in vitro and genetic aspects of treatments for …
TRANSCRIPT

IN VITRO AND GENETIC ASPECTS OF
TREATMENTS FOR FALCIPARUM MALARIA:
STUDIES OF CONVENTIONAL AND NOVEL DRUGS
WITH A PARTICULAR FOCUS ON
PAPUA NEW GUINEA
Rina Pok-Man Fu nee Wong
B. Sc. (First Hons)
School of Medicine & Pharmacology
This thesis is submitted to the University of Western Australia
for the degree of DOCTOR OF PHILOSOPHY OF MEDICINE
2011


DECLARATION
The research presented in this thesis is my own work unless otherwise stated. The majority of this work was undertaken in the School of Medicine and Pharmacology (Fremantle Unit), the University of Western Australia. Field components were carried out at collaborating institutions, specifically the Papua New Guinea Institute of Medical Research (PNGIMR), Madang, Papua New Guinea and Case Western Reserve University, Cleveland, Ohio, United States of America. This thesis has not been submitted for any other degree at this or any other tertiary institution.
• Patient recruitment, blood collection and P. falciparum screening were carried out by the nursing team at Alexishafen Health Centre, Madang, Papua New Guinea as part of an antimalarial treatment trial.
• Restriction fragment length polymorphism assays of P. falciparum field isolates were performed by laboratory staff at the PNGIMR as part of the same treatment trial.
Rina Pok-Man Fu nee Wong Perth, Australia 2011

ii
DECLARATION FOR THESIS CONTAINING PUBLISHED WORK AN D/OR WORK PREPARED FOR PUBLICATION
This thesis contains published work and/or work prepared for publication, some of which has been co-authored. The bibliographical details and where it appears in the thesis are outlined below. The candidate must attach to this declaration a statement for each publication detailing the percentage contribution by the candidate. This statement must be signed by all authors. If signatures from all the authors cannot be obtained, the statement detailing the candidate’s contribution to the published work must be signed by the coordinating supervisor.
• Harin, A. Karunajeewa, Ivo Mueller, Michele Senn, Enmoore Lin, Irwin Law, Servina P. Gomorrai, Olive Oa, Suzanne Griffin, Kaye Kotab, Penias Suano, Nandao Tarongka, Alice Ura, Dulcie Lautu, Madhu Page-Sharp, Rina P. M. Wong, Sam Salman, Peter Siba, Kenneth F. Ilett and Timothy M. E. Davis. (2008) A trial of combination antimalarial therapies in children from Papua New Guinea. N Engl J Med 359 (24) 2545-57. Precise Contributions: performed in vitro drug sensitivity testing and analysis, assisted with patient recruitment and sample processing. Overall Contribution: 5%.
• Rina P. M. Wong and Timothy M. E. Davis. (2009) Statins as potential antimalarial drugs: Low relative potency and lack of synergy with conventional antimalarial drugs. Antimicrob Agents Chemother 53 (5) 2212-2214. Precise Contributions: designed and performed all in vitro experiments, data analysis and interpretation, drafting of manuscript. Overall Contribution: 90%.
• Stephan Karl and Rina P.M. Wong (equal-first author), Tim St. Pierre, Timothy M.E. Davis. (2009) A comparative study of a flow-cytometry-based assessment of in vitro Plasmodium falciparum drug sensitivity. Malaria Journal 8, 294. Precise Contributions: designed and performed drug sensitivity assays suitable for three methods of growth response assessment, data collection for the reference isotopic and enzyme methods, data analysis and drafting of manuscript. Overall Contribution: 40%.
• Rina P. M. Wong, Dulcie Lautu, Livingstone Tavul, Sarah L. Hackett, Peter Siba, Harin A. Karunjeewa, Kenneth F Ilett, Ivo Mueller, Timothy M. E. Davis. (2010) In vitro sensitivity of Plasmodium falciparum to conventional and novel antimalarial drugs in Papua New Guinea. Trop Med Int Health 15(2) 342-349.

iii
Precise Contributions: designed and performed drug sensitivity assays, method validation and optimisation, assisted with sample collection and processing, data analysis and interpretation, drafting of manuscript. Overall Contribution: 80%.
• Rina P. M. Wong, Harin Karunajeewa, Ivo Mueller, Peter Siba, Peter A. Zimmerman, Timothy M. E. Davis. (2011) Molecular assessment of Plasmodium falciparum resistance to antimalarial drugs in Papua New Guinea using an extended ligase detection reaction-fluorescent microsphere assay. Antimicrob Agents Chemother 55(2) 798-805. Precise Contributions: performed molecular screening of Plasmodium species, and drug resistant genes, optimisation and extension of the LDR-FMA technique to include the screening of 10 additional SNPs in the pfmdr1 gene, data analysis and interpretation, determination of positive threshold parameters, drafting of manuscript. Overall Contribution: 85%.
• Rina P. M. Wong, Sam Salman, Kenneth F Ilett, Ivo Mueller, Timothy M. E. Davis. (2011) Desbutyl-lumefantrine is a metabolite of lumefantrine with potent in vitro antimalarial activity that may influence artemether-lumefantrine treatment outcome. Antimicrob Agents Chemother 55(3) 1194-1198. Precise Contributions: designed and performed in vitro antimalarial assays, drug interaction asssays, data analysis and interpretation, drafting of manuscript. Overall Contribution: 75%.
• Louise R. Whittell, Kevin T. Batty, Rina P. M. Wong, Erin Bolitho, Simon A. Fox, Timothy M. E. Davis, and Paul E. Murray. (2011) Synthesis and antimalarial evaluation of novel isocryptolepine derivatives. Bioorg Med Chem 19, 7519-7525. Precise Contributions: designed and performed in vitro antimalarial assays, data analysis and interpretation, manuscript preparation. Overall Contribution: 25%.
• Rina P. M. Wong and Timothy M. E. Davis. (2011) In vitro antimalarial efficacy and drug interactions of fenofibric acid. Antimicrob Agents Chemother (Manuscript submitted # AAC05076-11) Precise Contributions: designed and performed in vitro antimalarial assays, drug interaction asssays, data analysis and interpretation, drafting of manuscript. Overall Contribution: 90%.
• Rina P. M. Wong, Gavin R. Flematti and Timothy M. E. Davis. (2011) Detection of volatile organic compounds produced by Plasmodium falciparum in culture. Manuscript in preparation. Precise Contributions: designed culture-volatile compounds capture apparatus, performed experiments, GCMS and data analysis, and drafting of manuscript. Overall Contribution: 70%.
Candidate Signature: __________ Coordinating Supervisor Signature: __________

iv
PUBLICATIONS
• Harin, A. Karunajeewa, Ivo Mueller, Michele Senn, Enmoore Lin, Irwin Law, Servina P. Gomorrai, Olive Oa, Suzanne Griffin, Kaye Kotab, Penias Suano, Nandao Tarongka, Alice Ura, Dulcie Lautu, Madhu Page-Sharp, Rina P. M. Wong, Sam Salman, Peter Siba, Kenneth F. Ilett and Timothy M. E. Davis. (2008) A trial of combination antimalarial therapies in children from Papua New Guinea. N Engl J Med 359 (24) 2545-57.
• Rina P. M. Wong & Timothy M. E. Davis. (2009) Statins as potential antimalarial drugs: Low relative potency and lack of synergy with conventional antimalarial drugs. Antimicrob Agents Chemother 53 (5) 2212-2214.
• Stephan Karl and Rina P.M. Wong (equal-first author), Tim St. Pierre, Timothy M.E. Davis. (2009) A comparative study of a flow-cytometry-based assessment of in vitro Plasmodium falciparum drug sensitivity. Malaria Journal 8, 294-305.
• Rina P. M. Wong, Dulcie Lautu, Livingstone Tavul, Sarah L. Hackett, Peter Siba, Harin A. Karunjeewa, Kenneth F Ilett, Ivo Mueller, Timothy M. E. Davis. (2010) In vitro sensitivity of Plasmodium falciparum to conventional and novel antimalarial drugs in Papua New Guinea. Trop Med Int Health 15(2) 342-349.
• Rina P. M. Wong, Harin Karunajeewa, Ivo Mueller, Peter Siba, Peter A. Zimmerman, Timothy M. E. Davis. (2011) Molecular assessment of Plasmodium falciparum resistance to antimalarial drugs in Papua New Guinea using an extended ligase detection reaction-fluorescent microsphere assay. Antimicrob Agents Chemother 55(2) 798-805.
• Rina P. M. Wong, S. Salman, Kenneth F Ilett, Ivo Mueller, Timothy M. E. Davis. (2011) Desbutyl-lumefantrine is a metabolite of lumefantrine with potent in vitro antimalarial activity that may influence artemether-lumefantrine treatment outcome. Antimicrob Agents Chemother 55(3) 1194-1198.
• Louise R. Whittell, Kevin T. Batty, Rina P. M. Wong, Erin Bolitho, Simon A. Fox, Timothy M. E. Davis, and Paul E. Murray. (2011) Synthesis and antimalarial evaluation of novel isocryptolepine derivatives. Bioorg Med Chem 19, 7519-7525.
• Rina P. M. Wong and Timothy M. E. Davis. (2011) In vitro antimalarial efficacy and drug interactions of fenofibric acid. Antimicrob Agents Chemother (Manuscript submitted # AAC05076-11)
• Rina P. M. Wong, Gavin R. Flematti and Timothy M. E. Davis. (2011) Detection of volatile organic compounds produced by Plasmodium falciparum in culture. Manuscript in preparation.

v
CONFERENCE PRESENTATIONS
• Rina P. M. Wong & Timothy M. E. Davis. In vitro susceptibility and inter-relationships of nine standard and new antimalarials against Plasmodium falciparum isolates from Papua New Guinean children. (2008) Research Showcase: School of Medicine & Pharmacology, Perth, Australia. (Oral)
• Rina P. M. Wong & Timothy M. E. Davis. Malaria and statins. (2008) Annual Research Showcase: School of Medicine & Pharmacology, Perth, Australia. (Oral: Best Student Oral Award)
• Rina P. M. Wong & Timothy M. E. Davis. Statins and fibrates as potential antimalarial agents. (2009) The Australian Society for Medical Research, Medical Research Week Scientific Symposium, Perth, Australia. (Oral)
• Rina P. M. Wong, Harin Karunajeewa, Ivo Mueller, Peter Siba, Eric P. Carnevale, Peter A. Zimmerman and Timothy M. E. Davis. Drug Resistance Polymorphisms in Plasmodium falciparum from children in Papua New Guinea by a recently developed LDR-FMA technique. (2009) Combined Biological Sciences Meeting, Perth, Western Australia. (Oral: New Investigator Award)
• Rina P. M. Wong, Harin Karunajeewa, Ivo Mueller, Peter Siba, Eric P. Carnevale, Peter, A. Zimmerman and Timothy M.E. Davis. Novel molecular detection of drug resistance markers in Plasmodium falciparum from paediatric uncomplicated malaria in Papua New Guinea. (2010) 14th International Congress on Infectious Diseases, Miami, Florida, United States of America. (Oral)
• Rina P. M. Wong, S. Salman, Kenneth F. Ilett, Ivo Mueller and Timothy M. E. Davis. In vitro and in vivo evaluation of desbutyl-benflumetol, a promising antimalarial drug. (2010) XII International Congress on Parasitology, Melbourne, Australia. (Oral)
• Rina P. M. Wong, S. Salman, Kenneth F. Ilett, Ivo Mueller and Timothy M. E. Davis. Desbutyl-lumefantrine, a promising antimalarial drug. (2010) Combined Biological Sciences Meeting, Perth, Western Australia. (Poster: Best Postgraduate Poster Award)
• Rina. P. M. Wong – Three Minute Thesis Oration, Resistance of Plasmodium falciparum to antimalarials in Papua New Guinea. (2010) The Australian Society for Medical Research, Medical Research Week, Scientific Symposium, Perth, Australia (Song).

vi
• Rina. P. M. Wong – Three Minute Thesis Oration, Multi-resistant malaria: drugs, genes and sick babies. (2010) Three Minute Thesis Competition: The University of Western Australia, Perth, Australia (Finalist).
• Rina. P. M. Wong and Timothy. M. E. Davis. Fenofibric acid, metabolite of fenofibrate is a promising, novel antimalarial drug. (2011) Australian Society for Parasitology Annual Conference, Carins, Queensland, Australia. (Oral: Best Student Oral Prize).
• Rina. P. M. Wong, Gavin. R. Flematti and Timothy M. E. Davis. Detection of volatile organic compounds produced by Plasmodium falciparum in culture. (2011) The Australian Society for Medical Research, Medical Research Week, Scientific Symposium, Perth, Australia. (Oral).
• Rina. P. M. Wong and Timothy M. E. Davis. Lipid-modifying drugs as novel antimalarial therapy. (2011) BIT’s 1st Annual World Congress of Microbes: 1st Annual Symposium of Antiparasites, Beijing, China. (Oral, submitted by Invitation).

vii
ABSTRACT
Malaria remains a significant global health problem. Plasmodium falciparum, the
predominant and most virulent infecting species, has developed resistance to most
antimalarial drugs. Drug sensitivity is monitored by i) in vivo (clinical) outcome, ii) in
vitro response of cultured parasites to a range of drug concentrations, and iii) presence
of resistance-associated molecular markers. Few studies have integrated these
approaches which can all contribute to the development of treatment regimens that
improve clinical outcome and delay spread of resistance.
Recent clinical studies have shown high rates of treatment failure in Papua New Guinea
(PNG), necessitating a proposed change from chloroquine (CQ) or amodiaquine (AQ)
plus sulfadoxine-pyrimethamine (SP) to artemisinin combination therapy (ACT). The in
vitro sensitivity of 64 P. falciparum isolates from Madang Province to CQ, AQ,
monodesethyl-amodiaquine (DAQ), piperaquine (PQ), naphthoquine (NQ), mefloquine
(MQ), lumefantrine (LM), dihydroartemisinin (DHA) and azithromycin was assessed
by colorimetric lactate dehydrogenase growth inhibition assay. Its non-isotopic, semi-
automated, high-throughput nature makes it suitable for field use in developing
countries. The mean [95% confidence interval] concentration required to inhibit
parasite growth by 50% (IC50) was 215 [175-254] nM for CQ; 82% of strains were CQ-
resistant. Except for azithromycin, the mean IC50s of the other drugs were <27 nM.
There were strong associations between the IC50s of 4-aminoquinoline (CQ, AQ, DAQ
and NQ), bisquinoline (PQ) and aryl-aminoalcohol (MQ) drugs, suggesting cross-
resistance. The only such correlation for LM was with MQ which, with the low
artemisinin IC50s, supports artemether-LM as new first-line therapy in PNG.
Parasite mutations compromising treatment effectiveness were also evaluated using a
modified high-throughput post-PCR multiplexed ligase detection reaction-fluorescent
microsphere assay. The assay was used to detect single nucleotide polymorphisms
(SNPs) in 402 P. falciparum isolates from PNG children participating in an antimalarial
treatment trial. There was fixation of pfcrt K76T, pfdhfr C59R and S108N, and pfmdr1
mutations (92%, 93%, 95% and 91%, respectively). Multiple mutations were frequent,

viii
88% of isolates possessed a quintuple mutation SVMNT+NRNI+KAA+YYSND in
codons 72-76 for pfcrt, 51, 59, 108, 164 for pfdhfr, 540, 581, 613 for pfdhps, and 86,
184, 1034, 1042, 1246 for pfmdr1, and four carried the K540E pfdhps allele. Pfmdr1
D1246Y was associated with PCR-corrected day 42 treatment failure in children
allocated PQ-DHA (P=0.004). Although the pfmdr1 NFSDD haplotype was found in
only four isolates, it has been associated with artemether-LM treatment failure in
Africa. The assay allowed large-scale assessment of resistance-associated SNPs that
reflected previous heavy 4-aminoquinoline/SP use in PNG. Since artemether-LM and
PQ-DHA will become first- and second-line treatment, respectively, in PNG,
monitoring pfmdr1 SNPs appears a high priority.
Desbutyl-lumefantrine (DBL) is a metabolite of LM. Its in vitro activity and
interactions were assessed from tritium-labelled hypoxanthine uptake in laboratory-
adapted P. falciparum. DBL was more potent than LM. Isobolographic analysis of
DBL-LM combinations showed no interaction but mild synergy with DBL-DHA. Mean
plasma DBL concentrations in 94 day-7 samples from an antimalarial treatment trial
predicted treatment response, suggesting that it could be a useful alternative to LM as
part of ACT.
Drugs licensed for other indications can sometimes have antimalarial properties, an
example being lipid-lowering therapy which is becoming affordable even in malaria-
endemic developing countries. In vitro drug sensitivity experiments confirmed
atorvastatin to have the highest activity of available statins against P. falciparum
regardless of strain CQ sensitivity but at an IC50 well above plasma concentrations after
therapeutic doses in vivo. Fibrates have a different mechanism of action to that of
statins. Fenofibric acid had a relatively low in vitro IC50, similar to those of
conventional antimalarial drugs. It may act by interfering with parasite P-glycoprotein
and ABC-1 mediated transport and/or via a putative peroxisome proliferator-activated
receptor-like protein, and could have an adjunctive role in combination antimalarial
therapy.
The detection of P. falciparum-specific volatile organic compounds (VOCs) in
breath/other samples as a way of enhancing diagnosis and therapeutic monitoring was

ix
explored using culture-capture apparatus. Optimised conditions supported cultures of
high parasitaemia (>20%) from which VOCs within the headspace and supernatant
were extracted using traditional (solvent) and novel (solid-phase) methods. Gas
chromatography-mass spectrometry data revealed the production of a variety of volatile
compounds but no unique malarial finger-prints. Future in vivo studies analysing the
breath of patients with severe malaria may yet reveal specific clinically-useful volatile
biomarkers.

x
ACKNOWLEDGEMENTS
My PhD journey would not have been successful without the unyielding support of my
family, friends and colleagues.
To my supervisor Prof. Tim Davis, I have admired your expertise in the field and your
driven-nature since day-one. You have taught me to think critically, work
independently as well as providing opportunities for me to do research as part of a team
abroad. You have amazing writing skills that I can only learn to mimic. Thank you so
much for supporting me through the highs and lows. I’m grateful for your financial
support which helped to sustain my newly established family. It is a privilege to be part
of your team and have the chance to travel/work in two very different worlds: Papua
New Guinea (PNG) and the United States of America. The experiences gained from
these collaborations are extraordinary and most unforgettable, “Tenkyu tru!” (Pidgin).
Special thanks to Drs Wendy Davis, Martin Firth and Shih Ching Fu for your time and
invaluable advice on statistical modelling and analyses for the molecular aspect of this
project. To Dr Pete Zimmerman, Laurie Gray and many colleagues at Case Western
Reserve University, thank you for sharing your cutting edge molecular techniques and
being so accommodating when I used your machines up to 12 hours a day! I treasure
the time I spent in your lab and thank you for giving me this wonderful opportunity.
This project would not have been possible without support from Drs Peter Siba, Pascal
Michon, Ivo Mueller, study volunteers and administrative staff from the PNG Institute
of Medical Research. Thanks to Livingstone Tavul and Dulcie Lautu for your technical
support for the in vitro drug assays. Dr Harin Karunajeewa who led a dedicated team of
nurses at the Alexishafen Health Clinic, a big thank you for collecting the field samples
and allowing me to take part in this amazing experience.
Dr Jane Allan, you have always been there for me (literally) to offer comforting words
when I’m disheartened and provided tips that often facilitated my trouble shooting.
Thank you for looking after the parasites when I was sick and for your continuous

xi
support. I really appreciate your friendly and approachable qualities; you are an
awesome lab manager. To my colleagues at the Medical Sciences Lab, Fremantle
Hospital – Janet, Ross, Debbie, Caryn, Stephan, Eng, Carly, Angela, Yoke Leng, Bruce,
Roheeth, Frances, Borut, Kristine, Janina, Anita, Gwen and Chun Wei, thank you for
your friendship and amazing support throughout my PhD journey.
To our school administrator, Brenda Riley, thank you for your time and care in ironing
out the many hiccups and providing a listening ear. Michelle England and “the nurses
upstairs”, thank you for spicing things up and taking my blood every few weeks!
I’m grateful and indebted to Mr Graham Icke, aka.‘Pop’, my honours supervisor and
mentor. Thanks for your encouragement and time in critiquing this thesis. Thanks to
Rolf and friends from church, for your encouragements throughout this journey.
Shih Ching Fu, my loving husband who is also under the pump with his own thesis, you
have been a tremendous support. Thanks for sacrificing countless weekends to
accompany me to the lab and giving your red cells for my parasites. Thank you for your
patience, encouragement, understanding and lending me your shoulders to cry on. To
my Mum (Yolanda) and Dad (Siu Kee), thank you for spurring me on when I’m
discouraged and pulling me back when I’m exhausted. You have supported me so much
through words, deeds and prayers. Louis, although you can’t talk, I know you are
always supporting your sister in your heart. Thank you for being understanding and I
will strive to spend more time with you.
Most of all I would like to thank God for blessing me with this amazing PhD experience
and the love from his son Jesus that has carried me through these years and beyond.
The studies herein are supported by grants from the WHO Western Pacific Region, the National
Health and Medical Research Council of Australia (grant no. 353663, T. Davis as CIA) and the
U.S. National Institutes of Health (grants AI52312 and TW 007872). R. P. M. Wong is
supported by the Australian Postgraduate Award, Ad hoc Scholarship (School of Medicine and
Pharmacology), the UWA Student Travel Award and the UWA PhD Completion Scholarship.

xii
PREFACE
For the in vitro work presented in Chapter 3 and published in Tropical Medicine and
International Health (2010), I went to Papua New Guinea (PNG) for a period of 5
months to collect and test field isolates of P. falciparum. I was based in the Vector
Borne Disease Unit in the PNG Institute of Medical Research at Yagaum Hospital,
Madang. I took primary responsibility in setting up drug sensitivity assays, data analysis
and interpretation of results. A small number of assays were set up by local colleagues
Dulcie Lautu and Livingstone Tavul in my absence. Two to three times each week, I
helped out at a remote Health Clinic at Alexishafen, where young children presenting
with fever were screened for malaria infection and enrolled into a standard treatment
trial. The samples included in this thesis were mainly derived from this cohort but a few
were from children who presented to Modilon Hospital, Madang Town.
I travelled to Case Western Reserve University, Cleveland, Ohio for the molecular
analysis of parasite DNA for drug-resistant markers as described in Chapter 4 and
published in Antimicrobial Agents and Chemotherapy (2011). I spent three and a half
months at the Centre for Global Health and Diseases under the supervision of Dr Peter
Zimmerman and learnt a post-PCR technique that has been developed there, namely the
Ligase Detection Reaction-Fluorescence Microsphere Assay (LDR-FMA). After
familiarisation of the laboratory and equipment, I performed LDR-FMA for
Plasmodium species, and single nucleotide polymorphisms (SNPs) detection in the
pfcrt, pfdhps, pfdhfr and pfmdr1 genes of blood samples from the treatment trial cohort.
Initial PCR primers had been designed for the pfmdr1 LDR-FMA by Eric Carnevale,
which required further modifications. I continued to develop and optimise the assay and
successfully applied this to the field samples. I took primary responsibility for the
generation and analysis of the molecular data. Since there were no standard cutoff
points for discriminating between positive and negative SNPs signals, I liaised with Dr
Martin Firth from the Mathematics department, UWA. With his advice, I was able to
establish a new way of calculating appropriate thresholds. The SNPs data were then
used to predict treatment failure rates using the clinical data from the trial. I received

xiii
advice from Dr Wendy Davis regarding statistical methods and software tools, and I
took responsibility for performing these analyses.
Due to direct and indirect evidence in the published literature that fibrates and statins,
as well as being lipid-altering drugs, might also have antimalarial properties, I initiated
the investigation into these drug classes with the support of my supervisor W/Prof. Tim
Davis. Fibrates showed antimalarial activity in the experiments I conducted. The in
vitro studies regarding desbutyl-lumefantrine (DBL) (Chapter 5 and published in
Antimicrobial Agents and Chemotherapy (2011), fibrates and statins (Chapter 6, in part
published in Antimicrobial Agents and Chemotherapy (2009)) were performed at the
Malaria Culture Facilities at the University Department of Medicine, Fremantle
Hospital. I took responsibility for parasite culture maintenance, the design of
experiments and in establishing a renewed approach to assess drug interactions. Due to
the labour intensive nature of this work, I was assisted by a research assistant, Miss
Jenny Wong on a casual basis. The pharmacokinetic component of the DBL work
(Chapter 5) was performed by my colleague and fellow PhD student Sam Salman.
The investigation of volatile organic compounds in P. falciparum was carried out in
collaboration with the Chemistry Department, UWA. I was under the supervision of Dr
Gavin Flematti, School of Biomedical, Biomolecular and Chemical Sciences. Together
we designed two prototypes of culture flasks that enabled the capture and analysis of
the head space atmosphere. I was responsible for optimising culture conditions in these
and performed various extractions followed by GC-MS analysis. I received technical
support with the use of equipments and machinery, and advice regarding the analysis of
chromatogram peaks from Dr Gavin Flematti.
The work presented in this thesis was performed within the time constraints of my PhD
enrolment.

xiv
ABBREVIATIONS
ABC ATP-binding cassette sub-family A member
ACPR Adequate clinical and parasitological response
ACR Adequate clinical response
ACTs Artemisinin based combination therapies
AL Artemether-lumefantrine
amu Atomic mass unit
ANOVA Analysis of variance
APAD 3-acetylpyridine adenine dinucleotide
ARMD Accelerated resistance to multidrug
ART-SP Artesunate-sulfadoxine-pyrimethamine
AQ Amodiaquine
AV Atorvastatin
AZ Azithromycin
bp base-pairs
CDC Centre for Disease Control and Prevention (Atlanta, USA)
cfu Colony-forming units
CI Confidence interval

xv
CO2 Carbon dioxide
CPM Counts per minute
CSP Circumsporozoite protein
CQ Chloroquine
CYC Cycloguanil
CQ-SP Chloroquine-sulfadoxine-pyrimethamine
dAQ Monodesethyl-amodiaquine
DBL Desbutyl-lumefantrine
DDT Dichloro-diphenyl-trichloroethane
DELI Double-site enzyme-linked pLDH immunodetection
d. H2O Distilled water
DHA Dihydroartemisinin
DHFR Dihydrofolate reductase
DHPS Dihydropteroate synthase
DMSO Dimethylsulphoxide
DNA Deoxyribonucleic acid
dNTPs Deoxynucleotide triphosphate
DVB Divinylbenzene

xvi
EDTA Ethylenediaminetetraacetic acid
ETF Early treatment failure
fmol femtomole
FI Fluorescent intensities
FIC Fractional inhibitory concentration
GC-MS Gas chromatography- mass spectrometry
H2O Water
HCl Hydrochloric acid
hct Haematocrit
HDL High-density lipoprotein
HEPES N-2-hydroxyethylipiperazine-N-2ethanesulfonic acid
HF Halofantrine
HMG-CoA 3-hydroxy-methyl-glutaryl coenzyme A
HPLC High performance liquid chromatography
HRP-2 Histidine-rich protein-2
hr Hour
HTPBS Human tonicity phosphate buffered saline

xvii
IC50 Drug concentration required to inhibit parasite growth by 50%
IC90 Drug concentration required to inhibit parasite growth by 90%
IC99 Drug concentration required to inhibit parasite growth by 99%
ICAM-1 Intercellular cell adhesion molecule-1
IFN-γ Interferon-gamma
IL-10 Interleukin-10
LCF Late clinical failure
LC-MS Liquid chromatography-mass spectrometry
LDH Lactate dehydrogenase
LDL Low-density lipoprotein
LDR Ligase detection reaction
LDR-FMA Ligase detection reaction-fluorescent microsphere assay
LM Lumefantrine
LPF Late parasitological failure
LTF Late treatment failure
mg Milligram
min Minute(s)

xviii
mL Millilitre
mM Millimolar
MOI Multiplicity of infection
MR4 Malaria research and reference reagent centre
MQ Mefloquine
ng Nanogram
nm Nanometre
nM Nanomolar
NaCl Sodium chloride
NAD Nicotinamide adenine dinucleotide
NaOH Sodium hydroxide
NBF Nitro blue formazan
NBT Nitro blue tetrazolium
NQ Naphthoquine
O2 Oxygen
OD Optical density
PBS Phosphate saline buffer
PCR Polymerase chain reaction

xix
PDMS Polydimethylsiloxane
PfATP6 (see Pfserca)
Pfcrt Plasmodium falciparum chloroquine resistant transporter
Pfdhfr Plasmodium falciparum dihydrofolate reductase
Pfdhps Plasmodium falciparum dihydropteroate synthase
Pfmdr1 Plasmodium falciparum multidrug resistant-1
Pfserca Plasmodium falciparum sarco-endoplasmic reticulum calcium ATPase6
piRBC Packed infected red cells
pLDH Plasmodium lactate dehydrogenase
pmol Picomole
PNG Papua New Guinea
PPAR Peroxisome proliferator-activated receptor
PQ Piperaquine
PQ-DHA Piperaquine-dihydroartemisinin
PV Pravastatin
PYR Pyrimethamine
RBC Red blood cells
rDNA Ribosomal deoxyribonucleic acid

xx
RI Low grade resistance
RII Moderate resistance
RIII High grade resistance
rpm Revolutions per minute
RPMI Roswell Park Memorial Institute
RNA ribonucleic acid
rRNA ribosomal ribonucleic acid
RT Room temperature
RV Rosuvastatin
sec Second (s)
SD Standard deviation
SDS-PAGE Sodium dodecyl sulphate polyacrylamide gel electrophoresis
SNPs Single nucleotide polymorphisms
SP Sulfadoxine-Pyrimethamine (Fansidar)
SPME Solid phase micro-extraction
SV Simvastatin
TA Annealing temperature
TGF-β Transforming growth factor-β

xxi
TNF Tumour necrosis factor
v/v Percentage volume per volume
vs Versus
VOCs Volatile organic compounds
WARN World Antimalarial Resistance Network
WHO World Health Organisation
w/v Percentage weight per volume
3H Tritium
⁰ Degree
⁰C Celsius
% Percentage
α Alpha
β Beta
ΣFIC Sum of fractional inhibitory concentrations
µCi Microcurie
µg Microgram
µL Microlitre

xxii
µm Micron
µM Micromolar
Nucleotide Code
Adenine A
Cytosine C
Guanine G
Thymine T
Amino acid Single letter code
Alanine A
Arginine R
Asparagine N
Aspartic acid D
Cysteine C
Glutamic acid E
Glycine G
Isoleucine I
Leucine L
Lysine K
Methionine M
Phenylalanine F
Serine S
Threonine T
Tyrosine Y
Valine V

xxiii
TABLE OF CONTENTS
DECLARATION........................................................................................................................................ I
PUBLICATIONS .................................................................................................................................... IV
CONFERENCE PRESENTATIONS ......................................................................................................V
ABSTRACT............................................................................................................................................VII
ACKNOWLEDGEMENTS......................................................................................................................X
PREFACE...............................................................................................................................................XII
ABBREVIATIONS .............................................................................................................................. XIV
TABLE OF CONTENTS.................................................................................................................. XXIII
LIST OF TABLES .............................................................................................................................XXIX
LIST OF FIGURES ...........................................................................................................................XXXI
CHAPTER 1. GENERAL INTRODUCTION.........................................................................................2
1.1 INTRODUCTION..........................................................................................................................2
1.2 DISEASE DISTRIBUTION .............................................................................................................3
1.3 PLASMODIUM LIFE CYCLE AND BIOLOGY ..................................................................................4
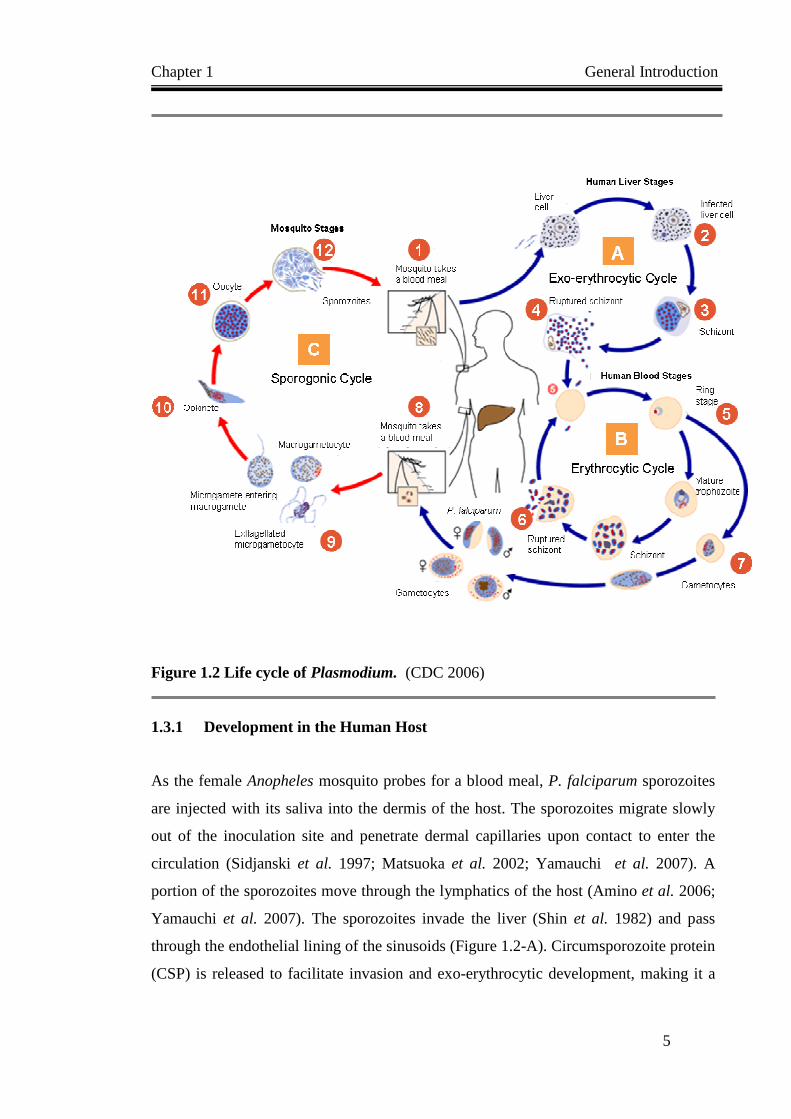
1.3.1 Development in the Human Host..........................................................................................5
1.3.2 Development in the Mosquito...............................................................................................7
1.4 PATHOLOGY...............................................................................................................................8
1.5 CLINICAL SIGNS AND SYMPTOMS ............................................................................................10
1.6 DIAGNOSIS...............................................................................................................................11
1.7 TRANSMISSION........................................................................................................................13
1.8 PREVENTION............................................................................................................................14
1.9 TREATMENT.............................................................................................................................14
1.10 MALARIA IN WESTERN PACIFIC...............................................................................................15
1.11 EMERGENCE OF ANTIMALARIAL RESISTANCE IN PAPUA NEW GUINEA ...................................16
1.12 ANTIMALARIAL CHEMOTHERAPY............................................................................................18
1.12.1 Quinoline Related Compounds......................................................................................19 1.12.1.1 Quinine .......................................................................................................................................... 19 1.12.1.2 Chloroquine ................................................................................................................................... 20 1.12.1.3 Amodiaquine.................................................................................................................................. 21 1.12.1.4 Mefloquine..................................................................................................................................... 23 1.12.1.5 Lumefantrine.................................................................................................................................. 24 1.12.1.6 Naphthoquine................................................................................................................................. 25

xxiv
1.12.1.7 Piperaquine.....................................................................................................................................26 1.12.2 Antifolate Combination Drugs...................................................................................... 26
1.12.3 Artemisinin and its Derivatives..................................................................................... 27
1.12.4 Antibiotics..................................................................................................................... 28
1.12.5 Summary of Antimalarial Activities .............................................................................. 31
1.13 LIPID-LOWERING AGENTS AS ANTIMALARIALS ...................................................................... 33
1.13.1 Statins ........................................................................................................................... 33
1.13.2 Fibrates......................................................................................................................... 35
1.14 ANTIMALARIAL DRUG RESISTANCE........................................................................................ 36
1.14.1 Definitions .................................................................................................................... 36
1.14.2 Treatment Failure and Drug Resistance....................................................................... 37
1.14.3 Emergence of Resistance to Principal Antimalarials ................................................... 38
1.14.4 Determinants of Antimalarial Resistance ..................................................................... 39
1.14.5 Mechanism of Resistance to 4-Aminoquinolines and Arylaminoalcohols .................... 42
1.14.6 Mechanism of Resistance to Antifolates........................................................................ 43
1.14.7 Mechanism of Resistance to Artemisinin and Derivatives............................................ 43
1.15 IN VITRO DETECTION OF RESISTANCE IN P. FALCIPARUM......................................................... 45
1.15.1 Schizont Maturation...................................................................................................... 46 1.15.1.1 Macro test.......................................................................................................................................46 1.15.1.2 Micro test........................................................................................................................................47
1.15.2 3H-Hypoxanthine Incorporation Assay......................................................................... 48
1.15.3 Plasmodium Lactate Dehydrogenase (pLDH) Detection ............................................. 48 1.15.3.1 Colourimetric pLDH microtests .....................................................................................................49 1.15.3.2 Immunocapture of pLDH ...............................................................................................................50
1.15.4 Histidine-Rich Protein II (HRP2) Assay....................................................................... 51
1.15.5 Dual Detection of HRP2 and PLDH............................................................................. 52
1.16 ASSESSMENT OF ANTIMALARIAL DRUG COMBINATIONS ........................................................ 52
1.17 IN VIVO DETECTION OF DRUG RESISTANCE IN P. FALCIPARUM................................................ 54
1.18 MOLECULAR MARKERS OF DRUG RESISTANCE....................................................................... 56
1.19 BREATH TEST FOR MALARIA .................................................................................................. 58
1.20 SCOPE OF THE STUDIES PRESENTED IN THIS THESIS................................................................ 59
CHAPTER 2. METHODS AND MATERIALS ................................................................................... 62
2.1 IN VITRO CULTURE TECHNIQUES............................................................................................... 62
2.1.1 Parasites............................................................................................................................ 62
2.1.2 Retrieval from Liquid Nitrogen ......................................................................................... 62
2.1.3 Maintenance of Cultures ................................................................................................... 63
2.1.4 Erythrocytes Preparation .................................................................................................. 63
2.1.5 Determination of Parasitaemia ......................................................................................... 64
2.1.6 Synchronisation of Parasite Forms ................................................................................... 64

xxv
2.1.7 Cryopreservation................................................................................................................66
2.2 DRUG SUSCEPTIBILITY ASSAYS...............................................................................................67
2.2.1 Drug/Compound Preparation ............................................................................................67
2.2.2 Preparation of parasitised cells .........................................................................................67
2.2.3 Controls..............................................................................................................................68
2.2.4 Plasmodium Lactate Dehydrogenase Assay.......................................................................70 2.2.4.1 Principle of pLDH Assay................................................................................................................. 70 2.2.4.2 Assay Set Up.................................................................................................................................... 70
2.2.5 3H-Hypoxanthine Incorporation Assay ..............................................................................71
2.3 MOLECULAR TECHNIQUES.......................................................................................................72
2.3.1 DNA Extraction..................................................................................................................73
2.3.2 Polymerase Chain Reaction (PCR)....................................................................................73 2.3.2.1 PCR for Plasmodium Species .......................................................................................................... 73 2.3.2.2 PCR for pfcrt, pfdhfr and pfdhps genes ........................................................................................... 74 2.3.2.3 Controls ........................................................................................................................................... 74
2.3.3 Detection of Amplified Products ........................................................................................75
2.3.4 Ligase Detection Reaction Fluorescent Microsphere Assay (LDR-FMA) .........................76 2.3.4.1 Ligase Detection Reaction for Plasmodium species ........................................................................ 76 2.3.4.2 Ligase Detection Reaction for pfcrt, pfdhfr, pfdhps SNPs............................................................... 77 2.3.4.3 Hybridisation and Reporter Labelling.............................................................................................. 78 2.3.4.4 Bio-plex Fluorescent Detection ....................................................................................................... 80
2.4 SOLID PHASE M ICRO-EXTRACTION (SPME) ............................................................................80
CHAPTER 3. IN VITRO SENSITIVITY OF P. FALCIPARUM TO NEW AND CONVENTIONAL
DRUGS IN PAPUA NEW GUINEA ......................................................................................................84
3.1 INTRODUCTION........................................................................................................................84
3.2 MATERIALS AND METHODS.....................................................................................................88
3.2.1 Study Site and Sample Collection.......................................................................................88
3.2.2 In vitro Culture of Parasite Isolates...................................................................................88
3.2.3 Drug Susceptibility Assays .................................................................................................89
3.2.4 Assay Validation.................................................................................................................90
3.2.5 Data Analysis .....................................................................................................................90
3.3 RESULTS..................................................................................................................................91
3.3.1 Comparison of pLDH and Isotopic Assays ........................................................................91
3.3.2 Effect of pLDH Reaction Duration on IC50 Values ............................................................91
3.3.3 Field Application of the pLDH Assay.................................................................................93
3.3.4 Antimalarial Susceptibility of PNG P. falciparum Isolates................................................97
3.3.5 Correlations of in vitro Responses to Nine Antimalarials..................................................98
3.4 DISCUSSION...........................................................................................................................100
CHAPTER 4. CHARACTERISATION OF DRUG RESISTANT POLYM ORPHISMS OF P.

xxvi
FALCIPARUM USING A NEW MOLECULAR ASSAY.................................................................. 106
4.1 INTRODUCTION ..................................................................................................................... 106
4.2 MATERIALS AND METHODS.................................................................................................. 108
4.2.1 Field Studies, P. falciparum isolates ............................................................................... 108
4.2.2 Genomic DNA.................................................................................................................. 109
4.2.3 Plasmodium Speciation ................................................................................................... 109
4.2.4 Detection of Drug Resistant Polymorphisms................................................................... 110
4.2.5 Data Analysis .................................................................................................................. 112
4.3 RESULTS............................................................................................................................... 113
4.3.1 Pfmdr1 LDR-FMA Development ..................................................................................... 113 4.3.1.1 PCR Optimisation...........................................................................................................................113 4.3.1.2 LDR Optimisation ..........................................................................................................................118 4.3.1.3 Optimised LDR-FMA for pfmdr1 and Multiplexed Detection of SNPs in pfdhfr, pfdhps and pfcrt
genes ..........................................................................................................................................................122 4.3.2 Assay Validation.............................................................................................................. 124
4.3.2.1 Comparison between LDR-FMA and RFLP speciation .................................................................124 4.3.2.2 Inter-assay concordance .................................................................................................................124 4.3.2.3 Identification of drug resistance alleles ..........................................................................................125
4.3.3 Field Application of the LDR-FMA ................................................................................. 126 4.3.3.1 Speciation and drug resistance genes in PNG field isolates............................................................126 4.3.3.2 Prevalence of polymorphic alleles in pfcrt, pfmdr1, pfdhfr and pfdhps .........................................127 4.3.3.3 Parasite drug resistance mutations and treatment outcome.............................................................130
4.4 DISCUSSION ...................................................................................................................... 131
CHAPTER 5. ANTIMALARIAL PROPERTIES OF DESBUTYL-LUME FANTRINE ............... 138
5.1 INTRODUCTION ..................................................................................................................... 138
5.2 MATERIALS AND METHODS.................................................................................................. 139
5.2.1 Parasite Cultures............................................................................................................. 139
5.2.2 Antimalarial Drugs.......................................................................................................... 139
5.2.3 In vitro Drug Susceptibility ............................................................................................. 140
5.2.4 Drug Interaction Studies ................................................................................................. 140
5.2.5 Study Site and Sample Collection.................................................................................... 141
5.2.6 Liquid Chromatography and Mass Spectrometry............................................................ 142
5.2.7 Statistical Analysis........................................................................................................... 143
5.3 RESULTS............................................................................................................................... 144
5.3.1 In vitro Antimalarial Potency of DBL ............................................................................. 144
5.3.2 DBL Interaction with Conventional Antimalarials.......................................................... 146
5.3.3 DBL Plasma Levels on Day 7 Post-Treatment ................................................................ 148
5.3.4 Influence of DBL Plasma Levels on Clinical Outcome ................................................... 148
5.4 DISCUSSION.......................................................................................................................... 150

xxvii
CHAPTER 6. STATINS AND FIBRATES: LIPID-MODIFYING DR UGS AS ANTIMALARIALS
.................................................................................................................................................................154
6.1 INTRODUCTION ......................................................................................................................154
6.1.1 Statins as Lipid-lowering and Antimicrobial Agents........................................................154
6.1.2 Fibrates as Potential Antimalarial Drugs........................................................................155
6.2 MATERIALS AND METHODS...................................................................................................156
6.2.1 In vitro Parasite Growth Inhibition..................................................................................156
6.2.2 Drug Interaction Studies ..................................................................................................157
6.2.3 Dosed Plasma Bioassay ...................................................................................................158
6.3 RESULTS................................................................................................................................160
6.3.1 In vitro Antimalarial Activities of Statins.........................................................................160
6.3.2 In vitro Antimalarial Activities of Fibrates ......................................................................160
6.3.3 Interaction of Atorvastatin with Conventional Antimalarials ..........................................164
6.3.4 Interaction of Fibrates with Conventional Antimalarials ................................................164
6.3.5 Bioassay of Atorvastatin...................................................................................................167
6.3.6 Bioassay of Fenofibric Acid .............................................................................................167
6.3.7 BLAST Analysis for PPAR-like Region in Plasmodium ...................................................170
6.3.8 BLAST Analysis for ABC-1 transporter in P. falciparum.................................................170
6.4 DISCUSSION...........................................................................................................................174
CHAPTER 7. CHARACTERISATION OF VOLATILE ORGANIC COM POUNDS OF P.
FALCIPARUM IN VITRO .....................................................................................................................178
7.1 INTRODUCTION......................................................................................................................178
7.2 MATERIALS AND METHODS...................................................................................................180
7.2.1 Parasites...........................................................................................................................180
7.2.2 Solid Phase Micro-Extraction (SPME) ............................................................................180
7.2.3 Solvent Extraction ............................................................................................................181
7.2.4 Thermal Desorption: Purge and Trap..............................................................................181
7.2.5 Gas Chromatography and Mass Spectrometry ................................................................183
7.2.6 Data Analysis ...................................................................................................................184
7.3 RESULTS................................................................................................................................184
7.3.1 Malaria VOCs Assay Development ..................................................................................184 7.3.1.1 Design of culture-capture apparatus............................................................................................... 184 7.3.1.2 Optimisation of culture conditions................................................................................................. 186
7.3.2 Analysis of VOCs..............................................................................................................187
7.4 DISCUSSION...........................................................................................................................190
CHAPTER 8. CONCLUDING DISCUSSION ....................................................................................196
8.1 OVERVIEW.............................................................................................................................196

xxviii
8.1.1 Major Findings and Contributions.................................................................................. 196
8.2 THE ROLE OF IN VITRO RESISTANCE AND PARASITE GENETIC MUTATIONS IN TREATMENT
OUTCOME............................................................................................................................................ 198
8.2.1 Limitations of PNG field studies...................................................................................... 200
8.3 UNCONVENTIONAL AND NOVEL ANTIMALARIAL AGENTS ...................................................202
8.3.1 Desbutyl-lumefantrine and its Potential Implementation ................................................202
8.3.2 Lipid-modifying Agents as Antimalarials ........................................................................ 204
8.4 A PILOT STUDY OF MALARIA VOCS..................................................................................... 206
8.5 CONCLUSION AND FUTURE DIRECTIONS............................................................................... 207
8.5.1 Directions for Future Research....................................................................................... 208
BIBLIOGRAPHY ................................................................................................................................. 209
APPENDICES ....................................................................................................................................... 259
APPENDIX A. ISOLATE INFORMATION .................... .................................................................. 261
APPENDIX B. RECIPES FOR SOLUTIONS.................................................................................... 263
Culture of P. falciparum ................................................................................................................ 263
LDH Assay ..................................................................................................................................... 268
Molecular Assays........................................................................................................................... 269
APPENDIX C. EFFECTS OF LDR ANNEALING TEMPERATURE AN D DILUTION............. 272
APPENDIX D. ISOBOLOGRAM ANALYSIS .................................................................................. 277
APPENDIX E. VOCS ANAYLSIS ...................................................................................................... 280

xxix
LIST OF TABLES
TABLE 1.1 ANTIMALARIAL DRUGS AND THEIR SPECIFIC ACTIVITIES. ..........................................................32
TABLE 1.2 DETERMINANTS OF ANTIMALARIAL RESISTANCE. ......................................................................40
TABLE 1.3 CLASSIFICATIONS OF IN VIVO ANTIMALARIAL SUSCEPTIBILITY OUTCOMES. ...............................55
TABLE 1.4 MOLECULAR MARKERS FOR ANTIMALARIAL DRUG RESISTANCE................................................58
TABLE 2.1 SOLVENTS AND OPTIMISED ASSAY CONCENTRATION RANGES FOR DRUG SUSCEPTIBILITY
TESTING.............................................................................................................................................69
TABLE 2.2 PCR PRIMER SEQUENCES AND THERMOCYCLING CONDITIONS FOR PFCRT, PFDHPS AND PFDHFR
TARGET SEQUENCES. .........................................................................................................................75
TABLE 2.3 LDR PRIMER SEQUENCES FOR PLASMODIUM SPECIES DIAGNOSIS...............................................77
TABLE 2.4 LDR PRIMER SEQUENCES FOR DRUG RESISTANCE MARKERS PFCRT, PFDHFR AND PFDHPS. .......79
TABLE 3.1 OVERVIEW OF IN VITRO DRUG SENSITIVITY FINDINGS IN PNG....................................................86
TABLE 3.2 IN VITRO SUSCEPTIBILITIES OF P. FALCIPARUM PNG ISOLATES AGAINST 4-AMINOQUINOLINES
AND OTHER ANTIMALARIAL DRUGS. ..................................................................................................98
TABLE 3.3 SPEARMAN CORRELATION CO-EFFICIENTS FOR ASSOCIATIONS BETWEEN IC50 VALUES..............99
TABLE 4.1 PRIMER SEQUENCES AND THERMOCYCLING CONDITIONS FOR PFMDR1 ASSAYS. ......................115
TABLE 4.2 OPTIMISED PCR CONDITIONS FOR PFMDR1 REGION 1. ……………………………………….120
TABLE 4.3 OPTIMISED PCR CONDITIONS FOR PFMDR1 REGION 2…….. ....................................................120
TABLE 4.4 LDR PRIMERS FOR P. FALCIPARUM PFMDR1 MOLECULAR MARKERS. .......................................121
TABLE 4.5 OPTIMISED LDR CONDITIONS FOR PFMDR1 REGION 1…………………………………… …..123
TABLE 4.6 OPTIMISED LDR CONDITIONS FOR PFMDR1 REGION 2…..........................................................123
TABLE 4.7 CONCORDANCE BETWEEN RFLP AND LDR-FMA DIAGNOSIS OF PLASMODIUM SPECIES IN PNG
FIELD SAMPLES. ...............................................................................................................................125
TABLE 4.8 LDR-FMA EVALUATION OF PFMDR1 SNPS IN LABORATORY-ADAPTED P. FALCIPARUM STRAINS.
........................................................................................................................................................126
TABLE 4.9 FLUORESCENCE DETECTION THRESHOLDS AND MAXIMA FOR P. FALCIPARUM PFDHPS, PFDHFR,
PFCRT AND PFMDR1 IN PNG FIELD SAMPLES. ..................................................................................128
TABLE 4.10 OCCURRENCE OF P. FALCIPARUM ISOLATES CARRYING MULTIPLE MUTATIONS ACROSS 4 GENES
ASSOCIATED WITH DRUG RESISTANCE. ............................................................................................130
TABLE 5.1 DRUG COMBINATION RATIOS FOR ISOBOLOGRAM ASSAYS. ......................................................140
TABLE 5.2 IN VITRO SENSITIVITY OF LABORATORY-ADAPTED P. FALCIPARUM TO DESBUTYL-LUMEFANTRINE
AND OTHER ANTIMALARIAL DRUGS. ................................................................................................144
TABLE 5.3 IN VITRO EFFICACY OF ANTIMALARIAL DRUG COMBINATIONS AGAINST P. FALCIPARUM CLONES
3D7 AND W2MEF AS ASSESSED BY ISOBOLOGRAPHIC ANALYSIS. ....................................................146
TABLE 6.1 INTERACTION RATIOS OF STATINS, FIBRATES AND CONVENTIONAL ANTIMALARIALS. .............158
TABLE 6.2 IN VITRO ACTIVITIES OF STATINS AGAINST CQ-SENSITIVE AND CQ-RESISTANT STRAINS OF P.
FALCIPARUM. ...................................................................................................................................161

xxx
TABLE 6.3 IN VITRO ACTIVITIES OF FIBRATES AGAINST CQ-SENSITIVE AND CQ-RESISTANT STRAINS OF P.
FALCIPARUM.................................................................................................................................... 162
TABLE 6.4 IN VITRO EFFICACY OF FIBRATES AND ANTIMALARIAL DRUG COMBINATIONS.......................... 166
TABLE 7.1 VOCS DETECTED IN CULTURE HEADSPACE. ............................................................................ 188

xxxi
LIST OF FIGURES
FIGURE 1.1 DISTRIBUTION OF MALARIA INCIDENCE BY COUNTRY. ...............................................................4
FIGURE 1.2 LIFE CYCLE OF PLASMODIUM. .....................................................................................................5
FIGURE 1.3 CHILD WITH MALARIA . .............................................................................................................11
FIGURE 1.4 ANOPHELES ALBIMANUS TAKING A BLOOD MEAL. ......................................................................13
FIGURE 1.5 MALARIA MORTALITY IN WESTERN PACIFIC. ...........................................................................15
FIGURE 1.6 REGIONS AND PROVINCES OF PAPUA NEW GUINEA. .................................................................16
FIGURE 1.7 ARTEMISIA AND CINCHONA. ....................................................................................................18
FIGURE 1.8 CHEMICAL STRUCTURE OF QUININE..........................................................................................19
FIGURE 1.9 CHEMICAL STRUCTURE OF CHLOROQUINE. ...............................................................................20
FIGURE 1.10 CHEMICAL STRUCTURE OF AMODIAQUINE. .............................................................................21
FIGURE 1.11 CHEMICAL STRUCTURE OF MEFLOQUINE. ...............................................................................23
FIGURE 1.12 CHEMICAL STRUCTURE OF LUMEFANTRINE. ...........................................................................24
FIGURE 1.13 CHEMICAL STRUCTURE OF NAPHTHOQUINE. ..........................................................................25
FIGURE 1.14 CHEMICAL STRUCTURE OF PIPERAQUINE. ..............................................................................26
FIGURE 1.15 CHEMICAL STRUCTURES OF SULFADOXINE (LEFT) AND PYRIMETHAMINE (RIGHT)..................26
FIGURE 1.16 CHEMICAL STRUCTURE OF ARTEMISININ (LEFT) AND ITS DERIVATIVES (RIGHT). ....................27
FIGURE 1.17 TARGETS OF ANTI-BACTERIAL DRUGS IN P. FALCIPARUM APICOPLAST....................................29
FIGURE 1.18 CHEMICAL STRUCTURE OF AZITHROMYCIN. ..........................................................................30
FIGURE 1.19 OVERVIEW OF ISOPRENOID BIOSYNTHESIS..............................................................................34
FIGURE 1.20 RIGHT-WARD SHIFT OF CONCENTRATION-EFFECT RELATIONSHIP DUE TO DRUG RESISTANCE. 37
FIGURE 1.21 EMERGENCE OF RESISTANCE TO PRINCIPAL ANTIMALARIAL DRUGS. ......................................39
FIGURE 1.22 REPRESENTATION OF ISOBOLES. .............................................................................................53
FIGURE 2.1 NALGENE DESICCATOR USED FOR P. FALCIPARUM CULTURE.....................................................63
FIGURE 2.2 GIEMSA-STAINED THIN SMEAR OF SYNCHRONISED P. FALCIPARUM CULTURE. ..........................65
FIGURE 2.3 PREPARATION OF A THIN SMEAR...............................................................................................66
FIGURE 2.4 LAYOUT OF A DRUG SUSCEPTIBILITY PANEL. ............................................................................68
FIGURE 2.5 COLOURIMETRIC DETECTION OF PLDH ACTIVITY . ...................................................................70
FIGURE 2.6 PLDH REACTION IN FIELD ISOLATES OF P. FALCIPARUM. ..........................................................71
FIGURE 2.7 TOMTEC HARVESTER 96 SYSTEM. ............................................................................................72
FIGURE 2.8 WOLSTEIN RESEARCH BUILDING, CWRU, CLEVELAND, OHIO, USA. .....................................73
FIGURE 2.9 ELECTROPHORESIS AND IMAGE PROCESSING FOR DNA VISUALISATION FOR EVALUATING PCR
AMPLIFICATION EFFICIENCY. .............................................................................................................76
FIGURE 2.10 BIO-PLEX ARRAY READER. .....................................................................................................80
FIGURE 2.11 SOLID PHASE MICRO-EXTRACTION SAMPLER. .........................................................................81
FIGURE 3.1 CANDLE JAR METHOD USED FOR P. FALCIPARUM CULTURE IN PNG..........................................89
FIGURE 3.2 COMPARISON OF PLDH AND 3H-HYPOXANTHINE INCORPORATION METHODS FOR ANALYSIS OF

xxxii
ANTIMALARIAL SENSITIVITY IN CULTURE -ADAPTED P. FALCIPARUM. ............................................... 92
FIGURE 3.3 EFFECT OF PLDH REACTION TIME ON IC50S IN PNG P. FALCIPARUM. ...................................... 94
FIGURE 3.4 SCATTER PLOT OF IC50S DETERMINED FROM THREE PLDH TIME POINTS.................................. 95
FIGURE 3.5 DISTRIBUTION OF 50% INHIBITORY CONCENTRATIONS (IC50) OF ANTIMALARIALS AGAINST PNG
P. FALCIPARUM ISOLATES.................................................................................................................. 96
FIGURE 4.1 PRINCIPLE OF LDR-FMA DIAGNOSIS OF DRUG RESISTANT POLYMORPHISMS. ....................... 111
FIGURE 4.2 PCR AMPLIFICATION OF PFMDR1 REGIONS 1 AND 2 IN 7G8 OVER A TEMPERATURE GRADIENT.
....................................................................................................................................................... 116
FIGURE 4.3 GEL SCAN OF PCR PRODUCTS GENERATED USING NEW PFMDR1 PRIMERS. ............................ 117
FIGURE 4.4 EFFECT OF PCR CYCLES ON PFMDR1 AMPLIFICATION............................................................ 119
FIGURE 4.5 PREVALENCE OF PFCRT, PFMDR1, PFDHFR, PFDHPS ALLELES IN P. FALCIPARUM-INFECTED
INDIVIDUALS FROM THE MADANG AND EAST SEPIK PROVINCES, PNG. ......................................... 129
FIGURE 4.6 FREQUENCY DISTRIBUTIONS OF PFCRT, PFDHPS, PFDHFR, PFMDR1 HAPLOTYPES IN P.
FALCIPARUM-INFECTED INDIVIDUALS FROM PNG FIELD SITES. ....................................................... 129
FIGURE 5.1 IN VITRO SUSCEPTIBILITY OF LABORATORY STRAINS OF P. FALCIPARUM TO CHLOROQUINE,
DESBUTYL-LUMEFANTRINE AND LUMEFANTRINE. .......................................................................... 145
FIGURE 5.2 ISOBOLOGRAMS ILLUSTRATING INTERACTIONS BETWEEN DESBUTYL-LUMEFANTRINE WITH
CONVENTIONAL ANTIMALARIALS . .................................................................................................. 147
FIGURE 5.3 BOXPLOTS SUMMARISING DAY 7 PLASMA LEVELS OF LUMEFANTRINE AND DESBUTYL-
LUMEFANTRINE. ............................................................................................................................. 149
FIGURE 6.1 IN VITRO SUSCEPTIBILITY OF LABORATORY STRAINS OF P. FALCIPARUM TO CHOLESTEROL-
LOWERING DRUGS AND CHLOROQUINE. .......................................................................................... 163
FIGURE 6.2 INTERACTION BETWEEN ATORVASTATIN AND CONVENTIONAL ANTIMALARIALS . .................. 165
FIGURE 6.3 ATORVASTATIN BIOASSAY. ................................................................................................... 168
FIGURE 6.4 FENOFIBRATE BIOASSAY........................................................................................................ 169
FIGURE 6.5 DISTRIBUTION OF PLASMODIUM BLAST HIT SEQUENCES ON HUMAN PPARΑ. ...................... 172
FIGURE 6.6 DISTRIBUTION OF P. FALCIPARUM BLAST HIT SEQUENCE ON HUMAN ABC-1....................... 173
FIGURE 7.1 EXTRACTION OF VOCS FROM CULTURE SUPERNATANT BY AN ORGANIC SOLVENT................ 182
FIGURE 7.2 PURGE AND TRAP SET-UP FOR THERMAL DESORPTION. .......................................................... 183
FIGURE 7.3 PROTOTYPE 1 CULTURE-CAPTURE APPARATUS WITH SPME.................................................. 185
FIGURE 7.4 DESIGN AND DIMENSIONS OF CULTURE CONTAINER (PROTOTYPE 2) FOR HEADSPACE CAPTURE.
....................................................................................................................................................... 186
FIGURE 7.5 CHROMATOGRAMS OF VOCS IN THE HEADSPACE OF CULTURED P. FALCIPARUM................... 189

CHAPTER 1
GENERAL INTRODUCTION

Chapter 1 General Introduction
2
CHAPTER 1. GENERAL INTRODUCTION
1.1 INTRODUCTION
The term malaria was derived from the Italian words “Mala aria” or bad air, as the
sickness was once thought to be associated with inhalation of foul smelling air near
swampy areas (Harrison 1978). The disease has been recognised for over 4000 years
and has significantly influenced human history (Cox 2002; Rich et al. 2009). Malaria
caused more casualties than those due to bullets during the World Wars and other
military campaigns, with famous victims including Alexander the Great, Genghis Khan,
Ho Chi Minh and George Clooney (MVI 2004; Kakkilaya 2008; News 2011). Malaria
inflicts greater detrimental impacts on the human population than other parasitic
diseases (CDC 2004). Close to 3 billion people are exposed to malaria world-wide, with
the disease accounting for 1 - 2 million deaths per year, most of which are in pregnant
women and children due to their low immunity (Bloland 2001; Snow et al. 2005; WHO
2008).
Five Plasmodium species are known to cause human malaria infections: P. falciparum,
P. vivax, P. malariae, P. ovale and P. knowlesi. The latter species originated from
macaque monkeys in Malaysian Borneo, and has been recently identified in naturally
acquired infections with mortality reported in South-East Asia and in European
travellers (Singh et al. 2004; Cox-Singh et al. 2008; Luchavez et al. 2008). P.
falciparum and P. vivax are the most common but P. falciparum is the most virulent
species. It is capable of invading erythrocytes (RBC) of all ages and RBC containing
mature forms sequester in the microvasculature of vital organs. P. vivax only infects
young RBC and may exhibit relatively weak cytoadherence (Carvalho et al. 2010). P.
falciparum has also developed rapid resistance to antimalarial drugs (Al-Yaman et al.
1996; Dondorp et al. 2009; Preechapornkul et al. 2009) and is responsible for
approximately 500 million cases annually and the highest morbidity of all infectious
diseases (Bloland 2001; Snow et al. 2005; WHO 2009).
The impact of malaria varies with local epidemiology. Developed countries such as
Australia, European countries and North America do not have local transmission apart

Chapter 1 General Introduction
3
from occasional autochthonous outbreaks, and virtually all cases are imported. (Gratz
2005; Berry et al. 2008). The most intense transmission occurs in sub-Saharan Africa
and other areas of the rural tropics such as Papua New Guinea (PNG) where young
children with limited immunity are at greatest risk of morbidity and death (Mueller et
al. 2003; Ouellette et al. 2003). Although both preventable and curable, malaria remains
a substantial social and economic burden in tropical regions (WHO 2009).
Mass treatment programs, variable drug compliance and counterfeit antimalarial drugs
have all contributed to widespread parasite drug resistance (White 2004; Newton et al.
2008). In particular, P. falciparum has developed resistance to multiple antimalarial
agents including, most recently, the potent artemisinin derivatives (Wongsrichanalai et
al. 1992a; Bloland 2001; Dondorp et al. 2009). Not only is the arsenal of effective
antimalarial drugs diminishing, our current understanding of their mechanisms of action
is still limited.
The present review outlines the basic epidemiology of falciparum malaria, and the
status of parasite drug resistance and its underlying mechanisms. It examines current
methods employed for the assessment of drug resistance and pharmacological strategies
for limiting its effects and spread, with particular reference to PNG. In addition, several
novel compounds with potential antimalarial properties are reviewed.
1.2 DISEASE DISTRIBUTION
According to a recent report, malaria is present in 109 countries, with the highest
transmission occurring in subtropical and tropical regions in sub-Saharan Africa,
Central and South America, the Middle East, the Indian subcontinent, South-East Asia
and Oceania (Figure 1.1) (WHO 2009). Transmission intensity and risk of infection are
dependent on climatic factors such as temperature, humidity, rainfall, altitude, and the
presence of the Anopheles mosquito vector. Typically, malaria transmission is rare in
the highlands at altitudes above 1500 m and in arid areas with <1,000 mm of rainfall
per year (Bloland 2001). However, these tropical areas suffer greater risks of epidemic
malaria especially with increased movement of people between these areas and
malarious lowlands (John et al. 2005; Mueller et al. 2005). Climatic conditions

Chapter 1 General Introduction
4
favourable to vector breeding coupled with the lack of, or low, immunity to malaria
within a local population have led to devastating epidemics with high mortality rates
(Fontaine et al. 1961; Mueller et al. 2005). Malaria intensity is generally higher in
regions adjacent to the equator where transmission is year-round. PNG is no exception,
with 1000-10000 reported cases per year (Figure 1.1). At temperatures below 16°C, P.
falciparum cannot complete a normal growth cycle in the mosquito host and hence
there is no transmission (Teklehaimanot et al. 2004).
Figure 1.1 Distribution of malaria incidence by country. (WHO 2004)
1.3 PLASMODIUM LIFE CYCLE AND BIOLOGY
The malaria parasite has developed an intricate relationship with its mammalian and
insect hosts. The sexual reproduction of the parasite takes place in the mosquito gut
whereas asexual replication occurs within RBC of the human host. Mammalian blood
stage of the P. falciparum life cycle starts with rupture and release of merozoites from
intra-hepatic schizonts into the blood stream (Figure 1.2).
Chapter 1 General Introduction
5
Figure 1.2 Life cycle of Plasmodium. (CDC 2006)
1.3.1 Development in the Human Host
As the female Anopheles mosquito probes for a blood meal, P. falciparum sporozoites
are injected with its saliva into the dermis of the host. The sporozoites migrate slowly
out of the inoculation site and penetrate dermal capillaries upon contact to enter the
circulation (Sidjanski et al. 1997; Matsuoka et al. 2002; Yamauchi et al. 2007). A
portion of the sporozoites move through the lymphatics of the host (Amino et al. 2006;
Yamauchi et al. 2007). The sporozoites invade the liver (Shin et al. 1982) and pass
through the endothelial lining of the sinusoids (Figure 1.2-A). Circumsporozoite protein
(CSP) is released to facilitate invasion and exo-erythrocytic development, making it a

Chapter 1 General Introduction
6
target for vaccine development (Cohen et al. 2009; Kester et al. 2009).
The asexual reproduction cycle of P. falciparum occurs at 48 hr intervals. It begins with
the release of merozoites from hepatic schizonts into the peripheral circulation (Figure
1.2-B). The merozoite is oval-shaped (1 to 1.5 µm) with a single nucleus and adjacent
cytoplasm. It is equipped with erythrocyte binding antigen (EBA 175), merozoite
surface protein-1 (MSP-1) and other proteins specific for adherence to the RBC
membrane (Camus et al. 1985; Perkins et al. 1988; Sam-Yellowe et al. 1988). During
invasion, the merozoite positions its apical end to form a tight junction with the RBC
membrane. Invasion is rapid, complete in 30 sec, and is facilitated by anterior
organelles such as polar rings, rhoptries and micronemes, and the posterior actin-
myosin network that drives the parasite into its host RBC (Kilejian 1976; Aikawa et al.
1978).
At entry, the outer structures of the merozoite degrade and the parasite rounds up as a
uninuclear trophozoite (Aikawa 1966). The young trophozoite is surrounded by a
parasitophorous vacuole membrane originated from the host cell. A vacuole develops,
forming the characteristic ‘ring stage’ of the malaria parasite. The trophozoite continues
to develop as it digests host cell cytoplasm and haemoglobin. This process however,
produces free haem by-products, the accumulation of which leads to toxicity and
parasite death. To circumvent this, the parasite polymerises the free haem into
haemazoin crystals, which is easily recognised microscopically as the ‘malaria pigment’
within the food vacuole in mature parasites (Dorn et al. 1995). The beginning of
parasitic nuclear division marks the beginning of the schizont stage. During schizogony,
rapid DNA synthesis and nuclear mitosis proceeds as the parasite becomes enlarged and
multinucleated. Mitochondrial budding occurs, and merozoite organelles reappear to
form 15 to 20 new merozoites within each schizont that are subsequently released for
re-invasion (Aikawa 1966).
Gametocytogenesis may take place instead of asexual replication where the merozoite
develops into gametocytes, sex cells that are subdivided into either microgametocyte
(male) or macrogametocyte (female). The events that trigger this process are not well
understood. The gametocyte develops through five morphologically distinctive stages to

Chapter 1 General Introduction
7
reach maturity (Hawking et al. 1971; Carter et al. 1979; Ponnudurai et al. 1982). It
features a single unagglomerated nucleus with the male being larger and has a paler
cytoplasm compared to the female gametocyte. It develops as a small rounded globule
to a triangular body taking up half the RBC to ellipsoidal and finally crescentic forms
(Figure 1.2-B7) (Jensen 1979) and is infective to its mosquito host.
1.3.2 Development in the Mosquito
Both sexual and asexual stages of malaria parasites are ingested through the blood meal
by a female Anopheles mosquito (Figure l.2-8). The presence of xanthurenic acid and
the decrease in temperature within the mosquito triggers gamete formation. The
macrogametocyte exits the RBC to form a macrogamete within 10 min, whereas
microgametogenesis is less rapid. Mitotic division begins as the nucleus divides into
eight portions. Each of these portions enters a projection on the surface of the
microgamete and breaks free as an individual microgamete in a process known as
exflagellation (Gao 1981; Aikawa et al. 1984). The microgamete promptly fertilises a
macrogamete forming a zygote (Figure 1.2-9). Within 24 hr of fertilisation, the zygote
develops into a slow moving ookinete (Gao 1981; Aikawa et al. 1984). With structures
similar to the merozoite stage (Garnham et al. 1962) the ookinete invades the microvilli
of the mosquito gut and secretes digestive enzymes to gain entry into the epithelial cell
(Huber et al. 1991). On reaching the cell basal membrane, it develops into a vegetative
oocyst (Figure 1.2-10) (Torii et al. 1992). After 6 days of repeated nuclear division, the
oocyst develops into a polypoid measuring 50 µm containing as many as 10,000
sporozoites (Aikawa 1988).
The motile sporozoite exits the oocyst and travels to the salivary glands of the
mosquito. It contains similar apical organelles as the merozoite stage (Aikawa 1988),
equipped with CSP and the thrombospondin-related anonymous protein (TRAP)
(Rogers et al. 1992) required for sporozoite formation (Menard et al. 1997) and cell
invasion in the mosquito salivary glands and hepatocytes of the human host,
respectively (Sultan et al. 1997). The infectivity of the sporozoite to the human host is
low then increases with residency in the mosquito salivary glands and gradually

Chapter 1 General Introduction
8
decreases with age. At the next feeding, sporozoites are injected subcutaneously into the
human host. From the site of inoculation, the sporozoites then migrate through the
circulation to the liver to begin the asexual cycle.
1.4 PATHOLOGY
P. falciparum is responsible for the most severe forms of malaria, causing diverse
clinical and pathological conditions in multiple organ systems. Several parasite factors
contribute to disease severity and manifestations. The predilection of P. falciparum
merozoites for RBC of all ages, augmented by the speed and abundance of schizogony,
makes them efficient in re-invasion. Progeny amplification at every 48 hr intervals
results in the rapid increase of parasite numbers to as many as 1013 per host
(Greenwood et al. 2008). Another distinguishing characteristic of P. falciparum is its
ability to bind to endothelium during the intra-erythrocytic stage of infection (Aikawa et
al. 1980). The sequestration of infected RBC in the microvasculature of organs
including the heart, liver and the brain in cerebral malaria (Sachanonta et al. 2008) can
alter or occlude blood flow and lead to potentially fatal complications (Luse et al.
1971). A number of studies have shown that P. falciparum infected RBC can bind
platelets to form erythrocyte clumps referred to as ‘platelet-mediated clumping’, a
cytoadherence phenomenon distinct from sequestration and rosette formation of
infected RBC, all of which have largely been associated with disease severity (Pain et
al. 2001; Miller et al. 2002; Chotivanich et al. 2004). However, a Malian study
demonstrated that platelet-mediated clumping of P. falciparum infected RBC is
primarily associated with high parasitaemia and not with severe clinical manifestations
of malaria (Arman et al. 2007) and the causality of malarial encephalopathy appears
multifactorial (Coltel et al. 2004; Mackintosh et al. 2004; Clark et al. 2009; Idro et al.
2010). A systemic host response relating to the release of inflammatory cytokines has
been proposed as a primary cause for disease (Clark et al. 2004; Clark et al. 2008). The
cyclic destruction of RBC via schizont rupture can result in anaemia and tissue anoxia.
The absence of reticulocytes during acute infection suggests defective erythropoiesis
which can further aggravate anaemia (Boonpucknavig et al. 1988).

