in vivo crispr screening unveils histone …in vivo crispr screening unveils histone demethylase utx...
TRANSCRIPT

In vivo CRISPR screening unveils histone demethylaseUTX as an important epigenetic regulator inlung tumorigenesisQibiao Wua,1, Yahui Tianb,1, Jian Zhanga,1, Xinyuan Tonga,1, Hsinyi Huanga, Shuai Lic, Hong Zhaob, Ying Tanga,Chongze Yuand,e, Kun Wangf, Zhaoyuan Fanga, Lei Gaob, Xin Hug,h, Fuming Lia, Zhen Qina, Shun Yaoa, Ting Chenc,Haiquan Chend,e, Gong Zhangb, Wanting Liub, Yihua Sund,e, Luonan Chena, Kwok-Kin Wongc, Kai Gei, Liang Chenb,2,and Hongbin Jia,j,2
aState Key Laboratory of Cell Biology, Innovation Center for Cell Signaling Network, CAS Center for Excellence in Molecular Cell Science, Shanghai Instituteof Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, Shanghai 200031, China; bInstitute of Life andHealth Engineering, Jinan University, Guangzhou, Guangdong 510632, China; cLaura & Isaac Perlmutter Cancer Center, NYU Langone Medical Center, NewYork, NY 10016; dDepartment of Thoracic Surgery, Fudan University Shanghai Cancer Center, Shanghai 200031, China; eDepartment of Oncology, ShanghaiMedical College, Fudan University, Shanghai 200031, China; fKey Laboratory of Molecular Imaging, Institute of Automation, Chinese Academy of Sciences,Beijing 100190, China; gThe University of Texas Graduate School in Biomedical Sciences, Houston, TX 77030; hDepartment of Genomic Medicine, TheUniversity of Texas MD Anderson Cancer Center, Houston, TX 77030; iLaboratory of Endocrinology and Receptor Biology, National Institute of Diabetes andDigestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892; and jSchool of Life Science and Technology, ShanghaiTech University,Shanghai 200120, China
Edited by Ronald A. DePinho, The University of Texas MD Anderson Cancer Center, Houston, TX, and approved March 19, 2018 (received for reviewSeptember 20, 2017)
Lung cancer is the leading cause of cancer-related death world-wide. Inactivation of tumor suppressor genes (TSGs) promoteslung cancer malignant progression. Here, we take advantage of theclustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-mediated somatic gene knockout in a KrasG12D/+ mouse modelto identify bona fide TSGs. From individual knockout of 55 potentialTSGs, we identify five genes, including Utx, Ptip, Acp5, Acacb, andClu, whose knockout significantly promotes lung tumorigenesis.These candidate genes are frequently down-regulated in humanlung cancer specimens and significantly associated with survival inpatients with lung cancer. Through crossing the conditional Utxknockout allele to the KrasG12D/+ mouse model, we further find thatUtx deletion dramatically promotes lung cancer progression. Thetumor-promotive effect of Utx knockout in vivo is mainly mediatedthrough an increase of the EZH2 level, which up-regulates theH3K27me3 level. Moreover, the Utx-knockout lung tumors are pref-erentially sensitive to EZH2 inhibitor treatment. Collectively, ourstudy provides a systematic screening of TSGs in vivo and identifiesUTX as an important epigenetic regulator in lung tumorigenesis.
tumor suppressor genes | CRISPR/Cas9 in vivo knockout | non-small cell lungcancer | UTX | EZH2 inhibitor
Lung cancer is one of the deadliest diseases worldwide, withvery high incidence and mortality (1). Tumor suppressor
genes (TSGs) play important roles in lung cancer initiation,progression, and even metastasis. Cancer genomic studies haveprovided a comprehensive spectrum of thousands of potentiallyimportant genetic alterations of TSGs (2, 3). Except for a fewwell-studied TSGs like RB1, TP53, STK11, and PTEN, most ofthese genetic aberrations still remain to be functionally validatedand characterized. To achieve this, the major existing challengeis to define bona fide TSGs, especially using in vivo systems.Genetically engineered mouse models (GEMMs) are often ap-
plied to validate those potentially important molecular alterationsin vivo. Systematic screening of TSGs using GEMMs seems im-practical due to the highly expensive and time-consuming process.This demands a more efficient and less costly technique. Recently,the clustered regularly interspaced short palindromic repeats(CRISPR)/Cas9 system has been proven to be a powerful genome-editing tool, making it possible for systematic TSG screening (4, 5).Indeed, recent studies have established highly efficient knockout ofsomatic genes in mouse cancer models (6, 7). The Cas9 nucleasecan be directed to a specific gene locus by single-guide RNA (sgRNA)
via 20 bases matched to targeted genomic loci and can create doublestrand breaks, which are then repaired by nonhomologous end-joining(8). Using this technique, insertions or deletions (in-dels) could beintroduced into the targeted locus and result in the loss of genefunction if it occurs in the gene-coding region. Previous studies haveshown that loss of LKB1 or TTF1 in mouse lungs via CRISPR/Cas9-mediated somatic genome editing significantly promotes lung cancermalignant progression (6, 9), similar to the observations fromGEMMs(10, 11). Thus, this type of somatic gene knockout technique makes itfeasible to efficiently and systematically identify potential TSGs in vivo.KRAS oncogenic mutations are frequently detected in human
lung cancer, and the KrasG12D/+ mouse model has been widely used
Significance
Tumor suppressor genes (TSGs) play important roles in lung cancerinitiation, progression, and even metastasis. Here, we take ad-vantage of the clustered regularly interspaced short palindromicrepeats/Cas9-mediated screening in vivo technique to identifymultiple tumor suppressor genes contributing to lung cancermalignant progression. Using genetically engineered mousemodels, we further confirm the tumor-suppressive role of epige-netic regulator UTX and provide therapeutic implications for UTX-deficient lung tumors. Thus, our work provides a systematicscreening of TSGs in vivo and demonstrates UTX functions as theimportant epigenetic regulator in lung tumorigenesis.
Author contributions: Q.W., Liang Chen, and H.J. designed research; Q.W., Y. Tian, J.Z.,X.T., H.H., S.L., H.Z., C.Y., K.W., L.G., F.L., Z.Q., S.Y., and T.C. performed research; H.C., G.Z.,W.L., Y.S., Luonan Chen, K.-K.W., and K.G. contributed new reagents/analytic tools; Q.W.,J.Z., X.T., H.Z., Y. Tang, Z.F., and X.H. analyzed data; and Q.W., Liang Chen, and H.J. wrotethe paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Published under the PNAS license.
Data deposition: The datasets reported in this paper have been deposited in the GeneExpression Omnibus (GEO) database, https://www.ncbi.nlm.nih.gov/geo (accession nos.GSE74095, GSE77684, and GSE93302) and in the Sequence Read Archive (SRA) (accessionno. SRP069330).1Q.W., Y. Tian, J.Z., and X.T. contributed equally to this work.2To whom correspondence may be addressed. Email: [email protected] or [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1716589115/-/DCSupplemental.
Published online April 9, 2018.
E3978–E3986 | PNAS | vol. 115 | no. 17 www.pnas.org/cgi/doi/10.1073/pnas.1716589115
Dow
nloa
ded
by g
uest
on
June
1, 2
020

for investigating the function of TSGs (12). Using CRISPR-mediatedsomatic gene knockout in the KrasG12D/+ mouse model, we havescreened a total of 55 potential TSGs individually. Our datashow that somatic knockout of any of five genes, including Utx,Ptip, Acp5, Acacb, and Clu, significantly promotes Kras-driven lungtumor progression. These five genes are frequently down-regulatedin human lung cancer specimens and significantly associated withpatient survival. Importantly, conditional knockout of the epige-netic regulator Utx dramatically accelerates lung tumorigenesis inthe KrasG12D/+ mouse model. Our data also demonstrates that lossof UTX results in increased EZH2 expression in Kras-driven lungcancer and EZH2 inhibitor preferentially suppresses the growth ofUtx-knockout lung tumors.
