http://parasol.tamu.edu using motion planning to map protein folding landscapes nancy m. amato...
Post on 20-Dec-2015
218 views
TRANSCRIPT

http://parasol.tamu.edu
Using Motion Planning to Map Protein Folding Landscapes
Nancy M. AmatoParasol Lab,Texas A&M University
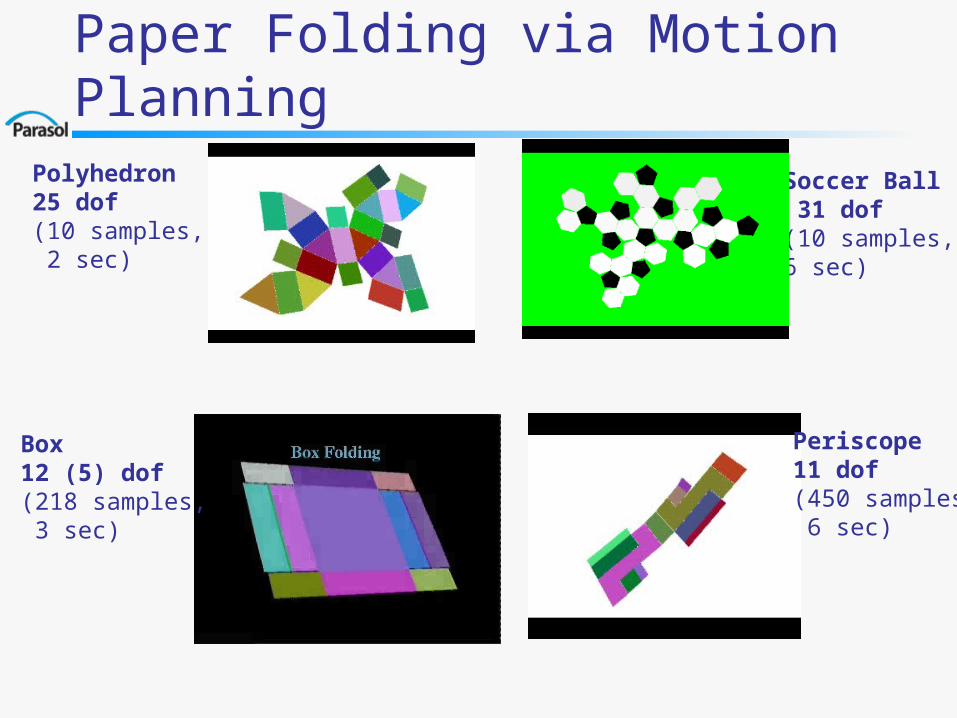
Soccer Ball 31 dof(10 samples, 6 sec)
Polyhedron25 dof(10 samples, 2 sec)
Paper Folding via Motion Planning
Box12 (5) dof(218 samples, 3 sec)
Periscope11 dof(450 samples, 6 sec)

Protein Folding via Motion PlanningFolding Paths for Proteins G & L
Protein LProtein G

Protein Folding We are interested in the folding process
– how the protein folds to its native structure
TTCCPSIVARSNFNVCRLPGTPEALCATYTGCIIIPGATCPGDYAN
Different from protein structure prediction – Predict native structure given amino acid sequence – Native 3D structure is important b/c influences function

Why Study Folding Pathways?
Importance of Studying Pathways– insight into protein interactions & function
– may lead to better structure prediction algorithms
– Diseases such as Alzheimer’s & Mad Cow related to misfolded proteins
Computational Techniques Critical– Hard to study experimentally (happens too fast)– Can study folding for thousands of already
solved structures – Help guide/design future experiments
normal - misfold
prion protein

Folding Landscapes
Each conformation has a potential energy– Native state is global
minimum Set of all conformations
forms landscape Shape of landscape
reflects folding behavior
Configuration space
Potential
Native state
Different proteins different landscapes different folding behaviors

Using Motion Planning to Map Folding Landscapes [RECOMB 01,02, 04; PSB 03]
A conformation
Configuration space
Potential
Use Probabilistic Roadmap (PRM) method from motion planning to build roadmap
Roadmap approximates the folding landscape– Characterizes the main
features of landscape– Can extract multiple
folding pathways from roadmap
– Compute population kinetics for roadmap
Native state

Related Work Folding landscape
Trajectory
(path #)
Path quality Time dependent
(running time)
Folding kinetics Native state needed
Molecular Dynamics
No Yes (1) good Yes.
(very long)
No No
Monte Carlo No Yes (1) good Yes
(very long)
No No
Statistical
Model
Yes No N/A No (short) Yes (only average)
Yes
Our PRM approach(RECOMB 01, 02,04, PSB 03)
Yes Yes
(many)
approximate No (short) Yes, multiple kinetics
Yes
Other PRM-Based approaches for studying molecular motions
– Other work on protein folding
([Apaydin et al, ICRA’01,RECOMB’02])
– Ligand binding
([Singh, Latombe, Brutlag, ISMB’99], [Bayazit, Song, Amato, ICRA’01])
– RNA Folding (Tang, Kirkpatrick, Thomas, Song, Amato [RECOMB 04])

