inhibition and reversal of nickel-induced transformation by the histone deacetylase inhibitor...
TRANSCRIPT

Inhibition and reversal of nickel-induced transformation by thehistone deacetylase inhibitor trichostatin A
Qunwei Zhang, Konstantin Salnikow, Thomas Kluz, Lung Chi Chen,Wei Cheng Su, and Max Costa*
Department of Environmental Medicine, NYU School of Medicine, 57 Old Forge Road, Tuxedo, NY 10987, USA
Received 18 April 2003; accepted 11 June 2003
Abstract
The carcinogenic process initiated by nongenotoxic carcinogens involves modulation of gene expression. Nickel compounds have lowmutagenic activity, but are highly carcinogenic. In vitro both mouse and human cells can be efficiently transformed by soluble and insolublenickel compounds to anchorage-independent growth. Because previous studies have shown that carcinogenic nickel compounds silencegenes by inhibiting histone acetylation and enhancing DNA methylation, we investigated the effect of enhancing histone acetylation on celltransformation. The exposure of nickel-transformed cells to the histone deacetylase inhibitor trichostatin A (TSA) resulted in the appearanceof significant number of revertants measured by their inability to grow in soft agar. Using the Affymetrix GeneChip we found that the levelof expression of a significant number of genes was changed (suppressed or upregulated) in nickel-transformed clones but returned to anormal level in revertants obtained following TSA treatment. Moreover, we found that treatment of cells with TSA inhibited the ability ofnickel to transform mouse PW cells to anchorage-independent growth. Treatment with TSA also inhibited the ability of nickel to transformhuman HOS cells, although to a lesser extent. In contrast, treatment with TSA was not able to revert established cancer cell lines as readilyas the nickel-transformed cells. These data indicated that modulation of gene expression is important for nickel-induced transformation.© 2003 Elsevier Inc. All rights reserved.
Keywords: Nickel; Trichostatin A; Gene expression; GeneChip; Soft agar; Cell transformation
Introduction
The low mutagenic and high carcinogenic activity ofnickel compounds suggests that epigenetic events might beinvolved in nickel carcinogenesis. Previous studies havedemonstrated that carcinogenic nickel (Ni) compounds werecapable of silencing genes and such silencing was impli-cated as an important event in the Ni-induced transforma-tion process (Broday et al., 1999; Klein et al., 1991; Lee etal., 1995; Lin et al., 1994; Salnikow et al., 1994; Sutherlandet al., 2001). Numerous studies have demonstrated thattransformation and carcinogenesis involve gene silencingpresumably by DNA methylation in the promoters of tumorsuppressor genes (Klein et al., 1991; Baylin and Herman,2001; Jones and Takai, 2001; Siegfried et al., 1999; Stirza-
ker et al., 1997). The mechanism for gene silencing causedby carcinogenic Ni compounds has been proposed to in-volve an increase in chromatin condensation which triggersde novo DNA methylation (Lee et al., 1995). A more de-tailed analysis of the molecular mechanisms of Ni-inducedgene silencing has implicated the ability of Ni to inhibithistone H4 acetylation (Broday et al., 2000), particularly inlysines positioned near a metal-anchoring histidine on theN-terminal tail of histone H4 (Zoroddu et al., 2001). Usingmodel peptides of the H4 N-terminal tail we have previ-ously shown that Ni can bind to this histidine and form avariety of complexes (Zoroddu et al., 2001). These com-plexes can structurally interfere with the ability of the his-tone acetylase enzyme to acetylate lysines of histone H4(Zoroddu et al., 2001). Ni can also bind to and changeconformation of other histones (Bal et al., 1998) indicatingpossible involvement of nickel in chromatin remodeling.Additionally, the ability of Ni to substitute for magnesium
* Corresponding author. Fax: �1-845-351-2118.E-mail address: [email protected] (M. Costa).
R
Available online at www.sciencedirect.com
Toxicology and Applied Pharmacology 192 (2003) 201–211 www.elsevier.com/locate/taap
0041-008X/$ – see front matter © 2003 Elsevier Inc. All rights reserved.doi:10.1016/S0041-008X(03)00280-1

and to increase chromatin condensation may contribute tothe gene silencing observed in Ni-exposed cells (Lee et al.,1995; Zoroddu et al., 2001).
Although numerous studies have implicated the role ofgene silencing in carcinogenesis (Klein et al., 1991; Baylinand Herman, 2001; Jones and Takai, 2001; Siegfried et al.,1999; Stirzaker et al., 1997), there are few studies that havedirectly addressed the role of histone acetylation in thecarcinogenesis process and also the role of histone deacety-lase inhibitors in reverting the transformed phenotype (Ar-cher and Hodin, 1999). Here we demonstrate that nickel cantransform mouse PW cells to an anchorage-independentgrowth and TSA can revert the transformed phenotype ofthese cells. Using the Affymetrix GeneChip, we show thatthe level of gene expression for over 1000 genes was re-verted to the level observed in the parental cell line follow-ing TSA exposure of nickel-transformed cells. Additionally,simultaneous exposure of cells to the histone deacetylaseinhibitor TSA and nickel resulted in the inhibition of Ni-induced transformation measured as anchorage-independentgrowth. In contrast, exposure to TSA did not revert theability of established human cancer cells to grow in softagar.
Materials and methods
Chemicals. Nickel subsulfide (Ni2S3) particles (INCO Ltd.,Toronto, Ontario) were ground and filtered to �5 �m sizeas described by Costa (1980). Nickel chloride (NiCl2) hexa-hydrate was purchased from Alfa Chemical (Ward Hill,MA). TSA was purchased from Sigma Chemical Company(St. Louis, MO).
Cell culture. The human osteoblastic cell line (HOS) TE 85was obtained from American Type Culture Collection(Rockville, MD) and grown in plastic flasks in minimalessential medium (�-MEM) containing 10% fetal bovineserum, 10 U/ml penicillin, and 10 �g/ml streptomycin.Mouse embryo fibroblasts, PW, were grown in plastic flasksin Dulbecco’s modified Eagle’s minimal essential medium(DMEM) containing 10% fetal bovine serum, 10 U/ml pen-icillin, and 10 �g/ml streptomycin. Human lung cancerA549 or H460 cells were grown in F12 K nutrient mixture(Kaighn’s modification) or in DMEM containing 10% fetalbovine serum, 10 U/ml penicillin, and 10 �g/ml streptomy-cin. Cells were incubated at 37°C in a humidified atmo-sphere of 5% CO2 and 95% air.
Cell transformation protocols. One million HOS or PWcells were seeded into 75-cm2 tissue culture flasks andincubated overnight. Cells were then exposed to 0, 0.15, or0.30 �g/cm3 Ni2S3 or 0, 1, or 2 mM NiCl2 for 24 h. Afterthe exposure nickel containing medium was removed andcells were rinsed with the sterile saline A three times. Thenfresh medium was added and the cells were allowed to grow
for 2 days. The cultures were split and subjected to anotherround of treatment. This kind of treatment was repeated ninetimes. For the TSA exposure, 5 ng/ml or 25 ng/ml TSAwere added to the cells 4 h before each exposure. After theexposure cells were then plated in soft agar to determine thelevel of anchorage-independent growth. Cells (5 � 104)were plated in 5 ml 0.33% agar in complete media overlaidonto a solid layer of 0.5% agar in complete media. After 4weeks of growth the colonies were stained with INT solu-tion (Sigma), and counted using Kodak electrophoresis doc-umentation and analysis system (EDAS 290). In some in-stances selected individual agar colonies were isolated andexpanded into mass culture for further analysis of the trans-formed phenotype and gene expression.
Cytotoxicity of TSA on cells. To assess the cytotoxicity ofTSA on parental PW cells and Ni-transformed clones, cellswere seeded in 60-mm dishes (300 cells/dish) and allowedto attach overnight. After cells were exposed to 0, 1, 5, 10,25, 40, 50, 60, 80, and 100 ng/ml TSA for 24 or 72 h, TSAwas removed and cells were rinsed with saline A threetimes. Then cells were maintained in fresh DMEM. Mediumwas changed every 3–4 days. After 3 weeks the colonieswere stained and scored.
Effect of TSA on soft agar growth. One million of theappropriate cell type (Ni-transformed cells, A549 cells, orH460 cells) were seeded into 75-cm2 tissue culture flasksand allowed to attach overnight. Cells were exposed to 0, 5,or 25 ng/ml TSA for 24 h. Then the TSA-containing me-dium was removed and cells were rinsed three times withsterile saline A. Fresh medium was added and the cells wereallowed to grow to �80% confluence. The cultures weresplit again and cells were seeded at a density of one millioncells per flask. Then cells were treated with TSA for asecond time. This procedure was repeated three times. Aftera third round of TSA exposure the actively growing cultureswere tested for the anchorage-independent growth as de-scribed above.
