identification of the proximate and ultimate forms of the...
TRANSCRIPT

(CANCER RESEARCH 39. 4160-4165. October 1979]0008-54 72 / 79/0039-OOOOS02.00
Identification of the Proximate and Ultimate Forms of the Carcinogen15,16-Dihydro-11 -methylcyclopentaCaJphenanthren-l 7-one
Maurice M. Coombs,1 Anna-Maija Kissonerghis, Jeffrey A. Allen,2 and Colin W. Vose
Chemistry Laboratory. Imperial Cancer Research Fund. Lincolns Inn Fields. London. WC2A 3PX. England ¡M.M. C. A-M. K.. J. A. A.¡,and Department ofMetabolic Studies. G D. Searle and Co.. Ltd., Lane End Road. High Wycombe. HP124HL. Buckinghamshire. England ¡C.W. V.¡
ABSTRACT
Enzymatic hydrolysis of calf thymus DNA treated in vitro withthe carcinogen 15,16-dihydro-11-methylcyclopenta[a]phen-anthren-17-one and a microsomal enzyme system followed bycolumn chromatography disclosed two adduci fractions, A andB, eluting after the natural nucleosides. Isolation and hydrolysisof the DNA from the skin of mice treated topically with 15,16-dihydro-11-methylcyclopenta[a]phenanthren-1 7-one or frommouse embryo cells exposed to 15,16-dihydro-11-methylcy-clopenta[a]phenathren-1 7-one in culture gave mainly Adduci
B identical with that obtained in vitro. The seven main metabolites formed from 15,16-dihydro-11-methylcyclopenta[a]phenanthren-1 7-one with the microsomal system were isolatedby high-pressure liquid chromatography and were individuallyincubated with DNA and the activating system; the DNA wassubsequently recovered and analyzed. Adduct B arose from asingle metabolite identified on the basis of its ultraviolet andmass spectra together with its general chemical behavior as a3,4-dihydro-frans-3,4-dihydroxy derivative of 15,16-dihydro-1l-methylcyclopenta[a]phenanthren-1 7-one. In addition, this
metabolite was the most mutagenic in the Ames test, beingmore active than the carcinogen itself. This metabolite is therefore the proximate form of the carcinogen.
The ultraviolet spectrum of Adduct B resembled that of a1,2,3,4-tetrahydro derivative of 15,16-dihydro-11-methylcy-clopenta[a]phenanthren-17-one, while the Chromatographiemobility of the adduci on Sephadex LH-20 was markedlyincreased by inclusion of borate in the eluant, indicating a cis-diol system. Since the 3,4-diol is frans, this suggests furthermetabolism of the benzo ring. It is therefore proposed that theultimate carcinogen is a 1,2-dihydro-1,2-epoxy-3,4-dihydro-frans-3,4-diol of 15,16-dihydro-11-methylcyclopenta[a]phen-anthren-1 7-one.
INTRODUCTION
After metabolic activation, the strong carcinogen 15,16-di-hydro-11-methylcyclopenta[a]phenanthren-17-one (Structure
I) (9) (Chart 1) binds to DNA in vitro (16) and is mutagenic toSalmonella typhimurium TA 100 (13). Both these activities aresuppressed by the aryl hydrocarbon hydroxylase inhibitor 7,8-benzoflavone, which strongly inhibits formation of metabolitesderived from enzymatic attack on the aromatic rings, but notthose produced by hydroxylation at saturated carbon atoms(C-1 5 and C-16 méthylènegroups and 11-methyl group) (16).
Skin tumor production by topical application of this carcinogen(Structure I) is also reduced by a coadministration of 7,8-
1To whom requests for reprints should be addressed.' Recipient of a bursary award from the Imperial Cancer Research Fund.
Received March 12, 1979; accepted June 11, 1979.
benzoflavone (11). Taken together with the observed correlation between mutagenicity and carcinogenicity in the cyclo-penta[a]phenanthrene series (13), this suggests that the metabolite responsible for the carcinogenic activity of Structure Iis derived from the aromatic part of this molecule. The ability ofStructure I to bind to DNA in vitro after metabolic activationdecreases rapidly under ambient conditions and is lost immediately on acidification, suggesting that the active metabolite isprobably an aryl oxide (16). This paper describes experimentswhich have led to its identification as a 3,4-dihydroxy-1,2-epoxide of Structure I. A preliminary repo'l has appeared (10).
MATERIALS AND METHODS
Labeled Compounds. 11-mefriy/-1'lC-labeled carcinogen
(Structure I) (9.3 mCi/mmol) was synthesized by the methoddescribed previously (15). Generally tritiated Structure I (13.9Ci/mmol) was prepared by platinum metal-catalyzed exchangein 70% [3H]acetic acid (6). Tritium nuclear magnetic resonance
revealed that, with the exception of C-1 and C-16, all positionswere labeled as follows: C-2 and C-3, together, 23.6; C-4,18.2; C-6, 8.2; C-7, 4.5; C-12, 3.1; C-15, 16.4; 11-methyl,
25.5%. The structures of these compounds are shown in Chart1.
Enzymes. DNase I (DN-CL), snake venom phosphodiester-ase type II (P-6877), alkaline phosphatase (Escherichia coli)type III (P-4252), and RNase A (R5125) were obtained from
Sigma, London, England. Proteinase K (fungal) and NADPHwere from British Drug Houses, Poole, Dorset, England.
