insulin-like growth factor i is essential for postnatal...
TRANSCRIPT

Insulin-Like Growth Factor I Is Essential for PostnatalGrowth in Response to Growth Hormone
JUN-LI LIU* AND DEREK LEROITH
Clinical Endocrinology Branch, The National Institute of Diabetes and Digestive and Kidney Diseases,National Institutes of Health, Bethesda, Maryland 20892-1758
ABSTRACTInsulin-like growth factor I (IGF-I) is essential for cell growth and
intrauterine development while both IGF-I and GH are required forpostnatal growth. To explore the possibility of direct GH action onbody growth, independent of IGF-I production, we have studied theeffects of GH in an IGF-I-deficient mouse line created by the Cre/loxPsystem. The IGF-I null mice are born with 35% growth retardationand show delayed onset of peripubertal growth, grow significantlyslower, and do not attain puberty. Their adult body weight was ap-proximately one third and body length about two thirds that of theirwild-type litter mates. Injection of recombinant human GH (rhGH, 3
mg/kg, twice daily, sc) between postnatal day 14 (P14) to P56 failedto stimulate their growth as measured as both body weight andlength. In contrast, wild-type mice receiving the same doses of rhGHexhibited accelerated growth starting at P21 that continued until P56,when their body weight was increased by 30% and length by 12%compared with control mice treated with diluent. Despite the lack ofresponse in growth, IGF-I null mice have normal levels of GH receptorexpression in the liver and increased liver Jun B expression and liversize in response to rhGH treatment. Our results support an essentialrole for IGF-I in GH-induced postnatal body growth in mice. (Endo-crinology 140: 5178–5184, 1999)
GH IS a small protein hormone of 191 amino acids thatcauses growth of almost all tissues of the body by
increasing cell size and cell number and by promoting dif-ferentiation of bone and muscle cells (1). Deficiency in eitherGH or the GH receptor causes severe postnatal growth re-tardation and proportionate dwarfism in humans and mice(2, 3). Insulin-like growth factor I (IGF-I), expressed by manycells and tissues throughout development, is an essentialfactor in cell growth, intrauterine development, and post-natal growth (4–7). IGF-I deficiency in humans and micecauses severe intrauterine and postnatal growth retardation,perinatal lethality, and developmental defects in the bone,muscle, and reproductive systems.
Several modes of GH action on postnatal body growthhave been proposed (8, 9). GH may act on a major targetorgan, namely the liver, to stimulate the synthesis and se-cretion of IGF-I, which reaches its skeletal targets as a trueendocrine reagent (the somatomedin hypothesis) (10). GHmay stimulate longitudinal bone growth directly throughlocal production of IGF-I (modified somatomedin hypothe-sis) (11–13). In addition, GH may have a direct mitogenicaction on target tissues such as the chondrocyte precursorcells, or GH action may be mediated by growth factors otherthan IGF-I (14–16). For example, GH was found to stimulatesynthesis of the bone morphogenetic proteins in the presenceof IGF-I antiserum (14). A recent report from our laboratoryhas demonstrated that liver-derived “endocrine” IGF-I is notessential for postnatal growth and has challenged the clas-
sical somatomedin hypothesis (17). In this study, to test thedirect effect of GH, we created viable dwarf mice with IGF-Ideficiency by the Cre/loxP system and studied the effect ofGH administration on their body growth. Our results dem-onstrate an essential role for IGF-I in GH-induced postnatalbody growth in mice and hence support the modified so-matomedin hypothesis.
Materials and MethodsAnimal production
IGF-I null mice were generated while studying EIIa-cre-induced generecombination in IGF-I floxed (flanked by loxP repeats) mice (18).Briefly, mice (F1) carrying one allele of igf-1/flox and EIIa-cre transgene(L/1, Cre/1) were intercrossed to generate homozygous igf-1/flox micethat were also EIIa-cre positive (F2, L/L, Cre/1) (Fig. 1). At that stage,Cre-induced recombination of the igf-1 locus had occurred (18). F2 micewere further intercrossed to generate mice (F3, L/2) with an allele ofigf-1/flox and an allele of igf-1 null (germ line transmission from F2)which had segregated the Cre transgene. These L/2 mice were assignedto multiple mating pairs to generate IGF-I null (2/2) mice. Among thethree genotypes generated [L/L, L/2, and 2/2] and used in the currentstudy, L/L and L/2 are virtually normal in IGF-I gene expression,development, and growth and are treated as wild-type mice. On theother hand, 2/2 (IGF-I null) mice have no IGF-I expression and dem-onstrate a severe defect in intrauterine development and postnatalgrowth. The animals were kept in a designated breeding room and wereobserved closely throughout the experiment. Genotyping by PCR andSouthern blot analysis has been reported (18). The Animal Care and UseCommittee of the NIDDK, National Institutes of Health (Bethesda, MD)approved all of the animal manipulations.
Growth response to GH
Wild-type (WT) and IGF-I null mice were treated with recombinanthuman GH (rhGH, Genentech, Inc., South San Francisco, CA), 3 mg/kgtwice daily, sc, from postnatal day 14 (P14) to P56. Control mice of bothgenotypes received an equal volume of normal saline (diluent). Theirbody weights were measured daily. At the end of the study (8 weeks ofage), mice were anesthetized using 0.2% avertin and bled via periorbitalpuncture; their body length (nose to anus) was measured, and organs
Received June 10, 1999.Address all correspondence and requests for reprints to: Dr. Derek
LeRoith, CEB/National Institute of Diabetes and Digestive and KidneyDiseases, Building 10, Room 8D12, National Institutes of Health, 10Center Drive, Bethesda, Maryland 20892-1758. E-mail: [email protected].
* Supported by a fellowship from the Medical Research Council ofCanada.