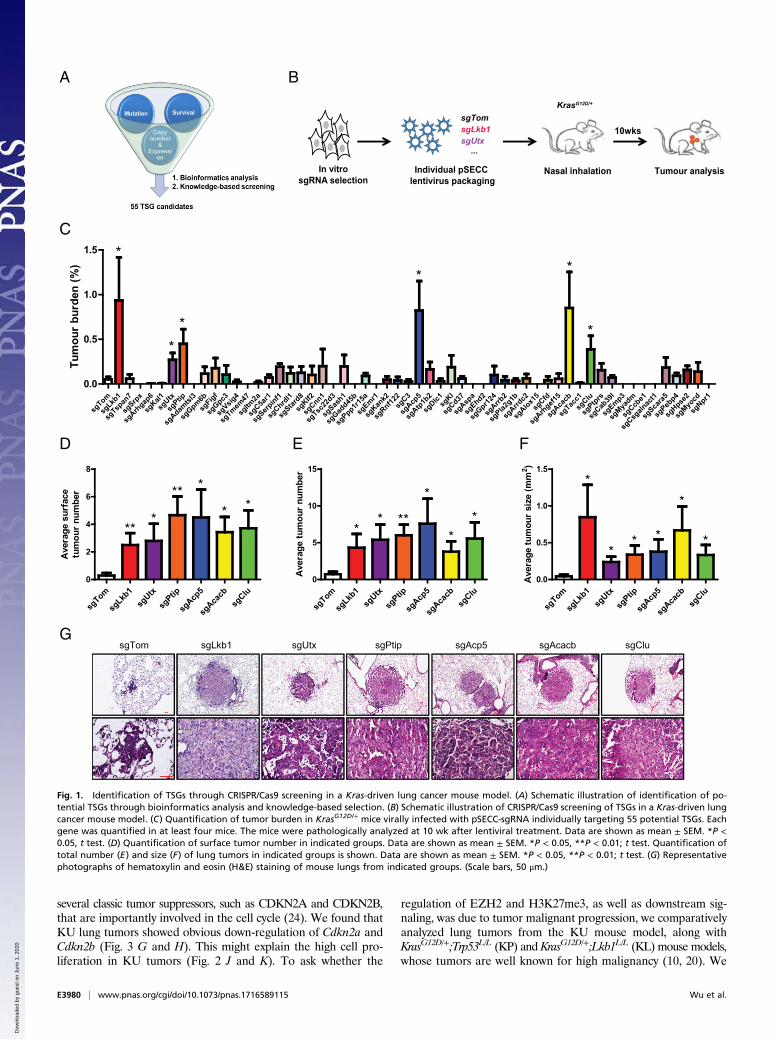
ResultsIdentification of TSGs via CRISPR Screening in Vivo. To performCRISPR/Cas9-mediated screening in vivo, we first generated alist of potential TSG candidates based on integrative analyses ofgene expression profiling, copy number variation from Chineselung cancer microarray data, and survival correlation analysesfrom the prediction of clinical outcomes from genomic profiles(PRECOG) dataset (https://precog.stanford.edu/). Based on theliterature and gene mutation status from the catalogue of somaticmutations in cancer (COSMIC) website (cancer.sanger.ac.uk/cosmic),we further narrowed down the list to 55 potential TSGs with relativeunknown function (Table S1). We found that 30 of these geneswere located in a recurrent genomic deletion locus (Table S1).Further analyses using the Broad lung adenocarcinoma dataset(13) showed that the inactivating mutations of these genes tended tobe concurrent with KRAS mutations (Table S1). We therefore chosethese 55 candidate genes for further individual CRISPR/Cas9 screeningin the KrasG12D/+ mouse model (Fig. 1A). To ensure optimal genedisruption in vivo, we designed three sgRNAs for each geneand performed in vitro screening first using the LentiCRISPRv2plasmid (Cas9-sgRNA-Puro system) in KrasG12D/+ mouse embryonicfibroblasts (MEFs) (14) (Fig. 1B and Table S2).With this, we were able to select the most efficient sgRNA with
minimal off-targeting effect for further study and then cloned theoptimal sgRNA into the pSECC plasmid (Cas9-sgRNA-Cre sys-tem), which could allow us to produce the lentiviruses for specificknockout of individual genes with simultaneous Cre expressionin the KrasG12D/+ mouse model as previously described (6)(Fig. 1B and Table S2). We have previously shown that somaticloss-of-function mutation of LKB1 is frequently detected in humanlung cancer and knockout of Lkb1 significantly promotes tumorprogression in the Kras-driven mouse model (10). Thus, we usedLkb1 as our positive control for this in vivo screening of potentialTSGs (Fig. 1B and Table S2). We treated the KrasG12D/+ mousemodel with nasal inhalation of 2 × 104 pfus of lentiviruses tar-geting either Lkb1 or Tomato (serving as a negative control) (6)and analyzed lung tumor formation at 10 wk after lentiviral in-fection (Fig. 1B and Table S2). Consistent with a previous report(9), pSECC-sgLkb1 lentiviral infection significantly promoted anincrease of the burden, number, and size of lung tumors in contrastto pSECC-sgTom lentiviral infection (Fig. 1 C–F). Moreover, lungadenocarcinoma was detectable in the pSECC-sgLkb1 group,whereas the pSECC-sgTom group mainly showed early lung cancerlesions like atypical adenomatous hyperplasia (AAH) (Fig. 1G).These data supported that this system could potentially be appliedto TSG screening in vivo.Following the same protocol, we then performed the individ-
ual knockout of these 55 candidate genes and analyzed the lungtumor formation (Table S3). Comparative analyses showed thatdeletion of any of five genes, including Utx, Ptip, Acp5, Acacb,and Clu, significantly increased the lung tumor burden (Fig. 1C).Consistently, lung tumors detectable on the lung surface werefound to be significantly increased (Fig. 1D). This was furthersupported by the notable increase of average tumor number and
size through detailed pathological analyses (Fig. 1 E–G). GenomicDNA sequencing data showed the clear genome editing in targetedalleles without notable off-targeting effects (Fig. S1 and Tables S4and S5). Through real-time PCR quantification analyses of pairedChinese lung cancer and pathologically normal lungs, we foundthat the expression of these five genes was significantly decreasedin cancer specimens (Fig. 2A and Fig. S2 A–D). Moreover, lowexpression of each TSG was associated with poor survival of patientswith lung cancer (Fig. 2 B and C and Fig. S2 E–H). Taken together,these data identified five TSGs that might contribute to lung tumor-igenesis through in vivo CRISPR screening.
