptor-binding sites are located both pm- and postsynaptically.
The N-methyl-D-aspartate (NMDA) receptor is a subtype of glutamate receptors which mediate excitatory synaptic transmission.9 The NMDA receptor-channel complex is subject to several regu- lation sites. Thus, the NMDA response is potentiated by glycine24 which acts at an allosteric site and is blocked in a voltage-dependent manner by Mg*+ 32 or by N-f I-phenylcyclohexyl)piperidine (PCP),20 which bind to specific sites within the channeLis These observations are consistent with autoradiographic and biochemical findings, which indicate that [)H]glycine6 and [3H]-N-( I-[2-thienyl]cyclohexyl)pip- eridine ([‘H]TCP),29 a PCP analogue, have a distribu- tion very similar to that of NMDA-receptor sites.29.30 It is now well established that the highest density of NMDA binding sites is found, in the stratum radi- atum of CAl, at the level of the terminal field of Schaeffer collaterals and in the molecular layer of the fascia dentata30 at the level of the terminal field of the perforant pathway. Physiological experiments indi- cate that, in these regions, the NMDA receptor- channel complex plays an important role in the long-term potentiation and memory processes.8,‘9
*To whom correspondence should be addressed, Abbreviulions: MK801, (+)-5-methyl-IO,1 l-dihydro-5H di-
~nzo[a,dJcyclohepten-5,10-imine; NMDA, N-methyl- n-aspartate; PCP, N-( l-phenyl~yclohexyI)pi~ridine; TCP, N-(I-~2-thienyl]cyclohexyl)pi~ridine.
Several lines of evidence also suggest that the activa- tion of the NMDA receptor-channel complex plays an impo~ant role in anoxic-ischemic brain damage, notably in the CA1 region of the hippocampus, the most vulnerable structure in the brain to anoxia- ischemia. To better understand the mechanisms un- derlying the long-term effect of NMDA receptor activation and its role in anoxic changes, it is im- portant to determine precisely whether these recep- tors are located pre- or postsynaptically. In the present study we have used experimental procedures which selectively destroy the various neuronal popu- lations of the hippocampal formation and quan- titative autoradiography to determine the localization of NMDA and its regulation binding sites. Pre- liminary results of this study have been reported in brief elsewhere.“J2
EXPERIMENTAL PROCEDURES
Male Wistar rats (ZOO-25Og) were used in these experi- ments. They had access to food and water ad lib&m and were housed in individual cages under diurnal lighting conditions, with lights on from 0800 to 2000 h. The regional distribution of the NMDA receptor-channel complex in CA1 was studied following two types of lesions. (1) In a series of 11 rats, the pyramidal cells of CA1 were destroyed by a bilateral hemispheric &hernia, according to a pre- viously described method.” In brief, the vertebra1 arteries were first eiectrocauterized under anaesthesia (chloral hy- drate 7%) and a clamp set on both carotid arteries. The next day, when the animal had recovered from the procedure, the
605

606 V. CREPEL et al.
carotid arteries were clamped in the unanesthetized rat for 30 min; this produced a general sedation. (2) In a series of 10 rats the pyramidal cells of CA3 and their Schaeffer collaterals were destroyed in one hemisphere, by an injection of kainate into the amygdala (1.2 pg in 0.3 ~1 of phosphate buffer, pH 7.4) using a previously described procedure.’ Three or 10 days after ischemia or kainate injection, the extent and the specificity of the lesion was determined by Nissl (Cresyl Violet), Fink/Heimer13 or Gallyas16 staining. To quantitate the degree of neuronal damage 10 days after the treatment, the number of cells per region (CA1 and CA3 fields) was counted in 10 separate coronal sections per animal, at the septal level of the hippocampus. The mean values were expressed as percentage + S.E.M.
For autoradiographic study, the rats were killed 10 days after ischemia (n = 5) or kainate injection (n = 5). Five additional non-treated rats were used as controls. Brains were removed and frozen in isopentane at - 50°C. Coronal sections (20 pm) of the hippocampus were cut in a crystat (-20°C) and mounted onto gelatin-coated slides.