Isolation of revertants. Nickel-transformed clones grown insoft agar were cultured in complete medium and were ex-posed to TSA for three rounds as described above. Threehundred cells from each nickel-transformed clone wereplated in 100-mm dishes to let single cells form clones.Approximately 10 clones were isolated from each dish andgrew in the flask. When cells grew to �70% confluence,part of them were immediately frozen and part of them wereused to test its ability to grow in soft agar as describedabove. A few clones with the lowest clonability in soft agarwere considered as revertants.
Analysis of cell growth rate. Cells 5 � 103 were plated intoeach well of six-well plates. Cell number was counted everyday for 5 days using Coulter Counter ZM (Coulter Elec-tronics Ltd, England).
202 Q. Zhang et al. / Toxicology and Applied Pharmacology 192 (2003) 201–211
RNA isolation for RT–PCR or GeneChip analysis. TotalRNA was isolated from parental PW cells, one nickel-transformed clone, and a revertant obtained from thatnickel-transformed clone. This RNA was used for RT–PCRor to prepare polyA mRNA. For GeneChip analysis double-stranded cDNA was synthesized from polyA mRNA with acDNA synthesis kit (Superscript cDNA Synthesis System;Invitrogen, Carlsbad, CA) by using an oligo(dT)24 primerwith a T7 RNA polymerase promoter site added to its 3�end. The isolated cDNA was used for in vitro transcription(T7 Megascript System; Ambion, Austin, TX) in the pres-ence of biotin-11-CTP and biotin-16-UTP (Enzo Diagnos-tics). A total of 25–50 �g of the cRNA product in buffer (40mM Tris–acetate, pH 8.1, 100 mM potassium acetate, 30mM magnesium acetate) was fragmented at 94°C for 35min. The prepared probe was used for hybridization as ahybridization mix with herring sperm DNA (0.1 mg/ml;Sigma). The test 3 chip served for evaluation of probequality as directed by the manufacturer (Affymetrix, SantaClara, CA).
Aliquots of the cRNA hybridization mixtures (10 �gcRNA in 200 �l hybridization mix) were hybridized to amouse GeneChip array and the chips were washed andscanned (Hewlett Packard, GeneArray scanner G2500A)according to procedures developed by the manufacturer(Affymetrix). The data were analyzed using GeneSpring5.01 software (Silicon Genetics, Redwood City, CA).
Analyses of gene expression level by RT–PCR. The cDNAwas synthesized from 1 �g of total RNA using SuperscriptII kit (Invitrogen) and was amplified by PCR as follows:94°C for 30 s, annealing for 30 s (pair of primers andannealing temperature for each gene was shown below), andextension at 72°C for 30 s. The final extension was 10 minat 72°C. The following primers and annealing conditionswere used:
GAPDH: forward primer 5�-CGGAGTCAACGGATTTG-GTCGTAT-3�, reverse primer 5� AGCCTTCTCCATGGTG-GTGAAGAC-3�, annealing temperature 56°C, 23 cycles;repetin: forward primer 5�-GCTTCCAGCTCATTCTCCA-AC-3�, reverse primer 5�-CATCCCAGGAGACACCCA-TTC-3�, annealing temperature 54°C, 28 cycles. CCR-6: for-ward primer 5�-GAATGAATTCCACAGAGTCCT-3�, re-verse primer 5�-AGATGATGATGGAGATGAACC-3�, an-nealing temperature 54°C, 30 cycles; basic domain/leucinezipper transcription factor: forward primer 5�-CCTGTACTG-GATGGCGAGCAA-3�, reverse primer 5�-GACCCGCCAG-GACTCACAGAA-3�, annealing temperature 62°C, 30 cycles;embigin: forward primer 5�-CCCACAGATCCAACTTTTA-CA-3�, reverse primer 5�-TGAGGGCATCTTTGTCTTTTA-3�, annealing temperature 54°C, 27 cycles; and protein kinaseC zeta: forward primer 5�-AGGGACGAAGTGCTCAT-CATT-3�, reverse primer 5�-GGAAGTTTTCTCTGCCT-CTGC-3�, annealing temperature 57°C, 25 cycles. PCR prod-ucts were resolved in 1% agarose gel. The images were ob-
tained with the Kodak electrophoresis documentation and anal-ysis system (EDAS 290).
Statistical analysis. Statistical significance of data presentedas mean � SE was analyzed by one-way analysis of vari-ance (ANOVA).
Results
Nickel-induced transformation changes gene expression
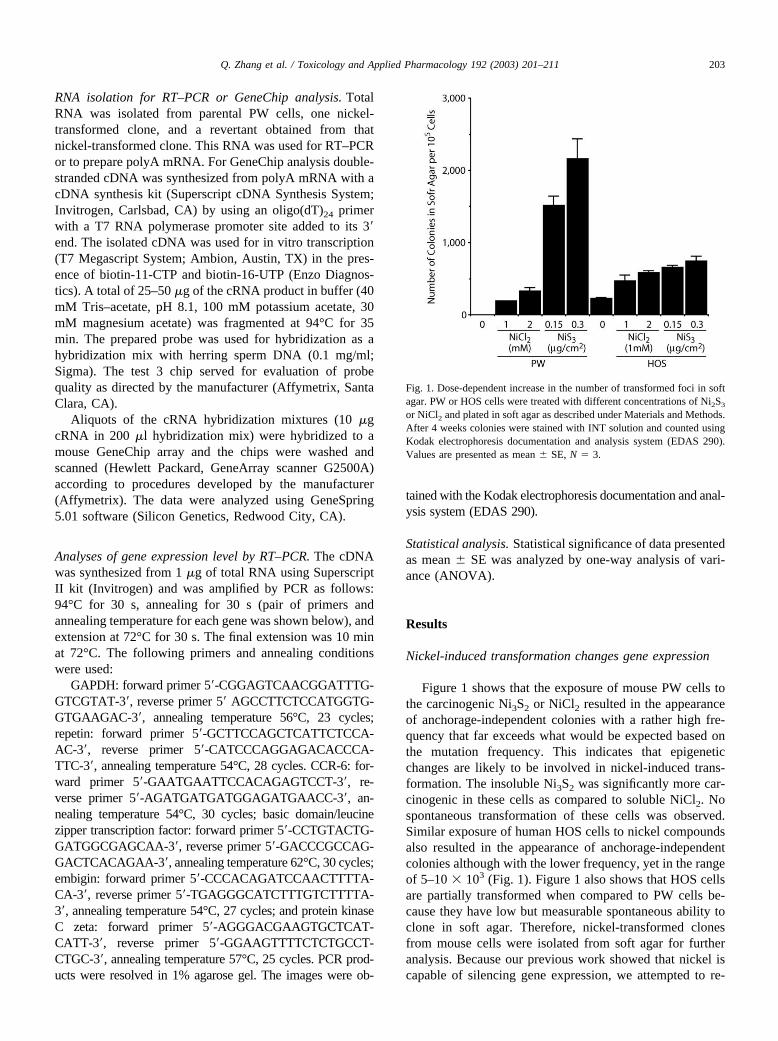
Figure 1 shows that the exposure of mouse PW cells tothe carcinogenic Ni3S2 or NiCl2 resulted in the appearanceof anchorage-independent colonies with a rather high fre-quency that far exceeds what would be expected based onthe mutation frequency. This indicates that epigeneticchanges are likely to be involved in nickel-induced trans-formation. The insoluble Ni3S2 was significantly more car-cinogenic in these cells as compared to soluble NiCl2. Nospontaneous transformation of these cells was observed.Similar exposure of human HOS cells to nickel compoundsalso resulted in the appearance of anchorage-independentcolonies although with the lower frequency, yet in the rangeof 5–10 � 103 (Fig. 1). Figure 1 also shows that HOS cellsare partially transformed when compared to PW cells be-cause they have low but measurable spontaneous ability toclone in soft agar. Therefore, nickel-transformed clonesfrom mouse cells were isolated from soft agar for furtheranalysis. Because our previous work showed that nickel iscapable of silencing gene expression, we attempted to re-
Fig. 1. Dose-dependent increase in the number of transformed foci in softagar. PW or HOS cells were treated with different concentrations of Ni2S3
or NiCl2 and plated in soft agar as described under Materials and Methods.After 4 weeks colonies were stained with INT solution and counted usingKodak electrophoresis documentation and analysis system (EDAS 290).Values are presented as mean � SE, N � 3.
