intravenous anaesthetic agents incudes benzodiazipenes, opiods,tiva ,neurolept analgesia...
TRANSCRIPT


Diazepam (Valium) was synthesized by Sternbachin 1959
Lorazepam a 2′ chloro-substitution product of oxazepam , was synthesized in 1971
Fryer and Walser's in 1976 synthesized midazolam
Midazolam was the first benzodiazepine that was produced primarily for use in anesthesia


Three benzodiazepine receptor agonists are commonly used in anesthesia are :-
short-lasting (midazolam),
intermediate-lasting (lorazepam),
long-lasting (diazepam)
relatively small and are lipid soluble at physiologic pH
Midazolam is the most lipid soluble of the three drugs



Binding of drug facilitates the actions of GABA ie principal inhibitory neurotransmitter in the CNS
Enhance the affinity of the receptors for GABA
enhanced opening of chloride gating channels leading to increased chloride conductance
hyperpolarization of the postsynaptic cell membrane, rendering postsynaptic neurons more resistant to excitation

Results into anxiolysis,sedation, anterograde amnesia, alcohol potentiation and anticonvulsant and skeletal muscle relaxant effects
Alpha-1 containing GABAA receptors are the most abundant receptor subtypes
Sedative effects of benzodiazepines reflect activation of alpha-1 subunits of GABAA
receptors
Anxiolytic activity is due to alpha-2 subunit activity


Midazolam is a water-soluble benzodiazepine with an imidazole ring in its structure that accounts for stability in aqueous solutions
Compared with diazepam, midazolam is two to three times more potent
has an affinity for the benzodiazepine receptor that is approximately twice that of diazepam
Compared with other benzodiazepines, the amnestic effects of midazolam are more potent than its sedative effects


Midazolam undergoes rapid absorption from the gastrointestinal tract and prompt passage across the blood-brain barrier
Only about 50% of an orally administered dose of midazolam reaches the systemic circulation, reflecting a substantial first-p ass hepatic effect
extensively bound to plasma proteins
elimination half-time of midazolam is 1 to 4 hours,
Short duration of action of a single dose of midazolam is due to its lipid solubility, leading to rapid redistribution from the brain to inactive tissue sites as well as rapid hepatic clearance

Midazolam is rapidly metabolized by hepatic and small intestine cytochrome P-450 (CYP3A4) enzymes
principal metabolite of midazolam, 1-hydroxym idazolam,has approximately half the activity of the parent compound
metabolite is rapidly conjugated to glucuronide and subsequently cleared by the kidneys
Metabolism of is slowed in the presence of drugs (cimetidine, erythromycin, calcium channel blockers,antifungal drugs) that inhibit cytochrome P-450 enzymes resulting in unexpected CNS depression
hepatic clearance rate of midazolam is five times greater than that of lorazepam and ten times greater than that of diazepam

Central Nervous System
Produces decreases in cerebral metabolic oxygen requirements(CMR0 2 ) and cerebral blood flow
Cerebral vasomotor responsiveness to carbon dioxide is preserved during midazolamanesthesia
show little or no change in intracranial pressure (ICP)
Midazolam is a potent anticonvulsant effective in the treatment of status epilepticus

produces dose-dependent decreases in ventilation
Transient apnea may occur after rapid injection of large doses of midazolam
depress the swallowing reflex and decrease upper airway activity
Cardiovascular System:-
decrease in systemic blood pressure and increase in heart rate
Cardiac output is not altered by midazolam

Induction
0.05-0.15 mg/kg
Maintenance 0.05 mg/kg
Sedation 0.5-1 mg repeated 0.07 mg/kg IM

1)Preoperative Medication:- most commonly used oral preoperative Midazolam 0.5 mg/kg administered orally 30
minutes before induction of anesthesia, provides reliable sedation and anxiolysis in children without producing delayed awakening
2) Intravenous Sedation:- Midazolam in doses of 1.0 to 2.5 mg i/v(onset within 30 to60 seconds) is effective for sedation duringregional anesthesia as well as for brief therapeutic procedures

