issn 1311-8641 Българска Неврология bulgarian...
TRANSCRIPT

Българска НеврологияBulgarian Neurology
Сдружение "Българско дружество по неврология"Official Journal of The Bulgarian Society of Neurology
ТОМ 11 / БРОЙ 1МАЙ, 2011
VOLUME 11 / NUMBER 1MAY, 2011
СЪДЪРЖАНИЕ
ОБЗОРИ
ИДИОПАТИЧНИ ГЕНЕРАЛИЗИРАНИ ЕПИЛЕПСИИ-ОСНОВНИ ГЕНЕТИЧНИ ХАРАКТЕРИСТИКИС.Желязкова , И.Търнев . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
КОМПРЕСИОННИ МОНОНЕВРОПАТИИ В ГОРНИ КРАЙНИЦИ: КРИТЕРИИ ЗА ДИАГНОЗА И ЛЕЧЕНИЕД. Богданова, И. Нисимов, И. Миланов . . . . . . . . . . . . . . . 7
ДИАГНОСТИЦИРАНЕ И ЛЕЧЕНИЕ НА ПАЦИЕНТИ С ТРАНЗИТОРНИ ИСХЕМИЧНИ АТАКИД. Масларов . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
СЪВРЕМЕННА КОНЦЕПЦИЯ ЗА ХРОНИЧНО ЛЕЧЕНИЕ ПРИ ПАРКИНСОНОВА БОЛЕСТД. Богданова, Н. Семерджиева, И. Миланов . . . . . . . . . . 22
LACOSAMIDE (VIMPAT) - ЕДНА НОВА ВЪЗМОЖНОСТ ЗА УСПЕШНО ЛЕЧЕНИЕ НА ПАРЦИАЛНИ ЕПИЛЕПСИИМ. Рашева . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
ОРИГИНАЛНИ СТАТИИ
ТРОМБОЛИЗА ПРИ ИСХЕМИЧЕН МОЗЪЧЕН ИНСУЛТ – СИГУРНОСТ И ЕФЕКТИВНОСТ. ОПИТЪТ НА КЛИНИКАТА ПО НЕВРОЛОГИЯ ПРИ УМБАЛ “ЦАРИЦА ЙОАННА - ИСУЛ” СОФИЯЕ. Василева, М. Клисурски, М. Миланова, Е. Ваврек, Н. Симеонов, Г. Благоев, Т. Стефанов, М. Радева, Я. Христов, И. Стайков, Ф. Алексиев, С. Исаков, Д. Цолова, Н. Тотева, М. Минчева, Н. Колева, П. Илиева, М. Даскалов, П. Стаменова . . . . . . . . . . . . . . 33
ИНТРАВЕНОЗНО ПРИЛАГАНЕ НА LEVETIRACETAM (KEPPRA) ПРИ ЕПИЛЕПТИЧЕН СТАТУСЗ.Захариев, Е. Витева, А. Джуркова . . . . . . . . . . . . . . . . . 37
ЕПИДЕМИОЛОГИЧНИ ПРОУЧВАНИЯ НА ЕПИЛЕПСИЯТА Пл. Антимов, И. Търнев . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
CONTENTS
REVIEWS
IDIOPATHIC GENERALIZED EPILEPSIES – BASIC GENETIC CHARACTERISTICSS. Zhelyazkova. I. Tournev. . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
ENTRAPMENT NEUROPATHIES OF UPPER EXTREMITIES: DIAGNOSIS AND TREATMENTD. Bogdanova, I. Nisimov, I. Milanov . . . . . . . . . . . . . . . . . . . 7
DIAGNOSTICS AND TREATMENT OF PATIENTS WITH TRANSIENT ISCHEMIC ATTACKSD. Maslarov . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
NEW CONCEPT FOR CHRONIC TREATMENT IN PARKINSON’S DISEASED.Bogdanova, N. Semerdjieva, I. Milanov . . . . . . . . . . . . . . 22
LACOSAMIDE (VIMPAT) – A NEW POSSIBILITY FOR SUCCESSFUL TREATMENT OF PARTIAL EPILEPSIESM. Rascheva. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
ORIGINAL PAPERS
THROMBOLYSIS IN ACUTE ISCHEMIC STROKE – SAFETY AND EFFICACY. THE EXPERIENCE OF THE CLINIC OF NEUROLOGY AT THE UNIVERSITY HOSPITAL “QUEEN GIOVANNA – ISUL”, SOFIAE. Vassileva, М. Klissurski, М. Milanova, Е. Vavrek, N. Simeonov, G. Blagoev, Т. Stefanov, М. Radeva, J. Hristov, I. Staikov, F. Alexiev, S. Isakov, D. Tzolova, N. Toteva, М. Mincheva, N. Koleva, P. Ilieva, М. Daskalov, P. Stamenova. . . . . . . . . . . . . . . . . . . 33
INTRAVENOUS ADMINISTRATION OF LEVETIRACETAM (KEPPRA) IN STATUS EPILEPTICUSZ. Zahariev, E. Viteva, A. Djurkova . . . . . . . . . . . . . . . . . . . . 37
EPIDEMIOLOGICAL STUDIES OF THE EPILEPSYPl. Antimov, I. Tournev . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
ISSN 1311-8641
(1,1) -1- BG Neurology_Cover_01_2011.indd 12.5.2011 ã., 07:40:52(1,1) -1- BG Neurology_Cover_01_2011.indd 12.5.2011 ã., 07:40:52
Българска НеврологияBulgarian Neurology
Сдружение "Българско дружество по неврология"Official Journal of The Bulgarian Society of Neurology
РЕДАКЦИОННА КОЛЕГИЯ
Алексиев А. София Балдаранов Д. София Божинов Ст. Плевен Божинова В София Василева Е. София Василева Т. Пловдив Ганева Г. София Георгиев Д. София Герасимов Б. София Гозманов Г. Пловдив Григорова О. София Делева Н. Варна Захариев З. Пловдив Колев О. СофияКолев П. СофияМанчев И. Ст.Загора Масларов Д. София Миланов И. София Миланова М. София Минчев Д. Варна Петров И. София Петрова Ю. София Райчев И. София Райчева М. СофияСтaйков И. СофияСтаменов Б. Плевен Титянова Е. София Трайков Л. София Търнев И. София Хавезова Л. Варна Хараланов Л. София Чалъкова Н. Пловдив Чернинкова С. София Шотеков П. София Янчева С. София
EDITORS
Alexiev A. Sofia Baldaranov D. SofiaBojinov St. Pleven Bojinoba V. SofiaVassileva E. SofiaVassileva T. PlovdivGaneva G. SofiaGeorgiev D. SofiaGerassimov B. SofiaGozmanov G. PlovdivGrigorova O. SofiaDeleva N. Varna Zahariev Z. PlovdivKolev O. SofiaKolev P. SofiaManchev I. St. ZagoraMaslarov D. SofiaMilanov I. SofiaMilanova M. SofiaMinchev D. Varna Petrov I. SofiaPetrova U. SofiaRaychev I. SofiaRaycheva M. SofiaStaikov I. SofiaStamenov B. PlevenTitianova E. SofiaTraykov L. SofiaTarnev I. SofiaHavezova L. Varna Haralanov L. SofiaChalakova N. PlovdivCharninkova S. SofiaShotekov P. SofiaYancheva S. Sofia
Publ
ishe
r Co
lor
Stud
io®
ГЛАВНИ РЕДАКТОРИ: И. Миланов, П. Стаменова, И. Велчева СЕКРЕТАР:М. Даскалов
EDITOR IN CHIEF:I. Milanov, P. Stamenova, I. Velcheva
SECRETARY:M. Daskalov

УКАЗАНИЕ ЗА АВТОРИТЕ
Българска Неврология е официален орган на Сдружение „Българско дружество по неврология“ и публикува статии от всички области на неврологията. Списанието съдържа следните рубрики:
● Редакционна статия с текст до3 страници. Възлага се от Редколегията.
● Оригинални статии - до 8 страници, включително табли-ци, фигури, книгопис. Към тези статии се изисква резюме на бъл-гарски и английски език общо до 40 реда, отпечатани на отделни страници.
● Резюмето трябва да съдържа заглавие, имената на автори-те, информация за целта и обекта на проучването, методиките, получените резултати. Посочват се до 6 ключови думи.
● Оригиналните статии включват кратък увод, контин-гент, методи, резултати, обсъждане и книгопис.
● Заглавната страница съдържа пълно и съкратено заглавие, имената на авторите с инициалите им, техните академични сте-пени и месторабота, адрес за кореспонденция с телефон, факс и е-mail.
● Кратки научни съобщения до 3 страници ● Обзорни статии до 10 страници ● Информации и рецензии на книги . Съдържа информация за
конгреси и конференции, нови книги, актуална лекарствена инфор-мация, предстоящи събития.
● Страници на читателя. Поместват се писма до редакто-ра, в които се коментират публикувани материали, кратки описа-ния на случаи и дискусии по актуални проблеми .
● Статиите трябва да бъдат представени на CD/USB, запи-сани на редакторска програма Word 6/Windows 96 или Windows Microsoft Word 97/2000XP, с един екземпляр разпечатка.
● Таблиците се представят на отделен лист, номерирани, като в текста се отбележи мястото им. Таблиците да имат крат-ко заглавие.
● Илюстрациите (фигури, диаграми, формули) трябва да са готови за непосредствено полиграфично възпроизвеждане. Тексто-вете под фигурите се представят на отделен лист
● Книгописът да се отпечата на отделен лист. Авторите да се подреждат по азбучен ред, като в началото се изброяват източ-ниците на кирилица, а след тях на латиница. Заглавията да се пред-ставят изцяло. В текста цитираните автори да се представят с пореден номер от книгописа. Данните в книгописа се представят по следния начин:
● Статия от списание : Автор /и/. Заглавие на статията. Заглавие на списанието / съкратено по Index medicus/, том, годи-на на издаване, номер на книжката, страница / от-до/. Пример: Andersen, G., Vestergaard, K., Lauritzen, L. Effective treatment of poststroke depression with the selective serotonin reuptake inhibitor citalopram. Stroke, 25, 1994, 6, 1099-1104.
● Книга: Автор /и/ . Заглавие. Подзаглавни данни. Местоиз-даване, издатество, година на издаване, страница /от-до/.Пример : Calligaro,K., DeLaurentis, D., Baker, W. Management of Extracranial Cerebrovascular Disease.Philadelphia-New York, Lippincott - Raven Publishers, 1997, 217.
● Публикации от сборник : Автор /и/. Заглавие: - В: Заглавие на сборника. Издател /и/. Местоиздаване, година на издаване, стра-ница / от-до/. Пример : Binnie, C., Jeavsons , .M. Photosensitive epilep-sies. In : Epileptic syndromes in infancy, childhood and adolescence , eds. J.Roger, M. Bureau, Ch.Dravet, F.E. Dreifuss, A.Perret , P.Wolf. London, John Libbey & Company, Ltd, 1992, 299-305.
● Авторите подписват декларация, че предоставения мате-риал не е предложен за печат на друго място.
● Предоставените материали и описаните в тях изследвания трябва да отговарят на етичните стандарти
● При съавторство утвърдената за печат статия трябва да бъде подписана от всички автори.
Ръкописите в два екземпляра (или един екземпляр и CD/USB) изпращайте на адрес:
София 1504, Ул. 4 Бяло море " № 8МБАЛ " Царица Йоанна" - ЕАД Клиника по неврология Проф. П.Стаменова
INSTRUCTION FOR AUTHORS
Bulgarian Neurology is the official journal of the Bulgarian Society of Neurology. It will consider for publication papers in neurology and related areas in the following categories:
● Editorials, consisting of up to 3 pages, when approved by the Editorial Board
● Original papers - up to 8 pages, including tables, figures and ref-erences. An abstract in Bulgarian and English up to 40 rows on a separate sheet is required. The abstract should contain title, authors, objective, background, methods, results and conclusions plus up to 6 keywords.
● The original papers include short introduction, material, methods results, discussion and references.
● The title page should carry the full title, the short running title, the authors with their initials, academic degrees, institutional affiliations, address for correspondence, with telephone, fax and e-mail
● Short communications and case reports up to 3 pages ● Review articles up to 10 pages ● Book reviews and information. It includes information for new
books, congresses and conferences, new drugs, future events in neurol-ogy
● Letters to the Editor with comments on previously published papers, short case reports and discussion on current problems
● The manuscripts should be submitted on CD/USB, using Word 6/Windows 96 or Windows Microsoft Word 97/2000XP with a printed copy.
● The tables should be presented on separate sheets with a short heading
● The illustrations /figures, diagrams, formulas/ should be ready for reproduction.
Explanatory legends should be provided on a separate sheet of paper. References should be presented in alphabetic order on a separate
sheet with all authors` names and full title of papers. In the text the authors should be indicated by the number from the reference list .
● The reference should be presented as follows: ● Journal paper: (1) author(s), (2) title, (3) journal name ( as abbrevi-
ated in Index Medicus ). (4) volume, (5) year of publication, (6) journal number, (7) inclusive pages. Example: Andersen, G., Vestergaard, K., Lauritzen, L. Effective treatment of poststroke depression with the selective serotonin reuptake inhibitor citalopram. Stroke, 25, 1994, 6, 1099-1104.
● Book: (1) author(s), (2) title, (3) city of publication, (4) publisher, (5) year of publication , (6) pages. Example: Calligaro,K., DeLaurentis, D., Baker, W. Management of Extracranial Cerebrovascular Disease. Philadelphia-New York, Lippincott - Raven Publishers, 1997, 217.
● References to books: (1) authors(s), (2) chapter title, (3) title of book, (4) editor(s), (5) city of publication, (6) publisher, (7) year of publication, (8) specific pages. Example: Binnie, C., Jeavsons, M. Photosensitive epilepsies. In: Epileptic syndromes in infancy, childhood and adolescence, eds. J.Roger, M. Bureau, Ch.Dravet, F.E. Dreifuss, A.Perret, P.Wolf. London, John Libbey & Company, Ltd, 1992, 299-305.
● The articles are considered for publication on the understanding that the presented material has not been published or is being submitted elsewhere before appearing in BULGARIAN NEUROLOGY.
● Authors should indicate that ethical approval of the study was granted and that informed consent was given.
● All authors should approve and sign the final manuscript.Manuscript with one copy (or a CD/USB and a copy) should be sent
to the following address:
Sofia 1504, Bulgaria, 8, Bialo more str.Department of Neurology University Hospital „Queene Jovanna“Prof. P. Stamenova
Българска Неврология Bulgarian Neurology

Българска Неврология Bulgarian Neurology
СЪДЪРЖАНИЕ
ОБЗОРИ
ИДИОПАТИЧНИ ГЕНЕРАЛИЗИРАНИ ЕПИЛЕПСИИ-ОСНОВНИ ГЕНЕТИЧНИ ХАРАКТЕРИСТИКИ С.Желязкова , И.Търнев . . . . . . . . . . . . . . . . . . . . . . . . . 1
КОМПРЕСИОННИ МОНОНЕВРОПАТИИ В ГОРНИ КРАЙНИЦИ: КРИТЕРИИ ЗА ДИАГНОЗА И ЛЕЧЕНИЕ Д. Богданова, И. Нисимов, И. Миланов . . . . . . . . . . . . 7
ДИАГНОСТИЦИРАНЕ И ЛЕЧЕНИЕ НА ПАЦИЕНТИ С ТРАНЗИТОРНИ ИСХЕМИЧНИ АТАКИ Д. Масларов . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
СЪВРЕМЕННА КОНЦЕПЦИЯ ЗА ХРОНИЧНО ЛЕЧЕНИЕ ПРИ ПАРКИНСОНОВА БОЛЕСТ Д. Богданова, Н. Семерджиева, И. Миланов . . . . . . . 22
LACOSAMIDE (VIMPAT) - ЕДНА НОВА ВЪЗМОЖНОСТ ЗА УСПЕШНО ЛЕЧЕНИЕ НА ПАРЦИАЛНИ ЕПИЛЕПСИИ М. Рашева . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
ОРИГИНАЛНИ СТАТИИ
ТРОМБОЛИЗА ПРИ ИСХЕМИЧЕН МОЗЪЧЕН ИНСУЛТ – СИГУРНОСТ И ЕФЕКТИВНОСТ. ОПИТЪТ НА КЛИНИКАТА ПО НЕВРОЛОГИЯ ПРИ УМБАЛ “ЦАРИЦА ЙОАННА - ИСУЛ” СОФИЯ Е. Василева, М. Клисурски, М. Миланова, Е. Ваврек, Н. Симеонов, Г. Благоев, Т. Стефанов, М. Радева, Я. Христов, И. Стайков, Ф. Алексиев, С. Исаков, Д. Цолова, Н. Тотева, М. Минчева, Н. Колева, П. Илиева, М. Даскалов, П. Стаменова . . . . . . . . . . . 33
ИНТРАВЕНОЗНО ПРИЛАГАНЕ НА LEVETIRACETAM (KEPPRA) ПРИ ЕПИЛЕПТИЧЕН СТАТУС З.Захариев, Е. Витева, А. Джуркова . . . . . . . . . . . . . . 37
ЕПИДЕМИОЛОГИЧНИ ПРОУЧВАНИЯ НА ЕПИЛЕПСИЯТА Пл. Антимов, И. Търнев . . . . . . . . . . . . . . . . . . . . . . . . 42
НЕВРОПРОТЕКТИВЕН ЕФЕКТ НА γ-АМИНОМАСЛЕНА КИСЕЛИНА В МОДЕЛ НА МОЗЪЧНА ИСХЕМИЯ IN VITRO В. Цанкова , С. Митушева . . . . . . . . . . . . . . . . . . . . . . 49
АПОЛИПОПРОТЕИН Е И НЕВРОПСИХИАТРИЧНИ СИМПТОМИ ПРИ БОЛНИ С БОЛЕСТ НА АЛЦХАЙМЕР Ш. Мехрабиан, А. Йорданова, М. Райчева, М. Петрова, Л. Трайков . . . . . . . . . . . . . . . . . . . . . . . . 54
ИНФОРМАЦИИ
ХII КОНГРЕС ПО НЕВРОЛОГИЯ С МЕЖДУНАРОДНО УЧАСТИЕ 19-21 МАЙ 2011 ХОТЕЛ САМОКОВ – БОРОВЕЦ . . . . . . . . . . . . . . . . . 59
IN MEMORIAM ПРОФ. ВЕНЦИСЛАВ БОСНЕВ (1928- 2011) . . . . . . 61
CONTENTS
REVIEWS
IDIOPATHIC GENERALIZED EPILEPSIES – BASIC GENETIC CHARACTERISTICS S. Zhelyazkova. I. Tournev. . . . . . . . . . . . . . . . . . . . . . . . . 1
ENTRAPMENT NEUROPATHIES OF UPPER EXTREMITIES: DIAGNOSIS AND TREATMENT D. Bogdanova, I. Nisimov, I. Milanov . . . . . . . . . . . . . . . . 7
DIAGNOSTICS AND TREATMENT OF PATIENTS WITH TRANSIENT ISCHEMIC ATTACKS D. Maslarov . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
NEW CONCEPT FOR CHRONIC TREATMENT IN PARKINSON’S DISEASE D.Bogdanova, N. Semerdjieva, I. Milanov . . . . . . . . . . . 22
LACOSAMIDE (VIMPAT) – A NEW POSSIBILITY FOR SUCCESSFUL TREATMENT OF PARTIAL EPILEPSIES M. Rascheva. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
ORIGINAL PAPERS
THROMBOLYSIS IN ACUTE ISCHEMIC STROKE – SAFETY AND EFFICACY. THE EXPERIENCE OF THE CLINIC OF NEUROLOGY AT THE UNIVERSITY HOSPITAL “QUEEN GIOVANNA – ISUL”, SOFIA E. Vassileva, М. Klissurski, М. Milanova, Е. Vavrek, N. Simeonov, G. Blagoev, Т. Stefanov, М. Radeva, J. Hristov, I. Staikov, F. Alexiev, S. Isakov, D. Tzolova, N. Toteva, М. Mincheva, N. Koleva, P. Ilieva, М. Daskalov, P. Stamenova. . . . . . . . . . . . . . . . 33
INTRAVENOUS ADMINISTRATION OF LEVETIRACETAM (KEPPRA) IN STATUS EPILEPTICUS Z. Zahariev, E. Viteva, A. Djurkova . . . . . . . . . . . . . . . . . 37
EPIDEMIOLOGICAL STUDIES OF THE EPILEPSY Pl. Antimov, I. Tournev . . . . . . . . . . . . . . . . . . . . . . . . . . 42
γ-AMINOBUTYRIC ACID NEUROPROTECTION IN AN IN VITRO MODEL OF RAT BRAIN ISCHEMIA V. Tzankova , S. Mitusheva . . . . . . . . . . . . . . . . . . . . . . . 49
APOLIPOPROTEIN E AND NEUROPSYCHIATRIC SYMPTOMS IN PATIENTS WITH ALZHEIMER’S DISEASE S. Mehrabian, A. Jordanova, M. Raycheva, M. Petrova, L. Traykov. . . . . . . . . . . . . . . . . . . . . . . . . . . 54
INFORMATION
XII NATIONAL CONGRESS OF NEUROLOGY WITH INTERNATIONAL PARTICIPATION 19th – 21th MAY 2011 HOTEL SAMOKOV BOROVETZ . . . . . . . . . . . . . . . . . 59
IN MEMORIAM PROFESSOR VENZISLAW BOSNEW (1928-2011) . . . 61

SUMMARYIDIOPATHIC GENERALIZED EPILEPSIES – BASIC
GENETIC CHARACTERISTICS.S.Zhelyazkova. I.Tournev
Idiopathic generalized epilepsies constitute nearly one third of all epilepsies and are genetically determined. They encompass a number of rare mendelian (monogenic epilepsies) and more common forms which are familial but manifest as complex, non-mendelian traits. In recent years, there have been major advances for the identification of the genes, mainly associated with rare monogenic syndromes, as they predominantly include genes encoding subunits of voltage- or -gated ion channels. A few mutations have also been found in the frequent classical forms of idiopathic generalized epilepsies. The aim of this review is to present the basic genetic features of the idiopatic generalized epilepsies.
KEY WORDS: idiopatic generalized epilepsies, genes, mode of inheritance.
РЕЗЮМЕ
Идиопатичните генерализирани епилепсии състав-ляват приблизително една трета от всички епилепсии и са генетично детерминирани. Към тях се отнасят малък брой редки форми, унаследяващи се по законите на Мендел (т.нар моногенни епилепсии) и по-чести форми, които също са фамилни, но показват комплексно унас-ледяване. През последните няколко години беше постиг-нат значителен напредък за идентифициране на гените свързани главно с моногенните синдроми, като това са основно гени кодиращи субединици на волтаж-зависими и лиганд-зависими йонни канали. Няколко мутации бяха открити и при честите класически форми на идиопа-тични генерализирани епилепсии. Целта на настоящия обзор е да се представят основните генетични характе-ристики на идиопатичните генерализирани епилепсии.
КЛЮЧОВИ ДУМИ: идиопатични генерализирани епи-лепсии, гени, начин на унаследяване
Идиопатичните генерализирани епилепсии (ИГЕ) съставляват приблизително около 20-30% от всички епилепсии (24) и засягат ~ 0.3% от хората в общата популация (40). Те се характеризират с комбинация от типични абсанси, миоклонични и генерализирани тонич-но- клонични пристъпи, генерализирани комплекси ост-рие-бавна вълна в електроенцефалограмата и липса на мозъчни лезии или метаболитни нарушения (35).
През 1950 година Lennox (48) и Metrakos (54) в своите изследвания първи предоставят научни доказателства за ролята на генетичните фактори в етиологията на идиопатичните генерализирани епилепсии. Те устано-вяват че, рискът за развитие на епилепсия сред родс-твеници на пациенти с епилепсия е 1-1.5 пъти по-голям в сравнение с общата популация. Тези резултати са потвърдени от изследвания при близнаци, като конкор-дантността при монозиготните близнаци е около 70%, а тази при двузиготните е много по-ниска (8,47). Според
Helbig и съавтори (35) генетичният риск за родстве-ници от първа степен на пациенти с епилепсия варира между 4% и 9%, а наследственост се установява в 80% от случаите.
Благодарение на молекулярно-генетичните изслед-вания в последните няколко години се идентифицираха генните мутации отговорни за някои по-редки моно-генни форми на идиопатични генерализирани епилепсии. Повечето открити гени кодират субединици на вол-тажно-зависими и лиганд-зависими йонни канали (61). Въпреки интензивните проучвания, гените предразпо-лагащи към най-честите идиопатични генерализирани епилепсии все още не са известни. Генетичната архи-тектура на ИГЕ най-вероятно представлява биологичен континуум, в който малки фракции се унаследяват моногенно, докато по-голяма част от ИГЕ показват олиго или полигенна предиспозиция (13).
Идиопатичните генерализирани епилепсии могат да бъдат разделени на две главни групи: класически, сред които най-чести са детската и ювенилна абсансни епи-лепсии, ювенилната миоклонична епилепсия (ЮМЕ) и такава само с ГТКП (35) и наскоро описаната генерали-зирана епилепсия с фебрилни пристъпи + (GEFS+). През последните няколко години бяха идентифицирани и други нови фамилни епилептични синдроми с преобладаващи миоклонични пристъпи в клиничната картина и разделе-ни на основата на начина на унаследяване и установените нови хромозомни локуси. Такива са бенигнената фамилна миоклонична епилепсия при възрастни и фамилната дет-ска миоклонична епилепсия. На табл. 1 са представени идентифицираните гени и генни локуси при различните форми идиопатични генерализирани епилепсии.
ГЕНЕРАЛИЗИРАНА (ГЕНЕТИЧНА) ЕПИЛЕПСИЯ С ФЕБРИЛНИ ПРИСТЪПИ + (GEFS+)
Генерализираната епилепсия с фебрилни пристъпи + (GEFS+) е фамилен епилептичен синдром с начало в детска възраст и изразена фенотипна хетерогенност както между отделните фамилии, така и между отдел-ните членове на една и съща фамилия. Описан е за първи път през 1997 година при австралийско семейство с 28 засегнати членове с различен по тежест фенотип, вариращ от фебрилни пристъпи, през генерализира-ни епилепсии до най-тежкият спектър на синдрома (миоклонично-астатична епилепсия и синдром на Dravet) (62). Унаследяването е автозомно-доминантно с непълна пенетранност. По-късно семейства с GEFS+ са описа-ни по целия свят, включително Сърбия, Тунис, Южна Америка и много азиатски държави, но при повечето от тях молекулярният дефект не е установен (60).
Най-честият GEFS+ фенотип включва: класически фебрилни пристъпи (49%) – конвулсивни
атаки, при температура над 380 С, с начало около 3 месечна възраст и край около 6 годишна възраст.
„фебрилни пристъпи „+” (24%), които представ-ляват фебрилни пристъпи продължаващи над 6
Българска Неврология Bulgarian Neurology 1Май 2011
ОбзорИДИОПАТИЧНИ ГЕНЕРАЛИЗИРАНИ ЕПИЛЕПСИИ-ОСНОВНИ
ГЕНЕТИЧНИ ХАРАКТЕРИСТИКИ
С.Желязкова1, И.Търнев1,2
1Клиника по неврология ,УМБАЛ Александровска, МУ-София2Нов български университет, Катедра по Когнитивна наука и психология

Българска Неврология Bulgarian Neurology 2
годишна възраст или такива, които се съчетават с афебрилни пристъпи
леки до тежки генерализирани епилепсии (13%)(62,67)– абсанси, миоклонични или атонични пристъпи, класическа миотонично-астатична епи-лепсия (МАЕ) и синдром на Dravet.
В последствие се описват фамилии с GEFS+, при които са налице и фокални пристъпи с или без пред-шестващи фебрилни пристъпи (1,59), като най-честия произход на тези пристъпи е от темпоралния или фрон-талния дял. Ето защо напоследък се предлага наимено-ванието GEFS+ да се замени с ”генетична епилепсия с фебрилни пристъпи +”(63).
Диагнозата се поставя при наличие на повече от един индивид от дадена фамилия, чиито епилептични пристъпи отговарят на специфичните субсиндроми или фенотипове в спектъра на GEFS+.
При пациентите с по-лека фенотипна изява, елект-роенцефалограмата обикновено е нормална. При тези с по-тежък фенотип, ЕЕГ находката зависи от вида на пристъпите. Напр. при фокални пристъпи в ЕЕГ се откриват епилептиформени разряди произхождащи от определена мозъчна област. Бързи генерализирани остри вълни могат да се видят при миоклонично-астатична епилепсия, а генерализирани остри вълни и огнищни раз-ряди – при пациенти със синдром на Dravet (20).
Магнитно-резонансното изследване на главен мозък почти винаги е нормално. В случаите с фокални прис-
тъпи, понякога се установява хипокампална склероза, включително при пациенти с доказана мутация в натри-евия канал, като при тях винаги е налице предшестваща анамнеза за фебрилни пристъпи (46).
Гените при GEFS+ са идентифицирани чрез изследване на големи фамилии с автозомно - доминантно унаследя-ване. По-късно се установява, че GEFS+ по-често има комплексно унаследяване, при което наред с влиянието на различни гени, значение за развитие на заболяването оказват и фактори на околната среда (62).
Понастоящем са идентифицирани мутации в няколко гена, които кодират различни субединици на волтажно – зависими и лиганд-зависими йонни канали.
Първата описана мутация е при Австралийско семейство с GEFS+ и скаченост с хромозома 19q13.1 и представлява точкова мутация C121W в SCN1B гена, кодиращ β1- субединицата на волтажно-зависимия натриев канал (76). Бета-1 субединицата на волтаж-но-зависимия натриев канал има важна модулираща роля за кинетиката на йонния канал и се разполага от двете страни на порообразуващата алфа-субединица. До настоящия момент са описани общо четири различни мутации в SCN1B гена – три missence (погрешносмисле-ни) и една вътрегенна делеция. Една от missencе мута-циите – описаната по-горе точкова мутация C121W се съобщава при още три други несвързани фамилии (65,4,34) и може би се дължи на мутация с ефект на родоначалника. Функционалните изследвания показват, че мутациите в SCN1B гена водят до загуба на функция на натриевия канал.
Най-често откриваните мутации при GEFS+ за тези в SCN1A гена. Той кодира порообразуващата алфа-1 субединица на натриевия канал, която се състои от четири хомоложни домена (DІ-ІV). Първите мутации в този ген са описани при две френски фамилии, картирани на 2q24 хромозома (6,55). Понастоящем са установени повече от 15 различни missence мутации, локализирани по цялата дължина на гена. Те се асоциират с пълния спектър на GEFS+ фенотипа, включващ МАЕ, Синдром на Dravet, парциални пристъпи, фебрилни гърчове, феб-рилни гърчове плюс и други типове генерализирани епи-лептични пристъпи (1,25,26). Проведените изследвания за установяване на функционалният ефект на SCN1A мутациите показват различни резултати, като някои от мутациите водят до загуба на функция на йонния канал, докато други променят или засилват тази фун-кция (49,50).
Друга група от гени асоциирани с GEFS+ са тези кодиращи субединици на GABAA рецептора – лиганд-зависим хлорен канал, който медиира бързата синаптич-на инхибиция. Мутации в гама–2 субединицата, GABRG2 на този рецептор са идентифицирани при семейства с GEFS+, а така също и при фамилии с детска абсансна епилепсия (5,75). Функционални изследвания показват, че мутациите в GABRG2 водят до нарушена функция и абнормен транспорт, поради невъзможност на рецеп-тора да достигне клетъчната повърхност. Dibbens и сътр. (18) описват различни промени в GABARD гена, кодиращ делта-субединицата на GABAA рецептора при малки фамилии с GEFS+. Тези промени водят до пони-жение на GABA медиирания йонен поток и повишена възбудимост, което показва централната роля на GABA за инхибицията.
Описаните гени и мутации се идентифицират само при 20% от големите фамилии с GEFS+, като 10% от тях са мутации в SCN1A гена (76). Това означава че все още има много главни гени, които предстоят да бъдат
Таблица 1. Идиопатични генерализирани епилепсии и идентифицирани гени и локуси.
Епилептичен синдром Локус ГенGEFS+ 19q13.1 SCN1B 2q24 SCN1A 5q31.1-33.1 GABARG2 2p24 - 1p36.3 GABRDРанна тежка миоклонична епилепсия 2q24 SCN1A (синдром на Dravet)Х-свързани инфантилни спазми Xp21 ARX Xp22 CDKL5Бенигнена фамилна миоклонична 8q24 - епилепсия при възрастниФамилна детска миоклонична 16р13 - епилепсияЮвенилна миоклонична епилепсия 6p12-11 EFHC1 15q14 - 6p21 BRD2 5q34-35 GABARA1 2q22-23 CACNB4Детска абсансна епилепсия 8q24 - 5q31.1-33.3 GABRG2 3q26 CLCN2 5q34-35 GABRA1Ювенилна миоклонична епилепсия 6p12-11 EFHC1 15q14 - 6p21 - 5q34-35 GABRA1 2q22-23 CACNB4Различни фенотипове 8q24 - 14q23 - 9q32-33 - 10q25-26 3q26 CLCN2Модифицирано по Weber et al.2008 и Deprez et al.2009.
Май 2011

Българска Неврология Bulgarian Neurology 3Май 2011
открити. Нещо повече, броят на гените допринасящи за комплексното унаследяване все още не е известен, нито какъв е ефекта на всеки отделен ген. Дали GEFS+ фенотипът е резултат от действието на пет или десет гена, как генните мутации и комбинации между тях влияят на фенотипа и по какъв начин факторите на околната среда допринасят за фенотипната хетеро-генност са въпроси на които предстои да се отговори в бъдеще. Поради тази причина, въпреки, че е възможно провеждане на молекулярно-генетично изследване, на този етап то не е информативно за това каква ще бъде тежестта на фенотипната изява при отделния паци-ент, нито за изхода на заболяването.
Прогнозата на заболяването обикновено е добра, тъй като повечето от фенотиповете на GEFS+ са бениг-нени. Изключение правят пациентите с миоклонично-астатична епилепсия и синдром на Dravet, при които наред с персистиращи епилептични пристъпи може да има вариабилен по тежест интелектуален дефицит и различни неврологични симптоми.
РАННА ТЕЖКА МИОКЛОНИЧНА ЕПИЛЕПСИЯ (SMEI, СИНДРОМ НА DRAVET)
Описана е за първи път от Charlotte Dravet през 1982 година, която идентифицира група от деца с полимор-фен епилептичен синдром с начало в ранна детска въз-раст, ранно нормално развитие и неблагоприятен ход на протичане с когнитивен регрес и зачестяване на епилеп-тичните пристъпи (21). Dravet установява, че при около 50% от случаите е налице фамилна анамнеза за епилепсия или фебрилни гърчове.
Синдромът се среща рядко. Hurst (38) посочва често-та 1/ 40 000 деца на възраст под 7 години в общата попу-лация, според Yakoub и сътр. (80) честотата е малко по-висока – 1/20 000-1/30 000. Мъжкият пол се засяга малко по-често от женския (22).
Фенотипът при ранната тежка миоклонична епилеп-сия е комплексен, като за поставяне на диагнозата зна-чение оказват различни фактори като възраст на начало на специфични епилептични пристъпи, характерния ход на психомоторно развитие и характерни ЕЕГ прояви. Диагнозата е като пъзел, при който клинициста трябва да комбинира отделните парчета за да определи дали детето има този специфичен електроклиничен синдром или не (64).
Описани са няколко различни клинични форми на този синдром:
1. Класическа форма- при която, при изходно нормал-но дете, на около 6 месечна възраст се развива хемикло-ничен или генерализиран фебрилен епилептичен статус (22). Следват повтарящи се конвулсивни епизоди с честота веднъж на 1-2 месеца. Те могат да бъдат хеми-конвулсивни и да се локализират в левите или десните крайници при различните пристъпи, което прави веро-ятността да се обуславят от едностранна хемисфери-ална лезия много малка. Пристъпите често се провоки-рат от фебрилитет и могат да бъдат продължителни. Между 1 и 4 годишна възраст се появяват и други типо-ве епилептични пристъпи, като най-честите са миок-лонични (80%), комплексни парциални пристъпи (46%) и атипични абсанси (40%). Могат да се наблюдават и атонични пристъпи. Обикновено пристъпите са резис-тентни на антиепилептични медикаменти и могат да се влошат от прием на Карбамазепин и Ламотрижин (31). Психомоторното развитие през първата година от живота обикновено е нормално, следва задръжка, може
да е налице и регрес, често на фона на повтарящи се епи-лептични статуси. С израстването на детето могат да се появят пирамидни белези и атаксия (22).
Въпреки честите пристъпи, ЕЕГ изследването в пър-вите 2 години от живота обикновено е нормално. След това могат да се установят генерализирани комплекси острие-бавна вълна, мултифокални разряди, фоточувс-твителност. МРТ изследването обикновено е нормално, рядко се откриват неспецифични промени като лека локална или генерализирана корова атрофия.
2. Гранична форма (Borderline Dravet syndrome) – терминът е въведен от японски автори за означаване на тези случаи, при които не се открива пълният спектър от симптоми на заболяването (27,43). Например все още не е напълно изяснено дали миоклоничните пристъ-пи са основен компонент от синдрома на Dravet или не. Възможно ли е лечението с Карбамазепин и Ламотрижин да е довело до екзацербация на миоклоничните пристъпи и това да е причина за високата им честота при този синдром. Други примери са пациенти с нормално инте-лектуално развитие и такива, при които ЕЕГ изслед-ването никога не е показвало генерализирани комплекси острие-бавна вълна (41,34).
3. Резистентна детска епилепсия с генерализирани тонично-клонични пристъпи (ICE-GTC) – подгрупа на граничната форма на синдрома, описана от японски и немски изследователи. Възрастта на начало на конвул-сивните атаки, които се провокират от фебрилитет е на същата възраст както при класическата форма, както и хода на развитие на синдрома. Най-важната отличителна черта обаче, е липсата на други видове епилептични пристъпи като миоклонични, комплексни парциални или абсанси (58, 20).
Молекулярно генетичните изследвания показват, че при 70-80% от случаите със синдром на Dravet се уста-новяват мутации в SCN1A гена (9,72,34). Описани са truncation мутации в 40% от случаите, missence мута-ции (40%) и малки вътрегенни мутации и такива в сплайс местата (34). Наскоро са установени и екзонни делеции и дупликации при 10% от децата с този синд-ром, при които конвенционалния мутационен анализ е негативен (56). Тези големи делеции могат да обхващат целия SCN1A ген или дори съседни гени, което в някои случаи може да доведе и до поява на дисморфични белези (76,70).
Приблизително 95% от мутациите в SCN1A гена възникват „de novo” (34). Фамилни мутации се иденти-фицират в около 5% от случаите, като обикновено те са missence, рядко се съобщават и наследствени truncation мутации (57,27). При другите членове от изследваните семейства се описва лек GEFS+ фенотип, което показва, че фамилната SCN1A missence мутация е само един от няколко възможни фактора, допринасящи за развитие на заболяването в това семейство. Най-вероятно различни модифициращи гени оказват влияние за развитие на най-тежкият фенотип при даден индивид от фамилията- синдрома на Dravet.
В литературата, вкл. и от нашия екип са описани няколко семейства, при които е установен мозаицизъм при един от родителите на деца със синдром на Dravet, като този родител или е здрав или има лек GEFS+ фено-тип (напр. само фебрилни гърчове)(3,29,19). Счита се, че наличието на мозаицизъм при един от родителите има важно значение за генетичното консултиране (64).
Мутации в SCN1A гена се откриват и при гранични-те форми на синдрома (34,28). Установено е, също така, че едни и същи мутации в SCN1A гена могат да обус-

Българска Неврология Bulgarian Neurology 4Май 2011
ловят развитието както на граничните форми, така и на класическата форма на синдрома на Dravet при различ-ните индивиди (34,28). Ето защо дори при генетичното изследване да се установи мутация в SCN1A гена, гено-тип-фенотип корелациите остават неясни (64).
Лечението на пациентите със синдром на Dravet се провежда с различни антиепилептични медикаменти като Валпроат, Клобазам, Топирамат и Леветирацетам (10). Кетогенната диета също може да бъде ефективна в някои случаи. Оптимизирането на лечението при тези пациенти е изключително важно, тъй като би могло да доведе до подобряване на когнитивните функции (64).
Х-СВЪРЗАНИ ИНФАНТИЛНИ СПАЗМИ
Терминът инфантилни спазми е по-общ и се използва за означаване на епилептични синдроми започващи в определена възраст и манифестиращи се с епилептични спазми (17). По-голяма част (70-80%) от инфантилните спазми са симптоматични и са резултат от различни причини като неонатална асфиксия, менингоенцефали-ти, туберозна склероза и хромозомни аномалии. Описани са и идиопатични форми с мултифакторна генетична предиспозиция (79). Най-често идиопатичните форми са спорадични и генетичният риск за близките е по-малък от 1% (23). В някои семейства е налице Х-свързано унаследяване, като са описани две форми на инфантилни спазми – рецесивна и доминантна.
При рецесивната форма се засяга мъжкия пол, уста-новяват се епилептични спазми, умствено изоставане и нормална находка при невроизобразяващите изследвания. Жените, хетерозиготни носителки са обикновено асим-птомни. Генетичният анализ в 3.5% от пациентите идентифицира мутации в ARХ-гена и вътрегенни екс-панзии в първата или втора полиаланинова част на про-теина, като вероятно дължината на експанзията коре-лира с тежестта на фенотипа (45). В едно семейство с missense мутация в ARХ гена са описани миоклонични и генерализирани тонично-клонични пристъпи като преоб-ладаващи в клиничната картина (66). При пациентите с рецесивна форма на инфантилни спазми често се развива тежка генерализирана дистония. Генетичен анализ за мутации в ARХ гена, се препоръчва при всички индивиди от мъжки пол с епилептични спазми, умствено изоста-ване и дистония.
Х-доминантната форма на инфантилни спазми е описана за първи път при две пациентки с тежки епи-лептични спазми с ранно начало и изоставане в психо-моторното развитие. И двете пациентки са носителки на балансирана Х-автозомна транслокация, уврежда-ща CDKL5 (cyclin-dependent kinase like 5) гена (42). Понастоящем фенотипният спектър асоцииран с мута-ции в CDKL5-гена е разширен и включва миоклонична енцефалопатия, атипичен синдром на Rett и вариант на синдрома на Rett с ранно начало на епилептичните прис-тъпи. Пристъпите започват преди 3 месечна възраст, най-често са генерализирани (тонични спазми, миокло-нични и генерализирани тонично-клонични пристъпи), могат да се съчетават с фокални пристъпи и са трудни за лечение. Всички пациенти са с умствено изоставане и нормална невроизобразяваща находка, като при някои от тях има и симптоми наподобяващи синдром на Rett-стереотипни движения на ръцете и апраксия на ръката. Повечето мутации в CDKL5-гена възникват de novo (30), описват се главно при жени, което предполага, че са летални за плодовете от мъжки пол по време на вът-реутробното развитие (78). Честотата на мутациите
в CDKL5-гена е 17% при пациентки с епилепсия с ранно начало, умствено изоставане и неизвестна етиология и 10% при такива с епилептични спазми (2). Генетично изследване се препоръчва при пациентки с тежка, резис-тентна на лечение епилепсия с начало през първите 3 месеца от живота, без установени метаболитни или структурни дефекти. При наличие на епилептични спаз-ми и симптоми наподобяващи синдром на Rett, генетич-ното изследване е задължително (30).
БЕНИГНЕНА ФАМИЛНА МИОКЛОНИЧНА ЕПИЛЕПСИЯ ПРИ ВЪЗРАСТНИ
Описана е за първи път от японски автори при семейства с множество засегнати индивиди от няколко последователни поколения и проявяващ се клинично с фин тремор на ръцете, миоклонии и редки тонично-кло-нични пристъпи. Електрофизиологичните изследвания при тези пациенти доказват, че треморът представ-лява кортикален рефлексен миоклонус и за това за означаването му се предлага терминът „кортикален тремор”(39).
Към настоящия момент в литературата са описани над 50 японски и 10 европейски родословия с този синд-ром (69). В различните изследвания, по-голяма част от пациентите са с хомогенна клинична характеристика и малки различия само по отношение тежестта на миок-лониите и типа на епилептични пристъпи.
Заболяването обикновено започва през второто десе-тилетие с лек тремор на ръцете и прогресира до редки тонично-клонични пристъпи и кортикален миоклонус към третото и четвъртото десетилетие от живо-та. Кортикалният тремор е акционен и постурален, аритмичен, фин и обхваща главно дисталните части на ръцете. Засилва се от емоции и умора и лесно може да се сбърка с есенциален тремор. Прогресия е възможна след 70 годишна възраст (70). В допълнение към тре-мора се наблюдават периодични дистални, сегментни, аритмични миоклонии в горните крайници, засилващи се от дадена поза и при движение (70). Тонично-клонични епилептични пристъпи обикновено се появяват по-късно от тремора, като пика е към 30 годишна възраст. Те са сравнително редки и се провокират от сънна деприва-ция, емоционален стрес и фотостимулация (39,73).
При ЕЕГ изследването често се установяват генера-лизирани пароксизми, особено при пациенти, които не получават антиконвулсантна терапия (70). Може да се наблюдава фотомиоклоничен отговор, както и огнищна пароксизмална активност (32).
МРТ изследването на главен мозък обикновено е нор-мално или показва леки, неспецифични абнормности. При МРТ спектроскопия се установява повишено съотно-шение холин/креатин в мозъчната кора в сравнение със здрави контроли (69).
Заболяването се унаследява автозомно-доминантно с висока пенетранност. Понастоящем, при японски и италиански фамилии, са идентифицирани два локуса, картирани на хромозоми 8q23.3-q24.1 и 2p11.1-q12.2 (73,15,32). С ефект на родоначалника би могло да се обяс-ни големия брой семейства с това заболяване от една и съща топографска област на Япония и Южна Италия (73). При други описани семейства не се установява скаченост с тези хромозомни области, което показва, наличието на поне още един трети локус отговорен за заболяването (16).
Прогнозата е добра, като епилептичните пристъпи се повлияват добре от лечението, въпреки, че са описани

Българска Неврология Bulgarian Neurology 5Май 2011
и случаи резистентни на терапия с антиепилептични медикаменти (32).
ФАМИЛНА ДЕСКА МИОКЛОНИЧНА ЕПИЛЕПСИЯ
През 2001г. de Falco и сътрудници (14) описват голяма италианска фамилия с миоклонични и тонично-клонични пристъпи с начало през първата година от живота, нор-мално психомоторно развитие и доброкачествен ход. Миоклоничните пристъпи персистират до късна въз-раст, засилват се при умора или сънливост и се прово-кират от интензивни и продължителни стимули или от повтарящи се движения. Интерикталната EEГ находка е неспецифична при повечето пациенти. Клиничната картина е сходна при различните засегнати индивиди, а унаследяването е автозомно-рецесивно. При анализа за скаченост е идентифициран локус на 16р13 хромозома.
КЛАСИЧЕСКИ ИДИОПАТИЧНИ ГЕНЕРАЛИЗИРАНИ ЕПИЛЕПСИИ
Детската абсансна епилепсия, ювенилната абсансна епилепсия, ювенилната миоклонична епилепсия и епи-лепсията с генерализирани тонично-клонични припадъци при събуждане са класическите и най-чести типове идиопатични генерализирани епилепсии.
Абсансите при детската абсансна епилепсия започ-ват обикновено около 6 годишна възраст, те са типич-ни, най-често с миоклонии на клепачите, с кратка про-дължителност (около 10 секунди) и се появяват на серии до 100 пристъпа дневно. В по-късна възраст могат да се добавят и генерализирани тонично-клонични пристъпи. Абсансите при ювенилната абсансна епилепсия са подоб-ни, но по-редки от тези при детската форма и започват около пубертета.
Миоклоничните пристъпи са характерен клиничен белег на ювенилната миоклонична епилепсия и ангажират главно горните крайници на фона на запазено съзнание. Могат да са леки и да не се разпознаят. Заболяването започва около пубертета, като пристъпите обикновено се появяват сутрин след събуждане и се провокират от сънна депривация. Генерализирани тонично-клонични пристъпи са налице при 75% от пациентите.
Епилепсията с генерализирани тонично-клонични пристъпи при събуждане започва между 9-24 годишна възраст. Основният тип са ГТКП, появяващи се в рамките на 1 час след събуждане независимо кое време е през деня, като са възможни абсанси и миоклонични пристъпи.
Всички тези синдроми могат да се припокриват при отделните пациенти или семейства и обикновено се асоциират с генерализирани пароксизми от комплекси острие-бавна вълна или полиспайк-бавна вълна в елект-роенцефалограмата. МРТ изследването е в норма.
За разлика от моногенните синдроми дискутирани по-горе, всички те показват комплексно унаследяване, като са описани само малък брой фамилии с множес-тво засегнати членове и ясно автозомно-доминантно унаследяване. Cossette и сътрудници (11) описват френ-ско-канадска фамилия с ювенилна миоклонична епилеп-сия, унаследяваща се по автозомно-доминантен начин. Фенотипът е хомогенен, с начало между 5 и 16 годишна възраст на миоклонични пристъпи и ГТКП. При малък брой от пациентите се съобщават и абсанси. При гене-тичният анализ се установява скаченост с хромозома 5q34 и мутация Ala322Asp в GABRA1 гена. Мутация в същият ген се описва и при дете с детска абсансна епилепсия (51). В няколко редки фамилии с фебрилни
пристъпи и детска абсансна епилепсия се откриват мутации с главен ефект в GABRG2 гена (43). Мутации в CLCN2 гена се идентифицира при семейства с ИГЕ и хетерогенен фенотип (12). Няколко семейства глав-но с ювенилна миоклонична епилепсия се асоциират с мутации в EFHC1 гена, кодиращ протеин с неизвестна функция (68). Редки варианти в CACNA1H гена се асоци-ират с детска абсансна епилепсия и други генерализирани епилептични фенотипове (37). Има данни, че варианти на GABRD, ME2, BRD2 и NEDD4L гени са свързани с предразположение към ИГЕ (35).
Helbig и съавтори доказват, че наличието на 15q13.3 микроделеция представлява рисков фактор за развитие на най-честите ИГЕ, като тази делеция се открива при 1% от пациентите и липсва при контроли (36). ИГЕ суб-синдромите, свързани с такава абнормност в 15q13.3 са детска абсансна епилепсия, ювенилна абсансна епилепсия и ювенилна миоклонична епилепсия (36).
В заключение: Молекулярно-генетичните изследвания на добре известните епилептични синдроми доведе до идентифицирането на отговорните за някои от тях гени, но също допринесе за откриването на нови епи-лептични синдроми, които понастоящем са включени в предложените от ILAЕ корекции в терминологията и класификацията на епилепсиите (6). Прогресът в раз-криване генетичната етиология на епилепсиите е от огромно значение за изясняване тяхната патогенеза и осигуряването на невробиологични критерии за класи-фицирането им.
ЛИТЕРАТУРА1. Abou-Khalil B, Ge Q, Desai R, et al. Partial and generalized
epilepsy with febrile seizures plus and a novel SCN1A mutation. Neurology 2001; 57: 2265–72.
2. Archer HL, Evans J, Edwards S, et al. CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. J Med Genet 2006;43:729–734.
3. Azmanov D, Zhelyazkova S, Dimova P, Radionova M, Bojinova V, Florez L, Smith S, Tournev I, Jablensky A, Mulley J, Scheffer I, Kalaydjieva L, Sander JW. Mosaicism of a missense SCN1A mutation and Dravet syndrome in a Roma/Gypsy family. Epileptic Disord 2010; 12 (2): 117-24
4. Audenaert D, Claes L, Ceulemans B, Lofgren A, Van Broeckhoven C, De Jonghe P. A deletion in SCN1B is associated with febrile seizures and early-onset absence epilepsy.Neurology 2003;61:854–6.
5. Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud’homme JF, et al. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet 2001;28:46–8
6. Baulac S, Gourfinkel-An I, Picard F, et al.A second locus for familial generalized epilepsy with febrile seizures plus maps to chromosome 2q21-q33. Am J Hum Genet 1999;65:1078–85.
7. Berg AT , Berkovic SF , Brodie MJ, Buchhalter J, Cross JH, Boas WV, Engel J, French J, Glauser TA, Mathern GW, Moshe SL, Nordli D, Plouin P, Scheffer IE. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 2010; 51 (4): 676 - 685.
8. Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins: genetics of the major epilepsy syndromes. Ann Neurol 1998; 43: 435–45.
9. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 2001;68:1327–32.
10. Coppola G, Capovilla G, Montagnini A, Romeo A, Spano M, Tortorella G, et al. Topiramate as add-on drug in severe myoclonic epilepsy in infancy: an Italian multicenter open trial. Epilepsy Res 2002;49:45–8
11. Cossette P, Liu L, Brisebois K, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet 2002; 31: 184–89.

