introduction aux reactions …blogperso.univ-rennes1.fr/pierre.van-de-weghe/public/...introduction...
TRANSCRIPT

INTRODUCTION AUX REACTIONSINTRODUCTION AUX REACTIONSSTEREOSELECTIVES.STEREOSELECTIVES.
Aspects générauxAspects généraux ..
Introduction aux réactions stéréosélectives
Aspects générauxAspects généraux ..
Maîtrise Option Chimie Organique
2005
"L’univers est dissymétrique“
Louis Pasteur (1893)
Contact: Pr. Pierre van de Weghe, UMR 6226 Sciences Chimiques de RennesEquipe Produits Naturels, Synthèses, Chimie Médicinale (bât 5, rdc)[email protected]://blogperso.univ-rennes1.fr/pierre.van-de-weghe/index.php/tél : 02 23 23 38 03

Introduction aux réactions stéréosélectives
Pré requis
� relations de stéréochimie (diastéréiosomérie, énantiomérie)
� détermination des configurations absolues des centres stéréogènes (R ou S)
� détermination des excès diastéréoisomériques et énantiomériques
� analyse conformationnelle (systèmes acycliques et cycliques)
� réactivité de base des grandes fonctions de la chimie organique� réactivité de base des grandes fonctions de la chimie organique
Ouvrages conseillés pour étoffer vos connaissances liées à ce cours
C. Rabiller Stéréochimie et chiralité en chimie organique, DeBoeck université.
Carey – Sundberg Tomes 1 & 2
M. Nogradi Stereoselective Synthesis, VCH.
E.L. Eliel, S.H. Wilen, M.P. Doyle Basic Organic Stereochemistry, Wiley Interscience.
J. Seyden-Penne Synthèse et catalyse asymétriques, CNRS Editions.

Introduction aux réactions stéréosélectives
Généralités
Chiralité en chimie
� molécules chirales: composés différents de leur image dans un miroir.� nomenclature des énantiomères: application de la règle de Cahn-Ingold-Prelog.
H3C H
CO2H
OH
(S)-acide lactique
1
2
3 4
CH3C
H HCH3
allène (R)
CO2HOH
HOCO2H
(R,R)-acide tartrique(S)-acide lactique allène (R) (R,R)-acide tartrique
QUIZZ: parmi les composés suivants, indiquer ceux qui présentent une isomérie optique (composés chiraux).
NEt
CH3
Phbutan-2-olPh
PEt
CH3
HO2C
H
CH3
H
NO2
NO2
NO2
HO2C
CO2H
NO2
CH3CH3
CO2H

Introduction aux réactions stéréosélectives
� excès énantiomérique – excès diastéréoisomérique.
� il y a stéréoisomérie quand deux composés ont la même formule plane (énantiomérie et diastéréoisomérie).Pour un substrat possédant deux atomes de carbone chiraux:
R,R
S,S
R,S
S,R
diastéréoisomérie
énantiomérie
OHO
OH
OH
thréose
OHO
OH
OH
érythrose
OHO
OH
OH OHO
OH
OH
� excès énantiomérique – excès diastéréoisomérique.
e.e. =[R] - [S]
[R] + [S]x 100 e.d. =
[R,R] - [S,R]x 100
[R,R]+ [S,R]
� prochiralité:
B H
A
H
B H
A
D
B D
A
Hcarboneprochiral
OH3CPh
H3CPh OH
H
H3CPh H
OH
faces prochirales
ré
si

Introduction aux réactions stéréosélectives
QUIZZ: relation de topicité: indiquer la relation existant entre les groupes soulignés (énantiotopes etc…).
O
H3C CH3 H3C CH3
O
O
H3C
H3C
O
H3C CH3
H3C CH3
QUIZZ: on considère le substrat ci-dessous. Indiquer la configuration absolue des centres chiraux. La fonctionaldéhyde subit une réaction de réduction, le composé obtenu est-il chiral? Justifiez votre réponse.
OH OHO
OH OHH
Introduction aux réactions stéréosélectives
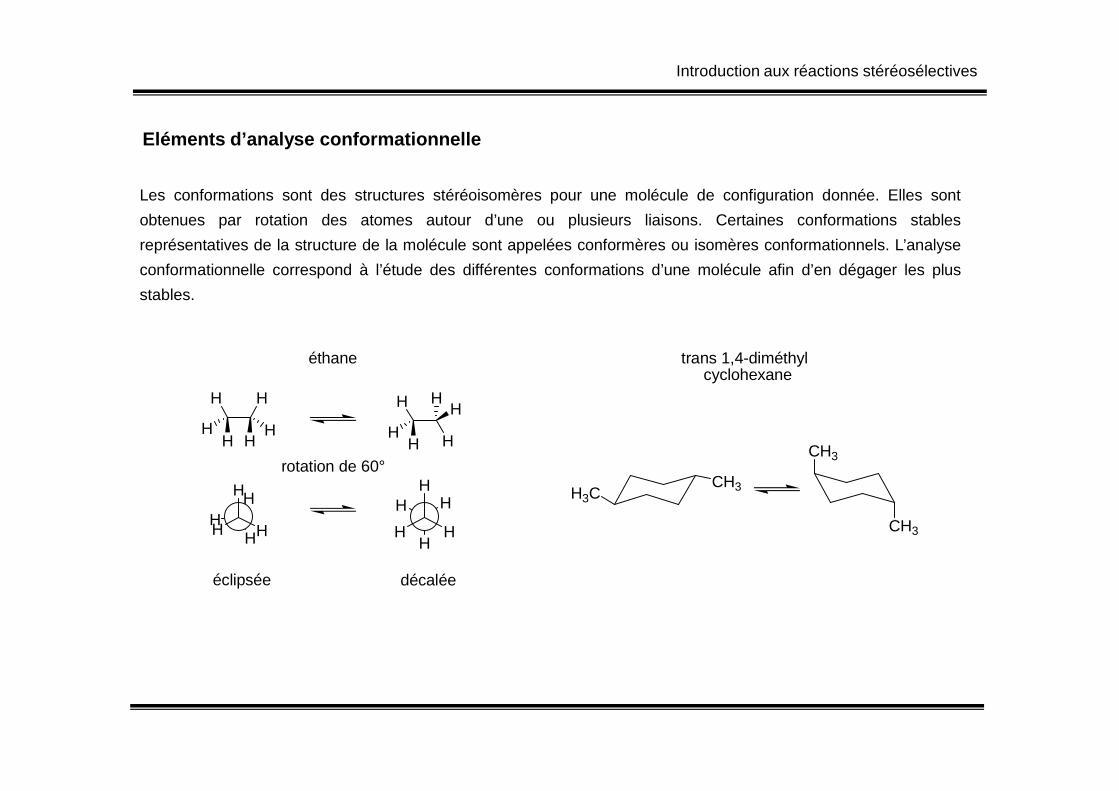
Eléments d’analyse conformationnelle
Les conformations sont des structures stéréoisomères pour une molécule de configuration donnée. Elles sont
obtenues par rotation des atomes autour d’une ou plusieurs liaisons. Certaines conformations stables
représentatives de la structure de la molécule sont appelées conformères ou isomères conformationnels. L’analyse
conformationnelle correspond à l’étude des différentes conformations d’une molécule afin d’en dégager les plus
stables.
HH H H
éthane trans 1,4-diméthyl cyclohexane
HH
HH H
HH
H HH
HH
rotation de 60°
HH
HH
HHH
H HH
HH
éclipsée décalée
CH3
H3CCH3
CH3

Introduction aux réactions stéréosélectives
Nomenclature de Klyne et Prelog: rotation autour d’ une liaison C-C simple
Angle de torsion (ω) désignation symbole
-30°à +30° synpériplanaire sp a
+30°à +90° +synclinal sc b
+90°à +150° +anticlinal +ac
+150 à -150° antipériplanaire ap c
-150° à -90° -anticlinal -ac-150° à -90° -anticlinal -ac
-90°à -30° -synclinal -sc
a: syn ou éclipsée sont couramment employés pour ω = 0°b: gauche est utilisé pour ω = 60°c: anti est utilisé pour ω = 180°
+sp
+sc
+ac
+ap
-sp
-sc
-ac
-ap
0°
+30°
+90°
+150°
+180°

Introduction aux réactions stéréosélectives
Conformation des molécules acycliques
Cas de l’éthane
L’éthane possède un nombre infini de conformations en raison de la libre rotation de la liaison C-C, cependant il n’y a
que trois minima. L’éthane possède donc trois conformères stables dégénérés (c’est-à-dire de même énergie) comme
l’atteste le graphe suivant.
La conformation éclipsée représente donc l’état
énergétique le plus élevé. En effet dans cette
conformation la distance séparant deux atomes
d’hydrogène situés sur chacun des deux atomes de
HH
HH H
H
gêne stérique importante
d’hydrogène situés sur chacun des deux atomes de
carbone est égale à 2,3 Å et le rayon de l’atome
d’hydrogène est de 1,2 Å. Il en résulte une très forte
gêne stérique.
Introduction aux réactions stéréosélectives
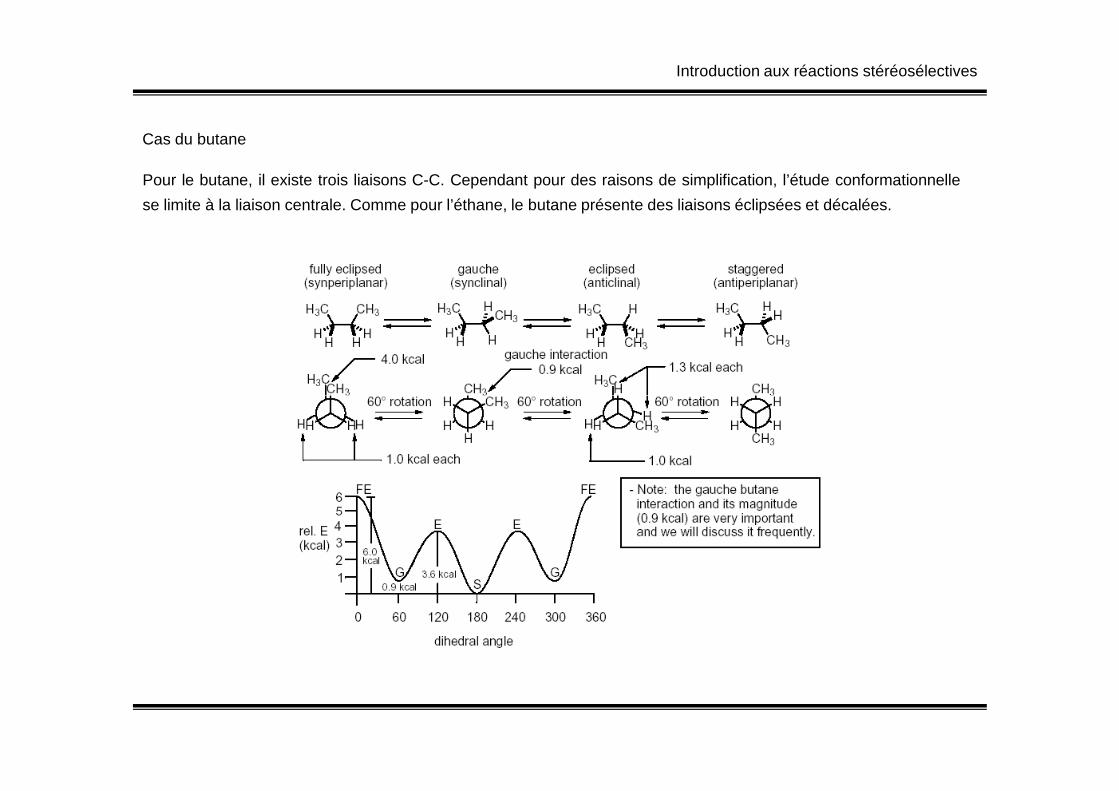
Cas du butane
Pour le butane, il existe trois liaisons C-C. Cependant pour des raisons de simplification, l’étude conformationnelle
se limite à la liaison centrale. Comme pour l’éthane, le butane présente des liaisons éclipsées et décalées.
Introduction aux réactions stéréosélectives
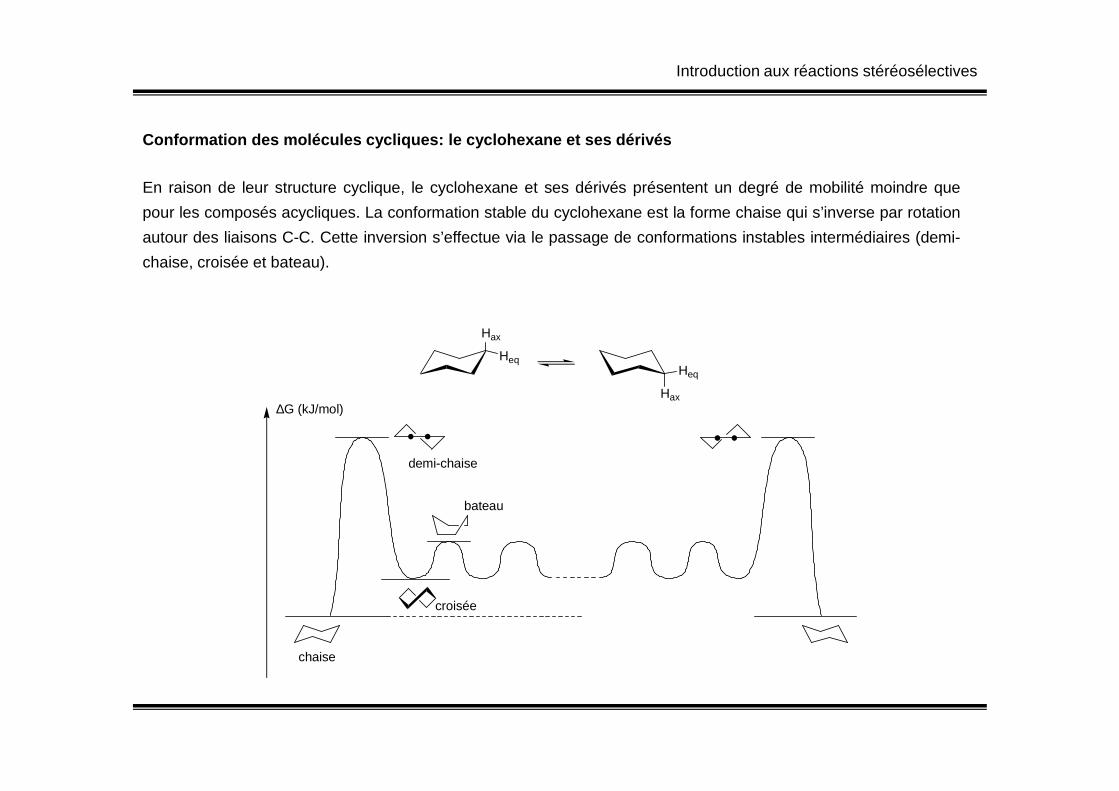
En raison de leur structure cyclique, le cyclohexane et ses dérivés présentent un degré de mobilité moindre que
pour les composés acycliques. La conformation stable du cyclohexane est la forme chaise qui s’inverse par rotation
autour des liaisons C-C. Cette inversion s’effectue via le passage de conformations instables intermédiaires (demi-
chaise, croisée et bateau).
Conformation des molécules cycliques: le cyclohexan e et ses dérivés
Hax
Heq
Hax
Heq
Hax
chaise
demi-chaise
bateau
croisée
∆G (kJ/mol)
Introduction aux réactions stéréosélectives
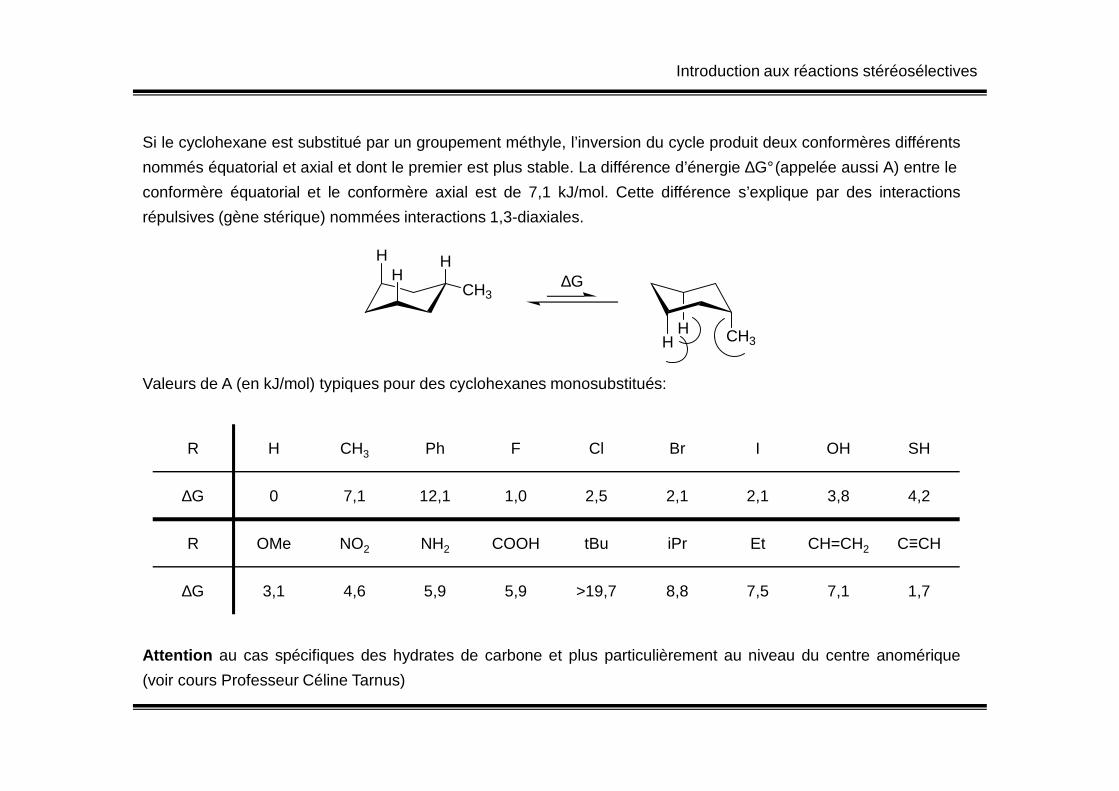
Si le cyclohexane est substitué par un groupement méthyle, l’inversion du cycle produit deux conformères différents
nommés équatorial et axial et dont le premier est plus stable. La différence d’énergie ∆G°(appelée aussi A) entre le
conformère équatorial et le conformère axial est de 7,1 kJ/mol. Cette différence s’explique par des interactions
répulsives (gène stérique) nommées interactions 1,3-diaxiales.
Valeurs de A (en kJ/mol) typiques pour des cyclohexanes monosubstitués:
CH3
CH3∆G
HH
HH
H
R H CH3 Ph F Cl Br I OH SH
∆G 0 7,1 12,1 1,0 2,5 2,1 2,1 3,8 4,2
R OMe NO2 NH2 COOH tBu iPr Et CH=CH2 C≡CH
∆G 3,1 4,6 5,9 5,9 >19,7 8,8 7,5 7,1 1,7
Attention au cas spécifiques des hydrates de carbone et plus particulièrement au niveau du centre anomérique
(voir cours Professeur Céline Tarnus)

Introduction aux réactions stéréosélectives
QUIZZ: représentez chacun des conformères possibles du composé ci-dessous et donnez graphiquement le profilénergétique.
H3C
CH3
CH3
QUIZZ: pour chacun des composés ci-dessous donnez la conformation la plus stable en justifiant votre choix.
CO2CH3
Cl
OHO
CH3
CH3
Introduction aux réactions stéréosélectives
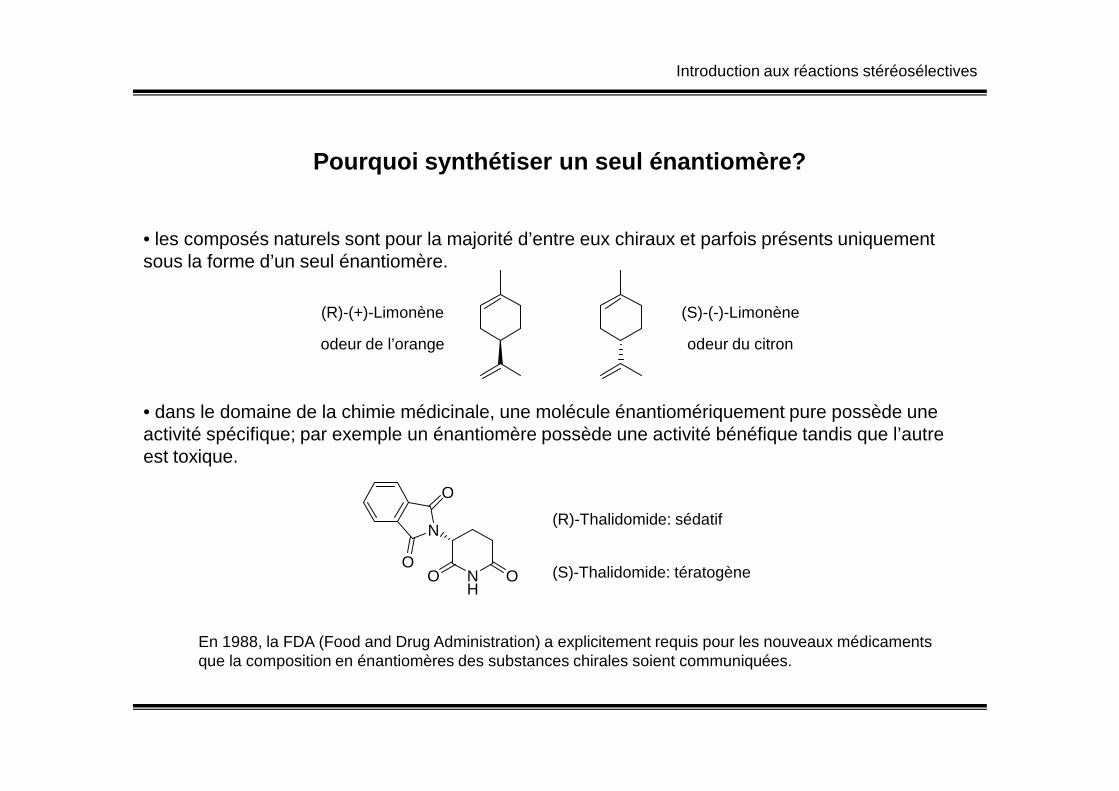
Pourquoi synthétiser un seul énantiomère?
• les composés naturels sont pour la majorité d’entre eux chiraux et parfois présents uniquement sous la forme d’un seul énantiomère.
(R)-(+)-Limonène
odeur de l’orange
(S)-(-)-Limonène
odeur du citron
• dans le domaine de la chimie médicinale, une molécule énantiomériquement pure possède une activité spécifique; par exemple un énantiomère possède une activité bénéfique tandis que l’autreest toxique.
NH
N
O OO
O
(R)-Thalidomide: sédatif
(S)-Thalidomide: tératogène
En 1988, la FDA (Food and Drug Administration) a explicitement requis pour les nouveaux médicamentsque la composition en énantiomères des substances chirales soient communiquées.
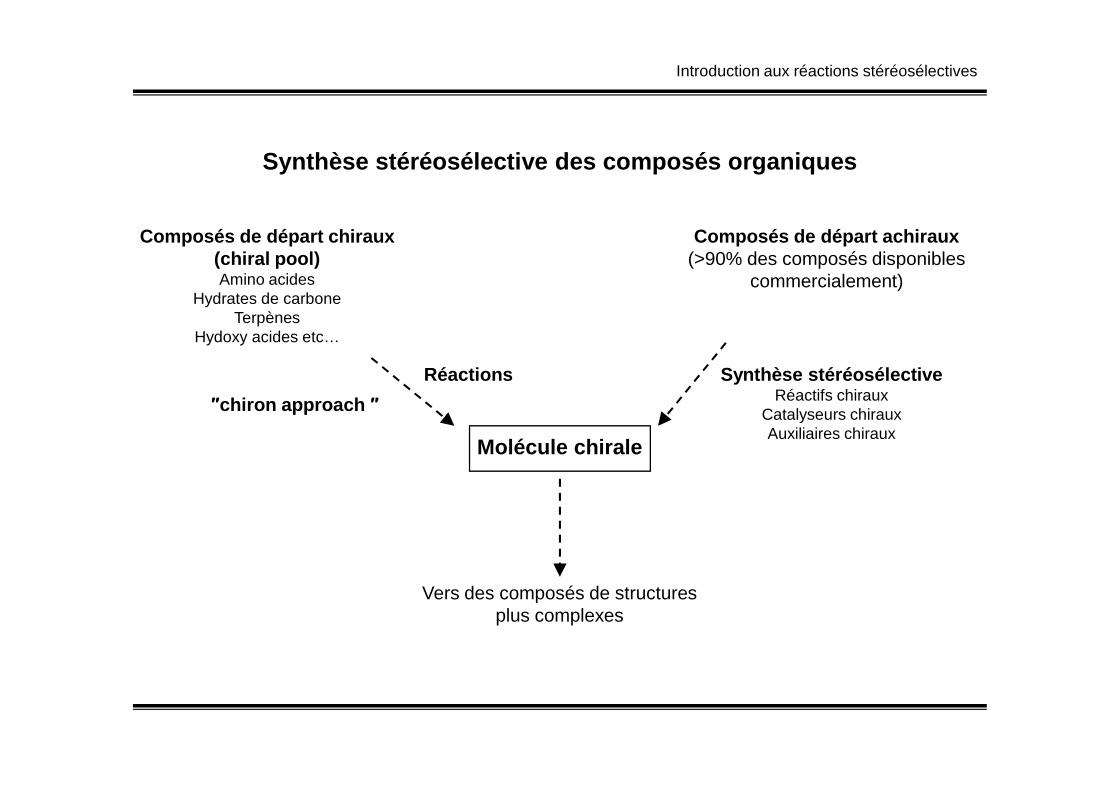
Introduction aux réactions stéréosélectives
Synthèse stéréosélective des composés organiques
Réactions
Composés de départ chiraux(chiral pool)Amino acides
Hydrates de carboneTerpènes
Hydoxy acides etc…
Composés de départ achiraux(>90% des composés disponibles
commercialement)
Synthèse stéréosélectiveRéactifs chiraux
″chiron approach ″
Molécule chirale
Réactifs chirauxCatalyseurs chirauxAuxiliaires chiraux
Vers des composés de structuresplus complexes
″chiron approach ″
Introduction aux réactions stéréosélectives
∆∆∆∆∆∆∆∆G
∆G
O Mg
R1
OMe
Me2N
Me
Ph
O
RH
Mg
R1
OMe
Me2N
Me
Ph
état de transitionhypothétique
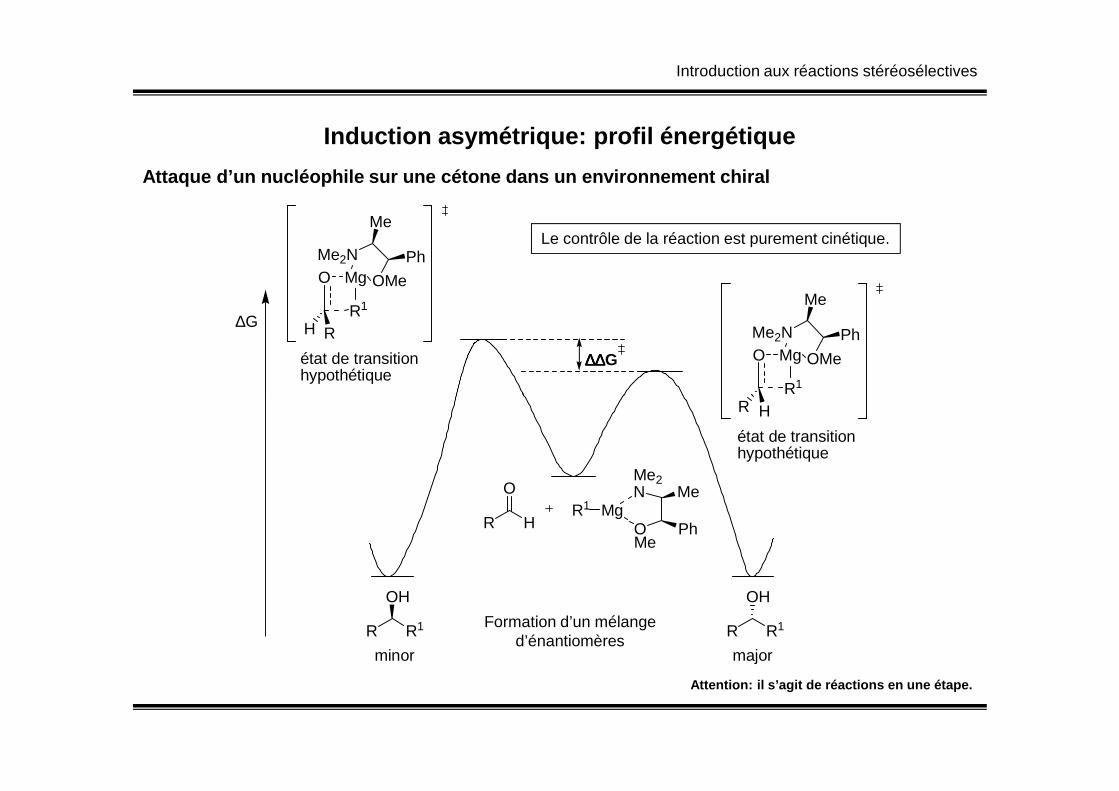
Induction asymétrique: profil énergétique
Attaque d’un nucléophile sur une cétone dans un env ironnement chiral
Le contrôle de la réaction est purement cinétique.
R H
O
Ph
MeMe2N
MgOMe
R1
R R1
OH
R R1
OH
majorminor
HRR
état de transitionhypothétique
Formation d’un mélanged’énantiomères
Attention: il s’agit de réactions en une étape.

