human embryonic stem cell model of ethanol-mediated early developmental toxicity
TRANSCRIPT

Experimental Neurology 234 (2012) 127–135
Contents lists available at SciVerse ScienceDirect
Experimental Neurology
j ourna l homepage: www.e lsev ie r .com/ locate /yexnr
Human embryonic stem cell model of ethanol-mediated early developmental toxicity
Rodney Nash, Malini Krishnamoorthy, Andrew Jenkins, Marie Csete ⁎Department of Anesthesiology, Emory University School of Medicine, Atlanta GA 30322, USA
⁎ Corresponding author at: UCSD Anesthesiology, 2092103, USA.
E-mail address: [email protected] (M. Csete).
0014-4886/$ – see front matter © 2011 Elsevier Inc. Alldoi:10.1016/j.expneurol.2011.12.022
a b s t r a c t
a r t i c l e i n f oArticle history:
Received 9 August 2011Revised 25 November 2011Accepted 14 December 2011Available online 29 December 2011Keywords:Astrocyte differentiationDevelopmental neurotoxicityFetal alcohol syndromeNeuronal differentiation
Background: Fetal alcohol syndrome is an important clinical problem. Human embryonic stem cells (hESC)have not been widely used to study developmental alcohol toxicity. Here we document the phenotype ofhESC exposed to clinically-relevant, low dose ethanol (20 mM).Methods: All cultures were maintained in 3% O2 to reflect normal physiologic conditions. UndifferentiatedhESC were expanded with basic fibroblast growth factor (bFGF), with or without ethanol, then differentiatedwithout ethanol. Proliferation and apoptosis in response to ethanol were assayed, and PCR used to examineexpression of GABA receptor subunits. Whole cell patch clamping was used to examine GABAA receptor func-tion in undifferentiated hESC. Immunocytochemistry and western blotting were used to follow differentia-tion of early neurons, astrocytes, and oligodendrocytes,Principal findings: Exposure to 20 mM ethanol resulted in larger colonies of undifferentiated hESC despite an in-
crease in apoptosis, because proliferation of the undifferentiated cells (and neuroblasts) was significantly increased.Differentiation of hESC (following a week of ethanol exposure) resulted in decreased expression of GFAP (by west-ern) compared to unexposed cells, suggesting that astrocyte differentiation was reduced, while markers of oligo-dendrocyte and neuron differentiation were unchanged. At the message level, undifferentiated hESC express allGABAA receptor subunits, but functional receptors were not found by whole cell patch clamping.Conclusion: Our results in hESC suggest a complex mix of ethanol-induced phenotypic changes when ethanol ex-posure occurs very early in development. Not only increased apoptosis, but inappropriate proliferation and lossof trophic astrocytes could result from low-dose ethanol exposure very early in development. More generally,these studies support a role for hESC in developing hypotheses and focusing questions to complement animal stud-ies of developmental toxicities.© 2011 Elsevier Inc. All rights reserved.
Introduction
Human embryonic stem cells (hESC) have received considerable at-tention as potential sources of cells for transplant therapies of degenera-tive, inherited or traumatic disease processes. Surprisingly, though hESCare a unique tool for studying the molecular mechanisms of toxicity tothe early human embryo, hESC have not been highly exploited for thesekinds of studies. Further, in the few examples in which hESC are used tostudy ethanol-mediated developmental defects, the ethanol doses usedare very high compared to those encountered clinically. Here we reportthe phenotype of undifferentiated hESC exposed to clinically relevant,low dose (20 mM) ethanol. For reference, 22 mM is 0.10% ethanol.
Animal models of fetal alcohol toxicity emphasize that ethanol tox-icity to developing neurons leads to massive apoptosis mediated inlarge part through gamma-aminobutyric acidA (GABAA) and N-methylD-aspartate (NMDA) receptors (Ikonomidou et al., 2000). Here weshow that apoptosis of undifferentiated hESC (representing an earlierstage of development than the focus of most animal studies) in
0 W. Arbor Dr., San Diego CA
rights reserved.
response to low dose ethanol exposure is independent of GABAA
receptor function. Further the phenotype of hESCs exposed to lowdoses of ethanol (≤20 mM) is dominated by a significant increase inproliferation, such that despite increased apoptosis, colony size isincreased in the presence of ethanol. Exposure of undifferentiatedhESCs to ethanol for a week also leads to significant changes insubsequent differentiation patterns even when that differentiation iscarried out without ethanol in the medium. These results suggest thatvery early embryonic exposure to ethanol can have a significantimpact on later brain development, even if maternal ethanol intakeceases. Overall the results suggest a more complex picture of ethanoltoxicity than simple direct toxicity, and highlight the important placeof hESC models in helping to refine and complement animal modelsof drug-mediated developmental toxicities.
Results
Undifferentiated hESC apoptosis is increased as a function of exposure to20 mM ethanol
Fig. 1 shows representative images of hESC after terminal deoxy-nucleotidyl transferase dUTP nick end labeling (TUNEL) done after a

20 mM0 mM
A B
Fig. 1. TUNEL staining of undifferentiated hESCs expanded for 1 week in growth medium (A) or in growth medium with 20 mM ethanol (B). Without ethanol 1.0+0.1% of Oct-4-immunoreactive cells were TUNEL-positive vs. 4.0+0.2% of hESC after the ethanol exposure (pb0.01). Blue=DAPI nuclear stain; Red=Oct-4, Green=TUNEL. See Table 1 for col-lected apoptosis data statistics.
128 R. Nash et al. / Experimental Neurology 234 (2012) 127–135
week of culture in growth medium with or without 20 mM ethanol.Significantly more Oct-4-positive cells (marking undifferentiatedhESC) were TUNEL-labeled after ethanol exposure than without, inboth H1 and H9 cells (Table 1). Similarly immunocytochemistry foractivated caspase-3 showed the same pattern of increased apoptosisafter ethanol exposure, though the absolute numbers of activatedcaspase-3-positive cells were less than the numbers that wereTUNEL-positive (Table 1).
