hydrogen-bond mediation of supramolecular aggregation in neutral bis-(c6f5)pt complexes with...
TRANSCRIPT

DO
I: 1
0.1
03
9/b
40
72
84
g
T h i s j o u r n a l i s © T h e R o y a l S o c i e t y o f C h e m i s t r y 2 0 0 4 2 7 3 3D a l t o n T r a n s . , 2 0 0 4 , 2 7 3 3 – 2 7 4 0
Dalton
ww
w.rsc.o
rg/d
alton
F U L L P A P E R
Hydrogen-bond mediation of supramolecular aggregation in neutral bis-(C6F5)Pt complexes with aromatic H-bond donating ligands. A synthetic and structural study†
José M. Casas, Beatriz E. Diosdado, Larry R. Falvello, Juan Forniés,* Antonio Martín and Angel J. RuedaDepartamento de Química Inorgánica, Instituto de Ciencia de Materiales de Aragón,Universidad de Zaragoza-C.S.I.C., E-50009, Zaragoza, Spain
Received 13th May 2004, Accepted 29th June 2004First published as an Advance Article on the web 23rd July 2004
Six pentafluorophenylplatinum(II) complexes containing proton acceptor atoms (F) and pyridine-like aromatic ligands able to act as proton donors have been synthesized and characterized, with emphasis on the factors that mediate their supramolecular aggregation in the solid state—hydrogen bonds and – interactions. The crystal structure analyses of the mononuclear complexes cis-[Pt(C6F5)2(napy)] (1), cis-[Pt(C6F5)2(CH2napy)] (3), cis-[Pt(C6F5)2(2-ammpy)] (5), and cis-[Pt(C6F5)2(2-bipym)] (6) reveal the influence of D–HPt and D–HF (D = C, N) hydrogen bonding on the organization of molecules into stacks, which can be further interconnected to generate channels. The prevalence of hydrogen bonding over – interactions between aromatic rings in establishing the nature of the observed supramolecular aggregation is demonstrated.
IntroductionHydrogen bonding, the most important of the non-covalent inter-actions in molecular materials, is well known to produce deviations from the behaviors that would be predicted on the basis of other non-covalent and bonding considerations, exerting its greatest influence in the liquid and solid states. The concept of hydrogen bonding has been expanded in recent years to include not only the interactions traditionally considered to fall into this category—namely of the type D–HA (D, A = N, O, F)—but also others such as C–HF, C–HO, O–HM and N–HM, which can be detected in the solid state and even in solution.1–7 The contribution of hydrogen bonding, despite its relative weakness in comparison to covalent and ionic bonding, can play a decisive role in the final organization of molecules and ions in the solid state; and the difficulty of evaluat-ing all of the possibilities for a given system impedes the prediction of the details of the resulting crystal structure.8 Hydrogen bonding has become a key concept in the emerging field of crystal engineer-ing because of the combination of strength with directionality attributable to this class of interaction.9 A CSD based investigation on X–HM hydrogen bonds has been carried out recently.10
The influence of – stacking, another of the important weak interactions, has lately been studied with increasing interest. Any of several interaction geometries are possible with aromatic rings, but the most important of these is the face-to-face arrangement, the principal contact topology observed in transition metal com-plexes with several organic ligands containing aromatic rings.11,12 Nitrogen-containing aromatic heterocycles are common ligands and have been demonstrated to be suitable for forming complexes that display this face-to-face interaction, often with the rings parallel to each other but slightly staggered.11
Both of these classes of non-covalent interaction, hydrogen bond-ing and – stacking, may be present simultaneously in the solid state, mediating the aggregation of molecules or ions into extended structures possessing a specific order and arrangement.12 This fact makes these interactions useful tools in supramolecular chemistry and crystal engineering; and a putative control of these forces is used in attempts to prepare multidimensional arrays directed towards properties such as luminescence13–15 or magnetism,15–17 or with possi-ble utility in fields such as host–guest exchange18,19 or catalysis.20,21
In this report we present the synthesis of a series of neutral organoplatinum(II) complexes containing substituents capable of tak-ing part in hydrogen bonding as well as in – interactions. The pre-sence of pentafluorophenyl groups as non-labile ligands bonded to platinum provides a substantial number of potential hydrogen-bond acceptors. We have used bidentate pyridine-like ligands to complete the coordination sphere about the metal, at the same time providing hydrogen-bond donors as well as aromatic rings. These bidentate ligands are well known to be able to bridge transition metal atoms, forming di- and polynuclear complexes; but these entities are also able to behave as simple chelating ligands, and in some cases behave this way preferentially. In the solid state, these sorts of complexes form hydrogen bonding arrays and – interactions that mediate aggregation patterns which may include channels or well defined layers and which may be able to accommodate solvent molecules.
Results and discussionSynthesis and characterization of [Pt(C6F5)2(napy)] (1)
The reaction of napy (1,8-naphthyridine) with cis-[Pt(C6F5)2(thf)2] generates a yellow complex of stoichiometry [Pt(C6F5)2(napy)]x (x = 1 or 2) (1) (see Chart 1). The C, H and N analysis, the infrared spectra and the 1H and 19F NMR spectra of complex 1 are in good agreement with the expected stoichiometry, being compatible with both the mono- and dinuclear species (see Chart 1). Since there is only one set of signals due to C6F5 or napy ligands in the NMR spec-tra it is clear that only one compound is present in solution. How-ever the FAB+ mass spectrum shows a peak corresponding to the cation [Pt(C6F5)2(napy)]+ suggesting a mononuclear formula for this complex. This was not the expected result, since the napy or napy-like ligands have frequently been used as dinucleating bridging ligands to force the proximity of the metal centers22–30 and only in a few cases do these ligands behave as chelating.31–35 The formation of a very strained four-membered ring in the chelate is in all likelihood the reason for the small number of complexes of this type.
The structure analysis of complex 1 by X-ray diffraction con-firms the chelating coordination of the napy ligand.
Fig. 1 depicts the structure of the neutral platinum complex. Crystal data and refinement parameters are given in Table 1, and selected bond distances and angles are given in Table 2. The napy ligand chelates the platinum center and two pentafluorophenyl groups complete the four-coordinate environment of the metal. The Pt–N and Pt–C distances are in the usual range found for other platinum complexes containing the same or analogous ligands.36–40
† Electronic supplementary information (ESI) available: Structure analysis of [Pt(C6F5)2(bipy)·0.5CH2Cl2. See http://www.rsc.org/suppdata/dt/b4/b407284g/
Dow
nloa
ded
by C
ape
Bre
ton
Uni
vers
ity o
n 21
/04/
2013
10:
44:3
7.
Publ
ishe
d on
23
July
200
4 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
4072
84G
View Article Online / Journal Homepage / Table of Contents for this issue
2 7 3 4 D a l t o n T r a n s . , 2 0 0 4 , 2 7 3 3 – 2 7 4 0 D a l t o n T r a n s . , 2 0 0 4 , 2 7 3 3 – 2 7 4 0 2 7 3 5
distorted square planar environment around the platinum atom. This stress is also reflected in the neutral ligand, which shows Pt–N–C(19) angles lower than those expected for sp2 hybridization [93.6(6)° and 94.8(6)°]. A useful indicator to quantify this type of distortion is the misdirected valence parameter41 which gives angles of 26.2° and 27.3° for N(1) and N(2) respectively. The pentafluoro-phenyl rings form dihedral angles of 62.6(6)° and 66.4(5)° with the platinum coordination plane, at the low end of the usual range for square planar pentafluorophenyl derivatives.5,6,42–44 The disposition of the pentafluorophenyl rings with respect to the platinum coordi-nation plane impedes the formation of the binuclear derivative for steric reasons (see Chart 1).
