i o ed. tyrosine sulfation modulates activity of tick ... table of contents expression of madanin-1...
TRANSCRIPT

In the format provided by the authors and unedited.
© 2017 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2744
NATURE CHEMISTRY | www.nature.com/naturechemistry 1
1
Supplementary Information
Tyrosine Sulfation Modulates Activity of Tick-Derived Thrombin
Inhibitors
Robert E. Thompson,1‡ Xuyu Liu,1‡ Jorge Ripoll-Rozada,2,3‡ Noelia Alonso-García,2,3 Benjamin L. Parker,4 Pedro José Barbosa Pereira,2,3 Richard J. Payne1
1School of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia
2IBMC - Instituto de Biologia Molecular e Celular, Universidade do Porto, 4200-135 Porto, Portugal
3Instituto de Investigação e Inovação em Saúde, Universidade do Porto, 4200-135 Porto, Portugal
4Charles Perkins Centre, The University of Sydney, NSW 2006, Australia
Correspondence and requests for materials should be addressed to Richard J. Payne (email: [email protected])
Tyrosine sulfation modulates activity of tick-derived thrombin inhibitors

2
Table of Contents
Expression of madanin-1 and chimadanin in insect cells 3
Liquid chromatography tandem mass spectrometry of insect cell culture media 3
General procedures for peptide and protein synthesis 6
Synthesis of madanin-1 peptide fragments 8
Synthesis of madanin-1 thioesters 12
General procedure for the kinetically-controlled one-pot ligation-desulfurization synthesis
of madanin-1 sulfoforms 19
Synthesis of chimadanin peptide fragments 27
General procedure for one-pot ligation-desulfurization synthesis of chimadanin sulfoforms 34
Screening for selectivity of serine protease inhibition 49
Thrombin amidolytic activity assay 49
Thrombin Time (TT) Assay 51
Cleavage of unsulfated and disulfated madanin-1 by human α-thrombin 51
Crystallization of a disulfated madanin-1•human α-thrombin complex 53
Data collection and processing 53
Structure determination and refinement 53
References 55

3
Supplementary Methods
Expression of madanin-1 and chimadanin in insect cells
Synthetic genes encoding madanin-1 or chimadanin from Haemaphysalis longicornis fused to the honeybee (Apis
mellifera) mellitin secretion signal and with optimized codon usage for expression in insect cells were obtained from a
commercial supplier (GenScript). The synthetic genes were cloned into the BamHI and HindIII restriction sites of the
pKL acceptor vector (EMBL, Grenoble1) and the integrity of the resulting constructs was verified by automatic DNA
sequencing. Bacmid preparation, transfection, and protein expression were performed according to the protocol described
by Fitzgerald et al.1 Briefly, Escherichia coli DH10EmbacY competent cells were transformed with the insert-containing
acceptor vectors and positive colonies (confirmed by PCR) were used for bacmid preparation. Trichoplusia ni High Five
cells (ThermoFisher Scientific), maintained at a cell density of 0.5-1 x 106 cells/mL at 27 °C with 80 r.p.m. shaking in
Express Five serum-free medium (ThermoFisher Scientific) supplemented with 18 mM of L-glutamine, were transfected
with each of the bacmids using X-tremeGENE HP DNA transfection reagent (Roche) and kept in the dark at 27 °C
without agitation. Virus-containing culture supernatants were collected after 50-60 h and stored at 4 °C (initial virus, V0).
For protein expression, 25 mL of Trichoplusia ni High Five cells at 0.5 x 106 cells/mL were inoculated with 3 mL of
corresponding V0 virus stock and incubated at 27 °C with 80 r.p.m. shaking. Cultures were diluted at 24 h intervals to
below 1 x 106 cells/mL until cell proliferation arrest occurred, after which aliquots of 1x106 cells were collected every 12
h and used to monitor the fluorescent signal of the co-expressed reporter, YFP. A fluorescent signal intensity plateau was
reached 48-60 h after proliferation arrest. At this point, the culture supernatants (10 mL for chimadanin and 40 mL for
madanin-1) were filtered (0.22 μm), lyophilised and stored at 4 °C until further use.
Liquid chromatography tandem mass spectrometry of insect cell culture media
25% of the lyophilised culture supernatants of madanin-1 and chimadanin were resuspended in 1 mM ammonium
bicarbonate pH 7.9 (5 mL). An aliquot (250 L) was removed and centrifuged at 14,000 x g for 5 min at 4 oC to remove
any insoluble material. The supernatant was filtered through a 30 kDa molecular weight cut-off filter and the filtrate
directly loaded onto a C18 solid-phase extraction micro column (3M empore). The column was washed with 10%
acetonitrile in water and eluted with 60% acetonitrile in water followed by vacuum centrifugation. The purified extract
was resuspended in water and analyzed on a Dionex 3500RS nanoUHPLC coupled to either a Q-Exactive plus or Orbitrap
Fusion mass spectrometer in positive mode. The mixture was separated using an in-house packed 75 μm x 40 cm pulled
column (1.9 μm particle size, C18AQ; Dr Maisch, Germany) with a gradient of 2 – 40% acetonitrile containing 0.1%
formic acid over 60 min at 250 nL min-1. An MS1 scan was acquired from 450 – 1650 m/z (140,000 resolution [Q-
Exactive Plus] or 120,000 resolution [Orbitrap Fusion]; 1e6 AGC [Q-Exactive Plus] or 5e5 AGC [Orbitrap Fusion], 100
ms injection time) followed by MS/MS data-dependent acquisition with HCD and detection in the orbitrap on the Q-
Exactive Plus (35,000 resolution, 1e5 AGC, 120 ms injection time, 32 NCE, 2.0 m/z quadrupole isolation width) or
EThcD and detection in the orbitrap on the Orbitrap Fusion (60,000 resolution, 2e5 AGC, 120 ms injection time, calibrated
charge-dependent ETD reaction times [2+ 121 ms; 3+ 54 ms; 4+ 30 ms; 5+ 20ms; 6+ 13 ms; 7+; 10 ms], 25 NCE for

4
HCD supplemental activation, 2.0 m/z quadrupole isolation width). All data were processed with Proteome Discoverer
(v2.1) using the Byonic node (v1.0.334)2 and manual analysis / annotation using GPMAW (Lighthouse Data, Denmark).
The precursor MS, HCD MS/MS and EThcD MS/MS tolerance were set to 20 ppm with no enzyme searching. The
peptides were searched with the following variable modifications; oxidation of methionine, deamidation of asparagine or
glutamine, sulfation of tyrosine and the modification of serine or threonine residues with a database of 78 O-linked glycan
compositions available through Byonic. A precursor isotope off set was enabled to account for incorrect precursor
monoisotopic reporting (+/- 2.0 Da).
Supplementary Figure 1. Analysis of insect cell culture medium containing baculovirus-assisted expression of
chimadanin by liquid chromatography tandem mass spectrometry. A) Average precursor MS1 scan highlighting
chimadanin sulfation isoforms. B) Tandem mass analysis with higher collisional dissociation (HCD) of disulfated
chimadanin. All ions are annotated within a tolerance of 20 ppm.
5
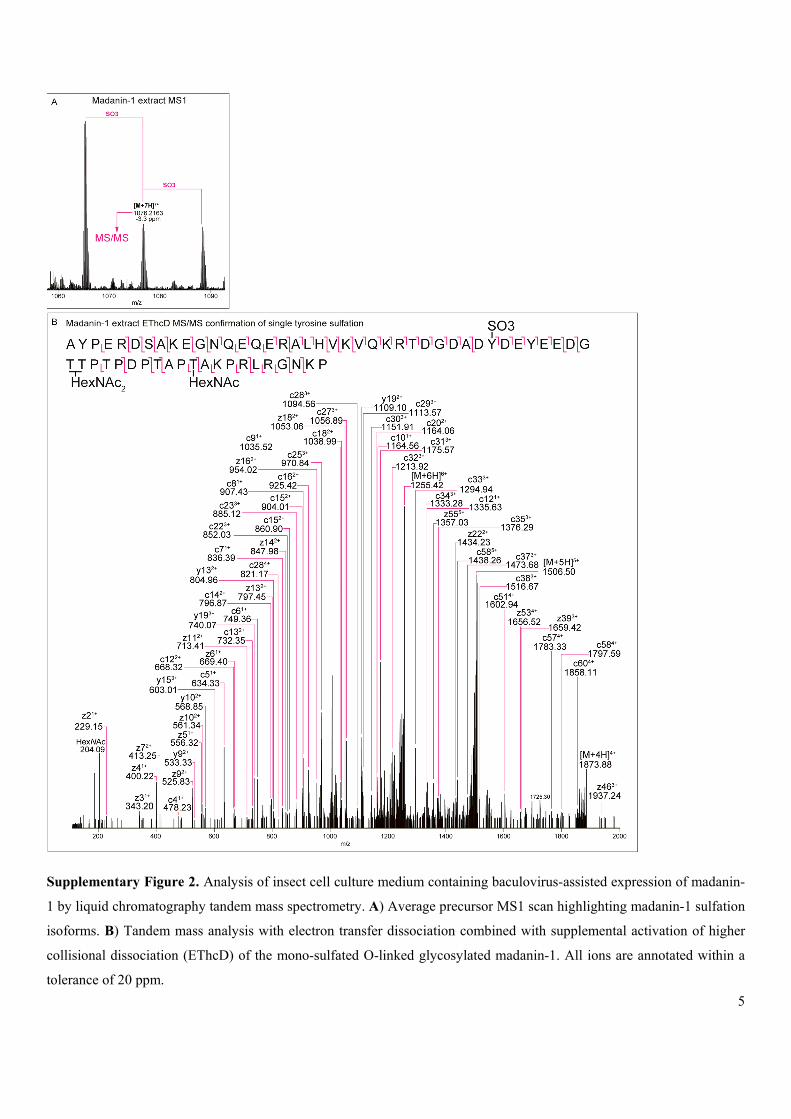
Supplementary Figure 2. Analysis of insect cell culture medium containing baculovirus-assisted expression of madanin-
1 by liquid chromatography tandem mass spectrometry. A) Average precursor MS1 scan highlighting madanin-1 sulfation
isoforms. B) Tandem mass analysis with electron transfer dissociation combined with supplemental activation of higher
collisional dissociation (EThcD) of the mono-sulfated O-linked glycosylated madanin-1. All ions are annotated within a
tolerance of 20 ppm.