0013-7227/99/$03.00/0 Vol. 140, No. 11Endocrinology Printed in U.S.A.Copyright © 1999 by The Endocrine Society
5178
by on March 19, 2007 endo.endojournals.orgDownloaded from

(brain, heart, liver, kidney, and spleen) were removed and weighed.Liver RNA was extracted, using RNAzol B reagent (Tel-Test, Friends-wood, TX), to determine the level of GH receptor messenger RNA(mRNA) using ribonuclease (RNase) protection assay.
Effect of GH on Jun B gene expression
To study the effect of GH on the expression of immediate early genes,liver expression of Jun B mRNA was analyzed. Mice of wild-type andknockout genotypes, at 6 weeks of age, were fasted overnight andinjected with rhGH (3 mg/kg, ip) or diluent. After 30 min, they wereanesthetized and bled. Total RNA was extracted from the liver to de-termine the level of Jun B mRNA by RNase protection assay.
RNase protection assay
The expression of IGF-I from various tissues and expression of Jun Band GH receptor from the liver were studied using the RNase protectionassay (19). 32P-labeled riboprobes were made from mouse IGF-I exon 4(18), pTRI-Jun (B)-mouse (Ambion, Inc., Austin, TX), and mouse GHreceptor exon 4 (BamHI/AvaI fragment, provided by Dr. John JKopchick, Ohio University, Athens, OH) (2).
Hormone assay, serum chemistry, and statistics
The serum concentration of total IGF-I (Diagnostics Systems Labo-ratories, Inc. Webster, TX), insulin (Linco, St-Charles, MO), and GH(Amersham Pharmacia Biotech, Arlington Heights, IL) were determinedby RIA kits available commercially. Glucose was measured with a Glu-cometer Elite (Bayer Corp., Elkhart, IN). Serum chemistry was per-formed by the Pathology Department at the Clinical Center of the NIH.Statistical significance was determined by the two-tailed t test using theprogram SigmaStat 2.03 (Access Softek, Inc., San Rafael, CA).
ResultsCharacterization of the IGF-I deletion mutant
To study IGF-I-independent, direct GH action on bodygrowth, we generated IGF-I null mice by inbreeding multiplepairs of L/2 mice (with an allele of igf-1/flox and an allele ofigf-1 null) (Fig. 1). Southern blot analysis was performed ontail DNA to identify the genotype of the offspring (data notshown). RNase protection assay on several tissues was em-ployed to assess the level of IGF-I gene expression and serumIGF-I level was determined by RIA. As shown in Fig. 2,wild-type mice have varying amounts of IGF-I mRNA in theliver, heart, and kidney, while IGF-I null mice have unde-tectable IGF-I mRNA. Their circulating IGF-I concentrationwas below the detection limit (Table 1). As such, the genedeletion was confirmed at the level of tissue DNA, mRNA,and circulating peptide.
As a consequence of a diminished negative feedback con-trol by IGF-I, we anticipated finding an increased release ofGH from the pituitary in IGF-I null mice. Indeed, in 5-week-old mice, serum GH concentration increased 12.4-fold (Table1). Consistent with GH hypersecretion, adult IGF-I null miceshow specific and significant increases in the relative weightof the following organs: brain (1113%), liver (141%), heart(137%), and kidney (121%). No change was found in theweight of the spleen (Table 2).
The serum glucose level (24 h fasted) in IGF-I null mice wasdecreased by 50% (P , 0.001), whereas serum insulin con-centrations tended to be higher in these mice (Table 1). Thisfinding is similar to a report on severe IGF-I deficient (midi)mice created by an insertional mutation (20). In the standardserum chemistry assay, there was a dramatic 58% reductionin alkaline phosphatase (Table 1), associated with the reduc-tion in bone growth.
Postnatal survival and growth rates
While the overall phenotype of the IGF-I null mice gen-erated in this study remains similar to those previously re-ported (4, 7), demonstrating severe defects in prenatal de-velopment and postnatal growth, our IGF-I null mice
FIG. 1. Breeding strategy for the production of IGF-I null mice. Seedetails in text of Animal Production in Materials and Methods.
FIG. 2. IGF-I gene expression in wild-type (lanes 1–4) and IGF-I nullmice (lanes 5–8). Total RNA was prepared from liver (10 mg), heart(50 mg), and kidney (10 mg) of adult mice and analyzed by RNaseprotection assay using probes for IGF-I exon 4 and 18S ribosome RNA.