It is interesting to note that the mononuclear complex is orga-nized in the crystal lattice into a supramolecular array held together by intermolecular C–HF hydrogen bonding between fluorine atoms of the pentafluorophenyl groups and hydrogen atoms of the napy ligand. – interactions between the aromatic rings of the napy are also present, contributing to the formation of a stack of interacting mononuclear complexes. These stacks are held together by two short hydrogen bonds (F(6B)H(18A), F(12A)H(17B), 2.41 and 2.50 Å, respectively) and by the partial overlapping of the napy rings with a centroid–centroid distance of 3.49 Å for the best overlapped rings, which is at the low end of the range defined for this type of interaction (3.4 to 3.8 Å).11 Two molecules of the stack and the main interactions between them are represented in Fig.2. The PtPt distance is 5.771(1) Å, excluding any kind of interac-tion between the platinum atoms, which show a zig-zag disposition. The individual complexes are slightly rotated with respect to each other so as to avoid unfavorable steric interactions between their pentafluorophenyl groups.
In addition, these stacks are interconnected by hydrogen bonds involving the para- and some of the meta- fluorine atoms as proton acceptors, with short intermolecular C–HF distances [F(4C)H(15A) 2.377 Å, F(4C)H(16A) 2.543 Å, F(10A)H(16E) 2.458 Å, F(11A)H(13E) 2.481 Å]. The result-ing aggregation of four stacks of molecules generates a channel, also extending along the c-axis, as can be seen in Fig 3. Inside
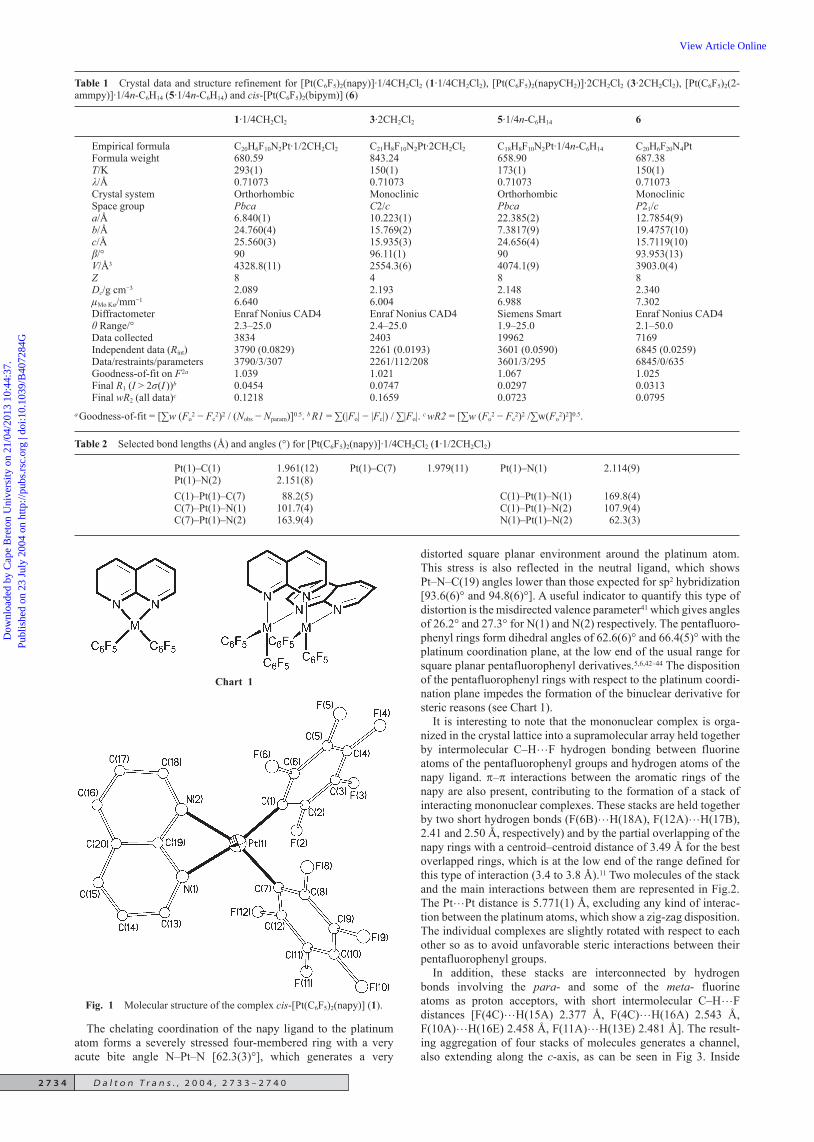
Table 1 Crystal data and structure refinement for [Pt(C6F5)2(napy)]·1/4CH2Cl2 (1·1/4CH2Cl2), [Pt(C6F5)2(napyCH2)]·2CH2Cl2 (3·2CH2Cl2), [Pt(C6F5)2(2-ammpy)]·1/4n-C6H14 (5·1/4n-C6H14) and cis-[Pt(C6F5)2(bipym)] (6)
1·1/4CH2Cl2 3·2CH2Cl2 5·1/4n-C6H14 6
Empirical formula C20H6F10N2Pt·1/2CH2Cl2 C21H8F10N2Pt·2CH2Cl2 C18H8F10N2Pt·1/4n-C6H14 C20H6F20N4PtFormula weight 680.59 843.24 658.90 687.38T/K 293(1) 150(1) 173(1) 150(1)/Å 0.71073 0.71073 0.71073 0.71073Crystal system Orthorhombic Monoclinic Orthorhombic MonoclinicSpace group Pbca C2/c Pbca P21/ca/Å 6.840(1) 10.223(1) 22.385(2) 12.7854(9)b/Å 24.760(4) 15.769(2) 7.3817(9) 19.4757(10)c/Å 25.560(3) 15.935(3) 24.656(4) 15.7119(10)/° 90 96.11(1) 90 93.953(13)V/Å3 4328.8(11) 2554.3(6) 4074.1(9) 3903.0(4)Z 8 4 8 8Dc/g cm−3 2.089 2.193 2.148 2.340Mo K/mm−1 6.640 6.004 6.988 7.302Diffractometer Enraf Nonius CAD4 Enraf Nonius CAD4 Siemens Smart Enraf Nonius CAD4 Range/° 2.3–25.0 2.4–25.0 1.9–25.0 2.1–50.0Data collected 3834 2403 19962 7169Independent data (Rint) 3790 (0.0829) 2261 (0.0193) 3601 (0.0590) 6845 (0.0259)Data/restraints/parameters 3790/3/307 2261/112/208 3601/3/295 6845/0/635Goodness-of-fit on F 2a 1.039 1.021 1.067 1.025Final R1 (I > 2(I ))b 0.0454 0.0747 0.0297 0.0313Final wR2 (all data)c 0.1218 0.1659 0.0723 0.0795
a Goodness-of-fit = [∑w (Fo2 − Fc
2)2 / (Nobs − Nparam)]0.5. b R1 = ∑(|Fo| − |Fc|) / ∑|Fo|. c wR2 = [∑w (Fo2 − Fc
2)2 /∑w(Fo2)2]0.5.
Chart 1
Fig. 1 Molecular structure of the complex cis-[Pt(C6F5)2(napy)] (1).
Table 2 Selected bond lengths (Å) and angles (°) for [Pt(C6F5)2(napy)]·1/4CH2Cl2 (1·1/2CH2Cl2)
Pt(1)–C(1) 1.961(12) Pt(1)–C(7) 1.979(11) Pt(1)–N(1) 2.114(9)Pt(1)–N(2) 2.151(8)C(1)–Pt(1)–C(7) 88.2(5) C(1)–Pt(1)–N(1) 169.8(4)C(7)–Pt(1)–N(1) 101.7(4) C(1)–Pt(1)–N(2) 107.9(4)C(7)–Pt(1)–N(2) 163.9(4) N(1)–Pt(1)–N(2) 62.3(3)
The chelating coordination of the napy ligand to the platinum atom forms a severely stressed four-membered ring with a very acute bite angle N–Pt–N [62.3(3)°], which generates a very
Dow
nloa
ded
by C
ape
Bre
ton
Uni
vers
ity o
n 21
/04/
2013
10:
44:3
7.
Publ
ishe
d on
23
July
200
4 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
4072
84G
View Article Online

2 7 3 4 D a l t o n T r a n s . , 2 0 0 4 , 2 7 3 3 – 2 7 4 0 D a l t o n T r a n s . , 2 0 0 4 , 2 7 3 3 – 2 7 4 0 2 7 3 5
these channels there are disordered CH2Cl2 molecules in the ratio complex/solvent of 4 : 1.