6
General procedures for peptide and protein synthesis
Spectroscopic grade dimethylformamide (DMF) was obtained from Merck. Amino acids, coupling reagents and resins for
Fmoc-solid-phase peptide synthesis (SPPS) were obtained from either Novabiochem or Bachem. SPPS was performed in
polypropylene syringes equipped with Teflon filters, purchased from Torviq. Analytical reversed-phase high-performance
liquid chromatography (HPLC) was performed on a Waters System 2695 separations module with a 2996 photodiode
array detector and an Alliance series column heater set at 30 °C. A Waters Sunfire 5 μm, 2.1 × 150 mm column (C18) was
used unless otherwise indicated at a flow rate of 0.2 mL min−1 using a mobile phase of 0.1% TFA in water (Solvent A)
and 0.1% TFA in acetonitrile (Solvent B) in a linear gradient as indicated. Peptides and proteins above 3 kDa in size were
chromatographed on a Waters XBridge-BEH300 5 μm, 2.1 × 150 mm column (C18). Peptides and proteins bearing free
sulfotyrosine residues were chromatographed on a Waters XBridge-BEH300 5 μm, 2.1 × 150 mm column (C18) using a
mobile phase of 0.1 M NH4OAc (Solvent A) and acetonitrile (Solvent B) using the indicated gradient. Results were
analyzed with Waters Empower software and retention times (Rt min) of pure peptides and proteins are reported with the
gradients specified.
Preparative and semi-preparative reversed-phase HPLC was performed using a Waters 600E Multisolvent Delivery
System with a Rheodyne 7725i Injection valve (4 mL loading loop) and Waters 500 pump with a Waters 490E
programmable wavelength detector operating at 214, 230, 254 or 280 nm. Preparative reversed-phase HPLC was
performed using a Waters Sunfire C18 column (5 μm, 19 × 150 mm) at a flow rate of 7 mL min−1. Semi-preparative
reversed-phase HPLC was performed using a Waters Sunfire C18 column (5 μm, 10 × 250 mm) at a flow rate of 4 mL
min−1. Peptides and proteins above 3 kDa in size were chromatographed on a Waters XBridge-BEH300 (5 μm, 10 × 250
mm) semi-preparative column (C18). Peptides and proteins bearing free sTyr residues were purified on a Waters XBridge-
BEH300 (5 μm, 10 × 250 mm) semi-preparative column (C18) operating at a flow rate of 4 mL min−1 using a mobile
phase of 0.1 M NH4OAc (Solvent A) and acetonitrile (Solvent B) using the indicated gradient. After lyophilisation,
peptides were isolated as TFA, formate or acetate salts depending on the chromatographic eluent.
High-performance liquid chromatography–mass spectrometry (HPLC-MS) was conducted on a Shimadzu 2020 LC–MS
instrument consisting of a LC-M20A pump and a SPD-20A UV/Vis detector coupled to a Shimadzu 2020 mass
spectrometer. Separations were performed on either a Waters Sunfire C18 column (5 µm, 2.1 × 150 mm), or a XBridge-
BEH300 C18 column (5 µm, 2.1 × 150 mm) operating at a flow rate of 0.2 mL min−1 using a mobile phase of 0.1% formic
acid in water (Solvent A) and 0.1% formic acid in acetonitrile (Solvent B) in a linear gradient as indicated.
Fmoc-Strategy SPPS General Procedures (25 μmol scale)
Resin Loading: 2-Chlorotrityl chloride resin (1.22 mmol/g loading) was swollen in dry CH2Cl2 for 30 min then washed
with CH2Cl2 (5 × 3 mL). A solution of a given amino acid (Fmoc-Xaa-OH) (0.5 mmol/g resin) and iPr2NEt (2.0 equiv.
relative to resin functionalization) in CH2Cl2 (final concentration 0.1 M of amino acid) was added and the resin shaken at
room temperature for 16 h. The resin was washed with DMF (5 × 3 mL) and CH2Cl2 (5 × 3 mL). The resin was then
treated with a solution of CH2Cl2/CH3OH/iPr2NEt (17:2:1, v/v/v, 3 × 3 mL × 5 min) for 1 h and washed with DMF (5 × 3

7
mL), CH2Cl2 (5 × 3 mL), and DMF (5 × 3 mL). The resin was subsequently submitted to iterative peptide assembly via
Fmoc-SPPS (see below).
Iterative Peptide Assembly (Fmoc-SPPS)
Deprotection: The resin was treated with piperidine/DMF (1:4, v/v, 3 mL, 3 × 5 min), filtered and then washed with
DMF (5 × 3 mL), CH2Cl2 (5 × 3 mL) and DMF (5 × 3 mL).
Coupling (standard Fmoc-protected amino acids): A solution of protected amino acid (4 equiv.), PyBOP (4 equiv.) and
NMM (8 equiv.) in DMF (final concentration 0.1 M) was added to the resin. After 1 h, the resin was filtered and washed
with DMF (5 × 3 mL), CH2Cl2 (5 × 3 mL) and DMF (5 × 3 mL).
Coupling [Fmoc-Tyr(SO2OCH2C(CH3)3)-OH]: A solution of amino acid (1.2 equiv.), HATU (1.2 equiv.) and NMM
(2.4 equiv.) in DMF (final concentration 0.1 M) was added to the resin. After 18 h, the resin was filtered and washed with
DMF (5 × 3 mL), CH2Cl2 (5 × 3 mL) and DMF (5 × 3 mL).
Coupling [Boc-Asp(STmob,OtBu)-OH]: A solution of amino acid (1.5 equiv.), PyBOP (3.0 equiv.), and NMM (3.0
equiv.) in DMF (final concentration 0.1 M) was added to the resin (1.0 equiv.) and shaken at room temperature for 16 h.
The resin was filtered and washed with DMF (5 × 3 mL), CH2Cl2 (5 × 3 mL) and DMF (5 × 3 mL).
Capping: Acetic anhydride/pyridine (1:9, v/v, 3 mL) was added to the resin. After 3 min the resin was filtered and
washed with DMF (5 × 3 mL), CH2Cl2 (5 × 3 mL) and DMF (5 × 3 mL).

8
Synthesis of madanin-1 peptide fragments
Peptide fragments 1 and 8 were synthesized using previously reported methods.3
Supplementary Figure 3. Synthesis of Madanin-1(29–47) alkyl thioester precursor (S5) through Fmoc-SPPS followed by
solution-phase thioesterification.
S1
S2
S3 S4
S5

9
Peptide S1 (100 μmol) bound to 2-chlorotrityl chloride resin was prepared by standard Fmoc SPPS (by Mimotopes Pty
Ltd.) and was elongated according to Fmoc-Strategy SPPS as outlined in the general procedures to give resin-bound
fully-protected peptide S2 (Supplementary Fig. 3). Mild acidic liberation of the fully-protected peptide from the resin was
achieved using HFIP/CH2Cl2 (1:4, v/v) to give protected peptide S3, which was subsequently treated with PyBOP
(260 mg, 0.5 mmol), iPr2NEt (88 μL, 0.5 mmol), and ethyl 3-mercaptopropionate (380 μL, 3 mmol) in DMF (10 mL) at
0 °C for 3 h to afford crude thioester S4. The crude reaction mixture was then concentrated in vacuo for 16 h. A solution
of TFA/iPr3SiH/H2O (95:2.5:2.5, v/v/v, 20 mL) was added and the solution stirred for 90 min at room temperature. The
reaction mixture was then concentrated to ~ 2 mL and crude peptide thioester S5 precipitated from cold Et2O (45 mL).
HPLC purification (0 to 25% B over 60 min, 0.1% TFA) afforded peptide thioester S5 (22 mg, 5% yield). Analytical
HPLC: Rt 14.6 min (0 to 40% B over 30 min, 0.1% TFA, = 214 nm); Calculated Mass [M+3H]3+: 1128.6, [M+4H]4+:
846.7, [M+5H]5+: 677.5, [M+6H]6+: 564.8, [M+7H]7+: 484.2, Mass Found (ESI) 1129.5 [M+3H]3+, 847.5 [M+4H]4+, 678.2
[M+5H]5+, 565.4 [M+6H]6+, 484.8 [M+7H]7+.
14.6 min
0
0.25
0.5
0.75
1
1.25
1.5
0 5 10 15 20 25 30
Ab
so
rban
ce (
AU
)
Retention Time (min)
484.8[M+7H]7+
565.4[M+6H]6+
678.2[M+5H]5+
847.5[M+4H]4+ 1129.5
[M+3H]3+
0
20
40
60
80
100
0 500 1000 1500 2000
Re
lati
ve A
bu
nd
an
ce
(%
)
m/z

10
Supplementary Figure 4. A) Synthesis of Madanin-1(29–47) TFET thioester (1) through thiol exchange with TFET. B)
Crude HPLC chromatogram (λ = 230 nm) of thiol–thioester exchange reaction after 2h.
Conversion of peptide thioester S5 to the activated trifluoroethyl thioester 1 was achieved through incubation of peptide
thioester S5 (16 mg, 3.7 μmol) in buffer (10 mL, 6 M Gn·HCl, 100 mM Na2HPO4, pH 7.5, not degassed) with 5 vol.%
TFET at 30 °C (Supplementary Fig. 4A). After 2 h, HPLC–MS analysis indicated complete exchange to activated
thioester 1 (Supplementary Fig. 4B).
HPLC purification (0 to 25% B over 60 min, 0.1% TFA) of the reaction mixture afforded peptide thioester 1 (11.9 mg,
75% yield). Analytical HPLC: Rt 14.7 min (0 to 40% B over 30 min, 0.1% TFA, = 214 nm); Calculated Mass
[M+3H]3+: 1122.6, [M+4H]4+: 842.2, [M+5H]5+: 673.9, [M+6H]6+: 561.8, [M+7H]7+: 481.7, Mass Found (ESI) 1123.6
[M+3H]3+, 842.9 [M+4H]4+, 674.6 [M+5H]5+, 562.4 [M+6H]6+, 482.2 [M+7H]7+.
14.7 min
0
0.25
0.5
0.75
1
0 5 10 15 20 25 30
Ab
sorb
ance
(A
U)
Retention Time (min)
S5 1
1
TFET
A
B