Introduction aux réactions stéréosélectives
Induction asymétrique: profil énergétique
Attaque d’un nucléophile sur une cétone chirale
ET1
ET2
∆G
∆∆∆∆∆∆∆∆G
O
HR1
R2 R3
+ Nuc
OH
NucR1
R2 R3
OH
NucR1
R2 R3
+
Dia 1 Dia 2
A0
Dia 1
Dia 2
L’énergie de formation du Dia 1 est inférieureà celle du Dia 2, cela signifie que le produit themor--dynamique de la réaction est le Dia 1. L’énergie del’état de transition ET1 est inférieure à celle de l’étatde transition ET2, le produit obtenu sous contrôle ci--nétique sera donc Dia 1. Dans le cas présent, leproduit cinétique est le même que le produitthermodynamique (dans le cas de l’existence d’unéquilibre entre les deux dia).
Attention: il s’agit de réactions en une étape.
Introduction aux réactions stéréosélectivesIntroduction aux réactions stéréosélectives
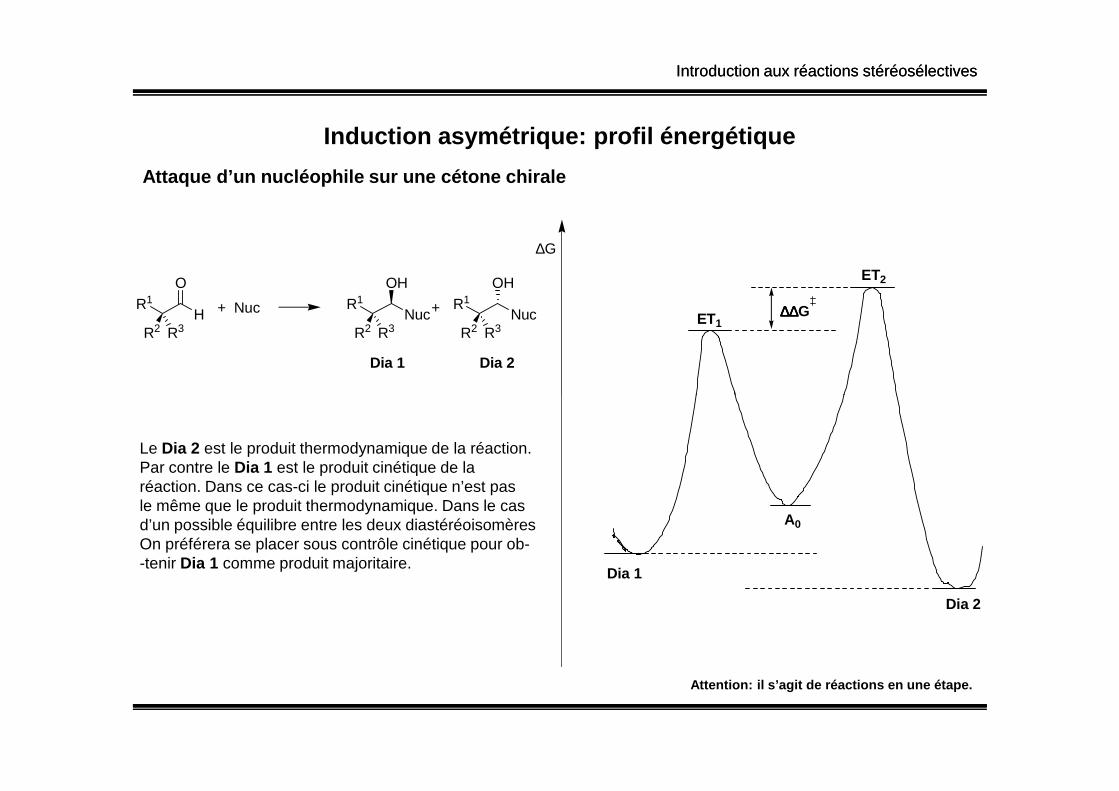
Induction asymétrique: profil énergétique
Attaque d’un nucléophile sur une cétone chirale
O
HR1
R2 R3
+ Nuc
OH
NucR1
R2 R3
OH
NucR1
R2 R3
+
Dia 1 Dia 2
ET1
ET2
∆G
∆∆∆∆∆∆∆∆G
A0
Dia 1
Dia 2
Le Dia 2 est le produit thermodynamique de la réaction.Par contre le Dia 1 est le produit cinétique de laréaction. Dans ce cas-ci le produit cinétique n’est pasle même que le produit thermodynamique. Dans le casd’un possible équilibre entre les deux diastéréoisomèresOn préférera se placer sous contrôle cinétique pour ob--tenir Dia 1 comme produit majoritaire.
Attention: il s’agit de réactions en une étape.
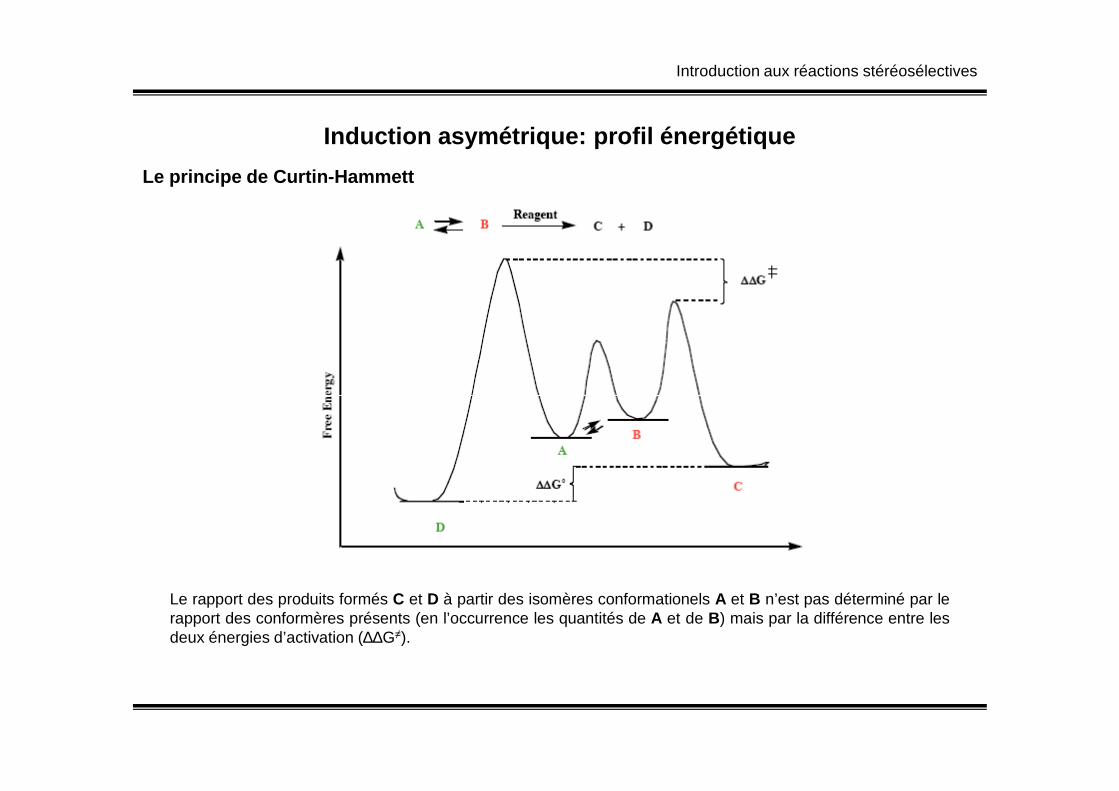
Introduction aux réactions stéréosélectives
Induction asymétrique: profil énergétique
Le principe de Curtin-Hammett
Le rapport des produits formés C et D à partir des isomères conformationels A et B n’est pas déterminé par lerapport des conformères présents (en l’occurrence les quantités de A et de B) mais par la différence entre lesdeux énergies d’activation (∆∆G≠).
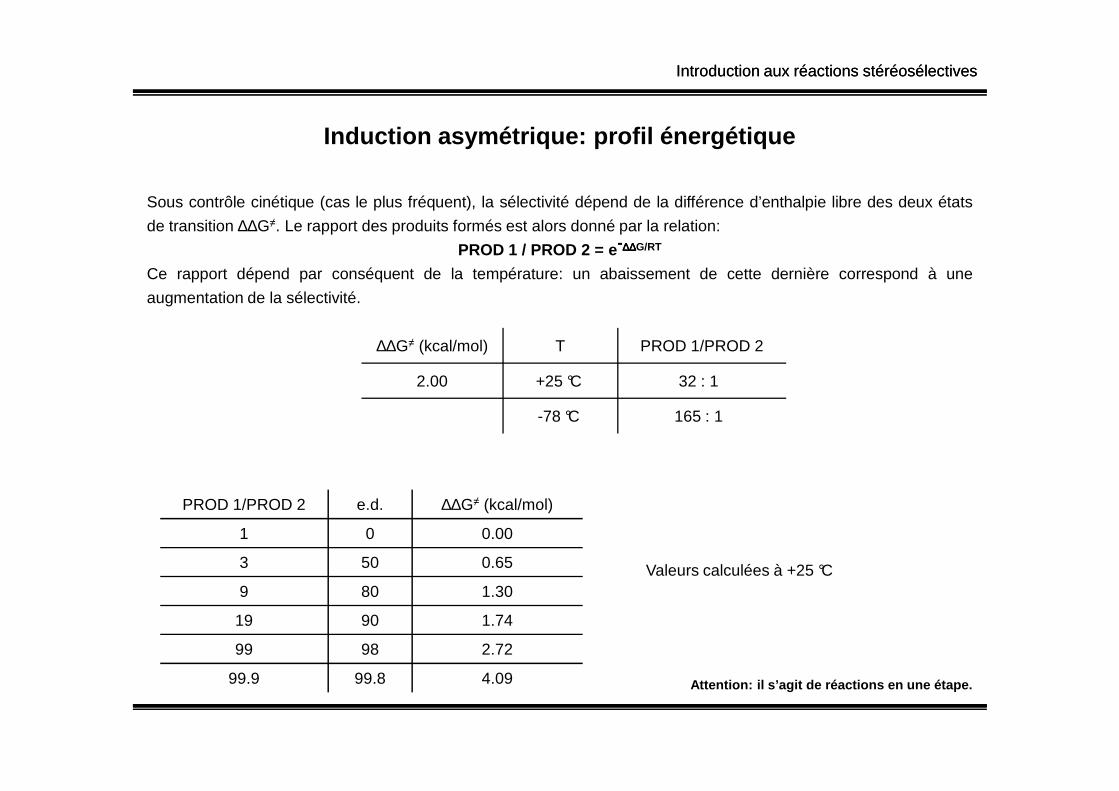
Introduction aux réactions stéréosélectivesIntroduction aux réactions stéréosélectives
Induction asymétrique: profil énergétique
Sous contrôle cinétique (cas le plus fréquent), la sélectivité dépend de la différence d’enthalpie libre des deux états
de transition ∆∆G≠. Le rapport des produits formés est alors donné par la relation:
PROD 1 / PROD 2 = e-∆∆∆∆∆∆∆∆G/RT
Ce rapport dépend par conséquent de la température: un abaissement de cette dernière correspond à une
augmentation de la sélectivité.
∆∆G≠ (kcal/mol) T PROD 1/PROD 2
2.00 +25 °C 32 : 1
-78 °C 165 : 1
PROD 1/PROD 2 e.d. ∆∆G≠ (kcal/mol)
1 0 0.00
3 50 0.65
9 80 1.30
19 90 1.74
99 98 2.72
99.9 99.8 4.09
Valeurs calculées à +25 °C
Attention: il s’agit de réactions en une étape.
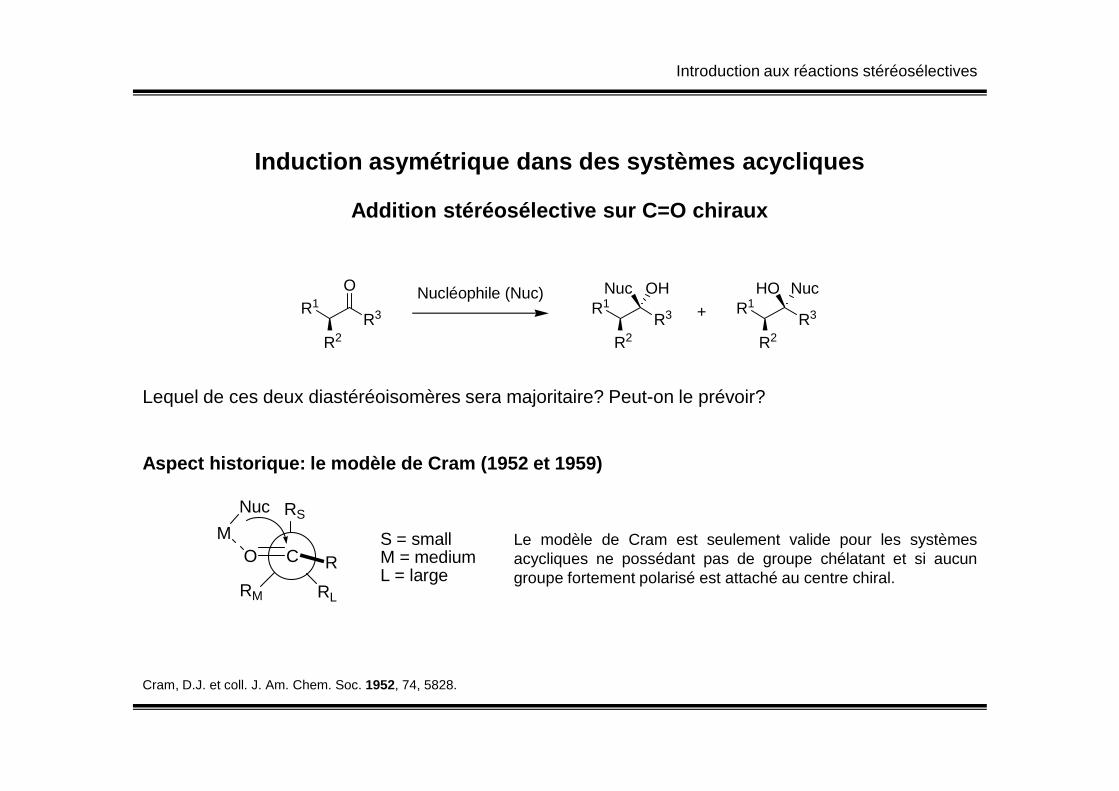
Introduction aux réactions stéréosélectives
Induction asymétrique dans des systèmes acycliques
Addition stéréosélective sur C=O chiraux
R1
R3
O
R2
Nucléophile (Nuc)R1
R3
R2
R1
R3
R2
+
OHNuc NucHO
Lequel de ces deux diastéréoisomères sera majoritaire? Peut-on le prévoir?Lequel de ces deux diastéréoisomères sera majoritaire? Peut-on le prévoir?
Aspect historique: le modèle de Cram (1952 et 1959)
O C R
RS
RM RL
M
Nuc
S = smallM = mediumL = large
Le modèle de Cram est seulement valide pour les systèmesacycliques ne possédant pas de groupe chélatant et si aucungroupe fortement polarisé est attaché au centre chiral.
Cram, D.J. et coll. J. Am. Chem. Soc. 1952, 74, 5828.

Introduction aux réactions stéréosélectives
Le modèle de Cram chélaté Le modèle dipolaire (règle de Cornfoth)
R
OR'O
RLRS
MNuc
attaque parla face avant
R
R'O
RLRS
NucOM
O
RX
RLRS
-δ
-δNucM
RX
RLRS OM
Nuc
attaque parla face avant
En 1967, Karabastos a suggéré une approche alternative, toujours basée sur l’état de transition du dérivé carbonyléEn 1967, Karabastos a suggéré une approche alternative, toujours basée sur l’état de transition du dérivé carbonyléde départ mais en tenant compte des nouvelles connaissances dans ce domaine. Contrairement au modèle de Cram(Newman décalé), le modèle de Karabastos utilise la conformation de Newman éclipsée.
O
RRSRL
RM
M
NucRL
RRM
RS
NucOM Cependant aucun de ces modèles n’est vraiment
satisfaisant. Ils ne permettent pas de prédire lerésultat de l’addition d’un nucléophile sur lescyclohexanones et ne tiennent pas compte de la taillede R sur la sélectivité.
Cornforth et coll. J. Chem. Soc. 1959, 112. Karabatsos, G.J. et coll. J. Am. Chem. Soc. 1967, 89, 1367.

Introduction aux réactions stéréosélectives
Le modèle de Felkin-Anh (1968 et 1977)
RL R
O
RM
O R
RL
HRM
RL
Nuc
Favorable RL RRM
OHNuc
NucHOR est le groupe le plus encombrant
OR
RL
HRM
Nuc
DéfavorableRL R
RM
NucHORL est le groupe le plus encombrantou le plus électronégatif.
Le nucléophile s’approche en respectant la trajectoire de Dunitz-Burgi (109°)
Le modèle de Felkin-Anh est un modèle stéréoélectronique utilisable pour prédire l’addition de nucléophiles surles carbonyles, cependant les conditions de réactions peuvent favoriser la formation d’un produit dont le résultatstéréochimique résulte d’un contrôle par chélation. Dans ce cas-ci le modèle de Cram chélaté peut s’appliquer.
Felkin, H. et coll. Tetrahedron Lett. 1968, 2199. Anh, N.T. Nouv. J. Chim. 1977, 1, 61. Anh, N.T. Top. Curr. Chem. 1980, 88, 1.
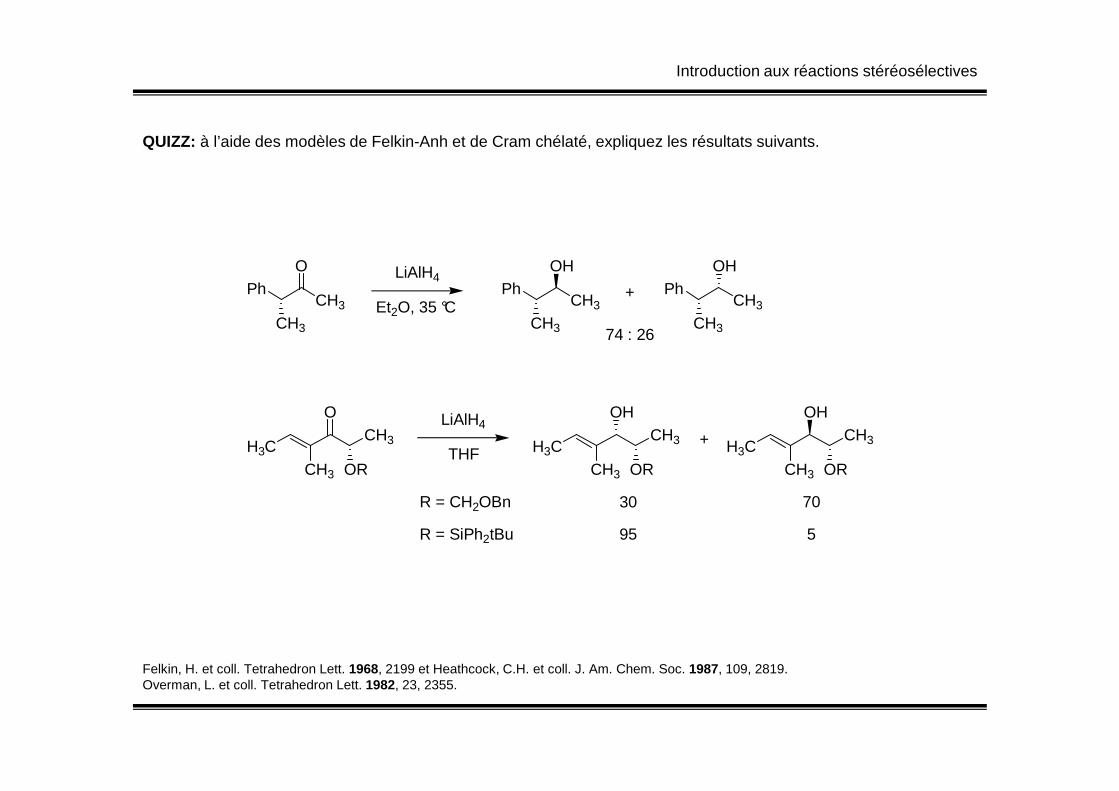
Introduction aux réactions stéréosélectives
QUIZZ: à l’aide des modèles de Felkin-Anh et de Cram chélaté, expliquez les résultats suivants.
PhO
CH3
CH3
LiAlH4
Et2O, 35 °CPh
OH
CH3
CH3
PhOH
CH3
CH374 : 26
+
H3CCH3 OR
OCH3 H3C
CH3 OR
OHCH3 H3C
CH3 OR
OHCH3
LiAlH4
THF+
R = CH2OBn 30 70
R = SiPh2tBu 95 5
Felkin, H. et coll. Tetrahedron Lett. 1968, 2199 et Heathcock, C.H. et coll. J. Am. Chem. Soc. 1987, 109, 2819.Overman, L. et coll. Tetrahedron Lett. 1982, 23, 2355.

Introduction aux réactions stéréosélectives
Attaque d’un nucléophile sur une cétone cyclique
La conformation chaise de la cyclohexanone place le carbonyle dans un environnement
asymétrique. On observe dans ce cas que les nucléophiles de petites tailles préfèrent
approcher le carbonyle par une attaque axiale. Par contre l’approche de nucléophiles plus
volumineux se fait par une attaque équatoriale (dans le cas d’une attaque axiale les gros
nucléophiles rencontrent les atomes d’hydrogène axiaux en position 3 et 5 sur leur trajectoire).
O
attaque axiale
attaque équatoriale
Avec la 4-tertbutylcyclohexanone, l’attaque équatoriale prédomine mais la sélectivité est faible. Lorsque les effets
stériques sont plus importants, l’addition peut devenir exclusivement équatoriale.
O
65%
35%
O
100%
0%
CH3
H3C
H3C
stéréosélectivité de l’addition du méthyl lithium.

