inaugural - dissertationarchiv.ub.uni-heidelberg.de/volltextserver/18552/1/dissertation...
TRANSCRIPT

INAUGURAL - DISSERTATION
zur Erlangung der Doktorwürde
der Naturwissenschaftlich-Mathematischen Gesamtfakultät
der Ruprecht - Karls - Universität
Heidelberg
vorgelegt von Diplom-Biologe Thorsten Ruppert
aus Frankenthal (Pfalz) Tag der mündlichen Prüfung: ................................................

Molekularbiologische Ursachen der chronischen tubulointerstitiellen Nephritis bei
Methylmalonazidurie-Patienten
Gutachter: Herr Prof. Dr. rer. nat. Gert Fricker
Herr Prof. Dr. med., Prof. h.c. (RCH) Georg F. Hoffmann

Abstract The aim of this study was to investigate the renal pathogenesis of chronic tubulointerstitial nephritis in patients of methylmalonic aciduria, which causes chronic kidney failure. As a research model we chose proximal tubule epithelial cells of patients of methylmalonic aciduria. Since in some studies a multiple disturbance of mitochondrial energy metabolism has been shown to be the cause of organ failure in patients of methylmalonic aciduria, we focused first on a description of the energy metabolism of the proximal tubule epithelial cells. The decreased activity of phosphofructokinase 1 results in less flux and thus causes a deterioration of glycolysis. In context of the unsuccessful isolation of mitochondria, the decreased activity of mitochondrial aconitase, 2-oxoglutarate-dehydrogenesis complex and cytochrome c oxidase suggest a mitochondrial defect in proximal tubule epithelial cells of patients of methylmalonic aciduria that manifested structurally and functionally. The mitochondrial defect led further to increased ROS production, which could be increased even more by metabolic stress due to the application of glucose, pyruvate, methylmalonic acid, propionic acid and isoleucine. A higher amount of reduced glutathione and a lower amount of free glutathione due to higher concentration of ROS strengthened the hypothesis of increased oxidative stress in proximal tubule epithelial cells of patients of methylmalonic aciduria. Higher concentrations of ROS resulted in concert with increased expression of pIRE1 and degenerated endoplasmic reticulum with enlarged lumen, which were recognizable on electron micrographs, in stress of endoplasmic reticulum in proximal tubule epithelial cells of patients of methylmalonic aciduria. Reduced activity of mTORC1 (mammalian target of rapamycin complex 1) under metabolic stress and disturbances in the energy metabolism in proximal tubule epithelial cells of patients of methylmalonic aciduria indicated a positive regulation of autophagosome formation. In concert, structures surrounded by a double membrane were recognizable on electron micrographs, and the increased expression of P62 and LC3-II under metabolic stress led to the adoption of increased macro-autophagy. This could be confirmed by a higher amount of autolysosomes. It is known that inflammation of a tissue is always accompanied by the infiltration by cells of the immune system. With respect to cytokines required for recruitment of the immune system the expression of interleukin-8 was higher and its apical and basolateral secretion was stronger in proximal tubule epithelial cells of patients of methylmalonic aciduria. The strong correlation between autophagy and the secretion of interleukin-8 in proximal tubule epithelial cells of patients of methylmalonic aciduria pointed out that autophagy is a direct cause of the increased transcription of interleukin-8 or promotes indirectly causes of the transcription of interleukin-8. This can lead to an infiltration of the interstitium of the proximal tubule by neutrophils, macrophages and T cells. The inflammatory response is triggered further and leads over a long period to failure of kidney function.

Übersicht Ziel dieser Arbeit ist die Untersuchung der renalen Pathogenese der chronischen tubulointerstitiellen Nephritis bei Methylmalonazidurie-Patienten, die zur Entwicklung eines chronischen Nierenversagens führt. Als Untersuchungsmodell wählten wir proximale Tubulusepithelzellen von Methylmalonazidurie-Patienten. Da in einigen Studien eine multiple Störung des mitochondrialen Energiestoffwechsels als Ursache des Organversagens bei Methylmalonazidurie-Patienten gezeigt wurde, fokussierten wir uns zunächst auf eine Beschreibung des Energiemetabolismus der proximalen Tubulusepithelzellen. Die verringerte Aktivität der Phosphofruktokinase 1 führt zu einem geringeren Flux und damit zu einer Beeinträchtigung der Glykolyse. In Zusammenhang mit der erfolglosen Isolierung von Mitochondrien, ließ die verminderte Aktivität der mitochondrialen Aconitase, des 2-Oxoglutarat-Dehydrogenasekomplexes und der Cytochrom c-Oxidase auf einen mitochondrialen Defekt in proximale Tubulusepithelzellen von Methylmalonazidurie-Patienten schließen, der sich strukturell und funktionell manifestierte. Der mitochondriale Defekt führte weiterhin zu erhöhter ROS-Produktion, die sich durch metabolischen Stress aufgrund der Applikation von Glukose, Pyruvat, Methylmalonsäure, Propionsäure und Isoleucin steigern ließ. Erhöhtes Glutathiondisulfid und verringertes freies Glutathion aufgrund höherer ROS-Konzentration stärkten die Hypothese erhöhten oxidativen Stresses in proximalen Tubulusepithelzellen von Methylmalonazidurie-Patienten. Die höhere ROS-Konzentration führte im Zusammenspiel mit erhöhter Expression von pIRE1 und degeneriertem endoplasmatischen Retikulum mit vergrößertem Lumen, die auf elektronenmikroskopischen Bildern erkennbar waren, zu Stress des Endoplasmatischen Retikulums in proximalen Tubulusepithelzellen von Methylmalonazidurie-Patienten. Verringerte Aktivität von mTORC1 (mammalian target of rapamycin complex 1) unter metabolischem Stress und die Störungen im Energiemetabolismus deuteten bei proximalen Tubulusepithelzellen von Methylmalonazidurie-Patienten auf eine positive Regulation der Autophagosombildung hin. In diesem Zusammenhang führten von einer Doppelmembran umgebene Strukturen, die auf elektronenmikroskopischen Bildern zu sehen waren, und die erhöhte Expression von P62 und LC3-II unter metabolischem Stress zur Annahme erhöhter Makro-Autophagie. Diese ließ sich durch mehr Autolysosomen bestätigen. Es ist bekannt, dass die Inflammation eines Gewebes immer mit der Infiltrierung durch Zellen des Immunsystems einhergeht. Die Expression der zur Rekrutierung des Immunsystems notwendigen Zytokine war bei proximalen Tubulusepithelzellen von Methylmalonazidurie-Patienten in Form von Interleukin-8 höher, die apikale und basolaterale Sekretion fiel stärker aus. Die starke Korrelation zwischen Autophagie und der Sezernierung von Interleukin-8 bei proximalen Tubulusepithelzellen von Methylmalonazidurie-Patienten deutete darauf hin, dass Autophagie eine direkte Ursache der erhöhten Transkription von Interleukin-8 ist oder indirekt Ursachen der Transkription von Interleukin-8 fördert. Dies kann zu einer Infiltration des Interstitiums des proximalen Tubulus durch Neutrophile, Makrophagen und T-Zellen führen. Die inflammatorische Reaktion wird somit weiter getriggert und führt über längere Zeit zum Versagen der Nierenfunktionen.

Eidesstattliche Versicherung
Hiermit versichere ich eidesstattlich, dass ich die vorliegende Dissertation
selbstständig verfasst und keine anderen als die von mir aufgeführten Hilfsmittel
verwendet habe.
Heidelberg, Januar 2015
Thorsten Ruppert

Danksagung Ich danke:
Herrn Prof. Dr. rer. nat. Gert Fricker und Herrn Prof. Dr. med., Prof. h.c. (RCH) Georg
Hoffmann für die Supervision des Promotionsvorhabens.
Herrn Priv.- Doz. Dr. rer. nat. Sven Sauer für die Bereitstellung des sehr komplexen
und interessanten Themas, die sehr gute persönliche und fachliche Betreuung, die
hilfreichen Denkanstöße und Diskussionen und seine Geduld.
Herrn Prof. Dr. med. Stefan Kölker und Herrn Priv.- Doz. Dr. phil. nat. Jürgen Okun
für die fruchtbaren Diskussionen.
Meinen (ehemaligen) Kolleginnen und Kollegen Silvana, Verena, Shoko, Tunc,
Roland 1, Roland 2, Thorsten 2, Sonja und Helga, sowie den Kolleginnen und
Kollegen der Nachbararbeitsgruppe.
Meinen Eltern für ihre unendliche Unterstützung, Rückendeckung, Vertrauen und
Geduld.
Bianca Baumert und ihren Eltern für ihre Motivation, Unterstützung und Geduld.

Teile dieser Arbeit wurden als Kongressbeiträge vorgestellt: Ruppert T, Opp S, Suormala T, Okun JG, Kölker S, Morath MA, Sauer SW (2011). Isolation and characterization of proximal tubule epithelial cells from urine of methylmalonic aciduria patients. Poster, SSIEM Genf, Schweiz. Ruppert T, Opp S, Suormala T, Okun JG, Kölker S, Morath MA, Sauer SW (2012). Isolation und Charakterisierung von proximalen Tubulusepithelzellen aus dem Urin von Methylmalonazidurie-Patienten. Poster, GPN Heidelberg, Deutschland. Ruppert T, Tuncel AT, Opp S, Suormala T, Okun JG, Kölker S, Morath MA, Sauer SW (2012). Nephrotoxicity of maleic acid and methylmalonic acid - on the difference of Fanconi Syndrome and tubulointerstitial nephritis. Poster, SSIEM Birmingham, England. Ruppert T, Tuncel AT, Opp S, Okun JG, Kölker S, Morath MA, Sauer SW (2013). Nephrotoxizität von Maleinsäure und Methylmalonsäure – Unterschied zwischen Fanconi-Syndrom und tubulointerstitieller Nephritis. Poster, APS Fulda, Deutschland. Ruppert T, Opp S, Tagscherer KE, Kastl L, Gröne HJ, Hoffmann GF, Kölker S, Okun JG, Morath MA, Sauer SW (2013). Activation of autophagic and proinflammatory cascades in proximal tubule of methylmalonic aciduria patients. Poster, ICIEM Barcelona, Spanien.


Inhaltsverzeichnis Verzeichnisse
1 Einleitung 11.1 Angeborene Störungen im zellulären Stoffwechsel 1
1.2 Organoazidurie 1
1.3 Methylmalonazidurie 2
1.4 Propionazidurie 4
1.5 Funktionsweise der Methylmalonyl-CoA-Mutase und beteiligte Co-Faktoren 4
1.6 Tiermodelle zur MMA 6
1.6.1 Die Mut+/- und Mut-/- Maus 6
1.6.2 Artifizielles Rattenmodell durch Applikation von Methylmalonsäure 8
1.7 Renale Manifestation bei MMA-Patienten 8
1.7.1 Aufbau und Funktionen der Niere 8
1.7.2 Eigenschaften und Funktionen von proximalen Tubulusepithelzellen 9
1.7.3 Inflammatorische Funktionen der Niere und Entwicklung einer chronischen
Nephritis 9
1.8 Mögliche Ursachen der cTIN bei MMA-Patienten 12
1.8.1 Stress des endoplasmatischen Retikulums - Unfolded Protein Response 13
1.8.2 Die Regulation von mTOR-Komplex 1 14
1.8.3 Aufbau von mTORC1 und Einfluss auf Autophagie 17
1.8.4 Autophagie 18
1.8.4.1 Makroautophagie 19
1.9 Fragestellung der Arbeit 21
2 Material 232.1 Chemikalien 23
2.2 Geräte 25
2.3 Verbrauchsmaterialien 25
2.4 Zellkulturmedien 26
2.5 Antikörper 26
2.6 ELISA-Kits 27
2.7 Lösungen für proteinbiochemische Methoden 27
3 Methoden 303.1 Zellkultur 30
3.1.1 Isolierung von humanen primären proximalen Tubulusepithelzellen (hPTEC) 30
3.1.2 Kultivierung von eukaryotischen Zellen 30
3.1.3 Kultivieren und Passagieren von PTEC 30

3.1.4 Kultivieren und Passagieren von immortalisierten Zellen 31
3.1.5 Ernten von immortalisierten Zellen 31
3.2 Molekularbiologische Methoden 31
3.2.1 Polymerase-Kettenreaktion 32
3.2.2 Agarosegelelektrophorese 32
3.2.3 Transformation von DNS in kompetente Bakterienzellen wofür 33
3.2.4 Analytische Isolierung von Plasmid-DNS mit dem „QIAprep Spin Miniprep Kit“ 33
3.2.5 Präparative Isolierung von Plasmid-DNS 33
3.2.6 Konzentrationsbestimmung von Nukleinsäuren 34
3.2.7 Herstellung kompetenter Bakterienzellen 34
3.2.8 Transfektion eukaryotischer Zellen zur Immortalisierung 34
3.3 Proteinbiochemische Methoden 35
3.3.1 Proteinbestimmung 35
3.3.2 SDS-PAGE (SDS-Polyacrylamid-Gelelektrophorese) 35
3.3.3 Färbung von Proteingelen durch Coomassie-Brillant Blau 36
3.3.4 Western Blot 36
3.3.5 Semi-Dry Blot 37
3.3.6 Entwicklung von Western Blots mittels Chemilumineszenz 37
3.3.7 Wiederverwendung von Western Blots 38
3.3.8 Enzyme linked immunosorbent assay (ELISA) 38
3.3.9 Expressionsnachweis von Proteinen mit Dotplots 38
3.4 Färbung der Zellen mit Acridinorange und FACS-Analyse 38
3.5 Spektrophotometrische Methoden 39
3.5.1 Subzellulare Fraktionierung durch differentielle Zentrifugation 40
3.5.2 Probenaufarbeitung für Enzyme des Krebszyklus und der Atmungskette 40
3.5.3 Bestimmung der Einzelenzymaktivitäten der Glykolyse 413.5.3.1 Hexokinase 41
3.5.3.2 Phosphofruktokinase 41
3.5.3.3 Triosephosphatisomerase 41
3.5.3.4 Glutaraldehydphosphatdehydrogenase 41
3.5.3.5 Phosphoglyzeratmutase 42
3.5.3.6 Enolase 42
3.5.3.7 Pyruvatkinase (hoch affin) 42
3.5.3.8 Pyruvatkinase (niedrig affin) 42
3.5.3.9 Laktatdehydrogenase 43
3.5.4 Bestimmung der Einzelenzymaktivitäten des Zitrat-Zyklus 43
3.5.4.1 Zitratsynthase 43
3.5.4.2 Mitochondriale Aconitase 43
3.5.4.3 Isozitratdehydrogenase 43

3.5.4.4 Fumarase 44
3.5.4.5 Malatdehydrogenase 44
3.5.4.6 Bestimmung der Aktivität von Pyruvatdehydrogenasekomplex (PDHc) und 2-
Oxoglutaratdehydrogenasekomplex (OGDHc) 44
3.5.5 Bestimmung der Aktivität der Enzyme der Atmungskettenkomplexe 443.5.5.1 Komplex I (NADH-Reduktase) 44
3.5.5.2 Komplex II (Succinat-Dehydrogenase) 45
3.5.5.3 Komplex III (Cytochrom c-Reduktase) 45
3.5.5.4 Komplex IV (Cytochrom c-Oxidase) 45
3.5.5.5 Komplex V (ATP-Synthase) 46
3.6 Bestimmung des mitochondrialen Sauerstoffverbrauchs 46
3.7 ROS-Nachweis durch reduziertes Glutathion 47
3.8 Färbung der Zellen mit H2DCFDA und ROS-Nachweis 47
3.9 Quantitative Analyse von MMA mittels MS/MS 48
3.10 Statistische Auswertung 48
4 Ergebnisse 494.1 Relevante Werte der Zellspender 49
4.2 Charakterisierung der verwendeten humanen PTEC 51
4.3 Produktion von Methylmalonsäure 55
4.4 Bioenergetik 56
4.4.1 Sauerstoffverbrauch der Mitochondrien 56
4.4.2 Glykolyse 57
4.4.3 Pyruvat-Dehydrogenasekomplex 59
4.4.4 Zitrat-Zyklus 60
4.4.5 Oxidative Phosphorylierung 61
4.5 Freisetzung reaktiver Sauerstoffspezies (ROS) 62
4.5.1 Nachweis von ROS durch die Oxidierung von reduziertem Glutathion 62
4.5.2 Nachweis von ROS durch 2’,7’-Dichloro-Dihydrofluoresceindiacetat 63
4.6 Subzellulare Strukturen der humanen PTEC im Elektronenmikroskop 65
4.7 Initiation, Entwicklung und Abbau von Autophagosomen bei PTEC
von MMA-Patienten 68
4.7.1 Regulation der Reifung der Phagophore durch mTORC1 68
4.7.2 Reifung der Phagophore 71
4.7.3 Lysosomaler Abbau des Autophagosoms 74
4.8 Stress des endoplasmatischen Retikulums - unfolded protein response 76
4.9 Inflammation 77
4.9.1 Expressionsvergleich löslicher Rezeptoren 77
4.9.2 Expressionsvergleich von Rezeptoren 79

4.9.3 Qualitativer Nachweis exprimierter Zytokine im Zellmedium 80
4.9.4 Quantitativer Nachweis von Interleukin-8 intrazellulär und im Zellmedium 81
4.10 Korrelationen der Expression von IL8, Generierung von ROS, erhöhter
Autophagie und verringerter Aktivität der Cytochrom c-Oxidase 83
5 Diskussion 855.1 Auswahl des verwendeten Zellkulturmodells 85
5.2 Charakterisierung des verwendeten Zellkulturmodells 86
5.3 PTEC von MMA-Patienten zeigen Defekte im Energiemetabolismus 87
5.4 PTEC von MMA-Patienten haben eine erhöhte ROS-Produktion 91
5.5 PTEC von MMA-Patienten zeigen erhöhte Makro-Autophagie 94
5.6 ER-Stress bei PTEC von MMA-Patienten verstärkt die Autophagie 97
5.7 PTEC von MMA-Patienten exprimieren Zytokine und Rezeptoren
zur Interaktion mit Immunzellen 98
5.8 Korrelation von Autophagie und IL-8-Expression bei PTEC von
MMA-Patienten 101
5.9 Ausblick 103
5.10 Zusammenfassung 105
6 Literaturverzeichnis 109
7 Anhang I7.1 Abkürzungsverzeichnis I
7.2 Tabellarische Auflistung von im Ergebnisteil nur graphisch dargestellter
Messwerte V
7.3 Western Blots der im Ergebnisteil graphisch dargestellten Expression
einzelner Proteine VII

Abbildungsverzeichnis
Abbildung (1) Die Umlagerung von D-Methylmalonyl-CoA zu L-Methylmalonyl-CoA 4
Abbildung (2) Schematische Darstellung der Umlagerungsreaktion von L-Methylmalonyl-CoA zu Succinyl-CoA 6
Abbildung (3) Die Aktivierung und Regulation von mTORC1 16
Abbildung (4) Lichtmikroskopische Aufnahmen aller verwendeten humanen PTEC 53
Abbildung (5) Expression von γ-Glutamyltransferase 1 (γGT1) und Aquaporin 1 (AQP1) der verwendeten PTEC 54
Abbildung (6) MMS-Konzentration im Medium von PTEC von Kontrollen und MMA-Patienten 55
Abbildung (7) Mitochondrialer Sauerstoffverbrauch der humanen PTEC 57
Abbildung (8) Enzymaktivitäten der glykolytischen Enzyme 58
Abbildung (9) Enzymaktivität des PDHc 59
Abbildung (10) Enzymaktivitäten des Zitrat-Zyklus 60
Abbildung (11) Enzymaktivitäten der Atmungskettenkomplexe von PTEC der Kontrollen, PA- und MMA-Patienten 61
Abbildung (12) Glutathion-Gehalt der PTEC von Kontrollen und MMA-Patienten 62
Abbildung (13) Entstehung von ROS 64
Abbildung (14) Elektronenmikroskopische Bilder der PTEC 67
Abbildung (15) Verlauf der Ratio der relativen Expression von pPRAS40/PRAS40 70
Abbildung (16) Verlauf der Ratio der relativen Expression von pmTO/mTOR 71
Abbildung (17) Relative Expression von LC3-II 72
Abbildung (18) Relative Expression von P62 74
Abbildung (19) Ratio der Acridinorangefärbung 75
Abbildung (20) Relative Expression von pIRE1 77
Abbildung (21) Relative Expression potentiell an der Interaktion mit Immunzellen beteiligter Proteine von PTEC der Kontrollen und MMA-Patienten 78

Abbildung (22) Expressionsvergleich der Immunrezeptoren CD46, CD55, CD59 und CD88 80
Abbildung (23) Relative Konzentration von IL-8 im Medium 81
Abbildung (24) Konzentration von Interleukin-8 in apikalem und basolateralem Medium 82
Abbildung (25) Zusammenwirkender Mechanismus der molekularbiologischen Ursachen der chronischen tubulointerstitiellen Nephritis in PTEC von MMA-Patienten 107
Tabellenverzeichnis
Tabelle (1) Zusammensetzung von Sammel- und Trenngelen für SDS-PAGE 36
Tabelle (2) Übersichtsdaten von Kontrollen, MMA- und PA-Patienten 49
Tabelle (3) Funktionen der untersuchten Immunrezeptoren CD46, CD55, CD59 und CD88 79
Tabelle (4) Schwellenwerte für die Stärke des Zusammenhangs zwischen 2 Variablen 83
Tabelle (5) Bestimmtheitsmaß und Zusammenhang der Korrelationsvariablen 84

1 Einleitung
1.1 Angeborene Störungen im zellulären Stoffwechsel
Als angeborene Störung im zellulären Stoffwechsel wird eine Gruppe von erblichen
Defekten bezeichnet, die bei Neugeborenen mit einer kumulativen Inzidenz von etwa
1:500 auftritt (Zschocke und Hoffmann, 2004). In der Medizin stellt die Erforschung
der angeborenen Stoffwechseldefekte ein relativ junges Gebiet dar, welches mit der
Entwicklung neuer diagnostischer Methoden etabliert wurde. Für Patienten sind die
Folgen unbehandelter Stoffwechselstörungen oft dramatisch, da häufig bereits kurz
nach der Geburt die klinische Symptomatik auftritt und sich ohne adäquate Therapie
rasch verschlechtert. Somit ist eine frühzeitige Diagnose der Störung und ihrer
unmittelbaren Behandlung von großer Wichtigkeit, da viele Defekte schon in den
ersten Lebensmonaten durch irreversible Schädigungen von Organen den
Krankheitsverlauf charakterisieren. Durch die Etablierung des
Neugeborenenscreenings konnten schon große Fortschritte zur Früherkennung von
angeborenen Stoffwechseldefekten erzielt werden. Da schon wenige Blutstropfen
ausreichen um mit der Massenspektrometrie Defekte zu erkennen und eine frühe
Behandlung zu ermöglichen, können so Organschäden oft verhindert werden. Zur
Entwicklung von Therapien für noch nicht oder schlecht behandelbare
Stoffwechselstörungen ist jedoch zudem die Entschlüsselung der zugrunde
liegenden Pathomechanismen notwendig.
1.2 Organoazidurie
Der Begriff Organoazidurie ist eine Sammelbezeichnung für eine heterogene Gruppe
von Stoffwechselerkrankungen, bei welchen der Abbau von Aminosäuren oder
Fettsäuren betroffen ist. Verschiedene Stoffwechselintermediate können nicht oder
nur unvollständig abgebaut werden und es kommt durch die Akkumulierung
organischer Säuren zu einer Intoxikation des gesamten Organismus, was auf
verschiedenen Ebenen Auswirkungen hat. Die Auswirkungen auf den
Gesamtorganismus sind im klinischen Bereich Hyperammonämie, Hypoglykämie,
Laktatazidose, Ketose oder Carnitinmangel. Typisch für den Verlauf der
Organoazidurien ist eine neonatale Stoffwechselkrise, welche wenige Stunden oder

Tage nach der Nahrungsaufnahme zu schweren metabolischen Entgleisungen
führen kann. Im weiteren Verlauf treten häufig wiederkehrende Krisen sowie mentale
oder Wachstumsretardierungen auf.
Die Organoazidurien lassen sich in sogenannte klassische Organoazidurien und
zerebrale Organoazidurien unterteilen. Zu den zerebralen Organoazidurien gehören
u.a. die Glutarazidurie Typ 1, D-2- und L-2-Hydroxyglutarazidurie. Die klassischen
Organoazidurien umfassen die Propionazidurie, Isovalerianazidurie und
Methylmalonazidurie.
1.3 Methylmalonazidurie
Zuerst beschrieben wurden die Auswirkungen der Methylmalonazidurie (MMA) 1967
von Oberholzer und Kollegen. Die MMA bilden eine Gruppe von ätiologisch
heterogenen, angeborenen Stoffwechseldefekten, welche sich im Abbau
verzweigtkettiger Aminosäuren, ungeradzahliger Fettsäuren und der Cholesterol-
seitenkette zeigen. Sie treten in einer kumulativen Inzidenz von 1:50.000 – 1:100.000
auf (Sniderman et al., 1999). Auf molekularer Ebene liegen die Ursachen der
autosomal-rezessiv vererbten Defekte der MMA bei einem Funktionsverlust des
mitochondrialen Enzyms Methylmalonyl-CoA-Mutase (MCM; EC 5.4.99.2, Ledley et
al., 1988), des Cobalamintransports oder der Synthese von 5´-
Deoxyadenosylcobalamin, welches als Cofaktor der MCM (Mahoney et al., 1975)
fungiert. Ist die MCM unmittelbar betroffen, liegt eine isolierte MMA vor, wobei der
Funktionsverlust komplett (mut0) oder partiell (mut-) sein kann. Bei einem Defekt im
Stoffwechsel des Cofaktors von MCM (CblA, CblB, CblC, CblD) kann die
Enzymaktivität durch Applikation von Hydroxycobalamin gesteigert werden.
Methylmalonyl-CoA wird von der MCM katalytisch zu Succinyl-CoA metabolisiert,
welches als Succinat den Zitrat-Zyklus speist und somit der Energieversorgung der
Zelle dient. Die Akkumulation von Methylmalonsäure (MMS) und den Metaboliten 3-
Hydroxypropionsäure, 2-Methylzitronensäure des alternativen Propionat-
Stoffwechselwegs tritt bei allen Formen der MMA auf (Fenton et al., 2001). Patienten
entwickeln häufig durch katabolen Stress metabolische Entgleisungen (z.B. im
Rahmen von fieberhaften Infektionskrankheiten), welche durch Laktatazidose,
Hypoglykämie, Hyperketose und Hyperammonämie gekennzeichnet sind. Bei
verzögertem oder ausbleibendem Therapiebeginn ist meist eine starke

Verschlechterung der Patienten zu beobachten, was über Multiorganversagen bis
zum Tod führen kann. Zudem treten schwere chronische Schädigungen des ZNS
und der Nieren, sowie anderer Organe (Herz, Pankreas, Auge) auf. Bei neonatalen
Patienten zeigen sich oft Episoden von Lethargie, Erbrechen, Hypotonie,
Hyperthermie, Atemproblemen und Thrombozytopenie.
Beim Pathomechanismus auf der Ebene des Energiemetabolismus zeigt MMS keine
direkte inhibitorische Effekte (Okun et al., 2002, Kölker et al., 2003). Weitere
Metabolite wie Propionyl-CoA und 2-Methylzitrat hemmen jedoch synergistisch den
mitochondrialen Energiestoffwechsel durch die Inhibition des Zitrat-Zyklus, des
Pyruvatdehydrogenase-Komplexes und der Atmungskette (Okun et al., 2002; Kölker
et al., 2003; Schwab et al., 2006; Morath et al., 2008).
Bei Patienten mit isolierter MMA, bei welchen die Krankheit durch einen partiellen
(mut-) oder kompletten (mut0) Mutasedefekt oder durch angeborene Defekte im
Cobalaminstoffwechsel (CblA, CblB) verursacht werden, entwickelt sich häufig eine
chronische tubulointerstitielle Nephritis (cTIN). Akuten schweren, metabolischen
Krisen wird mit einer Kurzzeit-Therapie oder einer Langzeittherapie mit metabolischer
Dauerbehandlung begegnet.
Die Kurzzeittherapie sieht eine Gabe von 1) L-Carnitin, 2) Antibiotika, sowie
zusätzlich 3) eine Gabe von Glukose und Insulin zur Energieversorgung, 4) einen
Stopp der Proteinzufuhr vor, um die Krise zu mildern. Das Ziel ist die Minimierung
des Katabolismus und die Förderung des Anabolismus durch Glukose bei
gleichzeitiger Reduzierung der Proteinzufuhr. Die Langzeit-Therapie umschließt 1)
eine proteinarme Diät, 2) eine Aminosäuremischung, die eine Zufuhr der
verzweigtkettigen Aminosäuren Leucin, Isoleucin, Valin und Threonin minimiert, 3)
eine Supplementation von L-Carnitin, das toxisches Acyl-CoA bindet und aus der
Zelle transportiert werden kann, 4) eine Supplementation von Hydroxycobalamin bei
cobalaminsensitiven Patienten, 5) Gabe von Antibiotika gegen propionsäurebildende
Bakterien im Darm (Morath et al., 2013).
Trotz allem führt beim Großteil der Patienten die cTIN zu einer chronischen
Niereninsuffizienz und weitergehend zur Dialysepflicht (Rötig et al., 1995; Buemi et
al., 1997; Hörster et al., 2009; Zwickler et al., 2008). Patienten mit mut0- und CblB-
Defekt zeigen die größte Anfälligkeit für die Entwicklung einer cTIN (Hörster et al.,
2007).

1.4 Propionazidurie
Patienten der Propionazidurie (PA) besitzen ähnliche Symptome wie Patienten der
MMA. Bei ihnen ist der Enzymdefekt in der Propionyl-CoA-Carboxylase lokalisiert,
die einen enzymatischen Schritt vor der Methylmalonyl-CoA-Mutase liegt. Die
Propionyl-CoA-Carboxylase verwendet Biotin als Co-Faktor und katalysiert die
Umwandlung von Propionyl-CoA zu D-Methylmalonyl-CoA (Deodato et al., 2006).
Systematische Studien über eine cTIN bei PA-Patienten wurden noch nicht
durchgeführt, es existieren nur vereinzelte Berichte und Fallbeschreibungen über
Patienten, die im Verlauf des Erwachsenenalters eine Niereninsuffizienz entwickeln
haben. Der Hauptunterschied zwischen dem Verlauf der MMA und der PA ist somit
das Auftreten der cTIN bei MMA-Patienten.
1.5 Funktionsweise der Methylmalonyl-CoA-Mutase und beteiligte Co-Faktoren
Die Methylmalonyl-CoA-Mutase (EC 5.4.99.2) (MCM) gehört zur Gruppe der
Isomerasen und katalysiert in den Mitochondrien die Umlagerungsreaktion von L-
Methylmalonyl-CoA zu Succinyl-CoA, das nach Abspaltung von CoA als Succinat in
den Zitrat-Zyklus einfließt. Die in Abbildung (1) gezeigte Reaktion ist reversibel, läuft
aber im Normalfall nur in eine Richtung.
Abbildung (1) Die Umlagerung von D-Methylmalonyl-CoA zu L-Methylmalonyl-CoA wird durch die Methylmalonyl-CoA Isomerase katalysiert. Im anschließenden Schritt katalysiert die Methylmalonyl-CoA Mutase mit dem Cofaktor Adenosylcobalamin die Isomerisierung von L-Methylmalonyl-CoA zu Succinyl-CoA.
Das 750 Aminosäuren große Protein wird beim Menschen vom Mut-Gen codiert und
ist auf Chromosom 6 lokalisiert.

Der zur Reaktion benötigte Cofaktor ist Adenosylcobalamin (AdoB12, Vitamin B12). Da
Cobalamin (Cbl) nicht von Menschen, Tieren und Pflanzen, sondern nur von
Bakterien gebildet werden kann, bezieht der Körper das benötigte Cobalamin von
Bakterien aus dem Verdauungstrakt. Cobalamin ist der einzige bekannte Naturstoff
mit einem Cobaltatom. Das sechswertige Cobaltatom ist von fünf Stickstoffatomen
umgeben und einem variablen Rest. Anhand des Rests lässt sich zwischen Cyano-
Cbl (R= -C≡N), Aquocobalamin (R= -OH2), Hydroxy-Cbl (R= -OH) und Methyl-Cbl
(R= -CH3) unterscheiden.
Der katalytische Mechanismus ist in Abbildung (2) dargestellt und beginnt mit der
homolytischen Spaltung der C-Co(III) Bindung, wobei Kohlenstoff- und Cobaltatom je
ein Elektron behalten. Das Cobaltatom wechselt daraufhin zwischen den zwei
Oxidationszuständen Co(III) und Co(II). Im katalytischen Prozess stellt AdoB12 somit
reversibel freie Radikale zur Verfügung, welche die Umlagerungsreaktion einleiten
(Abbildung (2)). Das freie Elektron des Ethylrests des AdoB12 zieht ein Elektron mit
Wasserstoffatom vom Methylrest des L-Methylmalonyl-CoA so dass ein Methylrest
entsteht. Das dadurch freigewordene Elektron des C-Atoms des entstandenen
Ethylrests von L-Methylmalonyl-CoA geht eine lose Bindung mit dem Co(II)-Atom von
AdoB12 ein, das dadurch in den Co(III)-Zustand übergeht. Das Elektronenpaar der
Kohlenstoffbindung zwischen C2- und C3-Atom wird zum Ethylrest gezogen (2). Das
freie Elektron springt vom Ethylrest zum Sauerstoff. Dadurch rückt ein
Elektronenpaar der Doppelbindung zwischen C3- und Sauerstoffatom zum C-Atom
des Ethylrests und die leichte Bindung geht vom Ethylrest des L-Methylmalonyl-CoA
und das Co(III)-Atom in den Co(II)-Zustand über und löst sich(3). Das Elektronenpaar
zwischen C2- und C3-Atom füllt die Doppelbindung zwischen C3-Atom und
Sauerstoffatom wieder auf. Das C2-Atom geht eine schwache Bindung mit dem
Co(II)-Atom ein, die Bindung zum C3-Atom besteht nur noch durch ein freies Elektron
(4). Das Wasserstoffatom welches zu Beginn vom Methylrest von L-Methylmalonyl-
CoA auf Adenosylcobalamin übertragen wurde, geht nun auf das C3-Atom von L-
Methylmalonyl-CoA über. Die Bindung zwischen Ethylrest und C3-Atom bildet sich
aus und das freie Elektron geht wieder auf das Co(II)-Atom des Adenosylcobalamin
über, so dass sich die Oxidationsstufe wieder in de Co(III)-Zustand ändert und das
Co(III)-Atom wieder eine Bindung mit dem Ethylrest des Adenosylcobalamins eingeht
und der Zyklus von neuem beginnen kann (5).

Abbildung (2) Schematische Darstellung der Umlagerungsreaktion von L-Methylmalonyl-CoA zu Succinyl-CoA durch die Methylmalonyl-CoA-Mutase und den Cofaktor Adenosylcobalamin.
1.6 Tiermodelle zur MMA
Das murine Mut-Gen liegt auf Chromosom 17 und weißt eine Homologie von 94 %
zum humanen Gen auf. Das Gen codiert für ein 748 Aminosäuren langes Protein mit
einer 30 Aminosäuren langen mitochondrialen Zielsequenz. Die murine und humane
MCM zeigen eine ähnlich hohe Aktivität. Die Aktivität wird indirekt über den 14C-
Einbau in Propionat ermittelt und liegt bei etwa 230 nmol/µmol Leucin (Wilkemeyer et
al., 1990).
1.6.1 Die Mut+/- und Mut-/- Maus
Zur Ursachenforschung der MMA existieren mehrere Mausmodelle mit
verschiedenen Charakteristiken. Zur Generierung von heterozygoten (Mut+/-) und
homozygoten (Mut-/-) Knock out-Mäusen kann das Mut-Gen auf unterschiedliche
Weise zerstört werden.

