implantable microprobe with arrayed microsensors for combined amperometric monitoring of the...
TRANSCRIPT

Journal of Electroanalytical Chemistry 682 (2012) 141–146
Contents lists available at SciVerse ScienceDirect
Journal of Electroanalytical Chemistry
journal homepage: www.elsevier .com/locate / je lechem
Implantable microprobe with arrayed microsensors for combinedamperometric monitoring of the neurotransmitters, glutamate and dopamine
Tina T.-C. Tseng 1, Harold G. Monbouquette ⇑Chemical and Biomolecular Engineering Department, University of California, Los Angeles, CA 90095, USA
a r t i c l e i n f o
Article history:Received 9 February 2012Received in revised form 12 July 2012Accepted 17 July 2012Available online 25 July 2012
Keywords:Microelectrode arrayCombined glutamate and dopaminemonitoringConstant potential amperometryBiosensor
1572-6657/$ - see front matter � 2012 Elsevier B.V. Ahttp://dx.doi.org/10.1016/j.jelechem.2012.07.014
⇑ Corresponding author. Tel.: +1 310 825 8946; faxE-mail address: [email protected] (H.G. Monbo
1 Present address: Department of Chemical Engineesity of Science and Technology, Taipei 10607, Taiwan.
a b s t r a c t
An implantable, micromachined microprobe with a microsensor array for combined monitoring of theneurotransmitters, glutamate (Glut) and dopamine (DA), by constant potential amperometry has beencreated and characterized. Microprobe studies in vitro revealed Glut and DA microsensor sensitivitiesof 126 ± 5 nA lM�1 cm�2 and 3250 ± 50 nA lM�1 cm�2, respectively, with corresponding detection limitsof 2.1 ± 0.2 lM and 62 ± 8 nM, both at comparable �1 s response times. No diffusional interaction of H2O2
among arrayed microelectrodes was observed. Also, no responses from the electroactive interferents,ascorbic acid (AA), uric acid (UA), DOPA (a DA catabolite) or DOPAC (a DA precursor), over their respectivephysiological concentration ranges, were detected. The dual sensing microbe attributes of size, detectionlimit, sensitivity, response time and selectivity make it attractive for combined sensing of Glut and DAin vivo.
� 2012 Elsevier B.V. All rights reserved.
1. Introduction
Studies of the interwoven roles of multiple neurotransmitterscan illuminate the mechanisms behind and the progression of neu-rological diseases and disorders. Investigation of the interplay be-tween the neurotransmitters, glutamate (Glut) and dopamine(DA), is of particular interest. For example, the loss of dopaminergicneurons, which dysregulates glutamatergic transmission, underliesthe symptoms observed in Parkinson’s disease [1]. Also, changes inrelative Glut and DA transmission in the basolateral amygdala andnucleus accumbens core may be associated with the shifts in re-ward seeking behavior leading to addiction [2]. Therefore, implant-able analytical tools for real-time, simultaneous monitoring of Glutand DA with high spatial resolution would be very useful for neu-roscience research. A number of important challenges are con-fronted in the design of sensing probes for neurotransmittersincluding subsecond response time, selectivity against the arrayof electroactive species present in brain extracellular fluid, and mi-cron-scale size to reduce tissue damage and provide spatial resolu-tion. Also low detection limit and high sensitivity are required todetect neurotransmitters commonly present in the nanomolar tomicromolar range. In particular, the background extracellular con-centration range of Glut is 610 lM [3] to 100 lM [4] after stimu-lation, while that of DA is 60.1 lM to only 1 lM after
ll rights reserved.
: +1 310 206 4107.uquette).ring, National Taiwan Univer-
stimulation [5]. These challenges in sensor design have made theinnovative development of new analytical tools for monitoringneurotransmitter levels in vivo an area of significant research andengineering activity for several decades.