Cas de la réaction d’allylation des dériv és carbonyl és
Introduction aux réactions stéréosélectives
Les réactions d’allylation des dérivés carbonylés sont très employées en synthèse organique. L’essentiel de ces
réactions impliquent l’addition d’allyles étain, de silicium ou de bore sur un aldéhyde. Ces réactions se font suivant un
mécanisme concerté et non par l’attaque d’un ˝ vrai ˝ nucléophile comme pour les organomagnésiens, lithiens etc…
MR H
O
+ R
OH
Dans le cas de l’addition d’allyles étain ou de silicium, la réaction est, en général, effectuée en présence d’un acide de
Lewis. Pour les allyles boranes, la réaction s’effectue sans acide de Lewis à basse température. Cette différence de
réactivité des allyles est fondamentale pour comprendre la stéréochimie du produit formé lors d’un processus
stéréosélectif.
M = Sn, Si, B
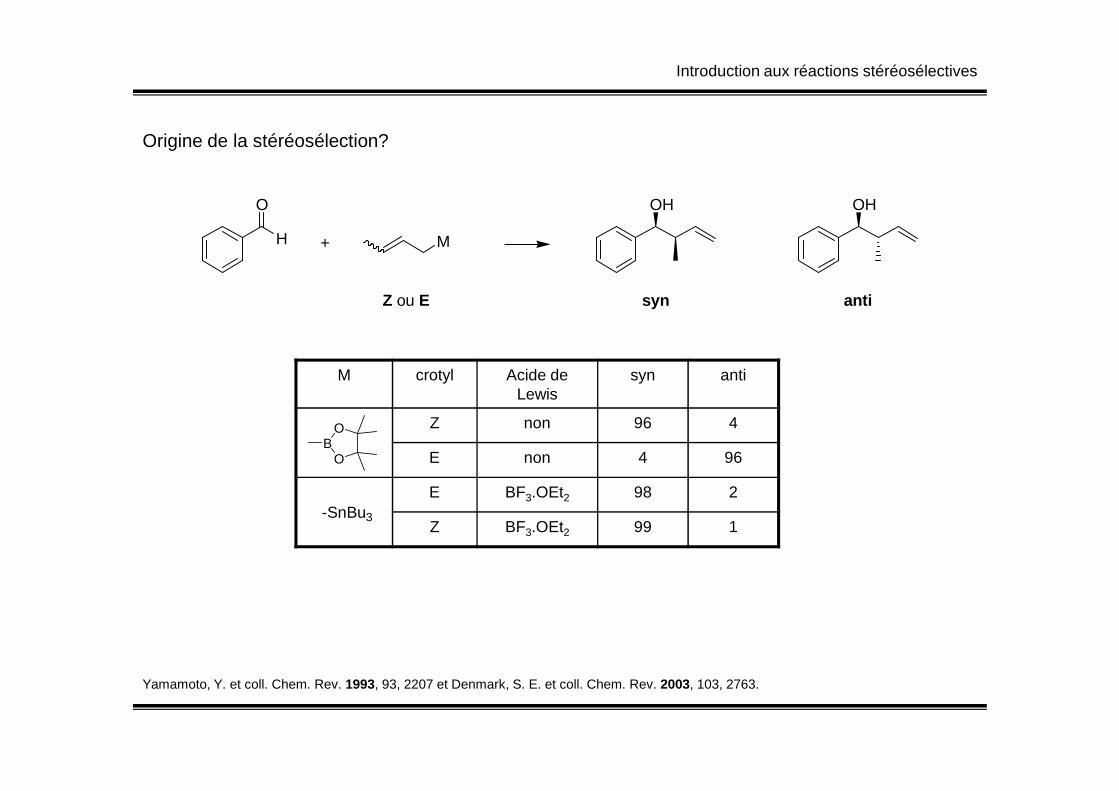
Introduction aux réactions stéréosélectives
H
O
+ M
OH OH
Z ou E syn anti
M crotyl Acide de Lewis
syn anti
Origine de la stéréosélection?
Lewis
Z non 96 4
E non 4 96
E BF3.OEt2 98 2
Z BF3.OEt2 99 1
OB
O
-SnBu3
Yamamoto, Y. et coll. Chem. Rev. 1993, 93, 2207 et Denmark, S. E. et coll. Chem. Rev. 2003, 103, 2763.
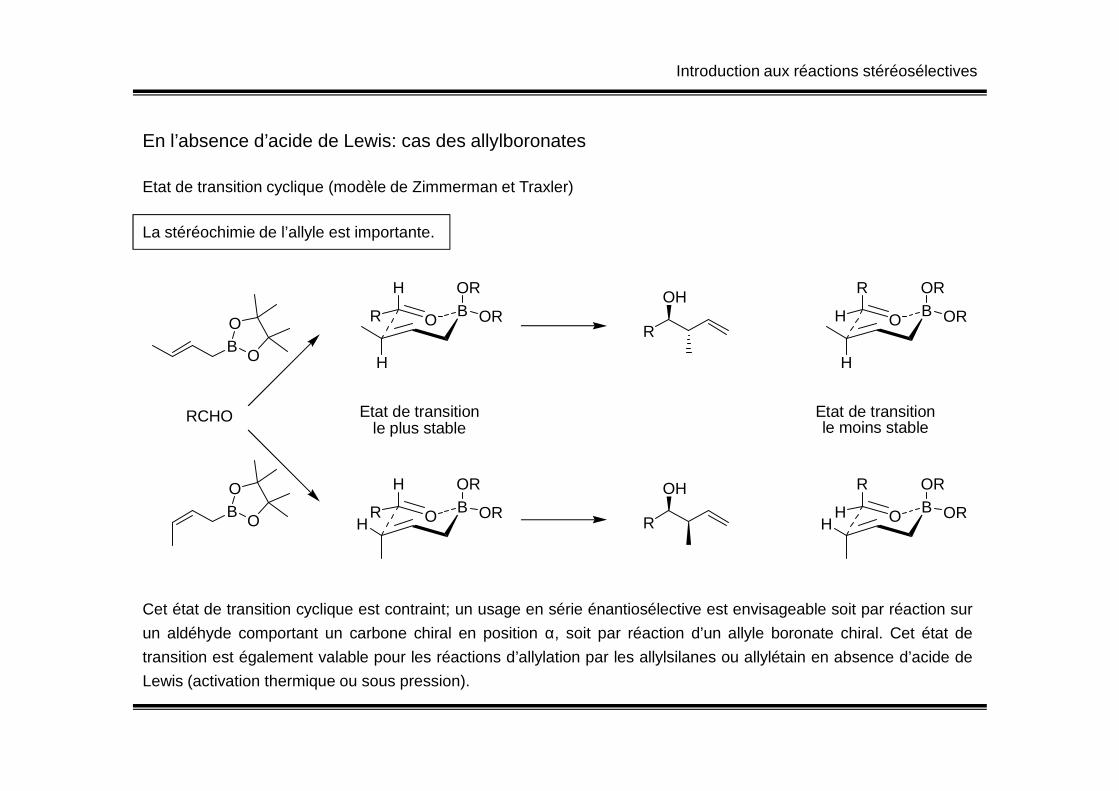
Introduction aux réactions stéréosélectives
En l’absence d’acide de Lewis: cas des allylboronates
Etat de transition cyclique (modèle de Zimmerman et Traxler)
B O
OB
H
H
R
OR
OROR
OHB
H
R
H
OR
ORO
La stéréochimie de l’allyle est importante.
Cet état de transition cyclique est contraint; un usage en série énantiosélective est envisageable soit par réaction sur
un aldéhyde comportant un carbone chiral en position α, soit par réaction d’un allyle boronate chiral. Cet état de
transition est également valable pour les réactions d’allylation par les allylsilanes ou allylétain en absence d’acide de
Lewis (activation thermique ou sous pression).
RCHO
B O
O
R
OHB
H
H
R
OR
ORO BH
R
H
OR
ORO
Etat de transitionle plus stable
Etat de transitionle moins stable

Introduction aux réactions stéréosélectives
En présence d’un acide de Lewis: cas des allylétains.
Etat de transition acyclique
H CH3
O
HR
F3B
SnBu3RCHO + SnBu3
BF3.OEt2
R
OH
La stéréochimie de l’allyle est sans importance.
H CH3
O
RH
F3B
SnBu3
R
OH
Quelle que soit la stéréochimie de l’allyle étain (ou silane), le produit de la réaction majoritaire sera de stéréochimie
syn. Cette réaction peut-être appliquée en série énantiosélective dans le cas d’un aldéhyde possédant un carbone
chiral en position α.

Introduction aux réactions stéréosélectives
Synthèses asymétriques diastéréosélectives
Principe général
XB
A+ Z* X
B
AZ*
B
AZ*
NucXH
B
A
Z*
NucXH
**clivage+ NucH
Le substrat est transformé en composé chiral par l’introduction d’une copule chirale. Ainsi, il subit une réaction
stéréosélective à l’aide d’un réactif achiral, induisant la formation majoritaire d’un diastéréoisomère. Après coupure
de la copule chirale, le produit de réaction est chiral dont l’un des énantiomères est majoritaire. Cette méthode pour
être utilisée efficacement doit respecter quelques impératifs:
� l’auxiliaire chiral doit être facile à additionner, facile à cliver (sans risquer de provoquer une racémisation du
produit) et si possible facile à récupérer (ou de faible coût, s’il est détruit après clivage)
� l’addition du nucléophile doit être hautement diastéréosélective
� chacun des énantiomères de l’auxiliaire chiral doit être disponible afin de permettre l’accès aux deux
énantiomères du produit de la réaction
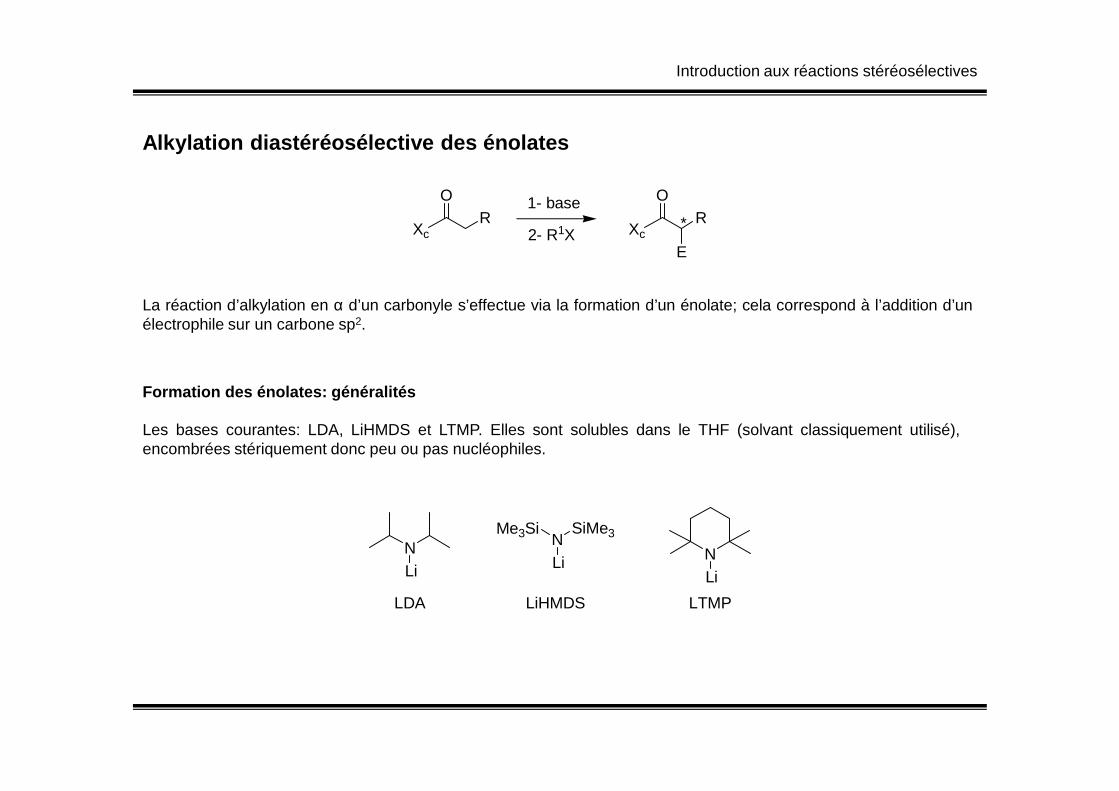
Introduction aux réactions stéréosélectives
Alkylation diastéréosélective des énolates
La réaction d’alkylation en α d’un carbonyle s’effectue via la formation d’un énolate; cela correspond à l’addition d’unélectrophile sur un carbone sp2.
Xc
OR
1- base
2- R1X Xc
OR
E
*
Formation des énolates: généralitésFormation des énolates: généralités
Les bases courantes: LDA, LiHMDS et LTMP. Elles sont solubles dans le THF (solvant classiquement utilisé),encombrées stériquement donc peu ou pas nucléophiles.
NLi
LDA
NSiMe3Me3Si
Li
LiHMDS
NLi
LTMP

Introduction aux réactions stéréosélectives
R
OCH3
R
OMCH3
R
OM
CH3
énolate Z
énolate E
(Z) vs (E)?
Les énolates (Z) sont thermodynamiquement lesplus stables. Plus R est gros, plus la formationde Z devient majoritaire.
Le modèle d’Ireland
• balance entre l’interaction 1,3-diaxiale et la contraintetorsionale.• normalement l’interaction diaxiale est la plusimportante par conséquent la déprotonation souscontrôle cinétique donne l’énolate E• si R devient trop gros, la contrainte torsionale prime,l’énolate Z devient prépondérant.• si l’interaction Li-oxygène est brisée, l’énolate Z estmajoritaire (la contrainte torsionale devientprépondérante).
HOLiN
H
CH3
R
HOLiN
H3C
H
R
énolate Z énolate E
interaction1,3-diaxiale
contraintetorsionale
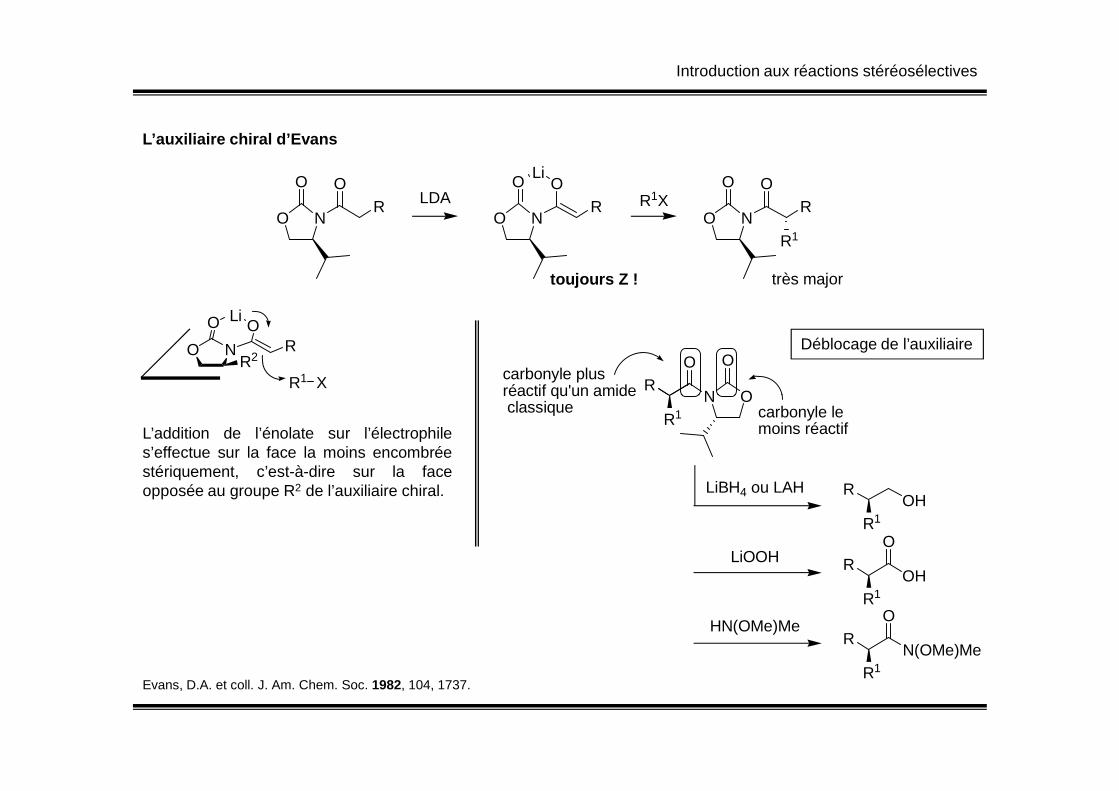
Introduction aux réactions stéréosélectives
L’auxiliaire chiral d’Evans
O
O
N
OR LDA
O
O
N
OR
Li
toujours Z !
R1XO
O
N
OR
R1
très major
NO
OLiO
RR2
R1 X
Déblocage de l’auxiliaire
RO
ON
Ocarbonyle plus réactif qu'un amide
L’addition de l’énolate sur l’électrophiles’effectue sur la face la moins encombréestériquement, c’est-à-dire sur la faceopposée au groupe R2 de l’auxiliaire chiral.
R1ONréactif qu'un amide
classique carbonyle le moins réactif
LiBH4 ou LAH ROH
R1
LiOOH ROH
R1
O
HN(OMe)MeR
N(OMe)MeR1
O
Evans, D.A. et coll. J. Am. Chem. Soc. 1982, 104, 1737.
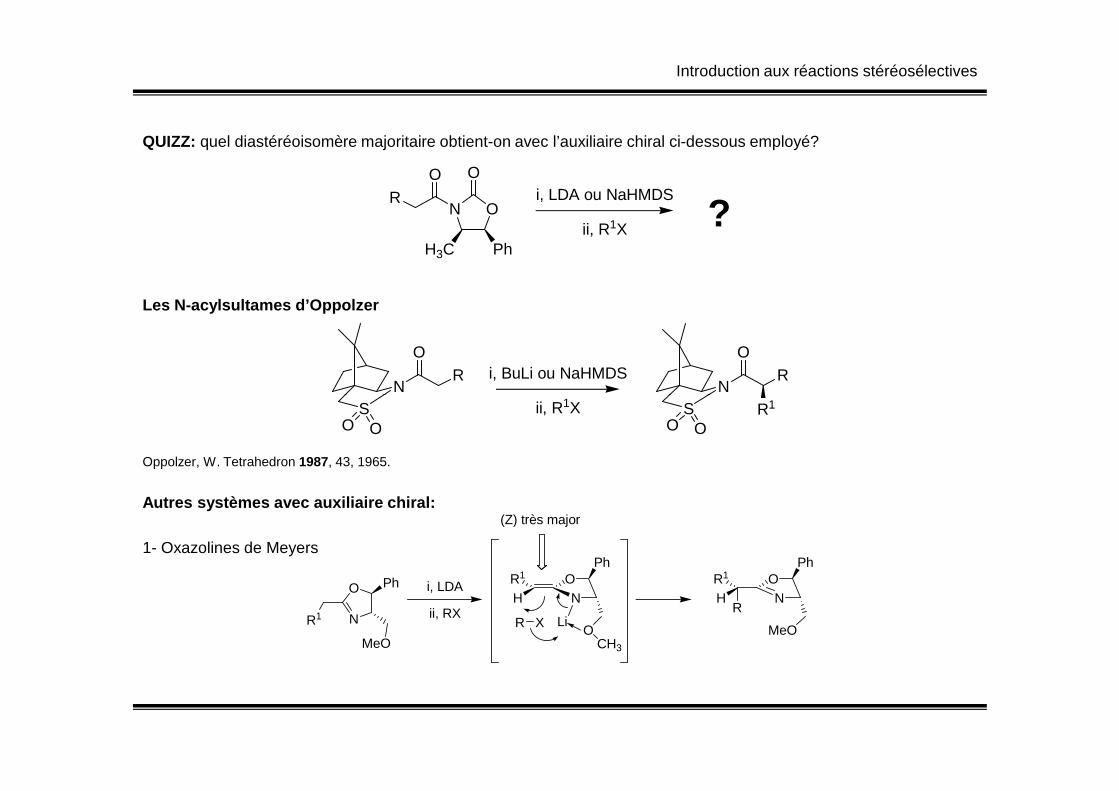
Introduction aux réactions stéréosélectives
QUIZZ: quel diastéréoisomère majoritaire obtient-on avec l’auxiliaire chiral ci-dessous employé?
RO
ON
O
H3C Ph
i, LDA ou NaHMDS
ii, R1X ?
Les N-acylsultames d’Oppolzer
N
OR i, BuLi ou NaHMDS
N
OR
Oppolzer, W. Tetrahedron 1987, 43, 1965.
SO O
ii, R1X SO O
R1
Autres systèmes avec auxiliaire chiral:
HR1 O
N
Ph
OCH3
LiR X
O
NR1
PhHR1 O
N
Ph
MeO
R
i, LDA
ii, RX
MeO
(Z) très major
1- Oxazolines de Meyers
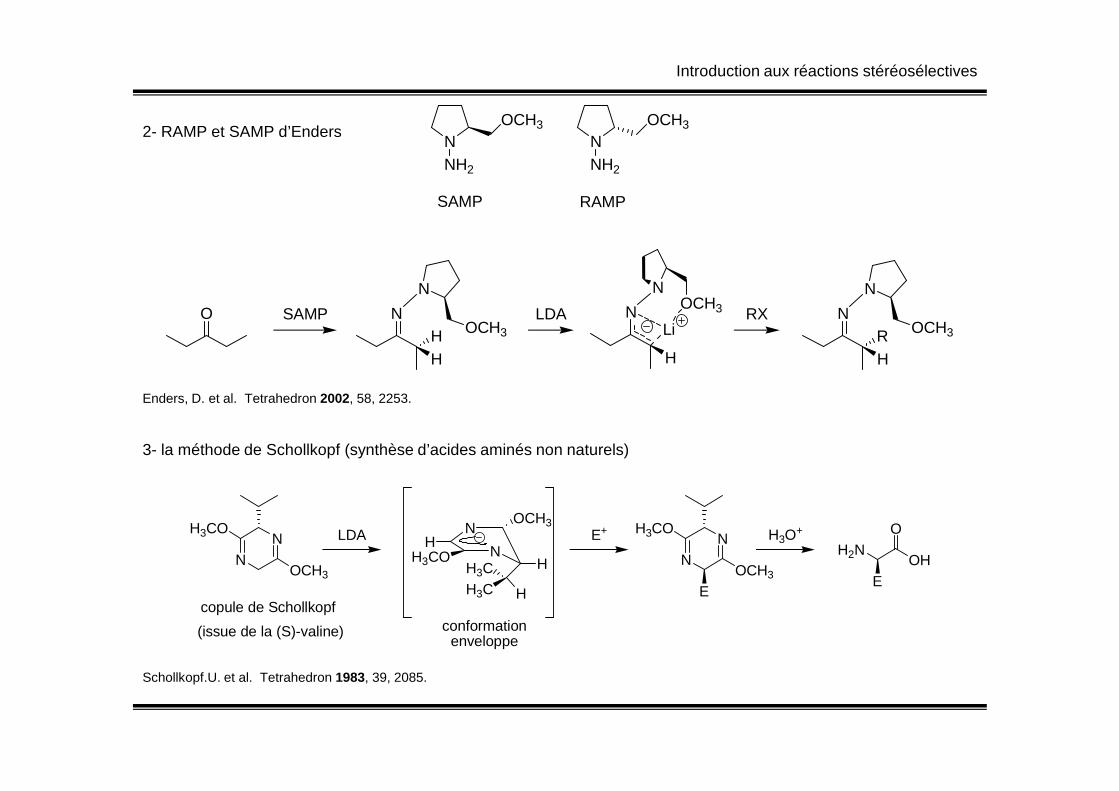
Introduction aux réactions stéréosélectives
2- RAMP et SAMP d’EndersN
OCH3
NH2
NOCH3
NH2
SAMP RAMP
O SAMP N
HH
N
OCH3LDA N
H
NOCH3
LiRX N
HR
N
OCH3
Enders, D. et al. Tetrahedron 2002, 58, 2253.Enders, D. et al. Tetrahedron 2002, 58, 2253.
3- la méthode de Schollkopf (synthèse d’acides aminés non naturels)
NN
H3CO
OCH3
copule de Schollkopf
(issue de la (S)-valine)
LDAN
NH
HH3CH3C H
H3CO
OCH3
conformationenveloppe
E+
NN
H3CO
OCH3
E
H3O+
E
H2NO
OH
Schollkopf.U. et al. Tetrahedron 1983, 39, 2085.
Introduction aux réactions stéréosélectives
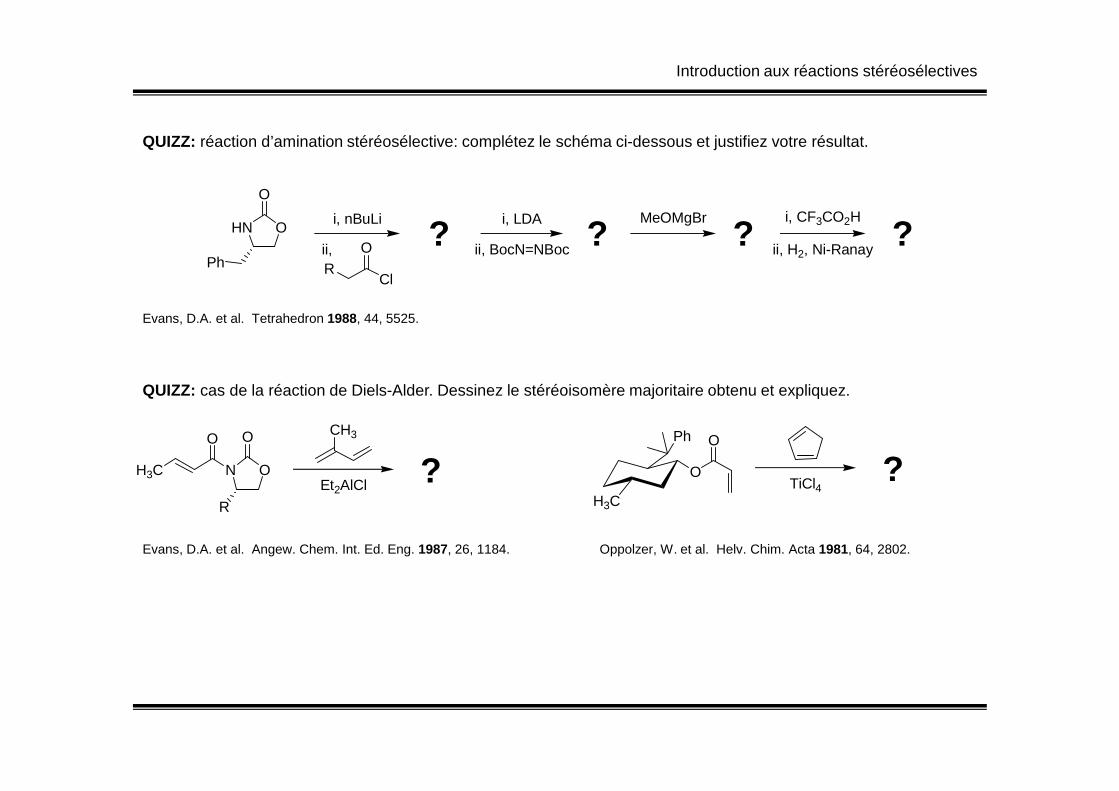
QUIZZ: réaction d’amination stéréosélective: complétez le schéma ci-dessous et justifiez votre résultat.
OHN
Ph
O
i, nBuLi
ii,R
Cl
O ??i, LDA
ii, BocN=NBoc
MeOMgBr
? ?i, CF3CO2H
ii, H2, Ni-Ranay
Evans, D.A. et al. Tetrahedron 1988, 44, 5525.
QUIZZ: cas de la réaction de Diels-Alder. Dessinez le stéréoisomère majoritaire obtenu et expliquez.QUIZZ: cas de la réaction de Diels-Alder. Dessinez le stéréoisomère majoritaire obtenu et expliquez.
Evans, D.A. et al. Angew. Chem. Int. Ed. Eng. 1987, 26, 1184. Oppolzer, W. et al. Helv. Chim. Acta 1981, 64, 2802.
O
O
NH3C
O
R
CH3
Et2AlCl ?H3C
Ph
O
O
TiCl4?