IGF-I IS ESSENTIAL FOR GH-STIMULATED GROWTH 5179
by on March 19, 2007 endo.endojournals.orgDownloaded from
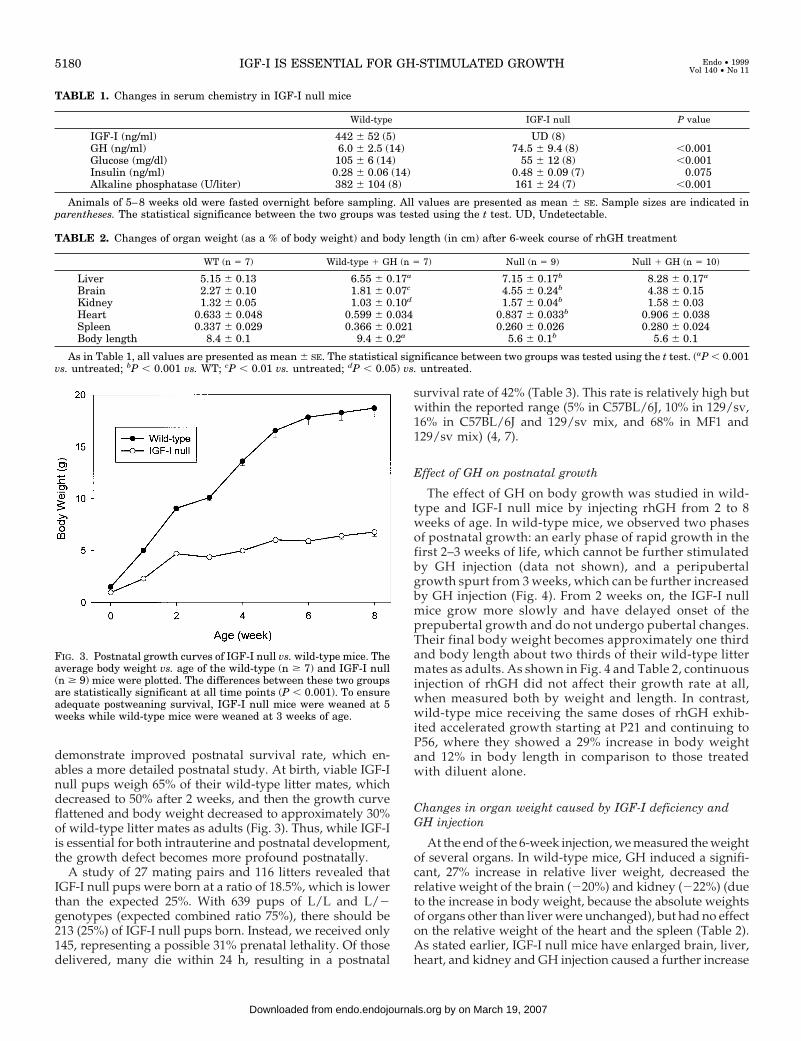
demonstrate improved postnatal survival rate, which en-ables a more detailed postnatal study. At birth, viable IGF-Inull pups weigh 65% of their wild-type litter mates, whichdecreased to 50% after 2 weeks, and then the growth curveflattened and body weight decreased to approximately 30%of wild-type litter mates as adults (Fig. 3). Thus, while IGF-Iis essential for both intrauterine and postnatal development,the growth defect becomes more profound postnatally.
A study of 27 mating pairs and 116 litters revealed thatIGF-I null pups were born at a ratio of 18.5%, which is lowerthan the expected 25%. With 639 pups of L/L and L/2genotypes (expected combined ratio 75%), there should be213 (25%) of IGF-I null pups born. Instead, we received only145, representing a possible 31% prenatal lethality. Of thosedelivered, many die within 24 h, resulting in a postnatal
survival rate of 42% (Table 3). This rate is relatively high butwithin the reported range (5% in C57BL/6J, 10% in 129/sv,16% in C57BL/6J and 129/sv mix, and 68% in MF1 and129/sv mix) (4, 7).
Effect of GH on postnatal growth
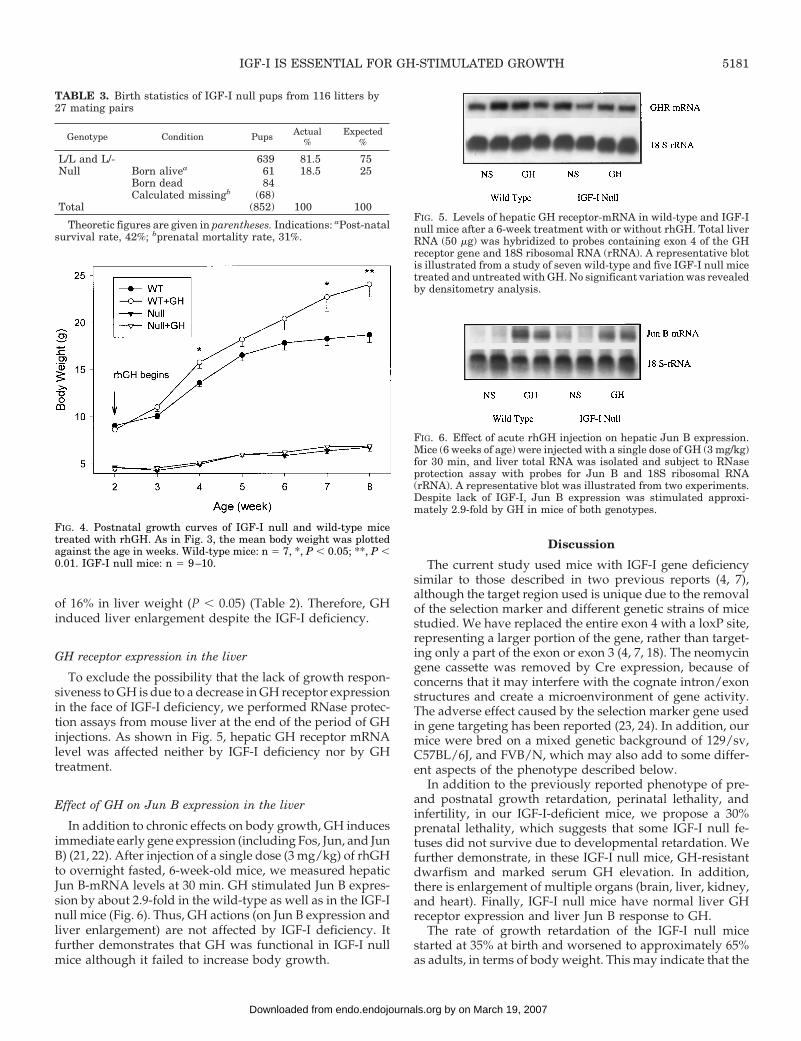
The effect of GH on body growth was studied in wild-type and IGF-I null mice by injecting rhGH from 2 to 8weeks of age. In wild-type mice, we observed two phasesof postnatal growth: an early phase of rapid growth in thefirst 2–3 weeks of life, which cannot be further stimulatedby GH injection (data not shown), and a peripubertalgrowth spurt from 3 weeks, which can be further increasedby GH injection (Fig. 4). From 2 weeks on, the IGF-I nullmice grow more slowly and have delayed onset of theprepubertal growth and do not undergo pubertal changes.Their final body weight becomes approximately one thirdand body length about two thirds of their wild-type littermates as adults. As shown in Fig. 4 and Table 2, continuousinjection of rhGH did not affect their growth rate at all,when measured both by weight and length. In contrast,wild-type mice receiving the same doses of rhGH exhib-ited accelerated growth starting at P21 and continuing toP56, where they showed a 29% increase in body weightand 12% in body length in comparison to those treatedwith diluent alone.