Durch Zerstörung des Mut-Gens im Bereich der Bindungsstelle für Coenzym A
wurden heterozygote Knock out-Mäuse (Mut+/-) generiert, die einen unveränderten
Phänotyp mit normaler Entwicklung haben und fruchtbar sind. Durch heterozygotes
Kreuzen erzeugte Nachkommen waren nach der Geburt unauffällig, erkrankten
jedoch nach 15 Stunden und verstarben nach 24 Stunden. MMS-Level im Urin und
die Werte von Propionylcarnitin waren erhöht. Frühes Versterben der Mäuse
verhinderte eine Beobachtung und Untersuchung von Langzeitfolgen (Peters et al.,
2003). Ein modifiziertes Knock out-Mausmodell, welches die humane MCM
hemizygot (Mut-/- Mut2h, 4h, 6h) oder homozygot (Mut-/- Mut2h/2h, 4h/4h, 6h/6h) exprimierte,
führte zum Überleben der neonatalen Periode und ermöglichte eine Beobachtung der
Entwicklung der MMA über eine längere Zeit. Durch die Expression der MCM wurde
der MMS-Spiegel auf einem tolerierbaren Niveau gehalten, die renale Dysfunktion
wurde jedoch nicht verbessert. Untersuchungen des proximalen Tubulus der Mäuse
zeigten vergrößerte Mitochondrien mit verminderter Cristae-Struktur, was auf eine
Dysfunktion der Mitochondrien hindeutete. Eine Inflammation oder cTIN konnte
jedoch nicht beobachtet werden (Peters et al., 2003).
Da die Mäuse ohne MCM die neonatale Phase nicht überlebten, war ein weiterer
Ansatz die stabile MCM-Expression in der Leber. Ein erhöhter MMS-Spiegel bei
tragenden Muttertieren von homozygoten (Mut-/-)-Mäusen, deutete auf pränatalen
Stoffwechsel und Intoxikationen der Föten hin. Depletionen von reduziertem
Glutathion und disulfidgebundenem Glutathion bei Hepatozyten wiesen auf radikale
Sauerstoffspezies (ROS) hin. In einigen Geweben mit hohem Energieumsatz (z.B.
proximale Tubuluszellen, Pankreaszellen und Leberzellen) waren
Megamitochondrien erkennbar, in anderen Geweben mit hohem Energieumsatz (z.B.
Herz- und Skelettmuskelzellen) nicht (Chandler et al., 2009).
Bei einem optimierten Modell von Peters und Kollegen wurde in (Mut-/-)-Mäusen die
MCM transgen unter dem Albuminpromotor stabil in der Leber exprimiert. Die
neonatale Mortalität konnte dadurch verhindert werden. Männliche Tiere verstarben
jedoch nach 2 Monaten, bei Weibchen trat nur ein Gewichtsverlust auf und die renale
Dysfunktion blieb bestehen. Nach proteinreicher Diät entwickelte sich eine
chronische Niereninsuffizienz und es kam zu einer Erhöhung der MMS-Werte im
Plasma und im Hirn, sowie zu einer Verminderung der glomerulären Filtrationsrate
(GFR). Zudem waren vergrößerte Mitochondrien des proximalen Tubulus mit
verkürzten Cristae und elektronendichten Partikel erkennbar; Glomeruli, Podozyten

und Hepatozyten waren unverändert. Antioxidationsmittel im Futter senkten den
Gewichtsverlust und die Verringerung der GFR. Dies deutete ebenfalls auf das
Vorkommen von ROS hin. Die Genexpression in Hinblick auf alternative,
lektinaktivierte und klassische Aktivierung des Komplementsystems in der Niere war
erhöht (Manoli et al., 2013).
1.6.2 Artifizielles Rattenmodell durch Applikation von Methylmalonsäure
Die subkutane Applikation von MMS bei Ratten stellt ein weiteres Tiermodell dar.
Nach 4 Wochen war ein Anstieg der proinflammatorischen Faktoren TNF-α und
TNF-β erkennbar. Dies deutete auf den Beginn einer Inflammation und auf
Differenzierungsprozesse im renalen Kortex hin. Beide Faktoren sind an der
Modulation der Zellproliferation und durch Stimulation der Zytokinsezernierung an der
Immunantwort beteiligt (Goyenechea et al., 2012).
1.7 Renale Manifestation bei MMA-Patienten
Bei MMA-Patienten (mut0, mut-, CblA, CblB) entwickelt sich im Verlauf der
Pathogenese eine chronische tubulointerstitielle Nephritis (cTIN) mit Ursprung im
Interstitium des proximalen Tubulus. Im Verlauf der Erkrankung wird die Funktion der
ganzen Niere geschädigt so dass es zu einer chronischen Niereninsuffizienz und
Dialysepflichtigkeit kommt (Rötig et al., 1995; Buemi et al., 1997; Hörster et al., 2007,
2009; Zwickler et al. 2008). Die Folge ist der Verlust einer oder beider Nieren, so
dass eine Transplantation notwendig wird.
1.7.1 Aufbau und Funktionen der Niere
Die Niere kommt in allen Wirbeltieren vor und ist ein paarweise angelegtes Organ.
Beim Menschen hat die Niere etwa die Größe und Form einer Bohne (engl.: kidney).
Sie setzt sich von außen nach innen durch die Nierenrinde, dem äußeren und dem
inneren Nierenmark zusammen.
Das Nephron ist die Grundfunktionseinheit der Niere und besteht aus dem in der
Rinde lokalisierten Glomerulus und dem Tubulus, der das äußere und teilweise das

innere Mark durchzieht. Beim Menschen besitzt jede Niere etwa 1-1,4 Millionen
Nephrone (Klinke & Silbernagl, Lehrbuch der Physiologie, 4. Auflage, 2003).
1.7.2 Eigenschaften und Funktionen von proximalen Tubulusepithelzellen
In der Niere ist das proximale Tubulusepithel für die Resorption filtrierter Substanzen
und die Sekretion von Abfallprodukten und Xenobiotika verantwortlich. Viele lösliche
Substanzen wie Phosphat, Urat, Glukose und Aminosäuren werden im Glomerulus
filtriert und im proximalen Tubulus durch elektrochemisch getriebenen
carriervermittelten Transport resorbiert (Madsen et al., 2008). Andere Bestandteile
des glomerulären Filtrats wie Albumin und Proteine mit geringem Molekulargewicht
werden durch rezeptorvermittelte Endozytose wieder aufgenommen (Gekle, 2005).
Die Exkretion metabolischer Abfallprodukte oder Medikamente und ihre
umgewandelten Moleküle erfolgt durch viele organische Ionentransporter. Sie
steuern den Influx aus dem Blut an der basolateralen Membran und den
anschließenden Efflux über die apikale Membran der proximalen
Tubulusepithelzellen (PTEC). Einige Transporter gehören zur solute carrier (SLC)
und ATP-binding cassette (ABC) Familie. Sie werden in PTEC exprimiert und
vermitteln die renale Ausscheidung verschiedener endogener und exogener
Moleküle. ABC-Transporter sind primäraktiv und verwenden ATP als Energiequelle
für den Efflux von Medikamenten. Viele Mitglieder der SLC-Familie sind
sekundäraktiv und koppeln den Transport ihrer Substrate an Diffusion oder Symport
bzw. Antiport mit anorganischen oder organischen Molekülen (Russel et al., 2002).
Durch primär aktiven Transport können sekundär aktive Transporter einen
Konzentrationsgradienten schaffen, der anschließend durch tertiär aktiven Transport
genutzt werden kann.
1.7.3 Inflammatorische Funktionen der Niere und Entwicklung einer chronischen Nephritis
Eine chronische Nephritis entwickelt sich über Monate oder Jahre, ist durch eine
fortschreitende Verringerung der GFR charakterisiert und mit vielen Symptomen
renaler tubulärer Dysfunktion verbunden. Primäre chronische interstitielle Nephritis ist
mit einer Infektion durch das Epstein-Barr Virus assoziiert, sekundäre chronische

interstitielle Nephritis mit renaler tubulärer Schädigung durch verschiedene andere
Ursachen wie Infektionen, Medikamente, Toxine, hämolytische/neoplastische
Erkrankungen, immunvermittelte Erkrankungen, genetische oder metabolische
Erkrankungen. Die häufigsten Ursachen sind Schädigungen durch Diabetes mellitus
Typ II (20 %; diabetische Nephropathie), Entzündung der Glomeruli (20 %;
chronische Glomerulonephritis), sowie tubulointerstitielle Nephritis (TIN; 15 %)
(Renz-Polster et al., 2011).
Eine TIN liegt bei einer entzündlichen Erkrankung des interstitiellen Gewebes des
proximalen Tubulus vor. Für die Entstehung der chronischen TIN (cTIN) mit
terminaler Niereninsuffizienz wurden zwei Modelle mit Schwerpunkt auf
Tubulointerstitium und Glomerulus entwickelt (Kriz und LeHir, 2005). Zum einen kann
ein Verlust an funktionellen Nephronen zu einer Überlastung noch funktionierender
Nephrone führen. Die Überlastung führt zu Ausfällen weiterer Nephrone und
schließlich zur Niereninsuffizienz. Zum anderen können verschiedene Einflüsse wie
verringertes interstitielles Volumen, Fibrose, eine Abnahme peritubulärer Kapillaren
und morphologische Veränderungen tubulärer Epithelzellen mit der Intensität der
cTIN korreliert werden (Bohle et al., 1990).
Der proximale Tubulus ist aktiv in die Modulation des Immunsystems durch die
Sekretion von Cytokinen und die Aktivierung des Komplementsystems, v.a. des C-
Systems involviert (Sacks et al. 1996; Daha und van Kooten, 2000; Quigg 2003;
Eardley und Cockwell 2005). Bei einer chronischen Entzündung des Interstitiums im
proximalen Tubulus kommt es zur Infiltration durch Makrophagen und anderen Zellen
des Immunsystems. Neutrophile, Basophile und T-Zellen können durch Sekretion
von Cytokinen wie Interleukin 8 (IL-8) durch Epithelzellen rekrutiert und aktiviert
werden.
Im Interstitium der Niere befinden sich auch unter physiologischen und nicht-
inflammatorischen Bedingungen Zellen des Immunsystems, wie dendritische Zellen
und Makrophagen, aber auch Lymphozyten, die jedoch nicht aktiv sind (Kaissling &
Le Hir, 1994; Kruger et al., 2004; Soos et al., 2006). Dendritische Zellen leiten die
primäre Immunantwort ein (Mellman et al., 2001). Ihre Vorläufer werden im
Knochenmark gebildet und in den Blutkreislauf entlassen. Von dort aus können sie
verschiedene Gewebe infiltrieren (Roake et al., 1995; del Hoyo et al., 2002). In
tubulointerstitiellen Regionen der humanen Niere konnten verschiedene Arten

dendritischer Zellen nachgewiesen werden, wobei myeloide dendritische Zellen in
höherer Anzahl vorkommen als plasmazytoide (Woltmann et al., 2007).
Zu Beginn der Inflammation kommt es zunächst zur Aktivierung residenter
Immunzellen im Interstitium der Niere, sowie zur weiteren Infiltration des Interstitiums
durch Lymphozyten, Monozyten und Makrophagen, je nach Ätiologie akkumulieren
auch Neutrophile, Eosinophile oder Plasmazellen. Die Folgen sind interstitielle
Fibrose, tubuläre Atrophie, abgeflachtes Epithel, tubuläre Dilation und Verdickung
der tubulären Basismembran. Indirekt kann es zum Verlust von Glomeruli kommen
(Glomerulosklerose). Hyperkalämie kann zur Bildung medullärer Mikrozysten führen
(Braden et al., 2005). Am Ende der chronischen Niereninsuffizienz tritt das
vollständige Versagen der Nierenfunktion ein und es kommt zur terminalen
Niereninsuffizienz (Baenkler et al.: Innere Medizin, Thieme Verlag. 2. Auflage 2009).
Die Niereninsuffizienz wird in fünf Stadien eingeteilt, die sich an der GFR orientieren.
In Stadium 1 beträgt die GFR noch mehr als 90ml/min und es kommt zu keinen
großen Auffälligkeiten. Kreatinin und Eiweißwerte können im Urin leicht erhöht sein.
In Stadium 2 sinkt die GFR auf 60 – 89 ml/min, es sind jedoch immer noch keine
großen Auffälligkeiten im Blut erkennbar, so dass nur gezielte Untersuchungen eine
gestörte Filterleistung erkennen lassen.
In Stadium 3 sinkt die GFR weiter bis auf 30 – 59 ml/min ab und die Blutwerte von
Kreatinin und Harnstoff steigen an. Die Folgen sind Bluthochdruck, Leistungsabfall
und rasche Ermüdbarkeit, sowie eine höhere Wahrscheinlichkeit für Herz-Kreislauf-
Erkrankungen. Um Nebenwirkungen zu vermeiden muss die Dosis von
Medikamenten, die normalerweise über die Nieren ausgeschieden werden, verringert
werden.
In Stadium 4 erreicht die GFR nur noch 15-29 ml/min, so dass stärkere Beschwerden
wie etwa Übelkeit, Erbrechen, Müdigkeit, Nerven- und Knochenschmerzen auftreten.
Zusätzlich können sich Ödeme im Gesicht oder an den Beinen bilden.
In Stadium 5 sinkt die GFR auf unter 15 ml/ml was auch als terminale
Niereninsuffizienz bezeichnet wird. Es kommt zum Ausfall der Nierenfunktion, das
Blut kann nicht mehr gereinigt werden und eine Vergiftung des Körpers mit
harnpflichtigen Substanzen tritt ein. Ohne Nierenersatzverfahren wie Blutwäsche
(Hämodialyse, HD), Bauchwäsche (Peritonealdialyse, PD) oder Nierentransplantation
kommt es zum Tod des Patienten.

1.8 Mögliche Ursachen der cTIN bei MMA-Patienten
Die renale Pathogenese der cTIN bei MMA-Patienten ist bisher noch weitgehend
unerforscht. Der Energiehaushalt ihrer Zellen ist auf mehreren Ebenen gestört (Okun
et al., 2002; Kölker et al., 2003; Schwab et al., 2006; Morath et al., 2008), was zu
Energiemangel führen kann. Patienten anderer Erkrankungen mit Defekten der
mitochondrialen Atmungskette entwickeln häufig eine chronische renale Schädigung.
Die verringerte mitochondriale Aktivität tritt in Verbindung mit erhöhten ROS-Werten
auf (Brasil et al., 2014). Durch mitochondrial generierte ROS erhöht sich die
Expression von TGF-β in PTEC und führt zu einer akuten tubulointerstitiellen
Nephritis (aTIN), welche sich zu einer cTIN entwickeln kann (Gentle et al., 2013).
ROS können zudem zu einer Entwicklung von Stress des endoplasmatischen
Retikulums (ER) führen, was eine Einleitung der unfolded protein response (UPR)
zur Folge hat. Eine weitere Ursache der Einleitung der UPR könnte der Verlust der
korrekten Faltung mit damit verbundenem kompletten (mut0) oder partiellen (mut-)
Aktivitätsverlust der MCM aufgrund der Mutation des Gens sein (Buck et al., 2012).
Fehlgefaltete Proteine werden im Proteasom der Zelle abgebaut. Eine genaue
Untersuchung von ER-Stress und UPR hat viele Verbindungen zu wichtigen
entzündungs- und stressinduzierten Signalwegen aufgezeigt. Dazu gehören die
Aktivierung der JNK-AP1 und NF-kB-IKK Signalwege (Deng et al., 2004; Hu et al.,
2006) und die Produktion von ROS und Nitritoxid (Cullinan & Diehl, 2006; Gotoh &
Mori, 2006). In Zellkultur löst die experimentelle Induktion der UPR eine erhöhte
Expression der proinflammatorischen Moleküle IL-8, IL-6, MCP-1 und TNF-α aus (Li
et al., 2005).
Die Störung des Energiehaushalts kann weiterhin einen Einfluss auf den mTOR-
Signalweg (mammalian target of rapamycin) ausüben, welcher in 2 Komplexen
organisiert ist. mTORC1 ist ein Sensor für die Verfügbarkeit von Energie und
(verzeigtkettige) Aminosäuren. Bei MMA-Patienten ist der Abbau von
verzweigtkettigen Aminosäuren, ungeradzahligen Fettsäuren und Cholesterol gestört
(Sniderman et al., 1999). Durch ausreichend Energie und Aminosäuren wird
mTORC1 aktiviert, bei Mangel wird mTORC1 inaktiviert, was zur Stimulation von
Autophagie führen kann (Guertin und Sabatini, 2007). Zusätzlich können ROS und
ER-Stress zu erhöhter Autophagie führen (Yorimitsu et al., 2006; Djavaheri-Mergny
et al., 2007).

Der Einfluss von Autophagie auf inflammatorische Prozesse ist noch weitgehend
ungeklärt und wird kontrovers diskutiert. Einerseits übt Autophagie eine negative
Regulation auf die Prozession und Sekretion von IL-1β und IL-18, sowie die
Sekretion von IL-1α in Makrophagen und dendritischen Zellen aus. Andererseits
reguliert Autophagie die Transkription und Sekretion von TNF-α, IL-8 und eventuell
IL-6 positiv, was eine erhöhte Cytokinsekretion zur Folge haben kann (Harris, 2011).
Proximale Tubulusepithelzellen sezernieren im Fall einer Inflammation IL-8.
Daraufhin infiltrieren Zellen des Immunsystems das Interstitium (Baggiolini et al.,
1989 ; Larsen et al., 1989; Baggiolini & Clark-Lewis 1992; Gerritsma et al., 1996).
TNF-α ist für vielfältige Effekte verantwortlich, z.B. die Expression der
Adhesionsmoleküle ICAM-1, VCAM-1 und E-Selectin bei Endothelzellen oder die
Aktivierung von Leukozyten und die Induktion der Expression von MHC-I und MHC-II.
Weiterhin wird die Sekretion von Cytokinen wie MCP-1, RANTES, IL-1, IL-6, IL-8 und
dem Komplementfaktor C3, von Prostaglandinen, Leukotrienen, Nitritoxid und ROS
stimuliert (Ernandez & Mayadas, 2009). In Kombination führt dies zu einer Infiltration
des Interstitiums des proximalen Tubulus durch Neutrophile, Makrophagen und T-
Zellen. Dadurch wird die inflammatorische Reaktion weiter getriggert. Hält dieser
Zustand über längere Zeit an, versagt die Nierenfunktionen (Lubrano et al., 2001).
Wie weit die Störung der Energiehomöostase, ROS, ER-Stress, mTOR-Regulierung
und Autophagie zu einer Schädigung der Zellen beitragen, die zu einer Produktion
und Sekretion von Cytokinen führt, soll in dieser Arbeit geklärt werden.
1.8.1 Stress des endoplasmatischen Retikulums - Unfolded Protein Response
ER-Stress kann durch Energie- oder Nährstoffüberschuss ausgelöst werden und hat
direkten Einfluss auf Inflammationssignalwege, die daraufhin die Cytokinexpression
stimulieren. Bei MMA können Dysfunktionen in Mitochondrien zu ROS führen, die zu
diesem Zyklus beitragen können. Die Verbindung einer chronischen Inflammation
und dem ER liegt in der als unfolded protein response (UPR) bezeichneten Antwort
des ER auf Stresszustände. Die Stressantwort beinhaltet die Aktivierung von 3 als
Sensoren dienenden Transmembranproteinen, die jeweils eine Domäne im Lumen
des ER aufweisen und damit ungefaltete Proteine erkennen. Eine weitere Domäne
leitet Signale an transkriptorische oder translationale Moleküle weiter. Die Proteine
sind 1) IRE1 (inositol requiring enzyme, 2) PERK (PKR-like endoplasmic reticulum

kinase) und 3) ATF6 (activating transcription factor) (Schröder & Kaufman, 2005;
Cullinan & Diehl, 2006; Gotoh und Mori, 2006; Ron & Walter, 2007; Zhang &
Kaufman, 2008).
IRE1 besitzt eine Proteinkinaseaktivität und eine Endoribonukleaseaktivität (Mori et
al., 1993; Cox et al., 1993), PERK besitzt ebenfalls eine Proteinkinaseaktivität, mit
welcher es die α-Untereinheit des eukaryotischen Translations-Initiationsfaktors 2α
(eIF2α) phosphorylieren kann (Shi et al., 1998; Harding et al., 1999), ATF6 ist ein
Transkriptionsfaktor mit Leucinzipper-Domäne (Haze et al., 1999). Normalerweise
liegen diese drei ER-Stresssensoren in inaktivem Zustand vor und sind mit dem ER-
Chaperon BiP (auch GRP78) assoziiert. Bei beginnendem ER-Stress sondert sich
BiP durch Bindung an ungefaltete Polypeptidketten und Proteine ab, wodurch es zur
Freisetzung und damit verbundener Aktivierung der ER-Stresssensoren kommt
(Bertolotti et al., 2000). Zu Beginn der UPR wird PERK durch Oligomerisierung und
Autophosphorylierung aktiviert und phosphoryliert daraufhin eIF2α, wodurch die
Assemblierung der ribosomalen 80 S Untereinheit und somit die Proteinsynthese
inhibiert wird. Die Autophosphorylierung von IRE1α führt zur Aktivierung seiner
RNAse-Aktivität worauf ein 26 Basen großes Intron aus der mRNS des
Transkriptionsfaktors X-Box-binding-Proteins 1 (XBP1) gespleißt wird und diesen
aktiviert (Schroder et al., 2005; Ron et al., 2007). ATF6 dissoziiert von BiP und wird
im Golgi-Apparat von der site-1 und site-2 Protease (S1P, S2P) geschnitten. Dies
führt zur Freisetzung eines aktiven ATF6-Fragments in das Zytosol, zur Migration
zum Zellkern und zur Aktivierung der Transkription (Haze et al., 1999; Ye et al.,
2000). Das ATF6-Fragment und aktiviertes XBP1 induzieren parallel die
Transkription von Genen, die für Chaperone und andere Enzyme der Proteinfaltung,
Reifung, Sekretion und ER-vermittelter Degradierung codieren (Yamamoto et al.,
2007; Wu et al., 2007).
1.8.2 Die Regulation von mTOR-Komplex 1
Bei der MMA akkumulieren Stoffwechselzwischenprodukte im Körper, da der Abbau
von verzweigtkettigen Aminosäuren, ungeradzahligen Fettsäuren und Cholesterol
gestört ist (Sniderman et al., 1999). Weiterhin ist der Energiehaushalt der Zellen auf
mehreren Ebenen gestört (Okun et al., 2002; Kölker et al., 2003; Schwab et al.,
2006; Morath et al., 2008), was zu einem Energiemangel führen kann.

Der Multiproteinkomplex mTORC1 (mammalian target of rapamycin complex 1)
verarbeitet zelluläre Signale und reagiert auf die Verfügbarkeit von
Wachstumsfaktoren, Sauerstoff, und vor allem auf die Verfügbarkeit von Energie und
Aminosäuren. In Abhängigkeit davon werden einige Prozesse beeinflusst, die das
Zellwachstum betreffen. Bei der Aktivitätsregulierung von mTORC1 ist der wichtigste
Sensor das Heterodimer TSC (tuberous sclerosis complex), das aus TSC1 und TSC2
gebildet wird. TSC1/2 wirkt für die GTPase Rheb (Ras-related homolog enriched in
brain) als aktivierendes Protein für die GTPase (GAP, GTPase-activating protein).
Die aktive Form von Rheb ist GTP gebunden und aktiviert mTORC1 direkt (Long et
al., 2005; Sancak et al., 2007). Umgekehrt inhibiert Rheb mTORC1, wenn es GDP
gebunden ist (Inoki et al., 2003; Tee et al., 2003).
Die Aktivierung von mTORC1 erfolgt weiterhin durch den Ras Signalweg und den
kanonischen Insulin Signalweg. In beiden Fällen führt eine Stimulierung zur
Phosphorylierung von TSC2 durch die Proteinkinase B (PKB bzw. AKT) (Potter et al.,
2002; Inoki et al., 2002), durch ERK1/2 (extracellular signal regulated kinase 1/2) (Ma
et al., 2005) und durch RSK1 (p90 ribosomal S6 Kinase 1) (Roux et al., 2004). Durch
die Phosphorylierung wird TSC1/2 inhibiert und mTORC1 somit aktiviert. Zusätzlich
kann durch Wachstumsfaktoren aktiviertes AKT mTORC1 unabhängig von TSC1/2
durch Phosphorylierung und Dissoziation von PRAS40 von mTORC1 aktivieren
(Wang et al., 2007; Sancak et al., 2007; van der Haar et al., 2007). Die volle Aktivität
von AKT benötigt seine Phosphorylierung an 2 Stellen: An Serin308 wird es von der
PDK1 (Phosphoinositid dependent Kinase 1) und an Serin473 durch mTORC2
phosphoryliert (Sarbassov et al., 2005). Gleichzeitig können Komponenten von
mTORC2 aber auch die Phosphorylierung von AKT oder manche seiner Substrate
hemmen (Jacinto et al., 2006; Guertin et al., 2006). Das Binden von Insulin an den
membranständigen Zellrezeptor fördert die Tyrosinkinase-Aktivität des Insulin-
Rezeptors, die Rekrutierung von IRS1 (insulin receptor substrate 1), die Produktion
von Phosphatidylinositol-(3,4,5)-triphosphat durch die Aktivierung von PI3K und die
Rekrutierung und Aktivierung von AKT an der Plasmamembran. In einigen Zellarten
führt die Aktivierung von mTORC1 zur Blockade der Achse von PI3K und AKT. Wird
S6K1 durch mTORC1 aktiviert, erfolgt durch die Phosphorylierung von IRS1 eine
Destabilisierung (Harrington et al., 2005). Dieser autoregulatorische Signalweg ist
eine S6K1 abhängige negative Rückregulierung und wird mit metabolischen
Erkrankungen und Tumorgenese in Verbindung gebracht (Manning, 2004).

Die Übertragung des Energiestatus der Zelle an mTORC1 wird durch die AMPK
(AMP-aktivierte Proteinkinase) gewährleistet (Hardie, 2007). Als Reaktion auf eine
niedrige ATP:ADP Ratio wird die AMPK aktiviert und phosphoryliert daraufhin TSC2,
das die Aktivität der GAP in Richtung Rheb erhöht und mTORC1 reduziert (Inoki et
al., 2003). Zusätzlich ist die AMPK als Antwort auf Energiedepletion in der Lage, die
Aktivität von mTORC1 durch Phosphorylierung zu vermindern (Gwinn et al., 2008).
Durch den Sauerstoffgehalt kann die Aktivität von mTORC1 durch verschiedene
Signalwege reguliert werden. Bei geringer Hypoxie aktiviert der Abfall an ATP die
AMPK, welche wie beschrieben TSC1/2 phosphoryliert und mTORC1 hemmt
(Arsham et al., 2003; Liu et al., 2006).
Die Verfügbarkeit von Aminosäuren reguliert mTORC1 positiv (Guertin und Sabatini,
2007) und unabhängig von TSC1/2 (Nobukuni et al., 2005). mTORC1 interagiert mit
den 4 verwandten GTPasen der Rag-Proteine und wird dadurch aktiviert. In der
Anwesenheit von Aminosäuren bindet Rag an Raptor und fördert die Verschiebung
von mTORC1 von verschiedenen Orten zum perinukleär gelegenen Aktivator Rheb
(Kim et al., 2008; Sancak et al., 2008).
Abbildung (3) Die Aktivierung und Regulation von mTORC1 hängt von verschiedenen Einflüssen wie Energiestatus der Zelle, Verfügbarkeit von Wachstumsfaktoren, Aminosäuren und Sauerstoff ab. Der Komplex ist an der Steuerung von Zellwachstum, Proteinbiosynthese, Lipidsynthese und der Regulation der Autophagie beteiligt.

1.8.3 Aufbau von mTORC1 und Einfluss auf Autophagie
mTORC1 ist für die positive Regulation von Proliferation und Zellwachstum
verantwortlich. Bei seiner Aktivierung kommt es zur Stimulation anaboler Prozesse
wie Proteinbiosynthese und Aufbau von Lipiden und Zellorganellen. Gleichzeitig
reduziert er die Einleitung kataboler Stoffwechselwege wie Autophagie (Guertin und
Sabatini, 2007).
Insgesamt bilden 5 Komponenten mTORC1 und steuern und beeinflussen die
Aktivität der TOR-Kinase, dazu zählen neben der TOR-Kinase noch Raptor
(regulatory-associated protein of mTOR), mLST8 (mammalian lethal with Sec13
protein), PRAS40 (proline-rich AKT substrate 40 kDa) und Deptor (DEP-domain-
containing mTOR interacting potein) (Peterson et al., 2009). Die Rolle jedes
einzelnen Proteins ist teilweise noch unverstanden. Raptor ist für die Regulation der
Aktivität des Komplexes verantwortlich, indem es der Assemblierung dient und die
Rekrutierung von Substraten beeinflusst (Hara et al., 2002; Kim et al., 2002). Die
Funktion von mLST8 ist unklar, da eine Deletion die Aktivität und Funktion von
mTORC1 nicht zu beeinflussen scheint (Guertin et al., 2006). Von PRAS40 und
Deptor hingegen ist bekannt, dass sie als Negativregulatoren eine Rolle spielen
(Peterson et al., 2009; Sancak et al., 2007; van der Haar et al., 2007). Ist die Aktivität
von mTORC1 reduziert, sind PRAS40 und Deptor näher am Komplex lokalisiert. Es
wurde eine direkte Funktion durch Inhibierung der Substratbindung postuliert (Wang
et al., 2007). Im Gegenzug kann mTORC1 PRAS40 und Deptor direkt
phosphorylieren, so dass ihre Bindung zum Komplex verringert und die Aktivierung
von mTORC1 gesteigert wird (Peterson et al., 2009; Wang et al., 2007).
Ein Hauptbestandteil der Funktion von mTORC1 ist die Regulation der Autophagie,
durch welche (defekte) Proteine oder Organellen in Autolysosomen abgebaut
werden. Dadurch kann bei Nährstoffmangel ein Abbau von Organellen und Proteinen
erfolgen um biologisches Material für die Biosynthese von Proteinen für die
Energieproduktion bereitzustellen. Die Entwicklung von Autophagie ist stark
abhängig von der Regulierung durch mTORC1. Aktivierung von mTORC1 hemmt
Autophagie, Inhibierung von mTORC1 fördert sie (Codogno und Meier, 2005). Der
Grund hierfür ist ein Komplex aus den 3 Proteinen ULK1 (unc-51-like kinase 1),
ATG13 (autophagy related gene 13) und FIP200 (focal adhesion kinase family-
interacting protein of 200 kDa), der auf Signale von mTORC1 reagiert und bei
Aktivierung die Reifung der Phagosome stimuliert (Jung et al., 2009; Ganley et al.,

2009; Hosokawa et al., 2009). Durch die Aktivierung von mTORC1 erfolgt die
Phosphorylierung von ULK1 und ATG13, was den Zerfall dieses Komplexes und die
Inhibition der Autophagie zur Folge hat. Umgekehrt führt eine Inhibition von mTORC1
zur Stimulation der Autophagie (Noda und Ohsumi, 1998; Kamada et al., 2000;
Loewith et al., 2002).
1.8.4 Autophagie
Autophagie kann die Expression von Cytokinen beeinflussen und stimulieren und
somit an inflammatorischen Prozessen wie sie z.B. bei einer chronischen Nephritis
auftreten, beteiligt sein. Umgekehrt sind auch einige Cytokine in der Lage, die Stärke
der Autophagie zu beeinflussen (Harris, 2011).
Autophagie ist der generelle Überbegriff für eine Vielzahl intrazellulärer Prozesse, bei
welchen zytosolische Bestandteile abgebaut werden. Auf zellulärer Ebene existieren
grundsätzlich zwei Abbauwege für Proteine: Der Ubiquitin Proteasom-Weg degradiert
kurzlebige Proteine selektiv (Edinger und Thompson, 2003; Kim et al., 2000;
Mizushima, 2007). Langlebigere Proteine werden in den Lysosomen abgebaut.
Lysosomen sind saure Zellkompartimente mit einem pH Wert von 5, welche sich vom
Golgi-Apparat abschnüren. Sie sind mit hydrolysierenden Enzymen angereichert, so
dass über die saure Hydrolyse der Abbau von Metaboliten erfolgt. So werden
Proteine, Nukleinsäuren und Lipide zu ihren Grundbausteinen Aminosäuren,
Nukleotiden und freien Fettsäuren abgebaut. Weiterhin werden auf diesem Weg
Toxine, Krankheitserreger, ROS, veränderte zytosolische Komponenten wie
Proteinaggregate, beschädigte und ungebrauchte Organellen und Proteine abgebaut
(Levine und Deretic, 2007; Levine und Kroemer, 2008; Mizushima, 2007). Der
Großteil intrazellulärer Proteine hat eine lange Halbwertszeit. Ändert sich die
Degradationsrate langlebiger Proteine aufgrund von Defekten, können sich
Proteinmasse und Proliferation von Zellen erheblich verändern.
Fehlerhafte Regulierung von Autophagie wird bei vielen Krankheiten mit der
jeweiligen Pathogenese in Zusammenhang gebracht und kann auch den
programmierten Zelltod einleiten. Häufig sind ausdifferenzierte (nichtproliferierende)
Zellen wie Neurone oder Muskelzellen betroffen, bei welchen die Akkumulation
beschädigten Materials eine Belastung der Zellfunktionen darstellt. (Maiuri et al.,
2007; Mizushima et al., 2008; Thorburn, 2008). Weiterhin kann die fehlerhafte

Regulierung der Autophagie bei (Embryonal)-Entwicklung, Wachstumskontrolle
(Proliferation, Änderung des Zellvolumens), Immunabwehr (Antigenpräsentation,
Entfernung intrazellulärer Krankheitserreger und toxischer Metabolite), Anpassung an
schlechte Umweltbedingungen (Nährstoff-, Sauerstoff- oder Energiemangel,
Temperatur, Zelldichte), Alterung und Zelltod (Cuervo et al., 2005; Maiuri et al., 2007;
Levine und Deretic, 2007; Levine, 2007; Thorburn, 2008; Mizushima et al., 2008), zu
schweren Krankheiten wie neurodegenerativen Erkrankungen, verschiedenen
Tumoren, muskulären Atrophien und Kardiomyopathien führen (Mizushima et al.,
2008; Levine und Kroemer, 2008). Der Großteil der intrazellulären Degradierung wird
von der Makroautophagie geleistet (Levine und Klionsky, 2004; Mizushima, 2007).
1.8.4.1 Makroautophagie
Die Einleitung der Makroautophagie kann direkt durch mTORC1-Inhibierung oder
indirekt über Dephosphorylierung von Atg13 erfolgen (Kamada et al., 2000; Kabeya
et al., 2005). Die Induktion der Autophagie erfolgt nach Aktivierung von Atg1 (Yang
und Klionsky, 2009), und es kommt zur Rekrutierung mehrerer Atg-Proteine zum Ort
der Assemblierung des Autophagosoms (PAS – phagophore assembly site) führt
(Suzuki et al., 2001; Kim et al., 2002). Die Membranherkunft des späteren
Autophagosoms ist noch weitgehend unbekannt. Neuere Erkenntnisse in
Säugerzellen weisen darauf hin, dass Mitochondrien an der Entstehung der
Membranen beteiligt sein können (Noda et al., 2000; He et al., 2006; Hailey et al.,
2010). Jedoch werden auch das ER und der Golgi Apparat oder auch eine komplette
Neubildung als Ursprung der Membran als mögliche Quellen diskutiert. Durch
Phosphatidylinositol-3-phosphat und die Rekrutierung weiterer Atg-Proteine und
deren Bindung an die PAS reift das Autophagosom weiter (Yang und Klionsky,
2009). Zur Vergrößerung und Fertigstellung des Autophagosoms werden zwei
Protein-Konjugationssysteme benötigt: Atg12-Atg5 und Atg8 (Kim et al., 2002). Das
integrale Membranprotein Atg8-PE lagert sich im Autophagosom in die innere und
äußere Membran ein. Das Atg12- als auch das Atg8-Konjugationssystem könnte
dabei als äußere Hülle fungieren, welche die Fusion des Autophagosoms mit der
Vakuole vor seiner Fertigstellung verhindert. Vor der Fusion mit dem Lysosom
dissoziieren beide Konjugationssysteme mit den meisten anderen Atg-Proteinen
wieder vom Autophagosom.

Ein Großteil der Proteine der Außenmembran wird wieder zu einer PAS recycelt. Das
Autophagosom gelangt als Einfach-Membran-Vesikel ins Lumen des Lysosoms, wo
es durch Hydrolasen abgebaut wird. Essenziell für den Abbau sind der saure pH-
Wert der Vakuole und die vakuolären Hydrolasen.

1.9 Fragestellung der Arbeit
Das Ziel der Arbeit ist, die der chronischen, tubulointerstitiellen Nephritis (cTIN) bei
Patienten mit Methylmalonazidurie zugrundeliegenden Mechanismen zu
entschlüsseln. Eine cTIN ist immer mit der Infiltration durch Immunzellen verbunden.
Aus dieser Tatsache wurde die Hypothese entwickelt, dass vor einer Infiltration des
Interstitiums des proximalen Tubulus eine vorausgehende Schädigung der hier
lokalisierten Zellen stattfinden muss. Diese Schädigung führt zu einer Rekrutierung
von Immunzellen, die daraufhin das Interstitium des proximalen Tubulus infiltrieren
und die Zellen schädigen, was am Ende zu einem Funktionsverlust der proximalen
Tubuluszellen führt und eine Niereninsuffizienz nach sich zieht.
Ausgehend von dieser Hypothese wird in dieser Arbeit untersucht, ob
1) sich der gestörte Energiemetabolismus und eine erhöhte ROS-Produktion bei
MMA-Patienten auf die Entwicklung einer cTIN auswirkt,
2) der mTOR-Signalweg an der Entwicklung der chronischen Inflammation des
proximalen Tubulus involviert ist,
3) eine veränderte Zytokinsekretion und eine hierdurch erhöhte Rekrutierung von
Immunzellen beteiligt ist.
Als Modell für die Untersuchung der zugrundeliegenden Mechanismen der cTIN
wurden humane immortalisierte Epithelzellen des proximalen Tubulus (PTEC) von
mut0- und CblB-Patienten als Modell verwendet. Als Kontrollen dienten PTEC von
gesunden Probanden sowie von Propionazidurie-Patienten.


