hit and run: x marks the spot!
TRANSCRIPT

nature structural & molecular biology volume 16 number 8 august 2009 801
Vikki M. Weake and Jerry L. Workman are at the Stowers Institute for Medical Research, Kansas City, Missouri, USA. e-mail: [email protected]
Hit and run: X marks the spot!Vikki M Weake & Jerry L Workman
Male fruitflies upregulate transcription of nearly all genes on their single X chromosome to equalize expression with the two X chromosomes in females. A new study shows that the distribution of the histone acetylation mark associated with this upregulation is much broader than that of the MSL complex responsible for depositing this mark.
compensated? Gelbart et al. examined the distribution of H4K16ac in male cultured D. melanogaster cells and in male third-instar larvae and observed a global enrichment of H4K16ac on the X chromosome relative to autosomes, both within intergenic and coding regions. The highest level of H4K16ac is associated with the middle and 3′ end of actively transcribed genes that are bound by the MSL complex. However, there is an elevated level of H4K16ac, but not other histone marks, on the remainder of the X chromosome. Similar findings have recently been independently reported in one of two additional studies examining H4K16ac distribution5,8.
Intriguingly, the current study identifies a class of genes associated with intermediate levels of H4K16ac but that have undetectable levels of MSL complex occupancy. However, as with authentic MSL target genes, H4K16ac at this second class of genes also requires an intact MSL complex. Following RNA interference (RNAi)-mediated knockdown of MSL2 or MOF in cultured cells, H4K16ac is lost from the entire X chromosome, even from this second class of sites that do not have detectable levels of associated MSL complex. Consistent with the cell culture results, H4K16ac is also absent from both MSL-bound and unbound sites on the X chromosome of mof mutant male larvae.
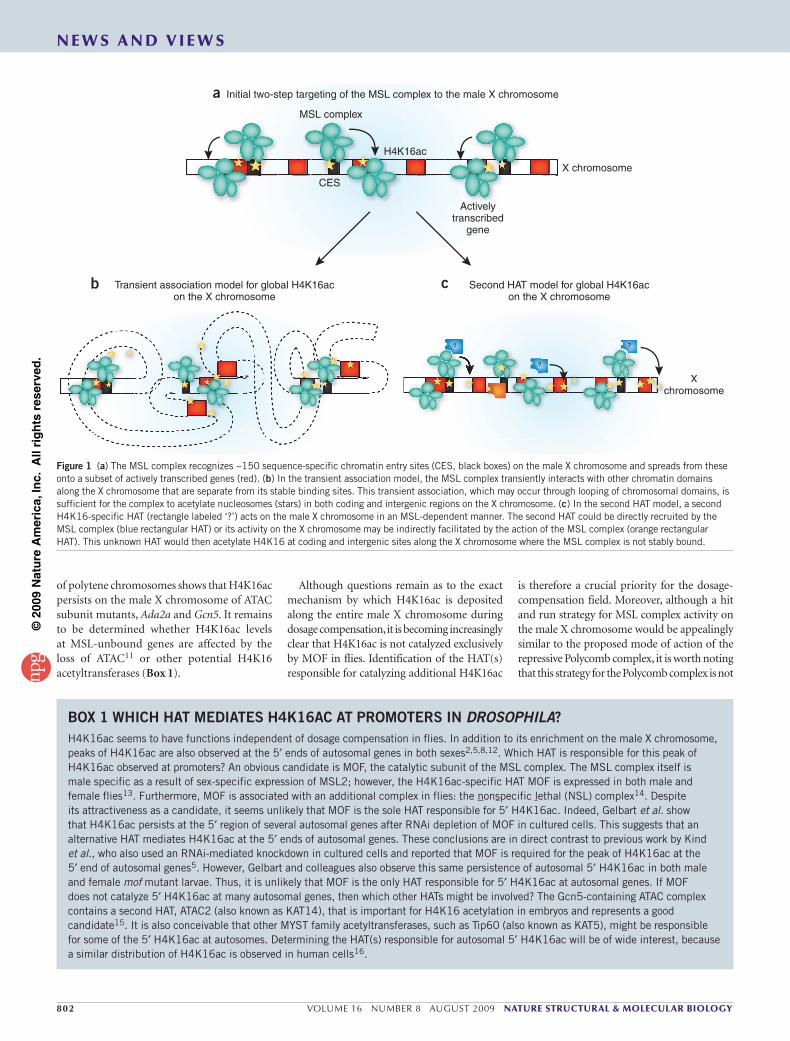
There are two alternative mechanisms by which H4K16ac might be deposited across the entire male X chromosome, even at those sites where the MSL complex is not detectably bound. We refer to these two distinct mechanisms as the ‘transient association’ and the ‘secondary HAT’ models, respectively. Gelbart et al. favor the first of these: the transient association model. In this model, after the MSL complex recognizes its ~150 CES on the X chromosome and spreads stably into nearby genes that are being actively transcribed (Fig. 1a), it also transiently associates with other portions of the X chromosome, including intergenic regions and additional transcribed genes using what the authors describe as a ‘hit and run’ approach (Fig. 1b). This transient association model is strongly reminiscent of that proposed