In Vitro Experiments with DNA. Binding of the carcinogen(Structure I) and its isolated metabolites to DNA in vitro andenzymatic hydrolysis of the DNA followed by separation of itsconstituents by chromatography on Sephadex LH-20 was car
ried out as described previously (16).Isolation of DNA from Mouse Skin. Male TO (Tyler s Origi
nal) mice, 7 to 9 weeks old, were used in groups of 15. Theirbacks were shaved with electric clippers, and 24 hr later G-3H-
labeled Structure I (400 /ig in 40 /il toluene) was applied toeach mouse; the area covered was approximately 2.5 x 2.5cm. Groups of mice were killed by cervical dislocation atselected times after treatment (Chart 3); the treated area ofskin was removed and frozen in liquid nitrogen in an earthenware mortar. The frozen skin fragments were ground to apowder by hand, using a pestle, and added in small portions toa lysing solution of proteinase K at 50°according to the general
method of Blin and Stafford (2) for isolation of DNA. Thisconsists of solubilization of the tissue, isolation of the nucleicacids by phenol extractions, reincubation with RNase, andreisolation of the DNA by further phenol extractions. The DNAsolution thus obtained was exhaustively dialyzed against tapwater and finally against distilled water before concentrationfor enzymatic hydrolysis.
4160 CANCER RESEARCH VOL. 39
on April 2, 2020. © 1979 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

Proximate-Ultimate Forms of a Cyclopenta[a]phenanthrene
12
Chart 1. Structures of the labeled forms of the carcinogen (Structure I) used.
Isolation of DNA from Mouse Embryo Cells. Secondarywhole-mouse-embryo cells were seeded at 5 x 106 cells/150-
mm tissue culture dish (Falcon Plastics, Oxnard, Calif.) inDulbecco's modification of Eagle's medium containing 10%
fetal calf serum (Gibco Bio-cult, Paisley, Scotland) and grownto confluency over 3 days. Each of 15 dishes (total 4 x 108cells) was treated with 50 jig of the G-3H-labeled carcinogen
(Structure I) in 50 fil of dimethyl sulfoxide to give a finalconcentration of 1.25 fig/ml, and incubation was continued fora further 48 hr. The medium was removed, the dishes werewashed twice with phosphate-buffered saline [containing NaCI(10 g), KCI (0.25 g), Na2HPO4 (1.44 g), KH2PO4 (0.25 g),CaCI2, 2H2O (0.17 g), and MgCI2, 6H2O (0.13 g) per liter ofsterile distilled water, pH 7.2], and the cells were scraped off,suspended in phosphate-buffered saline, and centrifuged at900 x g for 5 min. The cell pellet was washed twice withphosphate-buffered saline by resuspension and recentrifuga-
tion; it was finally added to the proteinase K lysing solution,and the DNA was isolated as already described.
in Vitro Metabolism and Separation of Metabolites. 11-mefA>y/-"'C-labeled Structure I (500 fig), microsomes from 10
g of rat liver, and NADPH (37.5 mg) in 0.1 M Tris buffer (10ml), pH 7.2, were shaken gently in air at 37°for 30 min, and
the metabolites formed were extracted with ethyl acetate essentially as already described (16).
Metabolites were separated by HPLC3 as described previ
ously (16) except that a Whatman Partisil M9 10/50 ODScolumn (Whatman Ltd., Maidstone, England) was used with agradient initially of 15% methanol in water changing to metha-nol alone at 1%/min and a flow rate of 120 ml/hr (Chart 4).
Identification of Metabolite (e). HPLC fractions containingMetabolite e were pooled and rerun, collecting only the centralpart of the peak. The aqueous methanolic solution thus obtained was evaporated to dryness in a vacuum at 35°,and the
residue was dissolved in a small volume of methanol. An aliquotcontaining about 50 /*g (based on its radioactivity) was evaporated in a small capillary tube for direct insertion into the inletof a A.E.I. MS 902 instrument, and the mass spectrum wasrecorded at 70 eV and 1500 (10% valley) resolving power(Table 2).
An aliquot was diluted with methanol for UV spectroscopy(Table 2; Chart 5), sodium borohydride (10 mg) was added,and the spectrum was rerecorded after 30 min. For dehydration, another aliquot (1 ml, 10 absorbance units) was heated ina boiling water bath for 30 min with 5 N sulfuric acid (4 ml).The cooled solution was extracted 3 times with ethyl acetate,and the latter was washed with saturated sodium hydrogencarbonate solution and with water and dried over anhydrous
1The abbreviation used is: HPLC, high-pressure liquid chromatography.
sodium sulfate. After evaporation to dryness in a vacuum, theresidue was redissolved in neutral methanol, and its UV spectrum was recorded before and after addition of N sodiumhydroxide (0.1 ml). A further aliquot (2 ml, 10 absorbanceunits) was stirred with 5% palladium-on-charcoal catalyst (1mg) in an atmosphere of hydrogen for 1 hr, and the UVspectrum was recorded after removal of the catalyst by cen-trifugation. This solution was further reduced with sodium borohydride as already described, and its spectrum was recorded.
The elution volume of Metabolite e (428 ml) run on a Seph-adex LH-20 column with Tris buffer (pH 8.7) was unchanged
when the eluting solvent was replaced by sodium borate buffer(pH 8.7).
