introduction, principle, instrumentation and applications of sds-page
TRANSCRIPT

INTRODUCTION, PRINCIPLE, INSTRUMENTATION & APPLICATIONS OF SDS-PAGE
MOHAMMED MOBEEN

CONTENTS• INTRODUCTION• PRINCIPLE• TYPES OF ELECTROPHORESIS• GEL ELECTROPHORESIS• 1-D GEL ELECTROPHORESIS• POLYACRYLAMIDE GEL• STACKING AND RESOLVING GELS• 2-D GEL ELECTROPHORESIS• AN OVERVIEW OF SDS-PAGE• ADVANTAGES & DISADVANTAGES• APPLICATIONS OF SDS-PAGE• CONCLUSIONS• REFERENCES

INTRODUCTION• Definition:
Electrophoresis is the process of moving charged molecules in solution by applying an electric field across the mixture. Because molecules in an electric field move with a speed dependent on their charge, shape, and size, electrophoresis has been extensively developed for molecular separations.
• Purpose for carrying out electrophoresis:1. To determine the number, amount and mobility of components in a given sample
or to separate them.2. To obtain information about the electrical double layers surrounding the
particles.3. Determination of molecular weight of proteins and DNA sequencing.

PRINCIPLE• Any charged ion or molecule migrates when placed in an electric field. The rate of
migration depend upon its net charge, size, shape and the applied electric current.• It can be represented by following equation:
where, v = velocity of migration of the moleculeE = electric field (in volts/cm)q = net electric charge on the molecule.F = frictional co-efficient (depends upon the mass and shape of the molecule).
• The movement of charged particle in an electric field is expressed in terms of electrophoretic mobility denoted by ‘µ’, where or
• For molecules with similar conformation, varies with size but not with shape. Thus electrophoretic mobility (µ) of a molecule is directly proportional to the charge density (charge/mass ratio).

TYPES OF ELECTROPHORESIS
ELECTROPHORESIS
FREE ELECTROPHORESIS
MICRO ELECTROPHO
RESISMOVING
BOUNDARY
ZONEELECTROPHORESIS
PAPER ELECTROPHOR
ESIS
CELLULOSE
ACETATE
ELECTROPHOR
ESIS
GELELECTROPHOR
ESIS

GEL ELECTROPHORESIS• It is a technique used for the separation of Deoxyribonucleic acid, Ribonucleic acid or
protein molecules according to their size and electrical charge using an electric current applied to a gel matrix.
• What is a gel? Gel is a cross linked polymer whose composition and porosity is chosen based on
the specific weight and porosity of the target molecules.• Types of gels used: Polyacrylamide gel, Agarose gel.• Types of gel electrophoresis:
Gel electrophoresis
One dimensional (1-D) gel electrophoresis
SDS-PAGE
Native PAGE
Isoelectric
focusing (IEF)
Two dimensional (2-D) gel electrophoresis
Separates proteins by isoelectric point in the first dimension and by
mass in the second dimension.

1-D GEL ELECTROPHORESIS• Procedure:
The samples are treated with SDS (sodium dodecyl sulfate), an anionic detergent which denatures the protein by breaking the disulfide bonds and gives negative charge to each protein in proportion to its mass.
Without SDS, different proteins with similar molecular weights would migrate differently due to differences in folding, as differences in folding patterns would cause some proteins to better fit through the gel matrix than others. SDS linearizes the proteins so that they may be separated strictly by molecular weight.
The SDS binds to the protein in a ratio of approximately 1.4 g SDS per 1.0 g giving an approximately uniform mass : charge ratio for most proteins, so that the distance of migration through the gel can be assumed to be directly related to only the size of the protein.
Proteins may be further treated with reducing agent, such as dithiothreitol (DTT) or TRP (Tributyl phosphine) to break any reformed disulfide bonds and then alkalated with iodoacetamide to prevent reformation of disulfide bonds.
A tracking dye like bromophenol blue may be added to the protein solution to track the progress of the protein solution through the gel during the electrophoretic run.

POLYACRYLAMIDE GEL• The gel used for SDS-PAGE is made out of acrylamide which form cross-linked
polymers of polyacrylamide. • Standard gels are typically composed of two layers, one top-most layer called the
stacking gel and a lower layer called separating or resolving gel.• The stacking layer contains a low percentage of acylamide and has low pH , while
the acrylamide concentration of the separating gel varies according to the samples to be run and has higher pH.
• The difference in pH and acrylamide concentration at the stacking and separating gel provides better resolution and sharper bands in the separating gel.
• This gel material can also withstand high voltage gradients, is amenable to various staining and destaining procedures, and can be digested to extract separated fractions or dried for autoradiography and permanent recording.