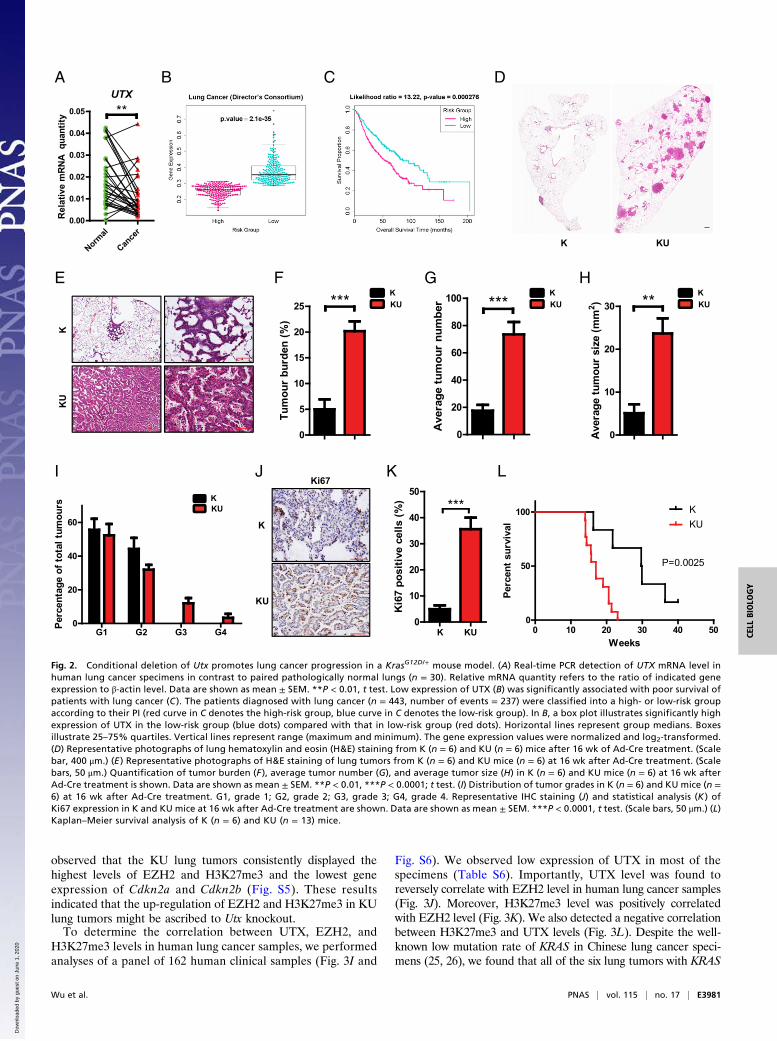
Utx Knockout Promotes Lung Tumor Progression in the KrasG12D/+
Mouse Model. Epigenetic regulation, such as H3K27 methyl-ation deregulation, is known to play important roles in cancermalignant progression (15). Interestingly, two identified TSGs,including UTX and PTIP, were located at the same histonemethyltransferase MLL3/MLL4 complex (16, 17). Importantly,UTX, also named KDM6A, is considered to be responsible forH3K27me3 demethylation (16, 17) and counteracts the methyl-transferase function of Polycomb Repressive Complex 2 (PRC2)during cancer malignant progression (15). We then focused onUTX and investigated the potential function of UTX in vivo. Forthis, we crossed the conditional UtxL/L allele (referred to as Umice) (18) with the KrasG12D/+ allele (referred to as K mice) andgenerated the compound alleles KrasG12D/+;UtxL/L (referred toas KU mice), which could become a homozygous deletion of Utxwhen the mice were given Cre recombinase adenovirus (Ad-Cre). We then delivered 2 × 106 pfus of Ad-Cre to the KU micevia nasal inhalation and analyzed lung tumor formation after16 wk of Ad-Cre treatment (19). Strikingly, homozygous deletionof Utx significantly promoted Kras-driven lung cancer progres-sion (Fig. 2 D and E). Statistical analyses showed that deletion ofUtx dramatically increased the Kras-driven lung tumor burden(Fig. 2F), as well as the tumor number and size (Fig. 2 G and H).Moreover, grade 3 and grade 4 lung tumors, including adeno-carcinoma and invasive adenocarcinoma (20, 21), were onlydetectable in the KU mouse model (Fig. 2I). Furthermore, theKU tumors also displayed high cell proliferation (Fig. 2 J and K).Consistently, the medium survival of the KU mouse model wasabout 17 wk, in comparison to 30 wk for the K mouse model(Fig. 2L). We further established mouse primary lung cancer cellsfrom lung tumors in the KrasG12D/+;Trp53L/L;UtxL/L (KPU) mousemodel (Fig. S3A). Using these KPU primary cancer cells, we foundthat deletion of Utx significantly promoted cell proliferation, whichcould be inhibited by reexpression of Utx (Fig. S3 B and C). Thesedata collectively supported the tumor-suppressive role of theepigenetic regulator UTX in lung tumorigenesis.
Utx Knockout Increases H3K27me3 Level Potentially Through EZH2 Up-Regulation. Given that UTX functions as histone H3K27me3demethylase (15), we next examined the H3K27me3 level in Utx-knockout lung tumors. As expected, Utx deletion resulted in asignificant increase of the H3K27me3 level in KU lung tumors(Fig. 3 A and B and Fig. S4 A and C). Previous studies haveindicated that UTX loss promoted the activity of the PRC2complex, the machinery mainly responsible for H3K27 methyl-ation, which contains the enzymatic subunit EZH2 and othercofactors, including SUZ12 and EED (15, 22). In considerationof the increased H3K27me3 level in KU lung tumors, we nexttested the expression of all the core PRC2 components, includingEZH1, EZH2, SUZ12, and EED, and another H3K27me3 de-methylase, KDM6B (17, 23). RNA sequencing data showed thatEZH2 is the only gene with significantly expressed up-regulationin KU lung tumors (Fig. 3C). We further confirmed this up-regulationof EZH2 through immunohistochemistry (IHC) staining andWesternblotting analyses (Fig. 3 D–F and Fig. S4 A and B). A previous studyhas demonstrated that H3K27 methylation silenced the expression of
Wu et al. PNAS | vol. 115 | no. 17 | E3979
CELL
BIOLO
GY
PNASPL
US
Dow
nloa
ded
by g
uest
on
June
1, 2
020
several classic tumor suppressors, such as CDKN2A and CDKN2B,that are importantly involved in the cell cycle (24). We found thatKU lung tumors showed obvious down-regulation of Cdkn2a andCdkn2b (Fig. 3 G and H). This might explain the high cell pro-liferation in KU tumors (Fig. 2 J and K). To ask whether the
regulation of EZH2 and H3K27me3, as well as downstream sig-naling, was due to tumor malignant progression, we comparativelyanalyzed lung tumors from the KU mouse model, along withKrasG12D/+;Trp53L/L (KP) and KrasG12D/+;Lkb1L/L (KL) mouse models,whose tumors are well known for high malignancy (10, 20). We
A B
G
C
D E F
sgLkb1 sgAcp5 sgAcacb sgClusgPtipsgUtx
10wks
Tumour analysisNasal inhalationIn vitrosgRNA selection
Individual pSECC lentivirus packaging
KrasG12D/+
sgTom
sgTom
sgLkb
1sg
Utx
sgPtip
sgAcp
5
sgAca
cbsg
Clu0
2
4
6
8
***
*
****
Ave
rag e
surf
ace
tum
our
num
ber
sgTom
sgLkb
1sg
Utx
sgPtip
sgAcp
5
sgAca
cbsg
Clu0
5
10
15
** ** *
*
*
Ave
rage
t um
o ur
num
ber
sgTom
sgLkb
1sg
Utx
sgPtip
sgAcp
5
sgAca
cbsg
Clu0.0
0.5
1.0
1.5*
*
*
** *A
vera
getu
mo u
rsi
ze(m
m2 )
sgTomsgLkb1
...sgUtx
sgTom
sgLkb
1
sgTsp
an7
sgSrp
x
sgArh
gap6
sgKal1
sgUtx
sgPtip
sgAdam
tsl3
sgGpm6b
sgFigf
sgGpc3
sgVsig
4
sgTmem
47
sgItm
2a
sgC5a
r1
sgSerp
inf1
sgChrd
l1
sgStar
d8
sgKlf2
sgCnn1
sgTsc
22d3
sgSas
h1
sgGad
d45b
sgPpp1r1
5a
sgEmr1
sgKan
k2
sgRnf12
2sg
C3
sgAcp
5
sgAtp1b
2
sgDlc1sg
Kl
sgCd37
sgAsp
a
sgEhd2
sgGpr12
4
sgArrb
2
sgPla2
g1b
sgArrd
c2
sgAlox1
5sg
Cfd
sgArh
gef15
sgAca
cb
sgTac
c1sg
Clu
sgPtprs
sgCab
39l
sgEmp3
sgMya
dm
sgCcb
e1
sgCsg
alnac
t1
sgSca
ra5
sgPeb
p4
sgHpse
2
sgMyo
cd
sgNpr1
0.0
0.5
1.0
1.5
**
*
*
**
Tum
our
burd
en(%
)
Fig. 1. Identification of TSGs through CRISPR/Cas9 screening in a Kras-driven lung cancer mouse model. (A) Schematic illustration of identification of po-tential TSGs through bioinformatics analysis and knowledge-based selection. (B) Schematic illustration of CRISPR/Cas9 screening of TSGs in a Kras-driven lungcancer mouse model. (C) Quantification of tumor burden in KrasG12D/+ mice virally infected with pSECC-sgRNA individually targeting 55 potential TSGs. Eachgene was quantified in at least four mice. The mice were pathologically analyzed at 10 wk after lentiviral treatment. Data are shown as mean ± SEM. *P <0.05, t test. (D) Quantification of surface tumor number in indicated groups. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01; t test. Quantification oftotal number (E) and size (F) of lung tumors in indicated groups is shown. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01; t test. (G) Representativephotographs of hematoxylin and eosin (H&E) staining of mouse lungs from indicated groups. (Scale bars, 50 μm.)
E3980 | www.pnas.org/cgi/doi/10.1073/pnas.1716589115 Wu et al.