Determination of Mutagenicity of Metabolites a to g. Metabolites a to g (Chart 4) were isolated by HPLC, and theirability to cause reversion to prototrophy in S. typhimuriumTA100 grown on a histidine-deficient medium was examined
essentially as described previously (13).Characterization of Adduct B. LH-20 fractions containing
this adduct (Chart 2) were pooled and concentrated in avacuum to 2 ml. The aqueous solution, XmaK260, 281 nm, wasreduced with sodium borohydride, Amax240, 285 nm; intensityat 260 nm was much decreased (for UV spectra of 1,2,3,4-tetrahydro derivatives of Structure I, see Table 2 and Chart 5).
The elution volume of Adduct B on a column of SephadexLH-20 equilibrated with Tris buffer, pH 8.7, was 256 ml. When
the buffer was replaced by sodium borate buffer, pH 8.7, thisvolume was reduced to 205 ml (Chart 6).
RESULTS
Binding of Structure I to DNA in Vitro and in Vivo. As shownpreviously (16), the 14C-labeled carcinogen (Structure I) binds
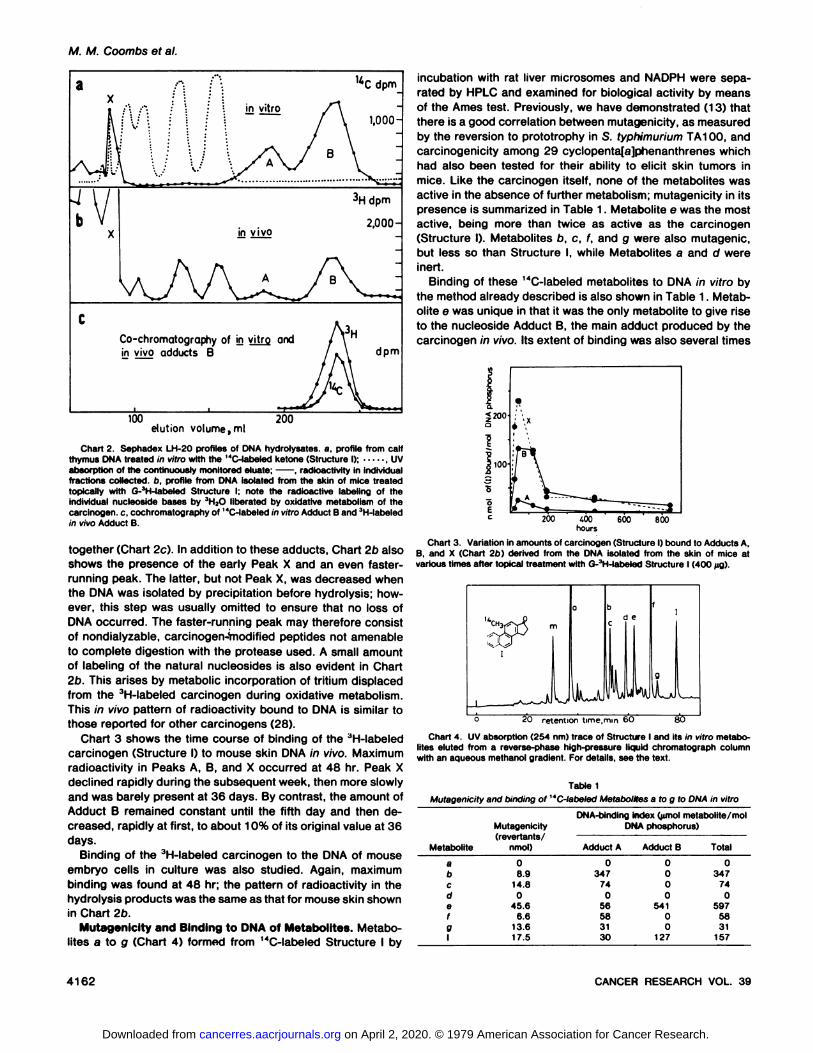
to DNA when the latter is present during the incubation with ratliver microsomes and NADPH. Enzymatic hydrolysis of thisDNA followed by chromatography of the resulting nucleosideson Sephadex LH-20 columns gives 2 radioactive peaks, A and
B, eluting after the natural nucleosides (UV trace) as shown inChart 2a. The same pattern was observed with calf thymus,salmon sperm, and E. coli DNA and with DNA prepared frommouse skin, provided in vitro incubations were used. In additionto Adducts A and B, a sharp coincident peak (Peak X) ofradioactivity and UV absorption always appeared at about 85ml, the approximate elution volume of polydeoxyribonucleo-tides. The size of this peak was noi altered by further incubationwith the various enzymes used in the isolation and hydrolysisof the DNA.