STACKING & RESOLVING GELS• Stacking gel:
The stacking gel is a large pore PAG (4%T). This gel is prepared with Tris/HCl buffer pH 6.8 of about 2.0 pH units lower than that of electrophoresis buffer (Tris/Glycine).
This gel is cast over the resolving gel. The height of the stacking gel region is always maintained more than double the height and th volume of the sample to be applied.
• Resolving gel: The resolving gel is a small pore polyacrylamide gel (3 -30% acrylamide
monomer) typically made using a pH 8.8 Tris/HCl buffer. In the resolving gel, macromolecules separate according to their size. Resolving
gels have an optimal range of separation that is based on the percent of monomer present in the polymerization reaction.

2-D GEL ELECTROPHORESIS• Two-dimensional gel electrophoresis, abbreviated as 2-DE or 2-D electrophoresis, is a
form of gel electrophoresis commonly used to analyze proteins. Mixtures of proteins are separated by two properties in two dimensions on 2D gels.
• 2-DE was first independently introduced by O'Farrell and Klose in 1975.• 2-D electrophoresis begins with 1-D electrophoresis but then separates the
molecules by a second property in a direction 90 degrees from the first. In 1-D electrophoresis, proteins (or other molecules) are separated in one dimension, so that all the proteins/molecules will lie along a lane but that the molecules are spread out across a 2-D gel. Because it is unlikely that two molecules will be similar in two distinct properties, molecules are more effectively separated in 2-D electrophoresis than in 1-D electrophoresis.
• The two dimensions that proteins are separated into using this technique can be isoelectric point, protein complex mass in the native state, and protein mass.
• Common techniques of 2-DE: IPG-DALT IEF SDS-PAGE

AN OVERVIEW OF SDS-PAGE• SDS-PAGE is a technique used by many researchers to separate mixtures of proteins
by their mass. • Sodium dodecyl sulfate (SDS) is a detergent that breaks up the interactions between
proteins.• SDS is an anionic detergent that disrupts secondary and non-disulfide-linked tertiary
structures and additionally applies a negative charge to each protein in proportion to its mass.
Fig. 1. An overview of SDS-PAGE

AN OVERVIEW OF SDS-PAGE• PRINCIPAL COMPONENTS OF SDS-PAGE:
The components of an SDS PAGE gel electrophoresis system are the following:1. A Slab holder for vertical or horizontal gels (thin, flat sheets of many individual
lanes)2. Polyacrylamide or agarose gels (cm x cm x mm); these are poured for each
analysis3. Gel is amended with SDS to dissociate & charge proteins.4. High voltage power supply (0.1-6 kV)5. A detection technique (dye staining, fluorescence, or autoradiography to
image separated bands) SDS-PAGE stands for Sodium Dodecyl Sulfate Poly-Acrylamide Gel Electrophoresis.
Sodium-Dodecyl Sulfate, (“SDS”), is an anionic detergent. It is composed of a hydrophilic group with a net negative charge and a long hydrophobic chain with neutral charge.
• SAMPLE PREPARATION: Samples may be any material containing proteins or nucleic acids. These may be
biologically derived, for example from prokaryotic or eukaryotic cells, tissues, viruses, environmental samples, or purified proteins.

AN OVERVIEW OF SDS-PAGE In the case of solid tissues or cells, these are often first broken down mechanically
using a blender (for larger sample volumes), using a homogenizer (smaller volumes), by sonicator or by using cycling of high pressure, and a combination of biochemical and mechanical techniques – including various types of filtration and centrifugation – may be used to separate different cell compartments and organelles prior to electrophoresis. Synthetic biomolecules such as oligonucleotides may also be used as analytes.
The sample to analyze is optionally mixed with a chemical denaturant if so desired, usually SDS for proteins or urea for nucleic acids.
SDS denatures secondary and non–disulfide–linked tertiary structures, and additionally applies a negative charge to each protein in proportion to its mass. Urea breaks the hydrogen bonds between the base pairs of the nucleic acid, causing the constituent strands to separate. Heating the samples to at least 60 °C further promotes denaturation.
In addition to SDS, proteins may optionally be briefly heated to near boiling in the presence of a reducing agent, such as dithiothreitol (DTT) or 2-mercaptoethanol (beta-mercaptoethanol/BME), which further denatures the proteins by reducing disulfide linkages, thus overcoming some forms of tertiary protein folding, and breaking up quaternary protein structure (oligomeric subunits). This is known as reducing SDS-PAGE.
A tracking dye may be added to the solution. This typically has a higher electrophoretic mobility than the analytes to allow the experimenter to track the progress of the solution through the gel during the electrophoretic run.