Although microdialysis has been used widely over many yearsfor the sampling of brain extracellular fluid in vivo [6] and its anal-ysis for neurotransmitters using associated analytical equipment(e.g., capillary electrophoresis coupled to laser-induced fluores-cence (CE–LIF) [7,8] and liquid chromatography coupled to tandemmass spectrometry (LC–MS/MS) [9,10]), the long analysis times (5–10 min) and relatively large probe sizes (P200 lm) gives this toolinadequate temporal and spatial resolution for many studies [7–11]. Micromachined microelectrode array microprobes with cus-tomizable microelectrode design of well-defined micron-size areacan provide better spatial resolution. Further, their smaller size re-sults in infliction of less tissue damage during implantation whilestill providing good mechanical strength [4,12–15]. A variety ofelectrochemical techniques may be used for sensing at the micro-electrode sites, yet constant potential amperometry offers the besttemporal resolution with sampling rates down to 1 ms [16].Although fast-scan cyclic voltammetry (FSCV) has proven to bean excellent electrochemical detection method for a number ofelectroactive neurotransmitters, principally DA [17,18], it requiresspecialized instrumentation. In contrast, constant potential amper-ometry requires only a standard potentiostat and straightforwardcollection and analysis of current signals obtained at a constant ap-plied potential. Constant potential amperometry therefore is thepreferred electroanalytical technique for monitoring neurotrans-mitter levels when the major electrooxidizable species are known

142 T.T.-C. Tseng, H.G. Monbouquette / Journal of Electroanalytical Chemistry 682 (2012) 141–146
and electroactive interferents either are selectively excluded fromthe sensing electrode surface or are below the detection limit[17,19].
Sensor selectivity against interferents can be achieved by mod-ifying the electrode surfaces with suitable permselective polymers.The perfluoronated ionomer, Nafion, commonly is used to excludeanionic interferents [20]. Overoxidized polypyrrole (OPPy) also hasbeen deposited on electrodes, commonly as a permselective cat-ion-exchange film [18,21]. OPPy films are created by first electrop-olymerizing pyrrole to form polypyrrole followed by itselectrooxidation at high potential in the absence of monomer.The electronegative carbonyl groups formed along the OPPy back-bone during electrooxidation can effectively attract cations and re-pel anions [22]. In fact, a thin OPPy film has been found to improvethe sensitivity of electrochemical DA sensors by enhancing DAadsorption at the electrode surface [18]. However, it also has beenreported that relatively thick, �100 nm OPPy films reject both neg-atively charged ascorbic acid and positively charged DA, but allowthe ready permeation of small neutral molecules, such as hydrogenperoxide (H2O2) [4,23–26], presumably because the incompleteelectrooxidation of thick polypyrrole films results in a deposit oflayered electropositive and electronegative character. Such thickOPPy films have proved invaluable in the construction of selectiveGlut sensors [4,23,25].
Most Glut sensor designs rely on the use of glutamate oxidase(GlutOx) as the selective sensing element. GlutOx catalyzes theoxidative deamination of Glut in the presence of oxygen to producea-ketoglutarate, ammonia, and H2O2 [27]. The neutral and electro-active H2O2 species can pass through permselective polymers de-signed to block charged electroactive interferents from anunderlying electrode and can be electrooxidized at constant poten-tial to give a current signal. Platinum (Pt) generally [24,28] is theelectrode material of choice for electrooxidation of H2O2 ratherthan other common materials, such as gold (Au), palladium (Pd),and glassy carbon (GC). Glut sensors based on cylindrical Pt micro-electrodes [25,29–31] or on Pt microelectrode array microprobes,including those micromachined from silicon wafers [4] and fromceramic substrates [13,23], have shown promising results in vivofor the selective monitoring of Glut in near real time.
However, the sensing of the electroactive DA species has beenreported to present a different set of sensor design issues, most of-ten cited are those related to electrode fouling and selectivity. Car-bon fiber (CF) microelectrodes have been shown to undergo littleor no fouling by the products of DA electrooxidation, and CF elec-trodes combined with FSCV have been employed with notable suc-cess to monitor DA release in vivo [32–34]; however as mentionedabove, FSCV requires specialized instrumentation and less straight-forward data analysis than constant potential amperometry. Theissue of electrode fouling when DA oxidation occurs at noble metalelectrode surfaces, such as Pt [35] and Au [36], has been mentionedfrequently, and therefore, the feasibility of using Pt electrodes foranalytical determination of DA has been viewed as questionable.Some alternative DA sensor designs have been explored that arebased on an electoenzymatic approach utilizing the enzymes,tyrosinase [37] and polyphenol oxidase [29]; but these enzymesare not selective for DA. Other promising DA sensors have beenconstructed with new materials, including graphene [38] and car-bon nanotubes [39,40], but some important sensor design issues(e.g., response time, selectivity, etc.) must be further investigated.DA microelectrode array sensors based on FSCV [15] and constantpotential amperometry [12] also have been reported recently.However, there remains a need for the development of a DA sensorwith the desired attributes of detection limit, sensitivity, selectivityand response time that can readily be incorporated in a commonprobe with the successful Glut sensors previously developed[4] that are based on straightforward, constant potential
amperometry. In this study, we describe a potentially implantablemicroprobe with OPPy/Nafion-modified Pt microelectrode arraymicrosensors for combined, near-real-time monitoring of non-electroactive Glut and electroactive DA with high sensitivity andselectivity as well as adequate detection limit.