Changes in organ weight caused by IGF-I deficiency andGH injection
At the end of the 6-week injection, we measured the weightof several organs. In wild-type mice, GH induced a signifi-cant, 27% increase in relative liver weight, decreased therelative weight of the brain (220%) and kidney (222%) (dueto the increase in body weight, because the absolute weightsof organs other than liver were unchanged), but had no effecton the relative weight of the heart and the spleen (Table 2).As stated earlier, IGF-I null mice have enlarged brain, liver,heart, and kidney and GH injection caused a further increase
FIG. 3. Postnatal growth curves of IGF-I null vs. wild-type mice. Theaverage body weight vs. age of the wild-type (n $ 7) and IGF-I null(n $ 9) mice were plotted. The differences between these two groupsare statistically significant at all time points (P , 0.001). To ensureadequate postweaning survival, IGF-I null mice were weaned at 5weeks while wild-type mice were weaned at 3 weeks of age.
TABLE 1. Changes in serum chemistry in IGF-I null mice
Wild-type IGF-I null P value
IGF-I (ng/ml) 442 6 52 (5) UD (8)GH (ng/ml) 6.0 6 2.5 (14) 74.5 6 9.4 (8) ,0.001Glucose (mg/dl) 105 6 6 (14) 55 6 12 (8) ,0.001Insulin (ng/ml) 0.28 6 0.06 (14) 0.48 6 0.09 (7) 0.075Alkaline phosphatase (U/liter) 382 6 104 (8) 161 6 24 (7) ,0.001
Animals of 5–8 weeks old were fasted overnight before sampling. All values are presented as mean 6 SE. Sample sizes are indicated inparentheses. The statistical significance between the two groups was tested using the t test. UD, Undetectable.
TABLE 2. Changes of organ weight (as a % of body weight) and body length (in cm) after 6-week course of rhGH treatment
WT (n 5 7) Wild-type 1 GH (n 5 7) Null (n 5 9) Null 1 GH (n 5 10)
Liver 5.15 6 0.13 6.55 6 0.17a 7.15 6 0.17b 8.28 6 0.17a
Brain 2.27 6 0.10 1.81 6 0.07c 4.55 6 0.24b 4.38 6 0.15Kidney 1.32 6 0.05 1.03 6 0.10d 1.57 6 0.04b 1.58 6 0.03Heart 0.633 6 0.048 0.599 6 0.034 0.837 6 0.033b 0.906 6 0.038Spleen 0.337 6 0.029 0.366 6 0.021 0.260 6 0.026 0.280 6 0.024Body length 8.4 6 0.1 9.4 6 0.2a 5.6 6 0.1b 5.6 6 0.1
As in Table 1, all values are presented as mean 6 SE. The statistical significance between two groups was tested using the t test. (aP , 0.001vs. untreated; bP , 0.001 vs. WT; cP , 0.01 vs. untreated; dP , 0.05) vs. untreated.
5180 IGF-I IS ESSENTIAL FOR GH-STIMULATED GROWTH Endo • 1999Vol 140 • No 11
by on March 19, 2007 endo.endojournals.orgDownloaded from
of 16% in liver weight (P , 0.05) (Table 2). Therefore, GHinduced liver enlargement despite the IGF-I deficiency.
GH receptor expression in the liver
To exclude the possibility that the lack of growth respon-siveness to GH is due to a decrease in GH receptor expressionin the face of IGF-I deficiency, we performed RNase protec-tion assays from mouse liver at the end of the period of GHinjections. As shown in Fig. 5, hepatic GH receptor mRNAlevel was affected neither by IGF-I deficiency nor by GHtreatment.
Effect of GH on Jun B expression in the liver
In addition to chronic effects on body growth, GH inducesimmediate early gene expression (including Fos, Jun, and JunB) (21, 22). After injection of a single dose (3 mg/kg) of rhGHto overnight fasted, 6-week-old mice, we measured hepaticJun B-mRNA levels at 30 min. GH stimulated Jun B expres-sion by about 2.9-fold in the wild-type as well as in the IGF-Inull mice (Fig. 6). Thus, GH actions (on Jun B expression andliver enlargement) are not affected by IGF-I deficiency. Itfurther demonstrates that GH was functional in IGF-I nullmice although it failed to increase body growth.
Discussion
The current study used mice with IGF-I gene deficiencysimilar to those described in two previous reports (4, 7),although the target region used is unique due to the removalof the selection marker and different genetic strains of micestudied. We have replaced the entire exon 4 with a loxP site,representing a larger portion of the gene, rather than target-ing only a part of the exon or exon 3 (4, 7, 18). The neomycingene cassette was removed by Cre expression, because ofconcerns that it may interfere with the cognate intron/exonstructures and create a microenvironment of gene activity.The adverse effect caused by the selection marker gene usedin gene targeting has been reported (23, 24). In addition, ourmice were bred on a mixed genetic background of 129/sv,C57BL/6J, and FVB/N, which may also add to some differ-ent aspects of the phenotype described below.