Anesthesia can be induced by administration of midazolam,0.1 to 0.2 mg/kg N, over 30 to 60 seconds
Elderly patients require less midazolam for the i/v induction
4) Maintenance of Anesthesia:- may be administered to supplement opioids,
propofol, and/or inhaled anesthetics during maintenance of anesthesia
5)Postoperative Sedation:- loading dose 0.5 to 4 mg i/v and maintenance
dose 1 to 7 mg/hr IV) to produce sedation in intubated patients

6)Paradoxical Vocal Cord Motion:-
Paradoxical vocal cord motion is a cause of non organic upper airway obstruction and stridor that may manifest postoperatively. Midazoalm 0.5 to 1 mg IV may be an effective treatment for paradoxical vocal cord motion
7) Midazolam is a potent anticonvulsant effective in the treatment of status epilepticus
8) Nausea and Vomiting Prophylaxis:-midazolam, may play in the prevention of PONV

highly lipid-soluble benzodiazepine with a more prolonged duration of action compared with midazolam
dissolved in organic solvents (propylene glycol,sodium benzoate) because it is insoluble in water
Dilution with water or saline causes cloudiness but does not alter the potency of the drug
Injection by either the iM or IV route may be painful

Diazepam is well absorbed from the gastrointestinal tract, with peak plasma levels usually achieved in 1hr
relatively lipid soluble and readily penetrates the blood–brain barrier
highly protein binding limits the efficacy of hemodialysis in the treatment of diazepam overdose.
Diazepam rapidly crosses the placenta, achieving fetal concentrations equal to and sometimes greater than those present in the maternal circulation

metabolized by hepatic microsomal enzymes using an oxidative pathway of N demethylation
metabolites of diazepam are desmethyldiazepam and oxazepam, with a lesser amount metabolized to temazepam
removed as a conjugate of glucuronic acid Cimetidine inhibits P-450 hepatic microsomal
enzymes and thus prolongs the elimination half-time of both diazepam and desmethyldiazepam
elimination half-time of diazepam is prolonged, ranging from 21 to 37 hours in healthy volunteers

Central Nervous System - reduce the CMRO2
and CBF similar to midazolam CNS intoxication can be expected at
diazepam plasma concentrations of > 1,000 ng/mL.
Respiratory system-Diazepam produces minimal depressanteffects on ventilation
Combination of diazepam with other CNS depressants (opioids, alcohol) or in patients with COPD may result in exaggerated or prolonged depression of ventilation

produces minimal decreases in systemic blood pressure, cardiac output, and systemic vascular resistance
transient depression of baroreceptor-mediated heart rate
Skeletal Muscle:-
diminishes the tonic facilitatory influence on
spinal gamma neurons, and, thus, skeletal muscle tone is decreased
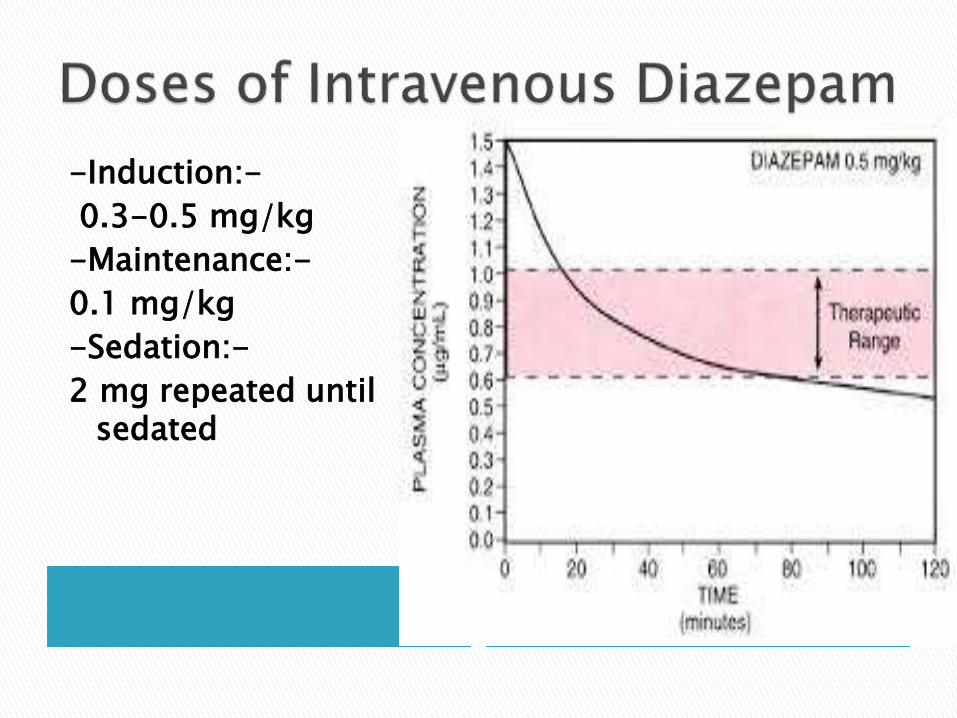
-Induction:-
0.3-0.5 mg/kg
-Maintenance:-
0.1 mg/kg
-Sedation:-
2 mg repeated until sedated