Proliferation of undifferentiated hESCs is significantly increased byexposure to 20 mM ethanol
Bromodeoxyurine (BrdU) uptake was used to quantify proliferationafter a week of continuous exposure to ethanol. BrdU immunoreactiv-ity was detected in 1.9±0.1% of undifferentiated H1 cells vs. 4.2±0.6%of H1 cells exposed to ethanol (pb0.01) after a 2-hour pulse. BrdU im-munoreactivity was detected in 3.4±0.2% of untreated undifferen-tiated H9 cells vs. 7.0±0.4% of H9 cells exposed to ethanol (pb0.01),Fig. 2. In earlier experiments, we found that treatment of theundifferentiated hES cells with 5 mM ethanol also resulted in signifi-cantly increased BrdU uptake to a similar extent as seen with 20 mMethanol (not shown).
Table 1TUNEL assays show low dose ethanol increases apoptosis in undifferentiated humanembryonic stem cells.
A.
hES line EtOH TUNEL% P value
H1 − 1.0±0.1 b0.001H1 + 4.0±0.2 b0.001H9 − 1.5±0.4 b0.001H9 + 7.9±1.3 b0.001
B.
hES line EtOH Act. Casp3% P value
H1 − 0.7±0.4 b0.01H1 + 1.4±0.1 b0.01H9 − 1.0±0.1 b0.01H9 + 2.0±0.8 b0.01
A. Percentage of TUNEL-positive undifferentiated hESCs assayed after a week of growthwith (+) or without (−) 20 mmol ethanol. Data are mean±S.D.B. Percentage of activated caspase-3-positive undifferentiated hESCs assayed after aweek of growth with or without 20 mmol ethanol. P value was derived using Chi2
testing.
Proliferation of human neural stem cells derived from hESC is alsoincreased by exposure to ethanol
In order to determine whether the increase in proliferation in re-sponse to ethanol was unique to the undifferentiated state of hESCs,neuroprogenitors derived from hESC were also cultured in the pres-ence of 20 mM ethanol for a week and BrdU uptake compared tountreated controls. The ethanol-exposed cells so overgrew themonolayer that accurate BrdU-positive cell counts were not possiblein the large three-dimensional colonies. By comparison the growth
Fig. 2. Proliferation was significantly increased in both H1 (shown) and H9 cellsexposed to 20 mM ethanol (A) for a week, compared to control cells (B). BrdU staining(green) was used to assess proliferation after a 2-hour pulse. Blue=DAPI. Scalebar=50 μm. See Table 2 for all proliferation data statistics.

129R. Nash et al. / Experimental Neurology 234 (2012) 127–135
of untreated neuroprogenitors was minimal, and cell density ofethanol treated cells was obviously greater than the density ofuntreated neuroprogenitors (Fig. 3).
Proliferation of hESCs is significantly increased by ethanol in the absenceof bFGF
When passaged onto Matrigel at very low density, the H1 hESC linedid not recover well (Fig. 4). Growth of this line at low (clonal) density,even in the presence of bFGF, is usually poor. But addition of ethanol tothe medium resulted in significant expansion of the cells under thesebFGF-depleted conditions. As expected when bFGF is withdrawn, mostof the proliferating cells also started to differentiate and expressedneuronal β-tubulin III (Fig. 4), supporting the pro-proliferative effect ofethanol observed in the neuroprogenitors derived from hESCs. Only afew small Oct-4-positive colonies expanded in the ethanol-treatedcultures.
Message for all GABAA receptors is expressed in undifferentiated hESCsand is unchanged by ethanol treatment
Because ethanol normally acts in part at GABA receptors in thebrain, and because murine ES cells express functional GABAA recep-tors (Andang et al., 2008) we investigated the expression and func-tion of GABA receptors in hESCs. Table 2 shows the results of nestedPCR reactions to describe the repertoire of GABA receptor subunitsexpressed in hESCs, and in neuroprogenitors derived from hESCs(NP). All GABAA receptor subunits are expressed at the messagelevel in the undifferentiated embryonic stem cells, and the size ofthe GABAA repertoire of receptors became smaller with differentia-tion to a neural stem cell stage. Message for the two GABAB isoformswas not detected in undifferentiated cells but the b isoform wasexpressed in the neural stem cells.
The first PCR reactions were performed using hESCs grown on fi-broblasts, so we independently measured the expression of GABA re-ceptors in hESCs grown on Matrigel, and confirmed the expressionpatterns shown in Table 2. We also assayed MEFs for expression ofthe GABA receptor subtypes and found that they express GABAA α1,α2, α3, α5, α6 and all three β subtypes. Furthermore, the MEFsexpressed (abundant) glutamic acid decarboxylase-67 (GAD-67),suggesting that they can make GABA. Undifferentiated hESCs alsoexpressed GAD-67 (not shown).
Because GABA receptor expression is labile in brains with clinicalethanol administration and withdrawal (Diaz et al., 2011) we exam-ined GABA receptor expression in hESCs after a week of exposure to20 mM ethanol. The pattern of expression of all subunits (as inTable 2) was unchanged. The abundance of the only GABA receptorsubtype we examined by western blotting, GABAA α1, was also
Fig. 3. Proliferation was significantly increased in neuroprogenitor cells derived from hESCsicant expansion of neuroprogenitors in response to ethanol was obvious, and the cells piled
unchanged after the week of ethanol exposure (Fig. 5). In order to de-termine that some message known to be labile to ethanol during de-velopment was changing with ethanol treatment (Ahlgren et al.,2002), we used semiquantitative PCR with varying cycle number toexamine sonic hedgehog (SHH) message. Very little SHH wasdetected in undifferentiated hESCs. SHH was more highly expressedin the neuroprogenitor cells and expression levels were reduced byincubation of these cells for a week in 20 mM ethanol (Fig. 5).