2. Experimental
2.1. Materials
Pyrrole, Nafion� (5%), glutaraldehyde solution (25%), bovine ser-um albumin (BSA) lyophilized powder, hydrogen peroxide solution(30%), L-glutamic acid, L-ascorbic acid, dopamine hydrochloride,3,4-dihydroxyphenylacetic acid, 3,4-dihydroxy-DL-phenylalanine,uric acid, (�)-epinephrine (+)-bitartrate salt, DL-norepinephrinehydrochloride, and serotonin hydrochloride were purchased fromSigma–Aldrich (St. Louis, MO). Isopropyl alcohol and sulfuric acid1 N solution were obtained from Fisher Scientific (Pittsburgh, PA).L-Glutamate oxidase (EC 1.4.3.11) from Streptomyces sp. X119-6,with a rated activity of 24.9 units per mg protein, produced by Ya-masa Corporation (Chiba, Japan), was obtained from Associates ofCape Cod, Inc. (Northstar BioProducts�, East Falmouth, MA). Siliconwafers (diameter: 4 inch; p-type boron doped; orientation h100i;thickness: 150 ± 15 lm) were purchased from Silicon Valley Micro-electronics (Santa Clara, CA). Ag/AgCl glass-bodied reference elec-trodes with 3 M NaCl electrolyte and a 0.5 mm diameter Pt wireauxiliary electrode were purchased from BASi (West Lafayette,IN). Sodium phosphate buffer (PBS) was composed of 50 mM so-dium phosphate (dibasic) and 100 mM sodium chloride (pH 7.4).
2.2. Instrumentation
Electrochemical experiments for sensor development and initialevaluation were performed using a Versatile Multichannel Poten-tiostat (model VMP3) equipped with the ‘p’ low current optionand N’Stat box driven by EC-LAB software (Bio-Logic USA, LLC,Knoxville, TN) in a three-electrode configuration consisting of thesensing electrode, a Pt wire auxiliary electrode, and a Ag/AgClglass-bodied reference electrode. Sensor calibration was conductedon a multichannel FAST-16 potentiostat (Quanteon, LLC, Lexington,KY) in two-electrode mode with a glass-bodied Ag/AgCl referenceelectrode.
2.3. Microprobe fabrication and sensor preparation
First, a 1 lm layer of silicon dioxide on a 4-inch silicon wafer(150 ± 15 lm in thickness) was grown by thermal oxidation. Aftera photolithographic patterning step, a Pt layer was deposited usingan electron-beam evaporator to define bonding pads, microelec-trode sites, and channel leads. Insulating layers were depositedusing plasma enhanced chemical vapor deposition. After a secondphotolithography step, the insulating layers were dry etched tocreate openings at the bonding pads and electrode sites. Finallyto make each probe releasable, the silicon substrate was etchedthrough using reactive ion etching after a third photolithographystep (see Fig. 1 for a microprobe fabrication overview). A 2 � 2microelectrode array was located at the tip (120 lm in widthand 150 lm in thickness) of the microprobe with 100 lm verticaland 40 lm lateral separations between the microelectrodes. Eachmicroelectrode had an average area of �5000 lm2.
Microelectrodes were rinsed with isopropyl alcohol followed byan electrochemical cleaning step with 1 N sulfuric acid beforeadministering modifications tailored to analyte sensing. Selectiveelectrodeposition of thin PPy films (2 mM Py in PBS, 20 mV/s,0.2–1.2 V, 2 cycles) was carried out at bottom sites for DA sensing,

Fig. 1. Micromachined microprobe fabrication overview.