Reactivity of [Pt(C6F5)2(napy)] towards CO and CH2N2
The highly strained four-membered ring in complex 1, produced by the forced chelating coordination of the napy ligand to the metal center, suggests the possibility of enhanced reactivity of the Pt–N bonds, specifically displacement and insertion reactions which would release one of them.
Stirring a solution of [Pt(C6F5)2(napy)] (1) under a CO atmos-phere for three hours produces decoordination of one nitrogen atom of the napy ligand, which results in a monodentate ligand, and the coordination of a -donor -acceptor ligand, namely carbon mon-oxide (see eqn. (1)).
[Pt(C6F5)2(napy)] + CO → cis-[Pt(C6F5)2(napy)(CO)] (1)
Complex (2), cis-[Pt(C6F5)2(napy)CO], was obtained as a stable white solid. The presence of CO was confirmed by a strong, sharp signal in the IR spectrum at 2108 cm−1 corresponding to (CO), indicating a perceptible degree of back-bonding. The 19F NMR spectrum, measured at room temperature, shows two signals for the para-fluorine atoms, consistent with the presence of two non-equi-valent pentafluorophenyl groups. The 1H NMR at room temperature shows six signals for the hydrogen atoms of the napy ligand; but these signals are not well resolved, probably because of a dynamic exchange between the two ligand contact points at this temperature. The 1H NMR spectrum obtained at −50 °C displays six well defined signals, thus indicating that at this temperature no exchange occurs on the NMR time scale.
The displacement of the N,N′ chelating ligand by CO is note-worthy, since such processes are not common and must be related in this case to the highly stressed nature of the four-membered ring in the starting complex. In fact, and as should be expected, cis-[Pt(C6F5)2(bipy)],45 a rather similar complex but with a five-membered chelate ring, does not react at all with CO under similar conditions or even with longer reaction times.
The reactivity with CO suggests other possibilities. Diazomethane, for example, has been widely used in insertion reactions at M–E bonds (with E = halide, hydride, alkyl or carboxylate) giving M–CH2 bond formation.46 However, insertion of methylene fragments into M–N bonds is not as well represented in the literature.47,48
The addition of an excess of H2CN2 to a solution of cis-[Pt(C6F5)2(napy)] (1) in CH2Cl2 under nitrogen produces a rapid change in the color of the solution from yellow to red. By work-ing up the solution a stable red solid (3) is obtained (see eqn. (2)). The IR spectrum indicates the presence of the napy fragment and also of two pentafluorophenyl groups in a cis- disposition. The 1H NMR spectrum provides the most important information about the insertion of the methylene group into the Pt–N bond, showing six signals for the six hydrogen atoms of the napy fragment and one additional singlet with platinum satellites [2J(195Pt, H) = 44.5 Hz ] at 5.4 ppm due to the –CH2– moiety. The 19F NMR spectrum at room temperature shows two signals for the para-fluorine atoms, indicat-ing the existence of two different pentafluorophenyl groups, as can be expected. The 13C NMR spectrum indicates that the –CH2– frag-ment [56.1 ppm; 1J(195Pt, C) = 595 Hz] is not bonded to the platinum center as a carbene ligand, but rather that an insertion reaction into one of the Pt–N bonds has taken place and that the ylide napyCH2 ligand has been formed.
[Pt(C6F5)2(napy)] + H2CN2 → cis-[Pt(C6F5)2(CH2napy)] + N2 (2)
This complex should display a molecular structure similar to that of 1 but with a less stressed, five-membered metallacycle. Since in complex 3 the –CH2– fragment of the zwitter-ionic ligand (napyCH2) is a poor proton donor we decided to study its X-ray structure not only to confirm the molecular connectivity but also to know how the molecules were assembled into an extended structure.
Fig. 4 depicts the molecular structure of complex 3. Crystal and structure refinement data are given in Table 1 and selected bond distances and angles are given in Table 3.
Fig. 2 View of the hydrogen-bonding interactions between a pair of mole-cules of complex (1).
Fig. 3 View of the channels created along the c-axis by the hydrogen-bonding interactions among four molecular stacks in the structure of complex (1), showing the partial overlap of aromatic rings that permits – interaction. Disordered CH2Cl2 solvent is in the middle of the channel.
Fig. 4 Molecular structure of cis-[Pt(C6F5)2(CH2napy)] (3).
The platinum center is located, as expected, in a distorted square planar environment formed by two pentafluorophenyl groups and the napyCH2 ligand. The angles between cis-ligands range from 79.4(9)° (for the chelate) to 108.9(5)°. The napyCH2 ligand is essentially planar and lies in the platinum coordination plane. The facile insertion of the methylene group into one of the Pt–N bonds of 1 forms a non-strained five membered ring [N(2)–Pt–C(7) is 79.4(9)°] and because of that the Pt–N(2) distance [1.939(11) Å]
Dow
nloa
ded
by C
ape
Bre
ton
Uni
vers
ity o
n 21
/04/
2013
10:
44:3
7.
Publ
ishe
d on
23
July
200
4 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
4072
84G
View Article Online

2 7 3 6 D a l t o n T r a n s . , 2 0 0 4 , 2 7 3 3 – 2 7 4 0 D a l t o n T r a n s . , 2 0 0 4 , 2 7 3 3 – 2 7 4 0 2 7 3 7
is perceptibly shorter than in 1 in which the stress of the four-membered ring forces the misdirection, and consequent weak-ness, of the Pt–N bonds. In 3 the misdirected valence parameter is reduced to 4.5°. However, the Pt–C(7) (methylene) distance is 2.27(3) Å, perceptibly longer than typical Pt–C(sp3) distances (aver-age value 2.081 Å).36
An interesting feature of the formation of complex 3 is that the insertion of the methylene fragment forms a zwitterionic ligand, with formal positive and negative charges at N(1) and C(7), respectively.
In the crystal complex 3 traps two molecules of CH2Cl2 per molecule of the complex, and in this case the interstitial entities are not disordered. Again, as in complex 1, hydrogen bonding is responsible for the supramolecular assembly observed in the solid state. In this case complex 3 and the solvent molecules create in-finite sheets (Fig. 5), interconnected by several types of hydrogen bonds [F(2)H(12) 2.352 Å and F(3)H(16B) 2.351 Å]. Adjacent sheets are also connected to each other through hydrogen bonds [F(3)H(7B) 2.452 Å and F(1)H(16A) 2.577 Å], but they are not perfectly parallel and do not present – interactions between the aromatic rings of adjacent napyCH2 ligands.
cis-[Pt(C6F5)2(thf)2] + C8H8N2 → cis-[Pt(C6F5)2(C8H8N2)] (3)
Unfortunately all attempts to obtain suitable crystals for X-ray were unsuccessful, not even by changing the solvents and the conditions used. For this reason we thought to prepare a complex containing the “Pt(C6F5)2” fragment and a neutral ligand similar to the 8-aminoquinoline (C8H8N2) that might crystallize better. We chose for this purpose the ligand 2-aminomethylpyridine (C6H8N2) with an identical atomic sequence Pt–N–C–C–N but with one less aromatic ring.
The reaction between [NBu4]2[Pt2(-Cl)2(C6F5)4] and the neutral ligand 2-aminomethylpyridine (2-ammpy) was carried out in CHCl3 and yielded a precipitate that was identified as the neutral complex [Pt(C6F5)2(2-ammpy)]x (x = 1 or 2) (5) by elemental analysis (see eqn. (4)). As was the case for compounds 2 and 3, the spectroscopic data confirm the presence of two different pentafluorophenyl groups and the bidentate coordination of the neutral ligand; however, they do not provide information on its coordination mode—chelating or bridging. Structure determination of complex 5 by X-ray diffraction confirmed the chelating coordination of the 2-aminomethylpyridine ligand.