In addition to the previously reported phenotype of pre-and postnatal growth retardation, perinatal lethality, andinfertility, in our IGF-I-deficient mice, we propose a 30%prenatal lethality, which suggests that some IGF-I null fe-tuses did not survive due to developmental retardation. Wefurther demonstrate, in these IGF-I null mice, GH-resistantdwarfism and marked serum GH elevation. In addition,there is enlargement of multiple organs (brain, liver, kidney,and heart). Finally, IGF-I null mice have normal liver GHreceptor expression and liver Jun B response to GH.
The rate of growth retardation of the IGF-I null micestarted at 35% at birth and worsened to approximately 65%as adults, in terms of body weight. This may indicate that the
FIG. 4. Postnatal growth curves of IGF-I null and wild-type micetreated with rhGH. As in Fig. 3, the mean body weight was plottedagainst the age in weeks. Wild-type mice: n 5 7, *, P , 0.05; **, P ,0.01. IGF-I null mice: n 5 9–10.
FIG. 5. Levels of hepatic GH receptor-mRNA in wild-type and IGF-Inull mice after a 6-week treatment with or without rhGH. Total liverRNA (50 mg) was hybridized to probes containing exon 4 of the GHreceptor gene and 18S ribosomal RNA (rRNA). A representative blotis illustrated from a study of seven wild-type and five IGF-I null micetreated and untreated with GH. No significant variation was revealedby densitometry analysis.
FIG. 6. Effect of acute rhGH injection on hepatic Jun B expression.Mice (6 weeks of age) were injected with a single dose of GH (3 mg/kg)for 30 min, and liver total RNA was isolated and subject to RNaseprotection assay with probes for Jun B and 18S ribosomal RNA(rRNA). A representative blot was illustrated from two experiments.Despite lack of IGF-I, Jun B expression was stimulated approxi-mately 2.9-fold by GH in mice of both genotypes.
TABLE 3. Birth statistics of IGF-I null pups from 116 litters by27 mating pairs
Genotype Condition Pups Actual%
Expected%
L/L and L/- 639 81.5 75Null Born alivea 61 18.5 25
Born dead 84Calculated missingb (68)
Total (852) 100 100
Theoretic figures are given in parentheses. Indications: aPost-natalsurvival rate, 42%; bprenatal mortality rate, 31%.
IGF-I IS ESSENTIAL FOR GH-STIMULATED GROWTH 5181
by on March 19, 2007 endo.endojournals.orgDownloaded from

fetal development of the IGF-I null mice is partially com-pensated by IGF-I produced from the placenta or possibly thepresence of IGF-II and insulin. The placenta produces a largenumber of hormones, including GH, IGF-I, and IGF-II, thatmay reach the fetus (25, 26). Deficiency in either IGF-II orinsulin contributes to intrauterine growth retardation. Birthweights of IGF-II knockout mice are 60% and IGF-I/II doubleknockout are only 30% of their wild-type litter mates (27, 28),while insulin-1 and -2 double knockouts also induce 15–20%growth retardation in mouse embryos (29). After birth, afurther growth retardation was observed, since the pupswere no longer influenced by placental IGF-I, mouse IGF-IIlevel diminishes quickly, and the role of insulin on postnatalgrowth is apparently very limited (29).
According to the somatomedin hypothesis, production ofIGF-I from the liver and other tissues is regulated by GHrelease from the pituitary. Like many other physiologicalsystems, overproduction of IGF-I sends a signal to inhibit GHproduction via a long loop (involving GHRH and soma-tostatin) and a short loop (directly on somatotrophs) negativefeedback mechanism (30–32). It is well established that ex-ogenous IGF-I, when administered in vivo, suppresses GHsecretion, while IGF-I deficiency is accompanied by GH hy-persecretion (20, 23, 33). In a separate study, when IGF-Iproduction was abolished exclusively from the liver (usingalbumin-Cre/loxP system), we effectively decreased serumIGF-I level by 75% without affecting the growth and devel-opment of extrahepatic tissues. Serum GH increased 6.4-foldin these mice, suggesting that circulating IGF-I affects GHsecretion (17). The current study abolished both endocrineand paracrine IGF-I production and thereby increased serumGH level even further to 12.4-fold. The fold increase in serumGH concentration correlates well with serum IGF-I levelsunder these circumstances. Therefore, our studies demon-strate the presence of a potent negative feedback on GHsecretion by IGF-I.
A central issue in the somatomedin hypothesis is that GHacts through IGF-I production from the liver or local tissuesin stimulating body growth. There have been suggestionsthat GH exerts some direct effects on target tissues that arenot mediated by IGF-I (15, 16, 25, 34). The current studydemonstrates that IGF-I null mice are selectively resistant toGH in terms of body growth, directly confirming that GHworks exclusively via IGF-I production in this aspect. This isconsistent with an earlier report on the ineffectiveness of GHon weight gain of IGF-I null mice (35). As for the role of IGF-Iin the circulation or as a paracrine growth factor, our studyon liver-specific IGF-I gene deletion demonstrated that liver-derived endocrine IGF-I is not essential for postnatal growth(17, 36). These studies, as well as those by other investigators(37), demonstrate that IGF-I, most probably produced bynonhepatic tissues, is essential for GH-stimulated postnatalgrowth.
One exception to the above conclusion is that the profounddefect in development and metabolism caused by IGF-I de-ficiency may have impaired the ability of GH and its othergrowth mediators to promote body growth. To address thisquestion, we have demonstrated that IGF-I null mice havenormal levels of GH receptor mRNA in the liver and thatrhGH caused a significant increase in liver size. Furthermore,
in an acute experiment, we tested the effect of rhGH on liverJun B expression. Early response genes encode for transcrip-tion factors including Fos, Jun, Jun B, and Myc, which can beinduced by GH and are involved in activating DNA syn-thesis, downstream gene expression, and cell proliferation(21, 22). As expected, Jun B expression was activated within30 min of an in vivo rhGH injection in wild-type mice. Theresponse in IGF-I null mice was virtually identical, furtherdemonstrating the presence of a functional GH signalingsystem. The current study therefore does not support directGH action on growth and indicates that those IGF-I-inde-pendent mechanisms of GH at least in mice, are not involvedin body growth regulation.