T.T.-C. Tseng, H.G. Monbouquette / Journal of Electroanalytical Chemistry 682 (2012) 141–146 143
while thick PPy films were electrodeposited (200 mM Py in stirredPBS, 0.85 V, �5 min) at top sites for Glut sensing. Electrodepositionwas followed by PPy over-oxidation at 989 mV (vs. Ag/AgCl) forP40 min (until a stable current response was reached). Microelec-trodes were dip-coated with 1% Nafion solution and then baked for3 min at 180 �C (repeated 8 times). GlutOx was selectively immo-bilized on the top left electrode site (Fig. 2a) using a microsyringeunder the microscope. The GlutOx solution for enzyme immobili-zation was prepared by mixing 2 lL GlutOx (250 unit/mL) with3 lL BSA solution (10 mg/mL) containing glutaraldehyde (0.125%
Fig. 2. (a) Scanning electron microscopy (SEM) image of selective GlutOx immo-bilization on the top left microelectrode site previously modified with a thick OPPyfilm and Nafion. (b) Schematic diagram of the final dual Glut/DA sensorconfiguration.
v/v). The resulting dual Glut/DA sensor microprobe was left todry overnight in a dessicator at 4 �C. The final sensor configurationis shown in Fig. 2b. Before making measurements, P30 min ofequilibrium time in sodium phosphate buffer (PBS) was requiredfor the current detected from the dual sensor to approach a con-stant baseline.
3. Results and discussion
3.1. Evaluation of DA fouling on Pt microelectrodes
Bare Pt microelectrode fouling was evaluated over a range of DAconcentrations by cyclic voltammetry. Although several prior stud-ies [35,36] indicated that the DA electrooxidation product canpolymerize and form an insulating film on noble metal electrodesurfaces, we found that fouling occurs only at excessively highDA concentrations. As shown in Fig. 3, repeated potential cyclingat bare Pt microelectrodes gave stable voltammograms in DA solu-tions up to at least 800 lM, which is far beyond the physiologicalconcentration of DA in the central nervous system [5]. At 4 mMDA, the previously reported fouling was observed, as evidencedby the decrease in DA oxidation peak amplitude with each succes-sive cycle. These results provided a promising basis for construc-tion of a useful Pt microelectrode sensor for DA in thephysiological concentration range.
3.2. DA sensor and combined sensing of Glut and DA
The microprobe constructed with DA sensors (Pt microelec-trodes modified with thin OPPy and Nafion) and control sensors
Fig. 3. Repeated cyclic voltammograms with bare Pt microelectrodes in dopaminesolutions of varied concentration (0 lM, 40 lM, 800 lM, and 4 mM in 50 mM PBS,pH 7.4). The scan rate was 100 mV/s conducted over the range, �0.2–0.8 V, for 20cycles.

Fig. 5. Combined sensing of Glut and DA at a constant potential of 0.7 V (vs. Ag/AgCl). The microprobe was tested with AA (250 lM), Glut (20 lM, 40 lM), DA(5 lM, 10 lM), H2O2 (10 lM), DA (60 lM), and Glut (140 lM), sequentially. Thefirst two injections resulting in Glut concentrations of 20 lM and 40 lM aredenoted as Glut0 and the latter injection at higher Glut concentration giving 140 lMis denoted as Glut’’. Similarly, the first two injections resulting in DA concentrationsof 5 lM and 10 lM are denoted as DA0 and the latter injection at higher DAconcentration giving 60 lM is denoted as DA00 .
144 T.T.-C. Tseng, H.G. Monbouquette / Journal of Electroanalytical Chemistry 682 (2012) 141–146
(Pt microelectrodes modified with thick OPPy and Nafion) was firsttested in vitro at 0.7 V (vs. Ag/AgCl) in stirred PBS. As shown inFig. 4, for DA sensors, no detectable response was observed inthe presence of the negatively charged electroactive interferent,ascorbic acid (AA), at up to 750 lM; in addition, the DA sensorsshowed highly sensitive responses with fast temporal resolution(�1 s) upon DA injections. These promising results suggested thatthe optimized combination of electronegative, thin OPPy and neg-atively charged Nafion not only can repel AA at physiological con-centrations (usually ranging from 10 to 200 lM) [41,42], but alsocan preconcentrate cationic DA at the electrode surface withoutsacrificing the DA sensing response time. On the other hand, atcontrol microelectrode sites, no response was observed in the pres-ence of either DA or AA suggesting that the combination of thickOPPy and negatively charged Nafion can reject effectively thesecharged species. This result is consistent with earlier reports thatrelatively thick OPPy can reject both positively charged DA andnegatively charged AA [4,23–26]. Both DA sensing sites and controlsites responded to H2O2 indicating that the small, neutral H2O2
molecule can pass through both permselective polymer layer com-binations and react at the Pt electrode surface at 0.7 V with goodsensitivity and response time.