Dow
nloa
ded
by g
uest
on
June
1, 2
020
observed that the KU lung tumors consistently displayed thehighest levels of EZH2 and H3K27me3 and the lowest geneexpression of Cdkn2a and Cdkn2b (Fig. S5). These resultsindicated that the up-regulation of EZH2 and H3K27me3 in KUlung tumors might be ascribed to Utx knockout.To determine the correlation between UTX, EZH2, and
H3K27me3 levels in human lung cancer samples, we performedanalyses of a panel of 162 human clinical samples (Fig. 3I and
Fig. S6). We observed low expression of UTX in most of thespecimens (Table S6). Importantly, UTX level was found toreversely correlate with EZH2 level in human lung cancer samples(Fig. 3J). Moreover, H3K27me3 level was positively correlatedwith EZH2 level (Fig. 3K). We also detected a negative correlationbetween H3K27me3 and UTX levels (Fig. 3L). Despite the well-known low mutation rate of KRAS in Chinese lung cancer speci-mens (25, 26), we found that all of the six lung tumors with KRAS
D
E F G H
K KU
I
A B
KKU
G1 G2 G3 G40
20
40
60
Perc
enta
geof
tota
ltum
ours
KK
U
0
10
20
30 **
Ave
rage
tum
our
siz e
(mm
2 )
KKU
UTX
Normal
Cance
r0.00
0.01
0.02
0.03
0.04
0.05 **
Rel
ativ
e m
RNA
quan
tity
L
0 10 20 30 40 500
50
100 KKU
P=0.0025
Weeks
Perc
ents
urvi
val
KJ
K
KU
Ki67
C
0
5
10
15
20
25 ***Tu
mou
rbu
rden
(%)
KKU
0
20
40
60
80
100 ***
Ave
rage
tum
our
num
ber
KKU
0
10
20
30
40
50***
Ki6
7po
sitiv
ece
lls(%
)
K KU
Fig. 2. Conditional deletion of Utx promotes lung cancer progression in a KrasG12D/+ mouse model. (A) Real-time PCR detection of UTX mRNA level inhuman lung cancer specimens in contrast to paired pathologically normal lungs (n = 30). Relative mRNA quantity refers to the ratio of indicated geneexpression to β-actin level. Data are shown as mean ± SEM. **P < 0.01, t test. Low expression of UTX (B) was significantly associated with poor survival ofpatients with lung cancer (C ). The patients diagnosed with lung cancer (n = 443, number of events = 237) were classified into a high- or low-risk groupaccording to their PI (red curve in C denotes the high-risk group, blue curve in C denotes the low-risk group). In B, a box plot illustrates significantly highexpression of UTX in the low-risk group (blue dots) compared with that in low-risk group (red dots). Horizontal lines represent group medians. Boxesillustrate 25–75% quartiles. Vertical lines represent range (maximum and minimum). The gene expression values were normalized and log2-transformed.(D) Representative photographs of lung hematoxylin and eosin (H&E) staining from K (n = 6) and KU (n = 6) mice after 16 wk of Ad-Cre treatment. (Scalebar, 400 μm.) (E ) Representative photographs of H&E staining of lung tumors from K (n = 6) and KU mice (n = 6) at 16 wk after Ad-Cre treatment. (Scalebars, 50 μm.) Quantification of tumor burden (F ), average tumor number (G), and average tumor size (H) in K (n = 6) and KU mice (n = 6) at 16 wk afterAd-Cre treatment is shown. Data are shown as mean ± SEM. **P < 0.01, ***P < 0.0001; t test. (I) Distribution of tumor grades in K (n = 6) and KU mice (n =6) at 16 wk after Ad-Cre treatment. G1, grade 1; G2, grade 2; G3, grade 3; G4, grade 4. Representative IHC staining (J) and statistical analysis (K ) ofKi67 expression in K and KU mice at 16 wk after Ad-Cre treatment are shown. Data are shown as mean ± SEM. ***P < 0.0001, t test. (Scale bars, 50 μm.) (L)Kaplan–Meier survival analysis of K (n = 6) and KU (n = 13) mice.
Wu et al. PNAS | vol. 115 | no. 17 | E3981
CELL
BIOLO
GY
PNASPL
US
Dow
nloa
ded
by g
uest
on
June
1, 2
020
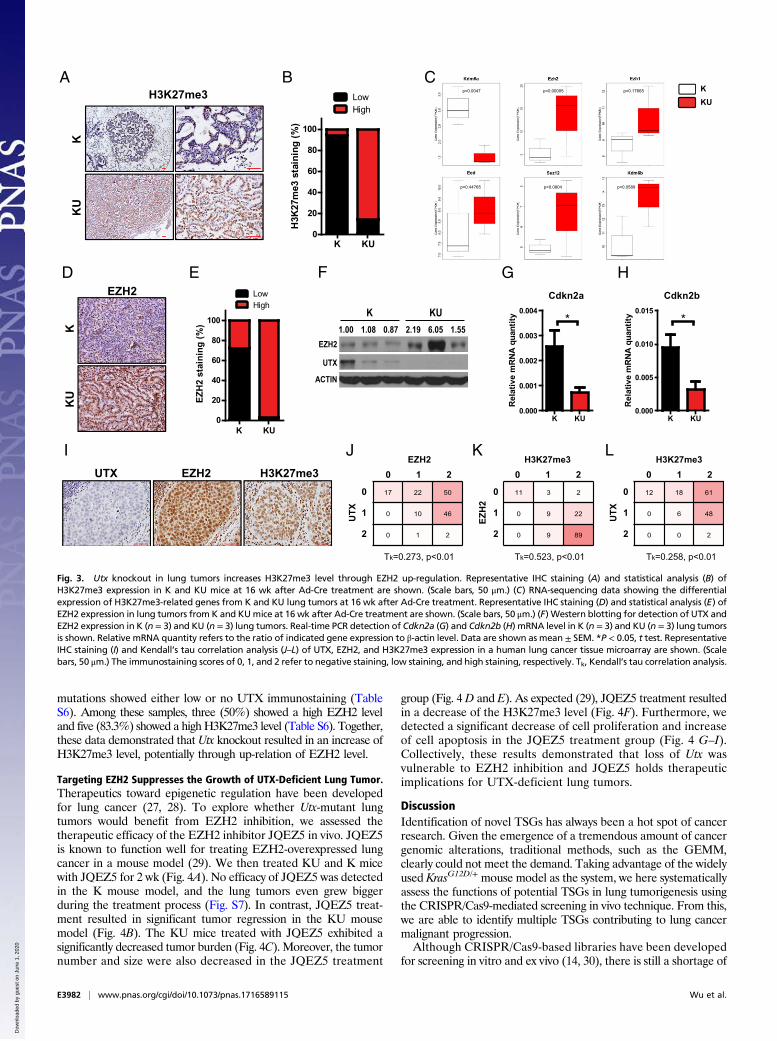
mutations showed either low or no UTX immunostaining (TableS6). Among these samples, three (50%) showed a high EZH2 leveland five (83.3%) showed a highH3K27me3 level (Table S6). Together,these data demonstrated that Utx knockout resulted in an increase ofH3K27me3 level, potentially through up-relation of EZH2 level.
