interactions of mitochondria-targeted and untargeted ... · interactions of mitochondria-targeted...
TRANSCRIPT

Interactions of Mitochondria-targeted and Untargeted Ubiquinoneswith the Mitochondrial Respiratory Chain andReactive Oxygen SpeciesIMPLICATIONS FOR THE USE OF EXOGENOUS UBIQUINONES AS THERAPIES ANDEXPERIMENTAL TOOLS*�
Received for publication, February 9, 2005Published, JBC Papers in Press, March 23, 2005, DOI 10.1074/jbc.M501527200
Andrew M. James‡, Helena M. Cocheme‡§, Robin A. J. Smith¶, and Michael P. Murphy‡�
From the ‡Medical Research Council Dunn Human Nutrition Unit, Wellcome Trust/MRC Building, Hills Road,Cambridge CB2 2XY, United Kingdom and the ¶Department of Chemistry, University of Otago,P. O. Box 56, Dunedin, New Zealand
Antioxidants, such as ubiquinones, are widely usedin mitochondrial studies as both potential therapiesand useful research tools. However, the effects of ex-ogenous ubiquinones can be difficult to interpret be-cause they can also be pro-oxidants or electron carri-ers that facilitate respiration. Recently we developed amitochondria-targeted ubiquinone (MitoQ10) that ac-cumulates within mitochondria. MitoQ10 has beenused to prevent mitochondrial oxidative damage andto infer the involvement of mitochondrial reactive ox-ygen species in signaling pathways. However, uncer-tainties remain about the mitochondrial reduction ofMitoQ10, its oxidation by the respiratory chain, and itspro-oxidant potential. Therefore, we compared MitoQanalogs of varying alkyl chain lengths (MitoQn, n �3–15) with untargeted exogenous ubiquinones. Wefound that MitoQ10 could not restore respiration inubiquinone-deficient mitochondria because oxidationof MitoQ analogs by complex III was minimal. ComplexII and glycerol 3-phosphate dehydrogenase reducedMitoQ analogs, and the rate depended on chain length.Because of its rapid reduction and negligible oxida-tion, MitoQ10 is a more effective antioxidant againstlipid peroxidation, peroxynitrite and superoxide. Par-adoxically, exogenous ubiquinols also autoxidize togenerate superoxide, but this requires their deproton-ation in the aqueous phase. Consequently, in the pres-ence of phospholipid bilayers, the rate of autoxidationis proportional to ubiquinol hydrophilicity. Superox-ide production by MitoQ10 was insufficient to damageaconitase but did lead to hydrogen peroxide produc-tion and nitric oxide consumption, both of which mayaffect cell signaling pathways. Our results comprehen-sively describe the interaction of exogenous ubiquino-nes with mitochondria and have implications for theirrational design and use as therapies and as researchtools to probe mitochondrial function.
Mitochondria are the major site of reactive oxygen speciesgeneration (ROS)1 within cells (1, 2). When ROS productionexceeds the capacity of detoxification and repair pathways,oxidative damage to protein, DNA, and phospholipid occurs,disrupting mitochondrial oxidative phosphorylation and lead-ing to cell damage and death. This contributes to a number ofhuman pathologies including Parkinson disease, Alzheimerdisease, Friedreich ataxia, ischemia-reperfusion injury, diabe-tes, and aging (2, 3). In addition to this pathological role, ROScan also act as redox signaling molecules (4, 5). Hence, mito-chondrial ROS production and oxidative damage are attractivetargets for pharmacological intervention for both therapeuticand investigative purposes (6–10).
Antioxidants have the potential to block oxidative damageand redox signaling, and exogenous ubiquinones have beenwidely used for this purpose in mitochondrial studies. Thesemolecules are based on the predominant human form of endog-enous ubiquinone, coenzyme Q10 (CoQ10; Fig. 1A), which issynthesized in the mitochondrial inner membrane and com-prises a ubiquinone head group attached to a tail of 10 five-carbon isoprenoid units (11). The ubiquinone moiety is redox-active, accepting two electrons and two protons in its reductionto a ubiquinol, while the extremely hydrophobic tail ensuresthat within the cell it is almost exclusively associated withphospholipid bilayers (Fig. 1B). The redox activity of theubiquinone moiety enables it to act as a mobile electron carrierin the mitochondrial inner membrane where it is reduced to aubiquinol by several membrane bound dehydrogenases andoxidized back to a ubiquinone by complex III. Furthermore, thereduced ubiquinol form of CoQ10 has an important protectivefunction as a chain breaking antioxidant, terminating lipid
* This work was supported by the Medical Research Council, Foun-dation for Research, Science and Technology New Zealand and byAntipodean Biotechnology. The costs of publication of this article weredefrayed in part by the payment of page charges. This article musttherefore be hereby marked “advertisement” in accordance with 18U.S.C. Section 1734 solely to indicate this fact.
� This article was selected as a Paper of the Week.§ Recipient of a Ph.D. Studentship from Research into Ageing, United
Kingdom.� To whom correspondence should be addressed. Tel.: 44-1223-
252900; Fax: 44-1223-252905; E-mail: [email protected].
1 The abbreviations used are: ROS, reactive oxygen species; �COQ2,coenzyme Q-deficient yeast strain; cyt c, cytochrome c; cyt cacet, acety-lated cytochrome c; BHM, bovine heart mitochondrial membranes;CoQ0, 2,3-dimethoxy-5-methyl-1,4-benzoquinone; CoQ1–10, coenzyme Qwith a tail of 1–10 isoprenoid units (2,3-dimethoxy-5-methyl-6-polypre-nyl-1,4-benzoquinone); CsA, cyclosporin A; DETA-NONOate, 3,3-bis(aminoethyl)-1-hydroxy-2-oxo-1-triazene; FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone; G3P, glycerol 3-phosphate; G3PDH,glycerol-3-phosphate dehydrogenase; MitoQ, ubiquinone linked to atriphenylphosphonium cation by an alkyl chain of unspecified length;MitoQ3–15, ubiquinone linked to a triphenylphosphonium cation by analkyl chain of 3–15 carbons; PA, cis-parinaric acid; PBS, phosphate-buffered saline; PTP, mitochondrial permeability transition pore; SOD,superoxide dismutase; tBHP, t-butyl-hydroperoxide; TPP, triphen-ylphosphonium cation; WT, wild-type yeast strain; TPMP, methyltri-phenylphosphonium; BSA, bovine serum albumin; HPLC, high per-formance liquid chromatography.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 280, No. 22, Issue of June 3, pp. 21295–21312, 2005© 2005 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org 21295
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

peroxidation in phospholipid bilayers (12, 13). Therefore,ubiquinone supplementation is an attractive therapeutic strat-egy in human pathology, as it could both stimulate oxidativephosphorylation by complementing any defects in respiration(14) and protect against oxidative damage (15). However, this
duality complicates experimental interpretation, as any effectsof CoQ10 can result from its interaction with oxidative phos-phorylation, oxidative damage, or redox signaling pathways.
While positive therapeutic effects have been observed withCoQ10 supplementation in humans (16–19), its oral bioavail-
FIG. 1. Ubiquinones used in this study. A, structures. B, octan-1-ol/PBS partition coefficients: a, calculated using Advanced ChemistryDevelopment (ACD) Software Solaris v4.67 as described in Smith et al. (31); b, determined experimentally in this study; c, determinedexperimentally in Kelso et al. (29); d, determined experimentally in Asin-Cayuela et al. (30). C, a schematic of MitoQ10 in a phospholipid bilayercomposed of C18 saturated fatty acids. The TPP cation and the alkyl chain favor the polar surface and hydrophobic core, respectively. The preferredposition of the uncharged but moderately hydrophilic ubiquinone moiety is less clear.
Exogenous Ubiquinones as Therapies and Tools21296
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

ability is poor due to its extreme hydrophobicity (Fig. 1B).Consequently, only a small fraction of orally administeredCoQ10 reaches the circulatory system, and augmentation ofmitochondrial CoQ10 content is lower still (20–22). Thereforethe beneficial effects of exogenous CoQ10 require high dosesand long term administration, and only subjects whose CoQ10
levels have been depleted by defective synthesis, age, or diseaseare responsive (18, 20–24). The negligible water solubility ofCoQ10 and its poor diffusion to mitochondria in cultured cellsalso hinder its usefulness as a tool to study mitochondrialoxidative damage and redox signaling in vitro. As a result thereis considerable interest in developing artificial ubiquinoneswith better bioavailability and pharmacokinetic properties.Idebenone is one such compound and it comprises a ubiquinonehead group attached to a ten carbon alkyl tail with a terminalhydroxyl (Fig. 1A) (25). Clinical trials with idebenone haveshown it can ameliorate cardiomyopathy in Friedreich ataxiapatients (26–28).
Although decreasing hydrophobicity improves overall bio-availability, it would also be of benefit to target ubiquinonesspecifically to mitochondria, as they are the main site of ubiqui-none utilization but represent only a small fraction of the cellvolume. Lipophilic cations, such as methyltriphenylphospho-nium (TPMP; Fig. 1A), are accumulated several hundred-foldby the large membrane potential (negative inside) generated bymitochondria during oxidative phosphorylation (6, 9). We haveexploited this property by covalently attaching a ubiquinonemoiety to the lipophilic triphenylphosphonium (TPP) cationgenerating a mitochondria-targeted ubiquinone (MitoQ10; Fig.1A), which is selectively accumulated within isolated mitochon-dria, and within mitochondria in cells and in vivo (9, 29–31).The interaction of amphipathic alkyltriphenylphosphoniumcations with phospholipid bilayers occurs as follows: the TPPlipophilic cation is bound as a monolayer in a potential well atabout the level of the phospholipid fatty acid carbonyls, whilethe hydrophobic alkyl chain is inserted into the hydrophobiccore of the membrane (31–34). This is illustrated for MitoQ10 inFig. 1C. Although the large ionic radius and hydrophobicity ofthe TPP cation allows molecules such as MitoQ10 to permeatephospholipid bilayers readily, their steady-state concentrationwithin the hydrophobic core of the membrane is low. Further-more, within energized mitochondria the membrane potentialcauses most MitoQ10 to be adsorbed to the matrix surface of theinner membrane. MitoQ10 is a particularly effective antioxi-dant against lipid peroxidation (29, 30) and has been used in arange of studies of mitochondrial dysfunction and oxidativestress where its mitochondrial localization has enabled the siteof intracellular redox signaling to be inferred (35–42). How-ever, the details of the mitochondrial processes affected byMitoQ10 remain unclear because it could act as an antioxidant(thereby blocking oxidative damage and redox signaling) or asan electron carrier in the respiratory chain (thereby stimulat-ing oxidative phosphorylation). A further consideration is that,while ubiquinols are potent antioxidants, there are also par-tially reduced and/or partially protonated intermediate formsthat can act as pro-oxidants through interacting with oxygen toform superoxide (O2
.) (13, 43, 44). The implications of autoxi-dation are unclear as stress response pathways that boostantioxidant defenses are often switched on by mild oxidativestress (a process termed hormesis). As a result autoxidationcould paradoxically contribute to conditioning and protectionagainst subsequent oxidative stress (4, 45, 46).
In summary, ubiquinone augmentation is an attractive ther-apy for degenerative diseases as it has the potential to bothstimulate oxidative phosphorylation and protect against lipidperoxidation. However this combination of effects makes inter-
pretation difficult, limiting the design of rational therapies.Furthermore, while MitoQ10 has been widely used to infer theexistence of mitochondrial ROS-dependent signaling path-ways, aspects of its mechanism of action remain uncertain. Toclarify these issues, we set out to determine how MitoQ10 anda number of related short-chain exogenous ubiquinones inter-act with both the mitochondrial respiratory chain and ROS. Forthis we used a range of MitoQ analogs that differ in the numberof carbons linking the ubiquinone to the TPP moiety (MitoQn;n � 3, 5, 10, or 15 CH2 groups) (29, 30) and compared theirinteractions with those of the untargeted short-chain ubiqui-none analogs, CoQ2, decylQ, and idebenone (Fig. 1A). This workhas led to a better understanding of how MitoQ and otherexogenous ubiquinones interact with the respiratory chain andROS and has considerable implications for their rational designand use as therapies and as tools to probe mitochondrial oxi-dative damage and redox signaling.
MATERIALS AND METHODS
Yeast Incubations—The Saccharomyces cerevisiae strains used wereWT (CEN.PK2–1C MATa his3 leu2 trp1 ura3) and �COQ2(CEN.PK2–1C coq2::HIS3), kindly supplied by Prof. Catherine Clarke,UCLA (47). Coq2 codes for the enzyme para-hydroxybenzoate:hexapre-nyl transferase, which catalyzes the transfer of the polyisoprenoidchain to 4-hydroxybenzoic acid in CoQ biosynthesis. �COQ2 is auxotro-phic for CoQ and fails to grow on non-fermentable carbon sources, suchas glycerol. Yeast were cultured in 10 ml of YPG (1% (w/v) yeast extract,2% (w/v) peptone, 3% (v/v) glycerol). The initial cell density was ad-justed to A600 �0.1. To achieve a reproducible transition to a respiratoryphenotype, the medium was supplemented with 0.05% (w/v) glucose.This allowed fermentative growth of �COQ2 up to A600 �0.4 after whichgrowth rapidly ceased unless ubiquinone supplementation restored ox-idative phosphorylation. Yeast were incubated in the dark at 30 °C withmechanical shaking at 250 rpm. Growth was monitored spectrophoto-metrically by measuring A600 over 120 h. For all yeast experiments,ubiquinones and other hydrophobic compounds were added in Me2SO to1% (v/v) of the total culture volume, which did not affect the growth ofthe WT and �COQ2 strains on glucose (data not shown).
�COQ2 yeast cultures for mitochondrial isolation were grown aero-bically at 30 °C to mid-logarithmic phase (A600 �1) in lactate medium(2% (v/v) DL-lactic acid, 0.3% yeast extract, 0.2% glucose, 0.05%CaCl2�2H2O, 0.05% NaCl, 0.06% MgCl2�6H2O, 0.1% KH2PO4, 0.1%NH4Cl (all w/v) (pH 5.5, NaOH). �COQ2 yeast can use D-lactate as arespiratory substrate because it donates electrons to oxidative phospho-rylation at complex IV via the reduction of cytochrome c (cyt c). For WTyeast, the level of glucose was kept at 0.05% (w/v). Mitochondria wereisolated according to published protocols (48, 49). The protein concen-tration was measured by the bicinchoninic acid assay using BSA as astandard (50). Aliquots of the mitochondrial preparation were mixedwith 10 mg�ml�1 fatty acid-free BSA as a cryoprotectant, snap-frozen ondry ice, and stored at �80 °C. Upon thawing the mitochondria retaineda membrane potential that was indistinguishable from that of freshlyisolated yeast mitochondria as confirmed by the uncoupler-sensitiveuptake of [3H]TPMP (data not shown) (29).
Oxygen consumption was measured with a Clark-type oxygen elec-trode (Rank Brothers, Bottisham, Cambridge, UK) in a 1-ml stirredchamber at 30 °C. Aliquots of frozen �COQ2 yeast mitochondria werethawed rapidly, washed, and resuspended in mannitol buffer (0.6 M
mannitol, 10 mM Tris maleate, 5 mM KPi, 0.5 mM EDTA (pH 6.8, KOH))at 0.2 mg protein�ml�1. The mitochondria were energized with 5 mM
glycerol 3-phosphate (G3P) and uncoupled with 1 �M carbonyl cyanidep-trifluoromethoxyphenylhydrazone (FCCP). Ubiquinones (1–20 �M inMe2SO) were titrated in successively, followed by 1 �M myxothiazol todetermine non-mitochondrial oxygen consumption. For some experi-ments mitochondria were sonicated (3 � 5 s, setting 4; Misonix XL-2020with microtip) in an ice bath prior to the measurement of respirationand addition of ubiquinone. Uptake of MitoQ analogs by yeast mito-chondria was measured using an electrode selective for TPP cations(30). For these experiments WT yeast mitochondria (0.4 mgprotein�ml�1) were incubated in 2.5 ml of mannitol buffer in the pres-ence of 2 �M MitoQ analog at 30 °C, and the uptake of MitoQ inresponse to energization with 5 mM ethanol was measured.
Mammalian Mitochondrial Preparations—Bovine heart mitochon-drial membranes (BHM) were prepared from isolated bovine heartmitochondria as described previously (51, 52). Rat liver mitochondria
Exogenous Ubiquinones as Therapies and Tools 21297
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