Whole cell patch clamping reveals absence of functional GABAA receptorsin undifferentiated hESCs
Given the expression of the entire repertoire of GABAA receptorsand protein-level expression of at least one major subunit, wecharacterized the undifferentiated cells electrophysiologically forresponse to GABA, using reported methodologies (Sebel et al.,2006). Whole cell current recording from undifferentiated hESCsfailed to yield any measurable response to GABA when applied at0.3–1000 μM (0/15 cells). In contrast, controls processed in parallel,HEK293 cells transfected with α1, β2 and γ2 cDNAs, producedrobust responses to GABA in 6/6 cells using the identicalexperimental setup (Fig. 6). The heterologously expressedreceptors were found to have normal sensitivity to GABA, with anEC50=18.2±2.3 μM; Hill coefficient, nH=1.9±0.2 and maximumcurrent, Imax=1.57±0.43 nA. The electrophysiologic silence ofhESC in response to GABA signaling suggests that the pro-apoptoticeffects of ethanol on hESC are not mediated through GABAA
receptor signaling.
The alcohol metabolizing enzyme cytochrome P450E1 (CYP450E1) is notexpressed in undifferentiated hESC
Ethanol toxicity in differentiated cells is also increased by the ac-tion of the cytochrome P450 enzyme, CYP2E1, (Comporti et al.,2010) via its toxic metabolite, acetaldehyde. The enzymes that medi-ate metabolism of ethanol to acetaldehyde (CYP450E1, as well as cat-alase and alcohol dehydrogenase) have been found in brain (Hipolitoet al., 2007). Using standard PCR, we found message for P450E1 inhuman brain control samples, but not in undifferentiated hESC(Fig. 7). Enzyme activity assays were not done.
Differentiation of early neurons, astrocytes, and oligodendrocytes afterethanol exposure in undifferentiated hESCs
Ethanol exposure was restricted to undifferentiated cells (20 mMfor 1 week), and differentiation then carried out without ethanol. Atthe end of the differentiation period, complex colonies of β-tubulinIII-positive neurons, GFAP-positive astrocytes, and O4-positive
. Untreated neuroprogenitors grew slowly and remained as monolayers (A). The signif-up in 3D clusters, making BrdU counts impossible (B).
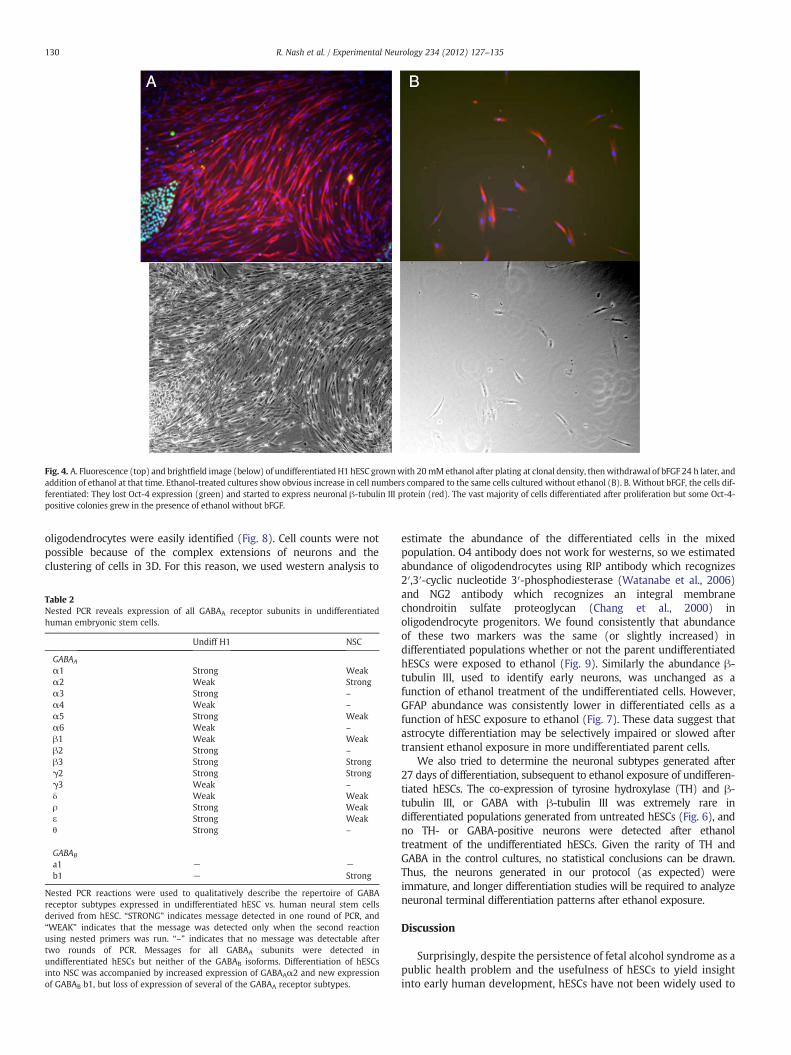
Fig. 4. A. Fluorescence (top) and brightfield image (below) of undifferentiated H1 hESC grownwith 20 mMethanol after plating at clonal density, thenwithdrawal of bFGF 24 h later, andaddition of ethanol at that time. Ethanol-treated cultures show obvious increase in cell numbers compared to the same cells cultured without ethanol (B). B. Without bFGF, the cells dif-ferentiated: They lost Oct-4 expression (green) and started to express neuronal β-tubulin III protein (red). The vast majority of cells differentiated after proliferation but some Oct-4-positive colonies grew in the presence of ethanol without bFGF.
130 R. Nash et al. / Experimental Neurology 234 (2012) 127–135
oligodendrocytes were easily identified (Fig. 8). Cell counts were notpossible because of the complex extensions of neurons and theclustering of cells in 3D. For this reason, we used western analysis to
Table 2Nested PCR reveals expression of all GABAA receptor subunits in undifferentiatedhuman embryonic stem cells.