Visualization of glutamate, NMDA, glycine and TCP binding sites was performed by quantitative auto- radiography, ujsing previously described methods with mi- nor modifications.6,30,40
L-[‘H]Glutamare binding assay30
The slices were preincubated or 20 min at 20°C in 50 mM Tris-acetate buffer (pH 7.2) to remove competing en- dogenous ligand. They were then incubated at 4°C for 45 min, in the same buffer, containing 150 nM r$H]glutamate (NEN, 54.2 Ci/mM), for total binding, Alternate sections were incubated in the same medium in the presence of 150pM cold glutamate (Sigma), to determine the non specific binding (which was a every case lower than 5%) or in the presence of 150 PM cold NMDA to displace glutamate from its NMDA-sensitive sites. Finally, the sec- tions were rinsed for 30 s in 50mM Tris-acetate buffer (pH 7.2) and then immersed in acetone containing 2.5% of glutaraldehyde.
[‘H]Glycine binding assay 6
The slices were preincubated for 45 min at 20°C in 50 mM Tris-acetate buffer (PH 7.4) to remove competing endogenous ligands. For total glycine binding, they were - - _ incubated at 4’C for 20 min, in the same buffer, containing 200 nM 13Hlrrlvcine (NEN. 41 CiimM). Alternate sections were incubii& in the same medium’ in the presence of 200 PM cold glycine (Sigma), to determine the non-specific binding (which was in every case lower than 15%) or in the presence of 1OpM strychnine to displace glycine from its strychnine-sensitives sites. After incubation, the sections were rinsed for 2 min in 50 mM Tris-acetate buffer (pH 7.4).
[‘H]N-( I-[2-Thienyl]cyclohexyl)piperidine binding assay”
The slices were incubated for 15 min at 2O”C, in 50 mM Tris-acetate (pH 7.4), to remove competing endogenous ligands. For total TCP binding, they were incubated at 4°C for 60 min, in the same buffer, containing 10 nM [‘H]TCP (CEA, 44 Ci/mM). Alternate sections were incubated in the same medium in the presence of 1OpM cold TCP, to determine non-specific binding (which was in every case lower than 5%). After incubation, the sections were rinsed for 2 min in 50 mM Tris-acetate buffer (pH 7.4).
After drying, the slides were put in an x ray cassette and apposed at a ‘H-sensitive LKB Ultrofilm. simultaneouslv with plastic tritium standards (Amersham).” Quantification was performed by computer-assisted microdensitometry (IMSTAR). At least 16 different sections from each animal and treatment were analysed; the differences were consid- ered significant when P i 0.01 (U-test of Mann and Whitney). After exposure the slices were stained with Cresyl Violet to determine the extent of the lesion.
RESULTS
Morphological eflects qf the anoxic-ischemic treat-
ment
In agreement with earlier studies,5,‘0,37 the CA1 area
was the most vulnerable region of the brain to the
ischemic treatment. Several features of this change, which have not been previously reported, deserve emphasis.
Three days after the insult, in the septal pole of the
hippocampus, the lesion involved specifically the subiculum, CA1 and the hilar zone. Fink-Heimer and Gallyas procedures revealed argyrophilic degen- erative neurons which were restricted to the pyr- amidal layer of CA1 and subicular cortex and to the polymorph hilar zone of the fascia dentata (Fig. ]A). Most pyramidal neurons of CA1 were heavily ar-
gyrophilic with the characteristic Golgi-type stain of apical and basal dendrites (Fig. IB). In the hilar region, a few cells were also stained with silver deposits (Fig. 1C). Interestingly, the soma of putative interneurons in the polymorph layer, which were not argyrophilic, were surrounded by a dense dotted pattern of silver deposits (Fig. 1 D); this is likely due to the degeneration of commissural inhibitory inter-
neurons.7,26 In agreement with a recent report,“’ we found a dense pattern of silver deposits in the outer part of the dentate molecular layer, corresponding to the terminal region of the perforant pathway; this likely correlates with a partial destruction of the median entorhinal cortex (Fig. 2C). Only occasional argyrophilic neurons were found in the rostra1 and medial pole of CA3 area. In contrast, in the caudal
pole of the hippocampus, the damage was not limited to the CA1 region and to the polymorph hilar zone of the fascia dentata. Argyrophilic degenerated neurons were conspicuous in all regions of the pyr- amidal layer of Ammon’s horn including CA22CA3 (Fig. 2A) and the granule layer of the fascia dentata (Fig. 2B). Therefore, with three days’ survival period, there is clearly a septotemporal gradient of destruc- tion produced by the anoxic-ischemic treatment.