Българска Неврология Bulgarian Neurology 6Май 2011
12. D’Agostino D, Bertelli M, Gallo S, et al. Mutations and polymorphisms of the CLCN2 gene in idiopathic epilepsy. Neurology 2004; 63: 1500–02
13. de Kovel C, Trucks H, Helbig I, Mefford H, Baker C, Leu C et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain 2010: 133; 23–32
14. de Falco FA, Majello L, Santangelo R, Stabile M, Dagna Bricarelli F, Zara F. Familial infantile myoclonic epilepsy. Clinical features in a large kindred with autosomal recessive inheritance. Epilepsia 2001;42:1541–1548.
15. de Falco FA, Striano P, de Falco A, Striano S, Santangelo R, Perretti A, Balbi P, Cecconi M, Zara F. Benign adult familial myoclonic epilepsy. Genetic heterogeneity and allelism with ADCME. Neurology 2003; 60:1381–1385.
16. Deng FY, Gong J, Zhang YC, Wang K, Xiao SM, et al. Absence of linkage to 8q23.3-q24.1 and 2p11.1-q12.2 in a new BAFME pedigree in China: indication of a third locus for BAFME. Epilepsy Res 2005;65:147–152
17. Deprez L, Jansen A, De Jonghe P. Genetics of epilepsy syndromes starting in the first year of life. Neurology 2009;72;273-281
18. Dibbens LM, Feng HJ, Richards MC, Harkin LA, Hodgson BL, Scott D, et al. GABRD encoding a protein for extra- or perisynaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet 2004;13:1315–9.
19. Depienne C, Arzimanoglou A, Trouillard O, Fedirko E, Baulac S, Saint-Martin C, et al. Parental mosaicism can cause recurrent transmission of SCN1A mutations associated with severe myoclonic epilepsy of infancy. Hum Mutat 2006;27:389.
20. Doose H, Lunau H, Castiglione E, Waltz S. Severe idiopathic generalized epilepsy of infancy with generalised tonic–clonic seizures. Neuropediatrics 1998;29:229–38.
21. Dravet C. Les epilepsies graves de l’enfant. La Vie Medicale 1978;8:543–8.
22. Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O. Severe myoclonic epilepsy in infancy (Dravet syndrome). In: Roger J,Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. Mountrouge: John Libbey Eurotext Ltd.; 2005. p.89–113.
23. Dulac O, Feingold J,Plouin P et al. Genetic predisposition to West syndrome. Epilepsia 2005;34(4), 732-7
24. Engel J Jr., A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: Report of the ILAE Task Force on Classifi cation and Terminology. Epilepsia 2001; 42: 796–803.
25. Escayg A, Heils A, Mac Donald BT et al. A nouvel SCN1A mutation associated with generalized epilepsy with febrile seizures plus- and prevalence of variants in patients with epilepsy. Am J Hun Genet. 2001;68(4);886-873
26. Escayg A, Mac Donald BT, Meisler MH, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+. Nat Genet. 2000;24(4):343-345
27. Fujiwara T,WatanabeM, Takahashi Y, Higashi T, Yagi K, Seino M. Long-term course of childhood epilepsy with intractable grandmal seizures. Jpn J Psychiatr Neurol 1992;46:297–302.
28. Fujiwara T, Sugawara T, Mazaki-Miyazaki E, Takahashi Y, Fukushima K, Watanabe M, et al. Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic–clonic seizures. Brain 2003;126(Pt 3):531–46.
29. Gennaro E, Santorelli FM, Bertini E, Buti D, Gaggero R, Gobbi G, et al. Somatic and germline mosaicisms in severe myoclonic epilepsy of infancy. Biochem Biophys Res Commun 2006;341:489–93.
30. Grosso S, Brogna A, Bazzotti S, Renieri A, Morgese G, Balestri P. Seizures and electroencephalographic findings in CDKL5 mutations: case report and review. Brain Dev 2007;29:239–242.
31. Guerrini R, Dravet C, Genton P, Belmonte A, Kaminska A, Dulac O. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia 1998;39s:508–12.
32. Guerrini R, Bonanni P, Patrignani A, Brown P, Parmeggiani L et al. Autosomal dominant cortical myoclonus and epilepsy (ADCME) with complex partial seizures and generalized seizures. A newly recognized epilepsy syndrome with linkage to chromosome 2p11.1-q12.2. Brain 2001; 124:2459–2475.
33. Harkin LA, Bowser DN, Dibbens LM, Singh R, Phillips F, Wallace RH, et al. Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet 2002;70:530–6.
34. Harkin LA, McMahon JM, Iona X, Dibbens L, Pelekanos JT, Zuberi SM, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 2007;130(Pt 3):843–52.
35. Helbig I, Scheffer IE, Mulley JC, Berkovic SF. Navigating the channels and beyond: unravelling the genetics of the epilepsies.
Lancet Neurology 2008 ; 7: 231-24536. Helbig I, Mefford HC, Sharp AJ et al. 15q13.3 microdeletions
increase risk of idiopathic generalized epilepsy. Nat Genet 2009: 41 (2): 160–162.
37. Heron SE, Khosravani H, Varela D, et al., Extended spectrum of idiopathic generalized epilepsies associated with CACNA1H functional variants. Ann Neurol 2007; 62: 560–68
38. Hurst DL. Epidemiology of severe myoclonic epilepsy of infancy. Epilepsia 1990; 31:397-400
39. Ikeda A, Kakigi R, Funai N, Neshige R, Kuroda Y, Shibasaki H. Сortical tremor: a variant of cortical reflex myoclonus. Neurology 1990;40:1561–1565.
40. Jallon P, Latour P.Epidemiology of idiopathic generalized epilepsies. Epilepsia 2005; 46(suppl 9):10–14.
41. Jansen FE, Sadleir LG, Harkin LA, Vadlamudi L, McMahon JM, Mulley JC, et al. Severe myoclonic epilepsy of infancy (Dravet syndrome): recognition and diagnosis in adults. Neurology 2006;67:2224–6.
42. Kalscheuer VM, Tao J, Donnelly A, et al. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am J Hum Genet 2003;72:1401–1411.
43. Kananura C, Haug K, Sander T, et al. A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol 2002; 59: 1137–41.
44. Kanazawa O. Refractory grand mal seizures with onset during infancy including severe myoclonic epilepsy in infancy. Brain Dev 2001;23:740–56.
45. Kato M, Saitoh S, Kamei A, et al. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am J Hum Genet 2007;81:361–366.
46. Kimura K, Sugawara T, Mazaki-Miyazaki E, Hoshino K, Nomura Y, Tateno A, et al. A missense mutation in SCN1A in brothers with severe myoclonic epilepsy in infancy (SMEI) inherited from a father with febrile seizures. Brain Dev 2005;27:424–30.
47. Kjeldsen MJ, Corey LA, Christensen K, Friis ML. Epileptic seizures and syndromes in twins: the importance of genetic factors. Epilepsy Res 2003; 55: 137–46.
48. Lenox WG. Heredity of epilepsy as told by relatives and twins. J.Am Med Assoc. 1951; 146:474-83.
49. Lossin C, Rhodes TH, Desai RR, et al. Epilepsy-Associated Dysfunction in the Voltage-Gated Neuronal Sodium Channel SCN1A. J Neurosci. 2003;23(36):11289-11295
50. Lossin C, Wang DW, Rhodes TH, et al. molecular basis of an inherited epilepsy. Neuron.2002;34(6):887-884.
51. Maljevic S, Krampfl K, Cobilanschi J, et al. A mutation in the GABA(A) receptor alpha(1)-subunit is associated with absence epilepsy. Ann Neurol 2006; 59: 983–87
52. Marini C, Harkin LA, Wallace RH et al. Childhood absence epilepsy and febrile seizure: a family with GABA(A) receptor mutation. Brain 2003; 126(Pt1): 230-240
53. Marini C, Scheffer IE, Crossland KM, et al. Genetic architecture of idiopathic generalized epilepsy: clinical genetic analysis of 55 multiplex families. Epilepsia 2004; 45: 467–78.
54. Metrakos K, Metrakos JD. Genetics of convulsive disorders.II.Genetic and electroencephalographic studies in centrencephalic epilepsy. Neurology.1961; 11:474-83.
55. Moulard B, Guipponi M, Chaigne D, et al. Identification of a new locus for generalized epilepsy with febrile seizures plus (GEFS+) on chromosome 2q24-q33. Am J Hum Genet 1999;65:1396–400.
56. Mulley JC, Nelson P, Guerrero S, Dibbens L, Iona X, McMahon JM, et al. A new molecular mechanism for severe myoclonic epilepsy of infancy: exonic deletions in SCN1A. Neurology 2006;67:1094–5.
57. Mulley JC, Scheffer IE, Petrou S, Dibbens LM, Berkovic SF, Harkin LA. SCN1A mutations and epilepsy. Hum Mutat 2005;25:535–42
58. Oguni H, Hayashi K, Awaya Y, Fukuyama Y, Osawa M. Severe myoclonic epilepsy in infants – a review based on the Tokyo women’s medical university series of 84 cases. Brain Dev 2001;23:736–48.
59. Ottman R, Lee JH, Hauser WA, Risch N. Are generalized and localization-related epilepsies genetically distinct? Arch Neurol 1998; 55: 339–44.
60. Pineda-Trujillo N, Carrizosa J, Cornejo W, Arias W, Franco C, Cabrera D, et al. A novel SCN1A mutation associated with severe GEFS+ in a large South American pedigree. Seizure 2005;14:123–8.
61. Reid CA, Berkovic SF, Petrou S. Mechanisms of human inherited epilepsies. Prog Neurobiol 2009; 87: 41–57.
62. Scheffеr IE, Berkovic SF. Generalized epilepsy with febrile seizures plus: a genetic disorder with heterogeneous clinical phenotypes.

Българска Неврология Bulgarian Neurology 7Май 2011
Brain 1997; 120: 479–90.63. Scheffer I, Berkovic S. Generalized (genetic) epilepsy with
febrile seizures plus. In: Engel JJ, Pedley T, editors. Epilepsy: a comprehensive textbook. Philadelphia: Lippincott, Williams & Wilkins; 2008. p. 2553–8.
64. Scheffer IE, Zhang YH, Jansen FE, Dibbens L. Dravet syndrome or genetic (generalized) epilepsy with febrile seizures plus? Brain Dev. 2009 May;31(5):394-400.
65. Scheffer IE, Harkin LA, Grinton BE, Dibbens LM, Turner SJ,Zielinski MA, et al. Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain 2007;130(Pt. 1):100–9.
66. Scheffer IE, Wallace RH, Phillips FL, et al. X-linked myoclonic epilepsy with spasticity and intellectual disability: mutation in the homeobox gene ARX. Neurology 2002; 59:348–356.
67. Singh R, Scheffer IE, Crossland K, Berkovic SF. Generalized epilepsy with febrile seizures plus: a common childhood-onset genetic epilepsy syndrome. Ann Neurol 1999; 45: 75–81
68. Stogmann E, Lichtner P, Baumgartner C, et al. Idiopathic generalized epilepsy phenotypes associated with diff erent EFHC1 mutations. Neurology 2006; 67: 2029–31.
69. Striano P, de Falco FA, Minetti C, Zara F. Familial benign nonprogressive myoclonic epilepsies. Epilepsia. 2009 May;50 Suppl 5:37-40
70. Striano P, Zara F, Striano S. Autosomal dominant cortical tremor, myoclonus and epilepsy: many syndromes, one phenotype. Acta Neurol Scand 2005;111:211–217.
71. Suls A, Claeys KG, Goossens D, Harding B, Luijk RV, Scheers S, et al. Microdeletions involving the SCN1A gene may be common in SCN1A-mutation-negative SMEI patients. Hum Mutat 2006;27:914–20.
72. Sun H, Zhang Y, Liang J, Liu X, Ma X, Qin J, et al. Seven novel SCN1A mutations in Chinese patients with severe myoclonic epilepsy of infancy. Epilepsia 2008;49:1104–7.
73. Uyama MD, Fu YH, Ptacek L. (2005) Familial adult myoclonic epilepsy (FAME). In Delgado-Escueta AV, Guerrini R, Medina MT, Genton P, Bureau M, Dravet C (Eds) Advances in neurology. Myoclonic epilepsies. Vol. 95. Ch. 22. Lippincott Williams & Wilkins, Philadelphia, pp. 281–288.
74. Wallace R, Wang D, Singh R et al. Febrile seizures and generalizes epilepsy associated with a mutation in the Na+-channel beta 1 subunit gene SCN1B. Nature Genet. 1998; 19(4), 366-370.
75 Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet 2001;28:49–52.
76. Wallace RH, Scheffer IE, Barnett S, et al. Neuronal sodium-channel alpha 1-subunit mutations in generalized epilepsy with febrile seizures plus. Am J Hun Genet. 2001; 68(4): 859-865
77. Wang JW, KurahashiH, IshiiA,Kojima T,OhfuM, Inoue T, et al. Microchromosomal deletions involving SCN1Aand adjacent genes in severemyoclonic epilepsy in infancy. Epilepsia 2008;49:1528–34.
78. Weaving LS, Christodoulou J,Williamson SL, et al.Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet 2004;75:1079–1093
79. Wong M, Trevathan E. Infantile spasms. Pediatr Neurol 2001;24:89–98.
80. Yakoub M, Dulac O, Jambaquel I et al. Early diagnosis of severe myoclonic epilepsy in infancy. Brain Dev. 1992; 14:299-303
Адрес за кореспонденция:Д-р С. ЖелязковаУМБАЛ „Александровска”Клиника по неврология,бул. „ Георги Софийски” 1, София 1431тел. 02/ 92 30 388
e-mail: [email protected]
SUMMARY
ENTRAPMENT NEUROPATHIES OF UPPER EXTREMITIES: DIAGNOSIS AND TREATMENT
D. Bogdanova, I. Nisimov, I. MilanovEntrapment neuropathy refers to a focal mononeuropathy
produced by chronic mechanical impingement of a nerve in normally narrow anatomical sites and is the most common form of focal neuropathy among the nontraumatic causes.
The disease is frequent. Carpal tunnel syndrome is the best example of entrapment neuropathy. The ulnar nerve is commonly affected. Entrapments of the deep ulnar branch and the suprascapular syndrome are rare.
Pathologically, entrapment neuropathies are characterized by paranodal demyelination in the early stages, complete demyelination in the full-blown stage and complete degeneration of fibers in the late stages. Waller degeneration in the nerve distal to the entrapment is a natural consequence of the late stage. The most common reasons are microtraumatization by repetitive movements, trauma osteoarthritis, inheritance, infection, etc. leading to direct trauma, compression, ischemia and inflammation.
The most prominent symptoms are pain, sensory
impairment, numbness, motor weakness and autonomic dysfunction in the distribution of the peripheral nerve.
The diagnosis is based on the clinical signs. The most precise paraclinical test is the electrodiagnosis – electroneuro- and electromyography.
Conservative treatment measures include splinting and oral or injected corticosteroids. Surgery is indicated in conservative management fail.
KEY WORDS: entrapment, neuropathy, upper extremities
РЕЗЮМЕ
Компрeсионните невропатии са фокални лезии на пери-ферните нерви, причинени от стесняване или механично разтягане на ствола на нерва във фиброзен или костно-фиброзен канал или от фиброзна тъкан. Състоянието е сравнително често и се характеризира се с болка и или загуба на функция, в резултат от хронично притискане (компресия).
Синдромът на карпалния канал е най-честата комп-ресионна невропатия, следвана от улнарна невропатия в областта на лакътя. По-редки форми на компресионни невропатии за ръцете са притискане на дълбокия клон на улнарния нерв и супраскапуларният синдром.
Най-честите причини за притискане на периферните нерви са травми, остеоартрит, микротравматизиране
ОбзорКОМПРЕСИОННИ МОНОНЕВРОПАТИИ В ГОРНИ КРАЙНИЦИ:
КРИТЕРИИ ЗА ДИАГНОЗА И ЛЕЧЕНИЕ
Д. Богданова, И. Нисимов*, И. Миланов
МБАЛНП “Св. Наум”, гр. София*Първа МБАЛ гр. София, Неврологична клиника

Българска Неврология Bulgarian Neurology 8Май 2011
от повтарящи се движения, наследственост и други, които водят до директна травма, притискане, разтяга-не, исхемия, инфекция или възпаление. Хроничното при-тискане води до исхемични промени, оток, структурни промени в мембраните и органелите на миелиновата обвивка и на аксона
Конкретната клинична картина зависи от засягане-то на определен нерв. Установяват се сетивни - болка и парестезии, хипо- и анестезии, двигателни -слабост и атрофии и автономни нарушения в зоната, инервирана от съответния периферен нерв.
Диагнозата и диференциалната диагноза на компре-сионните синдроми се основава основно на обективната клинична находка. Основно параклинично изследване е електромиографията.
Лечението бива консервативно и хирургично. При неуспех от медикаментозното лечение за 6-8 седмици се преминава към хирургично лечение
КЛЮЧОВИ ДУМИ: горни крайници, компресия, нев-ропатия
Компрeсионните невропатии са фокални лезии на периферните нерви, причинени от стесняване или механично разтягане на ствола на нерва във фиброзен или костно-фиброзен канал или от фиброзна тъкан. Характеризират се с болка и или загуба на функция, в резултат от хронично притискане (компресия)(1, 5).
Състоянието е сравнително често - около 10-20 % от случаите в неврохирургичните и неврологичните практики (6, 10, 17). Синдромът на карпалния канал е най-честата компресионна невропатия, следвана от улнарна невропатия в областта на лакътя (5, 16). Форми на редки компресионни невропатии за ръцете са компре-сия на дълбокия клон на улнарния нерв и супраскапулар-ният синдром (1).
Най-чести причини за притискане на периферните нерви са травми, неправилна поза, остеоартрит, мик-ротравматизиране от повторящи се движения, спорт, затлъстяване, наследственост и други, които водят до директна травма, притискане, разтягане, исхемия, инфекция или възпаление (6, 12, 17, 20). Хроничното при-тискане води до микроваскуларни (исхемични) промени, оток, дислокация на възлите на Ranvier, структурни промени в мембраните и органелите на миелиновата обвивка и на аксона (6, 12). Механичната деформация е най-голяма в повърхностните слоеве на нерва и в зоните между притиснатите и непритиснатите сегменти (20). Компресията води до нарушение на кръвоснабдяването на епиневриума и забавяне на вътреклетъчния аксонален транспорт. Ниска степен на компресия води до времен-ни промени в интраневралната микроциркулация, които вързо се възстановяват след лечение. Продължително високи нива на компресия нарушават аксоналния транс-порт и водят до функционални и структурни промени на периферните нерви и тялото на неврона, свързано с дълъг период на възстановяване (6, 12). Огнищната деми-елинизация е постоянна характеристика. Пълното възс-тановяване на функциите след хирургична декомпресия отразява процес на ремиелинизация (20). Непълното възстановяване при хронични и тежки случаи се дължи на Валерова дегенерация на аксоните и постоянни фиб-ротични изменения в нервно-мускулния апарат, които пречат на реинервацията (6, 17).
В зависимост от тежестта на засягането, увредата на периферните нерви бива: невропраксия – фокална или сегментна демиелинизация със запазване на аксона и въз-становяване за 2-12 седмици; аксонотмезис – аксонът е
прекъснат, но епиневриумът остана интактен и следва възстановяване с 1 мм дневно от мястото на увреда; невротмезис – нервът е прекъснат, липсва възстановя-ване. Най-честа при компресионните невропатии е ком-бинацията от невропраксия и аксонотмезис (1, 17).
Конкретната клинична картина зависи от засяга-нето на определен нерв. Най-общо се срещат въбуд-на сетивна симптоматика, като болка и парестезии, отпадна сетивна симптоматика – хипо- и анестезии и отпадна двигателна симптоматика, като слабост и атрофии (1, 5, 6, 17). При засягане на смесени нерви, както на медианния нерв (n. medianus), се прибавят и автономни нарушения – промени в цвета, сухота, заде-беляване на кожата, промени по ноктите, улцерации и други. Повечето компресионни невропатии в горни край-ници водят до двигателни и сетивни нарушения, с едно изключение – засягане на дълбокия клон на улнарния нерв (n. ulnaris)(само двигателни) в канала на Guyon (аналогич-на на сетивните нарушения при засягане на n. cutaneus femoris lateralis в долни крайници).
Компресионните невропатии в горни крайници имат обичайни места на засягане – такива са костно-фиброз-ните канали на китката, но могат да възникнат и на нео-бичайни места по тялото. Синдроми на двойно притис-кане на периферни нерви (т. нар. Double crush syndromes) се срещат често в практиката (18). Хипотезата е, че когато единични аксони са притиснати в един участък, функцията им се нарушава и те са предразпложение към увреда и в друг участък от нерва (18).
Основните клинични характеристики на по-честите компресионни невропатии в горни крайници са дадени на Таблица 1.
Неврогенният синдром на торакалния канал (thoracic outlet syndrome) е рядко заболяване (1, 3, 6, 11). Причинява се от абнормни фиброзни повлекла, притискащи брахи-алния плексус, често свързани с рудиментарно шийно ребро. Съществува и съдов (артериален и венозен) подвид на синдрома, съответно при притискане на a. subclavia и v. subclavia (5). Патогенезата не е изяснена, често симптоми възникват след травма с камшичен удар (1, 11, 17).
Пациентите имат оплаквания от изтръпване в ръце-те при абдукция и повдигане на рамената. Класическата картина е на слабост на малките мускули на ръката и сетивни нарушения по улнарната страна на ръката и предмишницата. Промяна в пулсовия обем при абдукция на мишницата се среща в 15% при здрави и не е индика-тор за заболяването (1, 10).
Диагностиката е насочена главно към изключване на други заболявания. Електромиография/електроневрогра-фия (ЕМГ/ЕНГ) се прилагат рутинно с цел да се изключи синдром на карпален канал или синдром на кубитален канал. В повечето случаи при синдром на торакалния канал не се установяват абнормности. Изследването е задължително при персистиране на симптомите повече от 3 месеца или липса на ефект от 8 седмици консерва-тивно лечение. ЕМГ критериите за неврогенен синдром на торакален канал са: Намалена амплитуда на сетив-ния отговор от n. ulnaris, или намалена амплитуда на моторния отговор от n. medianus, или удължени латентции на F-вълни от n. ulnaris или неврогенна увре-да в малките мускули на ръката при иглена ЕМГ (8, 17). Невроизобразяващи изследвания (рентгенография, КТ или МРТ) се налагат за изключване на допълнително шийно ребро.
До хирургично лечение се стига след 8 седмици неус-пешно консервативно лечение.

Българска Неврология Bulgarian Neurology 9Май 2011
Синдроми на притискане на периферни нерви на край-ниците на обичайни места. Синдром на карпален тунел (КТС).
КТС е най-честата компресионна невропатия с чес-тота между 125-515/100000 и е резултат от притискане на n. medianus от трансверзалния лигамент на китката (1, 16). Среща се в 2% от общата популация, в 14% при болни от захарен диабет без полиневропатия и в 30% при болни с диабетна полиневропатия (6,17).
Счита се, че е свързана в професионално натоварване на китката при машинописки, работещи с клавиатура и други. Наблюдава се и при намаление на напречния диаме-тър на карпалния тунел или увеличаване на неговия обем, както при разтягане на кости и лигаменти при фракту-ра или травма на предмишницата или ръката, свързано с генерализиран оток на ръката. Основен рисков фактор е захарният диабет. Бременността е предиспозиция, тъй като повишеният плазмен обем може да доведе до повишено налягане в карпалния тунел (в такива случаи симптомите изчезват след раждането( 4, 5, 16, 17).
Клинично се характеризира с тъпа и пареща болка в китката, която обхваща предмишницата до лакътя (1, 10). Болката типично се увеличава през нощта и често е свързана с парестезии на II и III пръст на ръката при събуждане. Обективно се установява намалена чувстви-телност по воларната повърхност на II и III пръст. При по-голяма давност има слабост и атрофия на m. abductor pollicis brevis с хипотрофия на thenar. M. flexor pollicis brevis има двойна инервация от n. medianus и n. ulnaris и не страда. По-късно се засяга и m. opponens pollicis. Форсирана флексия на китката води до парестезии и болка (симптом на Phalen). Леко почукване върху повърх-ността на нерва и retinaculum flexorum на китката води до парестезии (симптом на Tinel) (1, 5, 6, 10, 16).
При създаване на конкретни диагностични критерии за КТС се оказва, че няма златен стандарт при поста-вяне на диагнозата и че клинико-неврофизиологичната корелация дава най-точна информация (13).
Неврофизиологичните изследвания измерват нервна-та проводимост през карпалния тунел, изследването на сетивни влакна на n. medianus се сравнява с това на n. radialis и или n. ulnaris. Интерпретацията е сложна, особено при липса на патология и при съпътстваща патология. Основна абнормност са данните за огнищна демиелинизация. ЕНГ е особено необходима при разг-раничаване от дистална симетрична полиневропатия и т.нар. синдром на двойно притискане (double-crush синдром) при C7–С8 радикулопатия. Високата чувс-твителност (80%) и специфичност на ЕНГ и ЕМГ ги прави основен диагностичен метод при това заболяване. Най-чувствителна техника и китка-длан стимулация (61% чувствителност). Иглената ЕМГ има ограничена роля за диагнозата и потвърждава неврогенна увреда в засегнатите мускули. Има 3 основни показания: мускул-на слабост и хипотрофия на thenar, ЕНГ данни за друго заболяване и анамнеза за остра травма на ръката дис-тално (9, 15, 17).
Невроизобразяващите изследвания са чувствител-ни – рентгенография на карпалния тунел, доплерова сонография на китката и карпалния тунел, КТ и МРТ на китка. Магнитно-резонансната томография превъз-хожда останалите невроизобразяващи методи и дава възможност за оценка директно на n. medianus и обк-ръжаващите го тъкани. Друг метод на избор е допле-рова сонография (ултразвуково изследване) на китката, която е сравнима по диагностична стойност с електро-миографското изследване.
Лечението е консервативно с имобилизация и отст-раняване от вредна професионална дейност, които са ефективни при леки случаи без дефицит.
Решението за хирургично лечение зависи от тежест-та на симптомите, наличието на мускулна слабост, неуспех от консервативното лечение. Декомпресията на трансверзалния лигамент е метод на избор (12, 14,17).
Към компресионни невропатии на n. Ulnaris спадат синдром на кубиталния канал, притискане на дълбокия клон на нерва в областта на китката, притискане в канала на Гийон.
Синдром на кубитален канал. Това е втората по честота компресионна невропатия и се среща в 2.1% от общата популация. Резултат е от притискане на улнарния нерв в лакътя дистално от спиралния канал на humerus, под края на апоневрозата на m. flexor carpi ulnaris, т.е. в кубиталния канал. Развива се при дефор-мации на лакътната става след фрактури или след дълго притискане по време на операции. Често се наблюдава при алкохолици (5,6,16).
Етиологията включва травма, артрити, системни заболявания. Диференциална диагноза се прави с радику-лопатия C8-T1, синдром на торакален канал и компресия на нерва в китката. Патогенезата включва комбинация от демиелинизация и аксонална дегенерация (6, 17).
Клиничната картина включва болезнени парестезии в IV и V пръст на ръката, слабост и хипотрофия на интеросеалната мускулатура и hypothenar. Класически са симптомите на Froment (слабост на аддукция и опо-зиция на палеца) и слабост при флексия на IV и V пръст (1, 6, 10).
Основен диагностичен метод е ЕНГ/ЕМГ. Както при синдром на карпален тунел, скоростта на провеждане през лакътя е забавена. Основни ЕНГ абнормности са намалена амплитуда на сетивния отговор от n. ulnaris, намалена скорост на провеждане по сетивните влакна, неврогенна увреда (фибрилации) при иглена ЕМГ на mm. interossei (8, 17).
Лечението е консервативно. Резултатите от хирур-гично лечение не са обещаващи и включват медиал-на епикондилектомия, трансакция на апоневрозата на flexor carpi ulnaris, и транспозиция на n. ulnaris (17).
Компресионна невропатия на дълбокия клон на n. ulnaris в областта на китката. Засягат се обикнове-но хора с параплегия и употреба на бастун или пате-рици, които имат напречна част през дланта, при мотоциклетисти, оператори на пневматични машини. Класическате болни са млади хора, с болезнена атрофия на мускулите на hypothenar и mm. interossei, със запазване на мускулите на thenar. Различни по степен сетивни нару-шения в територията на n. ulnaris и болка по дланта, се срещат при болни, при които има рекурентен сетивен клон, излизащ в проксималната част на канала.(1,5, 10)
При синдром на канала на Гийон (Guyon)(1, 5, 6, 10) се притиска ствола на n. ulnaris в канала на Guyon в областта на китката. Причините могат да бъдат: конгенитални (аномалии на кости и мускули), травма, възпаление (ревматоиден артри), тумори, съдови (арте-риална тромбоза) и дегенеративни (остеоартрит).
Синдромът има 3 типа в зависимост от анато-мичното място на притискане: При тип I нервът се засяга проксимално или в канала и води до двигателни и сетивни нарушения – слабост на всички малки мускули, инервирани от n. ulnaris, сетивни нарушения в улнарна-та половина на палмарната повърхност на ръката и на IV пръст, но не и на гърба на ръката, която се инервира от n. cutaneus dorsalis. При тип II компресията е върху

Българска Неврология Bulgarian Neurology 10Май 2011
дълбокия клон и има само двигателни нарушения в муску-лите, инервирани от дълбокия клон на n. ulnaris. При тип III компресията е дистално в края на канала и има само сетивни нарушения по улнарната половина на дланта. Този синдром е най-рядък.
Тип I и II винаги са свързани с атрофия на m. interosseus dorsalis I. (1,5, 10).
Симптомите са подобни на тези при улнарна невро-патия в областта на лакътя, като няма сетивни нару-шения по гърба на ръката, тъй като дорзалния сетивен клон напуска предмишницата на 5-8 cm проксимално от китката. Болката, ако я има, се засилва при почукване върху пизиформената кост (симптом на Tinel), и може да ирадиира към предмишницата. Симптомът на m. palmaris brevis е полезен при диференциране с притис-кане на нерва в лакътя – когато болния абдуцира волево малкия пръст, има и симултантна контракция на m. palmaris brevis. Такова съкращение липсва при синдром на кубитален канал, но е запазено при притискане в китка-та(1, 5, 6, 17).
Диагнозата е клинична, но електромиография и опре-деляне на проводимостта е необходимо за локализиране на лезията и определяне степента на увреда. При тип I ЕНГ показва нормална скорост на провеждане по n. ulnaris и през лакътя и в сегмента китка-лакът, но има удължена дистална латенция при отвеждане от m. abductor digiti minimi и m. interosseus dorsalis I, удължена сетивна латенция и намален сетивен отговор от n. ulnaris. При тип II има нормална скорост на провеждане по двигателните влакна и сетивните влакна, нормална дистална латенция при отвеждане от m. abductor digiti minimi и удължена дистална латенция при отвеждане от m. interosseus dorsalis I, а при иглена ЕМГ има данни за денервация в m. interosseus dorsalis I (6, 17). Допълнителни изследвания са рентгенография на ръката и на китката, евентуално МРТ при суспектни туморни образования.
Пример за синдром на двойно притискане (double crush) тук е улнарна невропатия едновременно в лакътя и в китката (18).
Компресия на n. suprascapularis се среща често при спортисти – волейболисти, баскетболисти, гимнас-тици, вдигане на тежести. Клиничната картина е с внезапна поява на дълбока, тъпа болка в задната част на плещите и горната част на скапулата. Болката не достига шията и не ирадиира. Мускулна слабост и атро-фия се установява в m. supraspinatus (начало на абдукция на плешката) и m. infraspinatus (външна ротация на humerus) (1, 5, 6, 8, 10).
Компресионната невропатия на n. radialis е рядко състояние (0.6% от общата популация) и възниква в резултат от компресия на нерва в спиралния канал. Води до слабост с увисване на китката и сетивни нарушения по гърба на ръката, при запазена екстензия в лакътя (1, 5, 6, 10, 17)
Причините включват фрактури на humerus, травми върху постеро-латералната повърхност на предмишни-цата и мишницата, външна компресия на нерва в спирал-ния канал и на n. interosseus posterior (дистален моторен клон) в предмишницата.
При заден интеросеален синдром (n. interosseus posterior) се засягат предимно двигателни влакна. Симптомите са тъпа болка под лакътя, засилваща се при палпация на радио-nумералната става и слабост на екстензия в метакарпалните фалангеални стави, но не в интерфалангеалните стави. Втори и V пръст имат соб-ствени екстензорни сухожилия и са по-слабо засегнати от III и IV пръст. Прогресията е свързана със мускулна
слабост на всички ектензори на пръстите и на абдукция на палеца. Тъй като радиалните екстензори на китката са запазени поради по-проксимална инервация, мускул-ната слабост е предимно на улнарните екстензори на китката (1, 6).
Изолирана невропатия на повърхностния клон на n. radialis може да се предизвика от вътрешно притиска-не (синдром на Wartenberg), докато външна компресия на същия сетивен клон може да се получи от стегнат часовник или гривна около китката (5, 16).
Електроневрографията може да покаже намаление на амплитудата на сетивните отговорите от n. radialis спрямо контралатералния едноименен нерв, както и с сетивния отговор от n. medianus от същата страна (6, 8).
При наличие само на сетивни симптоми лечението е консервативно. При синдром на n. interosseus posterior, поради предимно двигателна симптоматика, лечени-ето изисква задължително хирургична декомпресия. Консервативното лечение не е успешно.
Някои заболявания с друга етио-патогенеза и пато-физиология могат да имитират компресионни невропа-тии и трябва да се имат предвид при диференциалната диагноза. С най-голямо значение тук са мултифокалната невропатия с блок в провеждането и миозит с вът-реклетъчни включвания (inclusion body myositis) (12). Мултифокалната невропатия с блок в провеждането, описана през 1988 година, е идиопатична чисто двига-телна невропатия, която се изявява като прогресираща слабост в горни крайници, обикновено асиметрична, без сетивна загуба (6). Повече от 80% от пациентите имат слабост на предмишницата и ръката, повече от 50% имат фасцикулации и мускулни крампи с лека хипотро-фия. Ходът е бавно прогресиращ и трябва да се диферен-цира от амиотрофична латерална склероза по липса на участие на централен двигателен неврон. Заболяването често може напълно да имитира синдром на карпален канал или компресия на n. radialis (6). Съществуват строги електродиагностични критерии за това заболя-ване, като най-важната ЕНГ находка е честичен блок в провеждането при изследване на двигателни нерви. Най-честа лабораторна находка е повишено серумно ниво на IgM антитела към ганглиозид GM1 в около половината от пациентите (6). Лечението е с интравенозен имуног-лобулин (6, 17).
Миозитът с включвания е възпалителна миопатия със засягане на пациенти на възраст над 50 години. Има бавно прогресиращ ход, асиметрична дистална слабост на мускулите на ръката може да имитира компресия на n. radialis. Засягат се и долни крайници. Електроневрографското изследване е нормално, но при иглена ЕМГ се установяват данни за миогенна увреда. Мускулна биопсия и изследване на серумна креатинфос-фокиназа показват миогенен тип увреда (6).
Диагнозата и диференциалната диагноза на компре-сионните синдроми се основава основно на обективната клинична находка.
Основен параклиничен метод на изследване е елект-ромиографията (ЕМГ/ ЕНГ). За типични случаи, като синдром на карпален канал и синдром на кубитален канал електроневрографията е силно чувствителна, но не е задължителна. При други невропатии, като синдром на n. interosseus posterior, синдром на притискане в канала на Guyon, притискане на n. suprascapulartis и други, ЕНГ абнормностите и наличието на неврогенна увреда в съответните мускули при иглена ЕМГ, са задължителни за потвърждаване на диагнозата (1,8, 17).
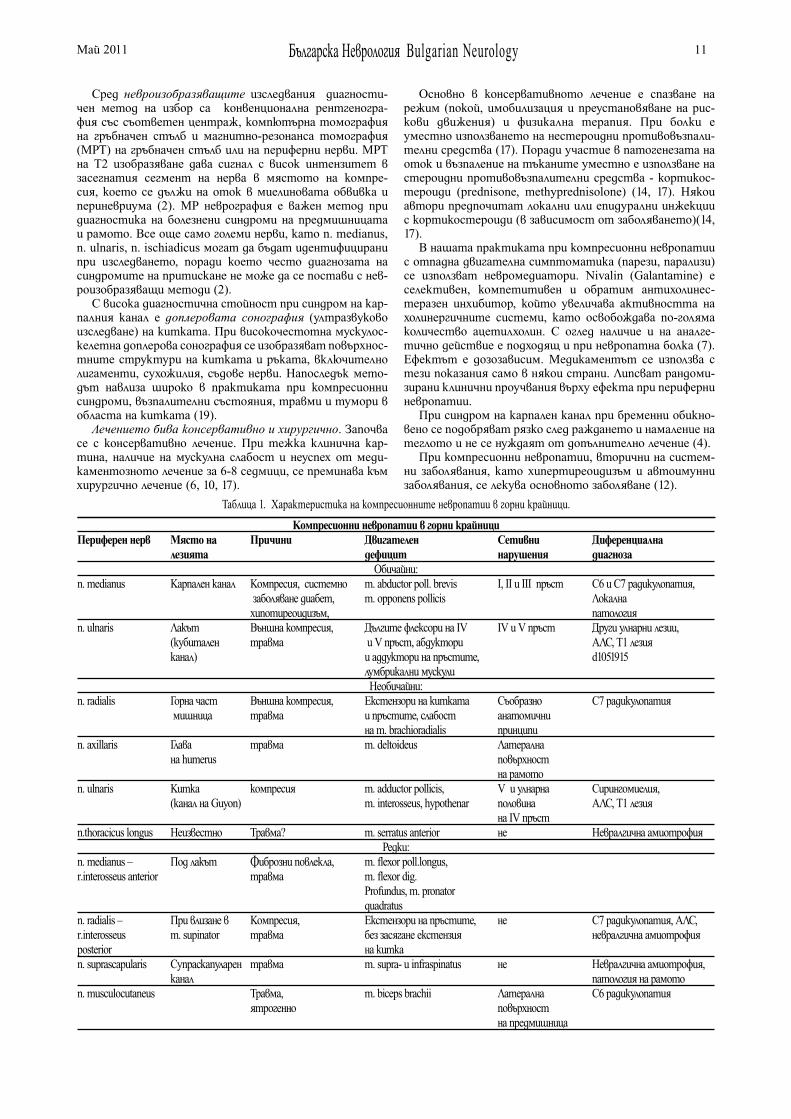
Българска Неврология Bulgarian Neurology 11Май 2011
Сред невроизобразяващите изследвания диагности-чен метод на избор са конвенционална рентгеногра-фия със съответен центраж, компютърна томография на гръбначен стълб и магнитно-резонанса томография (МРТ) на гръбначен стълб или на периферни нерви. МРТ на Т2 изобразяване дава сигнал с висок интензитет в засегнатия сегмент на нерва в мястото на компре-сия, което се дължи на оток в миелиновата обвивка и периневриума (2). МР неврография е важен метод при диагностика на болезнени синдроми на предмишницата и рамото. Все още само големи нерви, като n. medianus, n. ulnaris, n. ischiadicus могат да бъдат идентифицирани при изследването, поради което често диагнозата на синдромите на притискане не може да се постави с нев-роизобразяващи методи (2).
С висока диагностична стойност при синдром на кар-палния канал е доплеровата сонография (ултразвуково изследване) на китката. При високочестотна мускулос-келетна доплерова сонография се изобразяват повърхнос-тните структури на китката и ръката, включително лигаменти, сухожилия, съдове нерви. Напоследък мето-дът навлиза широко в практиката при компресионни синдроми, възпалителни състояния, травми и тумори в областа на китката (19).
Лечението бива консервативно и хирургично. Започва се с консервативно лечение. При тежка клинична кар-тина, наличие на мускулна слабост и неуспех от меди-каментозното лечение за 6-8 седмици, се преминава към хирургично лечение (6, 10, 17).
Основно в консервативното лечение е спазване на режим (покой, имобилизация и преустановяване на рис-кови движения) и физикална терапия. При болки е уместно използването на нестероидни противовъзпали-телни средства (17). Поради участие в патогенезата на оток и възпаление на тъканите уместно е използване на стероидни противовъзпалителни средства - кортикос-тероиди (prednisone, methyprednisolone) (14, 17). Някои автори предпочитат локални или епидурални инжекции с кортикостероиди (в зависимост от заболяването)(14, 17).
В нашата практиката при компресионни невропатии с отпадна двигателна симптоматика (парези, парализи) се използват невромедиатори. Nivalin (Galantamine) е селективен, компетитивен и обратим антихолинес-теразен инхибитор, който увеличава активността на холинергичните системи, като освобождава по-голяма количество ацетилхолин. С оглед наличие и на аналге-тично действие е подходящ и при невропатна болка (7). Ефектът е дозозависим. Медикаментът се използва с тези показания само в някои страни. Липсват рандоми-зирани клинични проучвания върху ефекта при периферни невропатии.
При синдром на карпален канал при бременни обикно-вено се подобряват рязко след раждането и намаление на теглото и не се нуждаят от допълнително лечение (4).
При компресионни невропатии, вторични на систем-ни заболявания, като хипертиреоидизъм и автоимунни заболявания, се лекува основното заболяване (12).
Таблица 1. Характеристика на компресионните невропатии в горни крайници.
Компресионни невропатии в горни крайнициПериферен нерв Място на Причини Двигателен Сетивни Диференциална лезията дефицит нарушения диагноза
Обичайни:n. medianus Карпален канал Компресия, системно m. abductor poll. brevis I, II и III пръст С6 и С7 радикулопатия, заболяване диабет, m. opponens pollicis Локална хипотиреоидизъм, патологияn. ulnaris Лакът Външна компресия, Дългите флексори на IV IV и V пръст Други улнарни лезии, (кубитален травма и V пръст, абдуктори АЛС, Т1 лезия канал) и аддуктори на пръстите, d1051915 лумбрикални мускули
Необичайни:n. radialis Горна част Външна компресия, Екстензори на китката Съобразно С7 радикулопатия мишница травма и пръстите, слабост анатомични на m. brachioradialis принципи n. axillaris Глава травма m. deltoideus Латерална на humerus повърхност на рамото n. ulnaris Китка компресия m. adductor pollicis, V и улнарна Сирингомиелия, (канал на Guyon) m. interosseus, hypothenar половина АЛС, Т1 лезия на IV пръст n.thoracicus longus Неизвестно Травма? m. serratus anterior не Невралгична амиотрофия
Редки:n. medianus – Под лакът Фиброзни повлекла, m. flexor poll.longus, r.interosseus anterior травма m. flexor dig. Profundus, m. pronator quadratus n. radialis – При влизане в Компресия, Екстензори на пръстите, не С7 радикулопатия, АЛС, r.interosseus m. supinator травма без засягане екстензия невралгична амиотрофия posterior на китка n. suprascapularis Супраскапуларен травма m. supra- и infraspinatus не Невралгична амиотрофия, канал патология на рамотоn. musculocutaneus Травма, m. biceps brachii Латерална С6 радикулопатия ятрогенно повърхност на предмишница