Chapter 1 General Introduction
9
Recent views regard the primary pathogenesis of malaria as similar to viral and
bacterial infectious diseases in which, the host’s unbridled response to the invading
organism rather than the organism itself is the cause of disease (Clark et al. 2000; Clark
et al. 2004). The importance of these “cytokine storms” in the pathogenesis of cerebral
malaria has been consistently championed by Clark since the late 1980s (Clark et al.
1987; Clark et al. 2008). Although this concept was criticised initially, it has gained
some support with the recognition that cytokines and immune effector cells may play
pivotal roles in severe malaria (Hunt et al. 2003; Kusi et al. 2008). The release of pro-
inflammatory cytokines such as tumour necrosis factor (TNF) by host cells as triggered
by malaria antigens has been considered a primary factor in the onset of pathology,
especially the pathogenesis of cerebral malaria (Gimenez et al. 2003; Hunt et al. 2003).
Clinical studies in African and PNG children have demonstrated a correlation between
circulating levels of TNF and disease severity as characterised by fever and parasite
density (Butcher et al. 1990; Kwiatkowski 1990a; Al-Yaman et al. 1998; Tchinda et al.
2007). Some clinical observations suggest circulating TNF levels predict death. Serum
levels of TNF were twice as high in cerebral malaria survivors, and up to ten-fold
higher amongst fatal cases compared to those with uncomplicated malaria
(Kwiatkowski et al. 1990b; Al-Yaman et al. 1998). Other evidence in support of the
importance of TNF include the in vivo prevention of the onset of neurological syndrome
in murine models by neutralisation of cytokines (Grau et al. 1987) and the absence of
cerebral malaria in transgenic murine models (Garcia et al. 1995).
The current understanding of severe malaria pathogenesis is that there is no longer a
clear-cut, single pathogenic correlate (Mackintosh et al. 2004; Haldar et al. 2007).
There is likely to be a complex interplay of inflammatory, pro-inflammatory cytokines
and cellular responses. TNF increases the expression of intercellular cell adhesion
molecule-1 (ICAM-1), particularly on brain endothelial cells and on some sequestered,
intravascular leukocytes in human and murine models (Turner et al. 1994). ICAM-1 is a
receptor for P. falciparum infected RBC, and both leukocytes and other parasitised
RBC have the potential to adhere to capillaries when its expression is up-regulated.
Therefore, subsequent blockages may be an outcome secondary to the surge of TNF
from host effector cells. Several findings from murine models have also shown the

Chapter 1 General Introduction
10
activation of platelets in contributing to the formation of neurovascular lesions seen in
cerebral cases (Grau et al. 1993a; Grau et al. 1993b). Other cytokines released by
natural killer T cells such as interferon-gamma (IFN-γ) and lymphotoxin (LT) have also
been implicated in driving the immunopathological progression to cerebral malaria
(Amani et al. 2000; Hansen et al. 2003), whilst the anti-inflammatory cytokines
interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β) appear to counter
this process (Ho et al. 1998). In uncomplicated malaria however, pro-inflammatory
cytokines particularly IFN-γ have been associated with a more favourable role, limiting
the progression to severe disease (Torre et al. 2002). The literature on IL-10 is generally
consistent on its host-protective effect by inhibiting the production of cytokines that are
suspected to cause complications of severe and cerebral malaria (Hunt et al. 2003).
However, a recent report indicated that IL-10 may indeed down-regulate pro-
inflammatory responses, but at the same time exacerbate the infection by inhibiting
anti-parasitic immune function. Hugosson et al found high baseline IL-10 levels were
associated with higher parasite densities post treatment after adjusting for initial
parasitaemia, age temperature and sex. The authors concluded that the induction of high
IL-10 production might be a direct or indirect mechanism whereby the parasite evades
host immune response (Hugosson et al. 2004). All in all, severe malaria is not a single
and discrete syndrome as coma develops through multiple pathways with many
mechanisms of brain injury (Idro et al. 2010).
1.5 CLINICAL SIGNS AND SYMPTOMS
The incubation period of malaria varies with the level of host immunity acquired
through prior exposure and the use of antimalarial prophylaxis. In non-immune
individuals, the time of infection to detectable parasitaemia ranges from 5 to 10 days,
and the development of symptoms from 6 to 14 days (Trampuz et al. 2003). Clinical
symptoms are primarily due to the destruction of RBC with the release of merozoites.
The presentation of malaria may be non-specific, often with symptoms resembling
those of a common cold. Infection with P. falciparum can be associated with well-
defined febrile paroxysms, consisting of fever, chills and rigors at regular intervals.
Other symptoms include headache, diaphoresis, nausea, vomiting, dizziness, malaise,
abdominal pain, mild diarrhoea and dry cough. Clinical signs include jaundice, pallor,

Chapter 1 General Introduction
11
tachycardia, orthostatic hypotension, hepatomegaly, splenomegaly, metabolic acidosis,
hypoglycaemia, seizures, coma, renal failure and cerebral oedema (Gilles 1988; Clark et
al. 2000; Trampuz et al. 2003; Mackintosh et al. 2004). Figure 1.3 demonstrates a child
infected by P. falciparum who was found to have splenomegaly and fever.
Figure 1.3 Child with malaria. Father comforting his malaria-infected son whilst
waiting outside the Alexishafen Health Care Centre, Madang Province, PNG.
1.6 DIAGNOSIS
Light microscopic examination of intracellular parasites on Giemsa stained thick and
thin blood smears remain the definitive method of diagnosis of malaria (Bloland 2001).
This method enables the quantification and identification of different Plasmodium
species, and the presence of mixed-species infection, diagnosis of which will have a
bearing on treatment selection. Parasite densities are liable to considerable fluctuation
due to the impact of chemo-suppression, development of host immunity, and their
cyclic multiplication patterns. In some instances, infection with P. falciparum may
produce scanty intra-erythrocytic stages with parasitaemia below detectable threshold,
as synchronous mature parasites are sequestered away from the peripheral circulation in
the microvasculature. In these circumstances, the identification of schizonts,

Chapter 1 General Introduction
12
gametocytes and malarial pigments in macrophages are of value (Shute 1988).
Before a blood slide is considered negative, at least 200 fields should be examined
under 1000 × magnification. With positive smears, the level of parasitaemia is reported
as a percentage of infected RBC or as the number of parasites per microliter (µL) of
blood. Microscopy diagnosis can be time consuming and requires trained personnel to
ensure consistent reliability. It has relatively low sensitivity compared to other
techniques, especially when parasitaemia is low and in areas of unstable, low
transmission (John et al. 2005; Coleman et al. 2006; Menge et al. 2008). Whilst the
availability of microscopy has been shown to reduce drug use in trial settings, in
practice blood film results are often disregarded by clinicians as presumptive clinical
diagnosis is customary and a cost saving approach in malaria endemic areas (Jonkman
1995; Barat et al. 1999; PNGDOH 2000).
Alternative diagnostic methods include rapid dipstick immunochromatographic assays
that detect species-specific parasite antigens targeting either histidine-rich protein-2
(HRP-2) by, for example, the Parasight®F test or Plasmodium lactate dehydrogenase
(pLDH) by the OptiMAL® test (Schiff et al. 1993; Makler et al. 1998). However,
persistence of the HRP-2 antigen in the circulation after parasite clearance can give rise
to false positives (Makler et al. 1998). The first generation of OptiMAL® tests were
prone to loss of sensitivity caused by humidity and high temperature. However, this
instability has been overcome in the second generation OptiMAL IT® tests (Moody et
al. 2002). Occasional cases of false-positive results for P. falciparum were observed in
samples containing high levels of heterophile antibodies (Moody et al. 2002). One field
evaluation of the OptiMAL® test showed 96% sensitivity, 100% specificity and with
100% and 97.5% positive and negative predictive values respectively compared to
conventional microscopy. However sensitivity decreased when parasitaemia was <300
parasites/µL whole blood (Zerpa et al. 2008). Although dipsticks tests may enhance the
speed of diagnosis, microscopy remains the most cost-effective diagnostic method in
malaria endemic areas.
The use of polymerase chain reaction (PCR) based assays for the detection of species-
specific Plasmodium genome is highly specific and sensitive, with a detection limit as

Chapter 1 General Introduction
13
low as 1 parasite/µL (Hanscheid et al. 2002). However, in addition to the cost of
reagents, this method requires a stable electricity supply and sophisticated equipment
that is often not available in field settings. PCR methods are more useful in
epidemiological studies and for the diagnosis of imported malaria in developed
countries (Berry et al. 2008; Menge et al. 2008).
1.7 TRANSMISSION
Malaria parasites are transmitted through the bite of infected female Anopheles
mosquitoes (Figure 1.4). On taking a blood meal as a requirement by the mosquito for
ovulation, malarial sporozoites harboured in the salivary glands of the insect are
inoculated into the capillary of the human host. At the same time, gametocytes from the
human host can be transferred into the mosquito to continue the sexual phase of its life
cycle (Section 1.3) (Garnham 1988). In this way, infected populations of both human
and Anopheles mosquitoes function as reservoirs for each other. Transmission can also
occur congenitally by transplacental transfer of infected RBC into the neonate, and
malaria in pregnancy contributes to a significant number of maternal and infant deaths
(Malhotra et al. 2006; Brabin 2007).
Figure 1.4 Anopheles albimanus taking a blood meal. (Cook et al. 2006)

Chapter 1 General Introduction
14
1.8 PREVENTION
In malaria endemic areas, the use of insect repellent and clothing that provides covering
are important measures to reduce the chance of mosquito biting. Anopheles generally
feeds in the evening, hence staying indoors and sleeping within insecticide-treated bed
nets are effective night-time preventive measures. The use of chemoprophylaxis is also
recommended for travellers to malaria endemic areas. Other community based
interventions include indoor residual spraying of insecticides, reducing mosquito
breeding sites such as stagnant water and improving drainage. Significant challenges in
mosquito control include increasing resistance to key insecticides such as DDT and
pyrethroids with the development of new pesticides proving costly and time consuming
(WHO 2009).
1.9 TREATMENT
The best treatment currently recommended for P. falciparum infection is artemisinin-
based combination therapy (ACT). Examples of ACTs include artemether-lumefantrine
(Coartem®), artesunate-SP, dihydroartemisinin-piperaquine (Duo-Cotecxin®) and
artemisinin-naphthoquine (ARCO®). However, the spread of parasite resistance is
undermining malaria control efforts and there are no effective alternatives to
artemisinins currently on the market or near completion of the drug development
process (WHO 2009). Recent clinical and molecular studies from the Cambodia-
Thailand border where artesunate-mefloquine is the standard treatment regimen suggest
the emergence of ACT-resistant parasite strains (Denis et al. 2006; Vijaykadga et al.
2006; Alker et al. 2007). The reported treatment failures were considered most likely
due to the high level of mefloquine resistance rather than to the artemisinin component,
as mefloquine was used for monotherapy long before the introduction of ACTs.
Molecular findings of increased pfmdr1 copy number in local P. falciparum isolates
attested to mefloquine resistant in conjunction with the loss of sensitivity in vivo (Price
et al. 2004; Wongsrichanalai and Meshnick 2008; Chaijaroenkul et al. 2010). However,
recent studies have also suggested that initial parasite clearance, the hallmark of
artemisinin efficacy, is delayed in this area. This suggests that resistance to this
valuable group of antimalarial drugs has now started to develop (Dondorp et al. 2010).

Chapter 1 General Introduction
15
1.10 MALARIA IN WESTERN PACIFIC
Ten of the thirty-seven Western Pacific countries are malaria endemic; China,
Philippines, Cambodia, Korea, Malaysia, Laos Peoples Democratic Republic, Viet
Nam, PNG, Solomon Islands and Vanuatu, accounting for 340,000 cases per year.
Recent statistics showed a substantial reduction in malaria cases and mortality from
760,000 cases in 1990 to half this figure in 2005. However, malaria still claims many
lives in the Pacific region (Figure 1.5). PNG has the highest mortality within the Pacific
region where the up-scaling of vector control has been sluggish (WHO and WPRO
2007).
Figure 1.5 Malaria mortality in Western Pacific. Malaria mortality rate (per 100,000
population) in selected countries in the Western Pacific Region, 1994-2007 (WHO
2009).

Chapter 1 General Introduction
16
1.11 EMERGENCE OF ANTIMALARIAL RESISTANCE IN
PAPUA NEW GUINEA
Similar to the situation in Africa, young children in PNG carry the major disease burden
due to their lack of immunity (Michon et al. 2007). In the 1970s, reports from a
missionary station in Milne Bay (Figure 1.6) described cases of P. falciparum infection
that apparently did not respond to chloroquine (CQ) treatment. A cascade of in vivo
trials soon followed (Saint-Yves 1971; Han et al. 1976). Investigations conducted in
Maprik and Popondetta reported on the retained effectiveness of CQ. These studies
however, encountered immense difficulties with patient recruitment as only a small
number (22 of 1000) of children screened were eligible, all of whom were aged 7 to 12
years old, and thus probably semi-immune. The early Maprik and Popondetta findings
may not therefore, have reflected true resistance, especially when younger children are
found to harbour resistant parasites (Michon et al. 2007).
Figure 1.6 Regions and provinces of Papua New Guinea. ENB = East New Britain,
WNB = West New Britain , EH = Eastern Highlands, SH = Southern Highlands, WH =
Western Highlands.

Chapter 1 General Introduction
17
Not long after the initial alert from Milne Bay, two cases of CQ treatment failure in
non-immune expatriates were confirmed in Port Moresby, the capital city of PNG. Both
patients had travelled to the Kiunga area of the Western Province. Six patients from the
same region who used CQ as a prophylaxis also suffered malaria that did not respond to
standard treatment (Grimmond et al. 1976). Subsequent studies confirmed the presence
of CQ-resistant P. falciparum particularly along the border with Indonesia and nearby
provinces in the Northern coast of PNG (Han 1978). The appearance of CQ-resistant P.
falciparum coincided with the decreased effectiveness of residual insecticide spraying
initiated in the 1950s which further exacerbated the spread of resistant parasites
(Colbourne et al. 1970). Three decades on, resistance to other 4-aminoquinolines
particularly amodiaquine (AQ) and quinine have been reported under vigilant in vivo
and in vitro surveillance (Schuurkamp and Kereu 1989; Trenholme et al. 1993; al-
Yaman et al. 1996; Marfurt et al. 2007). The rapid spread of resistant P. falciparum
across the provinces of PNG has been documented in clinical (Darlow et al. 1982;
Sapak et al. 1991; Genton et al. 2005; Marfurt et al. 2007) and in vitro studies
(summarised in Chapter 3).
In addition to a long history of monotherapy, mass dosing of pyrimethamine during the
era of malaria control/eradication has ensured constant drug pressure on the parasite
population thus selecting for resistance. A high level of resistance has been documented
in numerous in vivo (Schuurkamp and Kereu 1989; Karunajeewa et al. 2008; Darlow et
al. 1980) and in vitro studies (Reeder et al. 1996; Hombhanje 1998). In 2000, PNG
health authorities replaced CQ monotherapy with sulfadoxine-pyrimethamine (SP)
combination therapy as first-line treatment to improve clinical efficacy (Nsanzabana et
al. 2010) and to delay the development of drug resistance (Casey et al. 2004; Genton et
al. 2005). The year 2011 marked the beginning of another era in antimalarial drug usage
as PNG implemented artemether-lumefantrine as first-line treatment (Mueller, 2010,
personal communication).

Chapter 1 General Introduction
18
1.12 ANTIMALARIAL CHEMOTHERAPY
Quinine and artemisinin derivatives are, respectively, the most employed and most
active antimalarial compounds, both of which have a long history as ancient herbal
therapies. The famous sweet wormwood (Artemisia Annua) also known as Qinghaosu
(Figure 1.7) was recorded as early as 200 BC in a Chinese medical treatise; the “Fifty-
two Prescriptions” discovered in the Mawangdui Tomb of the Hunan Province. The
antimalarial application of Qinghaosu was also documented in "The Handbook of
Prescriptions for Emergencies" by Ge Hong in the 4th century. In other parts of the
world, Cinchona tree bark had been used by indigenous South American tribes for its
antipyretic properties. The use of Cinchona bark (Figure 1.7) for treating fever was
subsequently popularised by the 17th century Spanish Jesuit missionaries (Lee 2002).
Both natural sources served as prototypes of the potent antimalarials available today.
Despite tremendous resources devoted to the search for antimalarial drugs, only a
handful of compounds have been discovered. Despite extensive research, our
understanding of the molecular basis of antimalarial activity remains incomplete. The
following section outlines the history of current antimalarials, their proposed
mechanism of action and the development of drug resistance.
Figure 1.7 Artemisia and Cinchona. Artemisia annua (left) (NomadRSI 2002).
Cinchona bark (right) (KEW 2007).

Chapter 1 General Introduction
19
1.12.1 Quinoline Related Compounds
1.12.1.1 Quinine
Figure 1.8 Chemical structure of quinine. (O'Neill et al. 2006)
Quinine (Figure 1.8), derived from the bark of the Cinchona tree, was first employed as
an effective malaria treatment in South America in the 17th century. This alkaloid
compound was isolated in the early 1800s by Portuguese chemists and its synthesis was
described in 1944 (Turner et al. 1953). Under political and military influences,
Cinchona plantations outside Peru were established. The demand for quinine intensified
with the advent of World Wars I and II, as supplies were inadequate and unstable. The
associated neurotoxicity and other side-effects have provided an impetus to research
synthetic alternatives. The availability of better tolerated drugs such as CQ has reduced
its usage and slowed the development of widespread resistance to this drug
(Wongsrichanalai et al. 1992b).
Quinine is by far the most commonly used treatment for severe malaria in the world,
being the standard treatment for severe disease across Africa and other malaria endemic
regions (WHO 2000; Sinclair et al. 2011), however recent findings suggest that this is
likely to change in favour of artesunate (Checkley and Whitty, 2007; Dondorp et al.
2005; Dondorp and Day, 2007; Dondorp et al. 2010). Quinine is used with SP as a
second-line regimen for uncomplicated malaria and for the treatment of severe malaria
in PNG (Genton et al. 2005). Quinine has remained increasingly important for the
treatment of CQ-resistant P. falciparum in other parts of the world, particularly for

Chapter 1 General Introduction
20
treatment during pregnancy (SANDH 2007; Chico et al. 2010).
1.12.1.2 Chloroquine
Figure 1.9 Chemical structure of chloroquine. (O'Neill et al. 2006)
Originally known as Resochin, CQ (Figure 1.9) was first synthesised in Germany and
thought to be too toxic for further development. After World War II, re-evaluation
proved its clinical safety, high efficacy and low toxicity (Loeb et al. 1946). CQ has
since been used extensively, and has been the drug of choice for uncomplicated malaria
and chemoprophylaxis. In 1950s to 1960s, the WHO devised population-based dosing
regimens for the Global Eradication Programme in which over 84,000 tons of CQ was
supplied to Brazil, parts of Africa and Asia for incorporation in table salt as means of
chemoprophylaxis (Greenwood et al. 2008). Resistant P. falciparum emerged rapidly
and has since spread to many endemic areas of the world, compromising its usefulness
(Peters 1987; Fidock et al. 2004). In spite of this, CQ is still employed as first-line
treatment in PNG in combination with SP for the treatment of uncomplicated malaria
during pregnancy (Casey et al. 2004; Mueller et al. 2008) despite increasing evidence
for the loss of effectiveness (Marfurt et al. 2007; Karunajeewa et al. 2008). Although
generally perceived to have “timed out”, CQ remains effective in some parts of the
world. A study has shown that P. falciparum isolates from Malawi have regained their
sensitivity to CQ a decade after its withdrawal (Laufer et al. 2006; Nkhoma et al. 2007).
A similar trend to reduction in resistance to CQ was also evident in Kilifi, a coastal
town in Kenya, following its official withdrawal, although at a much slower rate (Mwai
et al. 2009a). Recent microsatellite analyses of resurgent CQ-susceptible parasites
revealed they are likely to be a re-expansion of pre-existing CQ-sensitive population,

Chapter 1 General Introduction
21
rather than a reverse mutation in a previously resistant parasite or a new selective sweep
(Laufer et al. 2010).
Extensive research has been devoted into elucidating CQ’s mechanism of action. Early
studies suggested the inhibition of DNA replication and RNA synthesis, albeit at rather
high concentrations (Allison et al. 1965; Thelu et al. 1994). Subsequent characterisation
of enzymes involved in DNA replication proved that they are unlikely targets of CQ
(Chavalitshewinkoon et al. 1993; White et al. 1993). To date, it is widely accepted that
CQ acts within the parasite food vacuole and exerts its antiplasmodial effect by
interfering with detoxification (Pagola et al. 2000; Ursos et al. 2002; Fidock et al.
2004). In uninfected RBC, the uptake of CQ is minimal (Macomber et al. 1966). In
parasitised RBC, its accumulation is several thousand-fold higher, localising within the
parasite food vacuole perhaps reflecting ion-trapping due to its dibasic nature
(Macomber et al. 1966; Aikawa 1972; Ginsburg et al. 1989; Hawley et al. 1996). As
the parasite digests haemoglobin, free haem (ferriprotoporphyrin IX) and reactive
oxygen species are produced as toxic by-products. To counter this, the parasite
polymerises the toxic haem into non-toxic haemazoin crystals (Figure 2.2) (Blauer et al.
1997). CQ acts by binding to these free haem moieties, thus interfering with
detoxification where toxic by-products accumulate, leading to parasite death (Fitch
1998; Foley et al. 1998; Pagola et al. 2000; Fidock et al. 2004).
1.12.1.3 Amodiaquine
Figure 1.10 Chemical structure of amodiaquine. (O'Neill et al. 2006)

Chapter 1 General Introduction
22
During World War II, the US government invested in a massive screening program
involving 300,000 compounds, and in 1955, AQ (Figure 1.10) was identified as one of
three compounds that were active against P. falciparum (Peters 1987).
AQ served as an alternative prophylaxis for falciparum malaria for over 40 years (Foley
et al. 1998). However, support for its prophylactic use was withdrawn by the WHO
after reports of fatal adverse reactions in the mid 1980s, and associated high incidence
of hepatitis and agranulocytosis (Greenwood 1995). However, the rapid spread of CQ
resistance has prompted the re-evaluation of AQ for therapeutic use. A subsequent
systematic review of AQ treatment in uncomplicated malaria found the rates of adverse
events were no higher than that of CQ and SP treatments in controlled trials, without
life-threatening events (Olliaro et al. 1996). In vitro studies have shown AQ to be more
potent than CQ against less susceptible strains of P. falciparum (Rieckmann 1971;
Siddiqui et al. 1972). Despite this, clinical resistance to AQ soon followed and AQ
monotherapy is not recommended (Al-Yaman et al. 1996). AQ has found its use as
first-line treatment and as seasonal intermittent preventative treatment for malaria as
part of ACT regimens (Adjei et al. 2008; Sokhna et al. 2008). AQ (Camoquine) has
been used as first-line treatment with SP, but more recently in combination with
artesunate for young children in PNG and Africa (PNGDOH 2000; Brasseur 2007;
Marfurt et al. 2007).
AQ and its active metabolite monodesethyl-AQ share structural similarity with CQ and
partial cross resistance has been reported (Basco and Ringwald 2003; Wong et al 2010).
AQ competitively inhibits CQ accumulation in the parasitic food vacuole (Fitch 1973).
As a diprotic weak base with lower pKa values compared to CQ, it may accumulate less
efficiently in the parasite food vacuole via ion-trapping. However, AQ is found in
higher concentrations than CQ in the parasite, indicative of enhanced uptake by an
additional pathway (Hawley et al. 1996). Several studies have demonstrated the ability
of AQ to bind to haem and inhibit haem polymerisation, analogous to CQ (Chou et al.
1993).

Chapter 1 General Introduction
23
1.12.1.4 Mefloquine
Figure 1.11 Chemical structure of mefloquine. (O’Neill et al. 2006)
Mefloquine (MQ) (Figure 1.11) was also selected from a screening program and
developed through global collaborative efforts. A distinguishing property of MQ is its
long terminal elimination half-life of 2 to 3 weeks, hence it can achieve clinical efficacy
with a single dose. It is well tolerated by adults and children. Common side-effects
include nausea, dizziness, headache, rash, pruritis, in some instances bradycardia and
the well publicised psychiatric disturbances (Palmer et al. 1993). It was first deployed
in Thailand in 1984, initially in combination with SP, but resistance developed
relatively quickly. It has been more recently incorporated with artesunate as part of
ACT. The relative high cost of MQ in addition to concerns regarding toxicity limits its
use in many resource-poor malaria endemic countries.

Chapter 1 General Introduction
24
1.12.1.5 Lumefantrine
Figure 1.12 Chemical structure of lumefantrine. (Basco et al. 1998)
Formerly known as benflumetol, lumefantrine is a 2, 3 benzindene that belongs to the
amino-alcohol class that also includes quinine, mefloquine and halofantrine (WHO
2006) (Figure 1.12). The compound was first synthesised in China and has high clinical
efficacy against multidrug resistant P. falciparum as a co-formulation with artemether
(Coartem®) (WHO 2006; Karunajeewa et al. 2008b). The oral formulation is
efficacious for the treatment of children with uncomplicated malaria and drug-related
adverse events are rare (Adjei et al. 2008; Karunajeewa et al. 2008b). Bioavailability of
lumefantrine is variable and its absorption is significantly increased when administrated
with fatty foods (Ezzet et al. 1998; White et al. 1999; Borrmann et al. 2010).
Lumefantrine-artemether is the current first-line treatment for uncomplicated malaria in
Africa (Kabanywanyi et al. 2007; Borrmann et al. 2010) and PNG (WHO 2007;
Schoepflin et al. 2010).

Chapter 1 General Introduction
25
1.12.1.6 Naphthoquine
Figure 1.13 Chemical Structure of Naphthoquine. (Wang et al. 2004)
Naphthoquine (NQ) (Figure 1.13) is a relatively new drug first registered in 1993 in
China. This drug exhibits potent schizontocidal effects in CQ-resistant parasites.
Clinical studies of NQ monotherapy carried out on Hainan Island, China reported 100%
and 96.7% cure rate that compared favourably with artemisinin monotherapy which had
a cure rate of 73.3% (Pang et al. 1999; Guo et al. 2000). No distinctive adverse
reactions have been associated with NQ use, although transient abdominal discomfort
has been reported in some patients. Elimination half-life of NQ ranges from 41 hr to
255 hr (Wang et al. 2004; Qu et al. 2010). The long half-life of NQ makes it a suitable
partner with artemisinin in the relatively new ARCOTM therapy that is undergoing
safety and efficacy trials in PNG (Hombhanje et al. 2009; Davis, unpublished). Parasite
susceptibility data outside of Chinese literature are limited, with only one study
performed in PNG field isolates (Wong et al. 2010), the findings of which will be
discussed in subsequent chapters.

Chapter 1 General Introduction
26
1.12.1.7 Piperaquine
Figure 1.14 Chemical Structure of Piperaquine. (Davis et al. 2005)
Piperaquine (PQ) (Figure 1.14) is a bisquinoline compound first synthesised from
screening programs in France and China during the 1960s. It was thought to offer little
advantage over CQ and hence development was not pursued except in China. PQ was
developed for clinical use and was the drug of choice in China and Indochina for two
decades until the emergence of resistance (Chen 1991; Fan et al. 1998). In the 1990s,
renewed interest has seen PQ as a long half-life partner in ACTs. Examples include
CV8® (PQ-dihydroartemisinin-trimethoprim-primaquine), Artecom® (PQ-primaquine)
and more recently Artekin® or Duo-Cotecxin® (PQ-dihydroartemisinin), the latter
producing high cure rates and tolerability in areas of CQ-resistant P. falciparum (WHO
2006; Karunajeewa et al. 2008b). A recent in vitro study from Cameroon has also
shown PQ to be highly active against both CQ sensitive and resistant strains of P.
falciparum isolates (Basco and Ringwald 2003) with a long elimination half-life of 33
days (Tarning et al. 2005).
1.12.2 Antifolate Combination Drugs
Figure 1.15 Chemical structures of sulfadoxine (left) and pyrimethamine (right).
(WHO 2006)

Chapter 1 General Introduction
27
Antifolate combinations comprise of inhibitors of Plasmodium dihydrofolate reductase
(dhfr) and dihydropteroate synthase (dhps), both of which are involved in parasite DNA
synthesis. Examples of pfdhfr inhibitors include pyrimethamine (Figure 1.15),
proguanil, chlorproguanil, and trimethoprim. Sulfa drugs that inhibit pfdhps include
sulfadoxine (Figure 1.15), dapsone, sulfalene and sulfamethoxazole. Genetic mutations
in parasite pfdhfr and pfdhps genes confer rapid resistance against these drugs,
particularly when used alone. When used in combination however, these drugs produce
a synergistic effect where they are effective even in the presence of parasite resistance
to individual components. Common antifolate combinations include SP, which is useful
in pregnant women for placental malaria, sulfamethoxazole-trimethoprim (co-
trimoxazole), sulfalene-pyrimethamine (metakelfin) and a more recent combination of
dapsone-chlorproguanil (LapDap) that is more affordable and provides higher cure rates
(Watkins et al. 1997) but which has now been withdrawn because of adverse effects
(haemolytic anaemia) in patients with glucose-6-phosphate dehydrogenase deficiency
(Bukirwa et al. 2004; Fanello et al. 2008).
1.12.3 Artemisinin and its Derivatives
Figure 1.16 Chemical structure of artemisinin (left) and its derivatives (right).
Artemether (R = OCH3); Artesunate (R = OCH2CH2CO2Na); Dihydroartemisinin (R =
OH).
Artemisinin and its derivatives (Figure 1.16) are frontline antimalarial treatments that
facilitate rapid parasitaemia clearance and prompt resolution of symptoms. Artemisinin
is a sesquiterpine lactone isolated from the plant Artemisia annua or Qinghaosu (Figure
1.7) which has been used for thousands of years by Chinese herbalists for curing febrile

Chapter 1 General Introduction
28
illnesses. Qinghaosu was re-discovered in 1972 from a mass screening project of
traditional medicinal herbs funded by the Chinese military (Tu You-you 1982; Zhang
2007). Artemisinin and its derivatives are the most potent antimalarials to date,
characterised by a rapid clearance of parasitaemia (Skinner et al. 1996). These have
proven to be efficacious and well tolerated as monotherapy although these are not
recommended to be used alone due to their short half-lives with associated high rates of
recrudescence related to rapid elimination. Examples of semi-synthetic artemisinin
derivatives include artemether and artesunate.
Since artemisinin and its derivatives are rapidly eliminated from the body and long
courses are required for high long-term cure rates, greater efficacy can be obtained by
partnering these potent but short-lived compounds with those with a longer half-life
such as in lumefantrine, MQ or AQ as ACTs. These strategies are now recommended in
Africa and PNG to delay the development of resistance (WHO 2006; Ekland et al.
2008; O'Neill et al. 2010). These regimens include artemisinin-NQ (Wang et al. 2004),
artesunate-AQ and artemether-lumefantrine (Adjei et al. 2008).
Dihydroartemisinin (DHA) is an active metabolite of artemether and artesunate. In
comparison to other derivatives, in vitro studies have shown DHA to act on all stages of
intra-erythrocytic P. falciparum, and is the most potent in achieving 100% inhibition
within 2 to 4 hr of exposure (Skinner et al. 1996).
1.12.4 Antibiotics
Several antibiotics are currently in use as prophylaxis and in combinations with other
antimalarials as treatment for uncomplicated P. falciparum infections (Nakornchai et al.
2006; Ejaz et al. 2007; Noedl et al. 2007). However, the exact mechanism and cellular
effects of these parasiticidal anti-bacterial drugs remain largely unknown. As with other
apicomplexa parasites such as Babesia spp, Toxoplasma gondii and Cyclospora
cayetanensis, Plasmodium species have an apicoplast similar to the plastid structure
found in algae and bacteria. This organelle is non-photosynthetic but contains essential
house-keeping machineries for DNA replication, transcription, translation and post-
translational modification, and various anabolic pathways (Goodman et al. 2007).

Chapter 1 General Introduction
29
Recent identification and characterisation of this unique apicomplexan structure has
revealed a number of putative targets with the potential to help explain the mysterious
activities of antibiotics against malaria (McFadden et al. 1996; Wilson et al. 1996;
Goodman et al. 2007b). The absence of a similar structure in mammalian cells renders
the Plasmodium apicoplast a valuable drug target.
A number of antibiotics have been shown to target the prokaryotic 70S ribosome in
Plasmodium apicoplast thus affecting protein synthesis (Figure 1.17). Examples include
azithromycin (AZ), clindamycin, tetracycline, doxycycline and thiostrepton. The
macrolides (AZ, clindamycin) block the peptide exit tunnel, preventing peptide
elongation, lincosamides (clindamycin) and tetracyclines interfere with the
translocation and binding of peptidyl tRNAs to the acceptor site, respectively
(Auerbach et al. 2002).
Figure 1.17 Targets of anti-bacterial drugs in P. falciparum apicoplast. Anti-
bacterial drugs targeting the “house-keeping” functions of the P. falciparum apicoplast.
(Modified and adapted from Goodman and McFadden, 2007)
An interesting phenomenon with antibiotics observed in apicomplexan parasite is the
delayed-death effect, where the drug has no apparent effect on the parasite as it
continues to grow and divide and its daughter cells re-invade a new host. It is not until
the second generation where the progenies fail to develop and eventually die (Fichera et

Chapter 1 General Introduction
30
al. 1995; Fichera et al. 1997). Various studies have shown the significant reduction of
IC50 values when in vitro antibiotic drug trials against P. falciparum were extended
from 48 hr to 96 hr (Yeo et al. 1995; Goodman et al. 2007b). It was hypothesised that
drugs targeting prokaryote-like translation, as those mentioned previously, would
produce this effect. Antibiotics that target DNA replication, transcription and interfere
with fatty acid and isoprenoid pathways such as fosmidomycin and triclosan, cause
immediate parasite death within the first intra-erythrocytic cycle (Jomaa et al. 1999;
Goodman et al. 2007a; Wiesner et al. 2008).
1.12.4.1 Azithromycin
Figure 1.18 Chemical Structure of Azithromycin.
Azithromycin (AZ) (Figure 1.18) is a broad spectrum macrolide derived from
erythromycin with more favourable pharmacokinetics and longer elimination half-life
(Girard et al. 1987). It is used clinically for the treatment of streptococcal,
staphylococcal, chlamydial and gonorrhoeal infections. Since the discovery of AZ’s
activity against CQ-resistant P. falciparum (Gingras et al. 1992; Gingras et al. 1993),
this drug has been studied with renewed interest in the campaign against multidrug
resistant malaria. Only recently has its mode of action been elucidated. According to
this report, AZ binds to the 50S ribosomal subunit in the apicoplast translation
machinery, interfering with protein synthesis and subsequently leads to a classic
delayed-death (Sidhu et al. 2007).
Despite its slow onset and considerably lower potency compared to other antimalarials
(Noedl et al. 2001; Noedl et al. 2007; Wong et al. 2010), the safety profile in young

Chapter 1 General Introduction
31
children and extensive experience with its use during pregnancy (Bace et al. 1999;
Arrieta et al. 2003; Sarkar et al. 2006) render it an attractive candidate to partner with
fast-acting conventional antimalarials. An earlier study with AZ-artesunate combination
in Thai adults with uncomplicated falciparum malaria resulted in an unexpectedly low
cure rate (Na-Bangchang et al. 1996). This was likely due to inadequate AZ dosage.
With a higher dose, a phase 2 clinical trial demonstrated the safe and efficacious use of
AZ-quinine and AZ-artesunate (Noedl et al. 2006).
1.12.5 Summary of Antimalarial Activities
The following table provides a list of common antimalarial drugs and their specific
activities (Table 1.1).

Chapter 1 General Introduction
32
Table 1.1 Antimalarial drugs and their specific activities. (WHO 2006)
Drug Class Compound Specific Activities 4-Aminoquinoline Chloroquine
Amodiaquine Naphthoquine
Blood schizontocides Interference of haem detoxification in parasite
food vacuole
8-Aminoquinoline Primaquine Tafenoquine
Intra-hepatic schizontocides Gametocytocides
Bis-quinoline Piperaquine Blood schizontocides
Arylaminoalcohol Quinine/Quinidine Mefloquine Halofantrine Lumefantrine
Mature trophozoites Blood schizontocides
Early stage gametocytocides
Antifolates Sulfadoxine Sulfamethoxazole
Dapsone Pyrimethamine
Proguanil Chlorproguanil Trimethoprim
Inhibitors of dihydropteroate synthase Inhibitors of dihydrofolate reductase
Slow-acting blood schizontocide Sporontocide
Possible activity against intra-hepatic forms
Artemisinin Artemisinin Artesunate
Dihydroartemisinin Arteether
Artemether
Fast-acting blood schizontocides Gametocytocides,
Inhibitors of calcium adenosine triphosphatase
Antibiotics Azithromycin Clindamycin Doxycycline Tetracycline
Ciprofloxacin Rifampicin
Thiostrepton
Slow-acting blood schizontocides Inhibitors of aminoacyl-tRNA binding during
protein synthesis
Naphthoquinones Atorvaquone Inhibits intra-hepatic forms Inhibits oocyte development in the mosquito
Interferes cytochrome electron transport

Chapter 1 General Introduction
33
1.13 LIPID-LOWERING AGENTS AS ANTIMALARIALS
Recent interest in alternative antimalarial drugs led to the discovery of P-glycoprotein,
an efflux transporter encoded by the multidrug resistant gene which actively removes
compounds from cells and prevents intracellular drug accumulation. Interestingly,
certain statins and fibrates have been shown to interact with P-glycoprotein in
mammalian cells expressing multidrug resistance (Wu et al. 2000; Ehrhardt et al.
2004). Although a P-glycoprotein homologue 1 has been identified in P. falciparum,
studies investigating the antimalarial activity of these lipid-lowering agents are limited.
1.13.1 Statins
Statins are cholesterol-lowering drugs that are considered first-line therapies for
reducing morbidity and mortality associated with atherosclerotic disease (Grundy et al.
2004; Wilt et al. 2004; Thavendiranathan et al. 2006). They are inhibitors of 3-hydroxy-
methyl-glutaryl coenzyme A (HMG-coA) reductase that decreases cholesterol synthesis
and plasma cholesterol and lipoprotein levels (Figure 1.19) (Maron et al. 2000).
Examples include atorvastatin (Lipitor®), rosuvastatin (Crestor®), pravastatin
(Pravacol®) and simvastatin (Zocor®). Despite a common mechanism of action, statins
differ in their chemical structures, pharmacokinetic profiles and lipid-modifying
efficacy. Pravastatin and simvastatin are derived from fungal metabolites and have
elimination half-lives of 1 to 3 hr. Atorvastatin and rosuvastatin are both synthetic
compounds, with elimination half-lives of 14 hr and 19 hr respectively. Rosuvastatin
(Crestor®) is a recently developed drug with the most potent LDL-cholesterol-lowering
properties, followed by atorvastatin, simvastatin and pravastatin (Soran et al. 2008).
In addition to their cholesterol-lowering efficacy and good clinical safety profile (Cilla
et al. 1996; Bellosta 2004), statins exhibit a wide range of antimicrobial activities
against bacteria, yeasts and protozoan including P. falciparum (Montalvetti 2000; Song
et al. 2003; Catron et al. 2004; Pradines et al. 2007; Wong et al. 2009). Atorvastatin
and lovastatin reduced the growth of Salmonella enterica in mouse model and in
cultured macrophages (Catron et al. 2004). They acted synergistically with fluconazole
to inhibit the eukaryotic sterol pathway, as demonstrated by the reduced growth of

Chapter 1 General Introduction
34
Candida albicans after exposure (Song et al. 2003). In addition, they reduced the
growth of Schistosoma mansoni, Trypanosoma cruzi and Leishmania species through
inhibition of 3HMG-CoA reductase (Chen et al. 1990; Urbina 1993; Andersson et al.
1996; Montalvetti 2000).
Figure 1.19 Overview of isoprenoid biosynthesis. In humans, the biosynthesis of
isopenteny-diphosphate and dimethylallyl-diphosphate and ultimately cholesterol
proceeds entirely via the mevanlonate pathway. In Plasmodium, these isoprenoid
building blocks are supplied by the 1-deoxy-D-xylulose-5-phosphate/2-C-methyl-
erythritol 4-phosphate (DOXP/MEP) pathway. Statins inhibit the mevalonate pathway
as indicated (Moreno et al. 2008).

Chapter 1 General Introduction
35
The antimalarial effects of statins have also been described. Lovastatin and simvastatin
inhibited intra-erythrocytic development of P. falciparum in vitro (Grellier et al. 1994).
In other recent studies, atorvastatin exhibited high in vitro activity against P. falciparum
(Pradines et al. 2007; Parquet et al. 2009; Wong et al. 2009). Despite these encouraging
findings, neither simvastatin nor atorvastatin given in high doses improved outcome in
P. berghei-infected mice (Bienvenu et al. 2008; Kobbe et al. 2008) and there was no
effect on parasitaemia (Bienvenu et al. 2008). No study to date has included the most
potent statin in clinical use, rosuvastatin (Soran et al. 2008). In addition, despite
demonstration of in vitro synergy between mevastatin and the P-glycoprotein inhibitor
tunicamycin against P. falciparum (Naik 2001), the interaction between statins and
conventional antimalarial drugs has not been assessed.
1.13.2 Fibrates
While statins decrease cholesterol synthesis, fibrates primarily intensify catabolism of
triglyceride-rich lipoproteins and increase HDL-cholesterol. Gemfibrozil, fenofibrate,
clofibrate are examples of fibrates in clinical use. The fibrates are agonists of
peroxisome proliferator-activated receptor alpha (PPARα). Fenofibrate has also been
shown to inhibit P-glycoprotein mediated transport with a similar potency to
simvastatin (Ehrhardt et al. 2004) and modulate the expression of the lipid-efflux
protein, ATP-binding cassette sub-family A member (ABC-1) (Arakawa et al. 2005).
If malaria pathology is attributable to systemic inflammation (Clark et al. 2008), the use
of fibrates to hinder the release of inflammatory cytokines may constitute a novel
treatment approach. A number of studies have investigated the anti-inflammatory
effects of fibrates. Gemfibrozil reduced TNF production in monocytes and down-
regulated the production of superoxide and expression of nuclear factor κB, both of
which regulate inflammation (Zhao et al. 2003; Calkin et al. 2006). Furthermore,
gemfibrozil and fenofibrate both inhibited TNF, IL-1 beta, IL-6 and nitric oxide
production (Xu et al. 2006). In a murine influenza model, where severe systemic
disease is thought to arise through overproduction of proinflammatory cytokines, Budd
et al. (2007) found gemfibrozil offered some protection against the virus, with an

Chapter 1 General Introduction
36
increased survival rate from 26% to 52% compared to vehicle-treated mice (Clark et al.
2004; Budd et al. 2007).
An early study exploring the effect of plasma free fatty acid concentrations and
temperature on parasitaemia in P. berghei infected mice used clofibrate as a lipid-
lowering agent. Coincidentally, the study reported on a significant retardation in the
development of parasitaemia in the clofibrate treatment group, suggesting a direct
inhibition on parasite growth (McQuistion 1979). A recent report highlighted the
potential use of PPAR agonists as a novel treatment for cerebral malaria (Balachandar
et al. 2011). However, no other study to date has investigated the effects of fibrates
against malaria.
The antimalarial activities of fibrates and statins, and their interactions with
conventional antimalarials will be explored in Chapter 6.
1.14 ANTIMALARIAL DRUG RESISTANCE
1.14.1 Definitions
The development of drug resistance has been a major obstacle in the battle against
malaria. The WHO defines antimalarial drug resistance as “the ability of parasite strains
to survive and/or multiply despite the administration and absorption of a drug given in
doses equal to or higher than those usually recommended but within tolerance of the
subject.” This was later modified in 1986 to specify that “the form of the drug active
against the parasite must be able to gain access to the parasite or the infected RBC for
the duration of the time necessary for its normal action” (Bruce-Chwatt 1986). This
refined definition takes into account the variations in drug pharmacodynamics and
pharmacokinetics between individuals, such as in the metabolism of sulfonamides and
sulfones (Trenholme 1975; WHO 2005). Drug resistance causes a right shift in the dose
response curve that can be observed in vitro (Figure 1.20).
Confirmation of drug resistance requires evidence of parasite recrudescence in a patient
(microscopically or by PCR) after receiving a treatment dose of antimalarial.

Chapter 1 General Introduction
37
Simultaneous demonstration of effective concentrations of the drug or its metabolite in
the blood using established laboratory methods such as HPLC is also required. Results
from in vitro drug susceptibility tests and detection of molecular markers associated
with drug resistance are additional indicators of resistance. In practice however, these
tests are seldom performed alongside in vivo studies. For this reason, inadequate
parasitological clearance is conventionally considered to be associated with resistance
(WHO 2005).
Figure 1.20 Right-ward shift of concentration-effect relationship due to drug
resistance. Changes in the concentration-effect relationship may occur as a parallel
shift (B) from the “normal” profile (A) or, the slope changes and/or a reduction in the
maximum parasite killing effect (C) (WHO treatment guideline 2006-Annex 6).
1.14.2 Treatment Failure and Drug Resistance
A distinction must be made between antimalarial drug resistance and treatment failure,
the latter of which is a failure to clear parasitaemia and/or resolve clinical symptoms
after treatment. While drug resistance may cause treatment failure, other factors may
contribute. These include incorrect dosage, patient non-compliance regarding duration
of dosing, poor drug quality, drug interactions, poor absorption or rapid elimination
(diarrhoea and vomiting), and misdiagnosis (WHO 2005). Many of these factors may
also accelerate the development and spread of drug resistance as parasites are exposed

Chapter 1 General Introduction
38
to suboptimal drug levels.
1.14.3 Emergence of Resistance to Principal Antimalarials
Despite an apparently replete list of antimalarials (Table 1.1), the emergence of P.
falciparum exhibiting resistance to multiple antimalarials has resulted in a gradual
degradation of the effectiveness of all available drugs. This along with the failure of
multifaceted approaches to eradicate and control the spread of malaria has seen
resurgence in the incidence of this life threatening infection on a global scale. For
comprehensive reviews on the evolution and selection of malarial drug resistance, see
(Plowe 2009) and (Mackinnon et al. 2010).
Resistance to CQ first observed in South-East Asia and South America about half a
century ago, now occurs almost everywhere that P. falciparum is transmitted. It is
important to recognise that the acquisition of resistance to quinine and CQ took many
years (278 and 12, respectively) compared with the rapid appearance of resistance to
proguanil, SP, MQ, and atovaquone, all within 5 years of their introduction
(Wongsrichanalai et al. 2002). Figure 1.21 illustrates the emergence of resistance to
principal antimalarials. Quinine, CQ and SP have remained effective for a considerable
length of time even after the initial reports of resistance. AQ was withdrawn by the
WHO in 1990 as denoted by the dotted line (Figure 1.21), but was re-introduced for
therapeutic usage after confirmation of its clinical safety and efficacy. Recent reports
from the Cambodia-Thailand border has indicated modest increase in P. falciparum
resistance to ACTs (artesunate-MQ) (Wongsrichanalai et al. 2008; Carrara et al. 2009;
Rogers et al. 2009).

Chapter 1 General Introduction
39
Figure 1.21 Emergence of resistance to principal antimalarial drugs. (Modified
and adapted from Ekland and Fidock 2008). Coloured bars represent antimalarial
regimen in combination and as monotherapy along with the year since its first
introduction and the first report of resistance. Rectangles signify the approximate time
when resistance has spread to various geographical regions. Timelines were derived
from (Al-Yaman et al. 1996; Wongsrichanalai et al. 2002; Ekland et al. 2008;
Wongsrichanalai et al. 2008) and references therein. ACTs = artemisinin-based
combination therapies; Atov/Prog = atovaquone/proguanil; AQ = amodiaquine; CQ =
chloroquine, Q = quinine; R = resistance; MQ = mefloquine; SP =
sulfadoxine/pyrimethamine; PNG = Papua New Guinea; E = East; S = South.
1.14.4 Determinants of Antimalarial Resistance
Numerous factors contribute to the advent, spread and intensification of malaria
resistance, although their relative contribution remains unknown. Gene mutations
conferring resistance occur naturally in low frequencies as parasite populations show

Chapter 1 General Introduction
40
heterogeneity. Therefore, selection of the most “fit” parasites occurs in the face of
chemotherapy, where parasites harbouring single or multiple point mutations that
provide survival advantage would recover and multiply (Wongsrichanalai et al. 2002;
Mackinnon et al. 2010). Other aspects attributing to resistance concern the dynamic
interplay of drug characteristics and usage, human behaviour and host factors, the
mosquito vector and environmental factors (Table 1.2).
Drug characteristics are important determinants of antimalarial resistance. Firstly, drugs
with a long elimination half-life, such as MQ and SP have the benefit of simpler, single-
dose regimen which can greatly improve compliance and dosing. However, the
subtherapeutic drug concentrations in plasma during slow elimination exert substantial
selection on parasites from new infections acquired after the initial treatment
(Wernsdorfer 1994; Bloland 2001). This is particularly common in areas with intense
malaria transmission (Wongsrichanalai et al. 2002).
Host factors such as immunity increase the efficacy of chemotherapy and may offset the
spread of resistance. A semi-immune individual may be cured by a drug despite partial
drug resistance of the infecting parasites. The specific immune response elicited by
repeated exposure to the pathogen is more effective in parasite clearance compared to
the non-specific response generated by those naïve to malaria. Introduction of resistant
parasites in migrant and refugee populations may thus increase the opportunity for
manifestation and spread of resistance (Wongsrichanalai et al. 2002).
Table 1.2 Determinants of antimalarial resistance. (Modified and adapted from
Wongsrichanalai et al. 2002*). pfcrt = Plasmodium falciparum chloroquine resistance
transporter; pfmdr1 = P. falciparum multidrug resistance 1; pfdhps, P. falciparum
dihydropteroate synthase; pfdhfr, P. falciparum dihydrofolate reductase; pfserca, P.
falciparum sarco-endoplasmic reticulum calcium ATPase6. Examples were obtained
from (Wernsdorfer et al. 1991** ; Jonkman 1995#; Molyneux et al. 1999§; PNGDOH
2000҂; Winstanley 2001^; Bloland 2001٭; Newton et al. 2008^^ ). (Wernsdorfer et al.
1991; Jonkman 1995; Molyneux et al. 1999; PNGDOH 2000; Winstanley 2001;
Newton et al. 2008).

Chapter 1 General Introduction
41
Factor and characteristic Example
Drug Half-life Resistance to chlorproguanil-dapsone (short half-life) develops more slowly than that to SP (long
half-life)^.
Dosing Use of subtherapeutic doses in self-treatment such as with antifolate drugs in Thailand in the 1970s; poor drug compliance; mass drug administration with subtherapeutic doses; use of chloroquinised
salt** .
Non-target drug pressure
Presumptive use of antimalarial drugs without laboratory diagnosis or for indications other than
malaria#,҂.
Pharmacokinetics Use of drug formulations with reduced bioavailability*.
Cross-resistance SP and sulfamethoxazole-trimethoprim*.
Quality Poor manufacturing practices with substandard content of active ingredients, intentional
counterfeiting, deterioration due to storage and handling^^.
Human Host immunity Non-immune, migrant gem-miners and resistance to mefloquine on the Thai-Cambodian border*.
Health Malnourished and HIV infected individuals have significantly poorer parasitological response٭.
Maintenance of resistant parasite
reservoir
Non-detection of drug failure*.
Parasite Genetic mutations Polymorphisms in the genes: pfcrt, pfmdr1, pfdhps, pfdhfr, pfserca*.
Transmission level
Whether low or high transmission has more influence on drug resistance is debatable;
prevalence of drug resistance is higher in regions of low transmission, whereas a model suggests the
benefits of transmission control in delaying resistance development§.
Vector and environment
Mosquito affinity of parasites
Increased infectivity and productivity of CQ-resistant parasites in Anopheles dirus and the
propagation of CQ resistance in South-East Asia and Western Oceania*.

Chapter 1 General Introduction
42
The interactions between parasite genetic polymorphisms have been identified to confer
or modulate antimalarial drug resistance. These involve genes that encode membrane
transporter proteins such as pfcrt and P-glycoprotein homologue 1 (pfmdr1) associated
with 4-aminoquinoline resistance. Mutations in the enzymes dihydrofolate reductase
and dihydropteroate synthase involved in folate synthesis, decreases sensitivity to
pyrimethamine and sulfadoxine respectively. The involvement of parasite genetics and
drug resistance are discussed in Section 1.18.
P. falciparum strains that already demonstrate resistance to a number of antimalarials
display a level of genetic plasticity that enables them to rapidly adapt to a new drug not
chemically related (Rathod et al. 1997; Nzila et al. 2010). Early reports using rodent
malaria have shown a strain resistant to one drug is more prone to give rise to resistant
lines to another drug, compared to strains that are fully susceptible (Powers et al. 1969;
Peters et al. 1976). Similar observations are evident during in vitro induction of
resistance, where the chance of generating parasite resistant lines increases with the
number of drug-resistant phenotypes. For instance, the ease of generating parasite
resistant line against atovaquone was greatest using the culture-adapted P. falciparum
strains W2 (cycloguanil, pyrimethamine and sulfadoxine resistant), followed by FCR3
(pyrimethamine and cycloguanil resistant), then 3D7 (sulfadoxine resistant) and the
least in the fully drug-susceptible D6 strain which failed to generate any drug resistant
parasite line (Rathod et al. 1997). This phenomenon is termed ‘accelerated resistance to
multidrug (ARMD)’, which is different to the known multidrug-resistant phenotype
(Rathod et al. 1997). The occurrence of ARMD may be due to low efficiency of DNA
repair mechanisms (Trotta et al. 2004), which can be attributed to the high mutation rate
during parasite multiplication. Hence, under drug pressure, parasites with the ARMD
phenotype have higher ability to produce a drug-resistant clone.
1.14.5 Mechanism of Resistance to 4-Aminoquinolines and Arylaminoalcohols
Much evidence attributes the activity of CQ to its capacity to concentrate itself from nM
concentrations in the extra-cellular environment to mM levels within the parasite food-
vacuole (Bray et al. 1998; Le Bras et al. 2003). Resistant parasites are found to
accumulate CQ less efficiently (Saliba et al. 1998). Verapamil is a modulator of P-

Chapter 1 General Introduction
43
glycoprotein in mammalian cells expressing multidrug resistance. The observation that
CQ resistance is reversible by verapamil has led to the discovery of an analogous efflux
protein in the food vacuole of P. falciparum. Mutations in the corresponding gene, the
pfmdr1 gene has been associated with in vivo CQ resistance which modulates in vitro
resistance (Foote et al. 1990; Reed et al. 2000; Nagesha et al. 2001). It also plays a
significant role in the parasite’s sensitivity to structurally related compounds such as
quinine and AQ. Genetic and field studies have linked parasite possession of the wild
type pfmdr1 gene and its amplification is associated with increased resistance to MQ,
lumefantrine, halofantrine and artemisinin (Price et al. 2004; Sidhu et al. 2006;
Chavchich et al. 2010). Interestingly, there is an inverse relationship between parasite
sensitivity to CQ and MQ (Duraisingh et al. 2005). Mutations in the pfcrt gene which
encodes for another transmembrane protein in the parasite digestive vacuole also confer
CQ resistance. Specific polymorphisms encoding for resistance in the pfmdr1 and pfcrt
genes are discussed in Section 1.18.
1.14.6 Mechanism of Resistance to Antifolates
Antifolate combination drugs such as SP act via sequential and synergistic inhibition of
two key enzymes involved in parasite folate synthesis. Dihydrofolate reductase and
dihydropteroate synthetase are encoded by the P. falciparum dihydrofolate reductase
(pfdhfr) and P. falciparum dihydropteroate synthase (pfdhps) genes respectively.
Mutation in pfdhfr reduces its affinity to pyrimethamine or related compounds, whereby
inhibition is attenuated (Le Bras et al. 2003). Similarly, mutation in the pfdhps is
associated with sulfadoxine resistance (Wang et al. 1997). Specific combinations of
mutations in both pfdhfr and pfdhps have been associated with varying degrees of
antifolate resistance; these are discussed in more detail in Section 1.18.
1.14.7 Mechanism of Resistance to Artemisinin and Derivatives
Although a number of putative targets have been proposed, the exact mechanisms of
action of artemisinin remain uncertain (O'Neill et al. 2010). Chavchich et al have
selected parasite lines that are resistant to artemisinin and artelinic acid under

Chapter 1 General Introduction
44
continuous drug pressure (Chavchich et al. 2010). The changes in parasite susceptibility
were accompanied by concomitant increase in pfmdr1 gene copy number and protein
expression. In additional to these molecular changes, the authors reported a reduction in
parasite sensitivity to MQ, quinine, lumefantrine and halofantrine, in concordance with
field observations (Sidhu et al. 2006). Besides pfmdr1 gene copy number, the
acquisition of artemisinin tolerance has been associated with parasite developmental
arrest and changes in transcriptomic modifications as a result of drug pressure
(Witkowski et al. 2010).
Studies of the cytotoxic effects of artemisinin suggested its mechanism of action may
be via interactions between the artemisinin endoperoxide bridge and haem-iron
(Kannan et al. 2005). Subsequent production of alkylated haem derivatives of
artemisinin (i.e. haemarts) has been proposed to cause parasite death due to its
interference with haemazoin formation as well as harmful effects due to free radicals
(Kannan et al. 2005). This hypothesis is consistent with studies demonstrating that
artemisinin activity can be enhanced by oxidising agents and attenuated by reducing
agents (Krungkrai et al. 1987). However, this theory has been challenged since
artemisinin is active against ring-stage parasites that do not harbour high concentrations
of haem (Olliaro et al. 2001). Wu et al proposed that artemisinin is activated on the
reductive cleavage of the peroxide bond by iron-sulfur redox centres common to
Plasmodium enzymes. As a result, alkylation of these enzymes may be responsible for
parasite death (Wu 2002a). This hypothesis is supported by the interactions between
radiolabelled artemisinin and various parasite proteins, highlighting the possibility that
parasite death may be due to endogenous protein alkylation and inactivation
(Bhisutthibhan et al. 2001). Some of the proposed target proteins for artemisinin
include those involved in the electron transport chain, cysteine protease, translationally
controlled tumour protein, and pfATP6 (Eckstein-Ludwig 2003; Li 2005). The latter is a
SERCA-type calcium ATPase where field and in vitro evidence show parasites
expressing the L263E mutation in pfATP6 have decreased sensitivity to artemisinin and
its derivatives (Uhlemann 2005; Krishna et al. 2006; Fidock et al. 2008).

Chapter 1 General Introduction
45
1.15 IN VITRO DETECTION OF RESISTANCE IN P.
FALCIPARUM
Vigilant detection and monitoring of antimalarial resistance is prudent for ensuring the
best choice of treatment within a given locality. In vitro testing of parasite susceptibility
is a valuable tool for resistance surveillance to complement clinical trials which are
much more time and resource-consuming. In vitro susceptibility testing involves the
short term culture of parasites isolated from an infected individual, and determining the
level of growth inhibition after exposing them to various drug concentrations.
Sensitivity is usually measured in terms of the concentration of drug required to inhibit
growth by 50% (IC50) (Rieckmann et al. 1978; WHO 2001; Noedl et al. 2007). The IC50
is subsequently compared against a threshold value for in vitro resistance, which is
uniquely determined for each drug. In the example of CQ for which the in vitro
resistance threshold is 100 nM, the value was determined by comparing IC50 values
obtained from 11 geographically distinct culture-adapted and field isolates (Cremer et
al. 1995). Most in vitro drug resistance thresholds were selected without information on
clinical outcome hence may not directly predict in vivo resistance (Ekland et al. 2008).
Nevertheless, IC50s have been used extensively as an international currency in the
assessment of drug susceptibility from different geographic regions.
The first in vitro drug sensitivity test was published in the late 1960s where infected
blood samples were treated with CQ, QN and cycloguanil and the extent of parasite
maturation in the presence of these drugs was assessed (Rieckmann et al. 1968). This
marked the beginning of short-term in vitro culture of malaria parasites and continuous
culture methods were described shortly after (Trager et al. 1976). Based on the new
milestones in malaria culture techniques, assessments of drug inhibition against all
developmental stages of parasites using 48 or 96 hr assays were developed (Trager
1978; Richards et al. 1979). These methods involved longer test periods which enabled
the testing of slower-acting antimalarials. Further development over the next few
decades produced diverse approaches including visual examination and counting of
mature parasite stages, determination of parasite enzyme activity, and sophisticated
assays that quantify the amount of newly synthesised DNA during parasite development

Chapter 1 General Introduction
46
by radio-isotope labelling (Rieckmann et al. 1978; Desjardin 1979; Makler et al. 1993a;
Radfar et al. 2009). The following sections provide an overview of the main types of in
vitro assays used for the assessment of P. falciparum growth inhibition in response to
different drug doses.
1.15.1 Schizont Maturation
1.15.1.1 Macro test
The WHO macro in vitro test is based on maturation of trophozoites and formation of
schizonts in the presence of different concentrations of drugs (WHO 1978). This assay
is supplied in a kit, primarily developed for the field assessment of P. falciparum
susceptibility to CQ. Briefly, 8 mL of venous blood are collected and transferred into a
sterile Erlenmeyer flask (25 mL) containing glass beads and stoppered. The sample is
defibrinated by physical rotation for 5 min. Into each test vial, 1 mL of blood is
aliquoted in the specified sequence for areas with suspected resistance: 2 control vials,
CQ vials containing 0.5, 1, 1.5, 2, 3, 0.25, 0.75, 1.25 nM and, for areas with no prior
indication of resistance 2 control vials, CQ vials containing 0.5, 1.0, 0.75, 0.25, 1.5, 2,
1.25, 3 nM. This sequence of testing increases the chance of parasite sensitivity being
assessed in the optimal range of drug concentrations in case of inadequate sample
volume. After gentle mixing the vials are closed and incubated in water at 38.5°C for 24
- 28 hr. Thick and thin blood smears are prepared after incubation, and the percentage
of schizonts in test wells relative to those of control wells are determined (Dulay et al.
1987).
The macro in vitro method is simple to use with little specialised equipment required. In
view of the fact that there are limited resources in malaria endemic areas, it has been
useful in field settings (Cattani et al. 1986). One limitation however, is the need for a
large-volume blood sample and waterbath space. Many large scale studies employ a
micro technique as a result (Rieckmann et al. 1978).