Ten days after the treatment, in the septal pole of the hippocampus, the Cresyl Violet staining con- firmed an almost complete pyramidal cell loss in the CA1 region (by 91% f 3; Fig. 4A); interneurons in oriens and radiatum were clearly less affected (Fig. 4B). There was an important gliotic reaction in the pyramidal, oriens, and radiatum layers of CA1 (Fig. 4B). In the rostra1 and medial pole of the hippocampus there was no conspicuous cell loss in the CA3 field; in contrast, in the caudal pole of the hippocampus, there was a clear neuronal loss of CA3-CA4 pyramidal neurons and granule cells.
Morphological e$fticts of the kainate treatment
In agreement with a earlier study,’ the regional
distribution of neuropathological sequelae, after kai- nate, was specifically localized to the limbic system.
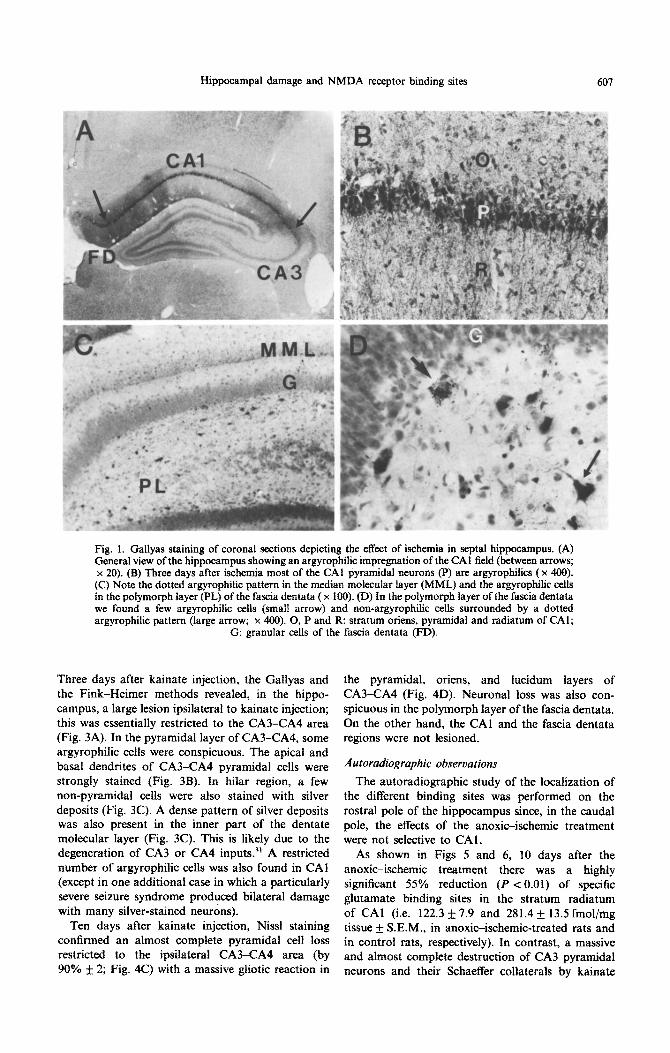
Hippocampal damage and NMDA receptor binding sites 601
Fig. 1. Gallyas staining of coronal sections depicting the effect of ischemia in septal hippocampus. (A) General view of the hippocampus showing an argyrophilic impregnation of the CA1 field (between arrows; x 20). (B) Three days after ischemia most of the CA1 pyramidal neurons (P) are argyrophilics ( x 400). (C) Note the dotted argyrophilic pattern in the median mokcuiar layer (MML) and the argyrophilic cells in the polymorph layer (PL) of the fascia dentata ( x 100). (D) In the polymorph layer of the fascia dentata we found a few argyrophilic cells (small arrow) and non-argyrophilic cells surrounded by a dotted argyrophilic pattern (large arrow; x 400). 0, P and R: stratum oriens, pyramidal and radiatum of CAl;
G: granular cells of the fascia dentata (FD).