2 Material
2.1 Chemikalien
2-Mercaptoethanol AppliChem, Darmstadt
6-Aminocapronsäure Sigma-Aldrich Chemie, Steinheim
Agarose Invitrogen, Karlsruhe
Ammoniumpersulfat Sigma-Aldrich Chemie, Steinheim
Acrylamid Bio-Rad, München
Bromphenolblau
BSA Sigma-Aldrich Chemie, Steinheim
Coomassie G-250 Serva, Heidelberg
Cytochrom c aus Pferdeherz Sigma-Aldrich Chemie, Steinheim
Cumarin Sigma-Aldrich Chemie, Steinheim
3-Dimethoxy-5-methyl-6-decyl– Sigma-Aldrich Chemie, Steinheim
1,4-benzochinon
D-Glukose Merck, Darmstadt
Dichlorophenolindophenol Sigma-Aldrich Chemie, Steinheim
Dimethylsulfoxid (DMSO) Sigma-Aldrich Chemie, Steinheim
Dulbecco’s modified Eagle medium PAA Laboratories GmbH, Linz
EDTA Sigma-Aldrich Chemie, Steinheim
Ethanol Carl Roth, Karlsruhe
Essigsäure Carl Roth, Karlsruhe
Glukose Sigma-Aldrich Chemie, Steinheim
Glycin Carl Roth, Karlsruhe
Glyzerin Sigma-Aldrich Chemie, Steinheim
Hepes Sigma-Aldrich Chemie, Steinheim
Insulin Sigma-Aldrich Chemie, Steinheim
Isopropanol Sigma-Aldrich Chemie, Steinheim
Kaliumchlorid Sigma-Aldrich Chemie, Steinheim
Kaliumdihydrogenphosphat Sigma-Aldrich Chemie, Steinheim
Kalziumchlorid Sigma-Aldrich Chemie, Steinheim
Kalziumsulfat Sigma-Aldrich Chemie, Steinheim
Kollagen

Laktatdehydrogenase Sigma-Aldrich Chemie, Steinheim
Laurylmaltosid Sigma-Aldrich Chemie, Steinheim
Luminol AppliChem, Darmstadt
Magnesiumchlorid Carl Roth, Karlsruhe
Methylmalonsäure Sigma-Aldrich Chemie, Steinheim
Magnesiumsulfat Merck, Darmstadt
Methanol Carl Roth, Karlsruhe
Natriumchlorid Carl Roth, Karlsruhe
Natriumdeoxycholat Sigma-Aldrich Chemie, Steinheim
Natriumhydrogencarbonat Merck, Darmstadt
Natriumazid Sigma-Aldrich Chemie, Steinheim
Natriumzyanid Sigma-Aldrich Chemie, Steinheim
Natriumhydroxid Carl Roth, Karlsruhe
N,N,N′,N′-Tetramethylethylenediamin Sigma-Aldrich Chemie, Steinheim
β-Nicotinamidadenindinukleotid Sigma-Aldrich Chemie, Steinheim
β-Nicotinamidadenindinukleotid-Phosphat Sigma-Aldrich Chemie, Steinheim
Nonidet™ P-40 Sigma-Aldrich Chemie, Steinheim
PBS PAA Laboratories, Linz
Phosphoenolpyruvat Roche Diagnostics, Mannheim
Pyruvatkinase Roche Diagnostics, Mannheim
Sodiumdodecylsulfat Serva, Heidelberg
Succinat Sigma-Aldrich Chemie, Steinheim
Tri Reagent Sigma-Aldrich Chemie, Steinheim
Tris Carl Roth, Karlsruhe
Triton X-100® (Alkylphenylpolyethylenglykol) Sigma-Aldrich Chemie, Steinheim
Trypanblau Sigma-Aldrich Chemie, Steinheim
Trypsin/EDTA PAA Laboratories GmbH, Linz
Turbofect Fermentas
Tween 20 Sigma-Aldrich Chemie, Steinheim
Vectashield-HardSet Vector Laboratoeries, Peterborough,
USA
Wasserstoffperoxid 30 % Merck, Darmstadt

2.2 Geräte
Blot-Kammer, Perfect Blue Semi-Dry Peqlab Biotechnologie, Erlangen
Elektrophoresekammer Hoefer Serva, Heidelberg
SE260 Mighty Small II)
Fluoreszenzmikroskop CTR 4000 Leica, Wetzlar
Lichtmikroskop Leica, Wetzlar
Magnetrührer IKA-Combimag RCT Ika-Werk, Staufen
Mehrkanalpipetten Brand, Wertheim
pH-Meter Mettler-Toledo, Giessen
Pinzetten neoLab, Heidelberg
Pipette 1-10 µl Eppendorf, Hamburg
Pipette 10-100 µl Eppendorf, Hamburg
Pipette 20-200 µl Eppendorf, Hamburg
Pipette 100-1000 µl Eppendorf, Hamburg
Pipette 1000-2500 µl Eppendorf, Hamburg
Pipettierhilfe (Automatikpipette) Integra Biosciences, Fernwald
Spectramax 340 PC Plus Microplate Reader Molecular Devices, Sunnyvale, USA
Tischzentrifuge “Biofuge Pico” Heraeus, Hanau
Thermomixer Eppendorf, Hamburg
Vakuumpumpe Laboport® KNF Neuberger, Freiburg
Vortex-Genie Bender-Hobein, Zürich
Waage Sartorius AG, Göttingen
Werkbank HERAsafe Heraeus, Hanau
Zählkammer nach Neubauer Renner, Darmstadt
Zentrifuge Beckman TJ-6 Beckman Instruments, München
2.3 Verbrauchsmaterialien
Blottingmembran PVDF GE Healthcare, München
Eingefriergefäße Cellstar® 1,5ml Greiner, Frickenhausen
Eukit® Sigma-Aldrich Chemie, Steinheim
Filtereinheit Steritop Millipore, Eschborn
Handschuhe Maimed GmbH, Neuenkirchen

Kryoröhrchen neoLab, Heidelberg
Magermilchpulver Carl Roth, Karlsruhe
Objekträger (24 x 50 mm) Carl Roth, Karlsruhe
Objektträger rund (18 mm) Carl Roth, Karlsruhe
Pasteurpipetten WU, Mainz
Pipettenspitzen Sarstedt, Nürnbrecht
(1-10µl, 10-200µl, 100-1000, 1-2,5ml)
PCR Gefäße Stein Labortechnik, Remchingen
Reaktionsgefäße (1,5 ml) Stein Labortechnik, Remchingen
Reaktionsgefäße Safe-Lock Stein Labortechnik, Remchingen
Sterile Einmalpipetten Corning Costar, Wiesbaden
(2 ml, 5 ml, 10 ml, 25 ml)
TransWell Filterplatten Corning Costar, Wiesbaden
Verschlussfolie Parafilm Brand, Wertheim
Whatman-Papier neoLab, Heidelberg
Zellkulturplatten 96 Well Sarstedt, Nürnbrecht
Zentrifugenröhrchen (15 ml, 50 ml) Sarstedt, Nürnbrecht
2.4 Zellkulturmedien
Dulbecco’s Modified Eagle Medium PAA Laboratories, Linz
high glucose
Dulbecco’s Modified Eagle Medium PAA Laboratories, Linz
low glucose
Ham’s F12 PAA Laboratories, Linz
Fötales Kälberserum Gibco
Trypsin-Lösung Gibco
Penicillin/Streptomycin Gibco
2.5 Antikörper
Anti GammaGlutamyltransferase GeneTex, Irvine, USA
Anti Aquaporin 1 Santa-Cruz, Heidelberg

Anti mTOR Cell Signaling, Frankfurt am Main
Anti Phospho-mTOR Cell Signaling, Frankfurt am Main
Anti Phospho-Raptor Cell Signaling, Frankfurt am Main
Anti PRAS40 Cell Signaling, Frankfurt am Main
Anti Phospho-PRAS40 Cell Signaling, Frankfurt am Main
Anti LC3 Sigma-Aldrich, Steinheim
Anti p62 Santa-Cruz, Heidelberg
Anti Tubulin Santa-Cruz, Heidelberg
Anti Phospho-IRE1 Abcam, Cambridge, UK
Anti Maus Santa-Cruz, Heidelberg
Anti Kaninchen Santa-Cruz, Heidelberg
2.6 ELISA-Kits
Multi-Analyte ELISArray Kit SA Biosciences, Qiagen
Eli-Pair IL8 Hölzel Diagnostika, Köln
2.7 Lösungen für proteinbiochemische Methoden
RIPA Puffer 150 mM Natriumchlorid
50 mM Tris/HCl pH 8
5 mM EDTA
1 % (w/v) Nonidet P40
0,1 % (w/v) SDS
0,5 % (w/v) Natriumdeoxycholat
Sammelgelpuffer (4 x) 0,25 M Tris/HCl pH 6,8
0,4 % (w/v) SDS
Trenngelpuffer (4 x) 1,5 M Tris/HCl pH 8,8
0,4 % (w/v) SDS

6 x Probenpuffer 480 mM Tris/HCl pH 6,8
45 % (w/v) Glyzerin
12 % (w/v) β-Mercaptoethanol
12 % (w/v) SDS
0,06 % (w/v) Bromphenolblau
Elektrophoresepuffer (10 x) 0,25 M Tris pH 8,3
1,92 M Glycin
1 % (w/v) SDS
TBS (10 x) 1,5 M Natriumchlorid
0,5 M Tris/HCl pH 7,5
TBST 1 x TBS
0,1 % Tween-20
Anodenpuffer Western Blot 750 mM Tris/HCl pH 7,4
20 % (v/v) Methanol
Kathodenpuffer Western Blot 25 mM Tris / HCl pH 9
40 mM 6-Aminocapronsäure
20 % (v/v) Methanol
Blockierungspuffer 1 x TBST
5 % (w/v) Magermilchpulver
Regenerierungspuffer 2 % (w/v) SDS
100 mM β-Mercaptoethanol
625 mM Tris/HCl pH 6,8
Enzym-Assay-Puffer 250 mM Saccharose
(EA-Puffer) 50 mM Kaliumchlorid
5 mM Magnesiumchlorid
20 mM Tris/HCl pH 7,4

Krebs-Ringer-Puffer 142 mM Natriumchlorid
3 mM Kaliumchlorid
1,5 mM Kaliumdihydrogenphosphat
100 mM Hepes
4 mM Glukose
1,2 mM Magnesiumchlorid
1,4 mM Kalziumchlorid

3 Methoden
3.1 Zellkultur
3.1.1 Isolierung von humanen primären proximalen Tubulusepithelzellen (hPTEC)
In der vorliegenden Arbeit wurden primäre humane proximale Tubulusepithelzellen
verwendet, die aus dem Urin von Patienten und Kontrollprobanden isoliert wurden.
Dazu wurde der Urin möglichst direkt in eine sterile Flasche mit DMEM
supplementiert mit 10% FKS, 100 U/ml Penicillin und 0.1 mg/ml Streptomycin
überführt und für 30 Minuten inkubiert. Anschließend wurde der Ansatz in sterile 50
ml Röhrchen (Falcon) überführt und bei 600 × g für 10 Minuten zentrifugiert. Das
Sediment wurde einmal mit DMEM inklusive genannter Supplemente gewaschen,
anschließend in PTEC-Medium resuspendiert und in mit Kollagen beschichteten
Kulturflaschen kultiviert.
Zusatz: Für die Gewinnung und Untersuchung von hPTEC aus Patientenurin liegt ein
positives Votum der Ethikkommission (S-074/2008) der medizinischen Fakultät
Heidelberg vor.
3.1.2 Kultivierung von eukaryotischen Zellen
Die Arbeiten mit eukaryotischen Zellen wurden unter einer Sicherheitswerkbank
(Modell SG 400, Baker USA) durchgeführt. Die verwendeten Medien und Lösungen
wurden entweder steril bezogen oder steril filtriert. Für Gebrauchsgegenstände
wurden sterilisierte Einmalartikel verwendet. Die Kultivierung der Zellen erfolgte bei
37°C in wasserdampfgesättigter 5% CO2-Atmosphäre im Brutschrank (Binder).
3.1.3 Kultivieren und Passagieren von PTEC
Um eine fortwährende Vitalität der Zellen zu gewährleisten, mussten adherente
Zellen bei Erreichen der Konfluenz verdünnt und neu ausgesät werden. Adherente
Zellen wurden durch Akkutasebehandlung vom Kulturflaschenboden gelöst und zum

weiteren Wachstum auf neue Kulturflaschen verteilt. Dazu wurde das Medium
entfernt und der Zellrasen mit PBS-Puffer pH 7,4 gewaschen. Anschließend wurden
die Zellen mit Akkutase-Lösung (0,5 mg/ml Akkumax, PAA) bedeckt und bei 37 °C
inkubiert. Nach einer Einwirkzeit von wenigen Minuten wurden die Zellen durch
Klopfen vom Gefäßboden gelöst. Die Inaktivierung der Akkutase erfolgte durch
Zugabe von frischem Medium. Anschließend wurden die Zellen resuspendiert und
auf neue Kulturflaschen verteilt.
3.1.4 Kultivieren und Passagieren von immortalisierten Zellen
Die Kultivierung immortalisierter Zellen wurde wie unter Abschnitt 3.1.3 beschrieben
mit folgenden Änderungen durchgeführt: Die Zellen wurden durch Behandlung mit
Trypsin-EDTA-Lösung (0,5 mg/mlTrypsin und 0,2 mg/ml EDTA-4 Na, Gibco) vom
Gefäßboden gelöst.
3.1.5 Ernten von immortalisierten Zellen
Zum Ernten von immortalisierten Zellen wurde das Medium abgesaugt und die Zellen
mit PBS-Puffer pH 7,4 gewaschen. Die Zellen wurden entweder mit einem
sterilisierten Zellschaber mechanisch oder durch Behandlung mit Trypsin-EDTA-
Lösung (0,5 mg/mlTrypsin und 0,2 mg/ml EDTA-4 Na, Gibco) vom Boden des
Kulturgefäßes gelöst und durch Zentrifugation bei 600 x g für 10 Minuten
sedimentiert. Nach erneutem Waschen mit PBS-Puffer pH 7,4 wurde das
Zellsediment mit für den jeweiligen Versuch adäquaten Puffer versetzt und weiter
verwendet.
3.2 Molekularbiologische Methoden
Die Durchführung gentechnologischer Arbeitsschritte erfolgte nach
molekularbiologischen Standardmethoden. Reagenzien und Proben für die Versuche
wurden auf Eis gekühlt. Alle verwendete Puffer, Medien und sonstige Lösungen
wurden, sofern nicht kommerziell erworben, mit destilierten Wasser angesetzt und
bei Bedarf durch Filtrieren sterilisiert. Die folgenden Vorschriften und Methoden

wurden, soweit nicht anders angegeben, dem Laborhandbuch "Molecularcloning"
(Sambrook und Russel, 2001) entnommen.
3.2.1 Polymerase-Kettenreaktion
Die Polymerase-Kettenreaktion (polymerase chain reaction, PCR) wurde dazu
verwendet, den zur Immortalisierung verwendeten Vektor pRNS1 zu amplifizieren.
Zur Durchführung einer PCR wurden in einem 0,2 ml Eppendorf-Reaktionsgefäß
folgende benötigten Reagenzien angesetzt:
X μl Matrizen-DNS
2,5 μl Primer 1
2,5 μl Primer 2
5 μl 10 x Reaktions-Puffer
1 μl 10 mM dNTPs
0,5 μl Polymerase
mit H2O steril. auf 50 μl aufgefüllt.
Zur Amplifikation der DNS-Fragmente wurde das folgende Protokoll angewandt:
98 °C Denaturierung 30 Sekunden
98 °C Denaturierung 5 Sekunden
TM + 2 °C Annealing 10 Sekunden 30-35 Zyklen
72 °C Elongation 10 Sekunden pro 1000 Basen
72 °C Elongation 2 Minuten
Zur Überprüfung der PCR wurde jeweils 1 μl des Ansatzes mittels Agarose-
Gelelektrophorese analysiert.
3.2.2 Agarosegelelektrophorese
Um die in der PCR erhaltenen DNS-Fragmente zu analysieren, wurde die
linearisierte DNS in Agarosegelen aufgetrennt. Es wurden 1,3 %-ige Agarosegele
verwendet, welche eine Auftrennung von DNS-Molekülen von 0,2 bis 7 kb Länge

ermöglichen. Ein entsprechender Größenstandard wurde zur Größenbestimmung der
DNS-Fragmente mitgeführt. Die elektrophoretische Trennung der DNS erfolgte bei
80–120 Volt.
3.2.3 Transformation von DNS in kompetente Bakterienzellen wofür
Der für die Immortalisierung der hPTEC benötigte Vektor pRNS1 wurde durch
Bakterien (Echerischia Coli) amplifiziert. Hierfür wurden die Bakterien zunächst mit
der Plasmid-DNS transformiert. Als Transformation wird die Aufnahme von Fremd-
DNS durch kompetente Bakterien bezeichnet. Dies wurde durch einen kurzen
Hitzeschock erreicht (Chung et al., 1989). Zur Transformation wurden 100 μl
kompetente Zellen auf Eis aufgetaut und mit 1μg Plasmid-DNS gemischt. Der Ansatz
wurde für weitere 30 Minuten auf Eis inkubiert und anschließend einem Hitzeschock
für eine Minute bei 42 °C unterzogen. Danach wurde der Ansatz für zwei Minuten auf
Eis inkubiert. Im Anschluss erfolgte die Zugabe von 900 μl LB-Medium und die
Inkubation für eine Stunde bei 37°C unter Schütteln. Von der Bakteriensuspension
wurden 150 μl auf LB-Agarplatten mit Ampicillin ausplattiert. Der restliche Ansatz
wurde für 5 Minuten bei 1000 x g zentrifugiert, der Überstand dekantiert, das
erhaltene Sediment im restlichen Medium resuspendiert und anschließend ebenfalls
ausplattiert. Die LB-Agarplatten wurden anschließend für 16 Stunden bei 37 °C im
Brutschrank inkubiert.
3.2.4 Analytische Isolierung von Plasmid-DNS mit dem „QIAprep Spin Miniprep Kit“
Um nach einer Transformation eines Ligationsansatzes Bakterienklone mit der
gewünschten pRNS1-DNS zu identifizieren, wurde von 3-5 Bakterienklonen eine
analytische Isolierung von Plasmid-DNS durchgeführt. Die Vorgehensweise erfolgte
nach Herstellerangaben.
3.2.5 Präparative Isolierung von Plasmid-DNS
Die für die Transformationen benötigten Mengen an pRNS1-DNS reichten durch
analytische Isolierung nicht aus und wurden durch präparative Isolierung gewonnen.

Hierzu wurde eine 200 ml Kultur mit 200 μl einer Vorkultur beimpft und für 16
Stunden bei 37 °C unter Schütteln inkubiert. Zur Reinigung größerer Mengen an
Plasmid-DNS wurde das „Qiagen™ HiPurePlasmid Maxiprep Kit“ nach
Herstellerangaben verwendet.
3.2.6 Konzentrationsbestimmung von Nukleinsäuren
Die Bestimmung der Konzentration und Reinheit einer DNS-Probe erfolgte
photometrisch.
3.2.7 Herstellung kompetenter Bakterienzellen
Die Transformation durch pRNS1-DNS erfolgte mit kompetenten Bakterien. Als
Kompetenz wird bei Bakterienzellen die Fähigkeit beschrieben, Fremd-DNS
aufzunehmen. Für die Herstellung von kompetenten Bakterienzellen der
unterschiedlichen Bakterienstämme wurde das Protokoll von Chung et al.
herangezogen (Chung et al., 1989). Vom gewünschten Bakterienstamm wurde
hierfür ein fraktionierter Bakterienausstrich angefertigt, von welchem eine einzelne
Bakterienkolonie in 5 ml LB-Medium ohne Antibiotikum überführt und für 16 Stunden
bei 37 °C unter Schütteln inkubiert wurde. Von dieser Vorkultur wurde ein Milliliter in
einen Erlenmeyer-Kolben mit 100 ml LB-Medium überführt und bis zu einer optischen
Dichte von 0,4–0,5 bei 578 nm ebenfalls bei 37°C unter Schütteln inkubiert. Danach
wurden die Bakterienzellen bei 4500 x g für 10 Minuten bei 4 °C sedimentiert. Der
Überstand wurde verworfen und das Bakteriensediment auf 5-10 ml Volumen mit
kaltem TSS-Puffer aufgefüllt und resuspendiert. Im Anschluss wurde die
Bakteriensuspension in 100 μl Aliquots in kalte Eppendorfgefäße pipettiert und in
flüssigem Stickstoff schockgefroren. Bis zur weiteren Verwendung wurden die
kompetenten Bakterienzellen bei -80 °C gelagert.
3.2.8 Transfektion eukaryotischer Zellen zur Immortalisierung
Unter dem Immortalisieren von Zellen wird das Einbringen von Fremd-DNS in die
Zelle verstanden, welche dann zeitweise oder dauerhaft exprimiert wird. Wird die

Fremd-DNS dauerhaft in das Genom der Zelle integriert, wurde eine stabile Zelllinie
generiert. Zur Transfektion der hPTEC mit Plasmid-DNS (pRNS1; Litzkas et al.,
1984; Small et al., 1982) wurde Transfect (Roche, Mannheim) verwendet. Diese
Methode orientiert sich an der Lipofektion, die von Felgner und Kollegen erstmals
beschrieben wurde (Felgner et al., 1987). Die Zellen wurden im Transfektionsansatz
für 24 Stunden inkubiert. Anschließend wurde DMEM mit 10 % FKS, 100 U/ml
Penicillin und 0.1 mg/ml Streptomycin zur Kultivierung verwendet.
3.3 Proteinbiochemische Methoden
3.3.1 Proteinbestimmung
Die Proteinkonzentrationen wurden mittels des DC Protein Assay Kit von Bio-Rad
(Katalognummer: 500-0112) bestimmt. Das Kit baut auf der von Lowry publizierten
colorimetrischen Methode auf (Lowry et al., 1951). Zur Quantifizierung wurde ein
Proteinstandard aus BSA-Lösungen unterschiedlicher Konzentrationen parallel
pipettiert. Zuvor wurden alle Ansätze in einer wässrigen, 1 %igen SDS-Lösung für die
Messung verdünnt. Nach 15 Minuten Inkubation auf einem Plattenschüttler wurde die
Extinktion bei 750 nm im Photometer gemessen und die jeweilige
Proteinkonzentration in Abhängigkeit von der BSA-Standardkurve berechnet.
3.3.2 SDS-PAGE (SDS-Polyacrylamid-Gelelektrophorese)
Um die Expression von Proteinen zu untersuchen, wurde die diskontinuierliche SDS-
Polyacrylamid-Gelelektrophorese nach Laemmli (Laemmli, 1970) eingesetzt.
Proteine wurden durch Zugabe von Natriumdodecylsulfat (sodiumdodecylsulfate,
SDS) und β-Mercaptoethanol denaturiert und was zum Verlust der Ladungs- und
Konformationsunterschiede. Somit bewegzen sie sich nur in Abhängigkeit ihrer
relativen molaren Masse in einem angelegten elektrischen Feld durch die Gelmatrix.
Je nach Bedarf wurden die Gele in verschiedenen Stärken verwendet (Tabelle 1).

Tabelle (1) Zusammensetzung von Sammel- und Trenngelen für SDS-PAGE. Das Sammelgel wurde stets 4,5 %ig angesetzt. Das Trenngel wurde je nach zu detekierender Proteingröße zwischen 7 und 15 % verwendet. SG: Sammelgelpffer pH 6,8; TG-Trenngelpuffer pH 8,8.
Sammelgel 4,5 % Trenngel 7 % 10 % 12 % 15 %
H2O [µl] 2100 H2O [µl] 6173 5950 4120 2913
SG-Puffer [µl] 450 TG-Puffer [µl] 3000 3000 3000 3000
AA/BA 40 %,
37,5 : 1 [µl]
AA/BA 40 %,
37,5 : 1
2777 3000 4830 6037
APS 10 %ig [µl] 40 APS 10 %ig [µl] 40 40 40 40
TEMED [µl] 4 TEMED [µl] 4 4 4 4
Die Proben wurden mit SDS-Probenpuffer im Verhältnis 1:6 verdünnt und für 5
Minuten bei 95 °C denaturiert. Um die Größe der Proben zu bestimmen, wurden
parallel dazu Proteine mit bekannten relativen Molekulargewichten aufgetrennt. Die
Elektrophorese wurde zunächst mit 80 Volt und ab Erreichen des Trenngels mit 130
Volt durchgeführt. Der Endpunkt der Elektrophorese war bei Auslaufen des im SDS-
Probenpuffer enthaltenen Bromphenolblaus aus dem Gel erreicht.
3.3.3 Färbung von Proteingelen durch Coomassie-Brillant Blau
Um die aufgetrennten Proteine nach der Elektrophorese sichtbar zu machen, wurde
das Gel mit PageBlueTM Protein Staining Solution (Fermentas, St. Leon-Roth)
gefärbt. Diese Methode basiert auf der Färbung durch Coomassie Brillant Blau
(Chrambach et al., 1967). Anschließend wurde es mit deionisiertem Wasser entfärbt,
bis die Proteinbanden gut sichtbar waren und dokumentiert.
3.3.4 Western Blot
Um ein relevantes Protein in einem Gemisch aus verschiedenen Proteinen gezielt
nachzuweisen, wurde für das gesuchte Protein ein spezifischer Antikörper
verwendet. Zunächst wurden die Proteine wie unter Abschnitt 3.3.2 beschrieben
elektrophoretisch aufgetrennt und anschließend auf eine Membran transferiert. Im
Anschluss wurde das Antigen (Protein) mit einem Antikörper auf der Membran
nachgewiesen.

Dieser Transfer erfolgte nach der diskontinuierlichen SDS-Gelelektrophorese durch
einen „Semi-Dry Blot“ auf eine PVDF-Membran (GE Healthcare, München).
3.3.5 Semi-Dry Blot
Das Blotten der Gele wurde mit einem Semi-Dry Blotsystem (Peqlab Biotechnologie
GmbH, Erlangen) durchgeführt. Der Vorteil dieser Methode liegt in der kurzen
Transferzeit von 90 Minuten gegenüber 180 Minuten beim Sandwich Blot. Der
Proteintransfer aus dem Gel auf die Membran erfolgte bei Gelen der Größe 9 x 9 cm.
Es wurde ein gefärbter Größenmarker verwendet, welcher auf der Membran sichtbar
war. Der Blot wurde so aufgebaut, dass ein luftblasenfreier Kontakt zwischen Gel und
Membran zustande kam. Pro Quadratzentimeter Membranfläche wurde eine
Spannung von 1 mA angelegt. Im Anschluss wurden die freien Bindestellen auf der
Membran für 1 Stunde in Blockierungspuffer geblockt und anschließend über Nacht
bei 4 °C mit dem Erstantikörper auf einem Drehrad inkubiert.
3.3.6 Entwicklung von Western Blots mittels Chemilumineszenz
Nichtgebundene Erst-Antikörper wurden entfernt, indem die Membran dreimal 10
Minuten mit TBS mit 0,1 % Tween (TBST) gewaschen wurde. Um die an die Proteine
gebundenen Erst-Antikörper zu detektieren, wurde ein peroxidasekonjugierter Zweit-
Antikörper verwendet. Nach einstündiger Inkubation mit dem Zweit-Antikörper wurde
die Membran zweimal 10 Minuten mit TBST und einmal 10 Minuten lang mit TBS
gewaschen. Anschließend wurde die Membran für 5 Minuten mit dem nach
Herstellerangaben frisch angesetzten ECL-Reagenz (Western Lighting
ChemilumineszenzPlus, NEN Perkin Elmer Life Sciences, Boston, USA) bedeckt.
Das Reagenz wurde entfernt und die entstandene Chemilumineszenz mit dem
Dokumentationssystem NIH image program (Peqlab) über einen Zeitraum von
maximal einer Stunde detektiert.

3.3.7 Wiederverwendung von Western Blots
Ein Western Blot kann nacheinander mit unterschiedlichen Erst-Antikörpern inkubiert
werden. Hierfür ist es notwendig, bereits gebundene Antikörper von der Membran zu
entfernen. Die Membran wurde viermal für 5 Minuten mit TBST gewaschen und im
Anschluss 30 Minuten lang bei 50 °C mit Regenerierungs-Puffer unter leichtem
Schütteln inkubiert. Das im Puffer enthaltene SDS und ß-Mercaptoethanol wurde
durch sechsmaliges Waschen für jeweils 5 Minuten mit TBST vollständig entfernt.
Die freiliegenden Proteinbindungsstellen werden erneut für eine Stunde mit
Blockierungspuffer gesättigt, so dass eine weitere Inkubation mit einem anderen
Erst-Antikörper möglich wurde.
3.3.8 Enzyme linked immunosorbent assay (ELISA)
Zum spezifischen Nachweis verschiedener Proteine wurde ein qualitativer (Mix-N-
Match ELISArray Kit, SA Bioscience) und quantitative (IL-8 human Eli-pair Kit,
abcam) ELISA im 96-Lochbodenplattenverfahren verwendet. Die Experimente
wurden nach Herstellerangaben durchgeführt.
3.3.9 Expressionsnachweis von Proteinen mit Dotplots
Um eine Übersicht von exprimierten Proteinen und Expressionsunterschieden
zwischen Kontrollen und Patientenzellen zu erhalten, wurde ein kommerzielles Kit
verwendet (Human Soluble Receptor Array Kit Non-Hematopoietic Panel, RnD
systems), welches auf dem Dotplot-Verfahren beruht. Die Experimente wurden nach
Herstellerangaben durchgeführt.
3.4 Färbung der Zellen mit Acridinorange und FACS-Analyse
Zur Bestimmung der Autophagierate wurden die Zellen mit dem Farbstoff
Acridinorange gefärbt. Acridinorange ist ungeladen, lipohil und kann durch einen
unspezifischen Permeations-Mechanismus biologische Membranen durchqueren
(Rundquist, 1984). Das Fluorophor hat die Eigenschaft einer schwachen Base und

kann bei saurem pH ein Proton aufnehmen, durch die Ladungsänderung wird die
freie Membranüberquerung unterbunden. Acridinorange mit gebundenem Proton
verbleibt in sauren Zellkompartimenten und reichert sich dort an. Bei der
Aufkonzentrierung des Farbstoffs in sauren Kompartimenten kommt es zu einer
Aufstapelung, die eine Lumineszenzverschiebung bewirkt. Die Anregung von
Monomeren mit blauem Laser (488 nm) führt zu grüner Fluoreszenz (525 nm; FL1).
Bei Multimeren verschiebt sich die Fluoreszenz in den roten Bereich (630 nm; FL3).
Die Intensität der roten Fluoreszenz ist proportional zum Volumen bzw. der Anzahl
saurer Kompartimente in der Zelle. Da es während dem Autophagieprozess zu einer
Zunahme von autophagosomalen und lysosomalen Kompartimenten kommt, lässt
sich direkt auf die Stärke der Autophagie rückschließen (Traganos, 1994).
Die Zellen wurden für 48 Stunden in 6 cm Schalen kultiviert und mit Acridinorange (1
µg/ml) in farblosem DMEM bei 37 °C für 20 Minuten inkubiert. Danach wurden die
Zellen gewaschen, geerntet und auf Eis gestellt. Die Messung der
Fluoreszenzintensität erfolgte durch FACS-Analyse (Canto II, Becton Dickinson) und
die Software CellQuestTMPro (Becton Dickinson). Als Grundautophagierate wurden 5
% der Kontrollzellinien festgelegt und die Patientenzelllinien wurden dazu in Relation
gesetzt. Durch Bildung des Quotienten von roter zu grüner Fluoreszenz (FL3/FL1)
konnte auf die Autophagierate geschlossen werden.
3.5 Spektrophotometrische Methoden
Die Aktivitätsmessung der Enzyme erfolgt wurde mit einem Computer gesteuerten
Spektrophotometer (Spectramax Plus Microplate Reader, Molecular Devices,
Sunnyvale, California, U.S.A.) durchgeführt. Zur Bestimmung der Aktivität wurde die
stabile Phase (Steady state) der Enzyme verwendet. Um störende
Umgebungseinflüsse wie z.B. Trübungen zu eliminieren wurden alle Enzymkinetiken
im Einstrahl-Zweiwellenlängenverfahren aufgenommen, mit Ausnahme von 5',5"-
Dithiobis-(2-Nitrobenzoat) (DTNB), welches im Einstrahl-Einlängenverfahren
aufgenommen wurde. Alle Messungen erfolgten mit einem finalen Volumen von 300
μl in temperierten 96-Lochbodenplatten (Greiner Bio-One). Die Bestimmung der
spezifischen Enzymaktivitäten (U/mg) erfolgte durch Proteinnormierung der
Enzymaktivität und des Extinktionskoeffizient des jeweils verwendeten Chromogens.
Es wurden folgende Extinktionskoeffizienten verwendet:

NAD/NADH: ε 340-400 nm = 6,1 mM-1 × cm-1
DCPIP: ε 610-750 nm = 22,0 mM-1 × cm-1
Cytochrom c: ε 540-550 nm = 19,0 mM-1 × cm-1
DTNB: ε 412 nm = 14,2 mM-1 × cm-1
3.5.1 Probenaufarbeitung für glykolytische Enzyme
Die Zellen wurden durch Abschaben geerntet und nach zweimaligem Waschen mit
PBS in EA-Puffer bestehend aus 250 mM Saccharose, 50 mM KCl, 5 mM MgCl2, 20
mM Tris/HCl, pH 7,4 aufgenommen und auf Eis gekühlt. Der Zellaufschluss erfolgte
mit Hilfe einer Spritze mit Kanüle.
3.5.1 Subzellulare Fraktionierung durch differentielle Zentrifugation
Die aufgeschlossenen Zellen lassen sich durch differentielle Zentrifugation in
Fraktionen bestehend aus Zellmembranen und Zelltrümmern, Zellkernen,
Zellorganellen und Zytosol aufteilen. Durch unterschiedliche Zentrifugations-
geschwindigkeiten und unterschiedliche Sedimentationsgeschwindigkeiten einzelnen
Komponenten, die auf den unterschiedlichen Dichten beruhen lassen sich die Zellen
physikalisch auftrennen. Die Zellkerne und große Membranbruchstücke besitzen die
höchste Dichte des Zellhomogenats, so dass diese durch Zentrifugation bei 600 x g
für 5 min aus dem Homogenat entfernt werden können. Die mitochondriale Fraktion
wurde durch Zentrifugation bei 5.200 x g für 10 min aus dem Überstand des ersten
Zentrifugationsschrittes isoliert und in EA-Puffer aufgenommen. Der nach dem
zweiten Zentrifugationsschritt erhaltene Überstand wurde bei 16.000 x g für 20
Minuten zentrifugiert, der Überstand bildete die zytosolische Fraktion.
3.5.2 Probenaufarbeitung für Enzyme des Krebszyklus und der Atmungskette
Aufgrund der Zerstörung der Mitochondrien beim Zellaufschluss mit einer Kanüle bei
PTEC von MMA-Patienten, erfolgte die Probenvorbereitung für die
Aktivitätsbestimmung der Enzyme des Zitrat-Zyklus und der Atmungskette durch
Permeabilisierung mit Digitonin. Die Zellen wurden durch Abschaben geerntet, in
PBS gewaschen und 20 Minuten auf Eis in EA-Puffer supplementiert mit 0,02 %
Digitonin (w/v) inkubiert.

3.5.3 Bestimmung der Einzelenzymaktivitäten der Glykolyse
Alle Einzelenzymmessungen der Glykolyse wurden in der zytosolischen Fraktion
durchgeführt. Es wurden je nach Enzym unterschiedliche Volumina verwendet und
zur Messung mit dem jeweiligen Reaktionspuffer auf 300 µl aufgefüllt.
3.5.3.1 Hexokinase
Die Aktivität der Hexokinase (HK) wurde in EA-Puffer supplementiert mit 0,2 mM
NADP, 2 mM Glukose, 2 mM ATP und 0,05 U/ml Glukose-6-
Phosphatdehydrogenase bei pH 7,4 und 25 °C gemessen. Die HK-Aktivität wurde als
NADP- Reduktion bei λ = 340-400 nm bestimmt.
3.5.3.2 Phosphofruktokinase
Die Aktivität der Phosphofruktokinase (PFK) wurde in EA-Puffer supplementiert mit
0,5 mM NADH, 4 mM Fruktose-6-Phosphat, 2 mM ATP, 0,2 U/ml Aldolase, 0,8 U/ml
Triosephosphatisomerase und 0,1 U/ml α-Glyzerophosphatdehydrogenase bei pH
7,4 und 25 °C gemessen. Die PFK-Aktivität wurde als NADH-Oxidation bei λ = 340-
400 nm bestimmt.
3.5.3.3 Triosephosphatisomerase
Die Aktivität der Triosephosphatisomerase (TPI) wurde in EA-Puffer supplementiert
mit 0,2 mM NADH, 4,9 mM DL-Glyzeraldehyd-3-Phosphat und 0,4 U/ml α-
Glyzerophosphatdehydrogenase bei pH 7,4 und 25 °C gemessen. Die TPI-Aktivität
wurde als NADH-Oxidation bei λ = 340-400 nm bestimmt.
3.5.3.4 Glutaraldehydphosphatdehydrogenase
Die Aktivität der Glutaraldehydphosphatdehydrogenase (GAPDH) wurde in EA-Puffer
supplementiert mit 83 mM Triethanolamin, 6,7 mM 3-Phosphoglyzerat, 3 mM L-
Cystein, 2 mM MgSO4, 0,1 mM NADH, 1,1 mM ATP und 10 U/ml 3-

Phosphoglyzeratphosphokinase bei pH 7,4 und 25 °C gemessen. Die GAPDH-
Aktivität wurde als NADH-Oxidation bei λ = 340-400 nm bestimmt.
3.5.3.5 Phosphoglyzeratmutase
Die Aktivität der Phosphoglyzeratmutase (PGM) wurde in EA-Puffer supplementiert
mit 1 mM 3-Phosphoglyzerat, 1 mM ADP, 0,5 mM NADH, 2 U/ml
Laktatdehydrogenase und 2 U/ml Pyruvatkinase bei pH 7,4 und 25 °C gemessen.
Die PGM-Aktivität wurde als NADH-Oxidation bei λ = 340-400 nm bestimmt.
3.5.3.6 Enolase
Die Aktivität der Enolase (E) wurde in EA-Puffer supplementiert mit 1 mM 2-
Phosphoglyzerat, 1 mM ADP, 0,5 mM NADH, 2 U/ml Laktatdehydrogenase und 2
U/ml Pyruvatkinase bei pH 7,4 und 25 °C gemessen. Die Enolase-Aktivität wurde als
NADH-Oxidation bei λ = 340-400 nm bestimmt.
3.5.3.7 Pyruvatkinase (hoch affin)
Die Aktivität der hoch affinen Pyruvatkinase (PK HA) wurde in der zytosolischen
Fraktion der Zellen in EA-Puffer supplementiert mit 1 mM ADP, 0,1 mM
Phosphoenolpyruvat, 0,5 mM NADH, 2 U/ml Laktatdehydrogenase bei pH 7,4 und 25
°C gemessen. Die PK HA-Aktivität wurde als NADH-Oxidation bei λ = 340-400 nm
bestimmt.
3.5.3.8 Pyruvatkinase (niedrig affin)
Die Aktivität der niedrig affinen Pyruvatkinase (PK LA) wurde in EA-Puffer
supplementiert mit 1 mM ADP, 10 mM Phosphoenolpyruvat, 0,5 mM NADH, 2 U/ml
Laktatdehydrogenase bei pH 7,4 und 25 °C gemessen. Die PK LA-Aktivität wurde als
NADH-Oxidation bei λ = 340-400 nm bestimmt.

3.5.3.9 Laktatdehydrogenase
Die Aktivität der Laktatdehydrogenase (LDH) wurde in EA-Puffer supplementiert mit 1
mM Natriumpyruvat und 0,5 mM NADH bei pH 7,4 und 25 °C gemessen. Die LDH-
Aktivität wurde als NADH-Oxidation bei λ = 340-400 nm bestimmt.
3.5.4 Bestimmung der Einzelenzymaktivitäten des Zitrat-Zyklus
Alle Einzelenzymmessungen des Zitrat-Zyklus wurden mit Ausnahme des 2-
Oxoglutarat-dehydrogenasekomplexes (OGDHc) mit Digitonin permeabilisierten
Zellen durchgeführt. Es wurden je nach Enzym unterschiedliche Volumina verwendet
und zur Messung mit dem jeweiligen Reaktionspuffer auf 300 µl aufgefüllt.
3.5.4.1 Zitratsynthase
Die Aktivität der Zitratsynthase (ZS) wurde in EA-Puffer supplementiert mit 0,5 mM
Oxalacetat, 0,3 mM Acetyl-CoA und 0,1 mM 5',5"-Dithiobis-(2-nitrobenzoate) (DTNB)
bei pH 7,4 und 25 °C gemessen. Die ZS-Aktivität wurde als DTNB-Reduktion bei λ =
512 nm bestimmt.
3.5.4.2 Mitochondriale Aconitase
Die Aktivität der Aconitase (A) wurde in EA-Puffer supplementiert mit 0,5 mM
Oxalacetat, 0,3 mM Acetyl-CoA und 0,1 mM 5',5"-Dithiobis-(2-nitrobenzoate) (DTNB)
bei pH 7,4 und 25 °C gemessen. Die A-Aktivität wurde als DTNB-Reduktion bei λ =
512 nm bestimmt.
3.5.4.3 Isozitratdehydrogenase
Die Aktivität der Isozitratdehydrogenase (IDH) wurde in EA-Puffer supplementiert mit
10 mM Natriumphosphat, 2 mM Zitrat und 0,2 mM NADH bei pH 7,4 und 25 °C
gemessen. Die CS-Aktivität wurde als NADH-Oxidation bei λ = 340-400 nm
bestimmt.