11
482.2[M+7H]7+
562.4[M+6H]6+
674.6[M+5H]5+
842.9[M+4H]4+
1123.6[M+3H]3+
0
20
40
60
80
100
0 500 1000 1500 2000
Rel
ati
ve A
bu
nd
ance
(%
)
m/z

12
Synthesis of madanin-1 thioesters
Supplementary Figure 5. Outline of the syntheses of madanin-1 (29-47) sulfopeptide thioesters 2-4.
2-Chlorotrityl chloride resin was loaded with Fmoc-Thr(tBu-OH (100 μmol, 0.5 mmol/g loading) and iterative Fmoc-
Strategy SPPS was carried out as outlined in the general procedures to give resin-bound peptide S3 (Supplementary Fig.
5). At this point the resin was split into 3 equal portions of 25 μmol and elongated in parallel, with appropriate installation
of either Fmoc-Tyr(tBu)-OH or Fmoc-Tyr[SO2OCH2C(CH3)3]-OH depending on the desired sulfoform being targeted.
After coupling of Boc-Asp(STmob,OtBu)-OH,4 each protected peptide (S6, S7 and S8) was liberated from resin using
HFIP/CH2Cl2 (1:4, v/v). The fully protected crude peptides S9-S11 were each treated with PyBOP (65 mg, 125 μmol),

13
iPr2NEt (22 μL, 125 μmol), and ethyl 3-mercaptopropionate (95 μL, 0.75 mmol) in DMF (1 mL) at −30 °C for 3 h. The
crude reaction mixtures were then concentrated and dried in vacuo for 16 h. A solution of TFA/iPr3SiH/H2O (95:2.5:2.5,
v/v/v, 10 mL) was added to each crude protected peptide thioester S12-S14 and stirred for 90 min at room temperature.
The deprotection mixtures were then concentrated to ~1 mL and precipitated from ice cold Et2O (45 mL), affording crude
peptide thioesters 2–4.
Madanin-1 (29-47)-sY32 peptide thioester (2)
HPLC purification of crude peptide thioester 2 (0 to 50% B over 60 min, 0.1% TFA) afforded pure peptide thioester 2 as
the TFA salt (7.6 mg, 12% yield). Analytical HPLC: Rt 24.5 min (0 to 50% B over 40 min, 0.1% TFA, = 214 nm);
Calculated Mass [M+2H]2+: 1215.4, [M+3H]3+: 810.6, Mass Found (ESI) 1215.6 [M+2H]2+, 810.8 [M+3H]3+. HRMS
(ESI) m/z calculated for C99H141N19Na2O46S322+ [M+2Na]2+: 1236.9112, found 1236.9111.
2

14
Madanin-1 (29-47)-sY35 peptide thioester (3)
HPLC purification of crude peptide thioester 3 (0 to 50% B over 60 min, 0.1% TFA) afforded pure peptide thioester 3 as
the TFA salt (10.2 mg, 16% yield). Analytical HPLC: Rt 24.5 min (0 to 50% B over 30 min, 0.1% TFA, = 214 nm);
Calculated Mass [M+2H]2+: 1215.4, [M+3H]3+: 810.6, Mass Found (ESI) 1215.5 [M+2H]2+, 810.9 [M+3H]3+. HRMS
(ESI) m/z calculated for C99H141N19Na2O46S322+ [M+2Na]2+: 1236.9112, found 1236.9110.
3

15
Madanin-1 (29-47)-sY32, sY35 peptide thioester (4)
HPLC purification of crude peptide thioester 4 (0 to 75% B over 60 min, 0.1% TFA) afforded pure peptide thioester 4 as
the TFA salt (10.1 mg, 15% yield). Analytical HPLC: Rt 22.5 min (0 to 70% B over 40 min, 0.1% TFA, = 214 nm);
Calculated Mass [M+2H]2+: 1290.4, [M+3H]3+: 860.6, Mass Found (ESI) 1290.3 [M+2H]2+, 860.9 [M+3H]3+.
4

16
Synthesis of madanin-1(48–60, A48C) (8)
Peptide 8, corresponding to Madanin-1(48–60, A48C), was synthesized via Fmoc-strategy SPPS on 2-chlorotrityl chloride
resin (25 μmol, 0.5 mmol/g loading) according to the general procedures. The fully assembled peptide was then cleaved
from the resin with concomitant global side-chain deprotection using a solution of TFA/iPr3SiH/H2O (95:2.5:2.5
v/v/v) to afford the crude peptide. Purification by HPLC (0 to 20% B over 40 min, 0.1% TFA) afforded peptide 20
(25 mg, 50% yield). Analytical HPLC: Rt 22.6 min (0 to 25% B over 40 min, 0.1% TFA, = 214 nm); Calculated Mass
[M+2H]2+: 719.4, [M+3H]3+: 479.9, [M+4H]4+: 360.2, Mass Found (ESI) 719.8 [M+2H]2+, 480.3 [M+3H]3+, 360.6
[M+4H]4+; HRMS (ESI) calculated for C61H109N22O16S+ [M+H]+, m/z = 1437.8107, found 1437.8089.
22.6 min
-0.25
0
0.25
0.5
0.75
1
1.25
1.5
0 5 10 15 20 25 30 35 40
Ab
so
rba
nc
e (A
U)
Retention Time (min)
360.6[M+4H]4+
480.3[M+3H]3+
719.8[M+2H]2+
0
20
40
60
80
100
0 250 500 750 1000 1250 1500 1750 2000
Re
lati
ve A
bu
nd
anc
e (%
)
m/z
8

17
Disulfated madanin-1 (22-54) fragment (S15)
Disulfated madanin-1 fragment S15 was synthesized on 2-chlorotrityl resin (12.5 µmol) according to the general Fmoc-
strategy SPPS procedures described above with the incorporation of Fmoc-Tyr[SO2OCH2C(CH3)3]-OH at Y32 and Y35.
The crude peptide was then deprotected and cleaved from the resin using TFA/iPr3SiH/H2O (90:5:5, v/v/v) for 2 h. The
crude deprotected peptide was precipitated from cold Et2O and purified by reverse-phase HPLC (0 to 70% B over 40 min,
0.1% TFA) to give the neopentyl-protected disulfated peptide which was immediately incubated in buffer (6 M Gn.HCl,
100 mM Na2HPO4, 25 mM TCEP, pH 7.0) for 48 h, at which point the neopentyl sulfate ester protecting groups had been
removed. The deprotected disulfated peptide was isolated by means of semi-preparative reverse phase HPLC (0 to 60% B
over 60 min) to give disulfated madanin-1 fragment S15 (5.0 mg, 9% yield). Analytical HPLC: Rt 12.8 min (0 to 50% B
over 30 min, λ = 230 nm). Calculated Mass [M-2H]2- : 1897.3, [M-3H]3- : 1264.5, [M-4H]4- : 948.1; Mass Found (ESI -
negative mode) 1897.6 [M-2H]2-, 1264.8 [M-3H]3-, 948.4 [M-4H]4-.
S15

18

19
General procedure for the kinetically-controlled one-pot ligation-desulfurization synthesis of madanin-1
sulfoforms
PTAKPRLRGNKPNH
O
HS
OHYPERDSAKEGNQEQERALHVKVQKRTDG ADYDEYEEDGTTPTPDPTNH
O
OH
O
HS
H
OR2
OR1
2: R1 = SO2OCH2C(CH3)3, R2 = H3: R1 = H, R2 = SO2OCH2C(CH3)34: R1,R2 = SO2OCH2C(CH3)3
PTAKPRLRGNKPH2N
O
HS
OH
YPERDSAKEGNQEQERALHVKVQKRTDG ADYDEYEEDGTTPTPDPTNH
O
OH
O
HS
H
OR2
OR1
S OEt
O
YPERDSAKEGNQEQERALHVKVQKRTDG
ADYDEYEEDGTTPTPDPTH2N
O
OH
O
HS
H
OR2
OR1
S OEt
O
S CF3
1
PTAKPRLRGNKPNH
OOHYPERDSAKEGNQEQERALHVKVQKRTDG ADYDEYEEDGTTPTPDPTN
HO
OH
O
H
OR2
OR1
Ligation 1
Desulfurization
Ligation 2
5: R1 = SO3, R2 = H6: R1 = H, R2 = SO37: R1,R2 = SO3
8
9: R1 = SO3, R2 = H10: R1 = H, R2 = SO311: R1,R2 = SO3
12: R1 = SO3, R2 = H13: R1 = H, R2 = SO314: R1,R2 = SO3
1 28
2947
1 28 29 47
48 60
32
35
32
35
32
351 28 29 47 48 60
32
351 28 29 47 48 60
Supplementary Figure 6. Schematic of the kinetically controlled one-pot ligation-desulfurization synthesis of madanin-1
sulfoforms 12-14 (Figure 2 in manuscript).
Peptide trifluoroethyl-thioester 1 and peptide thioesters 2–4 were reacted in ligation buffer at pH 7.4–7.5 for 1 h. After
this time, complete ligation between the peptide fragments had occurred, as judged by HPLC-MS analysis to afford

20
ligation products 5–7 (see Supplementary Fig. 7-9). Peptide fragment 8 was added to each reaction mixture along with 2
vol.% TFET in order to effect the second ligation reaction. After 12 h of reaction time, HPLC-MS analysis indicated that
in each case the second ligation reaction had proceeded efficiently to afford the corresponding ligation products 9-11 (see
Supplementary Fig. 7-9). Following the completion of the second ligation reaction (as judged by HPLC-MS), the
reactions were degassed prior to in situ desulfurization through argon sparging. Addition of TCEP (200 mM), glutathione
(40 mM) and VA-044 (20 mM) to the reaction mixtures containing ligation products 9–11 was followed by incubation at
37 °C. After 16 h of reaction time, HPLC-MS analysis of the desulfurization reactions indicated complete conversion to
the madanin-1 sulfoforms 12-14 (see Supplementary Fig. 7-9). Purification of the sulfoproteins was achieved using
reversed-phase HPLC, employing an ammonium acetate buffer as neutral eluent to prevent acidolytic desulfation of the
final products.
Madanin-1(sY32) – (12)
Madanin-1 sulfoform 12 was assembled from peptide trifluoroethyl-thioester 1 (1.9 mg, 0.45 μmol), peptide alkyl-
thioester 2 (1.0 mg, 0.5 μmol), and peptide 8 (1.0 mg, 0.5 μmol) according to the general procedure, monitoring the
progress using HPLC-MS (Supplementary Fig. 7). Following the assembly of 12, the reaction mixture was purified using
semipreparative HPLC (0 to 20% B over 80 min, eluent A = 0.1 M NH4OAc, B = MeCN), affording 12 as a white solid
after lyophilisation (0.8 mg, 28% yield from 1). Analytical HPLC: Rt 19.7 min (0 to 25% B over 30 min, A = 0.1 M
NH4OAc, B = MeCN, λ = 230 nm). Calculated Mass [M+6H]6+: 1142.2, [M+7H]7+: 979.2, [M+8H]8+: 856.9, [M+9H]9+:
761.8, [M+10H]10+: 685.7, [M+11H]11+: 623.5, [M+12H]12+: 571.6, Mass Found (ESI) 1142.8 [M+6H]6+, 979.6 [M+7H]7+,
857.3 [M+8H]8+, 762.3 [M+9H]9+, 686.1 [M+10H]10+, 623.9 [M+11H]11+, 572.0 [M+12H]12+.