were prepared by homogenization followed by differential centrifuga-tion as described previously (53). Rat heart mitochondria were preparedby tissue disruption using an Ultra-Turrax (5 s), followed by differentialcentrifugation as described previously (53). Protein concentration wasdetermined using the biuret assay with BSA as a standard (54).
Ubiquinone Reduction and Oxidation by Respiratory Complexes—Assays were based on previously described methods for measuringrespiratory complex activity (55). However, for all assays except com-plex III the spectrophotometric decrease in A275 (at this wavelengthubiquinone absorbs more strongly than ubiquinol) was monitored in-stead. Assays were performed in KPi buffer (50 mM KPi-KOH, 100 �M
EDTA, 100 �M diethylenetriaminepentaacetic acid (pH 7.8) unlessstated otherwise) at 37 °C. For complex I, the buffer was supplementedwith 100 �g protein�ml�1 BHM, 100 �M NADH, and 2 mM KCN, and thereaction was started by addition of 50 �M ubiquinone. For complex II,the buffer was supplemented with 100 �g protein�ml�1 BHM, 5 mM
succinate, 8 �g�ml�1 rotenone, and 2 mM KCN, and the reaction wasstarted by addition of 50 �M ubiquinone. For glycerol-3-phosphate de-hydrogenase (G3PDH), EDTA and diethylenetriaminepentaacetic acid(DTPA) were omitted as Ca2� may be required for activity (56), and thebuffer was supplemented with 200 �g protein�ml�1 BHM, 2 mM KCN,and 50 �M ubiquinone. The reaction was started with the addition of 10mM G3P. Rotenone (8 �g�ml�1) and malonate (20 mM) did not affect therate of ubiquinone reduction in the presence of G3P. For complex III,the buffer was supplemented with 50 �g protein�ml�1 BHM, 50 �M
bovine cyt c, 8 �g�ml�1 rotenone, and 2 mM KCN, plus or minus 400 nM
myxothiazol. The reaction was started with the addition of 50 �M
ubiquinol, and cyt c reduction was measured by an increase at A550 (55).The myxothiazol-insensitive rate was measured in parallel and sub-tracted as there is a significant non-enzymatic rate of cyt c reduction byubiquinols (43).
Measurement of the Ubiquinone Redox State—Spectrophotometricmeasurements were made at 275 nm. The ubiquinone redox state wasmeasured at 37 °C in KPi buffer supplemented with 100 �gprotein�ml�1 BHM, 8 �g�ml�1 rotenone, and 10 �M ubiquinone. BHMwere also supplemented with 5 �M bovine cyt c, as this can be lostduring membrane isolation. For succinate (5 mM) oxidation, fumarase(5 units�ml�1) was also added as fumarate absorbs at A275 (local maxi-mum at 240 nm). Furthermore, a background trace in the absence ofubiquinone was subtracted as fumarase does not completely abolishfumarate accumulation. For NADH oxidation, NADH absorption inter-fered with ubiquinone redox changes at A275, therefore an NADH re-generation system (5 mM lactate and 2 units�ml�1 lactate dehydrogen-ase) was used. Nucleotides were added as NAD� (50 �M) because unlikeNADH, it rapidly reached a steady-state NADH/NAD� ratio. Furtheradditions of lactate dehydrogenase had no effect on this ratio. A back-ground trace acquired in the absence of ubiquinone was subtracted. ForHPLC measurements, the incubation was as above, but fumarase wasomitted. Two min after the addition of succinate, 1 volume of dichlo-romethane:diethylether (1:2) was added, and the mixture was thenvortexed under argon. The phases were separated by centrifugation (1min at 1,000 � g), and the upper organic phase was removed to a testtube overgassed with N2. The lower aqueous phase was re-extracted asbefore with dichloromethane:diethylether (1:2), and the second organicphase was combined with the first. This was evaporated to drynessunder a flow of N2, then resuspended in 1 ml of 45% (v/v) acetonitrile,0.1% (v/v) trifluoroacetic acid. Samples were analyzed by reverse-phaseHPLC (Gilson 321 pump, 1 ml�min�1) on a C18 column (Jupiter 300 Å,Phenomenex), using a staged gradient (4.5% acetonitrile for 5 min,4.5–54% acetonitrile over 5 min, 54–72% acetonitrile over 20 min,72–90% acetonitrile over 5 min) containing 0.1% (v/v) trifluoroaceticacid and detected at 220 nm using a Gilson UV/VIS 151 spectropho-tometer. Peaks were assigned by comparison with ubiquinone or ubiqui-nol standards. Spectrophotometric measurements of ubiquinone in ratliver mitochondria were made at 275 nm. Mitochondria (200 �gprotein�ml�1) were incubated at 25 °C in a stirred cuvette containing250 mM sucrose, 5 mM Tris, 1 mM EGTA (pH 7.4, HCl) supplementedwith 8 �g�ml�1 rotenone and 5 �M ubiquinone. 5 mM succinate, 400 nM
FCCP, and 400 nM myxothiazol were added as indicated.Ubiquinol Oxidation by Peroxynitrite—Ubiquinol oxidation by
ONOO� was measured at 37 °C in KPi buffer supplemented with 100�g protein�ml�1 BHM, 8 �g�ml�1 rotenone, 5 �M cyt c, and 10 �M
ubiquinone. 5 mM succinate was added to energize the BHM, and twoadditions of 50 �M ONOO� were made. This was followed by 20 mM
malonate and a further addition of 50 �M ONOO�. Ubiquinone redoxchanges were measured at 275 nm. ONOO� has a strong absorbancemaximum at 302 nm (�302 � 1.67 mM�1�cm�1 (57)) leading to a transientspike in A275 until it has decayed. The decay back to base-line absorb-
ance took �8 s in KPi buffer. For cis-parinaric acid (PA; MolecularProbes, Eugene, OR) oxidation, the incubation was performed using aShimadzu RF-5301PC fluorimeter in a stirred 3-ml cuvette thermostat-ted at 37 °C. PA was excited at 324 nm, and its fluorescence wasmonitored at 413 nm. The assay was in KPi buffer supplemented with100 �g protein�ml�1 BHM, 8 �g�ml�1 rotenone, 5 �M cyt c, and 10 �M
ubiquinone. BHM were supplied with 5 mM succinate, then after 1 min2 �M PA was added, followed by 20 �M ONOO� and each minutethereafter. ONOO� was prepared as described previously (58).
Measurement of Reactive Oxygen Species and Autoxidation—Re-duced MitoQ10 was prepared as described previously (29). Ubiquinoloxidation by O2
. was measured spectrophotometrically at 275 nm in astirred 3-ml cuvette. Oxidation of reduced MitoQ10 (50 �M) by O2
. gen-erated from 0.015 unit�ml�1 xanthine oxidase and 5 mM acetaldehydewas measured at 37 °C in KPi buffer. After 18 min 100 units�ml�1 SODwas added. Oxidation of reduced MitoQ10 (50 �M) by O2
. generated fromKO2 was measured at 37 °C in PBS-saturated octan-1-ol or KPi buffer(pH 7.3). A saturated solution of KO2 (�10 mM) was prepared bydissolving 1.4 mg solid KO2 in 2 ml of 10 mM 18-crown-6 ether inMe2SO. A solution where KO2 had degraded to H2O2 was prepared bymixing �10 mM KO2 in 10 mM 18-crown-6 ether with 1 volume of H2Ofollowed by incubation for 2 min. Autoxidation of reduced MitoQ10 (50�M) was measured spectrophotometrically in KPi buffer (pH 6.8, 7.8,and 8.3) at 275 nm in a strirred 3-ml cuvette at 37 °C. O2
. generationfrom reduced MitoQ10 (50 �M) was measured using acetylated cyto-chrome c (cyt cacet) reduction, which was measured spectrophotometri-cally at 550 nm and 37 °C. KPi buffer was supplemented with 100 �gprotein�ml�1 BHM, 8 �g�ml�1 rotenone, 50 �M cyt cacet, 400 nM myx-othiazol, 2 mM KCN, 10 �M ubiquinone, and 5 mM succinate. Thereaction was repeated in the presence of 100 units�ml�1 SOD to deter-mine the SOD-sensitive rate. H2O2 production was measured using anApollo-4000 H2O2 electrode (World Precision Instruments) in an openstirred chamber at 37 °C. KPi buffer was supplemented with 200 �gprotein�ml�1 BHM, 8 �g�ml�1 rotenone, 5 �M cyt c, and 100 units�ml�1
SOD. To this, 400 nM myxothiazol, 50 �M CoQ2, and 5 mM succinatewere added as indicated. The electrode was calibrated with knownamounts of H2O2. NO� was measured using a ISO-NO NO� electrode(World Precision Instruments) in an open stirred chamber at 37 °C. KPi
buffer was supplemented with 20 �g protein�ml�1 BHM, 8 �g�ml�1
rotenone, 5 �M cyt c, and 10 �M CoQ2. 250 �M 3,3-bis(aminoethyl)-1-hydroxy-2-oxo-1-triazene (DETA-NONOate; 100 mM in 10 mM KOH), 5mM succinate, 400 nM myxothiazol, and 100 units�ml�1 SOD were addedas indicated. The electrode was calibrated with known amounts ofS-nitroso-N-acetyl penicillamine (SNAP) to saturated CuCl.
Aconitase inactivation within mitochondria was measured spectro-photometrically by a coupled enzyme assay linking isocitrate produc-tion by aconitase to NADP� reduction by isocitrate dehydrogenase (�340
NADPH � 6.22 mM�1�cm�1) (59, 60). Aliquots of frozen WT (EG103MAT� his3 leu2 trp1 ura3) yeast mitochondria (61) were thawed rap-idly, then washed in 0.6 M mannitol buffer and resuspended in thisbuffer at 0.3 mg of protein/ml. The mitochondria were incubated in thepresence of 5 mM G3P for 30 min in a shaking water bath at 30 °C.Samples were removed every 5 min, snap-frozen on dry ice, and thawedprior to assaying. Aconitase activity was assayed in a 96-well plate witha 10-�l sample added to 190 �l of assay buffer (50 mM Tris-HCl, 0.6 mM
MnCl2, 5 mM sodium citrate, 0.2 mM NADP�, 0.1% v/v Triton X-100, and0.4 unit�ml�1 isocitrate dehydrogenase (pH 7.4)) at 30 °C. A340 readingswere carried out at 15 s intervals over 7 min in an ELx808 UltraMicroplate Reader (Bio-Tek Instruments). Each time point was meas-ured in quintuplicate, and plotting the natural logarithm of activityversus time linearized the time course of aconitase inactivation. Theslope of the line corresponded to the pseudo-first order rate constant ofaconitase inactivation. The background rate of NADPH formation wasdetermined in the presence of fluorocitrate (100 �M), a competitiveinhibitor of aconitase, and was always less than 10% of the initial rate.
Efflux of H2O2 from isolated rat heart mitochondria was measuredusing a Shimadzu RF-5301PC fluorimeter in a stirred 3-ml cuvettethermostatted at 25 or 37 °C. Mitochondria (200 �g protein�ml�1) wereincubated in 120 mM KCl, 3 mM HEPES-KOH, 1 mM EGTA, and 0.01%(w/v) fatty acid-free BSA (pH 7.2), containing 4 units�ml�1 horseradishperoxidase, 50 �M Amplex Red (Molecular Probes) and 100 units�ml�1
SOD. 5 mM succinate and 8 �g�ml�1 rotenone were added as indicated.Amplex Red was excited at 560 nm, and its fluorescence was monitoredat 590 nm.
Partition Coefficients—Octan-1-ol/PBS partition coefficients (theconcentration of ubiquinone in octan-1-ol relative to the concentrationin PBS) were determined as described previously (30). Membrane/PBSpartition coefficients (the concentration of ubiquinone in membranes
Exogenous Ubiquinones as Therapies and Tools21298
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