Preoperative medication of adults
management of delirium tremens
treatment of local anesthetic-induced seizures
Production of skeletal muscle relaxation by diazepam is often used in the management of lumbar disc disease and may be in the rare patient who develops tetany


Lorazepam is a more potent sedative and amnesic than midazolam and diazepam
Pharmacokinetics:-
conjugated with glucuronic acid in the liver to form pharmacologically inactive metabolites that are excreted by the kidneys

The elimination halftime is 10 to 20 hours
Lorazepam has a slower onset of action than midazolam or diazepam because of its lower lipid solubility and slower entrance into the CNS
metabolism of lorazepam is less likely than that of diazepam to be influenced by alterations in hepatic function, increasing age, or drugs that inhibit P-450 enzymes such as cimetidine

Induction-0.1 mg/kgMaintenance-0.02 mg/kgSedation-0.25 mg repeated until sedated
Clinical use:- (a)IV induction of anesthesia, (b) IV sedation during regional anesthesia (c) use as an anticonvulsant (d) effective in limiting the incidence of
emergence reactions after administration of ketamine

Fatigue and drowsiness are the most common side effects in patients treated chronically with benzodiazepines.
Decreased motor coordination and impairment of cognitive function may occur, especially when benzodiazepines are used in combination with other CNS depressant drugs
Acute administration of benzodiazepines
may produce transient anterograde amnesia
Benzodiazepines may inhibit platelet-activating factor induced aggregation resulting in drug-induced inhibition of platelet aggregation

Respiratory depression:-most commonly assosiated with midazolam
venous irritation and thrombophlebitis:-
-lorazepam and diazepam mainly
Dependence:- Even therapeutic doses of benzodiazepines may produce dependence as evidenced by the onset of physical or psychologic symptoms after the dosage is decreased or the drug is discontinued
Withdrawal symptoms (irritability,insomnia, tremulousness)

first benzodiazepine antagonist approved for clinical use
it is a competitive antagonist at the benzodiazepine receptor
Antagonism is reversible and surmountable
Uses and Doses of Flumazenil
Reversal of benzodiazepines 0.2 mg repeated up to 3 mg
Diagnosis in coma 0.5 mg repeated up to 1 mg

term opioid refers broadly to all compounds related to opium(Papaver somniferum )
In 1806, Sertürner reported the isolation of a pure substance in opium that he named morphine after Morpheus, the Greek god of dreams
i/v used opioids are mainly –
fentanyl,
alfentanil,
sufentanil,
remifentanil

Receptor Clinical Effect
μ Supraspinal analgesia (μ 1 )Respiratory depression (μ 2 )Physical dependenceMuscle rigidity
κ SedationSpinal analgesia
δ AnalgesiaBehavioralEpileptogenic
σ DysphoriaHallucinationsRespiratorystimulation