203Q. Zhang et al. / Toxicology and Applied Pharmacology 192 (2003) 201–211
activate silenced genes using the histone deacetylase inhib-itor TSA. We suggested that this exposure could revertnickel-transformed cells to a normal phenotype. Fivenickel-transformed clones originated from mouse PW cellswere used for this experiment. Indeed three rounds of ex-posure to a nontoxic concentration of 5 ng/ml of TSAsignificantly decreased the ability of transformed coloniesobtained following exposure to Ni3S2 to grow in soft agar.And three rounds of exposure to 25 ng/ml of TSA furthersuppressed the ability of these clones to grow in soft agar(Table 1).
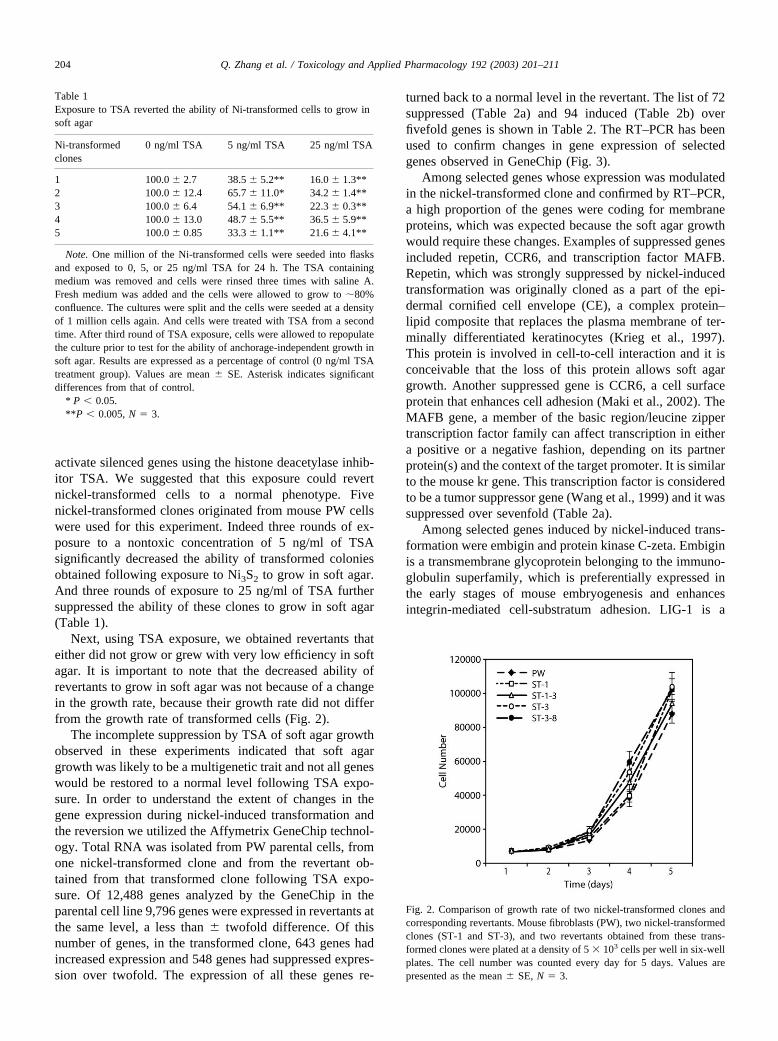
Next, using TSA exposure, we obtained revertants thateither did not grow or grew with very low efficiency in softagar. It is important to note that the decreased ability ofrevertants to grow in soft agar was not because of a changein the growth rate, because their growth rate did not differfrom the growth rate of transformed cells (Fig. 2).
The incomplete suppression by TSA of soft agar growthobserved in these experiments indicated that soft agargrowth was likely to be a multigenetic trait and not all geneswould be restored to a normal level following TSA expo-sure. In order to understand the extent of changes in thegene expression during nickel-induced transformation andthe reversion we utilized the Affymetrix GeneChip technol-ogy. Total RNA was isolated from PW parental cells, fromone nickel-transformed clone and from the revertant ob-tained from that transformed clone following TSA expo-sure. Of 12,488 genes analyzed by the GeneChip in theparental cell line 9,796 genes were expressed in revertants atthe same level, a less than � twofold difference. Of thisnumber of genes, in the transformed clone, 643 genes hadincreased expression and 548 genes had suppressed expres-sion over twofold. The expression of all these genes re-
turned back to a normal level in the revertant. The list of 72suppressed (Table 2a) and 94 induced (Table 2b) overfivefold genes is shown in Table 2. The RT–PCR has beenused to confirm changes in gene expression of selectedgenes observed in GeneChip (Fig. 3).
Among selected genes whose expression was modulatedin the nickel-transformed clone and confirmed by RT–PCR,a high proportion of the genes were coding for membraneproteins, which was expected because the soft agar growthwould require these changes. Examples of suppressed genesincluded repetin, CCR6, and transcription factor MAFB.Repetin, which was strongly suppressed by nickel-inducedtransformation was originally cloned as a part of the epi-dermal cornified cell envelope (CE), a complex protein–lipid composite that replaces the plasma membrane of ter-minally differentiated keratinocytes (Krieg et al., 1997).This protein is involved in cell-to-cell interaction and it isconceivable that the loss of this protein allows soft agargrowth. Another suppressed gene is CCR6, a cell surfaceprotein that enhances cell adhesion (Maki et al., 2002). TheMAFB gene, a member of the basic region/leucine zippertranscription factor family can affect transcription in eithera positive or a negative fashion, depending on its partnerprotein(s) and the context of the target promoter. It is similarto the mouse kr gene. This transcription factor is consideredto be a tumor suppressor gene (Wang et al., 1999) and it wassuppressed over sevenfold (Table 2a).
Among selected genes induced by nickel-induced trans-formation were embigin and protein kinase C-zeta. Embiginis a transmembrane glycoprotein belonging to the immuno-globulin superfamily, which is preferentially expressed inthe early stages of mouse embryogenesis and enhancesintegrin-mediated cell-substratum adhesion. LIG-1 is a
Fig. 2. Comparison of growth rate of two nickel-transformed clones andcorresponding revertants. Mouse fibroblasts (PW), two nickel-transformedclones (ST-1 and ST-3), and two revertants obtained from these trans-formed clones were plated at a density of 5 � 103 cells per well in six-wellplates. The cell number was counted every day for 5 days. Values arepresented as the mean � SE, N � 3.
Table 1Exposure to TSA reverted the ability of Ni-transformed cells to grow insoft agar
Ni-transformedclones
0 ng/ml TSA 5 ng/ml TSA 25 ng/ml TSA
1 100.0 � 2.7 38.5 � 5.2** 16.0 � 1.3**2 100.0 � 12.4 65.7 � 11.0* 34.2 � 1.4**3 100.0 � 6.4 54.1 � 6.9** 22.3 � 0.3**4 100.0 � 13.0 48.7 � 5.5** 36.5 � 5.9**5 100.0 � 0.85 33.3 � 1.1** 21.6 � 4.1**
Note. One million of the Ni-transformed cells were seeded into flasksand exposed to 0, 5, or 25 ng/ml TSA for 24 h. The TSA containingmedium was removed and cells were rinsed three times with saline A.Fresh medium was added and the cells were allowed to grow to �80%confluence. The cultures were split and the cells were seeded at a densityof 1 million cells again. And cells were treated with TSA from a secondtime. After third round of TSA exposure, cells were allowed to repopulatethe culture prior to test for the ability of anchorage-independent growth insoft agar. Results are expressed as a percentage of control (0 ng/ml TSAtreatment group). Values are mean � SE. Asterisk indicates significantdifferences from that of control.
* P � 0.05.**P � 0.005, N � 3.