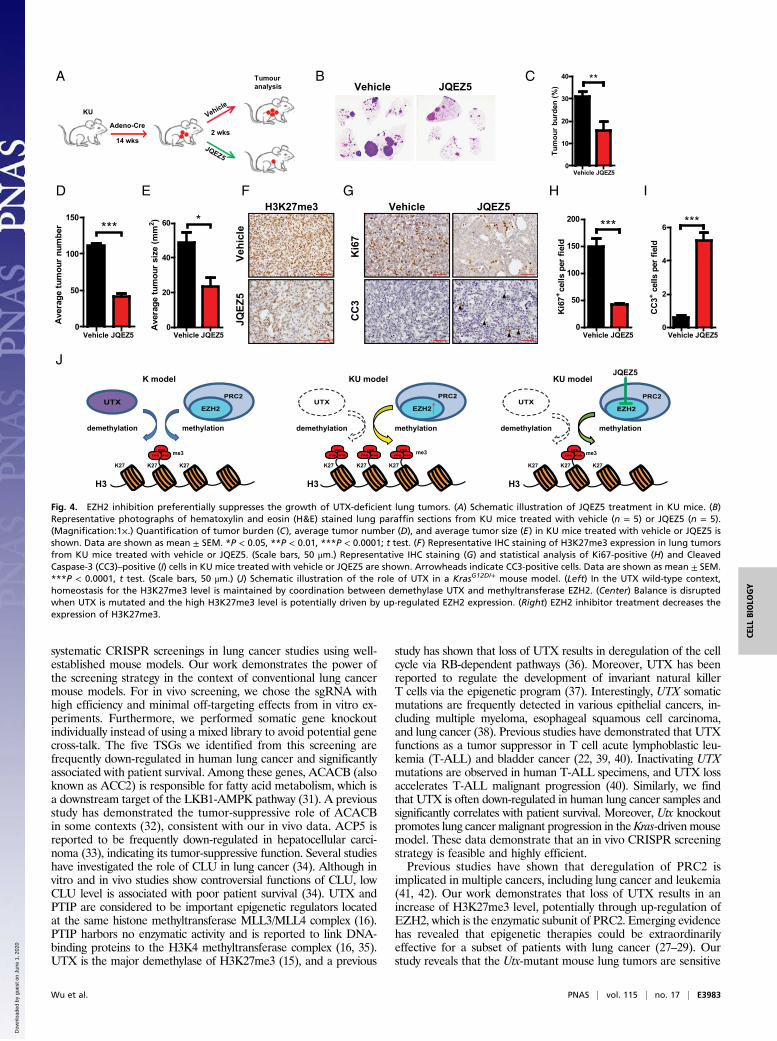
Targeting EZH2 Suppresses the Growth of UTX-Deficient Lung Tumor.Therapeutics toward epigenetic regulation have been developedfor lung cancer (27, 28). To explore whether Utx-mutant lungtumors would benefit from EZH2 inhibition, we assessed thetherapeutic efficacy of the EZH2 inhibitor JQEZ5 in vivo. JQEZ5is known to function well for treating EZH2-overexpressed lungcancer in a mouse model (29). We then treated KU and K micewith JQEZ5 for 2 wk (Fig. 4A). No efficacy of JQEZ5 was detectedin the K mouse model, and the lung tumors even grew biggerduring the treatment process (Fig. S7). In contrast, JQEZ5 treat-ment resulted in significant tumor regression in the KU mousemodel (Fig. 4B). The KU mice treated with JQEZ5 exhibited asignificantly decreased tumor burden (Fig. 4C). Moreover, the tumornumber and size were also decreased in the JQEZ5 treatment
group (Fig. 4D and E). As expected (29), JQEZ5 treatment resultedin a decrease of the H3K27me3 level (Fig. 4F). Furthermore, wedetected a significant decrease of cell proliferation and increaseof cell apoptosis in the JQEZ5 treatment group (Fig. 4 G–I).Collectively, these results demonstrated that loss of Utx wasvulnerable to EZH2 inhibition and JQEZ5 holds therapeuticimplications for UTX-deficient lung tumors.
DiscussionIdentification of novel TSGs has always been a hot spot of cancerresearch. Given the emergence of a tremendous amount of cancergenomic alterations, traditional methods, such as the GEMM,clearly could not meet the demand. Taking advantage of the widelyused KrasG12D/+ mouse model as the system, we here systematicallyassess the functions of potential TSGs in lung tumorigenesis usingthe CRISPR/Cas9-mediated screening in vivo technique. From this,we are able to identify multiple TSGs contributing to lung cancermalignant progression.Although CRISPR/Cas9-based libraries have been developed
for screening in vitro and ex vivo (14, 30), there is still a shortage of
CKKU
AK
KU
H3K27me3
GCdkn2a
K KU0.000
0.001
0.002
0.003
0.004*
Rel
ativ
e m
RN
A q
uant
ity
Cdkn2b
K KU0.000
0.005
0.010
0.015*
Rel
ativ
e m
RN
A q
uant
ity
LowHigh
K KU0
20
40
60
80
100
H3K
27m
e3 s
tain
ing
(%)
UTX
EZH2
ACTIN
K KU
EZH2FD
IH3K27me3UTX EZH2
J0 1 2
2
1
0
UTX
EZH2
Tk=0.273, p<0.01
17 22 50
0 10 46
0 1 2 2
1
0
0 1 2H3K27me3
UTX
Tk=0.258, p<0.01
12 18 61
0 6 48
0 0 22
1
0
0 1 2H3K27me3
EZH
2
Tk=0.523, p<0.01
11 3 2
0 9 22
0 9 89
KK
UB
E
0
20
40
60
80
100
EZH
2 st
aini
ng (%
)
K KU
LowHigh
H
p=0.0047 p=0.00005 p=0.17665
p=0.44765 p=0.0804 p=0.0589
LK
1.00 1.08 0.87 2.19 6.05 1.55
Fig. 3. Utx knockout in lung tumors increases H3K27me3 level through EZH2 up-regulation. Representative IHC staining (A) and statistical analysis (B) ofH3K27me3 expression in K and KU mice at 16 wk after Ad-Cre treatment are shown. (Scale bars, 50 μm.) (C) RNA-sequencing data showing the differentialexpression of H3K27me3-related genes from K and KU lung tumors at 16 wk after Ad-Cre treatment. Representative IHC staining (D) and statistical analysis (E) ofEZH2 expression in lung tumors from K and KUmice at 16 wk after Ad-Cre treatment are shown. (Scale bars, 50 μm.) (F) Western blotting for detection of UTX andEZH2 expression in K (n = 3) and KU (n = 3) lung tumors. Real-time PCR detection of Cdkn2a (G) and Cdkn2b (H) mRNA level in K (n = 3) and KU (n = 3) lung tumorsis shown. Relative mRNA quantity refers to the ratio of indicated gene expression to β-actin level. Data are shown as mean ± SEM. *P < 0.05, t test. RepresentativeIHC staining (I) and Kendall’s tau correlation analysis (J–L) of UTX, EZH2, and H3K27me3 expression in a human lung cancer tissue microarray are shown. (Scalebars, 50 μm.) The immunostaining scores of 0, 1, and 2 refer to negative staining, low staining, and high staining, respectively. Tk, Kendall’s tau correlation analysis.
E3982 | www.pnas.org/cgi/doi/10.1073/pnas.1716589115 Wu et al.
Dow
nloa
ded
by g
uest
on
June
1, 2
020
systematic CRISPR screenings in lung cancer studies using well-established mouse models. Our work demonstrates the power ofthe screening strategy in the context of conventional lung cancermouse models. For in vivo screening, we chose the sgRNA withhigh efficiency and minimal off-targeting effects from in vitro ex-periments. Furthermore, we performed somatic gene knockoutindividually instead of using a mixed library to avoid potential genecross-talk. The five TSGs we identified from this screening arefrequently down-regulated in human lung cancer and significantlyassociated with patient survival. Among these genes, ACACB (alsoknown as ACC2) is responsible for fatty acid metabolism, which isa downstream target of the LKB1-AMPK pathway (31). A previousstudy has demonstrated the tumor-suppressive role of ACACBin some contexts (32), consistent with our in vivo data. ACP5 isreported to be frequently down-regulated in hepatocellular carci-noma (33), indicating its tumor-suppressive function. Several studieshave investigated the role of CLU in lung cancer (34). Although invitro and in vivo studies show controversial functions of CLU, lowCLU level is associated with poor patient survival (34). UTX andPTIP are considered to be important epigenetic regulators locatedat the same histone methyltransferase MLL3/MLL4 complex (16).PTIP harbors no enzymatic activity and is reported to link DNA-binding proteins to the H3K4 methyltransferase complex (16, 35).UTX is the major demethylase of H3K27me3 (15), and a previous
study has shown that loss of UTX results in deregulation of the cellcycle via RB-dependent pathways (36). Moreover, UTX has beenreported to regulate the development of invariant natural killerT cells via the epigenetic program (37). Interestingly, UTX somaticmutations are frequently detected in various epithelial cancers, in-cluding multiple myeloma, esophageal squamous cell carcinoma,and lung cancer (38). Previous studies have demonstrated that UTXfunctions as a tumor suppressor in T cell acute lymphoblastic leu-kemia (T-ALL) and bladder cancer (22, 39, 40). Inactivating UTXmutations are observed in human T-ALL specimens, and UTX lossaccelerates T-ALL malignant progression (40). Similarly, we findthat UTX is often down-regulated in human lung cancer samples andsignificantly correlates with patient survival. Moreover, Utx knockoutpromotes lung cancer malignant progression in theKras-driven mousemodel. These data demonstrate that an in vivo CRISPR screeningstrategy is feasible and highly efficient.Previous studies have shown that deregulation of PRC2 is
implicated in multiple cancers, including lung cancer and leukemia(41, 42). Our work demonstrates that loss of UTX results in anincrease of H3K27me3 level, potentially through up-regulation ofEZH2, which is the enzymatic subunit of PRC2. Emerging evidencehas revealed that epigenetic therapies could be extraordinarilyeffective for a subset of patients with lung cancer (27–29). Ourstudy reveals that the Utx-mutant mouse lung tumors are sensitive
D
A C
Vehi
cle
JQEZ
5
H3K27me3E F
Vehicle JQEZ5
Ki6
7C
C3
B
HG
Adeno-Cre2 wks
Tumouranalysis
JQEZ5
KU
I
0
10
20
30
40 **
Tum
our
burd
en (%
)
Vehicle JQEZ5
0
50
100
150***
Ave
rage
tum
our
num
ber
Vehicle JQEZ50
20
40
60 *
Ave
rage
tum
our
size
(mm
2 )
JQEZ5
J
EZH2
PRC2UTX
methylationdemethylation