Chapter 1 General Introduction
47
1.15.1.2 Micro test
A scaled down modification of the macro test commonly known as the Rieckmann
microtechnique enables the assessment of parasite drug sensitivity using a small amount
of blood (Rieckmann et al. 1978). Briefly, finger-prick blood samples (100 µL) are
diluted 1:10 with complete culture medium and dispensed (50 µL) into 96-well plates
provided by the WHO that are pre-dosed with drugs at final concentrations from 0.25 to
16 pmol/well. Test plates are incubated in a candle jar to achieve a microaerophilic
atmosphere at 37°C for 24 to 36 hr depending on the rate of maturation in control wells.
Thick smears are subsequently prepared and the number of schizonts, which is defined
as intra-erythrocytic parasites with 3 or more nuclei, per 200 asexual parasites is
counted. Sensitivity is reported as the highest concentration of drug in which
schizogony occurred.
The in vitro microtechnique has been widely employed in field studies due to its
simplicity and requirement for minimal specialised equipment and technical personnel
(Kouznetsov et al. 1980; Trenholme et al. 1993; Noedl et al. 2001; Menezes et al.
2002). With further refinement, the microtechnique (Mark III) is now the WHO
standard assessment of P. falciparum antimalarial drug susceptibility (WHO 2001).
Despite these advantages, a number of limiting factors should be considered. Since the
sample is used directly in the test system, the parasite density in the inoculum
influences the susceptibility outcome (Kouznetsov et al. 1980; Ponnudurai et al. 1981).
Thaithong et al evaluated the effect of initial parasitaemia on parasite survival in the
presence of 5 antimalarials. The authors found that the actions of CQ, AQ, MQ and
quinine were significantly reduced in the presence of parasitaemia >1% (Thaithong et
al. 1983). In addition, the WHO in vitro micro kit only caters for a single test per
sample and not duplicates or triplicates as in the isotope incorporation assay and
Plasmodium lactate dehydrogenase measurement (Desjardin 1979; Makler et al.
1993b). In practical terms this is advantageous as schizont counting is labour intensive
and time consuming, especially when the method already requires the counting of 8
thick films per sample to determine sensitivity outcome. Another consideration
important for data interpretation is that schizont enumeration by microscopy tends to

Chapter 1 General Introduction
48
produce IC50 values that are two to three times higher than by the isotopic method
(Wernsdorfer et al. 1988).
1.15.2 3H-Hypoxanthine Incorporation Assay
The isotopic assay utilises the parasite’s requirement for hypoxanthine as a nucleic acid
precursor during its development (Desjardin 1979). The assay is applicable for both
field isolates and culture-adapted samples as detailed in Section 2.2.5. The isotopic
assay provides a semi-automated technique to assess drug susceptibility. It is
reproducible and sensitive, and is considered the reference method for drug sensitivity
assays (Makler et al. 1993b; Druilhe et al. 2001). Numerous in vitro studies have used
this method, and have circumvented the need for specialised equipment in the field by
transporting samples to a centralised laboratory for testing (Basco et al. 1998; Pradines
et al. 2006). This approach has proven successful in various African countries where
the high throughput assay enables more effective monitoring of resistance epidemiology
(Nzila-Mounda et al. 1998; Pradines et al. 1999a; Basco et al. 2003a; Jambou et al.
2005). However, the use of radioisotope involves high costs for consumables and
laboratory infrastructure, requiring supporting facilities such as safe disposal of
radioactive waste and reliable couriers. Technical training of research personnel to
perform meticulous handling of unsealed radio-isotopes is usually unavailable in
developing countries such as PNG. The high costs and technical constraints have
hindered the implementation of the isotopic method in many malaria-endemic
countries. Nonetheless, strategic collaboration between developing and developed
countries should enable the use of this sensitive method for the prudent ‘gold standard’
surveillance of P. falciparum drug resistance.
1.15.3 Plasmodium Lactate Dehydrogenase (pLDH) Detection
Plasmodium lactate dehydrogenase (pLDH) is the terminal enzyme of the anaerobic
Embden-Meyerhoff glycolytic pathway and is essential for energy production in
malaria parasites (Sherman 1979). Early interest in using pLDH as a marker for
parasitaemia and parasite growth stem from the favourable characteristics of the
enzyme. PLDH can be distinguished from human LDH based on its ability to rapidly

Chapter 1 General Introduction
49
utilise 3-acetyl pyridine adenine dinucleotide (APAD) as a coenzyme to convert lactate
to pyruvate at a rate 200-fold more effectively than the human isoforms (Sherman 1961;
Gomez et al. 1997). In addition, the clearance of pLDH from plasma is rapid (3 - 5
days) and correlates with parasitaemia in vitro and in vivo (Makler et al. 1993a). The
pLDH level declines rapidly when parasites are no longer metabolically viable (Piper et
al. 1999). These biochemical features constitute a sensitive and specific marker for the
determination of parasite growth. It has been applied to various assay formats for
assessing in vitro drug susceptibility as described in Section 2.2.4.
1.15.3.1 Colourimetric pLDH microtests
The original drug sensitivity assay using pLDH was described by Makler et al (Makler
et al. 1993a). The assay follows a similar set up for parasite drug exposure in the
isotopic assay, as described in Section 2.2.4. The reaction is allowed to develop at room
temperature (RT) and the consequential change in colour can be monitored visually or
measured spectrophotometrically at 650 nm (Figure 2.6). PLDH activity can also be
determined kinetically at 30 sec intervals for 30 min by the formation of reduced
APAD. The enzymatic approach has been successfully used in field studies (Makler et
al. 1993a; Basco et al. 1995; Wong et al. 2010). Inhibition profiles and IC50s obtained
by pLDH are comparable to those determined by the isotopic and microscopic assay
(Makler et al. 1993; Basco et al. 1995). For optimal sensitivity, however, the enzymatic
assay requires an initial parasitaemia between 1 and 2% at 1.5% haematocrit (hct), with
a detection limit of 0.4% parasitaemia. This range of initial parasitaemia is often too
high for most field isolates from sub-Saharan Africa where patients with acute
uncomplicated falciparum malaria usually present with parasitaemia <1% (Basco et al.
1995). The method is therefore not sensitive enough as a diagnostic method.
Nonetheless, it has been employed in monitoring therapeutic efficacy of drug treatments
in Chinese patients infected with P. falciparum and P. vivax (Wu et al. 2002b). A
modification to the colourimetric assay is the use of sodium-2,3-bis-[2-methoxy-4-
nitro-5-sulphophenyl]-2H-tetrazolium-5-carboxanilid (XTT) in place of NBT and the
reaction is followed by optical density (OD) measurement at 450 nm (Delhaes 1999).
However, this method requires even higher initial hct and parasitaemia, therefore, offers

Chapter 1 General Introduction
50
no advantage over the unmodified version.
1.15.3.2 Immunocapture of pLDH
Other variations based on immunocapture of pLDH have been developed with the use
of monoclonal antibodies (Makler et al. 1998; Kaddouri et al. 2006; Mayxay et al.
2007). This approach significantly improves assay sensitivity, as it is less prone to non-
specific reduction of NBT in the haemolysate and in the reagent mixture (Knobloch et
al. 1995; Oduola et al. 1997). In a Nigerian clinical study, Oduola et al (1997)
developed an enzyme-linked immunosorbent assay (ELISA) that combines the use of
antibody capture technique with APAD to enhance sensitivity and specificity of pLDH
detection. The immunocapture pLDH (IcpLDH) assay uses 96-well plate coated with
mouse monoclonal antibody specific to pLDH, to which the blood sample is added.
During incubation, pLDH is captured on the plate by antibodies, whilst non-specific
contents are washed off. NBT is subsequently added and end point absorbance is
measured. The authors observed a specific and immediate relationship between
dissipation of pLDH enzyme activities and resolution of infection in vivo (Oduola et al.
1997; Piper et al. 1999). Further refinement of this technique led to the development of
a double-site enzyme-linked pLDH immunodetection (DELI) assay (Moreno et al.
2001). Field trials in Thailand, Laos and Senegal demonstrated the DELI microtest to
be highly sensitive, allowing for the inclusion of isolates with parasitaemia as low as
0.005% (Moreno et al. 2001; Brockman et al. 2004; Mayxay et al. 2007). The IC50s
obtained using the DELI approach correlated well with the isotopic test, showing
similar proportions of drug resistant and sensitive isolates. It is also easier and faster to
implement than the isotopic test, and does not require sophisticated equipment (Moreno
et al. 2001; Kaddouri et al. 2008). A current drawback to its implementation is that
monoclonal antibodies towards pLDH are not commercially available, with its use
limited to collaborating laboratories. Overall, immunocapture pLDH colourimetric
assays are cost-effective and straight forward to perform, with great potential for world-
wide implementation for epidemiological monitoring of drug resistance.

Chapter 1 General Introduction
51
1.15.4 Histidine-Rich Protein II (HRP2) Assay
Histidine-rich protein II is a naturally occurring protein found in several cellular
compartments in the parasite including the cytoplasm. It has been implicated as an
important factor in the detoxification of haem and is readily recovered from plasma,
infected RBC membrane and culture supernatants (Howard et al. 1986; Sullivan et al.
1996; Lynn et al. 1999; Papalexis et al. 2001). The level of HRP2 in malaria cultures
increases with parasite development and multiplication (Desakorn et al. 1997), hence
making it an excellent indicator of parasite growth in response to antimalarial drugs. In
2002, a novel approach was described to assess drug sensitivity in Thai isolates by
measuring the level of HRP2 in an ELISA (Noedl et al. 2002). Briefly, parasitised RBC
samples were standardised (0.05% parasitaemia, 1.5% hct) and incubated with various
drug concentrations in 96-well plates similar to that in the pLDH assay. These were
subsequently frozen-thawed and the haemolysed samples transferred to commercial
ELISA plates pre-coated with mononclonal antibodies against HRP2. The plates were
incubated at RT for one hr and washed repeatedly to remove unbound content. Diluted
antibody conjugate was added to each well and incubated as previously. After several
washes, diluted chromogen was added for colour development in the dark for 15 min.
At the end of the reaction, a stop solution was added and OD measured at 450 nm for
IC50 determination (Noedl et al. 2002).
A number of field studies have implemented the new HRP2 microtest and found it
highly sensitive and simple to perform with results closely aligned to those obtained by 3H-hypoxanthine incorporation and the WHO schizont maturation test (Noedl et al.
2003; Attlmayr et al. 2005; Noedl et al. 2005). HRP2 assay is expensive to perform for
the purpose of in vitro drug susceptibility as three columns of the ELISA plates are
required for one drug to be tested in triplicate per sample. For multiple drug testing
required in large scale studies, the use of commercial kits (~AUD $100/plate) is costly
(Cellabs, Australia). More recently, an in-house HRP2 ELISA has been described using
commercially available monoclonal antibodies and is a cheaper alternative to using test
kits (~AUD $20/plate) (Noedl et al. 2005). The method requires additional coating of
test plates which is labour intensive and potentially introduces a bias if antibodies are

Chapter 1 General Introduction
52
not coated evenly across the wells. This in-house version produces similar results to
those by commercial kits (Noedl et al. 2005), and the availability of monoclonal
antibodies facilitates a more rapid implementation of a cost effective and sensitive in
vitro assay.
1.15.5 Dual Detection of HRP2 and PLDH
It is presently accepted that HRP2 levels reflect both past and current infections due to
its slow clearance (10 - 14 days) while pLDH levels reflect the current infection. The
concurrent measurement of these two bio-markers by means of a unified ELISA
approach has been recently described (Martin et al. 2009). The unified protocol enables
the direct comparison of both HRP2 and pLDH results and provide a more enhanced
assessment of parasite burden to include sequestered parasites, which would be
clinically relevant during pregnancy where microscopy can be unreliable (Martin et al.
2009).
1.16 ASSESSMENT OF ANTIMALARIAL DRUG
COMBINATIONS
Combination antimalarial therapies play a pivotal role in delaying the onset of
resistance to new agents and in reducing the effects of resistance to current agents
(White 1998). Effective combination drug regimens often achieve a therapeutic efficacy
greater than that achieved with monotherapy (Fivelman et al. 2004). In vitro drug
interaction studies provide essential information for the selection of optimal drug
combinations for further clinical trials. However, in vitro combination efficacy does not
necessarily translate to efficacy in vivo, as therapeutic efficacy is dependent on
pharmacokinetic characteristics of both drugs within the host (Fivelman et al. 2004).
The biological responses of two agents in combination can be assessed in vitro by the
construction of an isobologram, which graphically displays the effects of each agent
alone and in combination (Figure 1.22) (Berenbaum 1978; Czarniecki et al. 1984;
Chawira et al. 1987; Davis et al. 2006). The outcomes of drug interactions are either
synergistic, indifferent (no interaction) or antagonistic. The concentrations of agents,

Chapter 1 General Introduction
53
either in combination or alone, required to achieve 50% inhibition of parasite growth
are calculated and normalised to fractional inhibitory concentrations (FICs)
(Berenbaum 1978). The sum (Σ) of FICs can be calculated by the addition of FICs of
agents A and B, i.e. (IC50 of A in a mixture resulting in 50% inhibition/IC50 of A alone)
+ (IC50 of B in a mixture resulting in a 50% inhibition/IC50 of B alone) (Berenbaum
1978). When the ΣFIC equals 1.0, the combination is additive, or has no interaction. In
this case, the plotted points should fall close to the straight line drawn between the FICs
of 1.0 on the abscissa and ordinate (Figure 1.22). A ΣFIC of <1 indicates synergistic
interaction, the data points from which would form a concave isobole beneath the line
of additivity, and a ΣFIC of >1 is indicative of an antagonistic interaction represented
by a convex isobole (Berenbaum 1978; Chawira et al. 1987; Fivelman et al. 2004;
Davis et al. 2006). A more conservative interpretation of isobolographic results have
been recommended, where synergy is defined as ΣFIC values ≤ 0.5, antagonism as
ΣFIC values ≥ 4.0, and no interaction when ΣFIC >0.5 – 4.0 (Odds 2003).
Figure 1.22 Representation of isoboles.
The original checkerboard method by Berenbaum (1978) has been widely used to
evaluate antimalarial interactions (Hassan Alin et al. 1999; Skinner-Adams et al. 1999;
Gupta et al. 2002). An alternative fixed-ratio isobologram method formerly developed
for studies in bacteria has been applied for P. falciparum (Hall et al. 1983; Fivelman et
al. 2004; Wong et al. 2009). This newer approach demonstrated comparable findings
with the checkerboard method and is less labour intensive with fewer calculation steps

Chapter 1 General Introduction
54
(Fivelman et al. 2004). Both checkerboard and fixed-ratio methods require
predetermination of IC50 values of each drug alone. From these data, a starting
concentration for each agent is selected and drug mixtures are prepared in various ratios
of the initial concentrations and subjected to serial dilution. In the checkerboard
method, the concentration of agent A is fixed whilst that of agent B is varied and vice
versa. The fixed-ratio method on the other hand, uses serial dilutions of fixed ratios of
both agents, so that drug concentrations are varied at the same time over predetermined
sets of concentrations (Fivelman et al. 2004). The fixed-ratio method is more resilient
in terms of inter-day variations in IC50s where inaccurate initial IC50s may cause poor fit
of the sigmoidal growth response curve, resulting in clustering of data points at the
extremities of the isobole axes. In addition, dose-response curves from the fixed-ratio
approach are based on drug concentration ratios, each of which is constructed to range
from 100 to 0% parasite inhibition, thus enabling a more accurate regression curve fit
and IC50 determination (Fivelman et al. 2004).
1.17 IN VIVO DETECTION OF DRUG RESISTANCE IN P.
FALCIPARUM
The level of P. falciparum resistance to antimalarial drugs is often assessed by
therapeutic response (Wongsrichanalai et al. 2002). In vivo response involves the
assessment of clinical and parasitological response over a period of time post-treatment
(WHO 2006). Parasitological responses are classified by the clearance of parasitaemia
and are graded as sensitive (S) and three levels of resistance (RI, RII, and RIII) (Table
1.3). This classification system, though remaining valid in areas with low transmission,
may be difficult to apply in areas with intensive transmission, where new infections and
recrudescences cannot be differentiated on the basis of microscopy and complicates the
outcome (Wongsrichanalai et al. 2002). More recently, the WHO introduced a modified
system based on clinical symptoms and the level of parasitaemia (Table 1.3) (Bloland
2001). The previously established follow-up period of 14 days is considered insufficient
as a significant proportion of recrudescence often appeared after this period and shorter
observation periods have led to the overestimation of treatment efficacy (WHO 2006).
The current recommended duration of follow-up is a minimum of 28 days for most
antimalarial drugs in areas of intense as well as low to moderate transmission. Extended

Chapter 1 General Introduction
55
follow-up periods of 42 days and 63 days are recommended for slowly eliminated drugs
(i.e. lumefantrine and MQ, respectively) to effectively capture recrudescences (WHO
2006). Clinical studies however are costly and often confounded by poor compliance,
difficulty with recruitment and patient follow-up particularly in remote villages (Han et
al. 1976; Karunajeewa et al. 2008b).
Table 1.3 Classifications of in vivo antimalarial susceptibility outcomes. (Talisuna et
al. 2004)
Parasitological classifications Treatment failure classifications
Sensitive
Clearance of parasites after treatment without subsequent recrudescence within a defined period
Adequate clinical response (ACR)
Absence of parasitaemia on day 14, irrespective of fever status, without previously meeting any of the criteria for ETF or LTF
Absence of fever irrespective of the parasitaemia status without previously meeting any of the criteria for ETF or LTF
RI parasitological failure Initial clearance followed by recrudescence after day 7
Early treatment failure (ETF) Danger signs or severe malaria on day 1, 2, or 3 in the presence of parasitaemia Fever (axillary temperature, ≥37.5°C) persists on day 2 and the parasite density is greater than that on day 0. Fever and parasitaemia on day 3 Parasite density on day 3 is ≥25% of the day 0 parasite density
RII parasitological failure
Reduction of parasitaemia on day 2 to less than 25% of day 0 parasitaemia, but no complete clearance
RIII parasitological failure
On day 2, either no reduction of parasitaemia or reduction to a level equal to or greater than 25% of the day 0 parasitaemia
Late treatment failure (LTF) Danger signs or severe malaria develop in the presence of parasitaemia on any day from day 4 to day 14 Fever and parasitaemia on any day from day 4 to day 14, and yet the patient could not be classified as ETF

Chapter 1 General Introduction
56
1.18 MOLECULAR MARKERS OF DRUG RESISTANCE
Recent advances have provided powerful molecular tools to detect drug resistance
(Ekland et al. 2007). Molecular methods are renowned for their rapid quantitative
assessment for genetic markers of drug resistance, providing an attractive and relatively
inexpensive alternative to clinical approaches for monitoring the prevalence and spread
of resistant parasites (Ekland et al. 2008). The sequencing and annotation of the P.
falciparum genome in 2002 has greatly enhanced the identification of gene candidates
as genetic markers of drug resistance (Gardner et al. 2002). Conserved polymorphisms
within the genome including microsatellites (repeats of short nucleotide sequence),
single nucleotide polymorphisms (SNPs), and small insertions or deletions, act as
surrogate markers for drug resistance determinants (Ekland et al. 2007). The P.
falciparum genome contains an estimate of 25,000 to 50,000 SNPs that tend to cluster
in blocks known as haplotypes surrounded by recombination hotspots (Mu et al. 2005).
Since the number of polymorphisms is different within different genes, and by tracking
a signature set of SNP tags, various haplotypes associated with drug resistance can be
identified (Mehlotra et al. 2001; Carnevale et al. 2007; Ekland et al. 2007).
The identification of pfcrt as the primary determinant in CQ resistance has enabled the
development of a simple PCR assay that identifies the presence of resistant strains
(Fidock et al. 2000). Mutations in the pfcrt gene, particularly the substitution of lysine
(K) to threonine (T) at residue 76 (K76T), is central to CQ resistance. The K76T
polymorphism is consistently found in CQ-resistant strains regardless of geographic
origin. It can occur within different amino acid haplotypes between residues 72 to 76:
CVIET, CVMNT, CVMET, and SVMNT, all of which are associated with CQ
resistance (Fidock et al. 2000). Higher levels of CQ resistance can be attributed to
changes in the pfmdr1 gene in point mutation 86Y (Foote et al. 1990; Reed et al. 2000;
Babiker et al. 2001; Djimde et al. 2001; Pickard et al. 2003). Clinical resistance to AQ
has been associated with SNPs in pfcrt K76T, pfmdr1 N86Y, F184Y, D1246Y, in
particular the triple tyrosine pfmdr1 haplotype YYY at codons 86, 184 and 1246 has
been selected post AQ monotherapy (Humphreys et al. 2007; Tinto et al. 2008). The
predictive value of pfcrt and pfmdr1 polymorphisms for both CQ and AQ underscores
the similarity in their mode of action, and the predisposition of high level of CQ

Chapter 1 General Introduction
57
resistance in endemic regions may compromise the future use of AQ-artesunate as an
alternative ACT (Thwing et al. 2009). Allelic substitution experiments have also shown
both pfcrt and pfmdr1 polymorphisms contribute to quinine resistance (Reed et al.
2000; Sidhu et al. 2005). Another molecular determinant, the pfnhe1 gene located on
chromosome 13 which encodes a putative sodium/hydrogen exchanger was associated
with reduced quinine sensitivity in culture-adapted P. falciparum (Bennett et al. 2007).
Mutations in the pfmdr1 gene also confer resistance to multiple other antimalarials
including MQ, lumefantrine, halofantrine and quinine (Cowman et al. 1994; Peel et al.
1994; Reed et al. 2000). In a pfmdr1 gene allelic replacement study, Reed et al (2000)
demonstrated pfmdr1 mutations altered artemisinin susceptibility in parasite lines from
PNG (D10) and South America (7G8). This finding has serious implications for future
artemisinin effectiveness in endemic areas such as PNG. Polymorphic changes in the
pfdhfr and pfdhps genes encoding dihydrofolate reductase and dihydropteroate synthase
respectively are good indicators for pyrimethamine and sulfadoxine resistance. The
S108N mutation in pfdhfr has been implicated in pyrimethamine resistance with
additional mutations at positions 16, 50, 51, 59, 140 and 164 contributing to elevated
levels of resistance (Cowman et al. 1988; Peterson et al. 1988). Similarly, the A437G
mutation in pfdhps confers initial resistance to sulfadoxine. Other mutations including
K540E, A581G, S613A, S436A/F lead to higher resistance (Triglia and Cowman 1994;
Triglia and Cowman 1999). Table 1.4 provides a summary of molecular markers
associated with drug resistance.

Chapter 1 General Introduction
58
Table 1.4 Molecular markers for antimalarial drug resistance. (Triglia et al. 1994;
Reed et al. 2000; Chaijaroenkul et al. 2010).
Antimalarial Gene mutation Remarks
pfcrt 76T Strong association Chloroquine
pfmdr1 86Y, 184F, 1034C, 1042D, 1246Y
Possible association
Sulfadoxine-pyrimethamine
pfdhfr 108, 51, 59, 164 pfdhps 436, 437, 540, 581,
623
pfdhfr 108 essential for pyrimethamine resistance, degree of resistance
increases with additional mutations Leu51 and Arg59 or triple mutation. Absolute resistance conferred by the addition of Leu164, thus quadruple
mutation, irrespective of dhps mutations
Mefloquine, Quinine
pfmdr1 Not clearly understood, pfmdr1 copy number is associated with reduced sensitivity to mefloquine, quinine
Artemisinin pfmdr1 pfATPase6
pfmdr1 copy number is associated with reduced sensitivity to artesunate
1.19 BREATH TEST FOR MALARIA
Breath tests have been developed to assist in the early diagnosis of infections with
Mycobacterium tuberculosis and Helicobacter pylori (Lechner et al. 2005b; Phillips et
al. 2007). The procedure involves the trapping of a sample of a patient’s breath in a
sorbent column followed by extraction and analysis of the retained volatile organic
compounds (VOCs). The profile or ‘fingerprint’ of VOCs that is generated can be used
in determining whether a particular disease is present (Lechner et al. 2005a). Recent
developments of breath-collection apparatus allow portability of equipment and thus
testing may be performed promptly and more cost-effectively wherever the patient
presents (Phillips 1997; Lindstrom et al. 2002; Martin et al. 2010).
The identification of P. falciparum using conventional microscopy, antigen detection
and PCR are inadequate in circumstances where the peripheral parasitaemia is low. This
includes cases of severe malaria in which the majority of parasites are sequestered

Chapter 1 General Introduction
59
within the microvasculature (Sachanonta et al. 2008). In addition, antigen detection
methods and PCR in this context may not, if positive, differentiate between viable and
non-viable parasites, an important consideration for therapeutic monitoring. In such
instances, breath tests may provide a novel, non-invasive approach for the sensitive
detection of an active malaria infection.
1.20 SCOPE OF THE STUDIES PRESENTED IN THIS THESIS
The preceding literature review has provided an overview of malaria infection in
humans, the use of conventional and novel antimalarial agents in malaria management,
the role of in vitro and in vivo methods in the detection of parasite drug resistance, and
how these factors contribute to the understanding of malaria pathophysiology and
treatment outcome.
The proximity of PNG to Australia and the political and economic relationships
between the two countries mean that malaria control in PNG should be of concern to
Australia. A significant proportion of imported malaria in Australia originates in PNG
(Charles et al. 2005) while malaria outbreaks in the Torres Strait Islands are regularly
reported (Needham 2011). The studies reported in Chapters 3 and 4 of this thesis
include a multifaceted investigation into the efficacy of treatment for falciparum
malaria, including mechanisms underlying P. falciparum resistance, in PNG children.
Although this is an extremely important public health issue in PNG with clear
implications for Australians visiting PNG or living close to the southern part of the
country, there have been few studies of this type conducted to date.
The data from the present PNG paediatric malaria studies and the evolution of
resistance of P. falciparum and P. vivax to available antimalarial drugs in other parts of
the tropics underscore the need for the identification of novel treatments. This can
include compounds within existing or new classes of antimalarial drugs. Studies in
Chapters 5 and 6 of this thesis address both these possibilities, providing pre-clinical
and some clinical data that are essential to the further development and clinical
application of desbutyl-lumefantrine and fenofibrate. Desbutyl-lumefantrine is the
active metabolite of lumefantrine which is part of the recommended first-line ACT

Chapter 1 General Introduction
60
treatment of uncomplicated malaria in PNG. The impetus to examine lipid-modifying
drugs was provided by the recognition that non-communicable diseases, especially
cardiovascular disease, represent an increasing disease burden in developing countries
such as PNG (Ōtsuka et al. 2007). Drugs such as statins and fibrates will be
increasingly used as a result and antimalarial effects could provide significant health
and economic benefits to the individual and community in PNG and other countries
with similar epidemiology. This has been investigated in the case of the blood glucose-
lowering drug rosiglitazone (Boggild et al. 2009) but lipid-altering therapies have even
wider potential therapeutic application.
During the course of the foregoing studies, the deficiencies in available techniques for
diagnosis and antimalarial therapeutic monitoring provided the impetus to evaluating
VOCs as a surrogate sensitive biomarker of viable parasites in the circulation. At the
same time as data collection for the studies presented in Chapters 3 and 4 was
proceeding, a separate investigation done in Madang, PNG, by another group identified
the VOCs methyl nicotinate in the breath of tuberculosis patients (Syhre et al. 2009).
This suggested that a similar breath test might be feasible in malaria. A corollary of
parasite-specific VOCs production might be that these compounds have biological
activity including acting as anaesthetic agents in patients with cerebral sequestration
and consequent coma. The experiments in Chapter 7 detail attempts to determine, as the
first step in this line of investigation, whether cultured P. falciparum generates specific
VOCs as quantified using the most sensitive chemical assay techniques.
The main implications of these various studies, their inter-relationships and prospects
for future related research are discussed in Chapter 8.

CHAPTER 2
METHODS AND MATERIALS

Chapter 2 Methods and Materials
62
CHAPTER 2. METHODS AND MATERIALS
2.1 IN VITRO CULTURE TECHNIQUES
Culture techniques for P. falciparum described in the following section have been
adapted from previously published methods (Trager et al. 1976).
2.1.1 Parasites
PNG field isolates of P. falciparum were collected from children with uncomplicated
malaria (Chapter 3). Laboratory-adapted strains 3D7, Dd2, W2mef, K1 and E8B used in
other in vitro studies were obtained from The Walter and Eliza Hall Institute in
Melbourne and from Dr. Tina Skinner-Adams and colleagues of the Queensland
Institute of Medical Research, Australia.
2.1.2 Retrieval from Liquid Nitrogen
Intra-erythrocytic stages of P. falciparum preserved in cryo-suspensions were retrieved
from long term liquid nitrogen storage as described previously (Meryman et al. 1972;
Skinner-Adams 1999). Briefly, parasite stock vials were thawed at 37°C in a waterbath
and transferred into a centrifuge tube. Pre-warmed 12% NaCl solution (Appendix B)
was added drop-wise (1 drop/sec) with gentle mixing until a fifth of the original volume
was added (i.e. 200 µL to 1 mL cell suspension) and allowed to stand at RT for 3 min.
With gentle mixing, pre-warmed 1.6% NaCl solution (Appendix B) was added drop-
wise to reach 10 times the volume (i.e. 10 mL to 1 mL) then centrifuged (1200 rpm for
5 min). The supernatant was removed and pre-warmed 0.9% NaCl solution (Appendix
B) was added drop-wise to reach 10 times pellet volume whilst ensuring the RBC were
fully resuspended. After centrifugation, the supernatant was removed and the cells were
resuspended in complete media (Appendix B) at 5% haematocrit (hct) using RBC
(Section 2.1.4) and cultured in a microaerophilic environment (Section 2.1.3).

Chapter 2 Methods and Materials
63
2.1.3 Maintenance of Cultures
P. falciparum cultures were cultured in sterile lidded-dishes (5 mL, 10 mL or 20 mL) at
5% hct in a Nalgene desiccator (Figure 2.1). This provided a controlled microaerophilic
atmosphere for the culture of P. falciparum (Trager et al. 1976; Scheibel et al. 1979).
Non-infected RBC (Section 2.1.4) were added to the cultures at 2 - 3 day intervals to
maintain parasitaemia between 0.5 to 5%. To prevent accumulation of metabolic by-
products, high parasitaemia were reduced by expanding the culture to a larger dish or
by aspirating out part of the culture and re-adjusting the hct by replenishing with non-
parasitised RBC.
Figure 2.1 Nalgene desiccator used for P. falciparum culture. Parasite cultures were
maintained at 37⁰C in an airtight chamber (modified Nalgene desiccator cabinet,
Nalgene, U.S.A) flushed with a gas mixture of 1% (or 5%) O2 and 5% CO2 balanced in
N2 (BOC gases, Australia) for 90 sec daily or when opened. The oxygen concentration
within the chamber ranged between 3 - 9% over 24 hr as indicated by an O2 monitor
(ToxiRAE II, San Jose, CA USA).
2.1.4 Erythrocytes Preparation

Chapter 2 Methods and Materials
64
Blood from a healthy volunteer was collected into sodium heparin Vacuette® and
centrifuged (1300 rpm for 5 min). Buffy coat was removed and plasma was stored at -
20⁰C for later use as media supplement (Appendix B). Packed RBC was transferred into
a 50 mL tube topped with 3 times cell volume of media. After centrifugation (1300 rpm
for 15 min), the supernatant was discarded and the process repeated twice. After the
final wash, the cells were resuspended 1:1 (i.e. 50% RBC) in non-supplemented media.
The stock was refrigerated and used within 3 weeks for cultures and assays.
2.1.5 Determination of Parasitaemia
Parasite viability and development can be visually monitored by light microscopy
(Figure 2.2). A thin blood smear was prepared by taking 3 - 10 µL of RBC in an equal
volume of media and applied as a drop on a glass slide (Menzel-Glӓser, Lomb
Scientific) (Figure 2.3). After air drying, the thin smear was fixed in methanol and
stained for 10 min using freshly prepared 5% Giemsa Solution (Appendix B). After
rinsing under tap water, it was air-dried and examined under oil or anisole (Fluka)
immersion (100 × objective). The parasitaemia was determined by counting the number
of infected RBC in at least 1000 RBC and expressed as a percentage of total RBC.
2.1.6 Synchronisation of Parasite Forms
One developmental cycle prior to use, parasite cultures were synchronised by sorbitol
lysis to achieve ≥90% ring forms (Lambros 1979). RBC harbouring mature forms are
more fragile and lyse under osmotic stress. Briefly, cultures with >40% rings were
centrifuged and the pellet was resuspended in 10 mL of pre-warmed 5% sorbitol
(Appendix B) and incubated at 37°C for 10 min. This was centrifuged and resuspended
in complete media for culture.

Chapter 2 Methods and Materials
65
Figure 2.2 Giemsa-stained thin smear of synchronised P. falciparum culture.
Cultured parasites under anisole immersion (× 1000 magnification). From top to
bottom: rings, trophozoites and schizonts. Note the rupture of a schizont with two
newly released merozoites (M) about to invade an adjacent erythrocyte. Characteristic
malaria brown pigment or haemazoin crystals (H) were also observed within RBC or as
free crystals released into the culture space during schizont rupture.

Chapter 2 Methods and Materials
66
Figure 2.3 Preparation of a thin smear.
Thin smear is useful for detailed examination of parasite structures and developmental
stages. This was prepared by spreading a spot of blood along the edge of another glass
slide at a 45° angle and bringing it forward by a smooth gliding motion.
.
2.1.7 Cryopreservation
Field isolates or culture-adapted parasite strains were cryopreserved for future use.
Cryopreservation was performed on a routine basis to maintain stocks of various
parasite strains in liquid nitrogen. Cultures should be of high parasitaemia i.e. (>5%)
and mostly in the ring stage, as RBC infected with young parasites are more resilient to
lysis during the procedure. Briefly, after centrifugation (1300 rpm for 5 min) and
removal of the supernatant, the pellet was resuspended in an equal volume of cryo-
protective solution (Appendix B) and transferred into a cryopreservation vial (Nunc,
U.S.A.) and snap frozen in liquid nitrogen

Chapter 2 Methods and Materials
67
2.2 DRUG SUSCEPTIBILITY ASSAYS
2.2.1 Drug/Compound Preparation
Drug susceptibility assays were carried out in 96-well plates (Sarstedt) where parasites
were treated with low to high doses of drug. Most assays were carried out over 48 hr,
with the exception of AZ for 72 hr. A typical lay-out of a drug susceptibility panel is
shown in Figure 2.4.
The antimalarial drugs used in this study consisted of chloroquine (CQ), amodiaquine
(AQ) and its metabolite monodesethyl-amodiaquine (dAQ), piperaquine (PQ),
mefloquine (MQ), naphthoquine (NQ), lumefantrine (LM) and its metabolite desbutyl-
lumefantrine (DBL), dihydroartemisinin (DHA) and azithromycin (AZ). Details for
statins and fibrates are described in subsequent chapters. On solubilisation, stock
standards of 1 mM concentration were prepared in compatible solvents (Table 2.1) and
stored in aliquots protected from light at -20°C. Due to the slow-acting nature of AZ, it
was tested on a different panel. On the day of assay, drug stocks were thawed and
further diluted in media to prepare 5 µM working standards. These were used for two-
fold serial dilutions in media at double assay concentrations (Table 2.1) and dispensed
in triplicates at 100 µL per well for pLDH and 3H-hypoxanthine incorporation assays.
2.2.2 Preparation of parasitised cells
Prior to assay, parasite cultures were centrifuged with the supernatant removed and
packed RBC were used directly. For field isolates, whole blood samples were
centrifuged with plasma and buffy coat removed. RBC were washed 3 times in non-
supplemented media before use. Each drug panel comprised of four drugs tested in
triplicates hence a minimum of 10 mL suspension of cells standardised to 3% hct at 0.5
to 1% parasitaemia (depending on the method of growth assessment) was required. For
pLDH assessment, 100 µL per well of the suspension was added giving a final 1%
parasitaemia and 1.5% hct in a 200 µL mixture of drug/RBC/media. If growth was
assessed by 3H-hypoxanthine incorporation, 90 µL suspension/ well was used instead.

Chapter 2 Methods and Materials
68
Figure 2.4 Layout of a drug susceptibility panel.
The top row of the panel consisted of drug-free parasitised and non-parasitised controls.
Antimalarial drugs were serially diluted from the bottom row (highest concentration) to
the second row (lowest concentration) in triplicates.
2.2.3 Controls
For parasitised drug-free controls, 100 µL (or 90 µL for 3H-hypoxanthine
incorporation) of infected RBC suspension were added to 100 µL of assay media per
well. These wells should exhibit the highest parasite growth. For non-parasitised control
wells, uninfected RBC were used instead.

Chapter 2 Methods and Materials
69
Table 2.1 Solvents and optimised assay concentration ranges for drug
susceptibility testing.
Drug (Supplier) Solvent Assay Range
Chloroquine diphosphate (Sigma Chemicals, St Louis, USA)
Sterile d.H2O 12.5 nM – 1600 nM
Amodiaquine dihydrochloride (Sigma)
Sterile d.H2O 5 nM – 320 nM
Monodesethyl-amodiaquine (Sapec Fine Chemicals, Lugano, Switzerland)
Sterile d.H2O 5 nM – 320 nM
Naphthoquine phosphate (ZYF Pharm Chemical, Shanghai, China)
50% v/v ethanol 3.13 nM – 200 nM
Piperaquine tetraphosphate (Yick-Vic Chemicals and Pharmaceuticals, Hong Kong)
0.5% w/v lactic acid 6.25 nM – 400 nM
Mefloquine hydrochloride (Sigma)
70% v/v ethanol 0.78 nM – 200 nM
Dihydroartemisinin (Sigma) 70% v/v ethanol 0.78 nM – 51.2 nM
Lumefantrine (Novartis Pharma, Basel, Switzerland)
1:1:1 v/v/v mixture of linoleic acid, ethanol
and Tween 80
3.12 nM – 400 nM
Desbutyl-lumefantrine (Novartis Pharma, Basel, Switzerland)
1:1:1 v/v/v mixture of linoleic acid, ethanol
and Tween 80
3.12 nM – 400 nM
Azithromycin (Pfizer, NSW, Australia)
Sterile d.H2O 1250 nM – 160000 nM
Pravastatin sodium (Bristol-Myers Squibb, Australia)
DMSO 3.12 µM – 400 µM
Simvastatin (Alphapharm, QLD, Australia)
DMSO 3.12 µM – 400 µM
Rosuvastatin (AstraZeneca, NSW, Australia)
DMSO 3.12 µM – 400 µM
Atorvastatin calcium (Pfizer, NSW, Australia)
DMSO 3.12 µM – 400 µM

Chapter 2 Methods and Materials
70
2.2.4 Plasmodium Lactate Dehydrogenase Assay
2.2.4.1 Principle of pLDH Assay
P. falciparum multiplication correlates with a rise of pLDH activity thus this can be
used to assess parasite growth in response to drug treatment (Roth 1988; Makler et al.
1993a). A modification of the colourimetric method was used (Makler et al. 1993b;
Wong et al. 2010). PLDH is produced as a terminal glycolytic enzyme that converts
lactate to pyruvate in the presence of NAD+ (Figure 2.5). Activities of Plasmodium and
human isoforms can be distinguished by using 3-acetyl pyridine adeninedinucleotide
(APAD), an analogue of NAD+ that is specifically utilised by parasite LDH. Colour
intensity was then measured by absorbance. Inclusion of diaphorase in the solution
amplifies colour production (Figure 2.5).
Figure 2.5 Colourimetric detection of pLDH activity. As the reaction proceeds, the
APADH generated reduces nitro blue tetrazolium (NBT), a yellow compound, to nitro
blue formazan (NBF), a purple compound.
2.2.4.2 Assay Set Up
After incubation, the drug susceptibility plates were subjected to three cycles of freeze-
thawing to facilitate the release of pLDH. The haemolysate was homogenised by
pipetting up and down with a multichannel pipette. A 10 μL sample from each well was
added to 200 µL of Malstat solution (Appendix B), 10 µL of NBT solution (Appendix
B) and 10 µL of diaphorase solution (Appendix B). The pLDH reaction was allowed to
proceed at RT for 45 to 90 min to allow development of colour within the wells.

Chapter 2 Methods and Materials
71
Interference by air bubbles was circumvented by directing a blow-dryer over the plate.
Colour intensity was measured by absorbance at 650 nm (FLUOstar OPTIMA, BMG
biotechnologies) and the data were analysed by non-linear regression (Graphpad Prism
4.0) to construct dose-response curves for determination of drug-specific IC50.
Figure 2.6 pLDH reaction in field isolates of P. falciparum.
2.2.5 3H-Hypoxanthine Incorporation Assay
Hypoxanthine is required for DNA synthesis during parasite replication. The radio-
activity of the incorporated tritium (3H) reflects parasite sensitivity to antimalarial
compounds (Desjardin 1979). Briefly, drug susceptibility assays were set up at a final
mixture of 0.5% parasitaemia at 1.5% hct as previously described (Section 2.2). Each
well consisted of 90 µL of RBC suspension, 100 µL of drug diluted in media and 10 µL
of a 3H-hypoxanthine working solution (5 mg/mL) (Appendix B), resulting in a final
concentration of 0.5 µCi per well. After incubation, the plates were subjected to three
cycles of freeze-thawing and harvested onto a 96-well glass-fibre filtermat (Perkin
Elmer) using a Harvester 96 (Tomtec Incorporated, USA) (Figure 2.7). The filtermat

Chapter 2 Methods and Materials
72
was air-dried and sealed in a plastic envelope with 4 mL beta scintillant (Perkin Elmer)
and counted on a Wallac microbeta liquid scintillation counter (1450 Microbeta Plus).
The data generated were analysed by non-linear regression analysis (Graphpad Prism
4.0).
Figure 2.7 Tomtec Harvester 96 system.
2.3 MOLECULAR TECHNIQUES
The multiplex PCR ligase techniques for the detection of Plasmodium species and
genetic markers for drug resistance were recently developed by collaborators at Case
Western Reserve University (McNamara et al. 2004; Carnevale et al. 2007) (Figure
2.8). This platform enables high-throughput, sensitive and simultaneous diagnosis of
infection by different Plasmodium species. In a similar assay, this approach can
simultaneously detect a large number of SNPs from the pfcrt, pfdhfr, pfdhps, and
pfmdr1 genes that are associated with malarial drug resistance.

Chapter 2 Methods and Materials
73
2.3.1 DNA Extraction
P. falciparum DNA was isolated from whole blood from study participants or parasite
cultures using a DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s
protocol. The resulting DNA extracts (~200 µL) were stored at -20°C.
Figure 2.8 Wolstein Research Building, CWRU, Cleveland, Ohio, USA.
2.3.2 Polymerase Chain Reaction (PCR)
2.3.2.1 PCR for Plasmodium Species
A small-subunit ribosomal RNA gene fragment (491 to 500 bp) was amplified for
Plasmodium species diagnosis using oligoprimers and conditions previously described
(Mehlotra et al. 2000; McNamara et al. 2004). Briefly, PCR plates (ThermoGrid™ C-
18096, Denville Scientific Inc, USA) were irradiated with ultraviolet light in a
Stratalinker 2400 UV Crosslinker (Stratagene, CA, USA) before use to destroy any

Chapter 2 Methods and Materials
74
residual nucleic acid. Each well contained 25 µL of PCR master mix solution
(Appendix B) containing 67 mM Tris-HCl (pH 8.8), 6.7 mM MgSO4, 16.6 mM
(NH4)2SO4, 10 mM 2-mercapto-ethanol, 100 µM of dNTPs (Appendix B), 2.5 units of
thermo-stable DNA polymerase and 3 µL of genomic DNA sample. Sequences for
Plasmodium genus specific upstream and downstream primers were 5’-TTC AGA TGT
CAG AGG TGA AAT TCT-3’ and 3’-AAT TAG CAG GTT AAG ATC TCG TTC-3’
respectively (Integrated DNA Technologies, Iowa). The plates were sealed using
Microseal® ‘A’ film (Bio-Rad, USA) and mixed by centrifugation (3000 rpm for 30
sec) and amplification reactions were performed in a PTC-225 Peltier Thermal Cycler
(MJ Research, Iowa). The specific thermocycling conditions used were 92°C for 2 min
(1 cycle), 92°C for 30 sec and 63°C for 2 min (35 cycles), 63°C for 5 min (1 cycle)
(McNamara et al. 2004). PCR amplicons were stored at -20°C until assayed.
2.3.2.2 PCR for pfcrt, pfdhfr and pfdhps genes
The amplification of target sequences for P. falciparum pfcrt, pfdhfr, and pfdhps were
achieved using oligoprimers as described by Carnevale et al (2007). Primers and
thermocycling conditions for the amplification of pfcrt and pfdhfr were optimised to
eliminate the necessity of performing nested reactions (Table 2.2). The upstream and
downstream primers listed in (Table 2.2) were used to prepare master mixtures
(Appendix B) for each drug resistance gene.
2.3.2.3 Controls
Laboratory-adapted P. falciparum strains were obtained from the Malaria Research and
Reference Reagent Resource (MR4; ATCC, VA) and the haemolysate of various strains
(HB3, Dd2, 3D7, K1, 7G8, VS/1 and FCB) were kindly provided by Dr. Peter
Zimmerman (CWRU, Cleveland, Ohio, USA). DNA extracts from seven strains of P.
falciparum were included in each PCR run as batch controls in the Plasmodium species
and drug resistant SNPs assays. Distilled water used in the PCR reaction served as a
negative control for each amplification assay.

Chapter 2 Methods and Materials
75
Table 2.2 PCR primer sequences and thermocycling conditions for pfcrt, pfdhps
and pfdhfr target sequences. Conditions for pfdhfr and pfcrt (Carnevale et al. 2007)
were optimised to eliminate the necessity for performing nested reactions. apfcrt for
SNPs at codons 72 to 76. bdhfr fragment for SNPs at codons 51, 59, 108 and 164. cpfdhps gene fragment for SNPs at codons 540, 581, 613. dPCR programs were
preceded by an initial denaturation step at 95°C for 2 min.
Gene PCR Primer Sequence Thermocycling Condition
d
pfcrt a 5’ -TAATACGACTCACTATAGGGCCGTTA-3’ 5’-ATTAACCCTCACTAAAGGGACGGATG-3’
35 cycles of 95°C for 30 sec, 56°C for 30 sec, 60°C for 1 min
pfdhfr b 5’-TAACTACACATTTAGAGGTCTA-3’
5’-GTTGTATTGTTACTAGTATATAC-3’
35 cycles of 95°C for 30 sec, 56°C for 30 sec, 72°C for 1 min
pfdhps c 5’-AATGATAAATGAAGGTGCTAGT-3’
5’-ATGTAATTTTTGTTGTGTATTTA-3’
35 cycles of 95°C for 30 sec, 56°C for 30 sec, 60°C for 1 min
2.3.3 Detection of Amplified Products
To evaluate amplification efficiency, 5 µL of PCR product were premixed with 3 µL of
loading dye buffer and loaded (5 µL) on 2% agarose I gels (Appendix B). For a 96-well
gel, electrophoresis was performed at 290V for 30 min. The gel was subsequently
stained in SYBR Gold (Molecular Probes, Eugene, Oreg.) diluted 1:10,000 in 1 X TBE
buffer (Appendix B) on a rocking platform for 20 min and DNA products were
visualised on a Storm 860 PhosphorImager coupled with Image-Quant version 5.2
software (Molecular Dynamics, CA) (Figure 2.9).

Chapter 2 Methods and Materials
76
Figure 2.9 Electrophoresis and Image processing for DNA visualisation for
evaluating PCR amplification efficiency.
2.3.4 Ligase Detection Reaction Fluorescent Microsphere Assay (LDR-FMA)
The multiplex LDR-FMA facilitates both rapid and specific detection of Plasmodium
species in a single reaction, avoiding cumbersome, separate procedures (McNamara et
al. 2004; McNamara et al. 2006). Similarly, this platform has been adapted to analyse
simultaneously an assortment of SNPs associated with CQ and SP resistance (Carnevale
et al. 2007). The development of a LDR-FMA for pfmdr1 SNPs is described in Chapter
4.
2.3.4.1 Ligase Detection Reaction for Plasmodium species
Briefly, PCR products from the Plasmodium species assay were used directly for LDR
in a master mix solution (Appendix B) containing 20 mM Tris-HCl buffer (pH 7.6), 25
mM potassium acetate, 10 mM magnesium acetate, 1 mM NAD+, 10 mM dithiothreitol,
0.1% Triton X-100, 10 nM (200 fmol) of each LDR primer, 1 µL PCR product and 2
Units of Taq DNA ligase (New England Biolabs, MA). Thermocycling conditions
involved initial heating at 95°C for 1 min, followed by 32 cycles of denaturation at
95°C for 15 sec and annealing/ligation at 58°C for 2 min. LDR primer sequences for
species diagnosis are shown in Table 2.3 (McNamara et al. 2006).

Chapter 2 Methods and Materials
77
Table 2.3 LDR primer sequences for Plasmodium species diagnosis. aLowercased
nucleotides are nonspecific and were added to the rRNA gene LDR primers to reach a
desired specific length. Nucleotide W and R correspond to T or A and G or A
degeneracy, respectively. Pf1 and Pf2, P. falciparum 1 and 2; Pv, P. vivax; Pm, P.
malariae; Po, P. ovale; Common 1 and 2, common sequence. bID, identification. One
hundred unique Luminex microsphere sets are synthesised to exhibit unique
fluorescence. Each microsphere set is coupled to different anti-tag sequences. Anti-tag
sequences are complementary to species-specific tag sequences (adapted from
McNamara et al 2006).
Primer Sequence a ID
b
Pf1 5’-tacactttctttctttctttctttAAA AGT CAT CTT TCG AGG TGA CTT-3’ 12
Pf2 5’-ctatctatctaactatctatacaTGT AGC ATT TCT TAG GGA A TG TTG ATT TTA TAT-3’
78
Pv 5’-cttttcatcttttcatctttcaatAAA ATA AGA ATT TTC TCT TCG GAG TTT ATT C-3’
37
Pm 5’-ttacctttatacctttctttttacAAG AGA CAT TCT TAT ATA TGA GTG TTT CTT-3’
30
Po 5’-ctactatacatcttactatactttTAA GAA AAT TCC TTT CGG GGA AAT TTC-3’ 14
Common1 5’-phosphate-TAG AAT TGC TTC CTT CAG TAC CT T ATG-biotin-3’ 2
Common2 5’-phosphate-TTA GAT WGC TTC CTT CAG TRC CTT ATG-biotin-3’ 3
2.3.4.2 Ligase Detection Reaction for pfcrt, pfdhfr, pfdhps SNPs
Following the PCR amplifications of the pfcrt, pfdhps and pfdhfr genes carrying drug
resistance associated SNPs, the products were combined by transferring 5 µL of each
gene product into a fresh 96 well plate. Pooled PCR products were mixed by
centrifugation and 1 µL was used to prepare the LDR master mixture (Appendix B) and
sealed (sealing film B-1212-5, Denville Scientific Inc) prior to the multiplexed LDR.
Primer sequences for each drug resistance gene are detailed in Table 2.4 (Carnevale et
al. 2007). The LDR thermocycling conditions used were as for Plasmodium species.

Chapter 2 Methods and Materials
78
2.3.4.3 Hybridisation and Reporter Labelling
The second stage of the reaction initiates specific classification of the LDR products,
where hybridisation occurs between anti-tag oligonucleotides probes that are bound to
fluorescent microspheres (Luminex Corporation, TX) specific for the various tag
sequences at the 5’end of the LDR products. The hybridisation and reporter labelling
protocols are the same for Plasmodium species and drug resistance SNPs diagnosis.
This required adding 5 µL of LDR products to 60 µL of pre-warmed hybridisation
solution (Appendix B) containing 250 Luminex FlexMAP microspheres from each
allelic set (total of 18 alleles). The reaction mixture was heated to 95°C for 90 sec and
incubated at 37°C for 35 - 45 min. Reporter labelling of the conjugate followed with the
addition of 6 µL streptavadin-R-phycoerythrin dye (Molecular Probes, OR) diluted 1:50
v:v in TMAC (20 ng/µL) as it binds to the 3’biotin on the conserved sequence primers
at 37°C for 35 - 45 min in 96-well V-bottom plates (Costar 6511 M polycarbonate,
Corning Inc., Corning NY).

Chapter 2 Methods and Materials
79
Table 2.4 LDR primer sequences for drug resistance markers pfcrt, pfdhfr and
pfdhps. aLowercased nucleotides (24 bases) represent tag sequences added to the 5’
ends of each allele-specific LDR primer. bID, identification of Luminex microsphere. cCM, common sequence primer immediately downstream from the allele-specific
primer.
Gene Primer
c Sequence a ID
b
pfcrt CVMNK 5'-aatctacaaatccaataatctcatATTTAAGTGTATGTGTAATGAATAA-3' 60
CVIET 5'-tcataatctcaacaatctttctttAATTAAGTGTATGTGTAATTGAAAC-3' 68
SVMNT 5'-aatcctttctttaatctcaaatcaATTTAAGTGTAAGTGTAATGAATAC-3' 21
72-76 CM 5'-phosphate-AATTTTTGCTAAAAGAACTTTAAAC-biotin-3'
pfdhfr 51 I 5'-caatttcatcattcattcatttcaGAGTATTACCATGGAAATGTAT-3' 35
51 N 5'-ctactatacatcttactatactttGAGTATTACCATGGAAATGTAA-3' 14
51 CM 5'-phosphate-TTCCCTAGATATGAAATATTTT-biotin-3'
59 R 5'-cttttcatcttttcatctttcaatTCACATATGTTGTAACTGCACG-3' 37
59 C 5'-tacactttctttctttctttctttTCACATATGTTGTAACTGCACA-3' 12
59 CM 5'-phosphate-AAAATATTTCATATCTAGGGAAWTA-biotin-3'
108 T 5'-ctaactaacaataattaactaacTTGTAGTTATGGGAAGAACAAC-3' 80
108 S 5'-ctataaacatattacattcacatcTTGTAGTTATGGGAAGAACAAG-3' 69
108 N 5'-tcatcaatcaatctttttcactttTTGTAGTTATGGGAAGAACAAA-3' 59
108 CM 5'-phosphate-CTGGGAAAGCATTCCAAAAAAA-biotin-3'
164 I 5'-ctttctatctttctactcaataatGAAATTAAATTACTATAAATGTTTTATTA-3' 94
164 L 5'-ctatctttaaactacaaatctaacGAAATTAAATTACTATAAATGTTTTATTT-3' 100
164 CM 5'-phosphate-TAGGAGGTTCCGTTGTTTATC-biotin-3'
pfdhps 540 K 5'-ctacaaacaaacaaacattatcaaGGAAATCCACATACAATGGATA-3' 28
540 E 5'-ctttaatcctttatcactttatcaGAAATCCACATACAATGGATG-3' 17
540 CM 5'-phosphate-AACTAACAAATTATGATAATCTAG-biotin-3'
581 A 5'-aatctaacaaactcctctaaatacTTGATATTGGATTAGGATTTGC-3' 76
581 G 5'-ctttcaattacaatactcattacaTTGATATTGGATTAGGATTTGG-3' 43
581 CM 5'-phosphate-GAAGAAACATGATCAATCTATTA-biotin-3'
613 A 5'-tacactttaaacttactacactaaGATATTCAAGAAAAAGATTTATTG-3' 95
613 S 5'-aaacaaacttcacatctcaataatGGATATTCAAGAAAAAGATTTATTT-3' 48
613 CM 5'-phosphate-CCCATTGCATGAATGATCAAA-biotin-3'

Chapter 2 Methods and Materials
80
2.3.4.4 Bio-plex Fluorescent Detection
The SNP or species-specific LDR products with microsphere-labelled anti-tag probes
were detected by dual-fluorescence flow cytometry in the Bio-Plex array reader (Bio-
Rad Laboratories, CA). The chamber temperature was set to 37°C as reporter signals
were collected into allele-specific or species-specific classification bins via the Bio-Rad
software, Bio-Plex Manager 3.0 (Figure 2.10).
Figure 2.10 Bio-plex array reader. LDR-FMA Fluorescence signals of the
Plasmodium species and drug resistance SNPs are detected and sorted into
classification bins by the Bio-plex reader.
2.4 SOLID PHASE MICRO-EXTRACTION (SPME)
Solid phase micro-extraction (SPME) is a sensitive and simple method for the
extraction of volatile organic compounds (VOCs) from gaseous or liquid samples. This
technique was used to detect VOCs in the headspace of P. falciparum cultures. The
SPME fibre (SUPELCO™, Sigma Aldrich) is bonded to a plunger inside a protective
needle (Figure 2.11). The fibre was conditioned initially according to the
manufacturer’s instructions (PDMS phase 250°C for 0.5 hr and triple fibre phase 270°C

Chapter 2 Methods and Materials
81
for 1 hr. Before each analysis, the fibre was activated in the injector port of the gas
chromatography (GC) at 250°C for 5 min and repeated after each sampling. The SPME
fibre was introduced into the headspace of the flask by gently pushing the protective
needle through the septum that sealed the sample flask. The plunger was lowered to
expose the adsorbent fibre to the gaseous phase for one hour at 35⁰C. During this time,
equilibrium between the atmosphere and the fibre was achieved, and the volatile and
semi-volatile organic compounds were adsorbed onto the coating of the fibre. After
retracting the fibre and withdrawing the needle, the syringe sampler was taken to the
gas chromatography-mass spectrometry (GC-MS) for desorption and subsequent
analysis of VOCs. Details of VOCs experiments are described in Chapter 7.
Figure 2.11 Solid phase micro-extraction sampler.

Chapter 2 Methods and Materials
82

CHAPTER 3
IN VITRO SENSITIVITY OF
P. FALCIPARUM TO CONVENTIONAL
AND NEW DRUGS IN
PAPUA NEW GUINEA

Chapter 3 In vitro drug sensitivity of PNG field isolates
84
CHAPTER 3. IN VITRO SENSITIVITY OF P.
FALCIPARUM TO NEW AND CONVENTIONAL DRUGS
IN PAPUA NEW GUINEA
3.1 INTRODUCTION
Resistance of P. falciparum to CQ first emerged in PNG in the 1970’s (Saint-Yves
1971; Grimmond et al. 1976; Yung et al. 1976; Han 1978). As these treatment failures
were infrequent and low grade (RI), CQ or amodiaquine (AQ) were initially retained as
recommended therapy for uncomplicated malaria. However, because higher grade (RII
and RIII) in vivo resistance to CQ and AQ became more widespread (Darlow et al.
1981; Dulay et al. 1987; Schuurkamp et al. 1989; Sapak et al. 1991; Trenholme et al.
1993; Al-Yaman et al. 1996), the PNG Health Department added sulfadoxine-
pyrimethamine (SP) in 2000 to improve clinical efficacy (Casey et al. 2004). This
approach provided relatively brief respite. Further in vivo studies (Marfurt et al. 2007)
and a recently-published large-scale comparative efficacy trial (Karunajeewa et al.
2008a) demonstrated that neither CQ-SP nor AQ-SP met WHO criteria for retention as
first-line treatment in PNG (WHO 2006). As a result, the ACT artemether-lumefantrine
replaced these regimens in 2010. An overview of in vitro and treatment outcome studies
conducted in PNG is presented (Table 3.1)
Because of their relative simplicity and low cost compared with in vivo assessment, in
vitro tests of parasite drug sensitivity can serve as an early warning system for the
emergence of drug resistance (WHO 2005). This includes artemisinin and the longer
half-life partner components of ACT (Price et al. 2004; Ekland et al. 2008; Noedl et al.
2008). The antimalarial activity of the partner drug appears especially important for
selection of an appropriate ACT in countries such as PNG (Karunajeewa et al. 2008b),
but there is often a lack of in vitro data to facilitate this choice. In PNG, for example,
the most recent parasite sensitivity data are from the study period 1995 - 1996 for CQ,
AQ and antifolate drugs (Genton et al. 2005). In this study, the in vitro antimalarial
activities of a range of conventional and novel antimalarial drugs was assessed in P.
falciparum isolates collected from Madang Province where malaria transmission is

Chapter 3 In vitro drug sensitivity of PNG field isolates
85
hyperendemic (Cattani et al. 1986; Mueller et al. 2003) and where there has been
evidence of progression of parasite drug resistance (Darlow et al. 1981; Trenholme et
al. 1993; Al-Yaman et al. 1996; Al-Yaman et al. 1997; Casey et al. 2004; Marfurt et al.
2007).

Chapter 3 In vitro drug sensitivity of PNG field isolates
86
Table 3.1 Overview of in vitro drug sensitivity findings in PNG.
IVR = In vitro resistance reported as percentage, where no inhibitory concentrations were available. Reported median or mean IC50s of antimalarials;
chloroquine (CQ), amodiaquine (AQ), quinine (Q), mefloquine (MQ), halofantrine (HF) and Pyrimethamine (PYR), Cycloguanil (CYC); Schizont
Maturation = microtechnique where drug sensitivity was determined by schizont maturation. (Al-Yaman et al. 1996; Reeder et al. 1996; Hombhanje et
al. 1998a; Hombhanje 1998b; Genton et al. 2005; Mita et al. 2006b; Wong et al. 2010)

Chapter 3 In vitro drug sensitivity of PNG field isolates
87
Authors Province Study
Period
Samples
(n)
CQ AQ Q MQ HF PYC CYC Method
Mita et al
2006
East Sepik 2002-2003 57 IVR 82%
Mean
88 to 107nM
- - - - - - Schizont
Maturation
Genton et
al 2005
East Sepik 1994-1995 36 IVR 50% IVR 27% IVR 0% IVR 8% IVR 0% IVR 4% IVR 6% Schizont
Maturation
Hombhanje
1998b
Central 1995-1996 30 IVR 50%
Median
1150nM
- IVR 10%
Median
2760nM
IVR 0%
Median
350nM
IVR 0.7%
Median
1.0nM
- - Schizont
Maturation
Hombhanje
et al 1998a
Central Not stated 21 - - - - Median
1.5nM
- - Schizont
Maturation
al-Yaman
et al 1996
Madang 1990-1993 35 IVR 86% IVR 86% IVR 7% - - - - Schizont
Maturation
Reeder et al
1996
East Sepik 1990-1992 24 - - - - - IVR 38%
IVR 34%
Schizont
Maturation

Chapter 3 In vitro drug sensitivity of PNG field isolates
88
3.2 MATERIALS AND METHODS
3.2.1 Study Site and Sample Collection
The present study was carried out at the PNG Institute of Medical Research, Yagaum
Hospital, Madang, PNG. The study utilised blood samples taken in 2006 and 2007 from
children aged 6 months to 10 years as part of clinical studies conducted at the
Alexishafen Health Centre in Madang Province and at Modilon Hospital in Madang
town. In all cases, informed consent was obtained from the parents or legal guardians
before recruitment and blood sampling. Scientific and ethical approval for each study
was obtained from the Medical Research and Advisory Committee of the Ministry of
Health of PNG. Of the 64 samples collected, 45 (70.3%) were from children recruited
to a randomised trial of uncomplicated malaria (Karunajeewa et al. 2008b). The
remaining 18 samples (29.7%) were from pharmacokinetic studies conducted at
Alexishafen or studies of severe malaria in progress at Modilon Hospital.
3.2.2 In vitro Culture of Parasite Isolates
In vitro culture of P. falciparum isolates from paediatric patients followed similar
methods to those described in section 2.1.3, with modifications to suit field conditions.
After initial diagnosis of P. falciparum infection from a finger-prick blood smear, 4 mL
venous blood were collected into a heparin-containing tube and transported to the
laboratory within 24 hr. After centrifugation, both plasma and buffy coat were removed.
The packed RBC were washed twice in culture medium and blood smears were
prepared for confirmation of P. falciparum mono-infection and quantification of
parasitaemia by microscopy. Parasites were cultured at 37°C using a modified candle
jar method to create a CO2-rich microaerophilic atmosphere (Trager et al. 1976) (Figure
3.1). The composition of the culture medium was also different under field conditions
(Appendix B). Parasites were maintained in type O human RBC from a non-immune
individual at 5% hct in RPMI 1640 media supplemented with neomycin as well as
gentamycin. Since non-immune human plasma was not available, Albumax II (Gibco)
was used in the complete media (Appendix B).

Chapter 3 In vitro drug sensitivity of PNG field isolates
89
Figure 3.1 Candle jar method used for P. falciparum culture in PNG. A container
into which a lit candle is placed prior sealing the airtight lid. The flame burns until
extinguished by O2 deprivation, creating a CO2-rich microaerophilic environment.
3.2.3 Drug Susceptibility Assays
Stock solutions of CQ diphosphate (Sigma Chemicals, St Louis, USA), AQ
dihydrochloride (Sigma), monodesethyl-AQ (dAQ) (Sapec Fine Chemicals, Lugano,
Switzerland), piperaquine tetraphosphate (PQ) (Yick-Vic Chemicals and
Pharmaceuticals, Hong Kong), naphthoquine phosphate (NQ) (ZYF Pharm Chemical,
Shanghai, China), mefloquine hydrochloride (MQ) (Sigma), lumefantrine (LM)
(Novartis Pharma, Basel, Switzerland), dihydroartemisinin (DHA) (Sigma) and
azithromycin (AZ) (Pfizer, NSW, Australia) were prepared as described in Section
2.2.1.
Drug sensitivity was assessed in triplicate in 96-well plates, with each well containing
100 µL drug-containing media and 100 µL parasitised RBC suspension at 0.5 - 1.0%
parasitaemia and a final hct of 1.5% (Section 2.2). With the exception of AZ, which

Chapter 3 In vitro drug sensitivity of PNG field isolates
90
was incubated for 72 hr due to its slower antimalarial activity (Noedl et al. 2006), all
other plates were incubated for 48 hr at 37°C. The plates were then subjected to four
freeze-thaw cycles to achieve complete haemolysis. The haemolysate were kept frozen
until assayed. A modification of a Plasmodium lactate dehydrogenase (pLDH) detection
method was used to assess parasite growth as detailed in Section 2.2.4 (Makler et al.
1993a; Makler et al. 1993b).
3.2.4 Assay Validation
For validation purposes, the pLDH assay was compared to the reference 3H-
hypoxanthine incorporation method (Chulay et al. 1983) using culture-adapted CQ-
sensitive and CQ-resistant strains of P. falciparum 3D7, W2mef and E8B and a panel of
antimalarial drugs. Briefly, two sets of CQ, MQ and DHA drug dilutions in either
complete media or complete media without hypoxanthine as appropriate for the pLDH
and 3H-hypoxanthine assays, respectively. To allow for between-day variability in assay
performance the drug sensitivity of each P. falciparum strain was assessed
simultaneously using the two methods.
3.2.5 Data Analysis
Statistical analyses were performed using GraphPad PRISM version 4.0 (GraphPad
Software, CA) and Microsoft Excel for Windows. The concentration of drug required to
inhibit parasite growth by 50% (IC50) and 90% (IC90) for each antimalarial drug as
measured by pLDH assay were determined by non-linear regression analysis of
logarithmically-transformed dose-response curves using HN NonLin v.1.1, a free tool
for malaria in vitro drug sensitivity analysis (Noedl 2002). Comparisons between the
IC50 values in laboratory-adapted strains obtained by pLDH and 3H-hypoxanthine
incorporation assays were made using regression analysis (Bablok et al. 1988) and the
Bland and Altman method (Bland et al. 1986). One-way analysis of variance (ANOVA)
was used to compare differences between the IC50 values obtained from the three
different measurement time-points of the pLDH reaction. Associations between IC50
and IC90 values of drug pairs for evaluation of in vitro cross resistance were assessed
using Spearman’s rank correlation co-efficient. Because of the number of comparisons,

Chapter 3 In vitro drug sensitivity of PNG field isolates
91
a significant P-value <0.05 was used throughout. Correlations between the IC50s of CQ,
PQ and LM in a subset of the present patients have been reported previously
(Karunajeewa et al. 2008b).
3.3 RESULTS
3.3.1 Comparison of pLDH and Isotopic Assays
The pLDH assay was compared to the reference 3H-hypoxanthine incorporation method
in cultured-adapted P. falciparum (Figure 3.2). There was a significant linear
correlation between the data obtained by the two IC50 assay methods (r2=0.97, n=26;
P=0.001), with a slope and intercept ([95% confidence intervals]) of 1.13 [1.00 to 1.25]
and 7.83 [-3.3 to 18.98] nM, respectively. The Bland-Altman plot showed that the
pLDH assay sometimes significantly underestimated the IC50 at high values and
provided the least reliable estimations at IC50s >200 nM.
3.3.2 Effect of pLDH Reaction Duration on IC50 Values
For simplicity and efficiency, a single endpoint was used to interpret the results of the
pLDH assay; specifically measurement of the OD when substantial colour contrast had
developed during the enzyme reaction, rather than multiple measurements at 30 sec
intervals over 30 min as originally described (Makler et al. 1993a). In laboratory-
adapted P. falciparum, 45 to 60 min of incubation was adequate for visual evaluation of
dye reduction. At this point in the reaction, the colouration in drug-free wells is intense
(purple), reflecting heavy parasite growth, whilst non-parasitised control wells show
minimal colour change (pink) (see Figure 2.6). In field isolates however, the rate of
pLDH dye reduction varied between samples, with some requiring up to 2 hr to produce
maximal contrast.
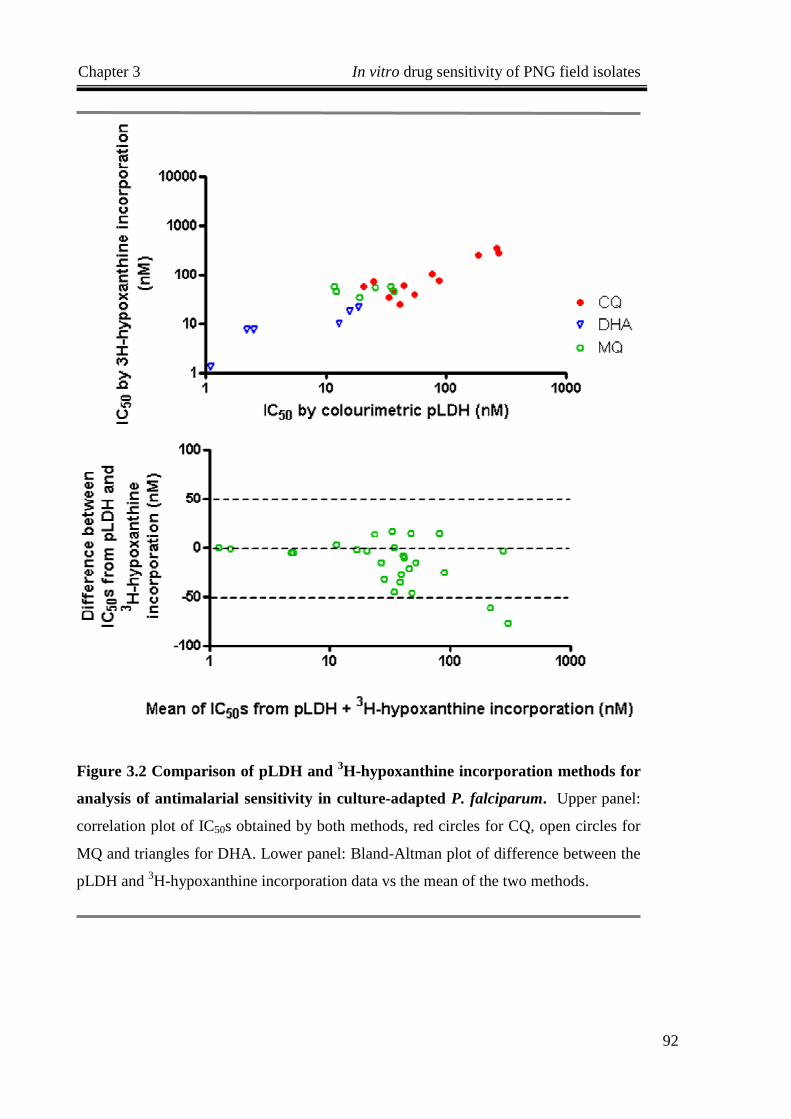
Chapter 3 In vitro drug sensitivity of PNG field isolates
92
Figure 3.2 Comparison of pLDH and 3H-hypoxanthine incorporation methods for
analysis of antimalarial sensitivity in culture-adapted P. falciparum. Upper panel:
correlation plot of IC50s obtained by both methods, red circles for CQ, open circles for
MQ and triangles for DHA. Lower panel: Bland-Altman plot of difference between the
pLDH and 3H-hypoxanthine incorporation data vs the mean of the two methods.