Introduction aux réactions stéréosélectives
Réaction d’aldolisation
R1
OMR2 + R3CHO
R3 R1
OOH
R2R3 R1
OOH
R2
+
syn anti
La réaction d’aldolisation fut découverte par Aleksandr Porfir’evich Borodin qui le premier a observé la formation d’un aldol, le 3-hydroxybutanal, à partir de l’acétaldéhyde sous l’influence d’un catalyseur comme l’acide chlorhydrique ou de dichlorure de zinc.
Généralisation et origine de la diastéréosélectivit é (le modèle de Zimmerman et Traxler)
Cas de l’énolate -(Z): Cas de l’énolate -(E):
R1
OMR2
+
R3CHO
MOH
R2 R3H O
R1
MOR3
R2 HH O
R1
FAVORABLE
DEFAVORABLE
R3 R1
OOH
R2
R3 R1
OOH
R2
syn
anti
Cas de l’énolate -(Z):
R1
OM
R3CHO
MOH
HR3
R2O
R1
MOR3
HH
R2O
R1
FAVORABLE
DEFAVORABLE
R3 R1
OOH
R2
R3 R1
OOH
R2
syn
anti
R2
Cas de l’énolate -(E):
+
Introduction aux réactions stéréosélectives
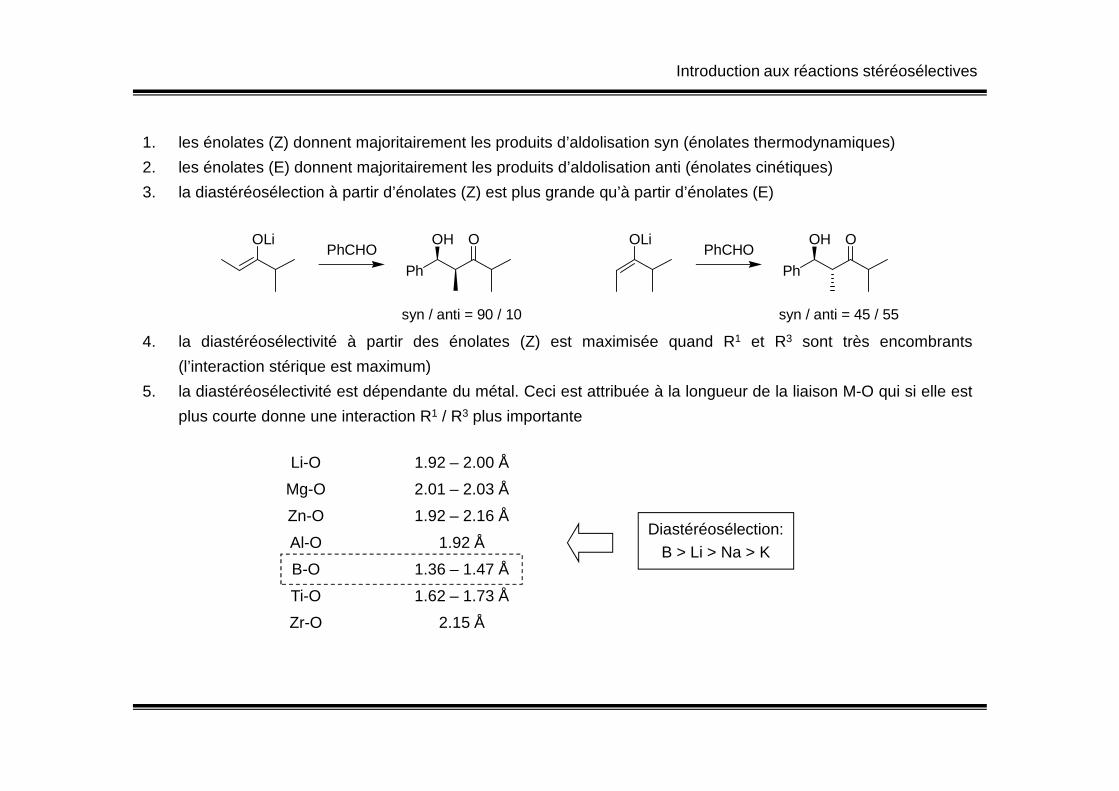
1. les énolates (Z) donnent majoritairement les produits d’aldolisation syn (énolates thermodynamiques)
2. les énolates (E) donnent majoritairement les produits d’aldolisation anti (énolates cinétiques)
3. la diastéréosélection à partir d’énolates (Z) est plus grande qu’à partir d’énolates (E)
4. la diastéréosélectivité à partir des énolates (Z) est maximisée quand R1 et R3 sont très encombrants
(l’interaction stérique est maximum)
5. la diastéréosélectivité est dépendante du métal. Ceci est attribuée à la longueur de la liaison M-O qui si elle est
OLiPhCHO
Ph
OOH
syn / anti = 90 / 10
OLiPhCHO
Ph
OOH
syn / anti = 45 / 55
5. la diastéréosélectivité est dépendante du métal. Ceci est attribuée à la longueur de la liaison M-O qui si elle est
plus courte donne une interaction R1 / R3 plus importante
Li-O 1.92 – 2.00 Å
Mg-O 2.01 – 2.03 Å
Zn-O 1.92 – 2.16 Å
Al-O 1.92 Å
B-O 1.36 – 1.47 Å
Ti-O 1.62 – 1.73 Å
Zr-O 2.15 Å
Diastéréosélection:B > Li > Na > K

Introduction aux réactions stéréosélectives
Effets du groupe R1 sur la diastéréosélection
R1 énolate (Z) énolate (E)
H 1.0 1.5
Et 9.0 1.5
iPr 9.0 1.0
Ph 7.0 -
tBu 70 -
mesityl > 50 < 0.02
ratio syn : anti
la diastéréosélection augmente avec l’augmentation de la taille de R1
R1
OLiH3C + PhCHO syn : anti aldol
mesityl > 50 < 0.02
Effets du groupe R2 sur la diastéréosélection
R2
tBu
OLi tBuCHO
Et2O, 20 °C tBu tBu
O
R2
OH
tBu tBu
O
R2
OH
anti syn
R2 = Me 0 100
= Et 0 100
= Pr 2 98
= iBu 3 97
= iPr 71 29
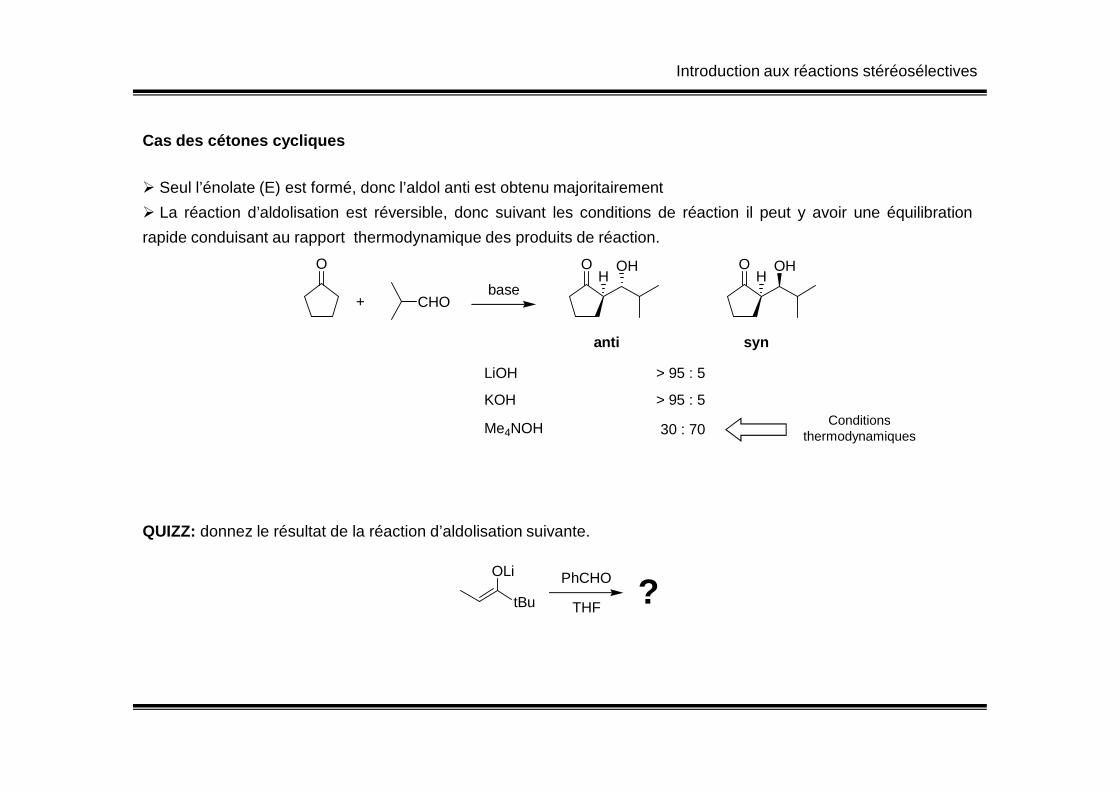
Introduction aux réactions stéréosélectives
Cas des cétones cycliques
� Seul l’énolate (E) est formé, donc l’aldol anti est obtenu majoritairement
� La réaction d’aldolisation est réversible, donc suivant les conditions de réaction il peut y avoir une équilibration
rapide conduisant au rapport thermodynamique des produits de réaction.
O
+ CHO
O OHH
O OHH
anti syn
base
LiOH > 95 : 5
KOH > 95 : 5KOH
Me4NOH 30 : 70
> 95 : 5Conditions
thermodynamiques
QUIZZ: donnez le résultat de la réaction d’aldolisation suivante.
tBu
OLi PhCHO
THF?

Introduction aux réactions stéréosélectives
Réaction d’aldolisation avec des aldéhydes chiraux
Quand un énolate est ajouté à un aldéhyde possédant deux faces diastéréotopiques (c’est-à-dire un aldéhyde chiral),
l’addition nucléophile de l’énolate suit le modèle de Felkin-Ahn.
Ph CHO
CH3
+
OM
R PhR
O
CH3
OHPh
R
O
CH3
OH
syn anti3 : 1
MO
R O
H O
Ph
MeH
H O
Ph
HMe
R
MO
R
major
minor
R
O
OHH
Ph
H Me
Ph
R
O
OHH
Me H
syn
anti
La diastéréosélectivité est invariante
quelque soit la taille de R.

Introduction aux réactions stéréosélectives
Réaction d’aldolisation avec des énolates chiraux: utilisation des auxiliaires chiraux d’Evans
Les auxiliaires chiraux d’Evans précédemment utilisés dans les réactions d’alkylation d’énolates ont été également
employés pour la réaction d’aldolisation. L’aldolisation dite d’Evans est devenue une des méthodes les plus courantes
de la synthèse organique moderne.
O
O
N
OR Bu2BOTf
iPr2NEt
O
O
N
OR
BBuBu
R1CHO
O
O
N
O
RR1
OH
ou
O
O
N
O
R1
OH
énolate ( Z) exclusivement(quelques soient les conditions réactionnelles)
O NR
R1
Résultats expérimentaux:
O
O
N
OMe
O
O
N
O
MeR
OH
O
O
N
O
MeR
OH
Bu2BOTf
RCHO
R = Bu 99.3 0.7
R = iPr 99.8 0.2
R = Ph >99.8 <0.2

Introduction aux réactions stéréosélectives
Rationalisation:
OH
OXc
RMe
syn aldol minor
BO
Me
H O
H
R
N
OO
O
H RH
CH3
O
NO
O
B
H
- interaction H – H- interaction stérique avec iPr- alignement des dipôles moins favorable
Evans, D.A. et al. Pure & Appl. Chem. 1981, 53, 1109 et Org. React. 1990, 68, 83.
OH
OXc
RMe
syn aldol obtenu
BOH
MeR
H O
O
NO
H
CH3
O
RH
O
B
ON
O
- l’auxiliaire chirale tourne (absence d’interaction H – H)- les dipôles sont non-alignés: plus favorable

Introduction aux réactions stéréosélectives
Ce qu’il faut retenir
� Reconnaître si une molécule est chirale ou non, si elle comporte des éléments de pro-chiralité.
� Etablir les relations stéréochimiques des différents centres stéréogéniques au sein d’une molécule.
� Déterminer la conformation d’une molécule dans l’espace (l’analyse conformationnelle permet de déduire la
conformation la plus stable d’un substrat).
� Connaître les principaux modèles d’addition d’un réactif sur un dérivé carbonylé: le modèle de Felkin-Ahn, de Cram
chélaté et le modèle cyclique de Zimmerman et Traxler.
� Etre capable de repérer dans un système de réaction si d’autres éléments extérieurs aux réactifs peuvent
influencer l’induction stéréochimique (présence ou non d’agent de chélation, le type de solvant ….).
Il existe de très nombreuses méthodes pour créer de la chiralité:
� réduction des oléfines .
� réduction des cétones (formation d’alcools chiraux).
� alkylation des dérivés carbonylés.
� oxydation des oléfines (époxydation, cis-dihydroxylation).
� formation par couplage de liaison C-C
� dédoublement enzymatique d’un mélange racémique.
… et autres systèmes.

INTRODUCTION AUX REACTIONSINTRODUCTION AUX REACTIONSSTEREOSELECTIVES.STEREOSELECTIVES.
Aspects générauxAspects généraux
Introduction aux réactions stéréosélectives
Aspects générauxAspects générauxsuitesuite
Maîtrise Option Chimie Organique
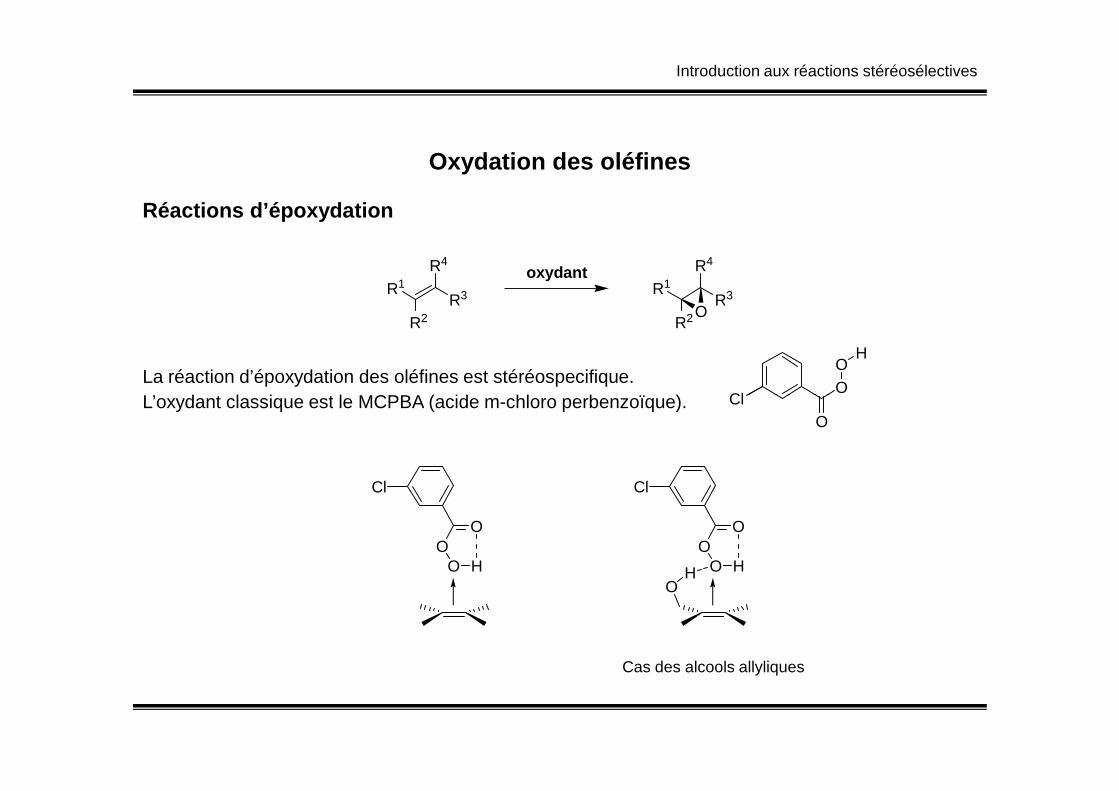
Introduction aux réactions stéréosélectives
Oxydation des oléfines
Réactions d’époxydation
R3
R4
R1
R2
oxydant
R3
R4
R1
R2O
ClOO
H
La réaction d’époxydation des oléfines est stéréospecifique.L’oxydant classique est le MCPBA (acide m-chloro perbenzoïque). Cl
O
OL’oxydant classique est le MCPBA (acide m-chloro perbenzoïque).
OO
O H
Cl
O
OO
O H
Cl
H
Cas des alcools allyliques
Introduction aux réactions stéréosélectives
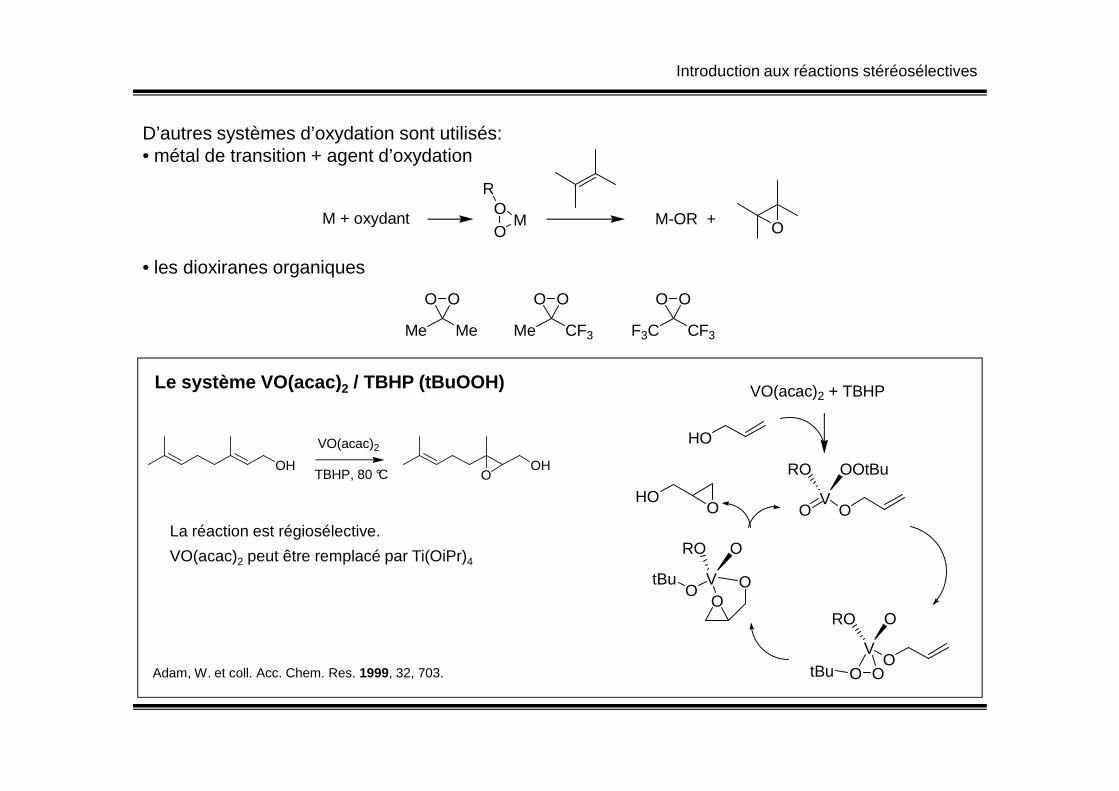
D’autres systèmes d’oxydation sont utilisés:• métal de transition + agent d’oxydation
• les dioxiranes organiques
M + oxydantOO
M
R
M-OR + O
O O O O O O
Me Me Me CF3 F3C CF3
Le système VO(acac) 2 / TBHP (tBuOOH) VO(acac)2 + TBHP
HO
OOV
ORO
OtBu
VO(acac)2 + TBHP
VO O
OOtBuRO
V
OO
ORO
OtBu
HOO
OH
VO(acac)2
TBHP, 80 °COH
O
Adam, W. et coll. Acc. Chem. Res. 1999, 32, 703.
La réaction est régiosélective.
VO(acac)2 peut être remplacé par Ti(OiPr)4
Introduction aux réactions stéréosélectives
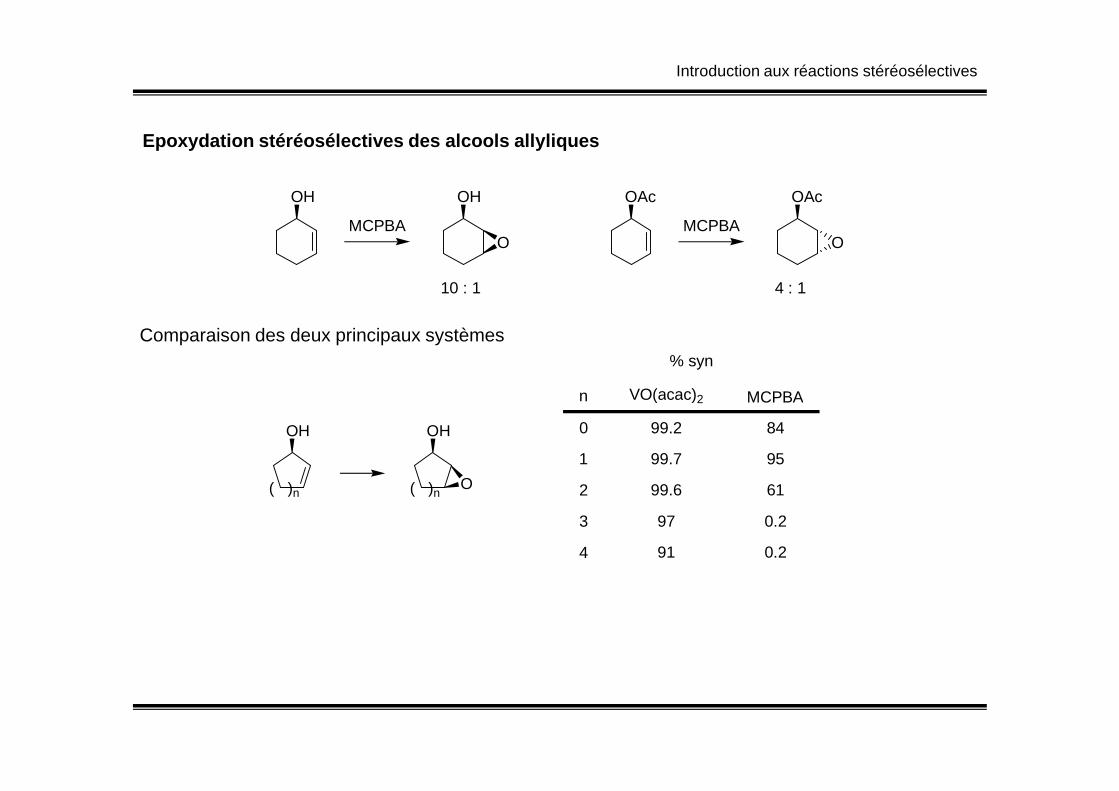
% syn
n VO(acac)2 MCPBA
Epoxydation stéréosélectives des alcools allyliques
OH
MCPBA
OH
O
10 : 1
OAc
MCPBA
OAc
O
4 : 1
Comparaison des deux principaux systèmes
OH
( )n
OH
( )nO
n VO(acac)2 MCPBA
0
1
2
3
4
99.2 84
99.7 95
99.6 61
97
91
0.2
0.2