relative to the concentration in PBS) were estimated by incubating 2 mlof PBS containing 250 �g protein�ml�1 BHM and 50 �M ubiquinone for5 min at 37 °C. BHM were pelleted by centrifugation (30 min at16,000 � g), after which the supernatant was removed to a test tube andback-extracted with 1 volume of octan-1-ol. One extraction was suffi-cient for all ubiquinones except MitoQ5 and MitoQ3, which were ex-tracted with 2 ml of octan-1-ol two and three times, respectively. Thepellet was fully aspirated to remove as much water as possible, andthen all ubiquinones were extracted four times with 100 �l of octan-1-ol.To estimate the partition coefficient, the inner membrane surface areaper unit volume of rat heart mitochondria (61 �m2��m3) was used alongwith values for membrane thickness (6 nm) and mitochondrial volume(0.6 �l�mg protein�1) (62). This gave an estimated membrane volume of0.22 �l�mg protein�1.
Thiol Oxidation—Thiol oxidation by H2O2 was measured at 37 °C inKPi buffer containing 10 mM glucose, 500 �M GSH, 200 �M NADPH, 0.4unit�ml�1 glutathione reductase, and 0.02 unit�ml�1 glutathione perox-idase. GSH oxidation was detected by glutathione reductase-dependentNADPH oxidation. After A340 stabilized, 0.006 unit�ml�1 glucose oxi-dase was added to generate a stable flux of H2O2. Subsequently ubiqui-none or NaBH4-reduced ubiquinol (50 �M) was added. The response waslinear with H2O2 over the range 1–100 �M and with glucose oxidasefrom 0.003 to 0.03 unit�ml�1. The rate of NADPH oxidation was low inthe absence of glutathione peroxidase and glutathione reductase. Glu-tathione peroxidase activity (0.02 unit�ml�1) was in excess of glucoseoxidase activity, yet kept to a minimum to allow for reaction of H2O2
with MitoQ10 and CoQ2.For opening of the mitochondrial permeability transition pore (PTP)
by t-butylhydroperoxide (tBHP) in isolated rat liver mitochondria, thefinal centrifugation during mitochondrial isolation was in 250 mM su-crose, 5 mM Tris (pH 7.4, HCl). Measurements of extra-mitochondrialCa2� were made in a stirred 3-ml cuvette thermostatted at 25 °C usinga Shimadzu RF-5301PC fluorimeter. Calcium green-5N (MolecularProbes), a membrane-impermeant Ca2� probe, was excited at 506 nm,and emission was monitored at 532 nm. Fluorescence was measured in250 mM sucrose, 5 mM Tris, 10 �M EGTA (pH 7.4, HCl) supplementedwith 0.5 mg protein�ml�1 rat liver mitochondria, 8 �g�ml�1 rotenone,and 1 �M calcium-green-5N. Mitochondria were supplied with 5 mM
succinate, and after 1 min 5 �M CaCl2 � 5 �M tBHP were added. 1–5 �M
MitoQ10 and/or 500 nM cyclosporin A (CsA) were also added to someincubations.
NMR—Reduced MitoQ10 was prepared by reaction with NaBH4 inmethanol under argon. Excess NaBH4 was quenched with 10% (v/v)methane sulfonic acid, and the resulting mixture was extracted withdichloromethane, washed with water, and the solvent evaporated invacuo. The 1H NMR spectra of the product indicated it was �90%reduced. Solutions of the ubiquinol (10 mM) in D2O � 10 mM H2O2
and/or 100 �M EDTA were prepared in 5-mm NMR tubes. Oxidation ofreduced MitoQ10 was monitored using 1H NMR of the ring methoxypeaks at 3.9 and 4 ppm for the ubiquinol and ubiquinone, respectively.This showed that 1 day exposed to air oxidized 3–6% of the ubiquinol,and this was identical in the presence or absence of H2O2.
Visualization of Respiratory Complexes—Structural files of complexII from Escherichia coli (Protein Data Bank code 1NEK) (63) andcomplex III from Bos taurus (Protein Data Bank code 1NTZ) (64) werevisualized with PyMOL (DeLano Scientific, San Carlos, CA). Structuralfiles for MitoQ analogs were generated with CORINA (Computer Chem-istry Center, University of Erlangen-Nuremberg, Erlangen, Germany).To position MitoQ derivatives relative to the enzymes, three carbonatoms from the ring of the ubiquinone head group were pair-fitted withthree corresponding atoms from the ubiquinone or inhibitor bound intheir active sites.
RESULTS
Mitochondrial Respiration in CoQ-deficient Yeast Is Restoredby Untargeted Exogenous Ubiquinones but Not by MitoQ10—The first question we addressed was whether MitoQ10 could actas an effective electron carrier in the mitochondrial respiratorychain. For this we used yeast that cannot synthesize CoQ(�COQ2); consequently oxidative phosphorylation is inactive,and this yeast strain cannot grow on non-fermentable media(47). This lack of growth could be rescued by supplementationwith exogenous CoQ6 (the endogenous form of CoQ in yeast)(Fig. 2A). Although supplemented growth was slower than forthe WT strain, the maximum cell density achieved was similar.Therefore exogenous ubiquinones can reach mitochondria
within cells and restore oxidative phosphorylation. The effec-tiveness of various CoQ analogs at restoring growth exhibited adependence on concentration and on the length of the isopre-noid tail, with CoQ6 being the most effective (Fig. 2B). Inter-estingly, ubiquinone analogs with a saturated alkyl tail did notrestore cell growth (Fig. 2C). MitoQ10 at concentrations of 100nM to 10 �M also failed to stimulate cell growth. WT yeastgrowth was inhibited at 10 �M MitoQ10, but as similar growthinhibition was observed with 10 �M decyl-TPP, but not with 10�M TPMP, toxicity is probably due to the higher local concen-tration of more hydrophobic TPP cations within phospholipidbilayers (data not shown). CoQ10, the predominant CoQ inhumans, did not support cell growth in non-fermentable mediaat 5 and 50 �M (data not shown). However, this was caused bypoor uptake of CoQ10 probably due to its insolubility, as CoQ-deficient yeast engineered to synthesize CoQ10 grow well onnon-fermentable substrates (65).
It is possible that MitoQ10, decylQ, and idebenone are actu-ally effective respiratory substrates but that their mitochon-drial accumulation by yeast is poor. Therefore we determinedwhether MitoQ10, decylQ, idebenone, and the CoQ analogscould stimulate uncoupled respiration by mitochondria isolatedfrom �COQ2 yeast. CoQ2 and decylQ were the most effective atrestoring respiration, followed by idebenone and CoQ1 (Fig.2D). Therefore the failure of decylQ and idebenone to comple-ment the respiratory defect in intact �COQ2 yeast (Fig. 2C)was a result of insufficient mitochondrial accumulation. Incontrast, MitoQ10 was still ineffective at stimulating respira-tion in isolated �COQ2 mitochondria, even though it was rap-idly accumulated by energized yeast mitochondria (data notshown). This confirms that MitoQ10 is an ineffective electroncarrier for respiration and explains its failure to restore yeastgrowth in non-fermentable media. Surprisingly, CoQ6 was alsoineffective at restoring respiration, despite being able to re-store growth. However, this was due to its slow uptake byisolated mitochondria as respiration more than doubled(�110% versus intact mitochondria with CoQ6) if mitochondriawere sonicated before CoQ6 additions were made (data notshown). Sonication did not increase respiration with CoQ0 orMitoQ10. Therefore, CoQ6 is presumably too hydrophobic todiffuse rapidly through the mitochondrial outer membrane andstimulate respiration in these short term experiments. In sum-mary we observe three classes of exogenous ubiquinone inter-action: CoQ1, CoQ2, and CoQ6, which all migrate to mitochon-dria in intact yeast and restore respiration; decylQ andidebenone, which can restore respiration but do not accumulatesufficiently in mitochondria in yeast cells; and MitoQ10, whichfails to restore respiration even after it accumulates in mito-chondria. We next focussed on determining why MitoQ10 wasunable to complement the respiratory defect in the CoQ-defi-cient mitochondria.
MitoQ Analogs Are Not Oxidized by Complex III but AreReduced by Complex II and Glycerol-3-Phosphate Dehydrogen-ase—The failure of MitoQ10 to stimulate respiration in CoQ-deficient mitochondria was general to both yeast and mam-mals, as it did not complement respiration in CoQ-deficienthuman fibroblasts, which could be rescued by decylQ.2 There-fore MitoQ10 is either poorly oxidized by complex III or poorlyreduced by mitochondrial ubiquinone reductases (Fig. 3A). Tofind out which we determined whether the reduced form ofMitoQ10 and other short-chain ubiquinols were oxidized bycomplex III in bovine heart mitochondrial membranes (BHM).The ubiquinol form of MitoQ10 was an ineffective substrate forcomplex III, while those of CoQ2, decylQ, and idebenone were
2 P. Rustin, personal communication.
Exogenous Ubiquinones as Therapies and Tools 21299
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

all rapidly oxidized (Fig. 3B). We then measured how effec-tively the oxidized forms of MitoQ10 CoQ2, decylQ, and ide-benone were reduced by complex I, complex II, and G3PDH.MitoQ10 was an ineffective substrate for complex I (Fig. 3C) butwas well reduced by complex II (Fig. 3D) and G3PDH (Fig. 3E).In contrast, CoQ2, decylQ, and idebenone were effective sub-strates for all three ubiquinone reductases (Fig. 3, C–E).
We next determined how the carbon chain length betweenthe TPP and ubiquinone moieties of MitoQ analogs affectedtheir interaction with the respiratory chain (Fig. 3, B–E). Noneof the MitoQ analogs reacted effectively with complexes I andIII. In contrast, MitoQ analogs did react with complex II andG3PDH in a manner that was sensitive to alkyl chain length,with the reduction rate slowing as the chain length decreasedfrom 10 to 3 carbons. Although increasing the carbon chainlength to 15 led to an apparent decrease in reduction raterelative to MitoQ10, this may be an artifact due to the lowsolubility of MitoQ15 as reduction of MitoQ15 at a lower con-centration (10 �M rather than 50 �M) by complex II is compa-rable with MitoQ10 (Fig. 4A).
Reduction of MitoQ10 by succinate is via complex II as it iscompletely inhibited by the competitive inhibitor malonate and
other electron carriers, such as O2., do not mediate it as reduc-
tion occurred at a similar rate under anaerobic conditions andin the presence of SOD (data not shown). Direct electron trans-fer from the reduced endogenous CoQ10 pool is also unlikely tocontribute to the reduction of MitoQ10 as electron transferbetween a ubiquinol and a ubiquinone occurs by sequentialdeprotonation/electron transfer reactions that cannot occurwithin the phospholipid bilayer (66). Therefore we concludethat the predominant sources of electrons for MitoQ10 in mito-chondria are the active sites of ubiquinone reductases. In sum-mary, MitoQ analogs cannot complement defects in respirationbecause they are poorly oxidized by complex III. The longerchain MitoQ analogs are extensively reduced by complex II andG3PDH but not by complex I.
MitoQ10 Remains Reduced under Conditions Where CoQ2,DecylQ, and Idebenone Are Oxidized—In addition to transfer-ring electrons in oxidative phosphorylation, the ubiquinol formof CoQ10 also acts as a chain breaking antioxidant in lipidperoxidation through donation of a hydrogen atom to a carbonor oxygen-centered radical (12). Therefore if an exogenouslyadded ubiquinone is to be an effective antioxidant, its redoxstate is critical. The poor reactivity of MitoQ10 with complex III
FIG. 2. Utilization of ubiquinone analogs by CoQ-deficient (�COQ2) yeast. Yeast growth was monitored by following A600 over time.Cultures were prepared in YPG � 0.05% glucose, and the initial cell density was adjusted to A600 �0.1. Supplementation of the glycerol mediumwith 0.05% glucose allows fermentative growth of �COQ2 yeast to A600 �0.4 (indicated by the horizontal line in B and C). A, growth of �COQ2 yeastcan be restored by supplementation with CoQ6. a–c, growth curves for WT yeast (a), �COQ2 yeast (b), and �COQ2 yeast supplemented with 50�M CoQ6 (c). Traces are means � ranges of duplicate determinations for a typical experiment, which was repeated three times. B, effect of CoQisoprenoid chain length on growth of �COQ2 yeast. A600 was measured after 120 h, by which time maximum cell density had been reached for allgrowth conditions. The values are the means � ranges of two independent experiments. C, MitoQ and non-isoprenoid untargeted ubiquinones areineffective at restoring growth in �COQ2 yeast. Conditions are the same as described for B. D, MitoQ10 cannot restore respiration in mitochondriaisolated from �COQ2 yeast. Mitochondria from �COQ2 yeast were incubated with 5 mM G3P and 1 �M FCCP. a–g, the myxothiazol-insensitiverespiration rate determined in the presence of the indicated concentrations of CoQ2 (a), decylQ (b), idebenone (c), CoQ1 (d), MitoQ10 (e), CoQ6 (f),and CoQ0 (g). Data are the means � S.D. of three independent experiments.
Exogenous Ubiquinones as Therapies and Tools21300
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from
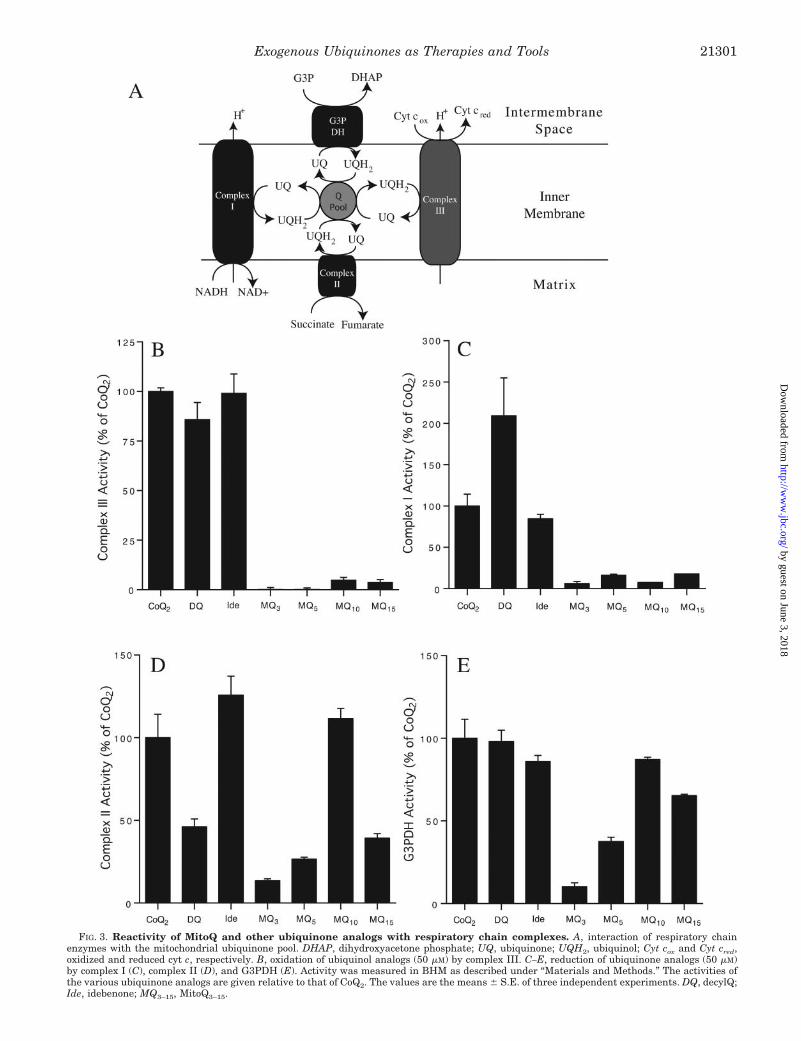
FIG. 3. Reactivity of MitoQ and other ubiquinone analogs with respiratory chain complexes. A, interaction of respiratory chainenzymes with the mitochondrial ubiquinone pool. DHAP, dihydroxyacetone phosphate; UQ, ubiquinone; UQH2, ubiquinol; Cyt cox and Cyt cred,oxidized and reduced cyt c, respectively. B, oxidation of ubiquinol analogs (50 �M) by complex III. C–E, reduction of ubiquinone analogs (50 �M)by complex I (C), complex II (D), and G3PDH (E). Activity was measured in BHM as described under “Materials and Methods.” The activities ofthe various ubiquinone analogs are given relative to that of CoQ2. The values are the means � S.E. of three independent experiments. DQ, decylQ;Ide, idebenone; MQ3–15, MitoQ3–15.
Exogenous Ubiquinones as Therapies and Tools 21301
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