The dual mode Glut/DA sensor was constructed by further mod-ifying one of the control sites described above with the GlutOximmobilization matrix. The resulting dual Glut/DA sensor wastested in vitro at +0.7 V (vs. Ag/AgCl) in stirred PBS. Again, in thepresence of AA at 250 lM no response was observed from all sen-sor sites (Fig. 5). Upon the addition of Glut, only the Glut sensor site(modified with thick OPPy, Nafion, and GlutOx) responded to Glutinjections with �1 s response time as indicated by the step signalshown; while in the presence of DA, only the DA sensor site (mod-ified with thin OPPy and Nafion) responded to DA additions. Thecontrol site (modified with thick OPPy and Nafion; without Glu-tOx) did not give any detectable responses to AA, Glut, and DA,only to H2O2. In summary: (1) the thick OPPy and Nafion modifiedpermselective films discriminated the small neutral H2O2 moleculefrom anionic and cationic electroactive species (i.e., AA and DA,respectively) [4,23–26] as desired, (2) the detected responses fromthe Glut sensor site upon Glut additions were from GlutOx-cata-lyzed generation of H2O2, (3) the thin OPPy and Nafion modifiedpermselective films on the DA sensor site excluded negativelycharged AA, but permitted access of positively charged DA[18,21], and (4) no diffusional interaction (or crosstalk) of H2O2
generated from the Glut sensor site to other closely arrayed micro-sensors (P40 lm separation) on the same probe occurred [43].This dual Glut/DA sensing data suggests the feasibility of combinedmonitoring of Glut and DA, provided the required sensitivity and
Fig. 4. Microprobe with two DA sensing sites and two control sites was tested withsteps in concentration of AA (250 lM, 500 lM, 750 lM), DA (5 lM, 10 lM), andH2O2 (10 lM, 20 lM) in 50 mM PBS, pH 7.4, at 0.7 V (vs. Ag/AgCl).
detection limit as well as selectivity against other electroactivespecies can be attained.
3.3. Dual Glut/DA sensor calibration curves
Typical calibration curves for the dual Glut/DA sensor are pre-sented in Fig. 6. Based on the calibration curve slope in the linearrange, the Glut microsensor exhibited a sensitivity of126 ± 5 nA lM�1 cm�2 and the DA microsensor had a sensitivity3250 ± 50 nA lM�1 cm�2 (n = 3). The detection limits at two timesthe level of noise were 2.1 ± 0.2 lM for Glut and 62 ± 8 nM for DA(n = 3). The dual sensor displayed a detection range of up toP630 lM for Glut and up to P40 lM for DA, which is suitablefor sensing physiological concentrations of both DA and Glut[3,5]. Although the DA detection limit is somewhat higher thanthat reported for FSCV (�25 nM), it still is sufficient to record thenaturally occurring transients in DA concentration of 0.1–>1 lMobserved in freely moving rats, for example [17]. Thus, the re-sponse time, sensitivity and detection limit of the combined micro-sensor were suggestive of its utility for study of the interplay ofGlut and DA transmission in vivo, provided sensor selectivityagainst a broader array of electroactive interferents could bedemonstrated.
3.4. Selectivity against electroactive interferents
The selectivity against interferents was evaluated for both theGlut and DA microsensors (with selectivity defined here as the ra-tio of sensitivity to the analyte divided by that for the interferent).The Glut microsensor showed excellent selectivity for Glut againstthe interferents tested, including AA, DA, DOPAC, DOPA, epineph-rine (EP), norepinephrine (NEP), uric acid (UA), and serotonin;whereas the DA microsensor showed excellent selectivity for DAagainst AA, DOPAC, DOPA, and UA at (or more than) typical phys-iological concentrations. Average selectivity ratios of Glut to AA(250 lM), DA (12.5 lM), DOPAC (50 lM), DOPA (50 lM), EP(12.5 lM), NEP (12.5 lM), UA (250 lM), and serotonin (5 lM)were all more than at least 1000:1 for the Glut microsensor (wherethe maximum tested concentration of the interferent is given inparentheses); and average selectivity ratios of DA to AA(250 lM), DOPAC (50 lM), DOPA (50 lM), and UA (250 lM) wereall more than at least 1000:1 for the DA microsensor as well.