3.5.4.4 Fumarase
Die Aktivität der Fumarase (Fum) wurde in EA-Puffer supplementiert mit 2 mM
Fumarat, 10 mM Kaliumphosphat, 0,33 mM NAD, 100 µM Acetyl-CoA, 660munits/ml
Malatdehydrogenase und 400munits/ml Zitratsynthase bei pH 7,4 und 37 °C
gemessen. Die F-Aktivität wurde als NAD-Reduktion bei λ = 340-400 nm bestimmt.
3.5.4.5 Malatdehydrogenase
Die Aktivität der Malatdehydrogenase (MDH) wurde in EA-Puffer supplementiert mit 2
mM Malat, 10 mM Kaliumphosphat, 0,33 mM NAD, 100 µM Acetyl-CoA und
400munits/ml Zitratsynthase bei pH 7,4 und 37 °C gemessen. Die MDH-Aktivität
wurde als NAD-Reduktion bei λ = 340-400 nm bestimmt.
3.5.4.6 Bestimmung der Aktivität von Pyruvatdehydrogenasekomplex (PDHc) und 2-Oxoglutaratdehydrogenasekomplex (OGDHc)
Die Aktivitätsbestimmung von PDHc und OGDHc erfolgte aufgrund der geringen
Aktivität radiometrisch. Die Zellen wurden für 48 Stunden in 6-Bodenlochplatten
kultiviert, geerntet und in KRB supplementiert mit Pyruvat oder 2-Oxoglutarat
überführt. Der Ansatz wurde nach Zugabe von 0,2 µC 14C-Pyruvat bzw. 14C-2-
Oxoglutarat für 10 Minuten inkubiert. Das 14C markierte CO2 wurde in Hyalin
gebunden und ausgezählt.
3.5.5 Bestimmung der Aktivität der Enzyme der Atmungskettenkomplexe
3.5.5.1 Komplex I (NADH-Reduktase)
Die Aktivität von Komplex I (CI) wurde in EA-Puffer supplementiert mit 0,2 mM
NADH, 0,05 % Laurylmaltosid (w/v), 60 µM 2,3-Dimethoxy-5-Methyl-6-Decyl–1,4-
Benzochinon (DBQ) und 2 mM NaN3 bei pH 7,4 und 30 °C gemessen. Die CI-
Aktivität wurde als NADH-Oxidation bei λ = 340-400 nm bestimmt.

3.5.5.2 Komplex II (Succinat-Dehydrogenase)
Die Aktivität von Komplex II (CII) wurde in EA-Puffer supplementiert mit 20 mM
Succinat, 0,05 % Triton X-100 (w/v), 40 µM 2,3-Dimethoxy-5-Methyl-6-Decyl–1,4-
Benzochinon (DBQ), 60 µM Dichlorphenolindophenol (DCPIP) und 2 mM NaN3 bei
pH 7,4 und 37 °C gemessen. Die CII-Aktivität wurde als DCPIP-Oxidation bei λ =
610-700 nm bestimmt.
3.5.5.3 Komplex III (Cytochrom c-Reduktase)
Die Aktivität von Komplex III (CIII) wurde in EA-Puffer supplementiert mit 50 μM
Decylubihydrochinon (DBH), 0,05 % Triton X-100 und 2 mM NaN3 bei pH 7,4 und 25
°C gemessen. In jedes Well wurden 2 μl oxidiertes Cytochrom c (10 mM in 20 mM
KPi und pH 7,4) vorgelegt und die CIII-Aktivität als Cytochrom c-Reduktion bei λ =
540-550 nm bestimmt.
Die Herstellung von DBQ aus durch Reduktion DBH erfolgte durch Zugabe von
Natrium-Dithionit (Brandt und Okun, 1997). Die Konzentrationsbestimmung von DBH
erfolgte bei 290-350 nm ε290-350 = 4,2 mM-1 × cm-1).
3.5.5.4 Komplex IV (Cytochrom c-Oxidase)
Die Aktivität von Komplex IV (CIV) wurde in EA-Puffer supplementiert mit 120 mM
KPi und 0,05 % Laurylmaltosid bei pH 7,4 und 25 °C gemessen. In jedes Well
wurden 2 μl reduziertes Cytochrom c (10 mM in 20 mM KPi und pH 7,4) vorgelegt
und die CIV-Aktivität als Cytochrom c-Oxidation bei λ = 540-550 nm bestimmt.
Die Herstellung von reduziertem aus oxidiertem Cytochrom c erfolgte durch Zugabe
von 0,3 ml einer 1 M Natriumdithionitlösung zu 2,7 ml einer 10 mM Cytochrom c-
Lösung in 20 mM KPi. Nach der Äquilibrierung einer Säule (Econo-Pac 10 DG
Columns) mit 100 ml wurde die Cytochrom c-Dithionitlösung auf die Säule gegeben.
Die Elution erfolgte mit stickstoffbegastem 20 mM KPi-Puffer und die reduzierte
Cytochrom c-Lösung wurde in 200 µl Fraktionen aufgefangen. Der Gehalt an

reduziertem Cytochrom c (ε 540-550 nm = 19,0 mM-1 × cm -1) in der Lösung wurde
spektroskopisch bestimmt.
3.5.5.5 Komplex V (ATP-Synthase)
Die Aktivität von Komplex V (CV) wurde in EA-Puffer supplementiert mit 250 µM
NADH, 1mM Phosphoenolpyruvat, 2,5 U/ml Laktatdehydrogenase, 2 U/ml
Pyruvatkinase, 2 mM ATP und 1 µM DQA bei pH 7,4 und 37 °C gemessen. Die CV-
Aktivität wurde als reverse Reaktion durch Hydrolyse von ATP zu ADP gemessen.
Die Pyruvatkinase setzt Phosphoenolpyruvat, zu Pyruvat um, dabei wird ATP aus
ADP gebildet. Die Laktatdehydrogenase katalysiert die Reaktion von Pyruvat zu
Laktat, die Enzymaktivität wurde durch die damit verbundene Oxidation von NADH
zu NAD bei λ = 340-400 nm bestimmt.
3.6 Bestimmung des mitochondrialen Sauerstoffverbrauchs
Zur Ermittlung des mitochondrialen Sauerstoffverbrauchs von Kontrollen und
Patientenzellen wurde ein Oroboros 1 oxygraph system der AG Mairbäurl (Paar,
Österreich) verwendet. Die Messung erfolgte für 2 Proben parallel, so dass immer
Kontrollen mit Patienten verglichen werden konnten. Die Elektrode im Oxygraph
erfasste die Sauerstoffkonzentration der Reaktionskammer sowie die Änderung der
Sauerstoffkonzentration über die Zeit. Die Zellen wurden geerntet, gewaschen und in
die Elektrodenkammern gegeben. Als Medium wurde DMEM + 10 % FKS, KRB mit 4
mM Glukose oder KRB mit 4 mM Pyruvat verwendet. Für die Initiation des Systems
und die Messung wurden die Zellen 20 Minuten unter leichtem Rühren in den
Kammern inkubiert, nach Zugabe von 10 mM Kaliumcyanid (KCN) wurde der
Sauerstoffverbrauch der Atmungskette unterbunden. Der folgende
Sauerstoffverbrauch umfasste alle Prozesse der Zelle außer der mitochondrialen
Atmung. Durch Bildung der Differenz von insensitiven (vor Zugabe KCN) und
sensitiven (nach Zugabe KCN) Sauerstoffverbrauch, wurde der mitochondriale Anteil
des Sauerstoffverbrauchs ermittelt (Krawczyk et al., 2010).
ΔO2 (sensitiv) = ΔO2 (insenstitv + sensitiv) - ΔO2 (insensitiv)

3.7 ROS-Nachweis durch reduziertes Glutathion
Zum Nachweis von ROS wurde der Gehalt an reduziertem und oxidiertem Glutathion
durch die Aktivität der Glutathionreduktase spektrophotometrisch ermittelt.
Reduziertes Glutathion (GSH) besitzt eine freie Thiolgruppe und kann durch
Elektronenabgabe an ROS oxidiert werden, wodurch sich zwei Glutathion-Moleküle
über eine Disulfidbrücke zu Glutathion-Disulfid (GSSG) verbinden.
Die Zellen von Kontrollen und Mut0-Patienten wurden für 48 Stunden in T25-
Kulturflaschen kultiviert. Anschließend wurden sie geerntet, gewaschen, in PBS
aufgenommen und auf Eis gestellt. Nach der Proteinbestimmung wurde der
Proteingehalt jeder auf den gleichen Wert eingestellt und durch Zugabe von
Sulfosalicylsäure deprotoniert. Die Proben wurden zentrifugiert und der Überstand
mit Triethylaminlösung und Vinylpyridinlösung für 1 Stunde bei Raumtemperatur
inkubiert. Danach konnten die Proben spektrophotometrisch gemessen werden. Zur
Einordnung des GSH und GSSG-Gehalts wurde eine Kalibrierreihe mit bekannter
Menge GSH parallel mitgemessen. Die Aktivität der Glutathionreduktase wurde in
einem Puffer bestehend aus 180 mM Dinatriumhydrogenphosphat, 6 mM EDTA, 0,3
mM NADPH und 0,6 mM DTNB bei 37 °C und einem pH-Wert von pH 7,5 gemessen.
Die Glutathionreduktase katalysiert die Reaktion von oxidiertem (GSSG) zu
reduziertem Glutathion (GSH), die Enzymaktivität wurde durch die damit verbundene
Oxidation von NADPH zu NADP bei λ = 340-400 nm bestimmt.
3.8 Färbung der Zellen mit H2DCFDA und ROS-Nachweis
Der Fluoreszenzfarbstoff 2’,7’-Dichloro-Dihydrofluoresceindiacetat (H2DCFDA) ist
zellpermeabel und ändert nach Abspaltung seiner Acetatgruppen durch intrazelluläre
Esterasen und Oxidation sein Absoprtionsspektrum. Es ist ein Derivat von
Fluorescein und besitzt Acetatgruppen, die nach Diffusion ins Zelllumen intrazellulär
von Esterasen abgespalten werden. ROS oxidieren die freigewordenen
Thiolgruppen, das Extinktionsmaximum ändert sich und die Fluoreszenz kann mit
einem Plattenfluorometer gemessen werden. Nach der Abspaltung der
Acetatgruppen ist der Farbstoff nicht mehr zellpermeabel und verbleibt in der Zelle.
Die Zellen wurden 48 Stunden in 6 Lochbodenplatten kultiviert und 4 Stunden in
Medium oder Krebs-Ringer-Puffer mit unterschiedlichen Supplementen inkubiert.

Nach 30 Minuten Färbung mit H2DCFDA wurde die Fluoreszenz mit einem
Plattenfluorometer in jedem Well an 9 Punkten gemessen und der Mittelwert gebildet.
3.9 Quantitative Analyse von MMA mittels MS/MS
MMS wurde im Medium von Kontrollen und Mut0-Zellen durch electrospray ionization
mass spectrometry (MS/MS) nachgewiesen. Dafür wurden die Zellen zunächst in
T75-Kulturflaschen für 96 Stunden kultiviert. Das Medium wurde abgenommen und
mit 1µl 3-fach deuterierter MMS als Standard versetzt (d3-MMS, 10 mM) versetzt.
Anschließend wurde das Medium mit Salzsäure angesäuert und gemischt um alle
Säuren in die organische Phase zu überführen. Durch Zugabe von Natriumchlorid
wurde der pH-Wert neutralisiert. Danach wurden die organischen Säuren mit
Ethylacetat extrahiert. Nach Zentrifugation konnte die organische von der wässrigen
Phase getrennt und in neue Pyrex-Röhrchen überführt werden. Das Eluat wurde bei
40 °C auf einem Heizblock unter Stickstoffbegasung eingedampft. Durch Zugabe
eines Lösungsmittels aus Ethanol/Acetonitril/Ameisensäure (50:50:1) (v/v) wurde das
Eluat wieder gelöst. Eine parallele Aufarbeitung erfolgte mit Medium das mit MMS
bekannter Konzentration zur Quantifizierung der Ergebnisse versetzt war. Nach der
Überführung in 96-Lochbodenplatten erfolgte die Analyse in einem Quattro Ultima
Micro API mass spectrometer (Micromass, Manchester, UK) mit einer Elektrospray
Ionenquelle und Micromass MassLynx data system statt. Durch einen Strom aus
Acetonitril/Wasser (60:40) (v/v) durch einen MS2777 autosampler (Waters SAS, St.
Quentin, Frankreich) und einer Flux Rheos 2000 HPLC Pumpe (Flux Instruments,
Basel, Suisse) wurde die Probe injiziert.
3.10 Statistische Auswertung
Bei Vergleichen aller Zelllinien (Kontrollen: n=3-9; PA: n=3; MMA: n=6) wurde eine
univariate Varianzanalyse (ANOVA) durchgeführt. Das Signifikanzniveau wurde auf 5
% festgesetzt. Als Kontraste wurden Ergebnisse von Kontrollen oder PA-Patienten
mit MMAzidurie-Patienten verglichen. Die statistische Auswertung der Ergebnisse
erfolgte mit der Software SPSS Version 22. Bei der Verwendung von 2 Zelllinien
wurde ein Student’s T-Test durchgeführt.

4 Ergebnisse
4.1 Relevante Werte der Zellspender
Die aus dem Urin isolierten humanen PTEC wurden von Kontrollen, MMA- und PA-
Patienten unterschiedlichen Alters und Geschlechts gewonnen. Für diese Arbeit
wurden Zellen von 9 Kontrollen, 3 PA-Patienten und 6 MMA-Patienten verwendet.
(Tabelle 2). Da sich die fünf mut0-Patienten und der CblB-Patient in allen
durchgeführten Versuchen nicht voneinander unterschieden, wurden sie unter der
Gruppe MMA zusammengefasst.
Tabelle (2) Übersichtsdaten von Kontrollen, MMA- und PA-Patienten die als Spender von PTEC dienten
PTEC Alter Geschlecht Mutation im Gen und Protein
MMS im Plasma über 2 Jahre [µmol/L]
Ctrl_1 37 W n.a. n.a.
Ctrl_2 29 M n.a. n.a.
Ctrl_3 22 W n.a. n.a.
Ctrl_4 24 M n.a. n.a.
Ctrl_5 12 M n.a. n.a.
Ctrl_6 7 M n.a. n.a.
Ctrl_7 6 M n.a. n.a.
Ctrl_8 11 W n.a. n.a.
Ctrl_9 10 M n.a. n.a.
PA_1 18 M n.a. n.a.
PA_2 11 M n.a. n.a.
PA_3 10 W n.a. n.a.
Mut0_1 22 W T862C Ser288Pro 4112 (± 1430)
Mut0_2 23 M C982T Leu328Phe 4239 (± 2845)
Mut0_3 18 W G607A Gly203Arg 1230 (± 414)
Mut0_4 11 W G607A Gly203Arg
C1105T Arg369Cys
3278 (± 1527)
Mut0_5 16 M T862C Ser288Pro
A1157G H386R
4579 (± 1527)
CblB_1 40 M C556T Arg186W 3143 (± 453)

Die Gruppe der MMA-Patienten bestand aus 5 mut0-Patienten und 1 Cobalamin B-
Patient (CblB). Die Mutationen der Patienten Mut0_1-5 befinden sich im Mut-Gen,
das für die MCM codiert. Die Mutation des Patienten CblB_1 befindet sich im MMAB-
Gen, das für die Cobalamin-Adenosyltransferase codiert. Werte für die MMS-
Konzentration im Blutplasma wurden nur von MMA-Patienten im Zuge ambulanter
oder stationärer Patientenbesuche mittels Tandem-MS erhoben. Das Plasma der
MMA-Patienten wurde im Zeitraum von 365 Tagen vor und nach Zellgewinnung auf
seine MMS-Konzentration untersucht. Die Höhe der MMS-Konzentration im Plasma
der Patienten Mut0_1 (4112 µmol/L), Mut0_2 (4239 µmol/L), Mut0_3 (1230 µmol/L),
Mut0_4 (3278 µmol/L), Mut0_5 (4579 µmol/L) und CblB_1 (3143 µmol/L) war von
starken Schwankungen gekennzeichnet.
Der Vergleich der MMS-Werte zeigt, dass die Menge an MMS bei 5 Patienten mit
Defekt der MCM und einem Patienten mit Defekt der Cobalamin-Adenosyltransferase
ein Jahr vor und nach Isolierung der PTEC stark erhöht war.

4.2 Charakterisierung der verwendeten humanen PTEC
Die verwendeten humanen PTEC sollten zunächst auf ihre Spezifität hin untersucht
werden. Es sollte überprüft werden, ob die Zellen der Kontrollen, MMA- und PA-
Patienten Charakteristiken von PTEC aufweisen. Dafür wurden sie auf Morphologie
und Expression von Markerproteinen hin getestet.
Epithelzellen weisen ein für sie typisches Wachstum in
Kopfsteinsteinpflasteranordnung auf. Alle verwendeten Zelllinien zeigten im
Lichtmikroskop eine kopfsteinpflasterartige Morphologie und Wachstum in
Einzelzellschicht (Abb. 4).
Ctrl_2 Ctrl_1
Ctrl_4 Ctrl_3

Ctrl_6 Ctrl_5
Ctrl_8 Ctrl_7
PA_1 Ctrl_9
PA_3 PA_2

Abbildung (4) Lichtmikroskopische Aufnahmen aller verwendeten humanen PTEC. Sie zeigten ein für Epithelzellen typisches Wachstum. Die Zellen der Kontrollen Ctrl_1-9, PA_1-3, Mut0_1-5 und CblB_1 wachsen alle in Kopfsteinpflasteranordnung.
Im Urogenitaltrakt des Menschen finden sich verschiedene Epithelzellen, die das
Interstitium zum Primärharn und Harn hin abschließen. Diese Zellen exprimieren
jeweils für sie charakteristische Proteine. Beim Epithel des proximalen Tubulus sind
dies das integrale Membranprotein Aquaporin 1 (AQP1) und das membranständige
Protein γ-Glutamyltransferase 1 (γGT1).
Mut0_1 Mut0_2
Mut0_4 Mut0_3
CblB_1 Mut0_5

Aquaporin 1 bildet eine Pore, die an der Regulation des Wassergehalts des
Primärharns im proximalen Tubulus beteiligt ist. Es wird beim Menschen im
proximalen, nicht aber im distalen Tubulus exprimiert (Bedford et al., 2003). Die γ-
Glutamyltransferase 1 überträgt den Glutamylrest von reduziertem Glutathion (GSH)
auf Peptide oder Wasser. Beide Proteine wurden in allen verwendeten Zelllinien
exprimiert (Abb. 5).
Abbildung (5) Expression von γ-Glutamyltransferase 1 (γGT1) und Aquaporin 1 (AQP1) der verwendeten PTEC. Im Western Blot zeigten alle verwendeten Zelllinien die Expression der Markerproteine AQP1 und γGT1. Der Nachweis von β-Aktin diente als innerer Standard.

4.3 Produktion von Methylmalonsäure
Neben Morphologie und der Expression von Markerproteinen sollten die Zellen in
Hinblick auf ihren enzymatischen Defekt der MCM bzw. des Cofaktors
Hydroxycobalamin hin untersucht werden. Dies wurde durch die Bestimmung der
Konzentration der MMS im Medium der Zellen überprüft. MMS entsteht direkt beim
Abbau von Propionsäure (PS) und wird normalerweise durch die MCM weiter zu
Succinat metabolisiert, um danach dem Zitrat-Zyklus zugeführt zu werden. Durch den
enzymatischen Defekt kommt es zur Akkumulation von MMS in der Zelle. Durch
Bindung der Säure an Carnitin kann die Zelle die Säure an ihre Umgebung abgeben,
um Schädigungen der Zelle und des Organismus zu verringern.
Im Medium der PTEC von MMA-Patienten war die MMS-Konzentration signifikant um
mehr als 100% gegenüber den Zellen der Kontrollen erhöht (p < 0,05; Abb. 6).
Abbildung (6) MMS-Konzentration im Medium von PTEC von Kontrollen und MMA-Patienten. PTEC von MMA-Patienten produzierten und sezernierten mehr MMS als PTEC von Kontrollen. Im Mittel lag die MMS-Konzentration mit 6 mmol/mg Protein gegenüber 2,5 mmol/mg Protein mehr als doppelt so hoch. *p < 0,05. (Ctrl: n=4, MMA: n=4).
*

4.4 Bioenergetik
Bei Patienten mit MMA wurde in unterschiedlichen Modellen gezeigt, dass es zu
einer Beeinträchtigung des Energiemetabolismus kommt, welche sich in einer
Unterversorgung vor allem der Zellen mit erhöhtem Energiebedarf äußert. Für
humane PTEC liegen bislang keine Daten hierzu vor. Um den Energiehaushalt
dieser Zellen zu überprüfen, wurden die Glykolyse, der Pyruvatdehydrogenase-
Komplex, der Zitrat-Zyklus und die Atmungskette untersucht.
4.4.1 Sauerstoffverbrauch der Mitochondrien
Der Sauerstoffverbrauch der mit Digitonin permeabilisierten Zellen wurde in 1)
DMEM, sowie in 2) Krebs-Ringer-Puffer mit Glukose und 3) Krebs-Ringer-Puffer mit
Pyruvat bestimmt. Die Bedingungen in DMEM stellen für die Zellen die
Idealbedingungen dar, unter welchen ihnen Energie in Form von Glukose,
Reduktionsmitteln und Aminosäuren zur Verfügung stehen. In KRB mit Glukose wird
speziell die Energiegewinnung durch die Glykolyse angesteuert, bei KRB mit Pyruvat
der Zitrat-Zyklus. Durch die verschiedenen Ansätze kann ein Defekt leichter der
Glykolyse, dem Zitrat-Zyklus oder der Atmungskette zugeordnet werden.
In DMEM zeigten Zellen von MMA- und PA-Patienten einen signifikant verringerten
Sauerstoffverbrauch um 70 % im Vergleich zu Kontrollen (p < 0,05; Abb. 7). Unter
Bedingungen, in welchen die Zellen alleine auf Glukose als Energieträger
zurückgreifen konnten, wiesen Zellen von MMA-Patienten einen signifikant
verringerten Sauerstoffverbrauch auf 30 % auf, bei PA-Patienten war die
Verringerung aufgrund der großen Streuung auf 26 % nicht signifikant (p < 0,05; Abb.
7). Bei Pyruvat als Energieträger war der Sauerstoffverbrauch von MMA-Patienten
gegenüber PA-Patienten signifikant um 50 % verringert, die Verringerung gegenüber
den Kontrollen war aufgrund der großen Streuung nicht signifikant (p < 0,05; Abb. 7).

Abbildung (7) Mitochondrialer Sauerstoffverbrauch der humanen PTEC in KRB mit 4 mM Glukose, KRB mit 4 mM Pyruvat und DMEM. *p < 0,05. (Ctrl: n=5, PA: n=3, MMA: n=6).
Da der verringerte mitochondriale Sauerstoffverbrauch, der bei PTEC von MMA-
Patienten unter DMEM, Glukose und Pyruvat als Energieträger und bei PA-Patienten
unter DMEM auftrat, wurde der Energiemetabolismus auf den Ebenen der Glykolyse,
des Zitrat-Zyklus und der Atmungskette untersucht.
4.4.2 Glykolyse
Zunächst wurden aufgrund des verringerten mitochondrialen Sauerstoffverbrauchs
bei PTEC von MMA-Patienten die Einzelenzymaktivitäten der Glykolyse untersucht.
Der Vergleich der Einzelenzymaktivität der glykolytischen Enzyme von Kontrollzellen,
Zellen von MMA- und PA-Patienten zeigte, dass bei PTEC der MMA-Patienten
gegenüber Kontrollen und PA-Patienten die Aktivität der Phosphofruktokinase (PFK)
signifikant auf 35 % verringert, die Aktivität der Triosephosphat-Isomerase (TPI)
signifikant um 30 % erhöht und die Aktivität der Glyceraldehydphosphat-
Dehydrogenase (GAPDH) signifikant um 15 % erhöht war (p < 0,05%; Abb. 8). Die
* * **

Aktivitäten von Phosphoglyceratmutase (PGM) und Enolase (E) war bei PTEC von
PA-Patienten im Vergleich zu MMA-Patienten signifikant verringert, die Aktivität der
beiden Enzyme bei PTEC von MMA-Patienten befanden sich jedoch etwa auf dem
Niveau von Kontrollzellen (p < 0,05; Abb. 8). Die anderen Enzyme der Glykolyse,
welche Hexokinase (HK), hoch affine und niedrig affine Pyruvatkinase (PK HA, PK
LA) sowie die Laktadehydrogenase (LDH) umfassen, waren nicht signifikant
verändert (Abb. 8).
Abbildung (8) Enzymaktivitäten der glykolytischen Enzyme. HK: Hexokinase; PFK: Phosphofruktokinase; TPI: Triosephosphatisomerase; GAPDH: Glyceraldehydphosphat-Dehydrogenase; PGM: Phosphoglyceratmutase; E: Enolase; PK HA: Pyruvatkinase tetramer; PK LA: Pyruvatkinase dimer; LDH: Laktatdehydrogenase in % der Kontrolle (*p < 0,05). (Ctrl: n=4, PA: n=3, MMA: n=5).
Insgesamt wurden 9 Enzymen der Glykolyse gemessen, bei 3 Enzymen wurden
signifikante Unterschiede zwischen Kontrollen und MMA-Patienten, sowie bei 5
Enzymen signifikante Unterschiede zwischen MMA- und PA-Patienten festgestellt.
*
* *
**
* **

4.4.3 Pyruvat-Dehydrogenasekomplex
Die Bestimmung der Aktivität des Pyruvat-Dehydrogenasekomplexes (PDHc) erfolgte
durch einen radiometrischen Ansatz. In der ersten Teilreaktion erfolgt eine
Decarboxylierung von Pyruvat. Durch Zugabe von radioaktiv markiertem Pyruvat zum
Versuchsansatz konnte das entstehende 14C-CO2 gemessen werden.
Die Aktivität des PDHc zeigte beim Vergleich bei PTEC der MMA-Patienten im
Vergleich zu PTEC von Kontrollen eine Tendenz zur Verringerung auf 65 %, die
aufgrund der großen Streuung der Kontrollen jedoch nicht signifikant war, bei PA-
Patienten fiel die Verringerung der Aktivität gegenüber PTEC von Kontrollen noch
etwas größer aus, war jedoch ebenfalls nicht signifikant (Abb. 9).
Abbildung (9) Enzymaktivität des PDHc der PTEC von PA- und MMA-Patienten als 14C-Produktion in % der Kontrolle (*p < 0,05). (Ctrl: n=9, PA: n=3, MMA: n=6).

4.4.4 Zitrat-Zyklus
Die Einzelenzymaktivitäten von Zitratsynthase (CS), mitochondrialer Aconitase (A),
Isozitrat-Dehydrogenase (IDH), Fumarase (Fum) und Malatdehydrogenase (MDH)
wurden spektrophotometrisch analog zur Glykolyse bestimmt (4.4.2), die
Aktivitätsbestimmung des 2-Oxoglutarat-Dehydrogenasekomplexes (OGDHc)
erfolgte analog zum PDHc radiometrisch (4.4.3).
Die Aktivität der A war bei PTEC von MMA-Patienten signifikant um 30 % verringert
(p < 0,05; Abb. 10). Die A besitzt ein Eisenatom im katalytischen Zentrum, welches
das Enzym anfällig für reaktive Sauerstoffspezies (ROS) werden lässt. Der OGDHc
war bei MMA-Patienten signifikant um 45 % verringert, die Verringerung bei PA-
Patienten um 50 % war nicht signifikant (p < 0,05; Abb. 10). Er katalysiert den
geschwindigkeitsbestimmenden Schritt im Zitrat-Zyklus.
Abbildung (10) Enzymaktivitäten des Zitrat-Zyklus. CS: Zitrat-Synthase; A: mitochondriale Aconitase; IDH: Isozitrat-Dehydrogenase; OGDHc: 2-Oxoglutarat-Dehydrogenasekomplex; Fum: Fumarase; MDH: Malat-Dehydrogenase in Prozent der Kontrolle (*p < 0,05). (Ctrl: n=9, PA: n=3, MMA: n=6)
* *

Die signifikant verringerten Aktivitäten der A und des OGDHc bei PTEC von MMA-
Patienten können auf eine Beeinträchtigung der Enzymaktivität, sowie einen
verlangsamten Ablauf des Zitrat-Zyklus hinweisen.
4.4.5 Oxidative Phosphorylierung
Die Aktivitätsbestimmung der Einzelenzymkomplexe der Atmungskette erfolgte in mit
Digitonin permeabilisierten PTEC. Zellen von PA-Patienten zeigten lediglich eine
nicht signifikante Verringerung der Aktivität der NADH-Dehydrogenase (Komplex I).
Bei PTEC von MMA-Patienten war eine signifikante Verringerung der Aktivität der
Cytochrom c-Oxidase (Komplex IV) gegenüber Kontrollzellen feststellbar (p < 0,05;
Abb. 11).
Abbildung (11) Enzymaktivitäten der Atmungskettenkomplexe von PTEC der Kontrollen, PA- und MMA-Patienten. CI: Komplex I; CII: Komplex II; CIII: Komplex III; CIV: Komplex IV; CV: Komplex V in % der Kontrolle (*p < 0,05). (Ctrl: n=9, PA: n=3, MMA: n=6).
*

4.5 Freisetzung reaktiver Sauerstoffspezies (ROS)
4.5.1 Nachweis von ROS durch die Oxidierung von reduziertem Glutathion
Die Gesamtmenge des interzellulären Glutathions war bei Kontrollen und MMA-
Patienten unverändert (Gesamt GSH). Die oxidierte Form von Glutathion, das
Glutathiondisulfid (GSSG), lag hingegen bei MMA-Patienten signifikant um 50 %
höher als bei Kontrollzellen vor, die reduzierte Form von Glutathion (GSH) lag
signifikant um 60 % geringer vor als bei Kontrollzellen (p < 0,05; Abb. 12).
Abbildung (12) Glutathion-Gehalt der PTEC von Kontrollen und MMA-Patienten in % der Kontrolle von Gesamt GSH: Gesamtglutathiongehalt; GSSG: disulfidgebundenes oxidiertes Glutathion; GSH: reduziertes Glutathion (*p < 0,05). (Ctrl: n=8, MMA: n=4).
* *

4.5.2 Nachweis von ROS durch 2’,7’-Dichloro-Dihydrofluoresceindiacetat
Zur Untersuchung der erhöhten ROS-Produktion bei PTEC von MMA-Patienten
wurde 2’,7’-Dichloro-Dihydrofluoresceindiacetat (H2DCFDA) verwendet.
Die Zellen wurden in William's E-Medium sowie in KRB mit verschiedenen
Supplementen kultiviert. Es sollte überprüft werden, ob die Entwicklung von ROS mit
einem Defekt des Energiemetabolismus verbunden sein könnte. KRB wurde mit
Glukose, Pyruvat oder Glutamin (4 mM) supplementiert, um den Energiegewinn
durch Glykolyse bzw. Zitrat-Zyklus und Atmungskette zu stimulieren. Desweiteren
wurde untersucht, ob sich die ROS-Produktion durch Inkubation mit PS, MMS oder
Isoleucin (Ile) (jeweils 5 mM) stimulieren ließ.
Williams E-Medium enthält viele Antioxidantien, die ROS teilweise entgegenwirken
können. Die Erhöhung der ROS war bei PTEC von MMA- und PA-Patienten nicht
signifikant verändert. In KRB, sowie in KRB supplementiert mit Glukose, Pyruvat und
Glutamin als Energiequelle war die ROS-Produktion von MMA-Patienten gegenüber
Kontrollen signifikant erhöht, in KRB supplementiert mit MMS oder Isoleucin als
Belastung war die ROS-Produktion gegenüber Kontrollen und PA-Patienten ebenfalls
signifikant erhöht (p < 0,05; Abb. 13).

Abbildung (13) Entstehung von ROS in PTEC von Kontrollen, PA- und MMA-Patienten in Williams E Medium, KRB, KRB mit Glukose, KRB mit Pyruvat, KRB mit Glutamin (Gln), KRB mit 5 mM Propionsäure (PS), KRB mit 5mM Methylmalonsäure (MMS), KRB mit 5mM Isoleucin (Ile) in % der Kontrolle. Alle Medien waren mit 10 µM H2DCFDA supplementiert (*p < 0,05). (Ctrl: n=3, PA: n=3, MMA: n=6).
* * * * * *
* **

4.6 Subzellulare Strukturen der humanen PTEC im Elektronenmikroskop
Neben Morphologie, Expression von Markerproteinen und Produktion von MMS
sollten die verwendeten Zellen auf Unterschiede und Auffälligkeiten in ihrer
Substruktur untersucht werden. Dafür wurden von 12 Zelllinien (6 Kontrollen: Ctrl_1,
Ctrl_2, Ctrl_3, Ctrl_5, Ctrl_6, Ctrl_7; 5 MMA-Patienten: Mut0_1, Mut0_2, Mut0_3,
Mut0_5, CblB_1; 1 PA-Patient: PA_1) elektronenmikroskopische Aufnahmen
angefertigt.
Auf den Bildern ist bei PTEC der MMA-Patienten die Akkumulation von
vesikelartigen, von einer Doppelmembran umgebenen Strukturen erkennbar, die auf
gebildete Autophagosome hinweisen (roter Pfeil). Weiterhin ist bei PTEC von MMA-
Patienten erkennbar, dass Teile des rauen endoplasmatischen Retikulums (ER)
ringförmige Strukturen bilden, die mit Mitochondrien assoziiert waren und ein
vergrößertes Lumen zeigten (Abb. 4.14, Mut0_1 – Mut0_5; CblB_1). Dies könnte ein
Hinweis auf Stress des ER sein (blauer Pfeil). Bei PTEC der Kontrollen und des PA-
Patienten sind weder von einer Doppelmembran umgebene Strukturen, noch
Auffälligkeiten des ER erkennbar (Abbildung 14, Ctrl_1 – Ctrl_7; PA_1). Die
Mitochondrien der MMA- und PA-Patienten erschienen im Vergleich zu Kontrollen
nicht unterschiedlich oder auffällig (Abb. 14).
Ctrl_1 Ctrl_2

Ctrl_5 Ctrl_3
Ctrl_7 Ctrl_6
Mut0_2 Mut0_1

Abbildung (14) Elektronenmikroskopische Bilder der PTEC von 6 Kontrollen, 5 Mut0-Patienten, einem CblB-Patienten und einem PA-Patient. Bei Zellen der MMA-Patienten sind von einer Doppelmembran umgebenen Vesikel erkennbar, die Autophagosome darstellen können (roter Pfeil). Das raue ER zeigt bei PTEC von MMA-Patienten Strukturveränderungen und ein vergrößertes Lumen (blauer Pfeil). Die Anfertigung der EM-Bilder erfolgte durch Frau Kaden in der Abteilung „Zelluläre und Molekulare Pathologie“ von Prof. Gröne im Deutschen Krebsforschungszentrum in Heidelberg.
Die von einer Doppelmembran umgrenzten vesikelartigen Strukturen und das
ausgedehnte Lumen des ER bei PTEC von MMA-Patienten können auf
autophagozytotische Prozesse und ER-Stress hinweisen.
Mut0_3 Mut0_5
PA_1 CblB_1

