in search of models for hepatic and placental...
TRANSCRIPT

IN SEARCH OF MODELS FOR HEPATIC AND PLACENTAL PHARMACOKINETICS
PÄIVIMYLLYNEN
Department of Pharmacologyand Toxicology,
University of Oulu
OULU 2003


PÄIVI MYLLYNEN
IN SEARCH OF MODELS FOR HEPATIC AND PLACENTAL PHARMACOKINETICS
Academic Dissertation to be presented with the assent ofthe Faculty of Medicine, University of Oulu, for publicdiscussion in the Auditorium of the Department ofPharmacology and Toxicology, on May 9th, 2003,at 12 noon.
OULUN YLIOPISTO, OULU 2003

Copyright © 2003University of Oulu, 2003
Supervised byProfessor Kirsi Vähäkangas
Reviewed byDocent Ulla EkbladProfessor Pauli Ylitalo
ISBN 951-42-7023-1 (URL: http://herkules.oulu.fi/isbn9514270231/)
ALSO AVAILABLE IN PRINTED FORMATActa Univ. Oul. D 724, 2003ISBN 951-42-7022-3ISSN 0355-3221 (URL: http://herkules.oulu.fi/issn03553221/)
OULU UNIVERSITY PRESSOULU 2003

Myllynen, Päivi, In search of models for hepatic and placental pharmacokinetics Department of Pharmacology and Toxicology, University of Oulu, P.O.Box 5000, FIN-90014University of Oulu, Finland Oulu, Finland2003
Abstract
Several in vitro methods using both human and animal tissues have been developed to study hepaticmetabolism and placental transfer. The pressure to minimize animal studies has increased during thepast few decades due to the public opinion and ethical considerations. However, these methods needfurther evaluation of their predictive power when applied in vivo. The aim of this work was to producenew information of the metabolism and transplacental passage of several anticonvulsants as well asto evaluate the usefulness of the placental perfusion method and several in vitro methods foranalyzing metabolism in the prediction of clinical pharmacokinetics.
Carbamazepine (CBZ) metabolism was studied using human and mouse liver microsomes, humanhepatocytes, human liver slices and yeast cells expressing recombinant enzymes. All test systemspredicted well the major metabolite carbamazepine-10,11-epoxide (CBZ-E). Also, minor metaboliteswere produced in slightly variable amounts in all systems except cells with recombinant enzymes. Allhuman liver systems demonstrated that CYP3A4 is the principal CBZ metabolising enzyme.However, our results on CBZ-treated mice suggested that the metabolism of CBZ to CBZ-E is mainlymediated by CYP1A1 in C57/BL6 mice. Autoinduction of CBZ metabolism was seen in hepatocytesand in incubations using microsomes from CBZ-treated mice. Human liver and mouse livermicrosomes metabolized oxcarbazepine (OCBZ) mainly to its active metabolite, 10-hydroxy-10,11-dihydro-carbamazepine (10-OH-CBZ). Also, 10,11-trans-dihydroxy-10,11-dihydro-carbamazepine(10,11-D) and an unknown metabolite were detected.
Placental transfer of lamotrigine (LTG) and diazepam (DZP) was considerable in the humanplacental perfusion system, indicating marked fetal exposure in vivo. The OCBZ, 10-OH-CBZ and10,11-D analyzed from maternal venous and cord blood also suggested significant fetal exposure. Theplacental perfusion system predicts well the transplacental passage of LTG and OCBZ and its majormetabolite. However, in vivo cord blood concentrations of DZP are higher than maternalconcentrations. Placental perfusion studies did not predict this. Still, even with its limitations, thehuman placental perfusion method provides information that can be used to evaluate the risk factorsassociated with drug use during pregnancy because understanding of specific transport characteristicsis a good basis for rational risk assessment.
In conclusion, all of the tested in vitro systems were useful in the prediction of at least someaspects of in vivo pharmacokinetics and metabolism, but validation and refinement are still essential,as is also the need to keep in mind the limitations characteristic of each in vitro method.
Keywords: anticonvulsants, materno-fetal exchange, metabolism, perfusion, pharmacokinetics, placenta


Acknowledgements
This work was carried out at the Department of Pharmacology and Toxicology, University of Oulu under supervision of Professor Kirsi Vähäkangas. I am grateful to my supervisor for introducing me to the fascinating world of science. Her experience and unending optimism have promoted this work in a most positive way.
I wish to express my sincere gratitude to Professor Olavi Pelkonen, Head of the Department of Pharmacology and Toxicology, who provided excellent conditions and an encouraging atmosphere for scientific work.
All the co-authors of the original publications deserve my deepest gratitude. My sincere thanks are due to Päivi Pienimäki, who first introduced to me the topic of transplacental drug transfer, as well as to Päivi Taavitsainen, PhD, whose help with the HPLC system has been valuable. My other co-authors, Professor Hannu Raunio, and Professor Pentti Jouppila and the participants of the EU Biomed2 project EUROCYP are also gratefully acknowledged.
My gratitude is extended to the official reviewers, Professor Pauli Ylitalo and Docent Ulla Ekblad, for their constructive review of the manuscript. Sirkka-Liisa Leinonen is acknowledged for correcting the language.
I wish to thank Merja Luukkonen and Ulla Hirvonen for their excellent technical assistance, Terttu Keränen and Marja Räinä for supplying spotless equipment and Raija Hanni for expert secretarial work. I am greatly thankful for the technical assistance of Kauno Nikkilä and Esa Kerttula, especially in keeping the astrup machine functioning. The friendship and collaboration of Miia Turpeinen, Jaana Rysä, Marja Luodonpää, and Raisa Serpi during the various phases of this project are appreciated. I wish to thank Markku Pasanen for giving constructive criticism and valuable suggestions. My sincere thanks are also due to all the other personnel in the Department of Pharmacology and Toxicology for creating a friendly atmosphere.
In particular, I want to acknowledge the co-operation of the whole nursing personnel in the delivery rooms and maternity wards. Without their co-operation, this work could never have been done.

All my friends are warmly acknowledged. I owe my gratitude especially to Saara-Mari Mähönen, Niina Korpela, Susanna Hannuksela and Jouni Jussila for their friendship throughout these years.
I thank my mother Raili and my sister Pirjo for support and encouragement. Finally, I want to thank my beloved husband Pasi for his love and support. Throughout these years, his help with computers and especially graphical processing has been indispensable.
This work was supported financially by the Foundation of Oulu University, the Research and Science Foundation of Farmos, the Finnish Medical Society, the Foundation of the Pharmacy of the University of Oulu, Finnish Epilepsy Research Foundation, The Finnish Foundation for Drug Research, Duodecim Society of Oulu, and the Finnish Cultural Foundation.
Oulu, April 2003 Päivi Myllynen

Abbrevations
CBZ carbamazepine 2-OH-CBZ 2-hydroxy-carbamazepine 3-OH-CBZ 3-hydroxy-carbamazepine 10-OH-CBZ 10-hydroxy-10,11-dihydro-carbamazepine CBZ-E carbamazepine-10,11-epoxide 9-AC 9-hydroxymethyl-10-carbamoyl acridan 10,11-D 10,11-trans-dihydroxy-10,11-dihydro-carbamazepine OCBZ oxcarbazepine LTG lamotrigine DZP diazepam DMD desmethyl-diazepam HPLC high-performance liquid chromatography CYP cytochrome P450 NADPH nicotinamide adenide dinucleotide phosphate UGTs uridine diphosphate (UDP)-glucuronosyltransferases


List of original articles
This thesis is based on the following articles, which are referred to in the text by their Roman numerals: I Myllynen P, Pienimäki P, Raunio H & Vähäkangas K (1998) Microsomal metabolism
of carbamazepine and oxcarbazepine in liver and placenta. Hum Exp Toxicol 17: 668-676.
II Pelkonen O, Myllynen P, Taavitsainen P, Boobis AR, Watts P, Lake BG, Price RJ, Renwick AB, Gómez-Lechón M-J, Castell JV, Ingelman-Sundberg M, Hidestrand M, Guillouzo A, Corcos L, Goldfarb PS & Lewis DFV (2001) Carbamazepine: a “blind” assessment of CYP-associated metabolism and interactions in human liver-derived in vitro systems. Xenobiotica 31: 321-343.
III Myllynen P, Pienimäki P, Jouppila P & Vähäkangas K (2001) Transplacental passage of oxcarbazepine and its metabolites in vivo. Epilepsia, 42(11): 1482-1485.
IV Myllynen P, Pienimäki P & Vähäkangas K (2003) Transplacental passage of lamotrigine in human placental perfusion system and in maternal and cord blood. Eur J Clin Pharmacol 58: 677-682.
V Myllynen P & Vähäkangas K (2003). An examination of whether human placental perfusion allows accurate prediction of placental diazepam transport. Submitted to J Pharmacol Toxicol Methods.


Contents
Abstract Acknowledgements List of abbreviations List of original articles Contents 1 Introduction ...................................................................................................................13 2 Review of the literature .................................................................................................15
2.1 Drug metabolism ....................................................................................................15 2.1.1 General aspects of drug metabolism................................................................15 2.1.2 Drug-metabolizing enzymes ............................................................................15 2.1.3 Induction and inhibition of drug metabolism...................................................16 2.1.4 Human in vitro models for the study of drug metabolism ...............................17
2.2 Pharmacokinetics during pregnancy .......................................................................18 2.3 Placental pharmacokinetics of xenobiotics .............................................................19
2.3.1 Placental anatomy and physiology ..................................................................19 2.3.2 Principles of placental drug transfer ................................................................21
2.3.2.1 Placental transporters ..........................................................................23 2.3.3 Placental metabolism of drugs.........................................................................24 2.3.4 Models for the study of placental drug metabolism and transfer.....................25
2.3.4.1 Human placental perfusion method.....................................................25 2.3.4.2 Other in vitro methods using human placental tissue ..........................27 2.3.4.3 Clinical studies ....................................................................................29 2.3.4.4 Animal studies .....................................................................................29
2.4 In vitro-in vivo correlation of different methods used as models of placental drug transfer and drug metabolism......................................................30 2.5 Anticonvulsants and epilepsy..................................................................................31 2.6 Pharmacokinetics of selected anticonvulsants ........................................................32
2.6.1 Carbamazepine ................................................................................................32 2.6.2 Oxcarbazepine .................................................................................................34 2.6.3 Lamotrigine .....................................................................................................36 2.6.4 Diazepam.........................................................................................................37

3 Aims of the study...........................................................................................................40 4 Materials and methods...................................................................................................41
4.1 Materials .................................................................................................................41 4.2 Methods to study drug metabolism.........................................................................41 4.3 Methods to study transplacental drug passage........................................................43
4.3.1 Samples from mothers on antiepileptic drug therapy ......................................43 4.3.2 Placental perfusion system ..............................................................................44
4.4 Analysis of drugs ....................................................................................................45 4.5 Statistical analysis...................................................................................................46 4.6 Ethical aspects ........................................................................................................46
5 Results ...........................................................................................................................48 5.1 Metabolism of carbamazepine and oxcarbazepine in liver and placenta ................48
5.1.1 Carbamazepine metabolism.............................................................................48 5.1.1.1 Metabolism of carbamazepine in human liver (papers I and II) ..........48 5.1.1.2 Identification of the CYP enzymes responsible for carbamazepine metabolite formation in various human liver in vitro systems (paper II)....................................................................50 5.1.1.3 Induction potential of carbamazepine (papers I and II) .......................50 5.1.1.4 Metabolism of carbamazepine in mouse liver (paper I) ......................50 5.1.1.5 Metabolism of carbamazepine in human placenta (paper I)................50
5.1.2 Oxcarbazepine metabolism (paper I) ...............................................................51 5.2 Placental perfusion experiments .............................................................................51
5.2.1 Placental perfusion method (papers IV and V) ................................................51 5.2.2 Antipyrine (papers IV and V) ..........................................................................51 5.2.3 Lamotrigine (paper IV)....................................................................................53 5.2.4 Diazepam (paper V).........................................................................................53
5.3 Clinical samples from maternal serum and cord blood...........................................53 5.3.1 Oxcarbazepine (paper III)................................................................................53 5.3.2 Lamotrigine (paper IV)....................................................................................54
6 Discussion......................................................................................................................55 6.1 Carbamazepine metabolism in different in vitro systems .......................................55 6.2 Oxcarbazepine metabolism in vitro in liver and placenta (paper I) ........................57 6.3 Binding of carbamazepine and oxcarbazepine to macromolecules (paper I)..........58 6.4 Distribution of lamotrigine and diazepam in placental perfusions (papers IV and V) ..................................................................................58 6.5 Placental transfer of anticonvulsants in a perfusion system and in clinical samples (papers III-V) ...........................................................................60
7 Summary and conclusions .............................................................................................63 References

1 Introduction
Drugs and other foreign compounds, often referred as to xenobiotics, do not belong to the normal compounds of the human body. Pharmacokinetics describes the processing of a drug by the body. Pharmacokinetics is often divided into 4 phases: absorption, distribution, metabolism and excretion. The rate and extent of each of these processes are influenced by biological, physiological and physiochemical factors as well as environmental and genetic and non-genetic host factors (Park et al. 1996, Kalow 2001). During the past few decades, the pressure towards minimizing animal studies has increased, and in vitro models using human tissues are usually less problematic ethically. The development and evaluation of experimental methods suitable for pharmacokinetic studies is one of the current challenges in drug development. Such methods will be essential for drug development and regulation.
The main mechanism of eliminating drug action is through metabolism. Drug-metabolizing enzymes can be divided into two large subgroups: functionalization and conjugation enzymes. Oxidative cytochrome P450 enzymes are the most important enzymes catalyzing drug metabolism (Lin & Lu 2001). Biochemical studies on the metabolism of various drugs by animals were started in the 1940s (Omura 1999). Nowadays, several human in vitro methods have been developed to study hepatic metabolism (Venkatakrishnan et al. 2001). In vitro studies are widely used to identify metabolic routes and possible interactions between drugs.
Only a few decades ago, it was still commonly believed that the placenta protects the fetus from harmful agents. However, the thalidomide catastrophe in the 1960s made it evident that drugs can cross the placenta and have unwanted effects on the fetus (Koren et al. 1998, Dally 1998). Until now, fewer than 30 drugs have been proved to be teratogenic in humans when used in clinically effective doses (Koren et al. 1998, Webster & Freeman 2001). Eleven of these drugs are included in two therapeutic groups of drugs, namely anticancer agents and anticonvulsants (Webster & Freeman 2001).
It is known that nearly all drugs cross the placenta at least to some extent (Pacifici & Nottoli 1995, Audus 1999). Until recently, passive diffusion was widely believed to be more or less the only mechanism of transplacental drug transport. During the past few years, however, several transporters relevant to placental drug distribution have been

14
discovered (Ganapathy et al. 2000). The target of pharmacotherapy is usually the mother, and transfer of the drug to the fetus is thus unwanted. Recently, however, pharmacological treatments for unborn babies have been also introduced (Ganapathy et al. 2000, Koren et al. 2002). It has been suggested that pharmacological manipulation of drug transporters in the placental tissue might help to optimize the transplacental pharmacotherapy in some cases (Ito 2001).
Nowadays, there is very little information available of the pharmacokinetics of drugs in the feto-placental unit. Detailed information about drug transport across the placenta would be valuable for the development of safe and effective treatments. For reasons of safety, human studies on placental transfer are restricted to a limited number of drugs. Interspecies differences limit the extrapolation of animal data to humans (Ala-Kokko et al. 2000). Several in vitro methods for the study of placental transfer have been developed over the past decades (Ala-Kokko et al. 2000). The placental perfusion method is the only experimental method that has been used to study human placental transfer of substances in organized placental tissue (Pienimäki 1996). Perfusion of a single cotyledon was introduced in the 1960s (Panigel 1962, see Pienimäki 1996 for the history of human placental perfusion). Although the placental perfusion method has been widely used to study of the transplacental passage of both endogenous and exogenous substances (Bourget et al. 1995, Pienimäki 1996, Ala-Kokko et al. 2000), validation for use as a preclinical tool is still lacking (Ala-Kokko et al. 2000).
The aim of this work was to produce new information on the metabolism and transplacental passage of several anticonvulsants as well as to evaluate the usefulness of the in vitro methods used to predict clinical pharmacokinetics.

2 Review of the literature
2.1 Drug metabolism
2.1.1 General aspects of drug metabolism
Genes of xenobiotic metabolizing enzymes exist in all eukaryotic and in most, if not all, prokaryotic cells (Nebert & Dieter 2000). These enzymes metabolize drugs and other compounds (Nebert & Russell 2002). Metabolizing enzymes convert most drugs into more water-soluble metabolites that can be excreted more rapidly (Krishna & Klotz 1994). The biological activity of drugs usually decreases during this process, but it can be also increased or altered. Therefore, metabolism sometimes leads to the transformation of an otherwise harmless substance into an active form, which may be more toxic than the parent compound (Park et al. 1995, Lin & Lu 1998).
In humans, the liver plays a major role in drug metabolism. However, all tissues express drug-metabolizing enzymes. In addition to the liver, other major sites for drug metabolism are the gastrointestinal tract, kidneys, lungs, skin and brain. (Krishna & Klotz 1994, Baron & Merk 2001, Doherty & Charman 2002). Also, it was shown over 30 years ago that the human placenta and the fetus are able to metabolize drugs and environmental chemicals during pregnancy (Welch et al. 1968, Pelkonen 1973, Hakkola et al. 1998, Ring et al. 1999, Pasanen 1999).
2.1.2 Drug-metabolizing enzymes
Drug-metabolizing enzymes have been divided into two large subgroups: functionalization (phase I) and conjugation (phase II) enzymes. Functionalization reactions consist of oxidation, reduction and hydrolysis. These reactions usually lead to a metabolite that is more polar than the parent compound. In a conjugation reaction, an

16
endogenous hydrophilic moiety is attached to a target molecule, producing a metabolite that is more water-soluble than the parent compound. Glucuronidation, sulphation, acetylation and conjugation to glutathione and amino acids are the major conjugation reactions (Krishna & Klotz 1994).
The most important enzyme system for drug metabolism is the cytochrome P450 (CYP) system (Lin & Lu 2001). Klingenberg first reported in 1958 the existence of an unknown carbon monoxide-binding pigment, which is nowadays called cytochrome P450 (see Omura 1999, for the history of cytochrome P450). Today, cytochrome P450 is used as the collective name for a large family of hemoproteins (Omura 1999). Cytochrome P450 enzymes have a wide variety of substrates, including numerous endogenous compounds, such as steroids, bile acids, fatty acids, prostaglandins as well as exogenous compounds (Nelson et al. 1996). The cytochrome P450 enzymes of eukaryotic organisms are all bound to membranes of endoplasmic reticulum or mitochondria, whereas most bacterial P450s are water-soluble (Omura 1999). The principal function of CYP enzymes is the mono-oxygenation of various substances. This reaction requires molecular oxygen and a supply of reducing equivalents from NADPH or NADH. A few P450s also catalyze the intramolecular transfer of the oxygen atom. Cytochrome P450 enzymes have been subdivided into families, subfamilies and isoforms (Nelson et al. 1996, Venkatakrishnan et al. 2001). Currently, more than 270 CYP gene families are known. Humans have 57 CYP genes and 33 pseudogenes arranged into 18 families and 42 subfamilies (Nebert & Russell 2002). The major human drug-metabolizing CYPs belong to the families 1, 2 and 3 (Venkatakrishnan et al. 2001). The most abundant cytochrome P450 enzyme in the human liver is CYP3A4, which contributes to the metabolism of approximately half of the drugs used nowadays (Guengerich 1999).
The regulation of drug metabolism is complex, being affected by both genetic and non-genetic host factors. The occurrence of polymorphic drug-metabolizing enzymes is a common cause for variation in drug metabolism (Kalow 2001, Guengerich 2002). Genetic factors may cause variability in an enzyme’s activity, function, stability and responsiveness to an inducer or regulator (Kalow 2001). Such variation in the metabolism of a drug may predispose patients to adverse effects (Pirmohamed & Park 2001, Park & Pirmohamed 2001). Other host factors, such as diseases, age, stress, obesity, physical exercise, smoking and pregnancy, also affect drug metabolism. In addition to host factors, a large number of environmental factors, such as environmental pollutants, occupational chemicals and other drugs, may affect drug metabolism (Park et al. 1996, Pelkonen et al. 1998, Frederiksen 2001, Lin & Lu 2001).
2.1.3 Induction and inhibition of drug metabolism
Drug interactions are a major concern in pharmacotherapy. Interactions may be pharmacokinetic or pharmacodynamic in origin, pharmacokinetic interactions being more common (Lin & Lu 2001). CYP enzymes are often rate-limiting enzymes in the biotransformation process, and due to this, they have an important role in the determination of in vivo kinetics and interactions (Pelkonen 2002). Induction or inhibition

17
of CYP enzymes is probably the most common cause of documented drug interactions (Lin & Lu 1998).
In drug metabolism research, the term ‘induction’ has been used to refer to an increase in the amount and/or activity of a drug-metabolizing enzyme. Induction occurs either due to increased transcription or translation or as a result of stabilization of enzymes, and it is a slow regulatory process (Lin & Lu 2001). Human drug-metabolizing enzymes can be induced by a large number of exogenous compounds, including drugs as well as endogenous factors (Pelkonen et al. 1998). Most CYP enzymes are inducible, but the extent of induction shows variation. Human CYP1A1/2, 2A6, 2C9, 2C19, 2E1 and 3A4 are known to be inducible (Lin & Lu 2001, Hollenberg 2002).
Drug metabolism by P450 can be inhibited through three different mechanisms: mutual competitive inhibition caused by co-administration of drugs metabolized by the same P450 isozyme, inactivation of the enzyme by the drug metabolite forming a complex with P450 or inhibition by the binding of imidazole or a hydrazine group to the haem portion (Ito et al. 1998b). Unlike enzyme induction, enzyme inhibition is an almost immediate reaction (Lin & Lu 2001, Hollenberg 2002). Wide interindividual variation in the responses to cytochrome P450 inhibition has been observed in vivo (Lin & Lu 2001).
2.1.4 Human in vitro models for the study of drug metabolism
Several in vitro methods have been developed to study human hepatic metabolism. The most commonly used models include liver microsomes, human hepatocytes, liver slices and purified or heterologously expressed drug-metabolizing enzymes (Venkatakrishnan et al. 2001).
Microsomes are the most widely used in vitro systems in drug metabolism research (Ekins et al. 2000). Microsomes contain several membrane-bound drug-metabolizing enzymes, including CYPs, flavin-containing mono-oxygenases and UDP-glucuronyltransferases (Venkatakrishnan et al. 2001). They are formed from smooth endoplasmic reticulum during tissue homogenization and ultracentrifugations. Microsomes generally produce primary metabolites from functionalization reactions (Pelkonen et al. 2001). Microsomes are useful for the study of metabolic routes and the production of metabolites (Pelkonen et al. 2001). Chemical or antibody inhibitors of drug-metabolizing enzymes can be used to study which enzyme or enzyme isoform is responsible for the production of a certain metabolite. The extent of inhibition achieved by a specific inhibitor in human liver microsomes reflects the relative contribution of the enzyme or isoform (Venkatakrishnan et al. 2001). Microsomes are easy to prepare and stable for extended periods when stored properly (Ekins et al. 2000). Potential problems in using microsomes in drug metabolism research include the lack of inhibitor specificity, the low inhibitory potency and the chromatographic interference of the inhibitor with the analytical assay used in the quantitation of a metabolite (Venkatakrishnan et al. 2001).
Human hepatocytes can be isolated from liver biopsies or transplantable livers (Li et al. 1997). One advantage of hepatocytes is that they are intact cells bearing intact plasma membranes, complete metabolic pathways, physiological cofactor-enzyme levels and

18
active gene expression (Li & Kedderis 1997). Because hepatocytes contain the full range of functionalization and conjugation enzymes, the whole metabolite pattern can be detected. Also, the induction of drug-metabolizing enzymes and possible toxic effects can be studied (Li et al. 1997). Prolonged storage of cryopreserved isolated hepatocytes is possible, but cryopreservation usually results in low cell recovery and alterations in functional activities. However, hepatocytes retain functional drug-metabolizing enzyme activities at least for a short time (Guillouzo et al. 1999).
Liver slices were used in metabolism studies in the early 20th century. The revival of this method was stimulated by the development of a culture method and a new tissues slicer, which produced slices of consistent dimensions with minimal cellular trauma (Ekins et al. 2000). Precision-cut liver slices contain the enzymes of the whole liver and the connections between individual cells and thus resemble the in vivo situation more closely than the other in vitro methods. In addition to metabolism studies, human liver slices have also been used to study the mechanisms of hepatic drug uptake (Olinga et al. 2001). Renwick and co-workers have shown that CYP enzyme activities also decrease during incubation in human liver slices (Renwick et al. 2000).
Nowadays, individual CYP enzymes are expressed transiently or stably in a variety of expression systems, including bacteria, yeast, insect and mammalian cells (Gonzalez & Korzekwa 1995, Ekins et al. 2000). Individual CYP isoforms can be used in reaction phenotyping to identify the isoforms responsible for the formation of certain metabolites (Venkatakrishnan et al. 2001).
2.2 Pharmacokinetics during pregnancy
Maternal physiological changes begin early in gestation and are most pronounced in the third trimester (Frederiksen 2001). Plasma volume increases by about 40-50 % during pregnancy and the concentration of plasma albumin decreases (Loebstein et al. 1997, Frederiksen 2001, Loebstein & Koren 2002). In contrast to albumin concentrations, the plasma total protein and α1-acid glycoprotein remain relatively unchanged (Frederiksen 2001). The total plasma concentrations of albumin-bound drugs decrease during pregnancy due to haemodilution (Dawes & Chowienczyk 2001). Due to distribution, metabolism and excretion, however, the free concentrations of drugs are usually not markedly influenced (Frederiksen 2001). Elevation of progesterone levels leads to a delayed gastric emptying and reduced small intestine motility (Frederiksen 2001, Dawes & Chowienczyk 2001, Loebstein & Koren 2002). This may lead to altered absorption of drugs, but the effects on total bioavailability are probably relatively small (Frederiksen 2001, Dawes & Chowienczyk 2001). Nausea and vomiting associated with early pregnancy may also prevent oral absorption (Dawes & Chowienczyk 2001, Loebstein & Koren 2002). The activity of hepatic drug-metabolizing enzymes changes during pregnancy because estrogens and progesterone induce some CYP enzymes and inhibit others (Loebstein et al. 1997, Dawes & Chowienczyk 2001, Loebstein & Koren 2002). Renal blood flow and the glomerulal filtration rate increase, leading to enhanced elimination of some drugs.