Three days after kainate injection, the Gallyas and the Fink-Heimer methods revealed, in the hippo- campus, a large lesion ipsilateral to kainate injection; this was essentially restricted to the CA3-CA4 area (Fig. 3A). In the pyramidal layer of CA3-CA4, some argyrophilic cells were conspicuous. The apical and basal dendrites of CA3-CA4 pyramidal cells were strongly stained (Fig. 3B). In hilar region, a few non-pyramidal cells were also stained with silver deposits (Fig. 3C). A dense pattern of silver deposits was also present in the inner part of the dentate molecular layer (Fig. 3C). This is likely due to the degeneration of CA3 or CA4 inputs.” A restricted number of argyrophilic cells was also found in CA1 (except in one additional case in which a particularly severe seizure syndrome produced bilateral damage with many silver-stained neurons).
Ten days after kainate injection, Nissl staining confirmed an almost complete pyramidal cell loss restricted to the ipsilateral CA3-CA4 area (by 90% + 2; Fig. 4C) with a massive gliotic reaction in
the pyramidal, oriens, and lucidum layers of CA3-CA4 (Fig. 4D). Neuronal loss was also con- spicuous in the polymorph layer of the fascia dentata. On the other hand, the CA1 and the fascia dentata regions were not lesioned.
The autoradio~aphic study of the localization of the different binding sites was performed on the rostra1 pole of the hippocampus since, in the caudal pole, the effects of the anoxic-ischemic treatment were not selective to CAI.
As shown in Figs 5 and 6, 10 days after the anoxic-ischemic trea~ent there was a highly significant 55% reduction (P < 0.01) of specific glutamate binding sites in the stratum radiatum of CA1 (i.e. 122.3 f 7.9 and 281.4 + 13.5fmol/mg tissue + S.E.M., in anoxic-ischemic-treated rats and in control rats, respectively). In contrast, a massive and almost complete destruction of CA3 pyramidal neurons and their Schaeffer collaterals by kainate

608 V. CREPEL et al.
Fig. 2. Photomicrographs showing the effect of ischemia in caudal hippocampus and in enthorinal cortex. (A) Horizontal sections of caudal hippocampus stained by Fink-Heimer procedure, showing argyrophilic neurons in the whole of Ammon’s horn (CA1 and CA3 area), as well as in the granule layer (G) and in the polymorph layer (PL) of the fascia dentata ( x 40). (B) View of the argyrophilic granular cells in the fascia dentata of the caudal hippocampus (arrow, x 200). (C) and (D) Entorinal cortex stained with the Gallyas procedure, showing argyrophilic neurons in the median part (between arrows, x 20 and x 200
respectively). FD: fascia dentata.
treatment had no significant effect on the distribution and density of specific glutamate binding sites in the stratum radiatum of CA1 (240 f 19.1 fmol/mg tissue + S.E.M.). Figure 6 shows that this obser- vation also applies to the NMDA subtype of glutamate binding sites. Thus, 10 days after the
anoxic-ischemic treatment, there was a decrease by about 55% of specific NMDA binding sites (i.e. 71.1 f 7.1 and 154.3 f 10 fmol/mg tissue + S.E.M., in anoxic-ischemic-treated rats and in control rats, respectively), whereas kainate treatment and its associated destruction of CA3 neurons did not cause a significant reduction of specific NMDA binding sites (146.7 f 16 fmol/mg tissue f S.E.M.).