21
Supplementary Figure 7. A) Crude HPLC-MS analysis of ligation reaction between trifluoroethyl thioester 1 and peptide
5 *
9
6+
7+ 8+
9+ 10+
11+
12+
12
#
6H+
7H+ 8H+
9H+
10H+
11H+
12H+
A
B
C

22
thioester 2 to give the corresponding ligation product 5 (λ = 230 nm). * = ligation product bearing internal thioester; i)
Mass calculated for 5 (100% relative abundance) [M+5H]5+: 1122.9, [M+6H]6+: 935.9, [M+7H]7+: 802.3, Mass found
(ESI) m/z 1123.1, 936.2, 802.5; B) Crude HPLC-MS analyses of the ligation reactions between peptide fragment 8 and
ligation product 5 to give the corresponding ligation products 9 (λ = 230 nm). Mass calculated for 9 (100% relative
abundance) [M+6H]6+: 1153.2, [M+7H]7+: 988.5, [M+8H]8+: 865.2, [M+9H]9+: 769.0, [M+10H]10+: 692.3, [M+11H]11+:
629.4, [M+12H]12+: 577.1, Mass found (ESI) m/z 1153.8, 988.8, 865.4, 769.3, 692.5, 629.6, 577.3; C) Crude HPLC-MS
analysis of the in situ desulfurization reaction of ligation product 9 after 3 h to give the corresponding madanin-1
sulfoform 12. # = TCEP sulfide; Mass calculated for 12 (100% relative abundance) [M+6H]6+: 1142.5, [M+7H]7+: 979.5,
[M+8H]8+: 857.2, [M+9H]9+: 762.0, [M+10H]10+: 685.9, [M+11H−SO3]11+: 616.3, [M+12H−SO3]
12+: 565.1, Mass found
(ESI) m/z 1142.7, 979.5, 857.4, 762.1, 868.1, 616.6, 565.3.
Madanin-1(sY35) – (13)
Madanin-1 sulfoform 13 was assembled from peptide trifluoroethyl-thioester 1 (1.9 mg, 0.45 μmol), peptide alkyl-
thioester 3 (1.0 mg, 0.4 μmol), and peptide 8 (1.0 mg, 0.5 μmol) according to the general procedure, monitoring the
progress using HPLC-MS (Supplementary Fig. 8). Following the assembly of 13, the reaction mixture was purified
though semipreparative HPLC (0 to 20% B over 80 min, eluent A = 0.1 M NH4OAc, B = MeCN) affording 13 as a white
solid after lyophilisation (1.5 mg, 54% yield from 1). Analytical HPLC: Rt 19.7 min (0 to 25% B over 30 min, A = 0.1 M
NH4OAc, B = MeCN, λ = 230 nm); Calculated Mass [M+6H]6+: 1142.2, [M+7H]7+: 979.2, [M+8H]8+: 856.9, [M+9H]9+:
761.8, [M+10H]10+: 685.7, [M+11H]11+: 623.5, Mass Found (ESI) 1142.7 [M+6H]6+, 979.6 [M+7H]7+, 857.3 [M+8H]8+,
762.2 [M+9H]9+, 686.2 [M+10H]10+, 623.9 [M+11H]11+.
19.7

23
Supplementary Figure 8. A) Crude HPLC-MS analysis of ligation reaction between trifluoroethyl thioester 1 and peptide
thioester 3 to give the corresponding ligation product 6 (λ = 230 nm). * = ligation product bearing internal thioester; Mass
calculated for 6 (100% relative abundance) [M+4H]4+: 1403.4, [M+5H]5+: 1122.9, [M+6H]6+: 935.9, [M+7H]7+: 802.3,
6 *
6+7+ 8+
9+
10+11+
12+
10
7+ 8+
9+
10+11+
12+
6+
13 #
A
B
C

24
[M+8H]8+: 702.2, Mass found (ESI) m/z 1403.6, 1123.1, 936.2, 802.6, 702.4; B) Crude HPLC-MS analyses of the ligation
reactions between peptide fragment 8 and ligation product 6 to give the corresponding ligation product 10 (λ = 230 nm).
Mass calculated for 10 (100% relative abundance) [M+6H]6+: 1153.2, [M+7H]7+: 988.5, [M+8H]8+: 865.2, [M+9H]9+:
769.0, [M+10H]10+: 692.3, [M+11H]11+: 629.4, [M+12H]12+: 577.1, Mass found (ESI) m/z 1153.6, 988.8, 865.4, 769.3,
692.5, 629.6, 577.3; C) Crude HPLC-MS analysis of the in situ desulfurization reaction of ligation product 10 after 3 h to
give the corresponding madanin-1 sulfoform 13. # = TCEP sulfide; Mass calculated for 13 (100% relative abundance)
[M+6H]6+: 1142.5, [M+7H]7+: 979.5, [M+8H]8+: 857.2, [M+9H]9+: 762.0, [M+10H]10+: 685.9, [M+11H]11+: 623.7,
[M+12H]12+: 571.8, Mass found (ESI) m/z 1142.7, 979.5, 857.1, 762.2, 686.1, 623.8, 572.0.
Madanin-1(sY32, sY35) – (14)
Madanin-1 sulfoform 14 was assembled from peptide trifluoroethyl-thioester 1 (3.1 mg, 0.72 μmol), peptide alkyl-
thioester 4 (1.8 mg, 0.66 μmol), and peptide 8 (1.6 mg, 0.79 μmol) according to the general procedure, monitoring the
progress using HPLC-MS (Supplementary Fig. 9). Following the assembly of 14, the reaction mixture was purified using
semipreparative HPLC (0 to 20% B over 80 min, eluent A = 0.1 M NH4OAc, B = MeCN), affording 14 as a white solid
after lyophilisation (1.8 mg, 40% yield from 4). Analytical HPLC: Rt 19.9 min (0 to 25% B over 30 min, A = 0.1 M
NH4OAc, B = MeCN, λ = 230 nm). Calculated Mass [M+6H]6+: 1155.5, [M+7H]7+: 990.5, [M+8H]8+: 866.9, [M+9H]9+:
770.7, [M+10H]10+: 693.7, Mass Found (ESI) 1156.0 [M+6H]6+, 991.1 [M+7H]7+, 867.4 [M+8H]8+, 771.1 [M+9H]9+,
694.1 [M+10H]10+.

25
11
*
7
6+
7+ 8+
9+ 10+
11+
12+
12+
14
6+
7+8+
9+ 10+
11+
#
A
B
C

26
Supplementary Figure 9. A) Crude HPLC-MS analysis of ligation reaction between trifluoroethyl thioester 1 and peptide
thioester 4 to give the corresponding ligation product 7 (λ = 230 nm). * = ligation product bearing internal thioester; i)
Mass calculated for 7 (100% relative abundance) [M+4H]4+: 1423.3, [M+5H]5+: 1138.9, [M+6H]6+: 949.2, [M+7H]7+:
813.8, Mass found (ESI) m/z 1423.5, 1139.2, 949.5, 813.9. B) Crude HPLC-MS analyses of the ligation reaction between
peptide fragment 8 and ligation product 7 to give the corresponding ligation product 11 (λ = 230 nm); Mass calculated for
11 (100% relative abundance), [M+6H]6+: 1166.5, [M+7H]7+: 999.9, [M+8H]8+: 875.1, [M+9H]9+: 777.9, [M+10H]10+:
700.3, [M+11H−SO3]11+: 629.4, [M+12H−SO3]
12+: 577.1, Mass found (ESI) m/z 1166.6, 1000.2, 875.3, 778.2, 700.5,
629.7, 577.3. C) Crude HPLC-MS analysis of the in situ desulfurization reaction of ligation product 11 after 3 h to give
the corresponding madanin-1 sulfoform 14. # = TCEP sulfide; Mass calculated for 14 (100% relative abundance)
[M+6H]6+: 1155.9, [M+7H]7+: 990.7, [M+8H]8+: 867.2, [M+9H]9+: 770.8, [M+10H]10+: 693.9, [M+11H]11+: 630.8, Mass
Found (ESI) m/z 1155.9, 991.0, 867.3, 771.1, 694.1, 631.1.