AN OVERVIEW OF SDS-PAGE
Fig. 2. Sample preparation for SDS-PAGE electrophoresis
• PREPARATION OF ACRYLAMIDE GELS: The gels typically consist of acrylamide, bisacrylamide, the optional denaturant (SDS
or urea), and a buffer with an adjusted pH. The solution may be degassed under a vacuum to prevent the formation of air bubbles during polymerization. Alternatively, butanol may be added to the resolving gel (for proteins) after it is poured, as butanol removes bubbles and makes the surface smooth.
A source of free radicals and a stabilizer, such as ammonium persulfate and TEMED are added to initiate polymerization. The polymerization reaction creates a gel because of the added bisacrylamide, which can form cross-links between two acrylamide molecules. The ratio of bisacrylamide to acrylamide can be varied for special purposes, but is generally about 1 part in 35.

AN OVERVIEW OF SDS-PAGE The acrylamide concentration of the gel can also be varied, generally in the range
from 5% to 25%. Lower percentage gels are better for resolving very high molecular weight molecules, while much higher percentages are needed to resolve smaller proteins.
Fig. 3. Mixture of acrylamide, bisacrylamide, the optional denaturant (SDS or urea), and a buffer with an adjusted pH, followed by addition of ammonium persulfate and TEMED
to initiate polymerization and subsequent stacking.
Gels are usually polymerized between two glass plates in a gel caster, with a comb inserted at the top to create the sample wells. After the gel is polymerized the comb can be removed and the gel is ready for electrophoresis.