Defects in GH receptor or downstream targets (such asSTAT5b, HNF-1a, and IGF-I) cause GH insensitivity (GHIS)or Laron Syndrome. Patients with GH receptor gene muta-tions show normal prenatal development, severe postnatalgrowth retardation, high serum GH, and low levels of serumIGF-I that cannot be corrected by GH injection (38). A secondclass of Laron type dwarfism, induced by HNF-1a deficiencyin mice, demonstrates normal GH receptor level, elevatedserum GH, and diminished IGF-I and insulin concentrations(23). These HNF-1a null mice are viable and sterile anddevelop non-insulin-dependent diabetes mellitus. Defi-ciency in STAT5b (signal transducer and activator of tran-scription 5b), another transcription factor downstream ofGH, also causes Laron type of dwarfism with virtually iden-tical features to HNF-1a (hepatocyte nuclear factor 1a) nullmice (39). Defects in IGF-I production are rare, probably dueto intrauterine lethality. The only case of IGF-I deficiencyreported features growth failure before and after birth, highGH level, and resistance to GH treatment (6). In agreementwith the report on patients, our data demonstrate a dramaticincrease in serum GH level and the inability of GH to pro-mote body growth in the face of IGF-I deficiency. It clearlydefines a new class of GH-resistant dwarfism (similar toGHIS) and proves that IGF-I is essential for GH-stimulatedpostnatal growth. This mouse model provides a valuable toolwith which to explore the relationship of the GH-IGF-I axisand to provide therapeutic modalities to treat such dwarfism.
The finding of multiple organ enlargement (relativeweight) in IGF-I null mice is an extension of previouslyobserved weight increases of the brain and the liver and maybe the result of a sustained GH hypersecretion (7, 40). Toexclude the influence of an overall change in body weight, weexpressed the organ weight as per body weight and foundthat IGF-I null mice have a 21–113% increase in the weightof the kidney, heart, liver, and brain. GH further inducedchanges in both wild-type and IGF-I null mice. In wild-typemice, chronic rhGH administration increased the weight ofthe liver by 27% but decreased the relative weights of thebrain and the kidney. In IGF-I null mice, a 15% increase inliver weight, caused by GH treatment, and no change in otherorgans were observed.
The relationship of GH with organ growth has been wellstudied. For example, prolonged excessive secretion of GHby GH3 tumor cells in rats induced widespread visceromeg-aly affecting the liver, kidney, spleen, heart, and adrenals thatwas associated with an increase in DNA synthesis (41); trans-genic mice overexpressing GH have selective splanchnomeg-
5182 IGF-I IS ESSENTIAL FOR GH-STIMULATED GROWTH Endo • 1999Vol 140 • No 11
by on March 19, 2007 endo.endojournals.orgDownloaded from

aly coupled with glomerular sclerosis and hepatomegaly(42); pulsatile administration of GH in broiler pullets in-duced hepatomegaly due largely to cellular hypertrophy(43); and GH-transgenic mice have accelerated liver, kidney,and skeletal growth (44). Because the IGF-I null mice have noIGF-I production and a marked increase in GH secretion, wepropose an IGF-I-independent, direct GH action on hepato-cyte growth and proliferation under conditions of bothIGF-I-deficient status and when GH was exogenously ad-ministered.
Analysis of a standard serum chemistry profile revealed amarked decrease in alkaline phosphatase and glucose con-centrations among IGF-I null mice. Alkaline phosphatasefrom the bone and other tissues is essential for normal skel-etal mineralization and growth (45–47). IGF-I has been foundto influence alkaline phosphatase activity as part of themechanism of stimulating bone formation. For example, lo-cally infused IGF-I into mouse femurs can stimulate expres-sion of alkaline phosphatase and bone formation; the age-dependent decline in bone formation can be attributed in partto a decline in local IGF-I expression and response (48); andIGF-I stimulates alkaline phosphatase activity and type Iprocollagen mRNA expression in osteoblastic cells in vitro(49). The current study demonstrates severe hypophosphata-sia due to IGF-I deficiency and suggests that alkaline phos-phatase activity may be an important downstream target ofIGF-I action on postnatal skeletal growth.
In conclusion, using the Cre/loxP system, we have gen-erated IGF-I null mice that have a 42% postnatal survival rateand enable an in-depth postnatal study. These mice exhibitedmultiple organ enlargement accompanied by decreased se-rum alkaline phosphatase and glucose concentrations. Theincrease in serum GH concentration and failure of GH instimulating body growth in these IGF-I null mice clearlydemonstrate GH resistance (with respect to body growth)and the role of IGF-I in feedback control of GH secretion.
Acknowledgments
The authors wish to thank Dr. John Kopchick (Ohio University,Athens, OH) for providing the GH receptor probe; Dr. Peter Rotwein(Oregon Health Sciences University, Portland, OR) for the IGF-I exon 4probe; Bernice Samuels (NIDDK, NIH, Bethesda, MD) for performinginsulin RIA; Genentech, Inc. (South San Francisco, CA) for providing therhGH; and Drs. Matthew Rechler, Mark Sperling, Michael Karas, LilianaUribe, and David Kleiner for helpful communications.