Българска Неврология Bulgarian Neurology 12Май 2011
В заключение компресионните невропатии в горни крайници възникват най-често на определи анатомични места. Диагнозата се базира на електромиографско-клинична корелация и познаване на анатомията и физи-ологията на периферните нерви. Лечението зависи от причините и от отговора на консервативно лечение.
ЛИТЕРАТУРА1. Миланов И. Болка в неврологичната практика. Медицина и
физкултура, София, 2009, 221-238.2. Andreisek G, Crook DW, Burg D, Marincek B, Weishaupt D.
Peripheral neuropathies of the median, radial, and ulnar nerves: MR imaging features. Radiographics, 26, 2006, 1267-1287
3. Ambrad-Chalela E., Thomas G. I., Johansen K.H.: Recurrent neurogenic thoracic outlet syndrome. Am J Surg, 187, 2004, 505-510.
4. Bahrami MH, Rayegani SM, Fereidouni M, Baghbani M. Prevalence and severity of carpal tunnel syndrome (CTS) during pregnancy. Electromyogr Clin Neurophysiol, 45, 2005,123–125
5. Conway SR. Jones HR Jr. Entrapment and compression neuropathies, In: Tollison CD ed. Handbook of pain management. 2nd ed. Baltimore: Williams & Wilkins., 1994, 470-483.
6. Corwin HM. Compression neuropathies of the upper extremity. Clin Occup Environ Med. 5, 2006, 333-352
7. Galantamine Gel for DPN. A Company with Attractive Products. Neuropathic Pain. www.sanochemia.at/.../SANOCHEMIA_Factbook_2009-11_EN_03.pdf
8. Herrmann D, Logigian E: Electrodiagnostic approach to the patients with suspected mononeuropathy of the upper extremity. Neurol Clin, 20, 2002, 451–478
9. Jablecki CK, Andary MT, Floeter MK, Miller RG, Quartly CA, Vennix MJ, Wilson JR. Practice parameter: Electrodiagnostic studies in carpal tunnel syndrome. Report of the American Association of Electrodiagnostic Medicine, American Academy of Neurology, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 11, 2002, 11, 1589-1592.
10. Miller TT, Reinus WR. Nerve entrapment syndromes of the elbow, forearm, and wrist. AJR Am J Roentgenol. 195, 2010, 3, 585-594.
11. Nannapaneni R., Marks S. M.: Neurogenic thoracic outlet syndrome. Br J Neurosurg 17, 2003, 17, 2, 144-148.
12. Poncelet AN. An algorithm for the evaluation of peripheral neuropathy. Am Fam Physician. 15, 1998, 4, 755-764.
13. Rempel D, Evanoff B, Amadio PC, de Krom M, Franklin G, Franzblau A, et al. Consensus criteria for the classification of carpal tunnel syndrome in epidemiologic studies. Am J Public Health, 88, 1998, 1447–1451.
14. Racasan O, Dubert T. The safest location for steroid injection in the treatment of carpal tunnel syndrome. J Hand Surg [Br], 30, 2005, 4, 412–414.
15. Sandin KJ, Asch SM, Jablecki CK, Kilmer DD, Nuckols TK; Carpal Tunnel Quality Group. Clinical quality measures for electrodiagnosis in suspected carpal tunnel syndrome. Muscle Nerve, 41, 2010, 4, 444-452
16. Serra J. Chapter 36 Painful entrapment disorders. Handb Clin Neurol, 81, 2006, 547-563.
17. Tapadia M, Mozaffar T, Gupta R. Compressive neuropathies of the upper extremity: update on pathophysiology, classification, and electrodiagnostic findings. J Hand Surg Am, 35, 2010, 668-677.
18. Upton AR, McComas AJ: The double crush in nerve entrapment syndromes. Lancet, 2, 1973, 359–362.
19. Wansaicheong G, Tsou I. Ultrasonography of the hand and wrist Singapore Med J, 50, 2009, 2, 219-222.
20. Warner MA. Perioperative neuropathies. Mayo Clin Proc, 73, 1998, 6, 567-574.
Адрес за кореспонденция:Д-р Десислава Богданова, дмМБАЛНП “Св. Наум”, Отделение по клинична неврофи-зиология,ул. Любен Русев №1, София 1113, България[email protected]
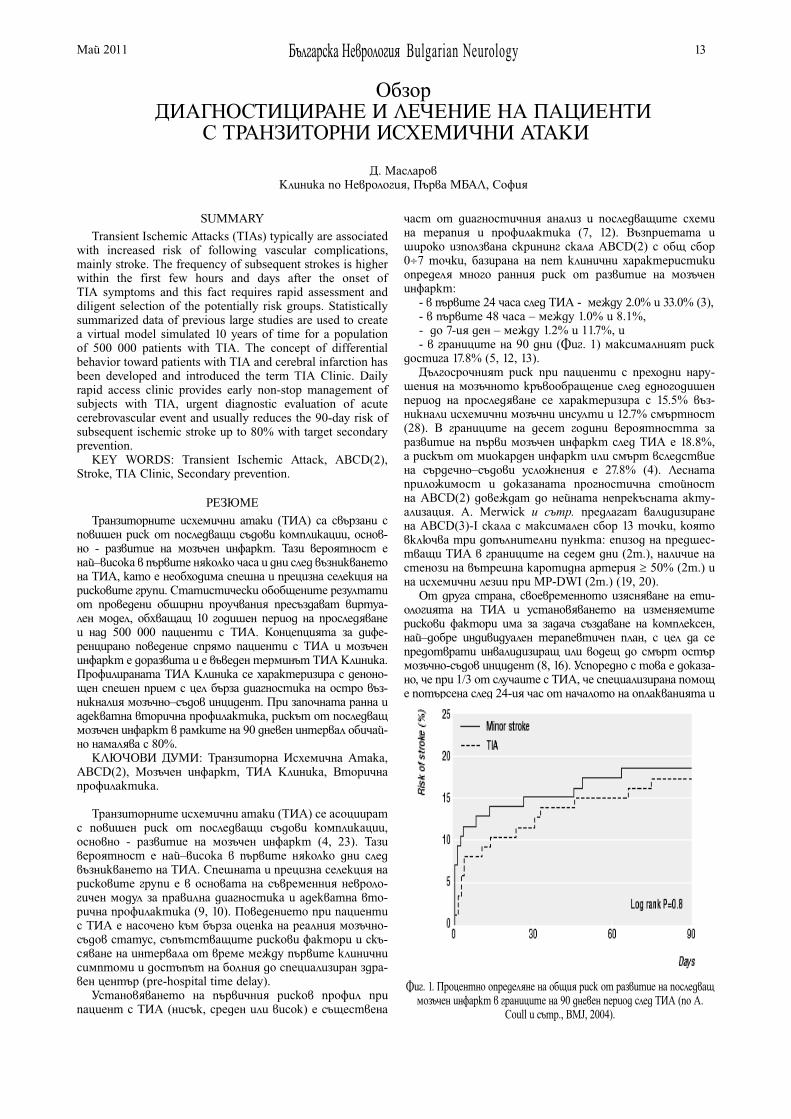
Българска Неврология Bulgarian Neurology 13Май 2011
SUMMARY
Transient Ischemic Attacks (TIAs) typically are associated with increased risk of following vascular complications, mainly stroke. The frequency of subsequent strokes is higher within the first few hours and days after the onset of TIA symptoms and this fact requires rapid assessment and diligent selection of the potentially risk groups. Statistically summarized data of previous large studies are used to create a virtual model simulated 10 years of time for a population of 500 000 patients with TIA. The concept of differential behavior toward patients with TIA and cerebral infarction has been developed and introduced the term TIA Clinic. Daily rapid access clinic provides early non-stop management of subjects with TIA, urgent diagnostic evaluation of acute cerebrovascular event and usually reduces the 90-day risk of subsequent ischemic stroke up to 80% with target secondary prevention.
KEY WORDS: Transient Ischemic Attack, ABCD(2), Stroke, TIA Clinic, Secondary prevention.
РЕЗЮМЕ
Транзиторните исхемични атаки (ТИА) са свързани с повишен риск от последващи съдови компликации, основ-но - развитие на мозъчен инфаркт. Тази вероятност е най–висока в първите няколко часа и дни след възникването на ТИА, като е необходима спешна и прецизна селекция на рисковите групи. Статистически обобщените резултати от проведени обширни проучвания пресъздават виртуа-лен модел, обхващащ 10 годишен период на проследяване и над 500 000 пациенти с ТИА. Концепцията за дифе-ренцирано поведение спрямо пациенти с ТИА и мозъчен инфаркт е доразвита и е въведен терминът ТИА Клиника. Профилираната ТИА Клиника се характеризира с деноно-щен спешен прием с цел бърза диагностика на остро въз-никналия мозъчно–съдов инцидент. При започната ранна и адекватна вторична профилактика, рискът от последващ мозъчен инфаркт в рамките на 90 дневен интервал обичай-но намалява с 80%.
КЛЮЧОВИ ДУМИ: Транзиторна Исхемична Атака, ABCD(2), Мозъчен инфаркт, ТИА Клиника, Вторична профилактика.
Транзиторните исхемични атаки (ТИА) се асоциират с повишен риск от последващи съдови компликации, основно - развитие на мозъчен инфаркт (4, 23). Тази вероятност е най–висока в първите няколко дни след възникването на ТИА. Спешната и прецизна селекция на рисковите групи е в основата на съвременния невроло-гичен модул за правилна диагностика и адекватна вто-рична профилактика (9, 10). Поведението при пациенти с ТИА е насочено към бърза оценка на реалния мозъчно-съдов статус, съпътстващите рискови фактори и скъ-сяване на интервала от време между първите клинични симптоми и достъпът на болния до специализиран здра-вен център (pre-hospital time delay).
Установяването на първичния рисков профил при пациент с ТИА (нисък, среден или висок) е съществена
част от диагностичния анализ и последващите схеми на терапия и профилактика (7, 12). Възприетата и широко използвана скрининг скала ABCD(2) с общ сбор 0÷7 точки, базирана на пет клинични характеристики определя много ранния риск от развитие на мозъчен инфаркт:
- в първите 24 часа след ТИА - между 2.0% и 33.0% (3),- в първите 48 часа – между 1.0% и 8.1%,- до 7-ия ден – между 1.2% и 11.7%, и- в границите на 90 дни (Фиг. 1) максималният риск
достига 17.8% (5, 12, 13).Дългосрочният риск при пациенти с преходни нару-
шения на мозъчното кръвообращение след едногодишен период на проследяване се характеризира с 15.5% въз-никнали исхемични мозъчни инсулти и 12.7% смъртност (28). В границите на десет години вероятността за развитие на първи мозъчен инфаркт след ТИА е 18.8%, а рискът от миокарден инфаркт или смърт вследствие на сърдечно–съдови усложнения е 27.8% (4). Лесната приложимост и доказаната прогностична стойност на ABCD(2) довеждат до нейната непрекъсната акту-ализация. A. Merwick и сътр. предлагат валидизиране на ABCD(3)-I скала с максимален сбор 13 точки, която включва три допълнителни пункта: епизод на предшес-тващи ТИА в границите на седем дни (2т.), наличие на стенози на вътрешна каротидна артерия ≥ 50% (2т.) и на исхемични лезии при МР-DWI (2т.) (19, 20).
От друга страна, своевременното изясняване на ети-ологията на ТИА и установяването на изменяемите рискови фактори има за задача създаване на комплексен, най–добре индивидуален терапевтичен план, с цел да се предотврати инвалидизиращ или водещ до смърт остър мозъчно-съдов инцидент (8, 16). Успоредно с това е доказа-но, че при 1/3 от случаите с ТИА, че специализирана помощ е потърсена след 24-ия час от началото на оплакванията и
ОбзорДИАГНОСТИЦИРАНЕ И ЛЕЧЕНИЕ НА ПАЦИЕНТИ
С ТРАНЗИТОРНИ ИСХЕМИЧНИ АТАКИ
Д. МасларовКлиника по Неврология, Първа МБАЛ, София
6��7�87� ��7��9�����5���������fd��������9���6��9����������5��d�?6�:���������9�� ������7�9�����VF�5����������5����5�ab��N��0��)"& ����������_�‹��`FFlO�
Фиг. 1. Процентно определяне на общия риск от развитие на последващ мозъчен инфаркт в границите на 90 дневен период след ТИА (по A.
Coull и сътр., BMJ, 2004).

Българска Неврология Bulgarian Neurology 14Май 2011
този удължен период се свързва с по–висок риск от разви-тие на последващ мозъчен инфаркт (6, 26).
Статистически обобщените резултати от проведе-ни обширни проучвания - Oxford Vascular Study (OXVASC), Newcastle Rapid Ambulance Protocol Study и QRESEARCH пресъздават виртуален модел, обхващащ 10 годишен период на проследяване и над 500 000 пациенти с ТИА. Те демонстрират икономическата, общомедицинска и психо-социална ефективност на стратегията за обо-собени специализирани диагностични центрове/клиники (еднодневни, при невъзможност – с еднократен или двукратен седмичен достъп), в сравнение с рутинната практика (18). Многобройни са публикациите, свърза-ни с положителни работни показатели и практически изводи в различни аспекти от създадените профилирани клиники във всички областни болници в САЩ и редица европейски държави (1, 2, 11, 14, 15, 17).
През януари 1999 год. във Великобритания, въз осно-ва на шестмесечен анализ и клиничен контингент от 128 пациенти с ТИА, се изнасят данни за подобрена ефективност по отношение на диагностично–тера-певтичния процес и предотвратени излишни хоспита-лизации в случаите на активно пренасочване на болни към специализирани Нервно-съдови клиники („one-stop“ NeuroVascular Clinic). Дават се препоръки подобни звена да бъдат асоциирани към отделения за мозъчно–съдова патология в болници в големи райони, като част от общата лечебна и превантивна стратегия (14). През 2000 год. вече се коментират задоволителните ико-номически резултати от предшестващи консултации и амбулаторно проведени изследвания (кръвни, ултраз-вукови и невроизобразяващи) при пациенти с остри мозъчно–съдови инциденти преди хоспитализацията им в специализирана клиника (CerebroVascular Disease Clinic) в болница St. George, Лондон (2).
По-късно концепцията за диференцирано спешно пове-дение спрямо пациенти с ТИА и мозъчен инфаркт се доразвива и се въвежда термина ТИА Отделение/Клиника. Редица проучвания демонстрират, че спе-циализираните ТИА Клиники осигуряват по–бърз и прецизен организационен процес, включващ клинични и допълнителни изследвания в сравнение със стандартни-те неврологични клиники, което е изключително важно за ранната и правилна вторична профилактика (1). През август 2006 год. се създава TIA Клиника към Центъра за Мозъчен инфаркт-Станфорд, Калифорния под ръко-водството на Greg Albers. Там вниманието е насочено към първоначална оценка и терапия на пациенти с пре-ходни нарушения, преценени на базата на клинични и неинвазивни изследвания като нискорискови за развитие на исхемичен мозъчен инсулт. Това са болни с предвари-телен общ резултат ABCD(2) ≤ 3т. Установено е, че приблизително 60 % от пациентите с ТИА не подлежат на хоспитализация и са обект на специален медицински режим в условията на спешност в профилирана ТИА Клиника (11).
Две независими и паралелно проведени проучвания потвърждават положителните резултати от функци-ониращи клиники за детайлна диагностика и поведение при пациенти с ТИА. SOS-TIA е представено от P. Lavallée и сътр. и обобщава сравнителни данни при безп-репятствен денонощен (24 часа/7 дни седмично) достъп на пациенти с внезапно възникнали мозъчни и/или рети-нални симптоми, преценени като исхемични и с пълно обратно развитие до момента на първичния преглед. Комплексната оценка включва неврологични, съдови и кардиологични изследвания и е извършена до 4-ия час от
постъпване в клиниката. Проследени са 1085 пациенти с вероятни ТИА, като крайните цели на проучването са: развитие на мозъчен инфаркт до 90 дни след инци-дента и мозъчен инсулт, миокарден инфаркт и/или други съдови компликации, водещи до смърт в границите на една година. При всички пациенти е започната вторична профилактика, при 5% - терапия с антикоагулант по повод предсърдно мъждене и 5% са подложени на спешна каротидна реваскуларизация. Честотата на развитие на последващ мозъчен инфаркт при диагностично уточнени пациенти, започнали адекватен терапевтично–профи-лактичен курс в границите на 90 дни след преживяна ТИА е 1.24% (13 исхемични мозъчни инсулти и 1 случай на мозъчен кръвоизлив), в сравнение с очакваната по - висо-ка честота от 5.96%, определена посредством ABCD(2) скала, т.е. практическият риск е с 80% по – нисък спря-мо теоретично определения (15).
Аналогични са и резултатите от проучването EXPRESS, като част от Oxford Vascular Study (OXVASC), изнесени през октомври 2007 год. в Lancet, където при проследяване на 1278 пациенти с ТИА се установява до 80% редукция на риска от последващ мозъчен инфаркт в 90 дневен интервал при започната ранна и адекватна вторична профилактика (24). Вследствие на това се намалява очаквания болничен престой, процента на инвалидизация и се отчита положителен икономически ефект (17).
Бързото диагностично и етиологично уточняване е съществен момент при първия контакт с пациент с предполагаема ТИА. Този процес включва задължителен алгоритъм от клинични и други изследвания, извършвани в амбулаторни условия и, при необходимост, насочване към Stroke Unit, с който може да бъде асоциирана ТИА Клиниката (21).
Селектирането на пациенти с вероятна ТИА се извършва въз основа на клиничната ABCD(2) скала.
Като високорискови за развитие на последващ мозъчен инфаркт се определят пациенти с резултат ABCD(2) ≥ 4т., особено с общ сбор между 6 и 7т. и с т. нар. крес-чендо ТИА – наличие на 2 или повече преходни наруше-ния в границите на една седмица, дори и при резултат ABCD(2) ≤ 3т. Някои автори дори ги подразделят като пациенти с много висок риск – ABCD(2) ≥ 6т. и пациен-ти с висок риск – ABCD(2) е 4÷5т. Пациентите, пре-ценени с висок риск подлежат на специализирана оценка и изследвания в първите 24 часа от появата на клинич-ните симптоми, незабавен прием на антиагрегантна терапия – аспирин 300 мг дневно и активна вторична профилактика, включително разглеждане на индивидуал-ните рискови фактори. Пациенти, които приемат пос-тоянна антикоагулантна терапия също следва да бъдат третирани като високорискови за развитие на мозъчен инсулт. Обикновено този контингент се насочва към Stroke Unit (27).
Групата пациенти с нисък риск класифицира тези с резултат АBCD(2) ≤ 3т. и/или с над едноседмична давност на оплакванията. При тях също се започва с незабавен прием на аспирин 300 мг дневно и насочена вторична профилактика, като интервалът за диагнос-тично уточняване може да бъде удължен в границите на седем дни от началото на симптомите (21, 22).
Освен класифицирането според АBCD(2) скала, е необходимо провеждането и на други диагностични изс-ледвания (25):
Артериално налягане, определяне на индекса на телесна маса, кръвни изследвания: кръвна захар, липиден и коагулационен статус;

Българска Неврология Bulgarian Neurology 15Май 2011
ЕКГ и ехокардиография – трансторакална и/или по преценка трансезофагеална;
КТ на главен мозък и Дуплекс ехография на каротид-ни артерии – според определени алгоритми тя не е задължителна при пациенти с ТИА във вертеброба-зиларния басейн;
В зависимост от последните две изследвания – насочване от т. нар. ТИА Координатор за осъ-ществяване на КТ/МРТ-ангиография и спешна кон-султация със съдов специалист (при наличие на симптомни високостепенни каротидни стенози).
Предлагаме да се използуват две основни форми за характеристика и класификация на пациенти с ТИА, в зависимост от това, дали са с нисък или с висок риск от развитие на последващ мозъчен инфаркт Приложение 4.
Групата с нисък риск – ABCD (2) ≤ 3т. подлежи на оценка и изследвания, които се отразяват в Протокол Приложение 1 и следва да бъдат осъществени в едносед-мичен интервал.
Групата с висок риск – ABCD(2) ≥ 4т. се насочват към Спешни отделения с попълнен Протокол Приложение 2, който придружава болния и още в първите 24 часа се уточняват диагностично, обикновено в Stroke Unit или в ТИА Отделение/Клиника и състоянието им се описва в Протокол Приложение 3.
Основните характеристики на ТИА Отделения/Клиники са:
- денонощен достъп на пациенти, без необходимост от предварително ангажиран час;
- асоциираност със Спешна зала и със Stroke Unit. По наше мнение ТИА Клиниките следва да бъдат звено към всеки пълноценно функциониращ Stroke Unit;
- максимално къс времеви интервал за диагностично уточняване на пациентите с ТИА – 12 - 24 часа от нача-лото на симптоматиката;
- ТИА Клиниките са еднодневни, с прием 24 часа/7 дни седмично. Освен икономическата ефективност се има в предвид и липсата на възникване на ятрогенни заболява-ния, свързани с всяка хоспитализация.
ЛИТЕРАТУРА:
1. Birns J, Vilasuso M, Cohen DL. One-stop clinics are more effective than neurology clinics for TIA. Age Ageing, 2006, 35(3):306-308.
2. Blight A, Pereira AC, Brown MM. A single consultation cerebro-vascular disease clinic is cost effective in the management of tran-sient ischaemic attack and minor stroke. JR Coll Physicians Lond. 2000 Sep-Oct; 34(5):452-5.
3. Chandratheva A, Mehta Z, Geraghty OC, et al. Oxford Vascular Study. Population – based study of risk and predictors of stroke in the first few hours after a TIA. Neurology. 2009, 72(22):1941-7.
4. Clark TG, Murphy MFG, Rothwell PM. Long term risks of stroke, myocardial infarction, and vascular death in “low risk” patients with a non-recent transient ischaemic attack. J Neurol Neurosurg Psychiatry 2003;74:577-580.
5. Coull AJ, Lovett JK, Rothwell PM. Population based study of early risk of stroke after transient ischaemic attack or minor stroke: implications for public education and organisation of services. BMJ Jan, 2004; 328:326.
6. Evenson KR, Rosamond WD, Morris DL. Prehospital and in-hospi-tal delays in acute stroke care. Neuroepidemiology. 2001, 20:65–76.
7. Fothergill A, Christianson TJ, Brown RD Jr, Rabinstein AA. Validation and refinement of the ABCD2 score: a population-based analysis. Stroke. 2009 Aug; 40(8):2669-73.
8. Giles MF, Rothwell PM. Transient ischaemic attack: clinical rel-evance, risk prediction and urgency of secondary prevention. Curr Opin Neurol. 2009 Feb; 22(1):46-53.
9. Gommans J, Barber PA, Fink J. Preventing strokes: the assessment and management of people with transient ischaemic attack. N Z Med J. 2009 Apr; 122(1293):3556.
10. Hill MD, Yiannakoulias N, Jeerakathil T, Tu JV, Svenson LW, Schopflocher DP. The high risk of stroke immediately after tran-
sient ischemic attack. A population-based study. Neurology. 2004; 62: 2015–2020.
11. Johnston SC, Albers GW, Gorelick PB, Cumbler E, Klingman J, Ross MA, Briggs M, Carlton J, Sloan EP, Vaince U. National stroke association recommendations for systems of care for TIA. Ann Neurol, 2010; doi:10.1002/ana.22332 [Pubmed].
12. Johnston SC, Rothwell PM, Nguyen-Huynh MN, Giles MF, Elkins JS, Bernstein AL, Sidney S. Validation and refinement of scores to predict very early stroke risk after transient ischaemic attack. The ABCD, California, and unified ABCD2 risk scores predicted stroke within 2, 7, and 90 days after TIA. Lancet 2007; 369:283–292.
13. Josephson SA, Sidney S, Pham TN, Bernstein AL, Johnston SC. Higher ABCD2 Score Predicts Patients Most Likely to Have True Transient Ischemic Attack. Stroke 2008;39;3096-3098.
14. Karunaratne PM, Norris CA, Syme PD. Analysis of six months’ referrals to a „one-stop“ neurovascular clinic in a district general hospital: implications for purchasers of a stroke service. Health Bull (Edinb). 1999 57(1):17-28.
15. Lavallée PC, Meseguer E, Abboud H, Cabrejo L, Olivot JM, Simon O, Mazighi M, Nifle C, Niclot P, Lapergue B, Klein IF, Brochet E, Steg PG, Lesèche G, Labreuche J, Touboul PJ, Amarenco P. A transient ischaemic attack clinic with round-the-clock access (SOS-TIA): feasibility and effects. The Lancet Neurology, 2007, Nov, Vol 6, Issue 11, pp 953-960.
16. Lindley RI. Patients With Transient Ischemic Attack Do Not Need To Be Admitted to Hospital for Urgent Evaluation and Treatment: Against. Stroke 2006;37;1139-1140.
17. Luengo-Fernandez R, Gray AM, Rothwell PM. Effect of urgent treatment for transient ischaemic attack and minor stroke on dis-ability and hospital costs (EXPRESS study): a prospective popu-lation-based sequential comparison. Lancet Neurol. 2009 Mar; 8(3):235-243.
18. Mant J, Ryan R, McManus R, Fletcher K, Kandiyali R, Barton P, Bryan S, Stevens A, Ford G, Rothwell P. What is the Optimum Model of Service Delivery for Transient Ischaemic Attack? Report for the National Co-ordinating Centre for NHS Service Delivery and Organisation R&D (NCCSDO). April, 2008 [Pubmed].
19. Merwick A, Albers GW, Amarenco P, Arsava EM, Ay H, Calvet D, Coutts SB, Cucchiara BL, Demchuk AM, Furie KL, Giles MF, Labreuche J, Lavallée PC, Mas JL, Olivot JM, Purroy F, Rothwell PM, Saver JL, Sheehan OC, Stack JP, Walsh C, Kelly PJ. Addition of brain and carotid imaging to the ABCD² score to identify patients at early risk of stroke after transient ischaemic attack: a multicentre observational study. Lancet Neurol. 2010 Nov, 9(11):1060-9.
20. Merwick A, Kelly PJ. Transient ischaemic attack clinics and man-agement of transient ischaemic attacks. Curr Opin Neur. 2011,Vol. 24, Issue 1, pp 50–58.
21. NICE. The diagnosis and acute management of stroke and transient ischaemic attacks. June, 2008. [Pubmed].
22. Power M, Blair P, Flynn P, Hunter AM, McCarron M, Russell C, Wiggam Iv, Brannigan P, Kerr P, Burns G, Herron M. Clinical Resource Efficiency Support Team (CREST). Guidelines for Investigation and Management of Transient Ischaemic Attack. October, 2006. [Pubmed].
23. Purroy F, Montaner J, Molina CA, Delgado P, Ribo M, Álvarez-Sabín J. Patterns and predictors of early risk of recurrence after transient ischemic attack with respect to etiologic subtypes. Stroke. 2007, 38(12):3225-3229.
24. Rothwell PM, Giles MF, Chandratheva A, Marquardt L, Geraghty O, Redgrave JNE, Lovelock CE, Binney LE, Bull LM, Cuthbertson FC, Welch SJV, Bosch S, Carasco-Alexander F, Silver LE, Gutnikov SA, Mehta Z. Effect of urgent treatment of transient isch-aemic attack and minor stroke on early recurrent stroke (EXPRESS study): a prospective population-based sequential comparison. The Lancet, 2007, Vol. 370; Issue 9596; pp 1432-1442.
25. Rothwell PM, Johnston SC. Transient Ischemic Attacks: Stratifying Risk. Stroke, Feb, 2006;37(2): 320 - 322.
26. Stead LG, Vaidyanathan L, Bellolio MF, Kashyap R, Bhagra A, Gilmore RM, Decker WW, Enduri S, Suravaram S, Mishra S, Nash D, Wood HM, Yassa AS, Hoff AM, Brown RD. Knowledge of signs, treatment and need for urgent management in patients presenting with an acute ischaemic stroke or transient ischaemic attack: a prospective study. Emerg Med J. 2008; 25(11):735-9.
27. Uchiyama S. Transient ischemic attack, a medical emergency. Brain Nerve. 2009, 61(9):1013-22.
28. Yang J, Fu J-H, Chen X-Y, et al. Validation of the ABCD2 Score to Identify the Patients With High Risk of Late Stroke After a Transient Ischemic Attack or Minor Ischemic Stroke. Stroke. 2010, 41(6):1298-1300.

Българска Неврология Bulgarian Neurology 16Май 2011
����� !"�!�$� ����
9��������������:;�����������;��������������������������������������������
<���=N�������6����:��������O
i9��������6����7���9�������6����������������d����������:���5���7�9�����f�6f�����?��<������6��<f�����6������'!(��)*+�!"����!�,*�*-�!��(��*����.���-����-�����*(.���!�"*�/�(�0!"��"1*�-��.�2�*"�+��!�"*�"3-��-��4"�
����35.*�4*��!�)�!"*��0*��"!(*5*."��-�/�'67�-��"�-*
�����������7���9�8
b?�8 ;€�8 j�6���98
�5������9�����8�?f<��9����B�
����������6���d�d������8
b?�8 ��9����:��������7�5��9�8
��9�8 h��8
���9��f9�8� ��:��������?�9?�9�8
�f�����9�����8�i7����8
���<�9��98����?����������:����98
������������������������;����������
������>����?
7-����2�.�����"*�!4�"�����.��0-��
.�)�����!�)��� ��!�!"8�"*9:.!��3�"��
"!��!�-*�*!�(*�'67���)*+�!"������!4.*�
4*��!�)�!"*��0�������"*��0�!�"*�
��/"!"�!�)����"�;!"�!�"*�4�*2"�(*�*�
�.�� !�!��!������)!+�*�����:�"!.����2 �
"*��*()��� !"�!�"*��!�!1�"<������������
�� �;
��9��������:�������������������������:��9����?�9?�8
;�����9�:�������9�������6��<f�������6�����
a���6�9������f������?��6��b6���������9���9�m�f�6�6�����������5�<���:�����?6�:��������
@����������������������������
=*��!���5!�! *��)�4��5"��������
/�"*���!�����)����.*>��(*5��3.*"�3 �
"*�)*+�!"�*8�"*��0�!�"*����-�.� �
1*-����8�*�!�2�����4�?2���
���5���5��������?������?�0_)�N`O�������6��7��������������9���6��9������?6�:���������9�
*AB�<C=�������e�6����m5��������5�����6<�9�9�6��������9��6f����9����9�����
j�6���98���p�rF��5��� ���E
��‚��rF��5��� ���F
��9���������������8
���9���p�ElF�GG/'�����5���9���p�VF�GG/'
���E
���9���‚�ElF�GG/'���5���9���‚�VF�GG/'
���F
�����:�����9���8
;5��9���������f�9�������@��7�9�
���`
€��������<c�����f�6�5����9�����5���7�9
���E
��<�� ���F��5��m�9����9���������:��9����?�9?�9���8
p�rF�?��<9� ���`��m5<�EF���TV�?��<9� ���E
‚�EF�?��<9� ���F
ƒ�>�����5��f�98����:��� ���E����� ���F
i���5���9��fd����f��N?�m5<�F���D�9:��O8

Българска Неврология Bulgarian Neurology 17Май 2011
�������D�����9�������;���?
������������������������;����E��
�����������8
���������������������������
���;F����?
� �������9���9��������7�9�����El� ��5��8
��7���9�9 � ���5�� � 5� � �� � ���:� � ?����?��� � f��6 � :��6 � ���7����6�����9������9���? �3.!"M%� �+*.� � �5����6���6�:������<�9� �ƒ�f������������?���������������5������m5��������a�����������?6���
����� � �� � 6� � ��7���9 � � � ���� � ����� � 9�� � ����:�� � �� � �5�� � ��� � ���:�������:���:��9��9��6f����9�8�
• 0_)�N`O�p�l�9��
• ����:������`��������:��ab�����6�����5����?���7�• ��9����<���9���9������• ���5���5��?�m5��������o:�9����������6?�������?�
��7���9�9 � 9��f�� � 5� � �� � 9��9��� � ��9 � ��������� � ��9��9� � ����9����6��f������ab����������������6�f���������?������������CFF�?����f��6�9�������f�m5������?����7�����9�2������������7�����9�9�d����d��9���������5������c��95����������7���9�9����5���5��������:����?�3.!"M%��+*.�������?�ab�������������������7���� � �������� � �5��m� � �� � 5�����9�:� � <9:������ � �������7�9�����`l�:����9���:��9������?�9?�9���9��
��7���9�9 � 9��f�� � 5� � �� � 9��9��� � ��9 � ��������� � ��9��9� � ����9����6��f������ab�����������������6�f���������?������������CFF�?��������5�9����DT2ETF�?��5�����������?��������9�c��������5����6�9��2�9�������f�m5������?����7�����9�2���������„�?�9������������������6���d����������B�������7���9�9 � ���5�� � 5� � �� � ���:� � ��? � ab� � ������� � � � �5��m� � ���5�����9�:� � <9:������ � � � �����7�9� � �� � D � 5�� � 9 � ��:��9 � �����?�9?�9���9��

Българска Неврология Bulgarian Neurology 18Май 2011
����� !"�!�%�
9��������������:;�����������;��������������������������������������������
<���=N�������6�����c��95������O
i9��������6����7���9�������6����������������d����������:���5���7�9�����f�6f�����?��<������6��<f�����6������'!(��)*+�!"����!�,*�*-�!��(��*����.���-����-�����*(.���!�"*�/�(�0!"��"1*�-��.�2�*"�+��!�"*�"3-��-��4"��
����35.*�4*��!�)�!"*��0*��"!(*5*."��-�/�'67�-��"�-*
�����������7���9�8
b?�8 ;€�8 j�6���98
�5������9�����8�?f<��9����B�
����������6���d�d������8
b?�8 ��9����:��������7�5��9�8
��9�8 h��8
���9��f9�8� ��:��������?�9?�9�8
�f�����9�����8�i7����8
���<�9��98����?����������:����98
������������������������;����������
������>����?
7-����2�.�����"*�!4�"�����.��0-��
.�)�����!�)��� ��!�!"8�"*9:.!��3�"��
"!��!�-*�*!�(*�'67���)*+�!"������!4.*�
4*��!�)�!"*��0�������"*��0�!�"*�
��/"!"�!�)����"�;!"�!�"*�4�*2"�(*�*�
�.�� !�!��!������)!+�*�����:�"!.����2 �
"*��*()��� !"�!�"*��!�!1�"<������������
�� �;
��9��������:�������������������������:��9����?�9?�8
;�����9�:�������9�������6��<f�������6�����
a���6�9������f������?��6��b6���������9���9�m�f�6�6�����������5�<���:�����?6�:��������
@����������������������������
=*��!���5!�! *��)�4��5"��������
/�"*���!�����)����.*>��(*5��3.*"�3 �
"*�)*+�!"�*8�"*��0�!�"*����-�.� �
1*-����8�*�!�2�����4�?2���
���5���5��������?������?�0_)�N`O�������6��7��������������9���6��9������?6�:���������9�
*AB�<C=�������e�6����m5��������5�����6<�9�9�6��������9��6f����9����9�����
j�6���98���p�rF��5��� ���E
��‚��rF��5��� ���F
��9���������������8
���9���p�ElF�GG/'�����5���9���p�VF�GG/'
���E
���9���‚�ElF�GG/'���5���9���‚�VF�GG/'
���F
�����:�����9���8
;5��9���������f�9�������@��7�9�
���`
€��������<c�����f�6�5����9�����5���7�9
���E
��<�� ���F��5��m�9����9���������:��9����?�9?�9���8
p�rF�?��<9� ���`��m5<�EF���TV�?��<9� ���E
‚�EF�?��<9� ���F
ƒ�>�����5��f�98����:��� ���E����� ���F
i���5���9��fd����f��N?�m5<�F���D�9:��O8

Българска Неврология Bulgarian Neurology 19Май 2011
�������D�����9�������;���8
������������������������;����E��
�����������8
���������������������������
���;F����?
� �������9���9��������7�9�����El� ��5��8
��7���9�9 � ���5�� � 5� � �� � ���:� � ?����?��� � f��6 � :��6 � ���7����6�����9������9���? �3.!"M%� �+*.� � �5����6���6�:������<�9� �ƒ�f������������?���������������5������m5��������a�����������?6���
����� � �� � 6� � ��7���9 � � � ���� � ����� � 9�� � ����:�� � �� � �5�� � ��� � ���:�������:���:��9��9��6f����9�8�
• 0_)�N`O�p�l�9��
• ����:������`��������:��ab�����6�����5����?���7�• ��9����<���9���9������• ���5���5��?�m5��������o:�9����������6?�������?�
��7���9�9 � 9��f�� � 5� � �� � 9��9��� � ��9 � ��������� � ��9��9� � ����9����6��f������ab����������������6�f���������?������������CFF�?����f��6�9�������f�m5������?����7�����9�2������������7�����9�9�d����d��9���������5������c��95����������7���9�9����5���5��������:����?�3.!"M%��+*.�������?�ab�������������������7���� � �������� � �5��m� � �� � 5�����9�:� � <9:������ � �������7�9�����`l�:����9���:��9������?�9?�9���9��
��7���9�9 � 9��f�� � 5� � �� � 9��9��� � ��9 � ��������� � ��9��9� � ����9����6��f������ab�����������������6�f���������?������������CFF�?��������5�9����DT2ETF�?��5�����������?��������9�c��������5����6�9��2�9�������f�m5������?����7�����9�2���������„�?�9������������������6���d����������B�������7���9�9 � ���5�� � 5� � �� � ���:� � ��? � ab� � ������� � � � �5��m� � ���5�����9�:� � <9:������ � � � �����7�9� � �� � D � 5�� � 9 � ��:��9 � �����?�9?�9���9��
Българска Неврология Bulgarian Neurology 20Май 2011
����� !"�!�&�
9���������������������;�>�����������������;�����G�>������H@������
�����������7���9�8 ������������:��d������8b?�8�������������������������������������������������������������������������������
;€�8……s……s……s……s……s……s……s……s……s……�bƒ�B�������������������
�5���8����������������������������������������������������������������������������������������������������������������������������������������������������������������������a��8��9�7s�f���������������������������������������������������������������
b?�8������������������������������������������������������������������������������ƒ��������������������������������������������������������������������������������5���8�����������������������������������������������������������������������������������������������������������������������������������������������������������������a�����8��9�7s�f����������������������������������������������������Z2G�* 8��������������������������������������������������������������������������
��9����:��������7�5��9�8 ��9����:�����������:���7�����s���������9��98��9�8��……s……s……�������������������������h��8��……8……����N`l�:�O ��9�8��……s……s……�������������������������h��8��……8……����N`l�:�O
��9����:���������:����8 ���:��d�����8��9�8��……s……s……�������������������������h��8��……8……����N`l�:�O ����:������������������������������������7�����9
��;����������c���7��9���������<�������:������9���8 ���9������?��6��s���<�������?�7��8
†�?�����6�s€����?�����6�
�� ���� ���
��9���������������8�������������s������������������
��5��m�9����9���������:��9����?�9?�9���8���������:��������?�������:��9����?�9?�9������5��m���‡ˆ�5���������������ˆ���
†�?�>����9�6�� ���<������6��<f�����6����� ƒ��<f�����6��9������� ������� �����5���7��s�9����� �9�������6�� ��6��9��� ���9�?������9���9�m �������������9�9��d��6�f�������8
q���������9��8 b6���5�����8
�����5���5��?�m5��������������� ����9��������>����9����������� ��b������������������������������������������� �������5���������9�������������� ����9�������������f�@���� ���6�:������<�9��������������������� ��†���������������������������������������� ��ƒ�>�����5��f�9����������������������� ���������5�?������������������������� �����5�:�����5�9�9�:��9�����
��ao9o��<c������� �����<?�7��������������>��
��9��s�h��8q�6<�9�98
������
����k;
���†���9���
���a�����7���5�
����������6�>��
������<��7����������9�9<�
��q��9������������f���5�f������7�������
��;�€����� ���a�s��qa�����������?6�������
�����������9�5�����9����
q��<���������?����?�5���?��9�8 ����m����?�5���?��9��9���:��9������?�9?�9�8

Българска Неврология Bulgarian Neurology 21Май 2011
0_)��N`O������8j�6���98 ‰sŠ�rF��5���
‚�rF��5���EF
ifd���6<�9�9�Š����9�
��9�������������������������:�����6?������
���9���‰sŠ�ElF����5���9���‰sŠ�VF��<��
��������EF
�����:���>����9����9��� ;5��9���������f�9������������@��7�9�������€��������<c����������f�6�5����9�����5���7�9��������<��
`
EF
��5��m�9����9��������:��9���?�9?�9���
‰sŠ�rF�?��<9�?�m5<�EF2TV�?��<9�‚�EF�?��<9�
��������`��������E��������F
ƒ�>�����5��f�9 ����:�������
��������E��������F
��:�����9������9�:��������������7���9���ab�8
��9����:��������:���������?����?�5���?��9�8��9�8��……s……s……�������������������������h��8��……8……����N`l�:�O�
��������CFF?������������������9�����6����������<�9������������:�����?�9?�������5��������?������������e�5f�������������5�?��`FF?�������9�����6�����6������?������������e����5�����DT?���?���9�9���lF?��e����7�����>���9����‚�C�W��9�>����9��6�����?�5���?��9�
�����������?���7���9���ab�8�2�5�����c������5��9����f�5��������5���9����7����6����������2�������9���?�9������7�5��9��e���6�f����f�m5�������9����EE`�2���7���9�9�5��f�5�����5�<m����������d����9������ab����������9�:���5�7������7�5��9��
Адрес за кореспонденция:Доц. д-р Д. Масларов, докторНеврологична КлиникаПърва МБАЛ-София, ЕАДБул. „Патриарх Евтимий” № 37София 1000e-mail: [email protected]