[NBu4]2[Pt2(-Cl)2(C6F5)4] + 2 C6H8N2 → 2 cis-[Pt(C6F5)2(C6H8N2)] (4)
Fig. 6 shows the structure of complex 5. Crystal and refinement data are given in Table 1, and selected bond distances and angles are given in Table 4. The 2-ammpy ligand chelates the platinum center through the nitrogen atoms, and two pentafluorophenyl groups complete the four-coordinate environment in a distorted square-planar disposition. The Pt–N and Pt–C distances are similar to those found in complexes described before. The angles around the platinum center show values in the range of 79.7(2)° to 96.4(2)°, with the N–Pt–N chelate angle formed by the neutral ligand show-ing the smallest value, which as expected is very similar to the corresponding angle in complex 3.
Crystals of complex 5 were obtained by slow diffusion of n-hexane into a dichloromethane solution at low temperature. In spite of the fact that 5 was crystallized under identical conditions to those
Table 3 Selected bond lengths (Å) and angles (°) for cis-[Pt(C6F5)2(napyCH2)]·2CH2Cl2 (3·2CH2Cl2)
Pt–N(2) 1.939(11) Pt–C(1) 2.022(12) Pt–C(7) 2.27(3)N(2)–Pt–C(1) 163.3(5) N(2)–Pt–C(1′) 108.9(5)C(1)–Pt–C(1′) 87.7(7) N(2)–Pt–C(7) 79.4(9)C(1)–Pt–C(7) 83.9(8) C(1′)–Pt–C(7) 170.7(8)
Symmetry transformation used to generate C(1′) equivalent atom is −x, y, 1/2 − z.
Fig. 5 View of the structure of cis-[Pt(C6F5)2(CH2napy)] (3), showing hy-drogen bonds between the complex and CH2Cl2 molecules, forming sheets.
Complex 3 presents only FH and ClH interactions in inter-molecular space, and not – stacking, since the insertion reaction produces a topological change between complexes 1 and 3. Even though both complexes were crystallized under analogous condi-tions (CH2Cl2/hexane at low temperature), their crystallization leads to different supramolecular arrangements. It can be seen that hydrogen bonds involving the F atoms are the exclusive weak forces mediating the solid state organization.
The differences between complexes 1 and 3 are the number of members in the chelate ring (4 or 5), the presence (in 3) or absence (in 1) of a positive formal charge on the chelating ligand and the presence (1) or absence (3) of – interactions between neighbor-ing aromatic rings. All of these factors should have concomitant effects, but we have tried to ascertain a principal reason for the lack of similarity in the supramolecular structures of 1 and 3 in the solid state. With this aim, we have used a similar ligand (8-amino-quinoline) which forms a five-membered chelate ring, employing the same atoms but in a different sequence (Pt–N–C–C–N instead of the Pt–N–C–N–C sequence of complex 3) and without any formal ligand charge.
Synthesis and characterization of cis-[Pt(C6F5)2(C8H8N2)] (4) and [Pt(C6F5)2(2-ammpy)] (5)
The reaction of 8-aminoquinoline with cis-[Pt(C6F5)2(thf)2] was car-ried out in CH2Cl2 and yielded a white solid precipitated that was identified as cis-[Pt(C6F5)2(C8H8N2)] by elemental analysis, IR and 1H and 19F NMR spectroscopic data (see eqn. (3)). Fig. 6 X-ray structure of one molecule of cis-[Pt(C6F5)2(2-ammpy)] (5).
Dow
nloa
ded
by C
ape
Bre
ton
Uni
vers
ity o
n 21
/04/
2013
10:
44:3
7.
Publ
ishe
d on
23
July
200
4 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
4072
84G
View Article Online

2 7 3 6 D a l t o n T r a n s . , 2 0 0 4 , 2 7 3 3 – 2 7 4 0 D a l t o n T r a n s . , 2 0 0 4 , 2 7 3 3 – 2 7 4 0 2 7 3 7
used for the crystallization of 1 and 3, complex 5 crystallizes with n-hexane as solvent, instead of CH2Cl2 as was the case with 1 and 3. Apropos of the discussion that follows, we note that the posi-tions of the amminic hydrogen atoms were located in difference density maps—i.e., determined experimentally—and not calculated geometrically.
Molecules of complex 5 are stacked in an arrangement similar to that described for Complex 1 (see Fig. 7). As we have seen, the mononuclear complex is slightly displaced with respect to its neighbors, thus avoiding steric crowding of the pentafluorophenyl groups of adjacent molecules. The stability of the molecular stack is exclusively due to hydrogen bonding interactions because, in spite of the fact that the neutral ligands (ammpy) lie in almost parallel planes at short distances to each other (around 3.7 Å), the aromatic rings are displaced in such a way that – interactions cannot be present. The main interactions along a stack involve the two hydrogen atoms of the amminic nitrogen –(N(2)) which act as proton donors to the platinum atom of the molecule of complex 5 lying above and to F(1) of the molecule lying below. These inter-molecular hydrogen bonds show very short distances [PtH(2B) 2.572 Å and F(1)H(2A) 2.108 Å], and good angles for this type of interaction [N(2)–H(2B)Pt 167.4° and N(2)–H(2B)F(1) 168.1°]. In addition, atom F(5) is involved in hydrogen bonding interactions along the stack, but with rather longer distances—F(5)H(2A), 2.534(4) Å and F(5)H(2B), 2.451(4) Å.
actions. The absence of this type of interaction in complex 5 may be a consequence of the stronger hydrogen bonding involving the amminic protons and, also, of the presence of only one aromatic ring contained in the coordination plane. The selection of a neutral ligand having two or more aromatic rings in the coordination plane but with the same atomic sequence (Pt–N–C–C–N) and without any amminic hydrogen atom could, we thought, help characterize the degree to which the – interaction influences the extended crystal structure. We used 2,2′-bipyrimidine (bipym) for this test, a ligand composed of two aromatic rings and containing four nitrogen atoms at the ortho positions. The topology of 2,2′-bipyrimidine causes it to act as a chelating ligand, creating a planar zone in the complex which in principal could establish – interactions and avoid the steric hind-rance of the hydrogen atoms that would be present if carbon atoms were located at the non-coordinated ortho positions of the rings.
Synthesis and characterization of [Pt(C6F5)2(bipym)] (6)
The addition of an equimolar amount of 2,2′-bipyrimidine (C8H6N4) to a dichloromethane solution of cis-[Pt(C6F5)2(thf)2] renders, after evaporation to dryness and addition of diethyl ether, a yellow solid which was identified (IR, 1H and 19F NMR spectroscopy, and C, H and N elemental analysis) as cis-[Pt(C6F5)2(bipym)] (6) (see eqn. (5)). Crystals of complex 6 suitable for X-ray analysis were obtained by slow diffusion of n-hexane into a dichloromethane solution of the complex.
cis-[Pt(C6F5)2(thf)2] + C8H6N4 → cis-[Pt(C6F5)2(C8H6N4)] (5)
The structure of complex 6 is shown in Fig. 9. Two slightly different molecules appear in the asymmetric unit. Crystal data and refinement parameters are given in Table 1, and selected bond distances and angles are given in Table 5. The platinum atoms of both independent neutral molecules are in similar distorted square-
Table 4 Selected bond lengths (Å) and angles (°) for cis-[Pt(C6F5)2(2-ammpy)]·1/4n-C6H14 (5·1/4n-C6H14)
Pt–C(1) 2.013(6) Pt–C(7) 2.014(6) Pt–N(1) 2.080(5)Pt–N(2) 2.095(5)C(1)–Pt–C(7) 87.7(2) C(1)–Pt–N(1) 175.5(2)C(7)–Pt–N(1) 96.4(2) C(1)–Pt–N(2) 96.0(2)C(7)–Pt–N(2) 175.8(2) N(1)–Pt–N(2) 79.73(18)
Fig. 7 View of the N–HPt hydrogen-bond interactions along a molecu-lar stack in the structure of (5).
There is no evidence of the presence of the PtH interaction or FH interaction in solution. The 1H NMR spectrum of complex 5 shows only one broad signal for both amminic hydrogen atoms with both side shoulders that are probably due to the coupling of both hydrogen atoms with the active isotope of platinum. This coupling has to be transmitted through the ring bond system and not caused by the hydrogen bond interaction.