for the repressive Polycomb complex, which shows sharp peaks of localization in contrast to the broad domains of H3K27me3, the modification deposited by the complex6. A proposed mechanism by which such transient association might occur would involve MSL complex, stably bound to its CES, looping to make transient contacts with nucleosomes at other sites9. These transient contacts might be enhanced by modifications associated with active transcription, such as H3K36me3 (ref. 4). Supporting this model, Gelbart et al. have reported a low level of MSL complex at some of these H4K16ac-enriched sites on the X chromosome that do not seem to stably associate with the complex.
Although the transient association model seems plausible, an alternative model cannot be excluded based on the current evidence. It is formally possible that a second HAT acetylates H4K16 at X chromosome sites that do not stably bind the MSL complex (Fig. 1c). In this second HAT model, the MSL complex first binds and acetylates H4K16 at its CES and spreads into nearby actively transcribed genes, continuing to acetylate H4K16. This initial MSL complex binding and/or activity facilitates the acetylation of the remaining H4K16 sites across the X chromosome by a second HAT. This model cannot be excluded, although in the absence of MSL2 and MOF, H4K16ac is lost from the entire X chromosome, as MSL2 is required for the MSL complex to recognize the CES10, its initial target sites on the X chromosome. Furthermore, the MSL complex is incapable of acetylating H4K16 in the absence of MOF. Thus, if the MSL complex and H4K16ac are essential for recruitment or activity of a second HAT, this HAT would be nonfunctional in the absence of these MSL subunits. Hence, the activity of a second HAT on the X could be MSL2- and MOF-dependent. The existence of other H4K16ac-specific HATs in flies, and the observation that MOF is not essential for 5′ H4K16ac at autosomal genes (see Box 1 for discussion), makes it difficult to dismiss this second HAT model. Furthermore, mutations in another HAT complex, ATAC, which acetylates H4K16, alter the global morphology of the male X chromosome11, although immunostaining
Dosage compensation, the process by which the transcription of all X-linked genes in males is upregulated twofold, is mediated by the male-specific lethal (MSL) complex in Drosophila melanogaster. The MSL complex, which is present only in males, is a histone acetyltransferase (HAT) complex composed of the MSL1, MSL2, MSL3, Maleless (MLE) and Males absent on the first (MOF; also known as KAT8) proteins, together with two noncoding roX (RNA on X) RNAs1. On page 825 of this issue, Gelbart et al. show that the histone acetyl mark deposited by the MSL complex, histone H4 Lys16 acetylation (H4K16ac), is associated with the majority of actively transcribed genes on the male X chromosome2. In contrast, the distribution of the MSL complex itself is more limited3–5. These findings support a model in which a transient (undetectable) association of the MSL complex with some genes on the male X chromosome is sufficient to deposit H4K16ac and cause dosage compensation. This model is reminiscent of that proposed for histone H3 Lys27 methylation (H3K27me) by the repressive Polycomb complex6 and suggests that such transient association might be a hallmark common to both activating and repressing chromatin-modifying complexes that spread over large genomic regions.