References
1. Isaksson OG, Eden S, Jansson JO 1985 Mode of action of pituitary growthhormone on target cells. Annu Rev Physiol 47:483–499
2. Zhou Y, Xu BC, Maheshwari HG, He L, Reed M, Lozykowski M, Okada S,Cataldo L, Coschigamo K, Wagner TE, Baumann G, Kopchick JJ 1997 Amammalian model for Laron syndrome produced by targeted disruption of themouse growth hormone receptor/binding protein gene (the Laron mouse).Proc Natl Acad Sci USA 94:13215–13220
3. Donahue LR, Beamer WG 1993 Growth hormone deficiency in ’little’ miceresults in aberrant body composition, reduced insulin-like growth factor-I andinsulin-like growth factor-binding protein-3 (IGFBP-3), but does not affectIGFBP-2, -1 or -4. J Endocrinol 136:91–104
4. Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A 1993 Mice carryingnull mutations of the genes encoding insulin-like growth factor I (Igf-1) andtype 1 IGF receptor (Igf1r). Cell 75:59–72
5. Baker J, Liu JP, Robertson EJ, Efstratiadis A 1993 Role of insulin-like growthfactors in embryonic and postnatal growth. Cell 75:73–82
6. Woods KA, Camacho-Hubner C, Savage MO, Clark AJ 1996 Intrauterine
growth retardation and postnatal growth failure associated with deletion of theinsulin-like growth factor I gene. N Engl J Med 335:1363–1367
7. Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S,Dalton D, Gillett N, Stewart TA 1993 IGF-I is required for normal embryonicgrowth in mice. Genes Dev 7:2609–2617
8. Carter-Su C, Schwartz J, Smith LS 1996 Molecular mechanism of growthhormone action. Annu Rev Physiol 58:187–207
9. Daughaday WH 1997 From sulphation factor to IGF-I, 40 years of research onthe regulation of cartilage growth. In: Takano K, Hizuka N, Takahashi S-I (eds)The 4th International Symposium on Insulin-like Growth Factors. Elsevier,Tokyo, Japan
10. Daughaday WH, Hall K, Raben MS, Salmon Jr WD, Brande JLvd, Wyk JJv1972 Somatomedin: proposed designation for sulphation factor. Nature235:107–109
11. Isaksson OG, Lindahl A, Nilsson A, Isgaard J 1987 Mechanism of the stim-ulatory effect of growth hormone on longitudinal bone growth. Endocr Rev8:426–438
12. Isaksson OG, Jansson JO, Gause IA 1982 Growth hormone stimulates lon-gitudinal bone growth directly. Science 216:1237–1239
13. Schlechter NL, Russell SM, Spencer EM, Nicoll CS 1986 Evidence suggestingthat the direct growth-promoting effect of growth hormone on cartilage in vivois mediated by local production of somatomedin. Proc Natl Acad Sci USA83:7932–7934
14. Li H, Bartold PM, Zhang CZ, Clarkson RW, Young WG, Waters MJ 1998Growth hormone and insulin-like growth factor I induce bone morphogeneticproteins 2 and 4: a mediator role in bone and tooth formation? Endocrinology139:3855–3862
15. Lindahl A, Nilsson A, Isaksson OG 1987 Effects of growth hormone andinsulin-like growth factor-I on colony formation of rabbit epiphyseal chon-drocytes at different stages of maturation. J Endocrinol 115:263–271
16. Billestrup N, Nielsen JH 1991 The stimulatory effect of growth hormone,prolactin, and placental lactogen on b-cell proliferation is not mediated byinsulin-like growth factor-I. Endocrinology 129:883–888
17. Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D 1999Normal growth and development in the absence of hepatic insulin-like growthfactor I. Proc Natl Acad Sci USA 96:7324–7329
18. Liu JL, Grinberg A, Westphal H, Sauer B, Accili D, Karas M, LeRoith D 1998Insulin-like growth factor-I affects perinatal lethality and postnatal develop-ment in a gene dosage-dependent manner: manipulation using the Cre/loxPsystem in transgenic mice. Mol Endocrinol 12:1452–1462
19. Werner H, Woloschak M, Adamo M, Shen-Orr Z, Roberts Jr CT, LeRoith D1989 Developmental regulation of the rat insulin-like growth factor I receptorgene. Proc Natl Acad Sci USA 86:7451–7455
20. Lembo G, Rockman HA, Hunter JJ, Steinmetz H, Koch WJ, Ma L, Prinz MP,Ross Jr J, Chien KR, Powell-Braxton L 1996 Elevated blood pressure andenhanced myocardial contractility in mice with severe IGF-1 deficiency. J ClinInvest 98:2648–2655
21. Slootweg MC, de Groot RP, Herrmann-Erlee MP, Koornneef I, Kruijer W,Kramer YM 1991 Growth hormone induces expression of c-jun and jun Boncogenes and employs a protein kinase C signal transduction pathway for theinduction of c-fos oncogene expression. J Mol Endocrinol 6:179–188
22. Rotwein P, Gronowski AM, Thomas MJ 1994 Rapid nuclear actions of growthhormone. Horm Res 42:170–175
23. Lee YH, Sauer B, Gonzalez FJ 1998 Laron dwarfism and non-insulin-depen-dent diabetes mellitus in the Hnf-1a knockout mouse. Mol Cell Biol18:3059–3068
24. Artelt P, Grannemann R, Stocking C, Friel J, Bartsch J, Hauser H 1991 Theprokaryotic neomycin-resistance-encoding gene acts as a transcriptional si-lencer in eukaryotic cells. Gene 99:249–254
25. Bauer MK, Harding JE, Bassett NS, Breier BH, Oliver MH, Gallaher BH,Evans PC, Woodall SM, Gluckman PD 1998 Fetal growth and placentalfunction. Mol Cell Endocrinol 140:115–120