FIG. 4. Ubiquinone redox state on reduction by mitochondrial membranes. A, MitoQ analogs, unlike other short-chain ubiquinones, arepredominantly in a reduced form in succinate-energized uninhibited BHM. BHM were supplemented with rotenone, cyt c, fumarase, and 10 �M
CoQ2 (a), MitoQ3 (b), MitoQ5 (c), MitoQ10 (d), or MitoQ15 (e). Succinate was added where indicated. The background rate in the absence ofubiquinone was subtracted. B, BHM were supplemented with rotenone, cyt c, and either ethanol (a), 10 �M CoQ2 (b), succinate, myxothiazol, and10 �M CoQ2 (c), or succinate and 10 �M CoQ2 (d), then incubated for 2 min, extracted, and run on an HPLC that measured A220. C, same as describedfor B but supplemented with 10 �M MitoQ10 instead of CoQ2. D, reduction state of CoQ2 or MitoQ10 on incubation with BHM. The ratios of theubiquinol to ubiquinone peak areas for MitoQ10 (filled bars) and CoQ2 (open bars) were determined by HPLC as shown in B and C. Data aremeans � S.E. of three independent experiments. E, MitoQ analogs and untargeted ubiquinones remain predominantly oxidized in the presence ofan NADH regeneration system. BHM were supplemented with cyt c, NAD�, lactate, and either 10 �M CoQ2 (a), MitoQ3 (b), MitoQ5 (c), MitoQ10(d), or MitoQ15 (e). Lactate dehydrogenase (LDH) and cyanide were added where indicated. A background rate in the absence of ubiquinone wassubtracted. F, the redox state of exogenous ubiquinones in intact liver mitochondria. Mitochondria (200 �g protein�ml�1) were supplemented withrotenone and a 5 �M concentration of either idebenone (a), CoQ2 (b), or MitoQ10 (c). Succinate, FCCP, and myxothiazol were added as indicated.A decrease in A275 indicates ubiquinone reduction, whereas an increase indicates ubiquinol oxidation.
Exogenous Ubiquinones as Therapies and Tools21302
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

implies that MitoQ10 may be persistently reduced and conse-quently a better antioxidant than CoQ2, decylQ, or idebenone.To investigate this, we measured the steady-state ubiquinone/ubiquinol ratio for exogenous ubiquinones in the presence ofBHM respiring on succinate (Fig. 4A). Under these conditionsCoQ2 remained largely oxidized (Fig. 4A, trace a), as did decylQand idebenone (data not shown). In contrast, MitoQ10 andMitoQ15 (Fig. 4A, traces d and e) were rapidly reduced. MitoQ3
and MitoQ5 (Fig. 4A, traces b and c) were slowly reduced,consistent with their lower rate of reduction by complex II (Fig.3D). For CoQ2 and MitoQ10, the relative amounts of ubiquinoland ubiquinone were determined by HPLC (Fig. 4, B and C),and the ratios of their peak areas are shown in Fig. 4D (openbars, CoQ2; filled bars, MitoQ10). CoQ2 and MitoQ10 were bothlargely in the oxidized form on incubation with BHM alone,but on addition of substrate, CoQ2 remained oxidized, whileMitoQ10 was reduced to its ubiquinol form. The complexIII-inhibitor myxothiazol caused CoQ2 to become reduced buthad no effect on MitoQ10 as it was already in the ubiquinol form.The ratio of reduced to oxidized idebenone was qualitativelysimilar to CoQ2 (data not shown), but co-eluting peaks present inBHM alone prevented precise quantification at 220 nm by HPLC.
That CoQ2, decylQ, and idebenone were all largely oxidizedon incubation with mitochondrial membranes was somewhatunexpected as endogenous CoQ10 is �75–90% reduced in iso-lated mitochondria during State 4, dropping to 50–60% re-duced in State 3 (67, 68). To determine whether this wasspecific to using succinate as an electron donor, we used NADHto drive reduction through complex I. Under these conditions,MitoQ3 and MitoQ5, as well as CoQ2, decylQ, and idebenone,remained predominantly in the oxidized form (Fig. 4E), whileMitoQ10 and MitoQ15 were slightly reduced (Fig. 4E, traces dand e). Ubiquinone reduction by NADH was possible as therespiratory inhibitor cyanide led to the rapid reduction of CoQ2,decylQ, and idebenone and to the gradual reduction of theMitoQ analogs, consistent with their slow reduction by complexI. Therefore the relatively oxidized steady state of the exoge-nous untargeted ubiquinones is independent of the electrondonor and is determined by the relative rates of electron entryto and efflux from the ubiquinone pool.
We next investigated whether the membrane potential inintact rat liver mitochondria largely prevented the oxidation ofexogenous ubiquinones by complex III. In contrast to BHM,CoQ2, idebenone, and MitoQ10 were all largely reduced in mi-tochondria energized with succinate (Fig. 4F). When the mem-brane potential was collapsed with the uncoupler FCCP, CoQ2
and idebenone rapidly became oxidized but were re-reduced onaddition of myxothiazol. DecylQ behaved in a manner similarto CoQ2 and idebenone (data not shown). Therefore the reducedstate of exogenous ubiquinones in coupled mitochondria wasdue to the membrane potential slowing their oxidation by com-plex III. This behavior was in marked contrast to MitoQ10 as itsnegligible oxidation by complex III meant that it remainedreduced even when the mitochondria were uncoupled. Interest-ingly, a partial decrease in the membrane potential upon ad-dition of ADP (State 3) did not lead to large scale oxidation ofCoQ2 (data not shown), thus ATP synthesis does not causeextensive oxidation of exogenous untargeted ubiquinones, butcomplete collapse of the membrane potential does.
In summary, the equilibrium redox state of an exogenousubiquinone is determined by its relative rates of reduction andoxidation (Fig. 3A). The oxidation of MitoQ10 by complex III isnegligible, while it is rapidly reduced by complex II, henceMitoQ10 is fully reduced under most conditions. This is not thecase for CoQ2, decylQ, and idebenone: although they are fullyreduced in coupled mitochondria, they are rapidly oxidized by
complex III when the membrane potential is low. This may becritical during pathological conditions where depolarization oc-curs such as during ischemic injury or following induction ofthe PTP.
The Ineffective Oxidation of MitoQ10 by Complex III En-hances Antioxidant Protection against Peroxynitrite—The anti-oxidant efficacy of MitoQ10 is due to its conversion to a ubiqui-nol, as the ubiquinone is inactive (29, 30). To see if the greatertendency of MitoQ10 to remain in the reduced form enhancedits antioxidant ability, we examined its interaction with thebiologically significant oxidant peroxynitrite (ONOO�), whichis produced in vivo by the reaction of O2
. with nitric oxide (NO�)(69). Among the oxidizing reactions of ONOO� is the one elec-tron oxidation of ubiquinol to a ubisemiquinone radical whichthen dismutates (70) (Reactions 1 and 2).
2UQH2 � 2ONOO� 3 2UQ. � 2H2O � 2NO�2
2UQ. � 2H� 3 UQH2 � UQ
REACTIONS 1 AND 2
An advantage of using ONOO� as the ubiquinol oxidant is thatat pH 7 it decays within a few seconds to unreactive endproducts (69), leading to a pulse of ubiquinol oxidation afterwhich regeneration of its antioxidant function by re-reductioncan be assessed. CoQ2 incubated with BHM respiring on suc-cinate remained in its ubiquinone form (Fig. 5A, trace a), whileMitoQ10 was reduced by the respiratory chain (Fig. 5A, trace b).MitoQ5 was also reduced but to a lesser extent due to its slowerreaction with complex II (Fig. 5A, trace c). Addition of ONOO�
led to a sharp upward spike in A275 for all three ubiquinones.For CoQ2 (Fig. 5A, trace a) the transient increase in A275 wassolely due to the absorbance of ONOO� itself (�302 � 1.67mM�1�cm�1 (57)), which decayed away over �8 s. For MitoQ5
(Fig. 5A, trace c) there was a transient spike in A275 due to bothONOO� itself and the formation of oxidized MitoQ5. After theONOO� had decayed away, A275 decreased as the ubiquinonewas slowly reduced back to the ubiquinol. For MitoQ10, therewas also a dramatic spike in A275, due to both ONOO� andubiquinol oxidation, but in this case the ubiquinone was re-duced back to the ubiquinol rapidly by complex II (Fig. 5A,trace b). This oxidation and re-reduction of MitoQ10 by ONOO�
could be repeated several times and two such reaction cyclesare shown in Fig. 5A. The re-reduction of the ubiquinone wasby complex II, as malonate prevented reduction of MitoQ5 andMitoQ10 after addition of ONOO� (Fig. 5A).
The slower reduction of MitoQ5 by complex II enabledONOO� decay and ubiquinone reduction to be easily distin-guished in Fig. 5A as a biphasic change in A275 after addition ofONOO�. The biphasic nature of MitoQ10 re-reduction afterONOO� addition was not obvious so the traces from Fig. 5Awere expanded to clearly show that the decay in ONOO� differsfrom the re-reduction of MitoQ10 (Fig. 5B). The base lines of thetraces have been aligned to emphasize the relative changes inA275. For CoQ2 � malonate (traces c and d) and for MitoQ10 �malonate (trace b), the addition of ONOO� leads to an increasein A275 that decays back to base line over �8 s due to thebreakdown of ONOO�. In contrast, addition of ONOO� toMitoQ10 in the presence of uninhibited BHM (trace a) is bipha-sic with an initial decay in A275 due to ONOO� that is followedby a slower decrease in A275 due to reduction of the ubiquinoneformed by ONOO� oxidation. Idebenone behaved in the sameway as CoQ2 (data not shown).
To confirm that the ubiquinol forms of idebenone and CoQ2
could also react with ONOO�, we repeated these experimentsin myxothiazol-inhibited BHM. Addition of succinate led to the
Exogenous Ubiquinones as Therapies and Tools 21303
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

complete reduction of idebenone (Fig. 5C, trace a) and MitoQ10
(Fig. 5C, trace b). ONOO� rapidly oxidized these ubiquinols,and this was reversed by the respiratory chain within �20 s ina malonate-sensitive fashion. CoQ2 behaved similarly to ide-benone (data not shown). Thus, the ubiquinol forms of allexogenous ubiquinones can be oxidized by ONOO� and thenrecycled by the respiratory chain.
We next tested whether MitoQ10 would be more protectiveagainst ONOO�-induced lipid peroxidation than CoQ2 and ide-benone in uninhibited BHM. For this we used PA, a conjugatedpolyunsaturated fluorescent fatty acid that loses its fluores-cence upon peroxidation. Sequential additions of ONOO� to
BHM respiring on succinate caused step decreases in PA fluo-rescence (Fig. 5D, trace c), and MitoQ10 protected against thisloss (Fig. 5D, trace a). Idebenone also protected against the lossof PA fluorescence (Fig. 5D, trace b); however, the protectionwas significantly less than that given by MitoQ10. The back-ground decay of PA was unaffected by ubiquinones, suggestingthat it is not related to lipid peroxidation (Fig. 5D, traces d–f).CoQ2 behaved like idebenone, while MitoQ10 in the absence ofsuccinate offered no protection (data not shown).
In summary, the reduced form of MitoQ10 is an effectiveantioxidant against ONOO�, and its slow oxidation by complexIII makes it a more effective antioxidant than untargeted
FIG. 5. Ubiquinone oxidation by ONOO�. A, MitoQ analogs are oxidized by ONOO� and re-reduced in uninhibited BHM. BHM weresupplemented with rotenone, cyt c, and either 10 �M CoQ2 (a), MitoQ10 (b), or MitoQ5 (c). Succinate, ONOO� (50 �M) and malonate were addedwhere indicated. The background change in absorbance due to fumarate production was subtracted. B, expansion of the period after ONOO�
addition in A. a–d, MitoQ10 before (a) and after malonate (b); CoQ2 before (c) and after malonate (d). C, short-chain ubiquinones are oxidized byONOO� in myxothiazol-inhibited BHM. BHM were supplemented with rotenone, myxothiazol, and either 10 �M idebenone (a), MitoQ10 (b), orMitoQ5 (c). Succinate, ONOO� (50 �M) and malonate were added where indicated. D, MitoQ10 prevents PA oxidation. BHM were supplementedwith rotenone, myxothiazol, cyt c, and either 10 �M MitoQ10 (a and d) or idebenone (b and e), or EtOH carrier (c and f). PA (2 �M) was added toall traces as indicated. ONOO� (50 �M) was added to traces a–c as indicated. Succinate was added 1 min prior to PA.
Exogenous Ubiquinones as Therapies and Tools21304
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