In addition to the interactions described so far, we also note the presence of other FH interactions, such as F(3)H(14A), 2.57 Å, F(7)H(17A), 2.48 Å and F(8)H(14A), 2.49 Å—probably weaker but not negligible because they contribute to the assembly of several rows into a supramolecular arrangement that renders infinite channels, quite similar to those described for complex 1. In this case the channels accommodate n-hexane molecules (Fig. 8).
The crystal structures of complexes 1 and 5 exhibit a special packing arrangement in which stacks of molecules generate channels in which solvent molecules such as CH2Cl2 or n-hexane can be located. In both complexes, supramolecular stacks are generated by association of molecules through hydrogen bonding and, in complex 1, with the additional contribution of – inter-
Fig. 8 View of the channels formed along the c-axis by the hydrogen bonding among four stacks of molecules of complex (5) without any – interactions. n-Hexane solvent is in the middle of the channel.
Dow
nloa
ded
by C
ape
Bre
ton
Uni
vers
ity o
n 21
/04/
2013
10:
44:3
7.
Publ
ishe
d on
23
July
200
4 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
4072
84G
View Article Online

2 7 3 8 D a l t o n T r a n s . , 2 0 0 4 , 2 7 3 3 – 2 7 4 0 D a l t o n T r a n s . , 2 0 0 4 , 2 7 3 3 – 2 7 4 0 2 7 3 9
planar environments, each formed by two pentafluorophenyl groups and one chelating 2,2′-bipyrimidine. The Pt–N distances are equal to within experimental error [range between 2.064(5) and 2.075(5) Å], and the same holds true for the Pt–C distances [2.005(6)–2.009(7) Å]. The values of the cis angles around the platinum atoms are similar for both [78.9(2)° to 97.6(2)°], with the greater deviations from perpendicularity corresponding to the N–Pt–N chelate angles. The coordination planes of the two molecules are nearly perpendicular to each other, forming a dihedral angle of 86.1(1)°. The extended coordination plane of the molecule contain-ing Pt(1) essentially bisects the coordination plane of Pt(2), lying between the pentafluorophenyl groups and bisecting the C–C single bond of the bipyrimidine ligand of the latter. In this arrangement, one of the hydrogen atoms, H(19), of the neutral ligand points towards the platinum center Pt(2) with a PtH(19) distance of 2.74(7) Å and a PtH(19)–C(19) angle of 159(5)°, which clearly indicates the presence of a platinum–hydrogen H-bonding interaction.
(a) No – interaction between the aromatic rings of the bipym ligands are observed.
(b) While the pyrimidinic rings of the bipym bonded to the proton acceptor molecule are nearly coplanar (dihedral angle 2.4(2)°), the analogous dihedral angle in the donor molecule is 6.8(2)°. In addition, three of the C6F5 rings form dihedral angles in the range of 73.0(2) to 77.3(2)° with their respective platinum coordination planes while the fourth ring (part of the acceptor molecule) forms a corresponding dihedral angle of 61.6(2)°, this way favoring the F(20)H(19) interaction.
It is also interesting that small differences can influence the entire structure of the compound in a molecular solid. The molecular structure of cis-[Pt(C6F5)2(bipy)], prepared thirty years ago,45 is very similar to the structure of 6; but no evidence of PtH inter-actions is observed.50
ConclusionsThe supramolecular structures observed in the solid state for compounds 1, 3, 5, and 6 stand out as unusual when taken together with the crystal structures of the other pentafluorophenyl platinum complexes that we have synthesized and structurally characterized to date. There are several differences that should be noted; the fore-most is the electrical neutrality of the present complexes, which eliminates the need for the usually bulky cations (NBu4
+or NEt4+,
or similar entities) required to crystallize anionic pentafluorophenyl complexes. The cations normally preclude the approach of the Pt-containing complexes to each other, thus eliminating the possibility of direct contacts between the complexes.
Thus, in these neutral complexes the absence of bulky cations in the crystal framework enables the molecules to approach each other and to establish intermolecular hydrogen bonds in a regular and periodic way, to form channels and holes.
As concerns the molecular building blocks of the observed ex-tended structures, the fact that half of the molecular complex consists of a neutral quasi-planar ligand which includes the hydrogen atoms and the aromatic systems, while the remainder is constituted by two anionic ligands containing proton acceptors, enhances the chances of establishing a significant superstructure strongly supported by hydrogen bonding. The presence of proton-donating –NH2 groups in the neutral ligand increases both the probability of forming hydrogen bridged interactions and the strength of the interactions formed. The capability of the platinum(II) center to act as proton acceptor (PtH) has been established, and this behavior of the metal center is likely the main factor in determining the supramolecular arrangement of the molecules in the solid state for complexes 5 and 6.
The structure of complex 6 demonstrates the relatively weak character of aromatic – interactions as compared to hydrogen bonding. These aromatic interactions may serve to reinforce the stability of structures based principally on other types of forces or interactions but they are not strong enough to neither determine the supramolecular arrangement in the solid state nor compete with hydrogen bonding capacity. Also complex 6 may serve to demon-strate that in addition to the major forces that establish the general features of the molecular shape, weaker forces may enter into play to effect finer tuning and thus to influence the resulting physical properties in the solid state.
Table 5 Selected bond lengths (Å) and angles (°) for cis-[Pt(C6F5)2(bipym)] (6)
Pt(1)–C(7) 2.007(6) Pt(1)–C(1) 2.008(6) Pt(1)–N(2) 2.071(5)Pt(1)–N(1) 2.074(5) Pt(2)–C(27) 2.005(6) Pt(2)–C(21) 2.009(7)Pt(2)–N(5) 2.064(5) Pt(2)–N(6) 2.075(5) C(19)–H(19) 0.87(7)Pt(2)H(19) 2.74(7)C(7)–Pt(1)–C(1) 87.8(3) C(7)–Pt(1)–N(2) 96.6(2)C(1)–Pt(1)–N(2) 175.5(2) C(7)–Pt(1)–N(1) 175.3(2)C(1)–Pt(1)–N(1) 96.5(2) N(2)–Pt(1)–N(1) 79.1(2)C(27)–Pt(2)–C(21) 89.6(3) C(27)–Pt(2)–N(5) 176.5(2)C(21)–Pt(2)–N(5) 93.8(2) C(27)–Pt(2)–N(6) 97.6(2)C(21)–Pt(2)–N(6) 172.7(2) N(5)–Pt(2)–N(6) 78.9(2)C(19)–H(19)–Pt(2) 159(5) H(19)–C(19)–N(4) 113(4)H(19)–C(19)–C(18) 124(4) N(4)–C(19)–C(18) 122.6(6)
Fig. 9 X-ray structure of one pair of molecules of cis-[Pt(C6F5)2(2-bipym)] (6), showing the C–HPt interaction.
This is a key point for understanding this structure. Complex 6 displays in the crystal two molecules of identical composition but with quite different behavior: One acts exclusively as a proton donor to the other, and the platinum center of the latter behaves as proton acceptor. This behavior is quite unusual and different from the case of complex 5, in which each one of the molecules form-ing rows acts simultaneously as proton donor and proton acceptor, depending on the group or the atom considered.49
Of all the hydrogen atoms of the bpym ligand, those located ortho to the non-coordinated N are, in principle, the most favored to interact with a basic center from another molecule, for both steric and electronic reasons. From the steric point of view, because these H atoms point outward, away from the bulky C6F5 groups, they are ipso facto, better situated to interact with a basic center. Further, these are the most acidic hydrogen atoms of the ligand, as indicated by the fact that they are shifted furthest downfield in the 1H NMR spectrum. Thus, one of these hydrogen atoms, H(19), interacts with Pt(2), while the chemically equivalent H(15) forms contacts with the uncoordinated N atoms of a third molecule [N(7)H(15) 2.59 Å, N(8)H(15) 2.63 Å]. Taken together, these interactions mediate the formation of four-molecule groupings.
Some small differences in the structures of the two molecules may be a consequence of the hydrogen bonding:
Dow
nloa
ded
by C
ape
Bre
ton
Uni
vers
ity o
n 21
/04/
2013
10:
44:3
7.