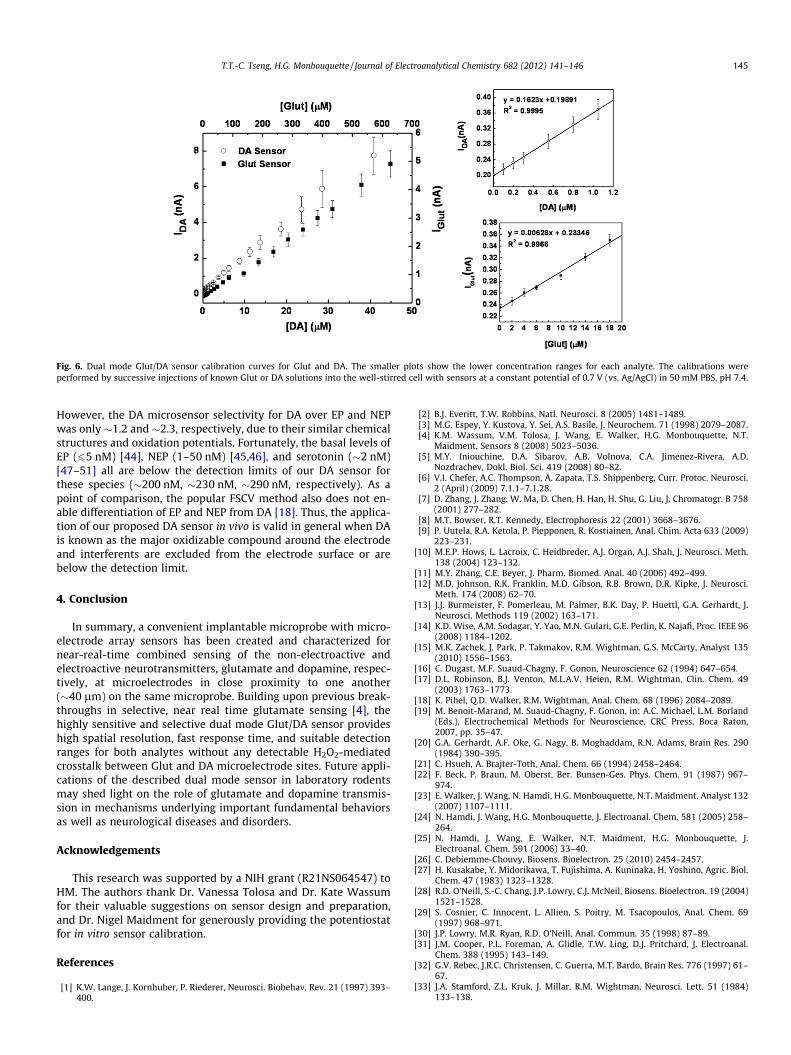
Fig. 6. Dual mode Glut/DA sensor calibration curves for Glut and DA. The smaller plots show the lower concentration ranges for each analyte. The calibrations wereperformed by successive injections of known Glut or DA solutions into the well-stirred cell with sensors at a constant potential of 0.7 V (vs. Ag/AgCl) in 50 mM PBS, pH 7.4.
T.T.-C. Tseng, H.G. Monbouquette / Journal of Electroanalytical Chemistry 682 (2012) 141–146 145
However, the DA microsensor selectivity for DA over EP and NEPwas only �1.2 and �2.3, respectively, due to their similar chemicalstructures and oxidation potentials. Fortunately, the basal levels ofEP (65 nM) [44], NEP (1–50 nM) [45,46], and serotonin (�2 nM)[47–51] all are below the detection limits of our DA sensor forthese species (�200 nM, �230 nM, �290 nM, respectively). As apoint of comparison, the popular FSCV method also does not en-able differentiation of EP and NEP from DA [18]. Thus, the applica-tion of our proposed DA sensor in vivo is valid in general when DAis known as the major oxidizable compound around the electrodeand interferents are excluded from the electrode surface or arebelow the detection limit.