Българска Неврология Bulgarian Neurology 22Май 2011
SUMMARY
NEW CONCEPT FOR CHRONIC TREATMENT IN PARKINSON’S DISEASE
D. Bogdanova, N. Semerdjieva, I. MilanovUniversity Hospital ‘St. Naum’, Sofia
Parkinson’s disease (PD) is a progressive neurodegenerative disorder of dopamine dysfunction characterized by motor, cognitive, behavioral, and autonomic symptoms. A real problem is converting from early to late stage of this disease and the development of complications and motor fluctuations.
It is thought that motor fluctuations in Parkinson’s disease (PD) to a large extent result from pulsatile dopaminergic stimulation due to the short half-life and erratic absorption of oral levodopa therapy.
Several novel treatment options that improve continuous dopaminergic stimulation in PD have recently become available, including transdermal patch and oral controlled-release formulations of dopamine agonists, the continuous enteral delivery of levodopa/carbidopa via a pump system and deep brain stimulation of the subthalamic nucleus, which targets the abnormal neuronal activity ownstream of the striatal dopamine deficiency.
The concept of continuous dopaminergic stimulation will remain a central topic of therapeutic research in PD in the near future.
KEY WORDS: dopamine, Parkinson’s Disease, treatment
РЕЗЮМЕ
Паркинсоновата болест (ПБ) е прогресиращо невроде-генеративно заболяване, свързано с допаминергична дис-функция и с поява на двигателни, когнитивни, поведен-чески и автономни симптоми. Основен проблем, който възниква при хроничното лечение на Паркинсоновата болест е преминаването от ранна към късна форма на заболяването и появата на усложнения и двигателни флуктуации.
През последното десетилетие беше доказано, че лево-допа-индуцираните двигателни усложнения при ПБ са свързани с непостоянната или пулсионна стимулация на допаминовите рецептори в стриатума. Двигателните усложнения могат да се повлияят или дори да се пре-дотвратят с помощта на дълготрайно допаминергично лечение, което теоретично осигурява по-продължител-на стимулация на стриарните допаминови рецептори.
Съществуват няколко основни възможности за ста-билизиране на фармакокинетиката на леводопа, за пре-дотвратяване на пулсионното й въвеждане и постигане на продължителна допаминергична стимулация, като използване на допаминови агонисти с контролирано осво-бождаване орално и като трансдермални пластири, про-дължително дуоденално освобождаване на леводопа чрез помпена система, използване на орални катехол-орто-метил-транферазни (КОМТ) инхибитори, субкутанна или венозна инфузия на лизурид (lisuride) и апоморфин (apomorphine). Друга, немедикаментозна възможност, е дълбоката мозъчна стимулация на субталамичните
ядра (ДМС), която променя абнормната невронална активност и коригира стриарния допаминов дефицит и е алтернатива при пациенти с тежки двигателни флук-туации или тремор.
КЛЮЧОВИ ДУМИ: допамин, лечение, Паркинсонова болест
Паркинсоновата болест (ПБ) е прогресиращо нев-родегенеративно заболяване, чиито основни двигател-ни симптоми – брадикинеза, ригидност и тремор, се повлияват ефективно от допаминергично лечение (1). Лечението е индивидуално при всеки пациент, но отго-варя на общи правила и принципи, които са установе-ни (2). Терапевтичните възможности непрекъснато се допълват, като се появяват нови медикаменти и нови идеи за контрол на заболяването (2).
ПБ е модел на прогресираща допаминергична дисфун-кция с двигателни, когнитивни, поведенчески и авто-номни симптоми в различните стадии на прогресия. При ранна ПБ основна стратегия е невропротекцията, която би била успешна чрез повлияване на етиологията и патогенезата на невродегенеративния процес (24). Много са опитите в това отношение, но все още липсва конкретна методология за оценка на невропротекцията при пациенти с ПБ (13). Бяха публикувани консенсусни правила за лечение на ранна Паркинсонова болест (12,16), които са познати и прилагани вече и в нашите условия (2) и на които няма да се спираме в настоящото изло-жение.
Както самото заболяване, така и неговото лечение-то е хронично и динамично, и налага постоянни промени съобразно клиничното протичане. Основен проблем, който възниква пред нас при хроничното лечение на Паркинсоновата болест е преминаването от ранна към късна ПБ и появата на усложнения и двигателни флук-туации – феномени на изчерпване на дозата, on-off фено-мени, поява на неволеви движения (1). Смята се, че до голяма степен усложненията се дължат на пулсовата допаминергична стимулация при лечение с орални лево-допа медикаменти, които имат къс полуживот и недоб-ра абсорбция (28). Осигуряването на по-продължителна допаминергична стимулация е основно за профилактика и за лечение на хипо- и хиперкинетичните двигателни флуктуации при ПБ.
Целта на този обзор е да се направи преглед на новите терапевтични възможности в комбинация с установе-ните схеми, с цел постигане на продължителна допами-нергична стимулация (ПДС) (20).
Съществуват няколко основни възможности за ста-билизиране на фармакокинетиката на леводопа, за пре-дотвратяване на пулсионното й въвеждане и пости-гане на продължителна допаминергична стимулация, като използаване на допаминови агонисти с контро-лирано освобождаване орално и като трансдермални пластири, продължително дуоденално освобождаване на леводопа чрез помпена система, използване на орал-ни катехол-орто-метил-транферазни (КОМТ) инхиби-тори, субкутанна или венозна инфузия на lisuride и
ОбзорСЪВРЕМЕННА КОНЦЕПЦИЯ ЗА ХРОНИЧНО ЛЕЧЕНИЕ
ПРИ ПАРКИНСОНОВА БОЛЕСТ
Д. Богданова, Н. Семерджиева, И. МилановМБАЛНП „Св. Наум”, София

Българска Неврология Bulgarian Neurology 23Май 2011
apomorphine(20,28). Друга, немедикаментозна възмож-ност, е дълбоката мозъчна стимулация на субталамич-ните ядра (ДМС), която променя абнормната невронал-на активност и коригира стриарния допаминов дефицит и е алтернатива при пациенти с тежки двигателни флуктуации или тремор (20, 28).
Леводопа (l-DOPA) е най-мощното допаминергичено лечение, от което рано или късно, повечето от паци-ентите с ПБ се нуждаят. Продължителното леводопа лечение, заедно с прогресията на заболяването, водят до постепенно изчерпване на ефекта и до поява на дви-гателни флуктуации, включващи т.нар. off-периоди (при изчерпване на ефекта от лекарството) и дискинезии (поява на дистонични или хореични неволеви движе-ния)(28). Фармакокинетичните свойства на леводопа, с полуживот 90-120 минути, имат голямо значение на продължителността на двигателния отговор. Някои периферни фактори повлияват плазмените нива на лево-допа - абнормната киселинност на стомаха, намаленият мотилитет на червата и аминокиселините в храната водят до нерегулярни флуктуации в плазмените нива, независимо от правилно дозирания орален прием на медикамент (25,28). При дегенерация на допаминер-гичните терминали в стриатума до <20%, леводопа прогресиращо започва да се метаболизира и в недопами-нергични клетки (например в серотонинерични неврони), което води до нерегулярно освобождаване на леводопа с липса на обратен захват от допаминергичните клет-ки. Като последица възникват големи флуктуации в екстрацелуларната концунтрация на допамин в струа-тума, паралелно на флуктуациите в плазмените нива на леводопа. Стриарните неврони, нормално свързани с относително стабилно синаптично освобождаване на допамин, стават „чувствителни”, тяхната импулсация се променя и това води до низходящи промени в невро-налната активност в базалните ганглии (28). Така при двигателните off-периоди има повишени абнормни зал-пове от субталамичното ядро, което има инхибиторно влияние върху изходящите ядра на базалните ганглии, вътрешната част на globus pallidus и ретикуларната част на substantia nigra. Повишеното освобождаване от тези ядра на гама-аминомаслена киселина (ГАМК) инхи-бира таламокортикалните и стволовите двигателни пътища и води до акинезия. Обратни са промените в патерна на невронална активност при дискинетичните on-периоди (28).
Освен двигателни усложнения, заместителната допаминергична терапия води също и до поведенчески нарушения като импулсивни нарушения на контрола (хиперсексуалност, патологично пазаруване, склонност към хазарт, абнормни репетитивни безцелни действия, натрапливо вземане на лекарства и други. Всички тези двигателни, когнитивни и поведенчески нарушения се припокриват в базалните ганглии (29). Това потвърж-дава нуждата от внимателно прецизиране на наличните методи на лечение, както и от упорито търсене на нови възможности.
Ранното лечение на експериментален MPTP модел на паркинсонизъм с допаминови агонисти (ДА) с дълъг полуживот отлага във времето появата на двигателни флуктуации, в сравнение с монотерапията с леводопа (28). Анализът на клиничните данни показва, че начал-ното лечение с т. нар. допаминови агонисти отлага във времето появата на двигателни усложнения, но е свързано с по-лош двигателен контрол и с по-висока честота на не-двигателни странични явления, в сравне-ние с лечението с леводопа (6). Допаминовите агонисти
pramipexole и ropinirole упражняват невропротекти-вен ефект в клетъчни култури и в експериментални животни чрез антиоксидативен и анти-апоптотич-ни механизми (14), доказани чрез невроизобразяващи методики (SPECT и PET). Проучването CALM-PD (27) сравнява pramipexole и леводопа за 4 години, измервайки загубата на неврони, изследвано чрез SPECT. Pramipexole значително редуцира невроналната загуба, в сравнение с леводопа (23). В проучването REAL-PET (31) се срав-няват ropinirole и леводопа в продължение на 2 години и при използване на PET. При лекуваните с ropinirole има много по-малка редукция на захвата на 18F-dopa (13.4%) в сравнение с лекуваните с леводопа (20.3%, p = 0.022) (31). Резултатите подсказват невропротективен ефект от допаминови агонисти или невротоксичност от леводопа, но е много възможно запазването на неврони при лечение с ДА да се дължи на съвсем друг фармакологичен ефект. В много от подобните проучвания няма данни за плацебо и липсва корелация между клинични и невроизобразява-щи данни, което не позволява окончателно заключение (23). В клиничната практика навлизат и нови формули на допаминови агонисти, като rotigotine трансдермални системи и ropinirole с контролирано освобождаване (23). Предимствата на тези нови форми също все още не са доказани в клинични проучвания.
Водноразтворим допамин, удобен за подкожно, веноз-но, сублингвално и назално приложение, e apomorphine (30). Редица клинични проучвания несъмнено показват, че аpomorphine има антипаркинсонов ефект, близък от леводопа (28). Продължителна допаминергична стимула-ция с апоморфинова инфузия постига редукция на диски-незиите - средно с 33%, редукция на off-фазата средно с 61% и повишаване на on-периода (30). Приложението обаче, е свързано с чести странични ефекти, предимно поради локално възпаление, формиране на подкожни ноду-ли, гадене, по-рядко ортостатична хипотония, сомно-лентност и халюцинации (30).
Друг начин да се удължи допаминергичния отговор е чрез инхибиране на ензима катехол-орто-метил тран-сфераза (КОМТ) с медикаменти като entacapone (14, 28). Заради късият си полуживот, entacapone изисква често дозиране- до 8 пъти дневно, като повечето паци-енти вземат entacapone със всяка доза леводопа (14). Друг медикамент е tolcapone, но той е рядко използван поради случаи на остра чернодробна недостатъчност (14). Теоретично КОМТ инхибиторите имат предим-ства над медикаментите на леводопа с контролирано освобождаване тъй като увеличават плазмената кон-центрация на леводопа без да удължават абсорбцията и без да увеличават времето до достигане на пикова кон-центрация. Фармокологично това води до удължаване на „on” времето без значимо влошаване на дискинезиите (14). През 2003 г е одобрена тройна комбинация, съдър-жаща леводопа, карбидопа и entacapone. В рандомизирано проучване върху 132 здрави контроли концентрацията на леводопа при тройната комбинация е подобна на тази при отделно прилагане на отделните компоненти, показващо еквивалентна фармакокинетика (14).
Използването на продължително допаминергична стимулация чрез използване на допаминови агонисти и чрез стабилизиране на периферните плазмени леводопа нива с помощта на КОМТ днес е основен прийом в лече-нието на ПБ.
През последните години беше въведен методът на продължителната дуоденална инфузия с освобождаване на леводопа/карбидопа под форма на гел в дуоденума чрез перкутанна тръба и помпа - лечението с duodopa (8).

Българска Неврология Bulgarian Neurology 24Май 2011
То осигурява по- постоянни плазмени нива на леводопа, в сравнение с оралните препарати, поради това, че зао-бикаля някои проблеми като изпразването на стомаха и абсорбцията в тънките черва. Методът вече е достъ-пен в България и е алтернатива на венозното приложе-ние на леводопа, което не е подходящо за практиката поради хидрофилността на медикамента, изискваща много голем обем течност за разтваряне (8). Duodopa e гел за интестинално приложение, съдържащ Levodopa: Carbidopa в съотношение 4:1. Системата се състои от портативна електронна помпа, чрез която автоматич-но се подава определената от специалист доза, и пер-манентна тръбна система, чрез която медикаментът се излъчва директно в duodenum/jejunum, посредством перкутанна ендоскопска гастростома. По този начин се осигурява бърза резорбция, независеща от стомаш-ния пасаж (8). Непрекъсната инфузия води до стабилна плазмена концентрация и подобряване на фармакоки-нетиката на леводопа (3, 4, 8). Методът се прилага като монотерапия. В редки случаи, при необходимост, може да бъдат приложени и други антипаркинсонови медикаменти. Обикновено в първите 6 месеца дозата се покачва, след което нуждите намаляват трайно с около 20% (3). При нужда от покачване на дозата, това може да стане с екстрадоза от 10-20 mg, което не причинява значителни пикове в кривата на концентрацията, за разлика от таблетната форма, която може рязко да повиши концентрацията и да доведе до дискинезии (3). Duodopa се прилага предимно през деня - средно 16 часа. При пациенти с нощна акинезия или нарушения на съня може да се приложи в 24 часова непрекъсната инфузия. В такива случаи количеството дуодопа приложено през нощта е около 2/3 (60-80%) от дневната доза. Според някои автори това може да доведе до непоносимост, психози или дискинезии, но по данни от последни про-учвания има пациенти които се повлияват добре от денонощно приложение (3, 4, 8).
Лечението с Duodopa води до намаляване на двига-телните флуктуации, редуцира времето с дискинезии и тежестта им и отчита значително подобрение в качеството на живот (92%) (3,4,8). Доказано е, че след 4-7 години лечение с Duodopa, моторните симптоми у болшинството пациенти търпят подобрение, в срав-нение с времето преди включването към терапията (4). Недостатъците са свързани с това, че е инвазивен метод, около оперативната раната може да има възпа-ление и енетуално перитонит (3,8). Едно от най-чес-тите усложнения е запушване, усукване или разкачване на тръбната система, което може да се предизвика от ускорена перисталтика, закачане на катетъра при бурни хиперкинези, неспазване правилата за употреба и др. Нежеланите лекарствени реакции са същите като при леводопа - дискинезии, халюцинации, ортостатична хипотония, замайване, ексцесивна сънливост (3, 4, 8).
Съвременен немедикаментозен метод за осигуря-ване на продължително допаминергична стимулация в базалните ганглии е дълбоката мозъчна стимулация (ДМС)(28). Свързана е с имплантиране на на електрод с четири контакта в определена таргетна зона в мозъ-ка (при ПБ това е nucleus subthalamicus), обикновено двустранно. Електродът е свързан със стимулатор, поставен подкожно в областта на гръдния кош (18). Високочестотна електрическа стимулация (>100Hz) на таргетната зона имитира ефект на лезия (18,28). Нискочестотна стимулация няма благоприятен ефект и дори може да задълбочи симптомите (28). Целта е повлияване на абнормната невронална активност на
базалните ганглии в условия на допаминергичен дефицит в стриатума. Въпреки, че конкретните механизми не са съвсем ясни, успехът на този метод е свързан с дви-гателния отговор на леводопа (28). Симптоматичния ефект на дълбоката мозъчна стимулация е потвърден в много клинични проучвания - в периода 1993-2004 са пуб-ликувани 34 статии с данни за общо 921 пациента (15). Прилагането на такава стимулация на практика замес-тва средно 60% от допаминергичното лечение и прибли-зително 10% от пациентите не се нуждаят изобщо от лечение с леводопа (15). Дискинезиите намаляват средно с 69.1%, продължителността на off–периодите - средно с 68.2% (15). Страничните ефекти са свръзани с хирур-гичния риск, предимно от хеморагия, което се установя-ва в 3.9% от пациентите (28). Психичните усложнения – обърканост, депресия, хипомания и други, са редки (28). При оценка на качеството на живот на пациенти с ПБ след ДБС, подобрението значително превишава риска от възможни странични ефекти (7). Много важен факт е, че споменатата „чувствителност” на допаминергични-те рецептори е обратима след ДМС – пациентите вече нямат толкова изразени леводопа-предизвикани диски-незии след 1 година лечение (5). Дали това е резултат от вторични промени в медикаментозното лечение или от продължителната електрическа стимулация, не е изяснено (28).
ПБ има закономерно прогресиращ ход, което се отра-зява във влошаване на двигателната активност, в час-тност на аксиалните симптоми (1,28). След ДМС пос-тепенно прогресират и когнитивните промени, апатия, промени в екзекутивните функции, което особено касае по-възрастните пациенти. Ретроспективно проучване показва, че пациенти над 70 годишна възраст имат вло-шаване на аксиалните симптоми дори при допълнителна леводопа терапия (28). Възрастните пациенти имат и по-малко подобрение в качеството на живот, незави-симо от подобрението на симптомите след ДМС (28). Идеалният кандидат за субталамична ДМС е пациент с идиопатична ПБ, с отличен ефект от леводопа, с тежък и неповлияващ се от лечение тремор или с двигателни флуктуации, всладствие леводопа лечение. Сред изключ-ващите критерии са деменция, психотични симптоми и депресия. Особено подходящи са млади пациенти с ПБ, които най-добре се вместват в критериите (28).
Основна дилема при съвременното хронично лечение на ПБ е изборът между лечението с дуодопа и с ДМС. И при двата метода основен принцип е добрия терапевти-чен отговор от леводопа. Липсват данни от клинични проучвания, сравняващи двата инвазивни метода. Все пак, клиничният опит показва, че определени субгру-пи пациенти са подходящи за едното или за другото лечение. Въпреки, че 30.9% от случайно подбрана група пациенти с ПБ имат двигателни усложнения, едва 1.6-4.5 % отговарят на критериите за лечение с ДМС (17). Основно ограничение е горната възрастова граница от 70 години. Като се има предвид, че именно това е прицелната възрастова група за заболяването, ограни-чението е проблем и показва нуждата от алтернативи при голяма група пациенти, при които двигателните усложнения не могат да се контролират с перорално лечение. Дуоденалната леводопа инфузия има по-малко ограничения по отношение на възраст, когнитивно и общо соматично състояние. Клиничния опит предполага сходен ефект, но по-леки и обратими странични ефек-ти при лечението с дуодопа (4). Преценката е строго индивидуална и ако пациентът покрива критериите и за двата метода на лечение, решаващи са субективни

Българска Неврология Bulgarian Neurology 25Май 2011
фактори като желанието на самия пациент и неговите близки (28).
Концепцията на продължителна допаминергична сти-мулация отговаря и на важни за неврологичната прак-тика въпроси, като кога да се започне лечение, как да се дозира леводопа, кога да се премине към продължителна допаминергична стимулация, как да се повлияят двига-телните флуктуации (25). Времето на започване на допа-минергичното лечение при ПБ е основен проблем в нев-рологията, за който все още липсва консенсус. Леводопа е най-ефикасният клинично медикамент, но времето на започване на лечението зависи от преценка на риска от поява на усложнения. Допреди години се предпочи-таше лечението да се отложи с оглед потенциалните дългосрочни негативни ефекти от допаминергичното лечение върху прогресията, идващо от хипотезата за невротоксичен ефект от леводопа поради продукцията на свободни радикали, които потенцират дегенератив-ния процес в базалните ганглии (10). Съществуват и доказателства против тази хипотеза (22). Дозите, при които леводопа потенциира клетъчната смърт, многок-ратно превишават терапевтичните дози при хора и при животни (19). Проучвания върху животни доказват, че дискинезиите са по-леки, когато приемите на леводопа са по-чести и при комбинирано лечение с инхибитор на КОМТ (28). Напоследък се утвърждава хипотезата, че ранното възстановяване на нормалната физиология на базалните ганглии с помощта на допаминови агонисти може да подпомогне компенсаторните механизми и да отложи необратимите промени в невроналните вери-ги на базалните ганглии, характеризиращи клиничната прогресия при ПБ (25, 28).
Съществуват и нови перспективи за продължителна допаминергична стимулация. Публикувани са данни за няколко инвазивни метода за клетъчна терапия с оглед на продължителна локална допаминергична стимулация в стриатума. Интересна възможност, клинично про-учвана в момента (фаза 2), е имплантация в putamen на човешки ретинални пигментни епителиални клетки (26). Тези клетки произвеждат леводопа, изолират се от човешка очна тъкан постмортем, култивират се и се имплантират в мозъка. Те не правят синаптичен контакт с околната тъкан, но играят роля на локал-ни леводопа „биореактори” (26). В отворено пилотно проучване при 6 пациента с напреднала ПБ и едност-ранна имплантация е установено средно подобрение от 48% при оценка на off – периодите след 1 година без повишаване на тежестта или продължителността на дискинезиите (26). В период на проучване (фаза 1) е инт-растриална инфузия на вирусен компонент, съдържащ ген за човешка ароматна аминокиселинна декарбокси-лаза (9). Трансплантационното лечение е в застой, след публикуване на данните от две независими проучвания, които не потвърждават ефект от трансплантиране на ембрионална мезенцефални клетки при ПБ (11,21). Тези методи са в експериментален стадий, но те потвърж-дават надежността на продължителната допаминер-гична стимулация като основна стратегия в лечението на Паркинсоновата болест.
В заключение, концепцията на продължителна допа-минергична стимулация при Паркинсонова болест цели избягване на непостоянната или пулсионна стимулация на допаминовите рецептори в стриатума и предотвра-тяване на двигателните усложнения. Тя включва изпол-зване на допаминови агонисти, продължително дуоде-нално освобождаване на леводопа чрез помпена система, използване на орални КОМТ инхибитори, субкутанна или
венозна инфузия на апоморфин, както и дълбока мозъчна стимулация на субталамичните ядра.
ЛИТЕРАТУРА1. Георгиев, Д., Миланов, И., Чипилски, Л. Актуални проблеми на
някои екстрапирамидни заболявания. София, 1996.2. Консенсус за диагностика и лечение на Паркинсоновата
болест. Двигателни нарушения 7, 2, 2010.3. Antonini A., Isaias I., Canesi M., et al. Duodenal levodopa infusion
for advanced Parkinson’s disease: 12th month treatment outcome. Mov Disord, 2007, 22, 1145-1149.
4. Antonini A., Mаnchini F.,Canesi M et al. Duodenal levodiopa infusion improve quality of life in advanced Parkinson’s disease. Neurodegenerative disease, 2008, 5, 244-246
5. Bejjani BP, et al. Levodopa-induced dyskinesias in Parkinson’s disease: is sensitization reversible? Ann Neurol, 2000, 47(5), 655–658.
6. Bode M. Dopamine agonist therapy in early Parkinson’s disease. A survey of a Cochrane Review. Ugeskr Laeger. 2009, 171(41), 2996-2999.
7. Deuschl G, et al. Deep brain stimulation: postoperative issues. Mov Disord, 2006, 21 (Suppl. 14), 219–237.
8. Devos D. Patient profile, indications, efficacy and safety of duodenal levodopa infusion in advanced Parkinson’s disease. Mov Disord, 2008, 24, 993-1000
9. Eberling JL, et al. Results from a phase I safety trial of hAADC gene therapy for Parkinson disease. Neurology, 2008, 70(21), 1980–1983.
10. Fahn S, Cohen G: The oxidant stress hypothesis in Parkinson’s disease: evidence supporting it. Ann Neurol 1992, 32, 804-812.
11. Freed CR, et al. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Engl J Med, 2001, 344(10), 710–719.
12. Goetz CG, Poewe W, Rascol O, Sampaio C: Evidence-based medical review update: pharmacological and surgical treatments of Parkinson’s disease: 2001 to 2004. Mov Disord 2005, 20, 523-539.
13. Hauser RA, Zesiewicz TA: Clinical trials aimed at detecting neuroprotection in Parkinson’s disease. Neurology 2006, 66(suppl 4), 58-68.
14. Jankovic J., Aguilar G. Current approaches to the treatment of Parkinson’s disease. Neuropsychiatr Dis Treat., 2008, 4(4), 743–757
15. Kleiner-Fisman G, et al. Subthalamic nucleus deep brain stimulation: summary and meta-analysis of outcomes. Mov Disord, 2006, 21(Suppl. 14), 290–304.
16. Miyasaki JM, Martin W, Suchowersky O. Practice parameter: initiation of treatment for Parkinson’s disease: an evidence-based review: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2002, 58, 11-17.
17. Morgante L, Morgante F, Moro E, Epifanio A. How many parkinsonian patients are suitable candidates for deep brain stimulation of subthalamic nucleus? Results of a questionnaire. Parkinsonism Relat Disord., 2007,13(8),528-531.
18. Moro E, et al. The impact on Parkinson’s disease of electrical parameter settings in STN stimulation. Neurology, 2002, 59(5), 706–713.
19. Murer MG, Dziewczapolski G, Menalled LB. Chronic levodopa is not toxic for remaining dopamine neurons, but instead promotes their recovery, in rats with moderate nigrostriatal lesions. Ann Neurol 1998, 43, 561-575.
20. Nutt JG. Continuous dopaminergic stimulation: Is it the answer to the motor complications of Levodopa? Mov Disord. 2007 Jan, 22(1), 1-9
21. Olanow CW, et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol, 2003, 54(3), 403–414.
22. Simuni T, Stern MB: Does levodopa accelerate Parkinson’s disease? Drugs Aging 1999, 14, 399-408.
23. Simuni, T., Lyons K., Pahwa R., Hauser R., Comella C., Elmer L., Weintraub D. Treatment of Early Parkinson’s Disease Part 2. Eur Neurol 2009, 61, 206-215
24. Shoulson I: Where do we stand on neuroprotection? Where do we go from here? Mov Disord 1998, 13, 46-48.
25. Stocchi, F. New Concepts in the Management of Parkinson’s Disease. Neurological Review, 3, 1, 2006, 38-39.
26. Stover NP, et al. Intrastriatal implantation of human retinal pigment epithelial cells attached to microcarriers in advanced Parkinson disease. Arch Neurol, 2005, 62(12), 1833–1837.
27. The Parkinson Study Group: Dopamine transporter brain imaging to assess the effects of pramipexole vs. levodopa on Parkinson disease progression. JAMA 2002, 287, 1653–1661.
28. Volkmann, J. Novel Treatment Options for Advanced Parkinson’s Disease. Neurological Review, 3, 1, 2006, 28-30.
29. Voon V et al. Chronic dopaminergic stimulation in Parkinson’s

Българска Неврология Bulgarian Neurology 26Май 2011
disease: from dyskinesias to impulse control disorders. Lancet Neurol, 2009, 8(12), 1140-1149.
30. Werner P.,Gregor K. Apomorphine: An underutilized therapy for Parkinson’s disease. Mov disord, 2001, 5, 5, 789-794.
31. Whone AL et al. Slower progression of Parkinson’s disease with ropinirole versus levodopa: The REAL-PET study. Ann Neurol 2003, 54, 93–101.
Адрес за кореспонденция:Д-р Десислава Богданова, дмМБАЛНП “Св. Наум”, ул. Любен Русев №1, София 1113, България[email protected]
Address for correspondence:Dessislava Bogdanova, MD, PhDUniversity Hospital St. Naum, Luben Russev Str 1, Sofia 1113, [email protected]
SUMMARY
LACOSAMIDE (VIMPAT) – A NEW POSSIBILITY FOR SUCCESSFUL TREATMENT OF PARTIAL EPILEPSIES
M. RaschevaMore than 30% of patients with epilepsy have inadequate
seizure control with available antiepileptic drugs (AEDs) or have adverse drug events (AEs). That is the reason for searching new AEDs with improved effectiveness. Lacosamide (LCM), a new AED, is approved for the add-on treatment of partial-onset seizures in adults. LCM acts with a new, different mechanism of action - selective enhancement of Na chanel slow inactivation without affecting fast inactivation. LCM has favourable farmacokinetic characteristics and potent anticonvulsant activity in various animal models including kindling-induced epileptogenesis. Both oral and intravenous formulations of LCM are being developed.
According to pooled data from three phase II/III studies LCM is effective as add-on therapy in adult patients (N=1308) with uncontrolled partial-onset seizures - the percentage of patients with >50% reduction of seizures frequency is greater in patients with add-on LCM than in add-on placebo – 34% for LCM 200 mg/day, 40% for 400 mg/day, 23% - for placebo group. The long-term efficacy is confirmed in extension phase of 3 phase II/III studies – responder rate is keeping on and even increases (the 50% responder rate is 46,6%). LCM is well tolerated.
AEs are generally mild to moderate and appear in the phase of titration. Dizziness is the most common. Open-label extension studies (observations more than 5,5 years) demonstrate the long term tolerability and safety of oral LCM. No new safety concerns are identified with long-term treatment. No clinically important changes are observed in emotional state and mood, cognition, body weight vital signs and laboratory. LCM demonstrates efficacy and safety regardles of wether or not it is added to AED regimens containing traditional sodium channel-blocking AEDs or AEDs with different mechanism of action.
In conclusion, add-on LCM offers promise in achieving significant and sustained seizure control among patients with poorly controlled partial-onset epilepsy. LCM is well tolerated over several years of treatment in optimal dose of 400 mg/day and may be a choice for better health quality of life in those patients.
KEY WORDS: lacosamide, partial onset seizures, add-on therapy, efficacy, tolerability, safety.
РЕЗЮМЕ
Над 30% от пациентите с епилепсия нямат адеква-тен контрол на епилептичните припадъци с достъп-ните антиепилептични медикаменти (АЕМ) или имат значителни нежелани странични действия (НСД). Това е причина за търсене на нови АЕМ с подобрена ефектив-ност.
Lacosamide (LCM), е нов АЕМ, одобрен като добавъч-на терапия при припадъци с парциално начало при паци-енти в зряла възраст. LCM е с нов, различен механизъм на действие (изборно увеличава бавната инактивация на натриевите канали без да въздейства върху бързата инактивация), има благоприятна фармакокинетика и е с мощна антиконвулсивна активност при различни модели на епилепсия при експериментални животни, включително при kindling- индуцирана епилептогенеза. Съществуват както перорална, така и интравенозна форма, които са биоеквивалентни при прилаганите лечебни дози. Според обединените данни от 3-те големи рандомизирани, двойно-слепи, контролирани клинични проучвания (RCT) във фаза II/III, LCM е ефективен като добавъчна терапия при пациенти над 16 г. (N=1308) с неконтролирани с парциално начало припадъци – процен-тът на пациентите с >50% редукция на припадъците е по-голям в групата с добавен LCM в сравнение с групата с добавен плацебо (34% за LCM 200 мг/дн, 40% за 400 мг/дн и 23% - за плацебо групата). Дългосрочната ефи-касност се потвърждава от продължителната фаза на 3-те големи проучвания – процентът на отговарящите на терапията е висок и дори нараства във времето (с 50% RR са 46,6% от случаите).
LCM се понася добре, НСД обикновено са леки до умерени и се срещат предимно в периода на титрация. Най-често НСД е замаяност. Отворените продължи-телни проучвания (наблюдение над 5,5 г.) демонстри-рат дългосрочна добра поносимост и безопасност на пероралния LCM. Не се описват нови НСД при дългос-рочното лечение. Не се наблюдават клинично значими промени в емоционалното състояние и настроението, в когницията, телесното тегло, жизнените показатели и лабораторните изследвания. LCM показва ефикасност и безопасност независимо от това дали се прилага като добавъчна терапия към АЕМ отнасяни към традици-онните, блокиращи натриевите канали или към АЕМ с различен механизъм на действие.
ОбзорLACOSAMIDE (VIMPAT) - ЕДНА НОВА ВЪЗМОЖНОСТ
ЗА УСПЕШНО ЛЕЧЕНИЕ НА ПАРЦИАЛНИ ЕПИЛЕПСИИ
М. Рашева – УМБАЛ «Царица Йоанна-ИСУЛ» София

Българска Неврология Bulgarian Neurology 27Май 2011
И като извод, добавъчната терапия с LCM обещава значим, поддържащ и продължителен контрол на припа-дъците при пациенти с неконтролирана парциална епи-лепсия. LCM се понася добре при продължаващо с години лечение, в доза 400 мг/дн и може да бъде средство на избор за подобряване на качеството на живот свързано със здравето.
KEY WORDS: lacosamide, partial onset seizures, add-on therapy, efficacy, tolerability, safety.
ВЪВЕДЕНИЕ
Епилепсията е едно от най-честите хронични нев-рологични нарушения, като в света са засегнати над 50 милиона души. Всяка година възникват над 80 нови случаи на 100000 от населението (38). По-висок е отно-сителният дял на пациентите с парциални епилепсии (33), а тяхната фармакотерапия е един от проблемите в клиничната неврология.
В развитите страни на пациентите с новодиагнос-тицирана епилепсия се включва лечение с антиепилепти-чен медикамент (АЕМ) (24) и монотерапията е успешна при повече от половината от случаите, включително при невисоки дози (34). При още 10% от новодиагности-цираните се постига контрол на припадъците с въвеж-дането на втори или трети АЕМ и само при няколко процента допълнително - при съчетано лечение. При над 30%, обаче, не се постига успех (24) и епилепсията се определя като рефрактерна или медикаментозно резистентна.
Според новата дефиниция на ILAE - резистентна е епилепсията “при неуспешни адекватни опити за лечение с 2 толерирани и съответно избрани и прилагани АЕМ, като монотерапия или в комбинация, за постигане на свобода от припадъци” (25). Причините за резистен-тност могат да бъдат както генетични, така и придо-бити (32). Съвременните хипотези, които обясняват резистентността са две. Според първата, определяна като целева или фармакодинамична, промените са на рецепторно ниво с нарушение на афинитета и ефикас-ността за свързване с целта поради намалена сензитив-ност за АЕМ. Втората, транспортна хипотеза, приема, че дотигащият до мозъка АЕМ е в недостатъчно коли-чество, поради слаба пропускливост на кръвно-мозъчна-та бариера и увеличен транспорт, извеждащ медикамен-та и непозволяващ достигането му до епилептичните неврони в достатъчна концентрация (8).
От клиничната практика и от рандомизираните кон-тролирани клинични проучвания (RCT) е добре известно, че медикаментозната резистентност не е постоянна и може да се променя, например при добавяне на нов АЕМ с уникален механизъм на действие. Оказва се, че в последните 26 г. комбинации с нови АЕМ са подобрили процента на продължителна свобода от пристъпи при новооткрити епилепсии от 64 до 68,4% (8). Според някои клинични проучвания възможност за свобода от епилептични припадъци с добавъчната терапия при кон-тингент с резистентни епилепсии се постига при 5% - 19% от случаите (9, 26).
Въпреки големия брой на достъпни нови АЕМ, неже-лани страничните действия (НСД) на някои от тях ограничават приложението им (14). Ензимни индуктори или блокери, както и различен процент на свързване с плазмените протеини могат да предизвикат НСД още при ниски нива на АЕМ (1). Комбинираното лечение с АЕМ с различен механизъм на действие е една добра терапевтична възможност. АЕМ от ново поколение са
насочени към подобряване и улесняване на приложението, намаляване на лекарствените взаимодействия, намаля-ване на НСД, откриване на АЕМ с уникален механизъм на действие, търсене на невропротективна активност. “Всеки появил се на пазара нов АЕМ е и нов шанс за паци-ента да се освободи от припадъците”- Elger, 2010.(цит. по 1). Следователно необходими са нови АЕМ, които да намаляват честотата и тежестта на епилептичните припадъци и да се понасят добре от пациентите.
LACOSAMIDE (VIMPAT ) – РАЗЛИЧЕН МЕХАНИЗЪМ НА ДЕЙСТВИЕ
Lacosamide (LCM) е нов АЕМ за допълващо лечение на припадъци с парциално начало, одобрен за приложение в Европейския съюз при пациенти над 16 г. и в САЩ - над 17 г. (13).
LCM е функционализирана аминокиселина, оптичен антипод на L-серин (1, 29) - Фиг 1.
LCM е АЕМ с нов, различен от другите АЕМ меха-низъм на действие (5). В норма волтаж-зависимите Na канали могат да се отворят чрез деполяризация на мемб-ранния потенциал (МР) под критично ниво. Отварянето причинява нахлуване на Nа йони вътреклетъчно. След мсек каналите се затварят и са в инактивирано бързо състояние. След реполяризация на МР каналите са в със-тояние на почивка и са отново достъпни за отваряне. Електрофизиологични изследвания in vitro доказват, че лека и продължителна деполяризация може да въведе Na канали в бавна инактивация, за сек до мин. LCМ действа селективно върху бавната инактивация на Na канали, която увеличава, без да засяга бързата инактивация, различно от carbamazepine (CBZ), phenitoin (PHT) и lamotrigine (LTG) - Cross et al. 2009 (13). По този начин хипервъзбудимите невронални мембрани биват стаби-лизирани, намалява продължителната наличност на Na канали готови за активация и следователно, намалява способността на епилептичните неврони да поддър-жат продължителни серии от репетитивни разряди. Успоредно с това физиологичната невронална възбуди-мост не се променя (3).
Нито LCM, нито метаболитите му се свързват значимо с някои от местата на свързване на другите АЕМ или аналгетици, няма въздействие върху обратния захват или метаболизма на големите невромедиатори като норадреналин, допамин, серотонин или гама амино-маслена киселина (GABA), също липсва потискане на GABA-трансаминаза (5).
Фиг. 1.

Българска Неврология Bulgarian Neurology 28Май 2011
LCM (VIMPAT) – БЛАГОПРИЯТНА ФАРМАКОКИНЕ-ТИКА
При перорално приложение LCM е с пълна и бърза абсорбция, без значение от храната (10, 15, 37).
LCM е с линейна фармакокинетика и бионаличност около 100% (6, 21).
cmax се достига за 1-4 ч. след перорален прием (21), а полуживотът на елиминация е 13 ч. Препаратът е при-ложим в 2-кратен дневен прием (6, 21).
Интравенозната форма на LCM е биоеквивалентна за C max и AUC в инфузия за 30 или 60 мин с пероралната форма при дози от 200 до 600 мг/дневно (23).
LCM е с незначително свързване с плазмените проте-ини (37). Поради това не се измества от други медика-менти (3).
Фармакокинетиката и на пероралната и на интра-венозна форма е дозозависима. Постоянна плазмена концентрация се постига за 3 дни (13).
LCM взаимодейства минимално с CYP-450 изоензими и не въздейства върху метаболизма на други медикамен-ти (metformin, digoxin, оmeprazole), както и те не про-менят неговите нива (3). Няма влияние върху нивата на пероралните противозачатъчни медикаменти. Макар че 2C19 чернодробен изоензим е отговорен за образуването на LCM-метаболита, приложението на CYP2C19 индук-тори или инхибитори не променя значимо фармакокине-тиката на LCM, т.е. налице е минимален метаболизъм чрез този изоензим.
Класическите ензимни индуктори (CBZ, PHT и РВ) при комбинирано лечение с LCM не въздействат клинич-но значимо върху неговата фармакокинетика. LCM не променя серумните нива на АЕМ - CBZ, LTG, valproate (VPA), oxcarbazepine (OXC) и неговия метаболит, на levetiracetam (LEV), topiramate (TPM), gabapentin (GP) и PHT (3, 13).
LCM се елиминира чрез бъбреците непроменен в >40%, а в < 30% - като неактивен метаболит O-desmethyl (7, 21).
При оценка на фармакокинетиката на LCM при спе-циални групи пациенти се доказва, че полът на пациента е без влияние, възраст >65г. не налага редуциране на дозата.
Лека до умерена бъбречна увреда – също не е показание за ниска доза. При тежка бъбречна увреда (клирънс на креатинина ≤30 mL/min) – максималната доза на LCM трябва да бъде 250 мг/дн. При пациенти на хемодиализа се препоръчва добавянето на 1/2 от определената дневна доза, непосредствено след приключване на хемодиализа-та.
При лека до умерена чернодробна увреда не е необ-ходимо адаптиране на дозата, няма проучвания при пациенти с тежка чернодробна увреда (13). Основните характеристики на LCM са представени на табл. 1.
ПРЕДКЛИНИЧЕН ПРОФИЛ НА LCM (VIMPAT) - МОДЕЛИ НА ЕПИЛЕПСИЯ ПРИ ЖИВОТНИ
LCM показва мощна антиконвулсивна активност в модели на парциални и резистентни припадъци, генера-лизирани тонично-клонични припадъци и епилептичен статус при експериментални животни (5, 36):
Ефективен е в превенцията на модел 6 хц психомо-торни припадъци, със синергични или допълващи ефекти с LЕV, CBZ, PHT, VPA, LTG, TPM, GP (16);
Защитава от припадъци в модел на аудиогенна епилеп-сия при мишки;
Предпазва от развитие на тонично-клонични припа-
дъци индуцирани от максимален електрошок;Повишава гърчовия праг при индуцирани от пенти-
лентетразол припадъци;Осъществява пълна превенция на тонични и частична
превенция на клонични конвулсии индуцирани от NMDA при мишки;
Ефективен в модели на амигдала и хипокамп- kindling, като е по-добър от PHT, CBZ, VPA и ethosuximide (ESM);
В предклинични изследвания има адитивен или синер-гичен ефект с LEV и CBZ;
Ефективен е също при пристъпи, предизвикани от самоподържащ се епилептичен статус при плъхове (5, 36). Хистологично изследване на хипокампа на трети-раните с LCM плъхове установява по-малко увреди в сравнение с контролните животни, данни, които пред-полагат невропротективен ефект на медикамента (5).
КЛИНИЧНИ ПРОУЧВАНИЯ – ЕФИКАСНОСТ НА LCM (VIMPAT)
Резултатите от ранните отворени проучвания показват, че LCM намалява честотата на епилептич-ните припадъци при пациенти с припадъци с парциално начало. Прилагaни са различни дози, което e причина за извършване на рандомизирани, контролирани проучва-яния (RCT), оценяващи оптималните дози за лечение според изискванията на регулаторните органи (3).
Три големи мултицентрови (в САЩ, Европа и Австралия), рандомизирани, двойно-слепи, фаза II/III, плацебо-контролирани проучвания оценяват ефикас-ността и безопасността на пероралния LCM, прилаган като добавъчна терапия във фиксирани дози към 1-3 АЕМ при пациенти с неконтролирани припадъци с пар-циално начало със или без вторична генерализация (4, 11, 19). Пациентите са между 16 и 70 г. Минималният срок на диагностицирана епилепсия с парциални прости, парциални комплексни и вторично генерализирани пар-циални припадъци е 2 г. при предшестващо неуспешно лечение с минимум 2 АЕМ (13). В предшестващия 8 седмичен период броят на припадъците не трябва да бъде по-малък от 4 за 28 дни или да липсва свободен от припадъци перод над 21 дни.
Пациентите са рандомизирани да получават като допълваща терапия LCM или плацебо. По време на форсиран период на титрация LCM се включва в доза 100 мг/дн и се увеличава всяка седмица със 100 мг/дн до достигане на целевата доза. Следва 12-седмичен период
Таблица 1. Основни фармакокинетични характеристики на LCM и приложение
Статус Одобрен от EMEA и FDA при пациенти с парциални епилепсии > 16 и 17 г.Механизъм на д-вие Селективно увеличава бавната инактивация на волтаж- зависимите Na каналиНачална доза 100 мг/дневноТитрация 100 мг/седмичноТерапевтична доза 200–400 мг/днПриеми/дневно 2/дневноПолуживот 13 часаВреме за C max 1–4 часаР.О. бионаличност (%) ~100 %Свързване с плазмените Под 15% протеини

Българска Неврология Bulgarian Neurology 29Май 2011
на подържащо лечение, завършващ с 2- седмичен тран-зиторен период на намаляване на дозата, в случай че пациентът не е влязъл във фазата на продължително отворено проучване.
Критерий за ефикасност е честотата на припадъ-ците, сравнявана с базовия период и RR (responder rate) - процентът на пациентите с редукция на припадъците над 50%, а безопасността е оценявана според съобщава-ните НСД, лабораторните данни, жизнените показате-ли, ЕКГ и теглото на пациента.
Общият брой на пациентите от 3-те проучвания е 1308 със средна продължителност на епилепсията 23,7 г. и среден брой на припадъците за 28 дни между 9,9 и 16,5. С 2-3 съпътстващи АЕМ са 84%, а ½ са лекувани преди включването неуспешно със 7 и повече АЕМ. Тези факти определят високата степен на резистентност на епи-лепсията при включените в проучването пациенти.
Резултатите от първото проучване (4) показват статистически значима редукция на честотата на при-падъците за 28 дни в сравнение с базовия период при доза на LCM 400 мг/дн (р=0,002) и 600 мг/дн (р=0,008) в срав-нение с плацебо. Също и броят на пациентите с редук-ция на припадъците над 50% за 28 дни е статистически значимо по висок в групата с добавъчна терапия с LCM 400 мг/дн (р=0,004) и 600 мг/дн (р=0,014) в сравнение с тези с плацебо.
Във второто проучване (19) са прилагани дози на LCM 200 и 400 мг/дн и редукция на средната честота на припадъците в сравнение с плацебо е налице и при двата дозови режима, (респективно p=0,022 и р=0,033), докато оценката на отговарящите на лечение с над 50% редук-ция на припадъците е статистически значима само при доза 400 мг/дн.
В третото проучване (11) дневните дози на LCM са 400 и 600 мг/дн. Резултатите са близки до първото и второ проучване, като и двете дозировки са със ста-тистически значим по-висок процент на отговарящи на терапията, респективно 38,3% и 41,2% от ITT (intention to treat) популацията.
Обобщените данни от 3-те проучвания показват, че LCM в сравнение с плацебо е свързан с по-голяма пропор-ция на пациентите отговарящи на лечението – 34% при доза 200 мг/дн и 40% - при доза 400 мг/дн, в сравнение с 23% при плацебо (13).
Няма значима разлика при ITT анализ в отговарящи-те на лечението с LCM 200 мг/дн в сравнение с плацебо в 2 от проучванията, изследвали ефикасността и на тази доза (4, 19), но при РР анализ (пациенти според про-токола) в първото проучване дозата 200 мг/дн е също статистически значимо ефективна (4).
Средната редукция на честотата на парциалните припадъци за 28 дни в сравнение с базовия период е значимо по-висока при 400 мг/дн LCM в сравнение с плацебо в ITT популацията и в 3-те проучвания. Статистически значима разлика за този параметър при доза 200 мг/дн LCM има в едното от 2-те проуч-вания прилагали тази доза (19), но при РР анализа и в двете проучвания дозата е ефективна в сравнение с плацебо (4, 19).
В отворените проучвания ефикасността е по-висока и редукцията на припадъците достига 47%, докато в плацебо-контролираните, както беше показано при пре-поръчаните дози от 400 мг/дн е 40%.
Ефектът е дозозависим - с >50% редукция на при-падъците отговарят 32.7% до 41.2% в зависимост от прилаганата доза LCM. (20).
ЕФИКАСНОСТ НА LCM (VIMPAT) ПРИ РАЗЛИЧНИ ВИДОВЕ ЕПИЛЕПТИЧНИ ПРИПАДЪЦИ
Въз основа на 935 участници в клиничната програма за LCМ, включваща 3-те RCT с фиксирани дози проуч-вания с 12-седмична фаза на подържащо лечение (Pooled Analysis of 3 Trials), се оценява ефикасността според типа на припадъците. Най-изразена е редукцията при комплексните парциални припадъци (КПП) и при вто-рично генерализираните парциални припадъци (ВГПП) – табл. 2.
Отговарящите на терапията с над 50% редукция на припадъците в зависимост от характера на припадъка е представена на табл. 3. Не са включени случаите с пар-циални прости пристъпи (в проучването според дизайна не могат да участват пациенти без моторни прояви, т.е. с парциални сензорни пристъпи, и броят не е доста-тъчен за релевантни изводи.
В проучването на Chung et al, 2010 (11) се подчертава отличния ефект от добавъчната терапия с LCM върху ВГТКП – средна редукция на честотата на припадъците с 59,4% при 400 мг дневна доза и с 93% при 600 мг/дн в сравнение с 14,3% в групата с плацебо; с отговарящи на терапията респективно 56% и 72,2% в сравнение с 33% при плацебо.
Според предклинични изследвания LCМ може да бъде ефективен и за ГТКП, но все още липсва достатъчно доказателен опит при хора за широк терапевтичен спек-тър на LCM. В тази насока са необходими нови клинични проучвания с оглед изясняване ефикасността на медика-мента при различни видове епилептични припадъци.
LCМ (VIMPAT) – ОПТИМИЗИРАНА КОМБИНИРАНА ТЕРАПИЯ
Общо прието е, че прибавянето на АЕМ с различен механизъм на действие е с по-добър потенциал за ефи-касност, както и с по-добра поносимост, отколкото комбинирането с АЕМ с еднакъв механизъм на действие (8, 14, 35).
Извършените обобщени анализи от 3-те фаза II/III рандомизирани, двойно-слепи плацебо-кинтролирани проучвания доказват, че LCM има допълнителна ефи-
Таблица 2.Средна редукция в честотата на припадъците/месечно.
Медикамент Комплексни парциални Вторично генерализирани LCM /плацебо припадъци (КПП) парциални припадъци (ВГПП)
Плацебо 22,4 32,5LCM 200 мг/дн 34,1 50,0LCM 400 мг/дн 40,8 55,6LCM 600 мг/дн 41,9 85,9
Таблица 3.Отговарящи на терапията с над 50% редукция на припадъците.
Медикамент КПП % ВГПП % LCM /плацебо
Плацебо 29.2 36.6LCM 200 мг/дн 35.8 50.9LCM 400 мг/дн 43.0 53.5LCM 600 мг/дн 41.8 64.7