19
Pharmacokinetic changes during pregnancy may alter the efficacy of drugs. For instance, the pharmacokinetics of several antiepileptic drugs may be altered during pregnancy (Yerby et al. 1990, Bardy et al. 1990, Lander & Eadie 1991, Bologa et al. 1991, Loebstein & Koren 2002). Some studies suggest that the decline in the serum levels of antiepileptic drugs correlates with an increase in seizure frequency (Al Bunyan 2001). Controversially, some studies suggest that, even though total plasma concentration decreases, the concentration of free drug is not significantly altered (Yerby et al. 1990, Dawes & Chowienczyk 2001, Loebstein & Koren 2002). However, therapeutic drug monitoring is recommended (Bruno & Harden 2002).
2.3 Placental pharmacokinetics of xenobiotics
2.3.1 Placental anatomy and physiology
In mammals, the placenta separates the fetal and maternal circulations. The placenta ensures the maintenance of pregnancy and fetal growth and development. It transfers oxygen, carbon dioxin, nutrients and waste products between the mother and the fetus. In addition to transport functions, the placenta has metabolic, endocrine and immunological functions (Page 1993).
The human placenta consists of 10-40 cotyledons. The exchange between the maternal and fetal circulations takes place in the chorionic villus, which is the functional unit of the human placenta. The villus consist of a central fetal capillary, stroma and an outer trophoblast layer (Kaufmann 1985, Page 1993) (Fig. 1). Trophoblastic cells are present as mononuclear cells called cytotrophoblasts and multinucleate cells called syncytiotrophoblasts (Enders & Blankenship 1999). The composition of the trophoblast layer in the human placenta changes during pregnancy. During the first trimester, the villi have a nearly complete cytotrophoblast layer underneath the syncytiotrophoblast layer. Later in the pregnancy, the cytotrophoblast layer becomes discontinuous (Jones & Fox 1991, Enders & Blankenship 1999). In addition to the trophoblast layer, the fetal and maternal circulations are separated by the trophoblastic basement membrane, connective tissue space, endothelial basement membrane and fetal capillary endothelium (Kaufmann 1985). In contrast to most other tissues, the endothelium in placental fetal vessels does not contain fenestrations (Enders & Blankenship 1999). At the end of the pregnancy, the minimal materno-fetal diffusion distance is about 4 µm (Kaufmann 1985).

20
Maternal blood inthe intervillousspaceSyncytiotrophoblast
Basal lamina andconnective tissueCytotrophoblast
Fetal blood
Capillaryendothelium
Maternal blood inthe intervillousspaceFetal blood
Syncytiotrophoblast
Cytotrophoblast
Basal lamina andconnective tissue
Umbilical arteriesUmbilical veinChorionic arteryand vein
Intervillous spaceVillous tree
Maternal arteryMaternal vein
A
C
B
Maternal cotyledon
Fetal cotyledon
Fig. 1. Human placental barrier between the fetal and maternal blood flows. (A) Schematic presentation of the cell layers separating the maternal and fetal circulations. (B) Structure of the terminal villus. (C) Schematic presentation of blood flow in a human placental cotyledon. The arrows indicate maternal blood flow. The picture is a reprint from International Journal of Obstetric Anesthesia, Vol 9, Ala-Kokko TI, Myllynen P, and Vähäkangas K: Ex vivo perfusion of the human placental cotyledon: implications for anesthetic pharmacology, pages 26-38, 2000 with permission from Elsevier Science.

21
Placental structure and functions show more marked interspecies diversity than any other mammalian organ (Faber et al. 1992, Leiser & Kaufmann 1994). The number of cell layers between the maternal and fetal circulations varies. According to the Grosser classification (Page 1993), placentas can be divided into epitheliochorial, endotheliochorial and hemochorial placentas based on the type of cells between the circulations (Table 1). Hemochorial placentas are further subdivided according to the number of trophoblastic layers (Page 1993). In a hemochorial placenta, maternal blood comes into direct contact with trophoblasts (Page 1993, Leiser & Kaufmann 1994). Placentas have also been divided according to the flow patterns (Table 1). Furthermore, differences between species have been described in placental permeability, transport activity and even metabolic activities (Enders & Blankenship 1999). The only placentas directly comparable to human placentas are those of great apes (Leiser & Kaufmann 1994).
Table 1. Comparison of placentas in different species. The table has been modified from Ala-Kokko and co-workers (2000), and it is based on articles by Page (1993), Leiser and Kaufmann (1994), Enders and Blankenship (1999), and Adamson and co-workers (2002).
Species Tissue layers between maternal and fetal circulations
Placental structure
Flow pattern
Human Hemomonochorial Villous Multivillous Rhesus monkey Hemomonochoria Villous Multivillous Guinea pig Hemomonochorial Labyrinthine Countercurrent Rabbit Hemodichorial Labyrinthine Countercurrent Rat Hemotrichorial Labyrinthine Countercurrent Mouse Hemotrichorial Labyrinthine Countercurrent Cat Endotheliochorial Lamellar Crosscurrent Pig Epitheliochorial Labyrinthine Double crosscurrent Sheep Epitheliochorial Folded Multivillous to
countercurrent
2.3.2 Principles of placental drug transfer
Chemical compounds cross the placenta mainly through simple passive diffusion (Audus 1999). Other possible mechanisms of transport also found in the placenta are facilitated diffusion, active transport, pinocytosis and filtration (Reynolds & Knott 1989, Pacifici & Nottoli 1995).
The important properties of drugs that determine the placental transfer by passive diffusion include molecular weight, pKa, lipid solubility and protein binding (Pacifici & Nottoli 1995, Audus 1999). Chemical compounds with molecular weight of over 500 D are transferred incompletely across the placenta (Pacifici & Nottoli 1995). Generally, the
Met
abol
ite
Feta
l Liv
er
Mat
erna
lTi
ssue
sM
ater
nal
Tiss
ues
Feta
lTi
ssue
sFe
tal
Tiss
ues
Free
Dru
g
Free
Dru
g
Mat
erna
lLi
ver
Mat
erna
lLi
ver
Amni
otic
Flu
id
Mat
erna
llyAd
min
iste
red
Dru
g
Met
abol
ite
Excr
etio
n
Boun
dD
rug
Boun
dD
rug
Met
abol
ite
Plac
enta
Stor
age
Stor
age
Fig.
2. S
chem
atic
pre
sent
atio
n of
dru
g di
spos
ition
in th
e m
ater
no-f
eto-
plac
enta
l uni
t. Pi
ctur
e m
odifi
ed fr
om M
irki
n (1
973)

23
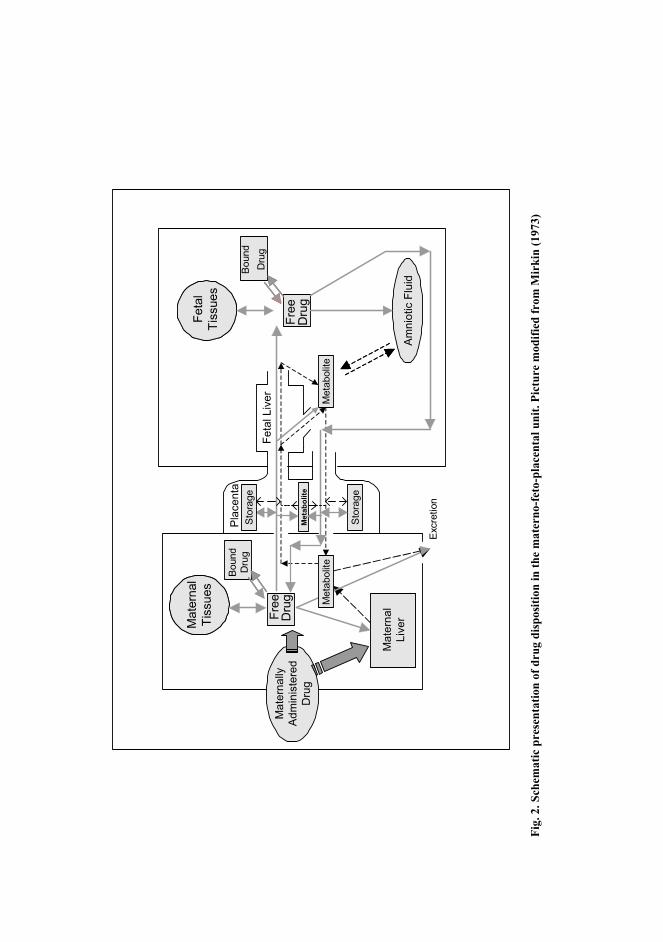
molecular weight of drugs is below this. However, some drugs, such as erythropoietin, do not cross the placenta in significant amounts due to their high molecular weight, as shown in vivo (Widness et al. 1991, Eichhorn et al. 1993) and in placental perfusion studies (Malek et al. 1994, Schneider & Malek 1995, Reisenberger et al. 1997).
Ionization also affects placental transfer. Unionized drugs cross the placenta more easily than ionized drugs (Reynolds & Knott 1989, Audus 1999). Fetal blood is more acidic than maternal blood, and drugs that are weak bases are therefore more ionized in the fetal circulation. This creates a concentration gradient of free drug towards the fetus, also called ion trapping. The concentration gradient is more pronounced in the presence of fetal acidosis (Reynolds & Knott 1989).
Only the free fraction of the drug crosses the placenta (Fig. 2). It has been shown in several in vitro studies that changes in protein concentrations affect placental transfer (Johnson et al. 1997, Johnson et al. 1999, He et al. 2000, Herman et al. 2000). It has been suggested that metabolites from functionalization reactions can be expected to cross the placenta more slowly than the parent compound, and that the placental transfer of conjugate metabolites is negligible (Reynolds & Knott 1989). However, Schenker and co-workers (1999) showed in a perfusion study that a glucuronide metabolite of olanzapine was transferred from the maternal to the fetal circulation, although the transferred amount was less than 10 % of the initial dose during 4-hour perfusion.
2.3.2.1 Placental transporters
During the past years, several placental transporter proteins have been identified (Knipp et al. 1999, Ganapathy et al. 2000, Ugele et al. 2002, St Pierre et al. 2002a, Young et al. 2003). Passive diffusion alone is not adequate to fulfil the fetal requirements for nutrients, and the placenta thus expresses transport proteins for several nutrients, including proteins for the transport of amino acids, fatty acids and glucose (Knipp et al. 1999). Transporters without known physiological substrates have also been identified (Ganapathy et al. 2000).
It was earlier believed that almost all drugs cross the placenta exclusively through passive diffusion. The relevance of several transporters to drug distribution in the placenta has been established recently (Ganapathy et al. 2000). Some of these transporters prevent the entry of xenobiotics into the fetoplacental unit (Ganapathy et al. 2000, Young et al. 2003). The most well-known of these is P-glycoprotein, which is an efflux pump that transports substrates from the intracellular to the extracellular compartment. P-glycoprotein has been detected in human placental trophoblasts from the first trimester to term (Tanabe et al. 2001, Young et al. 2003). In mouse, inhibition of the placental P-glycoprotein results in greatly induced transplacental passage of drugs into the fetus (Smit et al. 1999). Mice not expressing P-glycoprotein are more susceptible to cleft palate after the administration of a P-glycoprotein substrate L-652,280 (Lankas et al. 1998). The current hypothesis is that placental drug-transporting P-glycoprotein protects the developing embryo and fetus from toxic substances and suppresses teratogenesis. Other major drug efflux transporters identified from the placenta are multidrug

24
resistance-associated proteins (MRPs) and breast cancer-resistant protein (BCRP) (Young et al. 2003).
Several transporters facilitate the transfer of drugs to the fetal compartment (Ganapathy et al. 2000). For instance, some of the amino acid transporters may be involved in the transport of pharmacologically active drugs that structurally resemble amino acids. It is known that several therapeutic agents, such as the antiepileptic drug gabapentin, are substrates for specific amino acid transporters (Ganapathy et al. 2000, Ritchie & Taylor 2001, Uchino et al. 2002), and these transporters may therefore facilitate their transfer to the fetal circulation.
It is also possible that exogenous compounds interfere with the placental transfer of endogenous compounds (Ganapathy et al. 2000). For instance, cocaine, nicotine and cannabinoids inhibit amino acid transport in the placenta (Ganapathy et al. 1999). It was also shown in an in vitro perfusion study that cocaine and nicotine interfere with amino acid transfer in the human placenta (Pastrakuljic et al. 2000).
2.3.3 Placental metabolism of drugs
The placental metabolizing enzymes are already present in early pregnancy. In fact, it seems that the placenta expresses a wider variety of enzymes during the first trimester than at term (Hakkola et al. 1996a, Hakkola et al. 1996b). Both during the first trimester and at term, the placenta expresses several CYPs at mRNA levels, although only a few of them are functionally active (Pasanen 1999). Also, some enzymes responsible for conjugative reactions are expressed in the human placenta (Pasanen 1999, Collier et al. 2000, Smelt et al. 2000, Stanley et al. 2001, Collier et al. 2002a, Collier et al. 2002b). These enzyme activities are glutathione S-transferase, epoxide hydrolase, N-acetyltransferase, sulfotransferases and UDP-glucuronosyl transferase. The expression of these enzymes is more probably due to the placental endocrine functions than xenobiotic metabolism (Pasanen 1999). However, Schenker and co-workers (1999) showed that olanzapine is metabolized to N-glucuronide in human placental perfusion, and these enzymes are thus also capable of xenobiotic metabolism.
Environmental factors affect the activity of xenobiotic-metabolizing enzymes in the placenta. The induction of CYP1A1 by maternal cigarette smoking is well-known (Pasanen 1999). It has also been shown that maternal glucocorticoid therapy suppresses the activities of placental xenobiotic and steroid-metabolizing enzymes (Paakki et al. 2000).
Although placental metabolism is more restricted than liver metabolism, placental enzymes are capable of metabolizing several drugs and foreign chemicals (Hakkola et al. 1998, Pasanen 1999). Still, the metabolic activity of the placenta is probably more of toxicological interest than important for the distribution of drugs (Hakkola et al. 1998).

25
2.3.4 Models for the study of placental drug metabolism and transfer
2.3.4.1 Human placental perfusion method
The human placental perfusion system has been widely used to study transplacental passage of both endogenous and exogenous compounds (Omarini et al. 1992, Bourget et al. 1995, Pienimäki 1996, Ala-Kokko et al. 2000) (Table 2). The first placental perfusion experiments were made in the early 20th century (Schmitt 1922, see Pienimäki, 1996, for the history of human placental perfusion). The first perfusion of a single placental lobule was accomplished by Panigel (1962) in the 1960s. Nowadays, several variations of the human placental perfusion system of a single cotyledon exist. All dually perfused systems have separate fetal and maternal circulations. The flow of perfusate can be open or recirculating. In an open system, the samples are collected after one passage through the placenta. The perfusion systems also vary in terms of perfusate composition, gas mixture, flow rate, protein concentrations of the maternal and fetal circulations and perfusion pressure (Schneider & Huch 1985, Miller et al. 1989, Maguire et al. 1999, Ala-Kokko et al. 2000). The addition of a protein, most commonly albumin, makes it possible to evaluate the effect of protein binding on placental transfer (Johnson et al. 1997, Johnson et al. 1999, He et al. 2000, Herman et al. 2000). Most commonly, placentas from uncomplicated pregnancies have been used for perfusion studies, but placentas from complicated pregnancies have also been used to study potential alterations in placental function (Bourget et al. 1995, Osmond et al. 2000, Osmond et al. 2001, Clifton et al. 2001).
Although the placenta has fulfilled its purpose at delivery, it continues to be a viable tissue system under perfusion conditions for a short period. Most commonly, placental perfusions only last for a few hours, but perfusions for up to 48 hours have been described (Polliotti et al. 1996). A perfused human placenta is capable of producing placental secretory proteins, such as human chorionic gonadotrophin (hCG) and human placental lactogen (hPL). Also, oxygen and glucose consumption and lactate production are detectable (Challier et al. 1976, Dancis et al. 1979, Miller et al. 1985, Bloxam & Bullen 1986, Cannell et al. 1988, Hsieh et al. 1992). In addition to these indications of viable tissue, metabolism of both endogenous and exogenous compounds has been detected during human placental perfusions (Dodds et al. 1997, Sun et al. 1999, Nanovskaya et al. 2002). For instance, oxcarbazepine (10,11-dihydro-10-oxo-5H-dibenz[b,f]azepine-5-carboxamide; OCBZ) was metabolized to 10-hydroxy-10,11-dihydro-carbamazepine (10-OH-CBZ) under perfusion conditions in our laboratory (Pienimäki et al. 1997). We have also shown protein production after gene transfection in perfusions lasting for 16 hours (Heikkilä et al. 2002).
No uniform criteria for successful perfusions have been established so far. To confirm the viability of the perfused tissue, histological, biochemical (e.g. blood gas analysis, glucose consumption, hCG and hPL production) and physiological (e.g. perfusion pressure, flow rate, fetal volume loss) evaluations have been made (Cannell et al. 1988, Miller et al. 1989, Ala-Kokko et al. 2000). Several reference substances, such as antipyrine, have also been used to test viability and to normalize data between perfusions

26
(Wier & Miller 1985, Challier 1985, Henderson et al. 1992, Pienimäki 1996, Ala-Kokko et al. 2000).
Placental perfusion can be used to examine the transfer of even toxic substances without ethical concerns of maternal and fetal safety because the placenta is collected after birth. In addition to transfer, placental perfusion simultaneously allows the evaluation of a wide range of other functions (e.g placental metabolism, production and release of hormones and enzymes, transport of nutrients and waste products) (Slikker & Miller 1994). However, physiological conditions are not fully attainable in an isolated organ perfusion system, and the viability of the tissue is limited. Also, perfusions are done using term placentas, and extrapolation of the results to earlier periods is not possible (Bourget et al. 1995, Sastry 1999).
Table 2. Drugs studied in human placental perfusion systems during 1996-2002. For studies published before 1996, see reviews by Bourget and co-workers (1995) and Pienimäki (1996). T =transfer, M=metabolism.
Drug T/M Reference Antipsychotics
Olanzapine T, M Schenker et al. 1999 Antidepressants
Amitriptyline T Heikkinen et al. 2001 Nortriptyline T Heikkinen et al. 2001 Citalopram T, M Heikkine et al. 2002 Fluoxetin T, M Heikkine et al. 2002
Antithrombotic agents Enoxaparin T Lagrange et al. 2002 Fondaparinux T Lagrange et al. 2002
Antiepileptics Valproic acid T Barzago et al. 1996 Oxcarbazepine T, M Pienimäki et al. 1997 Carbamazepine M Pienimäki et al. 1997
Anesthetics Methohexital T Herman et al. 2000 Propofol T He et al. 2000, He et al. 2001, He et al. 2002
Local anesthetics Bupivacaine T Johnson et al. 1999 Ropivacaine T Johnson et al. 1999
Opioids Alfentanil T Giroux et al. 1997 Buprenorphine T, M Nanovskaya et al. 2002 Fentanyl T Giroux et al. 1997 Morphine T Kopecky et al. 1999 Sufentanil T, M Krishna et al. 1997, Johnson et al. 1997,
Giroux et al. 1997
27
Table 2. Continued
Drug T/M Reference Non-steroidal anti-inflammatory drugs
Indomethacin T, M Lampela et al. 1999 Sulindac T, M Lampela et al. 1999
Anti-infectious agents Azithromycin T Heikkinen et al. 2000 Erythromycin T Heikkinen et al. 2000 Roxithromycin T Heikkinen et al. 2000 Clarithromycin T Witt et al. 2003 Trovafloxacin T Casey & Bawdon 2000 Methohexital T Herman et al. 2000 Pyrimethamine T Peytavin et al. 2000
Protease inhibitors Saquinavir T Forestier et al. 2001 Ampenavir T Bawdon 1998 Ritonavir T Casey & Bawdon 1998 Azidothymidine T Boal et al. 1997, Olivero et al. 1999 Lamivudine T Bloom et al. 1997
Nucleoside inhibitors Abacavir T Bawdon 1998 Ritonavir T Casey & Bawdon 1998
Cardiovascular drugs Adenosine T, M Acevedo et al. 1997 Digoxin T Schmolling et al. 1996, Schmolling et al.
1997, Tsadkin et al. 2001 Glyceryl trinitrate T, M Bustard et al. 2002) Clonidine T Ala-Kokko et al. 1997 Dexmedetomidine T Ala-Kokko et al. 1997 Enalaprit T Reisenberger et al. 1996, Miller et al. 1998 Temocapril T Reisenberger et al. 1996 Hydralazine T Magee & Bawdon 2000
Hormones Cortisone T, M Sun et al. 1999 Cortisol T, M Dodds et al. 1997, Sun et al. 1999 Oxytocin T, M Malek et al. 1996 Erythropoietin T Reisenberger et al. 1997
Hyperthyroidism Methimazole T Mortimer et al. 1997 Propylthiouracil T Mortimer et al. 1997

28
Table 2. Continued
Drug T/M Reference Other compounds
Vitamin B12 T Perez-D'Gregorio & Miller 1998 Glucose T, M Nandakumaran et al. 1998, Schroder et al.
1999, Challis et al. 2000, Schneider et al. 2003
Carboxyfluorescain T Bajoria & Contractor 1997a, Bajoria & Contractor 1997b
Granulocyte-macrophage colony-stimulating factor
T Gregor et al. 1999
Nicotine T, M Sastry et al. 1998, Pastrakuljic et al. 1998, Pastrakuljic et al. 2000
Iron T Nandakumaran et al. 2002 Magnesium T Nandakumaran et al. 2002 Selenium T Nandakumaran et al. 2002
2.3.4.2 Other in vitro methods using human placental tissue
The in vitro models for the study of placental transfer and metabolism include cultured tissue slices, cultured syncytiotrophoblast tissue, trophoblast-derived cell cultures, microvillous membrane vesicles, isolated transporters and receptors and microsomes. Placental microsomes are prepared similarly to microsomes from other tissues. They have been used to study the metabolism of exogenous compounds (Bourget et al. 1995).
Human trophoblastic tissue preparations have been used to study the uptake of drugs and amino acids. This uptake represents transfer from maternal circulation into syncytiotrophoblasts. The effects of drugs on the release of hormones, peptides and biogenic amines have also been studied (Sastry 1999). One advantage of this approach is that cellular elements maintain their normal organization. However, the viability of these tissue preparations is restricted (Ringler & Strauss 1990).
Human trophoblast cell lines have been divided into three main groups according to the origin of the cells. Cell lines can be generated from normal tissue, malignant tissue or embryonal carcinomas that show evidence of trophoblast differentiation. Lines have been generated from villous explants or isolated single cells from chorionic villous samples from first-trimester or term placentas and choriodecidua (Bloxam et al. 1997, King et al. 2000). Isolated trophoblasts endogenously express multiple transporters, which makes it possible to study the metabolism and transport of various substances as well as the interactions between xenobiotics and natural substances (Sastry 1999, St Pierre et al. 2002b). However, cells are removed from their normal context, and this may cause them to alter their structure and function (Ringler & Strauss 1990). Thus, the adaptation of cells to an artificial environment and the unpolarized nature of cells in culture are disadvantages of these systems (St Pierre et al. 2002b).