In order to investigate the pattern of DNA binding in vivo,mice were treated, each on its dorsal region, with 400 /¿gofthe high-specific-activity 3H-labeled carcinogen. This is known
to be a powerful initiating dose which, when followed by twice-weekly promotion with croton oil, gave rise to tumors in 90%of the animals with a mean latent period of 35 weeks (8). After24 hr and at subsequent times up to 36 days mice were killed,DNA was isolated from the treated skin and was hydrolyzedand chromatographed as before. In each case, a pattern similarto that shown in Chart 2b was found, consisting of Adduct Band a trace of Adduct A. Identity of this in vivo 3H-labeledAdduct B with the 14C-labeled Adduct B formed in vitro was
established by cochromatography, when both isotopes eluted
OCTOBER 1979 4161
on April 2, 2020. © 1979 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
M. M. Coombs et al.
Co-chromatography of in vitro and
in vivo adducts B dpm
100elution volume, ml
200
Chart 2 Sephadex LH-20 profiles of DNA hydrolysates. a, profile from calfthymus DNA treated in vitro with the "C-labeled ketone (Structure I); , UV
absorption of the continuously monitored eluate; , radioactivity in individualfractions collected, b, profile from DNA isolated from the skin of mice treatedtopically with G-'H-labeled Structure I; note the radioactive labeling of theindividual nucleoside bases by JH2O liberated by oxidative metabolism of thecarcinogen, c. cochromatography of "C-labeled in vitro Adduci B and 3H-labeled
in vivo Adduci B.
together (Chart 2c). In addition to these adducts, Chart 2b alsoshows the presence of the early Peak X and an even faster-running peak. The latter, but not Peak X, was decreased whenthe DNA was isolated by precipitation before hydrolysis; however, this step was usually omitted to ensure that no loss ofDNA occurred. The faster-running peak may therefore consistof nondialyzable, carcinogen-Vnodified peptides not amenable
to complete digestion with the protease used. A small amountof labeling of the natural nucleosides is also evident in Chart2b. This arises by metabolic incorporation of tritium displacedfrom the 3H-labeled carcinogen during oxidative metabolism.
This in vivo pattern of radioactivity bound to DNA is similar tothose reported for other carcinogens (28).
Chart 3 shows the time course of binding of the JH-labeled
carcinogen (Structure I) to mouse skin DNA in vivo. Maximumradioactivity in Peaks A, B, and X occurred at 48 hr. Peak Xdeclined rapidly during the subsequent week, then more slowlyand was barely present at 36 days. By contrast, the amount ofAdduci B remained constant until the fifth day and then decreased, rapidly at first, to about 10% of its original value at 36days.
Binding of the 3H-labeled carcinogen to the DNA of mouse
embryo cells in culture was also studied. Again, maximumbinding was found at 48 hr; the pattern of radioactivity in thehydrolysis products was the same as that for mouse skin shownin Chart 2b.
Mutagenicity and Binding to DNA of Metabolites. Metabolites a to g (Chart 4) formed from "*C-labeled Structure I by
incubation with rat liver microsomes and NADPH were separated by HPLC and examined for biological activity by meansof the Ames test. Previously, we have demonstrated (13) thatthere is a good correlation between mutagenicity, as measuredby the reversion to prototrophy in S. typhimurium TA100, andcarcinogenicity among 29 cyclopenta[a]phenanthrenes whichhad also been tested for their ability to elicit skin tumors inmice. Like the carcinogen itself, none of the metabolites wasactive in the absence of further metabolism; mutagenicity in itspresence is summarized in Table 1. Metabolite e was the mostactive, being more than twice as active as the carcinogen(Structure I). Metabolites b, c, f, and g were also mutagenic,but less so than Structure I, while Metabolites a and d wereinert.
Binding of these 14C-labeled metabolites to DNA in vitro by
the method already described is also shown in Table 1. Metabolite e was unique in that it was the only metabolite to give riseto the nucleoside Adduct B, the main adduct produced by thecarcinogen in vivo. Its extent of binding was also several times
«200
¡100
200 ¿00hours
600 800
Chart 3. Variation in amounts of carcinogen (Structure I) bound to Adducts A.B, and X (Chart 2b) derived from the DNA isolated from the skin of mice atvarious times after topical treatment with G-3H-labeled Structure I (400 fig).
"CH
20 retention time,mm 6O 80
Chart 4. UV absorption (254 nm) trace of Structure I and its in vitro metabolites eluted from a reverse-phase high-pressure liquid Chromatograph columnwith an aqueous methanol gradient. For details, see the text.
Mutagenicity and binding of
Table 1'C-labeled Metabolites a to g to DNA in vitro
Metabolitea
bcde
/0
1Mutagenicity
(revertants/nmol)0
8.914.80
45.66.6
13.617.5DNA-binding
index (^mol metabolite/molDNAphosphorus)Adduct
A0347
740
56583130Adduct
B0
00
0541
00
127Total0
347740
5975831
157
4162 CANCER RESEARCH VOL. 39
on April 2, 2020. © 1979 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

Proximate-Ultimate Forms of a Cyclopenta[a]phenanthrene
greater than that of Structure I. Of the other metabolites,Metabolites b, c, f, and g all gave only Adduci A, while Metabolites a and d did not bind to DMA. The parallelism betweenmutagenicity and total amount of the metabolite bound to DMAis noticeable. The nature of these other metabolites is underinvestigation and will form the subject of a separate communication.
The Structure of Metabolite e (Table 2; Chart 5). Theretention volume of metabolite e suggested a diol, and this wasconfirmed by an accurate mass determination which showedits composition to be dsHieOs. The third oxygen atom ispresent as the original ketone function because mild reductioncaused a shift in the UV maximum from 273 to 250 nm. Thesespectra are unlike the spectra of Structure I and its reduction
Chart 5. UV absorption spectra. i. Carcinogen I ( ) and its bore-hydride reduction product, the corresponding 17-ol ( ); ii, 1,2,3.4-tetrahydro derivative of Structure I ( ) and its borohydride reduction product, the corresponding17-ol ( ); Hi, Metabolite e ( ) and its borohydride reduction product( ); iv. product of acid-catalyzed dehydration of Metabolite e in neutralmethanol ( ) and in alkaline methanol ( ); v. DMA Adduci B ( ) and itsborohydride reduction product ( ).