AN OVERVIEW OF SDS-PAGE
Fig. 4. Polymerization of acrylamide gel using a gel caster
• ELECTROPHORESIS: Various buffer systems are used in PAGE depending on the nature of the sample
and the experimental objective. The buffers used at the anode and cathode may be the same or different.
The gel is run usually for a few hours, though this depends on the voltage applied across the gel; migration occurs more quickly at higher voltages, but these results are typically less accurate than at those at lower voltages. After the set amount of time, the biomolecules have migrated different distances based on their size.
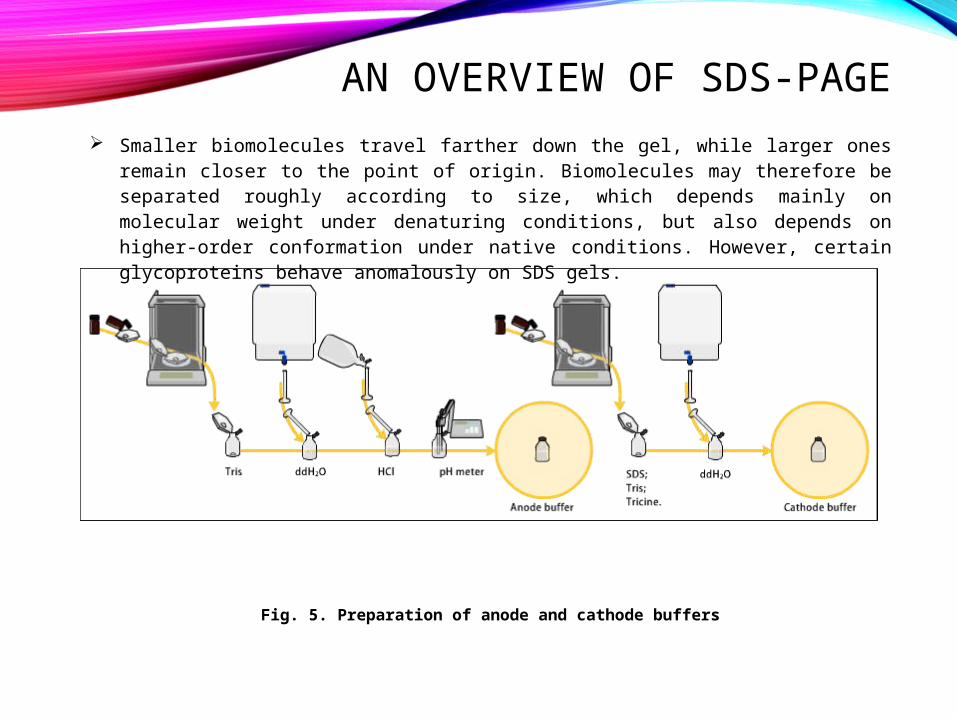
AN OVERVIEW OF SDS-PAGE Smaller biomolecules travel farther down the gel, while larger ones remain closer
to the point of origin. Biomolecules may therefore be separated roughly according to size, which depends mainly on molecular weight under denaturing conditions, but also depends on higher-order conformation under native conditions. However, certain glycoproteins behave anomalously on SDS gels.
Fig. 5. Preparation of anode and cathode buffers
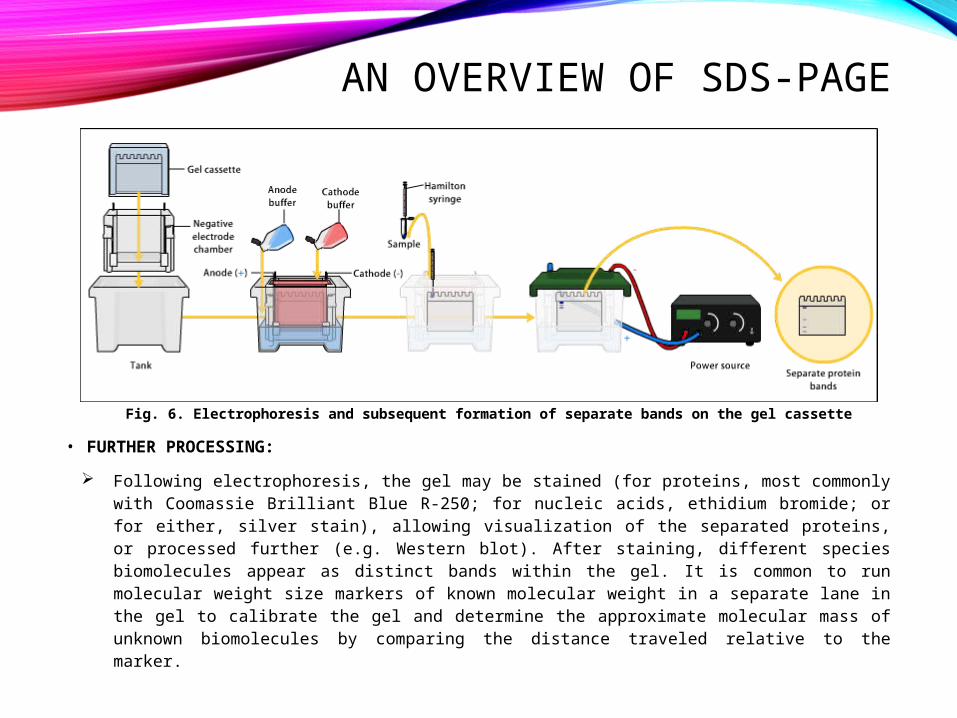
AN OVERVIEW OF SDS-PAGE
Fig. 6. Electrophoresis and subsequent formation of separate bands on the gel cassette
• FURTHER PROCESSING: Following electrophoresis, the gel may be stained (for proteins, most commonly
with Coomassie Brilliant Blue R-250; for nucleic acids, ethidium bromide; or for either, silver stain), allowing visualization of the separated proteins, or processed further (e.g. Western blot). After staining, different species biomolecules appear as distinct bands within the gel. It is common to run molecular weight size markers of known molecular weight in a separate lane in the gel to calibrate the gel and determine the approximate molecular mass of unknown biomolecules by comparing the distance traveled relative to the marker.