Undiff H1 NSC
GABAA
α1 Strong Weakα2 Weak Strongα3 Strong –
α4 Weak –
α5 Strong Weakα6 Weak –
β1 Weak Weakβ2 Strong –
β3 Strong Strongγ2 Strong Strongγ3 Weak –
δ Weak Weakρ Strong Weakε Strong Weakθ Strong –
GABAB
a1 − −b1 − Strong
Nested PCR reactions were used to qualitatively describe the repertoire of GABAreceptor subtypes expressed in undifferentiated hESC vs. human neural stem cellsderived from hESC. “STRONG” indicates message detected in one round of PCR, and“WEAK” indicates that the message was detected only when the second reactionusing nested primers was run. “–” indicates that no message was detectable aftertwo rounds of PCR. Messages for all GABAA subunits were detected inundifferentiated hESCs but neither of the GABAB isoforms. Differentiation of hESCsinto NSC was accompanied by increased expression of GABAAα2 and new expressionof GABAB b1, but loss of expression of several of the GABAA receptor subtypes.
estimate the abundance of the differentiated cells in the mixedpopulation. O4 antibody does not work for westerns, so we estimatedabundance of oligodendrocytes using RIP antibody which recognizes2′,3′-cyclic nucleotide 3′-phosphodiesterase (Watanabe et al., 2006)and NG2 antibody which recognizes an integral membranechondroitin sulfate proteoglycan (Chang et al., 2000) inoligodendrocyte progenitors. We found consistently that abundanceof these two markers was the same (or slightly increased) indifferentiated populations whether or not the parent undifferentiatedhESCs were exposed to ethanol (Fig. 9). Similarly the abundance β-tubulin III, used to identify early neurons, was unchanged as afunction of ethanol treatment of the undifferentiated cells. However,GFAP abundance was consistently lower in differentiated cells as afunction of hESC exposure to ethanol (Fig. 7). These data suggest thatastrocyte differentiation may be selectively impaired or slowed aftertransient ethanol exposure in more undifferentiated parent cells.
We also tried to determine the neuronal subtypes generated after27 days of differentiation, subsequent to ethanol exposure of undifferen-tiated hESCs. The co-expression of tyrosine hydroxylase (TH) and β-tubulin III, or GABA with β-tubulin III was extremely rare indifferentiated populations generated from untreated hESCs (Fig. 6), andno TH- or GABA-positive neurons were detected after ethanoltreatment of the undifferentiated hESCs. Given the rarity of TH andGABA in the control cultures, no statistical conclusions can be drawn.Thus, the neurons generated in our protocol (as expected) wereimmature, and longer differentiation studies will be required to analyzeneuronal terminal differentiation patterns after ethanol exposure.
Discussion
Surprisingly, despite the persistence of fetal alcohol syndrome as apublic health problem and the usefulness of hESCs to yield insightinto early human development, hESCs have not been widely used to

Fig. 5. A. Western blot for detection of GABAAα1 in hESCs not treated with ethanol (lane 2) and those treated with 20 mM ethanol for a week (lane 3), showing no change inabundance of the protein. Cdk2 blotting was used as an internal control. B. Message for sonic hedgehog (SHH) is reduced in neuroprogenitors after the cells were exposed to ethanolfor a week in culture. Undifferentiated hESCs express little SHH.
131R. Nash et al. / Experimental Neurology 234 (2012) 127–135
study developmental toxicity of ethanol. Our results suggest thatundifferentiated hESC (equivalent to the pre-implantation embryo)are sensitive to low doses of ethanol, and the ethanol phenotype ofpreimplantation embryo-stage cells is characterized by bothincreased proliferation and apoptosis of undifferentiated hESCs.Proliferation predominates in cell culture since hESC colony sizeincreases in the presence of ethanol.
20 mM ethanol was chosen for these studies since it is well withinthe range of blood levels that can result from binge drinking before awoman realizes she is pregnant, and peak blood alcohol levels withsuch bingeing are associated with neural deficits in exposed children(Streissguth et al., 1990). Alcohol passes freely across the placentainto the fetal circulation (Waltman and Iniquez, 1972). Blood alcohollevels twice this amount are common in trauma patients after bingedrinking (Savola et al., 2005). Nonetheless, a continuous concentra-tion of 20 mM ethanol (as in our study) is not the ideal way tomodel drinking patterns, and further examination of more realisticpatterns should be performed, as well as dose–response studies. Alco-hol consumption patterns among pregnant women differ in differentcultures and countries (Chambers et al., 2006), and are often difficultto quantify. Further, no consensus has been realized about the mini-mum safe alcohol consumption during (various stages of) pregnancy.hESC models, with more sophisticated modeling of blood alcohol
levels in the cultures, could potentially be used to help fill this infor-mation gap.
The dramatic phenotype of proliferation in response to ethanol inhESCs and neuroprogenitors was not expected. Both increases anddecreases in proliferation of stem cell populations in response to eth-anol have been reported. Consistent with our observation, culturedrat neuronal precursors from e15 embryo brains showed increasedproliferation in response to 26 mM ethanol (Santillano et al., 2005).In contrast, others have reported decreased proliferation of e13 ratneuronal precursors exposed to 25 mM ethanol, and ethanol interfer-ence with bFGF-induced proliferation (Ma et al., 2003). Similardisparities have been reported in a variety of CNS progenitor celltypes, and likely these differences are dependent in part on theprecise developmental age of the cells, brain location from whichthey were derived, ethanol concentrations, or other factors in cellhandling (Crews and Nixon, 2003).
We did not do in depth analysis of the apoptotic pathways in-duced by ethanol, but identified higher numbers of cells that wereTUNEL-labeled vs. stained for activated caspase-3. There are severalexplanations for this finding: First, in some cell types, TUNEL ishighly sensitive and endonuclease damage associated with primarynecrosis can be detected by TUNEL (Lecoeur, 2002). In ischemic ratbrain, others have noted that cells undergo a hybrid death (some

Fig. 6. A. Whole cell current recording from hESC, voltage clamped at −60 mV. The bars above the flat current trace indicate the duration of GABA application and are labeled withthe concentrations applied. The calibration bar represents 10 s and 200 pA. B. Whole cell current recording from HEK293 cells transfected with α1, β2, γ2 cDNAs and voltageclamped at −60 mV. The bars above the current trace indicate the duration of GABA applied and are labeled with the concentrations applied. The calibration bar represents 10 sand 500 pA.