All opioid receptors couple to G proteins; binding of an agonist to an opioid receptor causes membrane hyperpolarization
Acute effects are mediated by inhibition of adenyl cyclase (reductions in intracellular cyclic adenosine monophosphateconcentrations) and activation of phospholipase C
Opioid receptor activation inhibits the presynaptic release and postsynaptic response to excitatory neurotransmitters (eg, acetylcholine, substance P) from nociceptiveneurons

Rapid and complete absorption
After intravenous administration, the distribution half-lives of all of the opioids are fairly rapid (5–20 min).
low molecular weight and high lipid solubility of fentanyl also favor transdermal absorption the transdermal fentanyl “patch”
increased lipid solubility of fentanyl and sufentanil, are associated with a faster onset and shorter duration of action when administered iv

alfentanil has a more rapid onset of action and shorter duration of action than fentanyl(Sufentanyl-most potent)
After smaller doses of the lipid- soluble drugs (eg, fentanyl or sufentanil), redistribution alone is the driver for reducing blood concentrations, whereas after larger doses biotransformation becomes an important driver in reducing plasma levels below those that have clinical effects.
time required for fentanyl or sufentanilconcentrations to decrease by half is context sensitive

all opioids depend primarily on the liver for biotransformation and are metabolized by the cytochrome P (CYP) system
Small V d of alfentanil contributes to a short elimination half-life (1.5 h)
End products of fentanyl, sufentanil, and alfentanil are inactive
Norfentanyl, the metabolite of fentanyl has greatest importance in diagnosing fentanylabuse.
ester structure of remifentanil makes it susceptible to hydrolysis (in a manner similar to esmolol) by nonspecific esterases in red blood cells and tissue

A. Cardiovascular:-
Larger doses of fentanyl, sufentanil, remifentanil,and alfentanil are associated with a vagus nerve–mediated bradycardia
sufentanil and fentanyl can be associated with reduced cardiac output
mild myocardial depression

Opioids depress ventilation, particularly respiratory rate
Sequestration by stomach and reabsorbed opoidsmay produce Biphasic resp. depression(FentanylMost commonly)
The apneic threshold—rises, and hypoxic drive is decreased
Rapid administration of larger doses of opioids(particularly fentanyl, sufentanil,remifentanil, and alfentanil) can induce chest wall rigidity(Wooden chest syndrome) severe enough to prevent adequate bag-and-mask ventilation effectively --treated with neuromuscular blocking agents
B. Respiratory:-

High dose
Sleep
Old age
Central nervous system depressant
Inhaled anesthetics
alcohol, barbiturates, benzodiazepines
Renal insufficiency
Hyperventilation,
Respiratory acidosis

opioids reduce cerebral oxygen consumption, cerebral blood flow,cerebral blood volume,
Stimulation of the medullary chemoreceptor
trigger zone is responsible for opioid-induced nausea and vomiting
Repeated dosing of opioids will reliably produce tolerance, a phenomenon in which larger doses are required to produce the same response
Prolonged dosing of opioids can also produce “opioid-induced hyperalgesia”

slow gastrointestinal motility by binding to opioid receptors in the gut and reducing peristalsis
Biliary colic may result from opioid-induced contraction of the sphincter of Oddi
Patients receiving long-term opioid therapy (eg, for cancer pain)doesn’t become tolerant to constipation,so it lead to recent development of the peripheral opioidantagonists like methylnaltrexone and alvimopan

neuroendocrine stress response to surgical stimulation is measured in terms of the secretion of specific hormones, including catecholamines, antidiuretic hormone, and cortisol
Large doses of opioids (typically fentanyl or sufentanil) block the release of these hormones in response to surgery
F.Dependence-both physical &psychological
G.Tolerance-mainly pharmacodynamic ,seen with all actions except constipation & miosis

Sufentanil- Intraoperative anesthesia
- 0.25–20 mcg/kg iv
Alfentanil- Intraoperative anesthesia
Loading dose- 8–100 mcg/kg iv
Maintenance infusion- 0.5–3 mcg/kg/min iv
Remifentanil- Intraoperative anesthesia
Loading dose- 1.0 mcg/kg iv
Maintenance infusion- 0.5–20 mcg/kg/min iv
Postoperative analgesia/sedation- 0.05–0.3 mcg/kg/min i/v