Introduction aux réactions stéréosélectives
Epoxydation stéréosélectives des alcools allyliques
NHCl
MeO
Me OMeOMe
OMeOTBS
MeOHMe
Me
NHCl
MeO
Me OMeOMe
OMeOTBS
MeMe
Me
OOH
diastéréosélection > 20 : 1
Ti(OiPr)4, TBHP
Isobe et coll. J. Am. Chem. Soc. 1984, 106, 3252.
OHOH
O
VO(acac)2
TBHP, PhH
OHOH
O
O
OHOH
O
O
4.3 : 1
Sorensen, E.J. et coll. Angew. Chem. Int. Ed. 1999, 38, 971.
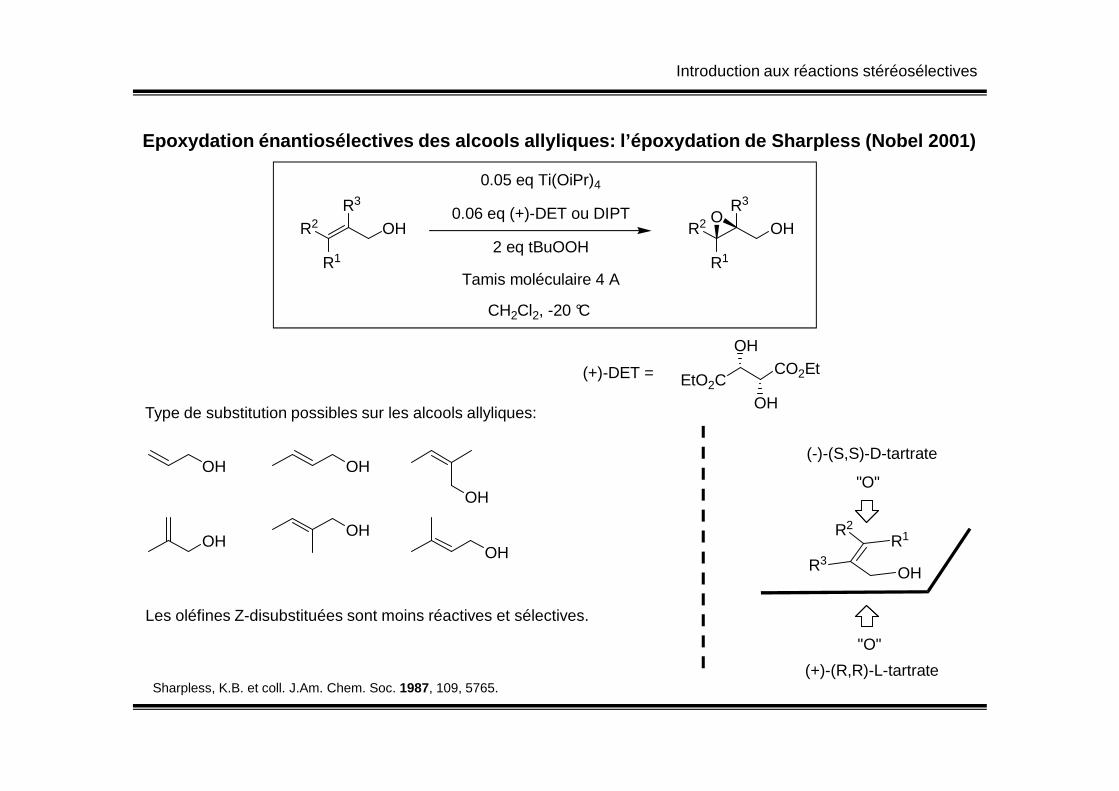
Introduction aux réactions stéréosélectives
Epoxydation énantiosélectives des alcools allylique s: l’époxydation de Sharpless (Nobel 2001)
R3
R2
R1
OH
0.05 eq Ti(OiPr)4
0.06 eq (+)-DET ou DIPT
2 eq tBuOOH
Tamis moléculaire 4 A
CH2Cl2, -20 °C
R3
R2
R1
OHO
EtO2CCO2Et
OH
OH
(+)-DET =
OH
R3
R1R2
OH
(-)-(S,S)-D-tartrate
(+)-(R,R)-L-tartrate
"O"
"O"
Sharpless, K.B. et coll. J.Am. Chem. Soc. 1987, 109, 5765.
OH OH
OH
OHOH
OH
Type de substitution possibles sur les alcools allyliques:
Les oléfines Z-disubstituées sont moins réactives et sélectives.
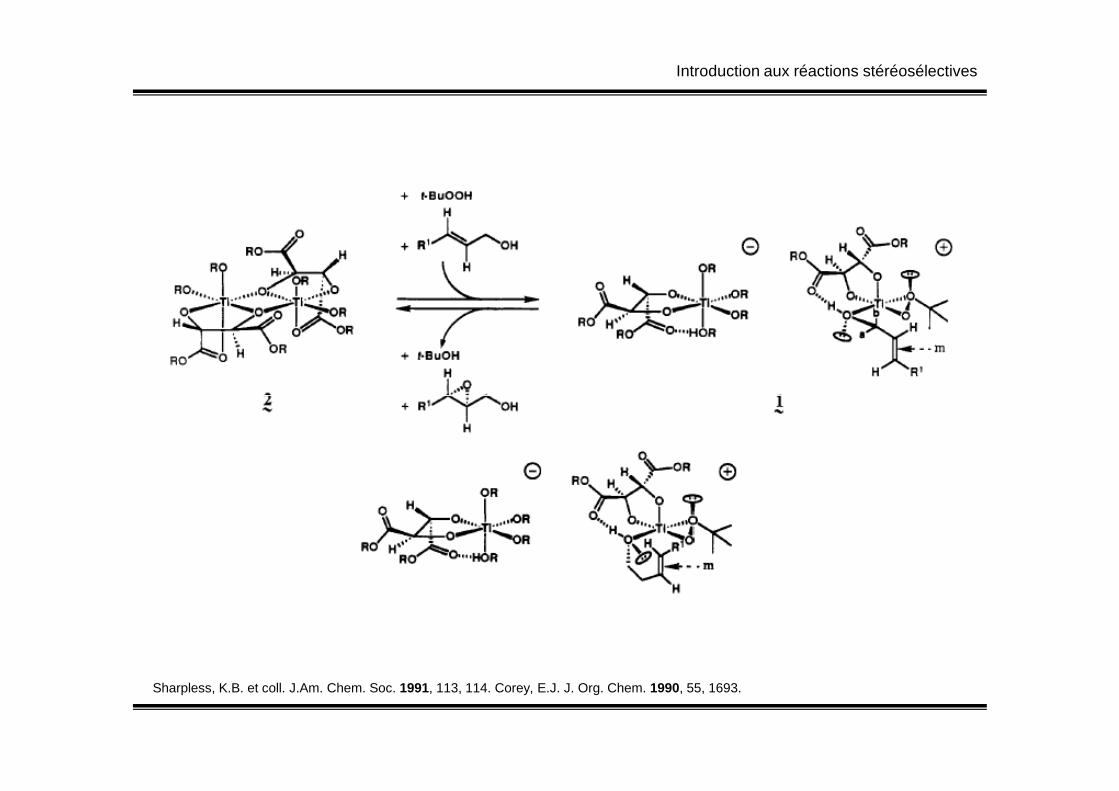
Introduction aux réactions stéréosélectives
Sharpless, K.B. et coll. J.Am. Chem. Soc. 1991, 113, 114. Corey, E.J. J. Org. Chem. 1990, 55, 1693.

Introduction aux réactions stéréosélectives
Exemples d’époxydation de Sharpless
OHO
(+)-DIPT
r = 65%
ee = 90%
OHO
(+)-DIPT
r = 89%
ee > 98%
Ph OHO
(+)-DET
r = 88%
ee = 95%
PrOH
O
(+)-DIPT
r = 79%
ee > 98%
PhMe
QUIZZ: indiquez le résultat de l’époxydation de Sharpless en vous aidant du modèle mnémotechnique.QUIZZ: indiquez le résultat de l’époxydation de Sharpless en vous aidant du modèle mnémotechnique.
Me
OH
(+)-DET(-)-DET
97%, 86% ee97%, 86% ee
? ?
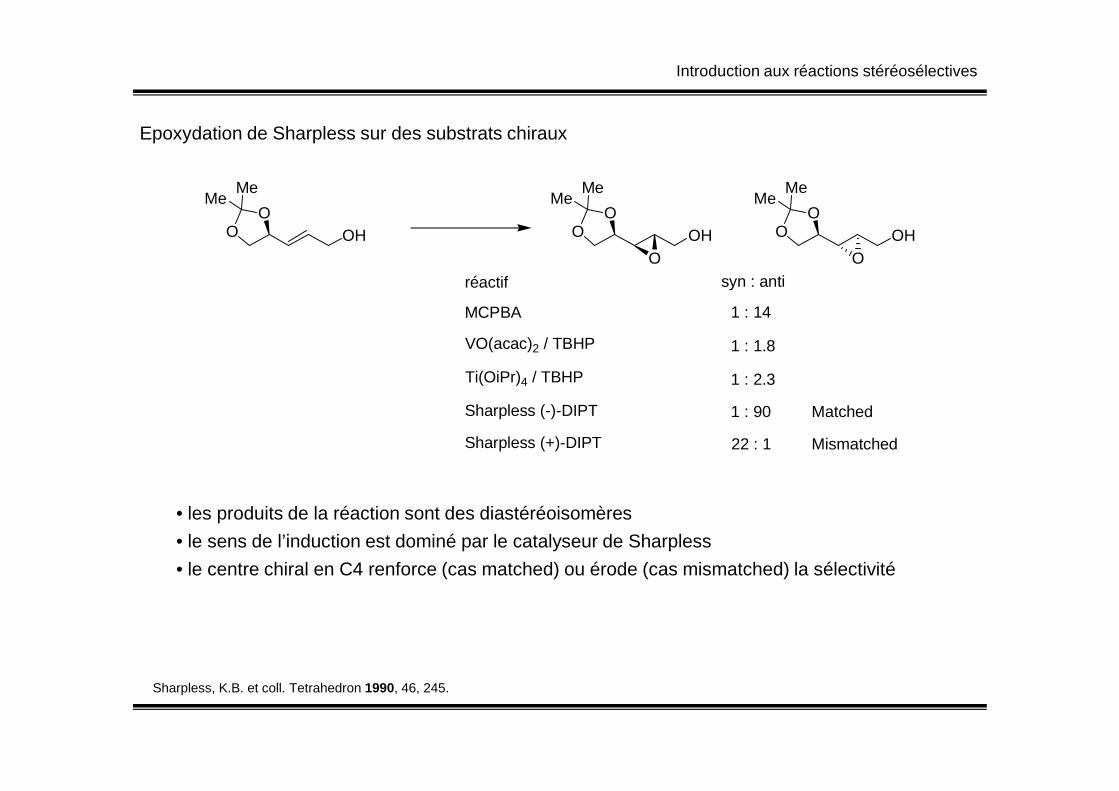
Introduction aux réactions stéréosélectives
Epoxydation de Sharpless sur des substrats chiraux
OHO
O
MeMe
OHO
O
MeMe
OHO
O
MeMe
O O
réactif syn : anti
MCPBA
VO(acac)2 / TBHP
Ti(OiPr)4 / TBHP
1 : 14
1 : 1.8
1 : 2.3
Sharpless (-)-DIPT
Sharpless (+)-DIPT
1 : 90
22 : 1
Matched
Mismatched
• les produits de la réaction sont des diastéréoisomères
• le sens de l’induction est dominé par le catalyseur de Sharpless
• le centre chiral en C4 renforce (cas matched) ou érode (cas mismatched) la sélectivité
Sharpless, K.B. et coll. Tetrahedron 1990, 46, 245.
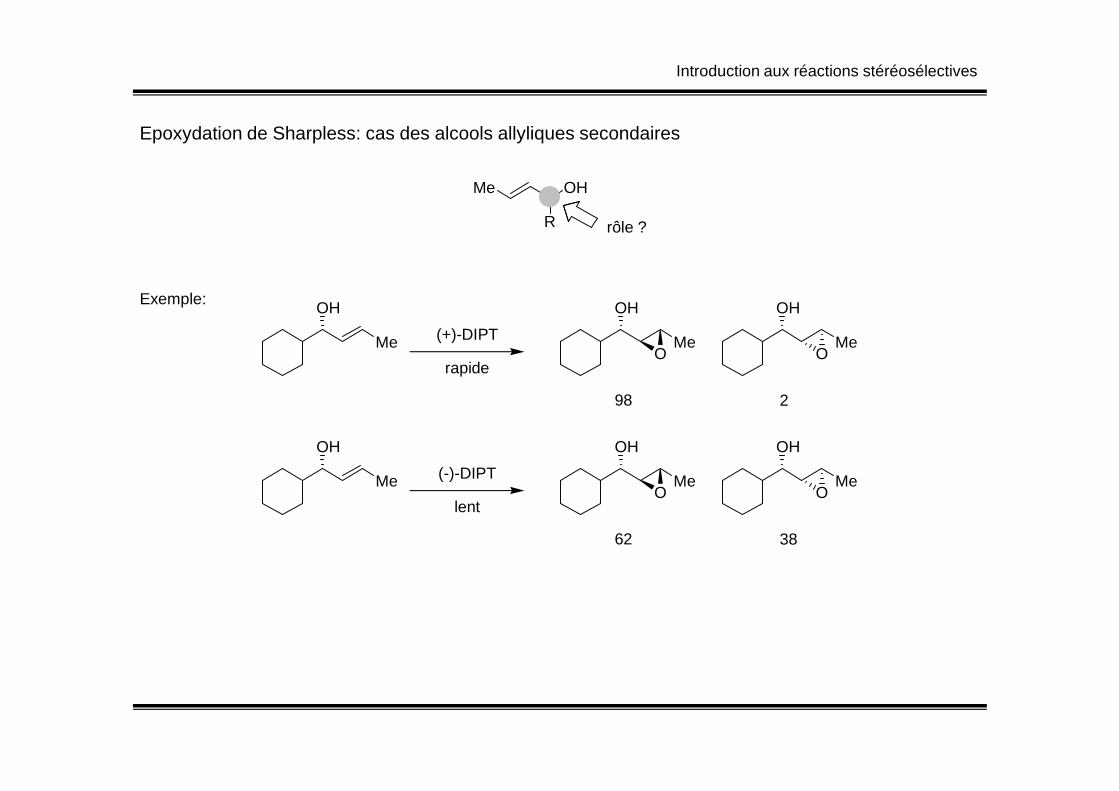
Introduction aux réactions stéréosélectives
Epoxydation de Sharpless: cas des alcools allyliques secondaires
Me OH
R rôle ?
Me
OH
Me
OH
Me
OH
O O(+)-DIPT
98 2
rapide
Exemple:
98 2
Me
OH
Me
OH
Me
OH
O O(-)-DIPT
62 38
lent
Introduction aux réactions stéréosélectives
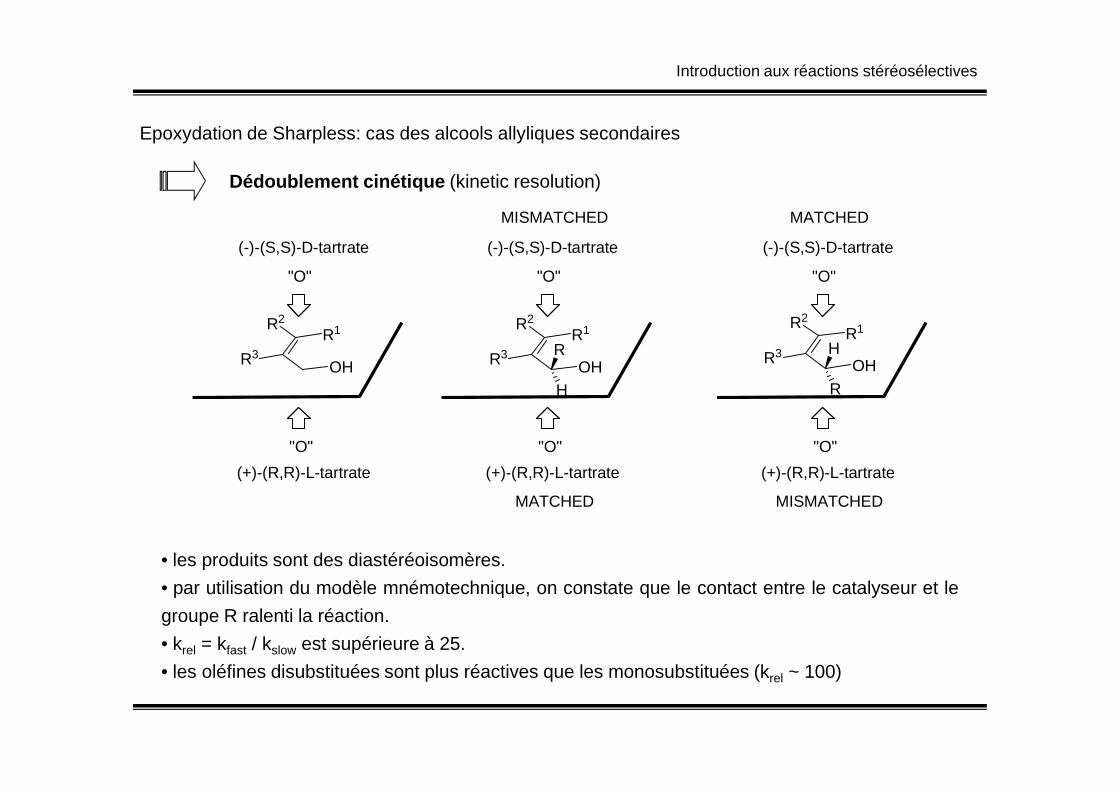
Epoxydation de Sharpless: cas des alcools allyliques secondaires
Dédoublement cinétique (kinetic resolution)
R3
R1R2
OH
(-)-(S,S)-D-tartrate
"O"
R3
R1R2
OH
(-)-(S,S)-D-tartrate
"O"
R3
R1R2
OH
(-)-(S,S)-D-tartrate
"O"
R
H
H
R
MATCHEDMISMATCHED
(+)-(R,R)-L-tartrate
"O"
(+)-(R,R)-L-tartrate
"O"
(+)-(R,R)-L-tartrate
"O"
H R
MISMATCHEDMATCHED
• les produits sont des diastéréoisomères.
• par utilisation du modèle mnémotechnique, on constate que le contact entre le catalyseur et le
groupe R ralenti la réaction.
• krel = kfast / kslow est supérieure à 25.
• les oléfines disubstituées sont plus réactives que les monosubstituées (krel ~ 100)
Introduction aux réactions stéréosélectives
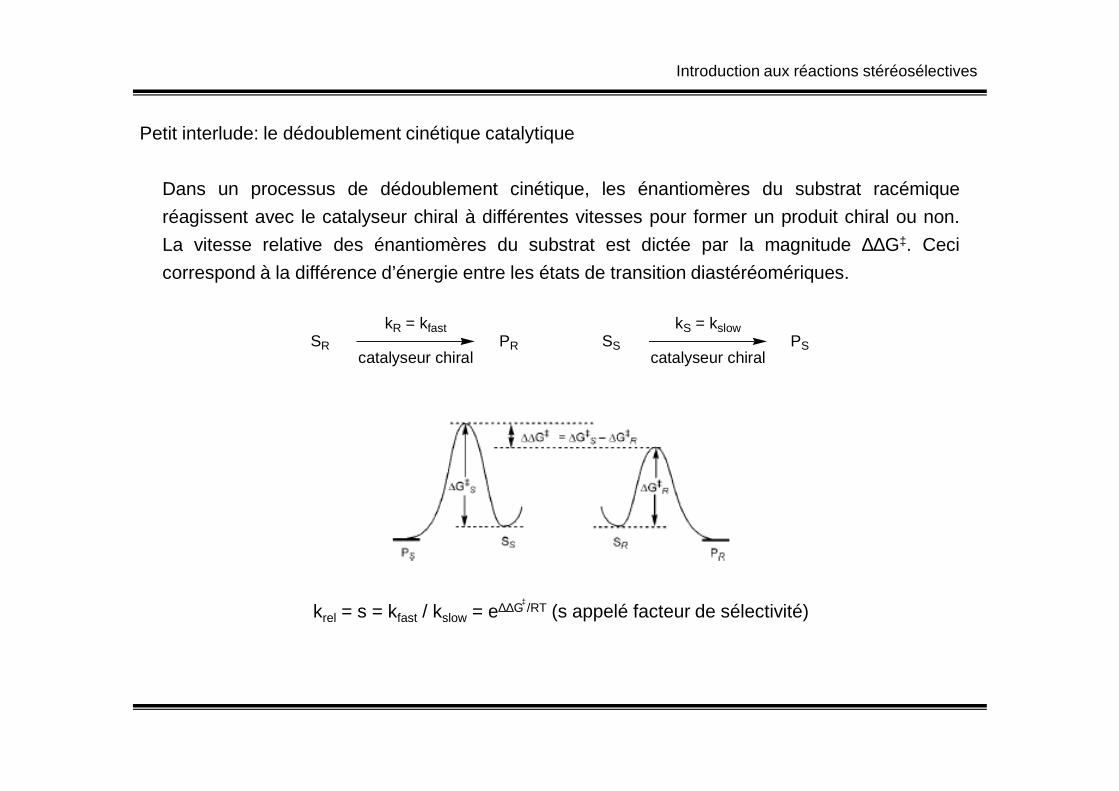
Petit interlude: le dédoublement cinétique catalytique
Dans un processus de dédoublement cinétique, les énantiomères du substrat racémique
réagissent avec le catalyseur chiral à différentes vitesses pour former un produit chiral ou non.
La vitesse relative des énantiomères du substrat est dictée par la magnitude ∆∆G‡. Ceci
correspond à la différence d’énergie entre les états de transition diastéréomériques.
SR
kR = kfast
catalyseur chiralPR
kS = kslow
catalyseur chiralSS PS
krel = s = kfast / kslow = e∆∆G /RT (s appelé facteur de sélectivité)‡
Introduction aux réactions stéréosélectives
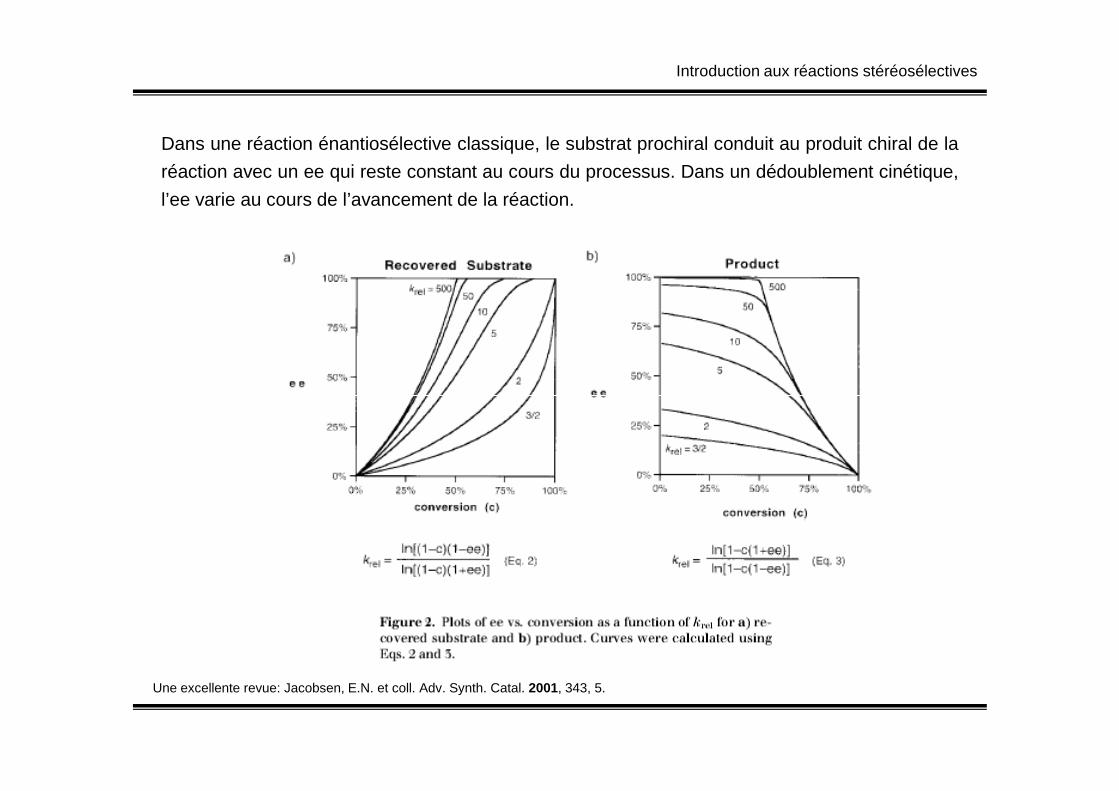
Dans une réaction énantiosélective classique, le substrat prochiral conduit au produit chiral de la
réaction avec un ee qui reste constant au cours du processus. Dans un dédoublement cinétique,
l’ee varie au cours de l’avancement de la réaction.
Une excellente revue: Jacobsen, E.N. et coll. Adv. Synth. Catal. 2001, 343, 5.
Introduction aux réactions stéréosélectives
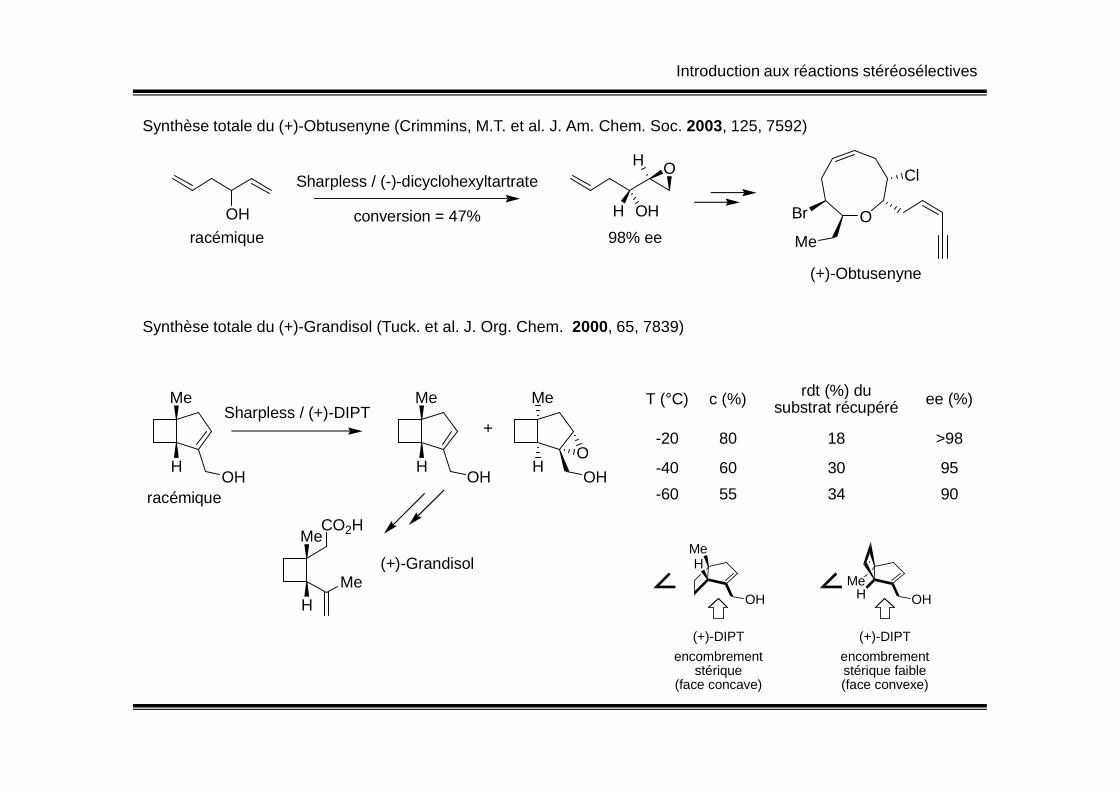
Synthèse totale du (+)-Obtusenyne (Crimmins, M.T. et al. J. Am. Chem. Soc. 2003, 125, 7592)
OH
racémique
Sharpless / (-)-dicyclohexyltartrate
H OH
OH
conversion = 47%98% ee
O
Cl
Br
Me
(+)-Obtusenyne
Synthèse totale du (+)-Grandisol (Tuck. et al. J. Org. Chem. 2000, 65, 7839)
Me Me Me T (°C) c (%) rdt (%) du ee (%)
OH
Me
H
racémique
Sharpless / (+)-DIPT
OH
Me
HOH
Me
HO
+
T (°C) c (%) rdt (%) du substrat récupéré ee (%)
-20 80 18 >98
-40 60 30 95
-60 55 34 90
Me
CO2HMe
H
(+)-Grandisol
OH
MeH
(+)-DIPT
encombrementstérique
(face concave)
OHHMe
(+)-DIPT
encombrementstérique faible(face convexe)
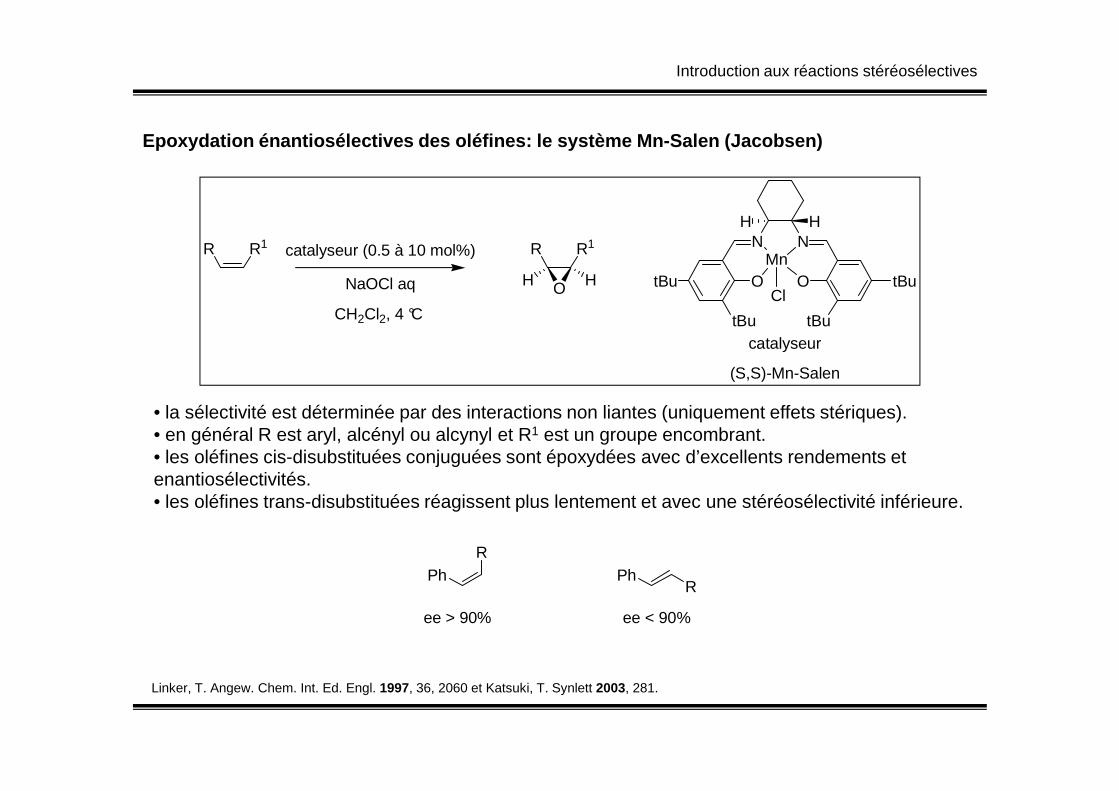
Introduction aux réactions stéréosélectives
Epoxydation énantiosélectives des oléfines: le syst ème Mn-Salen (Jacobsen)
R R1 catalyseur (0.5 à 10 mol%)
NaOCl aq
CH2Cl2, 4 °C
R R1
O HH
N N
O O
tBu
tBu
tBu
tBuMn
Cl
H H
catalyseur
(S,S)-Mn-Salen
Linker, T. Angew. Chem. Int. Ed. Engl. 1997, 36, 2060 et Katsuki, T. Synlett 2003, 281.
• la sélectivité est déterminée par des interactions non liantes (uniquement effets stériques).• en général R est aryl, alcényl ou alcynyl et R1 est un groupe encombrant.• les oléfines cis-disubstituées conjuguées sont époxydées avec d’excellents rendements et enantiosélectivités.• les oléfines trans-disubstituées réagissent plus lentement et avec une stéréosélectivité inférieure.
Ph PhR
R
ee > 90% ee < 90%
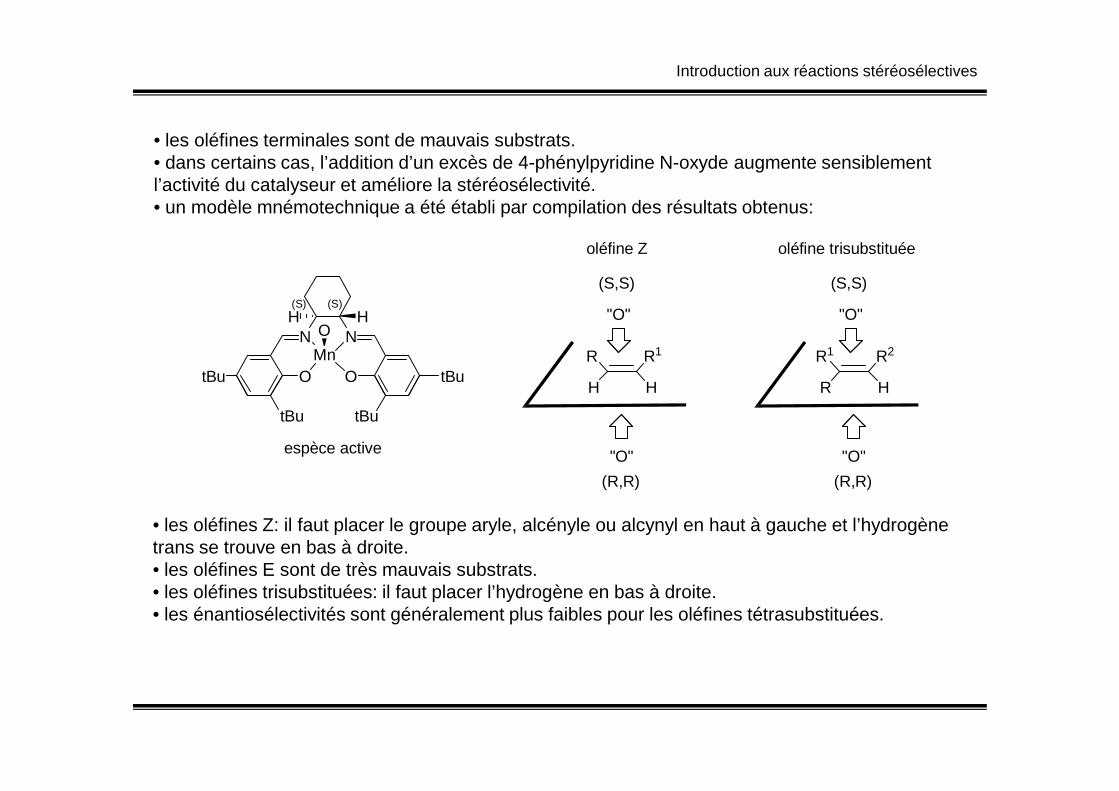
Introduction aux réactions stéréosélectives
• les oléfines terminales sont de mauvais substrats.• dans certains cas, l’addition d’un excès de 4-phénylpyridine N-oxyde augmente sensiblement l’activité du catalyseur et améliore la stéréosélectivité.• un modèle mnémotechnique a été établi par compilation des résultats obtenus:
N N
O OtBu tBuMn
H HO
(S)(S)
H
R1R
H
(S,S)
"O"
oléfine Z
H
R2R1
R
(S,S)
"O"
oléfine trisubstituée
tBu tBu
espèce active
HH
(R,R)
"O"
HR
(R,R)
"O"
• les oléfines Z: il faut placer le groupe aryle, alcényle ou alcynyl en haut à gauche et l’hydrogène trans se trouve en bas à droite.• les oléfines E sont de très mauvais substrats.• les oléfines trisubstituées: il faut placer l’hydrogène en bas à droite.• les énantiosélectivités sont généralement plus faibles pour les oléfines tétrasubstituées.