The prevailing model for targeting the MSL complex to the X chromosome involves a two-step process in which the complex first recognizes and binds to a few hundred sequence-specific chromatin entry sites (CES) on the X chromosome. Following this, the MSL complex spreads from these sites into flanking chromatin by associating predominantly with the middle and 3′ ends of actively transcribed genes1. However, despite the fact that nearly all transcribed genes on the male X chromosome are dosage compensated7, about a quarter of these genes do not stably recruit the MSL complex3,4. How then are these genes dosage
n e w s a n d v i e w s
©20
09 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
802 volume 16 number 8 august 2009 nature structural & molecular biology
is therefore a crucial priority for the dosage- compensation field. Moreover, although a hit and run strategy for MSL complex activity on the male X chromosome would be appealingly similar to the proposed mode of action of the repressive Polycomb complex, it is worth noting that this strategy for the Polycomb complex is not
Although questions remain as to the exact mechanism by which H4K16ac is deposited along the entire male X chromosome during dosage compensation, it is becoming increasingly clear that H4K16ac is not catalyzed exclusively by MOF in flies. Identification of the HAT(s) responsible for catalyzing additional H4K16ac
of polytene chromosomes shows that H4K16ac persists on the male X chromosome of ATAC subunit mutants, Ada2a and Gcn5. It remains to be determined whether H4K16ac levels at MSL-unbound genes are affected by the loss of ATAC11 or other potential H4K16 acetyltransferases (Box 1).
X chromosome
Activelytranscribed
gene
CES
MSL complex
a Initial two-step targeting of the MSL complex to the male X chromosome
b c
H4K16ac
Xchromosome
Transient association model for global H4K16acon the X chromosome
Second HAT model for global H4K16acon the X chromosome
somXosX
omhrochc
?
?
??
?
Figure 1 (a) The MSL complex recognizes ~150 sequence-specific chromatin entry sites (CES, black boxes) on the male X chromosome and spreads from these onto a subset of actively transcribed genes (red). (b) In the transient association model, the MSL complex transiently interacts with other chromatin domains along the X chromosome that are separate from its stable binding sites. This transient association, which may occur through looping of chromosomal domains, is sufficient for the complex to acetylate nucleosomes (stars) in both coding and intergenic regions on the X chromosome. (c) In the second HAT model, a second H4K16-specific HAT (rectangle labeled ‘?’) acts on the male X chromosome in an MSL-dependent manner. The second HAT could be directly recruited by the MSL complex (blue rectangular HAT) or its activity on the X chromosome may be indirectly facilitated by the action of the MSL complex (orange rectangular HAT). This unknown HAT would then acetylate H4K16 at coding and intergenic sites along the X chromosome where the MSL complex is not stably bound.
BoX 1 WHicH HAT MeDiATeS H4K16Ac AT proMoTerS iN Drosophila?H4K16ac seems to have functions independent of dosage compensation in flies. In addition to its enrichment on the male X chromosome, peaks of H4K16ac are also observed at the 5′ ends of autosomal genes in both sexes2,5,8,12. Which HAT is responsible for this peak of H4K16ac observed at promoters? An obvious candidate is MOF, the catalytic subunit of the MSL complex. The MSL complex itself is male specific as a result of sex-specific expression of MSL2; however, the H4K16ac-specific HAT MOF is expressed in both male and female flies13. Furthermore, MOF is associated with an additional complex in flies: the nonspecific lethal (NSL) complex14. Despite its attractiveness as a candidate, it seems unlikely that MOF is the sole HAT responsible for 5′ H4K16ac. Indeed, Gelbart et al. show that H4K16ac persists at the 5′ region of several autosomal genes after RNAi depletion of MOF in cultured cells. This suggests that an alternative HAT mediates H4K16ac at the 5′ ends of autosomal genes. These conclusions are in direct contrast to previous work by Kind et al., who also used an RNAi-mediated knockdown in cultured cells and reported that MOF is required for the peak of H4K16ac at the 5′ end of autosomal genes5. However, Gelbart and colleagues also observe this same persistence of autosomal 5′ H4K16ac in both male and female mof mutant larvae. Thus, it is unlikely that MOF is the only HAT responsible for 5′ H4K16ac at autosomal genes. If MOF does not catalyze 5′ H4K16ac at many autosomal genes, then which other HATs might be involved? The Gcn5-containing ATAC complex contains a second HAT, ATAC2 (also known as KAT14), that is important for H4K16 acetylation in embryos and represents a good candidate15. It is also conceivable that other MYST family acetyltransferases, such as Tip60 (also known as KAT5), might be responsible for some of the 5′ H4K16ac at autosomes. Determining the HAT(s) responsible for autosomal 5′ H4K16ac will be of wide interest, because a similar distribution of H4K16ac is observed in human cells16.