Chapter 3 In vitro drug sensitivity of PNG field isolates
93
To investigate whether the IC50 values calculated for antimalarial drugs were influenced
by the duration of pLDH incubation, dose-responses curves for six antimalarial agents
assessed against four field isolates were determined from OD measurements at 80, 120
and 180 min, and their respective IC50s compared (Figure 3.3). The majority of isolates
tested produced similar dose-response curves to each respective antimalarial drug over
a 3 hr period (data not shown). In some isolates (e.g. 106 and 112 in Figure 3.3), an
increase in CQ IC50 was observed with reaction time. This increase was due to greater
colour contrast as the reaction proceeded, causing changes in the slope of the dose-
response curve. Overall, there were no significant differences between IC50s calculated
from data measured at 80 to 180 min (n=93; P=0.60). When IC50s were plotted against
pLDH reaction time (Figure 3.4), the general distribution of data points was similar
between groups, with collective median IC50s of 15.7, 15.9 and 16.7 nM at 80, 120 and
180 min, respectively.
3.3.3 Field Application of the pLDH Assay
The in vitro activities of nine antimalarial drugs against P. falciparum isolates from
paediatric patients with uncomplicated malaria were evaluated. Blood samples with
parasitaemia ranged between 0.3 to 14% were included in the drug sensitivity assay.
Samples with inadequate RBC were only screened against 4-aminoquinolines drugs. Of
125 field isolates obtained, 64 (51%) were both cultured successfully and provided
valid data drug sensitivity data by pLDH assay. Loss of isolates reflected mainly
logistic issues with transportation, and laboratory-related problems including power
supply reliability, reagent availability, clotting of blood samples due to inadequate
mixing with anticoagulant and bacterial contamination. Experiments involving LM,
DHA and AZ were only undertaken during the latter part of the study period. The
distribution of IC50s in response to antimalarial drugs is illustrated in Figure 3.5.

Chapter 3 In vitro drug sensitivity of PNG field isolates
94
Figure 3.3 Effect of pLDH reaction time on IC50s in PNG P. falciparum. Four
samples of P. falciparum were tested against six antimalarial drugs; chloroquine (CQ);
piperaquine (PQ); desethyl-amodiaquine (dAQ); amodiaquine (AQ); naphthoquine
(NQ) and mefloquine (MQ). Optical measurements were taken at 80, 120 and 180 min
into the pLDH reaction. IC50s were calculated for each time point for comparison.

Chapter 3 In vitro drug sensitivity of PNG field isolates
95
Figure 3.4 Scatter plot of IC50s determined from three pLDH time points. IC50
values for CQ, PQ, AQ, dAQ and MQ at three different time points were compared in
18 field isolates of P. falciparum. Optical measurements were collected at 80, 120 and
180 min into the pLDH reaction from which IC50s were determined and plotted. The
median for each time point is shown.

Chapter 3 In vitro drug sensitivity of PNG field isolates
96
Figure 3.5 Distribution of 50% inhibitory concentrations (IC50) of antimalarials against PNG P. falciparum isolates.
Piperaquine (PQ), amodiaquine (AQ), desethyl-amodiaquine (dAQ), chloroquine (CQ), naphthoquine (NQ), mefloquine (MQ), dihydroartemisinin
(DHA), lumefantrine (LM) and azithromycin (AZ).

Chapter 3 In vitro drug sensitivity of PNG field isolates
97
3.3.4 Antimalarial Susceptibility of PNG P. falciparum Isolates
The mean IC50 and IC90 values for the nine antimalarial drugs are summarised in Table
3.2. The accepted in vitro resistance threshold for CQ (≥100 nM) is based on the in
vitro response of African isolates obtained from malaria-infected, non-immune
individuals taking CQ prophylaxis and semi-immune patients failing to respond to CQ
treatment (Le Bras et al. 1990; Cremer et al. 1995; Ringwald et al. 1996; Basco et al.
2002). This threshold was obtained using the reference 3H-hypoxanthine incorporation
method but has been employed in many subsequent studies using different techniques
for in vitro drug susceptibility assessment (Attlmayr et al. 2006; Kaddouri et al. 2006;
Mayxay et al. 2007; Nkhoma et al. 2007). Because there was close agreement between
the pLDH and 3H-hypoxanthine methods in the present study at this concentration, a
100 nM cut-point was used, with 82% of isolates tested exhibiting an IC50 above this
level. In vitro resistance thresholds for the other antimalarial drugs tested have not been
established through valid correlative in vivo studies.

Chapter 3 In vitro drug sensitivity of PNG field isolates
98
Table 3.2 In vitro susceptibilities of P. falciparum PNG isolates against 4-
aminoquinolines and other antimalarial drugs. IC50 = 50% inhibitory drug
concentrations and IC90 = 90% inhibitory drug concentrations, CI = confidence interval.
3.3.5 Correlations of in vitro Responses to Nine Antimalarials
Correlations between IC50 values for the panel of antimalarial drugs are shown in Table
3.3. Apart from LM, there were strong associations between the IC50s of 4-
aminoquinoline (CQ, AQ, DAQ and NQ), bisquinoline (PQ) and aryl-aminoalcohol
(MQ) compounds. Although the numbers of isolates tested were low, AZ activity did
not correlate significantly with that of any other drug. The artemisinin derivative DHA
IC50 values showed positive associations with those of the 4-aminoquinoline and related
compounds.
Isolates tested (n)
IC50 (nM) Mean (95% CI)
IC90 (nM) Mean (95% CI)
Chloroquine 63 215 (175 – 254) 431 (382 – 480)
Amodiaquine 64 26.3 (18.8 – 33.7) 54.3 (42.9 – 65.7)
Desethyl-amodiaquine 60 23.1 (19.4 – 26.8) 48.8 (38.7 – 58.8)
Piperaquine 57 15.5 (10.8 – 20.2) 48.8 (30.3 – 67.4)
Naphthoquine 41 10.3 (6.7 – 13.9) 31.0 (21.0 – 41.0)
Mefloquine 58 7.8 (5.8 – 9.8) 21.2 (16.2 – 26.2)
Lumefantrine 25 4.6 (1.1 – 8.0) 12.7 (5.7 – 19.7)
Dihydroartemisinin 30 3.5 (1.9 – 5.1) 11.1 (7.2 – 15.0)
Azithromycin 15 13,895 (6,277 – 21,513)
39,471 (21,300 – 57,642)

Chapter 3 In vitro drug sensitivity of PNG field isolates
99
Table 3.3 Spearman correlation co-efficients for associations between IC50 values.
The number of drug pairs analysed are shown in parentheses. *P<0.05, **P<0.01, ***P<0.001.
Chloroquine Amodiaquine Desethyl-
amodiaquine Piperaquine Naphthoquine Mefloquine Lumefantrine Dihydro-
artemisinin
Amodiaquine 0.46***
(60)
Desethyl-amodiaquine
0.45***
(58)
0.61***
(57)
Piperaquine 0.51***
(54)
0.59***
(53)
0.51***
(55)
Naphthoquine 0.56***
(37)
0.61***
(36)
0.64***
(37)
0.74***
(36)
Mefloquine 0.61***
(54)
0.52***
(57)
0.37**
(52)
0.52***
(48)
0.52**
(32)
Lumefantrine 0.09
(22)
0.18
(24)
0.35
(19)
0.17
(17)
0.25
(7)
0.47*
(23)
Dihydro- artemisinin
0.45*
(27)
0.37*
(29)
0.43*
(24)
0.22
(20)
0.84**
(10)
0.57**
(28)
0.31
(23)
Azithromycin 0.56
(12)
0.12
(13)
0.54
(9)
0.18
(9)
-0.50
(3)
0.24
(12)
-0.12
(12)
-0.29
(12)

Chapter 3 In vitro drug sensitivity of PNG field isolates
100
3.4 DISCUSSION
The high prevalence of in vitro CQ resistance in the present study accords with recent
local molecular and clinical data. A number of genotyping studies have reported near-
fixation of the CQ resistance-associated mutation pfcrt in PNG (Mehlotra et al. 2005;
Carnevale et al. 2007). In addition, the significant rates of in vivo CQ-SP treatment
failure in the Madang area (Marfurt et al. 2007; Karunajeewa et al. 2008b) are also
consistent with the present in vitro findings, although mutations associated with SP
resistance will have contributed (Casey et al. 2004; Mita et al. 2007; Saito-Nakano et
al. 2008).
The IC50s for AQ and its active metabolite dAQ in the present study were much lower
than in previous studies from PNG (Trenholme et al. 1993; Al-Yaman et al. 1996). This
is likely to reflect methodological differences, since microscopic assessment of schizont
maturation produces IC50 values several times higher than those derived from
radioisotope incorporation (Wernsdorfer et al. 1988) and, by implication, from the
pLDH assay. However, consistent with the present IC50 data, a recent study from
neighbouring East Sepik Province found a much lower prevalence of in vitro resistance
to AQ than CQ (Genton et al. 2005).
Clinical studies in PNG have found equivalent high treatment failure rates for AQ-SP
and CQ-SP (Marfurt et al. 2007; Karunajeewa et al. 2008b). However, AQ-SP is used
in younger children (those <19 kg in body weight) than CQ-SP under PNG national
treatment guidelines (PNGDOH 2000). This means that a lack of immunity may offset
relative parasite sensitivity to AQ in this age-group, producing comparable failure rates
to those with CQ-SP in older children. Indeed, AQ is more effective than CQ in African
children of similar age (Brasseur et al. 1999; Oduro et al. 2005; Pradines et al. 2006).
Nevertheless, there may not be a clear relationship between AQ in vitro parasite
sensitivity and clinical outcome (Trenholme et al. 1993; Pradines et al. 2006).
A valid in vitro resistance threshold for PQ remains to be confirmed. An IC50 <100 nM
has been used to identify sensitive strains of P. falciparum by radioisotope uptake
(Deloron et al. 1985; Basco 2003b; Mwai et al. 2009b), while Chinese investigators

Chapter 3 In vitro drug sensitivity of PNG field isolates
101
have reported resistant isolates with IC50 values >300 nM using the schizont maturation
microtechnique (Yang et al. 1999b; Lin et al. 2005). All PQ IC50 values in the present
study were <100 nM. Sixteen of the isolates were from children treated with DHA-PQ
in the recent comparative clinical trial (Karunajeewa et al. 2008b) and one (IC50 19.5
nM) was a late parasitological failure. These various data suggest that further in vivo-in
vitro correlation studies are needed to establish a clinically meaningful resistance
threshold for PQ.
MQ has not been used previously in PNG. All isolates had MQ IC50 values below the
resistance threshold of 108 nM established recently using in vivo responses, 3H-
hypoxanthine uptake and molecular characteristics including the number of copies of
the P. falciparum multidrug resistance 1 (pfmdr1) gene (Price et al. 2004). This
threshold was also employed in a study of field isolates from Laos which were assessed
using the pLDH method (Mayxay et al. 2007). The present in vitro MQ data are
consistent with the results of a recent molecular survey that reported an absence of
multiple copies of pfmdr1 gene in PNG isolates (Hodel et al. 2008).
Data from this current study are the first characterising the in vitro sensitivity of PNG
isolates to LM and NQ, drugs that have both recently become available as part of ACT
in PNG. A previously published resistance threshold for LM of >150 nM was based on 3H-hypoxanthine uptake studies in African isolates without in vivo correlation (Basco et
al. 1998). None of the PNG isolates had an IC50 value above this level. There are no
equivalent published cut-points for NQ but the median IC50 (10.3 nM) was less than
those reported for isolates from Southern China using the micro-test method (mean 88.5
nM for ‘artesunate-sensitive’ and 119.4 nM for ‘artesunate-resistant’ strains) (Yang et
al. 1999a). The IC50 values for DHA were all below the suggested cut-point of 10.5 nM
derived from 3H-hypoxanthine uptake studies in African isolates without in vivo
correlation (Pradines et al. 1998). The AZ IC50s from the present study (mean 13.9 µM)
are also largely below a previously reported range derived from Thai isolates and the
micro-test technique (mean 29.3 µM) (Noedl et al. 2001).
The positive inter-correlations between IC50 values for 4-aminoquinoline and related

Chapter 3 In vitro drug sensitivity of PNG field isolates
102
compounds have been reported in studies from other countries (Fan et al. 1998; Yang et
al. 1999a; Basco et al. 2003a; Pradines et al. 2006). These findings are consistent with
the observation that pfcrt alleles influence parasite susceptibility to drugs other than CQ
such as MQ (Sidhu et al. 2002; Johnson et al. 2004; Sidhu et al. 2005), but other
mechanisms such as common drug-specific effects on parasite haem polymerase may
be involved (Slater et al. 1992; Dorn et al. 1998). It is also possible that the positive
associations in the present study reflect general parasite fitness rather than shared
resistance determinants, but the lack of significant associations involving LM and AZ,
also reported by others (Noedl et al. 2001; Noedl et al. 2007), are against this. The IC50
values for PQ correlates with CQ, AQ and dAQ in the present study. However, this is
not the case in the African data set (Mwai et al. 2009b), raising questions about
mechanisms of action of PQ. In addition, the African study showed consistent inverse
relationship between LM sensitivities and CQ which has also be reported by others
(Pradines et al. 1999b; Price et al. 2006). These differences in the African and PNG
findings may be due to variations in methodologies (i.e. longer assay time of 84 hr,
adaption of field isolates to long-term culture prior to sensitivity testing) and different
histories of parasite drug exposure. As with most antibiotics with antimalarial activity,
the macrolide AZ is relatively weak and slow-acting, and best used as adjunctive
therapy (Anderson et al. 1995; Noedl et al. 2006; Noedl et al. 2007).
Lumefantrine and MQ are aryl-aminoalcohols with related chemical structures and a
similar mode of action (Peel et al. 1994; Basco et al. 1998; Pradines et al. 2006).
However, a significant correlation was found between the LM IC50s and those of MQ
but not with the other long half-life antimalarial drugs. This observation is in accord
with previous reports (Basco et al. 1998; Pradines et al. 2006) and is also consistent
with the significantly better clinical response to artemether-LM than DHA-PQ in a
recent comparative trial (Karunajeewa et al. 2008b). There were generally weak
associations between DHA IC50s and those of other drugs, consistent with the findings
of others (Basco et al. 2003a; Attlmayr et al. 2005; Pradines et al. 2006; Noedl et al.
2007). There is some evidence that pfcrt status influences the antimalarial activity of the
artemisinin derivatives (Sidhu et al. 2002), but the known association between P.
falciparum sensitivity to these drugs and pfmdr1 mutations and copy number (Sidhu et
al. 2005; Sidhu et al. 2006) does not apply in PNG (Hodel et al. 2008).

Chapter 3 In vitro drug sensitivity of PNG field isolates
103
PLDH assessment of P. falciparum drug sensitivity was originally described as a
kinetic assay requiring repeated measurement of absorbance (Makler et al. 1993a). This
method determines the mean Vmax (mOD/min) of pLDH activity reflecting parasite
growth (Makler et al. 1993a; Basco et al. 1995). A recent field study in Malawi
employed a similar single pLDH measurement as used in the present study (Druilhe et
al. 2001; Nkhoma et al. 2007). The IC50s determined by the kinetic approach correlated
positively with those determined by the reference isotopic method (Makler et al. 1993a;
Basco et al. 1995). In the interests of simplicity and efficiency in the field where a
spectrophotometer was not readily accessible, non-kinetic assessment of pLDH activity
was more convenient. The IC50s from this approach correlated well with the isotopic
method although it tended to overestimate the IC50s above 100 nM as shown in the
Bland-Altman analysis.
Unlike culture-adapted strains, P. falciparum isolates from patients varied in their in
vitro growth and consequent pLDH activity. A number of isolates failed to develop over
the 48 hr incubation period, hence little colour contrast was observed between drug-free
controls, antimalarial-dosed and non-parasitised control wells on pLDH assay. Some
isolates developed colour intensities more slowly than others, for which the OD at 180
min into the pLDH reaction was measured. The use of a pre-test was helpful in reducing
reagent wastage and for determining measurement time. This involved first testing the
haemolysate from one drug-free and one non-parasitised well from each sample and
time for colour development prior to running all three 96-well plates.
During validation of the modified pLDH method, efforts were made to ensure
comparability with the reference 3H-hypoxanthine incorporation technique. Factors
underlying between-day variations of in vitro estimates of drug sensitivity include the
time-dependent growth characteristics of the cultures. To minimise such effects on
assay performance, both assays were conducted in parallel and on the same day. In
order to reduce unnecessary transfer and handling of radioactive material, two sets of
drug dilutions were prepared for each method (one lacking hypoxanthine) rather than
taking an aliquot of cell suspension from the isotopic test wells for pLDH assessment,
as has been done previously (Makler et al. 1993a). Two data points showed

Chapter 3 In vitro drug sensitivity of PNG field isolates
104
unexpectedly high IC50s by the isotopic method for MQ (Figure 3.2), the influence of
these outliers may be reduced if more samples were examined for this drug. Agreement
between the two methods may be more substantial if the drug dilutions used or
haemolysate were from the same experiment set. Although it is worth noting that
previous reports have only compared correlations and not the agreement between the
two methods (Basco et al. 1995; Delhaes 1999).
The present study provides baseline data at a time when, as a result of the findings of a
large-scale clinical trial (Karunajeewa et al. 2008b), the treatment of uncomplicated
malaria in PNG will change from AQ-SP or CQ-SP to artemether-LM. Ongoing
assessment of in vitro sensitivity using the same techniques will facilitate assessment of
the adequacy of such treatment. Conventional monitoring involves the WHO micro-test
with labour-intensive visual enumeration of schizonts (Al-Yaman et al. 1996;
Hombhanje 1998b). The colourimetric pLDH assay allows prompt semi-automated
generation of parasite growth data from triplicate experiments involving multiple
antimalarial drugs. The IC50 values generated correlate well with those derived using 3H-hypoxanthine incorporation and there is no issue with disposal of radioisotopes, an
important consideration in countries such as PNG. Assays based on pLDH
quantification have been recently introduced for screening patient isolates against
multiple antimalarial drugs in Africa and Asia (Brockman et al. 2004; Kaddouri et al.
2006; Nkhoma et al. 2007). The assay may serve to monitor the possible reversal of CQ
resistance after its official withdrawal in PNG as evident in Malawi (Laufer et al. 2006).
Rational drug policy in countries such as PNG can only benefit from such convenient,
high-throughput in vitro testing, especially if this is done regularly so that emerging
resistance can be identified with relative confidence at an early stage.
This work for the most part has been published in Tropical Medicine and International
Health, titled “In vitro sensitivity of Plasmodium falciparum to conventional and novel
antimalarial drugs in Papua New Guinea”. Work regarding methodology validation has
been published in Malaria Journal, titled “A comparative study of a flow-cytometry-
based assessment of in vitro Plasmodium falciparum drug sensitivity”. A small portion
of data relating to drug sensitivity has been published in The New England Journal of
Medicine, titled “A trial of combination antimalarial therapies in children from PNG”.

CHAPTER 4
CHARACTERISATION OF DRUG
RESISTANT POLYMORPHISMS OF
P. FALCIPARUM USING A NEW
MOLECULAR ASSAY

Chapter 4 Molecular Characterisation of PNG isolates
106
CHAPTER 4. CHARACTERISATION OF DRUG
RESISTANT POLYMORPHISMS OF P. FALCIPARUM
USING A NEW MOLECULAR ASSAY
4.1 INTRODUCTION
Resistance of Plasmodium species to 4-aminoquinolines emerged in PNG in 1976 and
has since spread across the country (Grimmond et al. 1976; Marfurt et al. 2007). In
addition, mass dosing of pyrimethamine in the 1960’s conveyed continuous drug
pressure on the parasite population that led to the selection of resistant mutations. High-
level resistance has been documented both in vivo (Darlow et al. 1980; Marfurt et al.
2007; Karunajeewa et al. 2008b) and in vitro (Reeder et al. 1996; Mita et al. 2006a).
Chloroquine (CQ) or amodiaquine (AQ) monotherapy was retained as first-line
treatment for uncomplicated malaria until 2000 when sulfadoxine/pyrimethamine (SP)
was added to improve clinical efficacy (Casey et al. 2004). Despite initial success, cure
rates have since declined (Marfurt et al. 2007; Karunajeewa et al. 2008b).
Parasite drug resistance is largely assessed in three ways. The reference assessment is
by clinical efficacy trials where treatment outcome is monitored. However, they are
costly, time consuming, and patient recruitment and follow-up are often difficult (WHO
2005). Parasite drug sensitivity can be tested in vitro, but it can be labour intensive
particularly if multiple drugs are to be screened for each sample. With advancing
technology, molecular surveillance of malaria resistance is increasingly valuable as it
overcomes many challenges associated with clinical and in vitro approaches.
Single nucleotide polymorphisms (SNPs) in parasite genes determining drug effects can
underlie resistance. Mutations in the P. falciparum chloroquine transporter (pfcrt) gene,
in particular K76T, is central to determining the phenotype of CQ resistance and in
predicting treatment failure (Fidock et al. 2000; Basco et al. 2002). The pfcrt K76T
mutation is often associated within different amino acid haplotypes (CVIET, CVMNT,
CVMET, or SVMNT residues 72-76), however the roles of these haplotypes is not well

Chapter 4 Molecular Characterisation of PNG isolates
107
defined except that they are reflective of the parasite’s geographic origin (Fidock et al.
2000).
Higher-levels of CQ resistance result from other SNPs and is inversely associated with
the copy number of the multidrug resistance 1 (pfmdr1) gene (Foote et al. 1990; Reed et
al. 2000; Babiker et al. 2001; Pickard et al. 2003). Pfmdr1 gene polymorphisms also
confer resistance to other antimalarials including quinine, mefloquine, lumefantrine and
halofantrine (Cowman et al. 1994; Peel et al. 1994; Reed et al. 2000). Of particular
concern are the results of a pfmdr1 gene allelic replacement study in which various
polymorphisms reduced artemisinin susceptibility in cloned parasite lines (Reed et al.
2000). The study showed pfmdr1 polymorphisms at codons 86, 1034, 1042 and 1246
altered artemisinin susceptibility in D10 and 7G8, originating from PNG and South
America, respectively. This finding has serious implications for future prospect of
artemisinin effectiveness in endemic areas.
Polymorphic changes in the genes encoding dihydrofolate reductase (dhfr) and
dihydropteroate synthetase (dhps) underlie parasite resistance to pyrimethamine
(Cowman et al. 1988; Peterson et al. 1988) and sulfadoxine (Triglia et al. 1994; Triglia
et al. 1999), respectively. The S108N mutation in dhfr is a primary determinant of
pyrimethamine resistance and additional mutations at codons 16, 50, 51, 59, 140 and
164 cause higher-level resistance (Cowman et al. 1988; Peterson et al. 1988). Similarly,
polymorphisms involving S436A/F, A437G, and K540E in the pfdhps gene confer
initial mutation to sulfadoxine. Other genetic alterations such as A581G and S613A will
lead to higher-level resistance (Triglia et al. 1994; Triglia et al. 1999). Almost all
strains of P. falciparum from patients from Madang Province in PNG who fail CQ-SP
treatment carry pfcrt K76T and pfmdr1 N86Y, while pfdhfr C59R and S108N are also
found at moderate/high levels, reflecting the selective pressure from long periods of CQ
and pyrimethamine usage (Casey et al. 2004; Carnevale et al. 2007).
At present, most molecular techniques for SNP analysis are based on PCR restriction
fragment length polymorphism (RFLP), sequence-specific oligonucleotide probe
hybridisation (SSOPH) and direct sequencing. However, most such methods identify a

Chapter 4 Molecular Characterisation of PNG isolates
108
small number of candidate SNPs regarded as primary predictors of clinical resistance
(Ranford-Cartwright et al. 2002). Mutations that are not directly involved in resistance
but which may have compensatory or modulating effects that contribute to the overall
phenotype are often omitted. An approach based on DNA microarray allows parallel
detection of multiple SNPs (Crameri et al. 2007), but remains relatively expensive. An
alternative technique is a post-PCR ligase detection reaction-fluorescent microsphere
assay (LDR-FMA) that enables cost-effective evaluation of 22 SNPs (Carnevale et al.
2007).
In view of the importance of a low-cost system for large-scale monitoring of drug
resistance in developing countries, the present study further expanded this system to
detect an additional 10 different pfmdr1 allelic variants. In addition to assay
development, this new technique has been applied in a study of key drug resistance
mutations in P. falciparum field isolates from clinical studies conducted in PNG. The
prevalence of different allelic variants of the pfcrt, pfdhfr, pfdhps and pfmdr1 genes are
presented. Associations between these mutations and treatment outcome are also
examined.
4.2 MATERIALS AND METHODS
4.2.1 Field Studies, P. falciparum isolates
The present study utilised a subset of 402 samples for Plasmodium speciation from a
large-scale treatment trial in children aged 6 months to 5 years (mean 36 months) with
uncomplicated malaria (Karunajeewa et al. 2008b) (Australian New Zealand Clinical
Trials Registry ACTRN12605000550606). The study was conducted between 2005 and
2007 in Madang and East Sepik Provinces. Participants were assigned CQ-SP,
artesunate-SP (ART-SP), piperaquine-dihydroartemisinin (PQ-DHA) or artemether-
lumefantrine (AL). Children who had been treated with antimalarial drugs within the
previous 14 days were excluded. The samples used in the present study were those
collected at baseline prior to treatment allocation. Full details of the trial protocol have
been published previously (Karunajeewa et al. 2008b).

Chapter 4 Molecular Characterisation of PNG isolates
109
The number of samples that were assayed for pfcrt, pfdhps and pfdhfr genotypes from
the treatment groups CQ-SP, ART-SP, PQ-DHA and AL were 81, 86, 94 and 90,
respectively (total of 351) and for pfmdr1 were 63, 65, 79 and 72 respectively (total of
279) due to limited sample volume. Efficacy was assessed over 42 days using WHO
definitions (WHO 2003) with correction for re-infections by PCR genotyping
(Karunajeewa et al. 2008b), specifically adequate clinical and parasitological response
(ACPR), early treatment failure (ETF; an inadequate parasitological response and/or
worsening of clinical signs by day 3), late parasitological failures (LPF; emergent
parasitaemia between days 4 and 42), or late clinical failure (LCF; where LPF was
associated with fever). Informed consent was obtained from the parents/guardians
before recruitment. Scientific/ethical approvals for the main study and present sub-
study were obtained from the Medical Research and Advisory Committee of the
Ministry of Health of PNG, the University Hospitals Case Medical Centre and the
University of Western Australia Human Research Ethics Committee.
4.2.2 Genomic DNA
Laboratory-adapted P. falciparum strains including 3D7, Dd2, K1, 7G8 and HB3 were
provided by MR4, American Type Culture Collection. DNA was extracted from 200 µL
whole blood (field samples) or haemolysate (laboratory-adapted strains) using the
QIAmp 96 DNA blood kit or DNeasy Blood and Tissue Kit (Qiagen, CA) under the
manufacturer’s protocol (Section 2.3.1).
4.2.3 Plasmodium Speciation
Detection of Plasmodium species was by amplification of ssu rDNA by a modified
multiplex LDR-FMA (McNamara et al. 2006). Parasite genomic DNA served as
templates for the PCR primers flanking the small-subunit rRNA gene fragment (491-
500 base-pairs). This domain contains sequences conserved within the Plasmodium
genus and those that are species-specific (Section 2.3.2.1). All PCR reactions (25 µL)
were performed using a Peltier Thermal Cycler, PTC-225 (MJ Research, MA)
consisting of 3 µL genomic DNA in a master mix containing 3 pmol of appropriate

Chapter 4 Molecular Characterisation of PNG isolates
110
upstream and downstream primers (Section 2.3.2.1).
To evaluate amplification efficiency, the PCR products were visualised by
electrophoresis on 2% agarose gels stained with SYBR Gold and images were acquired
using a Storm 860 (Section 2.3.3). This was followed by species-specific ligase
detection reaction (LDR) as described previously (Section 2.3.4.1 and Table 2.3). LDR
utilises the ability of DNA ligase to preferentially join adjacent oligonucleotides to the
target PCR amplicon where there is a perfect complementation at the junction during
hybridisation.
The second step involves hybridisation of LDR products with anti-tag oligonucleotides
coupled with Plasmodium species-specific microspheres (Section 2.3.4.3). These
microspheres (Luminex Corporation, TX) are embedded with varying ratios of
red:infra-red fluorochromes and emit unique fluorescent ‘classification’ signatures. The
hybridised mixture is then labelled with a reporter dye (streptavidin-R-phycoerythrin,
Molecular Probes, OR) through binding to the biotin end of the LDR conjugate.
Fluorescent signals are sorted into allele-specific ‘bins’ by the bioplex array reader
(Bio-Rad Laboratories, CA).
4.2.4 Detection of Drug Resistant Polymorphisms
Amplification of target sequences for P. falciparum pfdhps, pfdhfr, pfcrt (Carnevale,
2007) and pfmdr1 were achieved using oligoprimers and conditions described in Table
2.2 and Section 4.3.1.1, respectively. Following PCR, the products were combined in a
multiplexed LDR (Table 2.4 and Section 4.3.1.2). Upstream LDR primers are allele-
specific and contain the complementary base to the SNP of interest at the 3’ end (Figure
4.1). The upstream primers were designed to have unique tag sequences of 24
nucleotides at the 5’ end that enables subsequent identification of specific SNPs.
Downstream LDR primers contain conserved sequence oligonucleotides and were 5’
phosphorylated and 3’ biotinylated.

Chapter 4 Molecular Characterisation of PNG isolates
111
Figure 4.1 Principle of LDR-FMA diagnosis of drug resistant polymorphisms.
Top: Main components in the reaction. Middle: During thermocycling, the LDR primers
hybridise to the PCR products with matching base-pairs. If there is a perfect match
between the junctions of adjacent LDR primers, the gap will be sealed by DNA ligase.
A single base mismatch will not result in ligation. Bottom: LDR products are hybridised
with anti-tag oligonucleotides coupled to microspheres that report signals specific to the
SNPs of interest. This is followed by labelling with streptavidin-R-phycoerythrin
(SAPE) through binding to biotin.

Chapter 4 Molecular Characterisation of PNG isolates
112
During thermocycling, the LDR primers hybridise to PCR products with matching base-
pairs. As a result, the LDR primers specific to the gene and SNP of interest are brought
together in close proximity. If there is a perfect match at the junction of the LDR
upstream and downstream primers, the nick will be sealed by a DNA ligase. A single
base mismatch will not result in ligation. Hence, this step is highly specific (Figure 4.1).
Details of the LDR for pfcrt, pfdhfr, and pfdhps are described in Section 2.3.4.2.
Recipes for PCR and LDR master mix solutions and respective primer sequences are
outlined in Appendix B.
4.2.5 Data Analysis
Statistical analysis was performed using GraphPad PRISM version 4.0 (GraphPad
Software, CA). Fluorescent signals from the field samples were classified positive or
negative for drug susceptibility markers according to thresholds determined by
standardised procedures. Fluorescent signals were first normalised to a mean of 10,000
and SD 1,000 arbitrary units for each codon by subtracting the calculated mean from
every signal within the corresponding codon, then multiplying by 1,000/codon-specific
SD, and finally adding 10,000. The same procedure was applied to fluorescent signals
from culture-adapted strains with known genotypes, thus providing controls within each
SNP assay. Once adjusted, codon-specific cut-points that applied to all control strains
were derived with a value that predicted the highest number of true positives as a
conservative cut-point for distinguishing positive signals from background
fluorescence.
A cut-point of >9600 had 97.5%, 98.8% and 98.6% accuracy for predicting true
positive alleles for codons 540, 581 and 613 in the pfdhps gene in control strains. The
>9600 cut-point also applied to codons 1042 and 1246 in the pfmdr1 gene, while >9800
accurately predicted known alleles at codon 86 in the pfmdr1 gene, codons 51, 59, 108
and 164 in the pfdhfr gene, and in the CVMNK, SVMNT, and CVIET pfcrt haplotypes.
A threshold of >10,000 applied to pfmdr1 codons 184 and 1034. By reversing the
normalisation process, the cut-points were made specific to each drug resistance
marker. A similar approach that uses polar-co-ordinates has also been used to determine
thresholds for the LDR-FMA system (DaRe et al. 2010).

Chapter 4 Molecular Characterisation of PNG isolates
113
Mixed strain infections can be identified when fluorescence signals from both alleles
(i.e. wild type and mutated) from the same codon occur above calculated cut-points.
Previous experiments have shown that strain-specific allele fluorescence signals are in
direct proportion to the ratio of the parasite strain densities within the sample
(Carnevale et al. 2007). Therefore, Day 28 and day 42 post-treatment blood samples
from patients from the clinical trial (Karunajeewa et al. 2008b) that were parasite
negative by both microscopy and PCR were assayed from which very low fluorescence
signals (<200) were found. These observations indicate that multiple P. falciparum
strains and non-falciparum DNA such as that from the human host do not interfere with
SNP detection by LDR-FMA. While haplotypes were assigned based on the dominant
allele signals at each locus, they have masked the presence of a minor clone in the case
of a multiple strain infection. Although multiplicity of infection (MOI) was not
calculated in the analysis of drug resistance haplotypes, efforts were made to exclude
samples showing mixed infection at more than two loci. In addition, previous studies
have shown that multiclonal infections are rare in PNG, with a mean MOI of 1.3 - 1.8
(Felger et al. 1994; Cortes et al. 2004).
Associations between parasite mutations and measures of treatment outcome were
assessed using Fisher’s exact test or ANOVA with Bonferroni post hoc adjustment for
multiple comparisons (SPSS v16.0, Chicago IL).
4.3 RESULTS
4.3.1 Pfmdr1 LDR-FMA Development
4.3.1.1 PCR Optimisation
Pfmdr1 SNPs are clustered in two regions approximately 2000 base-pairs (bp) apart.
Two sets of PCR primers (Integrated DNA Technologies) were designed for the
amplification of pfmdr1 polymorphisms at codons 86 and 184 (a total of 4 alleles)
designated as region 1, and polymorphisms at codons 1034, 1042 and 1246 (a total of 6
alleles) as region 2.

Chapter 4 Molecular Characterisation of PNG isolates
114
Previously designed PCR primers and conditions for pfmdr1 regions 1 and 2
(Carnevale, unpublished) were tested in seven laboratory strains of P. falciparum. This
involved two successive runs of PCRs with the second intended to amplify a target
sequence within the first-run product (i.e. nested PCR). Well defined PCR products of
expected size (~294 bp) were observed for 6 of the 7 control strains from pfmdr1 region
1. The nested 1 amplification of pfmdr1 region 2 was less successful, producing very
light bands. Non-specific amplification including multiple bands and smearing in the
nest 2 reaction was likely due to sub-optimal annealing temperature (TA).
To improve amplification specificity, gradient experiments were employed to select for
the optimal TA. This was tested using genomic DNA from 7G8 (Figure 4.2). PCR
amplification was successful across TA of 40°C to 60°C for region 1. For region 2
however, specificity increased when TA was >48°C. Since 56°C was the optimal TA for
the PCR amplification of other drug resistant genes (pfcrt, pfdhfr and pfdhps), it was
used for pfmdr1 for further validation.
The necessity of a nested PCR was assessed. Firstly, PCR products from the pfmdr1
region 2 nest 1 were used as a template for the nest 2 primers. Secondly, the region 2
nest 2 primers were used directly with genomic DNA. Multiple PCR product bands and
smearing resulted when nest 1 products were used (data not shown). However, nest 2
primers used directly with genomic DNA resulted in well defined bands of ~694bp,
which negated the need for a nested PCR for pfmdr1 region 2.
Despite acceptable PCR amplification, the products (required for subsequent LDR-
FMA) produced high background fluorescent intensities (FI), particularly for alleles
86Y/N and 1034S/C. The HB3 strain carries the pfmdr1 allele 86N (wild type);
however, FI for both 86N and 86Y were high at 18168 and 17758, respectively.
Similarly, in Dd2 which carries the 1034S allele, FI of 25537 and 13628 were obtained
for 1034S and 1034C, respectively. Ideally, the FI of the negative allele should be
<1500, as observed in other LDR-FMA assays (Carnevale et al. 2007). This high
background was suggestive of cross-reactivity or non-specific binding. Closer
examination of the oligonucleotide sequences of respective PCR and LDR primers

Chapter 4 Molecular Characterisation of PNG isolates
115
revealed an overlap of 19 bp. This may have caused partial amplification of the PCR
primers against LDR probes and contributed to the high background signals.
With the aim of enhancing the LDR-FMA FI specificity, new PCR primers were
designed (Table 4.1). Forward and reverse complementary oligonucleotides sequences
were selected from the P. falciparum genome (Genebank accession #X56851).
Amplification of pfmdr1 regions 1 and 2 using the new primers have proven successful
in control strains. Figure 4.3 illustrates well-defined amplicons of expected sizes from
both pfmdr1 regions in all control strains.
Table 4.1 Primer sequences and thermocycling conditions for pfmdr1 assays.
Initial and optimised conditions for the PCR amplification of pfmdr1 regions 1 and 2 (in
parentheses) are shown.
Set
Gene (region)
PCR Primer Sequence
pfmdr1(1) 754 forward 1048 reverse
5’-GTGTTTGGTGTAATATTAAAG-3’
5’-CAAACGTGCATTTTTTATTAATG-3’
pfmdr1(2) Nest 1 3439 forward 4489 reverse
5’-GATCCAAGTTTTTTAATACAGG-3’
5’-TTAGGTTCTCTTAATAATGCAC-3’
Initial
pfmdr1(2) Nest 2 3570 forward 4264 reverse
5’-TATTGTAAATGCAGCTTTATGG-3’
5’-CACTAACTATTGAAAATAAGTTTC-3’
pfmdr1(1) 681 forward 1119 reverse
5’-TGTATGTGCTGTATTATCAG-3’
5’-CTTATTACATATGACACCACA-3’
Optimised
pfmdr1(2) 3499 forward 4311 reverse
5’-TAGAAGATTATTTCTGTAATTTG-3’
5’-CAATGTTGCATCTTCTCTTCCA-3’

Chapter 4 Molecular Characterisation of PNG isolates
116
Figure 4.2 PCR amplification of pfmdr1 regions 1 and 2 in 7G8 over a temperature
gradient. PCR products of expected sizes (294 bp and 694 bp) were observed from
regions 1 and 2, respectively over a range of annealing temperature (TA). Specificity
was enhanced at TA >48°C for the region 2 reaction.

Chapter 4 Molecular Characterisation of PNG isolates
117
Figure 4.3 Gel scan of PCR products generated using new pfmdr1 primers.
PCR products of expected sizes (438 bp and 812 bp) were generated using new pfmdr1
primers. DNA ladder (HyperLadder™ IV, Bioline, London, UK) band size = 100 bp.
Upper: PCR amplicons of pfmdr1 region 1 from laboratory-adapted strains HB3, Dd2,
K1, 3D7, 7G8, empty, PNG 1917, Bk; water blank. Lower: PCR amplicons of pfmdr1
region 2 from control strains HB3, Dd2, K1, 3D7, 7G8, PNG1905, PNG1917 and water
blank.

Chapter 4 Molecular Characterisation of PNG isolates
118
Preliminary LDR-FMA data generated by using new PCR primers showed enhanced
clarity between known positive and negative alleles. A noticeable reduction in
background FI was evident in the HB3 strain, a carrier of the 86N allele. In this
example, signals correspond to 86Y and 86N alleles were 17758 and 18168 using initial
primers, compared with 5457 and 14393 using modified primers, respectively.
The effect of the number of PCR cycles in reducing background FI was also
investigated. DNA from control strains were subjected to PCR using 10, 15, 20, 25, 30,
35 and 40 amplification cycles. As expected, higher number of cycles produced more
PCR products in both regions (Figure 4.4). The optimal number of PCR cycles ranged
from 30 to 40 for pfmdr1 regions 1 and 2 (Figure 4.4).
Optimised pfmdr1 PCR conditions are summarised in Tables 4.2 and 4.3. For pfmdr1
region 1, the reaction begins by preheating at 95°C for 2 min, followed by 32 cycles of
94°C for 30 sec, annealing at 56°C for 30 sec, 72°C for 30 sec, followed by final
extension at 72°C for 4 min. Thermocycling conditions for the amplification of the
pfmdr1 region 2 were similar to that of region 1 except for a longer extension time at
72°C for 1 min and repeated for 40 cycles.
4.3.1.2 LDR Optimisation
Each allele-specific LDR primer was designed with a unique 24-bases tag sequence
added to its 5’ end. These tag sequences are complementary to the anti-tag sequences
that are bound to microspheres and each emits a distinctive classification code (Table
4.4).

Chapter 4 Molecular Characterisation of PNG isolates
119
Figure 4.4 Effect of PCR cycles on pfmdr1 amplification.
PCR amplification of pfmdr1 regions 1 (upper) and region 2 (lower) by different
number of cycles.

Chapter 4 Molecular Characterisation of PNG isolates
120
Table 4.2 Optimised PCR conditions for pfmdr1 region 1. Table 4.3 Optimised PCR conditions for pfmdr1 region 2.
PCR: pfmdr1 region 1
Volume (µL)
Cycling Program
Sterile distilled water 19.6 95°C – 2 min
10 x PCR Buffer 2.5 94°C – 30 sec
2.5mM dNTPs 2.0 56°C – 30 sec
72°C – 30 sec Forward primer (10pmol/µL) pfmdr1
(region 1) 681
0.3
X 32 cycles
72°C – 4 min Reverse primer (10pmol/µL) pfmdr1
(region 1) 1119
0.3
10°C
Mac Taq 0.3
Total volume per well 25.0
PCR: pfmdr1 region 2
Volume (µL)
Cycling Program
Sterile distilled water 19.6 95°C – 2 min
10 x PCR Buffer 2.5 94°C – 30 sec
2.5mM dNTPs 2.0 56°C – 30 sec
72°C – 1 min Forward primer (10pmol/µL) pfmdr1 (region 2) 3499
0.3
X 40 cycles
72°C – 4 min Reverse primer (10pmol/µL) pfmdr1 (region 2) 4311
0.3
10°C
Mac Taq 0.3
Total volume per well 25.0

Chapter 4 Molecular Characterisation of PNG isolates
121
Table 4.4 LDR primers for P. falciparum pfmdr1 molecular markers. aLowercase
nucleotides represent tag sequences added to the 5’ ends of each allele-specific LDR
primer. bID, microsphere fluorescence identification. Luminex microsphere sets are
synthesised to exhibit unique fluorescence. Each microsphere set is coupled to different
anti-tag sequences that are complementary to allele-specific tag sequences. cCom,
common (conserved) sequence primer positioned immediately downstream from the
allele-specific primer.
Gene
LDR Primer
Sequencesa
IDb
pfmdr1 region 1
86N
86Y
Com86
184Y
184F
Com184
5'tacactttctttctttctttctttTTGGTGTAATATTAAAGAACATGA-3’
5'atcatacatacatacaaatctacaTTGGTGTAATATTAAAGAACATGT-3’ 5’phosphate-ATTTAGGTGATGATATTAATCCTA-biotin-3’
5'tcaaaatctcaaatactcaaatcaGCCAGTTCCTTTTTAGGTTTATA-3’ 5'ctacaaacaaacaaacattatcaaGCCAGTTCCTTTTTAGGTTTATT-3’
5’phosphate-TATTTGGTCATTAATAAAAAATGCA-biotin-3’
10 12 -
18 28 -
pfmdr1 region 2
1034S
1034C
Com1034
1042N
1042D
Com1042
1246D
1246Y
Com1246
5'ttacctttatacctttctttttacATGCAGCTTTATGGGGATTCA-3’ 5'caatttcatcattcattcatttcaATGCAGCTTTATGGGGATTCT-3’ 5’phosphate-GTCAAAGCGCTCAATTATTTATT-biotin-3’
5'cttttcatcttttcatctttcaatCCAAACCAATAGGCAAAACTATT-3’ 5'ctttttcaatcactttcaattcatCAAACCAATAGGCAAAACTATC-3’
5’phosphate-AATAAATAATTGAGCGCTTTGAC-biotin-3’ 5'tcatcaatcaatctttttcactttAATATATGTGATTATAACTTAAGAG-3’
5'tcataatctcaacaatctttctttTAATATATGTGATTATAACTTAAGAT-3’ 5’phosphate-ATCTTAGAAACTTATTTTCAATAG-biotin-3’
30 35 -
37 54 -
59 68 -
A series of comprehensive experiments were set up to optimise pfmdr1 LDR
conditions. To test whether annealing temperature would improve signal clarity, parallel
LDR with TA at 58°C, 60°C and 62°C were performed. FI ratios between positive and
negative alleles were compared in control strains for different annealing temperatures
(Appendix C). For pfmdr1 region 1, 60°C produced the best clarity as higher
temperature dampened the FI for positive alleles. The opposite was observed in the
LDR for pfmdr1 region 2 in which higher TA enhanced signal contrast. Further

Chapter 4 Molecular Characterisation of PNG isolates
122
investigation for region 2 using annealing conditions at 62°C, 63°C, 64°C and 65°C,
indicated 65°C was optimal.
Secondly, the effect of PCR amplicon concentration on FI was investigated in P.
falciparum strains HB3, K1 and 7G8. Briefly, PCR products from pfmdr1 region 1,
region 2 and regions 1 plus 2 together, were subjected to LDR undiluted, diluted 1:2,
1:5, 1:10 and 1:100 in sterile distilled water. Although signal clarity was improved at
some dilutions, no consistent trend was found. In addition, DNA yield from field
samples would likely be lower than those derived from cultured parasites, hence
standardised dilutions may not be applicable.
Optimised LDR for pfmdr1 involves two separate reactions for each region. For pfmdr1
region 1, the reaction mixture was initially heated at 95°C for 1 min, followed by 32
cycles of denaturation at 95°C for 15 sec and annealing/ligation at 60°C for 2 min
(Table 4.5). Similar thermocycling conditions were used for LDR of pfmdr1 region 2,
with the exception of the final step performed at 65°C for 2 min (Table 4.6).
4.3.1.3 Optimised LDR-FMA for pfmdr1 and Multiplexed Detection of SNPs in pfdhfr,
pfdhps and pfcrt genes
The first step of the optimised LDR-FMA involves PCR amplification of the pfmdr1
gene from genomic DNA extracts. Briefly, sample DNA was centrifuged (3000 rpm for
30 sec) and 3 µL was combined with PCR master mix (25 µL/well) for pfmdr1 regions
1 and 2. The plate was sealed using Microseal® ‘A’ film (Bio-Rad, USA) and
centrifuged prior to PCR. Following PCR, the products were subjected to two separate
LDR for both pfmdr1 regions due to different TA requirements. LDR master mixes (14
µL/well) were prepared (Appendix B) and combined with 1 µL of PCR product from
corresponding regions. The plates were firmly sealed (sealing film B-1212-5, Denville
Scientific Inc), centrifuged and subjected to thermocycling (Table 4.5 and 4.6) in a
PTC-225 Peltier Thermal Cycler (MJ Research, Iowa). On completion, 2.5 µL of LDR
products from each pfmdr1 regions 1 and 2 were combined into 60 µL of pre-warmed
hybridisation solution (Appendix B) containing microspheres from each

Chapter 4 Molecular Characterisation of PNG isolates
123
Table 4.5 Optimised LDR conditions for pfmdr1 region 1. Table 4.6 Optimised LDR conditions for pfmdr1 region 2.
LDR: pfmdr1 region 1 Volume (µL) Cycling Program
Sterile distilled water 11.2 95°C – 1 min
Taq Ligase Buffer 1.5 95°C – 15 sec
Com86 0.2 60°C – 2 min
Com184 0.2 x 32 cycles of steps 2 and 3 only
86Ytag12 0.2
86Ntag10 0.2
184Ytag18 0.2
184Ftag28 0.2
Taq DNA Ligase 0.1
PCR product (region 1) 1
Total volume per well 15.0
LDR: pfmdr1 region 2 Volume (µL) Cycling Program
Sterile distilled water 10.6 95°C – 1 min
Taq Ligase Buffer 1.5 95°C – 15 sec
Com1034 0.2 65°C – 2 min
Com1042 0.2 x 32 cycles of steps 2 and 3 only
Com1246 0.2
1034Stag30 0.2
1034Ctag35 0.2
1042Dtag54 0.2
1042Ntag37 0.2
1246Dtag59 0.2
1246Ytag68 0.2
Taq DNA Ligase 0.1
PCR product (region 2) 1
Total volume per well 15.0

Chapter 4 Molecular Characterisation of PNG isolates
124
allelic set (total of 10 alleles). From this point on, the LDR products (5 µL) from pfcrt,
pfdhfr, and pfdhps genes can be multiplexed into a single well with the hybridisation
solution provided that it contains microspheres from their respective allelic set (total of
18 alleles). Hybridisation, reporter dye labelling and fluorescent signal measurement
were performed as described previously (Sections 2.3.4).
Further validation and application of the new LDR-FMA for pfmdr1 SNP screening of
culture-adapted and patient isolates of P. falciparum are described in the subsequent
section. Positive thresholds for each allele were established as detailed in Section 4.2.5.
4.3.2 Assay Validation
4.3.2.1 Comparison between LDR-FMA and RFLP speciation
Plasmodium speciation using LDR-FMA and the reference RFLP method were
compared in 374 samples. The sensitivity and specificity of LDR-FMA compared to the
reference RFLP method for P. falciparum speciation were 96.6% and 100%,
respectively. Concordances between the two methods are presented in Table 4.7. There
were high concordances for the diagnosis of P. falciparum (97%), P. vivax (90%) and
P. malariae (98%). 11 of 12 cases that were P. falciparum negative by LDR-FMA,
exhibited strong P. vivax fluorescent signals (>20,000), yet they have been classified as
P. falciparum positive by the RFLP approach. The remaining case proved to be a non-
falciparum mixed infection by the LDR-FMA characterised by strong positive signals
(23,145) for P. malariae and weak positive (667) for P. vivax. However, it was
classified as a P. falciparum mono-infection by the RFLP method. The LDR-FMA
method identified a total of 10 cases of P. malariae infections compared with 3 cases by
RFLP.
4.3.2.2 Inter-assay concordance
If a sample is positive for P. falciparum by LDR-FMA then the pfcrt, pfdhps, pfdhfr and
pfmdr1 genes should also be detectable. There were 304 samples in which speciation
and all four specific gene assays were performed using LDR-FMA. In 241 (79%), each
of the pfcrt, pfdhps, pfdhfr and pfmdr1 genes were identified. For discordant samples,

Chapter 4 Molecular Characterisation of PNG isolates
125
the pfdhps and pfdhfr assays were more often PCR negative in the amplification of
Plasmodium genome.
Table 4.7 Concordance between RFLP and LDR-FMA diagnosis of Plasmodium
species in PNG field samples.
P. falciparum
LDR (+)
LDR (-)
Total
RFLP & LDR-FMA Concordance
RFLP (+) 362 12 374
RFLP (-) 0 0 0
Total 362 12 374 96.8%
P. vivax
RFLP (+) 17 15 32
RFLP (-) 23 319 342
Total 40 334 374 89.8%
P. malariae
RFLP (+) 2 1 3
RFLP (-) 8 363 371
Total 10 364 374 97.6%
4.3.2.3 Identification of drug resistance alleles
The specificities of the pfcrt, pfdhfr and pfdhps assays have been reported previously
with 100% concordance with haplotypes from a variety of laboratory-adapted strains
(Carnevale et al. 2007). The specificities of the pfmdr1 LDR-FMA probes are shown in
Table 4.8. The results for the pfmdr1-specific LDR-FMAs show that allele-specific
background median fluorescence intensity (MFI) signals ranged from 168 to 6,464 and
positive allele-specific signals ranged from 1421 to 22,008. Strain-specific LDR-FMA
results were 100% concordant with published haplotypes (Mehlotra et al. 2008).

Chapter 4 Molecular Characterisation of PNG isolates
126
4.3.3 Field Application of the LDR-FMA
4.3.3.1 Speciation and drug resistance genes in PNG field isolates
In 402 samples with microscopy-confirmed P. falciparum, the Plasmodium species
LDR-FMA identified 28 patients with P. vivax co-infections and 13 with P. malariae
co-infections. No P. ovale co-infections were found. The maximum median FI obtained
for field isolates and thresholds used to determine the presence of an allele are shown in
Table 4.9.
Table 4.8 LDR-FMA evaluation of pfmdr1 SNPs in laboratory-adapted P.
falciparum strains. The fluorescence signal is expressed as median fluorescence
intensity (MFI) units. Boldfacing indicates positive allele-specific signals. apfmdr1
haplotypes for P. falciparum strains are for 3D7, NYSND; for 7G8, NFCDY; for Dd2,
YYSND; for HB3, NFSDD; K1, YYSND.
Fluorescence signal for the Pfmdr1a gene, codon and allele:
86 184 1034 1042 1246
Y N Y F S C D N D Y
3D7 2657 15306 17956 6464 21903 5568 225 21182 5243 327
7G8 1724 11351 1869 21092 1475 16556 6022 436 746 1421
Dd2 12684 3418 15465 5891 22008 5061 234 18333 4357 359
HB3 555 6381 530 10036 17881 1555 8238 271 2686 168
K1 19971 3482 17393 6055 21501 4519 199 18265 3476 543

Chapter 4 Molecular Characterisation of PNG isolates
127
4.3.3.2 Prevalence of polymorphic alleles in pfcrt, pfmdr1, pfdhfr and pfdhps
Overall, 9 mutant alleles were detected. These were at pfmdr1 codons N86Y (91%),
Y184F (2%), N1042D (2%), D1246Y (4%), pfcrt codons C72S and K76T (both 92%),
pfdhfr codons C59R (93%), S108N (95%) and pfdhps codon K540E (1.5%) (Figure
4.5). The pfcrt haplotype SVMNT was found to be at fixation in the sample (92%), with
only 7% of P. falciparum strains retaining the CQ-sensitive haplotype CVMNK. Two
children were infected with mixed strains carrying CVIET and SVMNT mutations. The
majority of the P. falciparum isolates carried the CQ resistance-associated YYSND
(93%) haplotype of the pfmdr1 gene (Figure 4.6).
One patient was infected with an isolate carrying a single pfdhfr S108N mutation.
However, most children (97%) had parasites with double mutations in the NRNI
haplotype corresponding to amino acids at codons 51, 59, 108 and 164. A few isolates
(3%) retained the wild haplotype NCSI. No isolates carried the pfdhfr I164L allele. The
pfdhps haplotype KAA which harbours a single mutation at codon 613 was
predominant (98%). Four isolates carried the pfdhps K540E mutation.
When the isolates with completely defined haplotypes were assessed for multiple-gene
mutations, it was found that 88% (100/113) of children were infected with P.
falciparum carrying quintuple mutations across the pfcrt, pfdhfr, pfmdr1 and pfdhps
genes characterised by the respective haplotypes SVMNT+NRNI+YYSND+KAA. Four
patients (one per treatment group) were infected with parasites carrying the six-fold
mutated haplotype SVMNT+NRNI+YYSND+EAA. One of these patients was treated
with PQ-DHA and had a LPF, whilst the other three had P. falciparum ACPR. Table
4.10 details the number of P. falciparum isolates with multiple combinations of
mutations across drug resistance genes.

Chapter 4 Molecular Characterisation of PNG isolates
128
Table 4.9 Fluorescence detection thresholds and maxima for P. falciparum pfdhps,
pfdhfr, pfcrt and pfmdr1 in PNG field samples. *Alleles not detected in the field
isolates.
Gene and allele Fluorescence intensity signal
dhps Threshold Maximum
540K 20,400
540E
1213
9,774
581A 21,876
581S*
1940
-
613A 21,403
613S*
2374
-
dhfr
51I* -
51N
4147
23,429
59R 23,368
59C
6087
23,490
108T* -
108S 16,258
108N
3348
22,198
164I 19,695
164L*
3914
-
pfcrt
CVIET * -
CVMNK 20,657
SVMNT
4791
22,523
pfmdr1
86Y 21,994
86N
6722
18,189
184Y 21,145
184F
7899
20,412
1034S 25,792
1034C*
11166
-
1042D 10,973
1042N
2836
23,200
1246D 9,921
1246Y
1410
3,174

Chapter 4 Molecular Characterisation of PNG isolates
129
Figure 4.5 Prevalence of pfcrt, pfmdr1, pfdhfr, pfdhps alleles in P. falciparum-
infected individuals from the Madang and East Sepik Provinces, PNG.
Figure 4.6 Frequency distributions of pfcrt, pfdhps, pfdhfr, pfmdr1 haplotypes in P.
falciparum-infected individuals from PNG field sites. The number of samples
demonstrating complete haplotype for each gene were 376, 254, 273 and 195,
respectively.

Chapter 4 Molecular Characterisation of PNG isolates
130
Table 4.10 Occurrence of P. falciparum isolates carrying multiple mutations across
4 genes associated with drug resistance. Wild-type alleles of pfcrt, pfdhfr, and pfmdr1
are indicated by boldfacing. Allele mutations are underlined. Pfcrt haplotypes
CVMNK, CVIET and SVMNT correspond to codons 72 to 76. Pfdhfr haplotypes
NCSI, NRNI and NCNI correspond to codons 51_59_108_164. Pfmdr1 haplotypes
NYSND, YYSND and NFSDD correspond to codons 86_184_1034_1042_1246.
pfcrt
pfdhfr
Double gene mutation
(pfcrt+pfdhfr only)*
Triple gene mutation (+pfmdr1)
NYSND YYSND NFSDD
NCSI 1 0 1 0
CVMNK with NRN I 14 1 1 0
NCNI* 0 0 0 0
NCSI 0 0 0 0
CVIET with NRNI 1^ 0 1^ 0
NCNI*
NCSI 2 0 2 1
SVMNT with NRNI 114 3 100^^ 3
NCNI 1 0 0 0
*Only assayed for pfcrt and pfdhfr due to inadequate sample volume.
^Sample with P. falciparum carrying both CVIET and SVMNT mutations.
^^Four of these isolates were also carrying the 540E mutation, whilst all other P.
falciparum sampled carried 540K allele of the pfdhps gene.
4.3.3.3 Parasite drug resistance mutations and treatment outcome
In evaluating the association of parasite genes with treatment outcome, the mean
number of parasite mutations in ACPR, ETF, LPF and LCF groups in 351 P.
falciparum cases was compared. Overall, there were 308 cases of ACPR, 3 cases of
ETF (one from CQ-SP; two from ART-SP), 3 cases of LCF (one case each from CQ-
SP, PQ-DHA and AL) and 37 cases of LPF (eleven from CQ-SP; twelve from ART-SP;
twelve from PQ-DHA and two from AL). There were 4 mutations in 41.6%, with 0.6%,
6.8%, 16%, 27.1%, 7.1%, 0.9% carrying no mutation and single, double, triple,

Chapter 4 Molecular Characterisation of PNG isolates
131
quintuple and sextuple mutations, respectively. There was no significant difference
between the mean number of mutations within the ACPR group (mean 3.3 [95% CI 3.1-
3.4]) and those in the ETF, LCF, LPF groups (means 2.3 [0-6.1], 3.0 [0.5-5.5] and 3.4
[3.0-3.8], respectively, P>0.05 in each case). For the purposes of subsequent analyses,
ETF, LCF and LPF were grouped as treatment failure.
The polymorphisms pfdhfr N51I, C59R, S108N and pfdhps K540E did not predict
treatment failure in 81 children allocated CQ-SP and in 86 allocated ART-SP (P>0.05).
In analyses of pfmdr1 N86Y, Y184F, N1042D and pfcrt K76T, pfmdr1 D1246Y was a
significant predictor of treatment failure in 79 children treated with PQ-DHA therapy;
40% (4 of 10) of such children carried D1246Y strains while only 3 of 69 (4%) who
responded to treatment harboured parasites with this mutation (P=0.004). In none of the
other three groups was there a significant association between the presence of D1246Y
and treatment failure (P≥0.10). Similar analyses did not reveal any associations
between haplotypes of pfcrt, pfdhfr and pfdhps and treatment failure.
4.4 DISCUSSION
The consequences of past antimalarial treatment policies in PNG were evident in these
data. Only 7% of the parasites carried the wild-type pfcrt CVMNK (codons 72 to 76)
associated with a CQ-sensitive phenotype. The high prevalence of the resistant K76T
polymorphism concurred with studies conducted between 2000 and 2005 in Madang
and East Sepik Provinces (Casey et al. 2004; Mehlotra et al. 2005; Schoepflin et al.
2008). The SVMNT haplotype (codons 72 to 76) is at fixation in the PNG parasite
population with an increase in prevalence from 83% from the early 1990s to 92.3% in
2003 - 2005 (Mehlotra et al. 2001; Marfurt et al. 2008; Schoepflin et al. 2008). As well
as being associated with CQ resistance, a transfection study has shown that parasites
carrying the SVMNT haplotype has reduced sensitivity to AQ and its metabolite (Sidhu
et al. 2002). Since AQ is recommended in place of CQ for treatment of malaria in PNG
children <19 kg (PNGDOH 2000), this may have implications for the most vulnerable
patients.
Another CQ-resistant haplotype, CVIET (codons 72 to 76) found commonly in Africa

Chapter 4 Molecular Characterisation of PNG isolates
132
and South-East Asia (Lim et al. 2003; Keen et al. 2007; Nsobya et al. 2007; Yang et al.
2007) was detected in two isolates as a mixed infection with the SVMNT strain,
confirming its recent emergence in PNG (DaRe et al. 2007). Differences between the
pfcrt intronic microsatellite diversity haplotypes of the SVMNT and CVIET parasites
from the province adjacent to Madang suggest that they have been imported (DaRe et
al. 2007), presumably by economic migrants from near-by Asian countries (Mehlotra et
al. 2001; Mehlotra et al. 2005; Mehlotra et al. 2008).
Although assessed in limited numbers of isolates (n=195) due to inadequate sample
volume, the CQ-sensitive pfmdr1 NYSND (codons 86_184_1034_1042_1246) was
present in 5.1%. Most others carried the N86Y polymorphism with the YYSND
haplotype (codons 86_184_1034_1042_1246) associated with CQ resistance. These
results reflect an increase of this haplotype since the mid 1990s (Mehlotra et al. 2005;
Mehlotra et al. 2008). It was also observed that four isolates (2%) carried the double
point mutations Y184F and N1042D, which confirmed the recent emergence of these
pfmdr1 polymorphisms in PNG (Marfurt et al. 2008). Although none of the four
patients with parasites carrying the pfmdr1 NFSDD haplotype (codons
86_184_1034_1042_1246) were assigned to the AL treatment group in this study. In a
Nigerian paediatric study (Happi et al. 2008), the presence of these SNPs with the wild-
type 86N allele was significantly associated with AL treatment failure. Since
introduction of this treatment in PNG is imminent, monitoring changes in the NFSDD
pfmdr1 haplotype should be a high priority.
It was found that signal intensities for the pfmdr1 1246 alleles were relatively weak
even in the controls. This was unlikely to reflect poor PCR amplification, as signal
intensities for codons 1034 and 1042 derived from the same region were unaffected.
Poor hybridisation with the LDR probe is possible. The D1246Y allele, present in 12%
of isolates in association with the wild-type allele 1246D, has not been previously
detected in PNG (Marfurt et al. 2008; Mehlotra et al. 2008). A recent African study
observed an increased prevalence of pfmdr1 N86Y and D1246Y alleles post AQ
exposure (Nawaz et al. 2009), but sporadic mutations at the 1246 codon have also been
documented in Thailand (Rungsihirunrat et al. 2009) and it may also have been
imported into PNG (Mehlotra et al. 2005). Both D1246Y and N86Y have been

Chapter 4 Molecular Characterisation of PNG isolates
133
associated with diminished in vitro sensitivity to CQ and AQ (Reed et al. 2000; Pickard
et al. 2003).
The concomitant occurrence of pfmdr1 N86Y and pfcrt K76T mutations was observed
in many isolates, with increased frequency compared to a decade earlier (Mehlotra et al.
2008). Concurrent mutations may reflect the involvement of CQ pressure in selecting
for the predominant pfmdr1 YYSND haplotype. This hypothesis was supported by the
selection of pfmdr1 N86Y allele after CQ and AQ therapy (Duraisingh et al. 1997;
Djimde et al. 2001). Concurrent mutations in pfcrt K76T, and pfdhfr C59R and S108N
were observed in 92.7% of the PNG isolates. Of those with pfmdr1 genotyping, 88.5%
also harboured N86Y mutations. Parasites with multiple mutations were more common
than found in another study conducted several years earlier in the same region
(Schoepflin et al. 2008).
The pfmdr1 haplotypes NFCDD, NFSND, and YFSND haplotypes (codons
86_184_1034_1042_1246) were not found in PNG isolates as found in those from
Thailand (Pickard et al. 2003), or the NFCDY and NFSDY haplotypes found in Brazil
and Colombia (Mehlotra et al. 2008). In vitro studies by Sá et al. showed 7G8 and
hybrid crosses with its pfmdr1 haplotype (NFCDY) resulted in higher 50% growth
inhibitory concentrations (IC50s) for AQ and its metabolite (Sá et al. 2009). However,
the “CDY” (i.e. S1034C, N1042D, D1246Y) mutation of the pfmdr1 gene has not been
detected in PNG. Collective polymorphisms in the 3’ coding region (N86Y and Y184F)
can confer resistance to quinine, mefloquine and halofantrine and modulate parasite
sensitivity to artemisinin drugs (Reed et al. 2000), and are found in South America,
Africa and Asia. Their geographical distribution suggests the requirement of drug
pressure for their maintenance and spread (Foote et al. 1990), as may occur with the
future use of ACTs in PNG. However, the frequency of the wild-type 86N may increase
with adoption of ACT as has been seen in African studies (Dokomajilar et al. 2006;
Humphreys et al. 2007).
This study demonstrated that 97% of the present isolates carried both the C59R and
S108N SNPs which constitute the major pfdhfr resistance alleles. Since the prevalence

Chapter 4 Molecular Characterisation of PNG isolates
134
of this double mutant in East Sepik Province was 72% - 91% in 2001 - 2003 (Mita et al.
2006b; Carnevale et al. 2007), this suggests that there has been continuing consistent
selection pressure for alleles conferring pyrimethamine resistance in northern PNG.
Carnevale et al. noted that a number of isolates from this area carried a single mutation
at codon 108 with the NCNI (n=13) and NCTI (n=1) haplotypes (Carnevale et al.
2007). In the present study, only one isolate had a single pfdhfr mutation with the NCNI
haplotype (codons 51_59_108_164) and, consistent with Marfurt et al. (Marfurt et al.
2008), the S108T mutant that was reported to be present in PNG in 1996 (Reeder et al.
1996) was not detected in the current study.
In the evaluation of pfdhps polymorphisms at codons 540, 581 and 613, only wild-type
alleles at loci 581 and 613 were detected. Four isolates were found to carry the K540E
variant first detected in studies in East Sepik and Simbu Provinces in 2002 - 2004 (Mita
et al. 2006b; Marfurt et al. 2008). Most of the present isolates (98.4%) carried the KAA
haplotype and 1.6% carried the EAA haplotype. SNPs at codons 436 and 437 were not
evaluated, as the respective LDR probes were unavailable at the time of study.
The lack of association between well known mutations (pfcrt K76T, pfdhfr S108N,
pfdhfr C59R, pfmdr1 N86Y) and treatment failure is largely due to fixation levels of
these mutations in Madang Province. Although carriers of multiple mutations were
more often observed in treatment failure cases, this did not reach statistical significance.
In the present study there was limited statistical power because of the relatively small
number of treatment failures (43 of 351), but the high baseline number of mutations per
isolate and the emergence of immunity in this age group in a high transmission setting
may also have attenuated the relationship. Although pfdhfr K540E has been shown to
predict SP treatment failure (Talisuna et al. 2004; Bacon et al. 2009), there was a low
prevalence of this allele in the isolates and it was unrelated to outcome. These data are
consistent with those of Marfurt et al. who showed that pfcrt K76T, and pfdhfr C59R
and S108N, did not predict treatment failure after CQ/SP treatment in a region north-
west of Madang (Marfurt et al. 2008), reflecting fixation of these mutations in PNG.
Interestingly, the association between pfmdr1 D1246Y allele with PQ-DHA and overall
treatment failure highlights the increasingly recognised role of this transporter gene in

Chapter 4 Molecular Characterisation of PNG isolates
135
modulating resistance to antimalarial drugs from different classes (Cowman et al. 1994;
Peel et al. 1994; Reed et al. 2000).
A novel approach to determine presence or absence of mutations using the LDR-FMA
system was adopted. Determination of signal positivity proved challenging. Initially,
allele-specific thresholds that have been established previously were applied for the
pfcrt, pfdhfr and pfdhps alleles (Carnevale et al. 2007). However, a number of samples
were plainly mis-classified when compared by visual discrimination via 2-dimensional
and 3-dimensional modelling. Various statistical models were tested but, in the absence
of valid malaria-negative samples, it was found that bimodal and gamma distributions
were inappropriate. Subsequent use of allelic signal ratios within each codon appeared
robust when applied to controls with known haplotypes, especially in cases where
strong positive signals were accompanied by high background. However, this approach
was based on the assumption of single strain infections, which can only be
circumvented by establishing a specific threshold. It was found that, through
normalisation of the data, application of gene-specific cut-points from the control
strains, and reversing the process to obtain codon-specific cut-offs, prediction of
positivity was relatively accurate. Unless better resolution of positive from negative
signal is possible, this method or related approaches (DaRe et al. 2010) can be adopted
for future LDR-FMA studies.
The multiplex LDR-FMA technique proved a cost-effective tool for epidemiologic
studies. Taking into consideration the reagents required for each step (DNA extraction,
plasticware, PCR and LDR primers, and Fleximap microspheres), the total cost per
sample for the analysis of 28 SNPs was AUD $4.14 ($0.15 per SNP), with over 60% of
the cost due to DNA extraction. This amounts to less than half the expense of the
recently-described microarray SNP detection approach (Crameri et al. 2007). However,
as with other expensive equipments in the PNG settings, maintenance of the bioplex
array reader and shipment of reagents maybe costly. Compared to PCR-based
approaches that have been used to evaluate polymorphisms in the pfdhfr, pfdhps, pfcrt
and pfmdr1 genes, most of which involve DNA probe hybridisation and post-PCR
RFLP methods (Duraisingh et al. 1998; Casey et al. 2004; Alifrangis et al. 2005; Farcas

Chapter 4 Molecular Characterisation of PNG isolates
136
et al. 2006; Veiga et al. 2006), the LDR-FMA system offers greater efficiency and
objectivity when analysing multiple mutations. Given these considerations and the
increase in key parasite mutations in resource-poor malaria-endemic countries such as
PNG, the LDR-FMA SNP assay represents an excellent tool for molecular surveillance.
The present LDR-FMA platform enables assessment of 18 SNPs in the pfcrt, pfdhps
and pfdhfr genes in a single-tube multiplex assay, a task that is beyond the capabilities
of existing real-time PCR and RFLP methodology. The inclusion of 10 additional SNPs
in the pfmdr1 gene can be easily accommodated through modified microsphere set
selection with all 28 SNPs having their own unique classification codes. Monitoring the
spread of resistance to CQ, sulfadoxine and pyrimethamine using this or equivalent
methodology is a high priority in countries such as PNG where these drugs are still
used. In addition, there will be an increasing need for monitoring pfmdr1 SNPs since
polymorphisms encompass diverse effects on parasite sensitivity to a range of
antimalarial drugs including those used in artemisinin-based combination therapy (Reed
et al. 2000).
This work has been published in Antimicrobial Agents and Chemotherapy, titled
“Molecular Assessment of Plasmodium falciparum Resistance to Antimalarial Drugs in
Papua New Guinea Using an Extended Ligase Detection Reaction Fluorescent
Microsphere Assay”.