K model
H3
K27
mememe
K27 K27
me3me
mememe
meme
H3
K27 K27 K27
mememe me3
KU model
methylationdemethylation
EZH2
PRC2UTX
Vehicle JQEZ5
0
50
100
150
200 ***
Ki6
7+ce
lls p
er fi
eld
JQEZ50
2
4
6 ***
CC
3+ce
lls p
er fi
eld
JQEZ5
H3
K27
mememe
K27 K27
me3
KU model
methylationdemethylation
EZH2
PRC2UTX
JQEZ5
14 wks
Vehicle
Vehicle Vehicle Vehicle
Fig. 4. EZH2 inhibition preferentially suppresses the growth of UTX-deficient lung tumors. (A) Schematic illustration of JQEZ5 treatment in KU mice. (B)Representative photographs of hematoxylin and eosin (H&E) stained lung paraffin sections from KU mice treated with vehicle (n = 5) or JQEZ5 (n = 5).(Magnification:1×.) Quantification of tumor burden (C), average tumor number (D), and average tumor size (E) in KU mice treated with vehicle or JQEZ5 isshown. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.0001; t test. (F) Representative IHC staining of H3K27me3 expression in lung tumorsfrom KU mice treated with vehicle or JQEZ5. (Scale bars, 50 μm.) Representative IHC staining (G) and statistical analysis of Ki67-positive (H) and CleavedCaspase-3 (CC3)–positive (I) cells in KU mice treated with vehicle or JQEZ5 are shown. Arrowheads indicate CC3-positive cells. Data are shown as mean ± SEM.***P < 0.0001, t test. (Scale bars, 50 μm.) (J) Schematic illustration of the role of UTX in a KrasG12D/+ mouse model. (Left) In the UTX wild-type context,homeostasis for the H3K27me3 level is maintained by coordination between demethylase UTX and methyltransferase EZH2. (Center) Balance is disruptedwhen UTX is mutated and the high H3K27me3 level is potentially driven by up-regulated EZH2 expression. (Right) EZH2 inhibitor treatment decreases theexpression of H3K27me3.
Wu et al. PNAS | vol. 115 | no. 17 | E3983
CELL
BIOLO
GY
PNASPL
US
Dow
nloa
ded
by g
uest
on
June
1, 2
020

to EZH2 inhibition, consistent with studies in T-ALL and bladdercancer (22, 29, 40). This is very helpful since direct targeting ofUTX to boost its activation seems impractical for now. In theKrasG12D/+ mouse model, homeostasis of the H3K27me3 level ismaintained by the coordination between the demethylase UTXand PRC2 complex containing methyltransferase EZH2 (Fig. 4J).However, this balance is disrupted when Utx is mutated andthe high H3K27me3 level is potentially driven by up-regulatedEZH2 expression through a currently unknown mechanism (Fig.4J). The increased H3K27me3 modification then silences severalTSGs, including CDKN2A and CDKN2B, and contributes to tumorprogression. Under the circumstance of UTX loss, EZH2 inhibi-tor works effectively to suppress lung tumor growth (Fig. 4J). Thehigh efficacy of EZH2 inhibitor in the KU mouse model providestherapeutic implications for human lung cancer with KRAS muta-tions exhibiting a low UTX level. However, KRAS tumors harboringwild-type UTX are more advanced after EZH2 inhibitor treatment,indicating the treatment would be dangerous for certain patients withlung cancer. In future studies, it will be interesting to ask whetherEZH2 inhibitor also works well to suppress UTX-low lung cancerindependent of the KRAS mutation status.
Materials and MethodsHuman Lung Cancer Specimen Analysis. Human lung cancer clinical sampleswere collected with the approval of the Institutional Review Committee ofShanghai Cancer Hospital at Fudan University as previously described (19).Written informed consent was obtained from all of the patients. All caseswere rereviewed by pathologists from the Department of Pathology atShanghai Cancer Hospital for confirmation of tumor histology and tumorcontent. The datasets used in this study have been deposited in the GeneExpression Omnibus (GEO) repository under accession nos. GSE74095 andGSE77684. Human Exon 1.0 profiling data of 76 Chinese lung adenocarci-nomas were quantified with Affymetrix Power Tools (v1.18.0) and summa-rized as gene-level expression (“rma-sketch”). Differential expression callsfor each tumor sample were obtained by comparison with normal samples,using the twofold expression change threshold. SNP 6.0 genotyping data ofthe same samples were preprocessed with PennCNV (v1.0.0) and annotated togene copy number estimates. To identify genes located in frequently deletedregions, raw copy number signals (log ratios) were first segmented with thecircular binary segmentation algorithm, and recurrent copy number alterationpeaks were called with GISTIC (v2.0.23). To prioritize deletions accompanied byan underexpression effect, concurrent samples between differential expres-sion calls and copy number alterations were counted and tested for enrich-ment with Fisher’s exact test. A set of 67 genes with concurrent allele loss anddown-regulated gene expression (five or more samples) was selected andmerged with another set of 43 genes that were identified (Z-scores < 0) to bethe favorable prognosis genes in eight lung cancer datasets according toPRECOG data analyses (Lung_cancer_ADENO_200, Lung_cancer_ADENO_206,Lung_cancer_ADENO_216, Lung_cancer_ADENO_221, Lung_cancer_SCC_234,Lung_cancer_SCC_243, Lung_cancer_SCC_251, and Lung_cancer_SCC_258). Thestatistical associations between genes and clinical outcomes were assessed byZ-scores generated by PRECOG (43). Based on the literature and COSMICmutational status, we further narrowed down the list to 55 potential TSGswith relative unknown function and high mutational frequency. Using theBroad lung adenocarcinoma dataset in cBioPortal (13), we analyzed the mu-tations of these genes for the tendency toward concurrence with KRAS mu-tations. Moreover, we also mapped these genes to the areas of recurrentdeletion. The clinical data for UTX were retrieved from the Director’s Chal-lenge (44). The univariate Cox regression analysis was applied to produce theprognostic index (PI). The patients were split into two risk groups based on PIrank. Then, the likelihood ratio test and log-rank test were performed on thePI to generate the corresponding P value and Kaplan–Meier curves from thetwo risk groups to compare the differences in survival. A human lung cancertissue microarray (detailed information is provided in Table S6) was providedby the Department of Pathology at Shanghai Cancer Hospital, Fudan Univer-sity, Shanghai, China.
CRISPR Plasmids Construction and Lentivirus Production. The pSECC andLentiCRISPRv2 plasmids were generously provided by F. J. Sanchez-Rivera, KochInstitute for Integrative Cancer Research at MIT, Cambridge, MA and T. Jacks,Koch Institute for Integrative Cancer Research at MIT, Cambridge, MA. All ofthe sgRNAs were designed using optimized CRISPR Design (crispr.mit.edu/)
except for sgTomato and sgLkb1, which were previously reported (6, 9) (thesequences of all sgRNAs for in vivo screening are shown in Table S2). The topthree exonic off-targeting sites were predicted using optimized CRISPR Design(Table S4). Cloning methods were optimized following established protocolsdescribed elsewhere (4, 14). Lentiviruses were produced by cotransfection ofHEK-293T cells with pSECC or LentiCRISPRv2 constructs and packaging vectors(psPAX2 and pMD2.G). For pSECC lentiviruses, supernatant was collected 48 hand 72 h posttransfection, concentrated by ultracentrifugation at 50,000 × gfor 2 h, and resuspended overnight in an appropriate volume of OptiMEM(Gibco).