27
Synthesis of chimadanin peptide fragments
Peptide fragments 15 and 19 were synthesized using previously reported methods.3
Supplementary Figure 10. Synthesis of peptide thioester 19 through Fmoc-SPPS.
Protected Peptide S16 corresponding to Chimadanin(2–20) was synthesized on 2-chlorotrityl chloride resin (25 μmol)
using standard Fmoc-SPPS (by Mimotopes Pty Ltd., Scheme S3). The peptide was Fmoc-deprotected with 20 vol.%
piperidine in DMF and L-pyroglutamate was subsequently coupled according to the general procedure for amino acid
coupling. The fully assembled protected peptide was then liberated from resin using 30 vol.% HFIP in CH2Cl2 (10 mL) to
give protected peptide S17. The crude peptide was subsequently treated with PyBOP (65 mg, 0.13 mmol, 5 equiv.),
iPr2NEt (22 μL, 16 mg, 0.13 mmol, 5 equiv.), and ethyl 3-mercaptopropionate (160 μL, 170 mg, 1.3 mmol, 50 equiv.) in
DMF (2 mL) at −30 °C for 3 h. At this point the reaction was quenched through addition of 0.1% TFA in CH2Cl2 (15 mL,
7.5 equiv. TFA) and the reaction mixture was then concentrated in vacuo. A solution of TFA/iPr3SiH/H2O/thioanisole
(85:5:5:5, v/v/v/v, 10 mL) was added to the crude residue at 0 oC and stirred at room temperature for 2 h. The deprotection
mixture was then concentrated under reduced pressure and the crude peptide thioester was precipitated from cold Et2O
(20 mL). The crude peptide thioester was purified by preparative HPLC (0 to 50% B over 100 min, 0.1% TFA) to afford
peptide thioester 15 as the TFA salt (28 mg, 43%). Analytical HPLC: Rt 24.2 min (0 to 40% B over 30 min, 0.1% TFA,
= 230 nm); Calculated Mass [M+2H]2+: 1113.1 (100.0%), [M+3H]3+: 742.4 (100.0%); Mass Found (ESI); [M+2H]2+:
1113.3, [M+3H]3+: 742.7; HRMS (ESI): 1112.5801 calculated for [C97H162N24O33S+2H]2+, found 1112.5804.
S16
S17
19

28
FmocN
O
NH
O
HN
O
NHBoc
N
O
HN
O
NPbfH2N
NH
NH
O
HN
O
N
O
HN
O
NHBoc
NH
O
TrtHN O
HN
O
NH
O
HN
O
NH
O
OtBuHN
O
NH
O
NHBoc
HN
O
NH
O
OtBu
O
HN
O
TrtHN O
NH
OH
N
O
NH
O
OtBu
O
HN
O
NH
O
OtBuHN
O
OtBu
NH
O
O OtBu
HN
O
OtBu
NH
O
NTrt
N
OHH2N
O
O OH
N
O
NH
O
HN
O
NH2
N
O
HN
O
NH2HN
NH
NH
O
HN
O
N
O
HN
O
NH2
NH
O
H2N O
HN
O
NH
O
HN
O
NH
O
OHHN
O
NH
O
NH2
HN
O
NH
O
OH
O
HN
O
H2N O
NH
OH
N
O
NH
O
OH
O
HN
O
NH
O
OHHN
O
OH
NH
O
HO O
HN
O
OH
NH
O
N
NH
MeSS
Cl
Cl
BocHN
O
O OtBu
OH
MeSS
IterativeDeprotectionCoupling
FmocSPPS
i) S18, PyBOP, NMM, DMF, 16hii) TFA/iPr3SiH/H2O (90:5:5, v/v/v)
O
Cl
S19
S18
15
Supplementary Figure 11. Synthesis of peptide 15.
2-Chlorotrityl chloride resin was loaded with Fmoc-His(Trt)-OH according to the general procedures (25 μmol,
0.5 mmol/g loading). Iterative Fmoc-Strategy SPPS was then conducted as outlined in the general procedures to afford
resin-bound protected peptide S18. After N-Fmoc deprotection through treatment with 20% piperidine in DMF, a solution
of protected amino acid S19 (15 mg, 38 μmol, 1.5 equiv.), PyBOP (20 mg, 38 μmol, 1.5 equiv.) and NMM (8.4 μL, 7.7
mg, 76 μmol, 3.0 equiv.) in DMF (0.4 mL, final concentration 0.1 M) was added to the resin and gently agitated. After 16
742.7 [M+3H]3+
1113.3 [M+2H]2+

29
h, the resin was washed with DMF (5 × 5 mL), CH2Cl2 (5 × 5 mL) and DMF (5 × 5 mL). The fully assembled peptide was
then cleaved from the resin with concomitant deprotection of the acid labile protecting groups using a solution of
TFA/iPr3SiH/H2O (90:5:5, v/v/v, 10 mL) for 2 h. The cleavage solution was then concentrated under reduced pressure and
the crude peptide precipitated from cold Et2O (20 mL) before purification by preparative HPLC (0 to 40% B over 40 min,
0.1% formic acid) to afford peptide 15 as the formate salt (23 mg, 30%). Analytical HPLC: Rt 18.2 min (0 to 40% B over
30 min, 0.1% TFA, = 230 nm); Calculated Mass [M+3H]3+: 1030.8 (100.0%), [M+4H]4+: 773.4 (100.0%), [M+5H]5+:
618.9; Mass Found (ESI) [M+3H]3+: 1031.1, [M+4H]4+: 773.6, [M+5H]5+:619.1; HRMS (ESI): 1030.4783 calculated for
[C131H201N39O44S2+3H]3+, found 1030.4786.

30
Synthesis of Chimadanin(21–41) sulfopeptide thioesters bearing N-terminal thiazolidine (16-18)
Supplementary Figure 12. Synthesis of chimadanin thioesters (16-18)
2-Chlorotrityl chloride resin was loaded with Fmoc-Gly-OH according to the general procedures (25 μmol, 0.5 mmol/g
loading). Iterative Fmoc-Strategy SPPS was then carried out as outlined in the general procedures to give fully assembled
resin bound peptides S20-S22, in which Fmoc-Tyr(SO3nP)-OH (21 mg, 38 μmol, 1.5 eq.) was coupled at the appropriate
position(s) using HATU (14 mg, 38 μmol, 1.5 eq.) and iPr2EtN (13 μL, 10 mg, 75 μmol, 3.0 eq.) as activating reagents in
DMF. After removal of the N-Fmoc protecting group with 20% piperidine in DMF, each protected peptide was liberated
from resin using 30% HFIP in CH2Cl2 (15 mL). After concentrated in vacuo, each crude, fully protected peptide was
subsequently treated with PyBOP (65 mg, 0.13 mmol, 5 equiv.), iPr2NEt (22 μL, 16 mg, 0.13 mmol, 5 equiv.), and ethyl
3-mercaptopropionate (160 μL, 168 mg, 1.3 mmol, 50 equiv.) in DMF (2 mL) at −30 °C for 3 h. After this time, each
reaction was quenched with 0.1% TFA in CH2Cl2 (15 ml, 7.5 equiv. of TFA) and the resulting mixture was concentrated
under N2 gas. A solution of TFA/iPr3SiH/H2O/thioanisole (85:5:5:5, v/v/v, 10 mL) was then added to each crude residue
at 0 °C and stirred at room temperature for 2 h. The deprotection mixture was then concentrated in vacuo and precipitated
from ice-cold Et2O (20 mL) to afford the crude peptide thioesters 16, 17 and 18.

31
Chimadanin (21-41) Peptide Thioester 16
Purification of the crude peptide by preparative reverse-phase HPLC (0 to 60% B over 60 min; 0.1% TFA) afforded pure
peptide 16 as a TFA salt after lyophilisation (8.0 mg, 11%). Analytical HPLC: Rt 25.7 min (0 to 50% B over 30 min,
0.1% TFA, = 230 nm); Calculated Mass [M+2H]2+: 1341.0 (100.0%), [M+3H]3+: 894.4 (100.0%); Mass Found (ESI);
[M+2H]2+: 1341.4, [M+3H]3+: 894.7.
25.7 min

32
Chimadanin (21-41) Peptide Thioester 17
Purification of the crude peptide by preparative reverse-phase HPLC (0 to 60% B over 60 min; 0.1% TFA) afforded pure
peptide 17 as a TFA salt after lyophilisation (11 mg, 15%). Analytical HPLC: Rt 22.6 min (0 to 50% B over 30 min, 0.1%
TFA, = 230 nm); Calculated Mass [M+2H]2+: 1341.0 (100.0%), [M+3H]3+: 894.4 (100.0%); Mass Found (ESI);
[M+2H]2+: 1340.9, [M+3H]3+: 894.5.
22.6 min

33
Chimadanin (21-41) Peptide Thioester 18
Purification of the crude peptide by preparative reverse-phase HPLC (0 to 60% CH3CN over 60 min; 0.1% TFA) afforded
pure peptide 18 as a TFA salt after lyophilisation (9.2 mg, 12%). Analytical HPLC: Rt 22.6 min (0 to 70% B over 30 min,
0.1% TFA, = 230 nm); Calculated Mass [M+2H]2+: 1416.1 (100.0%), [M+3H]3+: 944.4 (100.0%); Mass Found (ESI);
[M+2H]2+: 1416.3, [M+3H]3+: 944.8.
22.6 min

34
General procedure for One-pot Ligation-Desulfurization Synthesis of Chimadanin sulfoforms
Supplementary Figure 13. Schematic of the one-pot ligation-desulfurization synthesis of chimadanin sulfoforms 26-28
(Figure 3 in manuscript).

35
A solution of peptide 15 (1.2 equiv.) in ligation buffer (6 M Gn.HCl, 100 mM Na2HPO4, 25 mM TCEP, adjusted to pH
6.8) was added to peptide thioester 16, 17, or 18 (1.0 equiv.). The resulting solution was carefully readjusted to pH 6.8
with 2 M NaOH followed by the addition of TFET (2 vol.%) and the reaction incubated at 30 °C for 2 h. HPLC-MS
analysis indicated complete conversion to the ligated peptide 20, 21 or 22 (see Supplementary Fig. 14-16). At this point,
the reaction mixture was diluted with 120 µL of an aqueous solution of 0.2 M MeONH2.HCl in 6 M Gn.HCl, 100 mM
Na2HPO4, pH = 3.7) to achieve a final pH of 4.2. The solution was incubated at 30 °C and after 3 h, HPLC-MS analysis
indicated the majority of peptides 20, 21 or 22 had been converted to peptides S23, S24 or S25 respectively bearing an N-
terminal cysteine residue (see Supplementary Fig. 14-16). The reaction mixture was then readjusted to pH 6.8 through
careful addition of 2 M NaOH, followed by the addition of a solution of peptide 19 (1.3 equiv.) in buffer (6 M guanidine
HCl, 100 mM Na2HPO4, 150 µL) and TFET (2 vol%). After 18 h at 30 °C, HPLC-MS analysis showed a complete
conversion to the corresponding ligation product 23, 24 or 25 (see Supplementary Fig. 14-16). A neutral solution of TCEP
(0.5 M) in buffer (6 M guanidine HCl, 100 mM Na2PO4, 500 µL) was then added to give a 200 mM concentration of
TCEP and a 0.5 mM final concentration of the ligation product. The resulting solution was adjusted to pH 6.2-6.5 and
then degassed by sparging with argon for 10 min which also removed the excess TFET from the reaction mixture.
Glutathione (40 mM), and VA–044 (20 mM) were then added in solid form and the reaction mixture was gently agitated
and incubated at 37 °C for 5 h. After this time, HPLC-MS analysis showed a complete conversion to each chimadanin
sulfoform 26, 27 or 28 (see Supplementary Fig. 14-16).