ubiquinone analogs. Importantly, the respiratory chain canreduce MitoQ10 repeatedly recycling it back to its active anti-oxidant form after it has detoxified ONOO�.
Ubiquinols Are Oxidized by Superoxide—Ubiquinones andubiquinols as well as their partially protonated and reducedintermediates undergo a complex set of reactions with oxygenand O2
. (13). Oxygen can react with ubiquinols and ubisemiqui-nones to produce O2
.; conversely, O2. generation in the presence
of ubiquinols may result in O2. scavenging via semiquinone
formation and subsequent dismutation (Fig. 6A). As these re-actions can affect the steady-state concentration of O2
., they
have implications for the use of exogenous ubiquinones astherapies and for investigating ROS signaling pathways. In-deed, the potential for ROS generation, particularly from ide-benone autoxidation, has been raised as a potential concernabout the therapeutic use of ubiquinones (71). Therefore wehave analyzed the production and consumption of O2
. by exog-enous ubiquinones.
There is the possibility that ubiquinols can consume O2.,
probably by reaction with its protonated form (HO2� , pKa 4.8), in
a mechanism analogous to their chain terminating reaction inlipid peroxidation.
FIG. 6. Consumption of O2. by ubiquinols. A, proposed reactions through which ubiquinols both generate and consume O2
.. Ubiquinol (UQH2)directly consumes O2
./HO2� in the membrane or aqueous phase to generate H2O2. O2
. generation is dependent on deprotonation of ubiquinol to theubiquinolate anion (UQH�). UQ, ubiquinone; UQ., ubisemiquinone anion; UQH�, ubisemiquinone radical. The deprotonation reaction occurs in theaqueous phase and is thus inhibited by ubiquinol hydrophobicity. B, ubiquinols are slowly oxidized by O2
. in aqueous solution. Reduced MitoQ10(50 �M) was incubated in KPi buffer with 0.015 unit�ml�1 xanthine oxidase at: pH 7.8 (a) or pH 6.8 (b). Acetaldehyde (5 mM) and SOD (100units�ml�1) were added as indicated. C, ubiquinols are rapidly oxidized by O2
. in organic solvent. 25 �l of �10 mM KO2 in 10 mM 18-crown-6 ether(a) or 25 �l of 10 mM 18-crown-6 ether (b) or 50 �l of 1:1 �10 mM KO2 in 10 mM 18-crown-6 ether:H2O incubated for 2 min (c) was added to 50 �M
reduced MitoQ10 in 2.5 ml of PBS-saturated octan-1-ol. 25 �l of �10 mM KO2 in 10 mM 18-crown-6 ether was added to a 50 �M concentration ofthe reduced form of MitoQ10 in 2.5 ml of KPi (pH 7.4) (d). Inset, before and after the addition of KO2 to MitoQ10 in trace a. KO2 or equivalentadditions were made as indicated.
Exogenous Ubiquinones as Therapies and Tools 21305
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

UQH2 � HO�2 3 UQH� � H2O2
REACTION 3
To see if Reaction 3 could lead to a direct antioxidant effect ofexogenous ubiquinols on O2
. we first measured oxidation of theubiquinol form of MitoQ10 at 275 nm by O2
. generated fromacetaldehyde and xanthine oxidase in aqueous buffer. Genera-tion of O2
. caused slow oxidation of reduced MitoQ10 that couldbe fully blocked by SOD (Fig. 6B). Although O2
. can react withubiquinol in aqueous buffer, spontaneous dismutation to hy-drogen peroxide (H2O2) appears to dominate as the rate ofubiquinol oxidation was low relative to the rate of O2
. produc-tion. Reaction 3 is also likely to occur within phospholipidbilayers and could thereby provide a mechanism for detoxifyingHO2
� that is inaccessible to SOD. This would be expected to beimportant in tissues such as the heart, where cristae phospho-lipids occupy a volume similar to the aqueous mitochondrialmatrix (62). To investigate this we added �100 �M potassiumsuperoxide (KO2) to a 50 �M concentration of the reduced formof MitoQ10 in PBS-saturated octan-1-ol. KO2 rapidly oxidizedreduced MitoQ10 (Fig. 6C, trace a). Oxidation of reducedMitoQ10 was specific to O2
./HO2� as it was not oxidized by carrier
or by KO2 that was previously decomposed to H2O2 (Fig. 6C,traces b and c). Furthermore, reduced MitoQ10 in aqueousbuffer was not oxidized by KO2 due to its rapid dismutation toH2O2 (Fig. 6C, trace d). Therefore these results show thatubiquinols are likely to be effective scavengers of O2
. when itdiffuses into phospholipid bilayers as HO2
� . While several vari-ations of Reaction 3 with different protonation states of thereactants could contribute to this, their net effect would besimilar: ubiquinol oxidation, H2O2 generation, and a lowersteady-state concentration of O2
./HO2� .
Ubiquinol Autoxidation Requires Deprotonation and Is De-creased by Ubiquinol Hydrophobicity—While the ubiquinolform of MitoQ10 is not oxidized by complex III, like all otherexogenous ubiquinols it can be oxidized directly by oxygen toform O2
. in vitro. The transfer of electrons from ubiquinol to cytc has been studied extensively (43, 66, 72). From this it can beconcluded that the direct donation of an electron by ubiquinolto oxygen is unlikely (UQH2
�/UQH2, Em,7 � 850 mV (66)).Instead, electron transfer from ubiquinol requires an initialdeprotonation to a ubiquinolate anion (pKa 11.3 (66)), and thisis the likely electron donor to oxygen (UQH�/UQH�, Em,7 � 190mV (66); see Reactions 4 and 5).
UQH2 3 UQH� � H�
UQH� � O2 3 UQH� � O2.
REACTIONS 4 AND 5
The ubisemiquinone radical formed can also react with oxygento form O2
., or it can dismutate, but it is the initial reactionbetween the ubiquinolate and oxygen that is likely to be rate-limiting for autoxidation (Fig. 6A). To see if this model wouldaccount for ROS production by exogenous ubiquinols, we as-sessed the pH sensitivity of ubiquinol autoxidation by meas-uring the increase in ubiquinone absorption at 275 nm. Therewas negligible oxidation of the reduced form of MitoQ10 inaqueous solution at pH 6.8, but autoxidation increased a littleat pH 7.8 and dramatically at pH 8.3 (Fig. 7A). Consistent withFig. 6A, ubiquinol autoxidation was SOD-insensitive. To dem-onstrate that O2
. generation occurred during ubiquinol autoxi-dation we used acetylated cyt c (cyt cacet), which is readilyreduced by one electron transfer from O2
. (73) and ubisemiqui-nones (43, 72) but whose reduction and oxidation by the respi-ratory chain are limited (73). In aqueous buffer the ubiquinol,
but not the ubiquinone, forms of CoQ2 and MitoQ10 reduced cytcacet, and this reduction occurred primarily via O2
. as the ratewas 80–90% SOD-sensitive (data not shown). To demonstrateO
2
. generation during autoxidation of complex II-reduced Mito-Q10, we measured cyt cacet reduction in myxothiazol-inhibitedBHM (Fig. 7B). This showed that reduction of MitoQ10 bycomplex II also caused cyt cacet reduction and that this rateincreased with pH from pH 6.8 to 8.3. This pH dependence wasnot due to changes in MitoQ10 reduction by complex II as thisrate was identical at pH 6.8 and 8.3 (data not shown). There-fore O2
. production from exogenous ubiquinols producedchemically or by mitochondrial respiration is pH-dependent,consistent with ubiquinol deprotonation being critical forautoxidation.
As deprotonation creates two charged species, autoxidationwill predominantly occur in the aqueous phase rather thanwithin phospholipid bilayers and its rate should be inverselyproportional to ubiquinol hydrophobicity. To investigate thiswe measured ubiquinol autoxidation in myxothiazol-inhibitedBHM using a range of MitoQ and non-targeted ubiquinoneanalogs with a spectrum of hydrophobicities (Fig. 7C). All theexogenous ubiquinones reduced cyt cacet (Fig. 7C), and in allcases this rate of reduction was about �50% inhibitable bySOD (data not shown). The rate of O2
. production from autoxi-dizing untargeted ubiquinones was inversely proportional totheir octan-1-ol/PBS partition coefficients (Fig. 1B). The lowestlevels of O2
. were generated by the most hydrophobic ubiquinol,decylQ, with the most water-soluble, idebenol, producing themost O2
. and CoQ2 being intermediate. There was a similarinverse correlation with hydrophobicity for the MitoQ analogsfrom MitoQ5 to MitoQ15. MitoQ3 was an exception to this trend,possibly due to its slow reduction by complex II leading to alower ubiquinol concentration (Fig. 3C). While this inverserelationship between autoxidation and hydrophobicity heldwithin the two groups, the MitoQ analogs were less prone toautoxidation than more hydrophobic non-targeted ubiquinones(e.g. MitoQ10 versus idebenone). MitoQ analogs are chargedcations, while untargeted ubiquinones are neutral, so parti-tioning into octan-1-ol may not accurately reflect binding tophospholipid bilayers. Therefore we measured the relativebinding of MitoQ analogs and untargeted ubiquinones to mito-chondrial membranes (Fig. 7D) and found that increased bind-ing of MitoQ analogs to phospholipid bilayers could explaintheir lower rate of autoxidation when compared with equiva-lent untargeted ubiquinols.
In summary, all ubiquinols are autoxidized, but this requiresan initial deprotonation to a ubiquinolate anion, which is un-favorable within the hydrophobic environment of a phospho-lipid bilayer. Therefore there are two major determinants of thedegree to which ubiquinol will autoxidize: its extent of reduc-tion and its hydrophobicity. Furthermore, the charged TPPmoiety of MitoQ analogs may be a better way of loweringoverall hydrophobicity and improving pharmacokinetics with-out enhancing the tendency to autoxidation, as it leads toadsorption onto phospholipid bilayers and insertion of theubiquinol moiety into the hydrophobic core of the membrane.
Ubiquinol Autoxidation Produces Superoxide, Which CanDismutate to Hydrogen Peroxide or Consume Nitric Oxide—Inthe above analysis, O2
. generation during ubiquinol autoxida-tion was assessed using cyt cacet reduction, but cyt cacet can bereduced by O2
. and by the ubisemiquinone radical (43, 66, 72,73), both of which may form during ubiquinol autoxidation.Furthermore, even though the reduction of cyt cacet was �50%SOD-sensitive, it is still difficult to conclude a dominant role forO2
., as the steady-state concentration of the ubisemiquinoneradical decreases as a consequence of consuming O2
. by dismu-
Exogenous Ubiquinones as Therapies and Tools21306
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from
tation (74). In addition, cyt cacet itself may act as a O2. sink and
thus distort the relative rates and routes of ubiquinol autoxi-dation. Therefore to confirm that autoxidation of exogenous
ubiquinol produced O2., we used a H2O2 electrode to measure
the production of H2O2 from the dismutation of O2. (4). Meas-
urement of H2O2 is of further significance as MitoQ10 has been
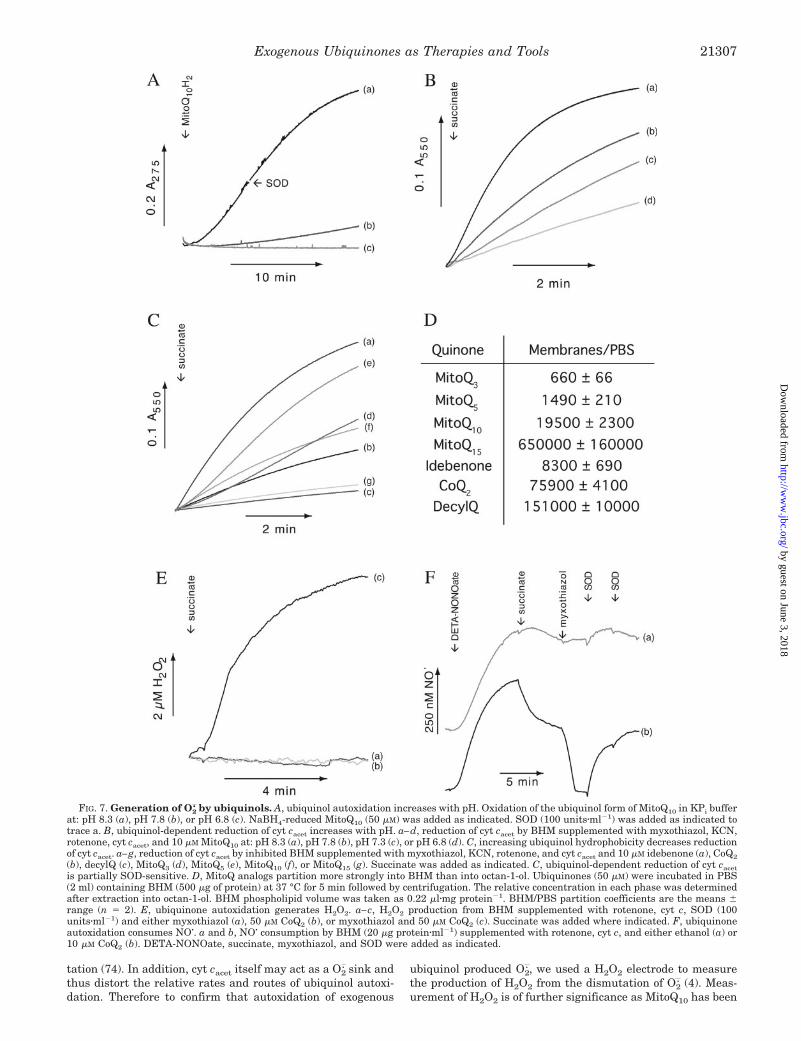
FIG. 7. Generation of O2. by ubiquinols. A, ubiquinol autoxidation increases with pH. Oxidation of the ubiquinol form of MitoQ10 in KPi buffer
at: pH 8.3 (a), pH 7.8 (b), or pH 6.8 (c). NaBH4-reduced MitoQ10 (50 �M) was added as indicated. SOD (100 units�ml�1) was added as indicated totrace a. B, ubiquinol-dependent reduction of cyt cacet increases with pH. a–d, reduction of cyt cacet by BHM supplemented with myxothiazol, KCN,rotenone, cyt cacet, and 10 �M MitoQ10 at: pH 8.3 (a), pH 7.8 (b), pH 7.3 (c), or pH 6.8 (d). C, increasing ubiquinol hydrophobicity decreases reductionof cyt cacet. a–g, reduction of cyt cacet by inhibited BHM supplemented with myxothiazol, KCN, rotenone, and cyt cacet and 10 �M idebenone (a), CoQ2(b), decylQ (c), MitoQ3 (d), MitoQ5 (e), MitoQ10 (f), or MitoQ15 (g). Succinate was added as indicated. C, ubiquinol-dependent reduction of cyt cacetis partially SOD-sensitive. D, MitoQ analogs partition more strongly into BHM than into octan-1-ol. Ubiquinones (50 �M) were incubated in PBS(2 ml) containing BHM (500 �g of protein) at 37 °C for 5 min followed by centrifugation. The relative concentration in each phase was determinedafter extraction into octan-1-ol. BHM phospholipid volume was taken as 0.22 �l�mg protein�1. BHM/PBS partition coefficients are the means �range (n � 2). E, ubiquinone autoxidation generates H2O2. a–c, H2O2 production from BHM supplemented with rotenone, cyt c, SOD (100units�ml�1) and either myxothiazol (a), 50 �M CoQ2 (b), or myxothiazol and 50 �M CoQ2 (c). Succinate was added where indicated. F, ubiquinoneautoxidation consumes NO�. a and b, NO� consumption by BHM (20 �g protein�ml�1) supplemented with rotenone, cyt c, and either ethanol (a) or10 �M CoQ2 (b). DETA-NONOate, succinate, myxothiazol, and SOD were added as indicated.
Exogenous Ubiquinones as Therapies and Tools 21307
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