CHAPTER 5
ANTIMALARIAL PROPERTIES OF
DESBUTYL-LUMEFANTRINE

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
138
CHAPTER 5. ANTIMALARIAL PROPERTIES OF
DESBUTYL-LUMEFANTRINE
5.1 INTRODUCTION
Desbutyl-lumfantrine (DBL), formerly known as desbutyl-benflumetol is a 2,3-
benzindene compound with antimalarial activity. Although previously considered only
a putative metabolite of lumefantrine because of a lack of supportive pharmacokinetic
data (Samal et al. 2005; Starzengruber et al. 2007), recent analytical developments have
enabled the reliable detection of relatively low concentrations of DBL in samples of
plasma from small numbers of patients treated with conventional doses of artemether-
lumefantrine combination therapy (McGready et al. 2006; Hatz et al. 2008; Ntale et al.
2008). The ratio of maximum plasma concentration (Cmax) of the parent compound to
that of the metabolite in this situation has varied substantially, from 6 (Ntale et al.
2008) to >270 (Hatz et al. 2008).
DBL is more potent in vitro against chloroquine (CQ)-resistant P. falciparum and P.
vivax field isolates than lumefantrine (Noedl et al. 2001; Kyavar et al. 2006;
Starzengruber et al. 2008). There is evidence of in vitro synergy between lumefantrine
and DBL against P. falciparum, but at ratios (999:1 and 995:5) that were presumably
selected at a time when plasma concentrations of DBL were assumed to be very much
lower than those of the parent compound (Starzengruber et al. 2007; Starzengruber et
al. 2008). Interactions between DBL and artemisinin were assessed in a study of
schizont maturation in Thai P. vivax field isolates in which antagonism was found at
low concentrations and apparent synergy at much higher concentrations (Kyavar et al.
2006). A subsequent similar field study confirmed concentration-dependent synergy in
strains of P. falciparum (Muller et al. 2008).
Because of its relative in vitro potency and evidence of low cardiac toxicity (Traebert et
al. 2004), DBL has been suggested as an antimalarial drug in its own right
(Starzengruber et al. 2007). There is, however, a need for further assessment of its
interactions with other antimalarial drugs, especially lumefantrine and the artemisinin
derivatives, in strains of P. falciparum of differing drug sensitivities. In addition, and

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
139
given that artemether-lumefantrine is recommended as first-line treatment for
uncomplicated malaria (WHO 2009), there is also a need to confirm the relative plasma
concentrations of lumefantrine and DBL together with their therapeutic implications.
The present chapter reports on in vitro antimalarial activity of DBL in comparison to
lumefantrine and CQ in CQ-resistant and CQ-sensitive laboratory-adapted strains of P.
falciparum. It also provides a comprehensive evaluation of DBL interaction with DHA
and lumefantrine across 17 combination-ratios. Plasma levels of DBL and lumefantrine
in 127 clinical samples and their relationship with artemether-lumefantrine treatment
outcome are also presented.
5.2 MATERIALS AND METHODS
5.2.1 Parasite Cultures
The laboratory-adapted P. falciparum strains 3D7 (CQ-sensitive) and W2mef (CQ-
resistant) were cultured as described previously (Section 2.1.3) (Trager et al. 1976;
Scheibel et al. 1979).
5.2.2 Antimalarial Drugs
Stock solutions of CQ diphosphate, piperaquine tetraphosphate (PQ), mefloquine
hydrochloride (MQ), lumefantrine, DBL and dihydroartemisinin (DHA) were prepared
as described in Section 2.2.1. Stocks and working standards of lumefantrine and DBL
were sonicated for 90 sec in an ultrasonic waterbath in pre-warmed media to facilitate
dissolution. On the day of assay, aliquots were thawed and further diluted in RPMI to a
working standard, and further two-fold serial dilutions in complete RPMI (without
hypoxanthine) at double assay concentrations were prepared for CQ (25 - 1600 nM),
PQ (6.25 - 400 nM), MQ (0.78 - 200 nM), DHA (0.78 - 51.2 nM), and lumefantrine and
DBL (3.12 - 400 nM).

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
140
5.2.3 In vitro Drug Susceptibility
Drug susceptibility was tested in triplicate as described in Section 2.2. To each well of a
96-well plate was added 100 µL of dosed or drug-free media without hypoxanthine, 90
µL of RBC suspension (final 0.5% parasitaemia, 1.5% hct) and 10 µL of 5 mg/ml 3H-
labelled hypoxanthine. After 48 hr incubation, the plates were harvested and processed
as detailed in Section 2.2.5.
5.2.4 Drug Interaction Studies
The traditional checkerboard method (Berenbaum 1978; Canfield et al. 1995; Hassan
Alin et al. 1999) and a recently described fixed-ratio approach (Fivelman et al. 2004)
were used to develop the following assay. Solutions (5 µM) of each drug were used to
prepare 17 fixed molar combination ratios in 12-well plates. The molar ratios tested for
lumefantrine:DBL and DHA:DBL are detailed in Table 5.1. Two hundred microlitres of
each drug combination were dispensed into row H of a 96-well plate. Rows A to G
were filled with 100 µL of media without hypoxanthine. Preparations of one drug alone
were assayed in triplicate, and other combination ratios in duplicates. Two-fold serial
dilutions were performed using an electronic multichannel pipette (Labnet excel, Fisher
Biotec, Australia) from rows H to B. Drug-free control wells were included (row A).
All wells contained a final volume of 200 µL including 3H-labelled hypoxanthine, were
standardised to 1.5% parasitaemia at 1.5% hct and were incubated for 48 hr and
processed as described previously (Section 2.2.5).
Table 5.1 Drug combination ratios for isobologram assays. Molar ratios and drug
concentrations of desbutyl-lumefantrine (DBL) paired with either lumefantrine (LM) or
dihydroartemisinin (DHA) are shown. Within-well concentration shown in the table is
that of the prepared drug combination. Further two-fold serial dilutions of each
combination were performed to give six additional tests at lower doses. For simplicity,
drug concentrations are displayed only for the first assay well (i.e. the one containing
the highest concentration of drugs) for a given combination.

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
141
Drug Combination
Molar Ratio Within-well assay concentration (nM) DBL LM DHA
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
1:0
0:1
1:1
1:3
1:10
1:30
1:100
1:300
1:1000
1:3000
3000:1
1000:1
300:1
100:1
30:1
10:1
3:1
100
0
100
33.3
10
3.33
1
0.33
0.1
0.033
100
100
100
100
100
100
100
0
400
400
400
400
400
400
400
400
400
0.133
0.4
1.33
4
13.33
40
133.3
0
100
100
100
100
100
100
100
100
100
.033
0.1
0.33
1
3.33
10
33.3
5.2.5 Study Site and Sample Collection
Plasma concentrations of DBL and lumefantrine were measured in samples taken from
children participating in an intervention trial carried out in the Madang Province of
Papua New Guinea (PNG) (Karunajeewa et al. 2008b). Children aged 0.5 to 5 years and
with uncomplicated falciparum or vivax malaria were randomised to one of four
treatments, one of which was artemether-lumefantrine (Coartem®, Novartis Pharma,
Basel, Switzerland) at a dose of 10 mg/kg lumefantrine twice daily for three days (total
dose of 60 mg/kg). Scientific and ethical approval was obtained from the Medical
Research and Advisory Committee of the Ministry of Health of PNG and informed
consent was obtained from the parents or legal guardians before recruitment and blood
sampling. A venous blood sample was taken at 7 day post-treatment for drug assay
since this is regarded as a useful surrogate marker of the area under the plasma
concentration-time curve and the time above the minimal inhibitory concentration for
long-acting antimalarial drugs (Price et al. 2007). Whole blood was centrifuged on site
and separated plasma was stored at -80°C until analysis. For the purposes of the present

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
142
study, therapeutic response was considered to be either an adequate parasitologic and
clinical response (ACPR) or treatment failure (early treatment failure, late parasitologic
failure or late clinical failure) during a follow-up of 42 days without PCR correction for
reinfection (WHO 2003; Karunajeewa et al. 2008b).
5.2.6 Liquid Chromatography and Mass Spectrometry
Lumefantrine was analysed using a validated high performance liquid chromatography
method (Mansor et al. 1996). Plasma samples of 1.0 mL were spiked with atovaquone
as an internal standard and mixed with 7 mL of hexane:diethylether (70:30). After
centrifugation, the organic layer was separated, evaporated and reconstituted with 200
µL methanol:acetic acid (98:2). Aliquots of 15 µL were injected onto a Phenomenex
C6-phenyl 4.6 x 150 mm column (Phenomenex, CA, USA). A mobile phase of
acetonitrile:0.05 M phosphate buffer at pH=2.0 (62:38) with 0.03 M sodium perchlorate
was pumped at 1 mL/min. Lumefantrine and atovaquone were measured at 335 nm and
quantified using Chemstation Software (Version 9, Agilent Technology, Waldbronn,
Germany). The linear range for lumefantrine was 20 - 20,000 ng/mL. Inter-day
variability was 4.94%, 4.93%, 7.16% and 11.23% and intraday variability 2.83%,
4.41%, 4.11%, 9.55% for 20,000, 2000, 200 and 20 ng/mL respectively.
DBL was analysed using a validated ultra high-performance liquid chromatography-
tandem mass spectrometry (UPLC-LCMS-MS) method using a hexyl-analogue as an
internal standard (IS). To facilitate protein precipitation, 40 µL of a 0.1 M ZnSO4 was
added to 20 µL sample and briefly vortexed. A 200 µL aliquot of acetonitrile containing
the internal standard was added before the sample was centrifuged and 10 µL of the
supernatant was injected onto a 2795/Quattro Premier XE UPLC-MS/MSWaters Corp,
MA) using a Waters XTerra MS C18, 2.5 x 50 mm 3.5 µm column. Gradient elution
was performed using aqueous 2 mM ammonium acetate/0.1% formic acid and
methanolic 2 mM ammonium acetate/0.1% formic acid as mobile phases at 0.4
mL/min. Transitions were monitored using positive electrospray ionisation with
multiple-reaction monitoring for DBL and the IS which were m/z 472.1/346.0 and
500.1/346.0, respectively. The linear range for DBL was 0.5 - 100 ng/mL with the
lower end taken as the limit of quantitation. Inter-day variability was 3.36%, 3.47%,

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
143
9.98% and 6.74% and intraday variability 2.47%, 3.46%, 8.16% and 3.48% for 50, 10,
1 and 0.5 ng/mL, respectively. When matrix effects were assessed, between-subject
variability was 3.37%, 4.47% and 9.43% at 50, 10 and 1 ng/mL, respectively.
5.2.7 Statistical Analysis
Drug susceptibility and interactions were analysed by non-linear regression of
logarithmically-transformed concentrations. The concentration that inhibited 50%
parasite growth (IC50) was determined for each drug, as was the concentration that
inhibited 99% of growth (IC99). The fractional inhibitory concentrations (FICs)
representing the concentration of each drug alone or in combination resulting in 50%
inhibition were used to construct isobolograms (Berenbaum 1978). Two analytical
approaches were employed (Davis et al. 2006). First, the sum of fractional inhibitory
concentrations (ΣFICs) was calculated using the formula (IC50 of A in a mixture
resulting in a 50% inhibition/IC50 of A alone) + (IC50 of B in a mixture resulting in a
50% inhibition/IC50 of B alone) (Berenbaum 1978). The combination has an indifferent
interaction when the ΣFIC is close to 1.0. A ΣFIC of <1 indicates synergy with data
points forming a concave isobole beneath the line of additivity (Figure 1.22). A ΣFIC of
>1 indicates antagonism as represented by a convex isobole (Berenbaum 1978; Chawira
et al. 1987; Fivelman et al. 2004; Davis et al. 2006). Second, the function (y = 1 - x/(x
+ (1-x)*exp(-I))) (Brueckner et al. 1991; Canfield et al. 1995) was fitted to the data
where y is the IC50 of drug A combined with drug B; x is the IC50 of drug B when
combined with drug A, and I is the interaction value. Positive I values indicate synergy,
negative values antagonism and values close to zero an indifferent interaction. Values
of I and FIC were considered significantly different from no interaction if both 95%
confidence intervals (CIs) of the estimates did not span zero and unity, respectively
(Davis et al. 2006).

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
144
5.3 RESULTS
5.3.1 In vitro Antimalarial Potency of DBL
The geometric mean IC50 and 95% CIs for DBL, lumefantrine and other drugs are
shown in Table 5.2. W2mef had, as expected, a higher CQ IC50 than 3D7 but there were
no other strain-specific differences. Consistent with the IC50 data, the geometric mean
IC99 values for DBL against 3D7 and W2mef were 78 and 56 nM, respectively, and, for
lumefantrine, 239 and 226 nM. Based on both the IC50 and IC99 values, the in vitro
activity of DBL was at least 3 times that of lumefantrine regardless of CQ sensitivity
(Figure 5.1).
Table 5.2 In vitro sensitivity of laboratory-adapted P. falciparum to desbutyl-
lumefantrine and other antimalarial drugs. IC50 values are geometric means. Data
represent at least six experiments performed in triplicate.
3D7 IC50 (nM) 95% CI
W2mef IC50 (nM) 95% CI
Chloroquine 29.6 15.3 - 57.5 171.4 144.4 - 203.4
Lumefantrine 65.2 42.3 - 100.8 55.5 40.6 - 75.7
Desbutyl-lumefantrine
9.0 5.7 - 14.4 9.5 7.5 - 11.9
Piperaquine 16.9 13.4 - 21.3 17.4 11.8 - 25.6
Dihydroartemisinin 4.2 2.5 - 7.0 3.1 1.8 - 5.3

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
145
Figure 5.1 In vitro susceptibility of laboratory strains of P. falciparum to
chloroquine, desbutyl-lumefantrine and lumefantrine.

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
146
5.3.2 DBL Interaction with Conventional Antimalarials
Isobolographic analysis of DBL and lumefantrine combinations showed no interaction
in both laboratory-adapted strains (see Table 5.3 and Figure 5.2). Drug interactions
between DBL and dihydroartemisinin were mildly synergistic as assessed from both I
and ΣFIC.
Table 5.3 In vitro efficacy of antimalarial drug combinations against P. falciparum
clones 3D7 and W2mef as assessed by isobolographic analysis. Data are the
interaction factor (I) and the summed fractional inhibitory concentration (ΣFIC) with
95% confidence intervals. The assessment of interaction is based on both I and ΣFIC
data (see text).
I (95% CI) ΣFIC (95% CI) Interaction
3D7
DBL-lumefantrine 0.41 (-0.24 to 1.05) 0.99 (0.93 to 1.05) Indifferent
DBL-dihydroartemisinin 0.99 (0.71 to 1.28) 0.92 (0.87 to 0.98) Mildly synergistic
W2mef
DBL-lumefantrine 0.79 (0.02 to 1.56) 1.06 (0.97 to 1.14) Indifferent
DBL-dihydroartemisinin 0.92 (0.73 to 1.10) 0.94 (0.90 to 0.99) Mildly synergistic

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
147
Figure 5.2 Isobolograms illustrating interactions between desbutyl-lumefantrine
with conventional antimalarials. Isobolograms showing effect of desbutyl-
lumefantrine in combination with lumefantrine against P. falciparum 3D7 (top left),
W2mef (top right). Effect of desbutyl-lumefantrine in combination with
dihydroartemisinin against P. falciparum 3D7 (bottom left) and W2mef (bottom right).
The isoboles are representative of three or four experiments in which each of the 17
drug ratios was tested in duplicate. Degree of interaction (I) with 95% CI are
represented by red and dotted lines, respectively.

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
148
5.3.3 DBL Plasma Levels on Day 7 Post-Treatment
DBL and lumefantrine were quantified in 94 available day 7 plasma samples from the
127 children with falciparum malaria recruited to the artemether-lumefantrine arm of
the intervention trial (Karunajeewa et al. 2008b). The mean (range) DBL concentrations
were 15.5 (0.6 - 58.2) ng/mL or 31.9 (1.3 - 123.1) nM – all children had a plasma
concentration above the 0.5 ng/mL lower limit of quantitation. For lumefantrine, the
mean (range) plasma concentration was 370 (26 - 1,720) ng/mL or 699 (49 - 3,251) nM.
The lumefantrine:DBL ratio ranged from 7.0 and 123.0 with a mean of 27.4.
5.3.4 Influence of DBL Plasma Levels on Clinical Outcome
The relationship between plasma lumefantrine and DBL concentrations, and treatment
outcome at 42 days is shown in Figure 5.3. The mean plasma concentrations of
lumefantrine in those children who failed treatment were lower than in those with an
ACPR, but this difference was not significant (P=0.22). In the case of DBL, there was a
similar result but at borderline significance (P=0.053). There was no difference in the
case of the lumefantrine:DBL ratio (P=0.97).

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
149
Figure 5.3 Boxplots summarising day 7 plasma levels of lumefantrine and
desbutyl-lumefantrine. Plasma lumefantrine (left panel) and desbutyl-lumefantrine
(right panel) concentrations in children who had an adequate clinical and parasitological
response (ACPR) or who failed treatment with artemether-lumefantrine

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
150
5.4 DISCUSSION
The present observations confirm and extend previous published data relating to in vitro
and in vivo aspects of the antimalarial activity of DBL. The two laboratory-adapted
strains of P. falciparum were more sensitive in vitro to DBL than lumefantrine,
consistent with previous studies using field isolates (Noedl et al. 2001; Starzengruber et
al. 2007; Starzengruber et al. 2008). This effect was independent of CQ sensitivity. The
lack of in vitro interaction between DBL and lumefantrine appears to contradict
previous reports of synergy in field isolates of P. falciparum using limited numbers of
drug combinations (Starzengruber et al. 2007; Starzengruber et al. 2008). However, the
synergy between DBL and DHA observed using a comprehensive method of
isobolographic assessment parallels data from studies of field isolates of P. vivax
(Kyavar et al. 2006) and P. falciparum (Muller et al. 2008). Both lumefantrine and
DBL were detected at mean concentrations that were well above the IC99 for both
laboratory-adapted strains of P. falciparum in day 7 plasma of children treated with
artemether-lumefantrine for uncomplicated malaria. The day 7 plasma DBL was a
stronger predictor of subsequent therapeutic outcome than plasma lumefantrine or the
plasma lumefantrine:DBL ratio. These various observations have potential clinical
implications.
When artemether-lumefantrine is administered to patients with malaria, plasma
lumefantrine concentrations rise after each of the six doses given over 3 days and then
decline with an elimination half-life of around 4 days (Ezzet et al. 1998). There are, as
yet, no equivalent pharmacokinetic data for DBL. The day 7 plasma concentrations
from this study show that both plasma lumefantrine and DBL concentrations remain
above the IC99 in most patients for at least three parasite life cycles. Even if there were
synergy between lumefantrine and DBL at these concentrations as found by others
(Starzengruber et al. 2007; Starzengruber et al. 2008), its relevance at this stage in
treatment is unclear as initial parasite clearance had occurred in all patients allocated
this therapy in the trial (Karunajeewa et al. 2008b) and the presence of one or other
compound at a concentration >IC99 should inhibit low-level (sub-microscopic) parasite
replication. In addition, the variable day 7 lumefantrine:DBL ratios in PNG children,
which are consistent with the marked apparent between-dose variability in lumefantrine

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
151
bioavailability (Ezzet et al. 1998; van Agtmael et al. 1998), would lead to inconsistent
interactions. Indeed, the data relating day 7 lumefantrine and DBL concentrations to
therapeutic outcome suggest that DBL has a stronger role than the parent compound in
suppressing recrudescence and/or preventing reinfection. This may reflect the fact that,
even at relatively low plasma concentrations, DBL has more potent antimalarial activity
than lumefantrine as exemplified by the present in vitro sensitivity data. The fact that
the lumefantrine:DBL ratio had no relationship with outcome is unlikely to translate
into a clinically important synergistic interaction.
Initial parasite clearance is considered due primarily to the artemisinin component of
ACT and, conversely, its prolongation is taken as evidence of artemisinin resistance
(Dondorp et al. 2010). It is, however, possible that the longer half-life partner and
interactions between the component drugs enhance the relatively rapid parasiticidal
effects of artemisinin derivatives. Synergy between artemether and lumefantrine has
been reported (Hassan Alin et al. 1999) and the present data and those of others
(Kyavar et al. 2006; Muller et al. 2008) suggest a similar interaction for DBL/DHA.
However, the clinical importance of such effects is unclear. The synergy observed in
the present study was mild. The relatively short half-lives of artemether and its active
metabolite DHA (1 - 2 hr) (Ezzet et al. 1998; Ezzet et al. 2000) limit the time-window
for such effects. Interactions can be antagonistic, such as that between artemisinin and
DBL at some concentrations (Kyavar et al. 2006) and between DHA and 4-
aminoquininolines and related drugs (Davis et al. 2006). The fact that a range of
different ACTs show equal efficacy (Sinclair et al. 2009) suggests that acute effects on
the parasite other than those from the artemisinin drug are minor.
According to the manufacturer, DBL is a degradation product of lumefantrine, and
Coartem® tablets contain <0.1% of DBL both at the time of manufacture, and at the
end of the expiry date (H. Gruninger, personal communication, Novartis Pharma). The
occurrence of such low relative concentrations of DBL in pure lumefantrine powder
was confirmed by UPLC-LCMS/MS (unpublished data), and excluded
DBL:lumefantrine at 1:10000 from the drug interaction analysis as a result. Although a
small amount of DBL is present at the time of Coartem® administration, it is

Chapter 5 Antimalarial Properties of Desbutyl-Lumefantrine
152
quantitatively far below the level detected in plasma. Thus, this exogenous contribution
is insufficient to influence the plasma DBL concentrations in the present study, with the
corollary that DBL is a true metabolite.
The present study confirms that DBL has potential as an antimalarial drug in its own
right. Its in vitro potency relative to the parent compound, its synergy with DHA, and
the positive relationship between day 7 plasma concentrations and ACPR suggest that it
could be a useful alternative to lumefantrine as part of ACT. Although the presence of
DBL at parasiticidal concentrations during conventional artemether-lumefantrine
therapy suggest that it is as safe as lumefantrine, further pharmacokinetic and safety
assessment after DBL administration would be required to facilitate the development of
optimal dose regimens. In contrast to N-desbutyl-halofantrine, the active metabolite of
halofantrine (Traebert et al. 2005), preliminary in vitro cardiotoxicity studies have not
raised any significant concerns (Traebert et al. 2004).
This work has been published in full in Antimicrobial Agents and Chemotherapy, titled
“Desbutyl-lumefantrine is a metabolite of lumefantrine with potent in vitro antimalarial
activity that may influence artemether-lumefantrine treatment outcome”.

CHAPTER 6
STATINS AND FIBRATES:
LIPID-MODIFYING DRUGS AS
ANTIMALARIALS

Chapter 6 Statins and Fibrates as Antimalarials
154
CHAPTER 6. STATINS AND FIBRATES: LIPID-
MODIFYING DRUGS AS ANTIMALARIALS
6.1 INTRODUCTION
The increase in non-communicable diseases of affluence in developing countries such
as PNG may see a significant shift from heavy use of anti-infective agents to therapies
for cardiovascular risk factor reduction (Martin et al. 1981; Iser et al. 1993; Hodge et
al. 1996; Lindeberg et al. 1997; Kende 2001; Lesley et al. 2001; Yamauchi et al. 2001;
Yamauchi et al. 2005). Drugs used for one indication can sometimes have wider
application and there is evidence that some cardiovascular therapies such as the anti-
diabetic drug rosiglitazone (Boggild et al. 2009) could have antimalarial properties.
This is of particular interest given the recent emergence of P. falciparum resistant to
artemisinin (Wongsrichanalai et al. 2008; Carrara et al. 2009; Dondorp et al. 2010).
Statins and fibrates are distinct classes of lipid-modifying drugs that reduce morbidity
and mortality associated with cardiovascular disease. This chapter investigates the
potential of these two groups of agents as novel antimalarials.
6.1.1 Statins as Lipid-lowering and Antimicrobial Agents
Statins are well tolerated and widely used for reducing cardiovascular morbidity and
mortality (Wilt et al. 2004; Thavendiranathan et al. 2006). They inhibit 3-hydroxy-3-
methylglutaryl coenzyme A (HMG-CoA) reductase (Figure 1.19), a key enzyme in
cholesterol biosynthesis, which consequently lower serum concentrations of low-
density lipoprotein–cholesterol. There has been interest in alternative applications of
statins, with studies demonstrating their abilities to inhibit the growth of bacteria
(Catron et al. 2004), yeasts (Song et al. 2003), and protozoa (Chen et al. 1990; Urbina
1993; Andersson et al. 1996; Montalvetti 2000). The first study of their antimalarial
effects found that lovastatin and simvastatin inhibited in vitro intra-erythrocytic
development of P. falciparum (Grellier et al. 1994). More recently, the in vitro
susceptibilities of P. falciparum to six statins revealed atorvastatin had the greatest
activity (Pradines et al. 2007). Despite these encouraging findings, neither simvastatin
nor atorvastatin in high doses improved the outcome in Plasmodium berghei-infected

Chapter 6 Statins and Fibrates as Antimalarials
155
mice (Bienvenu et al. 2008; Kobbe et al. 2008) and there was no effect on parasitaemia
(Bienvenu et al. 2008). However, no study to date has included the most-potent statin in
clinical use, rosuvastatin (Soran et al. 2008). In addition, despite the demonstration of
in vitro synergy between mevastatin and the glycoprotein inhibitor tunicamycin against
P. falciparum (Naik 2001), the interaction between statins and conventional
antimalarial drugs has not been evaluated. Therefore, the relative in vitro antimalarial
activity of rosuvastatin and atorvastain, against P. falciparum and the in vitro
interactions between statins and both chloroquine (CQ) and dihydroartemisinin (DHA)
are presented in this chapter.
6.1.2 Fibrates as Potential Antimalarial Drugs
Fibrates such as gemfibrozil and fenofibrate are agonists of peroxisome proliferator-
activated receptor alpha (PPARα). Although active via a different pathway to that of the
statins, fibrates exhibit potent lipid-modifying properties (Derosa et al. 2009). They
have well-characterised pharmacokinetic and pharmacodynamic profiles. An early
study on the effect of plasma free fatty acid concentrations and temperature on
parasitaemia in P. berghei-infected mice employed clofibrate as a lipid-lowering agent
(McQuistion 1979). Although not a primary interest of the study, clofibrate directly
inhibited the development of parasitaemia. However, the authors did not extend these
observations and no other subsequent study has examined the role of fibrates in the
treatment of malaria. It is possible that Plasmodium species have PPARα-like motifs
which could mediate such fibrate antimalarial effects.
There is further indirect evidence of a link between fibrates and malaria. In a
biochemical assay, Ehrhardt et al. assessed the in vitro influence of fibrates on P-
glycoprotein activity. Of gemfibrozil, fenofibric acid, clofibrate and fenofibrate, the
latter drug was the only member of the class to inhibit P-glycoprotein mediated
transport (Ehrhardt et al. 2004). A P-glycoprotein homologue 1 in P. falciparum has
been implicated in CQ and mefloquine resistance (Reed et al. 2000) and it is possible
that fenofibrate may decrease drug efflux mediated by this protein and thus reverse
resistance. Fibrates exhibit pleotropic effects that could also modify the effects of

Chapter 6 Statins and Fibrates as Antimalarials
156
malaria infection. An example of this is the attenuation of brain tissue injury associated
with inflammation (Bordet et al. 2006) and vascular occlusion (Guo et al. 2009). In a
murine influenza study, where severe systemic disease is thought to arise through
overproduction of proinflammatory cytokines, treatment with gemfibrozil doubled
survival compared to vehicle-treated mice (Budd et al. 2007). Similar host
inflammatory responses have been proposed for malaria pathology, particularly in coma
and other clinical manifestations associated with cerebral malaria (Clark et al. 2005;
Clark et al. 2008; Clark et al. 2009).
In the present study, the direct antimalarial activity of gemfibrozil, clofibrate,
fenofibrate, and its metabolite fenofibric acid against P. falciparum are investigated. In
vitro interactions between these fibrates and both CQ and DHA are also examined.
6.2 MATERIALS AND METHODS
6.2.1 In vitro Parasite Growth Inhibition
The laboratory-adapted P. falciparum strains 3D7 (Africa; CQ-sensitive), E8B (Brazil;
CQ-resistant), K1 (Thailand; CQ-resistant), W2mef and Dd2 (Indochina; CQ-resistant)
were maintained as previously described (Section 2.1). Stock solutions of CQ
diphosphate (Sigma Chemicals, St Louis, USA), atorvastatin (Waterstonetech, Carmel,
IN), rosuvastatin (Waterstonetech, Carmel, IN), fenofibrate (Sigma), clofibrate (Sigma),
gemfibrozil (Sigma-Aldrich) and fenofibric acid (Tyger Scientific Inc, NJ, USA) were
prepared in distilled water (CQ) and DMSO (statins and fibrates).
Serial dilutions of each drug were prepared in RPMI and added in triplicate to 96-well
plates with final concentrations of 12.5 to 1600 nM (CQ), 0.3 to 200 µM (atorvastatin)
and 0.6 to 400 µM (rosuvastatin, pravastatin and simvastatin), 6.2 to 800 µM
(gemfibrozil, clofibrate, fenofibrate) and 39 to 5000 nM (fenofibric acid). Synchronous
parasite suspensions (≥90% rings) were adjusted to 1.0% for non-isotopic and 0.5%
parasitaemia for isotopic assays and a final hct of 1.5% in the drug-parasite mixture.
Following 48 hr incubation, parasite growth was measured initially by a modification of
the pLDH assay as described previously (Section 2.2.4) (Makler et al. 1993a) and

Chapter 6 Statins and Fibrates as Antimalarials
157
subsequently by the 3H-hypoxanthine incorporation assay (Section 2.2.5) (Desjardin
1979). IC50s and IC90s were determined by non-linear regression analysis (Graphpad
Prism 4.0).
6.2.2 Drug Interaction Studies
A modified fixed-ratio isobologram method was used to assess drug interactions
(Fivelman et al. 2004). Inhibition assays using 3H-hypoxanthine incorporation (Section
2.) were first carried out to determine individual IC50s for statins, fibrates, CQ and
DHA (Desjardin 1979). These were used to establish test concentration ranges in the
combination assays. A total of six and eleven solutions containing fixed-ratios of
conventional antimalarials with statins and fibrates, respectively, were prepared as
shown in Table 6.1. The FICs of each drug in each combination determined from dose-
response curves were used to construct isobolograms from which the sum of each FIC
was calculated (Berenbaum 1978).

Chapter 6 Statins and Fibrates as Antimalarials
158
Table 6.1 Interaction ratios of statins, fibrates and conventional antimalarials.
Molar ratios and drug concentrations of atorvastatin or fibrates (gemfibrozil, fenofibrate
or fenofibric acid) paired with either chloroquine (CQ) or dihydroartemisinin (DHA)
are shown. Each within-well concentration shown is that of the prepared drug
combination. Further two-fold serial dilutions of each combination were performed to
give six additional tests at lower doses. For simplicity, drug concentrations are
displayed only for the first assay well (i.e. contains the highest concentration of drugs)
for a given combination. *Fenofibric acid concentrations are in nmol.
Drug pair Ratio Top concentrations within a combination
Statin/Fibrate (µM) CQ (nM) DHA (nM)
Atorvastatin-antimalarials
5:0
0:5
4:1
3:2
2:3
1:4
200
0
160
120
80
40
0
200
40
80
120
160
0
60
12
24
36
48
Fibrates-antimalarials
Gemfibrozil
Fenofibrate
Fenofibric acid*
1:0
0:1
1:1
1:3
1:30
1:300
1:3000
3:1
30:1
300:1
3000:1
1600
0
1600
533.3
53.3
5.33
0.533
1600
1600
1600
1600
2500*
0
2500
833.3
83.3
8.33
0.833
2500
2500
2500
2500
0
1600
1600
1600
1600
1600
1600
533.3
53.3
5.33
0.53
0
100
100
100
100
100
100
33.3
3.33
0.33
0.033
6.2.3 Dosed Plasma Bioassay
Due to the possibility that active metabolites and other in vivo factors might contribute
to enhanced antimalarial activity of fenofibrate and atorvastatin, such as those observed
for atorvaquone (Butcher et al. 2003; Edstein et al. 2005), bioassays were performed.
For atorvastatin bioassay, a pre-treatment venous blood sample was taken from a

Chapter 6 Statins and Fibrates as Antimalarials
159
healthy volunteer who was then given atorvastatin 80 mg (Lipitor™, Pfizer, NY) once
daily for 4 consecutive days. A second venous blood sample drawn 3 hr after the last
dose at the time of the predicted maximal plasma concentration (Cilla et al. 1996). The
samples were centrifuged promptly and aliquots of separated plasma stored at -20°C.
Aliquots of pre- and post-treatment plasma were used for atorvastatin assay using
HPLC (Nirogi et al. 2006).
For fenofibrate bioassay, a pre-treatment blood sample was taken from a healthy
volunteer who was then given fenofibrate at 145 mg (Lipidil ™, Solvay
Pharmaceuticals) once daily for 6 consecutive days. A second blood sample was drawn
on day 6 at the time of the predicted maximal plasma concentration of fenofibric acid at
steady state (i.e. 4 hr after the final dose) (Keating et al. 2002). The heparinised blood
samples were centrifuged promptly and aliquots of separated plasma were stored at -
20°C. Plasma fenofibric acid levels from pre- and post-treatment samples were
measured, after extraction in hexane/chloroform/isopropanol, (18:80:2, v/v/v), by
validated high performance liquid chromatography assay with tandem mass
spectrometric detection and 2-(2,4,5-trichlorophenoxy)-propionic acid as internal
standard (Laboratoires Fournier S.A., Daix, France) (Zhu et al. 2010). Approval for
these procedures was obtained from the South Metropolitan Area Health Service
Human Research Ethics Committee.
A modified microdilution isotopic technique (Desjardin 1979; Kotecka et al. 2003) was
used to determine the antimalarial activities of pre- and post-treatment plasma. For
atorvastatin bioassay, pre- and post-treatment plasma were spiked with CQ
(concentration range, 3.9 to 250 nM), and DHA (0.9 to 60 nM), or an equivalent
volume of drug-free RPMI. For fenofibrate bioassay, the drug levels in the post-
treatment plasma and inhibitory properties from preliminary in vitro data indicated that
these were adequate for a direct assessment using two-fold serial dilutions in drug-free
RPMI without the need to spike with an active antimalarial drug. In triplicate
experiments, aliquots of 100 µL of plasma were added to 90 µL of parasite suspension
(1% parasitaemia, 1.5% hct) and 10 µL of 3H-hypoxantine (0.5 µCi) in 96-well plates,

Chapter 6 Statins and Fibrates as Antimalarials
160
and the mixture was incubated, harvested and counted (Karl et al. 2009).
6.3 RESULTS
6.3.1 In vitro Antimalarial Activities of Statins
The in vitro inhibitory effects of atorvastatin, rosuvastatin, simvastatin, pravastatin and
CQ are summarised in Table 6.2. All statins showed antimalarial activity, but
atorvastatin was more potent than rosuvastatin. The IC50 and IC90 values for each statin
did not differ between CQ-sensitive and CQ-resistant strains and were well above those
for CQ, even against CQ-resistant strains. Simvastatin and pravastatin were weak
against P. falciparum (IC50>200 µM).
6.3.2 In vitro Antimalarial Activities of Fibrates
The in vitro inhibitory effects of gemfibrozil, clofibrate, fenofibrate, fenofibric acid and
CQ are summarised in Table 6.3. All fibrates showed antimalarial activity but
fenofibric acid was most potent. With the exception of gemfibrozil, the IC50 values of
other fibrates did not differ between CQ-sensitive and CQ-resistant strains and were
well above those for CQ.

Chapter 6 Statins and Fibrates as Antimalarials
161
Table 6.2 In vitro activities of statins against CQ-sensitive and CQ-resistant strains of P. falciparum. The means (and 95% CIs) shown are from
at least four independent triplicate experiments. *Inhibitory concentrations for chloroquine are in nanomoles. Data from CQ-resistant strains Dd2 and
E8B were similar and have been pooled
Strain Atorvastatin Rosuvastatin Pravastatin Simvastatin Chloroquine
IC50 (µM) IC90 (µM) IC50 (µM) IC90 (µM) IC50 (µM) IC50 (µM) IC50 (nM)
3D7 25 (16 - 39) 68 (38 - 121) 80 (47 - 137) 205 (113 - 373) >200 >200 30 (23 - 29)
Dd2/E8B 17 (8 - 37) 39 (24 - 62) 80 (37 - 174) 232 (133 - 403) >200 >200 271 (138 - 533)

Chapter 6 Statins and Fibrates as Antimalarials
162
Table 6.3 In vitro activities of fibrates against CQ-sensitive and CQ-resistant strains of P. falciparum. Data represent at least six experiments
performed in triplicate. *Inhibitory concentrations for chloroquine and fenofibric acid are in nanomoles.
3D7 IC50 (µM) IC90 (µM)
W2mef IC50 (µM) IC90 (µM)
Clofibrate 184 (98 - 345) 441 (264 - 736) 256 (197 - 332) 708 (514 - 976)
Gemfibrozil 311 (245 - 395) 541 (446 - 657) 245 (181 - 332) 511 (445 - 587)
Fenofibrate 69 (54 - 88) 228 (171 - 304) 72 (65 - 80) 178 (152 - 210)
Fenofibric acid 152 (109 - 212)* 250 (191 - 326)* 1120 (916 - 1369)* 2436 (1866 - 3179)*
Chloroquine 23 (15 - 34)* 48 (23 - 99)* 230 (202 - 261)* 495 (334 - 732)*

Chapter 6 Statins and Fibrates as Antimalarials
163
Figure 6.1 In vitro susceptibility of laboratory strains of P. falciparum to cholesterol-lowering drugs and chloroquine.

Chapter 6 Statins and Fibrates as Antimalarials
164
6.3.3 Interaction of Atorvastatin with Conventional Antim alarials
The potential use of atorvastatin as a partner drug with conventional antimalarials was
investigated by means of isobolograms. In vitro interactions between atorvastatin with
CQ and DHA are shown in Figure 6.2. The interaction was indifferent in each case with
a mean ΣFIC [95% confidence interval] of 1.05 [0.97 to 1.14] for atorvastatin-CQ and
1.08 [0.99 to 1.17] for atorvastatin-DHA.
6.3.4 Interaction of Fibrates with Conventional Antimalar ials
There were no synergistic combinations identified from the drug interaction studies
involving fibrates, CQ, DHA and atorvastatin. For fenofibric acid-CQ there was no
interaction present for 3D7 but antagonism for W2mef. In the case of gemfibrozil-CQ
against 3D7 there was antagonism and indifferent interaction in W2mef. The remaining
combinations including that for fenofibric acid-atorvastatin showed indifferent
interactions.

Chapter 6 Statins and Fibrates as Antimalarials
165
Figure 6.2 Interaction between atorvastatin and conventional antimalarials.
Isoboles showing the FICs of atorvastatin plotted against those for CQ (left-hand panel)
and DHA (right-hand panel). The isoboles are representative of three independent
experiments in which each of the 6 drug ratios was tested in duplicate. The degree of
interaction (I) with 95% CI is represented by grey and dotted lines, respectively.

Chapter 6 Statins and Fibrates as Antimalarials
166
Table 6.4 In vitro efficacy of fibrates and antimalarial drug combinations.
Fenofibrate, fenofibric acid and gemfibrozil in combination with CQ, DHA and
atorvastatin were assessed against P. falciparum clones 3D7, W2mef and Dd2 using
isobolographic analysis. Data are the interaction factor (I) and the summed fractional
inhibitory concentration (ΣFIC) with 95% CIs. The assessment of interaction is based
on both I and ΣFIC data (see text). Interaction between fenofibrate-dihydroartemisinin
was not determined.
I (95% CI) ΣFIC (95% CI) Interaction
3D7
Fenofibric acid-chloroquine 0.23 (-0.06 to 0.51) 0.99 (0.95 to 1.02) Indifferent
Fenofibric acid-dihydroartemisinin -0.26 (1.02 to 0.50) 1.11 (0.99 to 1.21) Indifferent
Fenofibric acid-atorvastatin -0.33 (-1.92 to 1.26) 1.19 (1.02 to 1.35) Indifferent
Fenofibrate-chloroquine -0.67 (-1.40 to 0.05) 1.08 (0.98 to (1.12) Indifferent
Gemfibrozil-chloroquine -1.53 (-2.44 to -0.62) 1.12 (1.01 to 1.23) Antagonistic
Gemfibrozil-dihydroartemisinin -1.34 (-3.34 to 0.66) 1.20 (0.95 to 1.45) Indifferent
W2mef/Dd2
Fenofibric acid-chloroquine -1.07 (-2.08 to -0.07) 1.19 (1.02 to 1.36) Antagonistic
Fenofibric acid-dihydroartemisinin -0.48 (-1.31 to 0.36) 1.11 (1.02 to 1.21) Indifferent
Fenofibric acid-atorvastatin -0.81 (-1.52 to -0.09) 1.08 (0.96 to 1.19) Indifferent
Fenofibrate-chloroquine -0.41 (-1.01 to 0.19) 1.01 (0.93 to 1.10) Indifferent
Gemfibrozil-chloroquine -0.55 (-1.80 to 0.73) 1.19 (0.99 to 1.40) Indifferent
Gemfibrozil-dihydroartemisinin -0.25 (-2.10 to 1.61) 1.15 (0.95 to 1.40) Indifferent

Chapter 6 Statins and Fibrates as Antimalarials
167
6.3.5 Bioassay of Atorvastatin
The ability of atorvastatin-containing plasma to inhibit P. falciparum in vitro was
assessed by bioassay. Plasma atorvastatin concentrations in the healthy volunteer were
undetectable at pre-treatment and were 118 µg/L (0.1 µM) post-treatment. Neither pre-
nor post-treatment plasma inhibited growth of 3D7 (Figure 6.3). The 3D7 IC50s for CQ
alone, CQ plus pre-treatment plasma, and CQ plus post-treatment plasma were similar
(20.5 nM, 20.5 nM, and 20.4 nM, respectively), as were those for DHA alone, DHA
plus pre-treatment plasma, and DHA plus post-treatment plasma (12.7 nM, 20.1 nM,
and 19.2 nM, respectively).
6.3.6 Bioassay of Fenofibric Acid
The antimalarial activity of fenofibric acid generated in vivo as a result of fenofibrate
dosing was assessed by bioassay (Figure 6.4). Plasma fenofibric acid concentrations in
the healthy volunteer were undetectable at pre-treatment and were 13.0 mg/L post-
treatment (i.e. plasma fenofibric acid concentration 40,816 nM). Post-treatment plasma
inhibited growth of 3D7 at dilutions between 8 to 1024-fold.

Chapter 6 Statins and Fibrates as Antimalarials
168
Figure 6.3 Atorvastatin bioassay. Dose response of laboratory-adapted P. falciparum
subjected to conventional antimalarials with pre- and post- Lipitor™ (atorvastatin)
treatment plasma. No significant differences were indicated in response to pre- and
post-treatment plasma added to chloroquine (CQ) (top) and to dihydroartemisinin
(DHA) (bottom).

Chapter 6 Statins and Fibrates as Antimalarials
169
Figure 6.4 Fenofibrate bioassay. Growth response of laboratory-adapted 3D7 to two-
fold dilutions of plasma pre- and post-treatment of Lipidil ™ (fenofibrate). Data points
were obtained from three independent bioassays performed in triplicate.

Chapter 6 Statins and Fibrates as Antimalarials
170
6.3.7 BLAST Analysis for PPAR-like Region in Plasmodium
To elucidate possible mechanisms underlying the activity of fenofibric acid against
malaria parasites, similarity searches were conducted between mRNA and protein
sequences of human PPARα and P. falciparum using basic local alignment search tool
(BLAST, NCBI). Nucleic acid alignments between human and Plasmodium mRNA
produced 102 blast hits of short sequences (<40 bp). One hit had non-random matches
(expected value (E) <0.02) identified as a region encoding for a P. falciparum 3D7
conserved Plasmodium protein, however its function is unknown.
Comparisons between the human PPARα protein (NP_001001928.1) and reference
proteins for P. falciparum taxid (5833) resulted in three blast hits (Figure 6.5).
Although the alignment scores were <40 for all three sequences, two were located at the
PPARα DNA binding domain and one at the ligand binding domain. The latter,
identified as a conserved Plasmodium protein (XP_001350490.2) is of particular
interest since it aligns with both the ligand binding site and the heterodimer interface,
with 17% coverage of the human PPARα protein sequence.
In addition, the same Plasmodium protein was one of five hits with other
apicomplexans in comparison with the human PPARα protein. There were two hits
from Babesia bovis showing 22 and 27% coverage of the human protein sequence (E =
1.4 and 1.9), and two from Toxoplasma gondii with 11 and 17% sequence coverage.
The region (959-1030) of the Plasmodium protein identified as Plasmodium 1 in Figure
6.5, showed similarity with hypothetical proteins in other Plasmodium species including
P. knowlesi, P. vivax, P. yoelii, P. chabaudi and P. berghei. It also weakly aligned with
the PPARα protein sequence from Rhesus Macaque, Common Chimpanzee, Guinea pig
and human.
6.3.8 BLAST Analysis for ABC-1 transporter in P. falciparum
Proteins within the ATP-binding cassette sub-family A member (ABC-1) mediate
cholesterol and lipid transport, are within the same superfamily of ATP-binding cassette

Chapter 6 Statins and Fibrates as Antimalarials
171
(ABC) transporters as the multidrug resistant P-glycoprotein. Nucleic acid alignments
between human and Plasmodium mRNA produced 103 blast hits of short sequences (up
to 50 bp). Similarity searches between reference protein sequences of human ABC-1
and P. falciparum revealed the presence of a parasite homolog of this lipid efflux pump.
There were 49 protein alignment hits, 13 of the P. falciparum nucleotide sequences
spanned across two regions (900 to 1116 and 1927 to 2110) with a coverage up to 19%
(E <0.006) of the human ABC-1 query sequence (Figure 6.6). Two significant
alignments were found in the P. falciparum ABC transporter protein (XP_001350233.1)
and the P. falciparum multi-drug resistance protein 2 (XP_001348629.1) with E values
of 8-16and 1-12, respectively.

Chapter 6 Statins and Fibrates as Antimalarials
172
Figure 6.5 Distribution of Plasmodium BLAST hit sequences on human PPARα. Protein-protein alignments between human PPARα (468bp) and
P. falciparum was analysed by BLAST. Three P. falciparum protein sequences (green) have 10-17% coverage of the human PPARα sequence (grey).
These specific hits span over the PPARα DNA binding (blue) and ligand binding (red) domains. Plasmodium 1: conserved Plasmodium protein (E =
0.18) (XP_001350490.2), Plasmodium 2: erythrocyte membrane protein, PfEMP1 (E = 1.6) (XP_001349513.1), Plasmodium 3: conserved
Plasmodium protein (E = 8.4) (XP_001349690.1).

Chapter 6 Statins and Fibrates as Antimalarials
173
Figure 6.6 Distribution of P. falciparum BLAST hit sequence on human ABC-1. Protein-protein alignments between human ABC-1 (2261 bp)
and P. falciparum was analysed by BLAST. Thirteen of forty-nine P. falciparum sequences from the reference nucleotide database aligned at the same
two regions (green) with an overall of 10-19% coverage of the human ABC-1 protein sequence (grey). These specific hits spanned over the ABC-1
conserved domains (CD, in red). Two significant alignments were the P. falciparum ABC transporter protein (E = 8-16) (XP_001350233.1), P.
falciparum multi-drug resistance protein 2 (E = 1-12) (XP_001348629.1), both have alignment scores of 50 to 200.

Chapter 6 Statins and Fibrates as Antimalarials
174
6.4 DISCUSSION
The present data confirm that atorvastatin inhibits the growth of P. falciparum in vitro
(Grellier et al. 1994; Pradines et al. 2007). Consistent with previous findings (Pradines
et al. 2007), pravastatin and simvastatin had minimal activity against P. falciparum.
The antimalarial activity of atorvastatin is greater than that of rosuvastatin, but the
atorvastatin IC50 (17 to 25µM; Table 6.2) is approximately 100 times above that
achievable in plasma with repeated maximal (80 mg) doses (0.1 to 0.3 µM) (Borek-
Dohalsky et al. 2006). As might have been predicted from this observation, and
consistent with the hypothesis that the metabolism of atorvastatin does not generate
compounds with antimalarial activity, the bioassay showed that therapeutic plasma
concentrations had no inhibitory effect against cultured P. falciparum. In addition, there
was no synergy with conventional antimalarial drugs.
The ability of the malaria parasite to synthesise cholesterol de novo (i.e. via the
mevalonate pathway) appears limited (Vial et al. 1984; Wunderlich et al. 1991), and the
presence of an HMG-CoA homolog was not revealed by BLASTX analysis of the P.
falciparum sequence with other protozoan HMG-CoA protein sequences (Pradines et
al. 2007). These observations and the greater antimalarial potency of atorvastatin versus
rosuvastatin (the reverse of their ability to inhibit cholesterol synthesis in humans
(Jones et al. 2003)) suggest an alternative, albeit low-potency, mechanism of
antimalarial action to inhibition of HMG-CoA reductase.
The present study demonstrates that gemfibrozil, fenofibrate and clofibrate have weak
activity relative to conventional antimalarial drugs in vitro. Nonetheless, their IC50s are
similar to antibiotics that are used for malaria prophylaxis and adjunctive therapy
(Nakornchai et al. 2006). This activity may have accounted for the lower infection rate
in the clofibrate-treated group of animals observed previously (McQuistion 1979),
although the inflammation modulating effects of clofibrate may have also contributed.
Fenofibric acid, the major metabolite of fenofibrate in vivo (Kirchgassler et al. 1998),
has greatest activity at nM media concentrations similar to those of conventional agents.
As might have been predicted from this observation, and consistent with the hypothesis
that the metabolism of fenofibrate would generate sufficient fenofibric acid with

Chapter 6 Statins and Fibrates as Antimalarials
175
antimalarial activity, the bioassay showed that therapeutic plasma concentrations (13
mg/L or 40,816 nM) had inhibitory effect against cultured P. falciparum with IC90s of
CQ-sensitive and resistant strains at 250 nM and 2436 nM, respectively.
An inhibitory effect was also observed in pre-treatment plasma against P. falciparum.
Plasma from healthy individuals can interfere with parasite survival via complement-
mediated cell lysis. Non-heat treated sera was shown to reduce parasite growth as much
as 25% compared to heat-treated control (Teja-Isavadharm et al. 2004). Therefore, heat
inactivation of pooled human plasma is required to reduce this growth inhibitory
activity prior supplementation in culture medium (Appendix B). Plasma fenofibric acid
is stable for up to 8 hr at room temperature (Dubey et al. 2010); however, its stability in
plasma at 50°C is unknown. Therefore, to minimise the risk of drug degradation,
plasma samples in the bioassays were not heat-treated. This may explain the apparent
dose-response of pre-treatment plasma against the parasite. Treatment of bioassay
plasma with non-heat approaches such as the use of Affingel Protein-A may reduce this
inhibition by removal of human immunoglobulin G (Teja-Isavadharm et al. 2004).
For a drug to be clinically useful, the plasma concentration achievable in vivo should be
a number of magnitudes higher than the in vitro inhibitory concentration. The steady-
state maximum plasma concentration for fenofibric acid is approximately 25.5 mg/L
(i.e. 80 µM) (Miller et al. 1998), whilst the maximum plasma concentration of
fenofibric acid is in the range 3 – 15 mg/L (9.4 – 47 µM) after an oral dose of
fenofibrate of 100 or 200 mg (Bhavesh et al. 2009) with levels >0.4 mg/L (>1.2 µM)
sustained over 2 days. The plasma fenofibric acid level available with repeated
therapeutic doses is approximately 100 times more than that required to inhibit 50% of
cultured P. falciparum. A newer formulation of micronised fenofibrate may further
enhance bioavailability (Keating et al. 2002).
Although its mode of action remains to be elucidated, fenofibric acid may act by
interfering with P-glycoprotein (Ehrhardt et al. 2004) and ABC-1 mediated transport,
and/or via a putative PPARα-like protein. BLAST analysis revealed partial similarities
between a conserved Plasmodium protein sequence and that of the PPARα ligand

Chapter 6 Statins and Fibrates as Antimalarials
176
binding domain in humans. Despite the low alignments score, there may be sufficient
similarities at key amino acid positions for the tertiary protein conformation to interact
with fenofibric acid, resulting in subsequent metabolic interference. In human and
murine cells, fenofibric acid interferes with the expression of ABC-1 (Jaye et al. 2003;
Arakawa et al. 2005), thus altering lipid accumulation. Fenofibric acid effects on the
Plasmodium ABC-1 homolog may disturb the development of P. falciparum by similar
mechanisms, depriving the growing parasite of lipid components of membranes and
other cellular structures.
In conclusion, this study confirmed statins to have antimalarial properties but at a
concentration higher than that achieved by therapeutic doses (Borek-Dohalsky et al.
2006). In addition, even at supra-therapeutic doses, no synergy with CQ or DHA was
observed. Although atorvastatin has been shown to prevent cytoadherence of P.
falciparum to endothelial cells in co-culture models (Taoufiq et al. 2011), outcome data
from animal models of severe malaria showed no protection (Bienvenu et al. 2008;
Kobbe et al. 2008). These in vivo studies together with the present in vitro findings and
do not support calls for clinical trials of statins as adjuvant antimalarial therapy
(Bienvenu et al. 2008).
The present experiments also revealed fenofibric acid to be the most active lipid-
modifying agent against P. falciparum in vitro. The favourable pharmacokinetics of
fenofibrate (Miller et al. 1998; Bhavesh et al. 2009) along with the high antimalarial in
vitro activity of its major metabolite fenofibric acid warrants confirmatory in vivo
investigation of this promising therapeutic application.
The statin component of this work has been published in Antimicrobial Agents and
Chemotherapy, titled “Statins as potential antimalarial drugs: Low relative potency and
lack of synergy with conventional antimalarial drugs”. The fenofibrate component is
undergoing review for a patent.

CHAPTER 7
DETECTION OF VOLATILE ORGANIC
COMPOUNDS OF PLASMODIUM
FALCIPARUM IN VITRO

Chapter 7 Volatile Organic Compounds
178
CHAPTER 7. CHARACTERISATION OF VOLATILE
ORGANIC COMPOUNDS OF P. FALCIPARUM IN VITRO
7.1 INTRODUCTION
The detection of viable parasite forms is an essential requirement for malaria diagnosis
and subsequent monitoring of the response to antimalarial therapy. For diagnosis,
microscopic examination of a peripheral blood smear remains the investigation of
choice in a wide variety of clinical situations. However, the sensitivity of microscopy is
limited even when expert microscopists view high quality slides. In addition, the
diagnosis may be missed in cases of severe falciparum malaria in which the majority of
parasites are sequestered within the microvasculature of major organs (Coltel et al.
2004; Safeukui et al. 2008) or in the placenta in infected expectant mothers (Duffy
2007; Goyal et al. 2009). Antigen detection kits can be used where reliable microscopy
is unavailable but they have even lower sensitivity and specificity (Section 1.6). PCR
increases diagnostic sensitivity but its timely availability is limited largely to
specialised laboratories in developed countries. In addition, the sensitivity of PCR
(down to 1 parasite/µL) means that even a child weighing only 15 kg and with a
circulating blood volume of approximately 1 litre who is PCR-negative may still
harbour up to a million malaria parasites. The monitoring of the response to antimalarial
therapy in individual patients depends on the availability of serial blood smears
complemented by PCR where available. Antigen detection methods cannot be used
because of the persistence of antigen after parasite clearance, while PCR does not
differentiate between DNA from viable and non-viable parasites.
There is a need for the development of alternative diagnostic tests that detect viable
parasites before and after treatment with greater specificity and sensitivity than
currently available methods. The human breath contains a large number of volatile
organic compounds (VOCs) derived from the blood by passive diffusion in the lungs
(Phillips et al. 1992c). VOCs in the breath are directly related to concentrations in blood
and other tissues as they flow from compartments with higher vapour pressure to those
with lower pressure (Phillips 1992b). Breath tests have been used to assist in the early
diagnosis of conditions such as heart disease, rheumatoid arthritis and lung cancer

Chapter 7 Volatile Organic Compounds
179
(Gordon et al. 1985; Humad et al. 1988; Weitz et al. 1991) as they detect increased
VOCs released as a result of disease-specific cellular injury. More recently, exogenous
VOCs produced by microorganisms such as Mycobacterium tuberculosis have been
found in the breath of infected patients (Phillips et al. 2007). Plasmodium species may,
in the same way, produce a characteristic VOCs ‘fingerprint’ that can facilitate
diagnosis and therapeutic monitoring. In the case of P. falciparum and perhaps P. vivax
(Anstey et al. 2007), the cytoadherence of mature parasite forms in the pulmonary
microvasculature may facilitate detection of Plasmodium-specific VOCs in breath
samples. The cause of altered consciousness in severe malaria remains unknown. VOCs
are used as general anaesthetics in clinical practice (Soukup et al. 2009) and it is
possible that coma complicating malaria may result from elaboration of VOCs by
malaria parasites in the cerebral microcirculation that have anaesthetic properties. In
any case, malaria may, through indirect pathogenic tissue effects such as oxidative
stress, alter the VOCs content of human breath in ways that are characteristic of the
infection.
A number of extraction techniques are used for the capture and analysis of VOCs from
human breath and the microbial culture atmosphere (Phillips 1997; Lechner et al.
2005a; Syhre et al. 2008; Martin et al. 2010; Risticevic et al. 2010). Sampling and
sample preparation involve pre-concentrating the analytes of interest by purge and trap,
headspace, liquid-liquid or solid phase extractions. These conventional techniques
consist of multiple labour-intensive procedures and/or require organic solvents. Solid
phase micro-extraction (SPME) is an adsorption/desorption technique that circumvents
most of the drawbacks to sample preparation (Risticevic et al. 2010). SPME can be
used to concentrate volatile and non-volatile compounds in liquid samples or headspace
without the use of solvents with sensitivities down to parts per trillion (Risticevic et al.
2010), where the target compounds are subsequently separated and quantified by gas
chromatography-mass spectrometry (GC-MS).
This chapter outlines the design and optimisation of a P. falciparum culture-sampling
system suitable for VOCs headspace capture and analysis. Mass spectra of VOCs
emitted by P. falciparum in vitro using GC-MS are also reported. Identification and

Chapter 7 Volatile Organic Compounds
180
evaluation of these chemical finger-prints may have useful indications for diagnosis and
may yield insights into the metabolism and pathogenicity of P. falciparum.
7.2 MATERIALS AND METHODS
7.2.1 Parasites
The laboratory-adapted P. falciparum strains 3D7 (chloroquine-sensitive) and W2mef
(chloroquine-resistant) were maintained in RPMI 1640 HEPES as previously described
(Section 2.1). Once the parasitaemia was >5%, synchronous cultures at the trophozoite
stage were transferred into custom-designed containers (Section 7.3.1) at 1% hct and
purged with a mixture of 1% O2 and 5% CO2 in nitrogen at 5 psi for 4 sec and 30 sec
for prototypes 1 and 2, respectively. Subsequent optimisation used 5% O2 and 5% CO2
in nitrogen at 15 psi for 40 sec in the prototype 2 culture-sampling apparatus. The
volume of media required to sustain high parasitaemia was calculated using the
formula: volume of media (mL)/24hr = 0.005 x (µL RBC pellet) x (% parasitaemia)
(Radfar et al. 2009). This equation takes into account the nutrient requirements for non-
parasitised as well as parasitised RBC. A control was set up with non-infected RBC
using similar conditions and incubated for 24 hr at 37°C.
7.2.2 Solid Phase Micro-Extraction (SPME)
After incubation, samples were double-contained and transported to the School of
Biomedical, Biomolecular and Chemical Sciences (UWA) for extraction and analysis.
Volatile and semi-volatile compounds within the headspace of non-parasitised control
and malaria cultures were pre-concentrated onto a polydimethylsiloxane (PDMS, 100
µM) coated SPME fibre (SUPELCO, Bellefonte, PA, USA, #57300-U or portable field
sampler #504823) for 1 hr in a heated waterbath (Section 2.4). In subsequent
experiments, a triple fibre, 50/30 µM Divinylbenzene/Carboxen/PDMS StableFlex
fibre™ (SUPELCO, Bellefonte, PA, USA, #57328-U) was also used. After sampling
was completed, the fibre was retracted and the SPME holder was manually loaded onto
the GC injector port where VOCs were desorbed for 5 min in splitless mode for 2 min.

Chapter 7 Volatile Organic Compounds
181
7.2.3 Solvent Extraction
Supernatants from 3D7 and W2mef cultures at high parasitaemia (5% to 13.2%) were
pooled together (100 mL and 150 mL, respectively). Cell pellets of each strain were
lysed by sonication (Microson™ ultrasonic cell disruptor, Misonix Inc, NY) for 30 sec
and diluted with distilled water prior to extraction. The aqueous supernatant was
transferred into a separation funnel and partitioned 3 times with ⅓ volume of an organic
solvent (hexane, dichloromethane or ethyl acetate). The procedure involved gentle
swirl-mixing with occasional depressurisation via the outlet valve of the funnel. After
repositioning the funnel to the retort stand, the aqueous and organic phases gradually
separated (Figure 7.1)
7.2.4 Thermal Desorption: Purge and Trap
For purge and trap extraction, non-parasitised control and P. falciparum infected RBC
were cultured in prototype 2 containers (Figure 7.2). Air was drawn through the side
inlets containing a loosely fitted cap and through the flask over the surface of the
samples and the VOCs were trapped using a Tenax™ trap (200 mg, SUPELCO,
Bellefonte, PA, USA). The headspace was collected for 1 hr at an airflow rate of 1.5
L/min using a portable air sampling pump (224-PCXR8, SKC Inc.) The trap was
inserted into a short path thermal desorption injector (TD-2, Scientific Instrument
Services, Inc.) and desorbed for 5 min at 200°C using a flow of helium (2 mL/min) into
the GC-MS injection port that was also set at 200°C. The desorbed VOCs were
collected on the column during the desorption process by cooling a small section of the
capillary column with an ethanol/dry ice bath (-20°C to -40°C). The column was then
equilibrated to 35°C and the temperature program on the GC-MS was started. The
compounds were separated using a 30 m × 0.25 mm i.d., 0.25 µm BPX-5 column
(SGE), which was set at 35°C for 2 min and increased at 7°C/min until 250°C, and held
for 10 min. The mass spectrometer was set to record between 45 and 400 amu.

Chapter 7 Volatile Organic Compounds
182
Figure 7.1 Extraction of VOCs from culture supernatant by an organic solvent.
The aqueous phase was set aside whilst the organic phase was collected into a clean,
acetone-rinsed flask. The aqueous phase was subjected to repeated extraction into an
organic solvent (a). The organic phase was then extracted against distilled water and
collected into a conical flask followed by drying over anhydrous magnesium sulphate.
After filtration (Whatman™ filter papers) (b), the extract was transferred into a round
bottom flask where the solvent was evaporated under reduced pressure by means of a
rotary evaporator (Rotavapor-R, Buchi Labortechnik, Flawil, Switzerland) (c). For
analysis by GC-MS, extracts were evaporated to dryness under nitrogen (d) and
resuspended in 100 µL of the extraction solvent.