204 Q. Zhang et al. / Toxicology and Applied Pharmacology 192 (2003) 201–211
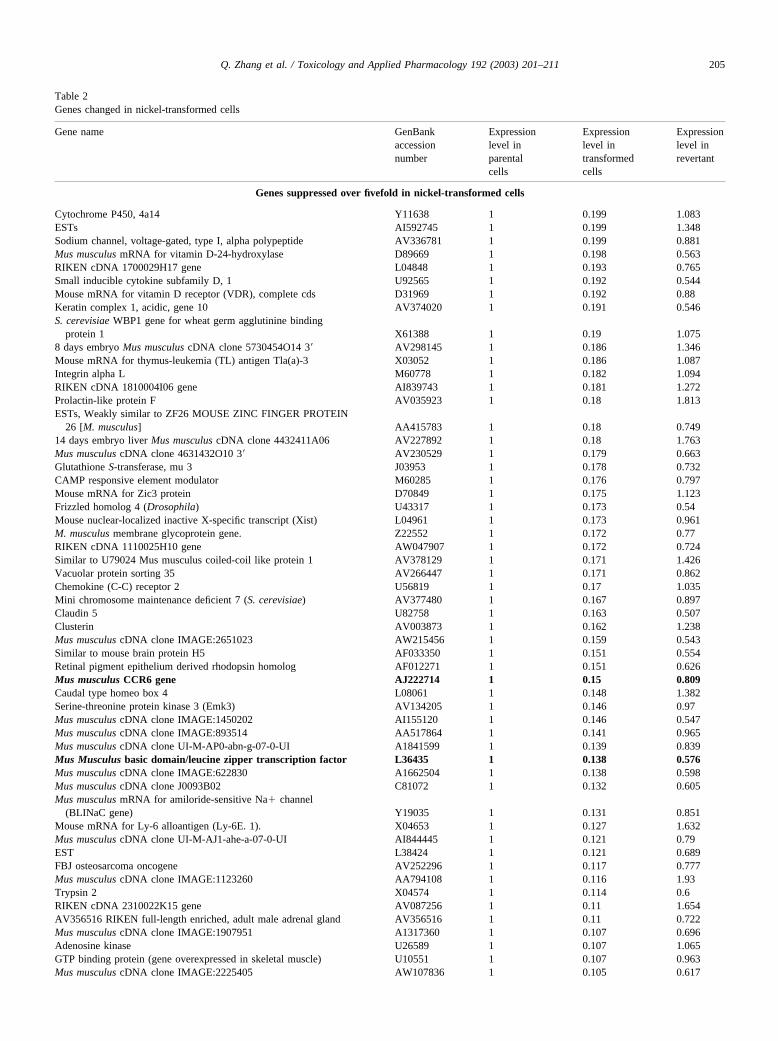
Table 2Genes changed in nickel-transformed cells
Gene name GenBankaccessionnumber
Expressionlevel inparentalcells
Expressionlevel intransformedcells
Expressionlevel inrevertant
Genes suppressed over fivefold in nickel-transformed cells
Cytochrome P450, 4a14 Y11638 1 0.199 1.083ESTs AI592745 1 0.199 1.348Sodium channel, voltage-gated, type I, alpha polypeptide AV336781 1 0.199 0.881Mus musculus mRNA for vitamin D-24-hydroxylase D89669 1 0.198 0.563RIKEN cDNA 1700029H17 gene L04848 1 0.193 0.765Small inducible cytokine subfamily D, 1 U92565 1 0.192 0.544Mouse mRNA for vitamin D receptor (VDR), complete cds D31969 1 0.192 0.88Keratin complex 1, acidic, gene 10 AV374020 1 0.191 0.546S. cerevisiae WBP1 gene for wheat germ agglutinine binding
protein 1 X61388 1 0.19 1.0758 days embryo Mus musculus cDNA clone 5730454O14 3� AV298145 1 0.186 1.346Mouse mRNA for thymus-leukemia (TL) antigen Tla(a)-3 X03052 1 0.186 1.087Integrin alpha L M60778 1 0.182 1.094RIKEN cDNA 1810004I06 gene AI839743 1 0.181 1.272Prolactin-like protein F AV035923 1 0.18 1.813ESTs, Weakly similar to ZF26 MOUSE ZINC FINGER PROTEIN
26 [M. musculus] AA415783 1 0.18 0.74914 days embryo liver Mus musculus cDNA clone 4432411A06 AV227892 1 0.18 1.763Mus musculus cDNA clone 4631432O10 3� AV230529 1 0.179 0.663Glutathione S-transferase, mu 3 J03953 1 0.178 0.732CAMP responsive element modulator M60285 1 0.176 0.797Mouse mRNA for Zic3 protein D70849 1 0.175 1.123Frizzled homolog 4 (Drosophila) U43317 1 0.173 0.54Mouse nuclear-localized inactive X-specific transcript (Xist) L04961 1 0.173 0.961M. musculus membrane glycoprotein gene. Z22552 1 0.172 0.77RIKEN cDNA 1110025H10 gene AW047907 1 0.172 0.724Similar to U79024 Mus musculus coiled-coil like protein 1 AV378129 1 0.171 1.426Vacuolar protein sorting 35 AV266447 1 0.171 0.862Chemokine (C-C) receptor 2 U56819 1 0.17 1.035Mini chromosome maintenance deficient 7 (S. cerevisiae) AV377480 1 0.167 0.897Claudin 5 U82758 1 0.163 0.507Clusterin AV003873 1 0.162 1.238Mus musculus cDNA clone IMAGE:2651023 AW215456 1 0.159 0.543Similar to mouse brain protein H5 AF033350 1 0.151 0.554Retinal pigment epithelium derived rhodopsin homolog AF012271 1 0.151 0.626Mus musculus CCR6 gene AJ222714 1 0.15 0.809Caudal type homeo box 4 L08061 1 0.148 1.382Serine-threonine protein kinase 3 (Emk3) AV134205 1 0.146 0.97Mus musculus cDNA clone IMAGE:1450202 AI155120 1 0.146 0.547Mus musculus cDNA clone IMAGE:893514 AA517864 1 0.141 0.965Mus musculus cDNA clone UI-M-AP0-abn-g-07-0-UI A1841599 1 0.139 0.839Mus Musculus basic domain/leucine zipper transcription factor L36435 1 0.138 0.576Mus musculus cDNA clone IMAGE:622830 A1662504 1 0.138 0.598Mus musculus cDNA clone J0093B02 C81072 1 0.132 0.605Mus musculus mRNA for amiloride-sensitive Na� channel
(BLINaC gene) Y19035 1 0.131 0.851Mouse mRNA for Ly-6 alloantigen (Ly-6E. 1). X04653 1 0.127 1.632Mus musculus cDNA clone UI-M-AJ1-ahe-a-07-0-UI AI844445 1 0.121 0.79EST L38424 1 0.121 0.689FBJ osteosarcoma oncogene AV252296 1 0.117 0.777Mus musculus cDNA clone IMAGE:1123260 AA794108 1 0.116 1.93Trypsin 2 X04574 1 0.114 0.6RIKEN cDNA 2310022K15 gene AV087256 1 0.11 1.654AV356516 RIKEN full-length enriched, adult male adrenal gland AV356516 1 0.11 0.722Mus musculus cDNA clone IMAGE:1907951 A1317360 1 0.107 0.696Adenosine kinase U26589 1 0.107 1.065GTP binding protein (gene overexpressed in skeletal muscle) U10551 1 0.107 0.963Mus musculus cDNA clone IMAGE:2225405 AW107836 1 0.105 0.617
205Q. Zhang et al. / Toxicology and Applied Pharmacology 192 (2003) 201–211
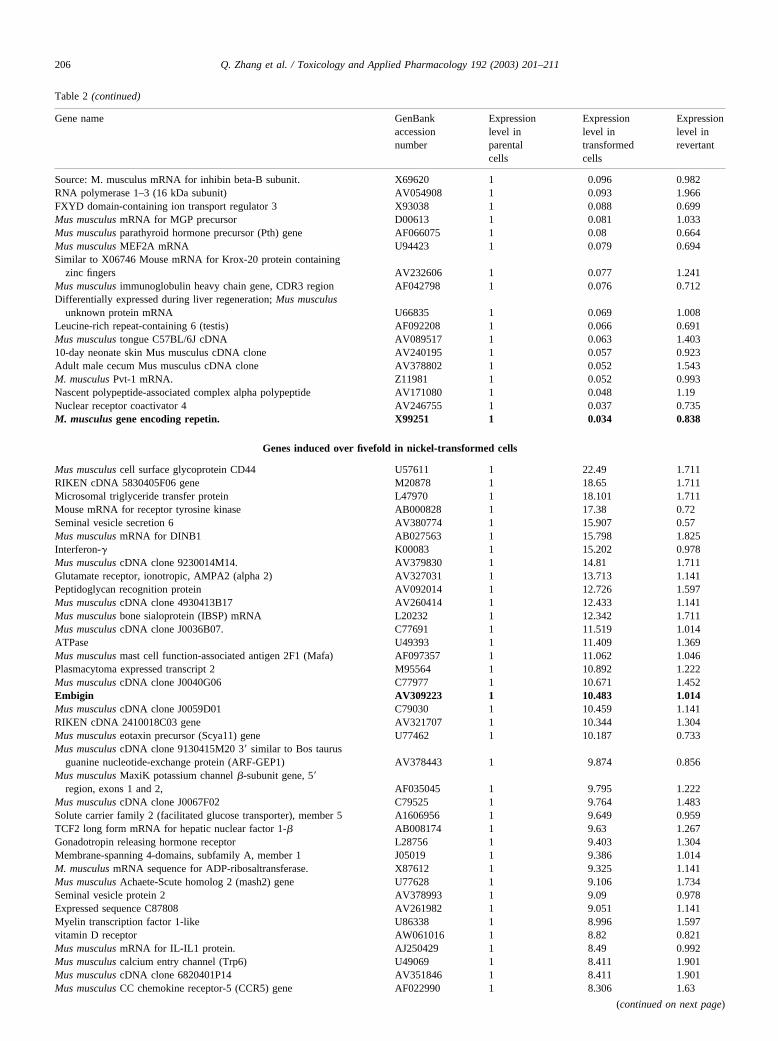
Table 2 (continued)
Gene name GenBankaccessionnumber
Expressionlevel inparentalcells
Expressionlevel intransformedcells
Expressionlevel inrevertant
Source: M. musculus mRNA for inhibin beta-B subunit. X69620 1 0.096 0.982RNA polymerase 1–3 (16 kDa subunit) AV054908 1 0.093 1.966FXYD domain-containing ion transport regulator 3 X93038 1 0.088 0.699Mus musculus mRNA for MGP precursor D00613 1 0.081 1.033Mus musculus parathyroid hormone precursor (Pth) gene AF066075 1 0.08 0.664Mus musculus MEF2A mRNA U94423 1 0.079 0.694Similar to X06746 Mouse mRNA for Krox-20 protein containing