132 R. Nash et al. / Experimental Neurology 234 (2012) 127–135
apoptotic pathways activated and also necrosis) such that TUNEL isnot specific to apoptosis (Wei et al., 2004). Similar to ourobservations in undifferentiated hESC, one study reported very little(2%) co-localization of activated caspase-3 staining and TUNEL inthe setting of a cell culture model of glutamate excitotoxicity. In thiscase the authors suggested that the two labels mark different partsof the dynamic process of apoptosis with activated caspase-3expression in early apoptosis and TUNEL detecting irreversiblydamaged cells at the end of apoptosis (Brecht et al., 2001). Thesedifferences between the two labels of apoptosis are cell-type andstress-type dependent.
In our study, both labels indicated increased apoptosis in responseto ethanol in undifferentiated hESC, but unlike ethanol-inducedapoptosis in later stages of neuronal development, apoptosis inundifferentiated hESC was independent of GABAA receptor activity. Inundifferentiated hESCs we demonstrated no chloride conductance inresponse to GABA, in contrast to findings in mouse ES cells whichhave functional GABAA receptors (Andang et al., 2008). Mouse andhuman ES cells represent slightly different stages of development,and the lack of functional GABAA receptors on undifferentiated hESCsmay reflect this difference.
Animal models suggest that developing neurons are most vulner-able to ethanol-induced apoptosis during synaptogenesis (e19 to p14in rats). In humans, the comparable period is from the third trimesterthrough several years of age (Dobbing and Sands, 1979). Thus when
1 2 3 4
338 bp
Fig. 7. CYP450E1 is expressed in whole human brain lysate (lane 1), but not in WA01undifferentiated hESC (lane 2) or the murine embryonic fibroblast feeder layer (lane3). Lane 4 is negative control (no RNA input), 100 bp ladder on left.
comparing the results of ethanol exposure to undifferentiated hESCs(the earliest stage of development) with various animal models, thedevelopmental timing of exposure is important to consider. Ourstudies model earlier stages of development than those the focus ofthese reports.
The waves of neuronal death that occur normally during develop-mental neurogenesis (at the time when neurons are reported to bevery vulnerable to ethanol toxicity in rodent models (Ikonomidou etal., 2000)) are largely dependent on neurotrophic signals. Earlier indevelopment, programmed cell death in undifferentiated neuronalprecursors is also a normal developmental process, but is distinctfrom the later death patterns that are due to inability to access neuro-trophins. The molecular controls over this earlier wave of apoptosisduring development are less well-characterized (Yeo and Gautier,2004). hESCs may be useful in exploring this early developmentalapoptosis, and molecular mechanisms underlying apoptosis of earlyundifferentiated hESCs deserve further study.
Importantly, our results suggest that ethanol exposure to undiffer-entiated cells has carry-over effects on later differentiation events,even when the ethanol is absent during differentiation, as astrocytegeneration appeared to be reduced after ethanol exposure. Otherinvestigators have reported a generalized differentiation defect incultured e15 rat neural stem cell populations, with astrogliogenesisparticularly affected by 50 mM ethanol (Vemuri and Chetty, 2005).At higher ethanol doses (100 mM) others have reported increasedastrogliogenesis in similar neural stem cells (Tateno et al., 2005),interpreted as a stress response to ethanol. In the brains of rodentsexposed to ethanol in utero, radial glial stem cells and thereforeneurons and glia generated from them, are reduced in number(Rubert et al., 2006). Recent work on human fetal derived, CD133+/nestin+neural stem cells suggests that early neuronaldifferentiation (and neuroblast proliferation) is not impaired by100 mM ethanol. In this same study, astrogliogenesis was notimpaired by the relatively high dose of ethanol, but oligodendrocytegeneration (using western quantitation with Gal-C antibody) wasreduced (Vangipuram and Lyman, 2010). Taken together, theliterature is difficult to summarize because species, stage ofdevelopment, ethanol dose and other handling issues vary widelyfrom study to study. These varied conclusions about the effect ofethanol on development suggest that progress in understanding

Fig. 8. Top left: GFAP (green) stain shows small cluster of differentiating astrocytes; red is stain for neurofilament light chain. Top right: Complex collection of neuronal extensions(red, β-tubulin III) and early oligodendrocytes (green, O4 antibody). In general differentiating astrocytes and oligodendrocytes were found in clusters (among differentiatingneurons) rather than distributed evenly through the cultures. Bottom left: Very rarely, a cell expressing tyrosine hydroxylase (green) was found after the 27 days of differentiation.Bottom right: Punctate GABA staining (green, indicated with arrows) was also very rare in differentiating neurons, whichwere generally immature at this timepoint (red,β-tubulin III).
133R. Nash et al. / Experimental Neurology 234 (2012) 127–135
ethanol toxicity will be greatly aided by better coordination betweenhuman ESCs and animal models, with particular attention toequivalency of developmental state and dosing.