29
Plasma membrane vesicles have been used to study placental transport. Plasma membrane vesicles have been mostly made of human placenta (Bissonnette 1982). Preparations can be isolated both from membranes of the brush border and from the basal surface of term trophoblasts (Boyd 1991). Specific transporter mechanisms located in the membranes facing the maternal or fetal side of the placenta can be examined separately (Bissonnette 1982). Microvesicles formed by plasma membrane have been used to measure amino acid uptake, for example (Sastry 1999).
During the recent years, it has also been possible to clone and express individual carriers in order to study placental transfer. Substrate specificity, inhibitor susceptibility, transport kinetics and regulation of carrier proteins can be studied. These data can be combined with immunological localization of carriers in placental tissue (St Pierre et al. 2002b).
2.3.4.3 Clinical studies
Clinical studies of drug transfer to the fetus are difficult for both ethical and technical reasons (Ala-Kokko et al. 2000), and clinical trials have traditionally not been conducted on pregnant women (Addis et al. 2000). Naturally, it is also impossible to study the transplacental passage of harmful or even potentially harmful agents in vivo in humans. The pharmacokinetics of drugs used during pregnancy can be studied from samples of maternal venous blood and umbilical blood taken after birth (Pacifici & Nottoli 1995). In addition, the information of drug levels in umbilical and maternal blood at birth is obtained from single-point measurements. Such clinical studies do not indicate, for instance, how long it takes to achieve complete equilibration between the mother and the fetus. Indirect information could be gained from the comparison of several patients with different intervals between drug intake by the mother and delivery, but such studies are scarce so far.
It is technically possible to take samples from the cord vein and artery under ultrasound control during pregnancy (Forestier et al. 1984, Pons et al. 1991, Kramer et al. 1995). However, such sampling must be indicated clinically. Sampling of fetal serum, tissues, coelomic and amniotic fluid has also been used to study the transfer of some drugs during legal pregnancy terminations (Pons et al. 1995, Shannon et al. 1998, Jauniaux & Gulbis 2000, Siu et al. 2002).
2.3.4.4 Animal studies
Placental transfer studies in animals have been done using whole animals, such as chronically cannulated sheep, as well as in situ and in vitro perfusions of animal placentas. The animal species most commonly used in perfusion studies is guinea pig, but sheep, rabbits, rats, monkeys, goats and pigs have also been used (Omarini et al. 1992). Animal placentas are more commonly perfused in situ than in vitro (Omarini et al. 1992).

30
In addition to invasive studies, positron emission tomography (PET) has been used to study the transplacental passage of drugs and nutrients (Berglund et al. 1989, Hartvig et al. 1989, Berglund et al. 1990). However, it is important to remember that the placenta shows great interspecies variation (Table 1). In fact, large interspecies differences have been shown in the placental permeability of hydrophilic molecules (Schneider 1991). The sheep placenta does not allow diffusion of hydrophilic molecules with a molecular weight > 400 daltons quite contrary to the guinea pig placenta, which allows diffusion of some molecules with molecular weight up to > 5000 daltons (Schneider 1991). It has also been shown that digoxin crosses the placenta in humans and rodents, while the ovine placenta is relatively impermeable to it (Nau 1986). Furthermore, placental gentamycin transfer differs in humans and goats (Nau 1986). In addition to permeability, other pharmacokinetic factors affecting the transplacental passage of drugs in whole animal models show interspecies diversity (e.g. protein binding, drug-metabolizing enzymes and fetal renal drug clearance) and affect the transfer of drugs into the fetus (Mihaly & Morgan 1983, Nau 1986). Therefore, extrapolation of animal data to the human model is difficult, and it has been suggested that the information obtained on any drug in a pregnant animal model should also be evaluated in a human model (Sastry 1999).
2.4 In vitro-in vivo correlation of different methods used as models of placental drug transfer and drug metabolism
In order to develop safe and effective drugs, it is important to know the exact pharmacokinetics as early as possible. In addition to species-related differences and ethical concerns, animal testing is expensive. Due to this, the development of in vitro model systems to predict the pharmacokinetics of drugs has become increasingly important (Davila et al. 1998, Ito et al. 1998b).
Prediction of the metabolite profile and the rate of metabolism are important aspects of in vitro-in vivo correlations (Lin 1998, Ito et al. 1998a). Prediction of drug interactions is also important. Currently, several methods using preclinical pharmacokinetic data and in vitro human metabolism data have been found useful in the prediction of these human pharmacokinetic parameters (Obach et al. 1997, Ito et al. 1998a, Ito et al. 1998b). Generally, data obtained with human primary hepatocytes and human liver slices have correlated well with the existing in vivo data (Li et al. 1997, Pelkonen et al. 2002). The qualitative metabolite profile obtained in vitro usually reflects quite accurately the in vivo metabolic pattern (Lin 1998). The extrapolation of in vitro data to in vivo conditions is not without problems, however. In the prediction of interactions, both the identity of the CYP isoform responsible for metabolism and the relative contribution of the metabolic pathway to overall elimination must be considered (Lin & Lu 1998). Several factors, including drug, inhibitor and protein concentrations and metabolic geno- and phenotypes, must be taken into account when assessing the clinical signifigance of findings (Lin & Lu 1998, Yuan et al. 1999). The prediction of interactions is even more complicated when multiple CYP enzymes take part in the metabolism (Obach et al. 1997). Also, all in vitro methods have their limitations. For instance, the enzymatic activities in both human liver

31
slices and hepatocytes tend to decrease during incubations (Guillouzo et al. 1999, Renwick et al. 2000, Pelkonen et al. 2002). Other pharmacokinetic factors often also affect the in vivo metabolism. For instance, hepatic blood flow limits the metabolism of some rapidly metabolized compounds, and due to this, in vitro metabolism may show larger interindividual variation than is observed in vivo (Kedderis 1997).
In contrast to the in vitro models for metabolism, only a few efforts have been made to compare in vitro and in vivo data on placental drug transfer. The comparison of in vitro data to clinical data is often difficult due to the lack of clinical data. The few examples from the literature suggest a similar pharmacokinetic profile in vivo and in the placental perfusion model (Omarini et al. 1992, Tuntland et al. 1999, Ala-Kokko et al. 2000). Also, some studies have compared data gained with the placental perfusion method with other models. Tuntland and co-workers compared several methods in the prediction of the mechanism, rate and extent of placental transfer of dideoxynucleoside drugs (Tuntland et al. 1999). Placental transfer of these drugs was found to be similar in human placental perfusion and in vivo in the pregnant macaque model. Dicke and co-workers (Dicke et al. 1988) compared placental transfer of four H2-receptor antagonists in perfused human and baboon placentas and found no significant differences.
2.5 Anticonvulsants and epilepsy
Epilepsy is one of the most common neurological diseases, affecting at least 50 million people worldwide (Scheuer & Pedley 1990). Because the treatment of epilepsy with classic antiepileptic drugs is ineffective in 25-30% of patients and some patients experience intolerable adverse effects, there has been a need for new antiepileptic drugs. The treatment options for epilepsy increased markedly after the introduction of several new drugs, including lamotrigine (3,5-diamino-6-[2,3-dichlorophenyl]-1,2,4-triazine, LTG) and OCBZ, in the 1990s (Pellock 2000). The new antiepileptic drugs are roughly comparable in efficacy to the older antiepileptic drugs, but they are tolerated better (Diaz-Arrastia et al. 2002).
About 0.5 to 1 % of pregnant women have epilepsy (Nulman et al. 1999, Morrell 2002). It is rather common that pregnant women with epilepsy discontinue or greatly reduce their prescribed medication without telling their clinician (Williams et al. 2002). So far, no agreement has been reached about the safest antiepileptic drug during pregnancy (Nulman et al. 1999). It is difficult to obtain a study population large enough for epidemiological studies on the teratogenicity of antiepileptic drugs because both epilepsy and major congenital malformations are fairly uncommon (Dolk & McElhatton 2002). However, it is commonly accepted that antiepileptic drugs bear some teratogenic potential (Morrell 1996, Samren et al. 1997, Holmes et al. 2001). The incidence of major malformations in infants born to mothers with epilepsy taking any one of the antiepileptic drugs is 4-6 % compared with 2-4 % in general populations (Morrell 2002). The most common abnormalities are neural tube defects, midface and digit hypoplasia, microcephaly and growth retardation (Holmes 2002). Lately, concerns have been raised about the in utero exposure to antiepileptic drugs possibly leading to long-lasting

32
neurodevelopmental or neurocognitive deficits (Dean et al. 2002, Holmes 2002, Morrell 2002). Several antiepileptic drugs are folic acid antagonists, and this has been suggested as the mechanism for the teratogenicity of antiepileptic drugs. Folic acid supplementation reduces the risk of neural tube defects in children of women without epilepsy and women who have previously given birth to a child with a neural tube defect. This has led to the recommendation that folic acid should be provided to pregnant women with epilepsy (Hernandez-Diaz et al. 2000, Morrell 2002).
2.6 Pharmacokinetics of selected anticonvulsants
2.6.1 Carbamazepine
Carbamazepine (5H-dibenz[b,f]azepine-5-carboxamide; CBZ) is a tricyclic lipophilic compound. It is widely used for the treatment of epilepsy alone or in combination with other antiepileptic drugs. It is has been shown to be effective in the treatment of simple and complex partial and generalized tonic-clonic seizures, but it is ineffective against generalized absence seizures (Gatti et al. 2001, Macdonald 2002b). It is also used commonly in the treatment of chronic pain syndromes and trigeminal neuralgia as well as in various psychiatric disorders (Beghi 2002, Trimble 2002). Current experimental evidence suggests inhibition of voltage-dependent sodium channels as a major mechanism of action for CBZ (Macdonald & Kelly 1995, Macdonald 2002b). In addition to the parent drug, the main metabolite of CBZ, carbamazepine-10,11-epoxide (CBZ-E), is pharmacologically active (Bertilsson 1978, Kerr & Levy 1995). Other metabolites of CBZ also possess anticonvulsant activity in rodents, but are not present in therapeutically relevant concentrations in patients on CBZ therapy (Kerr & Levy 1995).
CBZ, being a neutral, lipophilic compound, crosses membranes easily (Bertilsson 1978). It is relatively slowly absorbed from the gastrointestinal tract, and its oral bioavailability has been estimated to be over 70 % (Bertilsson 1978, Spina 2002). CBZ is bound both to albumin and, in a lesser degree, to α1-acid glycoprotein (Kodama et al. 1993a, Kodama et al. 1993b, Kodama et al. 1994). In humans, approximately 70 % of the dose is excreted in urine and the rest in feces (Bertilsson 1978). Less than 2 % of the drug is excreted as a parent compound (Spina 2002). CBZ is eliminated mainly hepatically, but its hepatic clearance is small in view of the hepatic blood flow (extraction ratio < 10 %) (Levy & Pitlick 1982). The plasma half-life of CBZ has been reported to range from 18 to 55 hours after a single oral dose (Spina 2002).
In vivo, the biotransformation of CBZ is complex. It is metabolized to over 30 metabolites both in rats and in humans (Lertratanangkoon & Horning 1982, Maggs et al. 1997). CBZ metabolism takes place mainly in the liver. Human fetal liver is also able to catalyze epoxidation of CBZ (Piafsky & Rane 1978). The metabolism of CBZ is dose-dependent in humans (Bernus et al. 1996), and it is metabolized along several major pathways, including the epoxide-diol pathway (quantitatively the most important), aromatic hydroxylations and conjugation reactions (Spina 2002). CBZ is oxidized to its

33
major metabolite, CBZ-E, and further hydrolyzed to 10,11-trans-dihydroxy-10,11-dihydro-carbamazepine (10,11-D) prior to excretion into urine (Spina 2002). Kerr and co-workers have shown that the principal catalyst of the epoxidation reaction is CYP3A4, and CYP2C8 is a minor enzyme involved in this reaction (Kerr et al. 1994). The formation of 10,11-D is catalyzed by epoxide hydrolaze (Faigle & Feldmann 1995). Less important routes of biotransformation are aromatic hydroxylations of the parent drug catalyzed by multiple CYPs leading to the formation of 2-hydroxy-carbamazepine (2-OH-CBZ) and 3-hydroxy-carbamazepine (3-OH-CBZ) (Pearce et al. 2002). Several glucuronide conjugates and other minor metabolites have also been identified (Lertratanangkoon & Horning 1982, Maggs et al. 1997, Spina 2002).
The plasma level/dose ratio and metabolism of CBZ show considerable interindividual variation (Gatti et al. 2001). This is due to such factors as the patient’s age, the daily dosage schedule, formulations and other concurrent medications (Lanchote et al. 1995, Svinarov & Pippenger 1996).
The CBZ drug interactions are mainly pharmacokinetic (Ketter et al. 1991a, Ketter et al. 1991b). It has been known for a long time that CBZ induces cytochrome P450 enzymes (Bertilsson 1978, Wurden & Levy 2002). CBZ is known to induce CYP3A4, but it is also likely to induce other CYP isoforms in vivo (Wurden & Levy 2002). Due to this, it is involved in numerous drug interactions. CBZ has been described to induce the biotransformation of several anticonvulsants, antidepressants, antipsychotics, oral contraceptives, oral anticoagulants, dihydropyridine calcium antagonists and chemotherapeutic agents (Wurden & Levy 2002). CBZ also induces its own metabolism (Mikati et al. 1989). The induction caused by CBZ is dose-dependent (Perucca et al. 1984).
Because CBZ metabolism is mediated through cytochrome P450, it is also induced or inhibited by many other drugs (Faigle & Feldmann 1995, Wurden & Levy 2002). The wide variety of drugs that induce CBZ metabolism include many other antiepileptic drugs, such as felbamate, phenytoin, phenobarbital, and primidone as well as other known enzyme inducers (Spina et al. 1991, Gatti et al. 2001). Erythromycin, fluconazole, ketokonazole and cimetidine are examples of known CYP3A4 inhibitors that reduce CBZ metabolism (Gatti et al. 2001, Wurden & Levy 2002). Inhibition of CYP 3A4 is the only isoform implicated in drug interactions resulting in inhibition of CBZ metabolism and increased CBZ concentrations (Wurden & Levy 2002).
Many CYP3A4 substrates are also substrates for P-glycoprotein, and their interactions may be partially mediated through this transport protein (Kim 2002). However, CBZ is not a substrate for P-glycoprotein according to a recent study (Owen et al. 2001). Valproic acid has been found to increase the free fraction of CBZ slightly, but clinically significant interactions due to altered protein binding are not likely (Wurden & Levy 2002).
CBZ has been used widely during pregnancy. Yerby and co-workers (1985) have suggested that the ratio of CBZ-E to CBZ concentrations rises during pregnancy. Tomson and co-workers (1994) similarly found a slight decrease in CBZ clearance during the last trimester. However, Bernus and co-workers (1995) found increased clearance of CBZ. Thus, the data on CBZ clearance during pregnancy are contradictory (Spina 2002). It is known that CBZ crosses the placenta in significant amounts. CBZ levels in the umbilical cord range from 50 to 80 % of the maternal concentrations (Spina 2002). However, the

34
free concentrations of CBZ, CBZ-E and 10,11-D have been similar (Yerby et al. 1985). The human placental perfusion study by Pienimäki and co-workers (1995b) also supports the assumption of considerable fetal exposure to CBZ.
A recent meta-analysis of prospective studies, including 1255 pooled CBZ-exposed cases, showed CBZ to be teratogenic (Matalon et al. 2002). CBZ therapy increased the rate of congenital malformation from 2.34 % among the control children to 6.7 % among the exposed children. The most common malformations were cardiovascular and urinary tract anomalies, neural tube defects and cleft palate. A combination of CBZ with other antiepileptic drugs is more harmful than CBZ monotherapy (Matalon et al. 2002). Although it has been suggested, on the basis of a case report, that CBZ causes eye malformations (Sutcliffe et al. 1998), a recent epidemiological study showed no clear evidence for an association between CBZ and eye malformations (Kroes et al. 2002). The mechanism of CBZ teratogenesis is not currently known, and putative mechanisms include inhibition of a rapid activating component of potassium channels, folic acid antagonism, reduction of serum all-trans and 13-cis retionoic acid concentrations and formation of reactive metabolites (Fex et al. 1995, Finnell et al. 1995, Amore et al. 1997, Azarbayjani & Danielsson 2002, Spina 2002).
2.6.2 Oxcarbazepine
Oxcarbazepine is a neutral lipophilic 10-ketoanalogue of CBZ. It is currently used in the treatment of epilepsy in both adults and children (Schmidt et al. 2001), also during pregnancy (Wellington & Goa 2001). OCBZ has been used for partial-onset seizures as monotherapy and adjunctive therapy (Schmidt et al. 2001).
The exact mechanism of action of OCBZ is unknown, but it is believed to involve blockade of voltage-dependent sodium channels (Kalis & Huff 2001). It is comparable in efficacy to the older antiepileptic drugs (Guerreiro et al. 1997, Schmidt et al. 2001, Schachter 2002).
OCBZ is completely and rapidly (≥96%) absorbed from the gastrointestinal tract (Tecoma 1999, Kalis & Huff 2001). After absorption, it is efficiently converted to its active metabolite 10,11-dihydro-10-hydroxy-carbamazepine (10-OH-CBZ) in humans (Schütz et al. 1986, Wellington & Goa 2001, Kalis & Huff 2001). This reaction is catalyzed by cytosolic arylketone reductase (Tecoma 1999, Wellington & Goa 2001, Kalis & Huff 2001). The plasma levels of 10-OH-CBZ are about 10-fold compared to those of the parent compound (Kalis & Huff 2001). OCBZ is detectable in blood in low concentrations and only for a few hours after the administration due to the rapid metabolism to 10-OH-CBZ (Dam & Owen 1995). OCBZ can thus be considered as a pro-drug for 10-OH-CBZ. Steady-state plasma concentrations of 10-OH-CBZ are achieved within 2-3 days in patients when OCBZ is given twice a day (Schmidt et al. 2001) The plasma protein binding of OCBZ is 60-67 % and that of 10-OH-CBZ 30-40 % (Kalis & Huff 2001, Schmidt et al. 2001). In plasma, 10-OH-CBZ is mainly bound to albumin (Schmidt et al. 2001). OCBZ is eliminated almost completely through the kidneys. More than 96% of the parent compound and metabolites end up in urine. 10-OH-CBZ is further

35
metabolized along two pathways in humans. Quantitatively the more important is the conjugation of 10-OH-CBZ to the glucuronide metabolite by glucuronyltransferase. The other pathway is oxidation to 10,11-D (Kalis & Huff 2001). Schütz and co-workers (1986) detected in two volunteers an unknown derivate of 10-OH-CBZ after a single oral dose. Later, an unknown trace metabolite was also detected in human placental perfusion studies and incubations using rat liver microsomes in our laboratory (Castrén et al. 1996, Pienimäki et al. 1997). However, it is not known whether these findings represent the same metabolite.
Unlike carbamazepine, oxcarbazepine has little, if any, effect on oxidizing cytochrome enzymes (Larkin et al. 1991). As a result, there are fewer drug interactions than with CBZ (Baruzzi et al. 1994, Sabers & Gram 2000, Wellington & Goa 2001, Kalis & Huff 2001). However, OCBZ may diminish the effectiveness of oral contraceptives (Jensen et al. 1992, Wellington & Goa 2001). It has been suggested that OCBZ could induce the CYP3A subfamily responsible for the metabolism of oral contraceptives and dihydropyridine calcium antagonists (Benedetti 2000). Phenobarbital has accelerated the biotransformation of OCBZ and 10-OH-CBZ slightly, but the magnitude of the change is unlikely to be clinically significant (Tartara et al. 1993). Also, concomitant use of OCBZ and CBZ leads to lowered 10-OH-CBZ levels (McKee et al. 1994). None of the known inhibitors of CYP3A interfering with CBZ metabolism (e.g. erythromycin, cimetidine, verapamil) inhibit the metabolism of OCBZ (Benedetti 2000).
A total of 47 pregnancies during OCBZ therapy had been reported into the Novartis database before August 31st, 1998 (Schmidt et al. 2001). Five malformations were reported and in 3 of these cases, the patients were also receiving other antiepileptics (Schmidt et al. 2001). Friis and co-workers (1993) described the pregnancy outcomes of 12 women with OCBZ medication during the first trimester of pregnancy from a larger cohort studied for the safety and efficacy of OCBZ. Nine mothers had a full-term baby without structural abnormalities, and three experienced a spontaneous abortion. Lindhout and Omtzigt (1994) report in their review article 11 prospectively monitored pregnancies. One case of spina bifida was diagnosed with concomitant therapy of OCBZ with valproate and clobazam. There is a case report of one child born with mild facial dysmorphism to a mother on OCBZ therapy throughout the pregnancy (Bülau et al. 1988). Later in life, however, these dysmorphic features disappeared and the child developed normally without any signs of mental retardation or neurological defects.
The teratogenic potential of OCBZ was studied in the preclinical phase of drug development. Three separate review articles refer to this data. According to a review by Dam and Jensen (1989), OCBZ showed no evidence of teratogenic potential in preclinical testing. According to more previous review, however, the rate of embryonic mortality in rats was increased over that of the control group with a 300 mg/kg dose (Dam & Owen 1995). Tecoma (1999) reports in her review that some antenatal and postnatal growth delay was observed in a dose-dependent fashion. OCBZ teratogenicity has also been studied in mice. No increase was found in the number of malformations beyond that seen in pair-fed, untreated control SWV mice (Bennett et al. 1996). However, more experience is needed before the teratogenic potetantial of OCBZ can be reliably estimated.
The pharmacokinetic data on OCBZ in humans during pregnancy is also limited. Bülay and co-workers (1988) reported concentrations from one mother and neonate, and

36
Pienimäki and co-workers (1997) reported the concentrations of three mothers. These cases indicate significant fetal exposure to OCBZ, the maternal and fetal concentrations being close to each other. In the human placental perfusion studies by Pienimäki and co-workers (1997), OCBZ crossed the placenta in significant amounts.
2.6.3 Lamotrigine
The first clinical trials with LTG began in 1984, and LTG was introduced for the treatment of epilepsy in Europe in 1991 (Richens 1994, Matsuo 1999). Originally, LTG was synthesized with the aim of producing new folate antagonists because of the former hypothesis that the anticonvulsant activity of some antiepileptic drugs (e.g. phenytoin) could be related to their ability to disturb folate metabolism. Although LTG has weak antifolate activity, subsequent studies indicated that it inhibits voltage-dependent sodium channels as its main mechanism of action (Rambeck & Wolf 1993). CBZ, OCBZ and LTG have shown similar potency in inhibiting the release of glutamate, possibly by blocking sodium channels in rat brain slices (Waldmeier et al. 1995). In animal models, LTG has a similar antiepileptic profile as phenytoin and CBZ. In humans, the efficacy of LTG is better than that of placebo and comparable to that of CBZ (Fitton & Goa 1995, Marson et al. 1997, Matsuo 1999, Kwan & Brodie 2001). According to recent reviews, LTG is effective for a broad range of seizure types both in children and in adults (Besag et al. 1995, Brodie et al. 1995) and moderately effective in bipolar disorders (Engle & Heck 2000, Hurley 2002). In addition, LTG has been studied in major depressive disorder, but the existing data are still inadequate for the evaluation of its efficacy for this purpose (Hurley 2002). LTG is also a promising candidate for the treatment of neuropathic pain (McCleane 2000, Simpson et al. 2000, Vestergaard et al. 2001).
LTG is rapidly absorbed after oral administration, and dosage and plasma concentrations show a linear correlation (Peck 1991). The bioavailability of LTG is about 98 %, and it is moderately bound to proteins (~55%) (Rambeck & Wolf 1993, Dickins & Chen 2002). Lamotrigine is mainly conjugated by N-glucuronidation and excreted renally (Cohen et al. 1987, Peck 1991, Sabers & Gram 2000, Benedetti 2000). The elimination half-life of LTG varies between 22 and 59 hours, depending on concurrent medications (Rambeck & Wolf 1993, Dickins & Chen 2002). Valproate inhibits LTG metabolism by competing for glucuronidation (Sabers & Gram 2000, Benedetti 2000). Concomitant administration of conventional antiepileptic drugs (CBZ, phenytoin, primidone, phenobarbital) reduces the half-life of lamotrigine (Benedetti 2000), and the discontinuation of such inducing medication increases the LTG concentration (Anderson et al. 2002). Also, OCBZ decreases lamotrigine concentrations by one third (Sabers & Gram 2000) and methsuximide by half (Besag et al. 2000), while felbamate and topiramate have no clinically significant effects on LTG concentrations (Colucci et al. 1996, Berry et al. 2002). Oral contraceptives have reduced LTG plasma levels, possibly through induction of glucuronide-conjugating enzymes (Sabers et al. 2001). LTG seems neither to induce nor to inhibit CYP enzymes (Benedetti 2000), and it has no effect on the pharmacokinetics of phenytoin, CBZ, phenobarbital or primidone (Jawad et al. 1987). As

37
monotherapy, LTG may enhance its own plasma clearance, suggesting weak induction of some UGTs (Benedetti 2000). The addition of LTG to valproate therapy has caused a small decrease in the mean plasma concentration of valproate, possibly due to the induction of some UGTs (Anderson et al. 1996).
Pharmacokinetic findings of LTG during pregnancy are limited. The pharmacokinetic parameters of LTG do not change during pregnancy in dogs (Matar et al. 1999). In humans, transplacental passage of LTG has been described in the literature in only 11 pregnancies (Tomson et al. 1997, Rambeck et al. 1997, Ohman et al. 2000). In these studies, the LTG concentrations in maternal venous blood have been similar to the cord blood concentrations, indicating extensive transfer of LTG. In animals, placental transfer of LTG has been studied in rats and rabbits, and it seems that transplacental passage of LTG is considerable (Parsons et al. 1995).
Because LTG is a weak dihydrofolate reductase inhibitor, the possibility of teratogenicity must be considered (Richens & Yuen 1991). The latest publication from the lamotrigine pregnancy registry (Tennis & Eldridge 2002) reports the outcomes of 334 pregnancies after first-trimester exposure. Three major birth defects (1.8 %) were reported from 168 mothers on monotherapy. Polytherapy increased the risk of major malformations. When LTG was combined with valproate, five (10 %) major birth defects were observed in 50 outcomes. Without valproate in LTG polytherapy, the major malformation rate was five (4.3 %) out of 116 pregnancies. Also, Ozkinay and co-worker (2003) report a case after LTG and valproate exposure in utero. The baby was born with dysmorphic features and 47,XXX karyotope. Whether the observed aberrations were due to LTG, valproate or coincidence is not known. A review by Morrell (2002) reports intrauterine growth retardation and delayed ossification in preclinical reproductive toxicity tests. In addition to this, one recent study in rats suggests teratogenic effects of LTG on the developing brain tissue (Marchi et al. 2001). Clearly, more clinical experience is needed before the safety of LTG can be fully established (Webster & Freeman 2001).
2.6.4 Diazepam
Benzodiazepines are used as anticonvulsants primarily to treat status epilepticus. They are also used for anxiety, as muscle relaxants and for their hypnotic activity (Macdonald 2002a). Diazepam (7-chloro-1,3-dihydro-1-methyl-5-phenyl-2H-1,4-benzodiazepin-2-one; DZP) is a long-acting benzodiazepine. Its mechanism of action is interaction with GABAA receptors and regulation of the single-channel properties of the receptor. In addition to central benzodiazepine receptors, peripheral benzodiazepine binding sites have been described (Fares & Gavish 1986, Zisterer & Williams 1997, Akinci & Schofield 1999). Peripheral benzodiazepine binding sites also exist in human term placental membranes (Fares & Gavish 1986).
The absorption, distribution and metabolism of DZP are well-known and have been presented in most textbooks of pharmacology. DZP is rapidly absorbed form the gastrointestinal tract and also from the rectum. The achieved plasma concentrations of

38
DZP differ widely, being up to 10-fold after a single oral dose. After administration, DZP distributes rapidly in lipoid tissue and crosses easily the blood-brain barrier. The apparent volume of distribution ranges from 1 to 2 liters/kg (Schmidt 1995). DZP is strongly bound to plasma proteins (97-99%), mainly to albumin (Anderson & Miller 2002). Humans excrete 62-73% of DZP in urine and approximately 10 % in feces (Schmidt 1995). No significant amounts are excreted in bile. Only a small percentage (2 to 3%) is excreted unchanged in urine (Anderson & Miller 2002). DZP is metabolized to several active metabolites. The demethylation reaction to desmethyl-diazepam (DMD) is catalyzed by CYP2C19 and CYP3A4. DMD is further hydroxylated to oxazepam by CYP2C19. Another route of DZP metabolism is hydroxylation by CYP3A4 to temazepam, which is further demethylated to oxazepam or excreted unchanged (Anderson & Miller 2002). The elimination half-life of DZP is 1 to 2 days (Anderson & Miller 2002). Pregnancy prolongs the elimination half-life to 2 to 3 days, probably because of a change in the volume of distribution (Schmidt 1995, Anderson & Miller 2002).
In humans, DZP crosses the placenta rapidly. In most studies, the cord blood concentrations at birth exceed the maternal concentrations (Table 3). In mice, hamsters and monkeys, efficient placental transfer, pronounced uptake by the fetus and long retention of DZP and its major metabolite in the fetal circulation have been found (Iqbal et al. 2002a).
The overall findings concerning teratogenicity, postnatal development and behavioral effects after DZP exposure during pregnancy or lactation are inconsistent (Iqbal et al. 2002a). Fetal developmental disturbances have been reported after DZP treatment during pregnancy in rats, hamsters, mice and cats (Iqbal et al. 2002a). In humans, some studies suggest that DZP causes malformations (e.g. oral clefts, central nervous system anomalies, cardiovascular anomalities), while others show no signs of teratogenicity (see McElhatton 1994, Iqbal et al. 2002a, Iqbal et al. 2002b, for reviews). So far, most reviewers have concluded that DZP is not teratogenic (Iqbal et al. 2002b). However, neonatal withdrawal syndrome and various acute toxic effects have been described in newborns when DZP has been used near term (Schmidt 1995, Iqbal et al. 2002b).