4.7 Initiation, Entwicklung und Abbau von Autophagosomen bei PTEC von MMA-Patienten
Um den Ursprung und die Entwicklung der mit einer Doppelmembran umschlossenen
vesikelartigen Strukturen zu untersuchen, welche bei PTEC von MMA-Patienten auf
elektronenmikroskopischen Bildern erkennbar waren, wurden die Zellen auf den drei
Ebenen 1) Regulation der Reifung der Phagophore, 2) Entwicklung der Phagophore
und 3) lysosomaler Abbau des Autophagosoms auf autophagozytotische Prozesse
hin untersucht.
Die statistische Signifikanz der Unterschiede zwischen den Zelllinien wurde
angegeben durch:
(CM* < 0,05): Kontrollen – MMA-Patienten
(CP* < 0,05): Kontrollen – PA-Patienten
(MP* < 0,05): MMA-Patienten – PA-Patienten
Die statistische Signifikanz der Unterschiede innerhalb der Zelllinien wurde
angegeben durch:
( *: p < 0,05): MMA-Patienten
( *: p < 0,05): PA-Patienten
4.7.1 Regulation der Reifung der Phagophore durch mTORC1
Zunächst wurde der Signalweg, der an der Steuerung der Reifung der Phagophore
beteiligt ist, untersucht. Die Regulierung der Reifung erfolgt über den
Multiproteinkomplex mTORC1. Die Aktivierung des mTOR-Signalweges wurde mit
Hilfe von Antikörpern gegen Proteine von mTORC1 (mTOR Regulation Antibody
Sampler Kit, Cell Signaling) bei Kontroll- und Patientenzellen im Western Blot
untersucht. Die Regulation von mTORC1 erfolgt über Phosphorylierung und
Dephosphorylierung der mTOR-Kinase, und durch am Komplex beteiligte
Regulatoren, wie z.B. PRAS40.
Das Protein PRAS40 ist dephosphoryliert an Raptor gebunden und inhibiert die
Assemblierung von mTORC1. Durch Phosphorylierung von PRAS40 (pPRAS40)

dissoziiert dieses von Raptor, die Assemblierung und die Aktivierung von mTORC1
wird gefördert.
Die Kinase mTOR ist an der Regulation von Autophagie durch Inhibierung des
Komplexes aus ULK1/FIP200/ATG13 beteiligt. Durch Phosphorylierung von mTOR
(pmTOR) wird mTORC1 aktiv und hemmt die Reifung der Phagophore.
Um die PTEC verschiedenen Stufen metabolischen Stresses auszusetzen, wurden
sie für 48 Stunden unter Bedingungen mit hohem (20 %), geringem (1 %) und sehr
geringem (0,5 %) FKS-Gehalt in DMEM kultiviert. Untersucht wurde der Verlauf der
Expressionslevel von Phospho-mTOR (Ser2448), mTOR, Phospho-PRAS40
(Thr246), PRAS40 und pRaptor (Ser792) in Abhängigkeit des FKS-Gehalts. Die
Werte der MMA- und PA-Patienten wurden auf die Kontrollen normiert dargestellt.
Unter Bedingungen mit 0,5 % FKS war die Ratio der relativen Expression von
pPRAS40/PRAS40 bei MMA-Patienten mit 180 % Expressionsstärke im Vergleich zu
Kontrollen am höchsten (CM*, MP*: p < 0,05), sie sank bei höheren
Supplementationen des Mediums mit 1 % FKS auf 140 % Expressionsstärke im
Vergleich zu Kontrollen (CM*, MP*: p < 0,05) und bei 20 % FKS auf 60 %
Expressionsstärke im Vergleich zu Kontrollen ab (CM*, MP*, *: p < 0,05). Somit
zeigte PRAS40 bei MMA-Patienten einen Zusammenhang mit dem FKS-Gehalt und
sank bei hohen FKS-Werten ab ( *: p < 0,05), was auf eine geringere Assemblierung
von mTORC1 hindeutete. Die Ratio der relativen Expression von pPRAS40/PRAS40
war unter Bedingungen mit 20 % FKS bei PA-Patienten mit 30 % Expressionsstärke
im Vergleich zu Kontrollen am geringsten (CP*, MP*: p < 0,05), sie stieg bei 1% FKS
auf 60 % Expressionsstärke im Vergleich zu Kontrollen an (CP*, MP*: p < 0,05), sank
jedoch bei 0,5 % FKS wieder auf 50 % Expressionsstärke im Vergleich zu Kontrollen
ab. (CP*, MP*: p < 0,05). Bei PRAS40 war kein Zusammenhang zwischen der
Phosphorylierung von PRAS40 und dem FKS-Gehalt erkennbar ( *: p < 0,05) (Abb.
15).
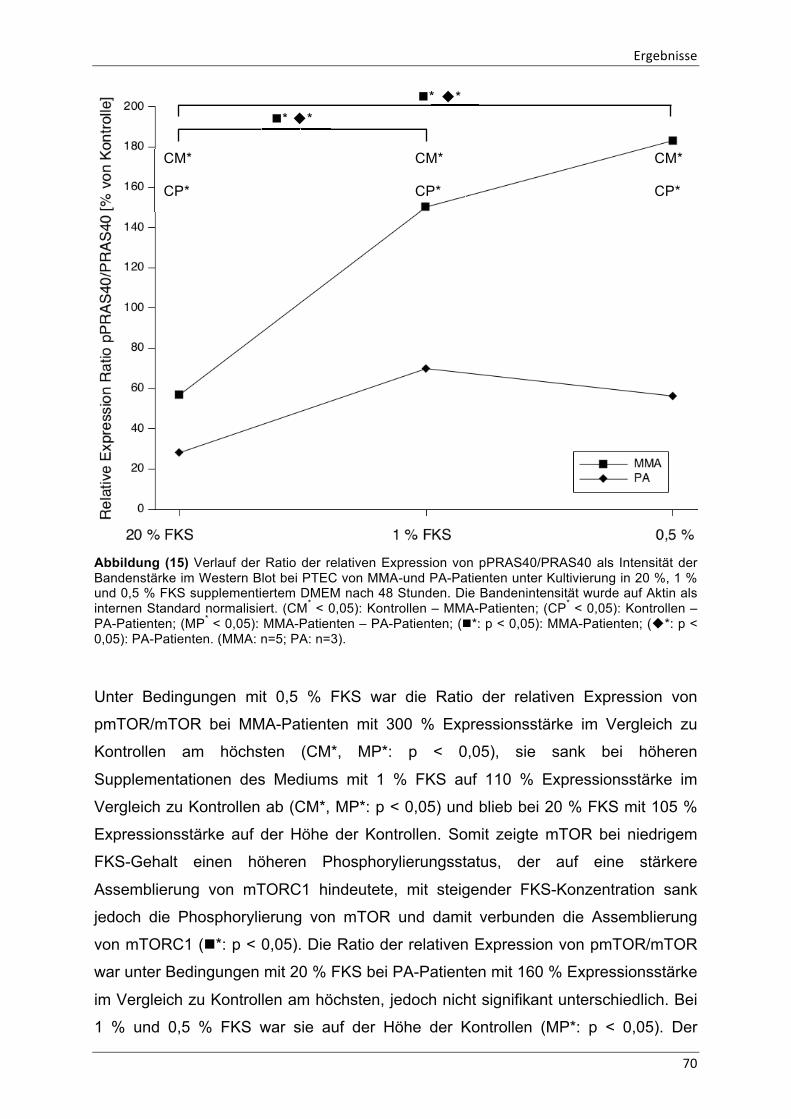
Abbildung (15) Verlauf der Ratio der relativen Expression von pPRAS40/PRAS40 als Intensität der Bandenstärke im Western Blot bei PTEC von MMA-und PA-Patienten unter Kultivierung in 20 %, 1 % und 0,5 % FKS supplementiertem DMEM nach 48 Stunden. Die Bandenintensität wurde auf Aktin als internen Standard normalisiert. (CM* < 0,05): Kontrollen – MMA-Patienten; (CP* < 0,05): Kontrollen – PA-Patienten; (MP* < 0,05): MMA-Patienten – PA-Patienten; ( *: p < 0,05): MMA-Patienten; ( *: p < 0,05): PA-Patienten. (MMA: n=5; PA: n=3).
Unter Bedingungen mit 0,5 % FKS war die Ratio der relativen Expression von
pmTOR/mTOR bei MMA-Patienten mit 300 % Expressionsstärke im Vergleich zu
Kontrollen am höchsten (CM*, MP*: p < 0,05), sie sank bei höheren
Supplementationen des Mediums mit 1 % FKS auf 110 % Expressionsstärke im
Vergleich zu Kontrollen ab (CM*, MP*: p < 0,05) und blieb bei 20 % FKS mit 105 %
Expressionsstärke auf der Höhe der Kontrollen. Somit zeigte mTOR bei niedrigem
FKS-Gehalt einen höheren Phosphorylierungsstatus, der auf eine stärkere
Assemblierung von mTORC1 hindeutete, mit steigender FKS-Konzentration sank
jedoch die Phosphorylierung von mTOR und damit verbunden die Assemblierung
von mTORC1 ( *: p < 0,05). Die Ratio der relativen Expression von pmTOR/mTOR
war unter Bedingungen mit 20 % FKS bei PA-Patienten mit 160 % Expressionsstärke
im Vergleich zu Kontrollen am höchsten, jedoch nicht signifikant unterschiedlich. Bei
1 % und 0,5 % FKS war sie auf der Höhe der Kontrollen (MP*: p < 0,05). Der
*** *** *
CM*
CP*
CM*
CP*
CM*
CP*

Phosphorylierungsstatus von mTOR, und die damit verbundene Assemblierung von
mTORC1, war bei PA-Patienten unabhängig vom FKS-Gehalt und nicht signifikant
unterschiedlich zu Kontrollen (Abb. 16).
Abbildung (16) Verlauf der Ratio der relativen Expression von pmTO/mTOR als Intensität der Bandenstärke im Western Blot bei PTEC von MMA- und PA-Patienten unter Kultivierung in 20 %, 1 % und 0,5 % FKS supplementiertem DMEM nach 48 Stunden. Die Bandenintensität wurde auf Aktin als internen Standard normalisiert. (CM* < 0,05): Kontrollen – MMA-Patienten; (CP* < 0,05): Kontrollen – PA-Patienten; (MP* < 0,05): MMA-Patienten – PA-Patienten; ( *: p < 0,05): MMA-Patienten; ( *: p < 0,05): PA-Patienten. (MMA: n=5; PA: n=3).
4.7.2 Reifung der Phagophore
Um zu überprüfen, ob mTORC1 tatsächlich zu einer Reifung der Phagophore und
damit verbundenen höheren Autophagie-Rate führt, wurde die Expression von zwei
Markerproteinen für die Reifung der Phagophore untersucht. Das microtubule light
chain protein 3 (LC3-II) und P62 binden an die reifende Phagophore und sind mit ihr
bis zur Degradierung in den Autolysosomen assoziiert.
**
CM*
MP*
CM*
MP*

Unter Bedingungen mit 0,5 % und 1 % FKS war die relative Expression von LC3-II
bei MMA-Patienten auf Höhe der Kontrollen, sie stieg bei 20 % FKS signifikant auf
150 % Expressionsstärke der Kontrollen an (CM*: p < 0,05). Somit zeigte die
Expression von LC3-II bei MMA-Patienten einen Zusammenhang mit dem FKS-
Gehalt und stieg bei hohen FKS-Werten an ( *: p < 0,05). Die erhöhte Expression
LC3-II bei hohem FKS-Gehalt deutete auf eine stärkere Reifung der Phagophore hin.
Die Expression von LC3-II war unter Bedingungen mit 20 % FKS bei PA-Patienten
mit 160 % Expressionsstärke im Vergleich zu Kontrollen am höchsten, jedoch nicht
signifikant unterschiedlich. Bei 1 % und 0,5 % FKS war sie auf der Höhe der
Kontrollen (MP*: p < 0,05). Der Phosphorylierungsstatus von mTOR und damit die
verbundene Assemblierung von mTORC1 war bei PA-Patienten unabhängig vom
FKS-Gehalt und nicht signifikant unterschiedlich zu Kontrollen (Abb. 17).
Abbildung (17) Relative Expression von LC3-II bei MMA- und PA-Patienten als Intensität der Bandenstärke im Western Blot unter Kultivierung in 20 %, 1 % und 0,5 % FKS supplementiertem DMEM nach 48 Stunden. Die Bandenintensität wurde auf Aktin als internen Standard normalisiert. (CM* < 0,05): Kontrollen – MMA-Patienten; (CP* < 0,05): Kontrollen – PA-Patienten; (MP* < 0,05): MMA-Patienten – PA-Patienten; ( *: p < 0,05): MMA-Patienten; ( *: p < 0,05): PA-Patienten. (MMA: n=5; PA: n=3).
*
CM*

Unter Bedingungen mit 0,5 % und 1 % FKS war die relative Expression von P62 bei
MMA-Patienten im Vergleich zu Kontrollen auf 60 % Expressionsstärke verringert
(CM*: p < 0,05), sie stieg bei 20 % FKS signifikant auf 200 % Expressionsstärke der
Kontrollen an (CM*: p < 0,05). Somit zeigte die Expression von P62 bei MMA-
Patienten einen Zusammenhang mit dem FKS-Gehalt und stieg bei hohen FKS-
Werten an ( *: p < 0,05). Die erhöhte Expression von P62 bei hohem FKS-Gehalt
deutete auf eine stärkere Reifung der Phagophore hin. Die Expression von P62 war
unter Bedingungen mit 20 % FKS bei PA-Patienten mit 180 % Expressionsstärke im
Vergleich zu Kontrollen am höchsten (CP*: p < 0,05), sie sank bei 1 % FKS auf 70 %
Expressionsstärke der Kontrollen ab (CP*: p < 0,05) und stieg bei 0,5 % FKS auf 120
% Expressionsstärke im Vergleich zu Kontrollen an (CP*: p < 0,05). Bei PA-Patienten
zeigte die Expression von P62 keinen Zusammenhang mit dem FKS-Gehalt da sie
bei der Änderung von 20 % auf 1 % FKS sank ( *: p < 0,05), bei 0,5 % FKS jedoch
wieder anstieg ( *: p < 0,05). Die erhöhte Expression von P62 bei hohem und sehr
niedrigem FKS-Gehalt deutete auf eine stärkere Reifung der Phagophore unter
diesen Bedingungen hin (Abb. 18).

Abbildung (18) Relative Expression von P62 bei MMA- und PA-Patienten als Intensität der Bandenstärke im Western Blot unter Kultivierung unter Kultivierung in 20 %, 1 % und 0,5 % FKS supplementiertem DMEM nach 48 Stunden. Die Bandenintensität wurde auf Aktin als internen Standard normalisiert. (CM* < 0,05): Kontrollen – MMA-Patienten; (CP* < 0,05): Kontrollen – PA-Patienten; (MP* < 0,05): MMA-Patienten – PA-Patienten; ( *: p < 0,05): MMA-Patienten; ( *: p < 0,05): PA-Patienten. (MMA: n=5; PA: n=3).
Die Analyse des Verlaufs der Expressionsstärken von LC3-II und P62 zeigte nur bei
MMA-Patienten erhöhte Werte mit steigendem FKS-Gehalt, bei PA-Patienten war
kein Zusammenhang zwischen der Expression und dem FKS-Gehalt erkennbar.
4.7.3 Lysosomaler Abbau des Autophagosoms
Aus der fertig gereiften Phagophore entstehen Autophagosome, die von Lysosomen
umschlossen und in den gebildeten Autolysosomen durch saure Hydrolyse
degradiert werden. Die Anzahl an Autolysosomen ist proportinal zur Höhe der
Autophagie. Die Ermittlung des Anteils an Autolysosomen und damit der
Autophagosome in Zellen von Kontrollen, MMA-Patienten und PA-Patienten erfolgte
mit dem Fluoreszenzfarbstoff Acridinorange. Dieser ändert in saurer Umgebung
** *
** *
*
CM*
CP*
CM*
CP*
CM*
CP*

durch einen pH-Umschlag sein Absorbtionsmaximum von orange zu grün. Je höher
die Konzentration an Autolysosomen ist, desto weiter verschiebt sich die Fluoreszenz
von orange (FL1) zu grün (FL3), so dass eine erhöhte Ratio von FL3/FL1 auf eine
erhöhte Autophagierate hinweist.
In FACS-Analysen zeigten PTEC von MMA-Patienten im Vergleich zu PTEC von
Kontrollen und PA-Patienten eine signifikant stärkere Verschiebung des
Absorptionsmaximums von FL1 zu FL3 um das 1,5 fache (p < 0,05; Abb. 19).
Abbildung (19) Ratio der Acridinorangefärbung von grüner zu orangener Emission (FL3/FL1) als relative Intensität (*p < 0,05). (Ctrl: n=4, PA: n=3, MMA: n=5). Die FACS-Analysen wurden von Frau Dr. Katrin Tagscherer aus der Abteilung „Molekulare Tumorpathologie“ des Deutschen Krebsforschungszentrums Heidelberg (DKFZ) durchgeführt.
Die höhere Ratio von FL3/FL1 ließ auf eine höhere Anzahl von Lysosomen und
Autophagosomen und damit verbundener Autophagie-Rate bei PTEC von MMA-
Patienten schließen.
*

4.8 Stress des endoplasmatischen Retikulums - unfolded protein response
Die Untersuchung der Antwort auf ER Stress in PTEC von MMA-Patienten erfolgte
durch Western Blot Analysen der Expression von phosphoryliertem IRE1 (pIRE1).
Unter Bedingungen mit 0,5 % und 1 % FKS war die relative Expression von pIRE1
bei MMA-Patienten auf Höhe der Kontrollen, sie stieg bei 20 % FKS signifikant auf
145 % Expressionsstärke der Kontrollen und PA-Patienten an (CM*, CP*: p < 0,05).
Die Expression von pIRE1 zeigte bei MMA-Patienten einen Zusammenhang mit dem
FKS-Gehalt und stieg bei hohen FKS-Werten an ( *: p < 0,05). Die erhöhte
Expression von pIRE1 bei hohem FKS-Gehalt deutete auf die Einleitung der UPR
und damit verbundenen stärkeren Stress des ER hin. Bei PA-Patienten war die
Expression von pIRE1 unter Bedingungen mit 20 %, 1 % und 0,5 % FKS auf Höhe
der Kontrollen und deutete auf keinen Zusammenhang mit dem FKS-Gehalt hin.
Somit fand sich kein Hinweis auf die Einleitung der UPR und damit assoziierten ER-
Stress (Abb. 20).

Abbildung (20) Relative Expression von pIRE1 als Intensität der Bandenstärke im Western Blot bei MMA-und PA-Patienten unter Kultivierung in 20 %, 1 % und 0,5 % FKS supplementiertem DMEM nach 48 Stunden. Die Bandenintensität wurde auf Aktin als internen Standard normalisiert. (CM* < 0,05): Kontrollen – MMA-Patienten; (CP* < 0,05): Kontrollen – PA-Patienten; (MP* < 0,05): MMA-Patienten – PA-Patienten; ( *: p < 0,05): MMA-Patienten; ( *: p < 0,05): PA-Patienten. (MMA: n=5; PA: n=3).
4.9 Inflammation
Bei einer Inflammation kommt es zu einer Infiltrierung des Gewebes mit aktivierten
Zellen des Immunsystems. Ihre Aktivierung erfolgt über die Wechselwirkung von
Signalmolekülen und Rezeptoren und kann das angeborene oder das erworbene
Immunsystem betreffen.
4.9.1 Expressionsvergleich löslicher Rezeptoren
Um an der Aktivierung des Immunsystems beteiligte Moleküle und Rezeptoren
einzugrenzen, die von PTEC exprimiert werden können, wurde ein screening mit
**
CM*
CP*
MP* MP*

einem Array durchgeführt, der verschiedene Rezeptoren auf Proteinbasis enthielt
(Soluble Receptor Array, R&D Systems).
Bei PTEC von MMA-Patienten wurde eine 3-4-fach erhöhte Expression von IL-8 im
Vergleich zum Mittelwert der Kontrollen festgestellt. Der Rezeptor IL-1RII (Interleukin-
1 Rezeptor Typ 2) war bei MMA-Patienten um den Faktor 5-8 höher exprimiert, der
Rezeptor IL-15R zeigte eine 1,5-fach erhöhte Expression. Das Adhäsionsprotein
VCAM-1 (vascular cell adhesion molecule 1) war um das 12.000 – 25.000-fache
erhöht und sorgt für die Bindung von Monozyten an andere Zellen (Abb. 21). Die
Ergebnisse ließen sich aufgrund der kleinen Gruppen (n = 2) nicht statistisch
belegen.
Abbildung (21) Relative Expression potentiell an der Interaktion mit Immunzellen beteiligter Proteine von PTEC der Kontrollen und MMA-Patienten. Relative Expression von CXCL8/IL8: Interleukin-8; IL-1RII: Rezeptor für Interleukin 1 Subtyp 2; IL-15 Rα: alpha-Untereinheit des Interleukin-15 Rezeptors in % der Kontrolle (Ctrl: n=2, MMA: n=2).

4.9.2 Expressionsvergleich von Rezeptoren
Zur Ermittlung einer möglichen Involvierung des Komplementsystems in die
Pathogenese der cTIN wurden die Expressionen von vier Rezeptoren des CD-Typs
(cluster of differentiation) verglichen. Die untersuchten Rezeptoren CD46, CD55,
CD59 und CD88 binden verschiedene Faktoren des Komplementsystems und
unterdrücken die Ausbildung des MAC (membrane attacking complex), so dass die
Zellen nicht lysiert werden (Tabelle 3).
Tabelle (3) Funktionen der untersuchten Immunrezeptoren CD46, CD55, CD59 und CD88
Rezeptor Bindungspartner Funktion
CD46 C3b, C4b Inhibiert Zelllyse durch binden der
Komplementfaktoren
CD55 C5 Inhibiert Zelllyse durch Unterbrechung der
Kaskade
CD59 C8, C9 Inhibiert Zelllyse durch Unterbrechung der
Kaskade
CD88 C5a Rezeptor für C5a
Der Vergleich der CD-Proteine zeigte keinen Unterschied zwischen Zellen von
Kontrollen und MMA-Patienten. Eine erhöhte Expression an Rezeptoren, die PTEC
vor Zellen des angeborenen Immunsystems und des MAC schützen, konnte nicht
nachgewiesen werden (Abb. 22).

Abbildung (22) Expressionsvergleich der Immunrezeptoren CD46, CD55, CD59 und CD88 der PTEC von MMA-Patienten in Prozent der Kontrolle (Ctrl: n=5, MMA: n=5).
4.9.3 Qualitativer Nachweis exprimierter Zytokine im Zellmedium
Um zu überprüfen, ob PTEC von MMA-Patienten neben IL-8 noch andere Zytokine
exprimieren und sezernieren, wurde ein ELISA für verschiedene Zytokine
durchgeführt. Zur qualitativen Analyse der Zytokinsekretion wurde das Medium der
Zellen auf TNF-ß, CCL2, CCL3, CCL5 und IL-8 überprüft. Die Zellen wurden dafür
auf TransWell-Filterplatten ausplattiert, nach Konfluenz wurde das Medium auf
Zytokine untersucht. Die Wachstumsseite der Zellen entsprach dabei der
basolateralen Seite, welche im Körper dem Interstitium zugewandt ist, wohingegen
die apikale Seite zum Lumen des proximalen Tubulus weist. Da die Sekretion zur
Aktivierung von Immunzellen vorwiegend ins Interstitium erfolgt und weniger über die
Harnseite, wurde das Medium der basolateralen Zellseite verwendet.
Zellen der MMA-Patienten zeigten gegenüber PTEC von Kontrollen und PA-
Patienten eine tendenziell erhöhte Expression von IL-8, die jedoch nicht signifikant
war (p < 0,05; Abb. 23). Die Expressionen von TNF-ß, CCL2, CCL3, CCL5 waren bei

allen Zellen unter der Nachweisgrenze der Herstellerangaben und sind nicht
aufgeführt.
Abbildung (23) Relative Konzentration von IL-8 im Medium der PTEC von Kontrollen, MMA- und PA-Patienten in Prozent der Kontrolle (Ctrl: n=3, PA: n=3, MMA: n=6).
4.9.4 Quantitativer Nachweis von Interleukin-8 intrazellulär und im Zellmedium
Zellen von MMA-Patienten sezernierten im Mittel mit etwa 2740 pg/mg signifikant fast
doppelt so viel IL-8 ins apikale Medium wie Zellen von Kontrollen und die signifikant
1,5-fache Menge wie PA-Patienten, die etwa 1520 bzw. 1850 pg/mg sezernierten. Im
basolateralen Medium konnten mit 4060 pg/mg bei Zellen von MMA-Patienten die
1,5-fache Menge bzw. die signifikant fast doppelte Menge an IL-8 nachgewiesen
werden wie bei Kontrollen und PA-Patienten, welche im Mittel 2760 bzw. 2100 pg/mg
IL-8 sezernierten. In den Zellen war die Konzentration mit 1200 pg/mg im Mittel bei
MMA-Patienten signifikant dreifach bzw. 1,5-fach höher als bei Kontrollen und PA-
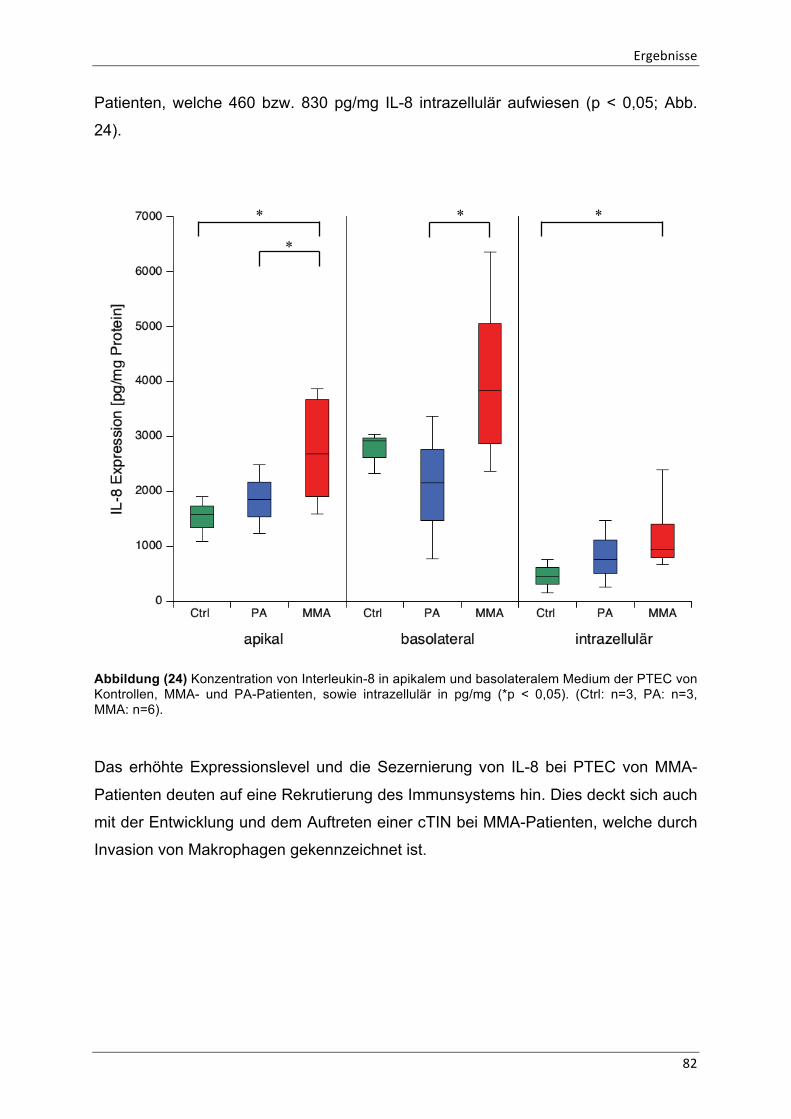
Patienten, welche 460 bzw. 830 pg/mg IL-8 intrazellulär aufwiesen (p < 0,05; Abb.
24).
Abbildung (24) Konzentration von Interleukin-8 in apikalem und basolateralem Medium der PTEC von Kontrollen, MMA- und PA-Patienten, sowie intrazellulär in pg/mg (*p < 0,05). (Ctrl: n=3, PA: n=3, MMA: n=6).
Das erhöhte Expressionslevel und die Sezernierung von IL-8 bei PTEC von MMA-
Patienten deuten auf eine Rekrutierung des Immunsystems hin. Dies deckt sich auch
mit der Entwicklung und dem Auftreten einer cTIN bei MMA-Patienten, welche durch
Invasion von Makrophagen gekennzeichnet ist.
* * *
*

4.10 Korrelationen der Expression von IL8, Generierung von ROS, erhöhter Autophagie und verringerter Aktivität der Cytochrom c-Oxidase
Zur Überprüfung eines direkten Zusammenhangs zwischen erhöhter IL8-Expression,
ROS und Autophagie bei PTEC von MMA-Patienten, wurde eine Korrelationsanalyse
durchgeführt. Als Variablen wurden „Erhöhte Autophagie“ (Ratio der Acridinorange-
Färbung), „Generierung von ROS“ (Färbung mit H2DCFDA), „Expression von IL8“ “
(IL-8 als Signalmolekül und Sezernierung zur basolateralen Zellseite) und
„verringerte Aktivität der Cytochrom c-Oxidase“ (Defekte Integrität der Mitochondrien)
verwendet.
Das Bestimmtheitsmaß R2 diente als Messgröße zur Einordnung der Hypothese, ob
die verwendeten Variablen in einem linearen Zusammenhang stehen. Die Werte für
R2 liegen zwischen 0 und 1, wobei der Zusammenhang größer wird, je näher er dem
Wert 1 kommt. Die Einordnung der Stärke der Korrelation erfolgt in schwacher,
mittlerer, starker und sehr starker Zusammenhang (Tabelle 4).
Tabelle (4) Schwellenwerte für die Stärke des Zusammenhangs zwischen 2 Variablen
Zusammenhang Korrelationseffizient R Bestimmtheitsmaß R2
Sehr stark 0,87 - 1 0,75 - 1
Stark 0,71 – 0,86 0,5 – 0,74
Mittel 0,5 – 0,7 0,25 – 0,49
Schwach 0 – 0,49 0 – 0,24
Die Korrelationsanalyse der Variablen „Generierung von ROS“, „Erhöhte
Autophagie“, „verringerte Aktivität der Cytochrom c-Oxidase“ und „Expression von
IL8“ bei PTEC von MMA-Patienten zeigte zwischen Autophagie und ROS, Aktivität
von Komplex IV der Atmungskette und IL8-Expression, sowie Aktivität von
Cytochrom c-Oxidase der Atmungskette und ROS nur einen schwachen
Zusammenhang. Die Korrelation von ROS und IL8-Expression, sowie der Aktivität
von Cytochrom c-Oxidase der Atmungskette und IL8-Expression einen mittelstarken
Zusammenhang. Ein starker Zusammenhang konnte zwischen Autophagie und
Expression von IL-8 festgestellt werden (Tabelle 5).

Tabelle (5) Bestimmtheitsmaß und Zusammenhang der Korrelationsvariablen
Korrelations-Variablen Zusammenhang Bestimmtheitsmaß R2
ROS und Expression von IL-8 Mittel 0,465458
Autophagie und ROS Schwach 0,216896
Autophagie und Expression von
IL-8
Sehr stark 0,80946
Aktivität Cytochrom c-Oxidase
und Autophagie
Schwach 0,0568924
Aktivität Cytochrom c-Oxidase
und Expression von IL-8
Mittel 0,285974
Aktivität Komplex IV und ROS Schwach 0,049656
Die Korrelationsanalyse macht einen Zusammenhang zwischen dem Auftreten von
erhöhter Autophagie und verstärkter Expression von IL-8 sehr wahrscheinlich. Da im
Experiment keine Interleukine oder anderen Botenstoffe eingesetzt wurden, kann
erhöhte Autophagie als mögliche Ursache für erhöhte Expression von IL-8 bei PTEC
von MMA-Patienten postuliert werden.

5 Diskussion
5.1 Auswahl des verwendeten Zellkulturmodells
Als Modell zur Untersuchung der Entstehung der chronischen tubulointerstitiellen
Nephritis diente ein Zellkultursystem aus immortalisierten humanen PTEC.
Der Vorteil des verwendeten Zellkulturmodells gegenüber bisher beschriebenen
Tiermodellen war der Gebrauch von humanen Zellen, da sich mit ihnen besser die
Charakteristiken und Ursachen der Erkrankung beim Menschen analysieren lassen,
als mit murinen Zellen oder Zellen anderer Organismen. Bei der Entwicklung einer
cTIN im Verlauf der MMA sind die PTEC der Patienten bereits in der Anfangsphase
von Schädigungen betroffen. Ihre Verwendung kann auf die Ursachen schließen
lassen, welche für die Entwicklung der Nephritis verantwortlich sind.
Nachteile des verwendeten Zellkulturmodells gegenüber Tiermodellen war die
Beschränkung der Ursachenanalyse der cTIN auf einen Zelltyp des betroffenen
Organs, da für die durchgeführten Experimente nur PTEC verwendet wurden.
Potenzielle Einflüsse durch die natürliche Umgebung der proximalen Tubuluszellen
der Niere und Wechselwirkungen mit anderen Zellen werden durch das verwendete
Modell folglich nicht erfasst. Tiermodelle lassen eine systemische Betrachtung der
Ursachen und der Pathologie zu, wodurch sich eine Beteiligung des Immunsystems
oder anderer Gewebetypen an der MMA noch umfassender analysieren lassen.
Die PTEC wurden aus dem Urin isoliert, somit war die Art der Zellgewinnung nicht
invasiv, für alle Probanden risikofrei und mit keinen unerwünschten Wirkungen
verbunden. Das Epithel erneuert sich ständig, PTEC werden mit dem Urin
ausgeschieden. Somit stehen theoretisch immer wieder neue primäre PTEC zur
Verfügung. Jedoch lässt sich aus organisatorischen Gründen nicht zu beliebigen
Zeitpunkten Patienten- und Probandenurin gewinnen. Die Limitierung in der Anzahl
der Zellen lag darüber hinaus ganz wesentlich im begrenzten Wachstum der
primären PTEC, welches sich auf höchstens 3-5 Passagen beschränkte.
Um diese Limitierung zu umgehen, wurden die PTEC mit dem Vektor pRNS1
transfiziert. Dieser enthält alle Teile des SV40-Genoms, die ori-Region besitzt jedoch
eine Deletion von 6 Nukleotiden um eine Selbstreplikation zu unterbinden (Small et
al., 1982; Litzkas et al., 1984). Dadurch erfolgte die Generierung sich fortwährend
teilenden Zelllinien, die sich bis über Passage 20 hinaus kultivieren ließen. Alle

Experimente wurden jedoch nur mit Zellen der Passagen 5 bis 10 durchgeführt, da
höher passagierte und damit ältere Zellen zu Veränderungen der Charakteristika, wie
z.B. DNS-Mutationen, neigen können (Hughes et al., 2007).
5.2 Charakterisierung des verwendeten Zellkulturmodells
Nach der Transfektion mit dem Vektor pRNS1 (Small et al., 1982; Litzkas et al.,
1984) wurden alle Zelllinien nach Passage 5 in Bezug auf 1) die typische
Morphologie von PTEC, 2) die Expression von Markerproteinen wie Aquaporin 1 und
γ-Glutamyltransferase 1, sowie 3) die Ausbildung von Mikrovilli und 4) tight junctions
hin untersucht.
Alle generierten Zelllinien von Patienten und Kontrollen wuchsen als
Einzelzellschicht. In elektronenmikroskopischen Aufnahmen waren die für
Epithelzellen des proximalen Tubulus charakteristischen Mikrovilli erkennbar, welche
nur an der apikalen Zelloberfläche ausgebildet werden und die Membranoberfläche
vergrößern.
Weiterhin wurden mit AQP1 und γGT1 Markerproteine nachgewiesen, die
charakteristisch für das Epithel des proximalen Tubulus sind und im Urogenitaltrakt
nur in diesen Zellen vorkommen. Das Transmembranprotein AQP1 wird in der Niere
nur in PTEC der Segmente S1-S3 des proximalen Tubulus an der apikalen
Membranseite der Mikrovilli exprimiert. Es ist an der Resorption von Wasser aus dem
Primärharn beteiligt, um es dem Körper wieder zurück zu führen (Sabolic et al., 1992;
Kim et al., 1999; Baer et al., 2006; Wilmers et al., 2010). Die γGT1 wird ebenfalls nur
im proximalen Tubulusepithel exprimiert (Helbert et al., 1997). Sie dient der
Übertragung des Glutamylrests des Tripeptids Glutathion auf Peptide, wodurch der
Abbau von GSH erfolgt. Da es keinen Transporter für Glutathion gibt, ist dies eine
Möglichkeit Cystein in die Zelle zu transportieren, wo es für den Glutathionaufbau
verwendet wird. Alle Zelllinien zeigten im Western Blot die für PTEC charakteristische
Expression von AQP1 und γGT1.
Die erhöhte MMS-Konzentration bei MMA-Patienten wurde auf zwei
unterschiedlichen Wegen nachgewiesen: 1) Im Plasma der MMA-Patienten lag die
MMS-Konzentration im untersuchten Zeitraum von einem Jahr vor und einem Jahr
nach Zellisolierung über dem Normalwert. Die Festlegung des Normalwerts erfolgte
über Literaturangaben, da kein Plasma der Kontrollen zur Verfügung stand. 2) Im

Medium der kultivierten PTEC von Patienten war die MMS-Konzentration mehr als
doppelt so hoch wie in kultivierten PTEC von Kontrollen. Beides belegte den
funktionellen Defekt der MMA-Patienten bzw. ihrer kultivierten PTEC, MMS in
Succinat umzuwandeln.
5.3 PTEC von MMA-Patienten zeigen Defekte im Energiemetabolismus
Bei der Pathogenese von MMA- und PA-Patienten spielt ein Defekt in der
Energiehomöostase nach bisherigen Erkenntnissen eine große Rolle. Eine
verringerte Leistung der am Energiemetabolismus beteiligten Enzyme kann eine
Ursache für Schädigungen der Zelle bis hin zum ihrem Untergang sein. Dabei ist
MMS nicht direkt verantwortlich, vielmehr führen die intrazellulär aus MMS gebildeten
Metabolite Malonsäure, Methyl-Zitrat und Propionyl-CoA zu Aktivitätseinbußen
einiger am Energiemetabolismus beteiligten Enzyme. Malonsäure führt in murinen
Neuronen des Striatums zu einer Inhibition von Komplex II der Atmungskette, Methyl-
Zitrat führt zur Inhibition der Zitrat-Synthase, Aconitase und Isozitrat-Dehydrogenase,
was einen verminderten Flux des Zitrat-Zyklus zur Folge hat (Okun et al., 2002;
Kölker et al., 2003), Propionyl-CoA hemmt die Aktivität von isoliertem PDHc (Schwab
et al., 2006; Morath et al., 2008). In der vorliegenden Arbeit konnten ebenfalls
verringerte Aktivitäten in einigen Enzymen des Energiemetabolismus gezeigt
werden. Der Vergleich des Energiemetabolismus auf den Ebenen der Glykolyse, des
Zitrat-Zyklus und der Atmungskette zeigte in allen Bereichen Veränderungen und
Beeinträchtigungen der Energieversorgung bei PTEC von MMA-Patienten, zudem
war der mitochondriale Sauerstoffverbrauch beeinträchtigt.
Auf Ebene der oxidativen Phosphorylierung war bei MMA-Patienten die Aktivität der
Cytochrom c-Oxidase verringert. Bei PTEC von MMA-Patienten war eine Präparation
von Fraktionen mit angereicherten intakten Mitochondrien durch differentielle
Zentrifugation nach Zelllyse nicht erfolgreich, da sich die Mitochondrien nicht
sedimentieren ließen. Bei PTEC von Kontrollen und PA-Patienten traten diese
Probleme nicht auf. Die in elektronenmikroskopischen Aufnahmen gefundenen
morphologischen Auffälligkeiten der PTEC von MMA-Patienten könnten zu fehlender
Integrität der Mitochondrien führen und für ihre Zerstörung bei Belastung durch
Zentrifugalkräfte verantwortlich sein. Die fehlende Integrität lässt in Verbindung mit

der verringerten Aktivität der Cytochrom c-Oxidase auf einen mitochondrialen Defekt
schließen, der die Funktion der Mitochondrien beeinträchtigt.
Beides wurde bereits in der Literatur bei der Präparation von Mitochondrien aus
Nieren von MCM-defizienten Knock out-Mäusen beschrieben. Chandler und Kollegen
konnten ebenfalls keine intakten und funktionellen Mitochondrien aus Mäusenieren
isolieren. Elektronenmikroskopische Aufnahmen zeigten ebenfalls morphologische
Auffälligkeiten, die sich jedoch unterschiedlich zur vorliegenden Arbeit äußerten.
Während bei humanen PTEC der vorliegenden Arbeit Strukturen erkennbar waren,
die auf Autophagie schließen lassen, war in den Zellen der Mäusenieren die
Auflösung der mitochondrialen Cristae-Struktur erkennbar. Diese wurde als Ursache
für das Präparationsproblem der Mitochondrien diskutiert. Die Bestimmung der
enzymatischen Aktivität der Atmungskettenkomplexe zeigte im Einklang mit der
vorliegenden Arbeit die verringerte Aktivität der Cytochrom c-Oxidase (Chandler et
al., 2009).
In Nierenzellen aus Gewebepunktionen eines MMA-Patienten konnten
Beeinträchtigungen der Atmungskettenkomplexe I – V gezeigt werden (de Keyzer et
al., 2009). In einer kürzlich erschienenen Arbeit wurde im Einklang mit den
Ergebnissen der vorliegenden Arbeit bei einem mut0-Patienten ebenfalls eine
Verringerung der Aktivität der Cytochrom c-Oxidase in Nierenzellen publiziert
(Zsengellér et al., 2014).
Es lässt sich postulieren, dass die MMA einen mitochondrialen Defekt verursacht, der
sich in einer verringerten Enzymaktivität der Cytochrom c-Oxidase manifestiert.
Auf Ebene der Glykolyse zeigten PTEC von MMA-Patienten eine stark verringerte
Enzymaktivität der Phosphofruktokinase 1 (PFK1), sowie eine Erhöhung der
Enzymaktivität der Triosephosphat-Isomerase (TPI) und der Glyceraldehydphosphat-
Dehydrogenase (GAPDH).
Der größte Unterschied zeigte sich im Abfall der Enzymaktivität der PFK1, die 45 %
der Aktivität der Kontrollen hatte. Sie katalysiert die Übertragung eines Phosphatrests
von ATP auf Fruktose-6-Phosphat, was zu Fruktose-1,6-bisphosphat und ADP führt.
Diese Reaktion stellt den geschwindigkeitsbestimmenden Schritt der Glykolyse dar,
und eine Verlangsamung wirkt sich stark auf die Gesamtrate der Glykolyse aus
(Yalcin et al., 2009b; Jenkins et al., 2011).
Die PFK1 ist ein Teil eines bifunktionellen Enzyms und katalysiert die Reaktion von
Fruktose-6-Phosphat und ATP zu Fruktose-1,6-bisphosphat und ADP. Die reverse