The specific glycine binding sites in the stratum radiatum in CA1 (Figs .i and 6) were signifi- cantly reduced 10 days after ischemia treat- ment (i.e. 155.1 + 26.4 and 318.45 f 15 fmol/mg tissue k S.E.M., in anoxic-ischemic-treated rats and
in control rats, respectively) but not after destruction of CA3 neurons and their Schaeffer collaterals by kainate treatment (285.3 + 15.5 fmol/mg tissue + S.E.M.). Figure 6 also shows that the ischemic treatment produced a decrease by about 60% of specific TCP binding sites (i.e. 26.65 f 1.25 and 67.9 k 2.9 fmol/mg tissue rf S.E.M., in anoxicc ischemic-treated rats and in control rats, re- spectively). In contrast, kainate treatment did not cause a significant reduction of specific TCP binding
sites (55.8 + 5.1 fmol/mg tissue + S.E.M.). In the molecular layer of fascia dentata, 10 days
after anoxic-ischemic treatment, there was a significant loss of the different markers of the NMDA receptor-channel complex, notably of TCP (50%) and NMDA-sensitive glutamate (40%) binding sites. In contrast, kainate treatment did not induce a significant change in the density of binding sites (Fig. 6).

Hippocampal damage and NMDA receptor binding sites 609
Fig. 3. Gallyas staining of coronal sections depicting the effect of kainate injection in the amygdala, in the hippocampus ipsilateral to the injection site. (A) General view of the hippocampus showing an argyrophilic impregnation of CA3 field (between arrows; x 20). (B) Three days after injection of kainate, most of the CA3 pyramidal neurons (P) are argyrophilics ( x 100). (C) In the fascia dentata, there are in the polymo~~ layer (PL) a few argyrophilic cells and in the inner part of the molecular layer (IML) a dotted argyrophilic pattern (x 200). 0, P and R: stratum oriens, pyramidal and radiatum of CA3; G:
granular cells of the fascia dentata (FD).
Interestingly, we found that in both control and treated cases the addition of an excess of strychnine (IO PM) to the incubation medium consistently in- duced a small but non-significant loss of specific glycine binding sites in the stratum radiatum of CA1 (by about 25%) and in the molecular layer of the fascia dentata (by about 20%); this may be due to a partial blockade of the opened NMDA channels by a high concentration of strychnine.3
As shown in Fig. 5, kainate treatment induces, ipsilateral to the lesion, a significant loss of glutamate and glycine binding sites (by 46.8% It 5 and 57.9 f 2, respectively) in the CA3 region; this correlates with the neuronal loss observed in this field.
DiSCUSSlON
The principal observation of the present study is that glutamate, NMDA, glycine and TCP binding sites are principally located postsynaptically on den- drites of the CA1 pyramidal neurons (over 50%). Since we found no evidence for presynaptic local-
ization (on the Schaeffer collaterals), the remaining binding sites after anoxia-i~hemia are likely to be iocated pos~ynaptically on CA1 interneurons which are relatively spared by ischemic treatment,2”6 or in glia. The remaining binding sites may also be located on intact synaptic complexes, i.e. including fragments of the postsynaptic membrane which could remain attached to the presynaptic boutons.*’ The present results are in agreement with a recent report4 which showed that anoxic-ischemic treatment, in gerbils, induced a loss of about 50% of MK-801 [( -t)-5-methyl-lo,1 I-dihydro-5H dibenzo[a,d]cyclo- hepten-5,10-imine] binding sites (another ligand for PCP binding sites), in the stratum radiatum of CA1 .