Modeling Proteins
One amino acid
Secondary Structure
helix sheet
+ + variable loops =
Tertiary Structure
Primary Structure
TTCCPSIVARSNFNVCRLPGTPEALCATYTGCIIIPGATCPGDYAN
We model an amino acid with 2 torsional degrees of freedom: – Standard practice by biochemists

• Sample using known native state– sample around it, gradually grow out – generate conformations by randomly
selecting phi/psi angles
• Criterion for accepting a node:– Compute potential energy E of each
node and retain it with probability:
Roadmap Construction: Node Generation
NNative state
Denser distribution around native state

Ramachandran Plots for Different Sampling Techniques
Uniform sampling Gaussian sampling
Iterative Gaussian sampling

Distributions for different types:Potential Energy vs. RMSD for roadmap nodes
all alpha alpha + beta all beta

Roadmap ConstructionNode Connection
1. Find k closest nodes for each roadmap node (k=20)• use Euclidean distance
Native state
Edge weight w(u,v) = f(E(C1), E(C2),… E(Cn))
c1 c2 c3 cn
…
lower weight more feasible
1 13 152 681
u v
2. Assign edge weight to reflect energetic feasibility:

PRMs for Protein Folding: Key Issues
• Energy Functions– The degree to which the roadmap accurately reflects
folding landscape depends on the quality of energy calculation.
– We use our own coarse potential (fast) and well known all atom potential (slow)
• Validation– In [ICRA’01, RECOMB ’01, JCB ’02], results validated with
experimental results [Li & Woodward 1999].

One Folding Path of Protein AA nice movie…. But so what?
B domain of staphylococcal protein A
Ribbon Model Space-fill Model

Roadmap AnalysisSecondary Structure Formation Order
Order in which secondary structure forms during folding
helix hairpin 1,2
Q: Which forms first?
[RECOMB’01, JCB’02, RECOMB’02, JCB’03, PSB’03]

Formation Time Calculation
Secondary structure has formed when x% of the native contacts are present– native contact: less than 7 A between C atoms in native state
native contact
1030
2040
50
time step at which each contact forms
If we pick x% as 60%, then at time step 30, three contacts present, structure considered formed

Contact Map
A contact map is a triangularmatrix which identifies all the native contacts among residues
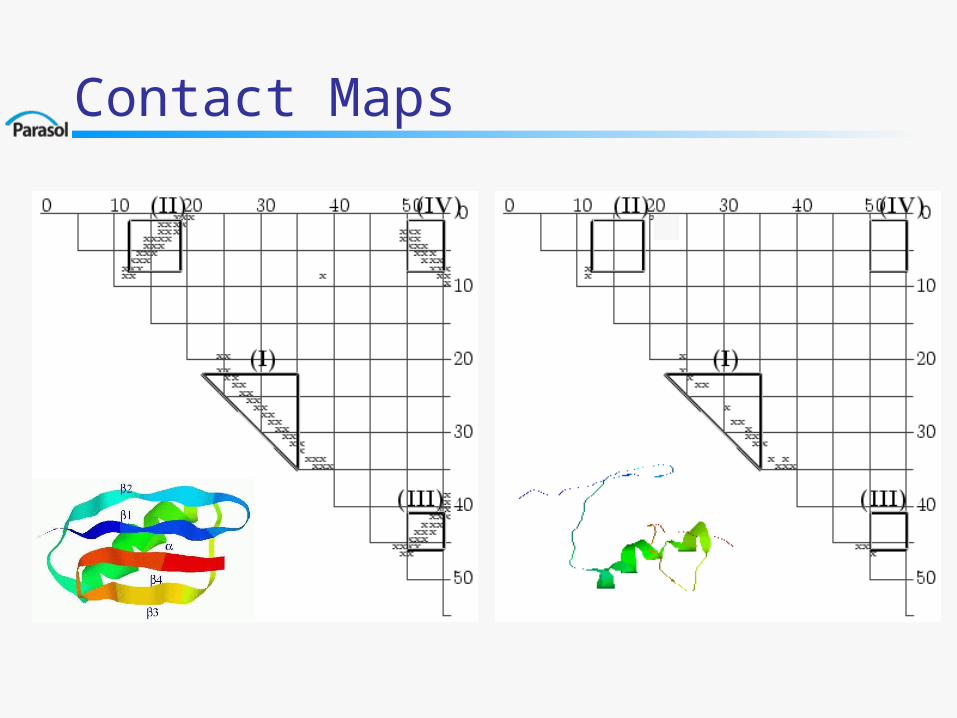
Contact Maps

Secondary Structure Formation Order:Timed Contact Map of a Path [JCB’02]
protein G (domain B1)
(IV: 1-4)
140 143
140 143 140
141 142 144
139 143 143 114
142135
131
1-4
3-4Average T = 142 Formation order:, 3-4, 1-2, 1-4
residue #
resid
ue #
1-2

Secondary Structure Formation Order:Timed Contact Map of a Path [JCB’02]
protein G (domain B1)
(IV: 1-4)
140 143
140 143 140
141 142 144
139 143 143 114
142135
131
1-4
3-4Average T = 142 Formation order:, 3-4, 1-2, 1-4
residue #
resid
ue #
1-2