Mouse Colony, Treatment, and Tumor Analysis. KrasG12D/+, Trp53L/L, Lkb1L/L,and UtxL/L mice were originally generously provided by T. Jacks, Koch In-stitute for Integrative Cancer Research at MIT, Cambridge, MA, R. Depinho,The University of Texas MD Anderson Cancer Center, Houston, and Kai Ge,National Institutes of Health, Bethesda. All mice were housed in a specificpathogen-free environment at the Shanghai Institute of Biochemistry andCell Biology and treated in strict accordance with protocols approved bythe Institutional Animal Care and Use Committee of the Shanghai Institutesfor Biological Sciences, Chinese Academy of Sciences. For CRISPR/Cas9screening, KrasG12D/+ mice at 6–8 wk of age were treated with 2 × 104 pfusof pSECC-sgRNA lentiviruses via nasal inhalation and analyses were per-formed after 10 wk. Either KrasG12D/+ or KrasG12D/+;UtxL/L mice at 6–8 wk ofage were treated with 2 × 106 pfus of Ad-Cre via nasal inhalation as previouslydescribed, unless especially noted (19), and analyses were performed after16 wk. Gross inspection and histopathological examination were performedduring the analyses of mice. Lung tumors were dissected for molecular andpathological analysis. Tumor burden and total number and size of lung tumors,as well as surface lung tumors, were analyzed as previously described (19). Tu-mor grades were analyzed as follows (20, 21): grade 1, AAH or AAH progressingto a small adenoma; grade 2, adenoma; grade 3, adenocarcinoma withpleomorphic nuclei or mixed cellular phenotypes; and grade 4, invasiveadenocarcinoma and adenocarcinoma with glandular/acinar architecture.
Deep Sequencing and Bioinformatics Analysis. The genomic DNA was isolatedfrom either freshly dissected lung tumors or lobes using a QIAamp DNA MiniKit (Qiagen). For each target gene or for potential off-target sites, a genomicregion containing the target sequence (the primers are shown in Table S5)was PCR-amplified (KOD-Plus-Neo DNA polymerase; TOYOBO) and gel-purified, followed by high-throughput sequencing analysis (HiSeq 2500;Illumina). Briefly, reference sequences of the genomic region containing thetarget sequence were indexed using the Burrows–Wheeler Aligner (BWA;version 0.7.12) (45), and sequences were aligned with the BWA-maximalexact match algorithm. Samtools (version 1.2) (46) and NGSUtils/BAMutilssoftware (version 0.5.7) (47) were used to detect total mutations and in-delsper position. The datasets used in this study have been deposited in theSequence Read Archive repository under accession no. SRP069330.
Cell Culture, Plasmids, and Lentiviral Infection. Non-small cell lung cancer(NSCLC) cell lines, KrasG12D/+ MEFs, KP cells, KPU cells, and HEK-293T cellswere all cultured in DMEM supplemented with 8% FBS. NSCLC cell lines andHEK-293T cells were obtained from the American Type Culture Collection.KrasG12D/+ MEFs were generated from KrasG12D/+ mice, KP cells were gen-erated from KrasG12D/+;Trp53L/L mice, and KPU cells were generated fromKrasG12D/+;Trp53L/L;UtxL/L mice. Genotyping was performed using the fol-lowing primers: Utx, U1 (5′-AACAAAAACCCAGGCTTTATTCAC-3′) and U2 (5′-AGTTTCAGGATACCTTTACTATAAG-3′). All cell lines used in this study havebeen tested for mycoplasma contamination.
The ORF of an indicated gene was built in expression vector pCDH-EF1-Puro.Lentiviruses were produced by cotransfection of HEK-293T cells with pCDH orLentiCRISPRv2 constructs and packaging vectors (psPAX2 and pMD2.G). Theprogeny viruses released from HEK-293T cells were filtered, collected, and usedto infect NSCLC cell lines, KPU cells, and KrasG12D/+ MEFs. Cells infected withlentiviruses were then selected by appropriate concentration of puromycin.
RT-PCR and Real-Time PCR. Total RNA was isolated using TRIzol reagent(Invitrogen) and retrotranscribed into first-strand cDNA using a RevertAidFirst Strand cDNA Synthesis Kit (Fermentas). The cDNAs were subjected toreal-time PCR with gene-specific primers on a 7500 Fast Real-Time PCR System(Applied Biosystems) using SYBR-Green Master PCR mix (Roche). GAPDH(human) and Actin (mouse) served as internal controls. Primers used for real-timePCR were as follows: UTX, 5′-GACATTGAGGGAAGCTCTCA-3′ and 5′-ACTTGCA-TCAGGTCCTCCAT-3′; PTIP, 5′-ACAATGCACTAGCCTCACACA-3′ and 5′-ACACTGAA-CGGACAGAATCAC-3′; ACP5, 5′-TGCAAGATGAGAATGGCGTG-3′ and 5′-CAA-AGCCACCCAGTGAGTCT-3′; ACACB, 5′-AGAAGACAAGAAGCAGGCAAAC-3′
E3984 | www.pnas.org/cgi/doi/10.1073/pnas.1716589115 Wu et al.
Dow
nloa
ded
by g
uest
on
June
1, 2
020

and 5′-GTAGACTCACGAGATGAGCCA-3′; CLU, 5′-CCAATCAGGGAAGTAAGTACGTC-3′ and 5′-CTTGCGCTCTTCGTTTGTTTT-3′; GAPDH, 5′-AGGTGAAGGTCGGAGTCAAC-3′and 5′-AGTTGAGGTCAATGAAGGGG-3′; Cdkn2a, 5′-CGCAGGTTCTTGGTCACTGT-3′and 5′-TGTTCACGAAAGCCAGAGCG-3′; Cdkn2b, 5′-CCCTGCCACCCTTAC-CAGA-3′ and 5′-CAGATACCTCGCAATGTCACG-3′; and Actin, 5′-CAGCCTTCC-TTCTTGGGTAT-3′ and 5′-GGTCTTTACGGATGTCAACG-3′.
Western Blotting. Western blotting was performed as previously described(19). Briefly, cells were lysed in lysis buffer and subjected to Western blotanalysis with following primary antibodies: UTX (33510; Cell Signaling), EZH2(D221769; Sangon), and ACTIN (A2228; Sigma).
In Vitro Functional Assays. For 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assays, cells were seeded on 96-well plates, and the viability ofcells wasmeasured byMTTdaily for 5 d. For soft agar assays, virally infected cellswere suspended in growth medium with 0.4% agar on top of 1% agar base insix-well plates. After 2–3 wk of culture, colonies were stained with 0.004%crystal violet and counted using ImageJ software (NIH). All experiments wereperformed in triplicate.
In Vivo Treatment Studies. KrasG12D/+ and KrasG12D/+;UtxL/L mice at 6–8 wk ofage were treated with 2 × 107 pfus of Ad-Cre via nasal inhalation. Fortreatment of KU mice with the EZH2 inhibitor JQEZ5, we randomized thesemice into two groups. One group was given the vehicle, while the othergroup was given JQEZ5. After 2 wk of treatment, mice were killed and lungtissues were collected and fixed. For treatment of K mice, we evaluatedthese mice by magnetic resonance imaging (MRI) to determine the tumorvolume. Mice received JQEZ5 by i.p. injection for 2 wk (75 mg/kg each day),as previously reported (29). JQEZ5 was initially dissolved in DMSO as a stockand diluted at a ratio of 1:10 in 10% (2-Hydroxypropyl)-β-cyclodextrin (H107;Sigma-Aldrich) for injection.
MRI Tumor Quantification. Mice were evaluated by MRI to determine thetumor volume. Tumor volume changes were analyzed by Sante DICOMViewer Free software.
IHC. IHC staining was performed as previously described (19). The followingantibodies were used: UTX (33510; Cell Signaling), H3K27me3 (A2363;ABclonal), EZH2 (D221769; Sangon), Ki67 (NCL-Ki67p; Leica Biosystems), andCleaved Caspase-3 (9661; Cell Signaling). With regard to statistical analysis ofH3K27me3 and EZH2 staining, “low” means the percentage of positive cells inone tumor <50% and “high”means the percentage of positive cells in one tumor>50%. Then, the numbers of high and low tumors were counted for analysis.
RNA Sequencing. Total RNA was isolated using TRIzol reagent from tumors ofKrasG12D/+ and KrasG12D/+;UtxL/L mice at 16 wk after Ad-Cre treatment. Se-quencing data have been deposited in the GEO database under the acces-sion no. GSE93302.
Statistical Analysis. All experimental data were analyzed by Student’s t test(two-tailed), and P < 0.05 was considered to be significant. All error barsindicate SEM.