AN OVERVIEW OF SDS-PAGE For proteins, SDS-PAGE is usually the first choice as an assay of purity due to its
reliability and ease. The presence of SDS and the denaturing step make proteins separate, approximately based on size, but aberrant migration of some proteins may occur. Different proteins may also stain differently, which interferes with quantification by staining. PAGE may also be used as a preparative technique for the purification of proteins. For example, quantitative preparative native continuous polyacrylamide gel electrophoresis (QPNC-PAGE) is a method for separating native metalloproteins in complex biological matrices.
Fig. 7. Two SDS-PAGE-gels after a completed run

AN OVERVIEW OF SDS-PAGE• Components of SDS-PAGE:
Chemical buffer: Stabilizes the pH value to the desired value within the gel itself and in the electrophoresis buffer. The buffer should also be unreactive and not modify or react with most proteins. Different buffers may be used as cathode and anode buffers, respectively, depending on the application. Multiple pH values may be used within a single gel, for example in DISC electrophoresis. Common buffers in PAGE include Tris, Bis-Tris, or imidazole.
Counter-ion: Balance the intrinsic charge of the buffer ion and also affect the electric field strength during electrophoresis. Highly charged and mobile ions are often avoided in SDS-PAGE cathode buffers, but may be included in the gel itself, where it migrates ahead of the protein.
Acrylamide (C3H5NO): When dissolved in water, slow, spontaneous autopolymerization of acrylamide takes place, joining molecules together by head on tail fashion to form long single-chain polymers. The presence of a free radical-generating system greatly accelerates polymerization. This kind of reaction is known as Vinyl addition polymerisation. A solution of these polymer chains becomes viscous but does not form a gel, because the chains simply slide over one another. Gel formation requires linking various chains together. Acrylamide is a neurotoxin. It is also essential to store acrylamide in a cool dark and dry place to reduce autopolymerisation and hydrolysis.
Bisacrylamide (N,N′-Methylenebisacrylamide) (C7H10N2O2): Bisacrylamide is the most frequently used cross linking agent for polyacrylamide gels. Chemically it can be thought of as two acrylamide molecules coupled head to head at their non-reactive ends. Bisacrylamide can crosslink two polyacrylamide chains to one another, thereby resulting in a gel.

AN OVERVIEW OF SDS-PAGE Sodium Dodecyl Sulfate (SDS) (C12H25NaO4S): SDS is a strong detergent agent
used to denature native proteins to unfolded, individual polypeptides. Without SDS, different proteins with similar molecular weights would migrate differently due to differences in mass-charge ratio, as each protein has an isoelectric point and molecular weight particular to its primary structure. This is known as Native PAGE. Adding SDS solves this problem, as it binds to and unfolds the protein, giving a near uniform negative charge along the length of the polypeptide.
Urea (CO(NH2)2): Urea is a chaotropic agent that increases the entropy of the system by interfering with intramolecular interactions mediated by non-covalent forces such as hydrogen bonds and van der Waals forces. Macromolecular structure is dependent on the net effect of these forces, therefore it follows that an increase in chaotropic solutes denatures macromolecules,
Ammonium persulfate (APS) (N2H8S2O8): APS is a source of free radicals and is often used as an initiator for gel formation. An alternative source of free radicals is riboflavin, which generated free radicals in a photochemical reaction.
TEMED (N, N, N′, N′-tetramethylethylenediamine) (C6H16N2): TEMED stabilizes free radicals and improves polymerization. The rate of polymerisation and the properties of the resulting gel depend on the concentrations of free radicals. Increasing the amount of free radicals results in a decrease in the average polymer chain length, an increase in gel turbidity and a decrease in gel elasticity. Decreasing the amount shows the reverse effect. The lowest catalytic concentrations that allow polymerisation in a reasonable period of time should be used. APS and TEMED are typically used at approximately equimolar concentrations in the range of 1 to 10 mM.