n e w s a n d v i e w s
©20
09 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.

nature structural & molecular biology volume 16 number 8 august 2009 803
9. Gu, W., Wei, X., Pannuti, A. & Lucchesi, J.C. EMBO J. 19, 5202–5211 (2000).
10. Lyman, L.M., Copps, K., Rastelli, L., Kelley, R.L. & Kuroda, M.I. Genetics 147, 1743–1753 (1997).
11. Carré, C. et al. EMBO Rep. 9, 187–192 (2008).12. Bell, O. et al. EMBO J. 26, 4974–4984 (2007).13. Hilfiker, A., Hilfiker-Kleiner, D., Pannuti, A. & Lucchesi,
J.C. EMBO J. 16, 2054–2060 (1997).14. Mendjan, S. et al. Mol. Cell 21, 811–823 (2006).15. Suganuma, T. et al. Nat. Struct. Mol. Biol. 15,
364–372 (2008).16. Wang, Z. et al. Nat. Genet. 40, 897–903 (2008).
1399–1410 (2009).2. Gelbart, M.E., Larschan, E., Peng, S., Park, P.J. &
Kuroda, M.I. Nat. Struct. Mol. Biol. 16, 825–832 (2009).
3. Alekseyenko, A.A., Larschan, E., Lai, W.R., Park, P.J. & Kuroda, M.I. Genes Dev. 20, 848–857 (2006).
4. Larschan, E. et al. Mol. Cell 28, 121–133 (2007).5. Kind, J. et al. Cell 133, 813–828 (2008).6. Schwartz, Y.B. & Pirrotta, V. Curr. Opin. Cell Biol. 20,
266–273 (2008).7. Gupta, V. et al. J. Biol. 5, 3 (2006).8. Schwaiger, M. et al. Genes Dev. 23, 589–601 (2009).
conserved in mammals. Instead, the mammalian Polycomb complex binds to a broad region that overlaps with H3K27me3 enrichment6. Thus, although important general principles on the spreading of histone modification marks are emerging, the specific mechanisms used by individual activating or repressive complexes may be diverse.1. Gelbart, M.E. & Kuroda, M.I. Development 136,
Derek F. J. Ceccarelli is in the Program in Molecular Biology and Cancer, Samuel Lunenfeld Research Institute, Mount Sinai Hospital, Toronto, Ontario, Canada. Frank Sicheri is in the Program in Molecular Biology and Cancer, Samuel Lunenfeld Research Institute, and the Department of Molecular and Medical Genetics, University of Toronto, Toronto, Ontario, Canada. email: [email protected] or [email protected]
Grb-ing hold of insulin signalingDerek F J Ceccarelli & Frank Sicheri
it is just as important to shut off a signaling pathway as it is to turn it on. A new study on the tandem ras-associating (rA) and pleckstrin-homology (pH) domains of Grb10 and Grb14 provides important insight into a multicomponent assembly for downregulating insulin receptor signaling.