Table 2
Characterization of the proximate carcinogen (Metabolite e)
Mass spectrum (only peaks greater than 10% of the base peak are listed)
m/e (relative abundance and tentative assignment)
Ci8H16O3 requires:Molecular ion, found:
Base peak:
M'280.1109M'280.1100 (28%)
279 (12%)264 (19%)263 (22%)262 (100%. M-H2O)234 (25%. M-H2O-CO)219 (16%, M-H2O-CO-CH3)191 (16%, M-H2O-CO-CH3-C2H4)
UV spectra (for experimental details, see text; see also Chart 5)
A™,dog £)
Metabolite e; 273 (4.68). 357 (3.39), 371 nm (3.40).NaBH4 reduction product; 250, 330 nm.Acid dehydration product; neutral, 271.5. 301 nm.Acid dehydration product; alkaline, 261, 325 nm.Hydrogénation product; 259 nm [cf. 1,2,3,4,15,16-hexahydro-11-methylcy-
clopenta[a]phenanthren-17-one, 260 (4.77). 287 (3.97), 297.5 nm (4.02)(7)].
NaBH»reduction product of hydrogénation product; 239. 279. 330 nm [cf.1.2,3,4,15,16-hexahydro-17-hydroxy-11 -methylcyclopentalajphenan-threne; 239, 288, 334 nm].
product (Xmax264 and 255 nm, respectively), indicating thatthe diol oxygens are situated in a conjugated ring. The intenseion in the mass spectrum of metabolite e at m/e 262, corresponding to a loss of water from the molecular ion, confirmsthe presence of a readily dehydrated vicinal diol system. TheUV spectrum of Metabolite e was different from those of theknown 1,2- (12), 6,7- (5), 11,12-, or 13,14-dihydro (4) derivatives of 15,16-dihydrocyclopenta[a]phenanthren-17-one; ittherefore probably represents the unknown 3,4-dihydro chro-mophore. Acid-catalyzed dehydration of Metabolite e gave a
phenol, the UV spectrum of which was not the same as thoseof the known 2- (14), 3-, 6-, and 7-phenols (5) of this series;the 4-phenol is the expected product from dehydration of the3,4-dihydro-3,4-diol, by comparison with the product (1-naph-thol) of dehydration of 1,2-dihydro-1,2-dihydroxynaphthalene(1 7, 19). The presence of the 3,4-dihydro system in Metabolite
e was confirmed by catalytic hydrogénation, the product ofwhich had the known chromophore of a 1,2,3,4-tetrahydro
compound (7). The frans stereochemistry expected of a diolformed metabolically with mammalian enzymes was also suggested by the failure to decrease the elution volume of thismetabolite by addition of sodium borate to the eluting buffer.Metabolite e is therefore the 3,4-dihydro-3,4-frans-dihydroxy
derivative and is the proximate carcinogen (Chart 7); the absolute stereochemistry, or indeed whether it is a single enan-
tiomorph, is not known at present.Identification of the Ultimate Carcinogen. The nature of the
reactive metabolite formed from the 3,4-dihydro-3,4-diol (Me
tabolite e) is suggested by consideration of the nucleosideAdduci B which arises from it. The UV spectrum of this materialis thai expecled of an adduci consisling of a 1,2,3,4-lelrahydro
derivative of Struclure I attached to a nucleoside base wilhoutconjugation of eilher chromophore. Adduci B eluled from aSephadex LH-20 column earlier and as a single peak when
borale buffer (23) was used in place of Tris buffer (Charl 6).This is indicalive of a c/s-diol system, and because the 3,4-diol
is frans the implication is thai Ihe adduci conlains a Ihirdhydroxyl group at C-2, cis with respect to that at C-3. This in
OCTOBER 1979 4163
on April 2, 2020. © 1979 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

M. M. Coombs et al.
dpm
•600 Borale,
pH 87kOO
Tris, pH 87
ISO 200 250 300Elution vol.ml
Chart 6. Effect of sodium borate on théelution volume of the nucleosideAdduct B.
CH
HO' T HO
OH OHproximate carcinogen
CH
OH
ultimate carcinogenChart 7 Proposed structures of the proximate and ultimate forms of the
carcinogen (Structure I).
turn implies metabolism of the 1,2-double bond in Metabolite
e.In view of this evidence and that for the involvement of an
aryl oxide mentioned in the introduction, it seems most probable that the reactive metabolite is an ani/-3,4-dihydro-3,4-frans-dihydroxyl-1,2-dihydro-1,2-epoxide of Structure I, analogousto the diol-epoxide now thought to be the active form ofbenzo(a)pyrene (29, 37). In the present case, the corresponding syn-diol epoxide is excluded because if adduci formationoccurs by nucleophilic attack at the benzylic C-1 positionaccompanied by inversion, but with retention of configurationat C-2 (23), this would lead to an all-frans structure. In the
present absence of knowledge of the absolute stereochemistryof the 3,4-diol (Metabolite e), we propose that the ultimate form
of Structure I is one, or both, of the optical isomers (Chart 7).Unequivocal proof of these structures awaits synthesis of the3,4-diol (Metabolite e) and its 1,2-oxide, work which is at
present in hand.