zinc fingers AV232606 1 0.077 1.241Mus musculus immunoglobulin heavy chain gene, CDR3 region AF042798 1 0.076 0.712Differentially expressed during liver regeneration; Mus musculus
unknown protein mRNA U66835 1 0.069 1.008Leucine-rich repeat-containing 6 (testis) AF092208 1 0.066 0.691Mus musculus tongue C57BL/6J cDNA AV089517 1 0.063 1.40310-day neonate skin Mus musculus cDNA clone AV240195 1 0.057 0.923Adult male cecum Mus musculus cDNA clone AV378802 1 0.052 1.543M. musculus Pvt-1 mRNA. Z11981 1 0.052 0.993Nascent polypeptide-associated complex alpha polypeptide AV171080 1 0.048 1.19Nuclear receptor coactivator 4 AV246755 1 0.037 0.735M. musculus gene encoding repetin. X99251 1 0.034 0.838
Genes induced over fivefold in nickel-transformed cells
Mus musculus cell surface glycoprotein CD44 U57611 1 22.49 1.711RIKEN cDNA 5830405F06 gene M20878 1 18.65 1.711Microsomal triglyceride transfer protein L47970 1 18.101 1.711Mouse mRNA for receptor tyrosine kinase AB000828 1 17.38 0.72Seminal vesicle secretion 6 AV380774 1 15.907 0.57Mus musculus mRNA for DINB1 AB027563 1 15.798 1.825Interferon-� K00083 1 15.202 0.978Mus musculus cDNA clone 9230014M14. AV379830 1 14.81 1.711Glutamate receptor, ionotropic, AMPA2 (alpha 2) AV327031 1 13.713 1.141Peptidoglycan recognition protein AV092014 1 12.726 1.597Mus musculus cDNA clone 4930413B17 AV260414 1 12.433 1.141Mus musculus bone sialoprotein (IBSP) mRNA L20232 1 12.342 1.711Mus musculus cDNA clone J0036B07. C77691 1 11.519 1.014ATPase U49393 1 11.409 1.369Mus musculus mast cell function-associated antigen 2F1 (Mafa) AF097357 1 11.062 1.046Plasmacytoma expressed transcript 2 M95564 1 10.892 1.222Mus musculus cDNA clone J0040G06 C77977 1 10.671 1.452Embigin AV309223 1 10.483 1.014Mus musculus cDNA clone J0059D01 C79030 1 10.459 1.141RIKEN cDNA 2410018C03 gene AV321707 1 10.344 1.304Mus musculus eotaxin precursor (Scya11) gene U77462 1 10.187 0.733Mus musculus cDNA clone 9130415M20 3� similar to Bos taurus
guanine nucleotide-exchange protein (ARF-GEP1) AV378443 1 9.874 0.856Mus musculus MaxiK potassium channel �-subunit gene, 5�
region, exons 1 and 2, AF035045 1 9.795 1.222Mus musculus cDNA clone J0067F02 C79525 1 9.764 1.483Solute carrier family 2 (facilitated glucose transporter), member 5 A1606956 1 9.649 0.959TCF2 long form mRNA for hepatic nuclear factor 1-� AB008174 1 9.63 1.267Gonadotropin releasing hormone receptor L28756 1 9.403 1.304Membrane-spanning 4-domains, subfamily A, member 1 J05019 1 9.386 1.014M. musculus mRNA sequence for ADP-ribosaltransferase. X87612 1 9.325 1.141Mus musculus Achaete-Scute homolog 2 (mash2) gene U77628 1 9.106 1.734Seminal vesicle protein 2 AV378993 1 9.09 0.978Expressed sequence C87808 AV261982 1 9.051 1.141Myelin transcription factor 1-like U86338 1 8.996 1.597vitamin D receptor AW061016 1 8.82 0.821Mus musculus mRNA for IL-IL1 protein. AJ250429 1 8.49 0.992Mus musculus calcium entry channel (Trp6) U49069 1 8.411 1.901Mus musculus cDNA clone 6820401P14 AV351846 1 8.411 1.901Mus musculus CC chemokine receptor-5 (CCR5) gene AF022990 1 8.306 1.63
(continued on next page)
206 Q. Zhang et al. / Toxicology and Applied Pharmacology 192 (2003) 201–211
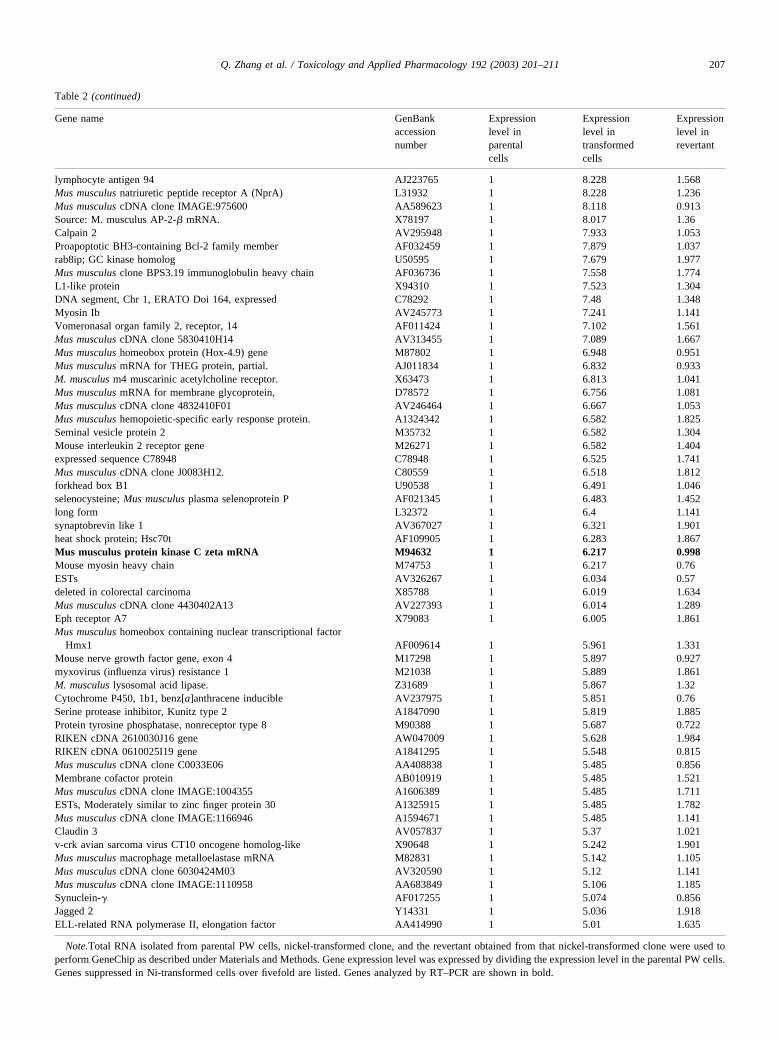
Table 2 (continued)
Gene name GenBankaccessionnumber
Expressionlevel inparentalcells
Expressionlevel intransformedcells
Expressionlevel inrevertant
lymphocyte antigen 94 AJ223765 1 8.228 1.568Mus musculus natriuretic peptide receptor A (NprA) L31932 1 8.228 1.236Mus musculus cDNA clone IMAGE:975600 AA589623 1 8.118 0.913Source: M. musculus AP-2-� mRNA. X78197 1 8.017 1.36Calpain 2 AV295948 1 7.933 1.053Proapoptotic BH3-containing Bcl-2 family member AF032459 1 7.879 1.037rab8ip; GC kinase homolog U50595 1 7.679 1.977Mus musculus clone BPS3.19 immunoglobulin heavy chain AF036736 1 7.558 1.774L1-like protein X94310 1 7.523 1.304DNA segment, Chr 1, ERATO Doi 164, expressed C78292 1 7.48 1.348Myosin Ib AV245773 1 7.241 1.141Vomeronasal organ family 2, receptor, 14 AF011424 1 7.102 1.561Mus musculus cDNA clone 5830410H14 AV313455 1 7.089 1.667Mus musculus homeobox protein (Hox-4.9) gene M87802 1 6.948 0.951Mus musculus mRNA for THEG protein, partial. AJ011834 1 6.832 0.933M. musculus m4 muscarinic acetylcholine receptor. X63473 1 6.813 1.041Mus musculus mRNA for membrane glycoprotein, D78572 1 6.756 1.081Mus musculus cDNA clone 