Publ
ishe
d on
23
July
200
4 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
4072
84G
View Article Online

2 7 3 8 D a l t o n T r a n s . , 2 0 0 4 , 2 7 3 3 – 2 7 4 0 D a l t o n T r a n s . , 2 0 0 4 , 2 7 3 3 – 2 7 4 0 2 7 3 9
Experimental sectionGeneral methods
C, H, and N analyses were carried out with a Perkin-Elmer 240B microanalyzer. IR spectra were recorded over the range of 4000–200 cm−1 on a Perkin-Elmer 883 spectrophotometer using Nujol mulls between polyethylene sheets. 1H and 19F NMR spectra at room temperature were recorded on a Bruker ARX-300 or a Unity-300 spectrometer in CDCl3 or acetone-d6 solutions. 1H, 19F NMR spectra at variable temperature were recorded in acetone-d6 or DMSO-d6 solutions on a Bruker ARX-300 spectrometer. Positive and negative ion FAB mass spectra were recorded on a VG-Auto-spec spectrometer operating at ca. 30 kV, using the standard cesium ion FAB gun and 3-nitrobenzyl alcohol as matrix.
[NBu4]2[Pt2(-Cl)2(C6F5)4],51 cis-[Pt(C6F5)2(thf)2]52 and 1,8-naphthyridine53 (napy) were prepared as described elsewhere. The aminomethylpyridine (2-ammpy) and 8-aminoquinoline ligands were used as purchased from Aldrich and also 2,2′-bipyrimidine (bipym) ligand was used as purchased from Lancaster.
cis-[Pt(C6F5)2(napy)] (1). To a solution of cis-[Pt(C6F5)2(thf)2] (0.103 g, 0.154 mmol) in CH2Cl2 (20 mL), an equimolar amount of the napy ligand was added (0.020 g, 0.154 mmol). The solution was stirred for 10 min and evaporated to dryness. The addition of 5 mL of CHCl3 rendered the product as a yellow solid which was filtered off and dried in air (85% yield). Found: C, 36.52; H, 1.12; N, 4.02%. C20H6F10N2Pt requires C, 36.43; H, 0.92; N, 4.42%. IR (cm−1): C6F5 X-sensitive mode,54 815 s, 806 s; others, 1635 w, 1607 m, 1499 vs, 1058 vs, 957 s; napy: 834 s. 1H NMR room temperature (CDCl3): 9.0 [d, 2H, o-H, 3J(o-H, m-H) = 4.9 Hz], 8.7 [d, 2H, p-H, 3J(p-H, m-H) = 8.4 Hz], 7.7 (dd, 2H, m-H). 19F NMR (CDCl3): −121.1 [d, 4F, o-F, 3J(195Pt, F) = 471.2 Hz], −163.9 (m, 4F, m-F), −160.9 (t, 2F, p-F). 195Pt NMR (CDCl3): −3018 [q, 3J(195Pt, o-F) = 471.2 Hz]. FAB+ MS: m/z 659 [Pt(C6F5)2(napy)]+.
cis-[Pt(C6F5)2(CO)(napy)] (2). A suspension of cis-[Pt(C6F5)2(napy)] (1) (0.250 g, 0.380 mmol) in freshly distilled CH2Cl2 (30 mL) was stirred for 3 h under a CO atmosphere. After that time a colorless solution was obtained and was then evaporated to dryness. To the residue 30 mL of n-hexane was added, render-ing a white solid which was filtered off, washed with n-hexane and finally dried in air (78% yield). Found: C, 36.87; H, 0.74; N, 3.87%. C21H6F10N2Pt requires C, 36.70; H, 0.88; N, 4.07%. IR (cm−1): C6F5 X-sensitive mode,54 809 s, 801 s; others, 1502 vs, 1056 s, 966 s; (CO) 2108 s, 2063 w; napy: 832 s, 635 w. 1H NMR at 218 K (CDCl3): 9.4 [br, 1H, o-H,], 9.0 [d, 1H, o-H, 3J(o-H, m-H) = 4.1 Hz], 7.8 [dd, 1H, m-H, 3J(m-H, p-H) = 7.7 Hz], 7.7 [dd, 1H, m-H, 3J(m-H, p-H) = 8.0 Hz], 8.6 [d, 1H, p-H], 8.4 [d, 1H, p-H]. 19F NMR at 218 K (CDCl3): −118.5 [d, 2F, o-F, 3J(195Pt, Fo) = 418.6 Hz], −120.1 [d, 2F, o-F, 3J(195Pt, Fo) = 373.7 Hz], −158.5 (t, 1F, p-F), −159.9 (t, 1F, p-F), −162.7 (m, 2F, m-F), −163.5 (m, 2F, m-F).
cis-[Pt(C6F5)2(napyCH2)] (3). To a solution of cis-[Pt(C6F5)2(napy)] (1) (0.200 g, 0.303 mmol) in freshly distilled CH2Cl2 (30 mL) under a nitrogen atmosphere, H2CN2 was added in 30% excess over the stoichiometric 1 : 1 ratio. The yellow solution turned red and was then quickly evaporated to dryness. To the resi-due 30 mL of n-hexane was added, rendering a red solid which was filtered off, washed with 5 ml of CHCl3 and finally dried in air (71% yield). Found: C, 33.81; H, 1.09; N, 3.61%. C21H8F10N2Pt·CHCl3 requires C, 33.33; H, 1.14; N, 3.53%. IR (cm−1): C6F5 X-sensitive mode,54 803 s, 781 s; others, 1500 vs, 1054 s, 952 s; napy: 842 m, 781 w. 1H NMR room temperature (CD2Cl2): 9.4 [dd, 1H, o-H, 3J(o-H, m-H) = 5.7 Hz], 9.2 [d, 1H, o-H, 3J(o-H, m-H) = 5.1 Hz, 3J(195Pt, H) = 29.7 Hz], 8.0 [dd, 1H, m-H, 3J(m-H, p-H) = 8.2 Hz], 7.8 [dd, 1H, m-H, 3J(m-H, p-H) = 8.4 Hz], 9.1 [dd, 1H, p-H, 3J(p-H, o-H) = 1.3 Hz], 9.0 [dd, 1H, p-H, 3J(p-H, o-H) = 1.5 Hz] 5.4 [s, 2H, –CH2, 2J(195Pt, H) = 44.5 Hz]. 19F NMR (CD2Cl2): −116.2 [d, 2F, o-F, 3J(195Pt, Fo) = 321.2 Hz], −117.0 [d, 2F, o-F, 3J(195Pt, Fo) = 542.5 Hz], −164.5 (t, 1F, p-F), −166.5 (t, 1F, p-F), −165.2 (m, 2F, m-F), −167.3 (m, 2F, m-F).