39
Tabl
e 3.
Stu
dies
of t
rans
plac
enta
l pas
sage
of d
iaze
pam
in v
ivo
in h
uman
s at t
erm
.
Dos
e n
Adm
inis
tratio
n in
terv
al
Cor
d or
feta
l cap
illar
y bl
ood
/ m
ater
nal r
atio
(m
ean±
STD
)
Cor
d or
feta
l cap
illar
y bl
ood/
m
ater
nal r
atio
(r
ange
)
Ref
eren
ces
0.2
mg/
kg iv
. 6
17-2
65 m
in
2.0
a,c
1.9
a,d
NA
K
anto
et a
l. 19
73
0.3
mg/
kg iv
. 30
3-
13 m
in s
0.57
±0.3
8b 0.
19-1
.0
Bak
ke e
t al.
1981
5
mg
iv.
5 90
-180
min
1.
73±0
.47a,
e 0.
92±0
.09a,
f
1.3-
2.4e
0.8-
1.0f
Rid
d et
al.
1989
10 m
g iv
. 6
12-2
15 m
in
1.32
±0.3
7a 0.
90-1
.94
Man
delli
et a
l. 19
75
10 m
g iv
11
15
-205
min
1.
45a
1.07
-2.5
9 G
ambl
e et
al.
1977
20
mg
iv.
30
3 m
in 4
s -1
3.5
min
0.
84a
0.33
b
0.49
-1.4
3 0.
16-0
.60
Har
am &
Bak
ke 1
980
30 m
g iv
. 73
55
s-13
.5 m
in
NA
0.
43-1
.48a
Bak
ke &
Har
am 1
982
30 m
g iv
. 33
55
s- 5
min
5 s
0.82
±0.3
5a 0.
19-1
.67
Har
am e
t al.
1978
20
0 m
g iv
. (r
epea
ted
dose
s)
1 >
600
min
4.
84a
- M
cCar
thy
et a
l. 19
73
10 m
g im
. 37
5-
401
min
1.
8a N
A
Erkk
ola
et a
l. 19
73
10 m
g im
. 16
30
-140
min
1.
4a,c
1.3a,
d N
A
Kan
to e
t al.
1973
10 m
g im
. 7
40-1
60 m
in
1.2±
0.59
a 0.
73-2
.5
Man
delli
et a
l. 19
75
10 m
g im
. x 2
4
480-
2160
h
2.05
±1.1
5a 1.
31-3
.77
Man
delli
et a
l. 19
75
5 m
g po
. 15
37
-165
min
2.
40±1
.65a,
d 0.
24-7
.8
Kan
to &
Sch
eini
n 19
87
15-2
0 m
g/ d
ay p
o.
4 R
epea
tedl
y fo
r 25
to
50 d
ays
1.50
±0.3
3a 1.
09-2
.00
Man
delli
et a
l. 19
75
a C
ord
bloo
d/m
ater
nal r
atio
; b Feta
l cap
illar
y bl
ood/
mat
erna
l rat
io; c um
bilic
al a
rtery
; d um
bilic
al v
ein;
NA
= no
t ava
ilabl
e; e
Tota
l dia
zepa
m; f u
nbou
nd d
iaze
pam

3 Aims of the study
Several human in vitro methods have been developed to study metabolism and placental transfer of drugs in humans. Although these methods are widely used, the evaluation of in vitro-in vivo correlations is still going on. The aim of this work was to produce new information on the metabolism and transplacental passage of several anticonvulsants as well as to evaluate the usefulness of in vitro methods in the prediction of clinical pharmacokinetics. The specific aims were as follows.
1. In the metabolism studies:
a. To characterize the metabolism of carbamazepine and oxcarbazepine in different tissues.
b. To evaluate the accuracy of different in vitro models in the prediction of in vivo carbamazepine metabolism.
c. To compare carbamazepine metabolism in mouse and human.
2. In the transplacental passage studies:
a. To set up an HPLC method for measuring lamotrigine and diazepam in perfusion medium.
b. To determine the transplacental passage of selected anticonvulsants, such as lamotrigine, oxcarbazepine and diazepam.
c. To test whether the human placental perfusion method could be used as a preclinical test for studying the materno-fetal transfer of new drugs

4 Materials and methods
4.1 Materials
The chemicals were usually obtained from commercial sources as mentioned in the original papers. Carbamazepine and oxcarbazepine and their metabolites were gifts from Novartis (Basle, Switzerland), while lamotrigine was a gift from Glaxo Wellcome (Cardiff, UK) and diazepam from Orion Pharma (Espoo, Finland). The structural and physiochemical properties for CBZ, OCBZ, LTG, DZP and antipyrine are presented in Table 4.
4.2 Methods to study drug metabolism
The models used to study CBZ and OCBZ metabolism are presented in Table 5. The detailed methods and the literature references are presented in the original publications, indicated with roman numerals in the Table. The incubation conditions as well as the inhibitors used in this work have also been presented in detail in the original publications.

42
Tabl
e 4.
Str
uctu
res
and
phys
ioch
emic
al p
rope
rtie
s of
car
bam
azep
ine,
oxc
arba
zepi
ne,
lam
otri
gine
, di
azep
am a
nd a
ntip
yrin
e (D
olle
ry
1991
, Pie
nim
äki 1
996,
Giro
ux e
t al.
1997
, Spi
na 2
002,
Dic
kins
& C
hen
2002
, Bia
ler 2
002)
.
Com
poun
d C
arba
maz
epin
e O
xcar
baze
pine
La
mot
rigin
e D
iaze
pam
A
ntip
yrin
e
Stru
ctur
e
N
ON
H2
N
O ON
H2
N
NN
NH
2N
H2
Cl
Cl
Cl
N
N
CH
3O
CHC
H
NN
O3
3
Empi
rical
form
ula
C15
H12
N2O
C
15H
12N
2O2
C9H
7Cl 2N
5 C
16H
13C
lN2O
C
11H
12N
2O
Mol
ecul
ar w
eigh
t 23
6.3
252.
3 25
6.1
284.
7 18
8.2
Che
mic
al n
atur
e N
eutra
l sub
stan
ce
Neu
tral s
ubst
ance
W
eak
base
W
eak
base
N
Aa
Solu
bilit
y in
wat
erb
0.24
g/l
0.13
g/l
0.17
g/l
Ver
y sl
ight
>1
g/1m
l
pKa
NA
a 10
.7
5.5
3.4
1.4
Parti
tion
coef
ficie
nt
2.4
5c 1.
31b
1.19
d H
igh
0.73
c
a Not
ava
ilabl
e; b So
lubi
lity
in w
ater
, 37°
C; c lo
g pa
rtitio
n be
twee
n n-
octa
nol/a
q pH
7.4
; d log
parti
tion
betw
een
n-oc
tano
l/aq
pH 7
.6

43
Table 5. In vitro research models used to study metabolism
Research model Drug studied
Paper Studies made
Human liver microsomes CBZ, OCBZ I, II Metabolite identification Inhibition of CYP-specific enzyme activities Inhibition of microsomal metabolism by CYP-selective inhibitors Metabolite profile catalysed by microsomes previously characterized for CYP content and activity (correlation study) Covalent binding to DNA
Human liver slices CBZ II Metabolism Cultured human hepatocytes CBZ II Metabolism
Autoinduction of CBZ metabolism 7-ethoxycoumarin O-deethylation activity Cytotoxicity
Recombinant CYP enzymes expressed in yeast cells (CYP1A1, 1A2, 2A6, 2B6, 2C9, 2D6, 2E1, 3A4)
CBZ II Pattern of enzymes metabolising CBZ
Mouse liver microsomes CBZ, OCBZ I Metabolite identification Autoinduction of CBZ metabolism 7-ethoxyresorufin O-deethylase and testosterone 6β-hydroxylase activities
Human placental microsomes CBZ, OCBZ I Metabolite identification after CBZ induction
4.3 Methods to study transplacental drug passage
4.3.1 Samples from mothers on antiepileptic drug therapy
Serum samples from maternal venous and mixed cord blood were obtained during delivery after informed consent. The pregnant mothers had been treated with OCBZ (12 patients) or LTG (2 patients) alone or in combination with other antiepileptic drugs. Placental tissue samples were also obtained from 8 out of 12 patients on OCBZ therapy.

44
4.3.2 Placental perfusion system
The perfusion method is based on the methods of Schneider and co-workers (1985), Miller and co-workers (1985), and Schenker and co-workers (1987) and Pienimäki (1996) modified it for the use in our laboratory, partly based on earlier perfusion studies conducted at the laboratory (Vähäkangas 1981). Placentas were obtained from the Department of Gynaecology and Obstetrics, Oulu University Hospital, immediately after either normal vaginal delivery or caesarean section performed at term. All pregnancies were uncomplicated, and all mothers were non-smokers.
Krebs-Ringer-phosphate buffer with heparin was injected within 10 minutes after delivery into the vessels of the placenta. An intact (peripheral) cotyledon with a single chorionic artery and vein was used in the placental perfusion experiment. Fetal vessels were cannulated and the lobe was placed in the perfusion apparatus (Fig. 3). On the maternal side, two cannulas were placed into the intervillous space through the basal
Maternalreservoir
Fetalreservoir
Pump
PLACENTALLOBULE
O /CO2 N /CO
Fetal arterial flow
Fetal venous flow
Maternal venous flow
Intervillous flow
Pump
2 2 2
Fig. 3. The dual human placental perfusion system. The picture is a reprint from International Journal of Obstetric Anesthesia, Vol 9, Ala-Kokko TI, Myllynen P, and Vähäkangas K: Ex vivo perfusion of the human placental cotyledon: implications for anesthetic pharmacology, pages 26-38, 2000 with permission from Elsevier Science.

45
plate. The perfusate consisted of Krebs-Ringer-phosphate-bicarbonate buffer with heparin (25 IU/ml), glucose (1g/L) and Dextran T 40 (30 g/L in fetal and 8.4 g/L in maternal perfusate). The final maternal volume was 200 ml and the fetal volume 100 ml. Both the maternal and the fetal sides were perfused, and the perfusates were recirculated (Fig. 3). Perfusion flow was 5 ml/ min on the fetal side and 9 ml/min on the maternal side. The maternal perfusate was gassed with 95% O2/5% CO2 and the fetal perfusate with 95% N2/5% O2 by a membrane oxygenator (Nevasaari 1976). Preperfusion lasted for 30-60 minutes. If the placenta was stable (leak less than 2 ml/h, fetal perfusion pressure less than 70 mmHg), study drugs were added to the maternal perfusate and the perfusions were continued for 2 hours. The study drugs and the reference compound antipyrine were added into the maternal perfusate simultaneously. Altogether 16 placentas were perfused with lamotrigine and 7 placentas with diazepam. As a control, one placenta was perfused only with antipyrine. pH, pO2 and pCO2 values were monitored every half an hour from the maternal inflow, maternal venous pool and fetal reservoir (ABL 300, Radiometer, Copenhagen, Denmark).
4.4 Analysis of drugs
The drug concentrations were analyzed using high-performance liquid chromatography (HPLC) in all studies. CBZ and OCBZ and their metabolites were analyzed as described previously (Pienimäki et al. 1995a). The same method was used to analyze LTG (Table 6, Fig. 4). For DZP, a HPLC method was set up. The conditions for analysis and the
Table 6. Isocratic HPLC assays used to determine the perfusate and plasma concentrations of drugs.
Drug Paper Column Detection Wave length (nm)
Mobile phase
CBZ I, II Superspher 60 RP-select B (125 x 4 mm i.d., 4 µm)
212 ACN 20%, 20mM KH2PO4
80%, TEA 0.05% OCBZ I, III Superspher 60 RP-select B (125
x 4 mm i.d., 4 µm) 212 ACN 20%, 20mM KH2PO4
80%, TEA 0.05% LTG IV Superspher 60 RP-select B (125
x 4 mm i.d., 4 µm) 212 ACN 20%, 20mM KH2PO4
80%, TEA 0.05% DZP V Waters Symmetry C18 (3.9 x 150
mm, 5 µm) 204 ACN 35%, 20mM KH2PO4
65%, TEA 0.05% ANT IV Superspher 60 RP-select B (125
x 4 mm i.d., 4 µm) 265 ACN 20%, 20mM KH2PO4
80%, TEA 0.05% ANT V Waters Symmetry C18 (3.9 x 150
mm, 5 µm) 255 ACN 35%, 20mM KH2PO4
65%, TEA 0.05%

46
extraction procedure are described in paper V and Table 6. The same HPLC analysis with different detection wavelengths could be used to determine antipyrine in these samples.
The method described by Lampela and co-workers (1999) was modified for the analysis of antipyrine from lamotrigine perfusions. The running conditions were similar to the CBZ and OCBZ method, with the exception that the UV-VIS detector was set at 265 nm. Extraction was done as described previously (Lampela et al. 1999).
4.5 Statistical analysis
Statistical analysis was performed using Student’s t-test, and p values of less than 0.05 were considered statistically significant. All calculations were done using two-tailed distributions. The maternal and fetal concentrations were compared using paired-sample analysis, but sample independence was assumed otherwise.
4.6 Ethical aspects
The Combined Ethical Committee of the University of Oulu and Oulu University Hospital approved the study protocol. Informed consent was obtained from the mothers participating in the studies III, IV and V. Human placenta is surplus material that would normally be disposed of after delivery. The collection of materials did not affect the management of deliveries. The human liver samples used in the studies I and II were obtained from livers that could not be used for transplantation or from patients undergoing cholecystectomy who had given informed consent. Appropriate local ethics committees had approved the collection of tissues (for details see original publications).
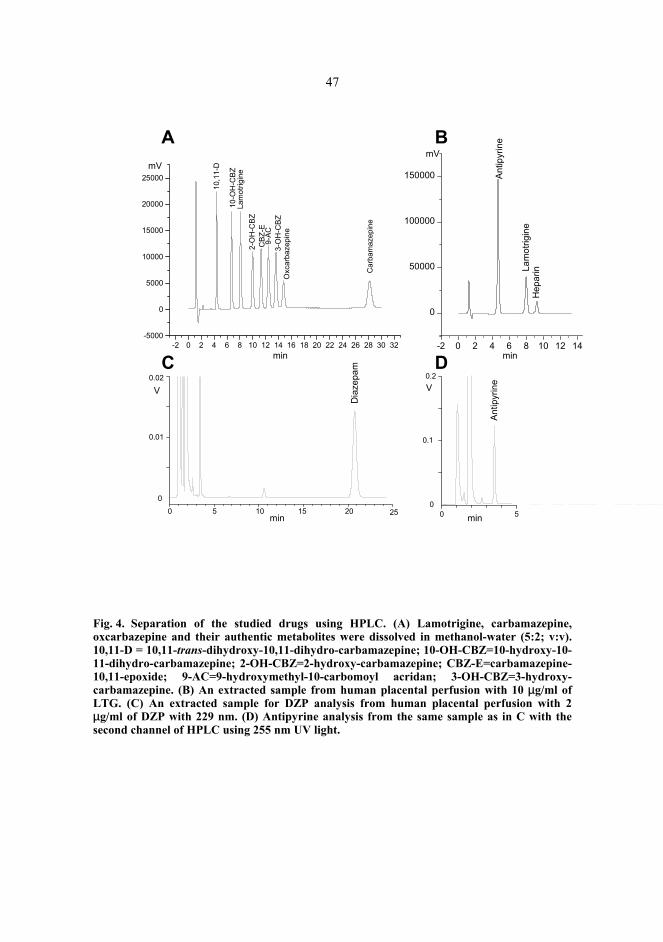
47
Dia
zepa
m
0 5 10 15 20 25min
V
0
0.01
0.02
-2 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32-5000
0
5000
10000
15000
20000
25000
Car
bam
azep
ine
Oxc
arba
zepi
ne
Lam
otrig
ine
3-O
H-C
BZ
10-O
H-C
BZ
10,1
1-D
2-O
H-C
BZ
9-AC
CBZ
-E
mV
min
A
C D
B
-2 0 2 4 6 8 10 12 14
0
50000
100000
150000 Antip
yrin
e
Lam
otrig
ine
Hep
arin
min
mV
Antip
yrin
e
0 50
0.1
0.2V
min
Fig. 4. Separation of the studied drugs using HPLC. (A) Lamotrigine, carbamazepine, oxcarbazepine and their authentic metabolites were dissolved in methanol-water (5:2; v:v). 10,11-D = 10,11-trans-dihydroxy-10,11-dihydro-carbamazepine; 10-OH-CBZ=10-hydroxy-10-11-dihydro-carbamazepine; 2-OH-CBZ=2-hydroxy-carbamazepine; CBZ-E=carbamazepine-10,11-epoxide; 9-AC=9-hydroxymethyl-10-carbomoyl acridan; 3-OH-CBZ=3-hydroxy-carbamazepine. (B) An extracted sample from human placental perfusion with 10 µg/ml of LTG. (C) An extracted sample for DZP analysis from human placental perfusion with 2 µg/ml of DZP with 229 nm. (D) Antipyrine analysis from the same sample as in C with the second channel of HPLC using 255 nm UV light.

5 Results
5.1 Metabolism of carbamazepine and oxcarbazepine in liver and placenta
5.1.1 Carbamazepine metabolism
5.1.1.1 Metabolism of carbamazepine in human liver (papers I and II)
The metabolites produced by human liver in vitro are presented in Table 7. CBZ-E was the major metabolite in all of the test systems used. Recombinant expressed human CYP enzymes catalyzed only the formation of CBZ-E by CYP3A4. All the other test systems also produced 3-OH-CBZ. Moreover, 9-hydroxymethyl-10-carbamoyl acridan (9-AC), 10-11-D, 10-OH-CBZ and 2-OH-CBZ were produced variably by different systems. Human hepatocytes produced one unidentified metabolite. Although the intrinsic clearance values for microsomes, liver slices and hepatocytes differed considerably from each other, they all predicted the known low clearance of CBZ (Paper II Table 3) (Spina 2002).

49
Tabl
e 7.
Car
bam
azep
ine
met
abol
ites
dete
cted
by
the
in v
itro
syst
ems
in t
his
stud
y. Th
e pe
rcen
tage
s in
dica
te t
he r
elat
ive
amou
nts
of
met
abol
ites o
ut o
f the
tota
l am
ount
of m
etab
olite
s.
Syst
em
Pa
per
CB
Z-E
3-O
H-C
BZ
9-A
C
10,1
1-D
IOL
10-O
H-C
BZ
2-O
H-C
BZ
Mou
se li
ver m
icro
som
es
I M
ajor
(7
0-80
%)
Med
ium
(2
0-30
%)
-a M
inor
b (<
1 %
) Tr
acec
-a
Hum
an li
ver m
icro
som
esd
I M
ajor
(6
3-90
%)
Min
or
(3-1
3%)b
Med
ium
(1
0-25
%)
-a Tr
acec
-a
Hum
an li
ver m
icro
som
ese
II
Maj
or
(>90
%)
Min
or
(<10
%)
-a Tr
ace
-a -a
Live
r slic
es
II
Maj
or
(70-
80 %
) M
inor
(1
-3 %
) M
ediu
m
(15-
25 %
) M
inor
(2
%)
-a M
inor
(1
-2 %
) H
epat
ocyt
es
II
Maj
or
(> 8
0 %
) M
inor
(1
%)
Med
ium
(5
-15%
) M
inor
(1
-2%
) -a
Trac
e
Rec
ombi
nant
enz
ymes
II
M
ajor
(1
00 %
) -a
a -a
a -a
Plac
enta
l mic
roso
mes
I -a
a -a
a -a
a
a = m
etab
olite
was
not
det
ecte
d.
b D
etec
ted
only
in 2
/8 in
cuba
tions
c T
race
= th
e m
etab
olite
was
det
ecte
d, b
ut th
e am
ount
was
bel
ow th
e qu
antit
atio
n lim
it
d Rep
rese
nts t
wo
diff
eren
t liv
ers
e Poo
led
mic
roso
mes
from
10
hum
an li
vers

50
5.1.1.2 Identification of the CYP enzymes responsible for carbamazepine metabolite formation in various human liver in vitro systems (paper II)
All the test systems studied identified the most important enzyme (CYP3A4) and the metabolic route (10,11-epoxidation). According to microsomal studies, CBZ-E is produced by CYP3A4, with possible minor contributions from CYP1A2, CYP2A6 and CYP2C8. 3-OH-CBZ appears to be produced by CYP2A6 and CYP2D6, with smaller contributions from CYP1A2, CYP2C8 and CYP3A4. 10,11-D appears to be produced by CYP2C8 and CYP2C19. In the recombinant system, only CYP3A4 formed appreciable amounts of CBZ-E, which was the only metabolite detected.
5.1.1.3 Induction potential of carbamazepine (papers I and II)
CBZ produced concentration-dependent induction in hepatocytes, the maximal induction being about 2-fold. CBZ also caused induction of its own epoxidation in mouse liver.
5.1.1.4 Metabolism of carbamazepine in mouse liver (paper I)
In CBZ incubations, mouse liver microsomes catalyzed the formation of CBZ-E, 10-OH-CBZ, 3-OH-CBZ and 10,11-D. There was also an unidentified peak present in the incubations with CBZ at the concentration 2 mM. CBZ-E was the most abundant metabolite. 9-AC was not detectable at all. The total amounts of CBZ-E were significantly higher in all CBZ-treated groups compared to the control groups. The metabolism of 3-OH-CBZ, however, was not increased by CBZ treatment.
5.1.1.5 Metabolism of carbamazepine in human placenta (paper I)
Incubations were done using microsomes prepared from placentas of mothers on CBZ therapy. A small amount of CBZ-E was detectable in the incubations, probably originating from in vivo metabolism by pregnant mothers because the same amount was also found in extracted non-incubated microsomes. No other metabolites of CBZ were detected. This is in accordance with the earlier perfusion studies by Pienimäki and co-workers (1995a, 1997), in which no metabolism was detected, either. Therefore, it is likely that the placenta does not contribute to CBZ metabolism even in an induced state in humans.