4832410F01 AV246464 1 6.667 1.053Mus musculus hemopoietic-specific early response protein. A1324342 1 6.582 1.825Seminal vesicle protein 2 M35732 1 6.582 1.304Mouse interleukin 2 receptor gene M26271 1 6.582 1.404expressed sequence C78948 C78948 1 6.525 1.741Mus musculus cDNA clone J0083H12. C80559 1 6.518 1.812forkhead box B1 U90538 1 6.491 1.046selenocysteine; Mus musculus plasma selenoprotein P AF021345 1 6.483 1.452long form L32372 1 6.4 1.141synaptobrevin like 1 AV367027 1 6.321 1.901heat shock protein; Hsc70t AF109905 1 6.283 1.867Mus musculus protein kinase C zeta mRNA M94632 1 6.217 0.998Mouse myosin heavy chain M74753 1 6.217 0.76ESTs AV326267 1 6.034 0.57deleted in colorectal carcinoma X85788 1 6.019 1.634Mus musculus cDNA clone 4430402A13 AV227393 1 6.014 1.289Eph receptor A7 X79083 1 6.005 1.861Mus musculus homeobox containing nuclear transcriptional factor
Hmx1 AF009614 1 5.961 1.331Mouse nerve growth factor gene, exon 4 M17298 1 5.897 0.927myxovirus (influenza virus) resistance 1 M21038 1 5.889 1.861M. musculus lysosomal acid lipase. Z31689 1 5.867 1.32Cytochrome P450, 1b1, benz[a]anthracene inducible AV237975 1 5.851 0.76Serine protease inhibitor, Kunitz type 2 A1847090 1 5.819 1.885Protein tyrosine phosphatase, nonreceptor type 8 M90388 1 5.687 0.722RIKEN cDNA 2610030J16 gene AW047009 1 5.628 1.984RIKEN cDNA 0610025I19 gene A1841295 1 5.548 0.815Mus musculus cDNA clone C0033E06 AA408838 1 5.485 0.856Membrane cofactor protein AB010919 1 5.485 1.521Mus musculus cDNA clone IMAGE:1004355 A1606389 1 5.485 1.711ESTs, Moderately similar to zinc finger protein 30 A1325915 1 5.485 1.782Mus musculus cDNA clone IMAGE:1166946 A1594671 1 5.485 1.141Claudin 3 AV057837 1 5.37 1.021v-crk avian sarcoma virus CT10 oncogene homolog-like X90648 1 5.242 1.901Mus musculus macrophage metalloelastase mRNA M82831 1 5.142 1.105Mus musculus cDNA clone 6030424M03 AV320590 1 5.12 1.141Mus musculus cDNA clone IMAGE:1110958 AA683849 1 5.106 1.185Synuclein-� AF017255 1 5.074 0.856Jagged 2 Y14331 1 5.036 1.918ELL-related RNA polymerase II, elongation factor AA414990 1 5.01 1.635
Note.Total RNA isolated from parental PW cells, nickel-transformed clone, and the revertant obtained from that nickel-transformed clone were used toperform GeneChip as described under Materials and Methods. Gene expression level was expressed by dividing the expression level in the parental PW cells.Genes suppressed in Ni-transformed cells over fivefold are listed. Genes analyzed by RT–PCR are shown in bold.
207Q. Zhang et al. / Toxicology and Applied Pharmacology 192 (2003) 201–211

novel integral membrane glycoprotein (1091 amino acids)containing an extracellular region (794 amino acids) with apotential signal peptide, 15 leucine-rich repeats, 3immnunoglobulin-like domains, and 7 potential N-glycosyl-ation sites, a transmembrane region of 23 amino acids, anda cytoplasmic region of 274 amino acids. This protein,therefore, is a new member of both the leucine-rich repeatand the immunoglobulin superfamilies (Suzuki et al., 1996).PKC-zeta was induced over sixfold. The overexpression ofthis PKC isoform suppresses the ability of Par-4 protein toinduce apoptosis (Diaz-Meco et al., 1996). It is of interestthat the blockade of PKC-zeta with dominant-negative mu-tants or antisense oligonucleotides is sufficient to promoteapoptosis and suppress soft agar growth (Barradas et al.,1999). Thus these results show that changes in gene expres-sion are important for nickel-induce transformation.
Inhibition of transformation of mouse PW or human HOScells by TSA.
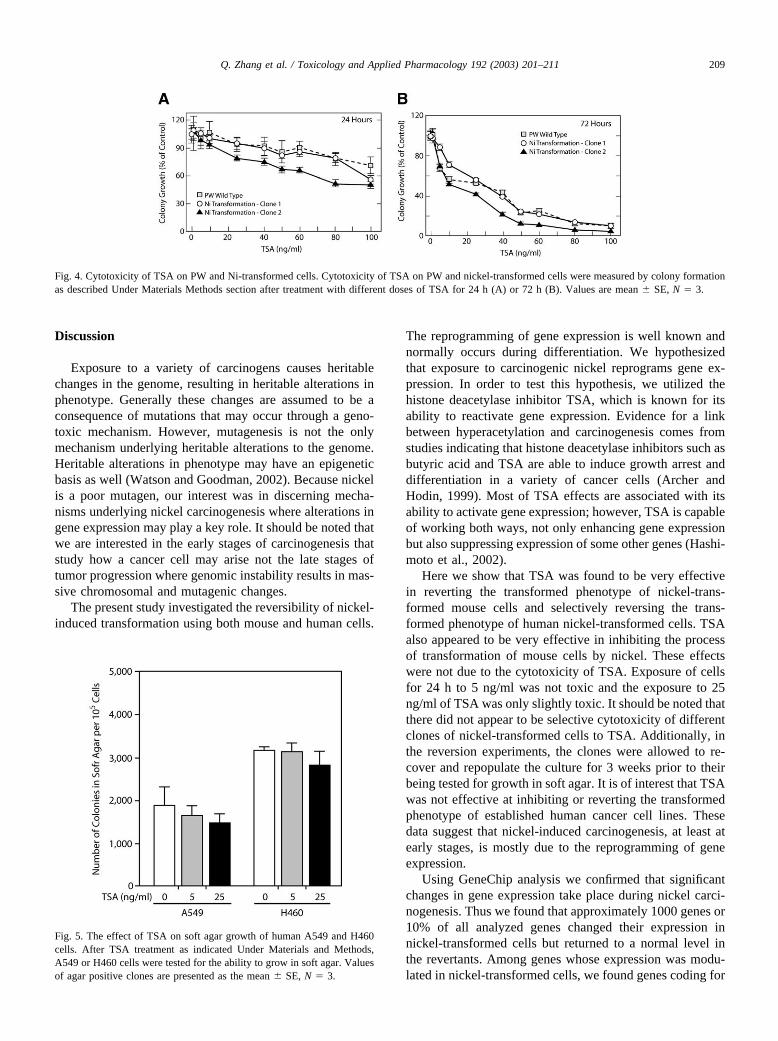
Because TSA was efficient in reverting nickel-trans-formed clones we decided to investigate whether treatmentwith TSA would prevent or reduce nickel-induced transfor-mation. Table 3 shows that the transformation of mouse PWcells by the carcinogenic Ni3S2 or NiCl2 is reduced bypretreatment with 5 or 25 ng/ml TSA in a dose-dependentmanner. The inhibition of soft agar growth was not due toany general cytotoxic effect of TSA because(a) even afterthe exposure to 25 ng/ml TSA for 24 h toxicity for all testedcells was approximately 10% less than untreated (Fig. 4Aand B) and (b) after the exposure cells were allowed torepopulate the culture (3 weeks) prior to being tested fortheir ability to grow in soft agar.