Българска Неврология Bulgarian Neurology 30Май 2011
касност при парциални епилепсии, когато се добавя към различни АЕМ. Най-висок е процентът на отговарящи на терапията при комбинация на LCM с VPA (в 48%) и с LEV (в 43%) и най-нисък с OXC ( в 30%) – Rosenfeld et al, 2009 (29). За съжаление малък брой от пациентите са само с един съпътстващ АЕМ, поради което е трудно да се оценят точно най-успешните специфични комби-нации (30).
Следващата задача е изясняване на ефикасността и поносимостта на LCM при комбинацията му с АЕМ, чието антиконвулсивно действие се свързва или не се свързва с блокиране на Na канали (30). Анализът (рost hoc) на обединените данни от фаза II/III RCT (N = 1308), сравнява ефективността:
1/. при пациентите с парциални епилепсии, провели добавъчна терапия с LCM (200, 400, 600 мг/дн или пла-цебо) към традиционните АЕМ, блокиращи Na канали (CBZ, LTG, OXC и PHT) с
2/. пациентите с добавъчна терапия LCM към АЕМ с други механизъми на действие.
От 1308 пациенти по-голямата част (82%) са при-лагали поне един от традиционните АЕМ, блокиращи Na канали. Повечето от пациентите са прилагали 4 или повече АЕМ преди включването в клиничното проуч-ване. Средният брой на АЕМ в комедикация с LCM по време на проучването е 2 или 3. Най-често прилагани са CBZ (в 35% от случаите), LTG (в 31%) и LEV (в 29%).
LCM в подгрупата като добавъчна терапия към АЕМ блокиращи Na канали показва статистически значима редукция на средната честота на припадъците за всички дози и значимо по-високи проценти на отговарящи на терапията с редукция на припадъците над 50% и над 75% за 400 мг/дн и за 600мг/дн в сравнение с плацебо. Резултатите са подобни на тези в обединените данни от фаза II/III популация. НСД са също сравними с тези от 3-те RCT (4, 11, 19).
В другата група пациенти, с комедикация на LCM с АЕМ различни от традиционните блокиращи Na канали (n = 231; 18%), се отчита впечатляващ и високо статис-тически значим дозозависим ефект при дози 400 и 600 мг/дн - редукция на средния процент на припадъци и уве-личаване на относителния дял на случаите отговарящи с над 50% и над 75% редукция на припадъците (RR над 50% се наблюдава при 62,3% от пациентите с 400 мг/дн LCM и при близо 80% от тези с доза 600 мг/дн).
Основният извод от този анализ е, че добавъчната терапия с LCM показва значима редукция на припадъци-те в сравнение с плацебо, независимо от съчетанието с АЕМ с различни механизми на действие, традиционни АЕМ, блокиращи Na канали или АЕМ неблокиращи Na канали. Получените резултати се обясняват с различния механизъм на действие на LCM. Подчертава се обаче, че директно сравнение между 2-те групи е невъзможно, тъй като в проучванията не се отчита действието на различните фактори за изхода, ограничен е броят на пациенти в някои групи, анализът е post-hoc, а ZNS, TPM, и VPA са с не един механизъм на действие. Препоръчват се допълнителни изследвания за оценка на индивидуални комбинации на LCM с АЕМ “нетрадиционни блокери на Na канали” с оглед установяване на адитивно или синер-гично действие (30).
LCM (VIMPAT) - ДЪЛГОСРОЧЕН ЕФЕКТ ОТ ЛЕЧЕНИ-ЕТО
Един от критериите за ефективност (ефикасност в съчетание с добра поносимост) е продължителността
на лечението. От общо 1319 лекувани пациенти с LCM 853 са продължили лечението за най-малко 1 година, 384 за най-малко 3 години и 67 - за най-малко 5 години
3.1% са свободни от припадъци от първата доза до поне 2 г.,
¼ от случаите са със 75% отговор, а ½ имат ефект от 50%.От всички 1319, получаващи LCM, 8%, 4% и 1% пре-
късват лечението поради липса на ефикасност съот-ветно в рамките на първата, третата и петата година (18). Ефектът не се изчерпва с времето – на 3 г. RR (отговарящи на лечението >50%) достига 60-70%, а на 5 г – 65-70%. Задържа се висок и процентът на отгово-рящите с >75% редукция на припадъците (около 40%) както на 3г., така и на 5 г. (18).
Резултатите от 3-те големи отворени проучвания с продължение на лечението над 5,5 г. показват че близо 77% продължават лечението над 1 г, 60,5% - над 2 г и 38% - над 3 г. със средна доза 400 мг/дн. Ефектът при това не се изчерпва с времето (13). Средният процент на редукция на честотата на припадъците в сравнение с базовия период е 46%, а с редукция над 50% са 46,6% от лекуваните (28).
Faught E, 2010 (17) представя крайните резултати от отвореното “extension” проучване оценяващо 5 г лечение с LCМ като добавъчна терапия при пациенти с парциални епилепсии (82% са с 2-3 съпътстващи АЕМ). Включени са пациенти, изпълнили фаза 3 клинично изс-ледване в САЩ, пожелали да продължат лечението. Причина за излизане от проучването е липса на ефикас-ност (26%) или НСД (11%). От всички пациенти 75% са изпълнили минимум 12 мес от наблюдението. Средната продължителност на лечението е 1075 дни. Промени в дозата са извършвани за 1 сед. с по 100 мг/дн (намаляване или увеличаване на дозата, макс до 800 мг). Средната доза в това проучване е 500 мг/дн, а 39% са на 600 мг/дн или повече. Трябва да се има предвид, че максималната препоръчвана доза в Европа и САЩ е 400 мг/дн. От 308 пациенти със средна продължителност на лечението 1075 дни със 75% отговор са 24,4%, а с 50% отговор – 48,2%. Това е един отличен резултат като се има предвид високата степен на резистентност на епилеп-сията в клиничния контингент на проучването.
В друго проучване извършено в Университетския епилептичен център в Марбург върху дългосрочната ефективност на LCM като добавъчна терапия при паци-енти с рефрактерна парциална епилепсия (39) се устано-вява профил на ефикасност, безопасност и поносимост почти идентичен с този описан в 3-те RCT (4, 11, 19).
СРАВНИТЕЛНА ОЦЕНКА НА LCM (VIMPAT) С НОВИ-ТЕ АЕМ.
Ефикасността на LCM е сравнима с тази на много от новите АЕМ, въпреки че не са завършeни все още проуч-ванията “head to head” със сравнителна монотерапия на медикамента с други АЕМ.
За сравняване ефикасността на различни АЕМ, като резултат от подобни RCT се прилага показател “ефи-касност на изпитвания АЕМ минус плацебо ефекта” с оглед по-реална преценка (12). Според този показател LCM се нарежда по ефикасност между OXC (доза 1200 мг/дн) и LEV (доза 3000 мг/дн), от една страна, като при тях тази разлика е 28% и LTG (доза 500 мг/дн) от друга страна с разлика 16%. За LCM при доза 400 мг/дн разли-ката е 18,4 % и за 600 мг/дн - 22,6%. Това е една много добра ефикасност при контингентът на проучванията с

Българска Неврология Bulgarian Neurology 31Май 2011
LCM с много висока рефрактерност на епилепсията.Обобщеният анализ от 3-те проучвания показва,
също още едно предимство на LCM - бърз антиконвулси-вен ефект. Разлика с плацебо в редукция на припадъците се наблюдава още от първата седмица на лечението при доза 100 мг/дн. Повечето от пациентите се подобряват преди достигане на целевата доза. Добре известно е от клиничната практика, че някои от АЕМ изискват про-дължителна и бавна титрация поради риска от НСД.
LCM е мощен АЕМ и действа и при тежки симп-томни епилептични припадъци, където опити с други АЕМ са без ефект. Специалисти от клиника по епи-лептология, ЕЕГ и неврология на съня към Харвардския МИ в Бостън установяват ефективност на LCM при лечението на пациенти в критично състояние с остри повтарящи се припадъци с периодична епилептиформена активност в ЕЕГ (PLED) при продължително монито-риране. От общо 17 пациенти, LCM при 12 е включен след втори или трети АЕМ и при 2 – след 4 и 5 неус-пешно прилагани АЕМ. При 15 от случаите след LCM не се е наложило въвеждане на друг АЕМ. При 12 настъпва клинично подобрение и отзвучаване на периодичните разряди в ЕЕГ. Не са наблюдавани значими НСД. При 11 пациенти лечението с LCM продължава и след изписва-нето от клиниката. (27).
LCM (VIMPAT) – ПОНОСИМОСТ И БЕЗОПАСНОСТ
В първото голямо проучване върху LCM се уточнява средната толерирана доза на медикамента при титра-ция 100 мг на седмица (31). Като най-чести НСД в него се съобщават замаяност, диплопия, умора и сънливост, а като еднократно наблюдавани - синкоп, астения, дис-пепсия, AV блок, тревожност. Само 12% излизат от проучването поради НСД. Средната толерирана доза е 300 мг/дн, а максималната – 600 мг.
Най-често срещаните НСД при лечение с LCM според обобщените данни от 3-те големи клинични проучвания при общоприетите дози от 200 до 400 мг/дневно са от страна на ЦНС и съответстват на описаните в първо-то проучване - замаяност, главоболие, гадене и дипло-пия. Те обикновено са леки до умерени и се наблюдават предимно във фазата на титрация ( 4, 11, 19). Някои от тях са дозозависими и могат да отзвучат при намаля-ване на дозата. Честотата и тежестта им намалява с времето.
НСД причиняващи излизане от проучването на повече от 1% от пациентите са диплопия, тремор, вертиго, гадене, повръщане, замаяност, нарушение на координаци-ята и нистагъм (15). Изтегляне от проучването поради странични ефекти според обединените данни от 3-те рандомизирани клинични проучвания се наблюдават в 8% при рандомизираните на доза 200 мг/дн, 17% - при тези на 400 мг/дн и 29% при 600 мг дневна доза в сравнение с 5% при плацебо групата (4, 11, 19). В страните от Европейския съюз медикаментът е одобрен за приложе-ние в доза до 400 мг/дн. Сериозните странични действия са редки – наблюдавани са в 3% - 7% в плацебо групата, в 3% - 9% при LCM 200 мг/дн, в 5% - 10% при LCM 400 мг/дн и в 3% - 10%- при тази с LCM 600 мг/дн.
В клиничната практика за разлика от RCT режимът на титриране е по-бавен и поради това НСД са по-редки и процентът на продължаващите лечението е по-голям. По-бавна титрация подобрява поносимостта и увелича-ва броят на оставащите в проучванията при АЕМ като PGB, GP и LCM (33).
Дългосрочните отворени продължителни проучвания
демонстрират подобен профил на леки до умерени НСД от ЦНС и гастроинтестиналната система с отпадане от лечението при средно 11% (28, 39). Не се съобщава за повишен риск от психични симптоми, нарушение на настроението, на когнитивните функции, промени в теглото или в лабораторните показатели (33).
В големите RCT в ЕКГ се установява леко удължаване на PR интервала , което не е клинично значимо, наблю-дава се само при 4 от 941 случаи в проучванията върху епилепсия (4, 11, 19), както и рядко (4/944 – 0,4%) - първа степен атрио-вентрикуларен блок (AV). Препоръчва се внимателно въвеждане на LCM при възрастни пациен-ти с диабет и сърдечно-съдова патология, особено при проводникови нарушения на сърцето – маркиран първа степен, втора и по-висока степен AV блок, миокардна исхемия, сърдечна недостатъчност (13). Подчертава се, че в тези случаи е наложителна ЕКГ преди включване на терапията и контролиране в хода на лечението.
В цитираните проучвания LCM е прилаган като доба-въчна терапия често към повече от 1 АЕМ и част от НСД могат да се дължат на комедикацията или евен-туалното взаимодействие между прилаганите АЕМ. В тези случаи ефектът може да бъде главно върху ЦНС и да се проявява например с умора, замаяност или сънли-вост. В проучването, сравняващо ефективността (ефи-касност + поносимост) на LCM в комбинация с АЕМ блокиращи Na канали и с АЕМ неблокиращи Na канали (30) се установява, че НСД не се отличават значимо от профила на тези, наблюдавани в 3-те големи RCT. Прави впечатление, обаче, че при пациентите от втората подгрупа (LCM+AEM неблокиращи Na канали) както НСД, така и напускане на проучването поради НСД е много рядко. В тази насока са необходими нови проучва-ния оценяващи комбинации LCM с отделни АЕМ с оглед уточняване възможността за адитивни и синергични въздействия.
Публикуваните данни върху ефикасността, поноси-мостта и безопасността на LCM при повече от 1300 пациенти (22) показват ефективна редукция на припа-дъците с парциално начало при резистентност на съв-ременните АЕМ като OXC, TPM и LEV. Висок процент от пациентите продължават с години лечението – 75% до 85%, което е доказателство за добра поносимост и безопасност на медикамента.
LCM е безопасен и при инцидентно приемане на ексце-сивна доза - описва се пациент с темпорална епилепсия, направил суициден опит с приемане на 12 грама LCM, в комбинация с висока доза GP, TPM и ZSM. Открит е в кома с повтарящи се ГТКП, аспирация, последваща пнев-мония, артериална хипотония и увеличен PR интервал в ЕКГ. След няколко дни при поддържащо лечение настъп-ва пълно възстановяване. Следователно интоксикация с висока доза LCM със свръхдоза и на други съпътстващи АЕМ може да се преживее без последици (2).
В заключение, LCM е нов АЕМ с нов механизъм на действие, c благоприятен фармакокинетичен профил, бързо начало на антиконвулсивния ефект и значима редукция на припадъците с парциално начало при дози 200 и 400 мг/дн при пациенти с терапевтична резис-тентност. Ефектът е продължителен и не се изчерпва с времето. LCM може да се комбинира успешно с АЕМ с различен механизъм на действие. LCM се понася добре с леки до умерени странични явления (световъртеж, главоболие, гадене), които при бавно титриране могат да се избегнат. Не дава седация, когнитивни дисфункции, обриви или нарушения в настроението, безопасен е и в дългосрочен аспект.

Българска Неврология Bulgarian Neurology 32Май 2011
ЛИТЕРАТУРА1. Лайтнер Х. Епилепсия. Комбинирана терапия. Неврология и
психиатрия 2010, 3, 22-23. 2. Bauer S, David RG, Mylius V et al, Lacosamide intoxication in
attempted suicide. Epilepsy Behav. 2010, 17, 549-51.3. Ben-Menachem E. Lacosamide: an investigational drug for
adjunctive treatment of partial-onset seizures. Drugs Today (Barc) 2008, 44, 35-40.
4. Ben-Menachem E, Biton V, Jatuzis D, et al. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia 2007 Jul; 48 (7): 1308-17.
5. Beyreuther BK, Freitag J, Heers C, et al. Lacosamide: a review of preclinical properties. CNS Drug Rev 2007, 13, 21-42.
6. Bialer M, Johannessen S I, Kupferberg H J, et al. Progress report on new AEDs. Epilepsy Res 2004, 61, 1-48.
7. Bialer M, Johannessen SI, Kupferberg HJ, et al. Progress report on new AEDs. . Epilepsy Res 2007, 73, 1-52.
8. Brodie M J, Antiepileptic drug therapy the story so far. Seizure 2010, 19, 650-55.
9. Callaghan BC, Anand K, Hesdorffer D, et al. Likelihood of seizure remission in an adult population with refractory epilepsy. Ann Neurol 2007,62,382-9.
10. Cawello W, Kropeit D, Schiltmeyer B et al. Food does not affect the pharmacokinetics of SPM 927. Epilepsia 2004, 45 Suppl.7, 307.
11. Chung S, Sperling MR, Biton V, et al. Lacosamide as adjunctive therapy for partial-onset seizures: a randomized controlled trial. Epilepsia 2010, 51, 958-67.
12. CramerJA, Fischer R, Ben-Menachem E. et al. New AEDs comparison of key clinical trials. Epilepsia, 1999, 40, 590-600.
13. Cross S A, Curran M P. Lacosamide in partial-onset seizures. Drugs 2009, 69, 449-459.
14. Deckers CL, Czuczwar SJ, Hekster YA, et al. Selection of antiepileptic drug polytherapy based on mechanisms of action: the evidence reviewed. Epilepsia 2000, 41, 1364-74.
15. Doty P, Rudd G D, Stoehr T et al, Lacosamide. Neurother 2007, 4, 145-148.
16. Duncan G E, Kohn H. The novel antiepileptic drug lacosamide blocks behavioral and brain metabolic manifestations of seizure activity in the 6 Hz psychomotor seizure model. Epilepsy Res. 2005, 67, 81-7.
17. Faught E. Long term efficacy of Lacosamide. American Epilepsy Society (AES) 64th Annual Meeting: Posters 1.249, 1.263, and 1.265. Presented December 4, 2010.
18. French J. et al. Platform presentation ECE 2010 (UCB Data on file Ref Type).
19. Halasz P, Kalviainen R, Mazurkiewicz-Beldzinska M, et al. Adjunctive lacosamide for partial-onset seizures: efficacy and safety results from a randomized controlled trial. Epilepsia 2009, 50, 443-53.
20. Harris JA, Murhy JA. Lacosamide: an adjunctive agent for partial-onset seizures and potential therapy for neuropathic pain. Ann Pharmacother 2009, 43, 1809-17.
21. Horstmann R, Bonn R, Cawello W, et al. Basic clinical pharmacologic investigations of the new antiepileptic drug SPM 927. Epilepsia 2002, 43 Suppl. 7: 188.
22. Krämer G, Heinzl S. Lacosamid. Stuttgart: Ligatur, 2008.23. Kropeit D, Schiltmeyer B, Cawello W, et al. Bioequivalence of
short time infusions compared to oral administration of lacosamide. Epilepsia 2004, 45 Suppl. 7:123.
24. Kwan P, Brodie MJ. Combination therapy in epilepsy: when and what to use. Drugs 2006; 66, 1817-29
25. Kwan P, Brodie MJ. Definition of refractory epilepsy: defining the indefinable? The Lancet Neurol 2010, 9, 27-29.
26. Luciano AL, Shorvon SD. Results of treatment changes in patients with apparently drug-resistsnt chronic epilepsy. Ann Neurol 2007,62,375-81.
27. Parkerson KA, Reinsberger C, Chou SH, et al. Lacosamide in the treatment of acute recurrent seizures and periodic epileptiform patterns in critically ill patientsEpilepsy Behav. 2011, 20, 48-51.
28. Rosenfeld W, Fountain N, Kaubrys G, et al. Lacosamide: an interim evaluation of the long-term safety and efficacy as oral adjunctive therapy in subjects with partial-onset seizures. Epilepsia 2007, 48, Suppl 6,318-319.
29. Rosenfeld WE, Rudd D, Hebert D, et al. Lacosamide efficacy is independent of concomitant AED(s) treatment. 61st American Academy of Neurology Annual Meeting; 2009 Apr 25-May 2; Seattle (WA).
30. Sake JK, Hebert D, IsojaЁrvi J et al, A Pooled Analysis of Lacosamide Clinical Trial Data Grouped by Mechanism of Action of Concomitant Antiepileptic Drugs. CNS Drugs 2010; 24, 1055-1068.
31. Sachdeo R, Montouris G, Beydoun A, et al. An open-label, maximum tolerated dose trial to evaluate oral SPM 927 as adjunctive therapy in patients with partial seizures. Neurology. 2003, 6 Suppl 1, A433.
32. Schmidt T, Locher W. Drug resistance in epilepsy. Epilepsia, 2005, 46, 858-77.
33. Schmitz В, Montouris G, Schauble B, Caleo S et al, Assessing the unmet treatment need in partial-onset epilepsy: Looking beyond seizure control Epilepsia, 2010, 51, 2231–2240.
34. Stephen LJ, Brodie MJ. Selection of AEDs in adults. Neurol Clin 2009, 27, 967-92.
35. St Louis EK. Truly ‘‘rational’’ polytherapy: maximizing efficacy and minimizing drug interactions, drug load, and adverse effects. Curr Neuropharmacol 2009, 7 (2), 96-105.
36. Stohr T, Kupferberg HJ, Stables JP, et al. Lacosamide, a novel anticonvulsant drug, shows efficacy with a wide safety margin in rodent models for epilepsy. Epilepsy Res 2007, 74, 147-154.
37. Thomas D, Scharfenecker U, Schiltmeyer B et al. Low potential for drug-drug interaction of lacosamide. Epilepsia 2006, 47 Suppl.4:200.
38. Wallace H, Shorvon S, Tallis R. Age-specific incidence and prevalence rates of treated epilepsy in an unselected population of 2052922 and age-specific fertility rates of women with epilepsy. Lancet 1998, 352, 1970-73.
39. Wehner L. Lacosamide as add-on therapy - long term efficacy in . Epilepsy Behav. 2009,16, 423-5.
Адрес за кореспонденция:Доц М. РашеваКлиника по неврология, УМБАЛ “Царица Йоанна- ИСУЛ”- София1527 София Ул. Бяло море 8, тел. (02)9432-365e-mail: [email protected]

Българска Неврология Bulgarian Neurology 33Май 2011
SUMMARY
THROMBOLYSIS IN ACUTE ISCHEMIC STROKE – SAFETY AND EFFICACY. THE EXPERIENCE OF
THE CLINIC OF NEUROLOGY AT THE UNIVERSITY HOSPITAL “QUEEN GIOVANNA – ISUL”, SOFIA
E. Vassileva, М. Klissurski, М. Milanova, Е. Vavrek, N. Simeonov, G. Blagoev, Т. Stefanov, М. Radeva, J. Hristov, I. Staikov, F. Alexiev, S. Isakov, D. Tzolova, N. Toteva, М.
Mincheva, N. Koleva, P. Ilieva, М. Daskalov, P. StamenovaThe aim of the study was to evaluate the efficacy and
safety of intravenous thrombolysis (TL) using Actilyse® in patients with acute ischemic stroke (AIS) and to discuss our first experience. A total of 30 patients with AIS were treated from August 2005 to November 2010, 22 males and 8 females, mean age 54.4 +12.5 years. The main risk factors were arterial hypertension (83.3%) and dyslipidemia (50%).
Mean door-to-needle time was 165 +15.5 min (120 - 180 min). Early signs of AIS on computed tomography (CT) before TL were found in 7 patients (23.3%). Ischemic lesions on control CT at 24 hour were seen in 23 patients (76,7%). Mean initial National Institute of Health Stroke Scale (NIHSS) score were 12.4 +3.6 (8-19) and NIHSS at 24 hour was 8.3 +7.1. Mean NIHSS of all patients on discharge between the 5th and 15th day was 4.6 +5.1. Mean modified Rankin scale (mRS) at 3 month was 2.3 +2.1. Fourteen of the patients (46.7%) had minimal disability or no deficit according to mRS 0-1. In 56.7% of them (17/30) mRS was 0-2, Severe disability (mRS 3-4) had 33.3% of patients (10/30). Symptomatic intracranial haemorrhages after use of t-PA were not observed. Mortality in our cohort was 13,3% (4/30).
In conclusion, our results were similar to some other early studies. They suggested that routine intravenous application of Actilyse®, up to the third hour after AIS in properly selected patients, is a safe therapy for all treated subjects, however, only in 46–57% of our cases TL was highly effective.
KEY WORDS: acute ischemic stroke, Actilyse®, thrombolysis, rt-PA.
РЕЗЮМЕ
Цел на изследването е да се проучи ефективността и сигурността от извършването на тромболитично лече-ние (ТЛ) с Actilyse® при болни остър исхемичен мозъчен инсулт (ИМИ). От август 2005 до ноември 2010 г. в нев-рологична клиника на УМБАЛ „Царица Йоанна - ИСУЛ”, София са лекувани 4850 болни с ИМИ. ТЛ е осъществена при 30 болни т.е. при 0,61% от тях - 22 мъже и 8 жени, на възраст от 20 до 75 год., при средна възраст 54.4 +12.5 г. Най-чести рискови фактори са артериалната хипер-тония (83,3%) и дислипидемията (50%).
Средното време между възникване на ИМИ и нача-лото на ТЛ (door-to-needle time) е 165 +15.5 min (от 120
до 180 min). Ранни белези на ИМИ въз основа на прове-дената КТ преди започване на ТЛ са установени при 7 болни (23,3%). Оформена исхемична лезия на 24-я час е открита при 23 болни (76,7%). Началният неврологичен дефицит при приемането по NIHSS e средно 12.4 +3.6 (8 до 19 т.). Средният дефицит по NIHSS на 24-я час е 8.3 +7.1, а при изписването, между 5 и 15-я ден - 4.6 +5.1. Степента на инвалидизация по mRS на третия месец e средно 2.3 +2.1 (0 до 4). 46.7 процента от болните (14/30) са без дефицит или с минимален дефицит - mRS 0-1. При 56.7% от тях (17/30) е наблюдавана mRS 0-2, Значим неврологичен дефицит (mRS 3-4) е регистриран при 33.3% (10/30). Симптоматични интракраниални кръвоизливи следствие на приложeнието на t-PA не са наблюдавани при нито един болен. Леталитетът е 13,3% (4/30 болни).
В заключение, нашите резултати са сходни с други по-ранни проучвания. Те показват, че рутинната упот-реба на Actilyse® в клиничната практика, при правилно подбрани болни с ИМИ, до 3-я час е безопасна при всички болни, но е високо ефективна терапия само при 46–57% от случаите.
КЛЮЧОВИ ДУМИ: актилизе, исхемичен мозъчен инсулт, тромболиза.
УВОД
Единственото доказано ефективно патогенетично лечение при болни с исхемичен мозъчен инсулт (ИМИ) до 3-4,5-ия час от началото на симптоматиката е извърш-ването на тромболитично лечение (ТЛ) с венозно прило-жение на rt-PA (alteplase, Actilyse®) в доза 0.9 mg/kg (10, 11, 14, 21, 22). Резултатите от първото рандомизиранo проучванe, относно тромболитичното лечение в ост-рата фаза на ИМИ са публикувани през 1995 година (21). През 1996 год. FDA одобрява и приема това лечение за ИМИ. В България ТЛ при болни с ИМИ се прилага от 2005 год (1, 2).
Целта на изследването е да се проучи ефективност-та и сигурността от извършването на ТЛ с Actilyse® при болни с остър ИМИ в клиничната практика и се дискутират получените резултати.
КОНТИНГЕНТ И МЕТОДИ
От август 2005 до ноември 2010 г. в неврологична клиника на УМБАЛ „Царица Йоанна - ИСУЛ”, София са лекувани 4850 болни с ИМИ. ТЛ е осъществено при 30 болни т.е. при 0,61% от тях. (таблица 1). Лечението е проведено съгласно Европейските препоръки - European Stroke Initiative recommendations for stroke management-update 2003 (11). Контингентът обхваща 22 мъже и 8 жени, на възраст от 20 до 75 год., при средна възраст 54.4 +12.5 г. Най-голям брой са болните във възрастовия
Оригинални статииТРОМБОЛИЗА ПРИ ИСХЕМИЧЕН МОЗЪЧЕН ИНСУЛТ –
СИГУРНОСТ И ЕФЕКТИВНОСТ. ОПИТЪТ НА КЛИНИКАТА ПО НЕВРОЛОГИЯ ПРИ УМБАЛ “ЦАРИЦА ЙОАННА - ИСУЛ” СОФИЯ
Е. Василева, М. Клисурски, М. Миланова, Е. Ваврек, Н. Симеонов, Г. Благоев, Т. Стефанов, М. Радева,
Я. Христов, И. Стайков, Ф. Алексиев, С. Исаков, Д. Цолова, Н. Тотева, М. Минчева, Н. Колева, П. Илиева, М. Даскалов, П. Стаменова
УМБАЛ “Царица Йоанна - ИСУЛ” София, Kлиника по нервни болестиМедицински Университет - София
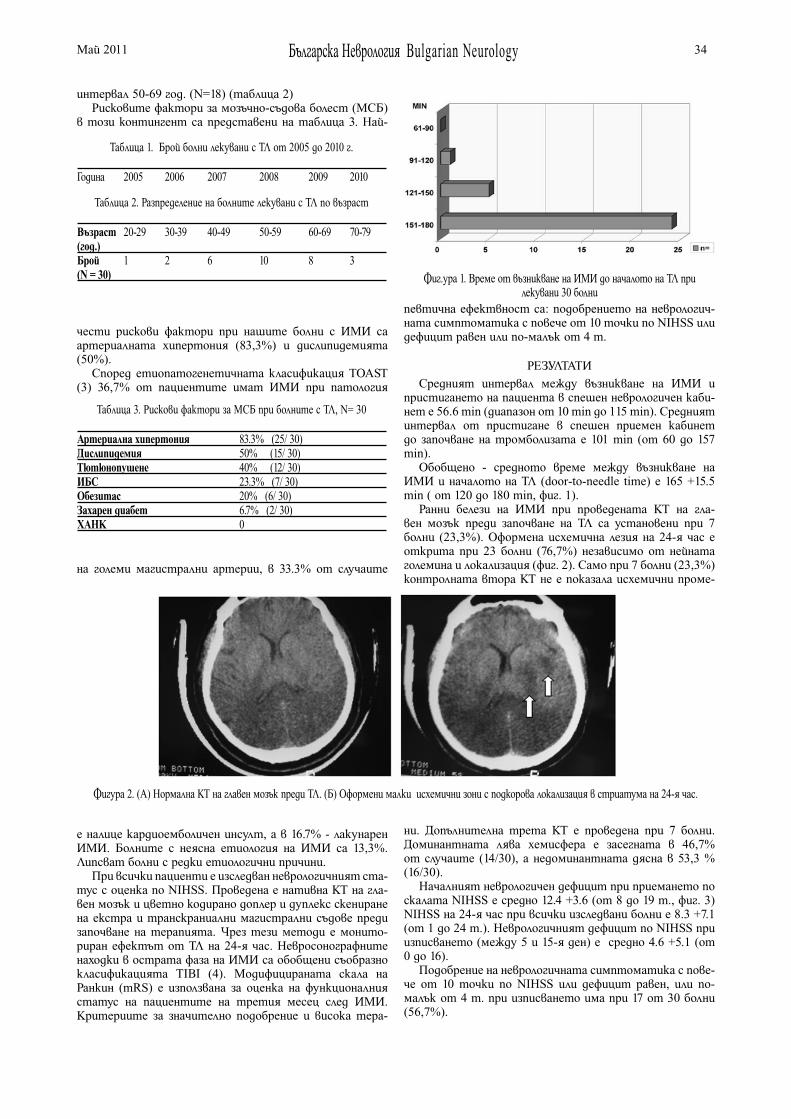
Българска Неврология Bulgarian Neurology 34Май 2011
интервал 50-69 год. (N=18) (таблица 2)Рисковите фактори за мозъчно-съдова болест (МСБ)
в този контингент са представени на таблица 3. Най-
чести рискови фактори при нашите болни с ИМИ са артериалната хипертония (83,3%) и дислипидемията (50%).
Според етиопатогенетичната класификация ТОАST (3) 36,7% от пациентите имат ИМИ при патология
на големи магистрални артерии, в 33.3% от случаите
е налице кардиоемболичен инсулт, а в 16.7% - лакунарен ИМИ. Болните с неясна етиология на ИМИ са 13,3%. Липсват болни с редки етиологични причини.
При всички пациенти е изследван неврологичният ста-тус с оценка по NIHSS. Проведена е нативна КТ на гла-вен мозък и цветно кодирано доплер и дуплекс скениране на екстра и транскраниални магистрални съдове преди започване на терапията. Чрез тeзи методи е монито-риран ефектът от ТЛ на 24-я час. Невросонографните находки в острата фаза на ИМИ са обобщени съобразно класификацията TIBI (4). Модифицираната скала на Ранкин (mRS) е използвана за оценка на функционалния статус на пациентите на третия месец след ИМИ. Критериите за значително подобрение и висока тера-
певтична ефектвност са: подобрението на неврологич-ната симптоматика с повече от 10 точки по NIHSS или дефицит равен или по-малък от 4 т.
РЕЗУЛТАТИ
Средният интервал между възникване на ИМИ и пристигането на пациента в спешен неврологичен каби-нет е 56.6 min (диапазон от 10 min до 115 min). Средният интервал от пристигане в спешен приемен кабинет до започване на тромболизата е 101 min (от 60 до 157 min).
Обобщено - средното време между възникване на ИМИ и началото на ТЛ (door-to-needle time) е 165 +15.5 min ( от 120 до 180 min, фиг. 1).
Ранни белези на ИМИ при проведената КТ на гла-вен мозък преди започване на ТЛ са установени при 7 болни (23,3%). Оформена исхемична лезия на 24-я час е открита при 23 болни (76,7%) независимо от нейната големина и локализация (фиг. 2). Само при 7 болни (23,3%) контролната втора КТ не е показала исхемични проме-
ни. Допълнителна трета КТ е проведена при 7 болни. Доминантната лява хемисфера е засегната в 46,7% от случаите (14/30), а недоминантната дясна в 53,3 % (16/30).
Началният неврологичен дефицит при приемането по скалата NIHSS e средно 12.4 +3.6 (от 8 до 19 т., фиг. 3) NIHSS на 24-я час при всички изследвани болни е 8.3 +7.1 (от 1 до 24 т.). Неврологичният дефицит по NIHSS при изписването (между 5 и 15-я ден) е средно 4.6 +5.1 (от 0 до 16).
Подобрение на неврологичната симптоматика с пове-че от 10 точки по NIHSS или дефицит равен, или по-малък от 4 т. при изписването има при 17 от 30 болни (56,7%).
Таблица 1. Брой болни лекувани с ТЛ от 2005 до 2010 г.
Година 2005 2006 2007 2008 2009 2010
Таблица 2. Разпределение на болните лекувани с ТЛ по възраст
Възраст 20-29 30-39 40-49 50-59 60-69 70-79 (год.)Брой 1 2 6 10 8 3 (N = 30)
Таблица 3. Рискови фактори за МСБ при болните с ТЛ, N= 30
Артериална хипертония 83.3% (25/ 30)Дислипидемия 50% (15/ 30)Тютюнопушене 40% (12/ 30)ИБС 23.3% (7/ 30)Обезитас 20% (6/ 30)Захарен диабет 6.7% (2/ 30)ХАНК 0
Фиг.ура 1. Време от възникване на ИМИ до началото на ТЛ при лекувани 30 болни
Фигура 2. (А) Нормална КТ на главен мозък преди ТЛ. (Б) Оформени малки исхемични зони с подкорова локализация в стриатума на 24-я час.
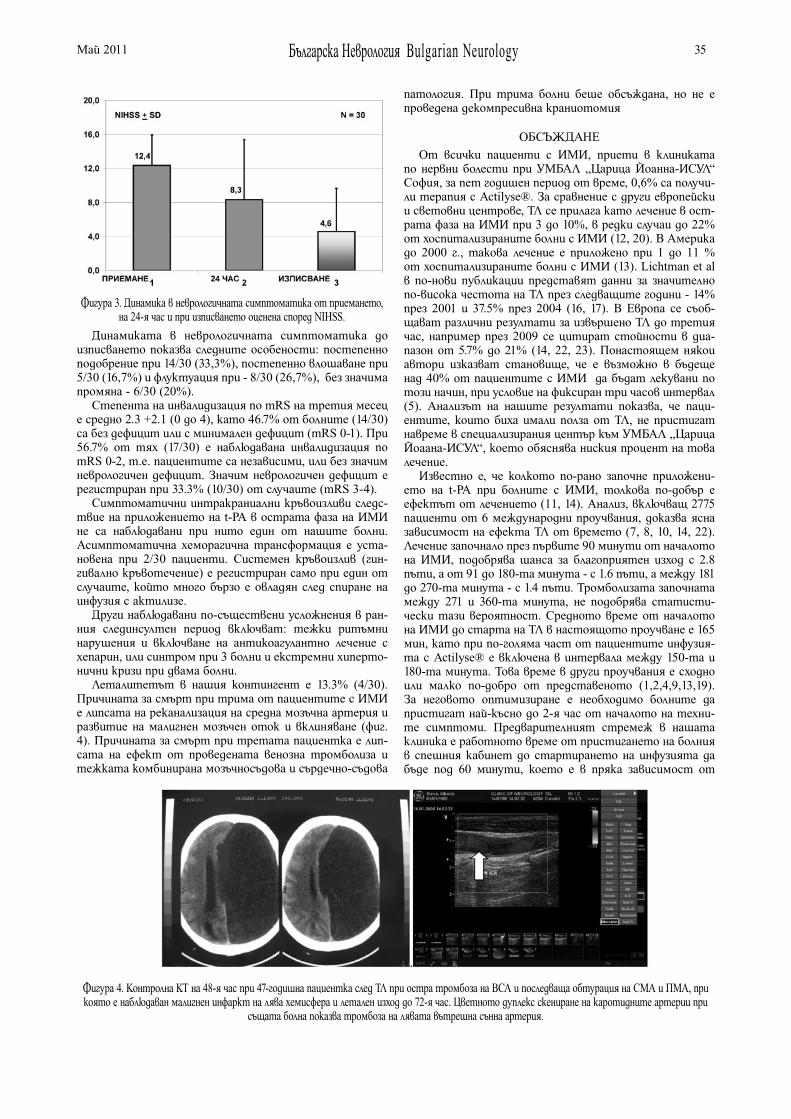
Българска Неврология Bulgarian Neurology 35Май 2011
Динамиката в неврологичната симптоматика до изписването показва следните особености: постепенно подобрение при 14/30 (33,3%), постепенно влошаване при 5/30 (16,7%) и флуктуация при - 8/30 (26,7%), без значима промяна - 6/30 (20%).
Степента на инвалидизация по mRS на третия месец e средно 2.3 +2.1 (0 до 4), като 46.7% от болните (14/30) са без дефицит или с минимален дефицит (mRS 0-1). При 56.7% от тях (17/30) е наблюдавана инвалидизация по mRS 0-2, т.е. пациентите са независими, или без значим неврологичен дефицит. Значим неврологичен дефицит е регистриран при 33.3% (10/30) от случаите (mRS 3-4).
Симптоматични интракраниални кръвоизливи следс-твие на приложeнието на t-PA в острата фаза на ИМИ не са наблюдавани при нито един от нашите болни. Асимптоматична хеморагична трансформация е уста-новена при 2/30 пациенти. Системен кръвоизлив (гин-гивално кръвотечение) е регистриран само при един от случаите, който много бързо е овладян след спиране на инфузия с актилизе.
Други наблюдавани по-съществени усложнения в ран-ния слединсултен период включват: тежки ритъмни нарушения и включване на антикоагулантно лечение с хепарин, или синтром при 3 болни и екстремни хиперто-нични кризи при двама болни.
Леталитетът в нашия контингент е 13.3% (4/30). Причината за смърт при трима от пациентите с ИМИ е липсата на реканализация на средна мозъчна артерия и развитие на малигнен мозъчен оток и вклиняване (фиг. 4). Причината за смърт при третата пациентка е лип-сата на ефект от проведената венозна тромболиза и тежката комбинирана мозъчносъдова и сърдечно-съдова
патология. При трима болни беше обсъждана, но не е проведена декомпресивна краниотомия
ОБСЪЖДАНЕ
От всички пациенти с ИМИ, приети в клиниката по нервни болести при УМБАЛ „Царица Йоанна-ИСУЛ” София, за пет годишен период от време, 0,6% са получи-ли терапия с Actilyse®. За сравнение с други европейски и световни центрове, ТЛ се прилага като лечение в ост-рата фаза на ИМИ при 3 до 10%, в редки случаи до 22% от хоспитализираните болни с ИМИ (12, 20). В Америка до 2000 г., такова лечение е приложено при 1 до 11 % от хоспитализираните болни с ИМИ (13). Lichtman et al в по-нови публикации представят данни за значително по-висока честота на ТЛ през следващите години - 14% през 2001 и 37.5% през 2004 (16, 17). В Европа се съоб-щават различни резултати за извършено ТЛ до третия час, например през 2009 се цитират стойности в диа-пазон от 5.7% до 21% (14, 22, 23). Понастоящем някои автори изказват становище, че е възможно в бъдеще над 40% от пациентите с ИМИ да бъдат лекувани по този начин, при условие на фиксиран три часов интервал (5). Анализът на нашите резултати показва, че паци-ентите, които биха имали полза от ТЛ, не пристигат навреме в специализирания център към УМБАЛ „Царица Йоаана-ИСУЛ”, което обяснява ниския процент на това лечение.
Известно е, че колкото по-рано започне приложени-ето на t-PA при болните с ИМИ, толкова по-добър е ефектът от лечението (11, 14). Анализ, включващ 2775 пациенти от 6 международни проучвания, доказва ясна зависимост на ефекта ТЛ от времето (7, 8, 10, 14, 22). Лечение започнало през първите 90 минути от началото на ИМИ, подобрява шанса за благоприятен изход с 2.8 пъти, а от 91 до 180-та минута - с 1.6 пъти, а между 181 до 270-та минута - с 1.4 пъти. Тромболизата започната между 271 и 360-та минута, не подобрява статисти-чески тази вероятност. Средното време от началото на ИМИ до старта на ТЛ в настоящото проучване е 165 мин, като при по-голяма част от пациентите инфузия-та с Actilyse® е включена в интервала между 150-та и 180-та минута. Това време в други проучвания е сходно или малко по-добро от представеното (1,2,4,9,13,19). За неговото оптимизиране е необходимо болните да пристигат най-късно до 2-я час от началото на техни-те симптоми. Предварителният стремеж в нашата клиника е работното време от пристигането на болния в спешния кабинет до стартирането на инфузията да бъде под 60 минути, което е в пряка зависимост от
Фигура 3. Динамика в неврологичната симптоматика от приемането, на 24-я час и при изписването оценена според NIHSS.
Фигура 4. Контролна КТ на 48-я час при 47-годишна пациентка след ТЛ при остра тромбоза на ВСА и последваща обтурация на СМА и ПМА, при която е наблюдаван малигнен инфаркт на лява хемисфера и летален изход до 72-я час. Цветното дуплекс скениране на каротидните артерии при
същата болна показва тромбоза на лявата вътрешна сънна артерия.
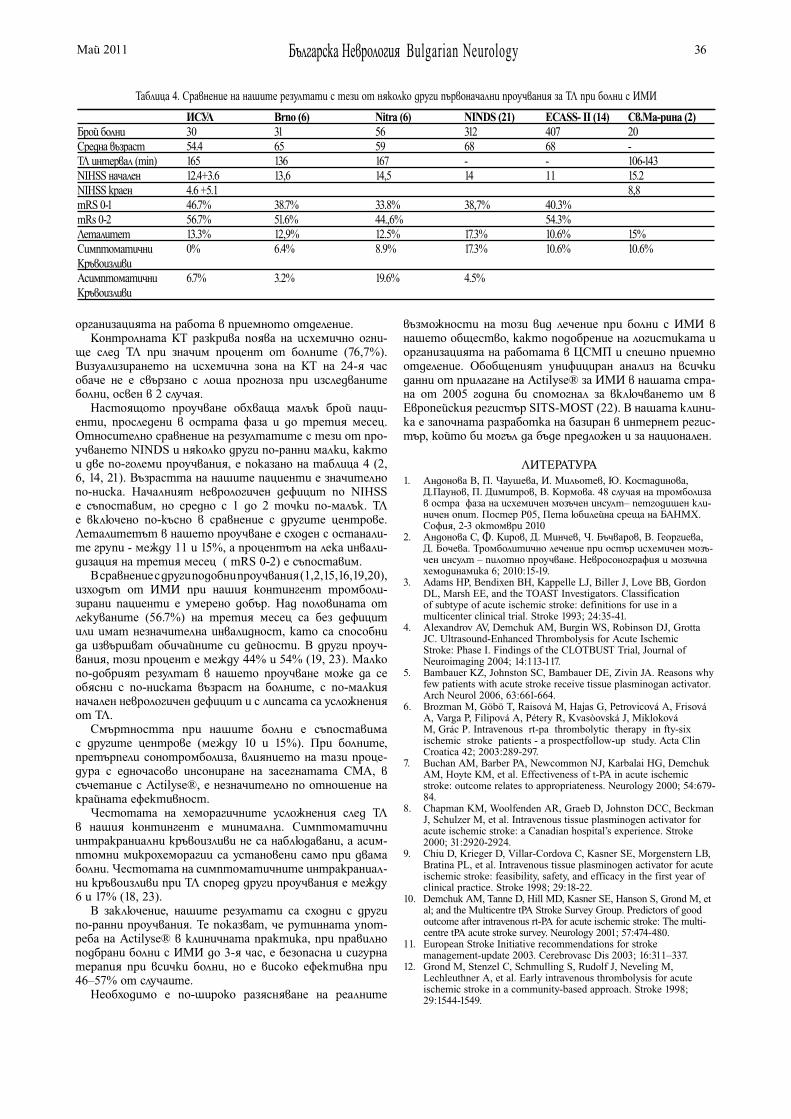
Българска Неврология Bulgarian Neurology 36Май 2011
организацията на работа в приемното отделение. Контролната КТ разкрива поява на исхемично огни-
ще след ТЛ при значим процент от болните (76,7%). Визуализирането на исхемична зона на КТ на 24-я час обаче не е свързано с лоша прогноза при изследваните болни, освен в 2 случая.
Настоящото проучване обхваща малък брой паци-енти, проследени в острата фаза и до третия месец. Относително сравнение на резултатите с тези от про-учването NINDS и няколко други по-ранни малки, както и две по-големи проучвания, е показано на таблица 4 (2, 6, 14, 21). Възрастта на нашите пациенти е значително по-ниска. Началният неврологичен дефицит по NIHSS е съпоставим, но средно с 1 до 2 точки по-малък. ТЛ е включено по-късно в сравнение с другите центрове. Леталитетът в нашето проучване е сходен с останали-те групи - между 11 и 15%, а процентът на лека инвали-дизация на третия месец ( mRS 0-2) е съпоставим.
В сравнение с други подобни проучвания (1,2,15,16,19,20), изходът от ИМИ при нашия контингент тромболи-зирани пациенти е умерено добър. Над половината от лекуваните (56.7%) на третия месец са без дефицит или имат незначителна инвалидност, като са способни да извършват обичайните си дейности. В други проуч-вания, този процент е между 44% и 54% (19, 23). Малко по-добрият резултат в нашето проучване може да се обясни с по-ниската възраст на болните, с по-малкия начален неврологичен дефицит и с липсата са усложнения от ТЛ.
Смъртността при нашите болни е съпоставима с другите центрове (между 10 и 15%). При болните, претърпели сонотромболиза, влиянието на тази проце-дура с едночасово инсониране на засегнатата СМА, в съчетание с Actilyse®, е незначително по отношение на крайната ефективност.
Честотата на хеморагичните усложнения след ТЛ в нашия контингент е минимална. Симптоматични интракраниални кръвоизливи не са наблюдавани, а асим-птомни микрохеморагии са установени само при двама болни. Честотата на симптоматичните интракраниал-ни кръвоизливи при ТЛ според други проучвания е между 6 и 17% (18, 23).
В заключение, нашите резултати са сходни с други по-ранни проучвания. Те показват, че рутинната упот-реба на Actilyse® в клиничната практика, при правилно подбрани болни с ИМИ до 3-я час, е безопасна и сигурна терапия при всички болни, но е високо ефективна при 46–57% от случаите.
Необходимо е по-широко разясняване на реалните
възможности на този вид лечение при болни с ИМИ в нашето общество, както подобрение на логистиката и организацията на работата в ЦСМП и спешно приемно отделение. Обобщеният унифициран анализ на всички данни от прилагане на Actilyse® за ИМИ в нашата стра-на от 2005 година би спомогнал за включването им в Европейския регистър SITS-MOST (22). В нашата клини-ка е започната разработка на базиран в интернет регис-тър, който би могъл да бъде предложен и за национален.
ЛИТЕРАТУРА1. Андонова В, П. Чаушева, И. Мильотев, Ю. Костадинова,
Д.Пaунов, П. Димитров, В. Кормова. 48 случая на тромболиза в остра фаза на исхемичен мозъчен инсулт– петгодишен кли-ничен опит. Постер Р05, Пета юбилейна среща на БАНМХ. София, 2-3 октомври 2010
2. Андонова С, Ф. Киров, Д. Минчев, Ч. Бъчваров, В. Георгиева, Д. Бочева. Тромболитично лечение при остър исхемичен мозъ-чен инсулт – пилотно проучване. Невросонография и мозъчна хемодинамика 6; 2010:15-19.
3. Adams HP, Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, Marsh EE, and the TOAST Investigators. Classification of subtype of acute ischemic stroke: definitions for use in a multicenter clinical trial. Stroke 1993; 24:35-41.
4. Alexandrov AV, Demchuk AM, Burgin WS, Robinson DJ, Grotta JC. Ultrasound-Enhanced Thrombolysis for Acute Ischemic Stroke: Phase I. Findings of the CLOTBUST Trial, Journal of Neuroimaging 2004; 14:113-117.
5. Bambauer KZ, Johnston SC, Bambauer DE, Zivin JA. Reasons why few patients with acute stroke receive tissue plasminogan activator. Arch Neurol 2006, 63:661-664.
6. Brozman M, Göbö T, Raisová M, Hajas G, Petrovicová A, Frisová A, Varga P, Filipová A, Pétery R, Kvasòovská J, Mikloková M, Grác P. Intravenous rt-pa thrombolytic therapy in fty-six ischemic stroke patients - a prospectfollow-up study. Acta Clin Croatica 42; 2003:289-297.
7. Buchan AM, Barber PA, Newcommon NJ, Karbalai HG, Demchuk AM, Hoyte KM, et al. Effectiveness of t-PA in acute ischemic stroke: outcome relates to appropriateness. Neurology 2000; 54:679-84.
8. Chapman KM, Woolfenden AR, Graeb D, Johnston DCC, Beckman J, Schulzer M, et al. Intravenous tissue plasminogen activator for acute ischemic stroke: a Canadian hospital’s experience. Stroke 2000; 31:2920-2924.
9. Chiu D, Krieger D, Villar-Cordova C, Kasner SE, Morgenstern LB, Bratina PL, et al. Intravenous tissue plasminogen activator for acute ischemic stroke: feasibility, safety, and efficacy in the first year of clinical practice. Stroke 1998; 29:18-22.
10. Demchuk AM, Tanne D, Hill MD, Kasner SE, Hanson S, Grond M, et al; and the Multicentre tPA Stroke Survey Group. Predictors of good outcome after intravenous rt-PA for acute ischemic stroke: The multi-centre tPA acute stroke survey. Neurology 2001; 57:474-480.
11. European Stroke Initiative recommendations for stroke management-update 2003. Cerebrovasc Dis 2003; 16:311–337.
12. Grond M, Stenzel C, Schmulling S, Rudolf J, Neveling M, Lechleuthner A, et al. Early intravenous thrombolysis for acute ischemic stroke in a community-based approach. Stroke 1998; 29:1544-1549.
Таблица 4. Сравнение на нашите резултати с тези от няколко други първоначални проучвания за ТЛ при болни с ИМИ
ИСУЛ Brno (6) Nitra (6) NINDS (21) ECASS- II (14) Св.Ма-рина (2) Брой болни 30 31 56 312 407 20Средна възраст 54.4 65 59 68 68 -ТЛ интервал (min) 165 136 167 - - 106-143NIHSS начален 12.4+3.6 13,6 14,5 14 11 15.2NIHSS краен 4.6 +5.1 8,8mRS 0-1 46.7% 38.7% 33.8% 38,7% 40.3% mRs 0-2 56.7% 51.6% 44.,6% 54.3% Леталитет 13.3% 12,9% 12.5% 17.3% 10.6% 15%Симптоматични 0% 6.4% 8.9% 17.3% 10.6% 10.6% Кръвоизливи Асимптоматични 6.7% 3.2% 19.6% 4.5% Кръвоизливи