Chapter 7 Volatile Organic Compounds
183
Figure 7.2 Purge and trap set-up for thermal desorption. The prototype 2 sampling
apparatus was kept warm (37°C) using a waterbath heated with a hotplate/magnetic
stirrer. A stirrer bar was used to equilibrate the temperature in the waterbath which was
measured with a thermometer. Air was drawn at a steady rate by vacuum through the
flask for 1 hr through a loose inlet cap (C). The headspace VOCs were passed through
an adsorptive trap (Tenax™) (B) fitted between Teflon tubing (A) where the VOCs were
retained.
7.2.5 Gas Chromatography and Mass Spectrometry
SPME and solvent extracted samples were subjected to GC-MS (Shimadzu GCMS-
QP2010, Kyoto, Japan). The separation of emitted components was achieved using a 30
m × 0.25 mm i.d., 0.1 µm Rt-Stabilwax (Restek, Bellefonte, PA) column in splitless
injection mode with ultra-high purity helium as the carrier gas at a constant flow rate of
1 mL/min. The initial oven temperature was set to 35°C and held for 5 min then ramped
at 7°C/min to 250°C at which compounds were desorbed. The desorption time for
SPME was 5 min, while for solvent injections 1 µL was injected. The ion source was
set at 200°C, and the spectrometer was set to record between 45 and 400 amu (Flematti

Chapter 7 Volatile Organic Compounds
184
et al. 2009).
7.2.6 Data Analysis
Data collection and mass spectra generation were performed using the GC-MS Real
Time Analysis software and Postrun application (GCMSsolution, version 2.40
Shimadzu Corporation). For compound identification, mass fragments of the target
molecule were screened interactively against commercial Mass Spectral Libraries
(NIST05, NIST05s, Gaithersburg, MD).
7.3 RESULTS
7.3.1 Malaria VOCs Assay Development
7.3.1.1 Design of culture-capture apparatus
Preliminary studies employed T25 flasks coupled with a rubber stopper with two inlets
for the culture and capture of headspace atmosphere (Skinner-Adams T, data
unpublished). However, the use of plastic containers and rubber introduces organic
contaminants which may interfere with the assay. Therefore, custom-designed glass
flasks were created for the in vitro capture of headspace VOCs experiments.
The prototype 1 sampling unit for VOCs analysis consisted of a 250 mL conical flask
modified with a B-40 joint (Figure 7.3). The flask was fitted with an custom built
aluminium stopper coupled with a delrin-polymer holder that served as the SPME
holder. A small machined section at the base of the delrin holder allowed the placement
of a polytetrafluoroethylene (PTFE) septum above a small opening in the metal stopper,
which was pierced during SPME sampling. The prototype 1 container allowed the
culture of 18 mL of parasite-cell-medium suspension. The culture flask and metal
stopper were autoclave-sterilised prior to use.
To maximise parasite mass and VOCs yield, a second design was proposed featuring a
shallow container with a large base-area (Figure 7.2 and Figure 7.4). The prototype 2

Chapter 7 Volatile Organic Compounds
185
sampling units were custom-made and equipped with two Duran screw thread tube
connections (GL14, Vel, Leuven, Belgium) useful for purge and trap, and SPME
sampling. A B-24 joint with a fitted glass stopper improved access to the parasite
cultures and media changing. This was particularly important during the optimisation
phase of the study in which parasite growth was monitored daily by microscopic
examination of blood smears. The new design allowed a larger volume set-up (50 mL)
of parasite-cell-media suspension. Cultures of high parasitaemia (>20%) were achieved
under the conditions specified in Section 7.2.1, as facilitated by the maximised
air/medium contact surface available for gas-exchange in the new design.
Figure 7.3 Prototype 1 culture-capture apparatus with SPME. The prototype 1
design consisted of a conical flask modified with a B-40 joint that served as a receptacle
for an aluminium stopper. A delrin polymer SPME holder was screwed into the
aluminium stopper and between the mating surfaces, a syringe piercable PTFE septum
was placed to seal the sample before analysis.

Chapter 7 Volatile Organic Compounds
186
Figure 7.4 Design and dimensions of culture container (prototype 2) for headspace
capture. The wide-base (154 cm2) and shallow design maximises culture efficiency
and total parasite yield. The B-24 joint allows for easy access during RPMI addition
and parasite sampling. Two side inlets facilitate the ease of low-oxygen gas-
replacement and act as an access point for ‘purge and trap’ procedures and SPME of the
headspace atmosphere.
7.3.1.2 Optimisation of culture conditions
Headspace capture requires an enclosed culture system, where the atmosphere within
the culture container must be optimised for parasite growth. The gas mixture of 1% O2
and 5% CO2 in nitrogen used for routine cultures (i.e. in a 28.4 L Nalgene dessicator)
was suboptimal to sustain P. falciparum growth in the custom-designed containers. On
macroscopic examination, RBC became much darker post incubation, likely attributable
to inadequate oxygenation. A series of down-sized, volume vs duration of gas injection
experiments were conducted with an oxygen monitor to determine optimal gas balance
(data not shown). Although conditions for normal parasite replication were achieved,
optimal growth was subsequently attained with a new gas mixture of 5% O2, and 5%
CO2 in nitrogen (Mlambo et al. 2007). This gas composition allowed complete
replacement of the air within the vessel with an atmosphere that is more suited for P.
falciparum culture. This adaptation negated the need to find the intricate balance of gas-
purging duration to lower the O2 content in the air as previously performed.

Chapter 7 Volatile Organic Compounds
187
Routine in vitro culture of P. falciparum usually maintains a maximum of 5%
parasitaemia at 5% hct. Prototype 2 container was more suitable for malaria culture than
its predecessor. The initial period for VOCs capture was over 48 hr (i.e. one parasite life
cycle), however, the system must remain enclosed during this time without the required
media changes. This approach produced stressed parasites as indicated by the presence
of gametocytes by microscopy. Therefore, subsequent capture experiments were set-up
over the later 24 hr of the developmental cycle where there is maximal parasite
metabolic activity. Synchronised P. falciparum were cultured at high parasitaemia and
pooled into prototype 2 containers at the trophozoite stage (>20% parasitaemia at 1%
hct). To promote liberation of VOCs from the parasite-media matrix to the headspace,
sample containers were incubated in a shaking incubator at slow rotation (40 rpm) for
24 hr.
7.3.2 Analysis of VOCs
This part of the present study employed a number of extraction approaches to capture
and analyse VOCs from the culture samples. Headspace VOCs were extracted by
SPME (PDMS and combined Carboxen/DVB/PDMS phases) and purge and trap
coupled with thermal desorption. Direct immersion of the SPME fibre, and traditional
extractions with organic solvents with various polarities were used to extract VOCs
trapped in the culture supernatant and cell lysate matrix. Overall, mass spectra of over
100 different compounds were detected in the headspace, supernatant and cell lysates of
both non-parasitised control and P. falciparum cultures (Appendix E). Most masses
represent hydrocarbons such as alkanes and alkenes, alcohols, benzene-derivatives and
organic acids. Table 7.1 presents a selection of commonly observed compounds in the
samples. Although minor differences in compound quantities were detected on
occasions, no unique biomarker for P. falciparum was identified. Similarities between
VOCs liberated from non-parasitised control and infected samples are demonstrated in
the chromatograms from various extraction methods (Figure 7.5).

Chapter 7 Volatile Organic Compounds
188
Table 7.1 VOCs detected in culture headspace.
Compound Intensity Relative abundance (%)
1-Hexanol, 2-ethyl-* 23,423,000 66
Heptane, 2,2,4,6,6-pentamethyl- 2,665,000 7.5
Benzene, 1,3-bis(1,1-dimethylethyl)- 2,532,000 7.1
Decane 1,794,000 5.1
1-Dodecanol 3,7,11-trimethyl- 1,005,000 2.8
Cyclohexanone 893,000 2.5
1-Octen-3-ol 649,000 1.8
Octane, 3,3-dimethyl- 634,000 1.8
Decane, 3,7-dimethyl- 334,000 0.9
Decane, 5,6-dipropyl- 298,000 0.8
2,4-Dimethyl-1-heptene 290,000 0.8
2,2,4,4-Tetramethyloctane 273,000 0.8
Decane, 3,7-dimethyl- 271,000 0.8
Undecane 218,000 0.6
Nonane, 4-methyl- 213,000 0.6
*Suspected organic contaminant.

Chapter 7 Volatile Organic Compounds
189
Figure 7.5 Chromatograms of VOCs in the headspace of cultured P. falciparum.
GC-MS total ion chromatograms; black line: VOCs detected in non-parasitised control;
pink line: VOCs detected in culture-adapted P. falciparum. Data collected from (a)
solid phase micro-extraction (SPME); (b) SPME-triple fibre; (c) purge and trap thermal
desorption; (d) solvent (dichloromethane) extraction of culture supernatant.

Chapter 7 Volatile Organic Compounds
190
7.4 DISCUSSION
Despite substantial attempts following a step-wise approach, the present in vitro study
revealed no specific patterns of VOCs released by P. falciparum cultures. Various
forms of solvent extractions (hexane, dichloromethane, ethylacetate) of supernatants
and cell lysates showed minimal differences compared to control non-parasitised
cultures. When used with a pre-concentration device such as SPME, GC-MS has
sufficient sensitivity in the low ppt range (Chambers et al. 2009; Chambers et al. 2010).
Despite this sensitivity, data from SPME using conventional (PDMS) and triple fibre
(PDMS+divinylbenzene+carbowax) revealed production of a variety of VOCs in
cultured P. falciparum but no unique compounds were identified. Thermal desorption
of purge and trap samples also showed no significant differences with a similar VOCs
profile to that observed with SPME, suggesting that both techniques were detecting the
majority of released VOCs.
Previous studies of VOCs generated by other infectious agents have shown positive
associations between VOCs liberated in vitro and those detected in the breath from
patients. This in vitro vs in vivo relationship is exemplified by studies of respiratory
infections with Aspergillus fumigatus (Syhre et al. 2008; Preti et al. 2009; Chambers et
al. 2010) and pulmonary tuberculosis (Phillips et al. 2007). There were, however, a
number of important differences between these studies and the present series of
experiments. Firstly, bacterial and fungal colonies are cultured on solid medium. Any
VOCs produced by bacteria and fungi are released directly into the culture headspace.
By contrast, P. falciparum are enveloped by two extra layers, being within RBC that
settle at the bottom of liquid medium in the culture plate. The possibility of loss of
VOCs of malarial origin in cell membranes and the culture medium prompted repeated
extractions of supernatant and cell lysate using various organic solvents. No obvious
differences between malaria and control cultures were found in these experiments.
Secondly, the biomass of bacteria and fungi in vitro assessed from colony-forming units
(cfu), viable bacterial or fungal cells per visible colony is much greater than that of P.
falciparum in a typical culture. The number cfu per visible colony is substantially
higher than the number of malaria parasites in a comparable volume of RBC. For

Chapter 7 Volatile Organic Compounds
191
Mycobacterium tuberculosis culture, an inoculum of 0.5 mL of a 1.0 McFarland
standard contains 1.5 x 108 cfu) (Phillips et al. 2007) compared to a 50 mL P.
falciparum culture of 20% parasitaemia at 1% hct which contains approximately 1.1 x
107 parasitised cells. In addition, the greater biomass for bacteria housed within a
smaller culture vessel (1 mL headspace) considerably increases the concentration and
thus likelihood of VOCs detection compared with the larger headspace (500 mL) of an
intra-erythrocytic parasite culture.
The analysis of VOCs released from in vitro malaria cultures presented a substantial
challenge because of the fastidious nature of P. falciparum in culture including
microaerophilic requirements, daily medium changes and the need for a closed-system
for headspace capture. Although the use of glass culture-capture apparatus minimised
the presence of contaminants related to the use of plastics, a number of external VOCs
were present. Compounds such as diethy-phthalate and ethylhexanol (Appendix E) may
have been derived from bis-2-ethyl-hexyl-phthalate, a common additive to plastics
which renders the plastic more flexible. Siloxane derivatives were also prevalent such
as decamethyl-cyclopentasiloxane and dimethyl-silanediol, and were most likely
derived from the PDMS coating of the SPME fibre or the GC column stationary phase.
A number of different gas mixtures have been used to support malaria culture.
Atmospheres with combinations of 0.5 to 21% O2 mixed with 1 to 7% CO2 diluted in
nitrogen and microbial gas sachets have been employed (Trager et al. 1976; Mirovsky
1989; Taylor-Robinson 1998; Onda et al. 1999; Radfar et al. 2009). High oxygen
concentrations are considered to cause deleterious effects on parasites and reduce yields
(Scheibel et al. 1979), however this has been debated (Radfar et al. 2009). A mixture of
5% O2 with 5% CO2 in nitrogen supports malaria growth better than 5% CO2 with 95%
air (Cohen et al. 1969), although both mixtures have been successful (Miyagami et al.
1985; Mirovsky 1989; Flores et al. 1997; Onda et al. 1999; Radfar et al. 2009).
Therefore, although gas composition is an important consideration, it should not be
singled out as the determining factor for successful cultures, particularly for high
parasitaemias. In addition to the use of premixed gas, the transition from 5% hct for
routine P. falciparum culture to a lower hct (1%) proved an important modification to

Chapter 7 Volatile Organic Compounds
192
support high parasitaemia for the VOCs experiments. An appropriate ratio of medium
to cell pellet volume prevents parasite toxicity and helps maintain viability (Radfar et
al. 2009).
As SPME is based on attaining equilibrium in the sample or headspace, the GC-MS
chromatographs may not directly correspond to the actual composition of the detected
compounds. Quantitative information would require inclusion of a calibration curve for
each component (Gorecki et al. 1997). The aim of the study was, however, to
investigate the qualitative capacity of SPME to extract and simultaneously concentrate
VOCs released at both low and high quantities thus enabling comparisons of GC-MS
profiles of different samples under similar experimental conditions.
There were slight inter-assay differences in relative abundance and retention time of
VOCs between controls and P. falciparum samples. The difference in relative
abundance probably reflects differences in metabolic requirements of batches of RBC
and intra-erythrocytic parasites. The minor differences in retention time were probably
due to use of the instrument for unrelated interspersed experiments with resulting shifts
in calibration. However, these differences were minor, inconsistent and did not underlie
unique signals or chemical patterns.
Although VOCs may be more readily detected in vivo in respiratory diseases (Gordon et
al. 1985; Chambers et al. 2009; Preti et al. 2009; Chapman et al. 2010), VOCs
generated as part of host response to a systemic disease may also serve as biomarkers.
Examples include increased production of pentane and carbon disulfide in the breath of
patients with schizophrenia (Phillips et al. 1993). Nevertheless, their specificity remains
questionable, as breath carbon disulfide has been detected in both smokers and non-
smokers (Phillips 1992a) and has been linked with myocardial infarction (Weitz et al.
1991). An assessment of a characteristic VOCs fingerprint in the context of malaria
(including severe and non-severe cases) was beyond the scope of the present in vitro
experiments.
The present study used optimised experimental conditions to enable the capture,
extraction and analysis of VOCs liberated from P. falciparum cultures. Even at high

Chapter 7 Volatile Organic Compounds
193
parasitaemia, VOCs unique to P. falciparum cultures were not detected using solvent
extraction, purge and trap-thermal desorption, or by SPME. GC-MS data revealed a
variety of VOCs but no unique malarial finger-prints. Future in vivo studies analysing
the breath of patients with severe malaria may yet reveal specific clinically-useful
volatile biomarkers. The in vivo parasite biomass is substantially greater than
achievable in vitro and there may be VOCs generated as a specific in vivo response to
the disease.

Chapter 7 Volatile Organic Compounds
194

CHAPTER 8
CONCLUDING DISCUSSION

Chapter 8 Concluding Discussion
196
CHAPTER 8. CONCLUDING DISCUSSION
8.1 OVERVIEW
With new national treatment regimens being introduced in PNG, this thesis presents a
timely assessment of the in vitro and genetic aspects underlying treatment efficacy in
PNG children with falciparum malaria. Through the implementation of two high-
throughput resistance surveillance techniques (pLDH and LDR-FMA), valuable
baseline data were obtained prior to the adoption of artemether-lumefantrine (AL) as
first-line treatment in PNG 2011. In addition, studies in this thesis explore novel
antimalarial agents that could become useful partners with artemisinin derivatives for
the treatment of uncomplicated falciparum malaria. The detection of P. falciparum-
specific volatile organic compounds (VOCs) in breath/other samples as a way of
enhancing diagnosis and therapeutic monitoring was also explored but this technique
does not appear to be a viable alternative to conventional methodologies for parasite
detection and enumeration.
8.1.1 Major Findings and Contributions
i. Most of the PNG isolates of P. falciparum tested (n=64) were resistant to
chloroquine (CQ) but not to other ACT partner drugs (lumefantrine (LM),
piperaquine (PQ), mefloquine (MQ) and naphthoquine (NQ)).
ii. In PNG P. falciparum isolates, strong associations were observed between their
in vitro responses to 4-aminoquinolines (CQ, amodiaquine (AQ) and NQ),
bisquinoline (PQ) and aryl-aminoalcohol (MQ) compounds suggesting cross-
resistance, but LM IC50s only correlated with that of MQ.
iii. There was fixation of pfcrt K76T, pfdhfr C59R and S108N, and pfmdr1
mutations in PNG isolates of P. falciparum (n=402). Multiple mutations were
frequent with 88% of isolates possessing quintuple mutations.

Chapter 8 Concluding Discussion
197
iv. The pfmdr1 D1246Y mutation was associated with PCR-corrected day 42 in
vivo treatment failure in children allocated dihydroartemisinin (DHA) plus PQ
(P=0.004).
v. A non-isotopic semi-automated, high-throughput drug assay (using pLDH) was
implemented for the first time in PNG. The assay can be adapted to a basic
laboratory setting and facilitates serial assessment of local parasite sensitivity so
that emerging resistance can be identified.
vi. A novel extension of a multiplex, high-throughput ligase detection reaction-
fluorescence microsphere assay (LDR-FMA) was developed and implemented
for the screening of pfmdr1 mutations in PNG P. falciparum isolates.
vii. Desbutyl-lumefantrine (DBL) was found to be several folds more potent than its
parent compound against reference strains of P. falciparum and was mildly
synergistic with DHA.
viii. Mean plasma DBL concentrations were lower in children who failed AL
treatment than in those with an adequate clinical and parasitological response
(ACPR; P=0.053 vs P>0.22 for plasma LM and plasma LM:DBL ratio).
ix. Atorvastatin was more active than rosuvastatin, pravastatin and simvastatin
against culture-adapted P. falciparum, although it had weak activity (mean IC50
≥17 µM) and an indifferent interaction with CQ and DHA.
x. Fenofibric acid was the most potent lipid-modifying drug against CQ-sensitive
and resistant P. falciparum (mean IC50s 152 and 1120 nM, respectively). As
confirmed by the bioassay, the plasma fenofibric acid level available with
therapeutic dosing was 100 times that required to inhibit growth of P.
falciparum by 50%.
xi. A culture-capture apparatus for P. falciparum headspace VOCs was developed.

Chapter 8 Concluding Discussion
198
Even at relatively high parasitaemia (>20%), in vitro parasite VOCs production
was undetectable, possibly due to loss of VOCs in the components of the
experimental system including erythrocytic forms and/or insufficient parasite
biomass.
The following sections highlight the major findings of the studies in this thesis and
discuss their relevance to the understanding of parasite resistance and their potential
applications. The first section examines the in vitro and genetic aspects of resistance in
PNG isolates of P. falciparum and considers their contribution to the prediction of
treatment efficacy. Limitations of these studies are also discussed. The second section
examines three different classes of agents as novel antimalarial treatments. The
screening of headspace biomarkers of P. falciparum cultures and insights into possible
underlying mechanisms of cerebral malaria and potential drug targets are also
discussed. Lastly, this chapter outlines directions for future research.
8.2 THE ROLE OF IN VITRO RESISTANCE AND PARASITE
GENETIC MUTATIONS IN TREATMENT OUTCOME
The results presented in this thesis provided timely baseline data on in vitro (Chapter 3)
and genetic (Chapter 4) aspects on drug resistance of P. falciparum isolated from
Madang children, prior to the change of national treatment policy from CQ-sulfadoxine
pyrimethamine (SP) to AL in 2011. In vitro drug sensitivity profiles of 64 isolates to
nine conventional (CQ, AQ, dAQ, DHA) and relatively novel (NQ, PQ, MQ, LM, AZ)
antimalarials were obtained. As expected from a history of heavy 4-aminoquinoline
usage, the majority (82%) of isolates were resistant to CQ in concordance with previous
PNG in vitro studies (Hombhanje 1998b; Mita et al. 2006a). The high level of in vitro
resistance was also consistent with near-fixation of the CQ resistance-associated pfcrt
allele as revealed by subsequent genotyping of parasite DNA (Chapter 4).
Strong associations were noted between the IC50s of 4-aminoquinoline (CQ, AQ, DAQ
and NQ), bisquinoline (PQ), and aryl aminoalcohol (MQ) compounds suggesting cross-
resistance, as observed in other countries (Fan et al. 1998; Basco et al. 2003a; Pradines
et al. 2006). These findings are consistent with the observation that pfcrt and pfmdr1

Chapter 8 Concluding Discussion
199
alleles influence parasite susceptibility to drugs other than CQ such as MQ (Sidhu et al.
2002; Johnson et al. 2004; Mita et al. 2006a). LM and NQ have recently become
available in PNG as part of ACTs. In vitro sensitivities of PNG field isolates to both of
these drugs are described for the first time and could be used as a baseline for future
monitoring of resistance to these agents.
Since microscopic assessment of parasite response to drug exposure is time-consuming
and laborious, a validated high-throughput enzyme method (pLDH) was modified and
implemented in the field laboratory setting (Chapter 3). This approach allowed for
testing of multiple antimalarial drugs in triplicate with relative ease. The non-isotopic
semi-automated nature of the pLDH assay facilitated convenient serial assessment of
local parasite sensitivity so that emerging resistance could be identified with relative
confidence at an early stage. Although the pLDH assay was readily adaptable in this
setting, its relative high cost (AUD $30/plate, assaying four drugs in triplicate)
compared to microscopy limits its use for routine screening. Alternative non-isotopic
growth detection methods such as Sybr Green staining for DNA, although non-specific
to Plasmodium, are much more affordable and would be more likely to be employed for
future routine sensitivity testing in PNG (Karl et al. 2009).
Surveillance for parasite drug resistance mutations is becoming an established tool in
predicting treatment effectiveness, especially as it overcomes the challenges and costs
associated with in vivo testing. To investigate underlying molecular mechanisms, a
post-PCR, multiplexed ligase detection reaction-fluorescent microsphere assay (LDR-
FMA) was used to detect relevant single nucleotide polymorphisms (SNPs) in P.
falciparum drug resistance genes (Chapter 4). This technique enables simultaneous
identification of 18 SNPs in pfcrt, pfdhfr and pfdhps (Carnevale et al. 2007), and was
extended for the purposes of the present studies to screen for 10 allelic variants in
pfmdr1. The method is cost-effective (the analysis of 28 SNPs per sample cost AUD
$4.14), has a high output format suitable for large-scale epidemiological studies and
extends current PCR-based methods.
Molecular findings from the studies in this thesis are consistent with previous heavy use

Chapter 8 Concluding Discussion
200
of 4-aminoquinolines and SP in PNG (Chapter 4). Field isolates collected from children
with falciparum malaria showed a fixation of pfcrt 76T, pfdhfr 59R and 108N and
pfmdr1 mutations (>90%), consistent with previous PNG studies (Carnevale et al. 2007;
Mita et al. 2006a; Mita et al. 2006b; Schoepflin et al. 2008). A high prevalence of
multiple mutations across these genes was noted, with majority of the isolates (88%)
harbouring a quintuple mutation SVMNT+NRNI+KAA+Y YSND in codons 72-76 for
pfcrt, 51, 59, 108, 164 for pfdhfr, 540, 581, 613 for pfdhps, and 86, 184, 1034, 1042,
1246 for pfmdr1.
In determining underlying molecular factors that may influence in vivo outcomes, the
presence of the pfmdr1 1246Y mutation was associated with treatment failure in all
treatment groups combined (i.e. CQ-SP, artesunate-SP, PQ-DHA and AL) and in the
PQ-DHA group in particular. However, the association between pfmdr1 1246Y and
treatment failure is likely to be location specific. Parasite mutations are largely selected
by drug pressure exerted over a period of time with genetic haplotypes reflecting
parasite lineage and type of treatment employed in the region (Wongsrichanalai et al.
2002; Mita et al. 2007; Plowe 2009). Although the present findings relating to pfmdr1
1246Y may not be generalisable to other countries even within Oceania, they may
inform future surveillance strategies in epidemiologically similar areas. This situation
also pertains to PNG. A Peruvian study has shown that the P. falciparum SNPs pfdhfr
164L and pfdhps 540E predict SP treatment failure (Bacon et al. 2009). Despite the
previous widespread use of SP, these two mutations are rare in PNG in the present and
other studies (Carnevale et al. 2007; Mita et al. 2007) but the Peruvian experience
would support active surveillance for these parasite mutations if SP remains part of
nationally recommended treatment regimens. Although the pfmdr1 haplotype NFSDD
was found in only four isolates, it has been associated with AL treatment failure in
Africa (Happi et al. 2008). Therefore, monitoring changes in the pfmdr1 gene should be
of high priority due to the recent introduction of this treatment in PNG.
8.2.1 Limitations of PNG field studies
The assessment of the relationship between parasite in vitro drug sensitivity and clinical
outcome was limited by a restricted number of isolates available for testing. Overall,

Chapter 8 Concluding Discussion
201
there were 40 isolates with in vitro drug sensitivity profiles from children allocated
randomly to one of the four treatment arms in the large comparative trial (Karunajeewa
et al., 2008). Sixteen were from children treated with DHA-PQ and eight were from
subjects who received each of CQ-SP, artesunate-SP and AL. Unfortunately, the in
vitro component of the trial started when most children had been recruited while a
number of isolates that were successfully tested in vitro were from children who were
excluded post-hoc from the trial because of protocol violations.
There were only three cases of treatment failure in this in vitro-in vivo dataset. Of these,
two were late parasitological failures (PCR-confirmed recrudescences on day 28 and or
day 42) in the CQ-SP group, and one in the DHA-PQ group. The CQ IC50s of isolates
from the CQ-SP treatment failures were 197 and 126 nM, within the range obtained for
successfully-treated children in the same group and for all isolates tested (Table 3.2).
Therefore, in vitro CQ sensitivity alone is unlikely to be a useful predictor of treatment
outcome in the PNG setting, especially when in vitro CQ resistance is at fixation. For
the single DHA-PQ treatment failure, the PQ IC50 was 19.5 nM. Subsequent molecular
analysis revealed that all three parasite isolates possessed the pfcrt K76T allele and the
pfdhfr N51 R59 N108 I164 double mutation. Albeit in small numbers in this subset, the
presence of pfcrt and pfdhfr (markers for 4-aminoquinoline and pyrimethamine
resistance) in association with treatment failure is consistent with previous findings
(Cowman et al. 1988; Peterson et al. 1991; Casey et al. 2004).
Although the measurement of IC50s provides an acceptable indicator of drug sensitivity
trends in parasite populations, an isolate showing in vitro resistance may not equate to
treatment failure and vice versa (Wongsrichanalai et al. 2002; Ekland et al. 2008). This
is mainly attributed to host immunity and other in vivo factors. Pharmacokinetic profiles
are required to confirm that an adequate drug concentration has been achieved to
substantiate true resistance and treatment failure. Although these data are available
through the main clinical trial, the very low number of children with in vitro drug
sensitivity data that failed treatment limited their application.
Despite these limitations, in vitro drug susceptibility testing remains an important part

Chapter 8 Concluding Discussion
202
of the monitoring of resistance in the ACT era. The emergence of parasite resistance to
individual drugs employed in ACTs may not be clinically apparent due to the
effectiveness of the partner drug (Laufer et al. 2007). Although candidate molecular
markers for artemisinin resistance have been proposed (Price et al. 1999; Jambou et al.
2005; Uhlemann 2005), these cannot be validated unless true clinical resistance to
artemisinin drugs occurs. Therefore, in vitro drug assays are at the front-line of
surveillance for resistance to artemisinin derivatives and ACTs.
8.3 UNCONVENTIONAL AND NOVEL ANTIMALARIAL
AGENTS
There is a pressing need for new drugs and drug combinations that facilitate prompt
resolution of the symptoms of malarial infection, improve treatment success rates, and
limit the development of parasite resistance. To address this need, the studies in this
thesis have been directed towards identifying a number of novel antimalarial drugs.
These include DBL, a metabolite of an ACT partner-drug, and two classes of lipid-
modifying drugs, statins (i.e. atorvastatin, rosuvastatin, pravstatin, simvastatin) and
fibrates (i.e. fenofibrate, fenofibric acid, gemfibrozil and clofibrate). Their antimalarial
activities and possible therapeutic implementation are discussed in the following
section.
8.3.1 Desbutyl-lumefantrine and its Potential Implementation
With the recent change to AL as first-line treatment for uncomplicated malaria in PNG,
it seems relevant to investigate the antimalarial activity of related drugs (Chapter 5).
Despite speculation that DBL is not a metabolite of LM (Starzengruber et al. 2007;
Starzengruber et al. 2008) and consistent with other reports (Ntale et al. 2008; Hodel et
al. 2009), data from this study confirmed that DBL is indeed a metabolite, being present
in significant concentrations in the plasma of children after AL treatment. The superior
antimalarial activity of DBL (mean IC50 9 nM) to LM (mean IC50 55 nM) reported in
this thesis (Table 5.2) is consistent with previous reports using field isolates (Noedl et
al. 2001; Starzengruber et al. 2008). In addition to its potent activity against P.
falciparum, DBL also effectively inhibits P. vivax (Pirker-Krassnig et al. 2004). This

Chapter 8 Concluding Discussion
203
broad-spectrum activity makes DBL a useful candidate for implementation in the field,
as co-endemicity of these two major Plasmodium species is common in Asia, the
Middle East and Oceania (Mehlotra et al. 2000; Dhangadamajhi et al.; Douglas et al.
2011; Wong et al. 2011).
The difference in antimalarial activity between the parent and metabolite is also
consistent with the relationship between plasma LM, plasma DBL and treatment
outcome (Figure 5.3). AL treatment failure cases had significantly lower plasma DBL
concentrations than ACPR cases. Although plasma LM concentrations were also lower
in treatment failure, this did not reach statistical significance. This suggests that DBL
has a stronger role than the parent compound in suppressing recrudescence/and or
reinfection. The in vitro synergy between DBL and DHA (arguably the most potent
antimalarial drug discovered to date) highlights DBL-DHA as a promising novel ACT.
With antimalarial activities greater than their respective parent compounds and pending
detailed pharmacokinetic characterisation, the DBL-DHA formulation may be able to
be given in a simpler dosing regimen than the recommended 6-doses of AL. African
and PNG studies show that the greatest cure rates after AL are when dosing is directly
supervised with fat supplementation given to improve drug absorption (Mutabingwa et
al. 2005; Piola et al. 2005; Karunajeewa et al. 2008b; Schoepflin et al. 2010). Poor
compliance with the number of doses and coadministered fat may lead to sub-curative
drug concentrations and treatment failure, with the likely development of parasite drug
resistance (Wongsrichanalai et al. 2002; White et al. 2009).
The lipophilicity of DBL means that, as with LM (Ezzet et al. 1998), its bioavailability
is likely to be enhanced by concomitant food intake, particularly fat. The fat content of
PNG meals tends to be relatively low, as local diets are typically based on a
carbohydrate staple and vegetables (Iser et al. 1993). Important sources of dietary fat
include oil crops and cooking oil (e.g. peanuts, mature coconuts, palm oil). Given the
abundancy and availability of fresh tuna in coastal PNG where malaria is holoendemic,
consumption of this oily fish should be encouraged as a dietary supplement with AL
dosing to maximise absorption and decrease the chance of recrudescence. Breast-

Chapter 8 Concluding Discussion
204
feeding mothers can use breast milk as a fat source if their young children are being
treated with AL. The education of patients and healthcare providers as to the
importance of taking therapy according to the prescribed regimen should increase
understanding and compliance. These considerations would extend to potential ACTs
involving DBL.
8.3.2 Lipid-modifying Agents as Antimalarials
Drugs licensed for other indications can sometimes have antimalarial properties, an
example being lipid-modifying therapy which is becoming affordable even in malaria-
endemic developing countries. Modernisation and lifestyle changes have seen an
increasing need for these agents to reduce cardiovascular diseases, (McMurry et al.
1991; Hodge et al. 1996; Gill 2001). Two classes of commonly used lipid-modifying
drugs, namely fibrates and statins, were tested against P. falciparum (Chapter 6).
The present in vitro drug sensitivity data have confirmed atorvastatin to have the
highest activity of available statins against P. falciparum regardless of strain CQ
sensitivity (Pradines et al. 2007), but at an IC50 well above plasma concentrations after
therapeutic doses in vivo. Although its mechanism of action against P. falciparum
remains unclear, atorvastatin may act via P-glycoprotein (Holtzman et al. 2006), an
efflux protein implicated in 4-aminoquinoline resistance.
Fibrates have different lipid-modifying and possibly antimalarial mechanisms of action
to those of statins. Gemfibrozil and clofibrate proved to have weak antimalarial activity,
whilst fenofibrate in the form of fenofibric acid had a relatively low in vitro IC50,
similar to those of conventional antimalarial drugs (Table 6.3). As evident in the
bioassay (Figure 6.4), plasma fenofibric acid levels generated in vivo after therapeutic
doses of Lipidil™ inhibited cultured P. falciparum (Figure 6.4). Although therapeutic
plasma levels of fenofibric acid after both single and repeated doses are well above the
concentrations required to produce 90% growth inhibition in CQ-resistant P. falciparum
(10-fold and 3-fold, respectively), it should not be used as antimalarial monotherapy but
in partnership with a more rapidly-acting antimalarial agent. The elimination half-life of
fenofibric acid is 20 hr (Bhavesh et al. 2009) which means it is cleared more promptly

Chapter 8 Concluding Discussion
205
than most 4-aminoquinolines (Krishna et al. 1996; Tarning et al. 2005; Qu et al. 2010).
Depending on the completeness of the initial parasiticidal effect, this may be
advantageous as an ACT partner drug with a shorter elimination half-life reduces the
time of parasite exposure to subcurative levels thus limiting subsequent selection of
resistant strains (Wongsrichanalai et al. 2002).
Fenofibric acid has indifferent in vitro interactions with DHA (Table 6.4) when
assessed using two mathematical analyses (Berenbaum 1978; Brueckner et al. 1991).
Visual assessment of isobolographic plots suggests, however, that the equations utilised
by these methods may not accurately characterise drug interactions with the result that
clinically important interactions may be missed. This visual-mathematical discordance
is further discussed in Appendix D.
Although its mode of action remains to be elucidated, fenofibric acid may act by
interfering with P-glycoprotein (Ehrhardt et al. 2004) and ABC-1 mediated transport,
and/or via a putative PPARα-like protein. P-glycoprotein prevents intracellular drug
accumulation which results in a multidrug resistance phenotype in both Plasmodium
and mammalian cells. Evidence from in vitro drug-uptake assays and confocal laser
scanning microscopy have confirmed fenofibrate as an inhibitor of P-glycoprotein
activity with a similar potency to verapamil at 7.1 and 4.7 µM, respectively (Ehrhardt et
al. 2004). Therefore, fenofibrate may reverse CQ resistance and produce synergistic
effects when combined with CQ. However, the present drug-interactions studies
revealed indifferent interactions in both CQ-sensitive and resistant P. falciparum.
Despite the superior antimalarial activity exhibited by fenofibric acid, biochemical data
show that it is neither a substrate nor an inhibitor of human P-glycoprotein (Ehrhardt et
al. 2004). The lack of synergistic interaction or potentiation of CQ by fenofibric acid in
this study supports previous observations (Ehrhardt et al. 2004). Fenofibric acid affects
the expression of ABC-1 lipid transport protein in mammalian cells (Jaye et al. 2003;
Arakawa et al. 2005) and may similarly affect the Plasmodium ABC-1 homolog. This
could inhibit the development of P. falciparum by depriving the growing parasite of
lipid components of membranes and other cellular structures. The relative importance
of the effects of fenofibric acid on the Plasmodium homolog of P-glycoprotein, ABC-1

Chapter 8 Concluding Discussion
206
and a putative PPARα-like protein, together with possible interactions between these
effects, cannot be ascertained from the present data.
8.4 A PILOT STUDY OF MALARIA VOCS
The use of a non-invasive breath test for the detection of falciparum malaria would
provide a unique tool for diagnosis and therapeutic monitoring that could be
conveniently carried out at the bed-side. This thesis describes, for the first time, a study
of the potential of P. falciparum cultures to release signature VOCs. In addition to
possibly permitting detection of viable parasites, these in vitro data could provide
insights into the metabolism and pathogenicity of the organism. VOCs are used as
general anaesthetics in clinical practice (Soukup et al. 2009) and it is possible that coma
complicating malaria may result from elaboration of VOCs by malaria parasites in the
cerebral microcirculation that have anaesthetic properties.
Challenges encountered in this proof of concept study began with the determination of a
microenvironment that optimally supports a high parasite yield within a system that also
permits the capture, extraction and analysis of headspace VOCs (Chapter 7). Some
important considerations for the design of the culture-capture apparatus included i) the
width and height of the culture flask, ii) the provision of sealable-access openings and
fittings adapted for VOCs extraction, iii) the parasite stage used and duration of
incubation, iv) the culture gas mixture and purging time, and v) initial parasitaemia and
haematocrit. Once optimised, VOCs were collected and concentrated by SPME, solvent
extraction and thermal desorption. Various steps were also taken to enhance the
liberation of VOCs from the liquid matrix and to attain gas-fibre equilibrium. Despite
substantial efforts to define the optimal conditions for the in vitro capture of P.
falciparum VOCs, GC-MS data revealed the production of a variety of volatile
compounds but no unique malarial finger-prints (Chapter 7).
A major limitation is the P. falciparum biomass achievable within the in vitro system.
Unlike bacterial or fungal colonies that are directly grown on solid medium with direct
exposure to the culture headspace, malaria parasites have little access to the headspace,
being enveloped within phosphobilayered-host cells within a liquid medium. The use of

Chapter 8 Concluding Discussion
207
a shaking incubator and the assessment of supernatant and cell lysate for VOCs
revealed minimal difference compared to control. Even with the attainment of a 20%
parasitaemia, the difference in VOCs is dependent on a minority of infected cells versus
the non-infected control cells. A recent publication described means to obtain a
synchronous culture of P. falciparum at 60% parasitaemia (Radfar et al. 2009).
However, the methodological requirements for achieving such high parasite yields are
not compatible with VOCs capture. This includes the need for medium changes at 12 hr
intervals, which would result in the escape of any VOCs produced. The use of large-
base culture containers and a low haematocrit are common key considerations noted in
this and other studies (Radfar et al. 2009).
Progress with culture techniques of other protozoans may yet promote novel approaches
to P. falciparum cultivation (Hijjawi et al. 2004; Hijjawi et al. 2010). Recent studies
have reported on the successful propagation of Cryptosporidium hominis, an intra-
cellular Apicomplexan that shares similar life-cycle stages with P. falciparum, in host
cell-free culture (Hijjawi et al. 2010). Such advancements, if possible for Plasmodium,
would bring a new dimension to malaria research.
8.5 CONCLUSION AND FUTURE DIRECTIONS
Despite past efforts to control malaria, resistance of P. falciparum to antimalarial drugs
continues to be a threat to the global community, especially in tropical developing
countries such as PNG. The response has been to change from monotherapy to
combination therapy and, more recently, to the wide deployment of ACTs to ensure
efficacy and retard the spread of parasite resistance (PNGDOH 2000; WHO 2007).
However, P. falciparum drug resistance reflects a complex interplay of parasite and
host factors (Wongsrichanalai et al. 2002; Mackinnon et al. 2010). The development of
novel antimalarials has been slow, despite a surge in research funding in recent years
(McCoy et al. 2009; Kazatchkine 2010; Muller et al. 2010). There remains an urgent
need for novel, effective and affordable antimalarial agents that can be used with
artemisinin derivatives to counter the rapid development of parasite resistance to
conventional drugs.

Chapter 8 Concluding Discussion
208
This thesis has contributed to the understanding of the in vitro and genetic aspects
underlying treatment outcome in PNG children. This was achieved via the culture and
testing of parasite sensitivity to antimalarial agents, molecular detection of parasite
mutations and their associations with treatment outcomes. The antimalarial efficacy
findings presented here may become useful for inclusion in the World Antimalarial
Resistance Network (WARN), a global database showcasing collaborative efforts in the
malaria research community to monitor and counter the development of resistance
(Plowe et al. 2007; Sibley et al. 2008; Sibley et al. 2010). Both molecular and in vitro
drug susceptibility profile from this study provided useful baseline data for monitoring
and evaluation of treatment regimens in coastal PNG and supports the need for adoption
of AL as first-line treatment for uncomplicated falciparum malaria.
8.5.1 Directions for Future Research
i. The present studies provide a platform for the continuation of parasite resistance
surveillance through the routine assessment of in vitro drug sensitivity and
molecular markers. The pLDH and LDR-FMA methods established from this
work enable the high-throughput monitoring of changes in parasite drug
sensitivity (particularly in pfmdr1 polymorphisms) under a new wave of ACT
selection. Ongoing assessment using the same techniques will facilitate
assessment of the adequacy of such treatment.
ii. The high in vitro antimalarial activity of fenofibric acid at media concentrations
which can be achieved in vivo may have therapeutic application. Confirmatory
preclinical studies in an animal model (perhaps the well recognised Plasmodium
berghei murine model) would be useful as the next step in assessing the in vivo
efficacy of fenofibric acid.
iii. The present studies have highlighted a number of technical challenges with in
vitro identification of VOCs from the headspace of P. falciparum cultures.
Future in vivo studies analysing the breath of patients with severe malaria may
yet reveal specific, clinically useful volatile biomarkers.

BIBLIOGRAPHY

Bibliography
210
Adjei, G. O., Kurtzhals, J. A., Rodrigues, O. P., Alifrangis, M., Hoegberg, L. C., Kitcher, E. D., Badoe, E. V., Lamptey, R. and Goka, B. Q. (2008). "Amodiaquine-artesunate vs artemether-lumefantrine for uncomplicated malaria in Ghanaian children: a randomized efficacy and safety trial with one year follow-up." Malar J 7: 127-137.
Aikawa, M. (1966). "The fine structure of the erythrocytic stages of three avian malarial parasites, Plasmodium fallax, P. lophurae, and P. cathemerium." Am J Trop Med Hyg 15(4): 449-71.
Aikawa, M. (1972). "High-resolution autoradiography of malarial parasites treated with 3H-chloroquine." Am J Pathol 67(2): 277-284.
Aikawa, M. (1988). Fine structure of malaria parasites in the various stages of development. Malaria. W.H.Wernsdorfer and S. I. McGregor. Edinburgh, London, Melbourne and New York, Churchill Livingstone. 1: 7-129.
Aikawa, M., Carter, R., Ito, Y. and Nijhout, M. M. (1984). "New observations on gametogenesis, fertilization and zygote transformation of Plasmodium gallinaceum." J Protozool 31: 403-413.
Aikawa, M., Miller, L. H., Johnson, J. and Rabbege, J. (1978). "Erythrocyte entry by malarial parasites. A moving junction between erythrocyte and parasite." J Cell Biol 77(1): 72-82.
Aikawa, M., Suzuki, M. and Gutierrez, Y., Eds. (1980). Pathology of malaria. Malaria. New York, Academic Press.
Al-Yaman, F., Genton, B., Mokela, D., Narara, A., Raiko, A. and Alpers, M. P. (1996). "Resistance of Plasmodium falciparum malaria to amodiaquine, chloroquine and quinine in the Madang Province of Papua New Guinea, 1990-1993." P N G Med J 39(1): 16-22.
Al-Yaman, F., Genton, B., Reeder, J. C., Anders, R. F. and Alpers, M. P. (1997). "Evidence that recurrent Plasmodium falciparum infection is caused by recrudescence of resistant parasites." Am J Trop Med Hyg 56(4): 436-439.
Al-Yaman, F. M., Genton, B. and Clark, I. A. (1998). "The ratio of reactive nitrogen intermediates to tumour necrosis factor and clinical outcome of falciparum malaria disease." Trans R Soc Trop Med Hyg 92(4): 417-420.
Alifrangis, M., Enosse, S., Pearce, R., Drakeley, C., Roper, C., Khalil, I. F., Nkya, W. M., Ronn, A. M., Theander, T. G. and Bygbjerg, I. C. (2005). "A simple, high-throughput method to detect Plasmodium falciparum single nucleotide polymorphisms in the dihydrofolate reductase, dihydropteroate synthase, and P. falciparum chloroquine resistance transporter genes using polymerase chain reaction- and enzyme-linked immunosorbent assay-based technology." Am J Trop Med Hyg 72(2): 155-162.

Bibliography
211
Alker, A. P., Lim, P., Sem, R., Shah, N. K., Yi, P., Bouth, D. M., Tsuyuoka, R., Maguire, J. D., Fandeur, T., Ariey, F., Wongsrichanalai, C. and Meshnick, S. R. (2007). "Pfmdr1 and in vivo resistance to artesunate-mefloquine in falciparum malaria on the Cambodian-Thai border." Am J Trop Med Hyg 76(4): 641-647.
Allison, J. L., O'Brien, R. L. and Hahn, F. E. (1965). "DNA: reaction with chloroquine." Science 149(688): 1111-1113.
Amani, V., Vigario, A. M., Belnoue, E., Marussig, M., Fonseca, L., Mazier, D. and Renia, L. (2000). "Involvement of IFN-gamma receptor-medicated signaling in pathology and anti-malarial immunity induced by Plasmodium berghei infection." Eur J Immunol 30(6): 1646-1655.
Amino, R., Thiberge, S., Martin, B., Celli, S., Shorte, S., Frischknecht, F., Menard, R. (2006). "Quantitative imaging of Plasmodium transmission from mosquito to mammal." Nat Med 12(2): 220-224.
Anderson, S. L., Berman, J., Kuschner, R., Wesche, D., Magill, A., Wellde, B., Schneider, I., Dunne, M. and Schuster, B. G. (1995). "Prophylaxis of Plasmodium falciparum malaria with azithromycin administered to volunteers." Ann Intern Med 123(10): 771-773.
Andersson, M., Low, P. and Bakhiet, M. (1996). "Lovastatin inhibits interferon-gamma-induced Trypanosoma brucei proliferation: evidence for mevalonate pathway involvement." J Interferon Cytokine Res 16(6): 435-439.
Anstey, N. M., Handojo, T., Pain, M. C., Kenangalem, E., Tjitra, E., Price, R. N. and Maguire, G. P. (2007). "Lung injury in vivax malaria: pathophysiological evidence for pulmonary vascular sequestration and posttreatment alveolar-capillary inflammation." J Infect Dis 195(4): 589-596.
Arakawa, R., Tamehiro, N., Nishimaki-Mogami, T., Ueda, K. and Yokoyama, S. (2005). "Fenofibric acid, an active form of fenofibrate, increases apolipoprotein A-I-mediated high-density lipoprotein biogenesis by enhancing transcription of ATP-binding cassette transporter A1 gene in a liver X receptor-dependent manner." Arterioscler Thromb Vasc Biol 25(6): 1193-1197.
Arman, M., Raza, A., Tempest, L. J., Lyke, K. E., Thera, M. A., Kone, A., Plowe, C. V., Doumbo, O. K. and Rowe, J. A. (2007). "Platelet-mediated clumping of Plasmodium falciparum infected erythrocytes is associated with high parasitemia but not severe clinical manifestations of malaria in African children." Am J Trop Med Hyg 77(5): 943-946.
Arrieta, A., Arguedas, A., Fernandez, P., Block, S. L., Emperanza, P., Vargas, S. L., Erhardt, W. A., de Caprariis, P. J. and Rothermel, C. D. (2003). "High-dose azithromycin versus high-dose amoxicillin-clavulanate for treatment of children with recurrent or persistent acute otitis media." Antimicrob Agents Chemother 47(10): 3179-3186.

Bibliography
212
Attlmayr, B., Kollaritsch, H., Wernsdorfer, W. H., Miller, R. S., Sirichaisinthop, J. and Noedl, H. (2005). "Drug sensitivity of Plasmodium falciparum along the Thai-Myanmar border using the new field-deployable HRP2 in vitro assay." Wien Klin Wochenschr 117(S4): 35-38.
Attlmayr, B., Thriemer, K., Haque, R., Wagatsuma, Y., Abdus Salam, M., Akhter, S., Fukuda, M., Schaecher, K., Miller, R. S. and Noedl, H. (2006). "In vitro antimalarial drug resistance in Southeastern Bangladesh." Wien Klin Wochenschr 118(19-20 Suppl 3): 58-61.
Auerbach, T., Bashan, A., Harms, J., Schluenzen, F., Zarivach, R., Bartels, H., Agmon, I., Kessler, M., Pioletti, M., Franceschi, F. and Yonath, A. (2002). "Antibiotics targeting ribosomes: crystallographic studies." Curr Drug Targets Infect Disord 2(2): 169-186.
Babiker, H. A., Pringle, S. J., Abdel-Muhsin, A., Mackinnon, M., Hunt, P. and Walliker, D. (2001). "High-level chloroquine resistance in Sudanese isolates of Plasmodium falciparum is associated with mutations in the chloroquine resistance transporter gene pfcrt and the multidrug resistance Gene pfmdr1." J Infect Dis 183(10): 1535-1538.
Bablok, W., Passing, H., Bender, R. and Schneider, B. (1988). "A general regression procedure for method transformation. Application of linear regression procedures for method comparison studies in clinical chemistry, Part III." J Clin Chem Clin Biochem 26(11): 783-790.
Bace, A., Zrnic, T., Begovac, J., Kuzmanovic, N. and Culig, J. (1999). "Short-term treatment of pertussis with azithromycin in infants and young children." Eur J Clin Microbiol Infect Dis 18(4): 296-298.
Bacon, D. J., Tang, D., Salas, C., Roncal, N., Lucas, C., Gerena, L., Tapia, L., Llanos-Cuentas, A. A., Garcia, C., Solari, L., Kyle, D. and Magill, A. J. (2009). "Effects of point mutations in Plasmodium falciparum dihydrofolate reductase and dihydropterate synthase genes on clinical outcomes and in vitro susceptibility to sulfadoxine and pyrimethamine." PLoS One 4(8): e6762.
Balachandar, S. and Katyal, A. (2011). "Peroxisome proliferator activating receptor (PPAR) in cerebral malaria (CM): a novel target for an additional therapy." Eur J Clin Microbiol Infect Dis 30(4): 483-498.
Ballereau, F. (1997). "Stability of essential drugs in the field: results of a study conducted over a two-year period in Burkina Faso." Am J Trop Med Hyg 57: 31-36.
Barat, L., Chipipa, J., Kolczak, M. and Sukwa, T. (1999). "Does the availability of blood slide microscopy for malaria at health centers improve the management of persons with fever in Zambia?" Am J Trop Med Hyg 60(6): 1024-1030.

Bibliography
213
Basco, L. K. (2003b). "Molecular epidemiology of malaria in Cameroon. XVI. Longitudinal surveillance of in vitro pyrimethamine resistance in Plasmodium falciparum." Am J Trop Med Hyg 69(2): 174-178.
Basco, L. K., Bickii, J. and Ringwald, P. (1998). "In vitro activity of lumefantrine (benflumetol) against clinical isolates of Plasmodium falciparum in Yaounde, Cameroon." Antimicrob Agents Chemother 42(9): 2347-2351.
Basco, L. K., Marquet, F., Makler, M. M. and Le Bras, J. (1995). "Plasmodium falciparum and Plasmodium vivax: lactate dehydrogenase activity and its application for in vitro drug susceptibility assay." Exp Parasitol 80(2): 260-271.
Basco, L. K., Ndounga, M., Ngane, V. F. and Soula, G. (2002). "Molecular epidemiology of malaria in Cameroon. XIV. Plasmodium falciparum chloroquine resistance transporter (PFCRT) gene sequences of isolates before and after chloroquine treatment." Am J Trop Med Hyg 67(4): 392-395.
Basco, L. K. and Ringwald, P. (2003a). "In vitro activities of piperaquine and other 4-aminoquinolines against clinical isolates of Plasmodium falciparum in Cameroon." Antimicrob Agents Chemother 47(4): 1391-1394.
Bellosta, S., Paoletti,R. and Corsini, A. (2004). "Safety of statins: focus on clinical pharmacokinetics and drug interactions." Circulation 109(23 Supplement 1): 50-57.
Bennett, T. N., Patel, J., Ferdig, M. T. and Roepe, P. D. (2007). "Plasmodium falciparum Na+/H+ exchanger activity and quinine resistance." Mol Biochem Parasitol 153(1): 48-58.
Berenbaum, M. C. (1978). "A method for testing for synergy with any number of agents." J. Infect Dis 137: 122-130.
Berry, A., Benoit-Vical, F., Fabre, R., Cassaing, S. and Magnaval, J. F. (2008). "PCR-based methods to the diagnosis of imported malaria." Parasite 15(3): 484-488.
Bhavesh, D. and Shah, S. (2009). "Determination of fenofibric acid in human plasma by ultra performance liquid chromatography-electrospray ionization mass spectrometry: application to a bioequivalence study." Biomed Chromatogr 23(9): 922-928.
Bhisutthibhan, J. and Meshnick, S. R. (2001). "Immunoprecipitation of [3H]dihydroartemisinin translationally controlled tumor protein (TCTP) adducts from Plasmodium falciparum-infected erythrocytes by using anti-TCTP antibodies." Antimicrob Agents Chemother 45: 2397-2399.
Bienvenu, A. L. and Picot, S. (2008). "Statins alone are ineffective in cerebral malaria but potentiate artesunate." Antimicrob Agents Chemother 52(11): 4203-4204.

Bibliography
214
Bland, J. M. and Altman, D. G. (1986). "Statistical methods for assessing agreement between two methods of clinical measurement." Lancet 1(8476): 307-310.
Blauer, G. and Akkawi, M. (1997). "Investigations of B- and beta-hematin." J Inorg Biochem 66(2): 145-152.
Bloland, P. B. (2001). "Drug resistance in malaria." Retrieved 3rd May 2009, from http://www.who.int/drugresistance/publications/WHO_CDS_CSR_DRS_2001_4/en/index.html.
Boggild, A. K., Krudsood, S., Patel, S. N., Serghides, L., Tangpukdee, N., Katz, K., Wilairatana, P., Liles, W. C., Looareesuwan, S. and Kain, K. C. (2009). "Use of peroxisome proliferator-activated receptor gamma agonists as adjunctive treatment for Plasmodium falciparum malaria: a randomized, double-blind, placebo-controlled trial." Clin Infect Dis 49(6): 841-849.
Boonpucknavig, V. and Boonpucknavig, S., Eds. (1988). The histopathology of malaria. Malaria, principles and practice of malariology. London, Churchill Livingstone.
Bordet, R., Ouk, T., Petrault, O., Gele, P., Gautier, S., Laprais, M., Deplanque, D., Duriez, P., Staels, B., Fruchart, J. C. and Bastide, M. (2006). "PPAR: a new pharmacological target for neuroprotection in stroke and neurodegenerative diseases." Biochem Soc Trans 34(6): 1341-1346.
Borek-Dohalsky, V., Huclova, J., Barrett, B., Nemec, B., Ulc, I. and Jelinek, I. (2006). "Validated HPLC-MS-MS method for simultaneous determination of atorvastatin and 2-hydroxyatorvastatin in human plasma-pharmacokinetic study." Anal Bioanal Chem 386(2): 275-285.
Borrmann, S., Sallas, W. M., Machevo, S., Gonzalez, R., Bjorkman, A., Martensson, A., Hamel, M., Juma, E., Peshu, J., Ogutu, B., Djimde, A., D'Alessandro, U., Marrast, A. C., Lefevre, G. and Kern, S. E. (2010). "The effect of food consumption on lumefantrine bioavailability in African children receiving artemether-lumefantrine crushed or dispersible tablets (Coartem) for acute uncomplicated Plasmodium falciparum malaria." Trop Med Int Health 15(4): 434-441.
Brabin, B. J. (2007). "Congenital malaria-a recurrent problem." Ann Trop Paediatr 27(2): 95-98.
Brasseur, P. (2007). "Tolerance of amodiaquine." Med Trop (Mars) 67(3): 288-290
Brasseur, P., Guigemde, R., Diallo, S., Guiyedi, V., Kombila, M., Ringwald, P. and Olliaro, P. (1999). "Amodiaquine remains effective for treating uncomplicated malaria in west and central Africa." Trans R Soc Trop Med Hyg 93(6): 645-650.
Bray, P. G., Mungthin, M., Ridley, R. G. and Ward, S. A. (1998). "Access to hematin: the basis of chloroquine resistance." Mol Pharmacol 54(1): 170-179.

Bibliography
215
Brockman, A., Singlam, S., Phiaphun, L., Looareesuwan, S., White, N. J. and Nosten, F. (2004). "Field evaluation of a novel colorimetric method-double-site enzyme-linked lactate dehydrogenase immunodetection assay-to determine drug susceptibilities of Plasmodium falciparum clinical isolates from northwestern Thailand." Antimicrob Agents Chemother 48(4): 1426-1429.
Bruce-Chwatt, L. J., Ed. (1986). Chemotherapy of malaria. Geneva, World Health Organization.
Brueckner, R. P., Milhous, W. K. and Canfield, C. J. (1991). "Quantitative isobolic analysis of antimalarial drug interactions. In 'Program and Abstract of the 40th Annual Meeting of the American Society of Tropical Medicine and Hygiene.'" Am J Trop Med Hyg 45(Supplement): 190.
Budd, A., Alleva, L., Alsharifi, M., Koskinen, A., Smythe, V., Mullbacher, A., Wood, J. and Clark, I. (2007). "Increased survival after gemfibrozil treatment of severe mouse influenza." Antimicrob Agents Chemother 51(8): 2965-2968.
Bukirwa, H., Garner, P. and Critchley, J. (2004). "Chlorproguanil-dapsone for treating uncomplicated malaria." Cochrane Database Syst Rev(4): CD004387.
Butcher, G. A., Garland, T., Ajdukiewicz, A. B. and Clark, I. A. (1990). "Serum tumor necrosis factor associated with malaria in patients in the Solomon Islands." Trans R Soc Trop Med Hyg 84(5): 658-61.
Butcher, G. A. and Sinden, R. E. (2003). "Persistence of atovaquone in human sera following treatment: inhibition of Plasmodium falciparum development in vivo and in vitro." Am J Trop Med Hyg 68(1): 111-114.
Calkin, A. C., Cooper, M. E., Jandeleit-Dahm, K. A. and Allen, T. J. (2006). "Gemfibrozil decreases atherosclerosis in experimental diabetes in association with a reduction in oxidative stress and inflammation." Diabetologia 49(4): 766-774.
Camus, D. and Hadley, T. J. (1985). "A Plasmodium falciparum antigen that binds to host erythrocytes and merozoites." Science 230(4725): 553-556.
Canfield, C. J., Pudney, M. and Gutteridge, W. E. (1995). "Interactions of atovaquone with other antimalarial drugs against Plasmodium falciparum in vitro." Exp Parasitol 80(3): 373-381.
Carnevale, E. P., Kouri, D., DaRe, J. T., McNamara, D. T., Mueller, I. and Zimmerman, P. A. (2007). "A multiplex ligase detection reaction-fluorescent microsphere assay for simultaneous detection of single nucleotide polymorphisms associated with Plasmodium falciparum drug resistance." J Clin Microbiol 45(3): 752-761.
Carrara, V. I., Zwang, J., Ashley, E. A., Price, R. N., Stepniewska, K., Barends, M., Brockman, A., Anderson, T., McGready, R., Phaiphun, L., Proux, S., van Vugt, M., Hutagalung, R., Lwin, K. M., Phyo, A. P., Preechapornkul, P., Imwong, M.,

Bibliography
216
Pukrittayakamee, S., Singhasivanon, P., White, N. J. and Nosten, F. (2009). "Changes in the treatment responses to artesunate-mefloquine on the northwestern border of Thailand during 13 years of continuous deployment." PLoS ONE 4(2): e4551.
Carter, R. and Miller, L. H. (1979). "Evidence for environmental modulation of gametocytogenesis in Plasmodium falciparum in continuous culture." Bull World Health Organ 57 Suppl 1: 37-52.
Carvalho, B. O., Lopes, S. C., Nogueira, P. A., Orlandi, P. P., Bargieri, D. Y., Blanco, Y. C., Mamoni, R., Leite, J. A., Rodrigues, M. M., Soares, I. S., Oliveira, T. R., Wunderlich, G., Lacerda, M. V., del Portillo, H. A., Araujo, M. O., Russell, B., Suwanarusk, R., Snounou, G., Renia, L. and Costa, F. T. (2010). "On the cytoadhesion of Plasmodium vivax-infected erythrocytes." J Infect Dis 202(4): 638-647.
Casey, G. J., Ginny, M., Uranoli, M., Mueller, I., Reeder, J. C., Genton, B. and Cowman, A. F. (2004). "Molecular analysis of Plasmodium falciparum from drug treatment failure patients in Papua New Guinea." Am J Trop Med Hyg 70(3): 251-255.
Catron, D. M., Lange, Y., Borensztajn, J., Sylvester, M. D., Jones, B. D. and Haldar, K. (2004). "Salmonella enterica serovar typhimurium requires nonsterol precursors of the cholesterol biosynthetic pathway for intracellular proliferation." Infect Immun 72(2): 1036-1042.
Cattani, J. A., Moir, J. S., Gibson, F. D., Ginny, M., Paino, J., Davidson, W. and Alpers, M. P. (1986). "Small-area variations in the epidemiology of malaria in Madang Province." P N G Med J 29(1): 11-7.
CDC. (2004). "The history of malaria, an ancient disease." Retrieved 23 February 2009, from http://www.cdc.gov/MALARIA/history/index.htm.
CDC. (2006). "Centers for Disease Control and Prevention." Retrieved 23 February 2009, from www.cdc.gov/malaria/biology/lifecycle.htm.
Chaijaroenkul, W., Wisedpanichkij, R. and Na-Bangchang, K. (2010). "Monitoring of in vitro susceptibilities and molecular markers of resistance of Plasmodium falciparum isolates from Thai-Myanmar border to chloroquine, quinine, mefloquine and artesunate." Acta Trop 113(2): 190-194.
Chambers, S. T., Bhandari, S., Scott-Thomas, A. and Syhre, M. (2010). "Novel diagnostics: progress toward a breath test for invasive Aspergillus fumigatus." Med Mycol 49 Suppl 1: S54-61.
Chambers, S. T., Syhre, M., Murdoch, D. R., McCartin, F. and Epton, M. J. (2009). "Detection of 2-pentylfuran in the breath of patients with Aspergillus fumigatus." Med Mycol 47(5): 468-476.

Bibliography
217
Chapman, E. A., Thomas, P. S. and Yates, D. H. (2010). "Breath analysis in asbestos-related disorders: a review of the literature and potential future applications." J Breath Res 4(3): 034001.
Charles, D. M., Hart, J., Davis, W. A., Sullivan, E., Dowse, G. K. and Davis, T. M. (2005). "Notifications of imported malaria in Western Australia, 1990-2001: incidence, associated factors and chemoprophylaxis." Med J Aust 182(4): 164-167.
Chavalitshewinkoon, P., de Vries, E., Stam, J. G., Franssen, F. F., van der Vliet, P. C. and Overdulve, J. P. (1993). "Purification and characterization of DNA polymerases from Plasmodium falciparum." Mol Biochem Parasitol 61(2): 243-253.
Chavchich, M., Gerena, L., Peters, J., Chen, N., Cheng, Q. and Kyle, D. E. (2010). "Induction of resistance to artemisinin derivatives in Plasmodium falciparum: Role of Pfmdr1 amplification and expression." Antimicrob Agents Chemother 54(6): 2455-2464.
Chawira, A. N. and Warhurst, D. C. (1987). "The effect of artemisinin combined with standard antimalarials against chloroquine-sensitive and chloroquine-resistant strains of Plasmodium falciparum in vitro." J Trop Med Hyg 90(1): 1-8.
Checkley, A. M. and Whitty, C. J. (2007). "Artesunate, artemether or quinine in severe Plasmodium falciparum malaria?" Expert Rev Anti Infect Ther 5(2): 199-204.
Chen, G. Z., Foster, L. and Bennett, J. L. (1990). "Antischistosomal action of mevinolin: evidence that 3-hydroxy-methylglutaryl-coenzyme a reductase activity in Schistosoma mansoni is vital for parasite survival." Naunyn Schmiedebergs Arch Pharmacol 342(4): 477-482.
Chen, L. (1991). "Recent studies on antimalarial efficacy of piperaquine and hydroxypiperaquine." Chin Med J (Engl) 104(2): 161-163.
Chico, R. M. and Chandramohan, D. (2010). "Quinine for the treatment of malaria in pregnancy." Lancet Infect Dis 10(3): 140-141.
Chotivanich, K., Sritabal, J., Udomsangpetch, R., Newton, P., Stepniewska, K. A., Ruangveerayuth, R., Looareesuwan, S., Roberts, D. J., White, N. J. (2004). "Platelet-induced autoagglutination of Plasmodium falciparum-infected red blood cells and disease severity in Thailand." J infect Dis 189(6): 1052-1055.
Chou, A. C. and Fitch, C. D. (1993). "Control of heme polymerase by chloroquine and other quinoline derivatives." Biochem Biophys Res Commun 195(1): 422-427.
Chulay, J. D., Haynes, J. D. and Diggs, C. L. (1983). "Plasmodium falciparum: assessment of in vitro growth by [3H]hypoxanthine incorporation." Exp Parasitol 55(1): 138-146.

Bibliography
218
Cilla, D. D., Jr., Whitfield, L. R., Gibson, D. M., Sedman, A. J. and Posvar, E. L. (1996). "Multiple-dose pharmacokinetics, pharmacodynamics, and safety of atorvastatin, an inhibitor of HMG-CoA reductase, in healthy subjects." Clin Pharmacol Ther 60(6): 687-695.
Clark, C. J., Mackay, G. M., Smythe, G. A., Bustamante, S., Stone, T. W. and Phillips, R. S. (2005). "Prolonged survival of a murine model of cerebral malaria by kynurenine pathway inhibition." Infect Immun 73(8): 5249-5251.
Clark, I. A. and Alleva, L. M. (2009). "Is human malarial coma caused, or merely deepened, by sequestration?" Trends Parasitol 25(7): 314-318.
Clark, I. A., Alleva, L. M., Budd, A. C. and Cowden, W. B. (2008). "Understanding the role of inflammatory cytokines in malaria and related diseases." Travel Med Infect Dis 6(1-2): 67-81.
Clark, I. A., Alleva, L. M., Mills, A. C. and Cowden, W. B. (2004). "Pathogenesis of malaria and clinically similar conditions." Clin Microbiol Rev 17(3): 509-539.
Clark, I. A., Cowden, W. B., Butcher, G. A. and Hunt, N. H. (1987). "Possible roles of tumor necrosis factor in the pathology of malaria." Am J Pathol 129(1): 192-199.
Clark, I. A. and Schofield, L. (2000). "Pathogenesis of malaria." Parasitol Today 16(10): 451-454.
Cohen, J., Nussenzweig, V., Nussenzweig, R., Vekemans, J. and Leach, A. (2009). "From the circumsporozoite protein to the RTS,S/AS candidate vaccine." Hum Vaccin 6(1): 90-96.
Cohen, S., Butcher, G. A. and Crandall, R. B. (1969). "Action of malarial antibody in vitro." Nature 223(5204): 368-371.
Colbourne, M. and Stevenson, K. A. P. (1970). The WHO anti-malarial programme in Papua New Guinea. Manila, World Health Organisation.
Coleman, R. E., Sattabongkot, J., Promstaporm, S., Maneechai, N., Tippayachai, B., Kengluecha, A., Rachapaew, N., Zollner, G., Miller, R. S., Vaughan, J. A., Thimasarn, K. and Khuntirat, B. (2006). "Comparison of PCR and microscopy for the detection of asymptomatic malaria in a Plasmodium falciparum/vivax endemic area in Thailand." Malar J 5: 121-128.
Coltel, N., Combes, V., Hunt, N. H. and Grau, G. E. (2004). "Cerebral malaria - a neurovascular pathology with many riddles still to be solved." Curr Neurovasc Res 1(2): 91-110.
Cook, J., Risley, P., Riley, E. and Corran, P. (2006). "Getting the bigger picture " Laboratory News Online, from www.labnews.co.uk/.../getting-the-bigger-picture.