In contrast to CAl, our observations suggest that there is a significant presynaptic component of NMDA binding sites in the molecular layer of fascia dentata. In fact, the anoxic-ischemic procedure in- duced a partial destruction of the entorhinal cortex which was associated with a 40% reduction of NMDA and a 35% reduction of TCP binding sites. We cannot, however, completely exclude that part of

Fig. 4. Cresyl Violet staining af coronal sections depicting the specific necrosis involving in the h~p~ampus, t0 days afer ischemia or kainate injection in the amygdala. (A) and (C) General view of the hippocampus showing the specific injury in CA1 and CA3 area, respectively (between arrows, x 20). (B) and (D) Note the disappearance of the most pyramidal cells, as well as the gliotic reaction in CA1 and CA3 area, respectively ( x 100). 0, P and K: stratum oriens, pyramidal and radiatum of CA1 and
CA3. FD: fascia dentata.
(3i-0 GLYCNE L-(3)-I) GLUTAMATE
Fig. 5. Autoradiographies of corona1 hippocampal sections performed with [3H]glycine and L-[‘HIglutamate in control, ischemic- and kainate-treated rats. Note the important reduction of labelling in the stratum radiatum (sr) of CAI, in hippocampus of ischemic rats. mol: moleculare layer of the fascia
dentata.

Hippocampal damage and NMDA receptor binding sites 611
300
IaJ
03
÷I
e.', 2 0 0
O3 ¢.0
..,-,
~I00 0
E
A) L-[3H] glutatamate
• 2 0 0
u4 0 3
t i q~
.-~ loo
O
E
CA1 MCIL
B) NMDA
iI CAI MOL
C) [3H] glycine D) [3H] TCP
4 0 0
L U
~6 +l 3 0 0
0~
o~ 200 ~a
~ l O 0
o i CA1
• 8 0 -
[d.I
+l 6 0 .
4O'
e0'
O MDL CAI MOL
Fig. 6. Mean density (fmol/mg tissue + S.E.M.) of specific L-[3H]glutamate (A), NMDA (B), [3H]glycine (C) and [3H]TCP (D) binding sites, in the stratum radiatum of CA1 and in the molecular layer (MOL) of the fascia dentata, of control (solid bars), kainate-treated (stippled bars) and ischemic (open bars) rat hippocampi. Note the significant loss of different NMDA receptor-channel complex binding sites in the stratum radiatum of CA1 and in the molecular layer of the fascia dentata, in hippocampus of ischemic
rats. *P < 0.01 (U-test of Mann and Whitney).
the reduction of binding sites produced by anoxia may be due to the destruction of hilar neurons 23 which project to the molecular layer. 31 However, a similar neuronal loss in kainic acid-treated rats 39 failed to induce any significant change in binding sites. Binding loss could also be due to a postsynaptic neuronal damage following the denervation of granular cells.
The N M D A receptor plays an important role in the ischemic neuronal cell less. The anoxic ischemic treatment induces a temporary increase in the release of the endogenous agonists of N M D A (glutamate and aspartate) 2'~8 in the vicinity of vulnerable CA1 pyramidal cells, and the t r e a t m e n t with N M D A - channel antagonist,14,33'35 as well as the lesion of excitatory afferent pathways, 22 protects against is-
chemic damage in the brain. Therefore, it has been suggested that, during anoxia-ischemia, the Schaeffer collaterals release glutamate or other N M D A - analogues in toxic amounts, which by means of N M D A receptors increase the cytosolic Ca 2+ contents 28 and subsequently activate ph0spholipases 27 and proteases, 38 leading to cell damage. Activation of the N M D A receptor is also required for the induction of long-term potentiation. 8'~9 The present results provide additional support for an important con- tribution of postsynaptic mechanisms in CA1, both for the long lasting enhancement of synaptic response in CA1 and for the anoxic-ischemic damage.
Acknowledgements--We are indebted to Dr Lazdunski for providing TCP, and to S. Lindenman and S. Guidasci for technical assistance and photography, respectively.
R E F E R E N C E S
1. Ben-Ari, Y., Tremblay E. and Ottersen O. P. (1980) Injections of kainic acid into the amygdaloid complex of the rat: an electrographic, clinical and histological study in relation to the pathology of epilepsy. Neuroseienee 5, 515-528.