36
Chimadanin-sY32 (26)
Chimadanin sulfoform 26 was assembled from peptide thioester 16 (1.1 mg, 0.38 μmol), peptide thioester 19 and peptide
15 according to the general procedure, monitoring the progress using HPLC-MS (Supplementary Fig. 14). Following the
assembly of 26, the reaction mixture was purified by reversed-phase semi-preparative HPLC (0 to 60% CH3CN over 60
min, 0.1 M aqueous NH4OAc solution/non-buffered CH3CN) to afford 26 as the ammonium salt (1.0 mg, 35% based on
the starting peptide 16, 1.1 mg, 0.38 µmol). Analytical HPLC: Rt 21.3 min (0 to 40% B over 30 min, 0.1% formic acid,
= 214 nm); Calculated Mass [M+4H]4+: 1884.4, [M+5H]5+: 1507.7, [M+6H]6+: 1256.6, [M+7H]7+: 1077.2 (100.0%),
[M+8H]8+: 942.70 (100.0%); Mass Found (ESI) [M+4H]4+: 1885.0, [M+5H]5+: 1508.1, [M+6H]6+: 1256.9, [M+7H]7+:
1077.4, [M+8H]8+: 942.8, HRMS (MALDI): 3768.2710 (100%) calculated for [C322H497N89O116S2 + 2H]2+, found
3768.2387.

37

38
Supplementary Figure 14. A) Crude HPLC-MS analysis of the ligation reaction between the C-terminal segment 15 and
peptide thioester 16 to give the corresponding ligation product 20 (λ = 230 nm). i) Mass calculated for 20 (100% relative
abundance) [M+5H]5+: 1104.9, [M+6H]6+: 920.9, Mass found (ESI) [M+5H]5+:1105.2, [M+6H]6+: 921.2. B) Crude HPLC-
MS analysis of the thiazolidine deprotection reaction of 20, to give the product S23 (λ = 230 nm). Mass calculated for S23
(100% relative abundance) [M+5H]5+: 1102.5, [M+6H]6+: 918.9, Mass found (ESI) [M+5H]5+: 1102.7, [M+6H]6+: 919.2.
C) Crude HPLC-MS analysis of the ligation reaction between the N-terminal segment 19 and deprotected product S23 to
give the corresponding ligation product 23 (λ = 230 nm). Mass calculated for 23 (100% relative abundance) [M+7H]7+:
1086.4, [M+8H]8+: 950.7, [M+9H]9+: 845.2 Mass found (ESI) [M+7H]7+: 1086.7, [M+8H]8+ : 950.9, [M+9H]9+: 845.5; D)
Crude HPLC-MS analysis of the in situ desulfurization reaction of ligation products 23 after 3 h to give the corresponding
chimadanin sulfoform 26. Mass calculated for 26 (100% relative abundance) [M+6H]6+: 1256.6, [M+7H]7+: 1077.2,
[M+8H]8+: 942.7, [M+9H]9+: 838.1, Mass found (ESI) [M+6H]6+: 1256.8, [M+7H]7+: 1077.5, [M+8H]8+: 942.9,
[M+9H]9+: 840.2.
Chimadanin-sY35 (27)
Chimadanin sulfoform 27 was assembled from peptide thioester 17 (1.0 mg, 0.34 μmol), peptide thioester 19 and peptide
15 according to the general procedure, monitoring the progress using HPLC-MS (Supplementary Fig. 15). Following the
assembly of 27, the reaction mixture was purified by reversed-phase semi-preparative HPLC (0 to 60% CH3CN over 30
min, 0.1 M aqueous NH4OAc solution/non-buffered CH3CN) to afford 27 as the ammonium salt (1.5 mg, 55% based on
the starting peptide 17, 1.0 mg, 0.36 µmol). Analytical HPLC: Rt 19.2 min (0 to 40% B over 30 min, 0.1% TFA, = 214
nm); Calculated Mass [M+6H]6+: 1256.6 (100%), [M+7H]7+: 1077.2 (100.0%), [M+8H]8+: 942.70 (100.0%), [M+9H]9+:
838.1 (100.0%); Mass Found (ESI) [M+6H]6+: 1258.4, [M+7H]7+: 1078.3, [M+8H]8+: 943.4, [M+9H]9+: 838.1; HRMS
(MALDI): 3768.2700 (100%) calculated for [C322H497N89O116S2 + 2H]2+, found 3769.2710.

39
19.2 min
838.1
[M+9H]9+
829.5
[M-SO3+9H]9+
943.4
[M+8H]8+
1078.3
[M+7H]7+
1258.4
[M+6H]6+
746.4
[M-SO3+10H]10+

40

41
Supplementary Figure 15. A) Crude HPLC-MS analysis of the ligation reaction between the C-terminal segment 15 and
peptide thioester 17 to give the corresponding ligation product 21 (λ = 230 nm). Mass calculated for 21 (100% relative
abundance) [M+5H]5+: 1104.9, [M+6H]6+: 920.9, [M+7H]7+: 789.5, Mass found (ESI) [M+5H]5+: 1105.2, [M+6H]6+:
921.2, [M+7H]7+: 789.8. B) Crude HPLC-MS analysis of the thiazolidine deprotection reaction of 21, to give the product
S24 (λ = 230 nm). Mass calculated for S24 (100% relative abundance) [M+4H]4+: 1377.8, [M+5H]5+: 1102.5, [M+6H]6+:
918.9, [M+7H]7+: 787.8, Mass found (ESI) [M+4H]4+: 1378.2, [M+5H]5+: 1102.9, [M+6H]6+: 919.2, [M+7H]7+: 788.0., C)
Crude HPLC-MS analysis of the ligation reaction between the N-terminal segment 19 and deprotected product S24 to give
the corresponding ligation product 24 (λ = 230 nm). Mass calculated for 24 (100% relative abundance) [M+7H]7+: 1086.4,
[M+8H]8+: 950.7, [M+9H]9+: 845.2, Mass found (ESI) [M+7H]7+: 1086.6, [M+8H]8+: 950.9, [M+9H]9+: 845.4. D) Crude
HPLC-MS analysis of the in situ desulfurization reaction of ligation product 24 after 3 h to give the corresponding
chimadanin sulfoform 27. Mass calculated for 27 (100% relative abundance) [M+6H]6+: 1256.6, [M+7H]7+: 1077.2,
[M+8H]8+: 942.7, [M+9H]9+: 838.1, Mass found (ESI) [M+6H]6+: 1258.4, [M+7H]7+: 1078.3, [M+8H]8+: 943.4,
[M+9H]9+: 838.0.
Chimadanin-sY28, sY31 (28)
Chimadanin sulfoform 28 was assembled from peptide thioester 18 (1.3 mg, 0.43 μmol), peptide thioester 19 and peptide
15 according to the general procedure, monitoring the progress using HPLC-MS (Supplementary Fig. 16). Following the
assembly of 28, the reaction mixture was purified by reversed-phase semi-preparative HPLC (0 to 60% CH3CN over 60
min, 0.1 M aqueous NH4OAc solution/non-buffered CH3CN) to afford 28 as the ammonium salt (1.0 mg, 31% based on
the starting peptide 18, 1.3 mg, 0.42 µmol). Analytical HPLC: Rt 19.0 min (0 to 40% B over 30 min, 0.1% TFA, = 214
nm); Calculated Mass [M+4H]4+: 1904.4 (100%), [M+5H]5+: 1523.7 (100%), [M+6H]6+: 1269.9 (100%), [M+7H]7+:
1088.6 (100.0%), [M+8H]8+: 952.7 (100.0%); Mass Found (ESI) [M+4H]4+: 1904.9, [M+5H]5+: 1524.0 [M+6H]6+ 1270.1,
[M+7H]7+: 1088.8, [M+8H]8+: 952.8; HRMS (MALDI): 3808.2493 (100%) calculated for [C322H497N89O119S3 + 2H]2+,
found 3808.2173.

42
19.0 min

43
Supplementary Figure 16. A) Crude HPLC-MS analysis of the ligation reaction between the C-terminal segment 15 and
peptide thioester 18 to give the corresponding ligation product 22 (λ = 230 nm). Mass calculated for 22 (100% relative
abundance) [M+7H]7+: 800.9, Mass found (ESI) [M+7H]7+: 801.3. B) Crude HPLC-MS analysis of the thiazolidine
deprotection reaction of 22, to give the product S25 (λ = 230 nm). Mass calculated for S25 (100% relative abundance),