DISCUSSION
Of the microsomal metabolites of 15,16-dihydro-11-methyl-cyclopenta[a]phenanthren-17-one (Structure I), the 3,4-dihy-dro-3,4-diol (Metabolite e) is the most active, being more
mutagenic and binding to DMA more strongly than does theparent carcinogen and being the only metabolite to give rise tothe in vivo Adduct B. Analogous activation of other phenan-threne derivatives, namely, benzo(a)pyrene (26, 29, 37),benz(a)anthracene (24, 30, 34, 35), 7-methylbenz(a)-anthracene (1, 26, 32), 7,12-dimethylbenz(a)anthracene(18, 21, 26, 27, 33), 3-methylcholanthrene (22, 31, 33), di-
benz(a,h)anthracene (36), chrysene (25), and 5-methylchry-
sene (20), has recently been reported. Further oxidation of thebay-region double bond in the 3,4-diol (Metabolite e) to a 3,4-diol-1,2-epoxide (Structure III) gives the ultimate carcinogen,
as in these other systems. In the case of the best studiedhydrocarbon, benzo(a)pyrene (23, 37), both syn- and anf/-diolepoxides are found; only the anti-diol-epoxide (Structure III) isformed from the carcinogen (Structure I). 7.12-Dimethylbenz[a]anthracene, with an analogous methyl group in the bay region,seems also to give mainly the anf/-isomer (18). While StructureI and the corresponding 11-methoxy-17-ketone are strongcarcinogens, the 11-ethyl-17-ketone is much less active, andthe 11-butyl-1 7-ketone is inactive as a carcinogen (9) and as
a mutagen (16). Possibly, these larger groups hinder approachof the oxidase to the 1,2-double bond and hence inhibit epox-
idation.In the present experiments, binding to DMA has been esti
mated from the sum of the radioactivity in Adducts A and B.However, there is evidence that the early Peak X also consistsat least partly of carcinogen-modified nucleotides (18, 28).
Although this peak is small in vitro (Chart 2a), in vivo it isappreciable, especially at early times (Chart 3). Peak X fromthe DMA of the skin of treated mice disappears more quicklyand completely than does Adduct B which is still easily demonstrable even after 36 days. In studying the binding of tritiated7,12-dimethylbenz[a]anthracene to the DNA of mouse embryo
cells in culture, Dipple and Nebzydoski (18) also observed thatthe peak eluting at 85 ml and the later adduci peaks weresimilar in amount at 24 hr but were at a ratio of 1:10 at 48 hr.
Nearly all potent polycyclic carcinogens possess a phenan-threne ring system together with additional, fused aromaticring(s). 15,16-Dihydro-11 -methylcyclopenta[a]phenanthren-17-one, with potency similar to that of benzo(a)pyrene, is
therefore one of the simplest molecules capable of beingactivated through a bay-region diol-epoxide to display high
carcinogenic activity. The importance of the carbonyl group atC-1 7 lies in its conjugation with the phenanthrene system, forit may be replaced by a carbon-carbon double bond at C-
16(1 7) with retention of strong activity, while the hydrocarbonwith a saturated 5-membered ring is only a weak carcinogen(9). The 11-methyl ketone (Structure I) is the most active of itspositional methyl isomers; the 7-methyl ketone is less active,and the 2-, 3-, 4-, 6-, and 12-methyl ketones, like the parentketone 15,16-dihydrocyclopenta[a]phenanthren-17-one, are
not carcinogens. Since all these compounds have similar bayregions, this of itself is not sufficient to ensure carcinogenicity;correct positional substitution by a small electron-releasing
group is also critical. The same pattern of activation by methylsubstitution is also seen among the methylbenz[a]anthracenesand methylchrysenes, which may be viewed as analogs ofStructure I in which conjugation by the 17-carbonyl group is
replaced by the conjugation of a further, fused benzene ring.Comparison of the metabolic products formed from StructureI with those from its inactive isomers may lead to a betterunderstanding of the role of the 11-methyl group in inducing
carcinogenic properties in these compounds.
REFERENCES
1. Baird, W. M., and Brookes, P. Isolation of the hydrocarbon-deoxyribonucle-oside products from the DNA of mouse embryo cells treated in culture with7-methylbenz[a)anthracene-:)H. Cancer Res., 33 2378-2383. 1973.
4164 CANCER RESEARCH VOL. 39
on April 2, 2020. © 1979 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

Proximate-Ultimate Forms of a Cyclopenta[a]phenanthrene
2. Blin, N., and Stafford. W. D. A general method of isolation of high molecularweight DMA from eukaryotes. Nucleic Acid Res., 3 2303-2308. 1976.
3. Chouroulinkov. !.. Gentil, A.. Tierney. B., Grover, P., and Sims, P. Themetabolic activation of 7-methylbenz[a]anthracene in mouse skin: high tumour-initiating activity of the 3,4-dihydrodiol. Cancer Lett.. 3. 247-253.1977.