shown to block a number of redox signaling and apoptoticpathways, but the mechanism remains unclear (35–37, 39–42).BHM alone did not produce H2O2 when inhibited by myxothia-zol (Fig. 7E, trace a) nor did BHM supplemented with CoQ2 andsuccinate (Fig. 7E, trace b). However, inhibition of ubiquinoloxidation with myxothiazol (Fig. 7E, trace c) or cyanide (datanot shown) led to a build up of ubiquinol and consequent H2O2
production. MitoQ10 also produced H2O2 but did so in theabsence of myxothiazol as it was already fully reduced (datanot shown). Therefore autoxidation of ubiquinols does produceO2
. that will dismutate to H2O2, and this could be important inredox signaling.
NO� is a signaling molecule produced by NO� synthases, andtheir activity may be associated with mitochondria and is im-portant for mitochondrial biogenesis (75–77). NO� interactswith mitochondria by reversibly inhibiting complex IV (78), aswell as affecting the function of proteins via S-nitrosylation ofcysteine residues (79). Therefore we next investigated the in-teraction of exogenous ubiquinols with NO�, which may reactdirectly with ubiquinols to produce NO� or with ubiquinol-generated O2
. to form ONOO� (80). For this the steady-stateNO� concentration produced by the NO� donor, DETA-NONO-ate, in the presence of BHM was measured using an NO�
electrode (Fig. 7F). DETA-NONOate gave a steady-state NO�
concentration of �400 nM after 8–10 min that persisted for afurther 10–20 min (Fig. 7F, trace a). BHM supplemented withCoQ2 consumed NO� at an increased rate upon addition ofsuccinate, and addition of myxothiazol led to the completedepletion of NO� that could be partially reversed by SOD (Fig.7F, trace b). MitoQ10 also consumed NO� to a similar extent, butas it is present in the reduced form, it consumed NO� rapidlyeven in the absence of myxothiazol (data not shown). Thereforethe autoxidation of exogenous ubiquinols generated by mito-chondrial respiration does lead to the formation of O2
. that canreact with NO� or dismutate to H2O2.
MitoQ Analogs Cause Efflux of Hydrogen Peroxide from Mi-tochondria, but Only MitoQ3 Damages Aconitase—The abovefindings show that all exogenous ubiquinols have the potentialto autoxidize and form O2
. in aqueous solution. To determinewhether ubiquinol autoxidation led to significant O2
. generationwithin intact mitochondria, we measured the efflux of H2O2
from isolated rat heart mitochondria in the presence of 1 �M
exogenous ubiquinol. H2O2 efflux is due to the dismutation ofO2
. to H2O2, which can then diffuse through the mitochondrialinner membrane (81) and be detected using Amplex Red andhorseradish peroxidase (Fig. 8A). A significant rate of H2O2
efflux was observed in the presence of rotenone and a 1 �M
concentration of either MitoQ3, MitoQ5, or MitoQ10 (Fig. 8A,traces b–d). As expected, the presence of exogenous SOD had noeffect on H2O2 efflux from MitoQ3 (data not shown). In BHMexogenous ubiquinols inhibit the horseradish peroxidase detec-
FIG. 8. MitoQ autoxidation causes H2O2 efflux from mitochon-dria but only MitoQ3 damages aconitase. A, MitoQ analogs gener-ate H2O2 within intact mitochondria. Isolated rat heart mitochondria(0.2 mg protein�ml�1) were incubated at 25 °C with rotenone, AmplexRed, horseradish peroxidase, SOD, and either ethanol (a), 1 �M MitoQ3(b), 1 �M MitoQ5 (c), 1 �M MitoQ10 (d), or 1 �M MitoQ15 (e). Succinatewas added where indicated. B, H2O2 generation from MitoQ3 compared
with other mechanisms of H2O2 production. Isolated rat heart mito-chondria (0.2 mg protein�ml�1) were incubated at 37 °C with AmplexRed, horseradish peroxidase, SOD, and either rotenone (a), ethanol (b),1 �M MitoQ3 (c), 1 mM Paraquat (d), or 100 �M Paraquat (e). Succinatewas added where indicated. C, aconitase inactivation was plotted as thenatural logarithm of activity versus time and given a linear fit. Isolatedwild-type yeast mitochondria (0.3 mg protein�ml�1) were incubated at30 °C with aliquots taken every 5 min for 30 min. White bar, Back-ground rate of aconitase inactivation in unenergized WT yeast mito-chondria. Black bars, energized with 5 mM G3P and incubated witheither 0–10 mM Paraquat, 1 �M FCCP, 1 �M MitoQ3, 1 �M MitoQ5, 1 �M
MitoQ10, or 1 �M decyl-TPP. Aconitase inactivation was plotted as timeversus the natural logarithm of activity and given a linear fit. The slopeof this line corresponds to the pseudo-first order rate constant of acon-itase inactivation. Data are the mean � S.D. of three independentexperiments. Statistical significance was calculated relative to G3Palone using a Student’s one-tailed t test. *, p 0.05; ***, p 0.001.
Exogenous Ubiquinones as Therapies and Tools21308
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from
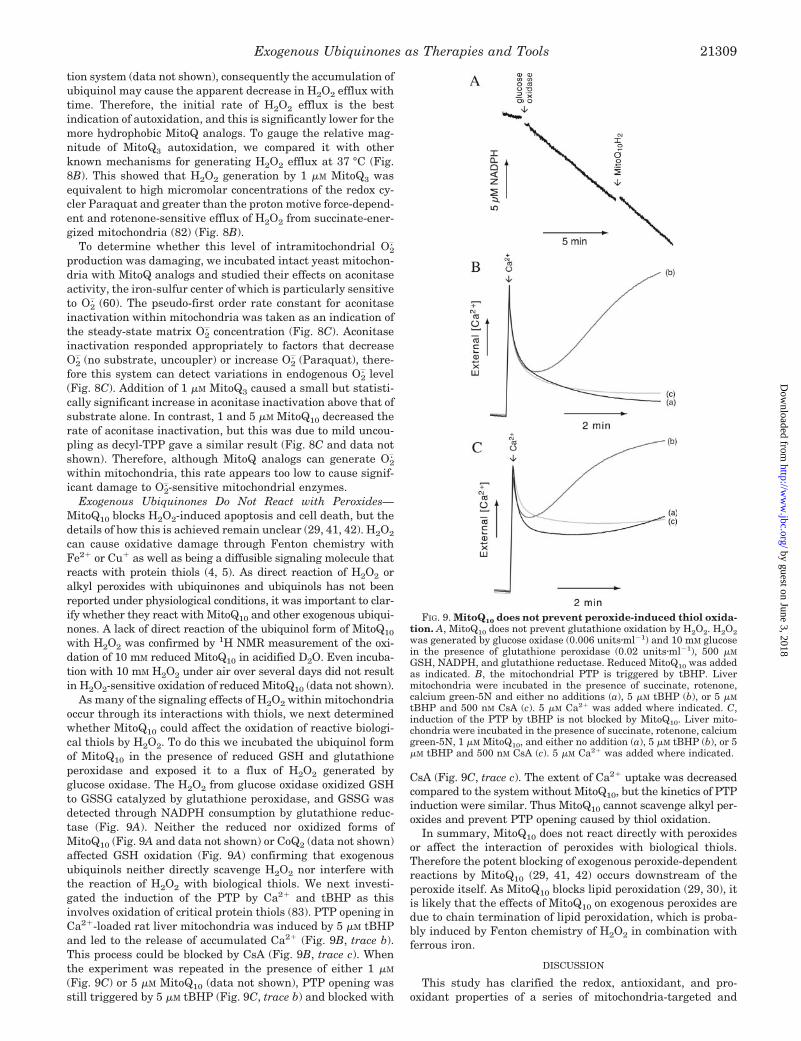
tion system (data not shown), consequently the accumulation ofubiquinol may cause the apparent decrease in H2O2 efflux withtime. Therefore, the initial rate of H2O2 efflux is the bestindication of autoxidation, and this is significantly lower for themore hydrophobic MitoQ analogs. To gauge the relative mag-nitude of MitoQ3 autoxidation, we compared it with otherknown mechanisms for generating H2O2 efflux at 37 °C (Fig.8B). This showed that H2O2 generation by 1 �M MitoQ3 wasequivalent to high micromolar concentrations of the redox cy-cler Paraquat and greater than the proton motive force-depend-ent and rotenone-sensitive efflux of H2O2 from succinate-ener-gized mitochondria (82) (Fig. 8B).
To determine whether this level of intramitochondrial O2.
production was damaging, we incubated intact yeast mitochon-dria with MitoQ analogs and studied their effects on aconitaseactivity, the iron-sulfur center of which is particularly sensitiveto O2
. (60). The pseudo-first order rate constant for aconitaseinactivation within mitochondria was taken as an indication ofthe steady-state matrix O2
. concentration (Fig. 8C). Aconitaseinactivation responded appropriately to factors that decreaseO2
. (no substrate, uncoupler) or increase O2. (Paraquat), there-
fore this system can detect variations in endogenous O2. level
(Fig. 8C). Addition of 1 �M MitoQ3 caused a small but statisti-cally significant increase in aconitase inactivation above that ofsubstrate alone. In contrast, 1 and 5 �M MitoQ10 decreased therate of aconitase inactivation, but this was due to mild uncou-pling as decyl-TPP gave a similar result (Fig. 8C and data notshown). Therefore, although MitoQ analogs can generate O2
.
within mitochondria, this rate appears too low to cause signif-icant damage to O2
.-sensitive mitochondrial enzymes.Exogenous Ubiquinones Do Not React with Peroxides—
MitoQ10 blocks H2O2-induced apoptosis and cell death, but thedetails of how this is achieved remain unclear (29, 41, 42). H2O2
can cause oxidative damage through Fenton chemistry withFe2� or Cu� as well as being a diffusible signaling molecule thatreacts with protein thiols (4, 5). As direct reaction of H2O2 oralkyl peroxides with ubiquinones and ubiquinols has not beenreported under physiological conditions, it was important to clar-ify whether they react with MitoQ10 and other exogenous ubiqui-nones. A lack of direct reaction of the ubiquinol form of MitoQ10
with H2O2 was confirmed by 1H NMR measurement of the oxi-dation of 10 mM reduced MitoQ10 in acidified D2O. Even incuba-tion with 10 mM H2O2 under air over several days did not resultin H2O2-sensitive oxidation of reduced MitoQ10 (data not shown).
As many of the signaling effects of H2O2 within mitochondriaoccur through its interactions with thiols, we next determinedwhether MitoQ10 could affect the oxidation of reactive biologi-cal thiols by H2O2. To do this we incubated the ubiquinol formof MitoQ10 in the presence of reduced GSH and glutathioneperoxidase and exposed it to a flux of H2O2 generated byglucose oxidase. The H2O2 from glucose oxidase oxidized GSHto GSSG catalyzed by glutathione peroxidase, and GSSG wasdetected through NADPH consumption by glutathione reduc-tase (Fig. 9A). Neither the reduced nor oxidized forms ofMitoQ10 (Fig. 9A and data not shown) or CoQ2 (data not shown)affected GSH oxidation (Fig. 9A) confirming that exogenousubiquinols neither directly scavenge H2O2 nor interfere withthe reaction of H2O2 with biological thiols. We next investi-gated the induction of the PTP by Ca2� and tBHP as thisinvolves oxidation of critical protein thiols (83). PTP opening inCa2�-loaded rat liver mitochondria was induced by 5 �M tBHPand led to the release of accumulated Ca2� (Fig. 9B, trace b).This process could be blocked by CsA (Fig. 9B, trace c). Whenthe experiment was repeated in the presence of either 1 �M
(Fig. 9C) or 5 �M MitoQ10 (data not shown), PTP opening wasstill triggered by 5 �M tBHP (Fig. 9C, trace b) and blocked with
CsA (Fig. 9C, trace c). The extent of Ca2� uptake was decreasedcompared to the system without MitoQ10, but the kinetics of PTPinduction were similar. Thus MitoQ10 cannot scavenge alkyl per-oxides and prevent PTP opening caused by thiol oxidation.
In summary, MitoQ10 does not react directly with peroxidesor affect the interaction of peroxides with biological thiols.Therefore the potent blocking of exogenous peroxide-dependentreactions by MitoQ10 (29, 41, 42) occurs downstream of theperoxide itself. As MitoQ10 blocks lipid peroxidation (29, 30), itis likely that the effects of MitoQ10 on exogenous peroxides aredue to chain termination of lipid peroxidation, which is proba-bly induced by Fenton chemistry of H2O2 in combination withferrous iron.
DISCUSSION
This study has clarified the redox, antioxidant, and pro-oxidant properties of a series of mitochondria-targeted and
FIG. 9. MitoQ10 does not prevent peroxide-induced thiol oxida-tion. A, MitoQ10 does not prevent glutathione oxidation by H2O2. H2O2was generated by glucose oxidase (0.006 units�ml�1) and 10 mM glucosein the presence of glutathione peroxidase (0.02 units�ml�1), 500 �M
GSH, NADPH, and glutathione reductase. Reduced MitoQ10 was addedas indicated. B, the mitochondrial PTP is triggered by tBHP. Livermitochondria were incubated in the presence of succinate, rotenone,calcium green-5N and either no additions (a), 5 �M tBHP (b), or 5 �M
tBHP and 500 nM CsA (c). 5 �M Ca2� was added where indicated. C,induction of the PTP by tBHP is not blocked by MitoQ10. Liver mito-chondria were incubated in the presence of succinate, rotenone, calciumgreen-5N, 1 �M MitoQ10, and either no addition (a), 5 �M tBHP (b), or 5�M tBHP and 500 nM CsA (c). 5 �M Ca2� was added where indicated.
Exogenous Ubiquinones as Therapies and Tools 21309
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