Bibliography
219
Cortes, A., Mellombo, M., Benet, A., Lorry, K., Rare, L. and Reeder, J. (2004). "Plasmodium falciparum: distribution of msp2 genotypes among symptomatic and asymptomatic individuals from the Wosera region of Papua New Guinea." Exp Parasitol 106: 22-29.
Cowman, A. F., Galatis, D. and Thompson, K. J. (1994). "Selection for mefloquine resistance in Plasmodium falciparum is linked to amplification of the pfmdr1 gene and cross-resistance to halofantrine and quinine." Proc Natl Acad Sci U S A 91: 1143-1147.
Cowman, A. F., Morry, M. J., Biggs, B. A., Cross, G. A. and Foote, S. J. (1988). "Amino acid changes linked to pyrimethamine resistance in the dihydrofolate reducatase-thymidylate synthase gene of Plasmodium falciparum." Proc Natl Acad Sci U S A 85(23): 9109-9113.
Cox-Singh, J., Davis, T. M., Lee, K. S., Shamsul, S. S., Matusop, A., Ratnam, S., Rahman, H. A., Conway, D. J. and Singh, B. (2008). "Plasmodium knowlesi malaria in humans is widely distributed and potentially life threatening." Clin Infect Dis 46(2): 165-171.
Cox, F. E. (2002). "History of human parasitology." Clin Microbiol Rev 15(4): 595-612.
Crameri, A., Marfurt, J., Mugittu, K., Maire, N., Regos, A., Coppee, J. Y., Sismeiro, O., Burki, R., Huber, E., Laubscher, D., Puijalon, O., Genton, B., Felger, I. and Beck, H. P. (2007). "Rapid microarray-based method for monitoring of all currently known single-nucleotide polymorphisms associated with parasite resistance to antimalaria drugs." J Clin Microbiol 45(11): 3685-3691.
Cremer, G., Basco, L. K., Le Bras, J., Camus, D. and Slomianny, C. (1995). "Plasmodium falciparum: detection of P-glycoprotein in chloroquine-susceptible and chloroquine-resistant clones and isolates." Exp Parasitol 81(1): 1-8.
Czarniecki, C. W., Fennie, C. W., Powers, D. B. and Estell, D. A. (1984). "Synergistic antiviral and antiproliferative activities of Escherichia coli-derived human alpha, beta, and gamma interferons." J Virol 49(2): 490-496.
DaRe, J. T., Kouri, D. P., Zimmerman, P. A. and Thomas, P. J. (2010). "Differentiating Plasmodium falciparum alleles by transforming Cartesian X,Y data to polar coordinates." BMC Genet 11: 57-69.
DaRe, J. T., Mehlotra, R. K., Michon, P., Mueller, I., Reeder, J., Sharma, Y. D., Stoneking, M. and Zimmerman, P. A. (2007). "Microsatellite polymorphism within pfcrt provides evidence of continuing evolution of chloroquine-resistant alleles in Papua New Guinea." Malar J 6: 34-44.
Darlow, B. and Vrbova, H. (1981). "Chloroquine-resistant Plasmodium falciparum malaria in Madang children." P N G Med J 24(2): 96-98.

Bibliography
220
Darlow, B., Vrbova, H., Gibney, S., Jolley, D., Stace, J. and Alpers, M. (1982). "Sulfadoxine-pyrimethamine for the treatment of acute malaria in children in Papua New Guinea. I. Plasmodium falciparum." Am J Trop Med Hyg 31(1): 1-9.
Darlow, B., Vrbova, H., Stace, J., Heywood, P. and Alpers, M. P. (1980). "Fansidar-resistant falciparum malaria in Papua New Guinea." Lancet 2: 1243.
Davis, T. M., Hamzah, J., Ilett, K. F., Karunajeewa, H. A., Reeder, J. C., Batty, K. T., Hackett, S. and Barrett, P. H. (2006). "In vitro interactions between piperaquine, dihydroartemisinin, and other conventional and novel antimalarial drugs." Antimicrob Agents Chemother 50(8): 2883-2885.
Davis, T. M., Hung, T. Y., Sim, I. K., Karunajeewa, H. A. and Ilett, K. F. (2005). "Piperaquine: a resurgent antimalarial drug." Drugs 65(1): 75-87.
Delhaes, L. (1999). "The microculture tetrazolium assay MTA: another colorimetric method of testing Plasmodium falciparum chemosensitivity." Ann Trop Med Parasitol 93(1): 31-40.
Deloron, P., Le Bras, J., Ramanamirija, J. A. and Coulanges, P. (1985). "Plasmodium falciparum in Madagascar: in vivo and in vitro sensitivity to seven drugs." Ann Trop Med Parasitol 79(4): 357-365.
Denis, M. B., Tsuyuoka, R., Poravuth, Y., Narann, T. S., Seila, S., Lim, C., Incardona, S., Lim, P., Sem, R., Socheat, D., Christophel, E. M. and Ringwald, P. (2006). "Surveillance of the efficacy of artesunate and mefloquine combination for the treatment of uncomplicated falciparum malaria in Cambodia." Trop Med Int Health 11(9): 1360-1366.
Derosa, G., Maffioli, P., Salvadeo, S. A., Ferrari, I., Gravina, A., Mereu, R., Palumbo, I., D'Angelo, A. and Cicero, A. F. (2009). "Fenofibrate, simvastatin and their combination in the management of dyslipidaemia in type 2 diabetic patients." Curr Med Res Opin 25(8): 1973-1983.
Desakorn, V., Silamut, K., Angus, B., Sahassananda, D., Chotivanich, K., Suntharasamai, P., Simpson, J. and White, N. J. (1997). "Semi-quantitative measurement of Plasmodium falciparum antigen PfHRP2 in blood and plasma." Trans R Soc Trop Med Hyg 91(4): 479-483.
Desjardin, R. E., Canfield, C.J.Haynes, J.D.Chulay, J.D. (1979). "Quantitative assessment of antimalarial activity in vitro by semiautomated microdilution technique." Antimicrob Agents Chemother 16: 710-718.
Dhangadamajhi, G., Rout, B. K., Kar, S. K. and Ranjit, M. R. (2010). "Genetic diversity of Plasmodium vivax in a hyperendemic area predominated by Plasmodium falciparum: A preliminary study." Trop Biomed 27(3): 578-584.

Bibliography
221
Djimde, A., Doumbo, O. K., Cortese, J. F., Kayentao, K., Doumbo, S., Diourte, Y., Dicko, A., Su, X., Nomura, T., Fidock, D. A., Wellems, T. E. and Plowe, C. V. (2001). "A molecular marker for chloroquine-resistant falciparum malaria." N Engl J Med 344(4): 257-263.
Dokomajilar, C., Nsobya, S. L., Greenhouse, B., Rosenthal, P. J. and Dorsey, G. (2006). "Selection of Plasmodium falciparum pfmdr1 alleles following therapy with artemether-lumefantrine in an area of Uganda where malaria is highly endemic." Antimicrob Agents Chemother 50(5): 1893-1895.
Dondorp, A. M. and Day, N. P. (2007). "The treatment of severe malaria." Trans R Soc Trop Med Hyg 101(7): 633-634.
Dondorp, A. M., Fanello, C. I., Hendriksen, I. C., Gomes, E., Seni, A., Chhaganlal, K. D., Bojang, K., Olaosebikan, R., Anunobi, N., Maitland, K., Kivaya, E., Agbenyega, T., Nguah, S. B., Evans, J., Gesase, S., Kahabuka, C., Mtove, G., Nadjm, B., Deen, J., Mwanga-Amumpaire, J., Nansumba, M., Karema, C., Umulisa, N., Uwimana, A., Mokuolu, O. A., Adedoyin, O. T., Johnson, W. B., Tshefu, A. K., Onyamboko, M. A., Sakulthae, T., Ngum, W. P., Silamut, K., Stepniewska, K., Woodrow, C. J., Bethell, D., Wills, B., Oneko, M., Peto, T. E., von Seidlein, L., Day, N. P., White, N. J. (2010). "Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial." Lancet 376(9753): 1647-1657.
Dondorp, A. M., Nosten, F., Stepniewska, K., Day, N., White, N. (2005). "Artesunate versus quinine for treatment of severe falciparum malaria: a randomised trial." Lancet 366(9487): 717-725.
Dondorp, A. M., Nosten, F., Yi, P., Das, D., Phyo, A. P., Tarning, J., Lwin, K. M., Ariey, F., Hanpithakpong, W., Lee, S. J., Ringwald, P., Silamut, K., Imwong, M., Chotivanich, K., Lim, P., Herdman, T., An, S. S., Yeung, S., Singhasivanon, P., Day, N. P., Lindegardh, N., Socheat, D. and White, N. J. (2009). "Artemisinin resistance in Plasmodium falciparum malaria." N Engl J Med 361(5): 455-467.
Dondorp, A. M., Yeung, S., White, L., Nguon, C., Day, N. P., Socheat, D. and von Seidlein, L. (2010). "Artemisinin resistance: current status and scenarios for containment." Nat Rev Microbiol 8(4): 272-280.
Dorn, A., Stoffel, R., Matile, H., Bubendorf, A. and Ridley, R. G. (1995). "Malarial haemozoin/beta-haematin supports haem polymerization in the absence of protein." Nature 374(6519): 269-271.
Dorn, A., Vippagunta, S. R., Matile, H., Jaquet, C., Vennerstrom, J. L. and Ridley, R. G. (1998). "An assessment of drug-haematin as a mechanism for inhibition of haematin polymerisation by quinoline antimalarials." Biochem Pharmacol 55: 727-736.

Bibliography
222
Douglas, N. M., Nosten, F., Ashley, E. A., Phaiphun, L., van Vugt, M., Singhasivanon, P., White, N. J. and Price, R. N. (2011). "Plasmodium vivax recurrence following falciparum and mixed species malaria: risk factors and effect of antimalarial kinetics." Clin Infect Dis 52(5): 612-620.
Druilhe, P., Moreno, A., Blanc, C., Brasseur, P. H. and Jacquier, P. (2001). "A colorimetric in vitro drug sensitivity assay for Plasmodium falciparum based on a highly sensitive double-site lactate dehydrogenase antigen-capture enzyme-linked immunosorbent assay." Am J Trop Med Hyg 64(5-6): 233-241.
Dubey, S., Tomar, M., Patni, A. K., Khuroo, A., Reyar, S. and Monif, T. (2010). "Rapid, sensitive and validated ultra-performance liquid chromatography/mass spectrometric method for the determination of fenofibric acid and its application to human pharmacokinetic study." E-Journal of Chemistry 7(1): 25-36.
Duffy, P. E. (2007). "Plasmodium in the placenta: parasites, parity, protection, prevention and possibly preeclampsia." Parasitology 134(13): 1877-1881.
Dulay, I. S., Gibson, F. D., Eyeson-Annan, M. B. and Narara, A. (1987). "Chloroquine resistance in Plasmodium falciparum and its geographical distribution in Papua New Guinea." P N G Med J 30(4): 281-290.
Duraisingh, M. T. and Cowman, A. F. (2005). "Contribution of the pfmdr1 gene to antimalarial drug-resistance." Acta Trop 94(3): 181-190.
Duraisingh, M. T., Curtis, J. and Warhurst, D. C. (1998). "Plasmodium falciparum: detection of polymorphisms in the dihydrofolate reductase and dihydropteroate synthetase genes by PCR and restriction digestion." Exp Parasitol 89(1): 1-8.
Duraisingh, M. T., Drakeley, C. J., Muller, O., Bailey, R., Snounou, G., Targett, G. A., Greenwood, B. M. and Warhurst, D. C. (1997). "Evidence for selection for the tyrosine-86 allele of the pfmdr1 gene of Plasmodium falciparum by chloroquine and amodiaquine." Parasitology 114(3): 205-11.
Eckstein-Ludwig, U. (2003). "Artemisinins target the SERCA of Plasmodium falciparum." Nature 424: 957-961.
Edstein, M. D., Kotecka, B. M., Anderson, K. L., Pombo, D. J., Kyle, D. E., Rieckmann, K. H. and Good, M. F. (2005). "Lengthy antimalarial activity of atovaquone in human plasma following atovaquone-proguanil administration." Antimicrob Agents Chemother 49(10): 4421-4422.
Ehrhardt, M., Lindenmaier, H., Burhenne, J., Haefeli, W. E. and Weiss, J. (2004). "Influence of lipid lowering fibrates on P-glycoprotein activity in vitro." Biochem Pharmacol 67(2): 285-292.
Ejaz, A., Haqnawaz, K., Hussain, Z., Butt, R., Awan, Z. I. and Bux, H. (2007). "Treatment of uncomplicated Plasmodium falciparum malaria with quinine-doxycycline combination therapy." J Pak Med Assoc 57(10): 502-505.

Bibliography
223
Ekland, E. H. and Fidock, D. A. (2007). "Advances in understanding the genetic basis of antimalarial drug resistance." Curr Opin Microbiol 10(4): 363-370.
Ekland, E. H. and Fidock, D. A. (2008). "In vitro evaluations of antimalarial drugs and their relevance to clinical outcomes." Int J Parasitol 38(7): 743-747.
Ezzet, F., Mull, R. and Karbwang, J. (1998). "Population pharmacokinetics and therapeutic response of CGP 56697 (artemether+benflumetol) in malaria patients." Br J Clin Pharmacol 46(6): 553-561.
Ezzet, F., van Vugt, M., Nosten, F., Looareesuwan, S. and White, N. J. (2000). "Pharmacokinetics and pharmacodynamics of lumefantrine (benflumetol) in acute falciparum malaria." Antimicrob Agents Chemother 44(3): 697-704.
Fan, B., Zhao, W., Ma, X. W., Huang, Z. M., Wen, Y. S., Yang, J. Q. and Yang, Z. X. (1998). "In vitro sensitivity of Plasmodium falciparum to chloroquine, piperaquine, pyronaridine and artesunate in Yuxi prefecture of Yunnan province." Chinese J Parasitol Parasit Dis 16: 460-462.
Fanello, C. I., Karema, C., Avellino, P., Bancone, G., Uwimana, A., Lee, S. J., d'Alessandro, U. and Modiano, D. (2008). "High risk of severe anaemia after chlorproguanil-dapsone+artesunate antimalarial treatment in patients with G6PD (A-) deficiency." PLoS One 3(12): e4031.
Farcas, G. A., Soeller, R., Zhong, K., Zahirieh, A. and Kain, K. C. (2006). "Real-time polymerase chain reaction assay for the rapid detection and characterization of chloroquine-resistant Plasmodium falciparum malaria in returned travelers." Clin Infect Dis 42(5): 622-627.
Felger, I., Tavul, L., Kabintik, S., Marshall, V., Genton, B., Alpers, M. P. and Beck, H. P. (1994). "Plasmodium falciparum: extensive polymorphism in merozoite surface antigen 2 alleles in an area with endemic malaria in Papua New Guinea." Exp Parasitol 79: 106-116.
Fichera, M. E., Bhopale, M. K. and Roos, D. S. (1995). "In vitro assays elucidate peculiar kinetics of clindamycin action against Toxoplasma gondii." Antimicrob Agents Chemother 39(7): 1530-1537.
Fichera, M. E. and Roos, D. S. (1997). "A plastid organelle as a drug target in apicomplexan parasites." Nature 390(6658): 407-409.
Fidock, D. A., Eastman, R. T., Ward, S. A. and Meshnick, S. R. (2008). "Recent highlights in antimalarial drug resistance and chemotherapy research." Trends Parasitol 24(12): 537-44.
Fidock, D. A., Nomura, T., Talley, R. A., Cooper, S. M., Dzekunov, M. T., Ferdig, L. M., Ursos, L. M., Sidhu, A. B., Naude, B., Deitsch, K. W., Su, X. Z., Wootton, P. D., Roepe, P. D. and Wellems, T. E. (2000). "Mutations in the P. falciparum

Bibliography
224
digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance." Mol Cell 6: 861-871.
Fidock, D. A., Rosenthal, P. J., Croft, S. L., Brun, R. and Nwaka, S. (2004). "Antimalarial drug discovery: efficacy models for compound screening." Nat Rev Drug Discov 3(6): 509-520.
Fitch, C. D. (1973). "Chloroquine-resistant Plasmodium falciparum: difference in the handling of 14C-amodiaquin and 14C-chloroquine." Antimicrob Agents Chemother 3(5): 545-8.
Fitch, C. D. (1998). "Involvement of heme in the antimalarial action of chloroquine." Trans Am Clin Climatol Assoc 109: 97-105.
Fivelman, Q. L., Adagu, I. S. and Warhurst, D. C. (2004). "Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum." Antimicrob Agents Chemother 48(11): 4097-4102.
Flematti, G. R., Ghisalberti, E. L., Dixon, K. W. and Trengove, R. D. (2009). "Identification of alkyl substituted 2H-furo[2,3-c]pyran-2-ones as germination stimulants present in smoke." J Agric Food Chem 57(20): 9475-9480.
Flores, M. V., Berger-Eiszele, S. M. and Stewart, T. S. (1997). "Long-term cultivation of Plasmodium falciparum in media with commercial non-serum supplements." Parasitol Res 83(7): 734-736.
Foley, M. and Tilley, L. (1998). "Quinoline antimalarials: mechanisms of action and resistance and prospects for new agents." Pharmacol Ther 79(1): 55-87.
Fontaine, R. E., Najjar, A. E. and Prince, J. S. (1961). "The 1958 malaria epidemic in Ethiopia." Am J Trop Med Hyg 10: 795-803.
Foote, S. J., Kyle, D. E., Martin, R. K., Oduola, A. M., Forsyth, K., Kemp, D. J. and Cowman, A. F. (1990). "Several alleles of the multidrug-resistance gene are closely linked to chloroquine resistance in Plasmodium falciparum." Nature 345(6272): 255-258.
Gao, X. (1981). "Morphological observations on the ookinete formation in rodent Plasmodium (Plasmodium berghei yoelii) in vitro." Acta Zool 27: 153-157.
Garcia, I., Miyazaki, Y., Araki, K., Araki, M., Lucas, R., Grau, G. E., Milon, G., Belkaid, Y., Montixi, C., Lesslauer, W. and et al. (1995). "Transgenic mice expressing high levels of soluble TNF-R1 fusion protein are protected from lethal septic shock and cerebral malaria, and are highly sensitive to Listeria monocytogenes and Leishmania major infections." Eur J Immunol 25(8): 2401-2407.

Bibliography
225
Gardner, M. J., Hall, N., Fung, E., White, O., Berriman, M., Hyman, R. W., Carlton, J. M., Pain, A., Nelson, K. E., Bowman, S., Paulsen, I. T., James, K., Eisen, J. A., Rutherford, K., Salzberg, S. L., Craig, A., Kyes, S., Chan, M. S., Nene, V., Shallom, S. J., Suh, B., Peterson, J., Angiuoli, S., Pertea, M., Allen, J., Selengut, J., Haft, D., Mather, M. W., Vaidya, A. B., Martin, D. M., Fairlamb, A. H., Fraunholz, M. J., Roos, D. S., Ralph, S. A., McFadden, G. I., Cummings, L. M., Subramanian, G. M., Mungall, C., Venter, J. C., Carucci, D. J., Hoffman, S. L., Newbold, C., Davis, R. W., Fraser, C. M. and Barrell, B. (2002). "Genome sequence of the human malaria parasite Plasmodium falciparum." Nature 419(6906): 498-511.
Garnham, P. C. C., Ed. (1988). Malaria parasites of man: life-cycles and morphology (excluding ultrastructure). Malaria, principles and practice of malariology London, Churchill Livingstone
Garnham, P. C. C., Bird, R. G. and Baker, J. R. (1962). "Electron microscope studies of motile stages of malaria parasites. III. The ookinetes of Haemamoeba and Plasmodium." Transactions of the Royal Society of Tropical Medicine and Hygiene 56: 116-120.
Genton, B., Baea, K., Lorry, K., Ginny, M., Wines, B. and Alpers, M. P. (2005). "Parasitological and clinical efficacy of standard treatment regimens against Plasmodium falciparum, P. vivax and P. malariae in Papua New Guinea." P N G Med J 48(3-4): 141-150.
Gill, T. P. (2001). "Cardiovascular risk in the Asia-Pacific region from a nutrition and metabolic point of view: abdominal obesity." Asia Pac J Clin Nutr 10(2): 85-89.
Gilles, H. M., Ed. (1988). The differential diagnosis of malaria. Malaria, principles and practice of malariology. London, Churchill Livingstone.
Gimenez, F., Barraud de Lagerie, S., Fernandez, C., Pino, P. and Mazier, D. (2003). "Tumor necrosis factor alpha in the pathogenesis of cerebral malaria." Cell Mol Life Sci 60(8): 1623-1635.
Gingras, B. A. and Jensen, J. B. (1992). "Activity of azithromycin (CP-62,993) and erythromycin against chloroquine-sensitive and chloroquine-resistant strains of Plasmodium falciparum in vitro." Am J Trop Med Hyg 47(3): 378-382.
Gingras, B. A. and Jensen, J. B. (1993). "Antimalarial activity of azithromycin and erythromycin against Plasmodium berghei." Am J Trop Med Hyg 49(1): 101-105.
Ginsburg, H., Nissani, E. and Krugliak, M. (1989). "Alkalinization of the food vacuole of malaria parasites by quinoline drugs and alkylamines is not correlated with their antimalarial activity." Biochem Pharmacol 38(16): 2645-2654.
Girard, A. E., Girard, D., English, A. R., Gootz, T. D., Cimochowski, C. R., Faiella, J. A., Haskell, S. L. and Retsema, J. A. (1987). "Pharmacokinetic and in vivo

Bibliography
226
studies with azithromycin (CP-62,993), a new macrolide with an extended half-life and excellent tissue distribution." Antimicrob Agents Chemother 31(12): 1948-1954.
Gomez, M. S., Piper, R. C., Hunsaker, L. A., Royer, R. E., Deck, L. M., Makler, M. T. and Vander Jagt, D. L. (1997). "Substrate and cofactor specificity and selective inhibition of lactate dehydrogenase from the malarial parasite P. falciparum." Mol Biochem Parasitol 90(1): 235-246.
Goodman, C. D. and McFadden, G. I. (2007a). "Fatty acid biosynthesis as a drug target in apicomplexan parasites." Curr Drug Targets 8(1): 15-30.
Goodman, C. D., Su, V. and McFadden, G. I. (2007b). "The effects of anti-bacterials on the malaria parasite Plasmodium falciparum." Mol Biochem Parasitol 152(2): 181-91.
Gordon, S. M., Szidon, J. P., Krotoszynski, B. K., Gibbons, R. D. and O'Neill, H. J. (1985). "Volatile organic compounds in exhaled air from patients with lung cancer." Clin Chem 31(8): 1278-1282.
Gorecki, T. and Pawliszyn, J. (1997). "Effect of sample volume on quantitative analysis by solid-phase microextraction. Part 1. Theoretical considerations." Analyst 122(10): 1079-1086.
Goyal, A., Goel, S. and Gowda, D. C. (2009). "Plasmodium falciparum: Assessment of parasite-infected red blood cell binding to placental chondroitin proteoglycan and bovine tracheal chondroitin sulfate A." Exp Parasitol 123(2): 105-110.
Gratz, N. G. (2005). Vector- and rodent-borne disease in Europe and North America: their distribution, public health burden and control, Cambridge University Press.
Grau, G. E., Fajardo, L. F., Piguet, P. F., Allet, B., Lambert, P. H. and Vassalli, P. (1987). "Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria." Science 237(4819): 1210-1212.
Grau, G. E. and Lou, J. (1993a). "TNF in vascular pathology: the importance of platelet-endothelium interactions." Res Immunol 144(5): 355-363.
Grau, G. E., Tacchini-Cottier, F., Vesin, C., Milon, G., Lou, J. N., Piguet, P. F. and Juillard, P. (1993b). "TNF-induced microvascular pathology: active role for platelets and importance of the LFA-1/ICAM-1 interaction." Eur Cytokine Netw 4(6): 415-419.
Greenwood, B. M., Fidock, D. A., Kyle, D. E., Kappe, S. H., Alonso, P. L., Collins, F. H. and Duffy, P. E. (2008). "Malaria: progress, perils, and prospects for eradication." J Clin Invest 118(4): 1266-1276.
Greenwood, D. (1995). "Conflicts of interest: the genesis of synthetic antimalarial agents in peace and war." J Antimicrob Chemother 36(5): 857-872.

Bibliography
227
Grellier, P., Valentin, A., Millerioux, V., Schrevel, J. and Rigomier, D. (1994). "3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors lovastatin and simvastatin inhibit in vitro development of Plasmodium falciparum and Babesia divergens in human erythrocytes." Antimicrob Agents Chemother 38(5): 1144-1148.
Grimmond, T. R., Donovan, K. O. and Riley, I. D. (1976). "Chloroquine resistant malaria in Papua New Guinea." P N G Med J 19(3): 184-185.
Grundy, S. M., Cleeman, J. I., Merz, C. N., Brewer, H. B., Jr., Clark, L. T., Hunninghake, D. B., Pasternak, R. C., Smith, S. C., Jr. and Stone, N. J. (2004). "Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines." Circulation 110(2): 227-239.
Guo, Q., Wang, G., Liu, X. and Namura, S. (2009). "Effects of gemfibrozil on outcome after permanent middle cerebral artery occlusion in mice." Brain Res 1279: 121-130.
Guo, W., Zheng, Q. and Li, G. Q. (2000). "A randomized-controlled study of naphthoquine and artesunate in the treatment of falciparum malaria." J Guangzhou Univ Trad Chin Med 17(3): 235-237.
Gupta, S., Thapar, M. M., Wernsdorfer, W. H. and Bjorkman, A. (2002). "In vitro interactions of artemisinin with atovaquone, quinine, and mefloquine against Plasmodium falciparum." Antimicrob Agents Chemother 46(5): 1510-1515.
Haldar, K., Murphy, S. C., Milner, D. A. and Taylor, T. E. (2007). "Malaria: mechanisms of erythrocytic infection and pathological correlates of severe disease." Annu Rev Pathol 2: 217-249.
Hall, M. J., Middleton, R. F. and Westmacott, D. (1983). "The fractional inhibitory concentration (FIC) index as a measure of synergy." J Antimicrob Chemother 11(5): 427-433.
Han, C. M. (1978). "Studies on the occurrence of a strain of chloroquine-resistant Plasmodium falciparum in Papua New Guinea." P N G Med J 21(4): 306-316.
Han, C. M. and Grimmond, T. R. (1976). "Chloroquine resistance trials in Papua New Guinea. 1. Maprik and Popondetta areas." P N G Med J 19(4): 236-242.
Hanscheid, T. and Grobusch, M. P. (2002). "How useful is PCR in the diagnosis of malaria?" Trends Parasitol 18(9): 395-398.
Hansen, D. S., Siomos, M. A., Buckingham, L., Scalzo, A. A. and Schofield, L. (2003). "Regulation of murine cerebral malaria pathogenesis by CD1d-restricted NKT cells and the natural killer complex." Immunity 18(3): 391-402.
Happi, C. T., Gbotosho, G. O., Folarin, O. A., Sowunmi, A., Hudson, T., O'Neil, M., Milhous, W., Wirth, D. F. and Oduola, A. M. (2008). "Selection of Plasmodium

Bibliography
228
falciparum Multidrug Resistance Gene 1 Alleles in Asexual stages and Gametocytes by Artemether-Lumefantrine in Nigerian Children with Uncomplicated Falciparum Malaria." Antimicrob Agents Chemother.
Harrison, G. (1978). Mosquitoes, malaria and man: A history of the hostilities since 1980. J. Murray. London.
Hassan Alin, M., Bjorkman, A. and Wernsdorfer, W. H. (1999). "Synergism of benflumetol and artemether in Plasmodium falciparum." Am J Trop Med Hyg 61(3): 439-445.
Hatz, C., Soto, J., Nothdurft, H. D., Zoller, T., Weitzel, T., Loutan, L., Bricaire, F., Gay, F., Burchard, G. D., Andriano, K., Lefevre, G., De Palacios, P. I. and Genton, B. (2008). "Treatment of acute uncomplicated falciparum malaria with artemether-lumefantrine in nonimmune populations: a safety, efficacy, and pharmacokinetic study." Am J Trop Med Hyg 78(2): 241-247.
Hawking, F., Wilson, M. E. and Gammage, K. (1971). "Evidence for cyclic development and short-lived maturity in the gametocytes of Plasmodium falciparum." Trans R Soc Trop Med Hyg 65(5): 549-559.
Hawley, S. R., Bray, P. G., Park, B. K. and Ward, S. A. (1996). "Amodiaquine accumulation in Plasmodium falciparum as a possible explanation for its superior antimalarial activity over chloroquine." Mol Biochem Parasitol 80(1): 15-25.
Hijjawi, N., Estcourt, A., Yang, R., Monis, P. and Ryan, U. (2010). "Complete development and multiplication of Cryptosporidium hominis in cell-free culture." Vet Parasitol 169(1-2): 29-36.
Hijjawi, N. S., Meloni, B. P., Ng'anzo, M., Ryan, U. M., Olson, M. E., Cox, P. T., Monis, P. T. and Thompson, R. C. (2004). "Complete development of Cryptosporidium parvum in host cell-free culture." Int J Parasitol 34(7): 769-777.
Ho, M., Schollaardt, T., Snape, S., Looareesuwan, S., Suntharasamai, P. and White, N. J. (1998). "Endogenous interleukin-10 modulates proinflammatory response in Plasmodium falciparum malaria." J Infect Dis 178(2): 520-525.
Hodel, E. M., Marfurt, J., Muller, D., Rippert, A., Borrmann, S., Muller, I., Reeder, J. C., Siba, P., Genton, B. and Beck, H. P. (2008). "Lack of multiple copies of pfmdr1 gene in Papua New Guinea." Trans R Soc Trop Med Hyg 102(11): 1151-1153.
Hodel, E. M., Zanolari, B., Mercier, T., Biollaz, J., Keiser, J., Olliaro, P., Genton, B. and Decosterd, L. A. (2009). "A single LC-tandem mass spectrometry method for the simultaneous determination of 14 antimalarial drugs and their metabolites in human plasma." J Chromatogr B Analyt Technol Biomed Life Sci 877(10): 867-886.

Bibliography
229
Hodge, A. M., Dowse, G. K., Erasmus, R. T., Spark, R. A., Nathaniel, K., Zimmet, P. Z. and Alpers, M. P. (1996). "Serum lipids and modernization in coastal and highland Papua New Guinea." Am J Epidemiol 144(12): 1129-1142.
Holtzman, C. W., Wiggins, B. S. and Spinler, S. A. (2006). "Role of P-glycoprotein in statin drug interactions." Pharmacotherapy 26(11): 1601-1607.
Hombhanje, F. W. (1998b). "In vitro susceptibility of Plasmodium falciparum to four antimalarial drugs in the Central Province of Papua New Guinea." P N G Med J 41(2): 51-58.
Hombhanje, F. W. and Kereu, R. K. (1998a). "In vitro susceptibility of Plasmodium falciparum isolates to halofantrine in the Central Province of Papua New Guinea." P N G Med J 41(1): 30-36.
Howard, R. J., Uni, S., Aikawa, M., Aley, S. B., Leech, J. H., Lew, A. M., Wellems, T. E., Rener, J. and Taylor, D. W. (1986). "Secretion of a malarial histidine-rich protein (Pf HRPII) from Plasmodium falciparum- infected erythrocytes." J Cell Biol 103: 1269-1277.
Huber, M., Cabib, E. and Miller, L. H. (1991). "Malaria parasite chitinase and penetration of the mosquito peritrophic membrane." Proc Natl Acad Sci U S A 88(7): 2807-2810.
Hugosson, E., Montgomery, S. M., Premji, Z., Troye-Blomberg, M. and Bjorkman, A. (2004). "Higher IL-10 levels are associated with less effective clearance of Plasmodium falciparum parasites." Parasite Immunol 26(3): 111-117.
Humad, S., Zarling, E., Clapper, M. and Skosey, J. L. (1988). "Breath pentane excretion as a marker of disease activity in rheumatoid arthritis." Free Radic Res Commun 5(2): 101-106.
Humphreys, G. S., Merinopoulos, I., Ahmed, J., Whitty, C. J., Mutabingwa, T. K., Sutherland, C. J. and Hallett, R. L. (2007). "Amodiaquine and artemether-lumefantrine select distinct alleles of the Plasmodium falciparum mdr1 gene in Tanzanian children treated for uncomplicated malaria." Antimicrob Agents Chemother 51(3): 991-997.
Hunt, N. H. and Grau, G. E. (2003). "Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria." Trends Immunol 24(9): 491-499.
Idro, R., Marsh, K., John, C. C. and Newton, C. R. (2010). "Cerebral malaria: mechanisms of brain injury and strategies for improved neurocognitive outcome." Pediatr Res 68(4): 267-274.
Iser, D. J. and Avera, K. (1993). "Has westernization influenced serum cholesterol levels in Bougainvillian males?" P N G Med J 36(4): 311-315.

Bibliography
230
Jambou, R., Legrand, E., Niang, M., Khim, N., Lim, P., Volney, B., Ekala, M. T., Bouchier, C., Esterre, P., Fandeur, T. and Mercereau-Puijalon, O. (2005). "Resistance of Plasmodium falciparum field isolates to in-vitro artemether and point mutations of the SERCA-type PfATPase6." Lancet 366(9501): 1960-1963.
Jaye, M., Duverger, N., Searfoss, G. and Minnich, A. (2003). Therapeutic uses of PPAR mediators. United States Patent Application Publication. United States. US 20030220373A1.
Jensen, J. B. (1979). "Observations on gametogenesis in Plasmodium falciparum from continuous culture." J Protozool 26(1): 129-132.
John, C. C., McHugh, M. M., Moormann, A. M., Sumba, P. O. and Ofulla, A. V. (2005). "Low prevalence of Plasmodium falciparum infection among asymptomatic individuals in a highland area of Kenya." Trans R Soc Trop Med Hyg 99(10): 780-786.
Johnson, D. J., Fidock, D. A., Mungthin, M., Lakshmanan, V., Sidhu, A. B., Bray, P. G. and Ward, S. A. (2004). "Evidence for a central role for PfCRT in conferring Plasmodium falciparum resistance to diverse antimalarial agents." Mol Cell 15(6): 867-877.
Jomaa, H., Wiesner, J., Sanderbrand, S., Altincicek, B., Weidemeyer, C., Hintz, M., Turbachova, I., Eberl, M., Zeidler, J., Lichtenthaler, H. K., Soldati, D. and Beck, E. (1999). "Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs." Science 285(5433): 1573-1576.
Jones, P. H., Davidson, M. H., Stein, E. A., Bays, H. E., McKenney, J. M., Miller, E., Cain, V. A. and Blasetto, J. W. (2003). "Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial)." Am J Cardiol 92(2): 152-60.
Jonkman, A. (1995). "Cost-saving through microscopy-based versus presumptive diagnosis of malaria in adult outpatients in Malawi." Bull World Health Organ 73: 223-227.
Kabanywanyi, A. M., Mwita, A., Sumari, D., Mandike, R., Mugittu, K. and Abdulla, S. (2007). "Efficacy and safety of artemisinin-based antimalarial in the treatment of uncomplicated malaria in children in southern Tanzania." Malar J 6: 146-150.
Kaddouri, H., Djimde, A., Dama, S., Kodio, A., Tekete, M., Hubert, V., Kone, A., Maiga, H., Yattara, O., Fofana, B., Sidibe, B., Sangare, C. P., Doumbo, O. and Le Bras, J. (2008). "Baseline in vitro efficacy of ACT component drugs on Plasmodium falciparum clinical isolates from Mali." Int J Parasitol 38(7): 791-798.
Kaddouri, H., Nakache, S., Houze, S., Mentre, F. and Le Bras, J. (2006). "Assessment of the drug susceptibility of Plasmodium falciparum clinical isolates from Africa by using a Plasmodium lactate dehydrogenase immunodetection assay

Bibliography
231
and an inhibitory maximum effect model for precise measurement of the 50-percent inhibitory concentration." Antimicrob Agents Chemother 50(10): 3343-3349.
Kakkilaya, B. S. (2008). "History of Malaria: Famous Victims." from http://www.malariasite.com/malaria/history_victims.htm.
Kannan, R., Kumar, K., Sahal, D., Kukreti, S. and Chauhan, V. S. (2005). "Reaction of artemisinin with haemoglobin: implications for antimalarial activity." Biochem J 385(2): 409-418.
Karl, S., Wong, R. P., St Pierre, T. G. and Davis, T. M. (2009). "A comparative study of a flow-cytometry-based assessment of in vitro Plasmodium falciparum drug sensitivity." Malar J 8: 294-304.
Karunajeewa, H. A., Ilett, K. F., Mueller, I., Siba, P., Law, I., Page-Sharp, M., Lin, E., Lammey, J., Batty, K. T. and Davis, T. M. (2008a). "Pharmacokinetics and efficacy of piperaquine and chloroquine in melanesian children with uncomplicated malaria." Antimicrob Agents Chemother 52(1): 237-243.
Karunajeewa, H. A., Mueller, I., Senn, M., Lin, E., Law, I., Gomorrai, P. S., Oa, O., Griffin, S., Kotab, K., Suano, P., Tarongka, N., Ura, A., Lautu, D., Page-Sharp, M., Wong, R., Salman, S., Siba, P., Ilett, K. F. and Davis, T. M. (2008b). "A trial of combination antimalarial therapies in children from Papua New Guinea." N Engl J Med 359(24): 2545-2557.
Kazatchkine, M. D. (2010). "Increased resources for the Global Fund, but pledges fall short of expected demand." Lancet 376(9751): 1439-1440.
Keating, G. M. and Ormrod, D. (2002). "Micronised fenofibrate: an updated review of its clinical efficacy in the management of dyslipidaemia." Drugs 62(13): 1909-1944.
Keen, J., Farcas, G. A., Zhong, K., Yohanna, S., Dunne, M. W. and Kain, K. C. (2007). "Real-time PCR assay for rapid detection and analysis of PfCRT haplotypes of chloroquine-resistant Plasmodium falciparum isolates from India." J Clin Microbiol 45(9): 2889-2893.
Kende, M. (2001). "Superiority of traditional village diet and lifestyle in minimizing cardiovascular disease risk in Papua New Guineans." P N G Med J 44(3-4): 135-150.
Kester, K. E., Cummings, J. F., Ofori-Anyinam, O., Ockenhouse, C. F., Krzych, U., Moris, P., Schwenk, R., Nielsen, R. A., Debebe, Z., Pinelis, E., Juompan, L., Williams, J., Dowler, M., Stewart, V. A., Wirtz, R. A., Dubois, M. C., Lievens, M., Cohen, J., Ballou, W. R. and Heppner, D. G., Jr. (2009). "Randomized, double-blind, phase 2a trial of falciparum malaria vaccines RTS,S/AS01B and RTS,S/AS02A in malaria-naive adults: safety, efficacy, and immunologic associates of protection." J Infect Dis 200(3): 337-346.

Bibliography
232
Kilejian, A. (1976). "Does a histidine-rich protein from Plasmodium lophurae have a function in merozoite penetration?" J Protozool 23(2): 272-277.
Kirchgassler, K. U., Schmitz, H. and Bach, G. (1998). "Effectiveness and tolerability of 12-week treatment with micronised fenofibrate 200 mg in a drugmonitoring
programme involving 9884 patients with dyslipidaemia." Clinical Drug Investigation 15: 197–204.
Knobloch, J. and Henk, M. (1995). "Screening for malaria by determination of parasite-specific lactate dehydrogenase." Trans Soc Trop Med Hyg 89: 269-270.
Kobbe, R., Schreiber, N., May, J. and Jacobs, T. (2008). "Simvastatin treatment shows no effect on the incidence of cerebral malaria or parasitemia during experimental malaria." Antimicrob Agents Chemother 52(4): 1583-1584.
Kotecka, B. M., Rieckmann, K. H., Davis, T. M., Batty, K. T. and Ilett, K. F. (2003). "Comparison of bioassay and high performance liquid chromatographic assay of artesunate and dihydroartemisinin in plasma." Acta Trop 87(3): 371-375.
Kouznetsov, R. L., Rooney, W., Wernsdorfer, W. H., El Gaddal, A. A., Payne, D. and Abdalla, R. E. (1980). "Use of the in vitro microtechnique for the assessment of drug sensitivity of Plasmodium falciparum in Sennar, Sudan." Bull World Health Organ 58(5): 785-789.
Krishna, S. and al., e. (2006). "Re-evaluation of how artemisinins work in light of emerging evidence of in vitro resistance." Trends Mol. Med. 12: 200–205.
Krishna, S. and White, N. J. (1996). "Pharmacokinetics of quinine, chloroquine and amodiaquine. Clinical implications." Clin Pharmacokinet 30(4): 263-299.
Krungkrai, S. R. and Yuthavong, Y. (1987). "The antimalarial action on Plasmodium falciparum of qinghaosu and artesunate in combination with agents which modulate oxidant stress." Trans R Soc Trop Med Hyg 81(5): 710-714.
Kusi, K. A., Gyan, B. A., Goka, B. Q., Dodoo, D., Obeng-Adjei, G., Troye-Blomberg, M., Akanmori, B. D. and Adjimani, J. P. (2008). "Levels of soluble CD163 and severity of malaria in children in Ghana." Clin Vaccine Immunol 15(9): 1456-1460.
Kwiatkowski, D. (1990a). "Tumour necrosis factor, fever and fatality in falciparum malaria." Immunol Lett 25(1-3): 213-216.
Kwiatkowski, D., Hill, A. V., Sambou, I., Twumasi, P., Castracane, J., Manogue, K. R., Cerami, A., Brewster, D. R. and Greenwood, B. M. (1990b). "TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria." Lancet 336(8725): 1201-1204.

Bibliography
233
Kyavar, L., Rojanawatsirivet, C., Kollaritsch, H., Wernsdorfer, G., Sirichaisinthop, J. and Wernsdorfer, W. H. (2006). "In vitro interaction between artemisinin and chloroquine as well as desbutyl-benflumetol in Plasmodium vivax." Wien Klin Wochenschr 118(19-20 Suppl 3): 62-69.
Lambros, C., Vanderberg, J.P. (1979). "Synchronization of Plasmodium falciparum erythrocytic stages in culture." J Parasitol. 65: 418-420.
Laufer, M. K., Djimde, A. A. and Plowe, C. V. (2007). "Monitoring and deterring drug-resistant malaria in the era of combination therapy." Am J Trop Med Hyg 77(6 Suppl): 160-169.
Laufer, M. K., Takala-Harrison, S., Dzinjalamala, F. K., Stine, O. C., Taylor, T. E. and Plowe, C. V. (2010). "Return of chloroquine-susceptible falciparum malaria in Malawi was a reexpansion of diverse susceptible parasites." J Infect Dis 202(5): 801-808.
Laufer, M. K., Thesing, P. C., Eddington, N. D., Masonga, R., Dzinjalamala, F. K., Takala, S. L., Taylor, T. E. and Plowe, C. V. (2006). "Return of chloroquine antimalarial efficacy in Malawi." N Engl J Med 355(19): 1959-1966.
Le Bras, J. and Durand, R. (2003). "The mechanisms of resistance to antimalarial drugs in Plasmodium falciparum." Fundam Clin Pharmacol 17(2): 147-153.
Le Bras, J. and Ringwald, P. (1990). "Plasmodium falciparum chemoresistance. The situation in Africa in 1989." Med Trop 50(1): 11-16.
Lechner, M., Fille, M., Hausdorfer, J., Dierich, M. P. and Rieder, J. (2005a). "Diagnosis of bacteria in vitro by mass spectrometric fingerprinting:a pilot study." Curr Microbiol 51(4): 267-269.
Lechner, M., Karlseder, A., Niederseer, D., Lirk, P., Neher, A., Rieder, J. and Tilg, H. (2005b). "H. pylori infection increases levels of exhaled nitrate." Helicobacter 10(5): 385-390.
Lee, M. R. (2002). "Plants against malaria. Part 1: Cinchona or the Peruvian bark." J R Coll Physicians Edinb 32(3): 189-196.
Lesley, J., Manning, L. A. and Ogle, G. D. (2001). "A survey of diabetes services in hospitals in Papua New Guinea." P N G Med J 44(3-4): 88-95.
Li, W. (2005). "Yeast model uncovers dual roles of mitochondria in action of artemisinin." PLoS Genet 1: e36.
Lim, P., Chy, S., Ariey, F., Incardona, S., Chim, P., Sem, R., Denis, M. B., Hewitt, S., Hoyer, S., Socheat, D., Merecreau-Puijalon, O. and Fandeur, T. (2003). "pfcrt polymorphism and chloroquine resistance in Plasmodium falciparum strains isolated in Cambodia." Antimicrob Agents Chemother 47(1): 87-94.

Bibliography
234
Lin, S. G., Liu, D. Q., Zhuo, K. R. and al., E. (2005). "Determination of sensitivity of Plasmodium falciparum to antimalarials in Ledong County, Hainan Province." China Tropical Medicine 5(8): 1707-1708.
Lindeberg, S., Berntorp, E., Nilsson-Ehle, P., Terent, A. and Vessby, B. (1997). "Age relations of cardiovascular risk factors in a traditional Melanesian society: the Kitava Study." Am J Clin Nutr 66(4): 845-852.
Lindstrom, A. B. and Pleil, J. D. (2002). "A review of the USEPA's single breath canister (SBC) method for exhaled volatile organic biomarkers." Biomarkers 7(3): 189-208.
Loeb, R. F., Clarke, W. M., Coateney, G. R., Coggeshall, L. T., Dieuaide, F. R., Dochez, A. R., Hakansson, E. G., Marshall, E. K., Marvel, S. C., McCoy, O. R., Sapero, J. J., Serbell, W. H., Shannon, J. A. and Carden, G. A. (1946). "Activity of a new antimalarial agent, chloroquine (SN 7618)." J Am Med Assoc 130: 1069-1070.
Luchavez, J., Espino, F., Curameng, P., Espina, R., Bell, D., Chiodini, P., Nolder, D., Sutherland, C., Lee, K. S. and Singh, B. (2008). "Human infections with Plasmodium knowlesi, the Philippines." Emerg Infect Dis 14(5): 811-813.
Luse, S. A. and Miller, L. H. (1971). "Plasmodium falciparum malaria. Ultrastructure of parasitized erythrocytes in cardiac vessels." Am J Trop Med Hyg 20(5): 655-60.
Lynn, A., Chandra, S., Malhotra, P. and Chauhan, V. S. (1999). "Heme binding and polymerization by Plasmodium falciparum histidine rich protein II: influence of pH on activity and conformation." FEBS Lett 459(2): 267-271.
Mackinnon, M. J. and Marsh, K. (2010). "The selection landscape of malaria parasites." Science 328(5980): 866-871.
Mackintosh, C. L., Beeson, J. G. and Marsh, K. (2004). "Clinical features and pathogenesis of severe malaria." Trends Parasitol 20(12): 597-603.
Macomber, P. B., O'Brien, R. L. and Hahn, F. E. (1966). "Chloroquine: physiological basis of drug resistance in Plasmodium berghei." Science 152(727): 1374-1375.
Makler, M., Piper, R. and Milhous, W. (1998). "Lactate dehydrogenase and the diagnosis of malaria." Parasitol Today 14(9): 376-377.
Makler, M. T. and Hinrichs, D. J. (1993a). "Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia." Am J Trop Med Hyg 48(2): 205-210.
Makler, M. T., Ries, J. M., Williams, J. A., Bancroft, J. E., Piper, R. C., Gibbins, B. L. and Hinrichs, D. J. (1993b). "Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity." Am J Trop Med Hyg 48(6): 739-741.

Bibliography
235
Malhotra, I., Mungai, P., Muchiri, E., Kwiek, J. J., Meshnick, S. R. and King, C. L. (2006). "Umbilical cord-blood infections with Plasmodium falciparum malaria are acquired antenatally in Kenya." J Infect Dis 194(2): 176-183.
Mansor, S. M., Navaratnam, V., Yahaya, N., Nair, N. K., Wernsdorfer, W. H. and Degen, P. H. (1996). "Determination of a new antimalarial drug, benflumetol, in blood plasma by high-performance liquid chromatography." J Chromatogr B Biomed Appl 682(2): 321-325.
Marfurt, J., Mueller, I., Sie, A., Maku, P., Goroti, M. and Reeder, J. (2007). "Low efficacy of amodiaquine or chloroquine plus sulfadozine-pyrimethamine against Plasmodium falciparum and P.vivax malaria in Papua New Guinea." Am J Trop Med Hyg 77: 947-954.
Marfurt, J., Muller, I., Sie, A., Oa, O., Reeder, J. C., Smith, T. A., Beck, H. P. and Genton, B. (2008). "The usefulness of twenty-four molecular markers in predicting treatment outcome with combination therapy of amodiaquine plus sulphadoxine-pyrimethamine against falciparum malaria in Papua New Guinea." Malar J 7: 61-71.
Martin, A. N., Farquar, G. R., Jones, A. D. and Frank, M. (2010). "Human breath analysis: methods for sample collection and reduction of localized background effects." Anal Bioanal Chem 396(2): 739-750.
Martin, F. I., Wyatt, G. B., Griew, A. R., Mathews, J. D. and Campbell, D. G. (1981). "Diabetic surveys in Papua New Guinea - results and implications." P N G Med J 24(3): 188-194.
Martin, S. K., Rajasekariah, G. H., Awinda, G., Waitumbi, J. and Kifude, C. (2009). "Unified parasite lactate dehydrogenase and histidine-rich protein ELISA for quantification of Plasmodium falciparum." Am J Trop Med Hyg 80(4): 516-522.
Matsuoka, H., Yoshida, S., Hirai, M., Ishii, A. (2002). "A rodent malaria, Plasmodium berghei, is experimentally transmitted to mice by merely probing of infective mosquito, Anopheles stephensi." Parasitol Int 51(1): 17-23.
Mayxay, M., Barends, M., Brockman, A., Jaidee, A., Nair, S., Sudimack, D., Pongvongsa, T., Phompida, S., Phetsouvanh, R., Anderson, T., White, N. J. and Newton, P. N. (2007). "In vitro antimalarial drug susceptibility and pfcrt mutation among fresh Plasmodium falciparum isolates from the Lao PDR (LAOS)." Am J Trop Med Hyg 76(2): 245-250.
McCoy, D., Kembhavi, G., Patel, J. and Luintel, A. (2009). "The Bill & Melinda Gates Foundation's grant-making programme for global health." Lancet 373(9675): 1645-1653.
McFadden, G. I., Reith, M. E., Munholland, J. and Lang-Unnasch, N. (1996). "Plastid in human parasites." Nature 381(6582): 482.

Bibliography
236
McGready, R., Stepniewska, K., Lindegardh, N., Ashley, E. A., La, Y., Singhasivanon, P., White, N. J. and Nosten, F. (2006). "The pharmacokinetics of artemether and lumefantrine in pregnant women with uncomplicated falciparum malaria." Eur J Clin Pharmacol 62(12): 1021-1031.
McMurry, M. P., Cerqueira, M. T., Connor, S. L. and Connor, W. E. (1991). "Changes in lipid and lipoprotein levels and body weight in Tarahumara Indians after consumption of an affluent diet." N Engl J Med 325(24): 1704-1708.
McNamara, D. T., Kasehagen, L. J., Grimberg, B. T., Cole-Tobian, J., Collins, W. E. and Zimmerman, P. A. (2006). "Diagnosing infection levels of four human malaria parasite species by a polymerase chain reaction/ligase detection reaction fluorescent microsphere-based assay." Am J Trop Med Hyg 74(3): 413-4121.
McNamara, D. T., Thomson, J. M., Kasehagen, L. J. and Zimmerman, P. A. (2004). "Development of a multiplex PCR-ligase detection reaction assay for diagnosis of infection by the four parasite species causing malaria in humans." J Clin Microbiol 42(6): 2403-2410.
McQuistion, T. E. (1979). "Effect of temperature and clofibrate on Plasmodium berghei infection in mice." Am J Trop Med Hyg 28(1): 12-14.
Mehlotra, R. K., Fujioka, H., Roepe, P. D., Janneh, O., Ursos, L. M., Jacobs-Lorena, V. and Zimmerman, P. A. (2001). "Evolution of a unique Plasmodium falciparum chloroquine-resistance phenotype in association with pfcrt polymorphism in Papua New Guinea and South America." Proc Natl Acad Sci U S A 98(22): 12689-12694.
Mehlotra, R. K., Lorry, K., Kastens, W., Miller, S. M., Alpers, M. P., Bockarie, M., Kazura, J. W. and Zimmerman, P. A. (2000). "Random distribution of mixed species malaria infections in Papua New Guinea." Am J Trop Med Hyg 62(2): 225-231.
Mehlotra, R. K., Mattera, G., Bhatia, K., Reeder, J. C., Stoneking, M. and Zimmerman, P. A. (2005). "Insight into the early spread of chloroquine-resistant Plasmodium falciparum infections in Papua New Guinea." J Infect Dis 192(12): 2174-2179.
Mehlotra, R. K., Mattera, G., Bockarie, M., Maguire, J., Baird, J. K., Sharma, V. P., Alifrangis, M., Dorsey, G., Rosenthal, P. J., Fryauff, D. J., Kazura, J. W., Stoneking, M. and Zimmerman, P. A. (2008). "Disconcordant patterns of genetic variation at two chloroquine resistance loci in worldwide populations of the malaria parasite Plasmodium falciparum." Antimicrob Agents Chemother 52(6): 2212-2222.
Menard, R., Sultan, A. A., Cortes, C., Altszuler, R., van Dijk, M. R., Janse, C. J., Waters, A. P., Nussenzweig, R. S. and Nussenzweig, V. (1997). "Circumsporozoite protein is required for development of malaria sporozoites in mosquitoes." Nature 385(6614): 336-340.

Bibliography
237
Menezes, C. M., Kirchgatter, K., Di Santi, S. M., Savalli, C., Monteiro, F. G., Paula, G. A. and Ferreira, E. I. (2002). "In vitro chloroquine resistance modulation study on fresh isolates of Brazilian Plasmodium falciparum: intrinsic antimalarial activity of phenothiazine drugs." Mem Inst Oswaldo Cruz 97(7): 1033-1039.
Menge, D. M., Ernst, K. C., Vulule, J. M., Zimmerman, P. A., Guo, H. and John, C. C. (2008). "Microscopy underestimates the frequency of Plasmodium falciparum infection in symptomatic individuals in a low transmission highland area." Am J Trop Med Hyg 79(2): 173-177.
Meryman, H. and Hornblower, M. (1972). "A method for freezing and washing red blood cells using high glycerol concentration." Transfusion 12: 145-156.
Michon, P., Cole-Tobian, J. L., Dabod, E., Schoepflin, S., Igu, J., Susapu, M., Tarongka, N., Zimmerman, P. A., Reeder, J. C., Beeson, J. G., Schofield, L., King, C. L. and Mueller, I. (2007). "The risk of malarial infections and disease in Papua New Guinean children." Am J Trop Med Hyg 76(6): 997-1008.
Middleton, B. S. (1989). "Analytical artifacts: GC, MS, HPLC, TLC, and PC." J Chromatogr Lib 44.
Miller, D. B. and Spence, J. D. (1998). "Clinical pharmacokinetics of fibric acid derivatives (fibrates)." Clin Pharmacokinet 34(2): 155-162.
Miller, L. H., Baruch, D. I., Marsh, K., Doumbo, O. K. (2002). "The pathogenic basis of malaria." Nature 415(6872): 673-679.
Mirovsky, P. (1989). "Continuous culture of Plasmodium falciparum asexual stages in "normal" air atmosphere." Folia Parasitol (Praha) 36(2): 107-112.
Mita, T., Kaneko, A., Hombhanje, F., Hwaihwanje, I., Takahashi, N., Osawa, H., Tsukahara, T., Masta, A., Lum, J. K., Kobayakawa, T., Ishizaki, T. and Bjorkman, A. (2006a). "Role of pfmdr1 mutations on chloroquine resistance in Plasmodium falciparum isolates with pfcrt K76T from Papua New Guinea." Acta Trop 98(2): 137-144.
Mita, T., Kaneko, A., Hwaihwanje, I., Tsukahara, T., Takahashi, N., Osawa, H., Tanabe, K., Kobayakawa, T. and Bjork, a., A. (2006b). "Rapid selection of dhfr mutant allele in Plasmodium falciparum isolates after the introduction of sulfadoxine/pyrimethamine in combination with 4-aminoquinolines in Papua New Guinea." Infection, Genetics and Evolution 6(6): 447-452.
Mita, T., Tanabe, K., Takahashi, N., Tsukahara, T., Eto, H., Dysoley, L., Ohmae, H., Kita, K., Krudsood, S., Looareesuwan, S., Kaneko, A., Bjorkman, A. and Kobayakawa, T. (2007). "Independent evolution of pyrimethamine resistance in Plasmodium falciparum isolates in Melanesia." Antimicrob Agents Chemother 51(3): 1071-1077.

Bibliography
238
Miyagami, T. and Waki, S. (1985). "In vitro cultivation of Plasmodium falciparum under aerobic atmosphere in a CO2 incubator." J Parasitol 71(2): 262-263.
Mlambo, G. and Kumar, N. (2007). "A modified Plasmodium falciparum growth inhibition assay (GIA) to assess activity of plasma from malaria endemic areas." Exp Parasitol 115(2): 211-214.
Molyneux, D. H., Floyd, K., Barnish, G. and Fevre, E. M. (1999). "Transmission control and drug resistance in malaria: a crucial interaction." Parasitol Today 15(6): 238-240.
Montalvetti, A., J. Pana-Diaz, R. Hurtado, L. M. Ruiz-Perez, and D. Gonzalez-Pacanowska. (2000). "Characterization and regulation of Leshmania major 3-hydroxy-methyl-glutaryl-CoA reductase." Biochem J 349: 27-34.
Moody, A. H. and Chiodini, P. L. (2002). "Non-microscopic method for malaria diagnosis using OptiMAL IT, a second-generation dipstick for malaria pLDH antigen detection." Br J Biomed Sci 59(4): 228-231.
Moreno, A., Brasseur, P., Cuzin-Ouattara, N., Blanc, C. and Druilhe, P. (2001). "Evaluation under field conditions of the colourimetric DELI-microtest for the assessment of Plasmodium falciparum drug resistance." Trans R Soc Trop Med Hyg 95(1): 100-103.
Moreno, S. N. and Li, Z. H. (2008). "Anti-infectives targeting the isoprenoid pathway of Toxoplasma gondii." Expert Opin Ther Targets 12(3): 253-263.
Mu, J., Awadalla, P., Duan, J., McGee, K. M., Joy, D. A., McVean, G. A. and Su, X. Z. (2005). "Recombination hotspots and population structure in Plasmodium falciparum." PLoS Biol 3(10): e335.
Mueller, I., Bockarie, M., Alpers, M. and Smith, T. (2003). "The epidemiology of malaria in Papua New Guinea." Trends in Parasitology 19(6): 253-259.
Mueller, I., Namuigi, P., Kundi, J., Ivivi, R., Tandrapah, T., Bjorge, S. and Reeder, J. C. (2005). "Epidemic malaria in the highlands of Papua New Guinea." Am J Trop Med Hyg 72(5): 554-560.
Mueller, I., Rogerson, S., Mola, G. D. and Reeder, J. C. (2008). "A review of the current state of malaria among pregnant women in Papua New Guinea." P N G Med J 51(1-2): 12-6.
Muller, G., Wernsdorfer, G., Sirichaisinthop, J., Starzengruber, P., Congpuong, K. and Wernsdorfer, W. H. (2008). "Synergism between monodesbutyl-benflumetol and artemisinin in Plasmodium falciparum in vitro." Wien Klin Wochenschr 120(19-20 Suppl 4): 80-4.
Muller, I. B. and Hyde, J. E. (2010). "Antimalarial drugs: modes of action and mechanisms of parasite resistance." Future Microbiol 5(12): 1857-1873.

Bibliography
239
Mutabingwa, T. K., Anthony, D., Heller, A., Hallett, R., Ahmed, J., Drakeley, C., Greenwood, B. M. and Whitty, C. J. (2005). "Amodiaquine alone, amodiaquine+sulfadoxine-pyrimethamine, amodiaquine+artesunate, and artemether-lumefantrine for outpatient treatment of malaria in Tanzanian children: a four-arm randomised effectiveness trial." Lancet 365(9469): 1474-1480.
MVI. (2004). "Fact sheet: malaria and the military." from http://www.malariavaccine.org/files/FS_Malaria-Military_9-15-04.pdf.
Mwai, L., Kiara, S. M., Abdirahman, A., Pole, L., Rippert, A., Diriye, A., Bull, P., Marsh, K., Borrmann, S. and Nzila, A. (2009b). "In vitro activities of piperaquine, lumefantrine, and dihydroartemisinin in Kenyan Plasmodium falciparum isolates and polymorphisms in pfcrt and pfmdr1." Antimicrob Agents Chemother 53(12): 5069-5073.
Mwai, L., Ochong, E., Abdirahman, A., Kiara, S. M., Ward, S., Kokwaro, G., Sasi, P., Marsh, K., Borrmann, S., Mackinnon, M. and Nzila, A. (2009a). "Chloroquine resistance before and after its withdrawal in Kenya." Malar J 8: 106-115.
Na-Bangchang, K., Kanda, T., Tipawangso, P., Thanavibul, A., Suprakob, K., Ibrahim, M., Wattanagoon, Y. and Karbwang, J. (1996). "Activity of artemether-azithromycin versus artemether-doxycycline in the treatment of multiple drug resistant falciparum malaria." Southeast Asian J Trop Med Public Health 27(3): 522-525.
Nagesha, H. S., Din, S., Casey, G. J., Susanti, A. I., Fryauff, D. J., Reeder, J. C. and Cowman, A. F. (2001). "Mutations in the pfmdr1, dhfr and dhps genes of Plasmodium falciparum are associated with in-vivo drug resistance in West Papua, Indonesia." Trans R Soc Trop Med Hyg 95(1): 43-49.
Naik, R. S., M. Venkatesan, and D. C. Gowda. (2001). "Plasmodium falciparum: the lethal effects of tunicamycin and mevastatin on the parasite are not mediated by inhibition of N-linked oligosaccharide biosynthesis." Exp Parasitol 98: 110-114.
Nakornchai, S. and Konthiang, P. (2006). "Activity of azithromycin or erythromycin in combination with antimalarial drugs against multidrug-resistant Plasmodium falciparum in vitro." Acta Trop 100(3): 185-191.
Nawaz, F., Nsobya, S. L., Kiggundu, M., Joloba, M. and Rosenthal, P. J. (2009). "Selection of parasites with diminished drug susceptibility by amodiaquine-containing antimalarial regimens in Uganda." J Infect Dis 200(11): 1650-1657.
Needham, E. (2011, 25 April 2011). "Travel ban between Torres Strait and Papua New Guinea because of malaria outbreak." Topnews Retrieved 27 April 2011 from http://topnews.us/content/239008-travel-ban-between-torres-strait-and-papua-guinea-because-malaria-outbreak.

Bibliography
240
News, B. (2011). "George Clooney recovering from malaria." Retrieved 21 Jan, 2011, from http://www.bbc.co.uk/news/entertainment-arts-12248440.
Newton, P. N., Fernandez, F. M., Plancon, A., Mildenhall, D. C., Green, M. D., Ziyong, L., Christophel, E. M., Phanouvong, S., Howells, S., McIntosh, E., Laurin, P., Blum, N., Hampton, C. Y., Faure, K., Nyadong, L., Soong, C. W., Santoso, B., Zhiguang, W., Newton, J. and Palmer, K. (2008). "A collaborative epidemiological investigation into the criminal fake artesunate trade in South East Asia." PLoS Med 5(2): e32.
Nirogi, R. V. S., Kandikere, V. N., Shukla, M., Mudigonda, K., Maurya, S., Boosi, R. and Anjaneyulu, Y. (2006). "Simultaneous quantification of atorvastatin and active metabolites in human plasma by liquid chromatography-tandem mass spectrometry using resuvastin as internal standard." Biomedical Chromatography 20: 924-936.
Nkhoma, S., Molyneux, M. and Ward, S. (2007). "In vitro antimalarial susceptibility profile and prcrt/pfmdr-1 genotypes of Plasmodium falciparum field isolates from Malawi." Am J Trop Med Hyg 76(6): 1107-1112.
Noedl, H. (2002). "HN-NonLin V1.1." from http://malaria.farch.net.
Noedl, H., Allmendinger, T., Prajakwong, S., Wernsdorfer, G. and Wernsdorfer, W. H. (2001). "Desbutyl-benflumetol, a novel antimalarial compound: in vitro activity in fresh isolates of Plasmodium falciparum from Thailand." Antimicrob Agents Chemother 45(7): 2106-2109.
Noedl, H., Bronnert, J., Yingyuen, K., Attlmayr, B., Kollaritsch, H. and Fukuda, M. (2005). "Simple histidine-rich protein 2 double-site sandwich enzyme-linked immunosorbent assay for use in malaria drug sensitivity testing." Antimicrob Agents Chemother 49(8): 3575-3577.
Noedl, H., Krudsood, S., Chalermratana, K., Silachamroon, U., Leowattana, W., Tangpukdee, N., Looareesuwan, S., Miller, R. S., Fukuda, M., Jongsakul, K., Sriwichai, S., Rowan, J., Bhattacharyya, H., Ohrt, C. and Knirsch, C. (2006). "Azithromycin combination therapy with artesunate or quinine for the treatment of uncomplicated Plasmodium falciparum malaria in adults: a randomized, phase 2 clinical trial in Thailand." Clin Infect Dis 43(10): 1264-1271.
Noedl, H., Krudsood, S., Leowattana, W., Tangpukdee, N., Thanachartwet, W., Looareesuwan, S., Miller, R. S., Fukuda, M., Jongsakul, K., Yingyuen, K., Sriwichai, S., Ohrt, C. and Knirsch, C. (2007). "In vitro antimalarial activity of azithromycin, artesunate, and quinine in combination and correlation with clinical outcome." Antimicrob Agents Chemother 51(2): 651-656.
Noedl, H., Se, Y., Schaecher, K., Smith, B. L., Socheat, D. and Fukuda, M. M. (2008). "Evidence of artemisinin-resistant malaria in western Cambodia." N Engl J Med 359(24): 2619-2620.