4. Coombs. M. M. Potentially carcinogenic cyclopenta[a]phenanthrenes. PartI. A new synthesis of 15.16-dihydro-17-oxo-cyclopenta[a]phenanthrene andthe phenanthrene analogue of 18-Norcestrone. J. Chem. Soc. C, 955-962.
1966.5. Coombs. M. M. Potentially carcinogenic cyclopenta[a]phenanthrenes. Part
III. Oxidation studies. J. Chem. Soc C. 2484-2488, 1969.
6. Coombs. M M. Tritium labeling of cyclopenta[a]phenanthrenes at very highspecific activity. J. Labeled Compd. Radiopharm., in press, 1979.
7. Coombs. M. M., and Bhatt. T. S. Potentially carcinogenic cyclopenta(a)-phenanthrenes. Part VI. 1,2,3,4-Tetrahydro-17-ketones. J. Chem. Soc. Per-kin Trans. I, 1251-1254, 1973.
8. Coombs. M. M., and Bhatt, T. S. Lack of initiating activity in mutagens whichare not carcinogenic. Br. J. Cancer, 38 148-150. 1978.
9. Coombs, M. M.. Bhatt, T. S.. and Croft. C. J. Correlation between carcino-genicity and chemical structure in cyclopenta[a]phenanthrenes CancerRes.. 33 832-837. 1973.
10. Coombs. M. M., Bhatt. T. S.. and Kissonerghis, A-M. Identification of theproximate form of the carcinogen 15,16-dihydro-11-methylcyclopenta[a]phenanthren-17-one. Br. J. Cancer, 38 192, 1978.
11. Coombs, M. M., Bhatt, T. S., and Vose, C. W. The metabolism, DNA binding,and carcinogenicity of 15,16-dihydro-11 -methylcyclopentalajphenanthren-17-one in the presence of a microsomal enzyme inhibitor. Cancer Res.. 35.305-309. 1975.
12. Coombs, M. M., and Crawley. F. E. H. Potentially carcinogenic cyclopenta[a)phenanthrenes. Part IX. Characterisation of a 5.10-epoxybenzocyclodeceneas a major urinary metabolite of the carcinogen 15,16-dihydro-11-methyl-cyclopenta(a]phenanthren-17-one. J. Chem. Soc. Perkins Trans. I. 2330-2335. 1974.
13. Coombs. M. M., Dixon. C.. and Kissonerghis. A-M. Evaluation of the muta-genicity of compounds of known carcinogenicity. belonging to the benz[a]anthracene, chrysene. and cyclopenta(a]phenanthrene-series. using Ames"
tests. Cancer Res . 36. 4525-4529, 1976.14. Coombs, M. M.. Hall, M., Siddle. V. A., and Vose, C. W. Potentially carci
nogenic cyclopenta[a]phenanthrenes. Part X. Oxygenated derivatives of thecarcinogen 15.16-dihydro-11-methylcyclopenta[a]phenanthren-17-one ofmetabolic interest. J. Chem. Soc. Perkins Trans. I, 265-270. 1975.
15. Coombs. M. M., Jaitly, S. B., and Crawley, F. E. H. Potentially carcinogeniccyclopenta[a]phenanthrenes. Part IV. Synthesis of 17-ketones by the Stobbereaction. J. Chem. Soc. C. 1266-1271. 1970.
16. Coombs, M. M., Kissonerghis, A.-M., and Allen, J. A. An investigation intothe binding of the carcinogen 15,16-dihydro-11-methylcyclopenta[a)phen-anthren-17-one to DNA in vitro. Cancer Res.. 36 4387-4393. 1976.
17. Corner. E. D. S., and Young. L. Biochemical studies of toxic agents. Themetabolism of naphthalene in animals of different species. Biochem. J.. 58.647-655. 1954.
18. Dipple. A., and Nebzydoski. J. A. Evidence for the involvement of a diol-epoxide in the binding of 7.12-dimethylbenz[a]anthracene to DNA in cells inculture. Chem.-Biol. Interact., 20. 17-26, 1978.
19. Fu, P. P.. Harvey. R. G., and Beland. F K. Molecular orbital theoreticalprediction of the isomerie products formed from reactions of arene oxidesand related metabolites of polycyclic aromatic hydrocarbons. Tetrahedron.34: 857-866. 1978.
20. Hecht. S. S., LaVoie, E.. Mazzarese, R., Amin. S.. Bedenko, V., andHoffmann, D. 1,2-Dihydro-1.2-dihydroxy-5-methylchrysene, a major activated metabolite of the environmental carcinogen 5-methylchrysene. CancerRes.. 38. 2191-2194. 1978.
21. lanovic, V., Geacintov. N. E.. Jeffrey, A. M.. Fu. P. P., Harvey, R. G.. and
Weinstein, I. B. Cell and microsome mediated binding of 7,12-dimethyl-benz[a]anthracene to DNA studied by fluorescence spectroscopy. CancerLett., 4: 131-140. 1978.
22. King. H. W.. Osborne. M. R., and Brookes. P. The identification of 3-methylcholanthrene-9.10-dihydrodiol as an intermediate in the binding of 3-methylcholanthrene to DNA in cells in culture. Chem.-Biol. Interact.. 20.367-371, 1978.