untargeted exogenous ubiquinones. While CoQ1, CoQ2, decylQ,CoQ6, and idebenone could all restore respiration in mitochon-dria lacking CoQ, MitoQ10 could not. The ability of the CoQanalogs to restore respiration was consistent with their fastreduction by ubiquinone reductases and their rapid oxidationby complex III. In contrast, none of the MitoQ analogs could actas electron carriers in respiration because they were not oxi-dized by complex III. While all the MitoQ analogs rapidlymigrate through the mitochondrial inner membrane (30), thehydrophobic core is still a significant activation energy barrierfor their transport, albeit a far lower one than that for equiv-alent hydrophilic cations (32, 33). Furthermore, althoughMitoQ10 has a similar octan-1-ol/PBS partition coefficient toidebenone, MitoQ analogs are amphipathic and consequentlynot evenly distributed within phospholipid bilayers (Fig. 1C)(32, 33). Thus for MitoQ analogs the steady-state concentrationof the ubiquinone moiety at a particular distance from themembrane surface will be related to the length of the alkylchain. This is in marked contrast to the untargeted ubiquino-nes, which will be freely soluble within the hydrophobic core.This affinity of MitoQ analogs for the surface of the phospho-lipid bilayer is simply demonstrated by its limited solubility incyclohexane, an organic solvent that mimics the membrane
core: MitoQ10 formed a separate, orange-red, oily phase, withonly some slight discoloration of the cyclohexane. In contrastidebenone was freely soluble up to at least 4 mM in cyclohexane,as were MitoQ10 and idebenone in octan-1-ol, a more polarsolvent that mimics the membrane surface. Therefore, the sig-nificantly decreased reactivity of all MitoQ analogs with com-plex III may be a consequence of the low concentration of theirubiquinone moieties in the active sites of complex III (Fig. 10A)(64, 84). These are near the center of the phospholipid bilayer,and for MitoQ analogs to dock into them, the TPP cation mustbe located in the hydrophobic core (Fig. 10A). In addition,enzyme shape and dimerization may further increase theeffective distance between these binding sites and the surfaceof the phospholipid bilayer (64, 84). Therefore the steady-stateconcentration of MitoQ analogs within the membrane core isdecreased relative to other ubiquinones, and this is likely toexplain their low reactivity with complex III.
A related explanation may account for the increasing reduc-tion of MitoQ analogs by complex II and G3PDH on increasingthe length of the carbon chain connecting the TPP and ubiqui-none moieties (Fig. 3, D and E). The ubiquinone reduction siteof complex II is close to the membrane surface, and it is possibleto dock the ubiquinone moiety of MitoQ10 into the active site
FIG. 10. Modeling of the binding ofMitoQ analogs to the active sites ofcomplexes II and III. A, MitoQ10 (red)bound to the ubiquinone reduction (left)and ubiquinol oxidation (right) bindingsites in complex III from B. taurus. B,MitoQ10 (red) bound to the ubiquinone re-duction site of complex II from E. coli. Cand D, binding of MitoQ3 (C) and MitoQ5(D) to the ubiquinone reduction site ofcomplex II. Val, Phe, Met, Ile, Leu, Tyr,and Trp amino acid residues were consid-ered hydrophobic and colored yellow toindicate the approximate position of thephospholipid bilayer; all other residuesare blue.
Exogenous Ubiquinones as Therapies and Tools21310
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

without the TPP cation dipping into the membrane (Fig. 10B).In contrast, the alkyl linkers for MitoQ3 and MitoQ5 appear tobe too short to span the distance from the active site to themembrane surface (Fig. 10, C and D). This offers a plausibleexplanation for the greater reactivity of MitoQ10 with complexII compared with MitoQ3 and MitoQ5, although with complex IIit is not possible to exclude a contribution from hydrophobicityor steric hindrance. If this explanation of the observed reactiv-ities is correct, it may lead to the development of alkyl TPPderivatives as useful probes of membrane proteins. For exam-ple, MitoQ10 was poorly reactive with complex I but did reactwell with G3PDH, suggesting that the ubiquinone binding siteof G3PDH may be closer to the membrane surface, while that ofcomplex I is nearer the hydrophobic core of the membrane.
The effects of MitoQ analogs are not due to their ability tocomplement respiration. In contrast, untargeted ubiquinonesshould be able to supplement respiratory defects in vivo pro-vided they can be delivered to mitochondria within cells. In ourexperiments only the ubiquinones with isoprenoid tails, CoQ6,CoQ2, and CoQ1, could restore non-fermentative growth inCoQ-deficient yeast, while the exogenous ubiquinones withsimple saturated carbon chains were ineffective. As this differ-ence is not due to poor electron transfer in the respiratorychain, there may be lower mitochondrial accumulation of thesaturated relative to the isoprenoid exogenous ubiquinones.This is not due to differences in passive diffusion as CoQ6 isseveral orders of magnitude more hydrophobic than decylQ,which in turn has an octan-1-ol/PBS partition coefficient 1,000-fold higher than CoQ1 (Fig. 1B) (31). Supporting this, thediffusion of CoQ6 through the mitochondrial outer membranewas far slower than for decylQ or idebenone (Fig. 2D). Asuptake does not appear to be passive, we speculate that inyeast there is a process for trafficking exogenous ubiquinonesfrom the plasma membrane to mitochondria and that it isselective for ubiquinones with isoprenoid tails. Such a processis suggested by some earlier work on yeast where accumulationof exogenous CoQ6 in mitochondria was deficient in somestrains (85). It is unclear whether a similar pathway wouldexist in mammalian cells; however, in a clinical case whereCoQ10 supplementation ameliorated the symptoms of CoQ de-ficiency, subsequent switching to idebenone caused clinical andmetabolic worsening, which disappeared on return to CoQ10
supplementation (86). As the final steps of CoQ synthesis are inmitochondria, it is not clear why a putative CoQ uptake systemshould exist. However, it is possible there is a pathway thatallows the export mitochondrial CoQ to the plasma membrane,and in the presence of exogenous CoQ it can operate in reverseto insert CoQ into mitochondria. This may explain why dietarysupplementation with CoQ10 leads to only small increases inmitochondrial CoQ10 concentration except when there is anunderlying CoQ10 deficiency (20–24, 87). Our results suggestthat short chain ubiquinone analogs containing isoprenoid sidechains may accumulate more effectively within mitochon-dria than saturated exogenous ubiquinones and are worth ex-ploring as potential therapies for human diseases with CoQdeficiencies.
The autoxidation of ubiquinols is well established, but itssignificance for exogenous short-chain ubiquinones is uncer-tain. Here we confirm that autoxidation of exogenous ubiqui-nones occurs by enzymatic reduction to the ubiquinol followedby deprotonation to the ubiquinolate anion, which reacts withoxygen to produce O2
.. Critically, ubiquinol autoxidation willnot occur when the ubiquinol head group is within the phos-pholipid bilayer and is therefore proportional to ubiquinol hy-drophilicity. However, while it was possible to detect autoxida-tion of ubiquinols in BHM and isolated mitochondria, the
autoxidation of MitoQ10 or idebenone did not lead to a signifi-cant increase in O2
.-dependent damage to matrix aconitase.This lack of autoxidation-dependent damage is consistent withclinical trials using idebenone that have shown it is well toler-ated for at least 2 years (28, 88–90), even though it has a hightendency to autoxidize in vitro (71). While the focus of studieson ROS is often their toxicity, it is possible that autoxidation ofexogenous ubiquinones generates subtoxic levels of ROS. Thismay even be beneficial as ROS can increase the expression ofantioxidant pathways via hormesis (4, 91). The complex natureof ROS interactions is illustrated by the paradoxical fact that20 �M H2O2, autoxidation-resistant ascorbate derivatives, andMitoQ10 all extend the lifespan of cultured cells and decreasetelomere shortening (39, 91). Therefore, it remains unclearwhether exogenous ubiquinone autoxidation is detrimentalin vivo as it is associated with a large increase in membraneassociated antioxidant capacity, against both lipid peroxidationand O2
., and could lead to the induction of antioxidant enzymesvia redox-sensitive transcription factors (92–94). Whether alarge increase in protection against lipid peroxidation is onbalance beneficial when coupled to a small increase in O2
. isbeyond the scope of this work and would require long termanimal studies looking at pathological markers. Even so, it isapparent that increasing hydrophobicity limits the “bad” aque-ous autoxidation of ubiquinols without diminishing its “good”organic phase antioxidant functions (Fig. 6). This could providea teleological explanation for the extreme hydrophobicity ofendogenous CoQ10 (Fig. 1B).
MitoQ10 has been shown to block a number of redox signalingpathways as well as cell death induced by exogenous H2O2 (29,41, 42); however, the details of the processes affected are un-certain. MitoQ10 does not react significantly with H2O2 or otheralkyl peroxides (Fig. 9) but is very effective against lipid per-oxidation (29, 30). This suggests that the effects of MitoQ10
against H2O2 are primarily due to blocking the consequences ofOH� presumably formed from iron-catalyzed Fenton chemistry.The presence OH� within mitochondria results in lipid peroxi-dation, which has a number of deleterious effects on mitochon-drial function and leads to breakdown products, such as thereactive aldehyde 4-hydroxy-2-nonenal, which may have a rolein cell damage and signaling (95). This work shows that whileMitoQ10 could interact with ROS in several ways in vivo, themost likely way is that it blocks the effects of exogenous H2O2
by acting downstream of H2O2 to inhibit peroxidative chainreactions.
Here we have investigated how a series of exogenous ubiqui-none analogs interact with oxidative phosphorylation and withROS metabolism. We found that while the type of reactionsthat MitoQ and untargeted ubiquinone analogs underwentwith ROS were similar, ubiquinone hydrophobicity dramati-cally affected the extent of these reactions. This work clearlyshows that the antioxidant reactions of exogenous ubiquinoneswill predominantly occur within phospholipid bilayers, whilethe pro-oxidant reactions require an aqueous environment.Therefore, the relative rates of these reactions can be fine-tuned by hydrophobicity, allowing a rational approach to thedesign of therapeutic ubiquinone-based antioxidants (e.g.MitoQ10) or mitochondria-targeted redox cyclers (e.g. MitoQ3).Furthermore, there was a sharp distinction between howMitoQ analogs and other short-chain ubiquinones interactedwith the mitochondrial respiratory chain. This occurred be-cause the TPP cation decreased the oxidation of MitoQ analogsby complex III. Thus MitoQ analogs function only as antioxi-dants, while other short-chain ubiquinones can also be oxida-tive phosphorylation substrates. When used in combinationthey may provide a way for separating cause and effect in
Exogenous Ubiquinones as Therapies and Tools 21311
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