2. Beneviste H., Drejer J., Schousbue A. and Diemer N. H. (1984) Elevation of extraeellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J. Neuroehem. 43, 136%1374.
3. Bertolino M. and Vicini S. (1988) Voltage-dependent block by strychnine of N-methyl-D-aspartic acid-activated cationic channels in rat cortical neurons in culture Molec. Pharmac. 34, 98-I03.

612 V. CREPEL rt crl.
4
5
6
7
8
9
10
11.
12.
13
14.
15. 16.
17.
18.
19.
20.
21.
22.
23.
24.
25. 26. 21.
28.
29.
30.
31.
32.
33.
34.
35.
36.
31.
38.
39.
40.
Bowery N. G., Wong E. H. F. and Hudson A. L. (1988) Q uantitative autoradiography of [‘H]MK-801 binding sites in mammalian brain Br. J. Phurmac. 93, 944954. Brierly J. B. (1977) Cerebral hypoxia. In Greerzfield’s Neuropathology (eds Blackwood W. and Corsellis J. A. N.), pp. 43-85. Arnold, Chicago. Bristow D. R., Bowery N. G. and Woodruff G. N. (1986) Light microscopic autoradiographic localization of [‘Hlglycine and [jH]strychnine binding sites in rat brain. Eur. J. Pharmac. 126, 303-307. Buzsiki G. and Eidelberg E. (198 I) Commissural projection to the dentate gyrus of the rat: evidence for feed-forward inhibition. Brain Res. 230, 346-350. Collingridge G. L., Kehl S. J. and McLennan H. (1983) Excitatory amino acids in synaptic transmission in the Scheaffer-commissural pathway of the rat hippocampus. J. Physiol., Lond. 334, 3346.
Cotman C. W. and Iversen L. L. (1987) Excitatory aminoacids in the brain focus on NMDA receptors. Trends Neurosci. 10, 263-302. Crain B. J., Westerkam W. D., Harrison A. H. and Nadler J. V. (1988) Selective neuronal death after transient forebrain ischemia in the mongolian gerbil: a silver impregnation study. Neuroscience 27, 387.-402. Crepe1 V., Represa A. and Ben-Ari Y. (1988) Effect of ischemia and intra-amygdaloid kainate injection on the density of NMDA binding sites in the hippocampal CA1 region. Eur. J. Pharmac. 151, 355-~356. Crepe1 V., Represa A., Beaudoin-Kais M. and Ben-Ari Y. (1988) Loss of NMDA-receptor channel complex in anoxic-ischemic hippocampal damage. Sot. Neurosci. Abstr. 14, 105.
Fink R. P. and Heimer L. (1967) Two methods for selective silver imprentation of degeneration axons and their synaptic endings in the central nervous system. Bruin Res. 4, 369-~374. Foster A. C., Gill R., Kemp J. A. and Woodruff G. N. (1987) Systemic administration of MK-801 prevents N-methyl-D-aspartate-induced neuronal degeneration in rat brain. Neurosci. Lerr. 76, 307-31 I. Foster A. C. and Fagg G. E. (1987) Taking apart NMDA receptors. Nature 329, 395-396. Gallyas F.. Wolff J. R., Biittcher H. and Zaborsky L. (1980) A reliable and sensitive method to localize terminal degeneration and lysosomes in the central nervous system. Stain Technol. 53, 299-306. Geary W. A., Toga A. W. and Wooten G. F. (1985) Q uantitative film autoradiography for tritium. Methodological considerations. Bruin Res. 337, 99-103. Hagberg H., Lehman A., Sandberg M., Nystrom B., Jacobson I. and Hamberg A. (1985) Ischemia induced shift of inhibitory and excitatory aminoacid from intra to extracellular compartments. J. cerebr. Blood Flow Merub. 5,413419. Harris E. W., Ganong A. H. and Cotman C. W. (1984) Long-term potentiation in the hippocampus involves activation in N-methyl-D-aspartate receptors. Brain Res. 323, 132.