23. King, H. W. S., Osborne, M. R., Beland, F. A.. Harvey, R. G.. and Brookes,P. (±)-7«,8/i-Dihydroxy-9/;, 10/i-epoxy-7.8.9,10-tetrahydrobenzo|a]pyreneis an intermediate in the metabolism and binding to DNA of benzo[a)pyreneProc. Nati. Acad. Sei. U. S. A.. 73. 2679-2681. 1976.
24. Levin, W., Thakker, D. R., Wood, A. W., Chang, R. L., Lehr, R. E.. Jerina, D.M., and Conney. A. H. Evidence that benzo|a]anthracene 3.4-diol-1,2-epox-ide is an ultimate carcinogen on mouse skin Cancer Res , 38. 1705-1710.1978.
25. Levin, W , Wood, A. W.. Chang, R L.. Yagi, H., Mäh,H D., Jerina, D. M..and Conney, A. H. Evidence for bay-region activation of chrysene to anultimate carcinogen. Cancer Res.. 38. 1832-1835, 1978.
26. Marquardt. H.. Grover. P. L., and Sims, P. In vitro malignant transformationof mouse fibroblasts by non-K-region dihydrodiols derived from 7-methyl-benz(a)anthracene. 7.12-dimethylbenz(a)anthracene. and benzo(a)pyrene.Cancer Res., 36. 2059-2064. 1976.
27. Malaveille, C., Bartsch. H.. Tierney. B.. Grover. P. L., and Sims. P. Micro-some-mediated mutagenicities of dihydrodiols of 7,12-dimethylbenz(a]an-thracene: high mutagenic activity of the 3,4-dihydrodiol. Biochem. Biophys.Res. Commun., 83 1468-1473, 1978.
28. Phillips. D. H.. Grover. P. L., and Sims. P. The covalent binding of polycyclichydrocarbons to DNA in the skin of mice of different strains. Int. J. Cancer,22. 487-494, 1978.
29. Sims, P., Grover, P. L., Swaisland. A.. Pal. K.. and Hewer, A Metabolicactivity of benzo[a)pyrene proceeds by a diol-epoxide. Nature (Lond.). 254.326-328, 1974.
30. Slaga, T. J.. Huberman. E., Selkirk, J. K., Harvey, R.. and Bracken, W MCarcinogenicity and mutagenicity of benz|a]anthracene diols and diol-epox-ides. Cancer Res.. 38 1699-1704. 1978.
31. Thakker, D. R., Levin, W.. Wood. A. W.. Conney. A. H.. Stoming. T. A., andJerina. D.M. Metabolic formation of 1,9.10-trihydroxy-9,10-dihydro-3-meth-ylcholanthrene: a potential proximate carcinogen from 3-methylcholan-threne. J. Am. Chem. Soc., 700 645-647. 1978
32. Tierney, R., Hewer. A.. Walsh, C., Grover, P. L., and Sims, P. The metabolicactivation of 7-methylbenz[a)anthracene in mouse skin Chem.-Biol. Interact., 78. 179-193, 1977.
33. Vigny, P., Duquesne, M., Coulomb. H.. Tierney, B., Grover, P. L.. and Sims.P. Fluorescence spectral studies on the metabolic activation of 3-methyl-cholanthrene and 7,12-dimethylbenz[a]anthracene in mouse skin. FEBSLett.. 82 278-282, 1977.
34 Wislocki, P. G.. Kapitulnik. K.. Levin. W.. Lehr. R.. Schaefer-Ridder, M.,Karle, J. M , Jerina, D. M.. and Conney, A. H. Exceptional carcinogenicactivity of benzo[a]anthracene 3.4-dihydrodiol in the newborn mouse andthe bay region theory. Cancer Res., 38 693-696. 1978.
35. Wood. A. W.. Levin, W.. Chang, R. L., Lehr, R. E.. Schaefer-Ridder. M..Karle. J. M.. Jerina. D. M.. and Conney. A. H. Tumorigenicity of fivedihydrodiols of benz|a]anthracene 3,4-dihydrodiol Proc. Nati. Acad Sci.U.S.A., 74. 3176-3179, 1977.
36. Wood. A. W.. Levin, W., Thomas, P E., Ryan. D.. Karle. J. M.. Yagi. H..Jerina. D. M.. and Conney. A H. Metabolic activation of dibenzo[a.ri]anthra-cene and its dihydrodiols to bacterial mutagens. Cancer Res , 38 1967-1973, 1978.
37. Yang. S K.. McCourt, D. W.. Roller. P P., and Gelboin. H V Enzymaticconversion of benzo(a]pyrene leading predominantly to the diol-epoxide r-7,(-8-dihydroxy-(-9,10-oxy-7,8,9,10-tetrahydrobenzo(a]pyrene through asingle enantiomer of r-7,f-8-dihydroxy-7,8-dihydrobenzo(alpyrene. Proc.Nati. Acad. Sei. U. S. A., 73 2594-2598. 1976
OCTOBER 1979 4165
on April 2, 2020. © 1979 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

1979;39:4160-4165. Cancer Res Maurice M. Coombs, Anna-Maija Kissonerghis, Jeffrey A. Allen, et al. ]phenanthren-17-one
aCarcinogen 15,16-Dihydro-11-methylcyclopenta[Identification of the Proximate and Ultimate Forms of the
Updated version
http://cancerres.aacrjournals.org/content/39/10/4160
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/39/10/4160To request permission to re-use all or part of this article, use this link
on April 2, 2020. © 1979 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from