51
5.1.2 Oxcarbazepine metabolism (paper I)
OCBZ was metabolized to 10-OH-CBZ. An unknown peak in HPLC between 10-OH-CBZ and OCBZ representing an unknown metabolite was detected. Mouse liver microsomes metabolized OCBZ to 10-OH-CBZ and an unknown metabolite. 10,11-D was found in incubations with control microsomes but not in microsomal incubations using CBZ-treated microsomes. Placental microsomes catalyzed OCBZ metabolism to 10-OH-CBZ and to an unknown metabolite.
5.2 Placental perfusion experiments
5.2.1 Placental perfusion method (papers IV and V)
The criteria for successful perfusion were as follows: 1) the leak from fetal to maternal circulation was less than 2 ml/h, and 2) the fetal perfusion pressure, monitored continuously, was below 70 mmHg at all times, as described previously by Pienimäki (1996). The viability of the perfused placental lobules was determined by pH, oxygen tensions and oxygen consumption (Table 8).
5.2.2 Antipyrine (papers IV and V)
Antipyrine in fetal circulation was detectable in the first samples 15 minutes after the addition of drugs in all perfusions. At this time point, the mean antipyrine concentration was 77.5 ± 13.2 µg/ml in the maternal perfusate and 16.1 ± 6.1µg/ml in the fetal perfusate, when the data from the 16 successful perfusions were pooled. These are expected concentrations based on the amount of drug added into the maternal circulation (100 µg/ml) at the beginning of the perfusions. The maternal and fetal concentrations equilibrated at 75 minutes in the pooled data (Fig. 5) as well as in the different subgroups. At 120 minutes, the mean fetal/maternal concentration ratio was 0.95 ± 0.14. The feto-maternal transfer percentage was around 30 % in all perfusions (Table 8). The perfusion subgroups did not differ significantly. Thus, the transfer of the reference substrate, antipyrine, was similar in the perfusions, which indicates that they were mutually comparable.

52
Table 8. Comparison of the weights of perfused lobules, fetal and maternal pH values, tissue oxygen consumptions, antipyrine transfer rates and drug recoveries in the perfusion studies. Means ±SD are given.
Parameter DZP perfusions
(n=7)
LTG perfusions
(n=8)
Control perfusion
(n=1) Weight of perfused lobule 14.5 ± 7.7 g 9.7 ± 2.8 ga 18.20 g Fetal pH at 120 minutes 7.46 ± 0.03 7.48 ± 0.07 7.33 Maternal pH at 120 minutes 7.57 ± 0.08 7.54 ± 0.09 7.55 Oxygen consumption (ml/min/g) 2.89 ± 2.67 4.62 ± 2.36 3.01 F/M ratio of antipyrine at 2 hours 1.0 ± 0.14 0.91 ± 0.14 0.86 Transfer % of antipyrine 31.5 ± 3.0 % 29.9 ± 3.2 % 30.2 % Recovery % of the studied drugb 37.6 ± 21% 81 ± 24 % NA aAverage of 7 perfusions, NA=not analyzed; b Recovery of the study drug found in the samples, end perfusates and perfused tissue (white and pink areas) from the dose added into the perfusion
µg/ml
0 20 40 60 80 100 1200
10
20
30
40
50
60
70
80
90
100
110
Antip
yrin
e co
ncen
tratio
n
Minutes
******
*** **** **
Fig. 5. Antipyrine transfer from maternal to fetal circulation. Combined data from LTG (n=8), DZP (n=7) and control (n=1) perfusions, which all contained 100 mg of antipyrine added into the maternal perfusate at point 0 in all perfusions. maternal concentration (µg/ml±SD); ■ ■ fetal concentration (µg/ml±SD). The maternal and fetal concentrations differed significantly until 75 minutes.

53
5.2.3 Lamotrigine (paper IV)
LTG was detectable in the fetal circulation at 15 minutes in all perfusions, indicating rapid transfer. The maternal and fetal concentrations reached equilibrium at 60 minutes with both concentrations used. The feto-maternal ratio was 1.26±0.20 with 10 µg/ml of LTG and 0.83±0.41 with 2.5µg/ml of LTG at the end of the perfusion. The transfer of LTG from the maternal to the fetal compartment at 120 minutes was 28.9±10.7% with 2.5µg/ml of LTG and 37.8±3.2% with 10 µg/ml of LTG (p>0.05). The amounts of LTG detected in five placentas (3 with 2.5 µg/ml LTG and 2 with 10 µg/ml LTG) varied from 4.1 % to 6.3% of the original dose added to the perfusion medium, and there were no indications of tissue accumulation.
5.2.4 Diazepam (paper V)
DZP was also detectable in the fetal circulation at 15 minutes in all perfusions, indicating rapid transfer. The maternal and fetal concentrations equilibrated at 60 minutes with both DZP concentrations. DZP concentrations were higher in the maternal than the fetal circulation throughout the perfusion with both initial concentrations. At the end of the perfusion, the feto-maternal ratio was 0.48±0.11 and the transfer from the maternal to the fetal compartment 18.4±3.6% with 2 µg/ml of DZP. The corresponding values for perfusions with 200 ng/ml of DZP were 0.55±0.10 and 20.5±3.1%. The average DZP concentrations in the perfused area of the placenta were 2-fold compared to the maternal perfusate and 3.6-fold compared to the fetal perfusate. The total recovery of DZP from the samples, perfusion fluid and perfused tissue remained low (Table 8).
5.3 Clinical samples from maternal serum and cord blood
5.3.1 Oxcarbazepine (paper III)
The concentrations of OCBZ, 10-OH-CBZ and 10,11-D were relatively similar in maternal and cord blood at delivery, and no statistically significant differences were detected. In some cases, however, the OCBZ concentrations in cord blood were two- to four-fold compared to the maternal venous concentrations. The tissue concentrations of OCBZ, 10,11-D and 10-OH-CBZ were close to the maternal and fetal serum concentrations, although the difference between the 10-OH-CBZ and serum concentrations was statistically significant.

54
5.3.2 Lamotrigine (paper IV)
The cord blood and maternal serum concentrations were close to each other in both of the studied cases with a cord blood-maternal concentration ratio of 1.02 in one pair and 1.55 in the other.

6 Discussion
6.1 Carbamazepine metabolism in different in vitro systems
All the liver-derived systems studied produced the major human in vivo metabolite CBZ-E as the major metabolite (Table 7). According to the literature, CBZ-E is also an important metabolite in mice (Bourgeois & Wad 1984, Finnell et al. 1986, Spina 2002). In man, CYP3A4 is the major catalyst of CBZ epoxidation with a minor contribution of CYP2C8 (Kerr et al. 1994, Valentine et al. 1996, Spina 2002). In accordance with this, all of our human liver systems showed CYP3A4 to be the principal metabolizing enzyme. Also, microsomal inhibitor and correlation studies suggested CYP2C8 as a minor contributor to the formation of CBZ-E. On the other hand, our results from C57/BL6 mice treated with CBZ implicated that, instead of CYP3A, CBZ metabolism to CBZ-E is mainly mediated by CYP1A1 in mouse. In contrast to our finding, Pirmohamed and coworkers (1992a) found that the marker enzyme acitivity for CYP3A, the microsomal oxidation of cortisol to 6ß-hydroxycortisol and CBZ epoxidation increased along with phenobarbital and dexamethasone treatment, indicating that CYP3A is responsible for CBZ-E formation in CBA/ca mice. Also, their immunoblotting experiments with antibodies raised against rat CYP3A support the notion that CBZ metabolism is mediated through CYP3A in CBA/ca mice. Thus, depending on the strain of mice, the similarity of the enzyme pattern for a particular metabolic pathway may vary.
10,11-D was found as a minor metabolite in a few incubations using mouse liver microsomes and in systems using human liver microsomes, liver slices and hepatocytes. 10,11-D is a known CBZ metabolite in vivo in humans as well as in mice (Bourgeois & Wad 1984, Spina 2002). Quantitatively, 10,11-D is more important in vivo than suggested by the different in vitro systems. However, the extraction percentage of 10,11-D was low from incubations, and this may have caused false negative findings. Furthermore, 10,11-D is produced from CBZ-E in a reaction catalyzed by epoxide hydrolaze, and CBZ-E must thus be produced first. Due to this, the relatively short incubation times may partially explain the low concentrations of 10,11-D.
Small quantities of 10-OH-CBZ were detectable in mouse and human liver microsomal incubations, but not in incubations done with liver slices, hepatocytes or

56
recombinant enzymes. This metabolite has rarely been described in the literature, but it has been detected as a minor metabolite of CBZ in humans (Faigle et al. 1976, Lertratanangkoon & Horning 1982).
Quantitatively, after CBZ-E, the second most important metabolite in human liver microsomes, liver slices and hepatocytes was 9-AC. Incubations with mouse liver microsomes and recombinant enzymes did not produce 9-AC. In humans, 9-AC is produced from the epoxide-diol pathway and CBZ itself (Spina 2002), and it is frequently detected in serum samples from patients on CBZ therapy, although the concentrations have been lower than the CBZ-E and 10,11-D concentrations (Eto et al. 1995, Pienimäki et al. 1995b, Pienimäki et al. 1997, Wad et al. 1997). 9-AC has also been detected as a CBZ metabolite in rats (Lertratanangkoon & Horning 1982, Castrén et al. 1996), but not in mouse (Amore et al. 1997). Thus, most of the human in vitro systems correctly predicted the production of 9-AC, although its proportion is smaller in vivo than in vitro. However, mouse and human liver systems differed in the production of this metabolite, suggesting inter-species differences in CBZ metabolism.
All systems, except recombinant enzymes, produced 3-OH-CBZ. In humans, both 2- and 3-OH-CBZ have been detected in vivo, but the hydroxylation of the aromatic rings of CBZ is quantitatively less important than the epoxide-diol pathway, as reviewed by Spina (2002). It was quantitatively more important in mouse liver microsomes than in systems using human liver-derived materials or in vivo. In addition to 3-OH-CBZ, liver slices and hepatocytes produced minor amounts of 2-OH-CBZ. Both 3-OH- and 2-OH-CBZ have been detected in mouse previously (Amore et al. 1997).
There was also an unidentified peak present in the incubations with mouse liver microsomes and human hepatocytes. This peak does not necessarily represent a new metabolite because more than thirty known CBZ metabolites exist (Lertratanangkoon & Horning 1982, Maggs et al. 1997), and only a small assortment of authentic reference metabolites were available for us (Pienimäki et al. 1995a).
The metabolic clearance of CBZ was calculated in various in vitro systems using human liver-derived materials. CBZ is typically a drug with slow clearance. The hepatic intrinsic clearances calculated were mostly systems clearly below 100 ml/min, predicting low extraction and slow clearance.
The induction potential of CBZ is well-known in many species, including man and rat (Tybring et al. 1981, Regnaud et al. 1988, Mikati et al. 1989, Wurden & Levy 2002). In this study, the CBZ induction potential was tested in mouse and isolated human hepatocytes. In hepatocytes, CBZ led to 2- to 3-fold induction of the marker activity 7-ethoxycoumarin O-deethylase, suggesting CYP induction. Also, autoinduction of CBZ metabolism to CBZ-E was found in C57/BL6 mice. Thus, the induction potential of CBZ was predicted by both of the tested systems. In the mouse model, however, the CYP3A-mediated testosterone 6ß-hydroxylase activity was not induced, even though the formation of CBZ-E was increased. Furthermore, the CYP1A1-mediated EROD activity was 3-fold in CBZ-treated compared to control microsomes, suggesting induction of CYP1A1 instead of CYP3A. Panesar and co-workers (1996) studied CBZ induction in rat. CBZ increased slightly the 3A levels in rats in their study. In the same study, 2B1 and 2B2 were highly inducible by CBZ. In addition to CBZ epoxidation, the induction of the hydroxylation pathway has shown species-related differences. In man, the pathway leading to the formation of 3-OH-CBZ is induced by other anticonvulsants but not by

57
CBZ alone (Eichelbaum et al. 1984, Eichelbaum et al. 1985). In our study, CBZ treatment did not cause any increase in the formation of 3-OH-CBZ in mice. However, Eichelbaum and coworkers (Eichelbaum et al. 1984) found CBZ to cause a 2- to 3-fold increase of 3-OH-CBZ in rat. According to our study, it seems that CBZ metabolism in mouse in this respect resembles more human than rat. However, it seems that CBZ has different potencies to induce CYP enzymes in different species, and that different CYP isoforms are induced in man and mice.
In conclusion, all of the test systems predicted well the major metabolite CBZ-E. The minor metabolites were produced in slightly variable amounts in all the other test systems except yeast cells with recombinant enzymes. It is possible that the inability of the recombinant system to detect other minor metabolites might be due to the slow metabolic turnover of CBZ. CBZ metabolism showed slight interspecies differences in the metabolite profile. More importantly, species differences were seen in the CYPs catalyzing the formation of the major metabolite as well as in the induction of CYP isoforms. However, the interspecies differences in CYP enzymes are well recognized (Shimada et al. 1997), and our finding is thus not surprising. Due to interspecies differences in the metabolism, detailed extrapolation of results on animals to humans is often difficult, and it is clear that studies on humans are needed to produce definite information on humans (Pelkonen & Breimer 1994, Lin 1998, Ekins et al. 2000). All human liver-derived test systems provided important qualitative and quantitative data on CBZ pharmacokinetics, and the next task should be the refinement of various systems as to their predictive power.
6.2 Oxcarbazepine metabolism in vitro in liver and placenta (paper I)
OCBZ was metabolized mainly to its active metabolite, 10-OH-CBZ, by human liver and mouse liver microsomes in this study similarly to the in vivo metabolism in humans (Schütz et al. 1986, Wellington & Goa 2001, Kalis & Huff 2001). 10,11-D was also present in some, but not all, incubations with mouse liver microsomes. This is probably due to the low extraction percentage of 10,11-D from incubation medium. In our study, there was an unknown peak present in HPLC in the OCBZ incubations. The very same peak was also present in the study by Castrén and co-workers (1996), who used phenobarbital-induced rat liver microsomes. They tried to further characterize the metabolite with HPLC-MS. However, the nature of the unknown metabolite was not revealed because it is either very labile or less polar than 10-OH-CBZ or OCBZ. Schütz and coworkers (1986) studied OCBZ after a single oral dose in two volunteers, and they found minor amounts of an unknown derivate of 10-OH-CBZ with an additional hydroxyl group. It remains open whether the metabolite found by us is the same metabolite as previously reported by Schütz and coworkers (1986).
The human placenta also metabolized OCBZ. In one of the two incubations, 10-OH-CBZ was found as a metabolite of OCBZ, and both incubations also contained an unknown peak in HPLC. In a previous study by Pienimäki and co-workers (1997), 10-

58
OH-CBZ was clearly detectable as a metabolite of OCBZ in five placental perfusions, and it thus seems that the placenta is capable of OCBZ metabolism.
6.3 Binding of carbamazepine and oxcarbazepine to macromolecules (paper I)
We found only minimal covalent binding of CBZ or OCBZ to DNA catalyzed by human liver microsomes. CBZ, CBZ-E, OCBZ and 10-OH-CBZ have not been found to bear mutagenic potential (Dam & Jensen 1989, Dam & Owen 1995, Sinues et al. 1995). However, it has been shown that reactive CBZ metabolites bind covalently to neutrophils and may thus be responsible for some of the observed adverse effects through direct cytotoxicity or immune response after hapten formation, as suggested by Furst and co-workers (Furst & Uetrecht 1993, Furst et al. 1995). Epoxide metabolites are generally chemically reactive. However, CBZ-E is highly stable with little electrophilic reactivity (Frigerio & Morselli 1975), and it is thus not likely to be responsible for the CBZ adverse effects. The formation of reactive metabolites of CBZ and their binding to proteins increase after phenobarbital induction and decrease after ketoconazole treatment, suggesting that cytochrome P450 enzymes are responsible for the formation of these metabolites (Pirmohamed et al. 1992b). Interestingly, the binding of OCBZ was greater by one order of a magnitude than that of CBZ. An exactly similar finding was obtained by Castrén and co-workers (1996) with rat liver microsomes. The significance of these findings, however, remains unclear because, as mentioned previously, no mutagenic potential of OCBZ or 10-OH-CBZ has been detected in bacterial and mammalian test systems (Dam & Jensen 1989) and the frequency of side effects is lower with OCBZ than with CBZ (Guerreiro et al. 1997, Beydoun 2000, Kalis & Huff 2001).
6.4 Distribution of lamotrigine and diazepam in placental perfusions (papers IV and V)
Although metabolism and binding to macromolecules can be studied in vitro, one needs whole organ systems to study the movements of molecules within tissues. Human placental perfusion provides a controlled way to study the transfer of drugs through the placenta (Omarini et al. 1992, Bourget et al. 1995, Tuntland et al. 1999, Ala-Kokko et al. 2000). Pienimäki and co-workers (1995, 1997) have earlier studied carbamazepine and oxcarbazepine in placental perfusion systems. In this work, we studied the transplacental passage of LTG and DZP using the same human placental perfusion method.
Significant amounts of DZP disappeared from the perfusates. This was not due to placental metabolism because similar disappearance was observed in the perfusions without placenta. Possible explanations are instability of DZP in the perfusion fluid or binding to the tubing in our perfusion system. Although the disappearance of DZP was

59
equivalent in both the maternal and fetal circulations and hence unlikely to affect the equilibration between the circulations, the results on DZP must be regarded as more preliminary than those on LTG.
The placental transfer of LTG and DZP is considerable in the human placental perfusion system. This implies marked fetal exposure in vivo. According to our perfusion data, significant amounts of both LTG and DZP can be detected in fetal circulation as soon as 15 minutes after drug administration into the maternal pool, suggesting rapid transfer of both compounds.
The placental transfers of both LTG and DZP were independent of the maternal concentrations, suggesting passive diffusion or nonsaturable active transport as a mechanism. Further, the DZP transfer percentage remained lower than the transfer percentage of antipyrine, as also suggested by an earlier perfusion study using an open perfusion system (Guerre-Millo et al. 1979). It is known that antipyrine crosses the placenta with passive diffusion (Challier et al. 1985), and our results thus suggest passive diffusion as the major mechanism of transfer. LTG in the present concentrations equilibrated slightly sooner than antipyrine, suggesting rapid initial transfer. The placental transfer of substances by passive diffusion is known to be affected by the physicochemical properties of the compounds (e.g. lipid solubility, molecular weight, protein binding, and degree of ionization) (Pacifici & Nottoli 1995), and our finding may reflect the difference in the lipid solubility of LTG and DZP as compared to antipyrine. On the other hand, during the past few years, several placental transporters have been identified, and they may participate in the transfer of drugs in addition to their physiological substrates (Ganapathy et al. 2000). Their putative role in the transfer processes was not studied in this work and hence cannot be ruled out.
We used protein-free perfusion medium, and our results thus reflect the placental passage of the unbound drug. LTG is only moderately bound to protein (55%), while DZP is highly protein-bound (Kanto et al. 1973, Garnett 2002). The protein binding of DZP is 86 % in fetal plasma and 96 % in maternal plasma (Kanto et al. 1973). Thus, protein binding theoretically affects DZP equilibration more than LTG equilibration. In an in vivo study, Ridd and co-workers (Ridd et al. 1989) measured both the total and the free drug concentrations of DZP. The total DZP concentration was higher in umbilical plasma than in maternal plasma, but the free DZP concentrations were similar on both sides of the placenta. This probably explains, at least to some extent, the observed difference between DZP transfer in the in vivo and the perfusion conditions. Addition of albumin to the perfusion system is possible, and it has been shown in several studies that changes in protein concentrations affect the placental transfer of drugs during perfusion (Johnson et al. 1997, Johnson et al. 1999, He et al. 2000, Herman et al. 2000).
Both LTG and DZP are weak bases with pKa values clearly below physiological pH. The pKa value is 5.5 for LTG and 3.4 for DZP (Table 4). Therefore, about 99 % of LTG and more than 99.9 % of DZP were non-ionized at perfusion conditions. Even though pH in the fetal circulation increased slightly during perfusion, such an increase at near-neutral pH does not significantly affect further the ionization and transfer of DZP or LTG. Nor was there any significant pH gradient between the maternal and fetal perfusion fluids. Maternal pH was up to 0.1 units higher than fetal pH, as it is also under in vivo conditions (Reynolds & Knott 1989).

60
Placental tissue accumulation was also studied. In contrast to LTG, DZP accumulated into placental tissue. In accordance with the earlier findings (Ala-Kokko et al. 1997), the DZP perfusion study indicates that the more lipophilic DZP probably accumulates into the placenta rather than crosses to the fetal side in placental perfusion, while LTG equilibrates quickly between the maternal and fetal perfusates. Controversially, earlier experiments done with the placental perfusion method suggested slight accumulation of 10-OH-CBZ into the placental tissue (Pienimäki et al. 1997). No such accumulation was observed in vivo. During OCBZ perfusions, however, the tissue was slightly oedematous, which was not the case in DZP perfusions (data not shown). Therefore, it is possible that the tissue oedema during the ex vivo perfusions favoured the accumulation of 10-OH-CBZ.
6.5 Placental transfer of anticonvulsants in a perfusion system and in clinical samples (papers III-V)
DZP was chosen to be studied because its pharmacokinetic characteristics are well-known in vivo in humans (Table 3), and it is therefore possible to compare in vivo and in vitro data. There are also available some data on the placental passage of LTG and OCBZ to enable comparison.
In our study, the placental perfusion system predicted well the transplacental passage of LTG. The LTG concentrations in the corresponding maternal and fetal circulations were similar in the placental perfusion system and the in vivo sample pairs (Tomson et al. 1997, Rambeck et al. 1997, Ohman et al. 2000). Also, the placental transfer of OCBZ and its major metabolites appears to be considerable, as suggested by earlier studies both in vivo and in vitro (Pienimäki et al. 1995b, Pienimäki et al. 1997). However, our in vitro perfusion study on DZP did not show a good correlation with the in vivo data obtained from the literature. In vivo, the cord blood concentrations of DZP are higher than the maternal concentrations (Table 3), and our placental perfusion system studies did not predict this.
The information of drug levels in fetal and maternal blood at birth provides only single-point measurements. Such clinical studies do not show, for example, how long it takes to achieve complete equilibration between the mother and the fetus. Indirect information could be gained from a comparison of several patients at different intervals between drug intake by the mother and delivery, but such studies are seldom done. There are no clinical data on the time course of the transplacental passage of OCBZ or LTG, and comparison is therefore impossible. However, clinical studies indicate very rapid transplacental passage of DZP (Table 3). Our placental perfusion experiments, however, indicated that equilibration takes longer than in vivo. In our studies, the drugs were added into the maternal reservoir and it takes some time before the drug reaches the placental tissue, and this may well explain the observed difference.
Although any comparison of in vitro and in vivo results is difficult, some studies have suggested a similar transfer profile in vivo and in a human placental perfusion system. Theophylline has been shown to exhibit a similar transfer profile in vivo and in a

61
perfusion study (Omarini et al. 1993). CBZ also seems to have a rather similar transfer profile in vivo and in a perfusion system (Pienimäki et al. 1995b, Pienimäki et al. 1997) similarly to LTG and OCBZ. According to the literature, it seems that the human placental perfusions system is capable of predicting quite well the low transfer of compounds into the fetal circulation. The transfer rates of the protease inhibitors ritonavir and saquinavir have been extremely low (Casey & Bawdon 1998, Forestier et al. 2001). A recent in vivo study also suggested low placental transfer of both drugs (Marzolini et al. 2002). Both saquinavir and ritonavir are known P-glycoprotein substrates. The placental transfer of other P-glycoprotein substrates, such as doxorubicin and cimetidine, has also been low both in perfusion studies and in vivo (Roboz et al. 1979, d'Incalci et al. 1983, Karp et al. 1983, Qvist et al. 1985, Ching et al. 1987, Schenker et al. 1987, Grohard et al. 1989, Pacifici & Nottoli 1995). Also, the placental transfer of erythropoietin, which is a macromolecule, has been found neligible in human placental perfusion systems and in vivo both in humans and in a sheep model (Widness et al. 1991, Eichhorn et al. 1993, Malek et al. 1994, Schneider & Malek 1995, Reisenberger et al. 1997).
It seems that a placental perfusion system is not able to predict the accumulation of drugs into the fetal circulation on all occasions. Allowing for the fact that our results represent the free drug concentrations, they indicate that the DZP transfer differs in the perfusion system from the in vivo situation in humans. Similarly to DZP, valproate concentrations in the fetus also exceed the maternal concentrations (Johannessen 1992, Pacifici & Nottoli 1995). Placental perfusion studies suggested equal concentrations of unbound drug in the maternal and fetal circulations (Johannessen 1992, Barzago et al. 1996). In vivo fetal protein binding is higher than maternal protein binding, and the half-life of valproate in fetus following placental transfer is considerably longer than in mother (Johannessen 1992). This explains the observed differences between the in vitro and in vivo findings. When the accumulation into the fetal circulation is not due to active transport in the placental tissue but more likely to non-placental pharmacokinetic factors, such as the distribution, excretion and metabolism of drugs in the mother and the fetus, it is natural that the perfusion system does not predict it. However, Pastrakuljic and co-workers (2000) have shown that amino acids that are substrates for placental transporters, and are thus actively transported into the fetal circulation, also accumulate into the fetal circulation in the placental perfusion system.
Protein binding plays an important role in the equilibration of the total drug concentrations in vivo (Reynolds & Knott 1989). The protein concentrations have been variable in different perfusions systems (Ala-Kokko et al. 2000). He and co-workers (2000) demonstrated that the propofol concentration increased significantly in the fetal circulation with an increasing albumin concentration. However, the free propofol concentration remained unchanged, suggesting that binding to fetal albumin is the decisive feature in placental transfer. It has also been shown that protein binding influences the placental transfer of ropivacaine, bupivacaine, methohexital, sufentanil and digoxin (Schmolling et al. 1996, Johnson et al. 1997, Johnson et al. 1999, Herman et al. 2000). The contribution of the protein concentrations in the placental perfusion system as well as in vivo must be taken into account when extrapolating perfusion data to the in vivo context.