Fentanil-100 times more potent than morphine
Can be given iv,i/m,transmucosal,epidural,intrathecally
Cardiac stable Maximum biphasic respiratory depression Alfentanil-compared to fantanil quick onset
and short acting & assosiated with highest incidence of muscle rigidity
Sufentanil-most potent & opoid of choice for inhibiting cardiovascular response to laryngoscopy and intubations

Metabolized by non specific esterase(pseudo cholinesterase)
Shortest acting so agent of choice for day care surgery
Opoid of choice for renal patients
Causes significant hypotension
Contains glycine so cann’t be used through spinal/epidural because it can cause motor weakness

Most commonly, an opioid is combined with another drug more likely to provide hypnosis and amnesia
combination of alfentanil and propofol produces excellent TIVA, Alfentanil provides analgesia and hemodynamic stability while blunting responses to noxious stimuli, On the other hand, propofolprovides hypnosis and amnesia and is antiemetic
Profound synergism also exists when more than two agents, such as propofol-alfentanil-midazolam, are combined
TIVA techniques are especially useful when delivery of inhaled agents is compromised

Loading Dose (µg/kg)
Maintenance Infusion Rate
Additional Boluses
Alfentanil 25-100 0.5-2 µg/kg/min
5-10 µg/kg
Sufentanil 0.25-2 0.5-1.5 µg/kg/hr
2.5-10 µg
Fentanyl 4-20 2-10 µg/kg/hr 25-100 µg
Remifentanil 1-2 0.1-1.0 µg/kg/min
0.1-1.0 µg/kg

Premedication before alfentanil-propofolanesthesia can prolong postoperative recovery and should be avoided
Induction of anesthesia with alfentanil (25 to 50 µg/kg) and propofol (0.5 to 1.5 mg/kg), followed by infusions of alfentanil at 0.5 to 1.5 µg/kg/min and propofol at 80 to 120 µg/kg/min, will produce anesthesia in patients ventilated with air and O2 with or without N2O for a variety of procedures
Maintenance infusions vary according to patient condition and surgical stimuli
optimal propofol concentration decreases in the order of fentanyl > alfentanil > sufentanil≫ remifentanil

Drug infusions should be terminated 10 to 20 minutes before the end of anesthesia if N2O is used,Otherwise, propofol infusions should be terminated 5 to 10 minutes before anticipated patient awakening. Alfentanilinfusion rates do not need to be less than 0.25 to 0.5 µg/kg/min until surgery is terminated

In patients undergoing ear-nose-throat surgery
children at risk for malignant hyperthermia (in whom volatile agents are to be avoided)
Patients undergoing brief radiologic or painful procedures when rapid recovery is needed (e.g., mri, bone marrow aspiration)
Neurosurgical procedures to assist with control of intracranial pressure and for cerebral metabolic protection
Other minor surgical procedures

1959 De Castro and Mundeleer derived the concept of Neuroleptanalgesia, which involved the combination of a major tranquilizer (usually the butyrophenone droperidol) and a potent opioid analgesic (fentanyl) to produce a detached, pain-free state of immobilization and insensitivity to pain
characterized by analgesia, absence of clinically apparent motor activity, suppression of autonomic reflexes, maintenance of cardiovascular stability, and amnesia
addition of an inhaled agent, usually N2O, improves amnesia and has been called Neuroleptanesthesia

Butyrophenones cause sedation, tranquility, immobility, antiemesis, an extrapyramidalsyndrome with face and neck dyskinesia, oculogyric crises, torticollis, agitation, and hallucinations so now a days it is rare used
Neuroleptanalgesia or neuroleptanesthesia is absolutely contraindicated in patients receiving monoamine oxidase inhibitors (MAOIs), in those who abuse drugs or alchohol, or in patients with Parkinson's disease.