Reaktion zu Fruktose-6-Phosphat und anorganischem Phosphor wird durch die
Fruktose-1,6-Bisphosphatase (F1,6BPase) katalysiert, die den anderen Teil des
bifunktionellen Enzyms darstellt. Der wichtigste Regulator dieses bifunktionellen
Enzyms ist das auf die PFK1 stimulierend wirkende Fruktose-2,6-bisphosphat. Die
PFK1 ist nur dann aktiv, wenn Fruktose-2,6-bisphosphat in ausreichender Menge zur
Verfügung steht, es hemmt die Aktivität der F1,6BPase (Hers und van Schaftingen,
1982; Voet and Voet, 1995).
Die Reaktion von Fruktose-6-Phosphat und ATP zu Fruktose-2,6-bisphosphat wird
durch die PFK2 katalysiert, die einen Teil eines weiteren bifunktionellen Enzyms, der
PFKFB bildet. Die reverse Reaktion von Fruktose-2,6-bisphosphat zu Fruktose-6-
Phosphat und anorganischem Phosphor wird durch die F2,6BPase katalysiert, die
den anderen Teil der PFKFB bildet. Wird die PFKFB phosphoryliert, ist die
F2,6BPase aktiviert und die Konzentration von Fruktose-2,6-bisphosphat sinkt durch
ihre Umsetzung zu Fruktose-6-Phosphat und anorganischem Phosphor. Bei
Dephosphorylierung ist die PFK2 aktiv und die Konzentration an Fruktose-2,6-
bisphosphat steigt (Kurland und Pilkis, 1995; Rider et al., 2004). Der Energiemangel
bei PTEC von MMA-Patienten könnte zur Bildung von cAMP und daraus
resultierender Aktivierung der Proteinkinase A führen. Diese kann die PFKFB
phosphorylieren und damit die F2,6BPase aktivieren. Dadurch sinkt die
Konzentration von Fruktose-2,6-bisphosphat, die Enzymaktivität der F1,6BPase
würde steigen und die Aktivität der PFK1 sinken.
Die geringere Aktivität der PFK1 bei PTEC von MMA-Patienten könnte weiterhin zu
höheren Konzentrationen von Fruktose-6-Phosphat, das durch die F1,6BPase
gebildet wird, führen. Fruktose-6-Phosphat könnte in Glukose-6-Phosphat
umgewandelt werden und in den Pentosephosphatweg einfließen. Dort wird durch
die Oxidaton von Glukose-6-Phosphat und NADP+ Ribose-5-Phosphat und NADPH
gebildet. Der Pentosephosphatweg liefert weiterhin Derivate, die als Grundgerüst für
DNS, RNS, sowie ATP, NADH und Coenzym A dienen. Das Reduktionsäquivalent
NADPH wird von der Glutathionreduktase als Cofaktor benötigt, um Glutathiondisulfid
zu Glutathion zu reduzieren. Reduziertes Glutathion dient der Zelle dazu, ROS zu
binden, wodurch wiederum Glutathiondisulfid entsteht. Dieses muss erneut zu
Glutathion reduziert werden, um weitere ROS zu binden (Patra & Hay, 2014). Die
verringerte Aktivität der PFK1 bei MMA-Patienten könnte die Hypothese stützen,
dass Glukose-6-Phosphat vermehrt dem Pentosephosphatweg als Substrat zur

Verfügung gestellt wird, um mehr Reduktionsäquivalente wie NADPH zu bilden.
Dadurch könnte Glutathiondisulfid schneller zu Glutathion reduziert werden, um ROS
schneller und effektiver zu binden.
Durch die verringerte Aktivität der PFK1 in PTEC von MMA-Patienten und die damit
verbundene Verlangsamung der Glykolyse ließe sich der geringere mitochondriale
Sauerstoffverbrauch in mit Glukose supplementiertem KRB erklären. Durch eine
Verlangsamung der Glykolyse wird weniger Acetyl-CoA in den Zitrat-Zyklus
eingebracht. Dies führt dazu, dass auch weniger Substrate für die oxidative
Phosphorylierung zur Verfügung stehen.
Im Vergleich der enzymatischen Aktivität des Zitrat-Zyklus waren bei PTEC von
MMA-Patienten die Aktivitäten der mitochondrialen Aconitase und des 2-Oxoglutarat-
Dehydrogenase-komplex (OGDHc) verringert. Die Aconitase katalysiert die
Umlagerung von Zitrat über cis-Aconitat zu Isozitrat. Ihr katalytische Zentrum enthält
ein Eisen-Schwefel-Cluster und reagiert empfindlich auf die Gegenwart von ROS, die
das Eisenatom oxidieren (Chinopoulos et al., 1999; Nulton-Persson und Szweda,
2001; Tretter und Adam-Vizi, 2005; Lushchak et al., 2014). Die Inhibition der
Aconitase durch ROS wirkt neurotoxisch (Cantu et al., 2009), zudem kann das
Enzym selbst ROS in Form von Wasserstoffperoxid (H2O2) und Superoxidradikal
generieren (Vasquez-Vivar et al., 2000).
Der OGDHc wandelt mit Hilfe von Coenzym A 2-Oxoglutarat unter oxidativer
Decarboxylierung zu Succinyl-CoA um und reduziert NAD+ zu NADH. Er ist ein
geschwindigkeitsbestimmendes Schlüsselenzym des Zitrat-Zyklus (Sauer et al.,
2005) und kann ebenfalls an der Produktion von ROS mitwirken oder als Ziel von
ROS in Erscheinung treten. ROS führen zur Oxidation des Eisen-Schwefel-Clusters
im katalytischen Zentrum und zur Verringerung der Enzymaktivtät (Starkov et al.,
2004; Tretter & Adam-Vizi, 2005).
Der mit Mikro-Respirometrie ermittelte Sauerstoffverbrauch ist spezifisch für
Mitochondrien, ermittelt wurde der Verbrauch pro Zeit. Durch Zugabe von
Kaliumzyanid konnte die Cytochrom c-Oxidase inhibiert werden und der
mitochondriale Sauerstoffverbrauch kam zum Erliegen. Durch Subtraktion des
verbliebenen Sauerstoffverbrauchs vom gesamten Sauerstoffverbrauch ließ sich der
mitochondriale Sauerstoffverbrauch ermitteln. Dieser war in Abhängigkeit des
Energieträgers sowohl bei PTEC von MMA- als auch von PA-Patienten verringert.
Die Supplementation von KRB mit Glukose stimulierte die Glykolyse im Zytosol, die

über die Atmungskette an den mitochondrialen Sauerstoffverbrauch gekoppelt war,
die Supplementation mit Pyruvat stimulierte den Zitrat-Zyklus in den Mitochondrien,
der ebenfalls über die Atmungskette an den mitochondrialen Sauerstoffverbrauch
gekoppelt war. In DMEM konnten die Zellen neben Glukose zusätzlich auch auf
Aminosäuren zur Energiegewinnung zurückgreifen.
Der mitochondriale Sauerstoffverbrauch von MMA-Patienten war bei Stimulation der
Glykolyse, des Zitrat-Zyklus und in DMEM reduziert. Bei PA-Patienten trat unter
Stimulierung der Glykolyse und in DMEM ebenfalls ein verminderter
Sauerstoffverbrauch auf, jedoch nicht bei der Stimulation des Zitrat-Zyklus. Dies
könnte auf einen wichtigen Unterschied im Pathomechanismus beider Erkrankungen
hinweisen und die Hypothese stützen, dass ein mitochondrialer Defekt bei MMA-
Patienten vorliegt.
5.4 PTEC von MMA-Patienten haben eine erhöhte ROS-Produktion
Die Defekte der Cytochrom c-Oxidase, der mitochondrialen Aconitase und des
OGDHc bei PTEC von MMA-Patienten könnten auf einer erhöhten ROS-Bildung
beruhen. Eine vermehrte ROS-Produktion führt zu einer Oxidation von Proteinen,
Lipiden und anderen Makromoleküle, wodurch diese ihre ursprüngliche Funktion
verlieren (Kowaltowski et al., 1999). Präparierte Zellen aus den Nieren von MCM
Knock-out Mäusen zeigten ebenfalls erhöhte ROS-Werte (Chandler et al., 2009).
Bei den PTEC der MMA-Patienten wurden Hinweise auf ROS auf zwei Ebenen
gefunden. Zum einen lag der oxidative Stressmarker Glutathion vermehrt in oxidierter
Form als Glutathiondisulfid vor, zum anderen war die ROS-Produktion erhöht, was
durch Oxidation einer reduzierten Form von Fluorescein gezeigt werden konnte, die
als Indikator für ROS diente.
Der Nachweis der ROS-Konzentration über die Bestimmung des Gehalts an
Glutathion und Glutathiondisulfid zeigte bei PTEC von MMA-Patienten verringertes
reduziertes Glutathion (GSH) und erhöhtes Glutathiondisulfid (GSSG), die
Gesamtmenge an zellulärem Glutathion war unverändert. Diese Veränderungen sind
bekannte gegenregulatorische Maßnahmen bei erhöhtem oxidativen Stress.
Glutathion dient der Zelle neben anderen Antioxidantien wie Vitaminen (z.B.
Ascorbinsäure) oder bestimmten Enzymen der Neutralisation von ROS. Sie sind in
der Lage, freie Elektronen aufzunehmen, so dass die Oxidation der für die Zelle

wichtigen Moleküle reduziert wird. Glutathiondisulfid muss jedoch immer wieder zu
Glutathion reduziert werden, da nur die reduzierte Form ROS binden kann. Dies
stützt die Hypothese, dass die PTEC von MMA-Patienten über die Stimulation des
Pentose-Phosphatwegs vermehrt NADPH bilden, um die Reduktion von Glutathion
aus Glutathiondisulfid zu beschleunigen. In Leberextrakten von Mut-defizienten
Mäusen war der Gehalt an reduziertem Glutathion ebenfalls geringer, im Gegensatz
zu den PTEC der MMA-Patienten in der vorliegenden Arbeit war der Gehalt an
Glutathion der Mäuse jedoch insgesamt erniedrigt (Treacy et al., 1996). Reaktives
Acryloyl-CoA, welches aus Propionyl-CoA entsteht, kann ebenfalls die Ursache für
ROS sein (Ando et al., 1972). Dies konnte in der vorliegenden Arbeit jedoch nicht
bestätigt werden, da PTEC von PA-Patienten durch den Defekt der Propionyl-CoA-
Decarboxylase ebenfalls erhöhte Werte an Propionyl-CoA aufweisen. Sie zeigten in
der vorliegenden Arbeit jedoch keine erhöhte ROS-Bildung gegenüber Kontrollen.
Der Nachweis der ROS-Produktion erfolgte durch das zellpermeable H2DCFDA
(2',7'-Dichloro-dihydrofluoresceindiacetat), dessen Diazetat-Rest intrazellulär von
Esterasen abgespalten wird und DCF•- entsteht, das nach Reaktion mit ROS
fluoresziert (Kalyanaraman et al., 2012).
Auf Ebene der ROS-Produktion wurden bei PTEC von MMA-Patienten bereits in KRB
ohne Supplementation mit potentiellen Stimulanzen der ROS-Bildung erhöhte Werte
festgestellt. Durch Supplementation mit Glutamin, PS, MMS und Isoleucin wurde die
ROS-Produktion noch zusätzlich gesteigert. Dies lässt darauf schließen, dass die
genannten Supplemente zu metabolischem Stress bei PTEC von MMA-Patienten
führten, der sich in einer erhöhten ROS-Produktion äußerte. Die Supplementation mit
Glukose bzw. Pyruvat führte zur höchsten gemessenen ROS-Produktion. Bei PTEC
von MMA-Patienten war weiterhin bei beiden Supplementen die Verwertung als
Energieträger aufgrund verringerter Enzymaktivitäten von PFK1, Aconitase und
OGDHc eingeschränkt, was sich im reduzierten mitochondrialen Sauerstoffverbrauch
äußerte (unter 5.3 beschrieben). Es lässt sich postulieren, dass die erhöhte ROS-
Produktion in PTEC von MMA-Patienten zu Defekten im Energiemetabolismus
beitragen und an der Inhibierung der mitochondrialen Aconitase, des OGDHc und
der Cytochrom c-Oxidase beteiligt sind.
Die häufigste Quelle für ROS-Bildung bei Säugetieren sind die Enzymkomplexe
NADH-Dehydrogenase (Komplex I) und Cytochrom c-Reduktase (Komplex III) der
mitochondrialen Atmungskette. Die erhöhte ROS-Bildung führt zur reduzierten

Aktivität der Cytochrom c-Oxidase (Komplex IV), was wiederum zu stärkerer
Reduktion der Redox-Zentren der NADH-Dehydrogenase und der Cytochrom c-
Reduktase führen kann. Dadurch kann die ROS-Produktion dieser Komplexe weiter
ansteigen (Dawson et al., 1993; Chen et al., 2003). Bei der ROS-Bildung an der
Atmungskette entsteht zunächst das Superoxid-Anion (O2•-) wenn molekularer
Sauerstoff ein Elektron aufnimmt. Bei der NADH-Dehydrogenase ist eines der Eisen-
Schwefel-Cluster N-1 oder N-2 die Hauptursache der Bildung von Superoxid-Anionen
(Genova et al., 2001; Kushnareva et al., 2002). Als Quelle von Superoxid-Anionen
der Cytochrom c-Reduktase konnte die Autoxidation von Ubisemiquinon an der
inneren und äußeren Seite der inneren Mitochondrienmembran identifiziert werden
(Boveris et al., 1976; Cadenas et al., 1977; Turrens et al., 1985; Han et al., 2001).
Superoxid ist nicht sehr reaktiv, bildet jedoch den Vorläufer mehrerer verschiedener
anderer Sauerstoff- und Stickstoffradikale. Es kann Cytochrom c im
Intermembranraum reduzieren oder durch Aufnahme von Protonen zu
Wasserstoffperoxid (H2O2) und molekularem Sauerstoff umgesetzt werden, sowie in
der Matrix und im Intermembranraum akkumulieren. Wasserstoffperoxid kann durch
unvollständige Übertragung der Elektronen von molekularem Sauerstoff auf Wasser
entstehen, die an der Atmungskette durch die Cytochrom c-Oxidase katalysiert wird.
Vermehrt auftretende reduzierte Metallionen wie Fe2+ oder Cu2+ begünstigen die
Umwandlung von dauerhaft erhöhten Konzentrationen des Superoxid-Anions zum
Hydroxyl-Radikal (OH•). Dieses kann wiederum durch Reaktion mit Stickstoffoxiden
zur Bildung von Peroxinitrit (ONOO-/ONOOH) führen (Beckman & Koppenol, 1996;
Radi et al., 2002). Hydroxyl-Radikale und Peroxynitrit sind starke Oxidationsmittel
und reagieren mit Nukleinsäuren und Proteinen, was deren Strukturzerstörung und
die Inhibition ihrer Funktionen zur Folge hat (Turrens, 2003).
Das aus H2DCFDA entstandene DCF•- bindet radikalisches Stickstoffdioxid (•NO2),
Hydroxyl-Radikale und Peroxinitrit, die im Verlauf der Reaktionen des Superoxid-
Anions mit Wasserstoffperoxid und Stickstoffoxiden entstehen, Wasserstoffperoxid
kann nicht gemessen werden, da es nicht mit DCF•- reagiert (Kalyanaraman et al.,
2012).
Den negativen Auswirkungen von ROS für die einzelne Zelle können auch positive
Auswirkungen für den Gesamtorganismus gegenüberstehen. ROS können eine
second-messenger Funktion übernehmen und die Aktivierung der Zytokinexpression
einleiten (Rada et al., 2011). Dies führt zur Aktivierung und Rekrutierung des

Immunsystems zum Ursprung der Inflammation (D'Autréaux & Toledano, 2007). Da
jedoch nur in Zellkultur gewachsene PTEC verwendet wurden, konnte die Wirkung
der Sezernierung von Zytokinen (IL-8) auf andere Gewebe nicht untersucht werden.
Somit erfolgte nur eine Abbildung des Zusammenhangs zwischen ROS-Bildung und
Zytokinsezernierung.
Weiterhin können unter Hungerbedingungen, und damit verbundenem
Nährstoffmangel, mitochondrial generierte ROS Einfluss auf autophagozytotische
Prozesse ausüben, indem sie die Bildung von Autophagosomen stimulieren (Scherz-
Shouval et al., 2007). Durch die Defekte im Energiemetabolismus in PTEC von MMA-
Patienten könnten Bedingungen auftreten, wie sie bei Energiemangel vorliegen und
die Bildung und Degradierung von Autophagosomen fördern.
Mitochondrien können an der Entstehung der Membranen von Autophagosomen
beteiligt sein (Hailey et al., 2010). Das erhöhte Vorkommen der Autophagosomen bei
PTEC von MMA-Patienten könnte zu einer Destabilisierung der Mitochondrien
beitragen und eine Ursache für ihre fehlende Integrität sein, die das Isolieren
mitochondrial angereicherter Fraktionen verhinderte (unter 5.3 beschrieben).
5.5 PTEC von MMA-Patienten zeigen erhöhte Makro-Autophagie
Auf elektronenmikroskopischen Aufnahmen waren bei PTEC der MMA-Patienten von
einer Doppelmembran umgebene Strukturen erkennbar, die am ehesten
Autophagosomen entsprechen. Da mTORC1, einer der Regulatoren der Makro-
Autophagie, sensibel auf den Energiestatus der Zelle reagiert und bei PTEC von
MMA-Patienten Defekte im Energiemetabolismus auf den Ebenen der Glykolyse, des
Zitrat-Zyklus und der mitochondrialen Atmungskette nachgewiesen werden konnten,
wurden die Zellen in Hinblick auf die Reifung der Phagophore und die Degradierung
der Autophagosome gezielt untersucht. Der Ablauf der Makroautophagie lässt sich in
die Schritte 1) Initiation und Nukleation, 2) Elongation, 3) Autophagosom-
Bildung/Maturation, 4) Fusion mit Lysosom, 5) Autolysosombildung und Cargo-
Degradation einteilen (Randall-Demllo et al. 2013).
Durch die Kultivierung der PTEC in Medium mit hohem FKS-Gehalt (20 %), sowie mit
geringem und sehr geringem FKS-Gehalt (1 % und 0,5 %), sollte metabolischer
Stress induziert werden. Die im FKS enthaltenen ungeradzahligen Fettsäuren

können von MMA-Patienten nicht vollständig degradiert werden und es kommt zur
Bildung potenziell toxischer Substanzen.
Der Nachweis der Makro-Autophagie erfolgte an der 1) Kontrollstelle durch
mTORC1, sowie auf Ebene der 2) Autophagosombildung/Maturation und 3) der
Menge an Autolysosomen. Um Fehler aufgrund unterschiedlicher Zellcharakteristika
zwischen den Gruppen Kontrollen, PA-Patienten und MMA-Patienten
auszuschließen, wurde der Verlauf der Expression an der 1) Kontrollstelle und der 2)
Autophagosombildung beteiligter Proteine innerhalb der Zell-Gruppen (kontrollen,
MMA-Patienten, PA-Patienten) in Abhängigkeit der FKS-Konzentration verglichen.
Zusätzlich wurde das Emissionsspektrum der Acridinorangefärbung untersucht, um
auf die Menge der 3) Autolysosomen zurückzuschließen.
1) Die Reifung der Phagophore wird durch mTORC1 reguliert. Die
Dephosphorylierung von mTOR ist mit der Dissoziierung und Inaktivierung von
mTORC1 verbunden, was zur Assemblierung des Multiproteinkomplexes aus
ULK1/FIP200/ATG13 führt, der die Reifung der Phagophore stimuliert (Codogno und
Meijer, 2005; Jung et a.l, 2009; Ganley et al., 2009; Hosokawa et al., 2009). Die
Assemblierung und Aktivierung von mTORC1 hängt von der Kinase mTOR ab, ihre
Aktivität wird durch dephosphoryliertes PRAS40 reduziert und mTORC1 dissoziiert.
Der Verlauf der Ratios von pPRAS40/PRAS40 und pmTOR/mTOR über die FKS-
Konzentration zeigte bei PTEC von MMA-Patienten eine Korrelation zwischen
sinkenden Ratios von pmTOR/mTOR und pPRAS40/PRAS40, sowie dem
ansteigenden FKS-Gehalt. Daraus lässt sich auf eine stärkere Dissoziierung und
Inaktivierung von mTORC1 und positiver Regulation der Reifung der Phagophore bei
steigendem metabolischen Stress schließen.
Bei PTEC von PA-Patienten war der Verlauf der Ratios über den FKS-Gehalt sehr
unregelmäßig und ließ keine Abhängigkeit erkennen, sie unterschieden sich in
diesem Punkt grundlegend von PTEC der MMA-Patienten.
2) Die Autophagosombildung/Maturation wurde anhand der Expressionsstärke von
LC3-II und P62, die mit der reifenden Phagophore assoziiert sind, untersucht (Choi &
Ryter, 2011). Bei PTEC von MMA-Patienten weist die steigende Expression von
LC3-II und P62 mit steigendem FKS-Gehalt auf eine erhöhte Autophagosombildung
und Maturation hin und dient als Ausdruck erhöhter Makro-Autophagie mit
zunehmendem metabolischen Stress.

Die Expression von LC3-II war bei PA-Patienten unabhängig vom FKS-Gehalt, P62
war bei hohem und sehr geringem FKS-Gehalt höher und bei geringem FKS-Gehalt
geringer exprimiert, eine Abhängigkeit vom FKS-Gehalt ließ sich somit nicht
feststellen. Auch dies war ein grundlegender Unterschied zwischen PTEC von MMA-
und von PA-Patienten.
3) Der Nachweis der Degradierung der Autophagosome in sauren Autolysosomen
bei PTEC von MMA-Patienten erfolgte durch den Nachweis der erhöhten Anzahl der
Autolysosomen. Das Lysosom umschließt das Autophagosom und bildet das
Autolysosom, das Lumen wird durch Protonenpumpen angesäuert und der Inhalt
durch saure Hydrolyse degradiert (Yang und Klionsky, 2009; Weidberg et al., 2011;
Rubinsztein et al., 2012). Die höhere Ratio von FL3/FL1 nach Färbung mit
Acridinorange bei PTEC von MMA-Patienten deutete auf eine höhere Anzahl von
Autolysosomen hin. Die erhöhte Anzahl von Autolysosomen kann zum einen auf
erhöhte Autophagie hinweisen, zum anderen aber auch aus einer verminderten
Degradierungsrate analog zum Vici-Syndrom resultieren. Bei dieser Erkrankung
akkumulieren Autolysosomen in der Zelle, da sie aufgrund eines Defekts im EPG5-
Gen nicht abgebaut werden können und die Patienten leiden unter schweren
Schädigungen (Cullup et al., 2013).
Insgesamt weisen die Ergebnisse auf eine erhöhte Autophagie bei PTEC von MMA-
Patienten hin. Von einer Doppelmembran umgebene Strukturen, wie sie auf EM-
Bildern von PTEC von MMA-Patienten erkennbar waren, treten bei Autophagosomen
auf (Tooze, 2013) und lassen in Zusammenhang mit den Verläufen der Expressionen
von pmTOR/mTOR, pPRAS40/PRAS40, LC3-II und P62, sowie der höheren Anzahl
von Autolysosomen ein erhöhtes Vorkommen von Makro-Autophagie postulieren.
In den bekannten Mausmodellen für die MMA konnte Autophagie bislang nicht
nachgewiesen werden (Peters et al., 2003; Chandler et al., 2009; Manoli et al.,
2013). Ein Grund hierfür könnte sein, dass der Zelltod der murinen Zellen eingetreten
ist, bevor es zur Einleitung autophagozytotischer Wege kam. Es wurden bei den
Knock out-Mäusen jedoch degenerierte Mitochondrien mit aufgelöster Cristae-
Struktur nachgewiesen (Chandler et al., 2009). Bei den PTEC der MMA-Patienten
war die Struktur und Morphologie der Mitochondrien nicht unterschiedlich im
Vergleich zu Kontrollen. Eine mögliche Erklärung hierfür könnte sein, dass in der
vorliegenden Arbeit reine PTEC verwendet wurden. Chandler und Kollegen
verwendeten nicht genauer spezifizierte Zellen aus dem Nierengewebe von Mäusen,

die einen Mix verschiedener Zellen darstellten (Chandler et al., 2009). Somit könnten
auch in der Niere von MMA-Patienten Schädigungen der Mitochondrien auftreten, die
durch die Verwendung reiner PTEC in der vorliegenden Arbeit jedoch nicht
abgebildet werden konnten.
Erhöhte Autophagie dient zudem als Schutzmechanismus gegen chronischen,
metabolischen mitochondrialen Stress bei PTEC und die Zelle versucht durch
Aminosäurekatabolismus die Energieversorgung durch die Mitochondrien zu
umgehen, um den mitochondrialen Stress zu verringern (Kimura et al., 2013). Es
lässt sich postulieren, dass bei PTEC von MMA-Patienten ein ähnlicher
Mechanismus wirken könnte. Der Abbau von verzweigtkettigen Aminosäuren kann im
Zuge katabolischen Stresses zu metabolischen Entgleisungen führen.
5.6 ER-Stress bei PTEC von MMA-Patienten verstärkt die Autophagie
Die Einleitung der Autophagie kann ebenfalls mit der Aktivierung von IRE1, sowie
elF-2α über PERK erfolgen (Kouroku et al., 2007; Fujita et al., 2007, Benbrook &
Long, 2012). IRE1, PERK und ATF6 sind Sensorproteine im ER, die bei Stress des
ER als Transkriptionsfaktoren wirken und die Transkription verschiedener Gene
veranlassen. Das auf EM-Bildern erkennbare vergrößerte Lumen des ER trat nur bei
PTEC von MMA-Patienten auf. Es bildete kreisförmige Strukturen und war mit
Mitochondrien assoziiert, beides sind Kennzeichen für ER-Stress. Bei MEF-Zellen ist
die Bildung von LC3-II konjugierten, autophagischen Vesikeln chemisch durch
Tunicamycin oder Thapsigargin induzierbar und von IRE1abhängig, jedoch nicht von
ATF6 und PERK (Ogata et al., 2006). Der Verlauf der Expression von pIRE1 zeigte
nur bei PTEC von MMA-Patienten einen Zusammenhang mit der Höhe des FKS-
Gehalts und stieg bei höheren FKS-Werten und damit steigendem metabolischen
Stress an. Bei PA-Patienten war die Höhe der Expression von pIRE1 unabhängig
von der Höhe des FKS-Gehalts, sie ließ somit keinen Zusammenhang erkennen und
stellt einen wichtigen Unterschied zwischen PTEC von MMA- und PA-Patienten dar.
Es lässt sich postulieren, dass metabolischer Stress bei PTEC von MMA-Patienten
zu ER-Stress führt.
Weiterhin lässt sich ein Zusammenhang zwischen ER-Stress und Autophagie bei
PTEC von MMA-Patienten postulieren. Dieser kann in der für das Autophagosom
benötigten Membran liegen. Die Membran kann ihren Ursprung in der Abschnürung

eines Teils des ER haben, wofür ER-Stress eine Voraussetzung ist (Hamasaki et al.,
2013; Tooze, 2013; Uemura et al., 2014).
Die in elektronenmikroskopischen Aufnahmen erkennbaren autophagozytotischen
Strukturen in PTEC von MMA-Patienten zeigen Ähnlichkeit zu Aufnahmen von
Aggresomen. Aggresom-ähnliche Strukturen sind im Zytosol vorkommende
Proteinaggregate, die eine weitere Art der Degradierung aggregierter Proteine
darstellen. Am Ende werden sie ebenfalls durch Autophagosome eingeschlossen
und lysosomal abgebaut (Garcia-Mata et al., 2002). In beiden Fällen ist das Protein
P62 mit den aggregierten Proteinen assoziiert. P62 dient als Regulator des
Proteinabbaus, vermittelt die Bildung aggresom-ähnlicher Strukturen und verbindet
Inflammation mit ER-Stress. Im Gegensatz zur Autophagie wird bei Aggresomen
jedoch nur P62 an die zu degradierenden Strukturen gebunden. Die Prozession von
LC3 zu LC3-I und anschließend zu LC3-II tritt nur bei Autophagie auf, was einer
Beteiligung von Aggresomen widersprechen würde und stärker für Autophagie als für
Aggresome bei PTEC von MMA-Patienten spricht (Liu et al., 2012).
Bei chronischem ER-Stress sind UPR und oxidativer Stress mit erhöhten Werten von
ROS durch UPR-stimulierten Hochregulation der Chaperone, die bei der Bildung von
Disulfidbindungen im ER-Lumen beteiligt sind, miteinander assoziiert (Cullinan and
Diehl, 2006). Die für die Bildung von Disulfidbindungen verantwortlichen Enzyme
verwenden Oxidations- und Reduktions-Reaktionen mit molekularem Sauerstoff als
letztem Elektronenempfänger (Haynes et al., 2004). Die in der vorliegenden Arbeit
beschriebene erhöhte Bildung von ROS bei PTEC von MMA-Patienten könnte neben
Defekten der Enzyme von Atmungskette und Zitrat-Zyklus somit zusätzlich durch
erhöhten ER-Stress begründet sein, der durch UPR hochregulierten Chaperone
verursacht werden könnte.
5.7 PTEC von MMA-Patienten exprimieren Zytokine und Rezeptoren zur Interaktion mit Immunzellen
Inflammationen sind durch die Infiltration des entzündeten Gewebes durch Zellen
des Immunsystems gekennzeichnet. Voraussetzung dafür ist die Kommunikation
zwischen den Zellen des Immunsystems und den Zellen des entzündeten Gewebes
über Zytokine und Rezeptoren. Es wurde zum einen untersucht, ob PTEC von MMA-
Patienten Rezeptoren zur Signalweiterleitung des angeborenen Immunsystems

exprimierten, zum anderen ob sie Rezeptoren oder Moleküle zur Interaktion und
Kommunikation mit Zellen des erworbenen Immunsystem exprimierten.
Auf der Ebene des angeborenen Immunsystems wurden Elemente untersucht, die an
der Weiterleitung der Signale des Komplementsystems an der Zielzelle beteiligt sind.
Ein Vergleich von Rezeptoren, die auf der Zelloberfläche Faktoren des
Komplementsystems binden, ergab keine Unterschiede zwischen Kontrollen und
MMA-Patienten. Eine Involvierung der angeborenen Immunantwort scheint somit bei
der durch MMA ausgelösten cTIN keine oder nur eine untergeordnete Rolle zu
spielen.
Auf der Ebene des erworbenen Immunsystems wurde bei PTEC von Kontrollen und
MMA-Patienten ein Expressionsvergleich mit löslichen Rezeptoren sowie mit
Zytokinen durchgeführt. Die Ergebnisse wurden nur durch je zwei Zelllinien
gewonnen und dienten dazu, mögliche Moleküle für die Immunzellaktivierung
qualitativ zu erfassen.
Der Expressionsvergleich löslicher Rezeptoren zeigte bei PTEC von MMA-Patienten
eine stark erhöhte Expression von VCAM-1 (vascular cell adhesion molecule 1)
gegenüber Kontrollen und ließ auf eine mögliche Interaktion der PTEC mit Zellen des
Immunsystems schließen. VCAM-1 ist ein Zelladhäsionsprotein und vermittelt die
Bindung von Monozyten an Epithelzellen (Alpers et al., 1993; Seron et al., 1991;
Brady, 1994; Hill et al., 1995). Die Expression von VCAM-1 in Epithelzellen der Niere
wird bei Inflammationen drastisch erhöht und korreliert mit der tubulointerstitiellen
Schädigung von infiltrierenden Leukozyten.
Die erhöhte Expression der löslichen Form von IL-1RII (Interleukin-1 Rezeptor Typ 2)
bei PTEC von MMA-Patienten ließ auf eine weitere mögliche Interaktion mit Zellen
des Immunsystems schließen. Nach subkutaner MMS-Applikation wiesen Ratten
erhöhte Mengen an Interleukin-1β im Blut auf, verursacht durch eine erhöhte Anzahl
an Neutrophilen und eine allgemeine Aktivierung des Immunsystems (Ribeiro et al.,
2013). IL-1RII ist in der Lage die Interleukine IL-1α, IL-1β sowie den Rezeptor IL-1RI
(Interleukin-1 Rezeptor Typ 1) zu binden, leitet jedoch keine Signale weiter
(Dinarello, 1996). Dadurch würden die Rezeptoren der PTEC von MMA-Patienten
diese Zytokine aus dem Blutkreislauf entfernen, so dass ihre Wirkung am Zielort
verringert werden würde. Eine verminderte Pathogenabwehr könnte die Folge sein
(Dinarello, 2005).