Animal models of fetal alcohol toxicity point to the importance ofneuronal apoptosis during development as a major contributor toclinical neurological and intellectual impairment in children exposedto ethanol during gestation. Our results showing increased prolifera-tion of hESCs and neuroprogenitors suggest that the final commonpathway of neuronal apoptosis in fetal alcohol toxicity may representmore than just direct toxicity. Inappropriate over-proliferation ofneuronal precursor cells could result in overproduction of neuronsthat cannot find trophic targets during critical periods of braindevelopment, thus increasing the numbers of neurons that areprogrammed for apoptosis. A second mechanism that is suggestedby our results is the relative lack of trophic astrocytes generatedafter ethanol exposure, again a potential contribution to neuronalapoptosis. In addition, we occasionally observed loss of adherencewhen cultured hESC were exposed to 50 mM or higher doses ofethanol (not shown). Loss of adhesion of developing cells to theirmatrix or loss of normal cell–cell contacts to transiently high dosesof ethanol could contribute to anoikis, though this phenotype wasnot observed with low-dose (5 to 20 mM) ethanol exposure inhESC. Ethanol exposure during development also impairs neuroblastmigration (Aronne et al., 2011) which we did not assay, but allthese defects could be modeled using hESCs and differentiated prod-ucts of hESCs.
A prominent feature of human fetal alcohol toxicity is microceph-aly, and neuronal apoptosis is considered a pathologic hallmark of thefetal alcohol toxicity seen in animal models. On the surface, our re-sults appear inconsistent with the human microcephaly phenotype,as proliferation of neural progenitors was increased by exposure to
ethanol. The model we used represented ethanol exposure veryearly in gestation, and the increased neuroblast proliferation doesnot preclude later neuronal toxicity with continued ethanol exposure,or a compensatory decrease in proliferation later in development ifethanol exposure is transient. In the end, the consequences of over-proliferation of neuronal precursors in response to ethanol will re-quire parallel animal model studies in which the transient exposureto the developing brain is designed to occur over an equivalent devel-opmental time period.
In general, these studies point to the usefulness of human embry-onic stem cells for studying toxicity to early developing humans me-diated by drugs of abuse (or by drugs that may be administeredduring early pregnancy, such as general anesthetics), as a comple-ment to animal models. Further, these studies raise questions thatcan be addressed in parallel animal models to further define the path-ophysiology of fetal alcohol toxicity.
Materials and methods
Cell culture
Use of human embryonic stem cells was approved by the EmoryUniversity HESCRO committee. H1 (male) human embryonic stemcells were used throughout and some studies as noted were alsodone using H9 (female) cells, purchased from WiCell. hESCs weregrown in 3% O2/5% CO2/92% N2 on mouse embryonic fibroblasts(MEFs, passage 3) isolated using the protocol below or on MEFsfrom Chemicon, Mountain View CA, or on Matrigel (BD Biosciences,Franklin Lakes NJ) as noted. MEF-conditioned medium was used forhESCs grown on Matrigel. hESC growth medium was DMEM-F12with 20% Knockout Serum Replacement (Invitrogen, Carlsbad CA)

Et OH Control Blank Brain
Lane 1 2 3 4
Tuj1-50 kD
GFAP-50 kD
RIP-78 kD
NG2-250 kD
Fig. 9. Several western blots are combined in this composite figure. Loading controls(cdk5) are not shown, but showed equal loading. All markers depicted were examinedin n=3 separate western assays. Western analysis of differentiated mixed cellpopulations consistently showed that treatment of undifferentiated hESC with ethanolbefore differentiation (lane 1) did not change β-tubulin III abundance (expressed in differ-entiating neurons), but did dramatically reduce GFAP protein levels, suggesting impairedastrocyte differentiation. Abundance of markers of early differentiating oligodendrocytes,NG2 and RIP, was also unchanged by ethanol treatment. In the bottom westerns, positivecontrol was human brain protein homogenate (lane 4). Lane 3 is empty.
134 R. Nash et al. / Experimental Neurology 234 (2012) 127–135
supplemented with L-glutamine, non-essential amino acids (NEAA),penicillin/streptomycin and 4 ng/ml basic fibroblast growth factor(bFGF) when grown on MEFs, and 8 ng/ml bFGF when grown onMatrigel. Cells were passaged at about 80% confluence. Medium waschanged daily during expansion, to account for evaporation of etha-nol from the culture medium even in humidified incubators.
For differentiation, cells were expanded as monolayer cultures for7 days then grown as embryoid bodies for 15 days. The embryoidbodies were then plated on poly-D-lysine and laminin-coated platesand cultured for another 12 days. Differentiation medium wasDMEM-F12 supplemented with 25 μg insulin, 20 nM progesterone,100 μM putrescine, 30 nM sodium selenite and 10 ng/ml bFGF, all ad-ditives from Sigma, St. Louis MO.
H1 and H9-derived neural progenitor cells were purchased fromAruna Biomedical, Athens GA and grown in Neurobasal Medium(Gibco, Invitrogen) supplemented with L-glutamine, B27, and leukemiainhibitory factor (LIF), on laminin-coated plates (10 μg/ml, Sigma) in3% O2. These neural stem cells do not express Oct-4; they uniformly ex-press nestin and Sox-2 which we confirmed immunohistochemically.
MEFs and MEF-conditioned medium
Mouse embryonic fibroblasts were purchased from Chemicon, Moun-tain View CA (catalog #PMEF-N). They were passaged using 0.25%trypsin-EDTA for 2 or 3 passages. Before hESCs were seeded, the MEFswere treated with mitomycin (MP Biomedicals, Irvine CA), 1 mg/ml for
2.5 h. Mitomycin-treatedMEFs were used tomakeMEF-conditionedme-dium, collected daily for a week, filtered and stored at −20 °C until use.
Terminal dUTP nick-end labeling (TUNEL) assays
Cells were fixed in paraformaldehyde (PFA) and then processedfor TUNEL assays according to the kit manufacturer's protocol (Pro-mega, Madison WI). For co-labeling of TUNEL with anti-Oct-4, thetwo primary and two secondary antibody incubations were carriedout simultaneously. The percentage of cells labeled with any givenantibody are expressed as mean±S.D., and percentages betweengroups compared using Chi2 analysis.
Bromodeoxyuridine (BrdU) uptake
Undifferentiated hESCs or hES-derived neural stem cells were in-cubated with 10 μM BrdU (Calbiochem). The hESCs were incubatedfor an hour, and neural stem cells for 4 h, then the cells were fixedin PFA. Immunohistochemical procedures for anti-BrdU labelingwere performed as noted below for immunohistochemistry. Primaryantibody was from Calbiochem, used at 1:100.