Българска Неврология Bulgarian Neurology 37Май 2011
13. Grotta JC, Burgin WS, El-Mitwalli A, Long M, Campbell M, Morgenstern LB, et al. Intravenous tissue-type plasminogen activator therapy for ischemic stroke: Houston experience 1996 to 2000. Arch Neurol 2001; 58:2009-2013.
14. Hacke W, Donnan G, Fieschi C, Kaste M, von Kummer R, Broderick JP, Brott T, Frankel M, Grotta JC, Haley EC Jr, Kwiatkowski T, Levine SR, Lewandowski C, Lu M, Lyden P, Marler JR, Patel S, Tilley BC, Albers G, Bluhmki E, Wilhelm M, Hamilton S; ATLANTIS Trials Investigators; ECASS Trials Investigators; NINDS rt-PA Study Group Investigators. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet. 2004; 363:768–774.
15. Katzan I, Furlan AJ, Lloyd LE, Frank JI, Harper DL, Hinchey JA, et al. Use of tissue-type plasminogen activator for acute ischemic stroke: the Cleveland area experience. JAMA 2000; 283:1151-1158.
16. Koennecke HC, Nohr R, Leistner S, Marx P. Intravenous tPA for ischemic stroke team performance over time, safety, and efficacy in a single-center, 2-year experience. Stroke 2001; 32:1074-1078.
17. Lichtman JH, Watanabe E, Allen NB, Jones SB, Dostal J, Goldstein LB. Hospital arrival time and intravenous t-PA use in US academic medical centers, 2001-2004. Stroke 2009; 40:3845-3850.
18. Lopez-Yunez AM, Bruno A, Williams LS, Yilmaz E, Zurrú C, Biller J. Protocol violations in community-based rt-PA
stroke treatment are associated with symptomatic intracerebral hemorrhage. Stroke 2001; 32:12-16.
19. Michael D. Hill, Alastair M. Buchan for The Canadian Alteplase for Stroke Effectiveness Study (CASES) Investigators Thrombolysis for acute ischemic stroke: results of the Canadian Alteplase for Stroke Effectiveness Study. CMAJ 2005; 172:1307-1312.
20. Smith RW, Scott PA, Grant RJ, Chudnofsky CR, Frederiksen SM. Emergency physician treatment of acute stroke with recombinant tissue plasminogen activator: a retrospective analysis. Acad Emerg Med 1999;6:618-25.
21. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med 1995; 333:1581-1587.
22. Wahlgren N, Ahmed N, Eriksson N. еt all. Safe Implementation of Thrombolysis in Stroke - MOnitoring STudy Investigators 2008; 39:3316-3322. Epub 2008 Oct 16 Multivariable analysis of outcome predictors and adjustment of main outcome results to baseline data profile in randomized controlled trials: Safe Implementation of Thrombolysis in Stroke-Monitoring Study (SITS-MOST).
23. Wang DZ, Rose JA, Honings DS, Garwacki DJ, Milbrandt JC. Treating acute stroke patients with intravenous tPA: the OSF Stroke Network experience. Stroke 2000; 31:77-81.
SUMMARY
INTRAVENOUS ADMINISTRATION OF LEVETIRACETAM (KEPPRA) IN STATUS EPILEPTICUS
Zahari Iv. Zahariev, Ekaterina Iv. Viteva, Albena G. Djurkova
The study of the efficacy of intravenous Keppra is demanded by the vital risk during status epilepticus and the small number of anticonvulsants for parenteral aministration, as well as by situations of impaired oral intake or contraindications for the use of classic anticonvulsants.
PURPOSE: Evaluation of the efficacy of intravenous Keppra for management of status epilepticus in a hospital setting in 10 cases.
POPULATION AND METHODS: We reported ten cases (4 women and 4 men) in which 1000 mg of Keppra was given IV over 15-20 minutes to control status epilepticus. We observed different types of seizures: myoclonic-astatic – in 1, simple partial versive – in 1, simple partial motor seizures - in 3, simple partial motor seizures with secondary generalization – in 2, complex partial seizures with secondary generalization – in 2, partial myoclonic seizures – in 1 case. In 2 of the patients we administered Keppra IV concomitantly with other anticonvulsants and in the rest – as single-agent therapy.
RESULTS: In all patients the status epilepticus was under control until the end of the infusion. Only in 4 patients there were no subsequent epileptic seizures and in the rest - we registered infrequent and milder, often abortive seizures in the following hours or days, which eventually completely stopped. In 5 patients we recommended oral therapy with Keppra as maintenance treatment. We did not observe side
effects in any of the patients.CONCLUSION: Intravenous Keppra is highly effective in
the management of status epilepticus irregardless of the type of seizures or their etiology.
KEY WORDS: satus epilepticus, Keppra, IV application
РЕЗЮМЕ
Проучването на ефективността на интравенозната форма на Keppra се налага от виталната застрашеност по време на епилептичния статус и малкия брой анти-конвулсанти с възможност за парентерално приложение, както и от наличието на ситуации, при които перорал-ният прием е невъзможен или има контраиндикации за класическите антиконвулсанти.
ЦЕЛ: Изследване на ефективността на интравенозно приложена Keppra при 10 случая за овладяване на епилеп-тичен статус в стационарни условия.
ПАЦИЕНТИ И МЕТОДИ: Описахме 10 случая (6 мъже и 4 жени), при които приложихме Keppra 1000 мг инт-равенозно за 15-20 мин. с цел овладяване на епилептичен статус. Седем от болните са със симптоматична, а останалите – с неизвестна етиология. Наблюдавахме различни епилептични пристъпи – миоклонично-аста-тични – при 1, прости парциални адверзивни – при 1, прости парциални моторни – при 3-ма, прости парциал-ни моторни с вторична генерализация – при 2-ма, КПП с вторична генерализация – при 1, ГТКП – при 1, парциални миоклонични – при 1. При 2-ма от пациентите прилагах-ме Keppra интравенозно едновременно с други антикон-вулсанти, а при останалите – самостоятелно.
Оригинални статииИНТРАВЕНОЗНО ПРИЛАГАНЕ НА LEVETIRACETAM (KEPPRA)
ПРИ ЕПИЛЕПТИЧЕН СТАТУС
Захари Ив. Захариев, Екатерина Ив. Витева, Албена Г. Джуркова
УМБАЛ „Св. Георги” – Пловдив, Клиника по нервни болести, Медицински Университет - Пловдив
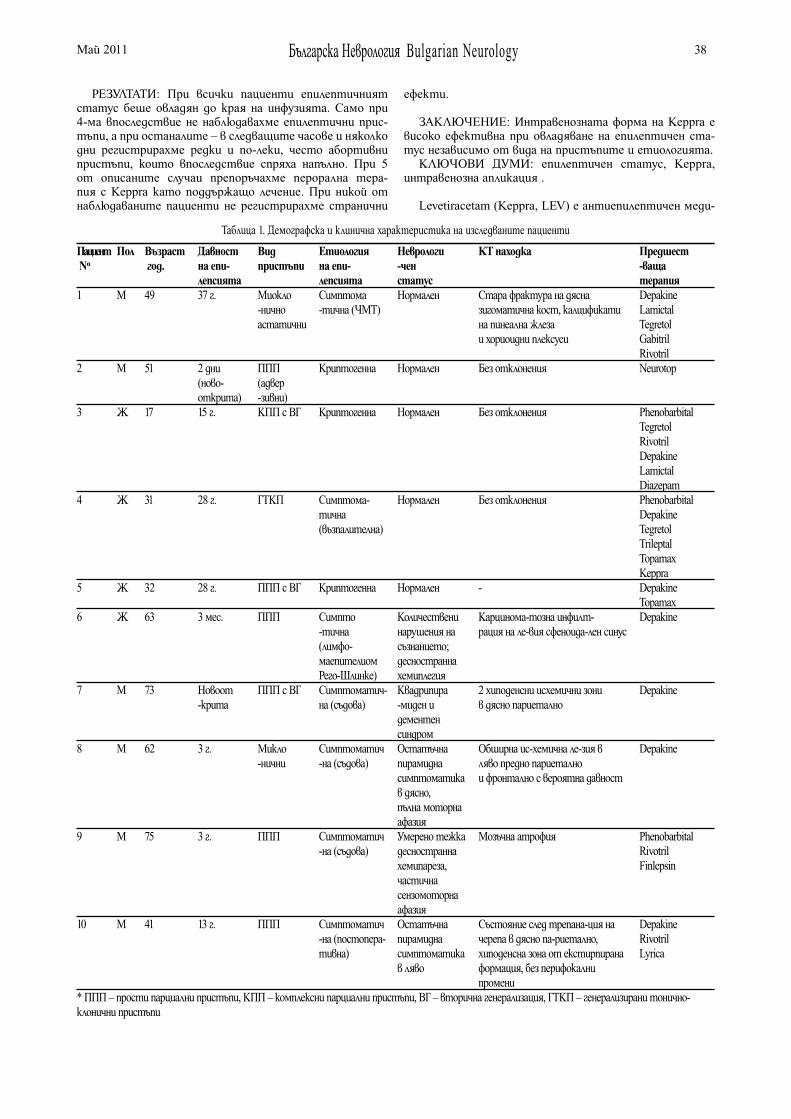
Българска Неврология Bulgarian Neurology 38Май 2011
РЕЗУЛТАТИ: При всички пациенти епилептичният статус беше овладян до края на инфузията. Само при 4-ма впоследствие не наблюдавахме епилептични прис-тъпи, а при останалите – в следващите часове и няколко дни регистрирахме редки и по-леки, често абортивни пристъпи, които впоследствие спряха напълно. При 5 от описаните случаи препоръчахме перорална тера-пия с Keppra като поддържащо лечение. При никой от наблюдаваните пациенти не регистрирахме странични
ефекти.
ЗАКЛЮЧЕНИЕ: Интравенозната форма на Keppra е високо ефективна при овладяване на епилептичен ста-тус независимо от вида на пристъпите и етиологията.
КЛЮЧОВИ ДУМИ: епилептичен статус, Keppra, интравенозна апликация .
Levetiracetam (Keppra, LEV) е антиепилептичен меди-
Таблица 1. Демографска и клинична характеристика на изследваните пациенти
Пациент Пол Възраст Давност Вид Етиология Неврологи КТ находка Предшест № год. на епи- пристъпи на епи- -чен -ваща лепсията лепсията статус терапия1 М 49 37 г. Миокло Симптома Нормален Стара фрактура на дясна Depakine -нично -тична (ЧМТ) зигоматична кост, калцификати Lamictal астатични на пинеална жлеза Tegretol и хориоидни плексуси Gabitril Rivotril2 М 51 2 дни ППП Криптогенна Нормален Без отклонения Neurotop (ново- (адвер открита) -зивни)3 Ж 17 15 г. КПП с ВГ Криптогенна Нормален Без отклонения Phenobarbital Tegretol Rivotril Depakine Lamictal Diazepam4 Ж 31 28 г. ГТКП Симптома- Нормален Без отклонения Phenobarbital тична Depakine (възпалителна) Tegretol Trileptal Topamax Keppra5 Ж 32 28 г. ППП с ВГ Криптогенна Нормален - Depakine Topamax6 Ж 63 3 мес. ППП Симпто Количествени Карцинома-тозна инфилт- Depakine -тична нарушения на рация на ле-вия сфеноида-лен синус (лимфо- съзнанието; маепителиом десностранна Рего-Шлинке) хемиплегия7 М 73 Новоот ППП с ВГ Симптоматич- Квадрипира 2 хиподенсни исхемични зони Depakine -крита на (съдова) -миден и в дясно париетално дементен синдром8 М 62 3 г. Микло Симптоматич Остатъчна Обширна ис-хемична ле-зия в Depakine -нични -на (съдова) пирамидна ляво предно париетално симптоматика и фронтално с вероятна давност в дясно, пълна моторна афазия 9 М 75 3 г. ППП Симптоматич Умерено тежка Мозъчна атрофия Phenobarbital -на (съдова) десностранна Rivotril хемипареза, Finlepsin частична сензомоторна афазия 10 М 41 13 г. ППП Симптоматич Остатъчна Състояние след трепана-ция на Depakine -на (постопера- пирамидна черепа в дясно па-риетално, Rivotril тивна) симптоматика хиподенсна зона от екстирпирана Lyrica в ляво формация, без перифокални промени * ППП – прости парциални пристъпи, КПП – комплексни парциални пристъпи, ВГ – вторична генерализация, ГТКП – генерализирани тонично-клонични пристъпи
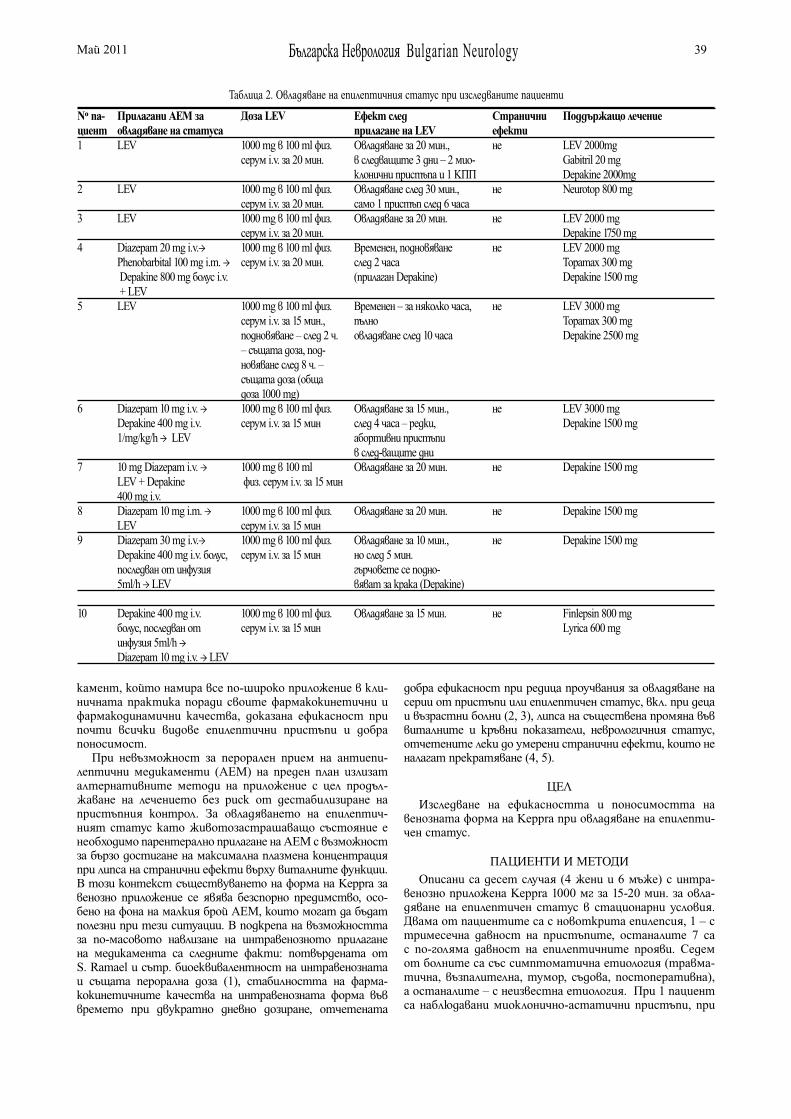
Българска Неврология Bulgarian Neurology 39Май 2011
камент, който намира все по-широко приложение в кли-ничната практика поради своите фармакокинетични и фармакодинамични качества, доказана ефикасност при почти всички видове епилептични пристъпи и добра поносимост.
При невъзможност за перорален прием на антиепи-лептични медикаменти (АЕМ) на преден план излизат алтернативните методи на приложение с цел продъл-жаване на лечението без риск от дестабилизиране на пристъпния контрол. За овладяването на епилептич-ният статус като животозастрашаващо състояние е необходимо парентерално прилагане на АЕМ с възможност за бързо достигане на максимална плазмена концентрация при липса на странични ефекти върху виталните функции. В този контекст съществуването на форма на Keppra за венозно приложение се явява безспорно предимство, осо-бено на фона на малкия брой АЕМ, които могат да бъдат полезни при тези ситуации. В подкрепа на възможността за по-масовото навлизане на интравенозното прилагане на медикамента са следните факти: потвърдената от S. Ramael и сътр. биоеквивалентност на интравенозната и същата перорална доза (1), стабилността на фарма-кокинетичните качества на интравенозната форма във времето при двукратно дневно дозиране, отчетената
добра ефикасност при редица проучвания за овладяване на серии от пристъпи или епилептичен статус, вкл. при деца и възрастни болни (2, 3), липса на съществена промяна във виталните и кръвни показатели, неврологичния статус, отчетените леки до умерени странични ефекти, които не налагат прекратяване (4, 5).
ЦЕЛ
Изследване на ефикасността и поносимостта на венозната форма на Keppra при овладяване на епилепти-чен статус.
ПАЦИЕНТИ И МЕТОДИ
Описани са десет случая (4 жени и 6 мъже) с интра-венозно приложена Keppra 1000 мг за 15-20 мин. за овла-дяване на епилептичен статус в стационарни условия. Двама от пациентите са с новоткрита епилепсия, 1 – с тримесечна давност на пристъпите, останалите 7 са с по-голяма давност на епилептичните прояви. Седем от болните са със симптоматична етиология (травма-тична, възпалителна, тумор, съдова, постоперативна), а останалите – с неизвестна етиология. При 1 пациент са наблюдавани миоклонично-астатични пристъпи, при
Таблица 2. Овладяване на епилептичния статус при изследваните пациенти
№ па- Прилагани АЕМ за Доза LEV Eфект след Странични Поддържащо лечение циент овладяване на статуса прилагане на LEV ефекти 1 LEV 1000 mg в 100 ml физ. Овладяване за 20 мин., не LEV 2000mg серум i.v. за 20 мин. в следващите 3 дни – 2 мио- Gabitril 20 mg клонични пристъпа и 1 КПП Depakine 2000mg2 LEV 1000 mg в 100 ml физ. Овладяване след 30 мин., не Neurotop 800 mg серум i.v. за 20 мин. само 1 пристъп след 6 часа 3 LEV 1000 mg в 100 ml физ. Овладяване за 20 мин. не LEV 2000 mg серум i.v. за 20 мин. Depakine 1750 mg4 Diazepam 20 mg i.v.→ 1000 mg в 100 ml физ. Временен, подновяване не LEV 2000 mg Phenobarbital 100 mg i.m. → серум i.v. за 20 мин. след 2 часа Topamax 300 mg Depakine 800 mg болус i.v. (прилаган Depakine) Depakine 1500 mg + LEV 5 LEV 1000 mg в 100 ml физ. Временен – за няколко часа, не LEV 3000 mg серум i.v. за 15 мин., пълно Topamax 300 mg подновяване – след 2 ч. овладяване след 10 часа Depakine 2500 mg – същата доза, под- новяване след 8 ч. – същата доза (обща доза 1000 mg) 6 Diazepam 10 mg i.v. → 1000 mg в 100 ml физ. Овладяване за 15 мин., не LEV 3000 mg Depakine 400 mg i.v. серум i.v. за 15 мин след 4 часа – редки, Depakine 1500 mg 1/mg/kg/h → LEV абортивни пристъпи в след-ващите дни7 10 mg Diazepam i.v. → 1000 mg в 100 ml Овладяване за 20 мин. не Depakine 1500 mg LEV + Depakine физ. серум i.v. за 15 мин 400 mg i.v. 8 Diazepam 10 mg i.m. → 1000 mg в 100 ml физ. Овладяване за 20 мин. не Depakine 1500 mg LEV серум i.v. за 15 мин9 Diazepam 30 mg i.v.→ 1000 mg в 100 ml физ. Овладяване за 10 мин., не Depakine 1500 mg Depakine 400 mg i.v. болус, серум i.v. за 15 мин но след 5 мин. последван от инфузия гърчовете се подно- 5ml/h → LEV вяват за крака (Depakine)
10 Depakine 400 mg i.v. 1000 mg в 100 ml физ. Овладяване за 15 мин. не Finlepsin 800 mg болус, последван от серум i.v. за 15 мин Lyrica 600 mg инфузия 5ml/h → Diazepam 10 mg i.v. → LEV

Българска Неврология Bulgarian Neurology 40Май 2011
1 - прости парциални адверзивни, при 3-ма - прости пар-циални моторни, при 2-ма – прости парциални моторни с вторична генерализация, при 1 – комплексни парциал-ни пристъпи (КПП) с вторична генерализация и при 1 – генерализирани тонично-клонични пристъпи (ГТКП), при 1 – парциални миоклонични пристъпи (Табл.1.)
РЕЗУЛТАТИ
При 2-ма от пациентите (случаи 4 и 7) прилагахме Керprа интравенозно едновременно с други антиконвул-санти (Депакин, Диазепам, Фенобарбитал). При 1 от тях (случай 7) не бяха наблюдавани повече пристъпи, докато при другия (случай 4) 2 часа по-късно започнаха отново ГТКП. При останалите 8 случая прилагахме Кеpрrа самостоятелно, като при 4 от тях - след пред-шестващо неефективно използване на Депакин с или без Диазепам интравенозно – Табл. 2. При 7 пациента инт-равенозната апликация доведе до овладяване на статуса до края на инфузията, при 1 пациент беше с кратък (5 мин.) и частичен ефект. При всички тях интравенозна-та апликация на 1000 мг Керprа в 100 мл Физиологичен серум за 15-20 мин. доведе до овладяване на епилептич-ния статус до края на инфузията. Впечатление прави бързото овладяване на статус от миоклонично-аста-тични пристъпи (случай 1). Интерес представлява слу-чай 5 – млада жена, при която пристъпите зачестяват от трети лунарен месец на бременността до епилепти-чен статус 10 дни преди термина. Приложената Керprа води до неговото овладяване, направено е секцио, плодът е нормален, жизнеспособен. С последващи двукратни апликации на същата доза поради 2 епизода от статуси пристъпите се овладяват напълно.
Само при 4 пациента впоследствие не наблюдавахме епилептични пристъпи, а при 6 – в следващите часове и няколко дни регистрирахме редки и по-леки, често абор-тивни пристъпи, които впоследствие спряха напълно, в повечето случаи - след повторна венозна апликация или включване на Керprа пер ос. При 5 от описаните случаи препоръчахме перорална терапия с Керprа като поддър-жащо лечение.
ОБСЪЖДАНЕ
От наличните в литературата данни става ясно, че най-често използваната доза за интравенозно приложе-ние на Керprа е 1000 мг за 15 мин., въпреки че някои от авторите са прилагали 500 мг с добър ефект (6), а други - по-високи дози (до 4000 мг) или при по-висока скорост (инфузия за 5 мин.) без съществени странични ефекти (7). Най-честите причини за включване на Керprа са: неовладяване на епилептичен статус или клъстери от пристъпи с парентерално прилагани класически анти-конвулсанти (бензодиазепини, валпроат, фенитоин, седа-тивни анестетици), противопоказания за някои от тях (чернодробна увреда, коагулопатия), невъзможност за перорален прием на медикамента. При всички проучва-ния пристъпите спират веднага след прекратяване на инфузията, което се обяснява с установеното дости-гане на максимална плазмена концентрация на 25-тата минута от началото на приложението. При 1 случай (8) в следващите 2 дни се наблюдават по-леки парциални пристъпи след спиране на придружаващия Тиопентал, но впоследствие те се овладяват напълно. От проведените проучвания достигаме до заключението за висока ефи-касност (≥ 50%) на интравенозно приложената Кеpрrа при парциален епилептичен статус, както и пълно овла-дяване при клъстери от пристъпи или невъзможност
за перорален прием (8-19). Редица автори съобщават за бързо купиране и на миоклоничен статус, вкл. и при пациент със Синдром на Lennox-Gastaut, при който тоничните пристъпи спират веднага след инфузията, а миоклоничните – постепенно в следващите 2 дни (20-23). Подобен е ефектът и при 2 деца на 20-месечна въз-раст с детска церебрална парализа (ДЦП) и резистентен на класическите антиконвулсанти миоклоничен статус (20). Налични са и съобщения, според които интравеноз-ното прилагане на Керprа води до овладяване и на гене-рализиран епилептичен статус от ГТКП веднага след инфузията (6, 16, 17).
Описвани са странични ефекти от интравенозното прилагане на Керprа, като те достигат честота до 44% (n = 25) при проучване на M. Baulac и сътр. (4). При всички пациенти те са леки към умерени, бързо преходни и не налагат спирането на медикамента (4, 7, 24). Най-често срещани са: главоболие, умора, сънливост, замай-ване (4, 5, 7, 25). По-рядко описвани са: намаляване на броя на тромбоцитите (26), левкопения и поведенчески нарушения при деца (27). Само при 1 пациент са наблю-давани гадене и повръщане след интравенозно прилагане на Керprа 1500 мг (17) и при 1 – преходно зачервяване на лицето (24). Най-общо авторите отчитат много добра поносимост при деца и при възрастни полиморбидни болни (2, 27).
ЗАКЛЮЧЕНИЕ
Oписаните от нас случаи с интравенозна апликация показват висока и бърза ефикасност на Кеpрrа при епи-лептичен статус от КПП с и без вторична генерализа-ция, прости парциални пристъпи, вкл. адверзивни кризи, миоклонични и миоклонично-астатични пристъпи, някои от тях неповлияни от парентерално приложени преди това класически антиконвулсанти (Диазепам, Депакин). Тези данни, липсата на странични ефекти, както и възможността за постигане на дългосрочен ефект с последващо продължаване на лечението с Кеppra пер ос, са в подкрепа на все по-масовото използване на медика-мента като основен или допълнителен при овладяване на епилептичен статус, независимо от вида пристъпи и тяхната етиология.
ЛИТЕРАТУРА1. Ramael, S., De Smedt, F., Toublanc, N., Otoul, C., Boulanger,
P., Riethuisen, JM., Stockis, A. Single-dose bioavailability of levetiracetam intravenous infusion relative to oral tablets and multiple-dose pharmacokinetics and tolerability of levetiracetam intravenous infusion compared with placebo in healthy subjects. Clin Ther., 28, 2006, 5, 734-44.
2. Beyenburg, S., Reuber, M., Maraite, N. Intravenous Levetiracetam for Epileptic Seizure Emergencies in Older People. Gerontology, 55, 2008, 1, 27-31.
3. Goraya, JS, Khurana, DS, Valencia, I., Melvin, JJ, Cruz, M., Legido, A., Kothare, SV. Intravenous levetiracetam in children with epilepsy. Pediatr Neurol., 38, 2008, 3, 177-80.
4. Baulac, M., Brodie, M., Elger, C., Krakow, K., Stockis, A., Meyvisch, P., Falter, U. Levetiracetam Intravenous Infusion as an Alternative to Оral Dosing in Patients with Partial-Onset Seizures. Epilepsia, 48, 2007, 3, 589-592.
5. Stockis, A., Toublanc, N., Ramael, S., De Smedt, F., Otoul, C., Boulanger, P., Riethuisen, J.-M. Safety, Tolerability and Pharmacokinetics of Levetiracetam Intravenous Infusion. The 1-st London Colloquium on Status Epilepticus, April 2007, Poster presentation, The Institute of child health, Guilford street, London, WC1N.
6. Dobis, C., Von Kegler, S. Intravenous Levetiracetam in Refractory Status Epilepticus: A Report of 3 Cases. The 1-st London Colloquium on Status Epilepticus, April 2007, Poster presentation, The Institute of child health, Guilford street, London, WC1N.
7. Troenaru, M., Ramael, S., Toublanc, N., Daoust, A., Otoul, C., Lu,

Българска Неврология Bulgarian Neurology 41Май 2011
Z., Stockis, A. Levetiracetam Intravenous Infusion : A Randomised Placebo-Controlled Safety Study. The 1-st London Colloquium on Status Epilepticus, April 2007, Poster presentation, The Institute of child health, Guilford street, London, WC1N.
8. Segers-Van Rijn, MWJ, Uges, JWF, Touw, DJ, Keunen, RWM. Intravenous Levetiracetam in Refractory Status Epilepticus: A Case Report. The 1-st London Colloquium on Status Epilepticus, April 2007, Poster presentation, The Institute of child health, Guilford street, London, WC1N.
9. Abend, NS, Florance, N., Finkel, RS, Licht, DJ, Dlugos, DJ. Intravenous Levetiracetam Terminates Refractory Focal Status Epilepticus. Neurocrit Care, 10, 2009, 1, 83-6.
10. Beerhorst, K., de Krom, MCTFM, van Kranen-Mastenbroek, VHJM. Intravenous Levetiracetam Administration during Continuous EEG Monitoring in the Treatment of Focal Status Epilepticus: A Case Report. The 1-st London Colloquium on Status Epilepticus, April 2007, Poster presentation, The Institute of child health, Guilford street, London, WC1N.
11. Bouma, P. Repeated Status Epilepticus – Safety Issues of Conventional AEDs and Successful Treatment with Levetiracetam IV Monotherapy: a Case Report. The 1-st London Colloquium on Status Epilepticus, April 2007, Poster presentation, The Institute of child health, Guilford street, London, WC1N.
12. Broessner, G., Dobesberger, J., Seppi, K., Walser, G., Ehling, R. Intravenous Levetiracetam in Seizure Emergency Situations Including Status Epilepticus. The 1-st London Colloquium on Status Epilepticus, April 2007, Poster presentation, The Institute of child health, Guilford street, London, WC1N.
13. Bunten, S., Kröger, B., Dempewolf, S., Rawert, H., Happe, S. Levetiracetam in the Treatment of Status Epilepticus. The 1-st London Colloquium on Status Epilepticus, April 2007, Poster presentation, The Institute of child health, Guilford street, London, WC1N.
14. Farooq, MU, Naravetla, B., Majid, A., Gupta, R., Pysh, JJ, Kassab, MY. IV levetiracetam in the management of non-convulsive status epilepticus. Neurocrit Care, 7, 2007, 1, 36-9.
15. Knake, S., Gruener, J., Hattemer, K., Klein, KM, Bauer, S., Oertel, WH, Hamer, HM, Rosenow, F. Intravenous levetiracetam in the treatment of benzodiazepine refractory status epilepticus. J Neurol Neurosurg Psychiatry, 79, 2008, 5, 588-9.
16. Merse, O., Oelmann, HD. Intravenous Levetiracetam in the Treatment of Acute Status Epilepticus: Three Case Studies. The 1-st London Colloquium on Status Epilepticus.
17. Möddel, G., Kellinghaus, C., Dziewas, R., Evers, S. Intravenous Levetiracetam /Keppra/ as a New Treatment Option for Status Epilepticus: First Clinical Experiences. The 1-st London Colloquium on Status Epilepticus, April 2007, Poster presentation, The Institute of child health, Guilford street, London, WC1N.
18. Rupprecht, S., Franke, K., Fitzek, S., Witte, OW, Hagemann,
G. Levetiracetam as a treatment option in non-convulsive status epilepticus. Epilepsy Res., 73, 2007, 3, 238-44.
19. Schulze-Bonhage, A., Hefft, S., Oehl, B. Termination of complex partial status epilepticus by intravenous levetiracetam - a case report. J Neurol Neurosurg Psychiatry, 80, 2009, 8, 931-3.
20. Haberlandt, E., Foerster, S., Baumgartner, S., Scholl-Buergi, S., Karall, D., Rauchenzauner, M. Myoclonic Status Epilepticus in Two Patients and Effective Treatment with Intravenous Levetiracetam. The 1-st London Colloquium on Status Epilepticus, April 2007, Poster presentation, The Institute of child health, Guilford street, London, WC1N.
21. Haberlandt, E., Sigl, SB, Scholl-Buergi, S., Karall, D., Rauchenzauner, M., Rostásy, K. Levetiractam in the treatment of two children with myoclonic status epilepticus. Eur J Paediatr Neurol., 13, 2009, 6, 546-9.
22. Reinshagen, A., Neubert, A., Hoffmann, F. Levetiracetam in Focal Myoclonic Status Epilepticus: a Case Report. The 1-st London Colloquium on Status Epilepticus, April 2007, Poster presentation, The Institute of child health, Guilford street, London, WC1N.
23. Reinshagen, A., Schreiber, K., Hoffmann, F. Levetiracetam in Complex Focal Myoclonic Status Epilepticus: a Case Report. The 1-st London Colloquium on Status Epilepticus, April 2007, Poster presentation, The Institute of child health, Guilford street, London, WC1N.
24. Uges, J., Van Huizen, M., Engelsman, J., Wilms, E., Touw, D., Peeters, E., Vecht, C. Safety and pharmacokinetics of intravenous levetiracetam infusion as add-on therapy in status epilepticus. Epilepsia, 50, 2009, 3, 415-21.
25. Patel, N., Laudan, I., Levin, J., Szaflarski, J., Wilker, A. The use of levetiracetam in refractory status epilepticus. Seizure, 15, 2006, 137-141.
26. Rüegg, S., Naegelin, Y., Hardmeier, M., Winkler, DT, Marsch, S., Fuhr, P. Intravenous levetiracetam: treatment experience with the first 50 critically ill patients. Epilepsy Behav., 12, 2008, 3, 477-80.
27. Michaelides, C., Thibert, RL, Shapiro, MJ, Kinirons, P., John, T., Manchharam, D., Thiele, EA. Tolerability and dosing experience of intravenous levetiracetam in children and infants. Epilepsy Res., 81, 2008, 2-3, 143-7.
Адрес за кореспонденция:Д-р Екатерина Иванова Витева дмМедицински Университет – Пловдив, Катедра по неврология4002 Пловдивбул. „Васил Априлов”15Аe-mail: [email protected]

Българска Неврология Bulgarian Neurology 42Май 2011
SUMMARY
EPIDEMIOLOGICAL STUDIES OF THE EPILEPSY Pl. Antimov1, I. Tournev1, 2
1Neurology Clinic, University Hospital “Alexandrovska”, Medical faculty
1,2New Bulgarian University, Chair of cognitive sciences and psychology
Epilepsy is one of the most common serious neurological conditions. The great number of epidemiological studies established considerable geographical variations in incidence of the epilepsy syndromes, often associated with genetic and social-economic features. According to International League Against Epilepsy (ILAE) one of the major shortcomings of the epidemiology of epilepsy is that most of the studies have been carried out in industrial countries. Epidemiological studies in the resource poor countries require a specific methodological approach because of the lack of precise medical records and facilities. Some of the risk factors are known, although the aetiology is not still precisely defined. Prospective population based epidemiological studies are appropriate to establish the magnitude of possible geographical differences in incidence. The International League Against Epilepsy strongly encourages realization of prospective epidemiological studies aiming to establish the magnitude of the geographical differences in incidence of epilepsy; the contribution on different risk factors to the geographic variation; incidence of the particular epilepsy syndromes.
KEY WORDS: epilepsy, epidemiological studies.
РЕЗЮМЕ
Епилепсията е едно от най-честите сериозни невро-логични заболявания. Множеството проведени епидеми-ологични проучвания на епилепсията показват значител-ни географски вариации в честотата на епилептичните синдроми, като често са свързани с генетични и соци-ално-икономически фактори. Според Международната лига срещу епилепсия (ILAE) една от основните слабос-ти на проведените досега епидемиологични проучвания на епилепсията е, че повечето проучвания са осъществе-ни в индустриалните държави. Епидемиологичните про-учвания в развиващите се страни изискват специален методологичен подход поради относителната липса на прецизна медицинска документация и регистри, както и недостига на скъпоструваща медицинска апаратура. Въпреки че са известни някои рискови фактори, по-голя-мата част от етиологичните причинители остават неизвестни. Особено подходящи за установяване връз-ката на различните етиологични и рискови фактори с географското разпределение на заболяването и опреде-ляне на степента на географските различия в разпрост-ранението му са проспективните population (community) based епидемиологични проучвания. Международната лига срещу епилепсия (ILAE) насърчава осъществяване-то на проспективни епидемиологични проучвания, целя-щи установяването на степента на съществуващите географски разлики в заболеваемостта от епилепсия, значението на различните етиологични и рискови фак-тори за географските вариации на заболяването и забо-
леваемостта на отделните епилептични синдроми.КЛЮЧОВИ ДУМИ: епилепсия, епидемиологични про-
учвания.
Епидемиологията (epi-на, demos-народ, logos-наука) е наука за разпространението на заболяване или физиоло-гично състояние в човешките популации и за факторите, които повлияват това разпространение (A.M.Lilienfeld). Според Кларк епидемиологията е област от науката, която се занимава с различни фактори и условия, опреде-лящи здравето, появата и разпространението на болес-тите, нетрудоспособността и смъртта между групи от индивиди. (31) (2)
На научно ниво медицинските проблеми се решават с помощта на три подхода: експериментален, клиничен и епидемиологичен. Епидемиологичният подход се използва за изучаване на количествената динамика на масовите заболявания и закономерностите на тяхното разпре-деление, както и за оценка ефективността на профи-лактичните мерки и изграждане на теорията на про-филактиката. Съществуват четири разновидности на епидемиологическия метод, които се дефинират като три типа епидемиология: дескриптивна (описателна), аналитична и експериментална. Някои автори описват и четвърти тип епидемиология – математическо моде-лиране. (2) (45)
Епидемиологичните проучвания на епилепсията обикно-вено използват дескриптивния и аналитичния метод. (45)
Болестността и заболеваемостта от епилепсия варират в различните държави. Заболеваемостта се дефинира като брой на нововъзникналите заболявания през годината. За епилепсията заболеваемостта се определя като брой заболели/100 000 население. В разви-тите страни заболеваемостта от епилепсия е около 50/100 000 за година (40 – 70/100 000/година). Този показател в развиващите се страни е в границите 100 – 190/100 000. Болестността представлява съвкупността от всички болни, нови и стари, през годината. За епилепсия се определя като брой болни на 1 000 души население. Най-често определяната болестност от епилепсия е 5 – 10/1000, като в тях не влизат фебрилните гърчове, единичните припадъци и неактивните случаи. Подобни стойности за болестност от епилепсия са получени също и в развиващите се страни. Приблизително 45 до 100 милиона души в света страдат от активна епи-лепсия. Несъответствието между високи стойности на заболеваемост и ниски стойности на болестност в развиващите се страни може да бъде обяснено отчасти с по-висока смъртност, отколкото в индустриални-те държави. Данните за смъртността при епилеп-тично болни в развиващите се държави са оскъдни. Съвкупността от нелекуваните и погрешно лекуваните болни съставлява т.нар. treatment gap. (45) (19) (35)
С п о р е д с т а н о в и щ е т о н а I LA E / I B E / W H O (Международната лига против епилепсия/Международно бюро по епилепсия/Световна здравна организация), отра-зено във въведението на глобалната кампания срещу епилепсия “Epilepsy out of the Shadows” около 85% от
Оригинални статииЕПИДЕМИОЛОГИЧНИ ПРОУЧВАНИЯ НА ЕПИЛЕПСИЯТА
Пл. Антимов1, И. Търнев1, 2
1Неврологична клиника, УМБАЛ Александровска, МФ – София2Нов български университет, Катедра по Когнитивна наука и психология