AN OVERVIEW OF SDS-PAGE• Chemicals used for processing and visualization:
Tracking dye: As proteins and nucleic acids are mostly colorless, their progress through the gel during electrophoresis cannot be easily followed. Anionic dyes of a known electrophoretic mobility are therefore usually included in the PAGE sample buffer. A very common tracking dye is Bromophenol blue (BPB, 3',3",5',5" tetrabromophenolsulfonphthalein). This dye is coloured at alkali and neutral pH and is a small negatively charged molecule that moves towards the anode. Being a highly mobile molecule it moves ahead of most proteins. As it reaches the anodic end of the electrophoresis medium electrophoresis is stopped. It can weakly bind to some proteins and impart a blue colour. Other common tracking dyes are xylene cyanol, which has lower mobility, and Orange G, which has a higher mobility.
Loading aids: Most PAGE systems are loaded from the top into wells within the gel. To ensure that the sample sinks to the bottom of the gel, sample buffer is supplemented with additives that increase the density of the sample. These additives should be non-ionic and non-reactive towards proteins to avoid interfering with electrophoresis. Common additives are glycerol and sucrose.
Coomassie Brilliant Blue R-250 (CBB)(C45H44N3NaO7S2): CBB is the most popular protein stain. It is an anionic dye, which non-specifically binds to proteins. Proteins in the gel are fixed by acetic acid and simultaneously stained. The excess dye incorporated into the gel can be removed by destaining with the same solution without the dye. The proteins are detected as blue bands on a clear background. As SDS is also anionic, it may interfere with staining process. Therefore, large volume of staining solution is recommended, at least ten times the volume of the gel.
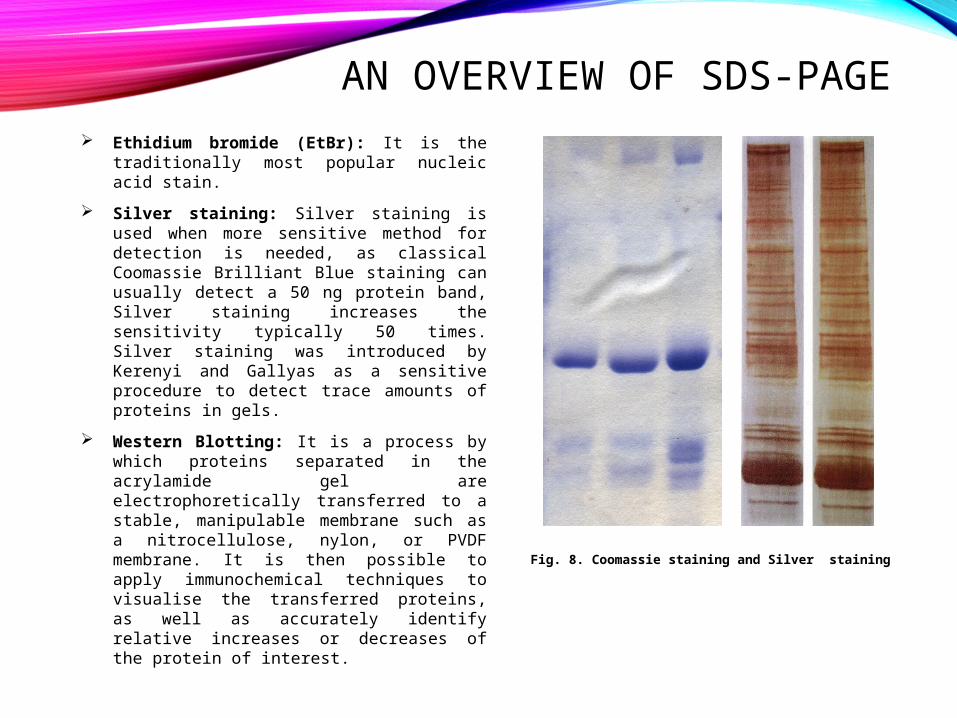
AN OVERVIEW OF SDS-PAGE Ethidium bromide (EtBr): It is the
traditionally most popular nucleic acid stain.
Silver staining: Silver staining is used when more sensitive method for detection is needed, as classical Coomassie Brilliant Blue staining can usually detect a 50 ng protein band, Silver staining increases the sensitivity typically 50 times. Silver staining was introduced by Kerenyi and Gallyas as a sensitive procedure to detect trace amounts of proteins in gels.
Western Blotting: It is a process by which proteins separated in the acrylamide gel are electrophoretically transferred to a stable, manipulable membrane such as a nitrocellulose, nylon, or PVDF membrane. It is then possible to apply immunochemical techniques to visualise the transferred proteins, as well as accurately identify relative increases or decreases of the protein of interest.
Fig. 8. Coomassie staining and Silver staining

AN OVERVIEW OF SDS-PAGE Ethidium bromide (EtBr): It is the traditionally most popular nucleic acid stain. Silver staining: Silver staining is used when more sensitive method for
detection is needed, as classical Coomassie Brilliant Blue staining can usually detect a 50 ng protein band, Silver staining increases the sensitivity typically 50 times. Silver staining was introduced by Kerenyi and Gallyas as a sensitive procedure to detect trace amounts of proteins in gels.
Western Blotting: It is a process by which proteins separated in the acrylamide gel are electrophoretically transferred to a stable, manipulable membrane such as a nitrocellulose, nylon, or PVDF membrane. It is then possible to apply immunochemical techniques to visualise the transferred proteins, as well as accurately identify relative increases or decreases of the protein of interest.