Table 3 also shows the transformation of HOS cells bycrystalline Ni2S3 was suppressed by TSA exposure. Al-though the effects were not as striking as in PW cells, therewas less growth in soft agar in the presence of TSA at thesame level of Ni2S3.
The effect of TSA on the proliferation of establishedcancer cells in soft agar
The previous figures and tables showed that TSA pre-treatment diminished the transformation induced by nickelin mouse and human cells. We next addressed what effectTSA exposure had on the ability of established cell lines togrow in soft agar.
Figure 5 shows that the effect of TSA was specific fornickel-transformed cells. In A549 cells, which proliferatedin soft agar, the exposure to TSA only slightly attenuatedtheir ability to grow in soft agar. Moreover, TSA had noeffect on the ability of H460 cells to proliferate in soft agar.Again toxic effect of TSA exposure was minimal for A549and H460 cells.
Fig. 3. RT–PCR analysis of expression of selected genes identified byGeneChip. Total RNA isolated from parental PW cells, a nickel-trans-formed clone and a revertant obtained from that nickel-transformed clonewas converted into cDNA and PCR was amplified using primers shownunder Materials and Methods. (a) Overexpression of embigin (1) andprotein kinase C-zeta (2) in nickel-transformed cells, and the expression ofglyceraldehyde phosphate dehydrogenase (3) was used to assure that equalamount of cDNA was used for amplification. (b) Suppression of repetin(1), CCR6 (2), and basic domain transcription factor (3) in nickel-trans-formed cells, and the expression of glyceraldehyde phosphate dehydroge-nase (4) was used to assure that equal amount of cDNA was used foramplification.
Table 3Suppression of nickel-induced transformation by TSA
Cell lines Treatment 0 ng/ml TSA 5 ng/ml TSA 25 ng/ml TSA
PW 1 mM NiCl2 100.2 � 13.4 76.1 � 9.5* 35.1 � 8.2**2 mM NiCl2 100.1 � 13.5 61.5 � 6.9* 53.0 � 10.4**0.15 �g/cm2 Ni2S3 99.9 � 12.9 47.1 � 10.3** 13.7 � 2.9**0.30 �g/cm2 Ni2S3 99.9 � 12.9 67.4 � 2.8* 38.7 � 1.7**
HOS 1 mM NiCl2 100.0 � 17.4 69.3 � 21* 39.7 � 5.7**2 mM NiCl2 100.0 � 4.3 59.5 � 3.1* 51.0 � 1.6**0.15 �g/cm2 Ni2S3 100.0 � 11.1 69.9 � 8.7* 63.7 � 14.7*0.30 �g/cm2Ni2S3 100.0 � 8.6 93.3 � 4.6 78.9 � 13.8*
Note. TSA (5 ng/ml or 25 ng/ml) was added to one million of PW or HOS cells 4 h before exposure to difference concentrations of Ni2S3 or NiCl2 asdescribed under Materials and Methods. Then cells were tested for the ability of anchorage-independent growth in soft agar. Results are expressed as apercentage of control (0 ng/ml TSA treatment). Values are mean � SE. Asterisk indicates significant differences from that of control.
* P � 0.05.** P � 0.005, N � 3.
208 Q. Zhang et al. / Toxicology and Applied Pharmacology 192 (2003) 201–211
Discussion
Exposure to a variety of carcinogens causes heritablechanges in the genome, resulting in heritable alterations inphenotype. Generally these changes are assumed to be aconsequence of mutations that may occur through a geno-toxic mechanism. However, mutagenesis is not the onlymechanism underlying heritable alterations to the genome.Heritable alterations in phenotype may have an epigeneticbasis as well (Watson and Goodman, 2002). Because nickelis a poor mutagen, our interest was in discerning mecha-nisms underlying nickel carcinogenesis where alterations ingene expression may play a key role. It should be noted thatwe are interested in the early stages of carcinogenesis thatstudy how a cancer cell may arise not the late stages oftumor progression where genomic instability results in mas-sive chromosomal and mutagenic changes.
The present study investigated the reversibility of nickel-induced transformation using both mouse and human cells.
The reprogramming of gene expression is well known andnormally occurs during differentiation. We hypothesizedthat exposure to carcinogenic nickel reprograms gene ex-pression. In order to test this hypothesis, we utilized thehistone deacetylase inhibitor TSA, which is known for itsability to reactivate gene expression. Evidence for a linkbetween hyperacetylation and carcinogenesis comes fromstudies indicating that histone deacetylase inhibitors such asbutyric acid and TSA are able to induce growth arrest anddifferentiation in a variety of cancer cells (Archer andHodin, 1999). Most of TSA effects are associated with itsability to activate gene expression; however, TSA is capableof working both ways, not only enhancing gene expressionbut also suppressing expression of some other genes (Hashi-moto et al., 2002).
Here we show that TSA was found to be very effectivein reverting the transformed phenotype of nickel-trans-formed mouse cells and selectively reversing the trans-formed phenotype of human nickel-transformed cells. TSAalso appeared to be very effective in inhibiting the processof transformation of mouse cells by nickel. These effectswere not due to the cytotoxicity of TSA. Exposure of cellsfor 24 h to 5 ng/ml was not toxic and the exposure to 25ng/ml of TSA was only slightly toxic. It should be noted thatthere did not appear to be selective cytotoxicity of differentclones of nickel-transformed cells to TSA. Additionally, inthe reversion experiments, the clones were allowed to re-cover and repopulate the culture for 3 weeks prior to theirbeing tested for growth in soft agar. It is of interest that TSAwas not effective at inhibiting or reverting the transformedphenotype of established human cancer cell lines. Thesedata suggest that nickel-induced carcinogenesis, at least atearly stages, is mostly due to the reprogramming of geneexpression.
Using GeneChip analysis we confirmed that significantchanges in gene expression take place during nickel carci-nogenesis. Thus we found that approximately 1000 genes or10% of all analyzed genes changed their expression innickel-transformed cells but returned to a normal level inthe revertants. Among genes whose expression was modu-lated in nickel-transformed cells, we found genes coding for
Fig. 4. Cytotoxicity of TSA on PW and Ni-transformed cells. Cytotoxicity of TSA on PW and nickel-transformed cells were measured by colony formationas described Under Materials Methods section after treatment with different doses of TSA for 24 h (A) or 72 h (B). Values are mean � SE, N � 3.
Fig. 5. The effect of TSA on soft agar growth of human A549 and H460cells. After TSA treatment as indicated Under Materials and Methods,A549 or H460 cells were tested for the ability to grow in soft agar. Valuesof agar positive clones are presented as the mean � SE, N � 3.
209Q. Zhang et al. / Toxicology and Applied Pharmacology 192 (2003) 201–211

proteins expressed on the cell membrane and responsible foradhesion. Other genes include transcription factors, regula-tory protein kinases, and antiapoptotic genes. Alteration inthe expression of these genes apparently provided a selec-tive advantage for cells growing in soft agar since we use itas an in vitro model. In general, the ability of cells to growin soft agar is well correlated with their tumorigenic poten-tial in vivo. This has been shown by numerous previousstudies (Costa, 1980; Steuer et al., 1977; Puutman et al.,1977; Mishra et al., 1978; Keshava, 2000). Thus the mod-ulation of cell proliferation in soft agar is an importantcancer phenotype that can be utilized to understand thecarcinogenesis process.