44
[M+4H]4+: 1397.8, [M+5H]5+: 1118.5, [M+6H]6+: 932.2, Mass found (ESI) [M+4H]4+: 1397.8, [M+5H]5+: 1118.9,
[M+6H]6+: 932.5. C) Crude HPLC-MS analysis of the ligation reaction between the N-terminal segment 19 and
deprotected product S25 to give the corresponding ligation product 25 (λ = 230 nm). Mass calculated for 25 (100%
relative abundance), [M-4H]4-: 1918.3, Mass found (ESI) [M-4H]4-: 1918.8. NB: the ESI spectrum of crude 25 in positive
mode led to large amounts of desulfated ions. D) Crude HPLC-MS analysis of the in situ desulfurization reaction of
ligation products 25 after 3 h to give the corresponding chimadanin sulfoform 28. Mass calculated for 28 (100% relative
abundance) [M+6H]6+: 1269.9, [M+7H]7+: 1088.6, [M+8H]8+: 952.7, [M+9H]9+: 846.9; Mass Found (ESI) [M+6H]6+:
1270.4, [M+7H]7+: 1089.1, [M+8H]8+: 953.0, [M+9H]9+: 847.1.
Loading
IterativeDeprotectionCouplingCapping
FmocSPPS
Deprotection:TFA/iPr3SiH/H2O(95:5:5 v/v/v)
O
O
O
BocHN
O
OtBu
O
NH
O
HN
O
OtBu
O
NH
O
O
HN
O
OtBu
O
NH
O
O OtBu
HN
O
O
NH
O
O OtBu
HN
O
O OtBu
NH
O
OtBu
O
HN
O
NH
O
OtBu
H
OtBu
OHN
O
NH
OtBu
OHN
N
O
NH
O
OtBu
N
O
NH
O
OtBu
H
HN
O
NHBoc
O
NH
OHN
H
OtBu
O
NH
O
NN
O
HN
O
NH
HN
NH
S OO
O
S OO
O
TmbS
FmocHN
O
NH
HN
NH
O
HO
H
Pbf
FmocHNOH
O
OS
O O
O
BocHNOH
O
OtBu
OSTmob
Fmoc-Tyr(SO2OCH2C(CH3)3)-OH Boc-Asp(STmob,OtBu)-OH
Pbf
O
O
29 54
O
H2N
O
OH
O
NH
O
HN
O
OH
O
NH
O
O
HN
O
OH
O
NH
O
O OH
HN
O
O
NH
O
O OH
HN
O
O OH
NH
O
OH
O
HN
O
NH
O
OH
H
OH
OHN
O
NH
OH
OHN
N
O
NH
O
OH
N
O
NH
O
OH
H
HN
O
NH2
O
NH
OHN
H
OH
O
NH
O
NN
O
HN
O
NH2HN
NH
S OO
O
S OO
O
HS
HOH
S26
S27
29 54
Supplementary Figure 17. Synthesis of peptide S27 through Fmoc-SPPS.
Protected peptide S26 corresponding to madanin-1 (29-54) was synthesized on Wang resin (62.5 μmol, 0.5 mmol/g
loading) using standard Fmoc-SPPS as outlined in the general procedures. The fully assembled peptide was then cleaved
from the resin with concomitant removal of the acid labile protecting groups using a solution of TFA/iPr3SiH/H2O
(90:5:5, v/v/v, 10 mL) for 2 h. The cleavage solution was then concentrated to ~1 mL and precipitated from cold Et2O (45
mL) before purification by preparative HPLC (20 to 60% B over 40 min, 0.1% TFA) to afford peptide S27 as the TFA salt
(36 mg, 16%). Analytical UPLC: Rt 4.0 min (20 to 80% B over 5 min, 0.1% TFA, λ = 214 nm). Calculated Mass
[M+2H]2+: 1593.1, [M+3H]3+: 1062.4, [M+4H]4+: 797.1; Mass found (ESI) [M+2H]2+: 1594.6, [M+3H]3+: 1063.3,
[M+4H]4+: 797.5.

45

46
Supplementary Figure 18. Schematic of the one-pot ligation-desulfurization synthesis of madanin-1 (1-54) S29
A solution of peptide thioester S5 (3.6 mol, 15.4 mg, 1.1 equiv.) in ligation buffer (650 L, 6 M Gn.HCl, 100 mM
Na2HPO4, 25 mM TCEP, adjusted to pH 7.2) was added to madanin-1 (29-54) S27 (3.3 mol, 11.5 mg, 1.0 equiv.). The
resulting solution was carefully readjusted to pH 7.0 with 2 M NaOH followed by the addition of TFET (13 L, 2 vol.%)
and the reaction incubated at 30 °C for 2 h 40 min. HPLC-MS analysis indicated complete conversion to the ligated
peptide S28 (see Supplementary Fig. 19). A solution of TCEP (0.5 M) and glutathione (0.2 M) in buffer (100 mM
Na2PO4, 6 M Gn.HCl, pH 6.1, 650 µL) was then added to give a 0.25 M concentration of TCEP, 0.1 M concentration of
glutathione and a 2.5 mM final concentration of the ligation product. The resulting solution was adjusted to pH 6.5 and
then degassed by sparging with Ar(g) for 10 min which also removed the excess TFET from the reaction mixture. VA–044
(2.1 mg, 5 mM) were then added in solid form and the reaction mixture was gently agitated and incubated at 37 °C for 16
h. After this time, HPLC-MS analysis showed a complete conversion to madanin-1 (1-54) S29 (see Supplementary Fig.
19).

47
Supplementary Figure 19. A) Crude UPLC-MS analysis of the ligation reaction between madanin 29-54 fragment S27
and peptide thioester S5 to give the corresponding ligation product S28 (λ = 230 nm). Mass calculated for S28 (100%
relative abundance) [M+4H]4+: 1574.4, [M+5H]5+: 1260.0, [M+6H]6+: 1050.1, [M+7H]7+: 900.3, [M+8H]8+: 787.9,
[M+9H]9+: 700.4; Mass found (ESI) [M+4H]4+: 1576.1, [M+5H]5+: 1261.1, [M+6H]6+: 1051.1, [M+7H]7+: 900.9,
[M+8H]8+: 788.3, [M+9H]9+: 700.8. B) Crude UPLC-MS analysis of the in situ desulfurization reaction of ligation product
S28 after 16 h to give the corresponding madanin (1-54) S29. Mass calculated for S29 (100% relative abundance)
[M+4H]4+: 1566.7, [M+5H]5+: 1253.6, [M+6H]6+: 1044.8, [M+7H]7+: 895.7, [M+8H]8+: 783.9, [M+9H]9+: 696.9; Mass
found (ESI) [M+4H]4+: 1568.2, [M+5H]5+: 1254.6, [M+6H]6+: 1045.6, [M+7H]7+:896.2, [M+8H]8+: 784.3, [M+9H]9+:
697.2.
Madanin-1 (1-54)-sY32, sY35 (S29)
Following the assembly of S29, the crude product was purified using semipreparative HPLC (0 to 30% B over 30 min,
0.1% TFA), affording S29 as a white solid after lyophilisation (14.6 mg, 61% yield from S29). Analytical UPLC: Rt 3.6

48
min (0 to 30% B over 5 min, 0.1% TFA, λ = 230 nm). Calculated Mass [M+4H]4+: 1566.7, [M+5H]5+: 1253.6, [M+6H]6+:
1044.8, [M+7H]7+: 895.7, [M+8H]8+: 783.9, [M+9H]9+: 696.9; Mass found (ESI) [M+4H]4+: 1567.9, [M+5H]5+: 1254.6,
[M+6H]6+: 1045.6, [M+7H]7+:896.3, [M+8H]8+: 784.3, [M+9H]9+: 697.3. HRMS (MALDI-TOF) m/z calculated for
C257H399N76O103S2+ [M+H]+: 6263.782, found 6264.983.

49
Screening for selectivity of serine protease inhibition
The inhibition activity of synthetic disulfated madanin-1 (1000-fold molar excess) was tested against α-thrombin
(Haematologic Technologies), coagulation factor Xa (New England Biolabs), trypsin (Sigma-Aldrich), and pancreatic
elastase (Sigma-Aldrich) using 100 µM of specific chromogenic substrate (Tos-Gly-Pro-Arg-p-nitroanilide (Chromozym
TH; Roche) for thrombin and trypsin; N-methoxycarbonyl-D-Cha-Gly-Arg-p-nitroanilide (Pefachrome FXa; Pentapharm)
for factor Xa, and N-Succinyl-Ala-Ala-Ala-p-nitroanilide (Sigma-Aldrich) for elastase). Trypsin and factor Xa (0.1 and
0.5 nM, respectively) were assayed in 50 mM Tris pH 8.0, 100 mM NaCl, 1 mg/ml bovine serum albumin at 37 °C. In the
case of factor Xa, 2mM CaCl2 was also added to the reaction mixture. The activity of elastase (10 nM) was measured in
50 mM Tris pH 8.0, 50 mM NaCl, 10 % (vol/vol) DMSO, 1 mg/ml bovine serum albumin at 25 °C. Reaction conditions
for thrombin are detailed in the specific section. All reactions were started by enzyme addition and monitored at 405 nm
for 30 minutes, or 10 minutes in the case of elastase and factor Xa, on a Synergy2 multi-mode microplate reader (BioTek).
The substrate hydrolysis rates of control reactions in the absence of madanin-1 were normalized to 100 %.
Supplementary Figure 20. Madanin-1 is a specific thrombin inhibitor. The inhibitory activity of disulfated madanin-1
(1000-fold molar excess) on the hydrolytic activity of four different serine proteinases was determined. Activity is
expressed as percentual residual hydrolytic activity, compared to controls in the absence of madanin-1. Error bars
represent SD of two independent experiments.
Thrombin amidolytic activity assay
The amidolytic activity of human α-thrombin (Haematologic Technologies) was followed spectrophotometrically using
Tos-Gly-Pro-Arg-p-nitroanilide (Chromozym TH; Roche) as chromogenic substrate. Experimental K0.5 (app) kinetic
constants were determined for human α or γ-thrombin (both at 0.2 nM) at substrate concentrations from 0 to 450 µM in 50
mM Tris pH 8, 50 mM NaCl, 1 mg/ml bovine serum albumin. Inhibition assays were performed in the same buffer with
0.14 nM human α-thrombin, 100 μM substrate and varying concentrations (0-29 µM) of inhibitor. For human γ-thrombin
(0.2 nM), inhibition assays were performed in the presence of 0-2 µM madanin-1 or 0-10.5 µM chimadanin. Inhibition
constants (Ki) were determined according to a tight-binding inhibitor model, using the Morrison equation5 with Prism
(GraphPad Software). In the case of disulfated madanin-1 (22-54) and (1-54) fragments, a tight-binding inhibitor model