62
In conclusion, the data obtained with a placental perfusion system are not equivalent to in vivo data on all occasions. However, even with its limitations, the human placental perfusion method provides information that can be used to evaluate the risk factors associated with drug use during pregnancy. Understanding of specific transport characteristics is a good basis for a rational risk assessment of drugs during pregnancy. Thus, perfusion data may serve as one method in a test battery for fetal risk assessment. At this point, no systematic evaluation has been done and no uniform criteria for successful perfusions have been established (Ala-Kokko et al. 2000). Also, due to individual variation, it is still unclear how many placentas should be used to get a representative picture of the interindividual variation. For a reasonable evaluation of the method as a tool in preclinical drug evaluation, further validation using drugs with different physiochemical properties is still needed.

7 Summary and conclusions
CBZ metabolism was studied with several in vitro methods. CBZ-E was the major metabolite in all test systems. 10,11-D was found as a minor metabolite in a few incubations using mouse liver microsomes and in systems using human liver microsomes, liver slices and hepatocytes. 9-AC was detected in the samples from incubations using human liver microsomes, liver slices and hepatocytes. All systems except recombinant enzymes produced 3-OH-CBZ. Liver slices and hepatocytes produced minor amounts of 2-OH-CBZ. Small quantities of 10-OH-CBZ were detectable in mouse and human liver microsomal incubations. There was also an unidentified peak present in the incubations with mouse liver microsomes and hepatocytes.
All human liver systems demonstrated that CYP3A4 is the principal CBZ-metabolising enzyme. Microsomal inhibitor and correlation studies implicated CYP2C8 as a minor contributor to the formation of CBZ-E. However, our results from CBZ- treated mice suggested thatCBZ metabolism to CBZ-E is mainly mediated by CYP1A1, rather than CYP3A, in C57/BL6 mice. Autoinduction of CBZ metabolism was seen in hepatocytes and in incubations using microsomes from CBZ-treated mice.
All test systems predicted well the major metabolite, i.e. CBZ-E. Minor metabolites were also produced in slightly variable amounts in all the other test systems except recombinant enzymes. It is possible that the inability of the recombinant system to detect other minor metabolites might be due to the slow metabolic turnover of CBZ. CBZ metabolism showed slight interspecies differences in the metabolite profile. More importantly, species-specific differences were seen in the CYPs catalyzing the formation of the major metabolite as well as in the induction of CYP isoforms, as also recognized previously. Due to species-specific differences in metabolism, detailed extrapolation of the results of animal tests to humans is often difficult, and studies on humans are needed to produce reliable information on humans.
OCBZ is metabolized to 10-OH-CBZ and an unknown metabolite by the human placenta. Human liver and mouse liver microsomes metabolized OCBZ mainly to its active metabolite, 10-OH-CBZ. 10,11-D and an unknown metabolite were also detected.
The placental transfers of LTG and DZP were considerable in the human placental perfusion system, indicating marked fetal exposure in vivo. Significant amounts of both

64
LTG and DZP could be detected in fetal circulation as soon as 15 minutes after drug administration into the maternal pool, suggesting rapid transfer of both compounds. The maternal and fetal concentrations of both LTG and DZP reached an equilibrium at 60 minutes. However, the DZP concentrations in the maternal circulation were higher than the fetal concentrations throughout the perfusions. The OCBZ, 10-OH-CBZ and 10,11-D values obtained from maternal venous and cord blood also suggest significant fetal exposure.
In our study, the placental perfusion system predicted well the transplacental passage of LTG. The LTG concentrations in the corresponding maternal and fetal circulations were similar in the placental perfusion system and in the in vivo sample pairs reported in this work and previously. Also, the placental transfer of OCBZ and its major metabolites appears to be considerable, as suggested by earlier studies both in vivo and in vitro. However, our study on DZP did not show a good correlation between the in vitro results and the in vivo data obtained from the literature. In vivo, the cord blood concentrations of DZP are higher than the maternal concentrations, and our placental perfusion system studies did not predict this. Protein binding partially explains the observed difference. Furthermore, the distribution, metabolism and elimination of DZP in the mother may be so fast that the transfer across the placenta is not quick enough to maintain the concentration equilibrium, and this may lead to higher fetal concentrations. The results obtained with the in vitro isolated organ perfusion method hence cannot be exactly similar to the in vivo situation.
The data obtained with a placental perfusion system are not equivalent to in vivo data on all occasions. However, even with its limitations, the human placental perfusion method provides information that can be used to evaluate the risk factors associated with drug use during pregnancy. Understanding of specific transport characteristics is a good basis for the rational risk assessment of drugs used during pregnancy. Thus, the human placentad perfusion system may serve as one method among a battery of tests for fetal risk assessment. At this point, no systematic evaluation has been made, and no uniform criteria for successful perfusions have been established. For a reasonable evaluation of the method as a tool in preclinical drug evaluation, further validation tests using drugs with different physiochemical properties are needed.
In conclusion, all of the tested in vitro systems are useful in the prediction of at least some aspects of in vivo pharmacokinetic and metabolic characteristics. Efforts are still needed for the validation and refinement of various systems. Also, the limitations of in vitro systems must be taken account when extrapolating data to the in vivo context.

References
Acevedo CG, Huambachano A, Perez E, Rojas S, Bravo I & Contreras E (1997) Effect of ethanol on human placental transport and metabolism of adenosine. Placenta 18: 387-392.
Adamson SL, Lu Y, Whiteley KJ, Holmyard D, Hemberger M, Pfarrer C & Cross JC (2002) Interactions between trophoblast cells and the maternal and fetal circulation in the mouse placenta. Dev Biol 250: 358-373.
Addis A, Sharabi S & Bonati M (2000) Risk classification systems for drug use during pregnancy: are they a reliable source of information? Drug Saf 23: 245-253.
Akinci MK & Schofield PR (1999) Widespread expression of GABA(A) receptor subunits in peripheral tissues. Neurosci Res 35: 145-153.
Al Bunyan MA (2001) Random total antiepileptic drug levels and seizure control during pregnancy. Saudi Med J 22: 355-359.
Ala-Kokko TI, Myllynen P & Vähäkangas K (2000) Ex vivo perfusion of human placental cotyledon implications for anesthetic pharmacology. International Journal of Obstetric Anesthesia 9: 26-38.
Ala-Kokko TI, Pienimäki P, Lampela E, Hollmen AI, Pelkonen O & Vähäkangas K (1997) Transfer of clonidine and dexmedetomidine across the isolated perfused human placenta. Acta Anaesthesiol Scand 41: 313-319.
Amore BM, Kalhorn TF, Skiles GL, Hunter AP, Bennett GD, Finnell RH, Nelson SD & Slattery JT (1997) Characterization of carbamazepine metabolism in a mouse model of carbamazepine teratogenicity. Drug Metab Dispos 25: 953-962.
Anderson GD, Gidal BE, Messenheimer JA & Gilliam FG (2002) Time course of lamotrigine de-induction: impact of step-wise withdrawal of carbamazepine or phenytoin. Epilepsy Res 49: 211-217.
Anderson GD & Miller JW (2002) Benzodiazepines. Chemistry, biotransformation and pharmacokinetics. In Levy R et al (eds) Antiepileptic drugs, 5th ed. Lippincott Williams & Wilkins, Philadelphia, p 187-205.
Anderson GD, Yau MK, Gidal BE, Harris SJ, Levy RH, Lai AA, Wolf KB, Wargin WA & Dren AT (1996) Bidirectional interaction of valproate and lamotrigine in healthy subjects. Clin Pharmacol Ther 60: 145-156.
Audus KL (1999) Controlling drug delivery across the placenta. Eur J Pharm Sci 8: 161-165. Azarbayjani F & Danielsson BR (2002) Embryonic arrhythmia by inhibition of HERG channels: a
common hypoxia-related teratogenic mechanism for antiepileptic drugs? Epilepsia 43: 457-468. Bajoria R & Contractor SF (1997a) Effect of surface charge of small unilamellar liposomes on
uptake and transfer of carboxyfluorescein across the perfused human term placenta. Pediatr Res 42: 520-527.
Bajoria R & Contractor SF (1997b) Effect of the size of liposomes on the transfer and uptake of carboxyfluorescein by the perfused human term placenta. J Pharm Pharmacol 49: 675-681.

66
Bakke OM & Haram K (1982) Time-course of transplacental passage of diazepam: Influence of injection-delivery interval on neonatal drug concentrations. Clin Pharmacokinet 7: 353-362.
Bakke OM, Haram K, Lygre T & Wallem G (1981) Comparison of the placental transfer of thiopental and diazepam in caesarean section. Eur J Clin Pharmacol 21: 221-227.
Bardy AH, Hiilesmaa VK, Teramo K & Neuvonen PJ (1990) Protein binding of antiepileptic drugs during pregnancy, labor, and puerperium. Ther Drug Monit 12: 40-46.
Baron JM & Merk HF (2001) Drug metabolism in the skin. Curr Opin Allergy Clin Immunol 1: 287-291.
Baruzzi A, Albani F & Riva R (1994) Oxcarbazepine: pharmacokinetic interactions and their clinical relevance. Epilepsia 35 Suppl 3: S14-S19.
Barzago MM, Bortolotti A, Stellari FF, Diomede L, Algeri M, Efrati S, Salmona M & Bonati M (1996) Placental transfer of valproic acid after liposome encapsulation during in vitro human placenta perfusion. J Pharmacol Exp Ther 277: 79-86.
Bawdon RE (1998) The ex vivo human placental transfer of the anti-HIV nucleoside inhibitor abacavir and the protease inhibitor amprenavir. Infect Dis Obstet Gynecol 6: 244-246.
Beghi E (2002) Carbamazepine. Clinical efficacy and use in other neurologic disorders. In Levy R et al (eds) Antiepileptic drugs, 5th ed. Lippincott Williams & Wilkins, Philadelphia, p 273-277.
Benedetti MS (2000) Enzyme induction and inhibition by new antiepileptic drugs: a review of human studies. Fundam Clin Pharmacol 14: 301-319.
Bennett GD, Amore BM, Finnell RH, Wlodarczyk B, Kalhorn TF, Skiles GL, Nelson SD & Slattery JT (1996) Teratogenicity of carbamazepine-10, 11-epoxide and oxcarbazepine in the SWV mouse. J Pharmacol Exp Ther 279: 1237-1242.
Berglund L, Andersson J, Lilja A, Lindberg BS, Gebre-Medhin M, Antoni G, Bjurling P, Langstrom B & Lundqvist H (1990) Amino acid transport across the placenta measured by positron emission tomography and analyzed by compartment modelling. J Perinat Med 18: 89-100.
Berglund L, Gebremedhin M, Lindberg B, Lilja A, Langstrom B & Lundqvist H (1989) Placental transfer of methionine during protein restriction in the rhesus monkey studied by position emission tomography. Placenta 10: 387-397.
Bernus I, Dickinson RG, Hooper WD & Eadie MJ (1996) Dose-dependent metabolism of carbamazepine in humans. Epilepsy Res 24: 163-172.
Bernus I, Hooper WD, Dickinson RG & Eadie MJ (1995) Metabolism of carbamazepine and co-administered anticonvulsants during pregnancy. Epilepsy Res 21: 65-75.
Berry DJ, Besag FM, Pool F, Natarajan J & Doose D (2002) Lack of an effect of topiramate on lamotrigine serum concentrations. Epilepsia 43: 818-823.
Bertilsson L (1978) Clinical pharmacokinetics of carbamazepine. Clin Pharmacokinet 3: 128-143. Besag FM, Berry DJ & Pool F (2000) Methsuximide lowers lamotrigine blood levels: A
pharmacokinetic antiepileptic drug interaction. Epilepsia 41: 624-627. Besag FM, Wallace SJ, Dulac O, Alving J, Spencer SC & Hosking G (1995) Lamotrigine for the
treatment of epilepsy in childhood. J Pediatr 127: 991-997. Beydoun A (2000) Safety and efficacy of oxcarbazepine: results of randomized, double-blind trials.
Pharmacotherapy 20: 152S-158S. Bialer M (2002) Oxcarbazepine. Chemistry, biotransformation, and pharmacokinetics. In Levy R et
al (eds) Antiepileptic drugs, 5th ed. Lippincott Williams & Wilkins, Philadelphia, p 457-469. Bissonnette JM (1982) Review article: membrane vesicles from trophoblast cells as models for
placental exchange studies. Placenta 3: 99-106. Bloom SL, Dias KM, Bawdon RE & Gilstrap LC, III (1997) The maternal-fetal transfer of
lamivudine in the ex vivo human placenta. Am J Obstet Gynecol 176: 291-293. Bloxam DL, Bax CM & Bax BE (1997) Culture of syncytiotrophoblast for the study of human
placental transfer. Part I: Isolation and purification of cytotrophoblast. Placenta 18: 93-98. Bloxam DL & Bullen BE (1986) Condition and performance of the perfused human placental
cotyledon. Am J Obstet Gynecol 155: 382-388. Boal JH, Plessinger MA, van den RC & Miller RK (1997) Pharmacokinetic and toxicity studies of
AZT (zidovudine) following perfusion of human term placenta for 14 hours. Toxicol Appl Pharmacol 143: 13-21.

67
Bologa M, Tang B, Klein J, Tesoro A & Koren G (1991) Pregnancy-induced changes in drug metabolism in epileptic women. J Pharmacol Exp Ther 257: 735-740.
Bourgeois BF & Wad N (1984) Individual and combined antiepileptic and neurotoxic activity of carbamazepine and carbamazepine-10,11-epoxide in mice. J Pharmacol Exp Ther 231: 411-415.
Bourget P, Roulot C & Fernandez H (1995) Models for placental transfer studies of drugs. Clin Pharmacokinet 28: 161-180.
Boyd CA (1991) Placental transport studied by means of isolated plasma membrane vesicles. Proc Nutr Soc 50: 337-343.
Brodie MJ, Richens A & Yuen AW (1995) Double-blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. UK Lamotrigine/Carbamazepine Monotherapy Trial Group. Lancet 345: 476-479.
Bruno MK & Harden CL (2002) Epilepsy in Pregnant Women. Curr Treat Options Neurol 4: 31-40.
Bülau P, Paar WD & von Unruh GE (1988) Pharmacokinetics of oxcarbazepine and 10-hydroxy-carbazepine in the newborn child of an oxcarbazepine-treated mother. Eur J Clin Pharmacol 34: 311-313.
Bustard MA, Farley AE & Smith GN (2002) The pharmacokinetics of glyceryl trinitrate with the use of the in vitro term human placental perfusion setup. Am J Obstet Gynecol 187: 187-190.
Cannell GR, Kluck RM, Hamilton SE, Mortimer RH, Hooper WD & Dickinson RG (1988) Markers of physical integrity and metabolic viability of the perfused human placental lobule. Clin Exp Pharmacol Physiol 15: 837-844.
Casey B & Bawdon RE (2000) Ex vivo human placental transfer of trovafloxacin. Infect Dis Obstet Gynecol 8: 228-229.
Casey BM & Bawdon RE (1998) Placental transfer of ritonavir with zidovudine in the ex vivo placental perfusion model. Am J Obstet Gynecol 179: 758-761.
Castrén K, Pienimäki P, Arvela P & Vähäkangas K (1996) Metabolites and DNA-binding of carbamazepine and oxcarbazepine in vitro by rat liver microsomes. Hum Exp Toxicol 15: 577-582.
Challier JC (1985) Criteria for evaluating perfusion experiments and presentation of results. Contrib Gynecol Obstet 13: 32-39.
Challier JC, Guerre-Millo M, Nandakumaran M, Gerbaut L & d'Athis P (1985) Clearance of compounds of different molecular size in the human placenta in vitro. Biol Neonate 48: 143-148.
Challier JC, Schneider H & Dancis J (1976) In vitro perfusion of human placenta. V. Oxygen consumption. Am J Obstet Gynecol 126: 261-265.
Challis DE, Pfarrer CD, Ritchie JW, Koren G & Adamson SL (2000) Glucose metabolism is elevated and vascular resistance and maternofetal transfer is normal in perfused placental cotyledons from severely growth-restricted fetuses. Pediatr Res 47: 309-315.
Ching MS, Mihaly GW, Morgan DJ, Date NM, Hardy KJ & Smallwood RA (1987) Low clearance of cimetidine across the human placenta. J Pharmacol Exp Ther 241: 1006-1009.
Clifton VL, Giles WB, Smith R, Bisits AT, Hempenstall PA, Kessell CG & Gibson PG (2001) Alterations of placental vascular function in asthmatic pregnancies. Am J Respir Crit Care Med 164: 546-553.
Cohen AF, Land GS, Breimer DD, Yuen WC, Winton C & Peck AW (1987) Lamotrigine, a new anticonvulsant: pharmacokinetics in normal humans. Clin Pharmacol Ther 42: 535-541.
Collier AC, Ganley NA, Tingle MD, Blumenstein M, Marvin KW, Paxton JW, Mitchell MD & Keelan JA (2002a) UDP-glucuronosyltransferase activity, expression and cellular localization in human placenta at term. Biochem Pharmacol 63: 409-419.
Collier AC, Tingle MD, Keelan JA, Paxton JW & Mitchell MD (2000) A highly sensitive fluorescent microplate method for the determination of UDP-glucuronosyl transferase activity in tissues and placental cell lines. Drug Metab Dispos 28: 1184-1186.
Collier AC, Tingle MD, Paxton JW, Mitchell MD & Keelan JA (2002b) Metabolizing enzyme localization and activities in the first trimester human placenta: the effect of maternal and gestational age, smoking and alcohol consumption. Hum Reprod 17: 2564-2572.

68
Colucci R, Glue P, Holt B, Banfield C, Reidenberg P, Meehan JW, Pai S, Nomeir A, Lim J, Lin CC & Affrime MB (1996) Effect of felbamate on the pharmacokinetics of lamotrigine. J Clin Pharmacol 36: 634-638.
d'Incalci M, Broggini M, Buscaglia M & Pardi G (1983) Transplacental passage of doxorubicin. Lancet 1: 75.
Dally A (1998) Thalidomide: was the tragedy preventable? Lancet 351: 1197-1199. Dam M & Jensen PK (1989) Potential antiepileptic drugs. Oxcarbazepine. In Levy R, Mattson R &
Meldrum B (eds) Antiepileptic drugs, 3rd ed. Raven Press, New York, p 913-924. Dam M & Owen A (1995) Other antiepileptic drugs. Oxcarbazepine. In Levy RH, Mattson RH &
Meldrum BS (eds) Antiepileptic drugs, 4 th ed. Raven Press, New York, p 987-995. Dancis J, Ghosh NK, Jansen V, Schneider H, Fallon RJ & Cox RP (1979) Secretory proteins in the
perfused human placenta. Biol Neonate 35: 188-193. Davila JC, Rodriguez RJ, Melchert RB & Acosta D, Jr. (1998) Predictive value of in vitro model
systems in toxicology. Annu Rev Pharmacol Toxicol 38: 63-96. Dawes M & Chowienczyk PJ (2001) Drugs in pregnancy. Pharmacokinetics in pregnancy. Best
Pract Res Clin Obstet Gynaecol 15: 819-826. Dean JC, Hailey H, Moore SJ, Lloyd DJ, Turnpenny PD & Little J (2002) Long term health and
neurodevelopment in children exposed to antiepileptic drugs before birth. J Med Genet 39: 251-259.
Diaz-Arrastia R, Agostini MA & Van Ness PC (2002) Evolving treatment strategies for epilepsy. JAMA 287: 2917-2920.
Dicke JM, Johnson RF, Henderson GI, Kuehl TJ & Schenker S (1988) A comparative evaluation of the transport of H2-receptor antagonists by the human and baboon placenta. Am J Med Sci 295: 198-206.
Dickins M & Chen C (2002) Lamotrigine. Chemistry, biotransformation, and pharmacokinetics. In Levy RH et al (eds) Antiepileptic drugs, 5th ed. Lippincott Williams & Wilkins, Philadelphia, p 370-379.
Dodds HM, Taylor PJ, Johnson LP, Mortimer RH, Pond SM & Cannell GR (1997) Cortisol metabolism and its inhibition by glycyrrhetinic acid in the isolated perfused human placental lobule. J Steroid Biochem Mol Biol 62: 337-343.
Doherty MM & Charman WN (2002) The mucosa of the small intestine: how clinically relevant as an organ of drug metabolism? Clin Pharmacokinet 41: 235-253.
Dolk H & McElhatton P (2002) Assessing epidemiological evidence for the teratogenic effects of anticonvulsant medications. J Med Genet 39: 243-244.
Dollery C (1991) Diazepam. In Dollery C (ed) Therapeutic drugs Volume 1. Churchill Linvingstone, Edinburgh, p D86-D91.
Eichelbaum M, Jensen C, von Sassen W, Bertilsson L & Tomson T (1984) In vivo and in vitro biotransformation of carbamazepine in man and rat. In Levy RH (ed) Metabolism of Antiepileptic drugs. Raven Press, New york, p 27-34.
Eichelbaum M, Tomson T, Tybring G & Bertilsson L (1985) Carbamazepine metabolism in man. Induction and pharmacogenetic aspects. Clin Pharmacokinet 10: 80-90.
Eichhorn KH, Bauer C, Eckardt KU, Zimmermann R, Huch A & Huch R (1993) Lack of associations between fetal and maternal serum-erythropoietin at birth. Eur J Obstet Gynecol Reprod Biol 50: 47-52.
Ekins S, Ring BJ, Grace J, McRobie-Belle DJ & Wrighton SA (2000) Present and future in vitro approaches for drug metabolism. J Pharmacol Toxicol Methods 44: 313-324.
Enders AC & Blankenship TN (1999) Comparative placental structure. Adv Drug Deliv Rev 38: 3-15.
Engle PM & Heck AM (2000) Lamotrigine for the treatment of bipolar disorder. Ann Pharmacother 34: 258-262.
Erkkola R, Kangas L & Pekkarinen A (1973) The transfer of diazepam across the placenta during labour. Acta Obstet Gynecol Scand 52: 167-170.
Eto S, Tanaka N, Noda H & Noda A (1995) 9-Hydroxymethyl-10-carbamoylacridan in human serum is one of the major metabolites of carbamazepine. Biol Pharm Bull 18: 926-928.