Polymerase chain reaction (PCR)
Nested PCR reactions were used to evaluate expression of theGABA receptor subtypes in undifferentiated hESC, neuroprogenitorsdifferentiated from hESC and in MEFs. Cells were collected in lysisbuffer, and total RNA extracted using the Qiagen RNeasy kit (Qiagen,Santa Clara CA), and quantified spectrophotometrically. cDNA wasprepared using 1 μg aliquots of total RNA and AMV reverse transcrip-tase (Promega), for use in the PCR assays. Taq polymerase for the PCRreactions was from Promega. Human brain cDNA (Clontech, Moun-tain View CA) was used as a positive control in all reactions, and allreactions were run with a negative control (no cDNA input).
The primers for all the GABAA receptors (external and internalsets) were from published protocols optimized for human cells(Alam et al., 2006). Primers for the two GABAB isoforms weredesigned using Vector NTI (open access software). Single (non-nested) PCR reactions used published primer sets for identificationof human sonic hedgehog (SHH) message (Ma et al., 2006) and forhuman cytochrome P2E1 (CYP2E1) (Hou et al., 2007). Semiquantita-tive PCR using variable cycle number was used to determine the rel-ative abundance of SHH in undifferentiated hES cells and inneuroprogenitors derived from them. Traditional non-nested PCRwas used to detect CYP2E1 message.
Western analysis
Cell pellets were stored at –80 °C in 20 mMHEPES, pH 7.6, 20% glyc-erol, 10 mMNaCl, 1.5 mMMgCl2, 0.2 mM EDTA, and 0.1% Triton X-100,with protease inhibitors (Complete; Boehringer Mannheim), homoge-nized, and incubated on ice for 1 h. After centrifugation, supernatantprotein concentration was assayed by BCA (Pierce, Rockford IL). Forwestern blots, proteins were separated on gradient (4–15%) acrylam-ide gels, 30 μg/lane. After transfer of proteins to nitrocellulose, themembrane was washed for 15 min in BioRad Tween-20 wash buffer,then blocked with 5% milk for an hour. Primary antibodies were incu-bated overnight at 4 °C, and after three 10-minute washes, secondaryantibodies were incubated for an hour at room temperature in 5%milk. The reactions were developed using enhanced chemilumines-cence (SuperSignal, Pierce). Primary antibody concentrations wereTuj1 1:2000, anti-GFAP 1:1000, anti-GABAAα1 (Abcam) 1:1000. Anti-tyrosine hydroxylase antibody (Pel-Freez Biologicals) was used at1:500, and secondary was HRP-conjugated goat anti-rabbit (Pierce) at1:5000.

135R. Nash et al. / Experimental Neurology 234 (2012) 127–135
Whole cell patch clamping
Undifferentiated hESCs were passaged and plated onto poly-D-lysine-coated glass coverslips and cultured for 24 h before transfer to a perfu-sion chamber for electrophysiologic characterization. HEK 293 cellswere transfected with human α1, β2, and γ2 GABAA receptor cDNAs aspreviously described (Sebel et al., 2006). Cells were whole cell voltageclamped at −60 mV with borosilicate glass microelectrodes containing145 nM N-methyl-D-glucamine HCl, 5 mM dipotassium ATP, 1.1 mMEGTA, 2 mM MgCl2, 5 mM HEPES/KOH, 0.1 mM CaCl2 adjusted to pH7.2. Cells were continuously superfused with an extracellular solutioncontaining 145 mM NaCl, 3 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2, 6 mMD-glucose, 10 mM HEPES/NaOH adjusted to pH 7.4. GABA (0.3, 1, 3, 10,30, 100, 300 and 1000 μM) was delivered to the cells via a 10-channelperfusion system (Sebel et al., 2006). The current peaks of the wholecell responses were used to establish the maximal current (Imax), effec-tive concentration for 50% activation (EC50) and Hill coefficient (nH) forthe actions of GABA as an agonist.
Immunohistochemistry
Standard immunohistochemical protocols were used on cells fixedin 4% paraformaldehyde (PFA) for 15 minutes at room temperature.Primary antibody sources and dilutions were α-Oct-4 (Santa Cruz,1:200), α-neuronal β-tubulin III (Tuj1 antibody from Covance,Berkeley CA, 1:2000), α-glial fibrillary acidic protein (GFAP, Cell Sig-naling Technology, Beverly MA, 1:100), O4 antibody to label imma-ture oligodendrocytes (Santa Cruz, 1:200) and α-activated caspase-3 (Cell Signaling 1:100), α-tyrosine hydroxylase (Pel-Freeze, RogersAR, 1:500), α-GABA (Sigma, 1:500). Secondary antibodies werefrom the Alexa Fluor series (Molecular Probes, Eugene OR). Mountingmedium was Vectashield with DAPI (4′,6-diamidino-2-phenylindole)from Vector Labs, Burlingame CA.
Acknowledgments
This work was supported by NINDS T32 award (RN), NIH RO1GM073959 (AJ), the Georgia Tech Center for the Engineering of LivingTissue (MC), and Emory Anesthesiology.
References
Ikonomidou, C., Bittigau, P., Ishimaru, M.J., Wozniak, D.F., Koch, C., et al., 2000. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 287,1056–1060.
Andang, M., Hierling-Leffler, J., Moliner, A., Lundgren, T.K., Castelo-Branco, G., et al.,2008. Histone H2AX-dependent GABA(A) receptor regulation of stem cell prolifer-ation. Nature 451, 460–464.
Diaz, M.R., Christian, D.R., Anderson, N.J., McCool, B.A., 2011. Chronic ethanol and with-drawal differentially modulate lateral/basolateral amygdale paracapsular and localGABAergic synapses. J. Pharmacol. Exp. Ther. 337, 162–170.