Introduction aux réactions stéréosélectives
Quelques exemples d’époxydation suivant les conditions de Jacobsen:
Me
OMe
Me(S,S)-Mn-Salen (0.03 eq)
NaOCl aq, CH2Cl2, 4 °C Me
OMe
Me
O
98%, 97% ee
Br
OMe
Me(S,S)-Mn-Salen (0.03 eq)
NaOCl aq, CH2Cl2, 4 °C Br
OMe
Me
OMe
MeMe
MeO
84%, 96% eeMe Me
PhMe
Ph
(R,R)-Mn-Salen (0.03 eq)
NaOCl aq, CH2Cl2, 4 °C
4-phénylpyridine N-oxyde
PhMe
PhO
87%, 88% ee
QUIZZ: retrouvez le dernier résultat ci-dessus à l’aide du modèle mnémotechnique.
Jacobsen, E.N. et coll. J. Am. Chem. Soc. 1991, 113, 7063; J. Org. Chem. 1994, 59, 4378; Tetrahedron Lett. 1995, 36, 5123.

Introduction aux réactions stéréosélectives
Réactions d’ouverture des époxydes méso par le système de Jacobsen
N N
O O
tBu
tBu
tBu
tBuCr
H H
Cl
(R,R)-Cr-salen
OR
R
1- catalyseur, Et2OMe3SiN3
2- H3O+R
R N3
OH
méso
N3
FmocNN3 Me N3
OH
72%, 81% ee
FmocNOH
80%, 95% ee
Me OH
65%, 82% ee
Jacobsen, E.N. et coll. J. Am. Chem. Soc. 1995, 117, 5897.Jacobsen, E.N. Acc. Chem. Res. 2000, 33, 421.
Jacobsen a également montré que son système catalytique permettait le dédoublementcinétique des époxydes racémiques terminaux par H2O, TMSN3, et des phénols (voir Science1997, 277, 936; J. Am. Chem. Soc. 1996, 118, 7420; J. Am. Chem. Soc. 1999, 121, 6086; Tetrahedron Lett.2000, 40, 7303).
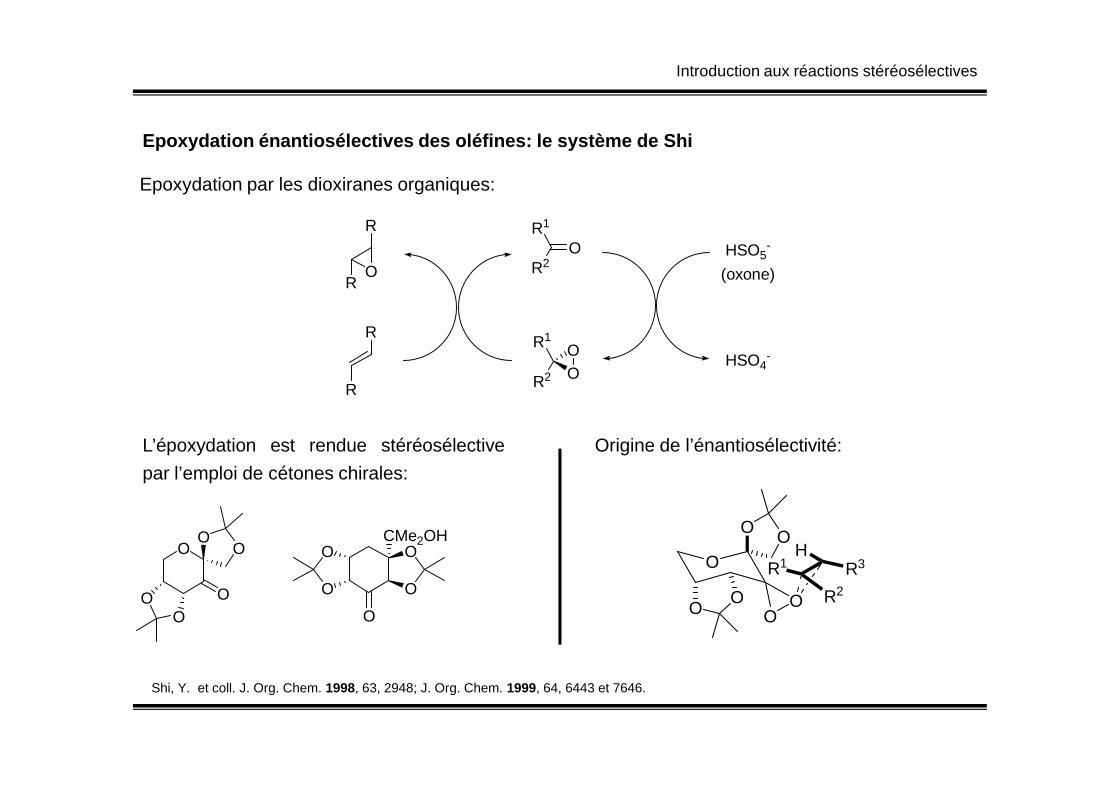
Introduction aux réactions stéréosélectives
Epoxydation énantiosélectives des oléfines: le syst ème de Shi
Epoxydation par les dioxiranes organiques:
OR1
R2
OOR1
R2
HSO5-
(oxone)
HSO4-
R
R
R
R
O
R
L’époxydation est rendue stéréosélective
par l’emploi de cétones chirales:
O
O
OO
OO
O
O O
O
O
CMe2OH
Shi, Y. et coll. J. Org. Chem. 1998, 63, 2948; J. Org. Chem. 1999, 64, 6443 et 7646.
Origine de l’énantiosélectivité:
O
O OO O
O O
R3R1
R2
H

Introduction aux réactions stéréosélectives
Quelques exemples d’époxydation suivant les conditions de Shi:
Ph
SiMe3
O
O
O O
OO
Oxone
H2O, solvantPh
SiMe3O
81%, 95% ee
Ph OH Ph OHOO
85%, 94% ee
OBz BzOO
82%, 93% ee
puis TBAFPh
OTBS
Ph
O
OH80%, 90% ee

Introduction aux réactions stéréosélectives
Réactions de cis -dihydroxylation
R1
R2R4
R3oxydant
HOOH
R3R4
R1 R2
La réaction d’hydroxylation des oléfines est stéréospecifique (cis-addition).Les oxydants sont de type O=M=O comme KMnO4, OsO4 . Le système le plus efficace consiste à
employer OsO4 en quantité catalytique en présence d’un réactif de réoxydation du métal comme la
N-méthyl morpholine oxyde (NMO).N-méthyl morpholine oxyde (NMO).
Le mécanisme de cette réaction est du type:
R
R
OsO
OOO
LO
OsOO
O
R
RL
hydrolyse
et NMOOsO
OOO
LHO
HO R
R

Introduction aux réactions stéréosélectives
Cis-dihydroxylation stéréosélectives des oléfines a cycliques
Me
OO
OsO4 cat, NMO
H2O, acétone, THF Me
OO
OH
OH 3.7 : 1
OO
OsO4 cat, NMO
H2O, acétone, THF
OO
Me Me
OH2
OH 7.6 : 1
OH
Me
O
Me
OOBn
SitButBu
OsO4 cat, NMO
H2O, acétone, THF
O
Me
OOBn
SitButBu
HOHO Me
60 : 1
Kishi, Y. et coll. Tetrahedron Lett. 1983, 24, 3943 et 3947; Evans, D.A. et coll. J. Org. Chem. 1990, 55, 1698.
Peut-on prévoir le résultat à l’avance?

Introduction aux réactions stéréosélectives
Quel modèle peut-on appliquer pour expliquer l’osmylation diastéréosélective?
HHMe
X
MeH
OsO4
Me
X Me
Me
X Me
OHOH
OsO4
X X OH
HMeH
X
MeHMe
X
Me
X
OHMe Me
OH
Le modèle proposé ici est purement empirique et ne peux s’appliquer que pour X = OR.
Kishi, Y. et coll. Tetrahedron Lett. 1983, 24, 3943 et Tetrahedron Lett. 1984, 24, 2247.
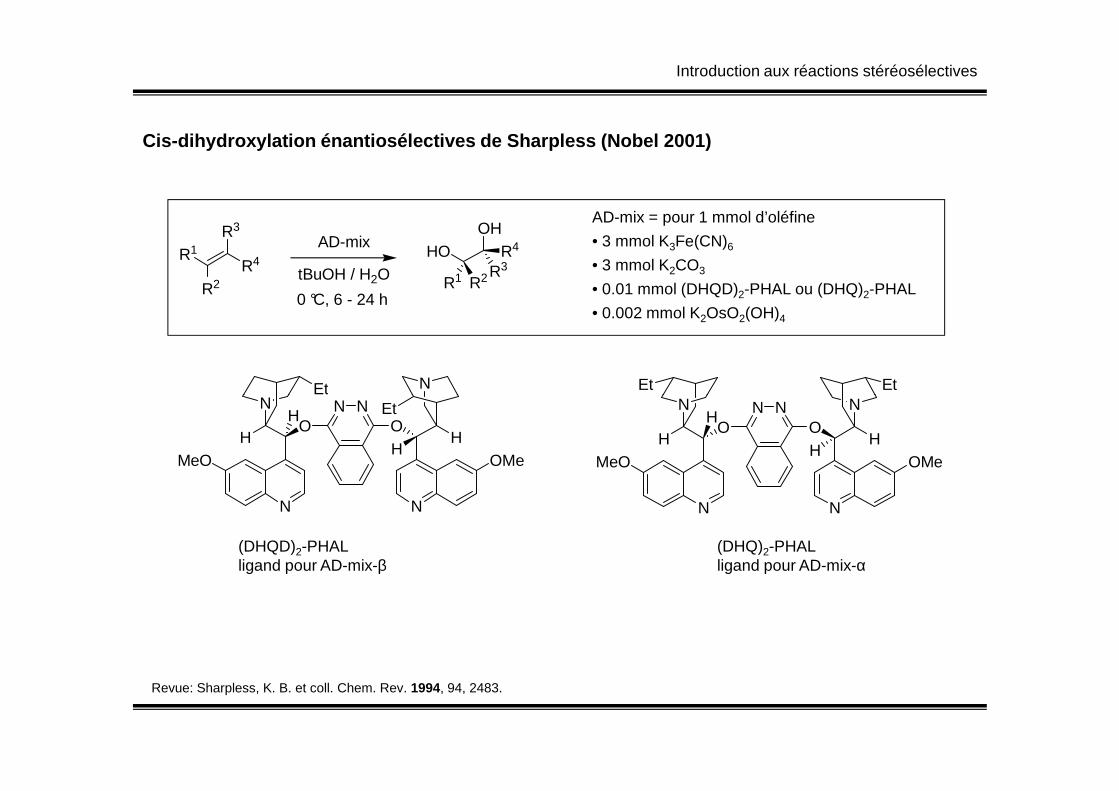
Introduction aux réactions stéréosélectives
Cis-dihydroxylation énantiosélectives de Sharpless (Nobel 2001)
AD-mix = pour 1 mmol d’oléfine
• 3 mmol K3Fe(CN)6
• 3 mmol K2CO3
• 0.01 mmol (DHQD)2-PHAL ou (DHQ)2-PHAL
• 0.002 mmol K2OsO2(OH)4
R1
R2R4
R3AD-mix
HOOH
R3R4
R1 R2tBuOH / H2O
0 °C, 6 - 24 h
NN
EtN N
EtEtNN
N
MeO
ON
Et
HH
N
OMe
Et
HOH
NN
N
MeO
OH
H
N
OMeH
OH
N N
(DHQD)2-PHAL (DHQ)2-PHALligand pour AD-mix-β ligand pour AD-mix-α
Revue: Sharpless, K. B. et coll. Chem. Rev. 1994, 94, 2483.
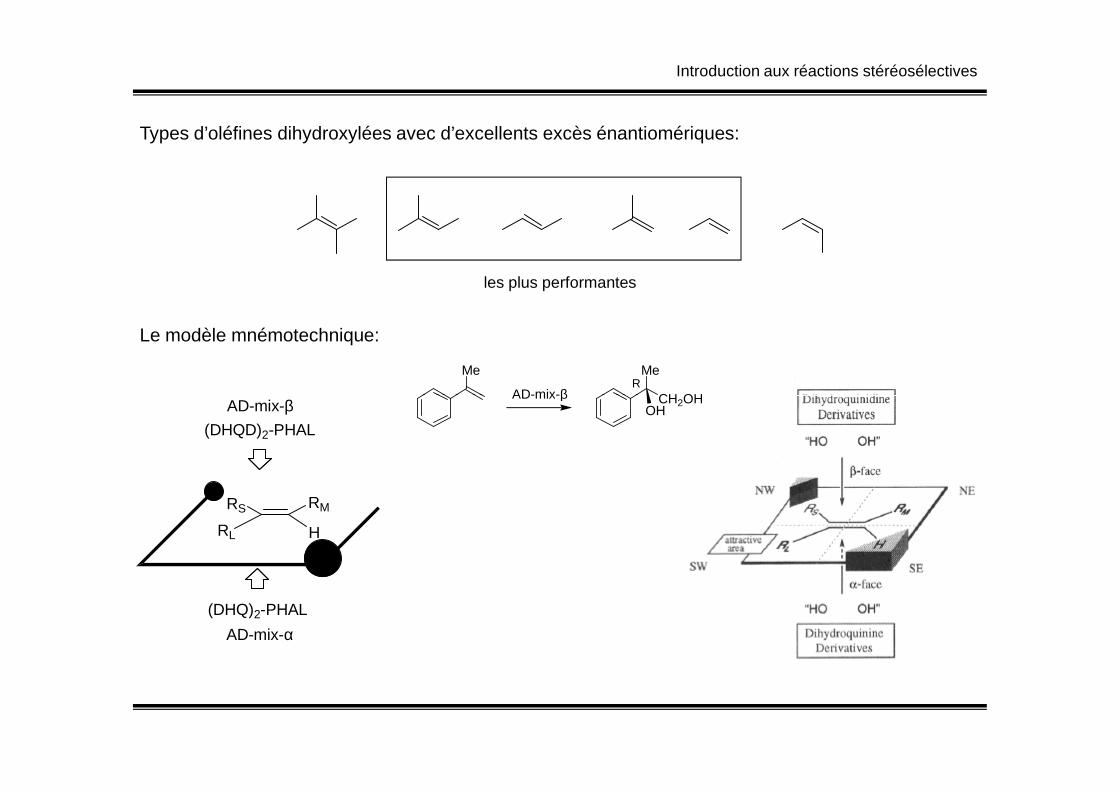
Introduction aux réactions stéréosélectives
Types d’oléfines dihydroxylées avec d’excellents excès énantiomériques:
les plus performantes
Le modèle mnémotechnique:
Me
AD-mix-β
Me
CH OHR
RL
RS RM
H
(DHQ)2-PHAL
AD-mix-α
(DHQD)2-PHAL
AD-mix-βAD-mix-β CH2OH
OH
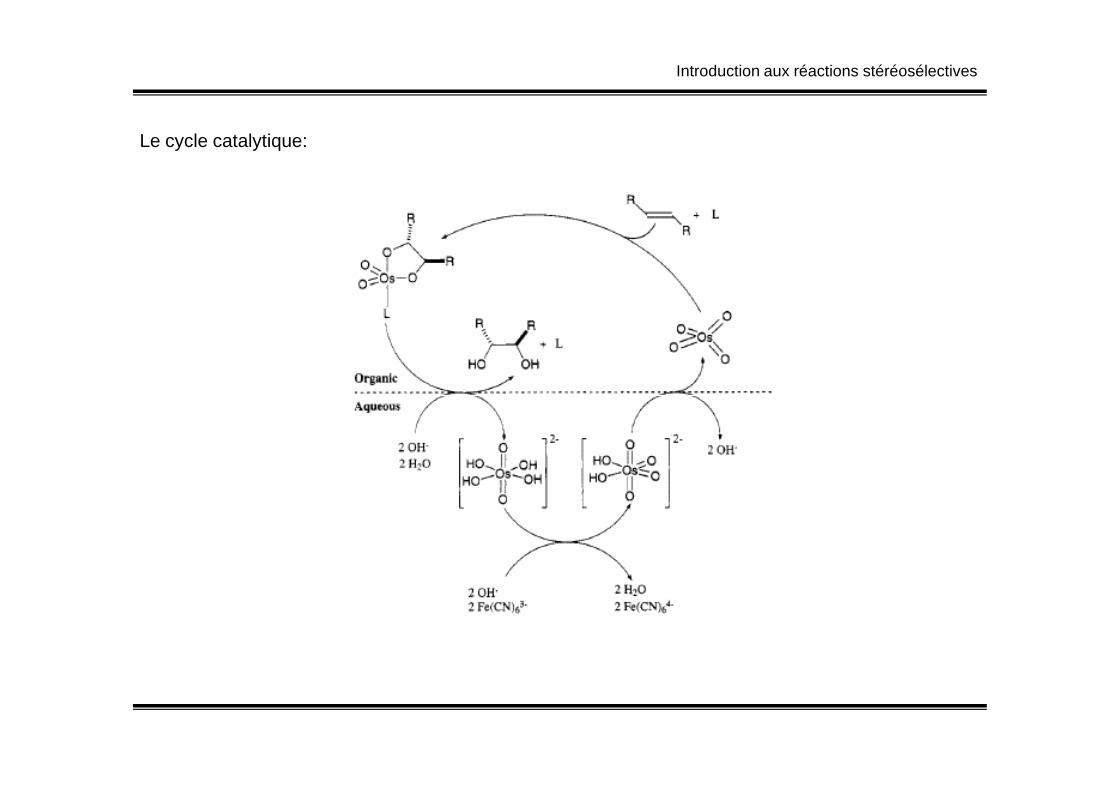
Introduction aux réactions stéréosélectives
Le cycle catalytique:

Introduction aux réactions stéréosélectives
Quelques exemples:
Me Me
Me
Me Me
Me
OHOH
AD-mix-β AD-mix-α
Me Me
Me
OHOH
98% ee 95% ee
C5H11CO2Et
C5H11CO2Et
OH
OHC5H11
CO2EtOH
OH
99% ee 96% ee
C8H17 C8H17 CH2OH
OH
C8H17 CH2OH
OH
84% ee 80% ee
QUIZZ: retrouvez les résultats ci-dessus à l’aide du modèle mnémotechnique.
Sharpless, K. B. et coll. J. Org. Chem. 1992, 57, 2768.
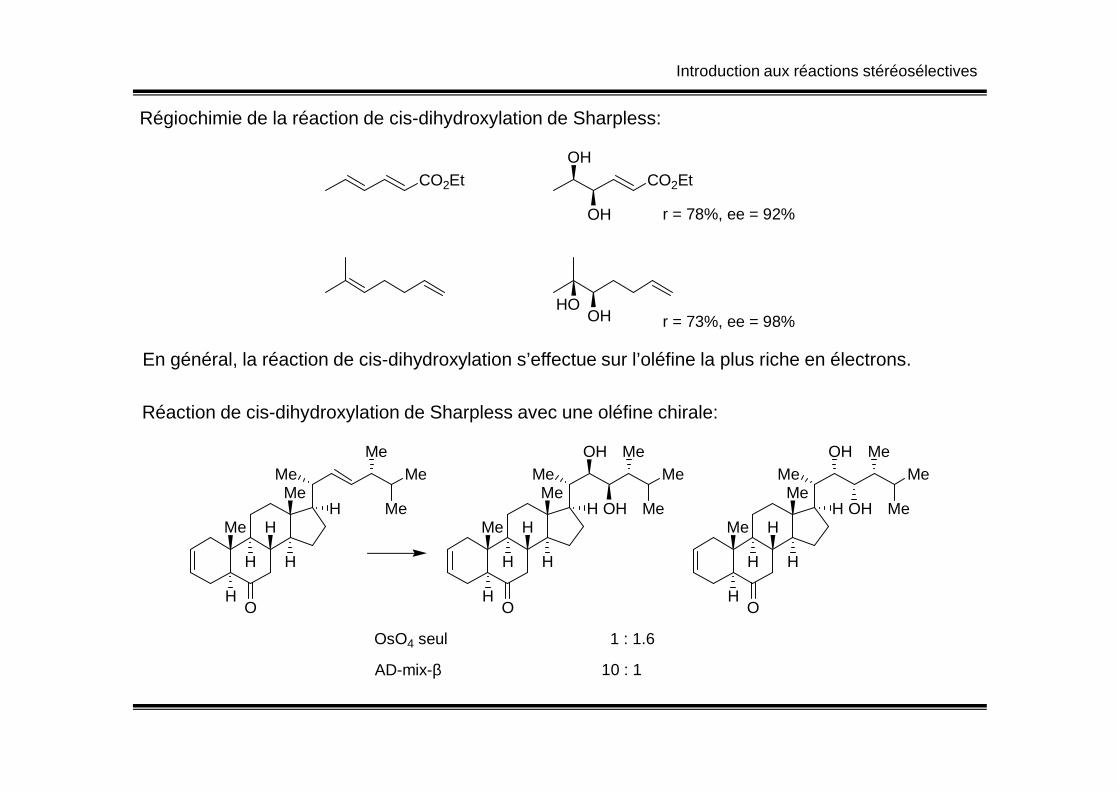
Introduction aux réactions stéréosélectives
Régiochimie de la réaction de cis-dihydroxylation de Sharpless:
CO2Et CO2EtOH
OH r = 78%, ee = 92%
OHHO
r = 73%, ee = 98%
En général, la réaction de cis-dihydroxylation s’effectue sur l’oléfine la plus riche en électrons.
Réaction de cis-dihydroxylation de Sharpless avec une oléfine chirale:
Me
Me
MeMe
H
O
Me
Me
HH
H
H
Me
Me
MeMe
H
O
Me
Me
HH
H
H
OH
OH
Me
Me
MeMe
H
O
Me
Me
HH
H
H
OH
OH
OsO4 seul 1 : 1.6
AD-mix-β 10 : 1
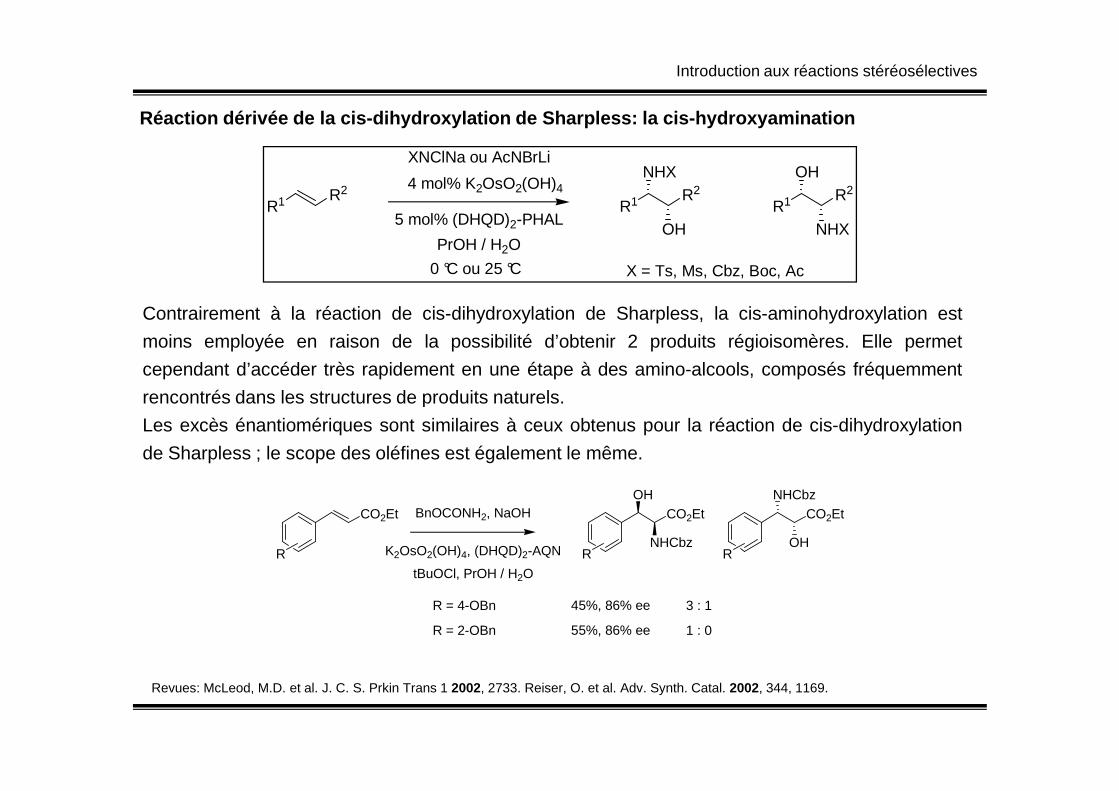
Introduction aux réactions stéréosélectives
Réaction dérivée de la cis-dihydroxylation de Sharp less: la cis-hydroxyamination
R1 R2
XNClNa ou AcNBrLi
4 mol% K2OsO2(OH)4
5 mol% (DHQD)2-PHAL
PrOH / H2O
0 °C ou 25 °C
R1 R2
R1 R2NHX
OH
OH
NHX
X = Ts, Ms, Cbz, Boc, Ac
Contrairement à la réaction de cis-dihydroxylation de Sharpless, la cis-aminohydroxylation est
moins employée en raison de la possibilité d’obtenir 2 produits régioisomères. Elle permet
cependant d’accéder très rapidement en une étape à des amino-alcools, composés fréquemment
rencontrés dans les structures de produits naturels.
Revues: McLeod, M.D. et al. J. C. S. Prkin Trans 1 2002, 2733. Reiser, O. et al. Adv. Synth. Catal. 2002, 344, 1169.
rencontrés dans les structures de produits naturels.
Les excès énantiomériques sont similaires à ceux obtenus pour la réaction de cis-dihydroxylation
de Sharpless ; le scope des oléfines est également le même.
CO2Et CO2Et CO2EtOH
NHCbz
NHCbz
OH
BnOCONH2, NaOH
K2OsO2(OH)4, (DHQD)2-AQN
tBuOCl, PrOH / H2O
R R R
R = 4-OBn 3 : 145%, 86% ee
R = 2-OBn 1 : 055%, 86% ee

Introduction aux réactions stéréosélectives
Hydrogénation des oléfines
Le mode principal de réduction des oléfines est la réaction d’hydrogénation catalysée par un métal
de transition. Les métaux les plus couramment employés sont le rhodium, le ruthénium et le
palladium. En présence d’un ligand chiral, la réaction d’hydrogénation devient alors
énantiosélective, c’est à dire qu’une face de l’oléfine est préférentiellement hydrogénée.
R2 R2
R1
R2
R3
R1
R2
R3H2
M / ligand chiral
Les meilleures inductions asymétriques sont obtenues à partir d’oléfines fonctionnalisées comme
les énamides et les dérivés carbonylés α,β-insaturés.

Introduction aux réactions stéréosélectives
Hydrogénation des énamides: formation d’acides amin és non naturels
Ph NHAc Rh(COD)2ClO4
ligand
H2
Ph NHAc
MeRh+
ClO4-
PPh2
PPh2O
O
DIOP
P Ph
OMeMeO DIPAMP
Knowles (1977)ee = 95%
PPh2
PPh2
BINAPNoyori (1980)
DIOP
Kagan (1972) PPh
ee = 51%
ee = 95% PPh2 ee = 98%
Il n’existe pas de système type pour effectuer une hydrogénation stéréosélective des énamides
(mais aussi de dérivés carbonylés α,β-insaturés). D’autres systèmes catalytiques sont utilisables
comme BINAP / Ru2Cl4 par exemples.
Revue: Knowles, N.S. Acc. Chem. Res. 1983, 16, 106.
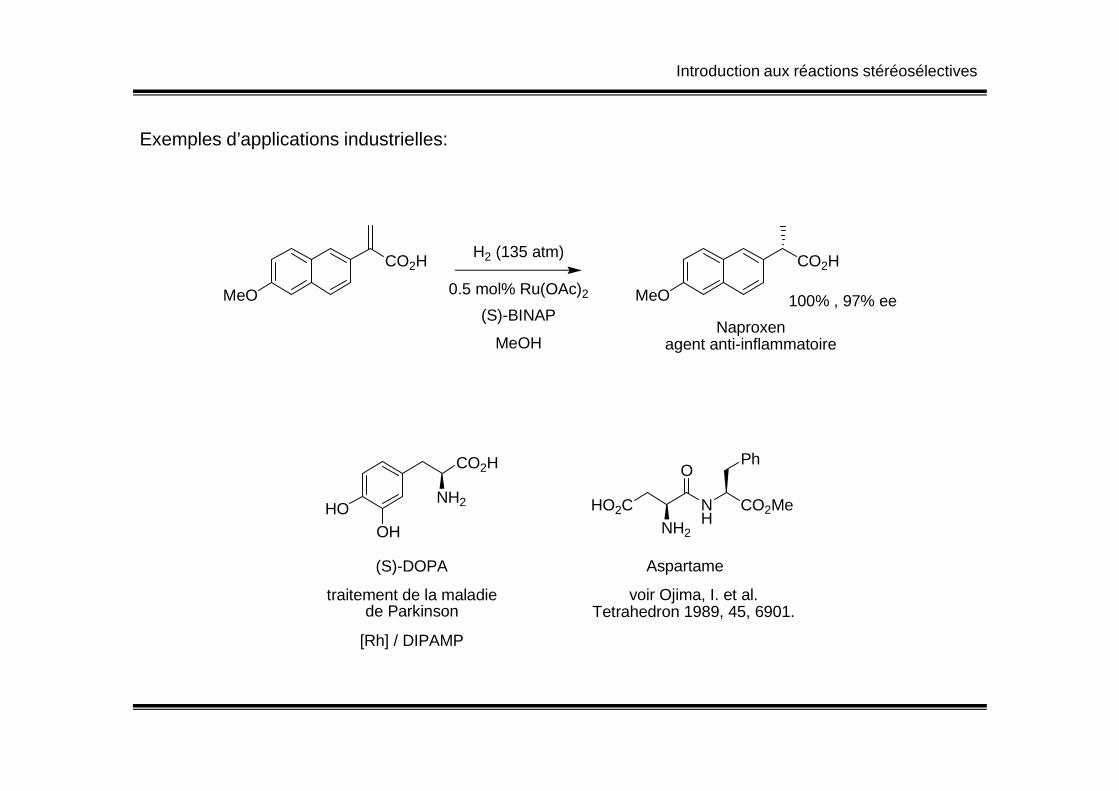
Introduction aux réactions stéréosélectives
Exemples d’applications industrielles:
MeO
CO2H
MeO
CO2HH2 (135 atm)
0.5 mol% Ru(OAc)2
(S)-BINAP
MeOH
100% , 97% ee
Naproxenagent anti-inflammatoire
HOOH
CO2H
NH2
(S)-DOPA
traitement de la maladiede Parkinson
[Rh] / DIPAMP
HO2C NH
O
CO2Me
Ph
NH2
Aspartame
voir Ojima, I. et al.Tetrahedron 1989, 45, 6901.
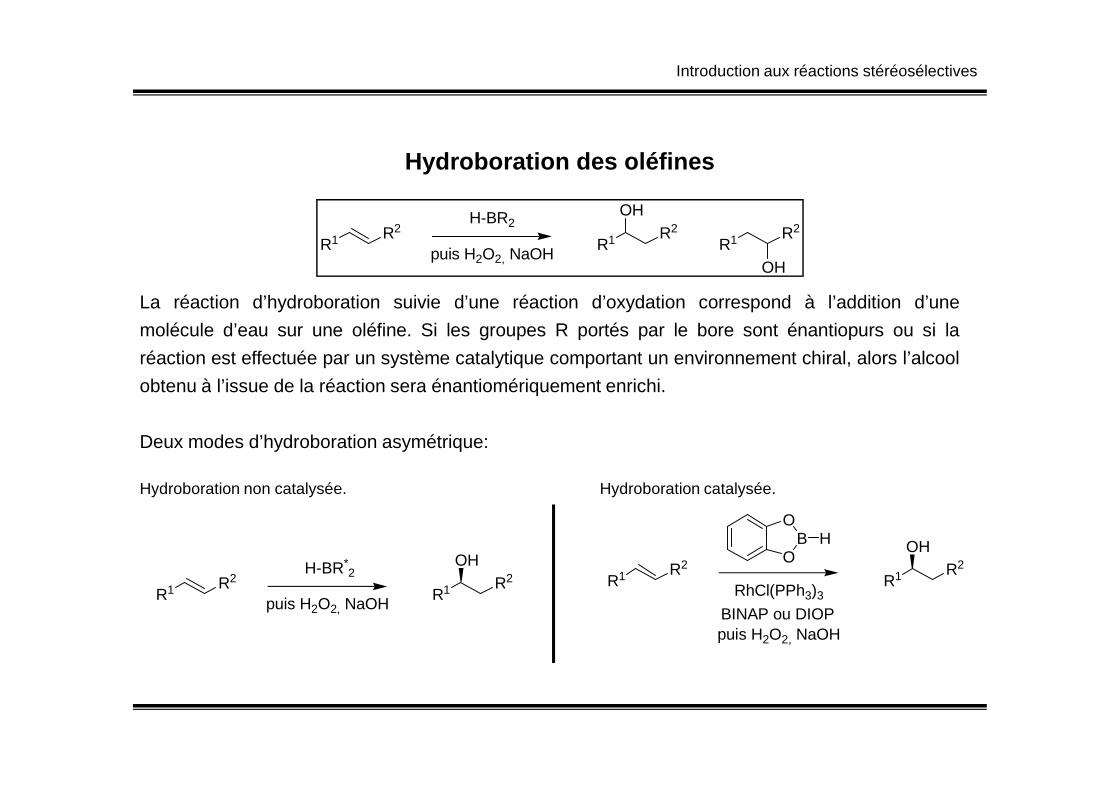
Introduction aux réactions stéréosélectives
Hydroboration des oléfines
R1 R2H-BR2
R1 R2
puis H2O2, NaOH
OH
R1 R2
OH
La réaction d’hydroboration suivie d’une réaction d’oxydation correspond à l’addition d’une
molécule d’eau sur une oléfine. Si les groupes R portés par le bore sont énantiopurs ou si la
réaction est effectuée par un système catalytique comportant un environnement chiral, alors l’alcool
obtenu à l’issue de la réaction sera énantiomériquement enrichi.obtenu à l’issue de la réaction sera énantiomériquement enrichi.
Deux modes d’hydroboration asymétrique:
R1 R2H-BR*
2
R1 R2
puis H2O2, NaOH
OHR1 R2
R1 R2
puis H2O2, NaOH
OHO
BO
H
RhCl(PPh3)3
BINAP ou DIOP
Hydroboration non catalysée. Hydroboration catalysée.

Introduction aux réactions stéréosélectives
Hydroboration non catalysée
Les différents réactifs chiraux:
BMe Me
H
BMe Me
H
MeBH2
HMe
H2BH
MeBHH
MeHB H
2 2
2,5-diméthylborolane
monoisopinocampheylboranediisopinocampheylboraneIpcBH2
Ipc2BH
Réactif de choix pour l’hydroborationdes oléfines Z-1,2-disubstituées.
Pour l’hydroboration d’oléfinesE-1,2-disubstituées ou trisubsti-tuées.
Pour l’hydroboration d’oléfinesZ ou E-1,2-disubstituées ou tri-substituées. Inefficace pour lesoléfines terminales.
O
Ipc2BH
puis H2O2, NaOH O
OH
r = 68-92%ee > 99%
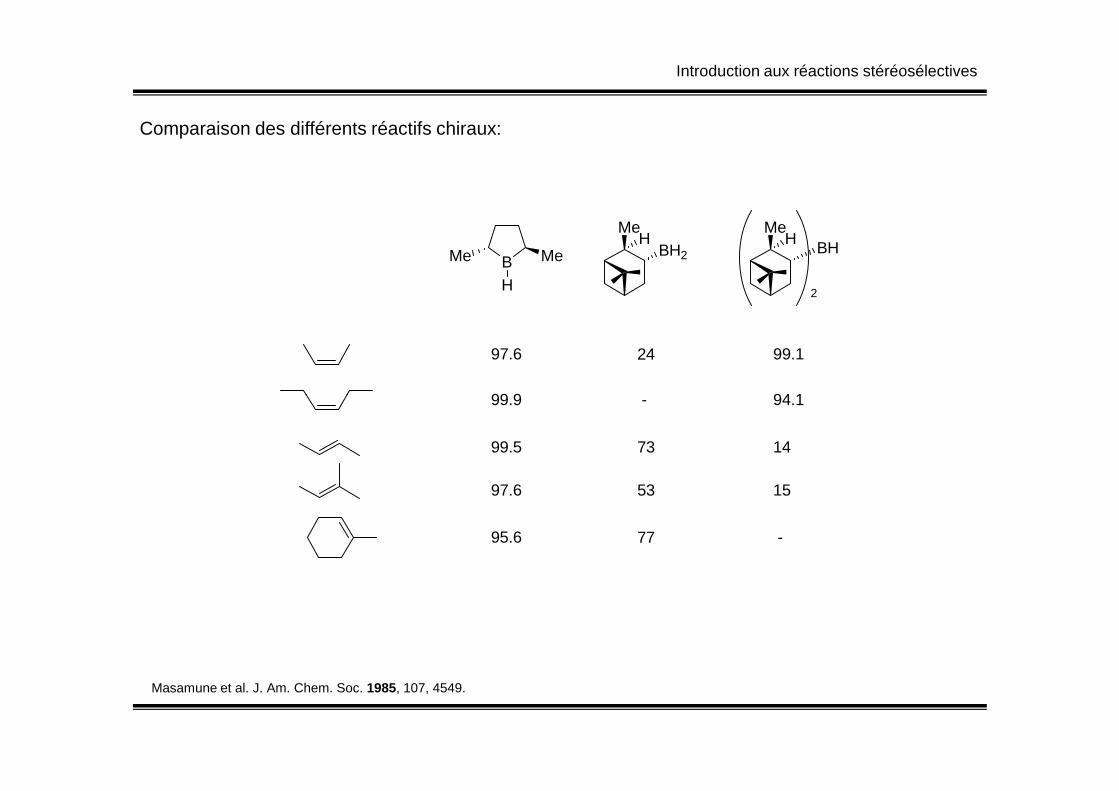
Introduction aux réactions stéréosélectives
Comparaison des différents réactifs chiraux:
BMe Me
H
MeBH2
HMe
BHH
2
97.6 99.124
99.9 94.1 -99.9 94.1 -
99.5 1473
97.6 1553
95.6 -77
Masamune et al. J. Am. Chem. Soc. 1985, 107, 4549.
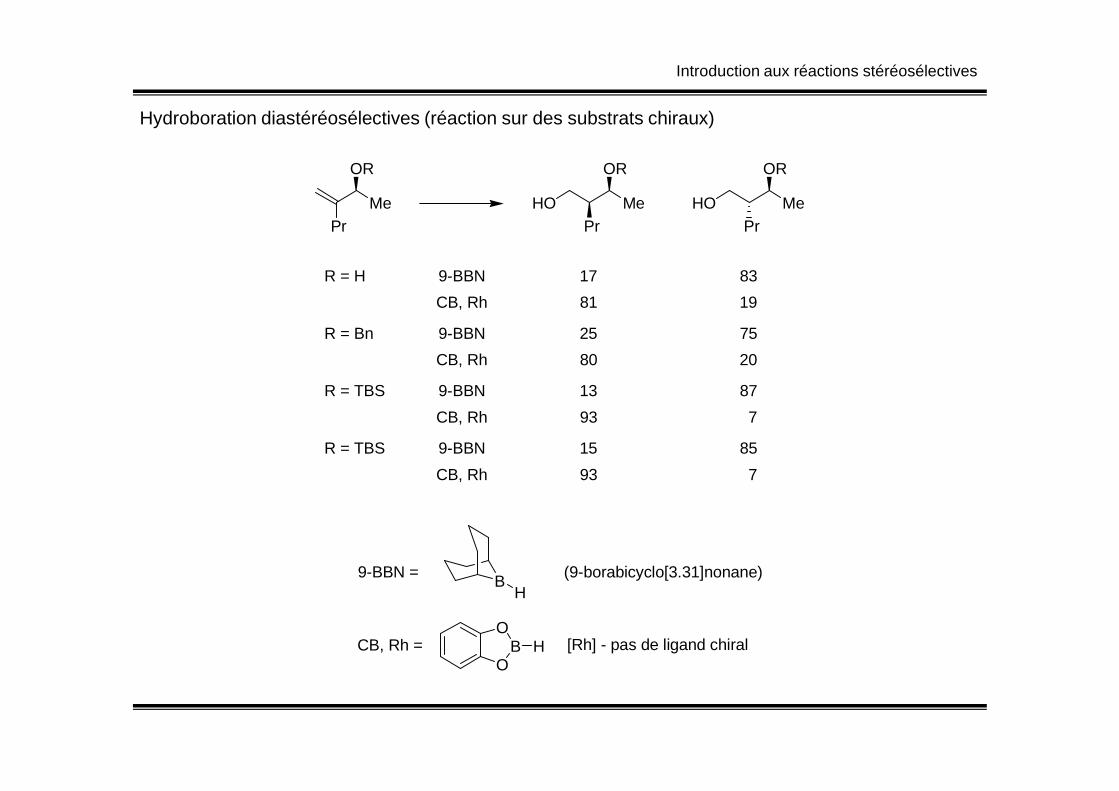
Introduction aux réactions stéréosélectives
Hydroboration diastéréosélectives (réaction sur des substrats chiraux)
PrMe
OR
PrMe
OR
HOPr
Me
OR
HO
R = H 9-BBN 17 83
CB, Rh 81 19
R = Bn 9-BBN 25 75
CB, Rh 80 20
R = TBS 9-BBN 13 87R = TBS 9-BBN 13 87
CB, Rh 93 7
R = TBS 9-BBN 15 85
CB, Rh 93 7
9-BBN =
CB, Rh =O
BO
H [Rh] - pas de ligand chiral
BH
(9-borabicyclo[3.31]nonane)

Introduction aux réactions stéréosélectives
Modèles pour justifier la stéréosélection:
RL
RRMH
HH
B H
modèle stérique
EWG (OR)
REDGH
HH
B H
modèle stéréoélectronique
RL
RRMH
HH
B H
modèle stérique
EDG
REWG (OR)H
HH
B H
modèle stéréoélectronique
modèle stérique modèle stéréoélectronique
modèle stérique modèle stéréoélectronique
hydroboration catalysée hydroboration non catalysée
EWG = groupe électroattracteur EDG = groupe électrodonneur

Introduction aux réactions stéréosélectives
Réduction des cétones prochirales
R1 R2
O "H "
R1 R2
HHO
R1 R2
OHHvs
Cette partie traite uniquement les réductions de cétones prochirales par des réactifs comportant un
environnement chiral. L’addition diastéréosélectives d’hydrures (c’est à dire les réactions sur desenvironnement chiral. L’addition diastéréosélectives d’hydrures (c’est à dire les réactions sur des
cétones comportant déjà un groupe stéréogène à proximité pouvant induire la stéréosélection) a
déjà été, en partie, traitée lors de l’étude du modèle de Felkin-Anh.
Il existe plusieurs systèmes de réduction des cétones prochirales:
• agents de réduction stoechiométriques chiraux (BINAL-H et boranes).
• source de réduction stoechiométrique en présence d’un catalyseur chiral (le système CBS).
• l’hydrogénation asymétrique catalysée (le système de Noyori).
• le transfert asymétrique d’hydrure.