ACKNOWLEDGMENTS. We thank Drs. T. Jacks and K. Wong for providing theKrasG12D/+ and Trp53L/L mice, Dr. R. Depinho for providing the Lkb1L/L mice. Wethank Drs. T. Jacks, W. Xue, and F. J. Sanchez-Rivera for providing the pSECCand LentiCRISPRv2 plasmids. This work was supported by the National BasicResearch Program of China (Grant 2017YFA0505501), Strategic Priority Re-search Program of the Chinese Academy of Sciences (Grant XDB19020201),National Natural Science Foundation of China (Grants 81325015, 81430066,91731314, 31621003, 31370747, 81402276, 81402371, 81401898, 81402498,81101583, and 81372509), Science and Technology Commission of ShanghaiMunicipality (Grant 15XD1504000), China Postdoctoral Science Foundation(Grant 2015M581673), Chinese Academy of Science Taiwan Young ScholarVisiting Program (Grant 2015TW1SB0001), National Science Foundation (Grant81472606), Science and Technology Program of Guangzhou (Grant 201803010124),and National Key R&D Program of China (Grant 2016YFC0905500).
1. Siegel R, Ma J, Zou Z, Jemal A (2014) Cancer statistics, 2014. CA Cancer J Clin 64:9–29.
2. Cancer Genome Atlas Research Network (2012) Comprehensive genomic character-ization of squamous cell lung cancers. Nature 489:519–525, and erratum (2012) 491:288.
3. Cancer Genome Atlas Research Network (2014) Comprehensive molecular profiling oflung adenocarcinoma. Nature 511:543–550, and erratum (2014) 514:262.
4. Cong L, et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science339:819–823.
5. Mali P, et al. (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826.
6. Sánchez-Rivera FJ, et al. (2014) Rapid modelling of cooperating genetic events incancer through somatic genome editing. Nature 516:428–431.
7. Xue W, et al. (2014) CRISPR-mediated direct mutation of cancer genes in the mouseliver. Nature 514:380–384.
8. Ran FA, et al. (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308.
9. Platt RJ, et al. (2014) CRISPR-Cas9 knockin mice for genome editing and cancermodeling. Cell 159:440–455.
10. Ji H, et al. (2007) LKB1 modulates lung cancer differentiation and metastasis. Nature448:807–810.
11. Snyder EL, et al. (2013) Nkx2-1 represses a latent gastric differentiation program inlung adenocarcinoma. Mol Cell 50:185–199.
12. Johnson L, et al. (2001) Somatic activation of the K-ras oncogene causes early onsetlung cancer in mice. Nature 410:1111–1116.
13. Imielinski M, et al. (2012) Mapping the hallmarks of lung adenocarcinoma withmassively parallel sequencing. Cell 150:1107–1120.
14. Shalem O, et al. (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells.Science 343:84–87.
15. Martinez-Garcia E, Licht JD (2010) Deregulation of H3K27 methylation in cancer. NatGenet 42:100–101.
16. Cho YW, et al. (2007) PTIP associates with MLL3- and MLL4-containing histoneH3 lysine 4 methyltransferase complex. J Biol Chem 282:20395–20406.
17. Hong S, et al. (2007) Identification of JmjC domain-containing UTX and JMJD3 ashistone H3 lysine 27 demethylases. Proc Natl Acad Sci USA 104:18439–18444.
18. Wang C, et al. (2012) UTX regulates mesoderm differentiation of embryonic stemcells independent of H3K27 demethylase activity. Proc Natl Acad Sci USA 109:15324–15329.
19. Li F, et al. (2015) LKB1 inactivation elicits a redox imbalance to modulate non-smallcell lung cancer plasticity and therapeutic response. Cancer Cell 27:698–711.
20. DuPage M, Dooley AL, Jacks T (2009) Conditional mouse lung cancer models usingadenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 4:1064–1072.
21. Jackson EL, et al. (2001) Analysis of lung tumor initiation and progression usingconditional expression of oncogenic K-ras. Genes Dev 15:3243–3248.
22. Ler LD, et al. (2017) Loss of tumor suppressor KDM6A amplifies PRC2-regulatedtranscriptional repression in bladder cancer and can be targeted through inhibitionof EZH2. Sci Transl Med 9:eaai8312.
23. Di Croce L, Helin K (2013) Transcriptional regulation by Polycomb group proteins. NatStruct Mol Biol 20:1147–1155.
24. Mills AA (2010) Throwing the cancer switch: Reciprocal roles of polycomb and tri-thorax proteins. Nat Rev Cancer 10:669–682.
25. Li F, et al. (2012) Identification of RET gene fusion by exon array analyses in “pan-negative” lung cancer from never smokers. Cell Res 22:928–931.
26. Wang R, et al. (2015) Comprehensive investigation of oncogenic driver mutations inChinese non-small cell lung cancer patients. Oncotarget 6:34300–34308.
27. Fillmore CM, et al. (2015) EZH2 inhibition sensitizes BRG1 and EGFR mutant lungtumours to TopoII inhibitors. Nature 520:239–242.
28. Mohammad HP, et al. (2015) A DNA hypomethylation signature predicts antitumoractivity of LSD1 inhibitors in SCLC. Cancer Cell 28:57–69.
29. Zhang H, et al. (2016) Oncogenic deregulation of EZH2 as an opportunity for targetedtherapy in lung cancer. Cancer Discov 6:1006–1021.
30. Chen S, et al. (2015) Genome-wide CRISPR screen in a mouse model of tumor growthand metastasis. Cell 160:1246–1260.
31. Kottakis F, Bardeesy N (2012) LKB1-AMPK axis revisited. Cell Res 22:1617–1620.32. Jeon SM, Chandel NS, Hay N (2012) AMPK regulates NADPH homeostasis to promote
tumour cell survival during energy stress. Nature 485:661–665.33. Chan KY, et al. (2005) Transcriptional profiling on chromosome 19p indicated frequent
downregulation of ACP5 expression in hepatocellular carcinoma. Int J Cancer 114:902–908.
34. Panico F, Rizzi F, Fabbri LM, Bettuzzi S, Luppi F (2009) Clusterin (CLU) and lung cancer.Adv Cancer Res 105:63–76.
35. Patel SR, Kim D, Levitan I, Dressler GR (2007) The BRCT-domain containing proteinPTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev Cell 13:580–592.
36. Wang JK, et al. (2010) The histone demethylase UTX enables RB-dependent cell fatecontrol. Genes Dev 24:327–332.
37. Beyaz S, et al. (2017) The histone demethylase UTX regulates the lineage-specific epi-genetic program of invariant natural killer T cells. Nat Immunol 18:184–195.
38. van Haaften G, et al. (2009) Somatic mutations of the histone H3K27 demethylasegene UTX in human cancer. Nat Genet 41:521–523.
39. Ntziachristos P, et al. (2014) Contrasting roles of histone 3 lysine 27 demethylases inacute lymphoblastic leukaemia. Nature 514:513–517.
40. Van der Meulen J, et al. (2015) The H3K27me3 demethylase UTX is a gender-specifictumor suppressor in T-cell acute lymphoblastic leukemia. Blood 125:13–21.
41. SerresiM, et al. (2016) Polycomb repressive complex 2 is a barrier to KRAS-driven inflammationand epithelial-mesenchymal transition in non-small-cell lung cancer. Cancer Cell 29:17–31.
42. Neff T, et al. (2012) Polycomb repressive complex 2 is required for MLL-AF9 leukemia.Proc Natl Acad Sci USA 109:5028–5033.
Wu et al. PNAS | vol. 115 | no. 17 | E3985
CELL
BIOLO
GY
PNASPL
US
Dow
nloa
ded
by g
uest
on
June
1, 2
020

43. Gentles AJ, et al. (2015) The prognostic landscape of genes and infiltrating immunecells across human cancers. Nat Med 21:938–945.
44. Shedden K, et al.; Director’s Challenge Consortium for the Molecular Classifica-tion of Lung Adenocarcinoma (2008) Gene expression-based survival predictionin lung adenocarcinoma: A multi-site, blinded validation study. Nat Med 14:822–827.
45. Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheelertransform. Bioinformatics 26:589–595.
46. Li H, et al.; 1000 Genome Project Data Processing Subgroup (2009) The SequenceAlignment/Map format and SAMtools. Bioinformatics 25:2078–2079.
47. Breese MR, Liu Y (2013) NGSUtils: A software suite for analyzing and manipulatingnext-generation sequencing datasets. Bioinformatics 29:494–496.
E3986 | www.pnas.org/cgi/doi/10.1073/pnas.1716589115 Wu et al.
Dow
nloa
ded
by g
uest
on
June
1, 2
020