4. Conclusion
In summary, a convenient implantable microprobe with micro-electrode array sensors has been created and characterized fornear-real-time combined sensing of the non-electroactive andelectroactive neurotransmitters, glutamate and dopamine, respec-tively, at microelectrodes in close proximity to one another(�40 lm) on the same microprobe. Building upon previous break-throughs in selective, near real time glutamate sensing [4], thehighly sensitive and selective dual mode Glut/DA sensor provideshigh spatial resolution, fast response time, and suitable detectionranges for both analytes without any detectable H2O2-mediatedcrosstalk between Glut and DA microelectrode sites. Future appli-cations of the described dual mode sensor in laboratory rodentsmay shed light on the role of glutamate and dopamine transmis-sion in mechanisms underlying important fundamental behaviorsas well as neurological diseases and disorders.
Acknowledgements
This research was supported by a NIH grant (R21NS064547) toHM. The authors thank Dr. Vanessa Tolosa and Dr. Kate Wassumfor their valuable suggestions on sensor design and preparation,and Dr. Nigel Maidment for generously providing the potentiostatfor in vitro sensor calibration.
References
[1] K.W. Lange, J. Kornhuber, P. Riederer, Neurosci. Biobehav. Rev. 21 (1997) 393–400.
[2] B.J. Everitt, T.W. Robbins, Natl. Neurosci. 8 (2005) 1481–1489.[3] M.G. Espey, Y. Kustova, Y. Sei, A.S. Basile, J. Neurochem. 71 (1998) 2079–2087.[4] K.M. Wassum, V.M. Tolosa, J. Wang, E. Walker, H.G. Monbouquette, N.T.
Maidment, Sensors 8 (2008) 5023–5036.[5] M.Y. Iniouchine, D.A. Sibarov, A.B. Volnova, C.A. Jimenez-Rivera, A.D.
Nozdrachev, Dokl. Biol. Sci. 419 (2008) 80–82.[6] V.I. Chefer, A.C. Thompson, A. Zapata, T.S. Shippenberg, Curr. Protoc. Neurosci.
2 (April) (2009) 7.1.1–7.1.28.[7] D. Zhang, J. Zhang, W. Ma, D. Chen, H. Han, H. Shu, G. Liu, J. Chromatogr. B 758
(2001) 277–282.[8] M.T. Bowser, R.T. Kennedy, Electrophoresis 22 (2001) 3668–3676.[9] P. Uutela, R.A. Ketola, P. Piepponen, R. Kostiainen, Anal. Chim. Acta 633 (2009)
223–231.[10] M.E.P. Hows, L. Lacroix, C. Heidbreder, A.J. Organ, A.J. Shah, J. Neurosci. Meth.
138 (2004) 123–132.[11] M.Y. Zhang, C.E. Beyer, J. Pharm. Biomed. Anal. 40 (2006) 492–499.[12] M.D. Johnson, R.K. Franklin, M.D. Gibson, R.B. Brown, D.R. Kipke, J. Neurosci.
Meth. 174 (2008) 62–70.[13] J.J. Burmeister, F. Pomerleau, M. Palmer, B.K. Day, P. Huettl, G.A. Gerhardt, J.
Neurosci. Methods 119 (2002) 163–171.[14] K.D. Wise, A.M. Sodagar, Y. Yao, M.N. Gulari, G.E. Perlin, K. Najafi, Proc. IEEE 96
(2008) 1184–1202.[15] M.K. Zachek, J. Park, P. Takmakov, R.M. Wightman, G.S. McCarty, Analyst 135
(2010) 1556–1563.[16] C. Dugast, M.F. Suaud-Chagny, F. Gonon, Neuroscience 62 (1994) 647–654.[17] D.L. Robinson, B.J. Venton, M.L.A.V. Heien, R.M. Wightman, Clin. Chem. 49
(2003) 1763–1773.[18] K. Pihel, Q.D. Walker, R.M. Wightman, Anal. Chem. 68 (1996) 2084–2089.[19] M. Benoit-Marand, M. Suaud-Chagny, F. Gonon, in: A.C. Michael, L.M. Borland
(Eds.), Electrochemical Methods for Neuroscience, CRC Press, Boca Raton,2007, pp. 35–47.
[20] G.A. Gerhardt, A.F. Oke, G. Nagy, B. Moghaddam, R.N. Adams, Brain Res. 290(1984) 390–395.