Българска Неврология Bulgarian Neurology 43Май 2011
епилептично болните в света не се лекуват или са неп-равилно лекувани. В някои развиващи се държави като Етиопия и Пакистан едва 2% от епилептично болните се лекуват. (44) (34)
Епилепсията е най-честото сериозно неинфекциозно неврологично заболяване в света (Fernandes and Sander, 1988) и второто по честота неврологично страдание след мозъчния инсулт в индустриалните държави. Тя може да бъде дефинирана като склонност за поява на непредизвикани епилептични припадъци. Въпреки че е едно от най-честите сериозни неврологични заболява-ния, съществуват редица трудности при епидемиоло-гичното и изучаване, до голяма степен поради хетеро-генната и природа. Епидемиологичните данни относно епилепсията имат съществено значение за клиницисти-те и здравните администратори, изграждащи страте-гии за борба с хроничните заболявания. (19) (40) (45)
Епилепсията е широко изучавано заболяване. Епидемиологични проучвания на епилепсията са пра-вени в над 30 държави в света, повечето от които икономически развити. Значително по-малко са епиде-миологичните проучвания на това заболяване, прове-дени в развиващите се държави като Чили, Еквадор, Танзания. Епидемиологични проучвания обикновено се базират на ретроспективни обзори на данните от различни медицински регистри или проучвания в общ-ността (community-based surveys). Клиничните данни за определена популация, обикновено събирани в клинични центрове, могат да бъдат използвани за епидемиологич-ни цели, което по-лесно се осъществява в развитите държави. Този подход обаче крие известни рискове, защото изследващият трябва да разчита на по-рано събрана информация и поставени диагнози. Проучвания, базирани на медицинска документация от Mayo Clinic или общопрактикуващите лекари във Великобритания например, използват такъв вид информация, която задо-волително се доближава до реалността. За съжаление в развиващите се страни тази процедура не може да бъде правдоподобна поради липсата на представителност и прецизност в регистрите или медицинската докумен-тация на определената популация. Независимо къде, теренните проучвания предлагат най-добрия подход за установяване на епидемиологията на заболяване в опре-делена популация. Такива проучвания вече са проведени в няколко латиноамерикански държави, наред с други като Китай, САЩ и Италия. Южноамериканските държави, в които са провеждани теренни проучвания, са Колумбия, Бразилия, Еквадор, Боливия, Мексико, Гватемала и Чили. Болестността от епилепсия в латиноамериканските държави се счита за висока като цяло, като в Бразилия резултатите са много хетерогенни: 13.3/1000 устано-вена от Marino Jr, et al., 36.8/1000 от Fernandes, et al., 1/1.000 от Almeida Filho, Fernandes & Sander; 7/1000, от Melo-Souza et al. (17)(18)(33)(37) Действително поради големината на бразилската популация и социално-иконо-мическото и културно разнообразие стойностите може да се различават в различните райони, а дори и в една и съща провинция, обикновено относно градски и провин-циални зони. (20)
При липсата на добра медицинска документация или научно-изследователски регистри е необходимо осъ-ществяването на community-based теренни проучва-ния на цялата популация за прецизното определяне на епилептично болните, което изисква употребата на специфичен и чувствителен скринингов инструмент и значителна логистична подкрепа (Schoenberg, 1982). (42)
Болшинството епилептично болни имат добра прог-
ноза по отношение на заболяването си, ако получат под-ходящо лечение. В развиващите се страни обаче недос-татъчното познаване на епилепсията, неадекватните медицински ресурси и оскъдното снабдяване с антиепи-лептични медикаменти възпрепятстват провеждането на подходящо лечение. В световен мащаб 60-90% от хората с епилепсия не получават никакво лечение или са неадекватно лекувани. (57)
ЕПИДЕМИОЛОГИЧНИ ПРОУЧВАНИЯ НА ЕПИЛЕП-СИЯТА В РАЗВИТИТЕ СТРАНИ
Голямо епидемиологично проучване на епилепсията е проведеното в Копаро, Италия през 1964 – 1978 година. То използва методите на дескриптивната епидемио-логия. Авторите събират данни за всички потенциал-но епилептично болни от регистрите на лекарите и социалните работници във всички здравни заведения на територия от 420 км2 за период от 18 години. Те също така интервюират учители от всички училища в района за идентифициране на недиагностицирани случаи. От събраните 498 суспектни веднага е прието, че 111 са болни, тъй като те се проследяват от някои от авто-рите на проекта. Останалите 387 са интервюирани и им е проведен неврологичен преглед в периода ноември 1980 – 1981 година. 43 отказват интервю, като проуч-ването при тях се основава единствено на наличната медицинска документация. От всички 498 изучавани болни, 173 са изключени, а за 325 е прието, че страдат от епилепсия. Използвани са диагностичните критерии на Hauser and Kurland (1975) (22), като са взети предвид само случаите на активна епилепсия, т.е. болни, които са имали два или повече нефебрилни и несимптом-ни епилептични припадъци или са на антиконвулсивна терапия. Епилептичните припадъци са класифицирани по клиничен тип по критериите на Международната лига против епилепсия (ILAE) от 1981 г. (23), които включват клинични и електроенцефалографски характе-ристики. Възможните рискови фактори са проучени в съответствие с критериите, предложени от Bergamini et al. (1977). (9) Проучването установява болестност от 6.2/1 000 (мъже и жени съответно 7.1 и 5.2). Най-висока болестност се установява във възрастовата група 10 – 19 години, където е 13.9/1 000. За отчетения период е установена годишна заболеваемост от 33.1/100 000 (мъже и жени съответно – 39.1 и 27.3). Най-високи стой-ности на заболеваемост се установяват в първите две десетилетия от живота. (21)
Установената в Копаро, Италия болестност от 6.2/1 000 е сходна с данните за 6.2 и 6.5/1000 в Рочестър, Минесота през 1965 и 1970 г. (5)(22), както и 6.9/1000 в Дания (1975 г.) (2) и по-висока от представената в Карлайл, Англия (1966 г.) (13) и Йерусалим, Израел (1968 г.) (29)
Друго голямо епидемиологично проучване по методи-те на дескриптивната епидемиология е проведено върху населението над петнадесет годишна възраст в източна Финландия, което наброява 251 715 души. То обхваща периода 1980 – 1985 година. Географската област, в която се провежда проучването включва четири града – Куопио, Иисалми, Суоненйоки и Варкаус, както и провин-циалните територии около тях. Населението е прибли-зително равно в градските и селските райони. Данните за епилептично болни или потенциално епилептично болни са събрани от различни източници за периода 1960 – 1982 г., като включват всички хоспитализирани и нехоспитализирани болни (inpatients и outpatients), както

Българска Неврология Bulgarian Neurology 44Май 2011
и данните от здравноосигурителните структури. На всички събрани пациенти е изпратено писмо с въпросник и покана за преглед.
Епилепсията е дефинирана като състояние с поне два непредизвикани епилептични припадъка с минимален интервал между тях поне 24 часа. Епилептичните про-мени в ЕЕГ не се приемат като задължително условие за поставяне на диагноза. В проучването са включени и пациентите със симптоматична епилепсия, причинена от етиологични причинители като мозъчни тумори и прогресивни неврологични заболявания, докато при-падъците в следствие на остри мозъчни заболявания, екзогенни и метаболитни нокси, при абстиненция и фебрилните гърчове са изключени от проучването. Епилепсията се определя като активна при наличие на припадък през последните пет години или когато бол-ният е на антиконвулсивна терапия. Епилептичните припадъци се класифицират съгласно дефинициите на Международната класификация на епилептичните при-падъци от 1981 г. От общо регистрираните 2 080 случай 1 833 (88%) са прегледани. От останалите 247 (12%) 71 са починали, 34 са се преместили в друг район и 142 не са отговорили. От прегледаните болни 1 607 (88%) са били диагностицирани с епилепсия по-рано, а останали-те 226 (12%) не са имали поставена диагноза епилепсия. Съгласно определената дефиниция 1 214 от диагности-цираните по-рано 1 607 болни имат епилепсия, както и шест от недиагностицираните. От 247те неявили се на клиничния преглед 155 са потвърдени като епилептично болни по данни от медицинската документация.
Проучването определя болестност на активна епи-лепсия от 6.29/1000 и заболеваемост – 24/100 000. Както и при повечето други епидемиологични изследва-ния, установява се по-голяма честота на епилепсията сред мъжете.
Методите на аналитичната епидемиология са изпол-звани при голямо проспективно случай-контрол про-учване (case-control study) в Швеция, целящо оценка на влиянието на пре- и перинатални рискови фактори за развитието на епилепсия. То е проведено в провинция Вестерботен, която се простира на 55 428 км2 с насе-ление 245 204 души, от които 50 692 са под 16 години. За целите на проучването са ангажирани всички лекари и медицински сестри, които работят с деца. Проучването включва всички деца от 0 до 15 години, живеещи в горепосочената област, получили първи непровокиран афебрилен епилептичен припадък през изучавания период от 01.11.1985 г. до 30.06.1987 г. За всеки един от регис-трираните случай се осъществява контролна група от две деца без анамнеза за припадъци на същата възраст и от същия пол, случайно подбрани сред децата, родени на същата дата в провинция Вестерботен. Дефинирането на термините е осъществено според критериите на СЗО и ILAE. За изучавания период са установени 75 деца с непровокирани припадъци, като 14 от тях са приети за неонатални и са изключени от изследването. В следващ етап на проучването е събрана медицинската докумен-тация за протичането на бременността и раждането на пациентите и контролната група. Проучването установява повишена заболеваемост при наличието на следните рискови фактори – вагинални кръвотечения на майката, тютюнопушене по време на бременност-та, гестационната възраст, раждане чрез секцио (като самото цезарово сечение не се явява рисков фактор, а е резултат от състояния, застрашаващи майката и плода). (52)
С оглед установяване на расовите различия при болес-
тността от епилепсия в САЩ е проведено транзвер-зално проучване по методите на аналитичната епиде-миология. Използвайки метода врата до врата (door to door) е изследвано цялото население на Копая Каунти, Мисисипи. Населението в областта е съставено от 50.1% бели, 49.1% афроамериканци и 0.8% други. При проучването не са използвани данни от здравните служ-би, а само въпросник и клиничен преглед на положител-ните от скрининга. Около 97% от домакинствата са взели участие в проекта. За поставянето на диагноза епилепсия е прието наличието на два непровокирани епи-лептични припадъка. Използвана е класификацията на ILAE. Приети са три нива на сигурност на диагнозата – сигурна, възможна и вероятна въз основа на данните, получени от източници извън изследването. За случаи на активна епилепсия са приети тези, които са на анти-конвулсивна терапия и са получили припадък през послед-ните три години или такива, които не се лекуват и са имали поне един епилептичен припадък през последната година. Клиничните прегледи са осъществени от чети-ри висококвалифицирани невролози от Университета на Мисисипи. Проучването установява болестност от 10.43/1 000, като активните форми са 6.78/1 000. 57% от случаите са диагностицирани по-рано от невролог или неврохирург, а 7% не са били диагностицирани преди, като 14% никога не са провеждали антиепилептично лечение. Активна епилепсия е отчетена при 0.5% от бялото и 0.8% от черното население. Епилепсията е по-честа сред мъжете – 0.8%, докато сред жените е 0.5%. (6)
ЕПИДЕМИОЛОГИЧНИ ПРОУЧВАНИЯ НА ЕПИЛЕП-СИЯТА В РАЗВИВАЩИТЕ СЕ СТРАНИ
Обширно невроепидемиологично изследване на епилеп-сията е проведено сред 72 121 жители в северен Еквадор с оглед определяне на заболеваемостта и болестността, както и демографските и географските вариации на заболяването. Изчисляването на показателите е напра-вено на три нива на събиране на информацията. Първото ниво обобщава информацията, получена при теренните проучвания (raw dataset). Второто ниво използва инфор-мация, добита с допълнителни диагностични процедури, като изключва епилептично болните, при които диаг-ностичната информация е недостатъчно изчерпателна (minimum estimated dataset). Третото ниво включва и фалшиво негативните от скрининга (maximum estimated dataset). Класифицирането на епилептичните пристъпи се основава на класификацията на ILAE. Проучването е осъществено в 7 етапа. Първият етап представлява door to door масов скрининг, осъществен от немеди-цински лица с помощта на въпросник, предварително валидизиран, със специфичност 92.9% и сензитивност 79.3%. Всички положителни от скрининга са прегледани от екип от провинциални лекари във втория етап, като случаите са категоризирани като – епилептични при-падъци, фебрилни гърчове, суспектни случаи и фалшиво положителни. В третия етап всички случаи, дефинирани като епилептични припадъци и суспектни са прегледа-ни от екип от невролози, като се използват същите категории. В четвъртия етап добитите данни се реви-зират от международна група невролози консултанти. Случайна група от 1.4% от негативните от скрининга са прегледани в петия етап от провинциалните лекари с оглед определяне на стойностите фалшиво отрицател-ни от първи етап. В шести етап 4.6% случайно подбрани фалшиво положителни от втори етап са прегледани

Българска Неврология Bulgarian Neurology 45Май 2011
от невролози с оглед определяне на степента фалшиво отрицателни от етап 2. Последният седми етап се състои от оценка на надеждността на неврологичната диагноза чрез повторен преглед на 33.9% от диагнос-тицираните епилептично болни. Проучването устано-вява болестност 12.2/1 000 и 19.5/1 000, като тази на активните форми е 6.7/1 000 и 8/1 000, в зависимост от нивото на оценяване. Заболеваемостта е в граници-те 122/100 000/година и 190/100 000/година. (42)
Друго голямо епидемиологично проучване в икономи-чески развиващите се държави е проведено в Танзания. То също използва методите на аналитичната епидеми-ология и е проведено като транзверзално изследване. Проучването е осъществено в областта Уланга, в която живеят 138 837 души, като около 18 000 от тях са изс-ледвани на случаен принцип. Областта Уланга се състои от пет географски и административно разграничени единици. Във всяка единица има от тринадесет до пет-надесет населени места. Изследването е проведено в единадесет случайно подбрани селища, по две от четири от административните единици и три от петата. Всички жители на избраните селища са включени в проучването. Всички селища се състоят от по десет домакинства, всяко от които има лидер, което предс-тавлява най-ниското ниво на администрация. По време на проучването лидерите са интервюирани за всеки един член от домакинството. Използван е скрининговият въпросник на СЗО за неврологични заболявания. Всички положителни от скрининга биват насочвани към невро-лога, който е част от екипа. За поставяне на диагноза са използвани класификацията на ILAE и критериите, използвани при други епидемиологични проучвания в Африка, Южна Америка и Азия (Shorvon and Farmer, 1988). Установената болестност на активна епилепсия е 10.2/1 000, като варира в широки граници в различните селища – от 5.1/1000 до 37.1/1 000. Заболеваемостта е 73.3/100 000. В 25.3% се установяват възможни етио-логични фактори, а в 38% има фамилност. За разлика от много други проучвания честотата при жените е по-висока, отколкото при мъжете. Това би могло да бъде обяснено с по-високата смъртност при мъжете, несвързана с епилепсия. (44)
Епидемиологично проучване от типа „врата до врата” е проведено в три района на два града в южна Бразилия с общо население от 96 300 души. Използван е валидизи-ран въпросник със сензитивност 95.8% и специфичност 97.8%. Скринираните с позитивен резултат от въпрос-ника са преглеждани и диагностицирани от невролог. Установена е болестност от 9.2/1 000. Болестността от активна епилепсия е била 5.4/1 000. Тя е по-висока в по-ниските социални класи (7.5/1 000, за разлика от по-високите, където е 1.6/1 000). 38% от болните с актив-на епилепсия не получават адекватно лечение, като 19% не приемат медикаменти. По същия метод са проведени епидемиологични проучвания и в São José do Rio Preto на популация от 17 293 души и Рио де Жанейро на 982 жители. В São José do Rio Preto е установена болестност от 18.2/1 000, от които 8.2/1 000 с активна епилепсия. Най-висока е честотата във възрастовата група над 65 години – 32.8/1 000. В проучването в Рио да Жанейро е установена болестност от 16.3/1 000 и болестност от активна епилепсия 5.1/1 000. (12)
В рамките на Националното епидемиологично про-учване на неврологичните заболявания (EPINEURO), проведено от септември 1995 година до август 1996, е включено и определяне на честотата на епилепсия в Колумбия. Проучването се осъществява в два етапа. В
първият се идентифицират пациентите, които веро-ятно страдат от неврологично заболяване, а във втория се диагностицират конкретните неврологични заболя-вания. Прегледани са 8 910 души, пропорционално подбра-ни от петте географски района на Колумбия (централен, югозападен, северозападен, източен и Карибски бряг). Проучването е осъществено по невроепидемиологичния протокол на СЗО. Установена е болестност от епилеп-сия 10.3/1 000. (43)
На базата на това проучване Научна група по нев-ронауки към университета Росарио в Богота оценя-ва според международните класификации и проследява за една година епилептично болните, установени при EPINEURO. Определена е болестност от 11.3/1 000 с малки вариации в различните географски райони, с изключение на източния, където болестността е била 23/1000. Болестността от активна епилепсия е 10.1/1 000. Жените имат малко по-голям (нестатистически значим) риск. Най-честия тип епилептични припадъци са фокалните, често с вторична генерализация, а от епи-лептичните синдроми – парциалните симптоматични / криптогенни епилепсии (80%). (55)
Епидемиологично проучване на епилепсията е прове-дено в Junín, град в провинция Буенос Айрес. Избрани са 17 049 жители от 5 839 домакинства. Използван е двуфазен метод. Първата фаза представлява скрининг на жителите от специално обучени немедицински лица. През втората фаза суспектните от първата се диаг-ностицират от невролог по дефинирани диагностични критерии. 14% са били новодиагностицираните случаи на епилепсия, а 78% са на антиепилептично лечение. Определена е болестност от 6.2/1 000, а на активна епилепсия 3.8/1 000. Генерализираните припадъци са били 58%, а парциалните 38%. (36)
Епидемиологическо проучване по метода „врата до врата” е проведено в област Саламà, състояща се пре-димно от провинциално население. Изследвани са 6 473 души, които съставляват 88% от населението на областта. В първия етап теренното проучване е осъ-ществено от работна група, включваща невролог, двама специалисти по обществено здраве и двама общопрак-тикуващи лекари и включва клинично диагностициране на болните. През втория етап на определените болни с активна епилепсия са осъществени видео ЕЕГ, КАТ на главен мозък и имунологични изследвания за цисти-церкоза. Типа на епилептичните припадъци и синдро-мите са определени съгласно класификацията на ILAE. Определена е болестност 23.3/1000, като активната епилепсия е 15.4/1 000 и заболеваемост 92.7/100 000. Най-чести са парциалните припадъци с или без вторична генерализация 92.2%. Симптомните епилепсии (62%) се дължат предимно на невроцистицеркоза (37%), перина-тални мозъчни травми (8%), посттравматични (3%) и постинсултни (2%). 8% са идиопатичните епилепсии, а 30% криптогенните. (35)
Епидемиологично проучване, целящо определяне на честотата на епилепсия е проведено в малък провин-циален район в Гватемала. Изследвани са 1 882 жители, които представляват 97.3% от местното население. Проучването е осъществено в два етапа – скрининг на населението за епилепсия с помощта на стандартните въпросници на СЗО и неврологичен преглед. Установена е болестност от 8.5/1 000, а на активна епилепсия 5.8/1 000. Най-честият тип припадъци са генерализираните тонично-клонични (50%), следвани от комплексните парциални (37.5%), простите парциални (6.2%) и генера-лизираните атонични пристъпи (6.2%). (26)

Българска Неврология Bulgarian Neurology 46Май 2011
На провинциална територия около Истанбул в регион Kucukcekmece от юни 1999 до февруари 2000 година е проведено епидемиологично проучване на популация от 2 187 души. На случен принцип са избрани 490 жилища, които са посетени. След попълването на въпросник на 58 души, суспектните за епилепсия, е снета анамнеза, неврологичен статус и ЕЕГ изследване. 17 души са диаг-ностицирани като епилептично болни. Определена е болестност от епилепсия 0.8%. 47% са генерализирани-те епилепсии, 41.2% парциалните, а в 11.8% припадъците не могат да бъдат класифицирани. Като рискови факто-ри се определят образователния статус, професията, фамилна анамнеза за епилепсия. При определянето на рисковите фактори е използвана контролна група здрави представители от същата популация.
По-голямо проучване на епилепсия е проведено в про-винциални райони около гр. Силиври от юни до октомври 1994 година. Проучени са 4 803 души от общата попула-ция от 70 394. Въпросникът, състоящ се от 15 въпроса, е разпространяван от личните лекари и интернисти. Сензитивността му е 99.9%, а специфичност – 76%. Всичките 415 положителни от скрининга са прегледани от невролози и са класифицирани. От 415те суспектни, 49 (24 жени и 25 мъже) са диагностицирани като епи-лептично болни. Определена е болестност от епилепсия 10.2/1 000, като при жените е 10.01/1 000, а при мъжете 10.39/1 000. 40.8% са генерализираните припадъци, 53.1% парциални и 6% не могат да бъдат класифицирани. Само 7.7% от парциалните епилепсии са дефинирани като вероятно симптомни. 44.98% са получавали антиепилеп-тично лечение, а 65.1% са посетили религиозни лечители в началото или по време на заболяването. (28)
Епидемиологично проучване на епилепсията сред град-ското население е осъществено в град Бурса. 2 116 души са интервюирани от септември 2004 до февруари2005 година. Изследваната група е избрана от населението на централния квартал на град Бурса съгласно статисти-чески метод, като са взети предвид социално-икономи-ческия клас, възраст и пол. Използвани са стандартизи-рани въпросници, с помощта на които специализанти по неврология са осъществили еднакво структурирани интервюта. От 199те положителни при тези интервю-та, 26 (11 жени и 15 мъже) са диагностицирани като епи-лептично болни. Болестността от активна епилепсия в централния квартал на Бурса е била 8.5/1 000, а общата болестност 12.2/1 000. Класифицирането на епилеп-тичните припадъци разкрива, че 30.7% от пациентите са имали парциални, а 65.3% генерализирани припадъци; припадъците, които не мога да се класифицират са били 3.8%. В 50% от случаите припадъците са дебютирали в първата декада на живота, а 34.6% редовно са приемали антиепилептични медикаменти. (14)
Сравнително теренно проучване на епилепсията, проведено по идентични протоколи, е осъществено в Турция и Пакистан. Основните особености в дизайна на това проучване са неселектираната популация що се отнася до предишни медицински изследвания и упот-ребата на стандартизираните протоколи (International Community-Based Epilepsy Research Group (ICBERG) protocols) за оценка на междукултурните различия. Проучени са 24 130 души в Пакистан и 11 497 в Турция (както в градски, така и в провинциални райони, във всички възрасти и от двата пола). Болестността от епилепсия в Пакистан е определена 9.98/1 000 и 7.0/1 000 в Турция (съответно 14.8/1 000 в провинциалните и 7.4/1 000 в градските територии и 8.8/1 000 в провин-циалните и 4.5/1 000 в градските зони). И в двете дър-
жави болестността от епилепсия е двойно по-голяма в провинциалните райони. Средната възраст на дебют на заболяването е била 13.3 години в Пакистан и 12.9 в Турция. Честотата на различните типове припадъци в градските и провинциалните райони е сходна, а именно за Пакистан и Турция респективно 80.5% и 65.4% са ГТКП, 5% и 7.4% простите парциални, 5% и 4.9% комплексните парциални. В Пакистан 27.5% от епилептично болните в градовете и 1.9% от болните в селата получават антиепилептично лечение, а в Турция – 30% (без същес-твена разлика между градско и провинциално население). Резултатите от проучването са сравними с тези от Еквадор, където е използван същия протокол. (7)
Проспективно лонгинтудинално проучване на епилеп-сията е проведено в провинциалните райони на Западен Бенгал, Индия. Целта на проучването е определяне на заболеваемостта от епилепсия. То е проведено в 12 селища на население от 20 966 души. Проучването е осъществено с помощта на местни представители и местното управление. То е осъществено на два етапа. При първият етап е осъществен скрининг тип „врата до врата” с помощта на валидизиран въпросник, пред-ложен от СЗО и преведен на местния език Bengali. Положителните от скрининга са преглеждани клинич-но във втория етап от невролози. ЕЕГ изследване не е провеждано рутинно за всички епилептично болни. Припадъците са класифицирани съгласно клиничните препоръки на ILAE от 1981 година. Такива повторни про-учвания са провеждани неколкократно през следващите 5 години за регистриране на новите случаи и проследява-не на старите. Установена е годишна заболеваемост от 36.66/100 000. Най-чести са генерализираните тонично клонични припадъци. (50)
Голямо епидемиологично проучване на епилепсията по типа „врата до врата” на популация от 55 616 души е проведено в провинциалните райони на Heilongjiang, Ningxia, Henan, Shanxi, Jiangsu през 2000 година, а малко по-късно и в Шанхай на население от 51 644 души. Проучваните общности са подбрани на случаен принцип. Участвали са лекари и здравни работници, обучени от екип на Пекинския Неврохирургичен Институт (Beijing Neurosurgical Institute) на стандартизирани техники. Скринирано е 94.6% от населението на тези области. Използван е въпросник със специфичност 78.5% и сензи-тивност 100%. Всеки скриниран с поне един положите-лен отговор от въпросника е преглеждан от невролог за потвърждаване или отхвърляне на диагнозата. Първото проучване установява болестност от 7.0/1 000 (4.5/1000 за активна епилепсия) население, а това в Шанхай 6.2/1 000; въпреки по-ниската стойност при второто раз-ликата не е статистически значима. Не се установява значителна разлика при жените и мъжете с епилепсия. В рамките на демонстрационен проект 6 месеца по-късно е проведено ново епидемиологично проучване в същите области. Установена е болестност 6.2/1 000, респективно 4.6/1 000 за активните форми. Прави впе-чатление разликата в нелекуваните адекватно болни с активна епилепсия (treatment gap), които при първото проучване са били 62.6%, докато при второто са с 12.9% по-малко – 49.8%.
Друго епидемиологично проучване е проведено в 6 града в Китай през 1983 година в популация от 63 195 души. Обхванато е цялото население на проучвани-те области. Проучването включва скрининг интервю заедно с клиничен преглед за установяване на често срещани неврологични заболявания, включително епи-лепсия. Всички суспектни за неврологични заболявания

Българска Неврология Bulgarian Neurology 47Май 2011
са преглеждани от старши невролози, които използвали стандартизирани диагностични критерии. Установена е болестност от епилепсия 4.4/1000. Най-чести са генерализираните конвулсивни припадъци. Основните предполагаеми рискови фактори за епилепсия са ЧМТ, невроинфекции и МСБ. (30)
В град Keelung в северен Тайван е проведено епиде-миологично проучване от 1 януари 2001 до 31 декември 2001 на население от 13 663 души над 30 години. То е организирано от службата за обществено здраве в града. Установена е болестност от активна епилепсия от 2.77/1 000, с най-висока честота е групата 40 – 49 годи-ни. Мъжете леко превалират над жените. Най-чести са били комплексните парциални припадъци – 46.0%. 24.3% са пациентите, които не се били диагностицира-ни преди. Главният рисков фактор за епилепсия е ЧМТ с честота 13.5%, следван от невроинфекциите 8.1%, мозъчния инсулт 5.4% и перинаталния инсулт 5.4%. общата болестност от епилепсия, включваща активни-те и неактивните форми, е 3.14/1 000. (15)
През 2005 година в провинциален район във Виетнам е проведено теренно проучване за определяне честотата на активна епилепсия. 47 269 души от приблизително 13 000 домакинства са скринирани за припадъци. От тях 2.8% са дали поне един положителен отговор на въпросника. Всички суспектни за епилепсия от скри-нинга са прегледани клинично от невролог за поставяне на диагноза. На всички епилептично болни е предложено ЕЕГ изследване. Установена е болестност от 4.4/1 000 (жени 3.7/1 000 и мъже 5.1/1 000). По-висока честота е установена при изследваните с по-нисше образование и несемейните. Честотата на епилепсия във Виетнам е съизмерима с тази в другите азиатски държави и по-ниска от тази в Южна Америка и Африка. (54)
Епидемиологичните проучвания в развиващите се страни изискват специален методологичен подход поради относителната липса на прецизна медицинска докумен-тация и регистри, както и недостига на скъпоструваща медицинска апаратура. (42) Според Международната лига срещу епилепсия (ILAE) една от основните слабос-ти на епидемиологичните проучвания на епилепсията е, че повечето от тях са осъществени в индустриалните държави. (24)
Множеството проведени епидемиологични проучвания на епилепсията показват значителни географски вариа-ции в честотата на епилептичните синдроми, като често са свързани с генетични и социално-икономически фактори. Различните рискови фактори се явяват отно-сително специфични за отделните възрастови групи, както и донякъде за географското разпространение на заболяването. (8)(46) Конгениталните и генетичните нокси, както и нарушенията в развитието на нервната система най-често се свързват с развитие на епилепсия в детска и млада възраст. Черепномозъчните травми, инфекциите на ЦНС и мозъчните тумори могат да се явят по всяка възраст, въпреки че туморите са обичай-ни след 40 годишна възраст. Мозъчносъдовата болест представлява най-честата причина за епилепсия при болни над 60 години. Относно географското разпределе-ние значение имат ендемични инфекции като малария, невроцистицеркоза, парагономиаза, токсокариаза. Тези инфекции са обичайни предимно за развиващите се дър-жави. Наличието на фамилна анамнеза за епилепсия пови-шава вероятността от развитие на заболяването при наличие на други рискови фактори, което предполага мул-тифакторна етиология на заболяването. (45) Въпреки че са известни някои рискови фактори, по-голямата част
от етиологичните причинители остават неизвестни. Особено подходящи за установяване връзката на различ-ните етиологични и рискови фактори с географското разпределение на заболяването и определяне на степен-та на географските различия в разпространението му са проспективните population based епидемиологични проучвания. (45) Международната лига срещу епилепсия (ILAE) насърчава осъществяването на проспективни епидемиологични проучвания, целящи установяването на степента на съществуващите географски разлики в заболеваемостта от епилепсия, значението на различ-ните етиологични и рискови фактори за географските вариации на заболяването и заболеваемостта на отдел-ните епилептични синдроми. (24)(45)
ЛИТЕРАТУРА1. Борисов, В Компендиум по социална медицина, стр. 21 – 25, 53 – 54. 2. Монев, В. Епидемиология на инфекциозните болести, стр. 28 – 36. 3. Anderson VE, Hauser WA, Rich SS. Genetic heterogeneity and
epidemiology of the epilepsies. Adv Neurol 1999; 79:59–73. 4. Anderson E, Berkovic S, Dulac O, et al. ILAE genetics commission
conference report: molecular analysis of complex genetic epilepsies. Epilepsia 2002; 43:1262–1267.
5. Annegers JF, Hauser WA. Unpublished findings cited in: Hauser WA. Epidemiology of epilepsy. In:Schoenberg BS, ed. Neurological epidemiology: principles und clinical applicutions. New York: Raven Press, 1978:313-39. (Advances in neurology; vol 19).
6. Armin F. Haerer, Dallas W. Anderson, and Bruce S. Schoenberg. Prevalence and Clinical Features of Epilepsy in a Biracial United States Population. Epilepsia. 27(1):6675, 1986.
7. Aziz H, Güvener A, Akhtar SW, Hasan KZ. Comparative epidemiology of epilepsy in Pakistan and Turkey: population-based studies using identical protocols. Epilepsia. 1997 Jun;38(6):716-22.
8. Bell GS, Sander JW. The epidemiology of epilepsy: the size of the problem. Seizure 2001; 10:306–314.
9. Bergamini L, Bergarnasco B, Benna P, Gilli P. Acquired etiological factors in 1,785 epileptic subjects: clinical-anamnestic research. Epilepsia 1977;18:437-44.
10. Bergen DC. Preventable neurological diseases worldwide. Neuroepidemiology 1998; 17:67–73.
11. Berkovic SF, Scheffer IE. Genetics of the epilepsies. Epilepsia 2001; 42 (Suppl 5):16–23.
12. Borges M.A. et al. Urban prevalence of epilepsy - Population study in Sao Jose do Rio Preto, a medium-sized city in Brazil. Arq. Neuropsiqulatr 2004;62(2-A);199-205
13. Brewis M, Poskanzer DC, Rolland C, Miller H. Neurological disease in an English city. Acra Neurol Scand (SuppTj 1966;24:9-89.
14. Calişir N, Bora I, Irgil E, Boz M. Prevalence of epilepsy in Bursa city center, an urban area of Turkey. Epilepsia. 2006 Oct;47(10):1691-9.
15. Chen CC, Chen TF, Hwang YC, Wen YR, Chiu YH, Wu CY, Chen RC, Chen TH, Liou HH. Population-based survey on prevalence of adult patients with epilepsy in Taiwan (Keelung community-based integrated screening no. 12). Epilepsy Res. 2006 Nov;72(1):67-74. Epub 2006 Aug 30.
16. Commission on Tropical Diseases of the International League Against Epilepsy. Relationship between epilepsy and tropical diseases. Epilepsia 1994; 35:89–93.
17. Fernandes J, Sander JW. (1998) Epidemiologia e história natural das epilepsias. In Costa J, Palmini A, Yacubian EM, Cavalheiro EA, (Eds) Fundamentos neurobiológicos das epilepsias. Aspectos clínicos e cirúrgicos. Lemos, São Paulo, pp. 3–20.
18. Fernandes JG, Schmidt MI, Tozzi S, Sander JWAS. Prevalence of epilepsy: the Porto Alegre study. Epilepsia 1992;33(Suppl 3):132.
19. Fong GCY, Mak W, Cheng TS, Chan KH, Fong JKY, Ho SL. A prevalence study of epilepsy in Hong Kong. Hong Kong Med J 2003;9:252-7.
20. Gomes Marleide da Mota, Regina Golnner Zeitoune, Leandro Albuquerque Lemgruber Kropf, Erica da Silva van Beeck. A house-to-house survey of epileptic seizures in an urban community of Rio de Janeiro, Brazil. Arq Neuropsiquiatr 2002;60(3-B):708-711.
21. Granieri Е., Giulio Rosati, Rosalia Tola, Mario Pavoni, Ezio Paolino, Luigi Pinna, and Vinceiiza C. Monetti. A Descriptive Study of Epilepsy in the District of Copparo, Italy, 1964-1978. Epilepsia, 24502-514, 1983.
22. Hauser WA, Kurland LT. Epidemiology of epilepsy in Rochester,

Българска Неврология Bulgarian Neurology 48Май 2011
Minnesota, 1935-1967. Epilepsia 1975;16: 1-66. 23. International League Against Epilepsy Commission on
Classification and Terminology. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsiа 1981;22:489-501.
24. ILAE Commission of Epidemiology and Prognosis. The epidemiology of the Epilepsies: future directions. Epilepsia 1997; 38:614–618.
25. Johnson MR, Sander JW. The clinical impact of epilepsy genetics. J Neurol Neurosurg Psychiatry 2001; 70:428–430.
26. Jorge E. Mendizabal and Luis F. Salguero. Prevalence of Epilepsy in a Rural Community of Guatemala. Epilepsia, 37(4):373-376, 1996
27. Juul-Jensen P, Ipsen J. Prevalence and incidence of epilepsy in greater Aarhus. Ugeskr Laeger1975; 137:2380-8.
28. Karaagaç N, Yeni SN, Senocak M, Bozluolçay M, Savrun FK, Ozdemir H, Cagatay P. Prevalence of epilepsy in Silivri, a rural area of Turkey. Epilepsia. 1999 May;40(5):637-42.
29. Leibowitz U, Alter M. Epilepsy in Jerusalem, Israel. Epilepsia 1968;9:87-105.
30. Li SC, Schoenberg BS, Wang CC, Cheng XM, Zhou SS, Bolis CL. Epidemiology of epilepsy in urban areas of the People’s Republic of China. Epilepsia. 1985 Sep-Oct;26(5):391-4.
31. Lilenfeld, A. M. Foundations of Epidemilolgy. Department of Epidemiology. The Johns Hopkins University. School of Hygiene and Public Health. New York. Oxford University Press. 1976. 3, 117-118.
32. MacDonald BK, Cockerell OC, Sander JW, et al. The incidence and lifetime prevalence of neurological disorders in a prospective community-based study in the UK. Brain 2000; 123:665–676.
33. Marino R Jr, Cukiert A, Pinho A, Pinho E. Aspectos epidemiológicos da epilepsia em São Paulo. Arq Neuropsiquiat 1982;44:243-254.
34. Meinardi, H., R. A. Scott, R. Reis, J. W. A. S. Sander on behalf of the ILAE Commission on the Developing World The Treatment Gap in Epilepsy: The Current Situation and Ways Forward. Epilepsia, 42(1):136–149, 2001.
35. Medina, Marko et al. Prevelance, Incidence, and Etiology of Epilepsies in Rural Honduras: The Salama Study. Epilepsia, 46(1):124-131.2005.
36. Melcon MO, Kochen S, Vergara RH. Prevalence and clinical features of epilepsy in Argentina. A community-based study. Neuroepidemiology. 2007;28(1):8-15. Epub 2006 Dec 8.
37. Melo-Souza SE, Tolentino MAS, Parada JCB, Oliveira ELG, Alves MAC. Epidemiologia da epilepsia em Goiânia-Brasil. Arq Neuropsiqiat 2000; 58(Suppl 2):30.
38. Molyneux ME. Impact of malaria on the brain and its prevention. Lancet 2000; 355:671–672.
39. Nicoletti A, Bartoloni A, Reggio A, et al. Epilepsy, cysticercosis, and toxocariasis: a population-based case-control study in rural Bolivia. Neurology 2002; 58:1256–1261.
40. Noronha Ana L. A., Moacir A. Borges, Lucia H. N. Marques, Dirce M. T. Zanetta, Paula T. Fernandes, Hanneke de Boer, Javier Espíndola, Claudio T. Miranda, Leonid Prilipko, Gail S. Bell, Josemir W. Sander, and Li M. Li. Prevalence and Pattern of Epilepsy Treatment in Different Socioeconomic Classes in Brazil. Epilepsia, 48(5):880–885, 2007.
41. Pal DK, Carpio A, Sander JW. Neurocysticercosis and epilepsy in developing countries. J Neurol Neurosurg Psychiatry 2000; 68:137–143.
42. Placencia, M., S. D. Shorvon, V. Paredes, C. Bimos, J. W. A. S. Sander, J. Suarez, and S. M. Cascante. Epileptic Seizures in an Andean Region of Ecuador Incidence and Prevalence and Regional Variation. Brain (1992), 115, 771-782.
43. Pradilla A., Gustavo; Vesga A., Boris E.; Leon-Sarmiento, Fidias E. and grupo geneco. National neuroepidemiological study in Colombia (EPINEURO). Rev Panam Salud Publica (online). 2003, vol.14, n.2, pp. 104-111. ISSN 1020-4989.
44. Rwiza HT, Kilonzo GP, Haule J, et al. Prevalence and incidence of epilepsy in Ulanga, a rural Tanzanian district: a community-based study. Epilepsia 1992; 33:1051–1056.
45. Sander, JW (2003) Epidemiology of epilepsy revisited. Curr Opin Neurol 16(2), 165-170.
46. Sander, J.W., M.C. Walker, J. E. Small Epilepsy 2007. Eleventh Epilepsy Teaching Weekend, St Anne’s College, Oxford.
47. Sander JW, Shorvon SD. Epidemiology of the epilepsies. J Neurol Neurosurg Psychiatry 1996; 61:433–443.
48. Sander JW. Some aspects of prognosis in the epilepsies: a review. Epilepsia 1993; 34:1007–1016.
49. Schoenberg BS (1982) Clinical neuroepidemiology in developing countries: neurology with few neurologists. Neuroepidemiology, 1, 137—142.
50. Shankar P. Saha et al. A Prospective incidence study of epilepsy in rural community of West Bengal, India. Neurology Asia 2008;13: 41-48
51. Shorvon SD, Farmer PJ. Epilepsy in developing countries: a review of epidemiological, sociocultural, and treatment aspects. Epilepsia 1988;29(suppl l);S36-54.
52. Sidenvall, R., J. Heijbel, H. K. Blomquist, L. Nystro¨m, and L. Forsgren. An Incident Case–Control Study of First Unprovoked Afebrile Seizures in Children: A Population-Based Study of Pre- and Perinatal Risk Factors. Epilepsia, 42(10):1261–1265, 2001.
53. Tapani Keranen, Paavo J. Riekkinen, and Matti Sillanpiiii. Incidence and Prevalence of Epilepsy in Adults in Eastern Finland. Epilepsia, 30(4):413421, 1989.
54. Tuan NA, Cuong le Q, Allebeck P, Chuc NT, Persson HE, Tomson T. The prevalence of epilepsy in a rural district of Vietnam: a population-based study from the EPIBAVI project. Epilepsia. 2008 Sep;49(9):1634-7. Epub 2008 May 20.
55. Velez, Alberto and Jorge Eslava-Cobos. Epilepsy in Colombia: Epidemiologic Profile and Classification of Epileptic Seizures and Syndromes. Epilepsia, 47(1):193–201, 2006
56. Waruiru CM, Newton CR, Forster D, et al. Epileptic seizures and malaria in Kenyan children. Trans R Soc Trop Med Hyg 1996; 90:152–155.
57. Wenzhi Wang, Jianzhong Wu, Xiuying Dai, Guangyu Ma, Bin, Yang, Taiping Wang, Chenglin Yuan, Ding Ding, Zhen Hong, Patrick Kwan, Gail S Bell, Leonid L Prilipko, Hanneke M de Boer, Josemir W Sander. Global campaign against epilepsy: assessment of a demonstration project in rural China. Bulletin of the World Health Organization; Type: Research Article DOI: 10.2471/BLT.07.047050.
58. Zarrelli MM, Beghi E, Rocca WA, et al. Incidence of epileptic syndromes in Rochester, Minnesota: 1980–1984. Epilepsia 1999; 40:1708–1714.