We hypothesized that modulation of gene expression bynickel will be also important for uncontrolled cell growth invivo. Moreover, considering the ability of nickel com-pounds to induce HIF-1 transcription factor and HIF-depen-dent genes (Keshava, 2000; Salnikow et al., 1999) we sug-gest that nickel is preparing cells for hypoxic conditions thatinevitably emerge at certain stages of tumor growth. Incontrast to nickel-transformed cells using TSA, we failed torevert established cancer cells. These data provide anotherargument that nickel is reprogramming gene expression andthese changes can be reverted at least at early stages ofnickel-induced transformation.
In conclusion, in the present study we have investigatedthe possible importance of the reprogramming of gene ex-pression by nickel in determining the ability of mouse andhuman cells to grow in soft agar. Exposure of nickel-trans-formed cells to TSA appeared to revert the transformedphenotype and suppress the ability of these cells to prolif-erate in soft agar. Moreover, treatment of cells with TSAinhibited nickel-induced transformation, indicating that re-programming of gene expression is part of nickel-inducedcarcinogenesis. It is conceivable that nickel exposure ischanging the level of histone acetylation and thus remodel-ing the chromatin template. TSA by inhibition of HDACactivities recovers the original transcriptional status. Morestudies on the correlation between chromatin acetylationlevels and alterations in gene expression during nickel-induced carcinogenesis are needed to confirm the role ofepigenetic changes in this process.
Acknowledgments
This work was supported by Grants ES05512, ES00260,ES10344, and T32ES07324 from the NIH/NIEHS andCA16087 from the NIH/NCI.
References
Archer, S.Y., Hodin, R.A., 1999. Histone acetylation and cancer. Curr.Opin. Genet. Dev. 9, 171–174.
Bal, W., Lukszo, J., Bialkowski, K., Kasprzak, K.S., 1998. Interactions ofNickel(II) with histones: interactions of Nickel(II) with CH3CO-Thr-
Glu-Ser-His-His-Lys-NH2, a peptide modeling the potential metalbinding site in the “C-Tail” region of histone H2A. Chem. Res. Toxi-col. 11 (9), 1014–1023.
Barradas, M., Monjas, A., Diaz-Meco, M.T., Serrano, M., Moscat, J., 1999.The downregulation of the pro-apoptotic protein Par-4 is critical forRas-induced survival and tumor progression. EMBO J. 18 (22), 6362–6369.
Baylin, S.B., Herman, J.G., 2001. Promoter hypermethylation-can thischange along ever designate true tumor suppressor gene function.J. Natl. Cancer Inst. 93, 664–665.
Broday, L., Cai, J., Costa, M., 1999. Nickel enhances telomeric silencingin Saccharomyces cerevisiae. Mutat. Res. 440, 121–130.
Broday, L., Peng, W., Kuo, M.H., Salnikow, K., Zoroddu, M., Costa, M.,2000. Nickel compounds are novel inhibitors of histone H4 acetylation.Cancer Res. 60, 238–241.
Costa, M., 1980. Metal carcinogenesis testing: Principles and in vitromethods. Humana Press Inc., Totowa, New Jersey.
Diaz-Meco, M.T., Municio, M.M., Frutos, S., Sanchez, P., Lozano, J.,Sanz, L., Moscat, J., 1996. The product of par-4, a gene induced duringapoptosis, interacts selectively with the atypical isoforms of proteinkinase C. Cell 86, 777–786.
Hashimoto, A., Suzuki, Y., Katsuno, T., Nakajima, H., Saito, Y., 2002.Caprylic acid and medium-chain triglycerides inhibit IL-8 gene tran-scription in Caco-2 cells: comparison with the potent histone deacety-lase inhibitor trichostatin A. Br. J. Pharmacol. 136 (2), 280–286.
Jones, P.A., Takai, D., 2001. The role of DNA methylation in mammalianepigenetics. Science 293, 1068–1070.
Keshava, N., 2000. Tumorigenicity of morphologically distinct trans-formed foci induced by 3-methylcholanthrene in BALB/c-3T3 cells.Mutat. Res. 477, 281–286.
Klein, C.B., Conway, K., Wang, X.W., Bhamra, R.K., Lin, X.H., Costa,M., 1991. Senescence of nickel-transformed cells by an X chromo-some: possible epigenetic control. Science 251, 796–799.
Krieg, P., Schuppler, M., Koesters, R., Mincheva, A., Lichter, P., Marks,F., 1997. Repetin (Rptn), a new member of the “fused gene” subgroupwithin the S100 gene family encoding a murine epidermal differenti-ation protein. Genomics 43 (3), 339–348.
Lee, Y.W., Klein, C.B., Kargacin, B., Salnikow, K., Kitahara, J., Dowjat,K., 1995. Carcinogenic nickel silences genes expression by chromatincondensation and DNA methylation: a new model for epigenetic car-cinogens. Mol. Cell. Biol. 15, 2547–2557.
Lin, X., Dowjat, W.K., Costa, M., 1994. Nickel-induced transformation ofhuman cells caused loss of phosphorylation of the retinoblastomaprotein. Cancer Res. 54, 2751–2754.
Maki, W., Morales, R.E., Carroll, V.A., Telford, W.G., Knibbs, R.N.,Stoolman, L.M., Hwang, S.T., 2002. CCR6 colocalizes with CD18 andenhances adhesion to activated endothelial cells in CCR6-transducedJurkat T cells. J. Immunol. 169 (5), 2346–2353.
Mishra, N.K., Wilson, C.M., Pant, K.J., Thomas, F.O., 1978. Simultaneousdetermination of cellular mutagenesis and transformation by chemicalcarcinogens in Fischer rat embryo cells. J. Toxicol. Environ. Health 4,79–91.
Puutman, D.L., Park, D.K., Rhim, S., Steuer, A.F., Ting, R.C., 1977.Correlation of cellular aggregation of transformed cells with theirgrowth in soft agar and tumorigenetic potential. Proc. Soc. Exp. Biol.Med. 155, 487–494.
Salnikow, K., An, W.G., Melillo, G., Blagosklonny, M.V., Costa, M.,1999. Nickel-induced transformation shifts the balance between HIF-1and p53 transcription factors. Carcinogenesis 20, 1819–1823.
Salnikow, K., Cosentino, S., Klein, C.B., Costa, M., 1994. Loss of throm-bospondin transcriptional activity in nickel-transformed cells. Mol.Cell. Biol. 14, 851–858.
Siegfried, Z., Eden, S., Mendelsohn, M., Feng, X., Tsuberi, B.Z., Cedar,H., 1999. DNA methylation represses transcription in vivo. Nat. Genet.22, 203–206.
210 Q. Zhang et al. / Toxicology and Applied Pharmacology 192 (2003) 201–211

Steuer, A.F., Rhim, J.S., Hentosh, P.M., Ting, R.C., 1977. Survival ofhuman cells in the aggregate form: potential index of in vitro celltransformation. J. Natl. Cancer Inst. 58, 917–921.
Stirzaker, C., Millar, D.S., Paul, C.L., Warnecke, P.M., Harrison, J., Vin-cent, P.C., 1997. Extensive DNA methylation spanning the Rb pro-moter in retinoblastoma tumors. Cancer Res. 57, 2229–2237.
Sutherland, J.E., Peng, W., Zhang, Q., Costa, M., 2001. The histonedeacetylase inhibitor trichostatin A reduces nickel-induced gene silenc-ing in yeast and mammalian cells. Mutat. Res. 479, 225–233.
Suzuki, Y., Sato, N., Tohyama, M., Wanaka, A., Takagi, T., 1996. cDNAcloning of a novel membrane glycoprotein that is expressed specificallyin glial cells in the mouse brain. LIG-1, a protein with leucine-rich
repeats and immunoglobulin-like domains. J. Biol. Chem. 271 (37),22522–22527.
Wang, P.W., Eisenbart, J.D., Cordes, S.P., Barsh, G.S., Stoffel, M., LeBeau, M.M., 1999. Human KRML (MAFB): cDNA cloning, genomicstructure, and evaluation as a candidate tumor suppressor gene inmyeloid leukemias. Genomics 59 (3), 275–281.
Watson, R.E., Goodman, J.I., 2002. Epigenetics and DNA methylationcome of age in toxicology. Toxicol Sci. 67 (1), 11–16, Review.
Zoroddu, M., Kowalik-Jankowaka, T., Kozlowski, H., Molinari, H., Salni-kow, K., Broday, L., 2001. Interaction of Ni(II) and Cu(II) with a metalbinding sequence of histone: AKRHRK, a model of the H4 tail. Bio-chem. Biophys. Acta 1475, 163–168.
211Q. Zhang et al. / Toxicology and Applied Pharmacology 192 (2003) 201–211