Secondary Structure Formation Order:Validation Sample Summary
PDB # of Residues
#order % of paths Secondary structure formation order Exp.
1GB1 56 2 66
34
,3-4,1-2,1-4
,1-2,3-4,1-4
Agreed
1BDD 60 1 100 2,3,1,2-3, 1-3 Agreed
1COA 64 2 90
10
, 3-4, 2-3, 1-4, -4
, 3-4, 2-3, -4, 1-4
Agreed
2AIT 74 66 9.1
7.4
4-5, 1-2 …
1-2, 4-5 …
Agreed
1UBQ 76 3 80
15
,3-4,1-2, 3-5,1-5
3-4, , 1-2, 3-5,1-5
Agreed
1BRN 110 4 75
8.3
1,2,3 …
1,3,2 …
Not sure

Detailed Study of Proteins G & L[PSB’03]
Protein G
Protein L
• Protein G & Protein L• Similar structure (1 helix, 2 beta strands), but 15% sequence identity• Fold differently
• Protein G: helix, beta 3-4, beta1-2, beta 1-4 [Kuszewski et al 1994, Orban et al. 1995]
• Protein L: helix, beta 1-2, beta 3-4, beta 1-4 [Yi & Baker 1996, Yi et al 1997]
• Can our approach detect the difference? Yes!• 75% Protein G paths & 80% Protein L paths have “right” order• Increases to 90% & 100%, resp., when use all atom potential
Protein G

Helix and Beta StrandsCoarse Potential [PSB’03]
Contacts SS Formation Order 20 40 60 80 100, 3-, -, - 76 66 77 55 58, -, -, - 23 34 23 45 42, 3-, -, - 85 78 77 62 67, 3-, -, - 11 11 9 8 8, -, -, - 4 10 14 29 24
Analyze First x% Contacts
all
hydrophobic
• Protein G:
• Protein L:
Contacts SS Formation Order 20 40 60 80 100, -, -, - 67 76 78 78 92, -, -, - 15 4 4 4 4, -, -, - 19 20 18 18 4, -, -, - 54 65 74 73 86, -, -, - 3 3 3 2 2, -, -, - 36 32 23 26 13
all
hydrophobic
Analyze First x% Contacts
12
3
4
12
3
4
3- 4 forms first) over 2k paths analyzed
1- 2 forms first) over 2k paths

• Protein G:
• Protein L:
Helix and Beta StrandsAll-atom Potential
12
3
4
12
3
4
3- 4 forms first)
1- 2 forms first)
Contacts SS Formation Order 20 40 60 80 100
, 3-, -, - 79 79 74 82 90
, -, -, - 21 21 26 18 10
, 3-, -, - 77 74 71 77 81
, 1-, -, - 23 26 29 23 19
Analyze First x% Contacts
all
hydrophobic
Contacts SS Formation Order 20 40 60 80 100
, -, -, - 100 100 100 100 100
, -, -, - 99 100 99 99 99
, -, -, - 1 0 1 1 1
all
hydrophobic
Analyze First x% Contacts

Summary: PRM-Based Protein Folding
• PRM roadmaps approximate energy landscapes• Efficiently produce multiple folding pathways
– Secondary structure formation order (e.g. G and L)– better than trajectory-based simulation methods, such as Monte
Carlo, molecular dynamics
• Provide a good way to study folding kinetics – multiple folding kinetics in same landscape (roadmap)– natural way to study the statistical behavior of folding– more realistic than statistical models (e.g. Lattice models, Baker’s
model PNAS’99, Munoz’s model, PNAS’99)

Population kinetics analysis on the roadmaps shows that heuristic 1 can efficiently describe the energy landscape using a small subset of nodes
Heuristics are used to approximate energy landscape using small roadmaps.
RNA Folding ResultsX. Tang, B. Kirkpatrick, S. Thomas, G. Song [RECOMB’04 ]
Our roadmaps contain many folding pathways.
Folding Steps
En
erg
y p
rofil
e
RNA energy landscape can be completely described by huge roadmaps.
Map1 (Complete): 142 Nodes Map2 (Heuristic 1): 15 Nodes Map3 (Heuristic 2): 33 Nodes
Folding Steps
Po
pu
latio
n
Folding Steps
Po
pu
latio
n
Folding Steps
Po
pu
latio
n

Ligand Binding[IEEE ICRA`01]
• Docking: Find a configuration of the ligand near the protein that satisfies geometric, electro-static and chemical constraints
• PRM Approach (Singh, Latombe, Brutlag, 1999)
– rapidly explores high dimensional space
– We use OBPRM: better suited for generating conformations in binding site (near protein surface)
• Haptic User interaction– haptics (sense of touch) helps user understand molecular interaction
– User assists planner by suggesting promising regions, and planner will post-process and ‘improve’

Contact Information
For more information, check out our website:
http://parasol.tamu.edu/~amato/
Credits:
My students: Guang Song (now a Postdoc at Iowa State), Shawna Thomas, Xinyu Tang
&
Ken Dill (UCSF) and Marty Scholtz (Texas A&M)