The spatial and temporal localization of proteins has an important role in the transduction of signals within a cell. In addition to the proper initiation of a signal-activating event, an equally important process is the appropriate termination of the signaling event. Protein kinases serve as important effector switches within signal- transduction pathways. Upon the binding of insulin, the kinase domain of the insulin receptor autophosphorylates, recruits and then phosphorylates effector proteins including insulin receptor substrates (IRS) 1–3 and Shc proteins. The IRS adaptor proteins in turn recruit and promote the activation of pathways, including the Ras-MAPK and phosphoinositide-3 kinase–Akt pathways, that elicit a range of metabolic responses to insulin, such as glucose uptake and glycogen synthesis1.
The transient nature of signaling processes requires downregulation of kinase activity and appropriate termination of insulin-mediated events. Grb10 and Grb14 are multidomain cytoplasmic adaptor proteins that serve to downregulate insulin receptor activity (reviewed in ref. 2). These Grb proteins comprise a Ras-association (RA), a pleckstrin homology
(PH) and a phosphotyrosine-binding SH2 domain, as well as a short element termed the between PH and SH2 (BPS) region. The BPS region in Grb14 directly inhibits the catalytic activity of the insulin receptor by binding as a pseudosubstrate in the kinase active site, whereas the C-terminal SH2 domain binds to the phosphorylated activation loop3. The RA and PH domains of Grb10 and Grb14 are also required for inhibition of insulin receptor signaling. Current findings from the Hubbard laboratory4, reported in this issue of Nature Structural & Molecular Biology, reveal how these two domains physically associate to form an integrated unit that interacts with phosphoinositides and the small GTPase Ras at membrane surfaces. The PH and RA domains share an extensive interface, including several conserved hydrophobic residues, which suggests this arrangement is not a condition of crystallization. Interestingly, the phosphoinositide- and GTPase-interaction sites are both fully accessible for interaction with their respective ligands in the reported crystal structure. The authors show that mutations that disrupt either phosphoinositide binding by the Grb14 PH domain or GTPase binding by the RA domain abrogate membrane localization and insulin receptor downregulation in cells.
On the basis of their findings, Depetris et al.4 propose a multicomponent assembly model that allows Grb10 and Grb14 proteins to fulfill their inhibitory function on insulin receptor signaling (Fig. 1). Assembly of a fully competent Grb10 or Grb14 repression complex with the insulin receptor involves (i) PH domain–mediated interaction with phosphoinositides, which serve to target Grb proteins to membrane surfaces, (ii) the RA domain, which directs Grb
proteins to activated GTPases that accumulate upon insulin receptor signaling, (iii) the BPS and SH2 domains of Grb proteins, which bind the insulin receptor kinase domain at two distinct sites, namely the substrate binding
PI
Grb10/14
GTP
N
C
Insulin
Insulinreceptor
P
P
P
RA
PH
Ras
BPSSH2
Figure 1 Multicomponent assembly for Grb10- and Grb14-mediated inhibition of insulin receptor signaling. Phosphoinositide (PI) interactions with the PH domain target Grb proteins to membrane surfaces. The structures of the RA-PH domains (PDB 3HK0)4 and a PH–phosphoinositide complex (PDB 1FGY)7 are known. The Grb RA domain interacts with activated GTPases. The RalGDS–Ras complex (PDB 1LFD)8 is representative of this interaction. The SH2 domain of Grb proteins engages with phosphotyrosine sites (P) on residues 1158 and 1162 on the insulin receptor kinase domain activation loop. The Grb BPS element engages the phosphotyrosine 1163 and also acts as a pseudosubstrate inhibitor within the peptide-binding groove of the kinase domain. Structures of the insulin receptor kinase–Grb14 BPS (PDB 2AUH)3 and insulin receptor kinase–SH2 (PDB 1RQQ)9 complexes are known. For the purpose of clarity, no dimer or multimer arrangement of subunits is represented in this model. N, N lobe of insulin receptor kinase domain; C, C lobe of insulin receptor kinase domain.
n e w s a n d v i e w s
©20
09 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.