ADVANTAGES & DISADVANTAGES• Advantages:
1. Migration of the molecules is proportional to their molecular weights.2. Highly sensitive test, separates molecules that have even a 2% difference in
mass.3. Requires very small amounts of samples.4. A stable chemically cross-linked gel is used.
• Disadvantages:1. Poor band resolution due to high alkaline operating pH.2. Acrylamide gel is a potent neurotoxin chemical.3. Gel preparation is difficult and takes a long time.4. Very costly.

APPLICATIONS OF SDS-PAGESDS-PAGE is used mainly for the following purposes:1. Measuring molecular weight.2. Peptide mapping.3. Estimation of protein size.4. Determination of protein subunits or aggregation structures.5. Estimation of protein purity.6. Protein quantitation.7. Monitoring protein integrity.8. Comparison of the polypeptide composition of different samples.9. Analysis of the number and size of polypeptide subunits.10. Post-electrophoresis applications, such as Western blotting.11. Staining of Proteins in Gels with Coomassie G-250 without Organic Solvent and Acetic
Acid.12. Pouring and Running a Protein Gel by reusing Commercial Cassettes.13. Selective Labelling of Cell-surface Proteins using CyDye DIGE Fluor Minimal Dyes.14. Detection of Protein Ubiquitination.15. SDS-PAGE/Immunoblot Detection of Aβ Multimers in Human Cortical Tissue Homogenates
using Antigen-Epitope Retrieval.

APPLICATIONS OF SDS-PAGE• Measuring Molecular Weight with SDS-PAGE:
The mobility (Rf) of a molecule in gel electrophoresis is determined by its free solution mobility, Y0 (= mobility in a gel of zero %) and the sieving action of the gel matrix. In denaturing protein electrophoresis, the addition of SDS to the electrophoresis buffer uniformly coats the proteins with negative charges, equalizing the charge to mass ratio for all proteins, thus making Y0 he same for all species. In this case, relative mobilities are determined solely by the sieving action of the gel. This sieving action is proportional to the molecular weight (MW) of the particular protein. Theoretical treatments suggest that the logarithm of R f should vary with MW, but most users use an empirical plot of the logarithm of MW vs Rf for several standards of known molecular weights to determine the molecular weights of unknowns.
In practice the proportionality of log(MW) vs R f holds true for most proteins, provided they are fully denatured, and provided the gel percentage has been chosen to match the molecular weight range of the sample. In fact, the actual plot of log(MW) vs Rf is sigmoidal, because at high MW, the sieving affect of the matrix is so large that molecules are unable to penetrate the gel, while at low MW, the sieving effect is negligible, and proteins migrate almost at their free mobility, which in SDS is independent of MW.
Given an appropriate selection of gel % and a protein which displays near-ideal behavior, molecular weights can be determined to within 5 - 10%. Molecular weights of non-ideal proteins can be determined by the use of Ferguson Plots, a technique which employs native protein electrophoresis.

APPLICATIONS OF SDS-PAGE
Fig. 9. A graph of logMW vs. Rf. Although it is sigmoidal, it is nearly linear for a range of molecular weights depending on the percentage monomer of the gel.
Determining Molecular Weight with Gradient SDS Gels: If a gradient of acrylamide concentration is introduced into SDS PAGE, larger ranges of proteins may be analyzed on the same gels, with greater resolution. The complexity of the relationship between migration and molecular weight is dependant upon the shape of the gradient. The overall equation is of the form log(MW) α log(P), where P is the concentration of acrylamide at the band position. A graph of log(MW) vs log(P) is linear, and allows the determination of MW's from a set of standard protein positions. For linear gradient gels, the percentage of acrylamide is proportional to the position in the gel, so log(MW) will be proportional to log(band position). Therefore, a graph of log(MW) vs log(R f) for a set of standards will be linear, and Rf values for unknowns can then be converted to MW values. On a 3 - 30% gradient gel, a range of proteins which differ in MW by up to 100 fold can be resolved and MW's determined.