Introduction aux réactions stéréosélectives
Le BINAL-HLe BINAL-H est un réactif stoechiométrique de réduction des dérivés carbonylés. Il est préparé in
situ par addition de LiAlH4 sur le (R)- ou (S)-binaphtol et d’éthanol :
LiAlH4 + EtOH +OHOH
THF
(R)- ou (S)-
OO
AlOEt
HLi
(R)- ou (S)-BINAL-H
Noyori, R. et al. J. Am. Chem. Soc. 1984, 106, 6709 et 6717.
OO
AlOEt
HLi
(R)-BINAL-H
OO
AlOEt
HLi
(S)-BINAL-H
En l’absence d’éthanol, le dihydrure de binaphtolate d’alumium lithium présente un très faible
pouvoir d’induction d’asymétrie (2% ee pour la réduction de l’acétophénone). Par ailleurs il a été
montré que le BINAL-H possède une bonne énantiosélectivité uniquement pour les cétones
insaturées.

Introduction aux réactions stéréosélectives
Origine de l’énantiosélectivité avec le BINAL-H.
R
O
Ph (S)-BINAL-H(R)-BINAL-HRPh
HO H
RPh
H OH
R %ee
Me > 95
n-alkyl > 95
iPr 71
tBu 44
Me
O
R (S)-BINAL-H MeR
H OH
R %ee
Ph > 95
CH3(CH2)3CH=CH 79
CH3(CH2)3CΞC 84
H
Al
OO Li
Et
O
OH
gêne stérique
O
Al H
O
O LiO
R
Un
Et
O
Al H
O
O LiO
Un
R
Et
vs vs
répulsionélectronique
Un = chaîne insaturée ou Ar
O
Al H
O
O LiO
Et
forme la plus stable
Cas du (S)- BINAL-H
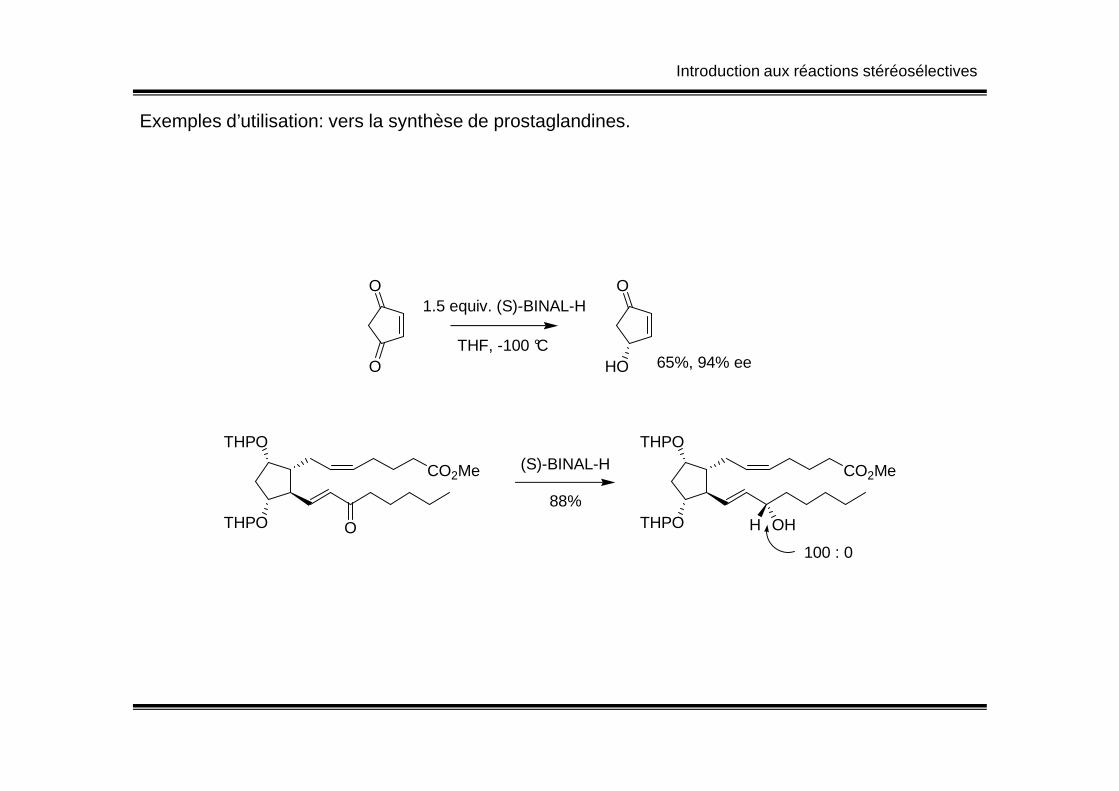
Introduction aux réactions stéréosélectives
Exemples d’utilisation: vers la synthèse de prostaglandines.
O
O
1.5 equiv. (S)-BINAL-H
THF, -100 °C
O
HO 65%, 94% ee
THPO
THPO
CO2Me
O THPO
THPO
CO2Me
H OH
(S)-BINAL-H
88%
100 : 0
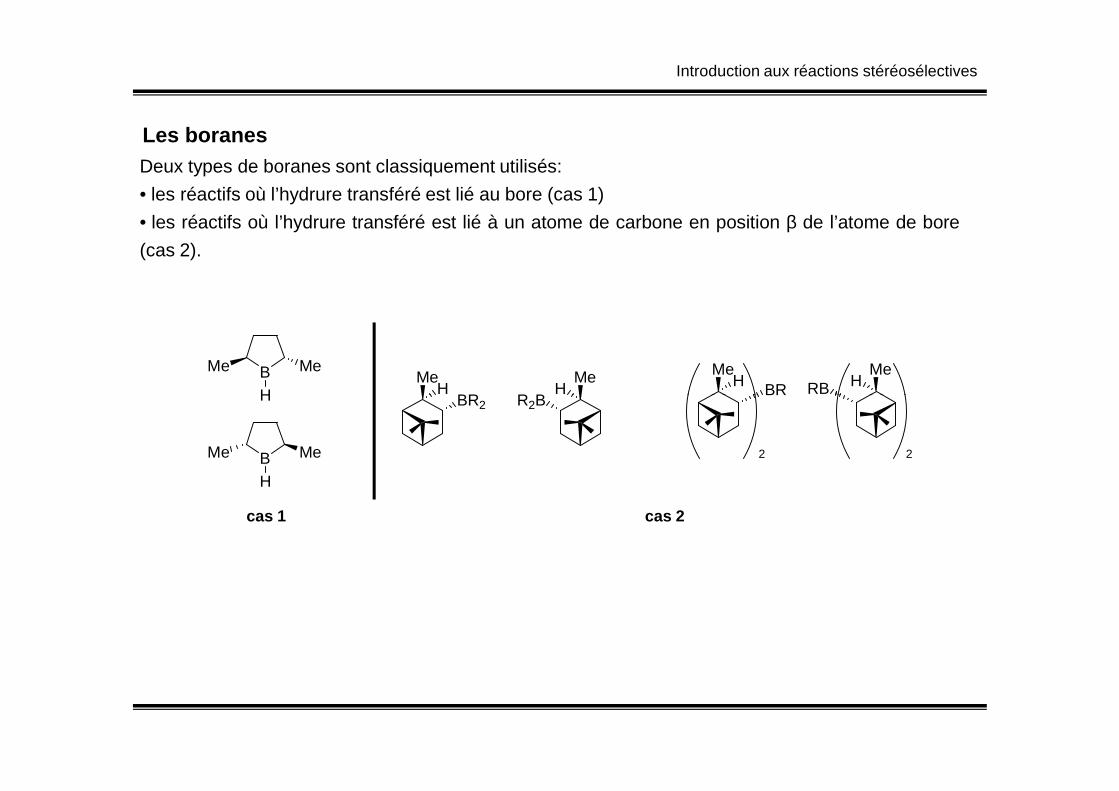
Introduction aux réactions stéréosélectives
Les boranesDeux types de boranes sont classiquement utilisés:
• les réactifs où l’hydrure transféré est lié au bore (cas 1)
• les réactifs où l’hydrure transféré est lié à un atome de carbone en position β de l’atome de bore
(cas 2).
BMe Me
HMe
BRH
MeR B
HMe
BRHMe
RB HH
BMe Me
H
BR2H
R2BH BR RB
2 2
cas 1 cas 2
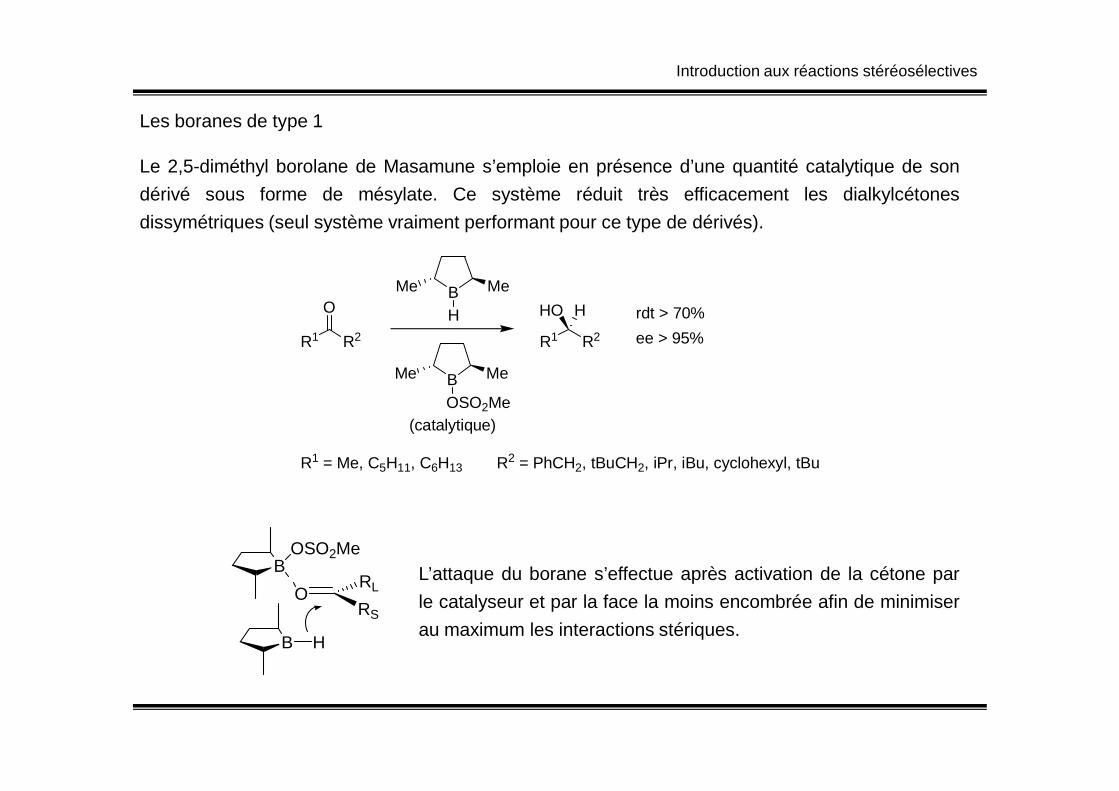
Introduction aux réactions stéréosélectives
Les boranes de type 1
Le 2,5-diméthyl borolane de Masamune s’emploie en présence d’une quantité catalytique de son
dérivé sous forme de mésylate. Ce système réduit très efficacement les dialkylcétones
dissymétriques (seul système vraiment performant pour ce type de dérivés).
R1 R2
O
R1 R2
HHOBMe Me
H
BMe Me
OSO Me
rdt > 70%
ee > 95%
OSO2Me(catalytique)
R1 = Me, C5H11, C6H13 R2 = PhCH2, tBuCH2, iPr, iBu, cyclohexyl, tBu
BOSO2Me
ORS
RL
B H
L’attaque du borane s’effectue après activation de la cétone par
le catalyseur et par la face la moins encombrée afin de minimiser
au maximum les interactions stériques.
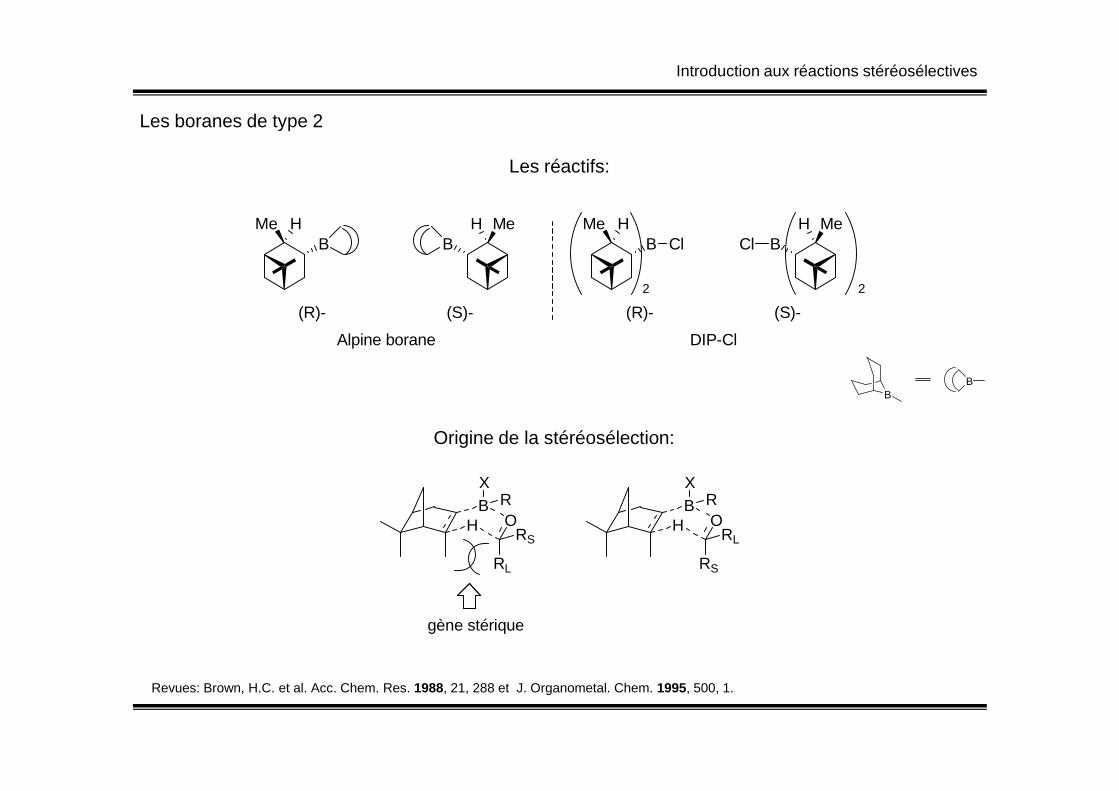
Introduction aux réactions stéréosélectives
Les boranes de type 2
Les réactifs:
BHMe
BH Me
(R)- (S)-
Alpine borane
BHMe
BH Me
(R)- (S)-
Cl Cl
DIP-Cl
2 2
BB
B
Revues: Brown, H.C. et al. Acc. Chem. Res. 1988, 21, 288 et J. Organometal. Chem. 1995, 500, 1.
HBX
RO
RL
RSH
BX
RO
RS
RL
gène stérique
Origine de la stéréosélection:
Introduction aux réactions stéréosélectives
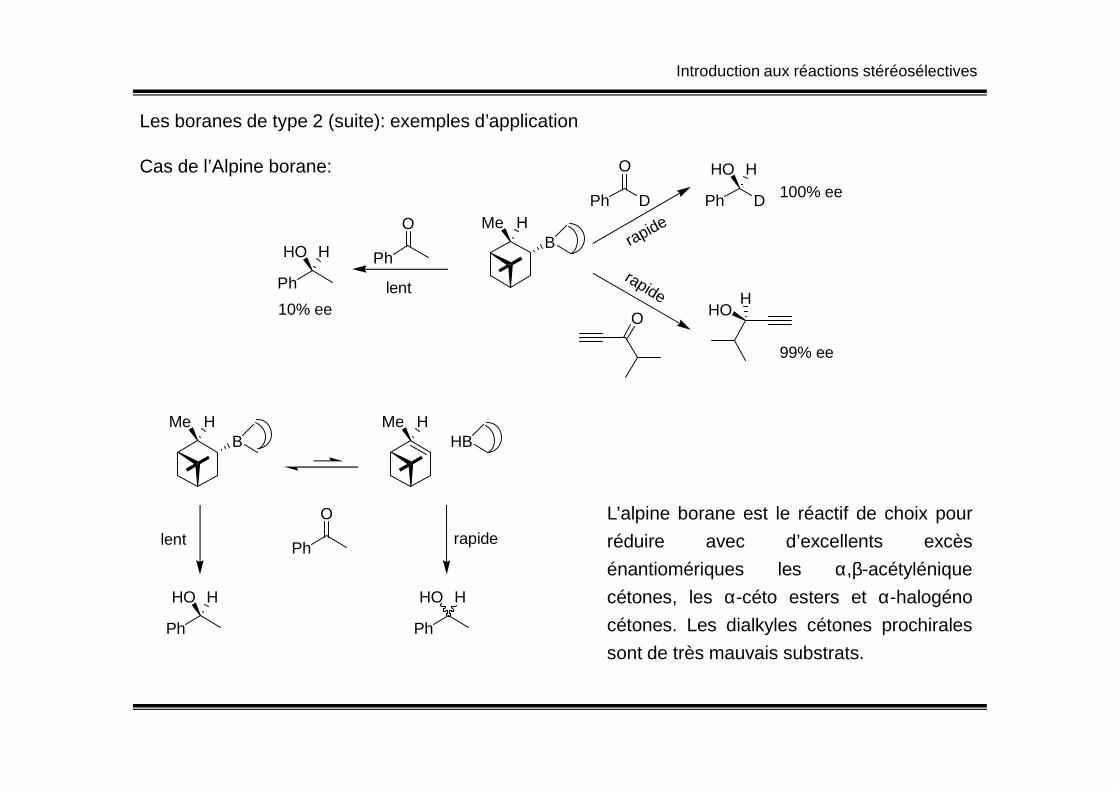
Les boranes de type 2 (suite): exemples d’application
Cas de l’Alpine borane:
BHMe
rapidePh D
O
Ph D
HHO100% ee
rapide
OH
HO
99% ee
lent
Ph
O
Ph
HHO
10% ee
L’alpine borane est le réactif de choix pour
réduire avec d’excellents excès
énantiomériques les α,β-acétylénique
cétones, les α-céto esters et α-halogéno
cétones. Les dialkyles cétones prochirales
sont de très mauvais substrats.
BHMe
HBHMe
lent rapidePh
O
Ph
HHO
Ph
HHO
Introduction aux réactions stéréosélectives
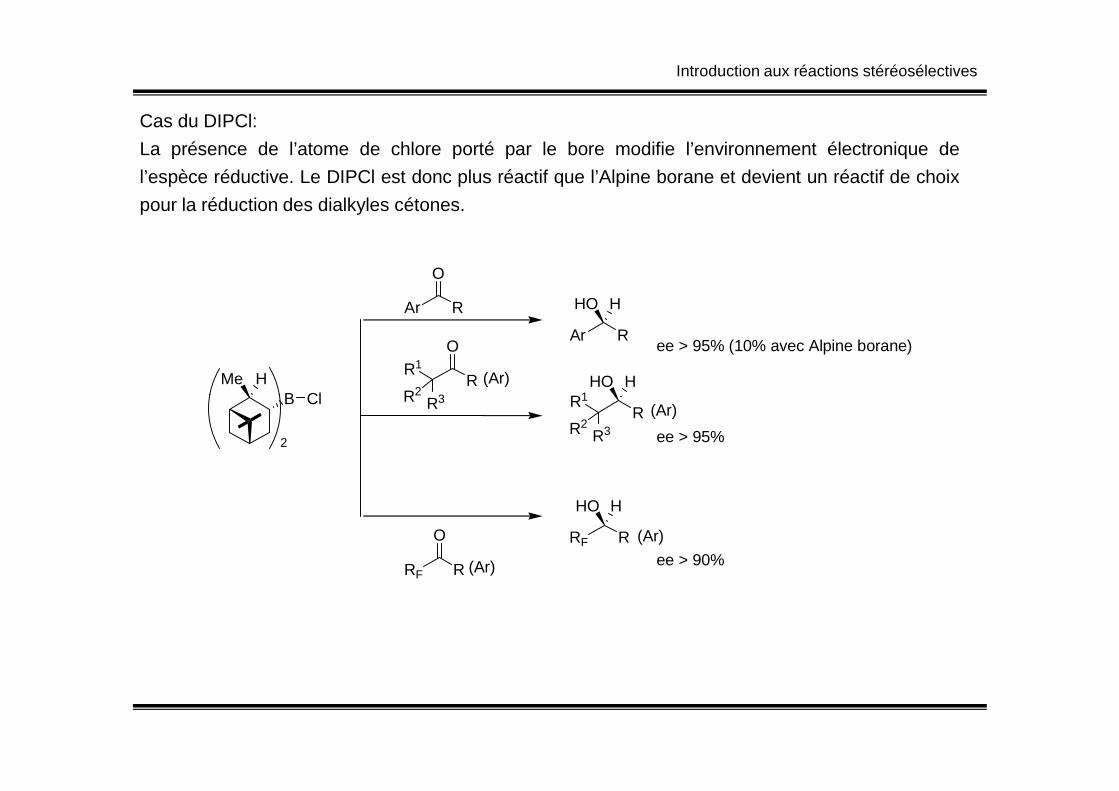
Cas du DIPCl:
La présence de l’atome de chlore porté par le bore modifie l’environnement électronique de
l’espèce réductive. Le DIPCl est donc plus réactif que l’Alpine borane et devient un réactif de choix
pour la réduction des dialkyles cétones.
BHMe
Cl
Ar R
O
Ar R
HHO
ee > 95% (10% avec Alpine borane)O
RR3
R1
R2R1
HHO(Ar)B Cl
2
RF R
O RF R
HHO
(Ar)
(Ar)
ee > 90%
R3R2
RR3
R1
R2ee > 95%
(Ar)
Introduction aux réactions stéréosélectives
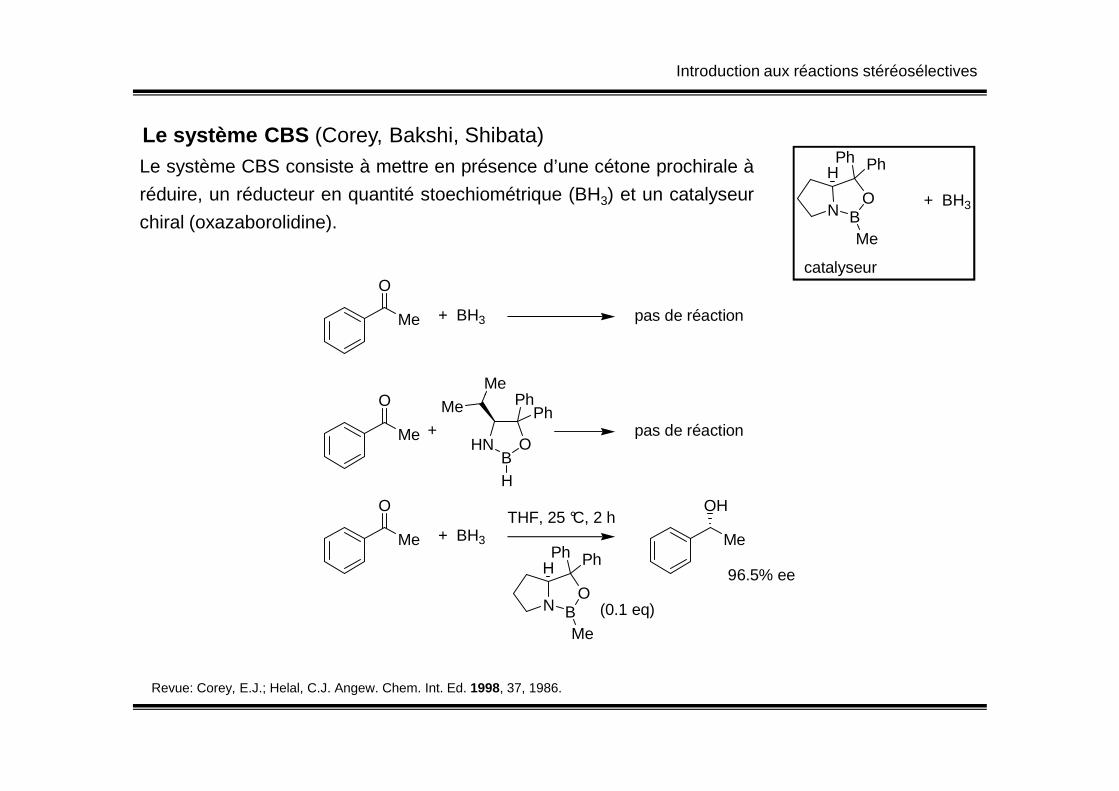
Le système CBS (Corey, Bakshi, Shibata)Le système CBS consiste à mettre en présence d’une cétone prochirale à
réduire, un réducteur en quantité stoechiométrique (BH3) et un catalyseur
chiral (oxazaborolidine).N B
O
Me
PhPhH
catalyseur
+ BH3
Me
O
+ BH3 pas de réaction
MePhO
Revue: Corey, E.J.; Helal, C.J. Angew. Chem. Int. Ed. 1998, 37, 1986.
HNB
O
Me
H
PhPh
Me
O
+ pas de réaction
Me
O
+ BH3
N BO
Me
PhPhH
(0.1 eq)
Me
OH
96.5% ee
THF, 25 °C, 2 h

Introduction aux réactions stéréosélectives
Origine de la stéréosélection
N
OB
Me
Ph
Ph
MeO
H2BH
favorable
Me
OH
OB
Ph H2BH
Me
OH
MeO
NB
MePh
défavorable
MeMe
Scope du réactif
Ar
O
R Ar1
O
Ar2
O
RR1
O
RR1
ee > 85% en général
R1 R2
Oee < 85%, sauf pour les cas R1 très encombrant stériquement (comme tBu, iPr)

Introduction aux réactions stéréosélectives
Exemple: synthèse du Prozac
O
Cl
N BO
Me
PhPhH
0.1 eq
0.6 eq BH3.THF
THF, 0 °C
Cl
HHO
quant.
94% ee
NH2Me
HO
F3C
Cl
(R)- fluotexine
Corey, E.J. et al. Tetrahedron Lett. 1989, 30, 5207.