-137. Honey C. R., Miljkovic Z. and MacDonald J. F. (1985) Ketamine and phencyclidine cause a voltage-dependent block of responses to r-aspartic acid. Neurosci. Lett. 61, 135-139. Johansen F. F., Jorgensen M. B. and Diemer N. H. (1983) Resistance of hippocampal CA1 interneurons to 20 min of transient cerebral ischemia in the rat. Acta neuroputh. 61, 135-140. Johansen F. F., Jorgensen M. B. and Diemer N. H. (1986) Ischemic CAI pyramidal cell loss is prevented by preischemic colchicine destruction of dentate gyrus granule cells. Brain Res. 377, 344347. Johansen F. F., Zimmer J. and Diemer N. H. (1987) Early loss of somatostatin neurons in dentate hilus after cerebral &hernia in the rat precedes CAI pyramidal cell loss. Actu neuroputh. 73, 1 lo- 114. Johnson J. W. and Ascher P. (1987) Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 325, 529-532. Kirino T. (1982) Delayed neuronal death in the gerbil hippocampal following ischemia. Bruin Res. 239, 57-69. Laurberg S. (1979) Commissural and intrinsic connections of the rat hippocampus. J. camp. Neurol. 184, 685-708. Mabe H., Nagai H., Umemuras S., Ohno M. and Takagi T. (1985) Effect of nimodipine on experimental cerebral ischemia. J. cerebr. Blood Flow Metub. 5, S341Hi342. MacDermott A. B., Mayer M. L., Westbrook G. L., Smith S. J. and Barker J. L. (1986) NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature 321, 519-~522. Maragos W., Penney J. and Young A. B. (1988) Anatomic correlation of NMDA and [3H]-TCP-labeled receptors in rat brain. J. Neurosci. 8, 493-501. Monaghan D. T. and Cotman C. W. (1985) Distribution of N-methyl-D-aspartate-sensitive L-[‘HIglutamate-binding sites in rat brain. J. Neurosci. 5, 2909-2919. Nadler J. V., Perry B. W., Gentry C. and Cotman C. W. (1980) Loss and reacquisition of hippocampal synapses after selective destruction of CA3%CA4 afferents with kainic acid. Brain Rex. 191, 387-403. Nowak L., Bregestovski P.. Ascher P., Herbert A. and Prochiantz A. (1984) Magnesium gates glutamate-activated channels in mouse central neurons. Nufure 307, 462465. Prince D. A. and Feeser H. R. (1988) Dextromethorphan protects against cerebral infarction in rat model of hypoxia-ischemia. Neurosci. Lerr. 85, 291-296. Pulsinelli W. A. and Brierly J. B. (1979) A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke 10, 267-272. Saver D., Nuglish J., Rossberg C., Mennel H-D., Beck T., Bielenger G. W. and Krieglestein J. (1988) Phencyclidine reduces postischemic neuronal necrosis in rat hippocampus without changing blood flow. Neurosci. Lelt. 91, 327-332. Schlander M., Hoyer S. and Frotscher M. (1988) Glutamate decarboxylase-immunoreactive neurons in the aging rat hippocampus are more resistant to ischemia than CA1 pyramidal cells. Neurosci. Lerr. 91, 241-246. Schmidt-Kastner R. and Hossman K. A. (1988) Distributrion of ischemic neuronal damage in the dorsal hippocampus of rat. Acta neuropath. 76, 41 l-421. Siejo B. K. and Wieloch T. (1985) Cerebral metabolism in ischemia: neurochemical basis for therapy. Br. J. Anuesrh. 57, 47-62. Sloviter R. S. and Damiano B. P. (1981) Sustained electrical stimulation of the perforant path duplicates kainate- induced electrophysiological effects and hippocampal damage in rats. Neurosci. Lefl. 24, 279-284. Vignon J., Chicheportiche R., Chicheportiche M., Kamenka J. M., Geneste P. and Lazdunski M. (1983) [‘HITCP: a new tool with high affinity for the PCP receptor in rat brain. Bruin Res. 280, 194197.
(Acceplrd 20 March 1989)