69
Faber JJ, Thornburg KL & Binder ND (1992) Physiology of placental tranfer in mammals. American Zoologist 32: 343-354.
Faigle JW, Brechbuhler S, Feldmann KF & Richter JW (1976) The biotransformation of carbamazepine. In Birkmayer W (ed) Epileptic seizures-Behaviour-Pain. Huber Publ, Berne, p 127-140.
Faigle JW & Feldmann KF (1995) Carbamazepine. Chemistry and biotransformation. In Levy R, Mattson R & Meldrum B (eds) Antiepileptic drugs, Fourth Edition ed. Raven Press, New York, p 499-513.
Fares F & Gavish M (1986) Characterization of peripheral benzodiazepine binding sites in human term placenta. Biochem Pharmacol 35: 227-230.
Fex G, Larsson K, Andersson A & Berggren-Soderlund M (1995) Low serum concentration of all-trans and 13-cis retinoic acids in patients treated with phenytoin, carbamazepine and valproate. Possible relation to teratogenicity. Arch Toxicol 69: 572-574.
Finnell RH, Bennett GD, Slattery JT, Amore BM, Bajpai M & Levy RH (1995) Effect of treatment with phenobarbital and stiripentol on carbamazepine-induced teratogenicity and reactive metabolite formation. Teratology 52: 324-332.
Finnell RH, Mohl VK, Bennett GD & Taylor SM (1986) Failure of epoxide formation to influence carbamazepine-induced teratogenesis in a mouse model. Teratog Carcinog Mutagen 6: 393-401.
Fitton A & Goa KL (1995) Lamotrigine. An update of its pharmacology and therapeutic use in epilepsy. Drugs 50: 691-713.
Forestier F, Daffos F & Capella-Pavlovsky M (1984) Low molecular weight heparin (PK 10169) does not cross the placenta during the second trimester of pregnancy study by direct fetal blood sampling under ultrasound. Thromb Res 34: 557-560.
Forestier F, de Renty P, Peytavin G, Dohin E, Farinotti R & Mandelbrot L (2001) Maternal-fetal transfer of saquinavir studied in the ex vivo placental perfusion model. Am J Obstet Gynecol 185: 178-181.
Frederiksen MC (2001) Physiologic changes in pregnancy and their effect on drug disposition. Semin Perinatol 25: 120-123.
Frigerio A & Morselli PL (1975) Carbamazepine: biotransformation. Adv Neurol 11: 295-308. Friis ML, Kristensen O, Boas J, Dalby M, Deth SH, Gram L, Mikkelsen M, Pedersen B, Sabers A,
Worm-Petersen J, Andersen D & Jensen PK (1993) Therapeutic experiences with 947 epileptic out-patients in oxcarbazepine treatment. Acta Neurol Scand 87(3):224-227
Furst SM, Sukhai P, McClelland RA & Uetrecht JP (1995) Covalent binding of carbamazepine oxidative metabolites to neutrophils. Drug Metab Dispos 23: 590-594.
Furst SM & Uetrecht JP (1993) Carbamazepine metabolism to a reactive intermediate by the myeloperoxidase system of activated neutrophils. Biochem Pharmacol 45: 1267-1275.
Gamble JAS, Moore J, Lamki H & Howard PJ (1977) A study of plasma diazepam levels in mother and infant. Br J Obstet Gynaecol 84: 588-591.
Ganapathy V, Prasad PD, Ganapathy ME & Leibach FH (1999) Drugs of abuse and placental transport. Adv Drug Deliv Rev 38: 99-110.
Ganapathy V, Prasad PD, Ganapathy ME & Leibach FH (2000) Placental transporters relevant to drug distribution across the maternal-fetal interface. J Pharmacol Exp Ther 294: 413-420.
Garnett WR (2002) Lamotrigine. Interactions with other drugs. In Levy RH et al (eds) Antiepileptic drugs, 5th ed. Lippincott Williams & Wilkins, Philadelphia, p 380-388.
Gatti G, Furlaut M & Perucca E (2001) Interindividual variability in the metabolism of anti-epileptic drugs and its clinical implications. In Pacifici GM& Pelkonen O (eds) Interindividual variability in human drug metabolim. Taylor & Francis, London and New York, p 157-180.
Giroux M, Teixera MG, Dumas JC, Desprats R, Grandjean H & Houin G (1997) Influence of maternal blood flow on the placental transfer of three opioids--fentanyl, alfentanil, sufentanil. Biol Neonate 72: 133-141.
Gonzalez FJ & Korzekwa KR (1995) Cytochromes P450 expression systems. Annu Rev Pharmacol Toxicol 35: 369-390.
Gregor H, Egarter C, Levin D, Sternberger B, Heinze G, Leitich H & Reisenberger K (1999) The passage of granulocyte-macrophage colony-stimulating factor across the human placenta perfused in vitro. J Soc Gynecol Investig 6: 307-310.

70
Grohard P, Akbaraly JP, Saux MC, Gimenez S, Robert J, Brachet-Liermain A & Leng JJ (1989) [Transplacental passage of doxorubicin]. J Gynecol Obstet Biol Reprod (Paris) 18: 595-600.
Guengerich FP (1999) Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol 39: 1-17.
Guengerich FP (2002) Update information on human P450s. Drug Metab Rev 34: 7-15. Guerre-Millo M, Rey E, Challier JC, Turquais JM, d'Athis P & Olive G (1979) Transfer in vitro of
three benzodiazepines across the human placenta. Eur J Clin Pharmacol 15: 171-173. Guerreiro MM, Vigonius U, Pohlmann H, de Manreza ML, Fejerman N, Antoniuk SA & Moore A
(1997) A double-blind controlled clinical trial of oxcarbazepine versus phenytoin in children and adolescents with epilepsy. Epilepsy Res 27: 205-213.
Guillouzo A, Rialland L, Fautrel A & Guyomard C (1999) Survival and function of isolated hepatocytes after cryopreservation. Chem Biol Interact 121: 7-16.
Hakkola J, Pasanen M, Hukkanen J, Pelkonen O, Mäenpää J, Edwards RJ, Boobis AR & Raunio H (1996a) Expression of xenobiotic-metabolizing cytochrome P450 forms in human full-term placenta. Biochem Pharmacol 51: 403-411.
Hakkola J, Pelkonen O, Pasanen M & Raunio H (1998) Xenobiotic-metabolizing cytochrome P450 enzymes in the human feto-placental unit: role in intrauterine toxicity. Crit Rev Toxicol 28: 35-72.
Hakkola J, Raunio H, Purkunen R, Pelkonen O, Saarikoski S, Cresteil T & Pasanen M (1996b) Detection of cytochrome P450 gene expression in human placenta in first trimester of pregnancy. Biochem Pharmacol 52: 379-383.
Haram K & Bakke OM (1980) Diazepam as an induction agent for caesarean section: a clinical and pharmacokinetic study of fetal drug exposure. Br J Obstet Gynaecol 87: 506-512.
Haram K, Bakke OM, Johannessen KH & Lund T (1978) Transplacental passage of diazepam during labor: influence of uterine contractions. Clin Pharmacol Ther 24: 590-599.
Hartvig P, Lindberg BS, Lilja A, Lundqvist H, Langstrom B & Rane A (1989) Positron emission tomography in studies on fetomaternal disposition of opioids. Dev Pharmacol Ther 12: 74-80.
He YL, Seno H, Sasaki K & Tashiro C (2002) The influences of maternal albumin concentrations on the placental transfer of propofol in human dually perfused cotyledon in vitro. Anesth Analg 94: 1312-4, table.
He YL, Seno H, Tsujimoto S & Tashiro C (2001) The effects of uterine and umbilical blood flows on the transfer of propofol across the human placenta during in vitro perfusion. Anesth Analg 93: 151-156.
He YL, Tsujimoto S, Tanimoto M, Okutani R, Murakawa K & Tashiro C (2000) Effects of protein binding on the placental transfer of propofol in the human dually perfused cotyledon in vitro. Br J Anaesth 85: 281-286.
Heikkilä A, Myllynen P, Keski-Nisula L, Heinonen S, Vähäkangas K & Yla-Herttuala S (2002) Gene transfer to human placenta ex vivo: a novel application of dual perfusion of human placental cotyledon. Am J Obstet Gynecol 186: 1046-1051.
Heikkine T, Ekblad U & Laine K (2002) Transplacental transfer of citalopram, fluoxetine and their primary demethylated metabolites in isolated perfused human placenta. BJOG 109: 1003-1008.
Heikkinen T, Ekblad U & Laine K (2001) Transplacental transfer of amitriptyline and nortriptyline in isolated perfused human placenta. Psychopharmacology (Berl) 153: 450-454.
Heikkinen T, Laine K, Neuvonen PJ & Ekblad U (2000) The transplacental transfer of the macrolide antibiotics erythromycin, roxithromycin and azithromycin. BJOG 107: 770-775.
Henderson GI, Hu ZQ, Johnson RF, Perez AB, Yang Y & Schenker S (1992) Acyclovir transport by the human placenta. J Lab Clin Med 120: 885-892.
Herman NL, Li AT, Van Decar TK, Johnson RF, Bjoraker RW, Downing JW & Jones D (2000) Transfer of methohexital across the perfused human placenta. J Clin Anesth 12: 25-30.
Hernandez-Diaz S, Werler MM, Walker AM & Mitchell AA (2000) Folic acid antagonists during pregnancy and the risk of birth defects. N Engl J Med 343: 1608-1614.
Hollenberg PF (2002) Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev 34: 17-35.
Holmes LB (2002) The teratogenicity of anticonvulsant drugs: a progress report. J Med Genet 39: 245-247.

71
Holmes LB, Harvey EA, Coull BA, Huntington KB, Khoshbin S, Hayes AM & Ryan LM (2001) The teratogenicity of anticonvulsant drugs. N Engl J Med 344: 1132-1138.
Hsieh TT, Chen KC & Chung PC (1992) In vitro perfusion study of the human placenta at term: preliminary results. J Formos Med Assoc 91: 799-803.
Hurley SC (2002) Lamotrigine update and its use in mood disorders. Ann Pharmacother 36: 860-873.
Iqbal MM, Sobhan T, Aftab SR & Mahmud SZ (2002a) Diazepam use during pregnancy: a review of the literature. Del Med J 74: 127-135.
Iqbal MM, Sobhan T & Ryals T (2002b) Effects of commonly used benzodiazepines on the fetus, the neonate, and the nursing infant. Psychiatr Serv 53: 39-49.
Ito K, Iwatsubo T, Kanamitsu S, Nakajima Y & Sugiyama Y (1998a) Quantitative prediction of in vivo drug clearance and drug interactions from in vitro data on metabolism, together with binding and transport. Annu Rev Pharmacol Toxicol 38: 461-499.
Ito K, Iwatsubo T, Kanamitsu S, Ueda K, Suzuki H & Sugiyama Y (1998b) Prediction of pharmacokinetic alterations caused by drug-drug interactions: metabolic interaction in the liver. Pharmacol Rev 50: 387-412.
Ito S (2001) Transplacental treatment of fetal tachycardia: implications of drug transporting proteins in placenta. Semin Perinatol 25: 196-201.
Jauniaux E & Gulbis B (2000) In vivo investigation of placental transfer early in human pregnancy. Eur J Obstet Gynecol Reprod Biol 92: 45-49.
Jawad S, Yuen WC, Peck AW, Hamilton MJ, Oxley JR & Richens A (1987) Lamotrigine: single-dose pharmacokinetics and initial 1 week experience in refractory epilepsy. Epilepsy Res 1: 194-201.
Jensen PK, Saano V, Haring P, Svenstrup B & Menge GP (1992) Possible interaction between oxcarbazepine and an oral contraceptive. Epilepsia 33: 1149-1152.
Johannessen SI (1992) Pharmacokinetics of valproate in pregnancy: mother-foetus-newborn. Pharm Weekbl Sci 14: 114-117.
Johnson RF, Cahana A, Olenick M, Herman N, Paschall RL, Minzter B, Ramasubramanian R, Gonzalez H & Downing JW (1999) A comparison of the placental transfer of ropivacaine versus bupivacaine. Anesth Analg 89: 703-708.
Johnson RF, Herman N, Arney TL, Johnson HV, Paschall RL & Downing JW (1997) The placental transfer of sufentanil: effects of fetal pH, protein binding, and sufentanil concentration. Anesth Analg 84: 1262-1268.
Jones CJ & Fox H (1991) Ultrastructure of the normal human placenta. Electron Microsc Rev 4: 129-178.
Kalis MM & Huff NA (2001) Oxcarbazepine, an antiepileptic agent. Clin Ther 23: 680-700. Kalow W (2001) Genetic factors that cause variability in human drug metabolism. In Pacifici GM&
Pelkonen O (eds) Interindividual variability in human drug metabolim. Taylor & Francis, London and New York, p 1-14.
Kanto J, Erkkola R & Sellman R (1973) Accumulation of diazepam and N-demethyldiazepam in the fetal blood during the labour. Ann Clin Res 5: 375-379.
Kanto J & Scheinin M (1987) Placental and blood-CSF transfer of orally administered diazepam in the same person. Pharmacol Toxicol 61: 72-74.
Karp GI, von Oeyen P, Valone F, Khetarpal VK, Israel M, Mayer RJ, Frigoletto FD & Garnick MB (1983) Doxorubicin in pregnancy: possible transplacental passage. Cancer Treat Rep 67: 773-777.
Kaufmann P (1985) Basic morphology of the fetal and maternal circuits in the human placenta. Contrib Gynecol Obstet 13: 5-17.
Kedderis GL (1997) Extrapolation of in vitro enzyme induction data to humans in vivo. Chem Biol Interact 107: 109-121.
Kerr BM & Levy RH (1995) Carbamazepine. Carbamazepine epoxide. In Levy R, Mattson R & Meldrum B (eds) Antiepileptic drugs, Fourth Edition ed. Raven Press, New York, p 529-541.
Kerr BM, Thummel KE, Wurden CJ, Klein SM, Kroetz DL, Gonzalez FJ & Levy RH (1994) Human liver carbamazepine metabolism. Role of CYP3A4 and CYP2C8 in 10,11-epoxide formation. Biochem Pharmacol 47: 1969-1979.

72
Ketter TA, Post RM & Worthington K (1991a) Principles of clinically important drug interactions with carbamazepine. Part I. J Clin Psychopharmacol 11: 198-203.
Ketter TA, Post RM & Worthington K (1991b) Principles of clinically important drug interactions with carbamazepine. Part II. J Clin Psychopharmacol 11: 306-313.
Kim RB (2002) Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metab Rev 34: 47-54.
King A, Thomas L & Bischof P (2000) Cell culture models of trophoblast II: trophoblast cell lines--a workshop report. Placenta 21 Suppl A: S113-S119.
Knipp GT, Audus KL & Soares MJ (1999) Nutrient transport across the placenta. Adv Drug Deliv Rev 38: 41-58.
Kodama Y, Kuranari M, Kodama H, Fujii I & Takeyama M (1993a) In vivo determinations of carbamazepine and carbamazepine-10, 11-epoxide binding parameters to serum proteins in monotherapy patients. J Clin Pharmacol 33: 851-855.
Kodama Y, Kuranari M, Tsutsumi K, Okamoto T, Kodama H, Yasunaga F, Fujii I & Takeyama M (1994) In vivo binding characteristics of carbamazepine and carbamazepine 10, 11-epoxide to serum proteins in monotherapy adult patients. Int J Clin Pharmacol Ther 32: 618-621.
Kodama Y, Tsutsumi K, Kuranari M, Kodama H, Fujii I & Takeyama M (1993b) In vivo binding characteristics of carbamazepine and carbamazepine-10,11-epoxide to serum proteins in paediatric patients with epilepsy. Eur J Clin Pharmacol 44: 291-293.
Kopecky EA, Simone C, Knie B & Koren G (1999) Transfer of morphine across the human placenta and its interaction with naloxone. Life Sci 65: 2359-2371.
Koren G, Klinger G & Ohlsson A (2002) Fetal pharmacotherapy. Drugs 62: 757-773. Koren G, Pastuszak A & Ito S (1998) Drugs in pregnancy. N Engl J Med 338: 1128-1137. Kramer WB, Saade G, Ou CN, Rognerud C, Dorman K, Mayes M & Moise KJ, Jr. (1995) Placental
transfer of sulindac and its active sulfide metabolite in humans. Am J Obstet Gynecol 172: 886-890.
Krishna BR, Zakowski MI & Grant GJ (1997) Sufentanil transfer in the human placenta during in vitro perfusion. Can J Anaesth 44: 996-1001.
Krishna DR & Klotz U (1994) Extrahepatic metabolism of drugs in humans. Clin Pharmacokinet 26: 144-160.
Kroes HY, Reefhuis J & Cornel MC (2002) Is there an association between maternal carbamazepine use during pregnancy and eye malformations in the child? Epilepsia 43: 929-931.
Kwan P & Brodie MJ (2001) Effectiveness of first antiepileptic drug. Epilepsia 42: 1255-1260. Lagrange F, Vergnes C, Brun JL, Paolucci F, Nadal T, Leng JJ, Saux MC & Banwarth B (2002)
Absence of placental transfer of pentasaccharide (Fondaparinux, Arixtra) in the dually perfused human cotyledon in vitro. Thromb Haemost 87: 831-835.
Lampela ES, Nuutinen LH, Ala-Kokko TI, Parikka RM, Laitinen RS, Jouppila PI & Vähäkangas KH (1999) Placental transfer of sulindac, sulindac sulfide, and indomethacin in a human placental perfusion model. Am J Obstet Gynecol 180: 174-180.
Lanchote VL, Bonato PS, Campos GM & Rodrigues I (1995) Factors influencing plasma concentrations of carbamazepine and carbamazepine-10,11-epoxide in epileptic children and adults. Ther Drug Monit 17: 47-52.
Lander CM & Eadie MJ (1991) Plasma antiepileptic drug concentrations during pregnancy. Epilepsia 32: 257-266.
Lankas GR, Wise LD, Cartwright ME, Pippert T & Umbenhauer DR (1998) Placental P-glycoprotein deficiency enhances susceptibility to chemically induced birth defects in mice. Reprod Toxicol 12: 457-463.
Larkin JG, McKee PJ, Forrest G, Beastall GH, Park BK, Lowrie JI, Lloyd P & Brodie MJ (1991) Lack of enzyme induction with oxcarbazepine (600 mg daily) in healthy subjects. Br J Clin Pharmacol 31: 65-71.
Leiser R & Kaufmann P (1994) Placental structure: in a comparative aspect. Exp Clin Endocrinol 102: 122-134.
Lertratanangkoon K & Horning MG (1982) Metabolism of carbamazepine. Drug Metab Dispos 10: 1-10.

73
Levy RH & Pitlick WH (1982) Carbamazepine. Interactions with other drugs. In Woodbury DM, Penry JK & Pippenger CE (eds) Antiepileptic drugs. Raven Press, New York, p 497-505.
Li AP & Kedderis GL (1997) Primary hepatocyte culture as an experimental model for the evaluation of interactions between xenobiotics and drug-metabolizing enzymes. Chem Biol Interact 107: 1-3.
Li AP, Maurel P, Gomez-Lechon MJ, Cheng LC & Jurima-Romet M (1997) Preclinical evaluation of drug-drug interaction potential: present status of the application of primary human hepatocytes in the evaluation of cytochrome P450 induction. Chem Biol Interact 107: 5-16.
Lin JH (1998) Applications and limitations of interspecies scaling and in vitro extrapolation in pharmacokinetics. Drug Metab Dispos 26: 1202-1212.
Lin JH & Lu AY (1998) Inhibition and induction of cytochrome P450 and the clinical implications. Clin Pharmacokinet 35: 361-390.
Lin JH & Lu AY (2001) Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu Rev Pharmacol Toxicol 41: 535-567.
Lindhout D & Omtzigt JG (1994) Teratogenic effects of antiepileptic drugs: implications for the management of epilepsy in women of childbearing age. Epilepsia 35 (Suppl 4):S19-28.
Loebstein R & Koren G (2002) Clinical relevance of therapeutic drug monitoring during pregnancy. Ther Drug Monit 24: 15-22.
Loebstein R, Lalkin A & Koren G (1997) Pharmacokinetic changes during pregnancy and their clinical relevance. Clin Pharmacokinet 33: 328-343.
Macdonald RL (2002a) Benzodiazepines. Mechanism of action. In Levy R et al (eds) Antiepileptic drugs, 5th ed. Lippincott Williams & Wilkins, Philadelphia, p 179-186.
Macdonald RL (2002b) Carbamazepine. Mechanism of action. In Levy R et al (eds) Antiepileptic drugs, 5th ed. Lippincott Williams & Wilkins, Philadelphia, p 227-235.
Macdonald RL & Kelly KM (1995) Antiepileptic drug mechanisms of action. Epilepsia 36 Suppl 2: S2-12.
Magee KP & Bawdon RE (2000) Ex vivo human placental transfer and the vasoactive properties of hydralazine. Am J Obstet Gynecol 182: 167-169.
Maggs JL, Pirmohamed M, Kitteringham NR & Park BK (1997) Characterization of the metabolites of carbamazepine in patient urine by liquid chromatography/mass spectrometry. Drug Metab Dispos 25: 275-280.
Maguire DJ, Cannell GR, Addison RS & Dawkins B (1999) In vitro placental perfusion. Practical parameters and procedural performance. Adv Exp Med Biol 471: 361-366.
Malek A, Blann E & Mattison DR (1996) Human placental transport of oxytocin. J Matern Fetal Med 5: 245-255.
Malek A, Sager R, Eckardt KU, Bauer C & Schneider H (1994) Lack of transport of erythropoietin across the human placenta as studied by an in vitro perfusion system. Pflugers Arch 427: 157-161.
Mandelli M, Morselli PL, Nordio S, Pardi G, Principi N, Sereni F & Tognoni G (1975) Placental transfer of diazepam and its disposition in the newborn. Clin Pharmacol Ther 17: 564-572.
Marchi NS, Azoubel R & Tognola WA (2001) Teratogenic effects of lamotrigine on rat fetal brain: a morphometric study. Arq Neuropsiquiatr 59: 362-364.
Marson AG, Kadir ZA, Hutton JL & Chadwick DW (1997) The new antiepileptic drugs: a systematic review of their efficacy and tolerability. Epilepsia 38: 859-880.
Marzolini C, Rudin C, Decosterd LA, Telenti A, Schreyer A, Biollaz J & Buclin T (2002) Transplacental passage of protease inhibitors at delivery. AIDS 16: 889-893.
Matalon S, Schechtman S, Goldzweig G & Ornoy A (2002) The teratogenic effect of carbamazepine: a meta-analysis of 1255 exposures. Reprod Toxicol 16: 9-17.
Matar KM, Nicholls PJ, Tekle A, Bawazir SA & Al Hassan MI (1999) Effects of pregnancy on the pharmacokinetics of lamotrigine in dogs. Epilepsia 40: 1353-1356.
Matsuo F (1999) Lamotrigine. Epilepsia 40 Suppl 5: S30-S36. McCarthy GT, O'Connell B & Robinson AE (1973) Blood levels of diazepam in infants of two
mothers given large doses of diazepam during labour. J Obstet Gynaecol Br Commonw 80: 349-352.

74
McCleane GJ (2000) Lamotrigine in the management of neuropathic pain: a review of the literature. Clin J Pain 16: 321-326.
McElhatton PR (1994) The effects of benzodiazepine use during pregnancy and lactation. Reprod Toxicol 8: 461-475.
McKee PJ, Blacklaw J, Forrest G, Gillham RA, Walker SM, Connelly D & Brodie MJ (1994) A double-blind, placebo-controlled interaction study between oxcarbazepine and carbamazepine, sodium valproate and phenytoin in epileptic patients. Br J Clin Pharmacol 37: 27-32.
Mihaly GW & Morgan DJ (1983) Placental drug transfer: effects of gestational age and species. Pharmacol Ther 23: 253-266.
Mikati MA, Browne TR & Collins JF (1989) Time course of carbamazepine autoinduction. The VA Cooperative Study No.118 Group. Neurology 39: 592-594.
Miller RK, Jessee L, Barrish A, Gilbert J & Manson JM (1998) Pharmacokinetic studies of enalaprilat in the in vitro perfused human placental lobule system. Teratology 58: 76-81.
Miller RK, Wier PJ, Maulik D & di Sant'Agnese PA (1985) Human placenta in vitro: characterization during 12 h of dual perfusion. Contrib Gynecol Obstet 13: 77-84.
Miller RK, Wier PJ, Shah Y, di Sant'Agnese PA & Perez-D'Gregorio R (1989) Criteria for in vitro dual perfusions in the human placental lobule: perfusions excess of 12 hours. In Genbacev O, Klopper A & Beaconsfield R (eds) Placenta as a model and source. Plenium Press, New York and London, p 27-38.
Mirkin BL (1973) Drug distribution in pregnancy. In Boreus L (ed) Fetal pharmacology. Raven Press, New York, p 1-27.
Morrell MJ (1996) The new antiepileptic drugs and women: efficacy, reproductive health, pregnancy, and fetal outcome. Epilepsia 37 Suppl 6: S34-S44.
Morrell MJ (2002) Antiepileptic drug use in women. In Levy R et al (eds) Antiepileptic drugs, 5th ed. Lippincott Williams & Wilkins, Philadelphia, p 132-148.
Mortimer RH, Cannell GR, Addison RS, Johnson LP, Roberts MS & Bernus I (1997) Methimazole and propylthiouracil equally cross the perfused human term placental lobule. J Clin Endocrinol Metab 82: 3099-3102.
Nandakumaran M, Dashti HM & Al Zaid NS (2002) Maternal-fetal transport kinetics of copper, selenium, magnesium and iron in perfused human placental lobule: in vitro study. Mol Cell Biochem 231: 9-14.
Nandakumaran M, Dev BR, Makhseed M & Sugathan TN (1998) Assessment of D-glucose transport kinetics in the perfused human placenta: an in vitro study. Acta Paediatr Jpn 40: 307-312.
Nanovskaya T, Deshmukh S, Brooks M & Ahmed MS (2002) Transplacental transfer and metabolism of buprenorphine. J Pharmacol Exp Ther 300: 26-33.
Nau H (1986) Species differences in pharmacokinetics and drug teratogenesis. Environ Health Perspect 70: 113-129.
Nebert DW & Dieter MZ (2000) The evolution of drug metabolism. Pharmacology 61: 124-135. Nebert DW & Russell DW (2002) Clinical importance of the cytochromes P450. Lancet 360: 1155-
1162. Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Feyereisen R, Waxman DJ, Waterman MR,
Gotoh O, Coon MJ, Estabrook RW, Gunsalus IC & Nebert DW (1996) P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 6: 1-42.
Nevasaari K (1976) A simple membrane oxygenator for the isolated rat liver perfusion. Experientia 32: 534-535.
Nulman I, Laslo D & Koren G (1999) Treatment of epilepsy in pregnancy. Drugs 57: 535-544. Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC, MacIntyre F, Rance DJ & Wastall P
(1997) The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther 283: 46-58.
Ohman I, Vitols S & Tomson T (2000) Lamotrigine in pregnancy: pharmacokinetics during delivery, in the neonate, and during lactation. Epilepsia 41: 709-713.