[21] C. Hsueh, A. Brajter-Toth, Anal. Chem. 66 (1994) 2458–2464.[22] F. Beck, P. Braun, M. Oberst, Ber. Bunsen-Ges. Phys. Chem. 91 (1987) 967–
974.[23] E. Walker, J. Wang, N. Hamdi, H.G. Monbouquette, N.T. Maidment, Analyst 132
(2007) 1107–1111.[24] N. Hamdi, J. Wang, H.G. Monbouquette, J. Electroanal. Chem. 581 (2005) 258–
264.[25] N. Hamdi, J. Wang, E. Walker, N.T. Maidment, H.G. Monbouquette, J.
Electroanal. Chem. 591 (2006) 33–40.[26] C. Debiemme-Chouvy, Biosens. Bioelectron. 25 (2010) 2454–2457.[27] H. Kusakabe, Y. Midorikawa, T. Fujishima, A. Kuninaka, H. Yoshino, Agric. Biol.
Chem. 47 (1983) 1323–1328.[28] R.D. O’Neill, S.-C. Chang, J.P. Lowry, C.J. McNeil, Biosens. Bioelectron. 19 (2004)
1521–1528.[29] S. Cosnier, C. Innocent, L. Allien, S. Poitry, M. Tsacopoulos, Anal. Chem. 69
(1997) 968–971.[30] J.P. Lowry, M.R. Ryan, R.D. O’Neill, Anal. Commun. 35 (1998) 87–89.[31] J.M. Cooper, P.L. Foreman, A. Glidle, T.W. Ling, D.J. Pritchard, J. Electroanal.
Chem. 388 (1995) 143–149.[32] G.V. Rebec, J.R.C. Christensen, C. Guerra, M.T. Bardo, Brain Res. 776 (1997) 61–
67.[33] J.A. Stamford, Z.L. Kruk, J. Millar, R.M. Wightman, Neurosci. Lett. 51 (1984)
133–138.

146 T.T.-C. Tseng, H.G. Monbouquette / Journal of Electroanalytical Chemistry 682 (2012) 141–146
[34] M.L.A.V. Heien, A.S. Khan, J.L. Ariansen, J.F. Cheer, P.E.M. Phillips, K.M. Wassum,R.M. Wightman, Proc. Natl. Acad. Sci. USA 102 (2005) 10023–10028.
[35] R.F. Lane, A.T. Hubbard, Anal. Chem. 48 (1976) 1287–1293.[36] T. Łuczac, Electrochim. Acta 53 (2008) 5725–5731.[37] J. Njagi, M.M. Chernov, J.C. Leiter, S. Andreescu, Anal. Chem. 82 (2010) 989–
996.[38] Y. Wang, Y. Li, L. Tang, J. Lu, J. Li, Electrochem. Commun. 11 (2009) 889–892.[39] B.E.K. Swamy, B.J. Venton, Analyst 132 (2007) 876–884.[40] S.B. Hocevar, J. Wang, R.P. Deo, M. Musameh, B. Ogorevc, Electroanalysis 17
(2005) 417–422.[41] A. Hallström, Å. Carlsson, L. Hillered, U. Ungerstedt, J. Pharm. Methods 21
(1989) 113–124.
[42] R. Spector, New Eng. J. Med. 296 (1977) 1393–1398.[43] J.E. Baur, H.M. Miller, M.A. Ritchason, Anal. Chim. Acta 397 (1999) 123–133.[44] B.R. Dev, P.A. Mason, C.R. Freed, J. Neurochem. 58 (1992) 1386–1394.[45] R. Zini, J.P. Tillement, D. Morin, Biologie Santé 1 (2000) 6–13.[46] P. Devoto, G. Flore, L. Pani, G.L. Gessa, Mol. Psychiatry 6 (2001) 657–664.[47] F. Béquet, D. Gomez-Merino, M. Berthelot, C.Y. Guezennec, Acta Physiol. Scand.
173 (2001) 223–230.[48] I.K. Wright, N. Upton, C.A. Marsden, Psychopharmacology 109 (1992) 338–346.[49] L.H. Parsons, G.F. Koob, F. Weiss, Behav. Brain. Res. 73 (1996) 225–228.[50] P. Kalén, R.E. Strecker, E. Rosengren, A. Björklund, J. Neurochem. 51 (1988)
1422–1435.[51] R. Invernizzi, S. Belli, R. Samanin, Brain Res. 584 (1992) 322–324.