Der Rezeptor IL-15R kann aus den Untereinheiten α oder β und γ aufgebaut sein.
Die Untereinheiten β und γ teilt sich der Rezeptor für IL-15 mit den Rezeptoren für IL-
2 (Anderson et al., 1995). Zu den IL-15 exprimierenden Zellen gehören
Makrophagen, Monozyten, dendritische Zellen, Keratinozyten, Fibroblasten und
Nervenzellen (Grabstein et al., 1994). IL-15 fungiert über den Rezeptor IL-15Rα in
der Niere als Überlebensfaktor von Epithelzellen (Giron-Michel et al., 2013).
Zytokine üben ihre Wirkung über Rezeptoren aus und vermitteln zwischen humoraler
und zellbasierter Immunantwort. Sie regulieren Reifung, Wachstum und Antwort
bestimmter Zellarten. Interleukine sind Peptidhormone und eine Untergruppe der
Zytokine. Sie tragen zur Kommunikation zwischen Immunzellen oder Immunzellen
mit anderen Zellen bei. Humane PTEC sind grundsätzlich in der Lage verschiedene
Zytokine wie CCL2 (MCP-1), CCL8 (MCP-2), CCL3 (MIP-1α), CCL4 (MIP-1β), CCL5
(RANTES), und CXCL8 (IL-8) zu bilden und zu sezernieren (Cockwell et al., 1998;
Rovin et al., 1996; Mezzano et al., 2000; Chakravorty et al., 2001; Tang et al., 2003).
Insbesondere IL-8 ist ein wichtiges Protein in der Immunantwort und dient der
Rekrutierung von neutrophilen Granulozyten und Makrophagen zum
Entzündungsherd (Baggiolini & Clark-Lewis, 1992). Während sich eine vermehrte
Sekretion von CCL2, CCL3, CCL4 und CCL5 nicht nachweisen ließ, sezernierten
Zellen von MMA-Patienten apikal und basolateral qualitativ mehr IL-8 ins Medium.
Die quantitative Analyse zeigte, dass die Menge an sezerniertem IL-8 gegenüber
Kontrollen (apikal, basolateral, intrazellulär) und PA-Patienten (apikal, basolateral)
erhöht war. Als stärkste Expressionsstimuli für IL-8 in PTEC-Kulturen in vitro wurden
IL-1α und TNF-α identifiziert (Gerritsma et al., 1996). Die Kultivierung der PTEC in
der vorliegenden Arbeit erfolgte in Medium ohne Supplementation von FKS oder
anderer Agenzien, da FKS ebenfalls zur Genexpression von IL-8 führen kann. In vivo
dient IL-8 als Signalmolekül und wirkt auf Zellen des Immunsystems chemotaktisch.
Die Menge an exprimiertem IL-8 von PTEC ist proportional zur chemotaktischen
Aktivierung von T-Zellen und ihrer Rekrutierung zum Ort der Inflammation (Demmers
et al., 2013). Die Signalentfaltung erfolgt über GTP-gekoppelte Rezeptoren, die
Zielzellen sind Neutrophile, Granulozyten und Makrophagen (Baggiolini & Clark-
Lewis, 1992). Bei intestinalen Epithelzellen führt erhöhte Autophagie zu stärkerer IL-
8 Sekretion (Li et al., 2011). Die Expression von IL-8 ist weiterhin durch ROS wie
Wasserstoffperoxid induzierbar (Vlahopoulos et al., 1999) und kann somit als
Weiterleitung von ROS-Signalen an Zellen des Immunsystems dienen, welches die

Rekrutierung von Neutrophilen, Basophilen und T-Zellen zum Herd der Inflammation
zur Folge hat.
5.8 Korrelation von Autophagie und IL-8-Expression bei PTEC von MMA-Patienten
Zwischen der Stärke der Autophagie und der Höhe des exprimierten IL-8 bestand in
PTEC von MMA-Patienten eine sehr starke Korrelation und deutete auf einen
Zusammenhang hin.
Autophagie und die Expression von Zytokinen können sich gegenseitig beeinflussen
und regulieren, Interleukine nehmen bei der Regulation von Autophagie kontroverse
Rollen ein. Zum einen wirkt IL-13 in der Epithelzelllinie HT29 als Inhibitor von durch
Nahrungsmangel ausgelöster Autophagie (Petiot et al., 2000; Arico et al., 2001),
andererseits übernimmt IL-8 die Rolle eines Aktivators der Autophagie indem es
mTORC1 hemmt und somit die Autophagie stimuliert. In diesem Zusammenhang
existiert ein Zusammenspiel zwischen ROS, Autophagie, Zytokinexpression und
Inflammation (Harris, 2011).
Autophagie hat einen unmittelbaren Einfluss auf die Transkription, Prozessierung
und Sekretion einer Reihe von Zytokinen. Die Störung der in allen Zellen auf einem
Grundniveau ablaufenden Autophagie steht in Verbindung mit erhöhter Sekretion der
proinflammatorischen Zytokine IL-1α, IL-1β und IL-18 (Saitoh et al., 2008; Crisan et
al., 2011; Harris et al., 2011; Nakahira et al., 2011; Zhou et al., 2011). Autophagie
reguliert die Transkription und Sekretion von TNF-α und IL-8 positiv (Harris, 2011).
Für einen direkten Zusammenhang zwischen erhöhter Autophagie und der
Expression und Sezernierung von IL-8 gibt es bisher noch keine Hinweise. Das
parallele Auftreten von Autophagie und akuter Nierenschädigung kann sich jedoch zu
einer chronischen Inflammation entwickeln (Tögel und Westenfelder, 2014).
In Patienten mit diabetischer oder nicht-diabetischer Nephritis im Endstadium kann
die Induktion der IL-8 Transkription durch ROS wie Wasserstoffperoxid erfolgen
(Lakshminarayanan et al., 1998). Der unmittelbare Einfluss von ROS auf die direkte
Aktivierung von NF-κB über Modifikation von Aminosäurenresten der einzelnen
Proteine P50, P52, P65, P100 und P105 von NF-κB kann je nach Zelltyp
unterschiedlich sein und NF-κB kann aktiviert oder deaktiviert werden (Morgan und
Liu, 2011). In der vom humanen proximalen Tubulus stammenden Zelllinie HK-2

führen Proteinbelastung und ROS über die Aktivierung von NF-κB zur Transkription
von Chemokinen (Morigi et al., 2002). Der Abbau von reduziertem Glutathion und die
anschließende Erhöhung von zytosolischem Glutathiondisulfid als Antwort auf
oxidativen Stress führen ebenfalls zu der Aktivierung von NF-κB (Raman und McNee,
2000). Die in der vorliegenden Arbeit verwendeten PTEC der MMA-Patienten wurden
alle von Spendern isoliert, die bereits eine starke Beteiligung der Nieren im
Krankheitsbild zeigten. Es wurden intrazellulär ebenfalls verringerte Konzentrationen
an reduziertem Glutathion und erhöhte Konzentrationen an zytosolischem
Glutathiondisulfid nachgewiesen, so dass hier auch eine Aktivierung von NF-κB mit
anschließender Transkription von IL-8 stattgefunden haben könnte.
Die Expressionsstimuli IL-1α und TNF-α wirken über ihre Rezeptoren und die
Aktivierung von NF-κB. In Rinderzellen stimulieren TNF-α und NF-κB die
Genxpression von IL-8 (Fitzgerald et al., 2007). In Fibroblasten führt erhöhte
Autophagie zur Aktivierung der IKK. Durch Aktivierung der IKK wird auch der NF-κB
Signalweg stimuliert und die Genexpression von Interleukinen (z.B. IL-8) kann
eingeleitet werden (Criollo et al., 2010). In PTEC von MMA-Patienten wäre beim
Zusammenhang zwischen Autophagie und der Expression von IL-8 der gleiche
Mechanismus vorstellbar.
Eine Rolle für das Zusammenspiel aus Autophagie, ROS und Inflammation könnte
das NLRP3-Inflammasom spielen, das auf ROS reagiert und zu Autophagie,
Apoptose, Fibrose und zur proinflammatorischen Zytokinausschüttung führen kann.
Dabei reichen bereits einzelne Komponenten des Inflammasoms aus, der komplette
Ablauf der Kaskade ist keine Voraussetzung (Shigeoka et al., 2010; Chang et al.,
2014).
Die erworbene Immunantwort wird durch die Interaktion zwischen dem T-Zell-
Rezeptor und Peptidsequenzen eingeleitet, die durch MHC-II Moleküle (major
histocompatibility class II) präsentiert werden. MHC-II Moleküle werden vor allem von
antigenpräsentierenden Zellen wie zirkulierenden Makrophagen oder dendritischen
Zellen gebildet, können jedoch auch von PTEC gebildet werden (Muller et al., 1989).
Die autophagische Antigenprozessierung stellt Peptide für die Präsentation auf MHC-
II Moleküle bereit. So können die nachgeschalteten immunologischen Effekte der
Präsentation von MHC-II Antigenen als ein Ergebnis der vorgelagerten Autophagie
gesehen werden (Leventhal et al., 2013). PTEC von MMA-Patienten könnten

ebenfalls durch autophagie-vermittelte MHC-II Bildung und IL-8 Sekretion mit T-
Zellen interagieren und die Inflammation einleiten.
5.9 Ausblick
Die in dieser Dissertation erarbeiteten Ergebnisse lassen auf einen Zusammenhang
zwischen der Entstehung und Stärke der Autophagie und der Expression von IL-8 als
Signalmolekül zur Rekrutierung des Immunsystems zum Herd der Inflammation bei
der Entstehung der cTIN bei MMA-Patienten schließen.
Das verwendete Zellkulturmodell lässt jedoch nur einen eingeschränkten Blickwinkel
auf einen Zelltyp des betroffenen Organs zu, da alle Versuche an PTEC durchgeführt
wurden.
Weiterführende Versuche könnten an PTEC zusätzlicher MMA- und PA-Patienten
erfolgen, um die jeweiligen Gruppen zu vergrößern und die in dieser Dissertation
erarbeiteten Ergebnisse zu stützen. Zusätzlich könnten Versuche an isolierten
humanen Zellen des Immunsystems von MMA-Patienten durchgeführt werden, um
die Aktivierbarkeit von T-Zellen, Neutrophilen und Makrophagen durch Kokultur mit
PTEC zu untersuchen.
Weiterhin könnte an einem neu entwickelten oder bereits etablierten MCM-knockout
Mausmodell eine systemische Betrachtung der gefundenen Ergebnisse durchgeführt
werden, in welche andere Gewebearten wie Skelettmuskelzellen, Herzmuskelzellen,
Leberzellen und verschiedene Zellen des Immunsystems mit einbezogen werden
könnten.
Im Rahmen dieser Dissertation wurden Zellen von MMA-Patienten verwendet, die
bereits eine cTIN und damit verbundene Schädigung der Niere entwickelt haben.
Weitere Untersuchungen bei PTEC von MMA-Patienten ohne cTIN könnte die
Hypothese unterstützen, dass ein Zusammenhang zwischen erhöhter Autophagie
und Sezernierung von IL-8 besteht. Als Hypothese ließe sich formulieren, dass nur
bei Patienten mit cTIN ein Zusammenhang zwischen erhöhter Autophagie und
Sezernierung von IL-8 besteht. Ergänzend dazu könnten Analysen anderer
Erkrankungen dienen, die ebenfalls eine cTIN entwickeln, um zu überprüfen, ob es
ein Pathomechanismus ist, der nur im Zuge der MMA auftritt, oder ob es ein Modell
darstellt, welches auch auf andere Erkrankungen zutrifft und allgemeiner gesehen
werden kann. Weiterhin könnte das NLRP3-Inflammasom als Schnittstelle zwischen

Autophagie, Apoptose und Fibrose dienen, die an der Sezernierung
proinflammatorischer Zytokine beteiligt ist. Dieser oder ein ähnlich funktionierender
Mechanismus wäre auch bei der cTIN von MMA-Patienten vorstellbar.
Ein weiterer wünschenswerter Aspekt wäre die Generierung eines Markers, der die
Entwicklung der cTIN schon früher prognostizierbar machen würde. Ein
naheliegender Kandidat dafür wäre IL-8, das bei PTEC von MMA-Patienten erhöht
war. Bei Hämodialysepatienten sind die Serumkonzentrationen der Zytokine
Interleukin 1, 2, 3, 4, 5, 6, 12, 13 und Tumornekrosefaktor J (TNFJ) erhöht (Kimmel
et al. 1998). IL-8 ist davon nicht betroffen, so dass es keine Verfälschungen durch
eine Behandlung geben würde. Die Bestimmung von IL-8 im Plasma wäre dennoch
nur bedingt als Marker für eine cTIN zu verwenden, da die MMA eine
multisystemische Erkrankung ist und IL-8 auch aus anderen Geweben stammen
kann. Der gemessene Plasma-Wert lässt sich nicht einzelnen Quellen zuordnen,
somit kann im Plasma gemessenes IL-8 nicht als Marker für eine cTIN verwendet
werden. Mit dem Urin ausgeschiedenes IL-8 könnte jedoch unter Umständen als
Marker dienen, es korreliert kaum mit dem Plasma-Wert (Jacobsen et al., 1996).
Aufgrund seiner geringen Größe könnte es den glomerulären Filter passieren. Da die
IL-8 Werte bei PTEC von MMA-Patienten auch intrazellulär erhöht waren, könnten
die in der vorliegenden Arbeit verwendete Methode der Isolierung von humanen
PTEC im Rahmen der Ambulanztermine der Patienten verwendet werden. Bei
stationären Klinikaufenthalten, wie sie z.B. nach metabolischen Krisen notwendig
werden, könnten die Zellen jedoch zu sehr geschädigt sein und zu falschen
Schlussfolgerungen führen. Die isolierten Zellen können für einige Passagen
kultiviert werden und reichen zur Bestimmung der Konzentration von IL-8 aus.

5.10 Zusammenfassung
Ziel dieser Arbeit ist die Untersuchung der renalen Pathogenese der chronischen
tubulointerstitiellen Nephritis bei Methylmalonazidurie-Patienten, die zur Entwicklung
eines chronischen Nierenversagens führt. Als Untersuchungsmodell wählten wir
proximale Tubulusepithelzellen von Methylmalonazidurie-Patienten. Da in einigen
Studien eine multiple Störung des mitochondrialen Energiestoffwechsels als Ursache
des Organversagens bei Methylmalonazidurie-Patienten gezeigt wurde, fokussierten
wir uns zunächst auf eine Beschreibung des Energiemetabolismus der proximalen
Tubulusepithelzellen. Die verringerte Aktivität der Phosphofruktokinase 1 führt zu
einem geringeren Flux und damit zu einer Beeinträchtigung der Glykolyse. In
Zusammenhang mit der erfolglosen Isolierung von Mitochondrien, ließ die
verminderte Aktivität der mitochondrialen Aconitase, des 2-Oxoglutarat-
Dehydrogenasekomplexes und der Cytochrom c-Oxidase auf einen mitochondrialen
Defekt in proximale Tubulusepithelzellen von Methylmalonazidurie-Patienten
schließen, der sich strukturell und funktionell manifestierte.
Der mitochondriale Defekt führte weiterhin zu erhöhter ROS-Produktion, die sich
durch metabolischen Stress aufgrund der Applikation von Glukose, Pyruvat,
Methylmalonsäure, Propionsäure und Isoleucin steigern ließ. Erhöhtes
Glutathiondisulfid und verringertes freies Glutathion aufgrund höherer ROS-
Konzentration stärkten die Hypothese erhöhten oxidativen Stresses in proximalen
Tubulusepithelzellen von Methylmalonazidurie-Patienten.
Die höhere ROS-Konzentration führte im Zusammenspiel mit erhöhter Expression
von pIRE1 und degeneriertem Endoplasmatischen Retikulum mit vergrößertem
Lumen, die auf elektronenmikroskopischen Bildern erkennbar waren, zu Stress des
Endoplasmatischen Retikulums in proximalen Tubulusepithelzellen von
Methylmalonazidurie-Patienten.
Verringerte Aktivität von mTORC1 (mammalian target of rapamycin complex 1) unter
metabolischem Stress und die Störungen im Energiemetabolismus deuteten bei
proximalen Tubulusepithelzellen von Methylmalonazidurie-Patienten auf eine positive
Regulation der Autophagosombildung hin. In diesem Zusammenhang führten von
einer Doppelmembran umgebene Strukturen, die auf elektronenmikroskopischen
Bildern zu sehen waren, und die erhöhte Expression von P62 und LC3-II unter
metabolischem Stress zur Annahme erhöhter Makro-Autophagie. Diese ließ sich
durch mehr Autolysosomen bestätigen.

Es ist bekannt, dass die Inflammation eines Gewebes immer mit der Infiltrierung
durch Zellen des Immunsystems einhergeht. Die Expression der zur Rekrutierung
des Immunsystems notwendigen Zytokine war bei proximalen Tubulusepithelzellen
von Methylmalonazidurie-Patienten in Form von Interleukin-8 höher, die apikale und
basolaterale Sekretion fiel stärker aus. Die starke Korrelation zwischen Autophagie
und der Sezernierung von Interleukin-8 bei proximalen Tubulusepithelzellen von
Methylmalonazidurie-Patienten deutete darauf hin, dass Autophagie eine direkte
Ursache der erhöhten Transkription von Interleukin-8 ist oder indirekt Ursachen der
Transkription von Interleukin-8 fördert. Dies kann zu einer Infiltration des Interstitiums
des proximalen Tubulus durch Neutrophile, Makrophagen und T-Zellen führen. Die
inflammatorische Reaktion wird somit weiter getriggert und führt über längere Zeit
zum Versagen der Nierenfunktionen.

Abbildung (25) Zusammenwirkender Mechanismus der molekularbiologischen Ursachen der chronischen tubulointerstitiellen Nephritis in PTEC von MMA-Patienten


6 Literaturverzeichnis
A
Alpers CE, Hudkins KL, Davis CL, Marsh CL, Riches W, McCarty JM, Benjamin CD,
Carlos TM, Harlan JM, Lobb R (1993). Expression of vascular cell adhesion
molecule-1 in kidney allograft rejection. Kidney Int 44: 805-16
Anderson DM, Kumaki S, Ahdieh M, Bertles J, Tometsko M, Loomis A, Giri J,
Copeland NG, Gilbert DJ, Jenkins NA, et al (1995). Functional characterization
of the human interleukin-15 receptor alpha chain and close linkage of IL15RA
and IL2RA genes. J Biol Chem 270: 29862-9
Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, Ogier-Denis E
(2001). The tumor suppressor PTEN positively regulates macroautophagy by
inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol
Chem 276: 35243-6
Arsham AM, Howell JJ, Simon MC (2003). A novel hypoxia-inducible factor-
independent hypoxic response regulating mammalian target of rapamycin and
its targets. J Biol Chem 278 (32): 29655-60
Ashford TP, Porter KR (1962). Cytoplasmic components in hepatic cell lysosomes. J
Cell Biol 12: 198-202
B
Baenkler HW, Goldschmitt H, Hahn JM, Hinterseer M (2009). Innere Medizin, Thieme
Verlag. 2. Auflage: 367-409
Back SH, Schröder M, Lee K, Zhang K, Kaufman RJ (2005). ER stress signaling by
regulated splicing: IRE1/HAC1/XBP1. Methods 35 (4): 395-416
Baggiolini M, Walz A, Kunkel SL. Neutrophil-activating peptide-1 / interleukin 8, a
novel cytokine that activates neutrophils (1989). J Clin Invest 84: 1045–9.
Baggiolini M, Clark-Lewis I (1992). Interleukin-8, a chemotactic and inflammatory
cytokine. FEBS Lett 307: 97-101
Baud L, Ardaillou R (1986). Reactive oxygen species: production and role in the
kidney. Am J Physiol 251: F765-76
Benbrook DM, Long A (2012). Integration of autophagy, proteasomal degradation,
unfolded protein response and apoptosis. Exp Oncol 34: 286-97

Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D (2000). Dynamic
interaction of BiP and ER stress transducers in the unfolded-protein response.
Nat Cell Biol 2 (6): 326-32
Bohle A, Mackensen-Haen S, VonGise, Grund KE, Wehrmann M, Batz C, Bogen
Schutz O, Schmitt H, Nagy J, Muller C (1990). The consequences of
tubuleinterstitial changes for renal function in glomerulopathies. A
morphometric and cytological analysis. Pathol Res Pract 186:135–144
Braden GL, O'Shea MH, Mulhern JG (2005). Tubulointerstitial diseases. Am J Kidney
Dis 46:560-72.
Brady HR (1994). Leukocyte adhesion molecules and kidney diseases. Kidney Int 45:
1285-300
Brasil S, Richard E, Jorge-Finnigan A, Leal F, Merinero B, Banerjee R, Desviat LR,
Ugarte M, Pérez B (2014). Methylmalonic aciduria cblB type: characterization
of two novel mutations and mitochondrial dysfunction studies. Clin Genet doi:
10.1111/cge.12426
Buck NE, Wood LR, Hamilton NJ, Bennett MJ, Peters HL (2012). Treatment of a
methylmalonyl-CoA mutase stopcodon mutation. Biochem Biophys Res
Commun 427 (4): 753-7
Buck NE, Dashnow H, Pitt JJ, Wood LR, Peters HL (2012). Development of
transgenic mice containing an introduced stop codon on the human
methylmalonyl-CoA mutase locus. PLoS One 7 (9): e 44974
Buemi M, Allegra A, Rotig A, Gubler MC, Aloisi C, Corica F, Pettinato G, Frisina N,
Niaudet P (1997). Renal failure from mitochondrial cytopathies. Nephron 76 (3): 249-53
C
Camougrand N, Kissová I, Velours G, Manon S (2004). Uth1p: a yeast mitochondrial
protein at the crossroads of stress, degradation and cell death. FEMS Yeast
Res 5: 133-40
Cantu D, Schaack J, Pate M (2009). Oxidative inactivation of mitochondrial aconitase
results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic
cultures. PLoS One 4: e7095
Cardoso SM, Pereira C und Oliveira R. (1999). Mitochondrial function is differentially
affected upon oxidative stress. Free Radic. Biol. Med. 26: 3-13.

Carr MW, Roth SJ, Luther E, Rose SS, Springer TA (1994). Monocyte
chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl
Acad Sci USA 91: 3652-6
Chang A, Ko K, Clark MR (2014). The emerging role of the inflammasome in kidney
diseases. Curr Opin Nephrol Hypertens 23: 204-10
Chakravorty SJ, Howie AJ, Girdlestone J, Gentle D, Savage CO (2001). Potential
role for monocyte chemotactic protein-4 (MCP-4) in monocyte/macrophage
recruitment in acute renal inflammation. J Pathol 194: 239-46
Chandler RJ, Zerfas PM, Shanske S, Sloan J, Hoffmann V, DiMauro S, Venditti CP
(2009). Mitochondrial dysfunction in mut methylmalonic acidemia. FASEB J 23
(4): 1252-61
Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL und Lesnefsky EJ (2003).
Production of reactive oxygen species by mitochondria: central role of complex
III. J. Biol. Chem. 278: 36027-31
Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Liu Y, Zheng P (2008). TSC-mTOR
maintains quiescence and function of hematopoietic stem cells by repressing
mitochondrial biogenesis and reactive oxygen species. J Exp Med 205: 2397-
408
Chinopoulos C, Tretter L, Adam-Vizi V (1999). Depolarization of in situ mitochondria
due to hydrogen peroxide-induced oxidative stress in nerve terminals:
inhibition of alpha-ketoglutarate dehydrogenase. J Neurochem 73: 220-8
Choi AJS, Ryter SW (2011). Autophagy in inflammatory diseases. Int J of Cell
Biology 2011: 732798
Cockwell P, Savage COS (1997). Chemokines and their receptors in leukocyte
trafficking and renal inflammation. Nephrol Dial Transplant 12: 1307-15
Cockwell P, Howie AJ, Adu D, Savage CO (1998). In situ analysis of C-C chemokine
mRNA in human glomerulonephritis. Kidney Int 54: 827-36
Cockwell P, Calderwood JW, Brooks CJ, Chakravorty SJ, Savage CO (2002).
Chemoattraction of T cells expressing CCR5, CXCR3 and CX3CR1 by
proximal tubular epithelial cell chemokines. Nephrol Dial Transplant 17: 734-
44
Codogno P, Meijer AJ (2005). Autophagy and signaling: their role in cell survival and
cell death. Cell Death Differ 12 Suppl. 2: 1509-1518

Cox JS, Shamu CE, Walter P (1993). Transcriptional induction of genes encoding
endoplasmic reticulum resident proteins requires a transmembrane protein
kinase. Cell 73 (6): 1197-206
Criollo A, Senovilla L, Authier H, Maiuri MC, Morselli E, Vitale I, Kepp O, Tasdemir E,
Galluzzi L, Shen S, Tailler M, Delahaye N, Tesniere A, De Stefano D, Younes
AB, Harper F, Pierron G, Lavandero S, Zitvogel L, Israel A, Baud V, Kroemer
G (2010). The IKK complex contributes to the induction of autophagy. EMBO J
29: 619-31.
Cuervo AM, Bergamini E, Brunk UT, Dröge W, Ffrench M, Terman A (2005).
Autophagy and aging: the importance of maintaining "clean" cells. Autophagy
1: 131-40
Cullinan SB, Diehl JA (2006). Coordination of ER and oxidative stress signaling: the
PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol 38:317–332.
Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P
(2007). mTOR controls mitochondrial oxidative function through a YY1-PGC-
1alpha transcriptional complex. Nature 450: 736-40
D
Daha MR, van Kooten C (2000). Is the proximal tubular cell a proinflammatory cell?
Nephrol Dial Transplant 15 (6): 41-3
D'Autréaux B & Toledano MB (2007). ROS as signalling molecules: mechanisms that
generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8: 813-24
del Hoyo GM, Martin P, Vargas HH, Ruiz S, Arias CF, Ardavin C (2002).
Characterization of a common precursor population for den- dritic cells.
Nature 415: 1043–7
De Keyzer Y, Valayannopoulos V, Benoist JF, Batteux F, Lacaille F, Hubert L,
Chrétien D, Chadefeaux-Vekemans B, Niaudet P, Touati G, Munnich A, de
Lonlay P (2009). Multiple OXPHOS deficiency in the liver, kidney, heart, and
skeletal muscle of patients with methylmalonic aciduria and propionic aciduria.
Pediatr Res 66: 91-5
Demmers MW, Baan CC, van Beelen E, Ijzermans JN, Weimar W, Rowshani AT
(2013). Differential effects of activated human renal epithelial cells on T-cell
migration. PLoS One 8: e64916

Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron
D (2004). Translational repression mediates activation of nuclear factor kappa
B by phosphorylated translation initiation factor 2. Mol Cell Biol 24: 10161–68
De Virgilio C, Loewith R (2006). Cell growth control: little eukaryotes make big
contributions. Oncogene 25: 6392-415
De Virgilio C, Loewith R (2006). The TOR signalling network from yeast to men. Int J
Biochem Biol 38: 1476-81
Deodato F, Boenzi S, Santorelli FM, Dionisi-Vici C (2006). Methylmalonic and
Propionic Aciduria. Am J Med Genett C 142C: 104–12
Di Mauro S und Wallace DC (1993). Mitochondrial DNA in Human Pathology. Raven
Press, New York
Dinarello CA (1996). Biologic basis for interleukin-1 in disease. Blood 87: 2095-147
Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM (2007). Linking of
autophagy to ubiquitin-proteasome system is important for the regulation of
endoplasmic reticulum stress and cell viability. Am J Pathol 171: 513–24
Djavaheri-Mergny M, Amelotti M, Mathieu J, Besançon F, Bauvy C, Codogno P
(2007). Regulation of autophagy by NFkappaB transcription factor and
reactives oxygen species. Autophagy 3: 390-2
E
Eardley KS, Cockwell P (2005). Macrophages and progressive tubulointerstitial
disease. Kidney Int 68 (2): 437-55
Edinger AL, Thompson CB (2003). Defective autophagy leads to cancer. Cancer Cell
4: 422-4
Ernandez T, Mayadas TN (2009). Immunoregulatory role of TNFa in inflammatory
kidney diseases. Kidney Int 76: 262-76
F
Fenton WAG, R.A.; Rosenblatt, D.S. (2001). Disorders of propionate and
methylmalonate metabolism. In Scriver CR, Beaudet AL, Valle AD, eds The
Metabolic and Molecular Bases of Inherited Disease McGraw-Hill, New York.
2001:2165-93
Fitzgerald DC, Meade KG, McEvoy AN, Lillis L, Murphy EP, MacHugh DE, Baird AW
(2007). Tumour necrosis factor-alpha (TNF-alpha) increases nuclear factor

kappaB (NFkappaB) activity in and interleukin-8 (IL-8) release from bovine
mammary epithelial cells. Vet Immunol Immunopathol 116: 59–68
Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, Sabatini DM (2006). mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms
define three distinct mTORC2s. Curr. Biol. 16, 1865-1870
Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, Hayashi YK, Momoi
T (2007). Two endoplasmic reticulum-associated degradation (ERAD) systems
for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and
autophagy/lysosome ERAD(II). Human Mol Genet 16: 618-29
G
Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X (2009).
ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for
autophagy. J. Biol. Chem. 284: 12297-12305
Garcia-Ruiz C, Colell A, Morales A, Kaplowitz N und Fernandez-Checa JC (1995).
Role of oxidative stress generated from the mitochondrial electron transport
chain and mitochondrial glutathione status in loss of mitochondrial function
and activation of transcription factor nuclear factor-kappa B: studies with
isolated mitochondria and rat hepatocytes. Mol. Pharmacol. 48: 825-34
Gekle M (2005). Renal tubule albumin transport. Annu Rev Physiol 67: 573-94
Gentle ME, Shi S, Daehn I, Zhang T, Qi H, Yu L, D’Agati, Schlondorff DO, Bottinger
EP (2013). Epithelial cell TGFβ signaling induces acute tubular injury and
interstitial inflammation. J AM Soc Nephrol 24: 787-99
Gerritsma JS, Hiemstra PS, Gerritsen AF, Prodjosudjadi W, Verweij CL, Van Es LA,
Daha MR (1996). Regulation and production of IL-8 by human proximal tubular
epithelial cells in vitro. Clin Exp Immunol 103: 289-94
Giron-Michel J, Azzi S, Ferrini S, Chouaib S, Camussi G, Eid P, Azzarone B (2013).
Interleukin-15 is a major regulator of the cell-microenvironment interactions in
human renal homeostasis. Cytokine Growth Factor Rev 24: 13-22
Gong W, Howard OM, Turpin JA, Grimm MC, Ueda H, Gray PW, Raport CJ,
Oppenheim JJ, Wang JM (1998). Monocyte chemotactic protein-2 activates
CCR5 and blocks CD4/CCR5-mediated HIV-1 entry/replication. J Biol Chem
273: 4289-92

Gotoh T, Mori M (2006). Nitric oxide and endoplasmic reticulum stress. Arterioscler
Thromb Vasc Biol 26: 1439-46.
Goyenechea E, Andrade F, de Las Heras J, Lage S, Prieto JÁ, Ruiz N, Aldámiz-
Echevarría L (2012). Expression of proinflammatory factors in renal cortex
induced by methylmalonic acid. Ren Fail 34 (7): 885-91
Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M,
Fitzgerald KJ, Sabatini DM (2006). Ablation in mice of the mTORC
components raptor, rictor, or mLST8 reveals that mTORC2 is required for
signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11 (6):859-71
Guertin DA, Sabatini DM (2007). Defining the role of mTOR in cancer. Cancer Cell
12 (1): 9-22
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk
BE, Shaw RJ (2008). AMPK phosphorylation of raptor mediates a metabolic
checkpoint. Mol Cell 30 (2): 214-26
H
Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK,
Lippincott-Schwartz J (2010). Mitochondria supply membranes for
autophagosome biogenesis during starvation. Cell 141: 656-67
Hamasaki M, Noda T, Baba M, Ohsumi Y (2005). Starvation triggers the delivery of
the endoplasmic reticulum to the vacuole via autophagy in yeast. Traffic 6: 56-
65
Hamasaki M, Shibutani ST, Yoshimori T (2013). Up-to-date membrane biogenesis in
the autophagosome formation. Cuur Opin Cell Biol 25: 455-60
Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J,
Yonezawa K (2002). Raptor, a binding partner of target of rapamycin (TOR),
mediates TOR action. Cell 110: 177-189
Hardie DG (2007). Role of AMP-activated protein kinase in the metabolic syndrome
and in heart disease. FEBS Lett 582 (1): 81-9
Harding HP, Zhang Y, Ron D (1999). Protein translation and folding are coupled by
an endoplasmic-reticulum-resident kinase. Nature 397 (6716): 271-4
Harrington LS, Findlay GM, Lamb RF (2005). Restraining PI3K: mTOR signalling
goes back to the membrane. Trends Biochem 30 (1): 35-42
Harris J (2011). Autophagy and cytokines. Cytokine 56: 140-4

Haynes CM, Titus EA, Cooper AA (2004). Degradation of misfolded proteins
prevents ER- derived oxidative stress and cell death. Mol Cell 15: 767-76
Haze K, Yoshida H, Yanagi H, Yura T, Mori K (1999). Mammalian transcription factor
ATF6 is synthesized as a transmembrane protein and activated by proteolysis
in response to endoplasmic reticulum stress. Mol Biol Cell 10 (11): 3787-99
He C, Song H, Yorimitsu T, Monastyrska I, Yen WL, Legakis JE, Klionsky DJ (2006).
Recruitment of Atg9 to the preautophagosomal structure by Atg11 is essential
for selective autophagy in budding yeast. J Cell Biol 175: 925-35
Hers HG, und van Schaftingen E (1982). Fructose 2,6-bisphosphate 2 years after its
discovery. Biochem J 206: 1–12
Herrero A und Barja G (2000). Localization of the site of oxygen radical generation
inside the Complex I of heart and nonsynaptic brain mammalian mitochondria.
J. Bioenerg. Biomembr. 32: 609–15.
Hill PA, Main IW, Atkins RC (1995). ICAM-1 and VCAM-1 in human renal allograft
rejection. Kidney Int 47: 1383-91
Hörster F, Baumgartner MR, Viardot C, Suormala T, Burgard P, Fowler B, Hoffmann
GF, Garbade SF, Kölker S, Baumgartner ER (2007). Long-term outcome in
methylmalonic acidurias is influenced by the underlying defect (mut0, mut-,
cblA, cblB).Pediatr Res 62 (2): 225-30
Hörster F, Garbade SF, Zwickler T, Aydin HI, Bodamer OA, Burlina AB, Das AM, De
Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002). TSC2 is phosphorylated and
inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4 (9): 648-57
Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume
T, Takehana K, Yamada N et al. (2009). Nutrient- dependent mTORC1
association with the ULK1-Atg13- FIP200 complex required for autophagy.
Mol. Biol. Cell 20: 1981-1991
Hu F, Liu F (2011). Mitochondrial stress: a bridge between mitochondrial dysfunction
and metabolic diseases? Cell Signa. 23: 1528–33.
Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH (2006). Autocrine tumor necrosis
factor alpha links endoplasmic reticulum stress to the membrane death
receptor pathway through IRE1alpha-mediated NF-kappaB activation and
downregulation of TRAF2 expression. Mol Cell Biol 26: 3071–84
Hume DA, Gordon S (1983). Mononuclear phagocyte system of the mouse defined
by immunohistochemical localization of antigen F4/80: Identification of

resident macrophages in renal medullary and cortical interstitium and the
juxtaglomerular complex. J Exp Med 157: 1704-09
Hurley JH, Schulmann BA (2014). Atomistic autophagy: the structures of cellular self-
digestion. Cell 157: 300-11
I
Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N,
Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y (2000). A ubiquitin-like
system mediates protein lipidation. Nature 408: 488-92
Inoki K, Zhu T, Guan KL (2003). TSC2 mediates cellular energy response to control
cell growth and survival. Cell 115 (5): 577-90
Inoki K, Li Y, Xu T, Guan KL (2003). Rheb GTPase is a direct target of TSC2 GAP
activity and regulates mTOR signaling. Genes Dev 17 (5): 1829-34
J
Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B
(2006). SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt
phosphorylation and substrate specificity. Cell 127 (1): 125-37
Jacinto E, Lorberg A (2008). TOR regulation of AGC kinases in yeast and mammals.
Biochem J 410: 19-37
Jacobson SH, Hylander B, Wretlind B, Brauner A (1996). Interleukin-6 and
interleukin-8 in serum and urine in patients with acute pyelonephritis in relation
to bacterial-virulence-associated traits and renal function. Nephron 67: 172–9.
Jenkins CM, Yang J, Sims HF, Gross RW (2011). Reversible high affinity inhibition of
phosphofructokinase-1 by acyl-CoA: A mechanism integrating glycolytic flux
with lipid metabolism. J Biol Chem 286: 11937–50
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH (2009). ULK-
Atg13-FIP200 complexes mediate mTOR signaling to the autophagy
machinery. Mol. Biol. Cell 20: 1992-2003
K
Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y (2005). Atg17
functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell
16: 2544-53

Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y (2000). Tor-
mediated induction of autophagy via an Apg1 protein kinase complex. J Cell
Biol 150: 1507-13
Kaissling B, Le Hir M (1994). Characterization and distribution of interstitial cell types
in the renal cortex of rats. Kidney Int 45: 709–20
Kanki T, Klionsky DJ (2008). Mitophagy in yeast occurs through a selective
mechanism. J Biol Chem 283: 32386-93
Kihara A, Noda T, Ishihara N, Ohsumi Y (2001). Two distinct Vps34
phosphatidylinositol 3-kinase complexes function in autophagy and
carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol 152: 519-
30
Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst
P, Sabatini DM (2002). mTOR interacts with raptor to form a nutrient-sensitive
complex that signals to the cell growth machinery. Cell 110: 163-175
Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL (2008). Regulation of TORC1
by Rag GTPases in nutrient response. Nat Cell Biol 10: 935-945.
Kim J, Klionsky DJ (2000). Autophagy, cytoplasm-to-vacuole targeting pathway, and
pexophagy in yeast and mammalian cells. Annu Rev Biochem 69: 303-42
Kim J, Huang WP, Stromhaug PE, Klionsky DJ (2002). Convergence of multiple
autophagy and cytoplasm to vacuole targeting components to a perivacuolar
membrane compartment prior to de novo vesicle formation. J Biol Chem 277:
763-73
Kim JE, Chen J (2004). Regulation of peroxisome proliferator-activated receptor-
gamma activity by mammalian target of rapamycin and amino acids in
adipogenesis. Diabetes 53: 2748-2756
Kim SK, Jung WH, Koo JS (2000). Expression of autophagy-related proteins in
phyllodes tumor. Int J Clin Exp Pathol 6: 2145-56
Kirkinezos IG, Moraes CT (2001). Reactive oxygen species and mitochondrial
diseases. Semin Cell Dev Biol 12: 449-57 Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, Ohsumi M,
Takao T, Noda T, Ohsumi Y. The reversible modification regulates the
membrane-binding state of Apg8/Aut7 essential for autophagy and the
cytoplasm to vacuole targeting pathway. J Cell Biol 151: 263-76

Klerk JB, Dionisi-Vici C, Geb S, Gökcay G, Guffon N, Maier EM, Morava E, Walter
JH, Schwahn B, Wijburg FA, Lindner M, Grünewald S, Baumgartner MR,
Kölker S (2009). Prediction of outcome in isolated methylmalonic acidurias:
combined use of clinical and biochemical parameters. J Inherit Metab Dis 32
(5): 630-9
Klinke R, Silbernagl S (2003). Lehrbuch der Physiologie, Thieme Verlag, 4. Auflage:
287-334
Klionsky DJ (2007). Autophagy: from phenomenology to molecular understanding in
less than a decade. Nat Rev Mol Cell Biol 8: 931-7
Kölker S, Schwab M, Hörster F, Sauer S, Hinz A, Wolf NI, Mayatepek E, Hoffmann
GF, Smeitink JA, Okun JG (2003). Methylmalonic acid, a biochemical hallmark
of methylmalonic acidurias but no inhibitor of mitochondrial respiratory chain. J
Biol Chem 278 (48): 47388-93
Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ,
Kominami E, Momoi T (2007). ER stress (PERK/eIF2alpha phosphorylation)
mediates the polyglutamine-induced LC3 conversion, an essential step for
autophagy formation. Cell Death Differ 14: 230-9
Kowaltowski AJ, Vercesi AE (1999). Mitochondrial damage induced by conditions of
oxidative stress. Free Radic Bio Med 26: 463-71
Kriz W, LeHir M (2005). Pathways to nephron loss starting from glomerular
diseases - insights from animal models. Kidney Int 67:404–419
Krüger T, Benke D, Eitner F, Lang A, Wirtz M, Hamilton-Williams EE, Engel D, Giese
B, Müller-Newen G, Floege J, Kurts C (2004). Identification and functional
characterization of dendritic cells in the healthy murine kidney and in
experimental glomerulonephritis. J Am Soc Nephrol 15: 613–21
Kuma A, Mizushima N, Ishihara N, Ohsumi Y (2002). Formation of the approximately
350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16
oligomerization, is essential for autophagy in yeast. J Cell Biol 277: 18619-25
Kurland IJ, Pilkis SJ (1995): Covalent control of 6-phosphofructo-2-kinase/fructose-
2,6-bisphosphatase: insights into autoregulation of a bifunctional enzyme.
Protein Sci. 4: 1023–37