75
Olinga P, Hof IH, Merema MT, Smit M, de Jager MH, Swart PJ, Slooff MJ, Meijer DK & Groothuis GM (2001) The applicability of rat and human liver slices to the study of mechanisms of hepatic drug uptake. J Pharmacol Toxicol Methods 45: 55-63.
Olivero OA, Parikka R, Poirier MC & Vähäkangas K (1999) 3'-azido-3'-deoxythymidine (AZT) transplacental perfusion kinetics and DNA incorporation in normal human placentas perfused with AZT. Mutat Res 428: 41-47.
Omarini D, Barzago MM, Bortolotti A, Lucchini G, Stellari F, Efrati S & Bonati M (1993) Placental transfer of theophylline in an in vitro closed perfusion system of human placenta isolated lobule. Eur J Drug Metab Pharmacokinet 18: 369-374.
Omarini D, Pistotti V & Bonati M (1992) Placental perfusion. An overview of the literature. J Pharmacol Toxicol Methods 28: 61-66.
Omura T (1999) Forty years of cytochrome P450. Biochem Biophys Res Commun 266: 690-698. Osmond DT, King RG, Brennecke SP & Gude NM (2001) Placental glucose transport and
utilisation is altered at term in insulin-treated, gestational-diabetic patients. Diabetologia 44: 1133-1139.
Osmond DT, Nolan CJ, King RG, Brennecke SP & Gude NM (2000) Effects of gestational diabetes on human placental glucose uptake, transfer, and utilisation. Diabetologia 43: 576-582.
Owen A, Pirmohamed M, Tettey JN, Morgan P, Chadwick D & Park BK (2001) Carbamazepine is not a substrate for P-glycoprotein. Br J Clin Pharmacol 51: 345-349.
Ozkinay F, Cogulu O, Gunduz C, Yilmaz D & Kultursay N (2003) Valproic acid and lamotrigine treatment during pregnancy. The risk of chromosomal abnormality. Mutat Res 534: 197-199.
Paakki P, Kirkinen P, Helin H, Pelkonen O, Raunio H & Pasanen M (2000) Antepartum glucocorticoid therapy suppresses human placental xenobiotic and steroid metabolizing enzymes. Placenta 21: 241-246.
Pacifici GM & Nottoli R (1995) Placental transfer of drugs administered to the mother. Clin Pharmacokinet 28: 235-269.
Page K (1993) The physiology of the human placenta. London: UCL Press Limited Panesar SK, Bandiera SM & Abbott FS (1996) Comparative effects of carbamazepine and
carbamazepine-10,11-epoxide on hepatic cytochromes P450 in the rat. Drug Metab Dispos 24: 619-627.
Panigel M (1962) Placental perfusion experiments. Am J Obstet Gynecol 84: 1664-1683. Park BK, Kitteringham NR, Pirmohamed M & Tucker GT (1996) Relevance of induction of human
drug-metabolizing enzymes: pharmacological and toxicological implications. Br J Clin Pharmacol 41: 477-491.
Park BK & Pirmohamed M (2001) Toxicogenetics in drug development. Toxicol Lett 120: 281-291.
Park BK, Pirmohamed M & Kitteringham NR (1995) The role of cytochrome P450 enzymes in hepatic and extrahepatic human drug toxicity. Pharmacol Ther 68: 385-424.
Parsons DN, Dickins M & Morley TJ (1995) Lamotrigine. Absorption, distribution, and excretion. In Levy RH, Mattson RH & Meldrum BS (eds) Antiepileptic Drugs, 4th ed. Raven Press, New York, p 877-881.
Pasanen M (1999) The expression and regulation of drug metabolism in human placenta. Adv Drug Deliv Rev 38: 81-97.
Pastrakuljic A, Derewlany LO, Knie B & Koren G (2000) The effects of cocaine and nicotine on amino acid transport across the human placental cotyledon perfused in vitro. J Pharmacol Exp Ther 294: 141-146.
Pastrakuljic A, Schwartz R, Simone C, Derewlany LO, Knie B & Koren G (1998) Transplacental transfer and biotransformation studies of nicotine in the human placental cotyledon perfused in vitro. Life Sci 63: 2333-2342.
Pearce RE, Vakkalagadda GR & Leeder JS (2002) Pathways of carbamazepine bioactivation in vitro I. Characterization of human cytochromes P450 responsible for the formation of 2- and 3-hydroxylated metabolites. Drug Metab Dispos 30: 1170-1179.
Peck AW (1991) Clinical pharmacology of lamotrigine. Epilepsia 32 Suppl 2: S9-12. Pelkonen O (1973). Studies on drug metabolizing enzymes in the human fetus. Oulu: Kirjapaino
osakeyhtiö Kaleva

76
Pelkonen O (2002) Human CYPs: in vivo and clinical aspects. Drug Metab Rev 34: 37-46. Pelkonen O, Boobis AR, Kremers P, Ingelman-Sundberg M & Rane A (2001) Interindividual
variation of P450 enzymes in vitro and its causes. In Pacifici GM& Pelkonen O (eds) Interindividual variability in human drug metabolim. Taylor & Francis, London and New York, p 269-332.
Pelkonen O & Breimer DD (1994) Role of environmental factors in the pharmacokinetics of drugs: Considerations with respect to animal models, P-450 enzymes, and probe drugs. Handbook of Experimental Pharmacology, Vol 100 Pharmacokinetics of drugs 110: 289-332.
Pelkonen O, Hukkanen J, Honkakoski P, Hakkola J, Viitala P & Raunio H (2002) In vitro screening of cytochrome P450 induction potential. In Pelkonen O, Baumann A & Reichel A (eds) Ernst Schering Research Foundation Workshop 37.Pharmacokinetic Challenges in Drug Discovery. p 105-137.
Pelkonen O, Mäenpää J, Taavitsainen P, Rautio A & Raunio H (1998) Inhibition and induction of human cytochrome P450 (CYP) enzymes. Xenobiotica 28: 1203-1253.
Pellock JM (2000) Treatment of epilepsy in the new millennium. Pharmacotherapy 20: 129S-138S. Perez-D'Gregorio RE & Miller RK (1998) Transport and endogenous release of vitamin B12 in the
dually perfused human placenta. J Pediatr 132: S35-S42. Perucca E, Hedges A, Makki KA, Ruprah M, Wilson JF & Richens A (1984) A comparative study
of the relative enzyme inducing properties of anticonvulsant drugs in epileptic patients. Br J Clin Pharmacol 18: 401-410.
Peytavin G, Leng JJ, Forestier F, Saux M, Hohlfeld P & Farinotti R (2000) Placental transfer of pyrimethamine studied in an ex vivo placental perfusion model. Biol Neonate 78: 83-85.
Piafsky KM & Rane A (1978) Formation of carbamazepine epoxide in human fetal liver. Drug Metab Dispos 6: 502-503.
Pienimäki P. (1996). Pharmacokinetics of carbamazepine and oxcarbazepine with special reference to placental transfer and metabolism. Acta Univ Oul D No. 396.
Pienimäki P, Fuchs S, Isojärvi J & Vähäkangas K (1995a) Improved detection and determination of carbamazepine and oxcarbazepine and their metabolites by high-performance liquid chromatography. J Chromatogr B Biomed Appl 673: 97-105.
Pienimäki P, Hartikainen AL, Arvela P, Partanen T, Herva R, Pelkonen O & Vähäkangas K (1995b) Carbamazepine and its metabolites in human perfused placenta and in maternal and cord blood. Epilepsia 36: 241-248.
Pienimäki P, Lampela E, Hakkola J, Arvela P, Raunio H & Vähäkangas K (1997) Pharmacokinetics of oxcarbazepine and carbamazepine in human placenta. Epilepsia 38: 309-316.
Pirmohamed M, Kitteringham NR, Breckenridge AM & Park BK (1992a) The effect of enzyme induction on the cytochrome P450-mediated bioactivation of carbamazepine by mouse liver microsomes. Biochem Pharmacol 44: 2307-2314.
Pirmohamed M, Kitteringham NR, Guenthner TM, Breckenridge AM & Park BK (1992b) An investigation of the formation of cytotoxic, protein-reactive and stable metabolites from carbamazepine in vitro. Biochem Pharmacol 43: 1675-1682.
Pirmohamed M & Park BK (2001) Genetic susceptibility to adverse drug reactions. Trends Pharmacol Sci 22: 298-305.
Polliotti BM, Holmes R, Cornish JD, Hulsey M, Keesling S, Schwartz D, Abramowsky CR, Huddleston J, Panigel M & Nahmias AJ (1996) Long-term dual perfusion of isolated human placental lobules with improved oxygenation for infectious diseases research. Placenta 17: 57-68.
Pons JC, Lebon P, Frydman R & Delfraissy JF (1995) Pharmacokinetics of interferon-alpha in pregnant women and fetoplacental passage. Fetal Diagn Ther 10: 7-10.
Pons JC, Taburet AM, Singlas E, Delfraissy JF & Papiernik E (1991) Placental passage of azathiothymidine (AZT) during the second trimester of pregnancy: study by direct fetal blood sampling under ultrasound. Eur J Obstet Gynecol Reprod Biol 40: 229-231.
Qvist N, Storm K & Holmskov A (1985) Cimetidine as pre-anesthetic agent for cesarean section: perinatal effects on the infant, the placental transfer of cimetidine and its elimination in the infants. J Perinat Med 13: 179-183.

77
Rambeck B, Kurlemann G, Stodieck SR, May TW & Jurgens U (1997) Concentrations of lamotrigine in a mother on lamotrigine treatment and her newborn child. Eur J Clin Pharmacol 51: 481-484.
Rambeck B & Wolf P (1993) Lamotrigine clinical pharmacokinetics. Clin Pharmacokinet 25: 433-443.
Regnaud L, Sirois G & Chakrabarti S (1988) Effect of four-day treatment with carbamazepine at different dose levels on microsomal enzyme induction, drug metabolism and drug toxicity. Pharmacol Toxicol 62: 3-6.
Reisenberger K, Egarter C, Kapiotis S, Sternberger B, Gregor H & Husslein P (1997) Transfer of erythropoietin across the placenta perfused in vitro. Obstet Gynecol 89: 738-742.
Reisenberger K, Egarter C, Sternberger B, Eckenberger P, Eberle E & Weissenbacher ER (1996) Placental passage of angiotensin-converting enzyme inhibitors. Am J Obstet Gynecol 174: 1450-1455.
Renwick AB, Watts PS, Edwards RJ, Barton PT, Guyonnet I, Price RJ, Tredger JM, Pelkonen O, Boobis AR & Lake BG (2000) Differential maintenance of cytochrome P450 enzymes in cultured precision-cut human liver slices. Drug Metab Dispos 28: 1202-1209.
Reynolds F & Knott C (1989) Pharmacokinetics in pregnancy and placental drug transfer. Oxf Rev Reprod Biol 11: 389-449.
Richens A (1994) Safety of lamotrigine. Epilepsia 35 Suppl 5: S37-S40. Richens A & Yuen AW (1991) Overview of the clinical efficacy of lamotrigine. Epilepsia 32 Suppl
2: S13-S16. Ridd MJ, Brown KF, Nation RL & Collier CB (1989) The disposition and placental transfer of
diazepam in cesarean section. Clin Pharmacol Ther 45: 506-512. Ring JA, Ghabrial H, Ching MS, Smallwood RA & Morgan DJ (1999) Fetal hepatic drug
elimination. Pharmacol Ther 84: 429-445. Ringler GE & Strauss JF, III (1990) In vitro systems for the study of human placental endocrine
function. Endocr Rev 11: 105-123. Ritchie JW & Taylor PM (2001) Role of the System L permease LAT1 in amino acid and
iodothyronine transport in placenta. Biochem J 356: 719-725. Roboz J, Gleicher N, Wu K, Chanihian P, Kerenyi T & Holland J (1979) Does doxorubicin cross
the placenta? Lancet 2: 1382-1383. Sabers A, Buchholt JM, Uldall P & Hansen EL (2001) Lamotrigine plasma levels reduced by oral
contraceptives. Epilepsy Res 47: 151-154. Sabers A & Gram L (2000) Newer anticonvulsants: comparative review of drug interactions and
adverse effects. Drugs 60: 23-33. Samren EB, van Duijn CM, Koch S, Hiilesmaa VK, Klepel H, Bardy AH, Mannagetta GB, Deichl
AW, Gaily E, Granstrom ML, Meinardi H, Grobbee DE, Hofman A, Janz D & Lindhout D (1997) Maternal use of antiepileptic drugs and the risk of major congenital malformations: a joint European prospective study of human teratogenesis associated with maternal epilepsy. Epilepsia 38: 981-990.
Sastry BV (1999) Techniques to study human placental transport. Adv Drug Deliv Rev 38: 17-39. Sastry BV, Chance MB, Hemontolor ME & Goddijn-Wessel TA (1998) Formation and retention of
cotinine during placental transfer of nicotine in human placental cotyledon. Pharmacology 57: 104-116.
Schachter S (2002) Oxcarbazepine. Clinical efficacy and use in epilepsy. In Levy R et al (eds) Antiepileptic drugs, 5th ed. Lippincott Williams & Wilkins, Philadelphia, p 470-475.
Schenker S, Dicke J, Johnson RF, Mor LL & Henderson GI (1987) Human placental transport of cimetidine. J Clin Invest 80: 1428-1434.
Schenker S, Yang Y, Mattiuz E, Tatum D & Lee M (1999) Olanzapine transfer by human placenta. Clin Exp Pharmacol Physiol 26: 691-697.
Scheuer ML & Pedley TA (1990) The evaluation and treatment of seizures. N Engl J Med 323: 1468-1474.
Schmidt D (1995) Benzodiazepines. Diazepam. In Levy RH, Mattson RH & Meldrum BS (eds) Antiepileptic drugs, Fourth edition ed. Raven Press, New York, p 705-724.

78
Schmidt D, Arroyo S, Baulac M, Dam M, Dulac O, Friis ML, Kälviäinen R, Kramer G, van Parys J, Pedersen B & Sachdeo R (2001) Recommendations on the clinical use of oxcarbazepine in the treatment of epilepsy: a consensus view. Acta Neurol Scand 104: 167-170.
Schmitt W (1922) Untersuchungen zur physiologie der Pazentargefäße. Z Biol: 75: 19-78. Schmolling J, Jung S, Reinsberg J & Schlebusch H (1996) Digoxin transfer across the isolated
placenta is influenced by maternal and fetal albumin concentrations. Reprod Fertil Dev 8: 969-974.
Schmolling J, Jung S, Reinsberg J & Schlebusch H (1997) Diffusion characteristics of placental preparations affect the digoxin passage across the isolated placental lobule. Ther Drug Monit 19: 11-16.
Schneider H (1991) The role of the placenta in nutrition of the human fetus. Am J Obstet Gynecol 164: 967-973.
Schneider H & Huch A (1985) Dual in vitro perfusion of an isolated lobe of human placenta: method and instrumentation. Contrib Gynecol Obstet 13: 40-47.
Schneider H & Malek A (1995) Lack of permeability of the human placenta for erythropoietin. J Perinat Med 23: 71-76.
Schneider H, Reiber W, Sager R & Malek A (2003) Asymmetrical transport of glucose across the in vitro perfused human placenta. Placenta 24: 27-33.
Schroder HJ, Dehne K, Andreas T, Rago S & Rybakowski C (1999) Diffusive transfer of water and glucose across the chorionic plate of the isolated human term placenta. Placenta 20: 59-63.
Schütz H, Feldmann KF, Faigle JW, Kriemler HP & Winkler T (1986) The metabolism of 14C-oxcarbazepine in man. Xenobiotica 16: 769-778.
Shannon C, Jauniaux E, Gulbis B, Thiry P, Sitham M & Bromley L (1998) Placental transfer of fentanyl in early human pregnancy. Hum Reprod 13: 2317-2320.
Shimada T, Mimura M, Inoue K, Nakamura S, Oda H, Ohmori S & Yamazaki H (1997) Cytochrome P450-dependent drug oxidation activities in liver microsomes of various animal species including rats, guinea pigs, dogs, monkeys, and humans. Arch Toxicol 71: 401-408.
Simpson DM, Olney R, McArthur JC, Khan A, Godbold J & Ebel-Frommer K (2000) A placebo-controlled trial of lamotrigine for painful HIV-associated neuropathy. Neurology 54: 2115-2119.
Sinues B, Gazulla J, Bernal ML, Lanuza J, Fanlo A, Saenz MA & Barolome M (1995) Six mutagenicity assays in exposure biomonitoring of patients receiving carbamazepine for epilepsy or trigeminal neuralgia. Mutat Res 334: 259-265.
Siu SS, Yeung JH & Lau TK (2002) An in-vivo study on placental transfer of naproxen in early human pregnancy. Hum Reprod 17: 1056-1059.
Slikker W Jr & Miller RK (1994) Placental metabolism and transfer. Role in developmental toxicology. In Kimmel CA& Buelke-Sam J (eds) Developmental Toxicology, 2nd ed. Raven Press, New York, p 245-283.
Smelt VA, Upton A, Adjaye J, Payton MA, Boukouvala S, Johnson N, Mardon HJ & Sim E (2000) Expression of arylamine N-acetyltransferases in pre-term placentas and in human pre-implantation embryos. Hum Mol Genet 9: 1101-1107.
Smit JW, Huisman MT, van Tellingen O, Wiltshire HR & Schinkel AH (1999) Absence or pharmacological blocking of placental P-glycoprotein profoundly increases fetal drug exposure. J Clin Invest 104: 1441-1447.
Spina E (2002) Carbamazepine. Chemistry, biotransformation, and pharmacokinetics. In Levy R et al (eds) Antiepileptic drugs, 5th ed. Lippincott Williams & Wilkins, Philadelphia, p 236-246.
Spina E, Martines C, Fazio A, Trio R, Pisani F & Tomson T (1991) Effect of phenobarbital on the pharmacokinetics of carbamazepine-10,11-epoxide, an active metabolite of carbamazepine. Ther Drug Monit 13: 109-112.
St Pierre MV, Hagenbuch B, Ugele B, Meier PJ & Stallmach T (2002a) Characterization of an organic anion-transporting polypeptide (OATP-B) in human placenta. J Clin Endocrinol Metab 87: 1856-1863.
St Pierre MV, Ugele B, Gambling L & Shiverick KT (2002b) Mechanisms of drug transfer across the human placenta-a workshop report. Placenta 23 Suppl A: S159-S164.

79
Stanley EL, Hume R, Visser TJ & Coughtrie MW (2001) Differential expression of sulfotransferase enzymes involved in thyroid hormone metabolism during human placental development. J Clin Endocrinol Metab 86: 5944-5955.
Sun K, Adamson SL, Yang K & Challis JR (1999) Interconversion of cortisol and cortisone by 11beta-hydroxysteroid dehydrogenases type 1 and 2 in the perfused human placenta. Placenta 20: 13-19.
Sutcliffe AG, Jones RB & Woodruff G (1998) Eye malformations associated with treatment with carbamazepine during pregnancy. Ophthalmic Genet 19: 59-62.
Svinarov DA & Pippenger CE (1996) Relationships between carbamazepine-diol, carbamazepine-epoxide, and carbamazepine total and free steady-state concentrations in epileptic patients: the influence of age, sex, and comedication. Ther Drug Monit 18: 660-665.
Tanabe M, Ieiri I, Nagata N, Inoue K, Ito S, Kanamori Y, Takahashi M, Kurata Y, Kigawa J, Higuchi S, Terakawa N & Otsubo K (2001) Expression of P-glycoprotein in human placenta: relation to genetic polymorphism of the multidrug resistance (MDR)-1 gene. J Pharmacol Exp Ther 297: 1137-1143.
Tartara A, Galimberti CA, Manni R, Morini R, Limido G, Gatti G, Bartoli A, Strada G & Perucca E (1993) The pharmacokinetics of oxcarbazepine and its active metabolite 10-hydroxy-carbazepine in healthy subjects and in epileptic patients treated with phenobarbitone or valproic acid. Br J clin Pharmac 36: 366-368.
Tecoma ES (1999) Oxcarbazepine. Epilepsia 40 Suppl 5: S37-S46. Tennis P & Eldridge RR (2002) Preliminary results on pregnancy outcomes in women using
lamotrigine. Epilepsia 43: 1161-1167. Tomson T, Lindbom U, Ekqvist B & Sundqvist A (1994) Disposition of carbamazepine and
phenytoin in pregnancy. Epilepsia 35: 131-135. Tomson T, Ohman I & Vitols S (1997) Lamotrigine in pregnancy and lactation: a case report.
Epilepsia 38: 1039-1041. Trimble M (2002) Carbamazepine. Clinical efficacy and use in psychiatric disorders. In Levy R et
al (eds) Antiepileptic drugs, 5th ed. Lippincott Williams & Wilkins, Philadelphia, p 278-284. Tsadkin M, Holcberg G, Sapir O, Hallak M, Huleihel M, Katz M, Polachek H, Mazor M & Ben Zvi
Z (2001) Albumin-dependent digoxin transfer in isolated perfused human placenta. Int J Clin Pharmacol Ther 39: 158-161.
Tuntland T, Odinecs A, Pereira CM, Nosbisch C & Unadkat JD (1999) In vitro models to predict the in vivo mechanism, rate, and extent of placental transfer of dideoxynucleoside drugs against human immunodeficiency virus. Am J Obstet Gynecol 180: 198-206.
Tybring G, von Bahr C, Bertilsson L, Collste H, Glaumann H & Solbrand M (1981) Metabolism of carbamazepine and its epoxide metabolite in human and rat liver in vitro. Drug Metab Dispos 9: 561-564.
Uchino H, Kanai Y, Kim dK, Wempe MF, Chairoungdua A, Morimoto E, Anders MW & Endou H (2002) Transport of amino acid-related compounds mediated by L-type amino acid transporter 1 (LAT1): insights into the mechanisms of substrate recognition. Mol Pharmacol 61: 729-737.
Ugele B, St Pierre MV, Pihusch M, Bahn A & Hantschmann P (2002) Characterization and identification of steroid sulfate transporters of isolated trophoblasts and human placental tissue. Am J Physiol Endocrinol Metab. 284: E390-398.
Vähäkangas K. (1981). Lung perfusion studies on benzo(a)pyrene metabolism. Acta Univ Oul D No. 77.
Valentine CR, Valentine JL, Seng J, Leakey J & Casciano D (1996) The use of transgenic cell lines for evaluating toxic metabolites of carbamazepine. Cell Biol Toxicol 12: 155-165.
Venkatakrishnan K, Von Moltke LL & Greenblatt DJ (2001) Human drug metabolism and the cytochromes P450: application and relevance of in vitro models. J Clin Pharmacol 41: 1149-1179.
Vestergaard K, Andersen G, Gottrup H, Kristensen BT & Jensen TS (2001) Lamotrigine for central poststroke pain: a randomized controlled trial. Neurology 56: 184-190.
Wad N, Guenat C & Kramer G (1997) Carbamazepine: detection of another metabolite in serum, 9 hydroxymethyl-10-carbamoyl acridan. Ther Drug Monit 19: 314-317.

80
Waldmeier PC, Baumann PA, Wicki P, Feldtrauer JJ, Stierlin C & Schmutz M (1995) Similar potency of carbamazepine, oxcarbazepine, and lamotrigine in inhibiting the release of glutamate and other neurotransmitters. Neurology 45: 1907-1913.
Webster WS & Freeman JA (2001) Is this drug safe in pregnancy? Reprod Toxicol 15: 619-629. Welch RM, Harrison YE, Conney AH, Poppers PJ & Finster M (1968) Cigarette smoking:
stimulatory effect on metabolism of 3,4-benzpyrene by enzymes in human placenta. Science 160: 541-542.
Wellington K & Goa KL (2001) Oxcarbazepine: an update of its efficacy in the management of epilepsy. CNS Drugs 15: 137-163.
Widness JA, Sawyer ST, Schmidt RL & Chestnut DH (1991) Lack of maternal to fetal transfer of 125I-labelled erythropoietin in sheep. J Dev Physiol 15: 139-143.
Wier PJ & Miller RK (1985) Oxygen transfer as an indicator of perfusion variability in the isolated human placental lobule. Contrib Gynecol Obstet 13: 127-131.
Williams J, Myson V, Steward S, Jones G, Wilson JF, Kerr MP & Smith PE (2002) Self-discontinuation of antiepileptic medication in pregnancy: detection by hair analysis. Epilepsia 43: 824-831.
Witt A, Sommer EM, Cichna M, Postlbauer K, Widhalm A, Gregor H & Reisenberger K (2003) Placental passage of clarithromycin surpasses other macrolide antibiotics. Am J Obstet Gynecol 188: 816-819.
Wurden CJ & Levy RH (2002) Carbamazepine. Interactions with other drugs. In Levy R et al (eds) Antiepileptic drugs, 5th ed. Lippincott Williams & Wilkins, Philadelphia, p 245-261.
Yerby MS, Friel PN, McCormick K, Koerner M, Van Allen M, Leavitt AM, Sells CJ & Yerby JA (1990) Pharmacokinetics of anticonvulsants in pregnancy: alterations in plasma protein binding. Epilepsy Res 5: 223-228.
Yerby MS, Friel PN & Miller DQ (1985) Carbamazepine protein binding and disposition in pregnancy. Ther Drug Monit 7: 269-273.
Young AM, Allen CE & Audus KL (2003) Efflux transporters of the human placenta. Adv Drug Deliv Rev 55: 125-132.
Yuan R, Parmelee T, Balian JD, Uppoor RS, Ajayi F, Burnett A, Lesko LJ & Marroum P (1999) In vitro metabolic interaction studies: experience of the Food and Drug Administration. Clin Pharmacol Ther 66: 9-15.
Zisterer DM & Williams DC (1997) Peripheral-type benzodiazepine receptors. Gen Pharmacol 29: 305-314.