Bibliography
241
Noedl, H., Wernsdorfer, W. H., Kollaritsch, H., Looareesuwan, S., Miller, R. S. and Wongsrichanalai, C. (2003). "Malaria drug-susceptibility testing. HRP2-based assays: current data, future perspectives." Wien Klin Wochenschr 115(S3): 23-27.
Noedl, H., Wernsdorfer, W. H., Krudsood, S., Wilairatana, P., Kollaritsch, H., Wiedermann, G. and Looareesuwan, S. (2001). "Antimalarial activity of azithromycin, artemisinin and dihydroartemisinin in fresh isolates of Plasmodium falciparum in Thailand." Acta Trop 80(1): 39-44.
Nsanzabana, C., Hastings, I. M., Marfurt, J., Muller, I., Baea, K., Rare, L., Schapira, A., Felger, I., Betschart, B., Smith, T. A., Beck, H. P. and Genton, B. (2010). "Quantifying the evolution and impact of antimalarial drug resistance: drug use, spread of resistance, and drug failure over a 12-year period in Papua New Guinea." J Infect Dis 201(3): 435-443.
Nsobya, S. L., Dokomajilar, C., Joloba, M., Dorsey, G. and Rosenthal, P. J. (2007). "Resistance-mediating Plasmodium falciparum pfcrt and pfmdr1 alleles after treatment with artesunate-amodiaquine in Uganda." Antimicrob Agents Chemother 51(8): 3023-3025.
Ntale, M., Ogwal-Okeng, J. W., Mahindi, M., Gustafsson, L. L. and Beck, O. (2008). "A field-adapted sampling and HPLC quantification method for lumefantrine and its desbutyl metabolite in whole blood spotted on filter paper." J Chromatogr B Analyt Technol Biomed Life Sci 876(2): 261-265.
Nzila-Mounda, A., Mberu, E. K., Sibley, C. H., Plowe, C. V., Winstanley, P. A. and Watkins, W. M. (1998). "Kenyan Plasmodium falciparum field isolates: correlation between pyrimethamine and chlorcycloguanil activity in vitro and point mutations in the dihydrofolate reductase domain." Antimicrob Agents Chemother 42(1): 164-169.
Nzila, A. and Mwai, L. (2010). "In vitro selection of Plasmodium falciparum drug-resistant parasite lines." J Antimicrob Chemother 65(3): 390-8.
O'Neill, P. M., Barton, V. E. and Ward, S. A. (2010). "The molecular mechanism of action of artemisinin--the debate continues." Molecules 15(3): 1705-1721.
O'Neill, P. M., Ward, S. A., Berry, N. G., Jeyadevan, J. P., Biagini, G. A., Asadollaly, E., Park, B. K. and Bray, P. G. (2006). "A medicinal chemistry perspective on 4-aminoquinoline antimalarial drugs." Curr Top Med Chem 6(5): 479-507.
Odds, F. C. (2003). "Synergy, antagonism, and what the chequerboard puts between them." J Antimicrob Chemother 52(1): 1.
Oduola, A. M., Omitowoju, G. O., Sowunmi, A., Makler, M. T., Falade, C. O., Kyle, D. E., Fehintola, F. A., Ogundahunsi, O. A., Piper, R. C., Schuster, B. G. and Milhous, W. K. (1997). "Plasmodium falciparum: evaluation of lactate

Bibliography
242
dehydrogenase in monitoring therapeutic responses to standard antimalarial drugs in Nigeria." Exp Parasitol 87(3): 283-289.
Oduro, A. R., Anyorigia, T., Hodgson, A., Ansah, P., Anto, F., Ansah, N. A., Atuguba, F., Mumuni, G. and Amankwa, J. (2005). "A randomized comparative study of chloroquine, amodiaquine and sulphadoxine-pyrimethamine for the treatment of uncomplicated malaria in Ghana." Trop Med Int Health 10: 179-184.
Olliaro, P., Nevill, C., LeBras, J., Ringwald, P., Mussano, P., Garner, P. and Brasseur, P. (1996). "Systematic review of amodiaquine treatment in uncomplicated malaria." Lancet 348(9036): 1196-1201.
Olliaro, P. L., Haynes, R. K., Meunier, B. and Yuthavong, Y. (2001). "Possible modes of action of the artemisinin-type compounds." Trends Parasitol 17(3): 122-126.
Onda, T., Lin, Q., Kano, S. and Suzuki, M. (1999). "Continuous in vitro culture of Plasmodium falciparum using microaerophilic gas generators and portable incubator." Kansenshogaku Zasshi 73(11): 1095-1098.
Ōtsuka, R. and Ulijaszek, S. J., Eds. (2007). Health change in the Asia-Pacific region: Biocultural and epidemiological approaches. Cambridge, Cambridge University Press.
Ouellette, M. and Ward, S. S. (2003). Drug resistance in parasites. Molecular Medical Parasitology, Elsevier Science Ltd.: 397-413.
Pagola, S., Stephens, P. W., Bohle, D. S., Kosar, A. D. and Madsen, S. K. (2000). "The structure of malaria pigment beta-haematin." Nature 404(6775): 307-310.
Pain, A., Ferguson, D. J., Kai, O., Urban, B. C., Lowe, B., Marsh, K., Roberts, D. J. (2001). "Platelet-mediated clumping of Plasmodium falciparum-infected erythrocytes is a common adhesive phenotype and is associated with severe malaria." Proc Natl Acad Sci U S A 98(4): 1805-1810.
Palmer, K. J., Holliday, S. M. and Brogden, R. N. (1993). "Mefloquine. A review of its antimalarial activity, pharmacokinetic properties and therapeutic efficacy." Drugs 45(3): 430-475.
Pang, X., Wang, G. and Xing, Q. (1999). "Hundred and one cases Plasmodium falciparum patients treated naphthoquine phosphate." Chinese J Parasitol Parasit Dis 17(1): 20.
Papalexis, V., Siomos, M. A., Campanale, N., Guo, X., Kocak, G., Foley, M. and Tilley, L. (2001). "Histidine-rich protein 2 of the malaria parasite, Plasmodium falciparum, is involved in detoxification of the by-products of haemoglobin degradation." Mol Biochem Parasitol 115(1): 77-86.
Parquet, V., Briolant, S., Torrentino-Madamet, M., Henry, M., Almeras, L., Amalvict, R., Baret, E., Fusai, T., Rogier, C. and Pradines, B. (2009). "Atorvastatin is a

Bibliography
243
promising partner for antimalarial drugs in treatment of Plasmodium falciparum malaria." Antimicrob Agents Chemother 53(6): 2248-2252.
Peel, S. A., Bright, P., Yount, B., Handy, J. and Baric, R. S. (1994). "A strong association between mefloquine and halofantrine resistance and amplification, overexpression, and mutation in the P-glycoprotein gene homolog (pfmdr) of Plasmodium falciparum in vitro. ." Am J Trop Med Hyg 51: 648-658.
Perkins, M. E. and Holt, E. H. (1988). "Erythrocyte receptor recognition varies in Plasmodium falciparum isolates." Mol Biochem Parasitol 27(1): 23-34.
Peters, W. (1987). Chemotherapy and drug resistance in malaria. London, Academic Press.
Peters, W. and Porter, M. (1976). "The chemotherapy of rodent malaria, XXVI. The potential value of WR 122,455 (a 9-phenanthrenemethanol) against drug-resistant malaria parasites." Ann Trop Med Parasitol 70(3): 271-281.
Peterson, D. S., Di Santi, S. M., Povoa, M., Calvosa, V. S., Do Rosario, V. E. and Wellems, T. E. (1991). "Prevalence of the dihydrofolate reductase Asn-108 mutation as the basis for pyrimethamine-resistant falciparum malaria in the Brazilian Amazon." Am J Trop Med Hyg 45(4): 492-497.
Peterson, D. S., Walliker, D. and Wellems, T. E. (1988). "Evidence that a point mutation in dihydrofolate reductase-thymidylate synthase confers resistance to pyrimethamine in falciparum malaria." Proc Natl Acad Sci U S A 85(23): 9114-9118.
Phillips, M. (1992a). "Detection of carbon disulfide in breath and air: a possible new risk factor for coronary artery disease." Int Arch Occup Environ Health 64(2): 119-123.
Phillips, M. (1992b). "Breath tests in medicine." Sci Am 267(1): 74-79.
Phillips, M. (1997). "Method for the collection and assay of volatile organic compounds in breath." Anal Biochem 247(2): 272-278.
Phillips, M., Cataneo, R. N., Condos, R., Ring Erickson, G. A., Greenberg, J., La Bombardi, V., Munawar, M. I. and Tietje, O. (2007). "Volatile biomarkers of pulmonary tuberculosis in the breath." Tuberculosis (Edinb) 87(1): 44-52.
Phillips, M. and Greenberg, J. (1992c). "Ion-trap detection of volatile organic compounds in alveolar breath." Clin Chem 38(1): 60-65.
Phillips, M., Sabas, M. and Greenberg, J. (1993). "Increased pentane and carbon disulfide in the breath of patients with schizophrenia." J Clin Pathol 46(9): 861-864.

Bibliography
244
Pickard, A. L., Wongsrichanalai, C., Purfield, A., Kamwendo, D., Emery, K., Zalewski, C., Kawamoto, F., Miller, R. S. and Meshnick, S. R. (2003). "Resistance to antimalarials in Southeast Asia and genetic polymorphisms in pfmdr1." Antimicrob Agents Chemother 47(8): 2418-2423.
Piola, P., Fogg, C., Bajunirwe, F., Biraro, S., Grandesso, F., Ruzagira, E., Babigumira, J., Kigozi, I., Kiguli, J., Kyomuhendo, J., Ferradini, L., Taylor, W., Checchi, F. and Guthmann, J. P. (2005). "Supervised versus unsupervised intake of six-dose artemether-lumefantrine for treatment of acute, uncomplicated Plasmodium falciparum malaria in Mbarara, Uganda: a randomised trial." Lancet 365(9469): 1467-1473.
Piper, R., Lebras, J., Wentworth, L., Hunt-Cooke, A., Houze, S., Chiodini, P. and Makler, M. (1999). "Immunocapture diagnostic assays for malaria using Plasmodium lactate dehydrogenase (pLDH)." Am J Trop Med Hyg 60(1): 109-118.
Pirker-Krassnig, D. K., Wernsdorfer, G., Sirichaisinthop, J., Rojanawatsirivet, C., Kollaritsch, H. and Wernsdorfer, W. H. (2004). "Comparative study on the in vitro activity of lumefantrine and desbutyl-benflumetol in fresh isolates of Plasmodium vivax from Thailand." Wien Klin Wochenschr 116(S4): 47-52.
Plowe, C. V. (2009). "The evolution of drug-resistant malaria." Trans R Soc Trop Med Hyg 103(S1): S11-4.
Plowe, C. V., Roper, C., Barnwell, J. W., Happi, C. T., Joshi, H. H., Mbacham, W., Meshnick, S. R., Mugittu, K., Naidoo, I., Price, R. N., Shafer, R. W., Sibley, C. H., Sutherland, C. J., Zimmerman, P. A. and Rosenthal, P. J. (2007). "World Antimalarial Resistance Network (WARN) III: molecular markers for drug resistant malaria." Malar J 6: 121-130.
PNGDOH (2000). Standard treatment of common illnesses of children in Papua New Guinea. P. N. G. D. o. Health, Government of Papua New Guinea
Ponnudurai, T., Leeuwenberg, A. D. and Meuwissen, J. H. (1981). "Chloroquine sensitivity of isolates of Plasmodium falciparum adapted to in vitro culture." Trop Geogr Med 33(1): 50-54.
Ponnudurai, T., Lensen, A. H., Leeuwenberg, A. D. and Meuwissen, J. H. (1982). "Cultivation of fertile Plasmodium falciparum gametocytes in semi-automated systems. 1. Static cultures." Trans R Soc Trop Med Hyg 76(6): 812-818.
Powers, K. G., Jacobs, R. L., Good, W. C. and Koontz, L. C. (1969). "Plasmodium vinckei: production of chloroquine-resistant strain." Exp Parasitol 26(2): 193-202.
Pradines, B., Hovette, P., Fusai, T., Atanda, H. L., Baret, E., Cheval, P., Mosnier, J., Callec, A., Cren, J., Amalvict, R., Gardair, J. P. and Rogier, C. (2006). "Prevalence of in vitro resistance to eleven standard or new antimalarial drugs

Bibliography
245
among Plasmodium falciparum isolates from Pointe-Noire, Republic of the Congo." J Clin Microbiol 44(7): 2404-2408.
Pradines, B., Mabika Mamfoumbi, M., Parzy, D., Owono Medang, M., Lebeau, C., Mourou Mbina, J. R., Doury, J. C. and Kombila, M. (1999a). "In vitro susceptibility of African isolates of Plasmodium falciparum from Gabon to pyronaridine." Am J Trop Med Hyg 60(1): 105-108.
Pradines, B., Tall, A., Fusai, T., Spiegel, A., Hienne, R., Rogier, C., Trape, J. F., Le Bras, J., Parzy, D. (1999b). "In vitro activities of benflumetol against 158 Senegalese isolates of Plasmodium falciparum in comparison with those of standard antimalarial drugs." Antimicrob Agents Chemother 43(2): 418-420.
Pradines, B., Tall, A., Parzy, D., Spiegel, A., Fusai, T., Hienne, R., Trape, J. F. and Doury, J. C. (1998). "In vitro activity of pyronaridine and amodiaquine against African isolates (Senegal) of Plasmodium falciparum in comparison with standard antimalarial agents." J Antimicrob Chemother 42: 333-339.
Pradines, B., Torrentino-Madamet, M., Fontaine, A., Henry, M., Baret, E., Mosnier, J., Briolant, S., Fusai, T. and Rogier, C. (2007). "Atorvastatin is 10-fold more active in vitro than other statins against Plasmodium falciparum." Antimicrob Agents Chemother 51(7): 2654-2655.
Preechapornkul, P., Imwong, M., Chotivanich, K., Pongtavornpinyo, W., Dondorp, A. M., Day, N. P., White, N. J. and Pukrittayakamee, S. (2009). "Plasmodium falciparum pfmdr1 amplification, mefloquine resistance, and parasite fitness." Antimicrob Agents Chemother 53(4): 1509-1515.
Preti, G., Thaler, E., Hanson, C. W., Troy, M., Eades, J. and Gelperin, A. (2009). "Volatile compounds characteristic of sinus-related bacteria and infected sinus mucus: analysis by solid-phase microextraction and gas chromatography-mass spectrometry." J Chromatogr B Analyt Technol Biomed Life Sci 877(22): 2011-2018.
Price, R. N., Cassar, C., Brockman, A., Duraisingh, M., van Vugt, M., White, N. J., Nosten, F. and Krishna, S. (1999). "The pfmdr1 gene is associated with a multidrug-resistant phenotype in Plasmodium falciparum from the western border of Thailand." Antimicrob Agents Chemother 43(12): 2943-2949.
Price, R. N., Hasugian, A. R., Ratcliff, A., Siswantoro, H., Purba, H. L., Kenangalem, E., Lindegardh, N., Penttinen, P., Laihad, F., Ebsworth, E. P., Anstey, N. M. and Tjitra, E. (2007). "Clinical and pharmacological determinants of the therapeutic response to dihydroartemisinin-piperaquine for drug-resistant malaria." Antimicrob Agents Chemother 51(11): 4090-4097.
Price, R. N., Uhlemann, A. C., Brockman, A., McGready, R., Ashley, E., Phaipun, L., Patel, R., Laing, K., Looareesuwan, S., White, N. J., Nosten, F. and Krishna, S.

Bibliography
246
(2004). "Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number." Lancet 364(9432): 438-447.
Price, R. N., Uhlemann, A. C., van Vugt, M., Brockman, A., Hutagalung, R., Nair, S., Nash, D., Singhasivanon, P., Anderson, T. J., Krishna, S., White, N. J., Nosten, F. (2006). "Molecular and pharmacological determinants of the therapeutic response to artemether-lumefantrine in multidrug-resistant Plasmodium falciparum malaria." Clin Infect Dis 42(11): 1570-1577.
Qu, H. Y., Gao, H. Z., Hao, G. T., Li, Y. Y., Li, H. Y., Hu, J. C., Wang, X. F., Liu, W. L. and Liu, Z. Y. (2010). "Single-dose safety, pharmacokinetics, and food effects studies of compound naphthoquine phosphate tablets in healthy volunteers." J Clin Pharmacol 50(11): 1310-1318.
Radfar, A., Mendez, D., Moneriz, C., Linares, M., Marin-Garcia, P., Puyet, A., Diez, A. and Bautista, J. M. (2009). "Synchronous culture of Plasmodium falciparum at high parasitemia levels." Nat Protoc 4(12): 1899-1915.
Ranford-Cartwright, L. C., Johnston, K. L., Abdel-Muhsin, A. M., Khan, B. K. and Babiker, H. A. (2002). "Critical comparison of molecular genotyping methods for detection of drug-resistant Plasmodium falciparum." Trans R Soc Trop Med Hyg 96(5): 568-572.
Rathod, P. K., McErlean, T. and Lee, P. C. (1997). "Variations in frequencies of drug resistance in Plasmodium falciparum." Proc Natl Acad Sci U S A 94(17): 9389-9393.
Reed, M. B., Saliba, K. J., Caruana, S. R., Kirk, K. and Cowman, A. F. (2000). "Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum." Nature 403(6772): 906-909.
Reeder, J., Rieckmann, K. H., Genton, B., Lorry, K., Wines, B. and Cowman, A. F. (1996). "Point mutations in the dihydrofolate reductase and dihydropteroate synthetase genes and in vitro susceptibility to pyrimethamine and cycloguanil of Plasmodium falciparum isolates from Papua New Guinea." Am J Trop Med Hyg 55(2): 209-213.
Rich, S. M., Leendertz, F. H., Xu, G., Lebreton, M., Djoko, C. F., Aminake, M. N., Takang, E. E., Diffo, J. L., Pike, B. L., Rosenthal, B. M., Formenty, P., Boesch, C., Ayala, F. J. and Wolfe, N. D. (2009). "The origin of malignant malaria." Proc Natl Acad Sci U S A.
Richards, W. H. and Maples, B. K. (1979). "Studies on Plasmodium falciparum in continuous cultivation. I. The effect of chloroquine and pyrimethamine on parasite growth and viability." Ann Trop Med Parasitol 73(2): 99-108.
Rieckmann, K. H. (1971). "Determination of the drug sensitivity of Plasmodium falciparum." Jama 217(5): 573-578.

Bibliography
247
Rieckmann, K. H., Campbell, G. H., Sax, L. J. and Mrema, J. E. (1978). "Drug sensitivity of Plasmodium falciparum. An in vitro microtechnique." Lancet 1(8054): 22-23.
Rieckmann, K. H., McNamara, J. V., Frischer, H., Stockest, T. A., Carson, P. E. and Powell, R. D. (1968). "Effects of chloroquine, quinine and cycloguanil upon the maturation of asexual erythrocytic forms of two strains of Plasmodium falciparum in vitro. ." Am J Trop Med Hyg 17: 661-671.
Ringwald, P., Bickii, J. and Basco, L. K. (1996). "In vitro activity of antimalarials against clinical isolates of Plasmodium falciparum in Yaounde, Cameroon." Am J Trop Med Hyg 55(3): 254-258.
Risticevic, S., Lord, H., Gorecki, T., Arthur, C. L. and Pawliszyn, J. (2010). "Protocol for solid-phase microextraction method development." Nat Protoc 5(1): 122-139.
Rogers, W. O., Malik, A., Mellouk, S., Nakamura, K., Rogers, M. D., Szarfman, A., Gordon, D. M., Nussler, A. K., Aikawa, M. and Hoffman, S. L. (1992). "Characterization of Plasmodium falciparum sporozoite surface protein 2." Proc Natl Acad Sci U S A 89(19): 9176-80.
Rogers, W. O., Sem, R., Tero, T., Chim, P., Lim, P., Muth, S., Socheat, D., Ariey, F. and Wongsrichanalai, C. (2009). "Failure of artesunate-mefloquine combination therapy for uncomplicated Plasmodium falciparum malaria in southern Cambodia." Malar J 8: 10-18.
Roth, E. F., Calvin,M-C.,Max-audit, I., Rosa, J. & Rosa, R. (1988). "The enzymes of glycolytic pathway in erythrocytes infected with Plasmodium falciparum malaria parasites." Blood 72: 1922-1925.
Rungsihirunrat, K., Chaijareonkul, W., Seugorn, A., Na-Bangchang, K. and Thaithong, S. (2009). "Association between chloroquine resistance phenotypes and point mutations in pfcrt and pfmdr1 in Plasmodium falciparum isolates from Thailand." Acta Trop 109(1): 37-40.
Sá, J. M., Twu, O., Hayton, K., Reyes, S., Fay, M. P., Ringwald, P. and Wellems, T. E. (2009). "Inaugural Article: Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine." Proc Natl Acad Sci U S A 106 (45) 18883-18889.
Sachanonta, N., Medana, I. M., Roberts, R., Jones, M., Day, N. P., White, N. J., Ferguson, D. J., Turner, G. D. and Pongponratn, E. (2008). "Host vascular endothelial growth factor is trophic for Plasmodium falciparum-infected red blood cells." Asian Pac J Allergy Immunol 26(1): 37-45.
Safeukui, I., Correas, J. M., Brousse, V., Hirt, D., Deplaine, G., Mule, S., Lesurtel, M., Goasguen, N., Sauvanet, A., Couvelard, A., Kerneis, S., Khun, H., Vigan-Womas, I., Ottone, C., Molina, T. J., Treluyer, J. M., Mercereau-Puijalon, O.,

Bibliography
248
Milon, G., David, P. H. and Buffet, P. A. (2008). "Retention of Plasmodium falciparum ring-infected erythrocytes in the slow, open microcirculation of the human spleen." Blood 112(6): 2520-2528.
Saint-Yves, I. F. (1971). "The alleged resistance of Plasmodium falciparum to chloroquine in the Milne Bay District." P N G Med J 14(3): 77-78.
Saito-Nakano, Y., Tanabe, K., Kamei, K., Iwagami, M., Komaki-Yasuda, K., Kawazu, S., Kano, S., Ohmae, H. and Endo, T. (2008). "Genetic evidence for Plasmodium falciparum resistance to chloroquine and pyrimethamine in Indochina and the Western Pacific between 1984 and 1998." Am J Trop Med Hyg 79(4): 613-619.
Saliba, K. J., Folb, P. I. and Smith, P. J. (1998). "Role for the Plasmodium falciparum digestive vacuole in chloroquine resistance." Biochem Pharmacol 56(3): 313-320.
Sam-Yellowe, T. Y., Shio, H. and Perkins, M. E. (1988). "Secretion of Plasmodium falciparum rhoptry protein into the plasma membrane of host erythrocytes." J Cell Biol 106(5): 1507-1513.
Samal, D., Rojanawatsirivet, C., Wernsdorfer, G., Kollaritsch, H., Sirichaisinthop, J. and Wernsdorfer, W. H. (2005). "Synergism of desbutyl-benflumetol and retinol against Plasmodium falciparum in vitro." Wien Klin Wochenschr 117 Suppl 4: 39-44.
SANDH. (2007). "Guidelines for the treatment of malaria in South Africa." from www.doh.gov.za/docs/factsheets/guidelines/malaria/treatment.
Sapak, P., Garner, P., Baea, M., Narara, A., Heywood, P. and Alpers, M. (1991). "Ineffectiveness of amodiaquine against Plasmodium falciparum malaria in symptomatic young children living in an endemic malarious area of Papua New Guinea." J Trop Pediatr 37(4): 185-190.
Sarkar, M., Woodland, C., Koren, G. and Einarson, A. R. (2006). "Pregnancy outcome following gestational exposure to azithromycin." BMC Pregnancy Childbirth 6: 18-22.
Scheibel, L. W., Ashton, S. H. and Trager, W. (1979). "Plasmodium falciparum: microaerophilic requirements in human red blood cells." Exp Parasitol 47(3): 410-418.
Schiff, C. J., Premji, Z. and Minjas, J. N. (1993). "The rapid manual Parasight®F test. A new diagnostic tool for Plasmodium falciparum infection." Trans R Soc Trop Med Hyg 87: 646-648.
Schoepflin, S., Lin, E., Kiniboro, B., DaRe, J. T., Mehlotra, R. K., Zimmerman, P. A., Mueller, I. and Felger, I. (2010). "Treatment with coartem (artemether-lumefantrine) in Papua New Guinea." Am J Trop Med Hyg 82(4): 529-534.

Bibliography
249
Schoepflin, S., Marfurt, J., Goroti, M., Baisor, M., Mueller, I. and Felger, I. (2008). "Heterogeneous distribution of Plasmodium falciparum drug resistance haplotypes in subsets of the host population." Malar J 7: 78-86.
Schuurkamp, G. J. T. and Kereu, R. K. (1989). "Resistance of Plasmodium falciparum to chemotherapy with 4-aminoquinolines in the Ok Tedi area of Papua New Guinea." P N G Med J 32: 33-44.
Sharkoor, O., Taylor, R. B. and Behrens, R. H. (1997). "Assessment of the incidence of substandard drugs in developing countries." Trop Med Int Health 2: 839-845.
Sherman, I. W. (1961). "Molecular heterogeneity of lactic dehydrogenase in avian malaria (Plasmodium lophurae)." J Exp Med 114: 1049-1062.
Sherman, I. W. (1979). "Biochemistry of Plasmodium (malarial parasites)." Microbiol Rev 43(4): 453-495.
Shin, S. C. J., Vanderberg, J. P. and Terzakis, J. A. (1982). "Direct infection of hepatocytes of Plasmodium berghei." J Protozool 29: 448-454.
Shute, G. T., Ed. (1988). The microscopic diagnosis of malaria. Malaria, principles and practice of malariology. London, Churchill Livingstone.
Sibley, C. H., Barnes, K. I., Watkins, W. M. and Plowe, C. V. (2008). "A network to monitor antimalarial drug resistance: a plan for moving forward." Trends Parasitol 24(1): 43-48.
Sibley, C. H., Guerin, P. J. and Ringwald, P. (2010). "Monitoring antimalarial resistance: launching a cooperative effort." Trends Parasitol 26(5): 221-224.
Siddiqui, W. A., Schnell, J. V. and Geiman, Q. M. (1972). "A model in vitro system to test the susceptibility of human malarial parasites to antimalarial drugs." Am J Trop Med Hyg 21(4): 393-399.
Sidhu, A. B., Sun, Q., Nkrumah, L. J., Dunne, M. W., Sacchettini, J. C. and Fidock, D. A. (2007). "In vitro efficacy, resistance selection, and structural modeling studies implicate the malarial parasite apicoplast as the target of azithromycin." J Biol Chem 282(4): 2494-2504.
Sidhu, A. B., Uhlemann, A. C., Valderramos, S. G., Valderramos, J. C., Krishna, S. and Fidock, D. A. (2006). "Decreasing pfmdr1 copy number in Plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin." J Infect Dis 194(4): 528-535.
Sidhu, A. B., Valderramos, S. G. and Fidock, D. A. (2005). "pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum." Mol Microbiol 57(4): 913-926.

Bibliography
250
Sidhu, A. B., Verdier-Pinard, D. and Fidock, D. A. (2002). "Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations." Science 298(5591): 210-213.
Sidjanski, S. and Vanderberg, J. P. (1997). "Delayed migration of Plasmodium sporozoites from the mosquito bite site to the blood." Am J Trop Med Hyg 57(4): 426-429.
Sinclair, D., Donegan, S., Lalloo, D. G. (2011). "Artesunate versus quinine for treating severe malaria." Cochrane Database Syst Rev (3) CD005967.
Sinclair, D., Zani, B., Donegan, S., Olliaro, P. and Garner, P. (2009). "Artemisinin-based combination therapy for treating uncomplicated malaria." Cochrane Database Syst Rev(3): CD007483.
Singh, B., Kim Sung, L., Matusop, A., Radhakrishnan, A., Shamsul, S. S., Cox-Singh, J., Thomas, A. and Conway, D. J. (2004). "A large focus of naturally acquired Plasmodium knowlesi infections in human beings." Lancet 363(9414): 1017-1024.
Skinner-Adams, T. (1999). The effects of antimalarial drugs on the replication, metabolism and cytoadherence of Plasmodium falciparum in vitro. University Department of Medicine. Perth, University of Western Australia.
Skinner-Adams, T. and Davis, T. M. (1999). "Synergistic in vitro antimalarial activity of omeprazole and quinine." Antimicrob Agents Chemother 43(5): 1304-1306.
Skinner, T. S., Manning, L. S., Johnston, W. A. and Davis, T. M. (1996). "In vitro stage-specific sensitivity of Plasmodium falciparum to quinine and artemisinin drugs." Int J Parasitol 26(5): 519-25.
Slater, A. F. G. and Cerami, A. (1992). "Inhibition by chloroquine of a novel haem polymerase enzyme activity in malaria trophozoites." Nature 355: 167-169.
Snow, R. W., Guerra, C. A., Noor, A. M., Myint, H. Y. and Hay, S. I. (2005). "The global distribution of clinical episodes of Plasmodium falciparum malaria." Nature 434: 214-217.
Sokhna, C., Cisse, B., Ba el, H., Milligan, P., Hallett, R., Sutherland, C., Gaye, O., Boulanger, D., Simondon, K., Simondon, F., Targett, G., Lines, J., Greenwood, B. and Trape, J. F. (2008). "A trial of the efficacy, safety and impact on drug resistance of four drug regimens for seasonal intermittent preventive treatment for malaria in Senegalese children." PLoS ONE 3(1): e1471.
Song, J. L., Lyons, C. N., Holleman, S., Oliver, B. G. and White, T. C. (2003). "Antifungal activity of fluconazole in combination with lovastatin and their effects on gene expression in the ergosterol and prenylation pathways in Candida albicans." Med Mycol 41(5): 417-425.

Bibliography
251
Soran, H. and Durrington, P. (2008). "Rosuvastatin: efficacy, safety and clinical effectiveness." Expert Opin Pharmacother 9(12): 2145-2160.
Soukup, J., Scharff, K., Kubosch, K., Pohl, C., Bomplitz, M. and Kompardt, J. (2009). "State of the art: sedation concepts with volatile anesthetics in critically Ill patients." J Crit Care 24(4): 535-544.
Starzengruber, P., Kollaritsch, H., Sirichaisinthop, J., Wernsdorfer, G., Congpuong, K. and Wernsdorfer, W. H. (2008). "Interaction between lumefantrine and monodesbutyl-benflumetol in Plasmodium falciparum in vitro." Wien Klin Wochenschr 120(Suppl 4): 85-89.
Starzengruber, P., Wernsdorfer, G., Parizek, M., Rojanawatsirivej, C., Kollaritsch, H. and Wernsdorfer, W. H. (2007). "Specific pharmacokinetic interaction between lumefantrine and monodesbutyl-benflumetol in Plasmodium falciparum " Wien Klin Wochenschr 119(Suppl 3): 60-66.
Sullivan, A. D. and Meshnick, S. R. (1996). "Haemozoin: identification and quantification." Parasitol Today 12(4): 161-163.
Sultan, A. A., Thathy, V., Frevert, U., Robson, K. J., Crisanti, A., Nussenzweig, V., Nussenzweig, R. S. and Menard, R. (1997). "TRAP is necessary for gliding motility and infectivity of Plasmodium sporozoites." Cell 90(3): 511-522.
Syhre, M., Manning, L., Phuanukoonnon, S., Harino, P. and Chambers, S. T. (2009). "The scent of Mycobacterium tuberculosis--part II breath." Tuberculosis (Edinb) 89(4): 263-266.
Syhre, M., Scotter, J. M. and Chambers, S. T. (2008). "Investigation into the production of 2-Pentylfuran by Aspergillus fumigatus and other respiratory pathogens in vitro and human breath samples." Med Mycol 46(3): 209-215.
Talisuna, A. O., Bloland, P. and D'Alessandro, U. (2004). "History, dynamics, and public health importance of malaria parasite resistance." Clin Microbiol Rev 17(1): 235-254.
Talisuna, A. O., Nalunkuma-Kazibwe, A., Langi, P., Mutabingwa, T. K., Watkins, W. W., Van Marck, E., Egwang, T. G. and D'Alessandro, U. (2004). "Two mutations in dihydrofolate reductase combined with one in the dihydropteroate synthase gene predict sulphadoxine-pyrimethamine parasitological failure in Ugandan children with uncomplicated falciparum malaria." Infect Genet Evol 4(4): 321-327.
Taoufiq, Z., Pino, P., N'Dilimabaka, N., Arrouss, I., Assi, S., Soubrier, F., Rebollo, A. and Mazier, D. (2011). "Atorvastatin prevents Plasmodium falciparum cytoadherence and endothelial damage." Malar J 10(1): 52-60.

Bibliography
252
Tarning, J., Lindegardh, N., Annerberg, A., Singtoroj, T., Day, N. P., Ashton, M. and White, N. J. (2005). "Pitfalls in estimating piperaquine elimination." Antimicrob Agents Chemother 49(12): 5127-5128.
Taylor-Robinson, A. W. (1998). "Cultivation of Plasmodium falciparum in vitro using microbiological gas sachets that generate microaerophilic conditions." Trans R Soc Trop Med Hyg 92(3): 357-358.
Tchinda, V. H., Tadem, A. D., Tako, E. A., Tene, G., Fogako, J., Nyonglema, P., Sama, G., Zhou, A. and Leke, R. G. (2007). "Severe malaria in Cameroonian children: correlation between plasma levels of three soluble inducible adhesion molecules and TNF-alpha." Acta Trop 102(1): 20-8.
Teja-Isavadharm, P., Peggins, J. O., Brewer, T. G., White, N. J., Webster, H. K. and Kyle, D. E. (2004). "Plasmodium falciparum-based bioassay for measurement of artemisinin derivatives in plasma or serum." Antimicrob Agents Chemother 48(3): 954-960.
Teklehaimanot, H. D., Lipsitch, M., Teklehaimanot, A. and Schwartz, J. (2004). "Weather-based prediction of Plasmodium falciparum malaria in epidemic-prone regions of Ethiopia I. Patterns of lagged weather effects reflect biological mechanisms." Malar J 3: 41-51.
Thaithong, S., Beale, G. H. and Chutmongkonkul, M. (1983). "Susceptibility of Plasmodium falciparum to five drugs: an in vitro study of isolates mainly from Thailand." Trans R Soc Trop Med Hyg 77(2): 228-231.
Thavendiranathan, P., Bagai, A., Brookhart, M. A. and Choudhry, N. K. (2006). "Primary prevention of cardiovascular diseases with statin therapy: a meta-analysis of randomized controlled trials." Arch Intern Med 166(21): 2307-2313.
Thelu, J., Burnod, J., Bracchi, V. and Ambroise-Thomas, P. (1994). "Identification of differentially transcribed RNA and DNA helicase-related genes of Plasmodium falciparum." DNA Cell Biol 13(11): 1109-1115.
Thwing, J. I., Odero, C. O., Odhiambo, F. O., Otieno, K. O., Kariuki, S., Ord, R., Roper, C., McMorrow, M., Vulule, J., Slutsker, L., Newman, R. D., Hamel, M. J. and Desai, M. (2009). "In-vivo efficacy of amodiaquine-artesunate in children with uncomplicated Plasmodium falciparum malaria in western Kenya." Trop Med Int Health 14(3): 294-300.
Tinto, H., Guekoun, L., Zongo, I., Guiguemde, R. T., D'Alessandro, U. and Ouedraogo, J. B. (2008). "Chloroquine-resistance molecular markers (Pfcrt T76 and Pfmdr-1 Y86) and amodiaquine resistance in Burkina Faso." Trop Med Int Health 13(2): 238-240.
Torii, M., Nakamura, K., Sieber, K. P., Miller, L. H. and Aikawa, M. (1992). "Penetration of the mosquito (Aedes aegypti) midgut wall by the ookinetes of Plasmodium gallinaceum." J Protozool 39(4): 449-454.

Bibliography
253
Torre, D., Speranza, F., Giola, M., Matteelli, A., Tambini, R. and Biondi, G. (2002). "Role of Th1 and Th2 cytokines in immune response to uncomplicated Plasmodium falciparum malaria." Clin Diagn Lab Immunol 9(2): 348-351.
Traebert, M. and Dumotier, B. (2005). "Antimalarial drugs: QT prolongation and cardiac arrhythmias." Expert Opin Drug Saf 4(3): 421-431.
Traebert, M., Dumotier, B., Meister, L., Hoffmann, P., Dominguez-Estevez, M. and Suter, W. (2004). "Inhibition of hERG K+ currents by antimalarial drugs in stably transfected HEK293 cells." Eur J Pharmacol 484(1): 41-48.
Trager, W. (1978). "Cultivation of parasites in vitro." Am J Trop Med Hyg 27(2 Pt 1): 216-222.
Trager, W. and Jensen, J. B. (1976). "Human malaria parasites in continuous culture." Science 193(4254): 673-675.
Trampuz, A., Jereb, M., Muzlovic, I. and Prabhu, R. M. (2003). "Clinical review: Severe malaria." Crit Care 7(4): 315-323.
Trenholme, K. R., Kum, D. E., Raiko, A. K., Gibson, N., Narara, A. and Alpers, M. P. (1993). "Resistance of Plasmodium falciparum to amodiaquine in Papua New Guinea." Trans R Soc Trop Med Hyg 87(4): 464-466.
Trenholme, M. G. (1975). "Host failure in treatment of malaria with sulfalene and pyrimethamine." Annals of Internal Medicine 82: 219-223.
Triglia, T. and Cowman, A. F. (1994). "Primary structure and expression of dihydropteroate synthetase gene of Plasmodium falciparum." Proc Natl Acad Sci U S A 91: 7149-7153.
Triglia, T. and Cowman, A. F. (1999). "The mechanism of resistance to sulfa drugs in Plasmodium falciparum." Drug Resist. 2: 15-19.
Trotta, R. F., Brown, M. L., Terrell, J. C. and Geyer, J. A. (2004). "Defective DNA repair as a potential mechanism for the rapid development of drug resistance in Plasmodium falciparum." Biochemistry 43(17): 4885-4891.
Tu You-you, N. M.-y., Zhong Yu-rong, Li Lan-na, Cui Shu-lian, Zhang Mu-qun, Wang Xiu-zhen, Ji Zheng and Liang Xiao-tian (1982). "Studies on the constituents of Artemisia annua Part II*." Planta Medica 44: 143-145.
Turner, G. D., Morrison, H., Jones, M., Davis, T. M., Looareesuwan, S., Buley, I. D., Gatter, K. C., Newbold, C. I., Pukritayakamee, S., Nagachinta, B. and et al. (1994). "An immunohistochemical study of the pathology of fatal malaria. Evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration." Am J Pathol 145(5): 1057-69.

Bibliography
254
Turner, R. B. and Woodward, R. B. (1953). The chemistry of the cinchona. New York, Academic Press.
Uhlemann, A. C. (2005). "A single amino acid residue can determine sensitivity of SERCAs to artemisinins." Nat Struct Mol Biol 12: 628-629.
Urbina, J. A., K. Lazardi, E. Marchan, G. Visbal, T. Aguirre, M.M. Piras, R. Piras, R. A. Maldonado, G. Payares, and W. de Souza. (1993). "Mevinolin (lovastatin) potentiates the antiproliferative effects of ketoconazole and terbinafine against Trypanosoma (Schizotrypanum) cruzi: in vitro and in vivo studies." Antimicrob Agents Chemother 37: 580-591.
Ursos, L. M. and Roepe, P. D. (2002). "Chloroquine resistance in the malarial parasite, Plasmodium falciparum." Med Res Rev 22(5): 465-491.
van Agtmael, M. A., Van Der Graaf, C. A., Dien, T. K., Koopmans, R. P. and van Boxtel, C. J. (1998). "The contribution of the enzymes CYP2D6 and CYP2C19 in the demethylation of artemether in healthy subjects." Eur J Drug Metab Pharmacokinet 23(3): 429-436.
Veiga, M. I., Ferreira, P. E., Bjorkman, A. and Gil, J. P. (2006). "Multiplex PCR-RFLP methods for pfcrt, pfmdr1 and pfdhfr mutations in Plasmodium falciparum." Mol Cell Probes 20(2): 100-104.
Vial, H. J., Philippot, J. R. and Wallach, D. F. (1984). "A reevaluation of the status of cholesterol in erythrocytes infected by Plasmodium knowlesi and P. falciparum." Mol Biochem Parasitol 13(1): 53-65.
Vijaykadga, S., Rojanawatsirivej, C., Cholpol, S., Phoungmanee, D., Nakavej, A. and Wongsrichanalai, C. (2006). "In vivo sensitivity monitoring of mefloquine monotherapy and artesunate-mefloquine combinations for the treatment of uncomplicated falciparum malaria in Thailand in 2003." Trop Med Int Health 11(2): 211-219.
Wang, J. Y., Cao, W. C., Shan, C. Q., Zhang, M., Li, G. F., Ding, D. B., Shi, Y. L. and Wu, B. A. (2004). "Naphthoquine phosphate and its combination with artemisinine." Acta Trop 89: 375-381.
Wang, P., Read, M., Sims, P. F. and Hyde, J. E. (1997). "Sulfadoxine resistance in the human malaria parasite Plasmodium falciparum is determined by mutations in dihydropteroate synthetase and an additional factor associated with folate utilization." Mol Microbiol 23(5): 979-86.
Watkins, W. M., Mberu, E. K., Winstanley, P. A. and Plowe, C. V. (1997). "The efficacy of antifolate antimalarial combinations in Africa: a predictive model based on pharmacodynamic and pharmacokinetic analyses." Parasitol Today 13(12): 459-464.

Bibliography
255
Weitz, Z. W., Birnbaum, A. J., Sobotka, P. A., Zarling, E. J. and Skosey, J. L. (1991). "High breath pentane concentrations during acute myocardial infarction." Lancet 337(8747): 933-935.
Wernsdorfer, W., H. (1994). "Epidemiology of drug resistance in malaria." Acta Trop 56: 143-156.
Wernsdorfer, W. H. and McGregor, I. A., Eds. (1988). Malaria: principles and practice of malariology. Drug sensitivity tests in malaria parasites. Edinburgh, Scotland., Churchchill Livingstone.
Wernsdorfer, W. H. and Payne, D. (1991). "The dynamics of drug resistance in Plasmodium falciparum." Pharmacol Ther 50(1): 95-121.
White, J. H., Kilbey, B. J., de Vries, E., Goman, M., Alano, P., Cheesman, S., McAleese, S. and Ridley, R. G. (1993). "The gene encoding DNA polymerase alpha from Plasmodium falciparum." Nucleic Acids Res 21(16): 3643-3646.
White, N. J. (1998). "Preventing antimalarial drug resistance through combinations." Drug Resist Updat 1(1): 3-9.
White, N. J. (2004). "Antimalarial drug resistance." J Clin Invest 113(8): 1085-1092.
White, N. J., Pongtavornpinyo, W., Maude, R. J., Saralamba, S., Aguas, R., Stepniewska, K., Lee, S. J., Dondorp, A. M., White, L. J. and Day, N. P. (2009). "Hyperparasitaemia and low dosing are an important source of anti-malarial drug resistance." Malar J 8: 253-270.
White, N. J., van Vugt, M. and Ezzet, F. (1999). "Clinical pharmacokinetics and pharmacodynamics and pharmacodynamics of artemether-lumefantrine." Clin Pharmacokinet 37(2): 105-125.
WHO (1978). Report of workshop on drug-resistant malaria, ICP/MPD/006. Manila, World Health Organization.
WHO (2001). In vitro micro-test (Mark III) for the assessment of the response of Plasmodium falciparum to chloroquine, mefloquine, quinine, amodiaquine, sulfadoxine/pyrimethamine and artemisinin. Instructions for use of the in vitro micro-test kit (Mark III), World Health Organisation Division of Control of Tropical Diseases.
WHO (2003). Assessment and monitoring of antimalarial drug efficacy for the treatment of uncomplicated falciparum malaria, World Health Organization, Geneva.
WHO (2004). Malaria cases (per 100,000) by country, latest available data, World Health Organization/Malaria Department Map Production.

Bibliography
256
WHO (2005). Susceptibility of Plasmodium falciparum to antimalarial drugs : report on global monitoring : 1996-2004. P. Ringwald. Geneva, Roll Back Malaria Department of the World Health Organization.
WHO (2006). Guidelines for the treatment of malaria. Geneva, World Health Organization.
WHO (2007). Global AMDP Database - WPRO., World Health Organization.
WHO (2008). World malaria report 2008. M. Aregawi, R. Cibulskis, M. Otten, R. Williams and C. Dye, World Health Organization.
WHO (2009). World Health Organization Global Malaria Programme. Malaria case management operations manual Geneva, World Health Organization.
WHO (2009). Western Pacific Region: Statistical Charts 2009: Annex Charts, World Health Organization.
WHO (2009). Malaria, Fact sheet No 94. Available at: http://www.who.int/mediacentre/factsheets/en/, World Health Organisation.
Wiesner, J., Reichenberg, A., Heinrich, S., Schlitzer, M. and Jomaa, H. (2008). "The plastid-like organelle of apicomplexan parasites as drug target." Curr Pharm Des 14(9): 855-871.
Wilson, R. J., Denny, P. W., Preiser, P. R., Rangachari, K., Roberts, K., Roy, A., Whyte, A., Strath, M., Moore, D. J., Moore, P. W. and Williamson, D. H. (1996). "Complete gene map of the plastid-like DNA of the malaria parasite Plasmodium falciparum." J Mol Biol 261(2): 155-172.
Wilt, T. J., Bloomfield, H. E., MacDonald, R., Nelson, D., Rutks, I., Ho, M., Larsen, G., McCall, A., Pineros, S. and Sales, A. (2004). "Effectiveness of statin therapy in adults with coronary heart disease." Arch Intern Med 164(13): 1427-1436.
Winstanley, P. (2001). "Modern chemotherapeutic options for malaria." Lancet Infect Dis 1: 1-19.
Witkowski, B., Lelievre, J., Lopez Barragan, M. J., Laurent, V., Su, X. Z., Berry, A. and Benoit-Vical, F. (2010). "Increased tolerance to artemisinin in Plasmodium falciparum is mediated by a quiescence mechanism." Antimicrob Agents Chemother 54(5): 1872-1877.
Wong, R. and Davis, T. M. (2009). "Statins as potential antimalarial drugs: low relative potency and lack of synergy with conventional antimalarial drugs." Antimicrob Agents Chemother 53(5): 2212-2214.
Wong, R. P., Karunajeewa, H., Mueller, I., Siba, P., Zimmerman, P. A. and Davis, T. M. (2011). "Molecular assessment of Plasmodium falciparum resistance to antimalarial drugs in Papua New Guinea using an extended ligase detection

Bibliography
257
reaction fluorescent microsphere assay." Antimicrob Agents Chemother 55(2): 798-805.
Wong, R. P., Lautu, D., Tavul, L., Hackett, S. L., Siba, P., Karunajeewa, H. A., Ilett, K. F., Mueller, I. and Davis, T. M. (2010). "In vitro sensitivity of Plasmodium falciparum to conventional and novel antimalarial drugs in Papua New Guinea." Trop Med Int Health 15(2): 342-349.
Wongsrichanalai, C. and Meshnick, S. R. (2008). "Declining artesunate-mefloquine efficacy against falciparum malaria on the Cambodia-Thailand border." Emerg Infect Dis 14(5): 716-719.
Wongsrichanalai, C., Pickard, A. L., Wernsdorfer, W. H. and Meshnick, S. R. (2002). "Epidemiology of drug-resistant malaria." Lancet Infect Dis 2: 209-218.
Wongsrichanalai, C., Webster, H. K., Wimonwattrawatee, T., Sookto, P., Chuanak, N., Thimasarn, K. and Wernsdorfer, W. H. (1992a). "Emergence of multidrug-resistant Plasmodium falciparum in Thailand: in vitro tracking." Am J Trop Med Hyg 47(1): 112-116.
Wongsrichanalai, C., Webster, H. K., Wimonwattrawatee, T., Sookto, P., Chuanak, N., Timasarn, K. and Wernsdorfer, W. H. (1992b). "In vitro sensitivity of Plasmodium falciparum isolates in Thailand to quinine and chloroquine, 1984-1990." Southeast Asian J Trop Med Public Health 23(3): 533-536.
Wu, X., Whitfield, L. R. and Stewart, B. H. (2000). "Atorvastatin transport in the Caco-2 cell model: contributions of P-glycoprotein and the proton-monocarboxylic acid co-transporter." Pharm Res 17(2): 209-215.
Wu, Y. (2002a). "How might qinghaosu (artemisinin) and related compounds kill the intraerythrocytic malaria parasite? A chemist's view." Acc Chem Res 35(5): 255-259.
Wu, Y. S., Li, M., Lu, J. H., Bi, H. X. and Li, Y. J. (2002b). "Plasmodium lactate dehydrogenase activity assay and its preliminary application in therapeutic efficacy monitoring of drugs." Di Yi Jun Yi Da Xue Xue Bao 22(1): 25-27.
Wunderlich, F., Fiebig, S., Vial, H. and Kleinig, H. (1991). "Distinct lipid compositions of parasite and host cell plasma membranes from Plasmodium chabaudi-infected erythrocytes." Mol Biochem Parasitol 44(2): 271-277.
Xu, J., Chavis, J. A., Racke, M. K. and Drew, P. D. (2006). "Peroxisome proliferator-activated receptor-alpha and retinoid X receptor agonists inhibit inflammatory responses of astrocytes." J Neuroimmunol 176(1-2): 95-105.
Yamauchi, L. M., Coppi, A., Snounou, G., Sinnis, P. (2007). "Plasmodium sporozoites trickle out of the injection site." Cell Microbiol 9(5): 1215-1222.

Bibliography
258
Yamauchi, T. and Umezaki, M. (2005). "Rural-urban migration and changing physical activity among Papua New Guinea highlanders from the perspective of energy expenditure and time use." Environ Sci 12(3): 155-166.
Yamauchi, T., Umezaki, M. and Ohtsuka, R. (2001). "Physical activity and subsistence pattern of the Huli, a Papua New Guinea Highland population." Am J Phys Anthropol 114(3): 258-268.
Yang, H., Gao, B. and Huang, K. (1999a). "Comparison of naphthoquine, metronidazole and norfloxacine to artesunate sensitive and resistant P. falciparum strains." Practical Parasitic Diseases 2(7).
Yang, H., Liu, D., Huang, K., Yang, Y., Yang, P., Liao, M. and Zhang, C. (1999b). "Assay of sensitivity of Plasmodium falciparum to chloroquine, amodiaquine, piperaquine, mefloquine and quinine in Yunnan province." Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi 17(1): 43-45.
Yang, Z., Zhang, Z., Sun, X., Wan, W., Cui, L., Zhang, X., Zhong, D., Yan, G. and Cui, L. (2007). "Molecular analysis of chloroquine resistance in Plasmodium falciparum in Yunnan Province, China." Trop Med Int Health 12(9): 1051-1060.
Yeo, A. E. and Rieckmann, K. H. (1995). "Increased antimalarial activity of azithromycin during prolonged exposure of Plasmodium falciparum in vitro." Int J Parasitol 25(4): 531-532.
Yung, A. P. and Bennett, N. M. (1976). "Chloroquine-resistant falciparum malaria acquired in Papua New Guinea." Med J Aust 2(22): 845.
Zerpa, N., Pabon, R., Wide, A., Gavidia, M., Medina, M., Caceres, J. L., Capaldo, J., Baker, M. and Noya, O. (2008). "Evaluation of the OptiMAL test for diagnosis of malaria in Venezuela." Invest Clin 49(1): 93-101.
Zhang, J.-f. (2007). The late report, the 523rd item, and the research record of artemisinin (article in Chinese), Yangcheng Evening News Publisher.
Zhao, S. P., Ye, H. J., Zhou, H. N., Nie, S. and Li, Q. Z. (2003). "Gemfibrozil reduces release of tumor necrosis factor-alpha in peripheral blood mononuclear cells from healthy subjects and patients with coronary heart disease." Clin Chim Acta 332(1-2): 61-67.
Zhu, T., Ansquer, J. C., Kelly, M. T., Sleep, D. J. and Pradhan, R. S. (2010). "Comparison of the gastrointestinal absorption and bioavailability of fenofibrate and fenofibric acid in humans." J Clin Pharmacol 50(8): 914-921.

APPENDICES


Appendix A: Isolate Information
261
APPENDIX A. ISOLATE INFORMATION
P. falciparum Isolate
Source Drug Sensitivity Comments
3D7 Netherlands CQ-sensitive Derived from NF54 isolated in Amsterdam; presumed of African origin, not confirmed
7G8 Brazil CQ-resistant Pyrimethamine-resistant
Derived from IMTM22 (Brazil)
Dd2 Indochina CQ-resistant Pyrimethamine-resistant
MQ-resistant
Derived from W2mef
E8B Australia CQ-resistant MQ-sensitive
Derived from ItG2F6 (Brazil)
HB3 Honduras CQ-sensitive Derived from I/CDC (Honduras)
K1 Thailand CQ-resistant Pyrimethamine-resistant
Kanchanaburi
PNG1905 Australia CQ-resistant Origin not confirmed
PNG1917 Australia CQ-resistant Papua New Guinea isolate
W2mef Indochina CQ-resistant Selected from W2 for resistance to mefloquine


Appendix B: Recipes and Solutions
263
APPENDIX B. RECIPES FOR SOLUTIONS
Culture of P. falciparum
5% Albumax II
� Albumax II (11021-045) (Gibco) 5.5 g
� Milli-Q water 110 mL
Filter sterilise using 0.2 µM membrane (Nalgene Filterware, USA).
Store at -20°C in 10 mL aliquots.
Complete Maintenance Medium (PNG)
� RPMI Medium 1640 powder (31800-089) (Gibco, Auckland, NZ) 5.735 g
� HEPES (Sigma-Aldrich) 4.47 g
� Hypoxanthine (Sigma) 22.5 mg
� 5% Albumax II (see section below) 50 mL
� 5% NaHCO3 (see section below) 21 mL
� Gentamycin sulfate (10mg/mL) (Calbiochem, Darmstadt, Germany) 5 mL
� Neomycin sulfate (10mg/mL) (Calbiochem, Darmstadt, Germany) 5 mL
� Milli-Q-water 500 mL (final)
Adjust to pH 7.3.
Filter sterilise using 0.2 µM membrane (Nalgene Filterware, USA).
Store at 4°C and use within 2 weeks, or supplement with L-glutamine after 2 weeks.

Appendix B: Recipes and Solutions
264
Complete Maintenance Medium
� RPMI Medium 1640 (R5886) (Gibco, liquid) 90 mL
� Human plasma (see section below) 10 mL
� L-glutamine solution (see section below) 1 mL
� Hypoxanthine solution (see section below) 1 mL
� Gentamycin (see section below) 100 µL
Store at 4°C and use within 2 weeks, or supplement with L-glutamine after 2 weeks.
Cryoprotective Solution
� Sorbitol (BDH, England) 37.8 g
� NaCl (BDH, Australia) 8.1 g
� Glycerol (BDH, Australia) 350 mL
� Milli-Q-water 900 mL (final)
Filter sterilise using 0.2 µM membrane (Nalgene Filterware, USA).
Store at 4°C.
Gentamycin Stock (50 mg/mL) (PNG)
� Gentamycin sulfate (Calbiochem) 1 g
� Milli-Q-water 20 mL
Protect from light, store at -20°C in 5 mL aliquots
Gentamycin Stock (40 mg/mL)
80 mg/2 mL (Delta West, Pharmacy IV injections).
Store at 4°C.

Appendix B: Recipes and Solutions
265
5% Giemsa Stain
� Giemsa stain (BDH, Australia) 0.5 mL
� PBS (pH 7.2) 9 mL
Stain is prepared fresh before use.
Human Plasma Inactivation
O+, A+, AB+ Plasma units (Fremantle Hospital, Transfusion Medicine) were pooled, defibrined and heat inactivated at 56°C in a waterbath as follows:
1. Plasma was added to sterile 500 mL conical flasks containing 1 - 2 cm of autoclaved (2 mm) glass beads, covered and shaken in a waterbath for 2 - 3 hr at 37°C.
2. Pool plasma and heat-inactivate at 56°C in waterbath for 40 min.
3. Use 10 mL of new plasma to make up test CM (100 mL) and test for support of 3D7 strain of P. falciparum cultures for 2 weeks prior use.
4. Store at -20°C.
Human Tonicity Phosphate Buffered Saline (HTPBS)
� NaCl (BDH, Australia) 7 g
� Na2HPO4 (BDH, Australia) 2.85 g
� NaH2PO4 (BDH, Australia) 0.625 g
� Milli-Q-water 1000 mL
Adjust to pH 7.3.
Filter sterilise using 0.2 µM membrane (Nalgene Filterware, USA).
Store at 4°C.

Appendix B: Recipes and Solutions
266
Hypoxanthine Solution (5 mg/mL)
� Hypoxanthine (Sigma, USA) 0.5 g
� Milli-Q-water 100 mL
Heat in microwave until dissolved.
Filter sterilise using 0.2 µM membrane (Nalgene Filterware, USA).
Store at 4°C.
L-Glutamine Solution (200mM)
� L-glutamine (Sigma) 2.92 g
� Milli-Q-water 100 mL
Filter sterilise using 0.2 µM membrane (Nalgene Filterware, USA).
Store at -20°C in 5 mL aliquots.
5% NaHCO3
� NaHCO3 (Sigma-Aldrich, St Louis, Mo, USA) 2.5 g
� Milli-Q-water 50 mL
Filter sterilise using 0.2 µM membrane (Nalgene Filterware, USA).
Store at -20°C in 10 mL aliquots.
PBS (6.7mM) for Giemsa Staining
� K2PO4 (BDH, Australia) 0.41 g
� Na2HPO4 (BDH, Australia) 0.65 g
� Milli-Q-water 1000 mL
Adjust to pH 7.1.
Filter sterilise using 0.2 µM membrane (Nalgene Filterware, USA) (optional).

Appendix B: Recipes and Solutions
267
12% Sodium Chloride (NaCl) Solution
� NaCl (BDH, Australia) 11.3 g
� HTPBS (see section above) 100 mL
Filter sterilise using 0.2 µM membrane (Nalgene Filterware, USA).
Store at 4°C.
1.6% Sodium Chloride (NaCl) Solution
� NaCl (BDH, Australia) 0.9 g
� HTPBS (see section above) 100 mL
Filter sterilise using 0.2 µM membrane (Nalgene Filterware, USA).
Store at 4°C.
0.9% Sodium Chloride (NaCl) Solution
� NaCl (BDH, Australia) 0.2 g
� Glucose (Sigma) 0.2 g
� HTPBS (see section above) 100 mL
Filter sterilise using 0.2 µM membrane (Nalgene Filterware, USA).
Store at 4°C.
5% Sorbitol for Synchronisation
� Sorbitol (BDH, England) 5 g
� Milli-Q-water 100 mL (final)
Filter sterilise using 0.2 µM membrane (Nalgene Filterware, USA).
Store at 4°C.

Appendix B: Recipes and Solutions
268
LDH Assay
Malstat Solution
� Trizma base (Sigma, Australia) 2.42 g
� Milli-Q-water 180 mL
� Adjust with HCl to pH 9.1
� Triton X-100 (Sigma, Australia) 0.4 mL
� Lithium-L-lactate (Sigma, Australia) 4 g
� APAD (Sigma, Australia) 132 mg
Protect from light and store at 4°C.
Use within one week.
Diaphorase Solution
� Diaphorase (Sigma-Aldrich, USA) 15 mg
� Milli-Q-water 15 mL
Protect from light and store at 4°C.
Nitro Blue Tetrazolium (NBT) Solution
� NBT chloride monohydrate (Sigma-Aldrich, USA) 15 mg
� Milli-Q-water 15 mL
Protect from light and store at 4°C.

Appendix B: Recipes and Solutions
269
Molecular Assays
2% Agarose Gel
� Agarose I ™ (Amresco, USA) 3 g
� 1 X TBE buffer 150 mL
Melt agarose in buffer before pouring into a large 96-well electrophoresis tray and allow 20 min for gel to set.
2.5mM dNTPs Working Solution
� 100 mM dGTP (Denville Scientific Inc, USA) 25 µL
� 100 mM dATP (Denville Scientific Inc, USA) 25 µL
� 100 mM dCTP (Denville Scientific Inc, USA) 25 µL
� 100 mM dTTP (Denville Scientific Inc, USA) 25 µL
� Nuclease free water 900 µL
1.5 X TMAC Hybrisation Buffer
� 5 M Tetramethyl ammonium chloride (TMAC) (Sigma, USA) 30 mL
� 1 M Tris pH 8.0 (Amresco, USA) 2.5 mL
� 0.5 M EDTA (Amresco, USA) 300 µL
� 20% Sodium dodecyl sulfate (Amresco, USA) 250 µL
� Sterile distilled water 16.95 mL

Appendix B: Recipes and Solutions
270
10 x Tris borate (TBE) Buffer
� Tris base 108 g
� Boric acid 55 g
� 0.5M EDTA 40 mL
� Milli-Q-water 1000 mL (final)
10 x PCR Buffer
� 1 M Tris pH 8.8 33.5 mL
� 1 M MgSO4 3.4 mL
� 1 M (NH4)2SO4 8.4 mL
� β-mercaptoethanol (14.3 M) 0.35 mL
� Nuclease-free water 4.4 mL
LDR Master Mix – Plasmodium Species
� Nuclease-free water 11.4 µL
� NEBuffer for Taq DNA (10X) (Biolabs, New England) 1.5 µL
� LDR primers for species assay (x 7) (Table 2.3) 0.15 µL/primer
� Taq DNA ligase (Biolabs, New England) 0.05 µL
� PCR product 1 µL

Appendix B: Recipes and Solutions
271
LDR Master Mix – Drug Resistance SNPs of pfcrt, pfdhfr, pfdhps genes
� Nuclease-free water 7.2 µL
� NEBuffer for Taq DNA (10X) (Biolabs, New England) 1.5 µL
� LDR primers for pfcrt, pfdhfr, pfdhps (x 18) (Table 2.4) 0.20 µL/primer
� Taq DNA ligase (Biolabs,New England) 0.10µL
� PCR product 1 µL
PCR Master Mix – Plasmodium Species, pfcrt, pfdhfr, pfdhps
� Nuclease-free water 19.6 µL
� 10 x PCR buffer 2.5 µL
� 2.5 mM dNTPs 2.0 µL
� Primers –upstream (Section 2.3.2) 0.3 µL/primer
� Primers – downstream (Section 2.3.2) 0.3 µL/primer
� MacTaq (DNA Taq polymerase, M&C Gene Technology, Beijing) 0.3 µL
Keep MacTaq on ice at all times.
SYBR Gold
� Cybergold (Molecular Probes, Eugene, OR) 15 µL
� 1 X TBE Buffer (see section above) 150 mL

Appendix C: LDR Optimisation
272
APPENDIX C. EFFECTS OF LDR ANNEALING TEMPERATURE AND DILUTION

Appendix C: LDR Optimisation
273

Appendix C: LDR Optimisation
274

Appendix C: LDR Optimisation
275

Appendix C: LDR Optimisation
276

Appendix D: Isobologram Analyasis
277
APPENDIX D. ISOBOLOGRAM ANALYSIS
The regression function used by Brueckner et al. is given below:
For determining the relative interaction value, I, this function is suitable when the FIC
values lie in the range [0, 1]. However, when values lie outside this range the fit is quite
poor. This is evident even from a simple visual comparison between the data points and
the regression curve of Figure (left) (denoted by blue circles and red line respectively).
Consequently, only marginal confidence may be attributed to the recovered I value of -
0.305. One straightforward way to improve the regression fit is by first normalising the
FIC values into the range [0, 1]. However, consideration must be given to the nature of
this scaling since it may introduce a bias into the recovered I value.
There are two approaches to applying the normalisation: either scale the data
independently along each axis direction (centre figure), or apply a single scale factor
globally (right figure). The former is done by finding the maximum FIC in each
direction and scaling the corresponding values by that maximum. The latter method
uses the global maximum over all the FICs regardless of axis and scaling all the values
by this factor. In both cases, the resultant values will lie in the unit square hence
suitable for regression using the Brueckner et al. fitting function.
Results of these normalisation methods and their subsequent regression curves are
shown in Figure-centre and right respectively. The original values are denoted as green
points and scaled values as blue circles. Notice that the resultant I values differ slightly
also, but this difference is much less than the difference with the I value found in Figure
(Left).

Appendix D: Isobologram Analyasis
278
Regarding which two approaches is more appropriate, the non-linear regression
function proposed by Brueckner et al should be considered. First of all, its curve has a
line of symmetry along X = Y. Therefore, if values are scaled by different factors in
different directions, then this might destroy any inherent symmetry in the data and
hence reduce the regression fit. This suggests that the second method of scaling all the
FICs by a single global factor is more appropriate. However, a second feature of
Brueckner’s fitting function is that it assumes isobologram start and end points at (0, 1)
and (1, 0) respectively. This is problematic for a global normalisation approach since it
might result in curves terminating before they reach these assumed end points.
In the examples shown in Figures (centre) and (right), the difference in the fitted curves
is negligible as indicated by the proximity of their respective I values. Furthermore, the
regression line still does not appear to capture the data-points lying in the centre of the
plot, away from each of the axes. This is explained by the clustering of data points at
the assumed endpoints of (1,0) and (0,1), that is, eight of the eleven data points lie near
the horizontal and vertical axes with only three in the centre of the plot. Since the least-
squares fitting method used in the Brueckner et al. regression gives equal weight to
every data-point then so long as the fitted curve terminates near (1,0) and (0,1) then the
fit is considered optimal. This suggests that to use Brueckner et al. fitting, it is
important to have sanitised data near the end points which is best offered by scaling the
data independent along each axis.

Appendix D: Isobologram Analysis
279
Figures (left): Isobologram using the raw FIC data Raw FICs, I = -0.305, (Centre) Isobologram using FICs which have been normalised along each
axis independently Scaled FICs (anisotropic), I = -1.088, (Right) Isobologram using FIC data normalised globally, Normalised FICs (isotropic), I = -
0.978.

280
APPENDIX E. VOCS ANAYLSIS
Compounds Common to both P. falciparum Culture and Non-parasitised Control SPME (Triple Fibre)
2,4-Dimethyl-1-heptene p-Trimethylsilyloxyphenyl-(trimethylsilyloxy)trimethylsilyacrylate*
Cyclotetrasiloxane, octamethyl-* Dodecane, 2,6,11-trimethyl-
Heptane, 2,2,4,6,6-pentamethyl- 1-Dodecanol, 3,7,11-trimethyl-
Nonane, 4-methyl- 3-Carene
Octane, 3,3-dimethyl- Furan, 2-pentyl-
Decane Cyclohexanone
Undecane, 3,6-dimethyl- Cyclohexanol
Decane, 3,7-dimethyl- Benzene, 1,3-bis(1,1-dimethylethyl)-
Dodecane 1-Octen-3-ol
1-Octene, 3,7-dimethyl- 1-Hexanol, 2-ethyl-
Decane, 5,6-dipropyl
Notes: Compounds have been identified by comparison of mass spectra with the NIST 2005 mass spectral database. Compounds in italic have been
tentatively identified (<90% match) and may indicate the structural class rather than the actual compound. *Siloxanes possibly derived from coating of
the SPME fibre or the GC column stationary phase (Middleton 1989).

281
Compounds Common to both P. falciparum Culture and Non-parasitised Control SPME (PDMS) Thermal Desorption (Purge and Trap) -
Dodecane Cyclohexanone
Benzene, (1-methylethenyl)- Benzene, 1-ethyl-3-methyl-
Cyclopentasiloxane, decamethyl-* Benzene, 1,2,3-trimethyl-
1-Heptanol, 2,4-diethyl- Decane
Benzene, 1,3-bis(1,1-dimethylethyl)- 1-Hexanol, 2-ethyl-
1-Hexanol, 2-ethyl- Silane, 9H-fluoren-9-yltrimethyl-*
Cyclohexasiloxane, dodecamethyl-* Cyclohexane, 1,2,3-trimethyl- (1.alpha., 2.alpha., 3.beta.)
Benzaldehyde, 2,5-bis[(trimethylsilyl)oxy]- 1-Hexene, 2,5,5-trimethyl-
Silanediol, dimethyl-* 2-Undecene, 6-methyl- (Z)-
Oxime-methoxy-phenyl- Cyclohexane, 1,2-dimethyl- cis-
1-Dodecanol Benzene, 1,3-bis(1,1-dimethylethyl)-
Diethyl Phthalate** Malonic acid, bis(2-trimethylsilylethyl ester)-
Notes: Compounds have been identified by comparison of mass spectra with the NIST 2005 mass spectral database. Compounds in italic have been
tentatively identified (<90% match) and may indicate the structural class rather than the actual compound. *Siloxanes possibly derived from coating of
the SPME fibre or the GC column stationary phase. **Known contaminants (Middleton 1989).

282
Compounds Common to both P. falciparum Culture and Non-parasitised Control Supernatant Extraction by SPME immersion Supernatant Extraction by Methanol and Sep-Pak
5-O-Methyl-d-gluconic acid dimethylamide Methane, (methylsulfinyl)(methylthio)-
Styrene Dodecane
Cyclohexanone N-Benzyl-2-aminocinnamate, methyl ester
Nonanal Hexadecanoic acid, methyl ester
Benzene, 1,3-bis(1,1-dimethylethyl)- 5-Thiazoleethanol, 4-methyl-
Acetic acid Silane, (2-methoxyethyl)trimethyl-*
1-Hexanol, 2-ethyl- 9-Octadecenoic acid (Z)-, methyl ester
Silanediol, dimethyl-* Octadecanoic acid, butyl ester
Oxime-methoxy-phenyl- n-Hexadecanoic acid
Propanoic acid, 2-methyl-2,2-dimethyl-1-(2-hydroxy-1-methylethyl) propyl ester Androst-2-en-17-one, (5.alpha.)-
Benzoic acid, 2,5-bis(trimethylsiloxy)-, trimethylsilyl ester Benzenesulfonamide, N-butyl-
1-Dodecanol 1H-Purine-2,6-dione, 3,7-dihydro-1,3,7-trimethyl-
Nonanoic acid Prasterone-3-sulfate
4H-Pyran-4-one, 2,3-dihydro-3,5-dihydroxy-6-methyl- 1,2-Benzenedicarboxylic acid, diisooctyl ester**
Octasiloxane, 1,1,3,3,5,5,7,7,9,9,11,11,13,13,15,15-hexadecamethyl-* Disiloxane, 1,1,3,3-tetramethyl-1,3-dioctadecyl-
Diethyl Phthalate** Cholest-5-en-3-ol (3.beta)-
Notes: Compounds have been identified by comparison of mass spectra with the NIST 2005 mass spectral database. Compounds in italic have been
tentatively identified (<90% match) and may indicate the structural class rather than the actual compound. *Siloxanes possibly derived from coating of
the SPME fibre or the GC column stationary phase. **Known contaminants (Middleton 1989).

283
Compounds Common to both P. falciparum Culture and Non-parasitised Control Dichloromethane (Supernatant) Petrol (Supernatant ) Petrol (Cell Lysate)
Cyclohexanol 4-Heptanol Cholesterol
Hexadecane 3-Heptanol 4-Heptanol
Oxalic acid, butyl 2-ethylhexyl ester 2-Heptanol 3-Heptanol
Octadecane 3,6-Heptanedione 2-Heptanol
Heneicosane Di-sec-butyl ether Nonanal
1-Dodecanol Tetrahydro-1,3-oxazine-2-thione 3,6-Heptanedione
Benzoic acid, 4-ethoxy-, ethyl ester Carbamodithioic acid, diethyl- methyl ester 1-Butoxy-2-ethylhexane
Nonanoic acid Disulfiram Octadecane
1-Tetradecanol 1-Hexadecanol 2,5-Dihydroxyheptane
1-Hexadecanol Behenyl chloride
Tridecanal 7-Oxabicyclo[4.1.0]heptane, 1-methyl-4-(2-methyloxiranyl)-
Pentadecanoic acid, methyl ester
1-Octadecanol Heneicosane
Diethyl Phthalate**
1-Hexadecanol
Tricosane
Notes: Compounds have been identified by comparison of mass spectra with the NIST 2005 mass spectral database. Compounds in italic have been
tentatively identified (<90% match) and may indicate the structural class rather than the actual compound. *Siloxanes possibly derived from coating of
the SPME fibre or the GC column stationary phase. **Known contaminants (Middleton 1989).