L
Lakshminarayanan V, Drab-Weiss EA, Roebuck KA (1998). H2O2 and tumor necrosis
factor-alpha induce differential binding of the redox-responsive transcription
factors AP-1 and NF-kappaB to the interleukin-8 promoter in endothelial and
epithelial cells. J Biol Chem 273: 32670-32678
Lam C, Desviat LR, Perez-Cerdá C, Ugarte M, Barshop BA, Cederbaum S (2011).
45-Year-old female with propionic acidemia, renal failure, and premature
ovarian failure; late complications of propionic acidemia? Mol Genet Metab
103: 338-40
Larsen CG, Anderson A, Appella E, Oppenheim JJ, Matsushima K (1989). The
neutrophil-activating protein NAP-1 is also chemotactic for T lymphocytes.
Science 243: 1464–6.
Ledley FD, Lumetta MR, Zoghbi HY, VanTuinen P, Ledbetter SA, Ledbetter DH
(1988). Mapping of human methylmalonyl CoA mutase (MUT) locus on
chromosome 6. Am J Hum Genet 42 (6): 839-46
Ledley FD, Lumetta M, Nguyen PN, Kolhouse JF, Allen RH (1988) Molecular cloning
of L-methylmalonyl-CoA mutase: gene transfer and analysis of mut cell lines.
Proc Nati Acad Sci USA 85 (10): 3518-21
Lenaz G, Bovina C, Formiggini G und Parenti Castelli G (1999). Mitochondria,
oxidative stress, and antioxidant defences. Acta Biochim. Polon. 46: 1-21.
Lenaz G (2001). The mitochondrial production of reactive oxygen species:
mechanisms and implications in human pathology. IUBMB Life 52: 159–64
Levine B, Klionsky DJ (2004). Development by self-digestion: molecular
mechanisms and biological functions of autophagy. Dev Cell 6: 463-77
Levine B, Deretic V (2007). Unveiling the roles of autophagy in innate and adaptive
immunity. Nat Rev Immunol 7: 767-77
Levine B, Kroemer G (2008). Autophagy in the pathogenesis of disease. Cell 132:
27-42
Levine B, Kroemer G (2008). Autophagic cell death: the story of a misnomer. Nat
Rev Mol Cell Biol 9: 1004-10
Li L, Chen Y, Gibson SB (2013). Starvation-induced autophagy is regulated by
mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal
25: 50-65

Li Y, Schwabe RF, DeVries-Seimon T, Yao PM, Gerbod-Giannone MC, Tall AR,
Davis RJ, Flavell R, Brenner DA, Tabas I (2005). Free cholesterol-loaded
macrophages are an abundant source of tumor necrosis factor-alpha and
interleukin-6: model of NF-kappaB- and map kinase-dependent inflammation
in advanced atherosclerosis. J Biol Chem 280: 21763-72.
Li YY, Ishihara S, Aziz MM, Oka A, Kusunoki R, Tada Y, Yuki T, Amano Y, Ansary
MU, Kinoshita Y (2011). Autophagy is required for toll-like receptor-mediated
interleukin-8 production in intestinal epithelial cells. Int J Mol Med 27: 337-44
Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC (2006). Hypoxia-
induced energy stress regulates mRNA translation and cell growth. Mol. Cell
21: 521-531
Liu XD, Ko S, Xu Y, Fattah EA, Xiang Q, Jagannath C, Ishii T, Komatsu M, Eissa NT
(2012). Transient aggregation of ubiquitinated proteins is a cytosolic unfolded
protein response to inflammation and endoplasmic reticulum stress. J Biol
Chem 287: 19687-98
Liu Y (2006). Rapamycin and chronic kidney disease: beyond the inhibition of
inflammation. Kidney Int 69: 1925-7
Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger
W, Jenoe P, Hall MN (2002). Two TOR complexes, only one of which is
rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 10:
457-68
Long X, Ortiz-Vega S, Lin Y, Avruch J (2005). Rheb binding to mammalian target of
rapamycin (mTOR) is regulated by amino acid sufficiency. J Biol Chem 280
(25): 23433-6
Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J (2005). Rheb binds and
regulates the mTOR kinase. Curr Biol 15 (8): 702-13
Lubrano R, Scoppi P, Barsotti P, Travasso E, Scateni S, Cristaldi S, Castello MA
(2001). Kidney transplantation in a girl with methylmalonic acidemia and end
stage renal failure. Pediatr Nephrol 16 (11): 848-51
Lushchak OV, Piroddi M, Galli F, Lushchak VI (2014). Aconitase post-translational
modification as a key in linkage between Krebs cycle, iron homeostasis, redox
signaling, and metabolism of reactive oxygen species. Redox Rep 19: 8-15

M
Madsen KM, Nielsen S, Tisher CC (2008). Proximal tubule. The Kidney, 8th ed.,
edited by Brenner BM, Philadelhia, WB Saunder, 2008, pp 39-50
Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP (2005).
Phosphorylation and functional inactivation of TSC2 by Erk implications for
tuberous sclerosis and cancer pathogenesis. Cell 121 (2): 179-93
Ma XM, Blenis J (2009). Molecular mechanisms of mTOR-mediated translational
control. Nat. Rev. Mol. Cell Biol. 10: 307-318
Mahoney MJ, Hart AC, Steen VD, Rosenberg LE (1975). Methylmalonic acidemia:
biochemical hetrogenity in defects of 5’-deoxyadenosylcobalamin synthesis.
Proc Nati Acad Sci USA 72 (7): 2799-803
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G (2007). Self-eating and self-killing:
crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8: 741-52
Manning BD (2004). Balancing Akt with S6K: implications for both metabolic
diseases and tumorigenesis. J Cell Biol 167 (3): 399-403
Manoli I, Sysol JR, Li L, Houillier P, Garone C, Wang C, Zerfas PM, Cusmano-Ozog
K, Young S, Trivedi NS, Cheng J, Sloan JL, Chandler RJ, Abu-Asab M,
Tsokos M, Elkahloun AG, Rosen S, Enns GM, Berry GT, Hoffmann V,
DiMauro S, Schnermann J, Venditti CP (2013). Targeting proximal tubule
mitochondrial dysfunction attenuates the renal disease of methylmalonic
acidemia. Proc Nati Acad Sci USA 110 (33): 13552-7
Maurer M, Stebut E (2004). Macrophage inflammatory protein-1. Int J Biochem Cell
Biol 36: 1882-6
Mayer C, Zhao J, Yuan X, Grummt I (2004). mTOR-dependent activation of the
transcription factor TIF- IA links rRNA synthesis to nutrient availability. Genes
Dev. 18: 423-434
McCord JM, Fridovich I (1988). Superoxide dismutase: the first twenty years (1968-
1988). Free Radic Biol Me. 5: 363–9.
McLaughlin BA, Nelson D, Silver IA, Erecinska M, Chesselet MF (1998).
Methylmalonate toxicity in primary neuronal cultures. Neuroscience 86 (1):
279-90
Mellman I, Steinman RM (2003). Dendritic cells: Specialized and regulated antigen
processing machines. Cell 106: 255–8

Mezzano SA, Droguett MA, Burgos ME, Ardiles LG, Aros CA, Caorsi I, Egido J
(2000). Overexpression of chemokines, fibrogenic cytokines, and
myofibroblasts in human membranous nephropathy. Kidney Int 57: 147-58
Mizushima N (2007). Autophagy: process and function. Genes Dev 21: 2861-73
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008). Autophagy fights disease
through cellular self-digestion. Nature 451: 1069-75
Mor I, Cheung EC, Vousden KH (2011). Control of Glycolysis through Regulation of
PFK1: Old Friends and Recent Additions. Cold Spring Harb Symp Quant Biol
76: 211-6
Morath MA, Okun JG, Müller IB, Sauer SW, Hörster F, Hoffmann GF, Kölker S
(2008). Neurodegeneration and chronic renal failure in methylmalonic aciduria-
a pathophysiological approach. J Inherit Metab Dis 31 (1): 35-43
Mori K, Ma W, Gething MJ, Sambrook J (1993). A transmembrane protein with a
cdc2+/CDC28-related kinase activity is required for signaling from the ER to
the nucleus. Cell 74 (4): 743-56
Muller CA, Markovic-Lipkovski J, Risler T, Bohle A, Muller GA (1989). Expression of
HLA-DQ, -DR, and -DP antigens in normal kidney and glomerulonephritis.
Kidney Int 35: 116-24
N
Narasimhan P, Sklar R, Murrell M, Swanson RA, Sharp FR (1996). Methylmalonyl-
CoA mutase induction by cerebral ischemia and neurotoxicity of the
mitochondrial toxin methylmalonic acid. J Neuroscience 16 (22): 7336-46
Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, Byfield MP, Backer
JM, Natt F, Bos JL, Zwartkruis FJ, Thomas G (2005). Amino acids mediate
mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-
kinase. Proc Natl Acad Sci USA 102 (40): 14238-43
Noda T, Ohsumi Y (1998). Tor, a phosphatidylinositol kinase homologue, controls
autophagy in yeast. J Biol Chem 273: 3963-6
Noda T, Kim J, Huang WP, Baba M, Tokunaga C, Ohsumi Y, Klionsky DJ (2000).
Apg9p/Cvt7p is an integral membrane protein required for transport vesicle
formation in the Cvt and autophagy pathways. J Cell Biol 148: 465-80
Nulton-Persson AC, Szweda LI (2001). Modulation of mitochondrial function by
hydrogen peroxide. J Biol Chem 276: 23357-61

O
Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T,
Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F,
Imaizumi K (2006). Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol 26: 9220-31
Okun JG, Hörster F, Farkas LM, Feyh P, Hinz A, Sauer S, Hoffmann GF, Unsicker K,
Mayatepek E, Kölker S (2002). Neurodegeneration in methylmalonic aciduria
involves inhibition of complex II and the tricarboxylic acid cycle, and
synergistically acting excitotoxicity. J Biol Chem 277 (17): 1674-80
P
Pahl HL, Baeuerle PA (1995). A novel signal transduction pathway from the
endoplasmic reticulum to the nucleus is mediated by transcription factor NF-
kappa B. EMBO J 14: 2580–8.
Paschen SA und Neupert W (2001). Protein import into mitochondria. IUBMB Life 52: 101-12
Patra KC, Hay N (2014). The pentose phosphate pathway and cancer. Trends
Biochem Sci. 39:347-354
Peters H, Nefedov M, Sarsero J, Pitt J, Fowler KJ, Gazeas S, Kahler SG, Ioannou
PA (2003). A knock-out mouse model for methylmalonic aciduria resulting in
neonatal lethality. J Biol Chem 278 (52): 52909-13
Peters HL, Pitt JJ, Wood LR, Hamilton NJ, Sarsero JP, Buck NE (2012). Mouse
models for methylmalonic aciduria. PLoS One 7 (7): e40609
Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS,
Sabatini DM (2009). DEPTOR is an mTOR inhibitor frequently overexpressed
in multiple myeloma cells and required for their survival. Cell 137: 873-886
Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P (2000). Distinct
classes of phosphatidylinositol 3'-kinases are involved in signaling pathways
that control macroautophagy in HT-29 cells. J Biol Chem 275: 992-8
Pompella A, Visvikis A, Paolicchi A, de Tata V, Casini AF (2003). The changing faces
of glutathione, a cellular protagonist. Biochem Pharmacol 66: 1499-503
Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung
YL, Schulze A (2008). SREBP activity is regulated by mTORC1 and
contributes to Akt-dependent cell growth. Cell Metab. 8: 224-236

Potter CJ, Pedraza LG, Xu T (2002). Akt regulates growth by directly phosphorylating
Tsc2. Nat Cell Biol 4 (9): 658-65
Proost P, Wuyts A, van Damme J (1996). The role of chemokines in inflammation. Int
J Clin Lab Res 26: 211-23
Q
Quigg RJ (2003). Complement and the kidney. J Immunol 171 (7): 3319-24
R
Rada B, Gardina P, Myers TG, Leto Tl (2011). Reactive oxygen species mediate
inflammatory cytokine release and EGFR-dependent mucin secretion in airway
epithelial cells exposed to Pseudomonas pyocyanin. Mucosal Immunol 4: 158-
71
Rahman I, MacNee W (2000). Regulation of redox glutathione levels and gene
transcription in lung inflammation: therapeutic approaches. Free Radic Biol
Med 28: 1405-1420
Ren F, Shu G, Liu G, Liu D, Zhou J, Yuan L, Zhou J (2014). Knockdown of
p62/sequestosome 1 attenuates autophagy and inhibits colorectal cancer cell
growth. Mol Cell Biochem 385: 95-102
Ren M, Guo Q, Guo L, Lenz M, Qian F, Koenen RR, Xu H, Schilling AB, Weber C,
Ye RD, Dinner AR, Tang WJ (2010). Polymerization of MIP-1 chemokine
(CCL3 and CCL4) and clearance of MIP-1 by insulin-degrading enzyme.
EMBO J 29: 3952-66
Renz-Polster, H., Krautzig, S., Braun, J. (Hrsg.): Basislehrbuch Innere Medizin.
Urban & Fischer, München 2011
Ribeiro LR, Della-Pace ID, de Oliveira Ferreira AP, Funck VR, Pinton S, Bobinski F,
de Oliveira CV, da Silva Fiorin F, Duarte MM, Furian AF, Oliveira MS,
Nogueira CW, Dos Santos AR, Royes LF, Fighera MR (2013). Chronic
administration of methylmalonate on young rats alters neuroinflammatory
markers and spatial memory. Immunobiology 218:1175-83
Richter JD, Sonenberg N (2005). Regulation of cap-dependent translation by eIF4E
inhibitory proteins. Nature 433: 477-480

Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L (2004). 6-
phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a
bifunctional enzyme that controls glycolysis. Biochem J. 381: 561-79.
Roake JA, Rao AS, Morris PJ, Larsen CP, Hankins DF, Austyn JM (1995). Dendritic
cell loss from nonlymphoid tissues after systemic administration of
lipopolysaccharide, tumor necrosis factor, and interleukin 1. J Exp Med 181:
2237–47
Rodriguez-Iturbe B, Garcia Garcia G (2010). The role of tubulointerstitial
inflammation in the progression of chronic renal failure. Nephron Clin Pract
116:c81–c88
Rötig A, Goutières F, Niaudet P, Rustin P, Chretien D, Guest G, Mikol J, Gubler MC,
Munnich A (1995). Deletion of mitochondrial DNA in patient with chronic
tubulointerstitial nephritis. J Pediatr 126 (4): 597-601
Rötig A, Lehnert A, Rustin P, Chretien D, Bourgeron T, Niaudet P, Munnich A (1995).
Kidney involvement in mitochondrial disorders. Adv Nephrol Necker Hosp 24:
367-78
Ron D, Walter P (2007). Signal integration in the endoplasmic reticulum unfolded
protein response. Nat Rev Cell Biol 8 (7): 519-29
Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J (2004). Tumor-promoting phorbol
esters and activated Ras inactivate the tuberous sclerosis tumor suppressor
complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci USA 101 (37):
13489-94
Rovin BH, Doe N, Tan LC (1996). Monocyte chemoattractant protein-1 levels in
patients with glomerular disease. Am J Kidney Dis 27: 640-6
Rubinsztein DC, Shpilka T, Elazar Z (2012). Mechanisms of autophagosome
biogenesis. Curr Biol 22: R29–R34
Russel FG, Masereeuw R, van Aubel RA (2002). Molecular aspects of renal anionic
drug transport. Annu Rev Physiol 64: 563-94
S
Sacks SH, Zhou W, Sheerin NS (1996). Complement synthesis in the injured kidney:
does it have a role in immune complex glomerulonephritis? J Am Soc Nephrol
7 (11): 2314-9

Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA,
Sabatini DM (2007). PRAS40 is an insulin-regulated inhibitor of the mTORC1
protein kinase. Mol Cell 25 (6): 903-15
Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini
DM (2008). The Rag GTPases bind raptor and mediate amino acid signaling
to mTORC1. Science 320: 1496-1501
Sarbassov DD, Ali SM, Sabatini DM (2005). Growing roles fort he mTOR pathway.
Curr Opin Cell Biol 176 (6): 596-603
Sarbassov DD, Sabatini DM (2005). Redox regulation oft he nutrient-sensitive raptor-
mTOR pathway and complex. J Biol Chem 280 (47): 39505-9
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005). Phosphorylation and
regulation of Akt/PKB by the rictor-mTOR complex. Science 307 (5712): 1098-
101
Schröder M, Kaufman RJ (2005). The mammalian unfolded protein response. Annu
Rev Biochem 74: 739-89
Sattler T, Mayer A (2000). Cell-free reconstitution of microautophagic vacuole
invagination and vesicle formation. J Cell Biol 151: 529-38
Schieke SM, Phillips D, McCoy JP Jr, Aponte AM, Shen RF, Balaban RS, Finkel T
(2006). The mammalian target of rapamycin (mTOR) pathway regulates
mitochondrial oxygen consumption and oxidative capacity. J Biol Chem 281:
27643-52
Schwab MA, Sauer SW, Okun JG, Nijtmans LG, Rodenburg RJ, van den Heuvel LP,
Dröse S, Brandt U, Hoffmann GF, Ter Laak H, Kölker S, Smeitink JA (2006).
Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role
for endogenous mitochondrial toxins. Biochem J 398 (1): 107-12
Seron D, Cameron JS, Haskard DO (1991). Expression of VCAM-1 in the normal and
diseased kidney. Nephrol Dial Transplant 6: 917-22
Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC (1998). Identification
and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit
kinase, PEK, involved in translational control. Mol Cell Biol 18 (12): 7499-509
Shigeoka AA, Mueller JL, Kambo A, Mathison JC, King AJ, Hall WF, Correia Jda S,
Ulevitch RJ, Hoffman HM, McKay DB (2010). An inflammasome-independent
role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J
Immunol 185: 6277-85

Sies H (1997). Oxidative stress: oxidants and antioxidants. Exp Physiol 82: 291-5
Sniderman LC, Lambert M, Giguère R, Auray-Blais C, Lemieux B, Laframboise R,
Rosenblatt DS, Treacy EP (1999). Outcome of individuals with low-moderate
methylmalonic aciduria detected through a neonatal screening program. J
Pediatr 134 (6): 675-80
Soos TJ, Sims TN, Barisoni L, Lin K, Littman DR, Dustin ML, Nelson PJ (2006).
CX3CR1+ interstitial dendritic cells form a contiguous network throughout the
entire kidney. Kidney Int 70: 591–96
Stack JH, DeWald DB, Takegawa K, Emr SD (1995). Vesicle-mediated protein
transport: regulatory interactions between the Vps15 protein kinase and the
Vps34 PtdIns 3-kinase essential for protein sorting to the vacuole in yeast. J Cell Biol
129: 321-34
Stack JH, Horazdovsky B, Emr SD (1995). Receptor-mediated protein sorting to the
vacuole in yeast: roles for a protein kinase, a lipid kinase and GTP-binding
proteins. Annu Rev Cell Dev Biol 11: 1-33
Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF
(2004). Mitochondrial alpha-ketoglutarate dehydrogenase complex generates
reactive oxygen species. J Neurosci 24: 7779-88
Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y (2001). The pre-
autophagosomal structure organized by concerted functions of APG genes is
essential for autophagosome formation. EMBO J 20: 5971-81
T
Talloczy Z, Jiang W, Virgin HW IV, Leib DA, Scheuner D, Kaufman RJ, Eskelinen E-
L, Levine B (2002). Regulation of starvation- and virus-induced autophagy by
the eIF2 α kinase signaling pathway. Proc Natl Acad Sci USA 99: 190-95
Tang S, Leung JC, Abe K, Chan KW, Chan LY, Chan TM, Lai KN (2003). Albumin
stimulates interleukin-8 expression in proximal tubular epithelial cells in vitro
and in vivo. J Clin Invest 111: 515-27
Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J (2003). Tuberous sclerosis
complex gene products, Tuberin and Hamartin, control mTOR signaling by
acting as a GTPase-activating protein complex toward Rheb. Curr Biol 13 (15):
1259-68

Thorburn A (2008). Studying autophagy's relationship to cell death. Autophagy 4:
391-4
Thorburn A (2008). Apoptosis and autophagy: regulatory connections between two
supposedly different processes. Apoptosis 13: 1-9
Tooze SA (2013). Current views on the spurce of the autophagosome membrane.
Essays Biochem 55: 29-38
Treacy E, Arbour L, Chessex P, Graham G, Kasprzak L, Casey K, Bell L, Mamer O,
Scriver CR (1996). Glutathione deficiency as a complication of methylmalonic
acidemia: response to high doses of ascorbate. J Pediatr 129: 445-8
Tretter L, Adam-Vizi V (2005). Alpha-ketoglutarate dehydrogenase: a target and
generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci 360: 2335-
45
Turrens JF (1997). Superoxide production by the mitochondrial respiratory chain.
Biosci. Report 17: 3–8
Tuttle DL, Lewin AS, Dunn WA Jr (1993). Selective autophagy of peroxisomes in
methylotrophic yeasts. Eur J Cell Biol 60: 283-90
U
Uemura T, Yamamoto M, Kametaka A, Sou YS, Yabashi A, Yamada A, Annoh H,
Kametaka S, Komatsu M, Waguri S (2014). A cluster of thin tubular structures
mediates transformation of the endoplasmic reticulum to autophagic isolation
membrane. Mol Cell Biol 34: 1695-706
Uttenweiler A, Schwarz H, Neumann H, Mayer A (2007). The vacuolar transporter
chaperone (VTC) complex is required for microautophagy. Mol Biol Cell 18:
166-75
V
Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH (2007). Insulin signalling to
mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 9: 316-323
Van Schaftingen E, Hers HG (August 1981). Phosphofructokinase 2: the enzyme
that forms fructose 2,6-bisphosphate from fructose 6-phosphate and ATP.
Biochem Biophys Res Commun 101: 1078–84
Van Schaftingen E, Davies DR, Hers HG (1982). Fructose-2,6-bisphosphatase from
rat liver. Eur J Biochem 124: 143-9

Vasquez-Vivar J, Kalyanaraman B, Kennedy MC (2000). Mitochondrial aconitase is a
source of hydroxyl radical. An electron spin resonance investigation. J Biol
Chem 275: 14064-9
Veenhuis M, Douma A, Harder W, Osumi M (1983). Degradation and turnover of
peroxisomes in the yeast Hansenula polymorpha induced by selective
inactivation of peroxisomal enzymes. Arch Microbiol 134: 193-203
Vlahopoulos S, Boldogh I, Casola A, Brasier AR (1999). Nuclear factor-kappaB-
dependent induction of interleukin-8 gene expression by tumor necrosis factor
alpha: evidence for an antioxidant sensitive activating pathway distinct from
nuclear translocation. Blood 94: 1878-89
Voet D, Voet JG (1995). Biochemistry. Wiley, New York. XVII, 1361 s. pp.
W
Wang CW, Klionsky DJ (2003). The molecular mechanism of autophagy. Mol Med 9:
65-76
Wang L, Harris TE, Roth RA, Lawrence JC Jr (2007). PRAS40 regulates mTORC1
kinase activity by functioning as a direct inhibitor of substrate binding. J Biol
Chem 282 (27): 20036-44
Weidberg H, Shvets E, Elazar Z (2011). Biogenesis and Cargo Selectivity of
Autophagosomes. Annual Review of Biochemistry 80, Kornberg RD, Raetz
CRH, Rothman JE, Thorner JW, eds. (Palo Alto, CA: Annual Reviews): pp.
125–156.
Wilkemeyer MF, Crane AM, Ledley FD (1990). Primary structure and activity of
mouse methylmalonyl-CoA mutase. Biochem J 27 (12): 449-55
Woltman AM, de Fijter JW, Zuidwijk K, Vlug AG, Bajema IM, van der Kooij SW, van
Ham V, van Kooten C (2007). Quantification of dendritic cell subsets in human
renal tissue under normal and pathological conditions. Kidney Int 71: 1001–8
Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau
GD, Kaufman RJ (2007). ATF6alpha optimizes long-term endoplasmic
reticulum function to protect cells from chronic stress. Dev Cell 13 (3): 351-64

X
Xu LL, Warren MK, Rose WL, Gong W, Wang JM (1996). Human recombinant
monocyte chemotactic protein and other C-C chemokines bind and induce
directional migration of dendritic cells in vitro. J Leukoc Biol 60: 365-71
Y
Yalcin A, Telang S, Clem B, Chesney J (2009). Regulation of glucose metabolism by
6-phosphofructo-2-kinase/fructose- 2,6-bisphosphatases in cancer. Exp Mol
Pathol 86: 174–9
Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K
(2007). Transcriptional induction of mammalian ER quality control proteins is
mediated by single or combined action of ATF6alpha and XBP1. Dev Cell 13
(3): 365-76
Yang Z, Klionsky DJ (2010). Mammalian autophagy: core molecular machinery and
signaling regulation. Curr Opin Cell Biol 22: 124-31
Yang Z, Klionsky DJ (2009). An overview of the molecular mechanism of autophagy.
Curr Top Microbiol Immunol 335: 1-32
Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, Brown MS, Goldstein JL
(2000). ER stress induces cleavage of membrane-bound ATF6 by the same
proteases that process SREBPs. Mol Cell 6 (6): 1355-64
Yorimitsu T, Nair U, Yang Z, Klionsky DJ (2006). Endoplasmic reticulum stress
triggers autophagy. J Biol Chem 281: 30299–304
Z
Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman
RJ (2005). Endoplasmic reticulum stress activates cleavage of CREBH to
induce a systemic inflammatory response. Cell 124: 587-99
Zhang K, Kaufman RJ (2008). From endoplasmic-reticulum stress to the
inflammatory response. Nature 454 (7203): 455-62
Zhang K, Kaufman RJ (2008). Identification and characterization of endoplasmic
reticulum stress-induced apoptosis in vivo. Methods Enzymol 442: 395-419
Zschoke J und Hoffmann GF (2004). Diagnose und Therapie erblicher Stoffwechsel-
krankheiten. Vademecum Metabolicum, 3.Auflage, Schattauer

Zsengellér ZK, Aljinovic N, Teot LA, Korson M, Rodig N, Sloan JL, Venditti CP, Berry
GT, Rosen S (2014). Methylmalonic acidemia: A megamitochondrial disorder
affecting the kidney. Pediatr Nephrol. May 28 PMID: 24865477
Zwickler T, Lindner M, Aydin HI, Baumgartner MR, Bodamer OA, Burlina AB, Das
AM, DeKlerk JB, Gökcay G, Grünewald S, Guffon N, Maier EM, Morava E,
Geb S, Schwahn B, Walter JH, Wendel U, Wijburg FA, Müller E, Kölker S,
Hörster F (2008). Diagnostic work-up and management of patients with
isolated methylmalonic acidurias in European metabolic centres. J Inherit
Metab Dis 31: 361-7

7 Anhang
7.1 Abkürzungsverzeichnis
A mitochondriale Aconitase
ABC ATP Bindekassette
AQP Aquaporin
ATF6 activating transcription factor
AdoB12 Adenosylcobalamin, Vitamin B12
ADP Adenosin 5'-Diphosphat
AMPK Adenosin 5'-Monophosphat Kinase
ATG13 autophagy related gene 13
aTIN akute tubulointerstitielle Nephritis
ATP Adenosin 5'-Triphosphat
BSA Albumin aus Rinderserum
Cbl Cobalamin
CCL Chemokin (C-C Motiv) Ligand
CD Cluster of Differentiation
CoA Coenzym A
CS Zitratsynthase
cTIN chronische tubulointerstitielle Nephritis
Da Dalton
DBH Decylubihydrochinon
DBQ 3-Dimethoxy-5-Methyl-6-Decyl–1,4-Benzochinon
DCF Dichloro-Dihydrofluorescein
DCPIP Dichlorophenolindophenol
DQA 2-n-Decylquinazolin-4-yl-Amin 2
DMEM Dulbeccos’s modified Eagle medium
DTNB 5',5"-Dithiobis-(2-Nitrobenzoat)
E Enolase
EDTA Ethylendiamin-N, N, N´, N´-Tetraessigsäure
ER Endoplasmatisches Retikulum
ERK extracellular signal regulated kinase

Fum Fumarase
FAD Flavinadenindinukleotid, oxidiert
FADH2 Flavinadenindinukleotid, reduziert
FKS Fötales Kälberserum
g Erdbeschleunigung
GAPDH Glutaraldehydphosphat-Dehydrogenase
MS Malonsäure
MS/MS Tandem-Massenspektrometrie
GCDH Glutaryl-CoA-Dehydrogenase
GDP Guanosin-Diphosphat
GFR glomeruläre Filtrationsrate
GSSG Glutathion-Disulfid
GSH Glutathion, reduziert
GTP Guanosin-Triphosphat
H2DCFDA 2’,7’-Dichloro-Dihydrofluoresceindiacetat
HK Hexokinase
IDH Isozitrat-Dehydrogenase
IKK IκBα-Kinase-Komplex
IL Interleukin
Ile Isoleucin
IRE1 inositol requiring enzyme 1
IRS1 Insulinrezeptorsubstrat 1
Kb Kilobasenpaar
KCN Kaliumcyanid
KPi K2HPO4
KRB Krebs-Ringer Puffern
LB Luria Broth
LC3 Microtubule light chain protein 3
LDH Laktatdehydrogenase
MCM Methylmalonyl-CoA Mutase
MCP Monocyte chemotactic protein
MHC Major Histocompatibility Complex
MDH Malat-Dehydrogenase
MMA Methylmalonazidurie

MMS Methylmalonsäure
mTOR mammalian target of rapamycin
mTORC1 mammalian target of rapamycin complex 1
n Anzahl
NaCN Natriumzyanid
NAD β-Nicotinamid Adenin Dinucleotid, oxidiert
NADH β-Nicotinamid Adenin Dinucleotid, reduziert
NADP Nikotinamidadenindinukleotidphosphat, oxidiert
NADPH Nikotinamidadenindinukleotidphosphat, reduziert
OGDHc 2-Oxoglutarat-Dehydrogenase-Komplex
p Wahrscheinlichkeit, dass die Nullhypothese richtig ist
PA Propionazidurie
PS Propionsäure
PBS Phosphat-gepufferte Salzlösung
PCR Polymerase Chain Reaction
PDHc Pyruvat-Dehydrogenase-Komplex
PE Phosphatidylenositol
PEP Phosphoenolpyruvat
PERK PKR-like endoplasmic reticulum kinase
PFK Phosphofruktokinase
PGM Phosphoglyzerat-Mutase
PK Pyruvatkinase
PRAS40 proline-rich AKT substrate 40 kDa
PS Propionsäure
(h)PTEC (humane) proximale Tubulusepithelzellen
PVDF Polyvinyliden-Fluorid
R Bestimmtheitsmaß
RANTES Regulated on activation, normal T cell expressed and secreted
Raptor Regulatory associated protein of mTOR
Rheb Ras-related homolog enriched in brain
RIPA (Puffer) Radio Immuno Protein Assay Puffer
ROS reaktive Sauerstoffspezies
RSK1 p90 ribosomal S6 Kinase 1
SDS Sodium Dodecylsulfat

SLC solute carrier
TNF Tumornekrosefaktor
TPI Triosephosphat-Isomerase
Tris Trishydroxymethyaminomethan
TSC tuberous sclerosis complex
U/mg Units pro mg [μmol min-1 mg-1], spezifische Aktivität
ULK1 unc-51-like kinase 1
upm Umdrehungen pro Minute
UPR unfolded protein response

7.2 Tabellarische Auflistung von im Ergebnisteil nur graphisch dargestellter Messwerte
Messgröße Maßeinheit Kontrolle MMA PA MMS im Medium Konzentration mmol/L/mg 2,79 ± 0,63 6,53 ± 1,84 n.a. Mt. Sauerstoffverbrauch O2-Änderung/Zeit DMEM ΔO2 (sensitiv)/mg 0,50 ± 0,06 0,14 ± 0,20 0,13 ± 0,14 KRB+Glukose ΔO2 (sensitiv)/mg 0,58 ± 0,22 0,26 ± 0,15 0,40 ± 0,52 KRB+Pyruvat ΔO2 (sensitiv)/mg 2,54 ± 2,21 0,98 ± 0,75 2,63 ± 1,17 Glykolyse Enzymaktivität HK mOD/min/mg 0,66 ± 0,14 0,56 ± 0,07 0,54 ± 0,03 PFK mOD/min/mg 2,57 ± 0,73 0,94 ± 0,61 2,59 ± 0,59 TPI mOD/min/mg 2,39 ± 0,29 3,17 ± 0,29 2,24 ± 0,40 PGM mOD/min/mg 12,35 ± 0,93 12,69 ± 0,76 8,13 ± 3,97 GAPDH mOD/min/mg 6,53 ± 0,49 7,47 ± 0,38 5,39 ± 0,40 E mOD/min/mg 13,8 ± 1,37 15,49 ± 1,09 9,56 ± 4,61 PK HA mOD/min/mg 1,51 ± 0,28 1,10 ± 0,37 1,10 ± 0,12 PK LA mOD/min/mg 11,33 ± 1,14 11,10 ± 0,79 10,74 ± 1,94 LDH mOD/min/mg 8,20 ± 0,58 11,37 ± 3,28 7,17 ± 4,12 PDHc 14C-Einbau/mg 24,53 ± 15,10 14,60 ± 4,72 18,61 ± 5,20 Zitrat-Zyklus Enzymaktivität CS mOD/min/mg 33,83 ± 16,88 20,91 ± 14,52 37,82 ± 15,07 mA mOD/min/mg 7,48 ± 2,02 5,22 ± 1,55 5,98 ± 0,94 IDH mOD/min/mg 28,13 ± 7,21 29,68 ± 6,11 36,08 ± 9,32 OGDHc 14C-Einbau/mg 38,49 ± 18,34 21,25 ± 11,00 22,37 ± 11,56 Fum mOD/min/mg 10,43 ± 5,96 11,97 ± 3,03 8,40 ± 4,84 MDH mOD/min/mg 38,51 ± 13,10 44,15 ± 12,61 36,58 ± 6,94 Atmungskette Enzymaktivität Komplex I mOD/min/mg 4,50 ± 1,52 4,37 ± 0,86 2,89 ± 0,84 Komplex II mOD/min/mg 0,51 ± 0,14 0,51 ± 0,11 0,41 ± 0,08 Komplex III mOD/min/mg 1,40 ± 0,22 1,48 ± 0,41 1,61 ± 0,43 Komplex IV mOD/min/mg 1,67 ± 0,49 1,10 ± 0,47 1,32 ± 0,34 Komplex V mOD/min/mg 13,30 ± 3,26 16,01 ± 4,07 12,17 ± 4,21 Glutathion-Gehalt Gesamt GSH nmol/mg 0,68 ± 0,33 0,57 ± 0,16 n.a. GSSG nmol/mg 0,36 ± 0,14 0,60 ± 0,14 n.a. Freies GSH nmol/mg 0,32 ± 0,29 0,04 ± 0,05 n.a. ROS-Produktion Intensität Williams E Intensität/mg 5,13 ± 0,40 10,15 ± 4,63 7,13 ± 1,75 KRB Intensität/mg 13,87 ± 2,21 39,38 ± 21,27 15,83 ± 9,04 KRB+Glukose Intensität/mg 0,57 ± 0,12 1,97 ± 1,07 1,07 ± 0,40 KRB+Pyruvat Intensität/mg 2,37 ± 0,71 8,65 ± 4,41 5,10 ± 1,35 KRB+Glutamin Intensität/mg 0,50 ± 0,10 1,40 ± 0,70 0,70 ± 0,26 KRB+PS Intensität/mg 7,20 ± 1,04 22,62 ± 15,78 6,17 ± 4,61 KRB+MMS Intensität/mg 13,87 ± 2,56 38,68 ± 21,93 18,93 ± 8,44 KRB+Ile Intensität/mg 10,37 ± 2,37 29,35 ± 16,40 12,60 ± 5,41

Messgröße Messeinheit Kontrolle MMA PA Autophagie Expressionsstärke pRAS40/PRAS40 20%FKS
Intensität/mg 1,55 ± 0,39 0,88 ± 0,36 0,43 ± 0,07
pRAS40/PRAS40 1%FKS Intensität/mg 0,39 ± 0,09 0,58 ± 0,13 0,27 ± 0,02 pRAS40/PRAS40 0,5%FKS
Intensität/mg 0,61 ± 0,18 1,12 ± 0,68 0,34 ± 0,12
pmTOR/mTOR 20%FKS Intensität/mg 0,12 ± 0,05 0,14 ± 0,03 0,18 ± 0,07 pmTOR/mTOR 1%FKS Intensität/mg 2,16 ± 0,75 2,84 ± 0,28 2,05 ± 0,46 pmTOR/mTOR 0,5%FKS Intensität/mg 0,76 ± 0,37 2,26 ± 1,13 0,63 ± 0,22 LC3-II 20% FKS Intensität/mg 1,30 ± 0,48 1,95 ± 0,52 1,26 ± 0,23 LC3-II 1% FKS Intensität/mg 1,60 ± 0,36 2,20 ± 0,85 1,75 ± 0,42 LC3-II 0,5% FKS Intensität/mg 4,09 ± 0,80 5,23 ± 1,54 4,43 ± 0,56 P62 20%FKS Intensität/mg 1,10 ± 0,28 2,24 ± 0,47 1,92 ± 0,13 P62 1%FKS Intensität/mg 2,94 ± 0,33 1,98 ± 0,66 2,09 ± 0,09 P62 0,5%FKS Intensität/mg 2,33 ± 0,27 1,81 ± 0,42 2,86 ± 0,36 pIRE1 20%FKS Intensität/mg 4,34 ± 1,60 6,23 ± 0,04 5,07 ± 0,55 pIRE1 1%FKS Intensität/mg 2,05 ± 0,44 1,94 ± 0,24 2,36 ± 0,24 pIRE1 0,5%FKS Intensität/mg 2,87 ± 0,67 2,34 ± 0,76 3,47 ± 0,60 AO Ratio Intensität FL3/FL1/mg 1,76 ± 0,35 2,46 ± 0,66 1,90 ± 0,36 Soluble Receptor Array Expressionsstärke Ctrl_1; Ctrl_7 MMA_1; MMA_2 IL-8 Intensität/mg 1194; 3886 9865 n.a. IL-1RII Intensität/mg 220; 23 595; 940 n.a. IL-15Ra Intensität/mg 189; 0 257; 189 n.a. VCAM-1 Intensität/mg 37; 0 2610; 4666 n.a. Immunrezeptoren Anzahl Moleküle CD46 Moleküle/Zelle 59684 ± 8792 58249 ± 18124 n.a. CD55 Moleküle/Zelle 27834 ± 11405 23046 ± 9139 n.a. CD59 Moleküle/Zelle 519648 ± 110158 530239 ± 139626 n.a. CD88 Moleküle/Zelle 16474 ± 1161 13984 ± 10822 n.a. Expression IL-8 Intensität IL-8 qualitativ Intensität/mg 4,53 ± 2,44 6,25 ± 2,26 4,09 ± 2,04 IL-8 quantitativ Konzentration Apikal pg/mg 1521 ± 410 2870 ± 1092 1854 ± 624 Basolateral pg/mg 2757 ± 382 4321 ± 1616 2098 ± 1295 Intrazellulär pg/mg 459 ± 302 1294 ± 692 828 ± 612

7.3 Western Blots der im Ergebnisteil graphisch dargestellten Expression einzelner Proteine