APPLICATIONS OF SDS-PAGE• Peptide Mapping:
Peptide mapping involves controlled cleavage of a pure protein with small amounts of a pure protease to generate peptides of characteristic, reproducible sizes. These peptides can be separated on PAGE to produce a "fingerprint" characteristic of the protein. Peptide mapping can map cleavage sites in an unknown protein, or it can identify an unknown protein based upon its fingerprint identity with a previously tested sample.
The polyacrylamide gel used can be either denaturing or non-denaturing, but SDS PAGE is most often used because it gives molecular weight information about the peptides produced. Small amounts of protease are used, so that minor variations in time and temperature of incubations will not overly perturb the results.
Proteins for peptide mapping can be taken from bands sliced out of electrophoresis gels, or purified by standard means. Protocols are provided for the mapping of a pure protein and a protein in an acrylamide gel.
Analytical gels for peptide mapping: The choice of gel system for the analysis of peptide mapping is dictated by the anticipated results. If a wide range of peptide sizes is anticipated, a< gradient gel may be required. For peptides over 7kd, standard Tris Glycine SDS PAGE gels will give superior results. Small peptides will require strongly denaturing fixatives to avoid loss of signal during staining. For extremely small peptides, analysis on native PAGE gels may be superior. In native protein PAGE, separation is based partly on charge to mass ratio. This can enable the resolution of peptides that would run too close to the SDS/dye front in SDS PAGE.

CONCLUSIONS• SDS-PAGE is a major tool that has wide applications apart from analytical sciences.• SDS-PAGE is a technique widely used in biochemistry, forensics, genetics, molecular
biology and biotechnology to separate biological macromolecules, usually proteins or nucleic acids, according to their electrophoretic mobility.
• This is an advancement in gel electrophoresis and the most widely used 1-D gel electrophoresis method.
• With the help of this, proteins can be separated from a mixture and individual proteins could be determined.

REFERENCES1. O'Farrell, PH (1975); High resolution two-dimensional electrophoresis of proteins; J.
Biol. Chem.; 250 (10): 4007–21.2. Mikkelsen, Susan; Cortón, Eduardo (2004); Bioanalytical Chemistry; John Wiley & Sons,
Inc.; p. 224.3. Shapiro AL, Viñuela E, Maizel JV Jr. (September 1967); Molecular weight estimation of
polypeptide chains by electrophoresis in SDS-polyacrylamide gels; Biochem Biophys Res Commun.; 28 (5): 815–820.
4. Weber K, Osborn M (August 1969); The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis; J Biol Chem.; 244 (16): 4406–4412.
5. Laemmli UK (August 1970); Cleavage of structural proteins during the assembly of the head of bacteriophage T4; Nature 227 (5259): 680–685.
6. Schägger H, von Jagow G (Nov 1987); Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa; Anal Biochem.; 166 (2): 368–379.
7. Davis BJ, Ornstein L (1959); A new high resolution electrophoresis method; The Society for the Study of Blood at the New York Academy of Medicine.
8. Raymond S, Weintraub L. (1959); Acrylamide gel as a supporting medium for zone electrophoresis; Science 130 (3377): 711.
9. Rüchel R, Steere RL, Erbe EF (1978); Transmission-electron microscopic observations of freeze-etched polyacrylamide gels; J Chromatogr.; 166 (2): 563–575.

REFERENCES10. Golgi C (1873); Sulla struttura della sostanza grigia del cervello; Gazzetta Medica
Italiana (Lombardia); 33: 244–246.11. Kerenyi L, Gallyas F (1973); Über Probleme der quantitiven Auswertung der mit
physikalischer Entwicklung versilberten Agarelektrophoretogramme; Clin. Chim. Acta; 47 (3): 425–436.
12. Switzer RC 3rd, Merril CR, Shifrin S (Sep 1979); A highly sensitive silver stain for detecting proteins and peptides in polyacrylamide gels; Anal Biochem.’ 98 (1): 231–237.
13. Hempelmann E, Schulze M, Götze O (1984); Free SH-groups are important for the polychromatic staining of proteins with silver nitrat; Neuhof V (ed)Electrophoresis '84; Verlag Chemie Weinheim; 1984: 328–330.
14. Grant G (Oct 2007); How the 1906 Nobel Prize in Physiology or Medicine was shared between Golgi and Cajal; Brain Res Rev; 55 (2): 490–498.

THANK YOU