26. Bassett NS, Breier BH, Hodgkinson SC, Davis SR, Henderson HV, Gluck-man PD 1990 Plasma clearance of radiolabelled IGF-1 in the late gestationovine fetus. J Dev Physiol 14:73–79
27. DeChiara TM, Efstratiadis A, Robertson EJ 1990 A growth-deficiency phe-notype in heterozygous mice carrying an insulin-like growth factor II genedisrupted by targeting. Nature 345:78–80
28. Louvi A, Accili D, Efstratiadis A 1997 Growth-promoting interaction of IGF-IIwith the insulin receptor during mouse embryonic development. Dev Biol189:33–48
29. Duvillie B, Cordonnier N, Deltour L, Dandoy-Dron F, Itier JM, MonthiouxE, Jami J, Joshi RL, Bucchini D 1997 Phenotypic alterations in insulin-deficientmutant mice. Proc Natl Acad Sci USA 94:5137–5140
30. Yamashita S, Melmed S 1987 Insulin-like growth factor I regulation of growthhormone gene transcription in primary rat pituitary cells. J Clin Invest79:449–452
31. Tannenbaum GS, Guyda HJ, Posner BI 1983 Insulin-like growth factors: a rolein growth hormone negative feedback and body weight regulation via brain.Science 220:77–79
32. Berelowitz M, Szabo M, Frohman LA, Firestone S, Chu L, Hintz RL 1981
IGF-I IS ESSENTIAL FOR GH-STIMULATED GROWTH 5183
by on March 19, 2007 endo.endojournals.orgDownloaded from

Somatomedin-C mediates growth hormone negative feedback by effects onboth the hypothalamus and the pituitary. Science 212:1279–1281
33. Hartman ML, Clayton PE, Johnson ML, Celniker A, Perlman AJ, Alberti KG,Thorner MO 1993 A low dose euglycemic infusion of recombinant humaninsulin-like growth factor I rapidly suppresses fasting-enhanced pulsatilegrowth hormone secretion in humans. J Clin Invest 91:2453–2462
34. Green H, Morikawa M, Nixon T 1985 A dual effector theory of growth-hormone action. Differentiation 29:195–198
35. Won W, Powell-Braxton L 1997 Insulin-like growth factor gene targeting. In:Takano K, Hizuka N, Takahashi S-I (eds) The 4th International Symposium onInsulin-like Growth Factors. Elsevier, Tokyo, Japan, pp 57–63
36. Sjogren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, TornellJ, Isaksson OG, Jansson JO, Ohlsson C 1999 Liver-derived insulin-like growthfactor I (IGF-I) is the principal source of IGF-I in blood but is not required forpostnatal body growth in mice. Proc Natl Acad Sci USA 96:7088–7092
37. Wang J, Zhou J, Powell-Braxton L, Bondy C 1999 Effects of Igf1 gene deletionon postnatal growth patterns. Endocrinology 140:3391–3394
38. Rosenfeld RG, Rosenbloom AL, Guevara-Aguirre J 1994 Growth hormone(GH) insensitivity due to primary GH receptor deficiency. Endocr Rev15:369–390
39. Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ,Davey HW 1997 Requirement of STAT5b for sexual dimorphism of bodygrowth rates and liver gene expression. Proc Natl Acad Sci USA 94:7239–7244
40. Beck KD, Powell-Braxton L, Widmer HR, Valverde J, Hefti F 1995 Igf1 genedisruption results in reduced brain size, CNS hypomyelination, and loss ofhippocampal granule and striatal parvalbumin-containing neurons. Neuron14:717–730
41. Prysor-Jones RA, Jenkins JS 1980 Effect of excessive secretion of growthhormone on tissues of the rat, with particular reference to the heart and skeletalmuscle. J Endocrinol 85:75–82
42. Quaife CJ, Mathews LS, Pinkert CA, Hammer RE, Brinster RL, Palmiter RD1989 Histopathology associated with elevated levels of growth hormone andinsulin-like growth factor I in transgenic mice. Endocrinology 124:40–48
43. Cravener TL, Vasilatos-Younken R, Andersen BJ 1990 Hepatomegaly in-duced by the pulsatile, but not continuous, intravenous administration ofpurified chicken growth hormone in broiler pullets: liver composition andnucleic-acid content. Poult Sci 69:845–848
44. Wanke R, Hermanns W, Folger S, Wolf E, Brem G 1991 Accelerated growthand visceral lesions in transgenic mice expressing foreign genes of the growthhormone family: an overview. Pediatr Nephrol 5:513–521
45. Whyte MP 1994 Hypophosphatasia and the role of alkaline phosphatase inskeletal mineralization. Endocr Rev 15:439–461
46. Narisawa S, Frohlander N, Millan JL 1997 Inactivation of two mouse alkalinephosphatase genes and establishment of a model of infantile hypophosphata-sia. Dev Dyn 208:432–446
47. Weiss MJ, Cole DE, Ray K, Whyte MP, Lafferty MA, Mulivor R, Harris H1989 First identification of a gene defect for hypophosphatasia: evidence thatalkaline phosphatase acts in skeletal mineralization. Connect Tissue Res 21:99–104; discussion 104–106
48. Wakisaka A, Tanaka H, Barnes J, Liang CT 1998 Effect of locally infused IGF-Ion femoral gene expression and bone turnover activity in old rats. J Bone MinerRes 13:13–19
49. Chihara K, Sugimoto T 1997 The action of GH/IGF-I/IGFBP in osteoblastsand osteoclasts. Horm Res 48 [Suppl] 5:45–49
5184 IGF-I IS ESSENTIAL FOR GH-STIMULATED GROWTH Endo • 1999Vol 140 • No 11
by on March 19, 2007 endo.endojournals.orgDownloaded from