Ahlgren, S.C., Thakur, V., Bronner-Fraser, M., 2002. Sonic hedgehog rescues cranial neu-ral crest from cell death induced by ethanol exposure. Proc. Natl. Acad. Sci. 99,10476–10481.
Sebel, L.E., Richardson, J.E., Singh, S.P., Bell, S.V., Jenkins, A., 2006. Additive effects ofsevoflurane and propofol on gamma-aminobutyric acid receptor function. Anes-thesiology 104, 1176–1183.
Comporti, M., Signorini, C., Leoncini, S., Gardi, C., Ciccoli, L., Giardini, A., Vecchio, D.,Arezzini, B., 2010. Ethanol-induced oxidative stress: basic knowledge. GenesNutr. 5, 101–109.
Hipolito, L., Sanchez, M.J., Polache, A., Granero, L., 2007. Brain metabolism of ethanoland alcoholism: an update. Curr. Drug Metab. 8, 716–727.
Watanabe, M., Sakurai, Y., Ichinose, T., Aikawa, Y., Kotani, M., Itoh, K., 2006. Monoclonalantibody Rip specifically recognizes 2′,3′-cyclic nucleotide 3′-phophodiesterase inoligodendrocytes. J. Neurosci. Res. 84, 525–533.
Chang, A., Nishiyama, A., Peterson, J., Prineas, J., Trapp, B.D., 2000. NG2-positive oligo-dendrocyte progenitor cells in adult human brain and multiple sclerosis lesions.J. Neurosci. 20, 6404–6412.
Streissguth, A.P., Barr, H.M., Sampson, P.D., 1990. Moderate prenatal alcohol exposure:effects on child IQ and learning problems at age 7.5 years. Alcohol. Clin. Exp. Res.14, 662–669.
Waltman, R., Iniquez, E.S., 1972. Placental transfer of ethanol and its elimination atterm. Obstet. Gynecol. 40, 180–185.
Savola, O., Niemela, O., Hillbom, M., 2005. Alcohol intake and the pattern of trauma inyoung adults and working aged people admitted after trauma. Alcohol Alcohol. 40,269–273.
Chambers, C.D., Kavteladze, L., Joutchenko, L., Bakhireva, L.N., Jones, K.L., 2006. Alcoholconsumption patterns among pregnant women in the Moscow region of theRussian Federation. Alcohol 38, 133–137.
Santillano, D.R., Kumar, L.S., Prock, T.L., Camarillo, C., Tingling, J.D., Miranda, R.C., 2005.Ethanol induces cell-cycle activity and reduces stem cell diversity to alter bothregenerative capacity and differentiation potential of cerebral cortical neuroe-pithelial precursors. BMC Neurosci. 6, 59.
Ma, W., Li, B.S., Maric, D., Zhao, W.Q., Lin, H.J., et al., 2003. Ethanol blocks both basic fi-broblast growth factor- and carbachol-mediated neuroepithelial cell expansionwith differential effects on carbachol-activated signaling pathways. Neuroscience118, 37–47.
Crews, F.T., Nixon, K., 2003. Alcohol, neural stem cells, and adult neurogenesis. AlcoholRes. Health 27, 197–204.
Lecoeur, H., 2002. Nuclear apoptosis detection by flow cytometry: influence of endog-enous endonucleases. Exp. Cell Res. 277, 1–14.
Wei, L., Ying, D.-J., Cui, L., Langsdorf, J., Yu, S.P., 2004. Necrosis, apoptosis and hybriddeath in the cortex and thalamus after barrel cortex ischemia in rats. Brain Res.1022, 54–61.
Brecht, S., Gelderblom, M., Srinivasan, A., Mielke, K., Dityateva, G., Herdegen, T., 2001.Caspase-3 activation and DNA fragmentation in primary hippocampal neurons fol-lowing glutamate excitotoxicity. Brain Res. Mol. Brain Res. 94, 25–34.
Dobbing, J., Sands, J., 1979. Comparative aspects of the brain growth spurt. Early Hum.Dev. 3, 79–83.
Yeo, W., Gautier, J., 2004. Early neural cell death: dying to become neurons. Dev. Biol.274, 233–244.
Vemuri, M.C., Chetty, C.S., 2005. Alcohol impairs astrogliogenesis by stem cells in ro-dent neurospheres. Neurochem. Int. 47, 129–135.
Tateno, M., Ukai, W., Yamamoto, M., Hashimoto, E., Ikeda, H., Saito, T., 2005. The effectof ethanol on cell fate determination of neural stem cells. Alcohol. Clin. Exp. Res.29, 225S–229S.
Rubert, G., Minana, R., Pascual, M., Guerri, C., 2006. Ethanol exposure during embryo-genesis decreases the radial glial progenitor pool and affects the generation of neu-rons and astrocytes. J. Neurosci. Res. 84, 483–496.
Vangipuram, S.D., Lyman, W.D., 2010. Ethanol alters cell fate of fetal human brain-derived stem and progenitor cells. Alcohol. Clin. Exp. Res. 34, 1574–1583.
Aronne, M.P., Guadagnoli, T., Fontanet, P., Evrard, S.G., Brusco, A., 2011. Effects of pre-natal ethanol exposure on rat brain radial glia and neuroblast migration. Exp. Neu-rol. 229, 364–371.
Alam, S., Laughton, D.L., Walding, A., Wolstenholme, A.J., 2006. Human peripheralblood mononuclear cells express GABAA receptor subunits. Mol. Immunol. 43,1432–1442.
Ma, X.L., Sun, H.J., Wang, Y.S., Huang, S.H., Xie, J.W., et al., 2006. Study of Sonic hedgehogsignaling pathway related molecules in gastric carcinoma. World J. Gastroenterol. 12,3965–3969.
Hou, D.F., Wang, S.L., He, Z.M., Yang, F., Chen, Z.C., 2007. Expression of Cyp2E1 inhuman nasopharynx and its metabolic effect in vitro. Mol. Cell. Biochem. 298,93–100.