50
could not be applied. Inhibition assays for human α-thrombin (0.5 nM) were performed at increasing substrate
concentrations (0-400 μM) and different concentrations of inhibitor (0-20 nM). Ki values were determined by fitting the
data to a competitive inhibition model (Supplementary Fig. 21). For the inhibitors produced in baculovirus-infected insect
cells, 50 μl of culture supernatant (undiluted and 5- and 10-fold dilutions) were used. The culture supernatant of cells
transfected with an empty bacmid was used as control (Supplementary Fig. 22). All reactions were initiated by addition of
thrombin and were carried out at least in duplicate at 37 °C in 96-well microtiter plates. Reaction progress was monitored
at 405 nm for 30 minutes on a Synergy2 multi-mode microplate reader (BioTek).
Supplementary Figure 21. Dose-response curves for the hydrolysis of chromogenic substrate (0 to 400 μM) in the
presence of increasing concentrations (0 to 20 nM) of A) synthetic disulfated madanin-1 (22-54) fragment or B) disulfated
madanin-1 (1-54) fragment. Kinetic parameters of human α-thrombin inhibition (Ki ± SEM) given are representative of
two independent experiments. Data were fitted to a competitive inhibition model using Prism (GraphPad Software).
Supplementary Figure 22. Inhibition of the amidolytic activity of human α-thrombin by madanin-1 and chimadanin
produced in an insect cell expression system. The amidolytic activity of human α-thrombin was measured in the presence
of three different dilutions of insect cell culture supernatants of Trichopulsia ni High Five insect cells transfected with
empty pKL acceptor vector (control), pKL-madanin-1 or pKL-chimadanin vectors. The activity of human α-thrombin in
the absence of supernatant media and in the same experimental conditions was considered as 100 %. Error bars represent

51
SD of two (for pKL-madanin-1or pKL-chimadanin) or six (for pKL) replicates.
Thrombin Time (TT) Assay
The anticoagulant activities of madanin-1 and chimadanin were determined by measuring their ability to prolong clotting
of human plasma. Human plasma (800 µl) was mixed with 200 µl of synthetic madanin-1 or synthetic chimadanin
solutions (0-5 µM final concentration in 20 mM Tris-HCl pH 8.0, 50 mM NaCl), and TT was measured at a commercial
diagnostics laboratory (BM Análises Clínicas) following standard protocols.
Cleavage of unsulfated and disulfated madanin-1 by human α-thrombin
The hydrolytic cleavage of synthetic unsulfated (S30) or disulfated madanin-1 (14) by thrombin between K21-V22 and
between R54-L55 was investigated as described for other thrombin substrates previously.12 Briefly, S30 or 14 (50 µM)
was incubated with human α-thrombin (1 µM) in 20 mM Tris-HCl pH 8.0, 150 mM NaCl at 37 °C (Supplementary Fig.
23). At specific time points, the reaction was analyzed directly by RP-HPLC. A fast amide cleavage between residues R54
and L55 of S30 (unsulfated) afforded fragments S31 and S32, while cleavage of 14 (disulfated) afforded S29 and S32. A
subsequent slow cleavage reaction between residues K21 and V22 of S31 (unsulfated) afforded fragments S33 and S34,
while slow cleavage of S29 (disulfated) afforded fragments S33 and S15. The relative proportions of amide bond cleavage
from both proteolysis events was quantified at different times through peak integration at λ = 280 nm (UV absorption by
free tyrosine residues Y1, Y32 and Y35) of intact inhibitors and peptide fragments (Supplementary Fig. 24).
Absorption at λ = 280 nm by sulfotyrosine residues in 14, S29 and S15 was negligible compared to free tyrosine and thus
fragment S15 containing no free tyrosine was omitted from these calculations.

52
OH OHH
OH H OH
YPERDSAKEGNQEQERALHVKVQKRTDGDADYDEYEEDGTTPTPDPTAPTAKPR-LRGNKPH
OR2
OR1
S30: R1,R2 = H14: R1,R2 = SO3
351
32
60
YPERDSAKEGNQEQERALHVK-VQKRTDGDADYDEYEEDGTTPTPDPTAPTAKPRLRGNKPRH
OR2
OR1
S31: R1,R2 = HS29: R1,R2 = SO3
351
32
60LRGNKP
5554
5554
FASTCleavage
VQKRTDGDADYDEYEEDGTTPTPDPTAPTAKPRLRGNKPR
OR2
OR1
35
32
54YPERDSAKEGNQEQERALHVKH
S34: R1,R2 = HS15: R1,R2 = SO3
1
OH
Thrombin (1 M)/20 mM Tris.HCl pH 8.0, 150 mM NaCl, 37 °C
21 22
H OH60
LRGNKP55
SLOWCleavage
21 22
S32
S33
Supplementary Figure 23. Schematic for the processing of unsulfated (S30) and disulfated madanin-1 (14) by thrombin
to give fragments S33, S32 and either S34 or S15.
Supplementary Figure 24. Time courses of thrombin-mediated proteolysis of unsulfated and disulfated madanin-1
between A) R54-L55 and B) K21-V22, as determined by RP-HPLC.

53
Crystallization of a disulfated madanin-1•human α-thrombin complex
Human α-thrombin (Haematologic Technologies) was mixed in 20 mM HEPES pH 7.5, 125 mM NaCl with a six-fold
molar excess of disulfated madanin-1 (22-54) fragment (S15) and incubated on ice for one hour. The resulting complex
was concentrated by ultrafiltration using a 2 kDa cut-off centrifugal device (Sartorius). An initial crystallization screen at
20 °C was performed at the High Throughput Crystallization Laboratory of the European Molecular Biology Laboratory
(Grenoble, France). Preliminary crystallization conditions were optimized in-house until single crystals belonging to the
orthorhombic space group P212121 were obtained after 8-10 days using the vapor diffusion sitting-drop method from drops
consisting of equal volumes (1 μL) of protein complex (at 5.5 mg/mL) and precipitant solution (0.1 M MMT pH 7, 30%
(wt/vol) PEG 1500) equilibrated against a 300 μL reservoir.
Data collection and processing
Crystals were cryoprotected by immersing them briefly in precipitant solution with a higher content (32% (wt/vol)) of
PEG 1500 and flash-cooled in liquid nitrogen. Diffraction data (2700 images in 0.05ᵒ oscillation steps and 0.02 s
exposure) were recorded on a Pilatus 6M detector (Dectris) using a wavelength of 0.9686 Å at beam line ID296 of the
European Synchrotron Radiation Facility (Grenoble, France). Data were integrated with XDS7 and reduced with utilities
from the CCP4 program suite.8 Data collection statistics are summarized in Supplementary Table 3. The diffraction
dataset9 was deposited with the Structural Biology Data Grid.10
Structure determination and refinement
The structure of the complex was solved by molecular replacement with Phaser11 using the coordinates of free wild-type
human α-thrombin (PDB entry 3U6912). Alternating cycles of model building with Coot13 and refinement with PHENIX14
were performed until model completion (refinement statistics are summarized in Supplementary Table 3). All
crystallographic software was supported by SBGrid.15 Refined coordinates and structure factors were deposited at the
PDB with accession number 5L6N.

54
Supplementary Table 3 – Crystallographic data processing and refinement statistics.
Data Collection
Beamline ESRF ID29
Wavelength (Å) 0.9686
Resolution range (Å) 44.55 - 1.63 (1.71 - 1.63)
Space group P212121
Unit cell dimensions (Å) a=44.5 b=80.2 c=91.4
Reflections (measured/unique) 204982/41771 (29537/5967)
Multiplicity 4.9 (5.0)
Completeness (%) 99.6 (98.5)
Mean (I) / σ (I) 13.6 (1.80)
Wilson B-factor 21.8
Rmerge (%) 6.4 (82.6)
CC1/2 0.999 (0.493)
Complexes per asymmetric unit 1
Matthews coefficient (Å3 Da-1)
Solvent content (%)
2.21
44.3
Refinement
Rwork 0.167
Rfree 0.199
Non-hydrogen macromolecule
atoms
2499
Non-hydrogen ligand atoms 15
Water molecules 233
Protein residues 308
r.m.s.d bond lengths (Å) 0.009
r.m.s.d bond angles (°) 0.90
Ramachandran favored (%) 97
Ramachandran outliers (%) 0
* Values in parentheses correspond to the outermost resolution shell.

55
References
1. Fitzgerald, D. J., Berger, P., Schaffitzel, C., Yamada, K., Richmond, T. J. & Berger, I. Protein complex expression by using multigene baculoviral vectors. Nat. Methods 3, 1021-1032 (2006).
2. Bern, M., Kil, Y. J. & Becker, C. Byonic: advanced peptide and protein identification software. Curr. Prot. Bioinform., 13.20. 11-13.20. 14 (2012).
3. Thompson, R. E., Liu, X., Alonso-García, N., Pereira, P. J. B., Jolliffe, K. A. & Payne, R. J. Trifluoroethanethiol: an additive for efficient one-pot peptide ligation− desulfurization chemistry. J. Am. Chem. Soc. 136, 8161-8164 (2014).
4. Thompson, R. E., Chan, B., Radom, L., Jolliffe, K. A. & Payne, R. J. Chemoselective peptide ligation–desulfurization at aspartate. Angew. Chem. Int. Ed. 52, 9723-9727 (2013).
5. Williams, J. W. & Morrison, J. F. The kinetics of reversible tight-binding inhibition. Methods Enzymol. 63, 437-467 (1979).
6. de Sanctis, D., et al. ID29: a high-intensity highly automated ESRF beamline for macromolecular crystallography experiments exploiting anomalous scattering. J. Synchrotron Radiat. 19, 455-461 (2012).
7. Kabsch, W. XDS. Acta Crystallogr. Sect. D: Biol. Crystallogr. 66, 125-132 (2010).
8. Winn, M. D., et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D: Biol. Crystallogr. 67, 235-242 (2011).
9. Ripoll-Rozada, J. & Pereira, P. J. B. X-Ray Diffraction data for: Human thrombin-sulfated madanin complex. PDB Code 5LN6. Structural Biology Data Grid, V1, http://dx.doi.org/10.15785/SBGRID/15324) (2016).
10. Meyer, P. A., et al. Data publication with the structural biology data grid supports live analysis. Nat. Commun. 7, 10882 (2016).
11. McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658-674 (2007).
12. Figueiredo, A. C., Clement, C. C., Zakia, S., Gingold, J., Philipp, M., & Pereira, P. J. B. Rational design and characterization of D-Phe-Pro-D-Arg-derived direct thrombin inhibitors. PLoS ONE 7, e34354 (2012).
13. Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta. Crystallogr. D Biol. Crystallogr. 66, 486-501 (2010).
14. Adams, P. D., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta. Crystallogr. D Biol. Crystallogr. 66, 213-221 (2010).
15. Morin, A., et al. Collaboration gets the most out of software. Elife 2, e01456 (2013).