cis-[Pt(C6F5)2(8-aminoquinoline)] (4). To a solution of cis-[Pt(C6F5)2(thf)2] (0.083 g, 0.123 mmol) in CH2Cl2 (15 mL), an equi-molar amount of 8-aminoquinoline (C9H8N2) was added (0.018 g, 0.123 mmol). Instantaneously a white precipitate was obtained and it was filtered off, washed with 5 mL of n-hexane and air dried (67% yield). Anal. Found: C: 37.40%, H: 0.93%, N: 4.35%. C21H8F10N2Pt requires C, 37.45; H, 1.19; N, 4.16%. IR (cm−1): C6F5 X-sensitive mode,54 812 vs, 801 vs; others, 1608 m, 1564 m,, 1504 vs, 1068 vs, 956 vs; C9H8N2: (N–H): 3297 s 1738 w, 1686 w, 1640 m, 1627 m, 1595 s, 1505 vs, 1320 s, 1220 m, 1037 s, 828 s, 786 s, 773 s, 763 s, 743 m. 1H NMR room temperature (acetone-d6): 8.9 (dd, 1H), 8.7 [d, 1H, 2J(195Pt, H) = 33.3 Hz], 8.1 (d + d overlapped, 2H), 7.8 (t, 1H), 7.7 (dd, 1H), 7.1 (s, 2H). 1H NMR at 188 K (acetone-d6): 8.9 (d, 1H), 8.7 (d, 1H), 8.1 (d, 1H), 8.0 (d, 1H), 7.8 (t, 1H), 7.7 (dd, 1H), 7.5 (s, 2H). 1H NMR at 323 K (acetone-d6): 8.8 (dd, 1H), 8.6 [d, 1H, 2J(195Pt, H) = 33.3 Hz], 8.1 (d, 2H), 7.8 (t, 1H), 7.7 (dd, 1H), 7.0 (s, 2H). 19F NMR room temperature (acetone-d6): −117.9 [d, 2F, o-F, 3J(195Pt, Fo) = 494.21 Hz], −118.5 [d, 2F, o-F, 3J(195Pt, Fo) = 474.4 Hz], −164.4 (t, 1F, p-F), −164.5 (t, 1F, p-F), −165.8 (m, 2F, m-F), −166.5 (m, 2F, m-F). 19F NMR at 188 K (acetone-d6): −119.2 [d, 2F, o-F, 3J(195Pt, Fo) = 438.3 Hz], −120.0 [d, 2F, o-F, 3J(195Pt, Fo) = 486.9 Hz], −164.7 (t, 1F, p-F), −164.9 (t, 1F, p-F), −166.2 (m, 2F, m-F), −166.7 (m, 2F, m-F). 19F NMR at 323 K (acetone-d6): −114.1 [dd, 2F, o-F, 3J(195Pt, Fo) = 467.1 Hz], −114.6 [dd, 2F, o-F, 3J(195Pt, Fo) = 479.8 Hz], −160.7 (t, 1F, p-F), −160.9 (t, 1F, p-F), −162.1 (m, 2F, m-F), −162.8 (m, 2F, m-F). FAB+ (m/z): [Pt(C6F5)2(C8H8N2)]: 673.
cis-[Pt(C6F5)2(2-ammpy)] (5). To a solution of [NBu4]2[Pt2(-Cl)2(C6F5)4] (0.489 g, 0.303 mmol) in CHCl3 (15 mL), 62.47 L (0.606 mmoles) of a 2-ammpy (C6H8N2) solution was added. The solution was stirred for 15 min and after that the resulting white solid was filtered off, washed with 15 mL of CHCl3 and n-hexane and finally dried in air (45% yield). Found: C, 33.92; H, 1.55; N, 4.14%. C18H8F10N2Pt requires C, 33.92; H, 1.26; N, 4.39%. IR (cm−1): C6F5 X-sensitive mode,54 807 s, 800 s; others, 1636 w, 1594 m, 1501 vs, 1064 vs, 956 vs; C6H8N2: (N–H) 3337 s, 3183 w, others, 1550 s, 1607 s, 1550 m, 1166 m, 973 m, 765 s. 1H NMR room temperature (acetone-d6): 8.2 (d, 1H, o-H, 3J(195Pt, H) = 15.6 Hz), 8.2 (t, 1H, m-H), 7.8 (d, 1H, m-H), 7.4 (m, 1H, p-H), 5.14 (broad s, 2H, –NH2), 4.7 (t, 2H, –CH2). 19F NMR (acetone-d6): −118.9 [d, 2F, o-F, 3J(195Pt, Fo) = 467.1 Hz], −119.0 [d, 2F, o-F, 3J(195Pt, Fo) = 461.5 Hz], −165.2 (t, 1F, p-F), −165.4 (t, 1F, p-F), −166.4 (m, 2F, m-F), −167.1 (m, 2F, m-F). FAB+ (m/z): [Pt(C6F5)2(C6H8N2)]: 637.
cis-[Pt(C6F5)2(bipym)] (6). To a solution of cis-[Pt(C6F5)2(thf)2] (0.115 g, 0.171 mmol) in CHCl3 (15 mL), an equimolar amount of bipym (C8H6N4) was added (0.028 g, 0.171 mmol). The solution was stirred for 15 min and evaporated to dryness. The addition of 10 mL of OEt2 rendered the product as a yellow solid which was filtered off, washed with 5 mL of n-hexane and air dried (69% yield). Found: C, 35.57; H, 1.12; N, 8.47%. C20H6F10N4Pt requires C, 34.95; H, 0.88; N, 8.15%. IR (cm−1): C6F5 X-sensitive mode,54 810 s, 801 s; others, 1585 m, 1558 m, 1504 vs, 1062 vs, 960 vs; C8H6N4: 1410 s, 752 m. 1H NMR room temperature (CD2Cl2): 9.3 (dd, 2H), 8.6 [dd, 2H, 3J(195Pt–H) = 28.9 Hz] 7.7 (m, 2H). 19F NMR (CD2Cl2): −118.5 [d, 4F, o-F, 3J(195Pt, Fo) = 454.1 Hz], −160.2 (t, 2F, p-F), −162.9 (m, 2F, m-F). FAB+ (m/z): [Pt(C6F5)2(C8H6N4)]: 687.
X-ray structure determinations of [Pt(C6F5)2(napy)]·1/4CH2Cl2 (1·1/4CH2Cl2), [Pt(C6F5)2(napyCH2)]·2CH2Cl2 (3·2CH2Cl2), [Pt(C6F5)2(2-ammpy)]·1/4n-C6H14 (5·1/4n-C6H14) and cis-[Pt(C6F5)2(bipym)] (6). Crystal data and other details of the structure analyses are presented in Table 1. Crystals suitable for X-ray diffraction studies were obtained by slow diffusion of n-hexane into solutions of 0.020 g of the complexes in 3 mL of CH2Cl2. Absorption corrections were applied for all structures based on azimuthal scan data (CAD-4) or by the multi-scan method (Smart).55–57 Lorentz and polarization corrections were applied for all the structures.
Dow
nloa
ded
by C
ape
Bre
ton
Uni
vers
ity o
n 21
/04/
2013
10:
44:3
7.
Publ
ishe
d on
23
July
200
4 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
4072
84G
View Article Online

2 7 4 0 D a l t o n T r a n s . , 2 0 0 4 , 2 7 3 3 – 2 7 4 0
The structures were solved by Patterson and Fourier methods, and refined using the program SHELXL-97.58 All non-hydrogen atoms were assigned anisotropic displacement parameters. All hydrogen atoms were placed at calculated positions and constrained to ride on their parent atoms with a constrained isotropic displacement parameter. For 5·1/4n-C6H14, the C–C distances in the n-hexane solvent molecule were restrained to sensible values. Full-matrix least-squares refinement on F 2 of these models converged to final residual indices given in Table 1.
The structure determination of 3·2CH2Cl2 merits further com-ment, as it presented a space group ambiguity. Refinement was con-ducted both with space group Cc, without disorder, and with space group C2/c, with the napyCH2 ligand disordered about a crystallo-graphic 2-fold axis upon which the Pt atom lies. The results of the latter refinement are reported here. The core of the napyCH2 group was idealized by fitting the napy fragment to a similar fragment59 extracted from the Cambridge Database (Refcode KERDAM),60 and was then refined as variable metric rigid group. The CH2 carbon atom of this moiety was refined freely. The refinement using space group Cc, without disorder, gave lower residuals but had poor convergence, left about half of the atoms non-positive-definite, and produced widely discrepant values of chemically equivalent bond distances.
CCDC reference numbers 238849–238853.See http://www.rsc.org/suppdata/dt/b4/b407284g/ for crystallo-
graphic data in CIF or other electronic format.
AcknowledgementsWe thank the Spanish Comisión Interministerial de Ciencia y Tecnología (CICYT) (Projects BQU2002-0-3997-C02-02, and BQU2002-00554) and DGA (Grupos Consolidados) for their financial support and for a grant to BED (CICYT).
References 1 L. Brammer, Dalton Trans., 2003, 3145. 2 L. Brammer, J. M. Charnock, P. L. Goggin, R. J. Goodfellow,
A. G. Orpen and T. F. Koetzle, J. Chem. Soc., Dalton Trans., 1991, 1789.
3 I. C. M. Wehman-Ooyevaar, D. M. Grove, H. Kooijman, P. van der Sluis, A. L. Spek and G. van Koten, J. Am. Chem. Soc., 1992, 114, 9916.