mitochondrial DNA diseases, Parkinson disease, and agingitself where both a decline in oxidative phosphorylation and anincrease in ROS may overlap to cause pathology.
Acknowledgments—We thank D. R. Gauntlett for technical assist-ance and Dr. Abdul-Rahman B. Manas for the supply of syntheticchemicals.
REFERENCES
1. Turrens, J. F. (2003) J. Physiol. (Lond.) 552, 335–3442. Duchen, M. R. (2004) Mol. Aspects Med. 25, 365–4513. McKenzie, M., Liolitsa, D., and Hanna, M. G. (2004) Neurochem. Res. 29,
589–6004. Droge, W. (2002) Physiol. Rev. 82, 47–955. Finkel, T. (2003) Curr. Opin. Cell Biol. 15, 247–2546. Murphy, M. P., and Smith, R. A. (2000) Adv. Drug Deliv. Rev. 41, 235–2507. Murphy, M. P. (2001) Exp. Opin. Biol. Ther. 1, 753–7648. Smith, R. A., Kelso, G. F., Blaikie, F. H., Porteous, C. M., Ledgerwood, E. C.,
Hughes, G., James, A. M., Ross, M. F., Asin-Cayuela, J., Cocheme, H. M.,Filipovska, A., and Murphy, M. P. (2003) Biochem. Soc. Trans. 31,1295–1299
9. Smith, R. A., Porteous, C. M., Gane, A. M., and Murphy, M. P. (2003) Proc.Natl. Acad. Sci. U. S. A. 100, 5407–5412
10. Wright, A. F., Jacobson, S. G., Cideciyan, A. V., Roman, A. J., Shu, X.,Vlachantoni, D., McInnes, R. R., and Riemersma, R. A. (2004) Nat. Genet.36, 1153–1158
11. Turunen, M., Olsson, J., and Dallner, G. (2004) Biochim. Biophys. Acta 1660,171–199
12. Frei, B., Kim, M. C., and Ames, B. N. (1990) Proc. Natl. Acad. Sci. U. S. A. 87,4879–4883
13. James, A. M., Smith, R. A., and Murphy, M. P. (2004) Arch. Biochem. Biophys.423, 47–56
14. Marriage, B. J., Clandinin, M. T., Macdonald, I. M., and Glerum, D. M. (2004)Mol. Genet. Metab. 81, 263–272
15. Quiles, J. L., Ochoa, J. J., Huertas, J. R., and Mataix, J. (2004) Exp. Gerontol.39, 189–194
16. Abe, K., Matsuo, Y., Kadekawa, J., Inoue, S., and Yanagihara, T. (1999)J. Neurol. Sci. 162, 65–68
17. Di Giovanni, S., Mirabella, M., Spinazzola, A., Crociani, P., Silvestri, G.,Broccolini, A., Tonali, P., Di Mauro, S., and Servidei, S. (2001) Neurology 57,515–518
18. Shults, C. W., Oakes, D., Kieburtz, K., Beal, M. F., Haas, R., Plumb, S., Juncos,J. L., Nutt, J., Shoulson, I., Carter, J., Kompoliti, K., Perlmutter, J. S.,Reich, S., Stern, M., Watts, R. L., Kurlan, R., Molho, E., Harrison, M., andLew, M. (2002) Arch. Neurol. 59, 1541–1550
19. Muller, T., Buttner, T., Gholipour, A. F., and Kuhn, W. (2003) Neurosci. Lett.341, 201–204
20. Zhang, Y., Turunen, M., and Appelkvist, E. L. (1996) J. Nutr. 126, 2089–209721. Kwong, L. K., Kamzalov, S., Rebrin, I., Bayne, A. C., Jana, C. K., Morris, P.,
Forster, M. J., and Sohal, R. S. (2002) Free Radic. Biol. Med. 33, 627–63822. Kamzalov, S., Sumien, N., Forster, M. J., and Sohal, R. S. (2003) J. Nutr. 133,
3175–318023. Matthews, R. T., Yang, L., Browne, S., Baik, M., and Beal, M. F. (1998) Proc.
Natl. Acad. Sci. U. S. A. 95, 8892–889724. Svensson, M., Malm, C., Tonkonogi, M., Ekblom, B., Sjodin, B., and Sahlin, K.
(1999) Int. J. Sport Nutr. 9, 166–18025. Suno, M., and Nagaoka, A. (1984) Jpn. J. Pharmacol. 35, 196–19826. Rustin, P., Rotig, A., Munnich, A., and Sidi, D. (2002) Free Radic. Res. 36,
467–46927. Hausse, A. O., Aggoun, Y., Bonnet, D., Sidi, D., Munnich, A., Rotig, A., and
Rustin, P. (2002) Heart 87, 346–34928. Mariotti, C., Solari, A., Torta, D., Marano, L., Fiorentini, C., and Di Donato, S.
(2003) Neurology 60, 1676–167929. Kelso, G. F., Porteous, C. M., Coulter, C. V., Hughes, G., Porteous, W. K.,
Ledgerwood, E. C., Smith, R. A., and Murphy, M. P. (2001) J. Biol. Chem.276, 4588–4596
30. Asin-Cayuela, J., Manas, A.-R. B., James, A. M., Smith, R. A., and Murphy,M. P. (2004) FEBS Lett. 571, 9–16
31. Smith, R. A., Kelso, G. F., James, A. M., and Murphy, M. P. (2004) MethodsEnzymol. 382, 45–67
32. Ketterer, B., Neumcke, B., and Laeuger, P. (1971) J. Membr. Biol. 5, 225–24533. Flewelling, R. F., and Hubbell, W. L. (1986) Biophys. J. 49, 531–54034. Ono, A., Miyauchi, S., Demura, M., Asakura, T., and Kamo, N. (1994) Bio-
chemistry 33, 4312–431835. Hwang, P. M., Bunz, F., Yu, J., Rago, C., Chan, T. A., Murphy, M. P., Kelso,
G. F., Smith, R. A., Kinzler, K. W., and Vogelstein, B. (2001) Nat. Med. 7,1111–1117
36. Echtay, K. S., Murphy, M. P., Smith, R. A., Talbot, D. A., and Brand, M. D.(2002) J. Biol. Chem. 277, 47129–47135
37. Bedogni, B., Pani, G., Colavitti, R., Riccio, A., Borrello, S., Murphy, M., Smith,R., Eboli, M. L., and Galeotti, T. (2003) J. Biol. Chem. 278, 16510–16519
38. Jauslin, M. L., Meier, T., Smith, R. A., and Murphy, M. P. (2003) FASEB J. 17,1972–1974
39. Saretzki, G., Murphy, M. P., and von Zglinicki, T. (2003) Aging Cell 2, 141–14340. Schafer, M., Schafer, C., Ewald, N., Piper, H. M., and Noll, T. (2003) Circ. Res.
92, 1010–101541. Chen, K., Thomas, S. R., Albano, A., Murphy, M. P., and Keaney, J. F., Jr.
(2004) J. Biol. Chem. 279, 35079–3508642. Dhanasekaran, A., Kotamraju, S., Kalivendi, S. V., Matsunaga, T., Shang, T.,
Keszler, A., Joseph, J., and Kalyanaraman, B. (2004) J. Biol. Chem. 279,37575–37587
43. Rich, P. R., and Bendall, D. S. (1979) FEBS Lett. 105, 189–19444. Nohl, H., Gille, L., and Kozlov, A. V. (1998) Free Radic. Biol. Med. 25, 666–67545. Kaiser, J. (2003) Science 302, 376–37946. Calabrese, E. J. (2004) EMBO Rep. 5, S37–S4047. Hsu, A. Y., Do, T. Q., Lee, P. T., and Clarke, C. F. (2000) Biochim. Biophys.
Acta 1484, 287–29748. Diekert, K., de Kroon, A. I., Kispal, G., and Lill, R. (2001) Methods Cell Biol.
65, 37–5149. Murphy, M. P., Echtay, K. S., Blaikie, F. H., Asin-Cayuela, J., Cocheme, H. M.,
Green, K., Buckingham, J. A., Taylor, E. R., Hurrell, F., Hughes, G., Miwa,S., Cooper, C. E., Svistunenko, D. A., Smith, R. A., and Brand, M. D. (2003)J. Biol. Chem. 278, 48534–48545
50. Smith, P. K., Krohn, R. I., Hermanson, G. T., Mallia, A. K., Gartner, F. H.,Provenzano, M. D., Fujimoto, E. K., Goeke, N. M., Olson, B. J., and Klenk,D. C. (1985) Anal. Biochem. 150, 76–85
51. Smith, A. L. (1967) Methods Enzymol. 10, 81–8652. Walker, J. E., Skehel, J. M., and Buchanan, S. K. (1995) Methods Enzymol.
260, 14–3453. Chappell, J. B., and Hansford, R. G. (1972) in Subcellular Components: Prep-
aration and Fractionation (Birnie, G. D., ed) 2nd Ed., pp. 77–91, Butter-worths, London
54. Gornall, A. G., Bardawill, C. J., and David, M. M. (1949) J. Biol. Chem. 177,751–766
55. James, A. M., Wei, Y. H., Pang, C. Y., and Murphy, M. P. (1996) Biochem. J.318, 401–407
56. Garrib, A., and McMurray, W. C. (1986) J. Biol. Chem. 261, 8042–804857. Hughes, M. N., and Nicklin, H. G. (1968) J. Am. Chem. Soc. A, 450–45258. Packer, M. A., and Murphy, M. P. (1994) FEBS Lett. 345, 237–24059. Rose, I. A., and O’Connell, E. L. (1967) J. Biol. Chem. 242, 1870–187960. Gardner, P. R. (2002) Methods Enzymol. 349, 9–2361. Gralla, E. B., and Valentine, J. S. (1991) J. Bacteriol. 173, 5918–592062. Tyler, D. D. (1992) The Mitochondrion in Health and Disease, pp. 41–93, VCH
Publishers Inc., New York63. Yankovskaya, V., Horsefield, R., Tornroth, S., Luna-Chavez, C., Miyoshi, H.,
Leger, C., Byrne, B., Cecchini, G., and Iwata, S. (2003) Science 299,700–704
64. Gao, X., Wen, X., Esser, L., Quinn, B., Yu, L., Yu, C. A., and Xia, D. (2003)Biochemistry 42, 9067–9080
65. Okada, K., Kainou, T., Matsuda, H., and Kawamukai, M. (1998) FEBS Lett.431, 241–244
66. Rich, P. R. (1984) Biochim. Biophys. Acta 768, 53–7967. Crane, F. L., and Barr, R. (1971) Methods Enzymol. C18, 137–16568. Takada, M., Ikenoya, S., Yuzuriha, T., and Katayama, K. (1984) Methods
Enzymol. 105, 147–15569. Beckman, J. S., Beckman, T. W., Chen, J., Marshall, P. A., and Freeman, B. A.
(1990) Proc. Natl. Acad. Sci. U. S. A. 87, 1620–162470. Schopfer, F., Riobo, N., Carreras, M. C., Alvarez, B., Radi, R., Boveris, A.,
Cadenas, E., and Poderoso, J. J. (2000) Biochem. J. 349, 35–4271. Genova, M. L., Pich, M. M., Biondi, A., Bernacchia, A., Falasca, A., Bovina, C.,
Formiggini, G., Castelli, G. P., and Lenaz, G. (2003) Exp. Biol. Med. (May-wood) 228, 506–513
72. Rich, P. R., and Bendall, D. S. (1980) Biochim. Biophys. Acta 592, 506–51873. Azzi, A., Montecucco, C., and Richter, C. (1975) Biochem. Biophys. Res. Com-
mun. 65, 597–60374. Winterbourn, C. C. (1981) Arch. Biochem. Biophys. 209, 159–16775. Palmer, R. M., Ferrige, A. G., and Moncada, S. (1987) Nature 327, 524–52676. Carreras, M. C., Franco, M. C., Peralta, J. G., and Poderoso, J. J. (2004) Mol.
Aspects Med. 25, 125–13977. Nisoli, E., Falcone, S., Tonello, C., Cozzi, V., Palomba, L., Fiorani, M., Pisconti,
A., Brunelli, S., Cardile, A., Francolini, M., Cantoni, O., Carruba, M. O.,Moncada, S., and Clementi, E. (2004) Proc. Natl. Acad. Sci. U. S. A. 101,16507–16512
78. Brown, G. C. (2001) Biochim. Biophys. Acta 1504, 46–5779. Liu, L., Yan, Y., Zeng, M., Zhang, J., Hanes, M. A., Ahearn, G., McMahon, T. J.,
Dickfeld, T., Marshall, H. E., Que, L. G., and Stamler, J. S. (2004) Cell 116,617–628
80. Poderoso, J. J., Carreras, M. C., Schopfer, F., Lisdero, C. L., Riobo, N. A.,Giulivi, C., Boveris, A. D., Boveris, A., and Cadenas, E. (1999) Free Radic.Biol. Med. 26, 925–935
81. St-Pierre, J., Buckingham, J. A., Roebuck, S. J., and Brand, M. D. (2002)J. Biol. Chem. 277, 44784–44790
82. Lambert, A. J., and Brand, M. D. (2004) Biochem. J. 382, 511–51783. Crompton, M. (1999) Biochem. J. 341, 233–24984. Xia, D., Yu, C. A., Kim, H., Xia, J. Z., Kachurin, A. M., Zhang, L., Yu, L., and
Deisenhofer, J. (1997) Science 277, 60–6685. Santos-Ocana, C., Do, T. Q., Padilla, S., Navas, P., and Clarke, C. F. (2002)
J. Biol. Chem. 277, 10973–1098186. Aure, K., Benoist, J. F., Ogier de Baulny, H., Romero, N. B., Rigal, O., and
Lombes, A. (2004) Neurology 63, 727–72987. Dallner, G., and Sindelar, P. J. (2000) Free Radic. Biol. Med. 29, 285–29488. Gillis, J. C., Benefield, P., and McTavish, D. (1994) Drugs Aging 5, 133–15289. Rustin, P., von Kleist-Retzow, J. C., Chantrel-Groussard, K., Sidi, D., Mun-
nich, A., and Rotig, A. (1999) Lancet 354, 477–47990. Buyse, G., Mertens, L., Di Salvo, G., Matthijs, I., Weidemann, F., Eyskens, B.,
Goossens, W., Goemans, N., Sutherland, G. R., and Van Hove, J. L. (2003)Neurology 60, 1679–1681
91. Yokoo, S., Furumoto, K., Hiyama, E., and Miwa, N. (2004) J. Cell. Biochem. 93,588–597
92. Meyer, M., Schreck, R., and Baeuerle, P. A. (1993) EMBO J. 12, 2005–201593. Borrello, S., and Demple, B. (1997) Arch. Biochem. Biophys. 348, 289–29494. Muller, J. M., Rupec, R. A., and Baeuerle, P. A. (1997) Methods 11, 301–31295. Petersen, D. R., and Doorn, J. A. (2004) Free Radic. Biol. Med. 37, 937–945
Exogenous Ubiquinones as Therapies and Tools21312
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Andrew M. James, Helena M. Cochemé, Robin A. J. Smith and Michael P. MurphyEXPERIMENTAL TOOLS
FOR THE USE OF EXOGENOUS UBIQUINONES AS THERAPIES ANDMitochondrial Respiratory Chain and Reactive Oxygen Species: IMPLICATIONS
Interactions of Mitochondria-targeted and Untargeted Ubiquinones with the
doi: 10.1074/jbc.M501527200 originally published online March 23, 20052005, 280:21295-21312.J. Biol. Chem.
10.1074/jbc.M501527200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/280/22/21295.full.html#ref-list-1
This article cites 92 references, 32 of which can be accessed free at
by guest on June 3, 2018http://w
ww
.jbc.org/D
ownloaded from