4 D. Zhao, F. T. Ladipo, J. Braddock-Wilking, L. Brammer and P. Sherwood, Organometallics, 1996, 15, 1441.
5 J. M. Casas, L. R. Falvello, J. Forniés, A. Martin and A. J. Welch, Inorg. Chem., 1996, 35, 6009.
6 J. M. Casas, J. Fornies and A. Martin, J. Chem. Soc., Dalton Trans., 1997, 1559.
7 L. Brammer, J. C. Mareque Rivas and D. Zhao, Inorg. Chem., 1998, 37, 5512.
8 J. D. Dunitz, Chem. Commun., 2003, 545. 9 D. Braga, F. Greponi and E. Tedesco, Organometallics, 1998, 17, 2669.10 D. Braga, F. Grepioni, E. Tedesco, K. Biradha and G. R. Desiraju,
Organometallics, 1997, 16, 1846.11 C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885.12 H. W. Roesky and M. Andruh, Coord. Chem. Rev., 2003, 236, 91.13 R. Büchner, C. T. Cunningham, J. S. Field, R. J. Haines, D. R. McMillin
and G. C. Summerton, J. Chem. Soc., Dalton Trans., 1999, 711.14 M. Kato, C. Kosuge, K. Morii, J. S. Ahn, H. Kitagawa, M. Tadaoki,
M. Matsushita, T. Kato, S. Yano and M. Kimura, Inorg. Chem., 1999, 38, 1638.
15 J. S. Miller, Angew. Chem. Int. Ed. Engl., 2003, 42, 27.16 J. S. Miller and A. J. Epstein, Angew. Chem. Int. Ed. Engl., 1994, 33,
385.17 E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García, J. Ensling and
P. Gütlich, Chem. Eur. J., 2000, 6, 552.18 C. J. Kepert and J. Rossinsky, J. Chem. Soc., Chem. Commun., 1999,
375.19 O. M. Yaghi, G. M. Li and H. Li, Nature, 1995, 378, 703.
20 H. Li, M. Eddaoudi, T. L. Groy and O. M. Yaghi, J. Am. Chem. Soc., 1998, 120, 8571.
21 K. Endo, T. Koike, T. Sawaki, T. Hayashida, H. Masuda and Y. Aoyama, J. Am. Chem. Soc., 1997, 119, 4117.
22 C. Mealli and F. Zanobini, Chem. Commun., 1982, 97.23 C. He and S. J. Lippard, J. Am. Chem. Soc., 2000, 122, 184.24 C. Mealli and D. Gatteschi, Inorg. Chem., 1974, 13, 1985.25 J. A. Cabeza, L. A. Oro, A. Tiripicchio and M. Tiripicchio Camellini, J.
Chem. Soc., Dalton Trans., 1988, 1437.26 A. Bencini, E. Berti, A. Caneschi, E. Giannasi and I. Invernizzi, Chem.-
Eur.J., 2002, 8, 3660.27 A. Dossing, S. Larsen, A. van Lelieveld and R. M. Bruun, Acta Chem.
Scand., 1999, 53, 230.28 A. Tiripicchio, M. Tiripicchio Camellini, R. Usón, L. A. Oro,
M. A. Ciriano and F. Viguri, J. Chem. Soc., Dalton Trans., 1984, 125.29 D. Gatteschi, C. Mealli and L. Sacconi, J. Am. Chem. Soc., 1973, 95,
2736.30 M. Maekawa, M. Munakata, S. Kitagawa, T. Kuroda-Sowa, Y. Suenaga
and M. Yamamoto, Inorg. Chim. Acta, 1998, 271, 129.31 H. Nakajima, H. Nagao and K. Tanaka, J. Chem. Soc., Dalton Trans.,
1996, 1405.32 M. J. Bermejo, J. I. Ruiz, X. Solans and J. Vinaixa, J. Organomet.
Chem., 1993, 463, 143.33 J. C. Dewan, D. L. Kepert and A. H. White, J. Chem. Soc., Dalton
Trans., 1975, 490.34 J. M. Epstein, J. C. Dewan, D. L. Kepert and A. H. White, J. Chem.
Soc., Dalton Trans., 1974, 1949.35 C. Wing-Han, P. Shie-Ming and C. Chi-Ming, J. Chem. Soc., Dalton
Trans., 1998, 2867.36 A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and
R. Taylor, J. Chem. Soc., Dalton Trans., 1989, S1.37 R. F. Usón, M. Tomás, J. M. Casas, F. A. Cotton, L. R. Falvello and
R. Llusar, Organometallics, 1988, 7, 2279.38 R. Usón, J. Forniés, M. Tomás, J. M. Casas and R. Navarro, J. Chem.
Soc. Dalton Trans., 1989, 169.39 J. M. Casas, J. Forniés, A. Martin and B. Menjon, Organometallics,
1993, 12, 4376.40 I. Ara, J. M. Casas, J. Fornies and A. J. Rueda, Inorg. Chem., 1996, 35,
7345.41 L. R. Falvello, M. A. Hitchman, F. Palacio, I. Pascual, A. J. Schultz,
H. Stratemeier, M. Tomás, E. P. Urriolabeitia and D. M. Young, J. Am. Chem. Soc., 1999, 121, 2808.
42 J. M. Casas, J. Forniés, A. Martin and A. J. Rueda, Organometallics, 2002, 21, 4560.
43 J. M. Casas, L. R. Falvello, J. Forniés, G. Mansilla and A. Martin, Poly-hedron, 1999, 18, 403.
44 R. Usón, J. Forniés, L. R. Falvello, M. Tomás, J. M. Casas and A. Martín, Inorg. Chem., 1993, 32, 5212.
45 R. Usón, J. Forniés, J. Gimeno, P. Espinet and R. Navarro, J. Organo-met. Chem., 1974, 81, 115.
46 M. F. Lappert and J. S. Poland, Adv. Organomet. Chem., 1970, 9, 397.47 I. Artaud, N. Gregorie, P. Leduc and D. Mansuy, J. Am. Chem. Soc.,
1990, 112, 6899.48 J. Setsune and D. Dolphin, Organometallics, 1984, 3, 440.49 J. M. Casas, L. R. Falvello, J. Forniés and A. Martin, Inorg. Chem.,
1996, 35, 56.50 An ortep plot of cis-[Pt(C6F5)2(bipy)] and crystallographic experimental
details are given in the ESI.† The shortest intermolecular PtH distance is 3.52 Å.‡
51 R. Usón, J. Forniés, M. Tomás and R. Fandos, J. Organomet. Chem., 1984, 263, 253.
52 R. Usón, J. Forniés, M. Tomás and B. Menjón, Organometallics, 1985, 4, 1912.
53 W. W. Pandler and T. J. Kress, J. Org. Chem., 1967, 32.54 R. Usón and J. Forniés, Adv. Organomet. Chem., 1988, 28, 188.55 In ‘SHELXTL: Release 5.05/VMS: 1996. Siemens Analytical X.-ray
Instruments, Inc., Madison, WI.’56 G. Kopfmann and R. Huber, Acta Crystallogr., 1968, A24, 348.57 In ‘G. M. Sheldrick, SADABS V2.03. University of Göttingen,
Germany.’58 In ‘SHELXL-97: Fortram program for crystal structure refinement’,
George M. Sheldrick, 1997.59 Z. Yin, G. Xinmin, T. Ning and T. Minyu, Bull. Chem. Soc. Jpn., 1989,
62, 1279.60 F. H. Allen, Acta Crystallogr., 2002, B58, 380.
‡ Crystal data and structure refinement for [Pt(C6F5)2(bipy)]0.5·CH2Cl2. CCDC reference number 238853. See http://www.rsc.org/suppdata/dt/b4/b407284g/ for crystallographic data in CIF or other electronic format.
Dow
nloa
ded
by C
ape
Bre
ton
Uni
vers
ity o
n 21
/04/
2013
10:
44:3
7.
Publ
ishe
d on
23
July
200
4 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
4072
84G
View Article Online