Българска Неврология Bulgarian Neurology 49Май 2011
SUMMARY
γ-AMINOBUTYRIC ACID NEUROPROTECTION IN AN IN VITRO MODEL OF RAT BRAIN ISCHEMIA
V. Tzankova and S. Mitusheva Medical University - Sofia, Faculty of Pharmacy,
Department of Pharmacology, Pharmacotherapy and Toxicology
Since increased glutamatergic activity is the major primary event that results in cell death following an acute hypoxic-ischemic stroke, γ-aminobutyric acid (GABA) might therefore be expected to be neuroprotective. The aim of the present study was to assess neuroprotection exerted by γ-aminobutyric acid (0.0001–100 mM) in rat cortical brain slices subjected to oxygen–glucose deprivation and reoxygenation in vitro. Neuronal injury and neuroprotection were assessed by measuring the release of glutamate and lactate dehydrogenase and tissue water content. Results demonstrate that γ-aminobutyric acid exerted neuroprotective effects according to a “U-shaped”, hormetic-like, concentration-response curve, with an efficacy window of 10-100 µM concentration. To clarify the mechanism of neuroprotecttion exert by GABA, the effects of GABAA receptor agonist muscimol and GABAB receptor agonist baclofen were also investigated. In conclusion, it was found that GABA-mediated neuroprotection during energy deprivation was essentially mediated by GABAA, while GABAB receptors were not involved.
KEY WORDS: γ-aminobutyric acid, lactate dehydrogenase, rat brain ischemia.
РЕЗЮМЕ
Повишената активност на възбудния невромеди-атор глутамат е една от основните причини, която води до клетъчна смърт след остър исхемичен инсулт. Предполага се, че основният потискащ невромедиатор в ЦНС- γ-аминомаслена киселина (ГАМК) има невроп-ротективен ефект спрямо глутамат-индуцираната невротоксичност. Целта на настоящото проучване е да се оценят невропротективните ефекти на γ-амино-маслената киселина (0.0001-100 mM) в in vitro модел на мозъчна исхемия при плъх, в условията на кислородно-глюкозен глад. Невроналното увреждане и невропротек-цията са оценени чрез определяне на освобождаването на глутамат и лактат дехидрогеназа, и задръжката на течности (едем) в мозъчните срезове. Резултатите показват, че γ-аминомаслената киселина проявява нев-ропротективните ефекти, следвайки “U”-образна кон-центрационна крива, характерна за феномена хормезис. Статистически значим ефект на невромедиатора бе наблюдаван в концентрационния интервал 10-100 µM. Беше установено, че ГАМК- медиираната невропро-текция в in vitro модел на мозъчна исхемия при плъх, в условията на кислородно-глюкозен глад се осъществява по механизми, свързани с ГАМКА, но не и с ГАМКВ рецепторите.
КЛЮЧОВИ ДУМИ: γ-аминомаслена киселина, лактат дехидрогеназа, мозъчна исхемия, плъх.
УВОД
Мозъчният инсулт е на трето място сред фактори-те, водещи до смъртност в развитите индустриални държави и е основна причина за дълготрайни увреждания (2). Концепциятата за разработване на невропротектив-ни средства е следствие от натрупаната в последните години информация за биохимичните механизми, проти-чащи в мозъка на експериментални животни при остър хипоксично – исхемичен епизод (6). При мозъчна исхемия се наблюдава фокално понижение на мозъчния кръвен ток, намаляване на енергийните източници кислород и глюкоза, което води до деполяризация в невроните и глиалните клетки и до освобождаването и натрупването на невро-медиатора глутамат в екстрацелуларното пространст-во (19). Невротоксичността на глутамат е установена експериментално след екзогенно приложение на невроме-диатора и включва процеси, като образуване на свободни радикали, увреждане на митохондриите, фрагментация на ДНК и клетъчна смърт (15, 20).
Гамааминомаслената киселина (γ-аминомаслена киселина, ГАМК) е най-често срещания инхибиторен невромедиатор в човешкия мозък (7). Прилагането на лекарства, които усилват невронално инхибиране медиирано от ГАМК - рецепторите е възможна стратегия за предотвратяване на невроналното увреждане, предизвикано от глутамат. Тъй като глутаматната и ГАМК- системата работят на противоположен принцип се предполага, че подобря-ването на дейността на ГАМК-ергичната система ще антагонизира невротоксичните ефекти от прекомерното освобождаване на глутамат. ГАМК и нейните агонисти притежават физиологични и фармакологични свойства да инхибират клетъчната активност в мозъка, и по този начин се предполага, че могат да намалят последствията от исхемичното увреждане (17, 21).
Цел на настоящото проучване е да се оцени невропро-тективния ефект на ГАМК и ГАМК-рецепторните аго-нисти мусцимол (ГАМКА агонист) и баклофен (ГАМКB агонист) в експериментален модел на мозъчна исхемия при плъх in vitro в условия на кислородно-глюкозен глад. Този модел е подходящ за проучаване на исхемия/репер-фузия предизвикано увреждане на мозъка и за оценка на невропротективните ефекти на много лекарства (3, 11). Kлетъчното увреждане и невропротекцията в мозъчната тъкан бяха оценени чрез определяне на освобождаването на глутамат и лактат дехидрогеназа (LDH) в среда от изкуствена гръбначно-мозъчната теч-ност (aCSF) след периода на реперфузия, както и чрез определяне на задръжката на вода от мозъчната тъкан в края на експеримента
МАТЕРИАЛ И МЕТОДИ
ЕКСПЕРИМЕНТАЛНИ ЖИВОТНИ И ПРИГОТВЯНЕ НА СРЕЗОВЕ ОТ МОЗЪК НА ПЛЪХ
Експериментите са проведени в съответствие с изискванията на националното и европейско законо-
Оригинални статииНЕВРОПРОТЕКТИВЕН ЕФЕКТ НА γ-АМИНОМАСЛЕНА
КИСЕЛИНА В МОДЕЛ НА МОЗЪЧНА ИСХЕМИЯ IN VITRO
В. Цанкова, С. МитушеваМедицински Университет – София, Фармацевтичен факултет, Катедра по”Фармакология, Фармакотерапия и
Токсикология”

Българска Неврология Bulgarian Neurology 50Май 2011
дателство и съответните наредби и ръководства за работа с лабораторни животни (86/609/EEC; National Research Council. The Guide for the Care and Use of Laboratory Animals. Washington, D.C., National Academy Press, 1996).
Използвани са мьжки пльхове от линията „Sprague-Dawley“ с тегло 350-400 g (Charles River, Italy). Животните са отглеждани при стандартни условия с достьп до храна и вода ad libitum. След изолиране, мозъ-кът се поставя в студен разтвор на aCSF (състав в mM: 120 NaCl, 2.5 KCl, 1.3 MgCl2, 1.0 NaH2PO4, 1.5 CaCl2, 26 NaHCO3, 11 glucose, наситен с карбоген 95% O2–5% CO2, pH на разтвора 7.4, осмолалитет 285–290 mOsmol). След дисекция, от кортекса се получават срезове с раз-мери 400 µm посредством Мanual Сhopper Stoelting Co., Wood Dale, IL, USA. След това срезовете се поставят в среда от оксигениран разтвор aCSF, в който са прибаве-ни аскорбинова киселина 400 µM и се темперират за 1 h при стайна температура с цел максимално възстановя-ване от травматичния стрес при подготовката (4).
МОДЕЛ НА ЕКСПЕРИМEНТАЛНА ИСХЕМИЯ IN VITRO
Накратко, мозъчните срезове (≈4–5, общо тегло 33.6±2.6 mg, n=10) се поставят в покрити инкубационни петрита, съдържащи 2 ml aCSF под непрекъснато обга-зяване с карбоген (95% O2/5% CO2) и се инкубират при 37 °C за период от 30 min. Следва периодът на кислород-но-глюкозен глад, който се провежда чрез инкубиране на срезовете за 30 min в разтвор на aCSF, в който глюко-зата е заместена с еквимоларен разтвор на захароза, а обгазяването с карбоген е заместено с 95% N2/5% CO2. След този етап на кислородно-глюкозен глад, разтворът е заменен с пресен, оксигениран aCSF (с карбоген) за следващ период от 90 min (реперфузия/ реоксиганация). В експериментите за оценка на ГАМК-индуцираната невропротекция, след OGD периода, ГАМК, мусцимол или баклофен бяха добавени в различни концентрации в оксигенирания aCSF разтвор.
ОЦЕНКА НА НЕВРОНАЛНОТО УВРЕЖДАНЕ
Невроналното увреждане бе количествено оценено чрез измерване на освободения глутамат и лактат дехидрогеназа (LDH) в разтвора aCSF след периода на 90 min реоксигенация. Глутамат бе определян флуоримет-рично excitation 366 nm; emission:
450 nm), използвайки превръщането на NAD+ в NADH посреством ензима glutamate
dehydrogenase (5). Активността на LDH е определяна спектрофотометрично чрез понижението в абсорбци-ята при 340 nm, което се получава при реакцията на окисление на NADH до NAD+ (8). Окислението се ката-лизира от LDH, a реакцията се извършва в присъствие на пируват. Количественото определяне на задръжката на вода в мозъчната тъкан (gH2O/g tissue dry weight) се изчислява по метода, описан от MacGregor et al. (12).
СТАТИСТИЧЕСКА ОБРАБОТКА НА РЕЗУЛТАТИТЕ
Всички експерименти са проведени чрез използва-нето на мозъчни срезове, приготвени от най-малко 4 животни. Представени са данните ± S.E.M., като с „n” е означен броят на пробите в съответния експеримент. Данните за глутамат и лактат дехидрогеназа (LDH) са представени съответно в nmol/mg тъкан и U/mg тъкан. Статистическата оценка на резултатите е извършена чрез one-way ANOVA followed by post-hoc Dunnett test
(GraphPad INSTAT v3.00, GraphPad Software, San Diego, CA, USA)
РЕЗУЛТАТИ
ЕФЕКТ НА ГАМК ВЪРХУ УВРЕЖДАНЕТО, ПРЕ-ДИЗВИКАНО ОТ КИСЛОРОДНО-ГЛЮКОЗЕН ГЛАД/ РЕПЕРФУЗИЯ В МОЗЪЧНИ СРЕЗОВЕ ОТ ПЛЪХ IN
VITRO
Мозъчните срезове (Контрола, CTRL) са инкубира-ни в разтвор aCSF за 120 min. В контролните проби, освобождаването на лактатдехидрогеназа (LDH) и глу-тамат в инкубационния разтвор бе съответно 5.53 ± 0.83 U/mg proteins (n = 12) и 0.31 ± 0.12 nmol/mg wet tissue (n = 8), а съдържанието на вода бе 10.01± 0.02 gH2O/g tissue dry weight (n = 10). В срезовете, подложени на кислородно-глюкозен глад (OGD) за 30 min, с последващ период на реоксигенация за 90 min, бе отчетено значи-телно освобождаване на LDH до 18.0 ± 5.2 U/mg proteins и на глутамат 0.69 ± 0.03 nmol/mg wet tissue (P<0.001 vs control, n=18) в инкубационната среда. Задръжката на вода в мозъчни срезове бе повишена значимо до 14.05 ± 0.53 g H2O/g tissue dry weight (P < 0.001 vs control, n= 25) (Фиг. 1 A, B, C).
За да установим дали ГАМК проявява протективен ефект върху невроналното увреждане, в условията на кислородно-глюкозен глад in vitro, ГАМК (0.0001 - 100 mМ) бе инкубирана в aCSF във фазата на реперфузия/ реоксигенация (90 min). Установено бе, че ГАМК понижа-ва концентрационно-зависимо количеството на задър-жаната от мозъчната тъкан вода (IC50 46.5 µM) (Фиг. 1А). Максимален ефект се наблюдава при концентрация на ГАМК 100 mM, при която редукцията в количест-вото на задържаната вода бе 79% (P<0.01 спрямо OGD периода n=16).
ГАМК значително понижава освобождаването на LDH в концентрации 0.01-10 mM, и на глутамат в кон-центрационния интервал 0.001-0.1 mM (Фиг. 1B и 1C). В по-ниските (0.0001-0.001 mM) и по-високи концент-рации на ГАМК (над 10 mM) не се наблюдава статис-тически значим ефект върху освобождаването на LDH. Интересно е да се отбележи, че тези ефекти на ГАМК следват “U-образна” концентрационна крива, характер-на за т.нар. постекспозиционен хормезис. По отношение на освобождаването на глутамат, най-ефективна бе концентрацията на ГАМК 0.01 mM, при която бе наблю-давано статистически значимо понижаване до 0.44±0.03 nmol/mg wet tissue (P<0.01, n=9), докато при концент-рация на ГАМК 0.1 mM освобождаването на LDH бе понижено до 12.5 ± 0.7 U/mg proteins (P<0.01, n=11-16). По отношение на намаляване на тъканния едем, ГАМК бе най-ефективна в концентрации 10 mM и 100 mM. Наблюдаваният ефект бе концентрационно-зависим, като понижаването на количеството на задръжката на вода, предизвикано в OGD-периода бе приблизително 50% при 10 mM ГАМК, P<0.05, n=20 и 78,8% при 100 mM ГАМК, P<0.05, n=10 (Фиг. 1А).
ЕФЕКТИ НА АГОНИСТИТЕ НА ГАМКA (МУСЦИ-МОЛ) И ГАМКB (БАКЛОФЕН) ВЪРХУ УВРЕЖДАНЕ-ТО, ПРЕДИЗВИКАНО ОТ КИСЛОРОДНО-ГЛЮКОЗЕН
ГЛАД/ РЕПЕРФУЗИЯ В МОЗЪЧНИ СРЕЗОВЕ ОТ ПЛЪХ IN VITRO
За да установим дали ефектите на ГАМК върху понижаване на освобождаването на LDH, глутамат или тъканния едем е медииран от механизъм, свързан
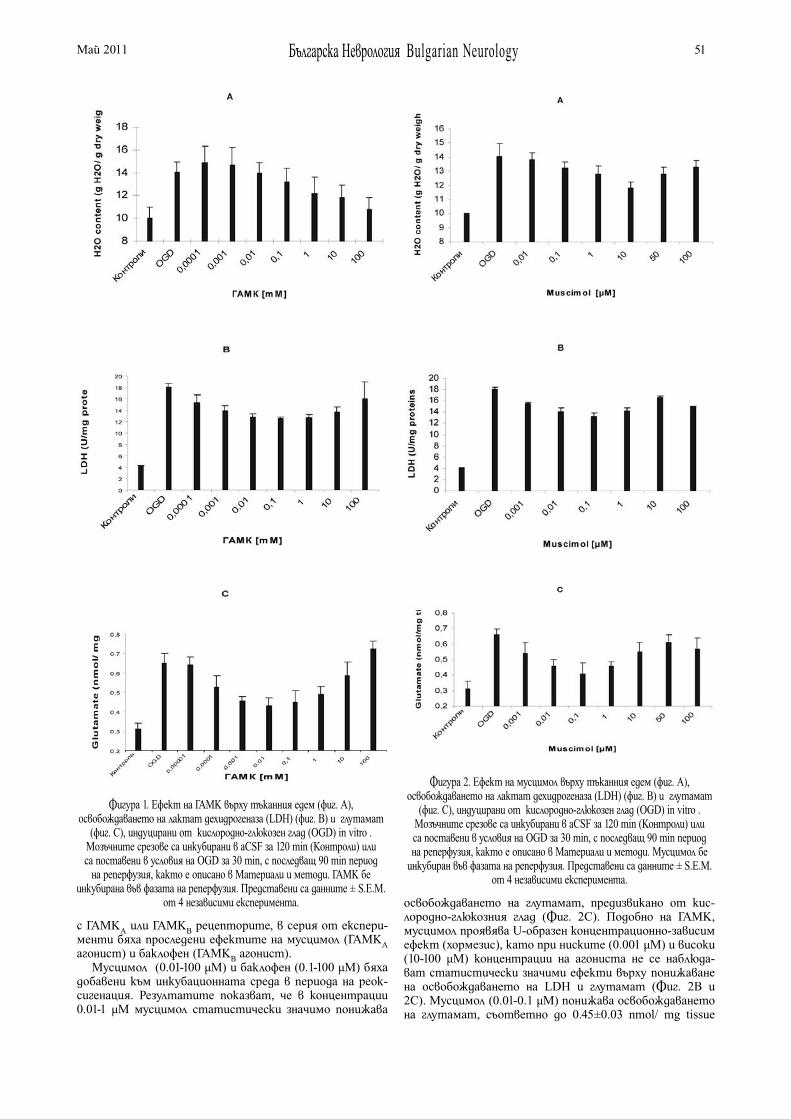
Българска Неврология Bulgarian Neurology 51Май 2011
с ГАМКA или ГАМКB рецепторите, в серия от експери-менти бяха проследени ефектите на мусцимол (ГАМКA агонист) и баклофен (ГАМКB агонист).
Мусцимол (0.01-100 µM) и баклофен (0.1-100 µM) бяха добавени към инкубационната среда в периода на реок-сигенация. Резултатите показват, че в концентрации 0.01-1 µM мусцимол статистически значимо понижава
освобождаването на глутамат, предизвикано от кис-лородно-глюкозния глад (Фиг. 2C). Подобно на ГАМК, мусцимол проявява U-образен концентрационно-зависим ефект (хормезис), като при ниските (0.001 µM) и високи (10-100 µM) концентрации на агониста не се наблюда-ват статистически значими ефекти върху понижаване на освобождаването на LDH и глутамат (Фиг. 2B и 2C). Mусцимол (0.01-0.1 µM) понижава освобождаването на глутамат, съответно до 0.45±0.03 nmol/ mg tissue
Фигура 1. Ефект на ГАМК върху тъканния едем (фиг. А), освобождаването на лактат дехидрогеназа (LDH) (фиг. B) и глутамат
(фиг. C), индуцирани от кислородно-глюкозен глад (OGD) in vitro . Мозъчните срезове са инкубирани в aCSF за 120 min (Контроли) или са поставени в условия на OGD за 30 min, с последващ 90 min период
на реперфузия, както е описано в Материали и методи. ГАМК бе инкубирана във фазата на реперфузия. Представени са данните ± S.E.M.
от 4 независими експеримента.
Фигура 2. Ефект на мусцимол върху тъканния едем (фиг. А), освобождаването на лактат дехидрогеназа (LDH) (фиг. B) и глутамат
(фиг. C), индуцирани от кислородно-глюкозен глад (OGD) in vitro . Мозъчните срезове са инкубирани в aCSF за 120 min (Контроли) или са поставени в условия на OGD за 30 min, с последващ 90 min период на реперфузия, както е описано в Материали и методи. Мусцимол бе
инкубиран във фазата на реперфузия. Представени са данните ± S.E.M. от 4 независими експеримента.
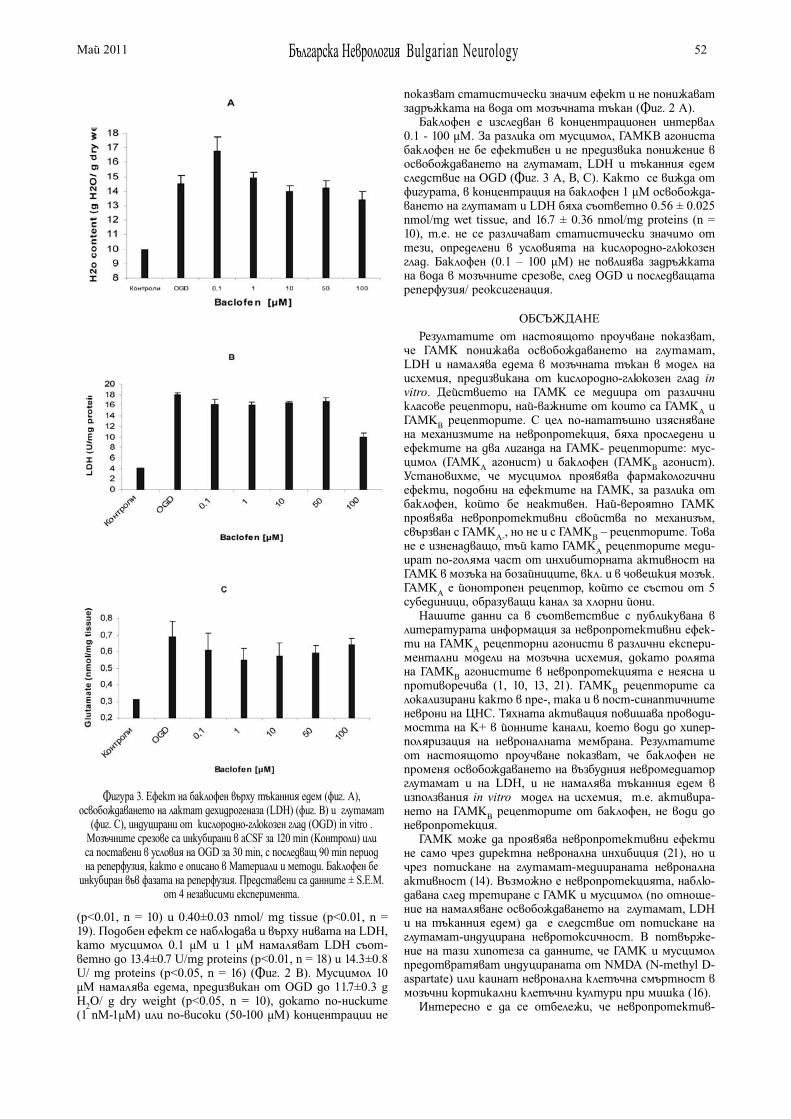
Българска Неврология Bulgarian Neurology 52Май 2011
(p<0.01, n = 10) и 0.40±0.03 nmol/ mg tissue (p<0.01, n = 19). Подобен ефект се наблюдава и върху нивата на LDH, като мусцимол 0.1 µM и 1 µM намаляват LDH съот-ветно до 13.4±0.7 U/mg proteins (p<0.01, n = 18) и 14.3±0.8 U/ mg proteins (p<0.05, n = 16) (Фиг. 2 B). Мусцимол 10 µM намалява едема, предизвикан от OGD до 11.7±0.3 g H2O/ g dry weight (p<0.05, n = 10), докато по-ниските (1 nM-1µM) или по-високи (50-100 µM) концентрации не
показват статистически значим ефект и не понижават задръжката на вода от мозъчната тъкан (Фиг. 2 A).
Баклофен е изследван в концентрационен интервал 0.1 - 100 µM. За разлика от мусцимол, ГАМКB агониста баклофен не бе ефективен и не предизвика понижение в освобождаването на глутамат, LDH и тъканния едем следствие на OGD (Фиг. 3 А, B, C). Както се вижда от фигурата, в концентрация на баклофен 1 µM освобожда-ването на глутамат и LDH бяха съответно 0.56 ± 0.025 nmol/mg wet tissue, and 16.7 ± 0.36 nmol/mg proteins (n = 10), т.е. не се различават статистически значимо от тези, определени в условията на кислородно-глюкозен глад. Баклофен (0.1 – 100 µM) не повлиява задръжката на вода в мозъчните срезове, след OGD и последващата реперфузия/ реоксигенация.
ОБСЪЖДАНЕ
Резултатите от настоящото проучване показват, че ГАМК понижава освобождаването на глутамат, LDH и намалява едема в мозъчната тъкан в модел на исхемия, предизвикана от кислородно-глюкозен глад in vitro. Действието на ГАМК се медиира от различни класoве рецептори, най-важните от които са ГАМКА и ГАМКB рецепторите. С цел по-нататъшно изясняване на механизмите на невропротекция, бяха проследени и ефектите на два лиганда на ГАМК- рецепторите: мус-цимол (ГАМКА агонист) и баклофен (ГАМКB агонист). Установихме, че мусцимол проявява фармакологични ефекти, подобни на ефектите на ГАМК, за разлика от баклофен, който бе неактивен. Най-вероятно ГАМК проявява невропротективни свойства по механизъм, свързван с ГАМКА-, но не и с ГАМКB – рецепторите. Това не е изненадващо, тъй като ГАМКА рецепторите меди-ират по-голяма част от инхибиторната активност на ГАМК в мозъка на бозайниците, вкл. и в човешкия мозък. ГАМКА е йонотропен рецептор, който се състои от 5 субединици, образуващи канал за хлорни йони.
Нашите данни са в съответствие с публикувана в литературата информация за невропротективни ефек-ти на ГАМКА рецепторни агонисти в различни експери-ментални модели на мозъчна исхемия, докато ролята на ГАМКВ агонистите в невропротекцията е неясна и противоречива (1, 10, 13, 21). ГАМКВ рецепторите са локализирани както в пре-, така и в пост-синаптичните неврони на ЦНС. Тяхната активация повишава проводи-мостта на K+ в йонните канали, което води до хипер-поляризация на невроналната мембрана. Резултатите от настоящото проучване показват, че баклофен не променя освобождаването на възбудния невромедиатор глутамат и на LDH, и не намалява тъканния едем в използвания in vitro модел на исхемия, т.е. активира-нето на ГАМКВ рецепторите от баклофен, не води до невропротекция.
ГАМК може да проявява невропротективни ефекти не само чрез директна невронална инхибиция (21), но и чрез потискане на глутамат-медиираната невронална активност (14). Възможно е невропротекцията, наблю-давана след третиране с ГАМК и мусцимол (по отноше-ние на намаляване освобождаването на глутамат, LDH и на тъканния едем) да е следствие от потискане на глутамат-индуцирана невротоксичност. В потвърже-ние на тази хипотеза са данните, че ГАМК и мусцимол предотвратяват индуцираната от NMDA (N-methyl D-aspartate) или каинат невронална клетъчна смъртност в мозъчни кортикални клетъчни култури при мишка (16).
Интересно е да се отбележи, че невропротектив-
Фигура 3. Ефект на баклофен върху тъканния едем (фиг. А), освобождаването на лактат дехидрогеназа (LDH) (фиг. B) и глутамат
(фиг. C), индуцирани от кислородно-глюкозен глад (OGD) in vitro . Мозъчните срезове са инкубирани в aCSF за 120 min (Контроли) или са поставени в условия на OGD за 30 min, с последващ 90 min период на реперфузия, както е описано в Материали и методи. Баклофен бе
инкубиран във фазата на реперфузия. Представени са данните ± S.E.M. от 4 независими експеримента.

Българска Неврология Bulgarian Neurology 53Май 2011
ният ефект на ГАМК не е в линейна зависимост от концентрацията на медиатора, а следва “U”-образна концентрационна крива, като статистически значим ефект бе наблюдаван в концентрационния интервал 1 µM – 1 mM. В по-ниските (0.0001-0.001 mM) и по-високи концентрации на ГАМК (100 mM) не се наблюдава ста-тистически значим ефект върху изследваните показа-тели. Този феномен, известен като «хормезис» се харак-теризира с бифазност по отношение на амплитудата и степента на отговора. Хормезисът е адаптивен отго-вор, следствие на директна индукция или e резултат от компенсаторни процеси, следствие нарушаване на хоме-остазата. Наблюдаваният в експериментите хормезис може да се обясни с различни механизми. Мозъчната исхемия е съпътствана от нарушение на невроналната йонна хомеостаза, включително значително увеличение на вътреклетъчните Na+, Cl- и Ca2+, както и с пови-шаване на извънклетъчния K+ (21). Установено е, че при кислородно-глюкозния глад, в невроните от хипокампус на плъх се наблюдава покачване на вътреклетъчните концентрации на Cl-, най-вероятно заради засилената активност на котранспортера за хлорни йони NKCC-1 (9, 18). Реперфузията, респективно реоксигенацията частично възстановяват вътреклетъчните нива на хлорни йони, но следва втора фаза на продължително покачване на вътреклетъчния Cl- (18). Възможно е пови-шените нива на ГАМК и мусцимол в синаптичната цепка време на реперфузията (при най-високите използвани концентрации) да водят до прекомерно активиране на ГАМК A рецепторите, което от своя страна води до високите вътреклетъчни концентрации на хлорни йони (13). В резултат се наблюдават деполяризацията на мембраната, невронална хипервъзбудимост и клетъчно увреждане. От друга страна, високите концентрации на ГАМК и на мусцимол могат да предизвикат понижаване на чувствителността на рецепторите (десензитиза-ция), което също може да предизвика деполяризация и невронално увреждане (13).
В заключение, резултатите от in vitro изследва-нето потвърждават хипотезата, че с повишаването на ГАМК- ергичната активност може да се повиши клетъчната невропротекция и да се предотврати нев-роналната смърт след исхемичен епизод, като невропро-тективните ефекти на ГАМК са медиирани от ГАМКА, но не и от ГАМКВ рецепторите.
Благодарности: Северина Митушева бе стипендиант по програма Erasmus във Факултета по Фармация на Университета в Сиена, Италия.
ЛИТЕРАТУРА1. Araki, T., H. Kato and K. Kogure. Comparative protective effects
of vinconate, baclofen and pentobarbital against neuronal damage following repeated brief cerebral ischemia in the gerbil brain. Res. Exp. Med. 191, 1991, 371-378.
2. Bonita, R. Epidemiology of stroke. Lancet, 339, 1992, 342–344.3. Budd SL. Mechanism of neuronal damage in brain hipoxia\
ischemia : focus on the role of mitochondrial calcium accumulation. Pharmacol Ther. 80, 1998, 203-29.
4. Brahma, B., R.E. Forman, E.E. Stewart, C. Nicholson and M.E. Rice. Ascorbate inhibits edema in brain slices. J. Neurochem. 74,
2000, 1263-1270.5. Eilers, H., C.H. Kindler and P.E. Bickler. Different effects of
volatile anesthetics and polyhalogenated alkanes on depolarization-evoked glutamate release in rat cortical brain slices. Anesthesia and Analgesia. 88, 1999, 1168-1174.
6. Green, A.R., Cross, A.J. Neuroprotective agents and cerebral ischaemia. Academic Press, London, 1997.
7. Green, A.R., Hainsworth, A.H., Jackson, D.M. GABA potentiation: a logical pharmacological approach for the treatment of acute ischaemic stroke. Neuropharmacology 39, 2000, 1483–1494.
8. Gay, R.J., R.B. McComb and G.N. Bowers Jr. Optimum reaction conditions for human lactate dehydrogenase isoenzymes as they affect total lactate dehydrogenase activity. Clin Chem 14, 1968, 740-753.
9. Galeffi, F., R. Sah, B.B. Pond, A. George and R.D. Schwartz-Bloom, Changes in intracellular chloride after oxygen-glucose deprivation of the adult hippocampal slice: Effect of diazepam. J. Neurosci. 24, 2004, 4478-4488.
10. Ito, H., Y. Watanabe, A. Isshiki and H. Uchino. Neuroprotective properties of propofol and midazolam, but not pentobarbital, on neuronal damage induced by forebrain ischemia, based on the GABAA receptors. Acta Anaesthesiol. Scandinavica. 43, 1994, 153-162.
11. Martin, L.J., Al-Abdulla N.A., Brambrink A.M., Kirsh J.R., Sieberr F.E., Portera-Cailliau C. Neurodegeneration in excitatory, global cerebral ischemia, and target deprivation:A perspective on the contributions of apoptosis and necrosis. Brain Res Bull. 46, 2001, 281-309.
12. MacGregor, D.G., M.V. Avshalumov and M.E. Rice. Brain edema induced by in vitro ischemia: Causal factors and neuroprotection. J. Neurochem 85, 2003, 1402-1411.
13. Muir, J.K., D. Lobner, H. Monyer and D.W. Choi, GABAA receptor activation attenuates excitotoxicity but exacerbates oxygen-glucose deprivation-induced neuronal injury in vitro. J. Cerebral Blood Flow Metab. 16, 1996, 1211-1218.
14. Moulder, K.L., J.P. Meeks and S. Mennerick. Homeostatic regulation of glutamate release in response to depolarization. Molecular Neurobiol. 33, 2006, 133-153.
15. Olney, J.W., Labruyere, J., Price, M.T. Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science 244, 1989, 1360–1362.
16. Ohkuma, S., S.H. Chen, M. Katsura, D.Z. Chen and K. Kuriyama, Muscimol prevents neuronal injury induced by NMDA. Jap. J. Pharmacol. 1994, 64, 125-128.
17. Pellegrini-Giampietro, D.E. The distinct role of mGlu1 receptors in postischemic neuronal death. Trends Pharmacol. Sci. 24, 2003, 461–470.
18. Pond, B.B., K. Berglund, T. Kuner, G. Feng, G.J. Augustine and R.D. Schwartz-Bloom, The chloride transporter Na(+)-K(+)-Cl- cotransporter isoform-1 contributes to intracellular chloride increases after in vitro ischemia. J. Neurosci. 26, 2006, 1396-1406.
19. Rossi, D.J., Oshima, T., Attwell, D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403, 2000, 316–321.
20. Saito, A., Maier, C.M., Narasimhan, P. Et al., Oxidative stress and neuronal death/ survival signaling in cerebral ischemia. Mol. Neurobiol. 31, 2005, 105–116.
21. Schwartz-Bloom, R.D. and R. Sah, Gamma- Aminobutyric acid (A) neurotransmission and cerebral ischemia. J. Neurochem. 77, 2001, 353-371.
Адрес за кореспонденция:В. ЦанковаКатедра по фармакология, фармакотерапия и токсико-логияФармацевтичен Факултет, МУ – СофияУл. Дунав № 2, СофияТел: +35929236524Еmail: [email protected]

Българска Неврология Bulgarian Neurology 54Май 2011
SUMMARY
APOLIPOPROTEIN E AND NEUROPSYCHIATRIC SYMPTOMS IN PATIENTS WITH ALZHEIMER’S
DISEASES. Mehrabian, A. Jordanova, M. Raycheva, M. Petrova,
L. TraykovDepartment of Neurology, “Alexandrovska” Hospital Medical
University, Sofia, BulgariaLaboratory of Molecular Pathology, Medical University,
Sofia, BulgariaAlzheimer’s disease (AD) is a neurodegenerative disorder
with cognitive and behavioural clinical manifestations. The presence of Apolipoprotein E (ApoE) - ε4 allele is associated with an increased risk for late-onset AD. Patients bearing ε4 allele show increased amyloid plaques, and neurofibrillary tangles and severe depletion of cholinergic markers in the frontal and temporal cortex and the hippocampus. There have been controversial results to date on the association between the ε4 allele and neuropsychiatric symptoms (NPS) in AD.
The aim of this study is to establish the role of Apo-ε4 allele on pattern of behaviour changes in patients with AD.
The study is carried out at the Clinic of Neurology of the University Hospital ‘Alexandrovska”-Sofia. We studied 122 patients with AD. The patients are allocated in 3 groups, depending on Apo-ε4 allele presence; I group- non ε4 allele-bearing patients (ε4-); II group- patients, bearing one ε4-allele (ε4+); III group- patients, bearing two ε4-alleles (ε4/ε4). ApoE genotyping was performed by the method of Hixson and Powers. The Neuropsychiatric Inventory (NPI) was used to evaluate the frequency and severity of NPS. There was a significant association between the ε4 allele and prevalence and severity of behavioural symptoms that was mainly attributable to delusions and agitation/aggression, which were more common and severer among ApoE-ε4 carriers.
In conclusion, the results support the hypothesis that the ApoE-ε4 is associated with severer pathologic changes of AD and we show that leads to a specific pattern of behaviour changes for bearing ε4 allele patients.
KEY WORDS: AD, Neuropsychiatric phenotype, Apolipoprotein E, NPI.
РЕЗЮМЕ
Болестта на Алцхаймер (БА) е невродегенеративно заболяване с клинични прояви на когнитивни и поведен-чески нарушения. При БА с късно начало, наличието на ε4 алелът на Аполипопротеин Е (АпоЕ) е свързано с увеличен риск от поява на БА. При пациентите, които са носители на ε4 алел се наблюдават по-голям брой амилоидни плаки, неврофибрилерни дегенерации и по-тежко снижение на холинергичната медиация във фронталния и темпоралния дялове и хипокампа. Проведените проучвания показват противоречиви резултати за връзката между ε4 алела и невропсихиатричните симптоми при БА.
Изследването има за цел да установи влиянието на ε4 алела върху поведенческите разстройства при БА.
Проучването е проведено в Клиниката по Неврология на УМБАЛ „Александровска”-София, при 122 дементно болни страдащи от БА. Болните са разпределени в три групи в зависимост от наличието на АпоЕ-ε4 алела: I група: болни, които не са носители на ε4 алел (ε4); II група: болни носители на един ε4 алел (ε4+); III група: болни ноители на два ε4 алела (ε4/ε4). АпоЕ генотипа на всеки пациент беще определян пo метода на Hixson and Powers. Разстройствата в поведението се преценяват със структурирано интервю за честота и тежест на симптомите чрез скалата: Невропсихиатричен въпрос-ник (NPI).
Съществува значима разлика между появата и тежестта на поведенческите нарушения и наличието на ε4 алел. Тази значимост основно се изразява в значимо по-честата поява на налудности, възбуда и агресия при групата болни носители на ε4 алел.
В заключение, нашите резултати подкрепят хипоте-зата, че АпоЕ-ε4 алела е свързан с по-тежки патологични промени, в резултат на което показваме наличието на специфичен профил на поведенчески разстройства за пациенти носители на ε4 алела.
КЛЮЧОВИ ДУМИ: БА, Невропсихиатричен профил, Аполипопопротеин Е, NPI.
ВЪВЕДЕНИЕ
Болестта на Алцхаймер (БА) е невродегенеративно заболяване с клинични прояви на когнитивни и поведен-чески нарушения. Генетични фактори и фактори от околната среда играят важна роля за експресията на фенотипа.
От 1991 година резултатите от генетичните про-учвания са довели до идентифициране на мутации и полиморфизми, които могат да причинят БА или да увеличат риска от развитие на заболяването. Мутации в три гена: ген за синтеза на Аmyloid Рrecursor Рrotein (APP, Goate, et al., 1991), Presenilin 1 (PSEN1, Sherrington, et al., 1995) и Presenilin 2 (PSEN2, Levy – Lаhad, et al., 1995a) за момента са асоциирани с БА с ранно нача-ло. При БА с късно начало, наличието на ε4 алелът на Аполипопротеин Е (АпоЕ) е свързано с увеличен риск от поява на БА (7).
АпоЕ ε4 може да модулира патологичните процеси, които са в основата на заболяването. Пациентите с БА носители на АпоЕ -ε4 алел имат по-тежки невропа-тологични промени с повишени кортикални и церебро-васкуларни отлагания, увеличен брой на неврофибрилни дегенерации (НФД), както и увеличен холинергичен дефи-цит. Някои от тези прояви имат дозов ефект спрямо ε4 генотипа (10, 20, 22). АпоЕ е важен за растежа на нев-роналните клетки и възстановяването им след увреж-дания. Wilson и съавт. (2002), както и други проучвания (20, 22) показват, че наличието на ε4 алел е лош пока-зател за ефективното невронално възстановяване, в сравнение с другите алели. Някои изследвания показват, че ефектът на ε4 алела е специфична по отношение на
Оригинални статииАПОЛИПОПРОТЕИН Е И НЕВРОПСИХИАТРИЧНИ СИМПТОМИ
ПРИ БОЛНИ С БОЛЕСТ НА АЛЦХАЙМЕР
Ш. Мехрабиан1, А. Йорданова2, М. Райчева1, М. Петрова1, Л. Трайков1
1Клиника по Неврология, МБАЛ “Александровска”, Медицински Университет, София2Лаборатория по Молекулярна Патология, Медицински Университет, София
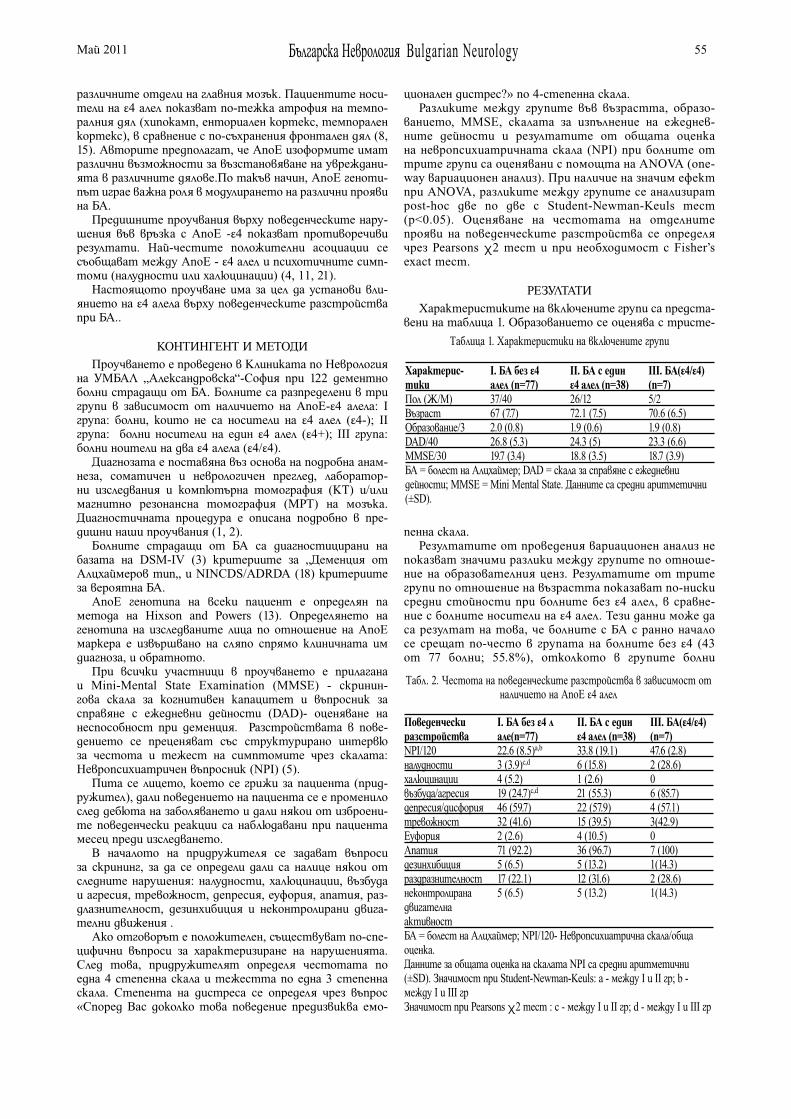
Българска Неврология Bulgarian Neurology 55Май 2011
различните отдели на главния мозък. Пациентите носи-тели на ε4 алел показват по-тежка атрофия на темпо-ралния дял (хипокамп, енториален кортекс, темпорален кортекс), в сравнение с по-съхранения фронтален дял (8, 15). Авторите предполагат, че АпоЕ изоформите имат различни възможности за възстановяване на увреждани-ята в различните дялове.По такъв начин, АпоЕ геноти-път играе важна роля в модулирането на различни прояви на БА.
Предишните проучвания върху поведенческите нару-шения във връзка с АпоЕ -ε4 показват противоречиви резултати. Най-честите положителни асоциации се съобщават между АпоЕ - ε4 алел и психотичните симп-томи (налудности или халюцинации) (4, 11, 21).
Настоящото проучване има за цел да установи вли-янието на ε4 алела върху поведенческите разстройства при БА..
КОНТИНГЕНТ И МЕТОДИ
Проучването е проведено в Клиниката по Неврология на УМБАЛ „Александровска”-София при 122 дементно болни страдащи от БА. Болните са разпределени в три групи в зависимост от наличието на АпоЕ-ε4 алела: I група: болни, които не са носители на ε4 алел (ε4-); II група: болни носители на един ε4 алел (ε4+); III група: болни ноители на два ε4 алела (ε4/ε4).
Диагнозата е поставяна въз основа на подробна анам-неза, соматичен и неврологичен преглед, лаборатор-ни изследвания и компютърна томография (КT) и/или магнитно резонансна томография (МРТ) на мозъка. Диагностичната процедура е описана подробно в пре-дишни наши проучвания (1, 2).
Болните страдащи от БА са диагностицирани на базата на DSM-IV (3) критериите за „Деменция от Алцхаймеров тип“ и NINCDS/ADRDA (18) критериите за вероятна БА.
АпоЕ генотипа на всеки пациент е определян па метода на Hixson and Powers (13). Определянето на генотипа на изследваните лица по отношение на АпoЕ маркера е извършвано на сляпо спрямо клиничната им диагноза, и обратното.
При всички участници в проучването е прилагана и Mini-Mental State Examination (MMSE) - скринин-гова скала за когнитивен капацитет и въпросник за справяне с ежедневни дейности (DAD)- оценяване на неспособност при деменция. Разстройствата в пове-дението се преценяват със структурирано интервю за честота и тежест на симптомите чрез скалата: Невропсихиатричен въпросник (NPI) (5).
Пита се лицето, което се грижи за пациента (прид-ружител), дали поведението на пациента се е променило след дебюта на заболяването и дали някои от изброени-те поведенчески реакции са наблюдавани при пациента месец преди изследването.
В началото на придружителя се задават въпроси за скрининг, за да се определи дали са налице някои от следните нарушения: налудности, халюцинации, възбуда и агресия, тревожност, депресия, еуфория, апатия, раз-длазнителност, дезинхибиция и неконтролирани двига-телни движения .
Ако отговорът е положителен, съществуват по-спе-цифични въпроси за характеризиране на нарушенията. След това, придружителят определя честотата по една 4 степенна скала и тежестта по една 3 степенна скала. Степента на дистреса се определя чрез въпрос «Според Вас доколко това поведение предизвиква емо-
ционален дистрес?» по 4-степенна скала.Разликите между групите във възрастта, образо-
ванието, MMSE, скалата за изпълнение на ежеднев-ните дейности и резултатите от общата оценка на невропсихиатричната скала (NPI) при болните от трите групи са оценявани с помощта на ANOVA (one-way вариационен анализ). При наличие на значим ефект при ANOVA, разликите между групите се анализират post-hoc две по две с Student-Newman-Keuls тест (p<0.05). Оценяване на честотата на отделните прояви на поведенческите разстройства се определя чрез Pearsons u2 тест и при необходимост с Fisher’s exact тест.
РЕЗУЛТАТИ
Характеристиките на включените групи са предста-вени на таблица 1. Образованието се оценява с тристе-
пенна скала.Резултатите от проведения вариационен анализ не
показват значими разлики между групите по отноше-ние на образователния ценз. Резултатите от трите групи по отношение на възрастта показават по-ниски средни стойности при болните без ε4 алел, в сравне-ние с болните носители на ε4 алел. Тези данни може да са резултат на това, че болните с БА с ранно начало се срещат по-често в групата на болните без ε4 (43 от 77 болни; 55.8%), отколкото в групите болни
Tаблица 1. Характеристики на включените групи
Характерис- I. БА без ε4 II. БА с един III. БА(ε4/ε4) тики алел (n=77) ε4 алел (n=38) (n=7)Пол (Ж/M) 37/40 26/12 5/2Възраст 67 (7.7) 72.1 (7.5) 70.6 (6.5)Образование/3 2.0 (0.8) 1.9 (0.6) 1.9 (0.8)DAD/40 26.8 (5.3) 24.3 (5) 23.3 (6.6)MMSE/30 19.7 (3.4) 18.8 (3.5) 18.7 (3.9)БА = болест на Алцхаймер; DAD = скала за справяне с ежедневни дейности; MMSЕ = Mini Mental State. Данните са средни аритметични (±SD).
Tабл. 2. Честота на поведенческите разстройства в зависимост от наличието на АпоЕ ε4 алел
Поведенчески I. БА без ε4 л II. БА с един III. БА(ε4/ε4) разстройства але(n=77) ε4 алел (n=38) (n=7)NPI/120 22.6 (8.5)a,b 33.8 (19.1) 47.6 (2.8)налудности 3 (3.9)c,d 6 (15.8) 2 (28.6)халюцинации 4 (5.2) 1 (2.6) 0възбуда/агресия 19 (24.7)c,d 21 (55.3) 6 (85.7)депресия/дисфория 46 (59.7) 22 (57.9) 4 (57.1)тревожност 32 (41.6) 15 (39.5) 3(42.9)Еуфория 2 (2.6) 4 (10.5) 0Апатия 71 (92.2) 36 (96.7) 7 (100)дезинхибиция 5 (6.5) 5 (13.2) 1(14.3)раздразнителност 17 (22.1) 12 (31.6) 2 (28.6)неконтролирана 5 (6.5) 5 (13.2) 1(14.3) двигателна активностБА = болест на Алцхаймер; NPI/120- Невропсихиатрична скала/обща оценка. Данните за общата оценка на скалата NPI са средни аритметични (±SD). Значимост при Student-Newman-Keuls: а - между I и II гр; b - между I и III гр Значимост при Pearsons u2 тест : c - между I и II гр; d - между I и III гр

Българска Неврология Bulgarian Neurology 56Май 2011
носители на един или два ε4 алела (9 от 45 болни; 20%). Статистическите анализи на стойностите от MMSE и скалата за изпълнение на ежедневните дейности не установяват разлика между групата на болните без ε4 алел, в сравнение с тези на носителите на един или два ε4 алела.
РЕЗУЛТАТИ ОТ НЕВРОПСИХИАТРИЧНОТО ПРОУЧВАНЕ
Резултатите от общата оценка на невропсихиат-ричната скала (NPI) при болните от трите групи са представени на табл. 2. Анализирането на данните с ANOVA и post-hoc показва, че в общия сбор на тесто-вете оценяващи поведенческите разстройства, има значима разлика между болните без ε4 и болните носи-тели на поне един ε4 алели. Не се установява дозов ефект при наличието на два алела.
Оценяване на честотата на отделните прояви на поведенческите разстройства се определя чрез Pearsons u2 тест и при необходимост с Fisher’s exact тест.
НАЛУДНОСТИ
Проучването на честотата на „налудностите” при трите групи показва значима разлика между: групата болни носители на един ε4 алел и групата болни без ε4 (u2 =4.1; p<0.04); групата без ε4 и групата с два ε4 алела (u2=5.2; p<0.02). Не се установява дозов ефект при нали-чието на два алела.
ХАЛЮЦИНАЦИИ
Проучването на честотата на „халюцинациите” при трите групи не показва значима разлика между: групата болни носители на един ε4 алел и групата болни без ε4 (u2 =0.4; p<0.5); групата болни носители на един ε4 алел и групата с два ε4 алела u2 =0.2; p<0.7); групата без ε4 алел и групата болни носители на два ε4 алела (u2 =0.4; p<0.5).
ВЪЗБУДА И АГРЕСИЯ
Проучването на честотата на „възбуда / агресия” при трите групи установява: значима разлика между група-та болни носители на един ε4 алел и групата на болните без ε4 (u2 =4.8; p<0.02); липса на значимост между група-та на болните носители на един ε4 алел и групата с два ε4 алела (u2 =0.5; p<0.5); значима разлика между групата без ε4 и групата с два ε4 алела (u2 =4.5; p<0.03).
ДЕПРЕСИЯ/ДИСФОРИЯ
Проучването на честотата на „депресия/дифория” при трите групи не показва значима разлика между: групата на болните носители на един ε4 алел и групата на болните без ε4 u2 =0.009; p<0.9); групата на болните носители на един ε4 алел и групата с два ε4 алела (u2 =3.7; p<0.9); групата без ε4 и групата с два ε4 алела u2 =0.005; p<0.9).
ТРЕВОЖНОСТ
Проучването на честотата на „тревожност” при трите групи не установява значима разлика между: групата на болните носители на един ε4 алел и групата на болните без ε4 (u2 =0.01; p<0.9); групата на болните носители на един ε4 алел и групата с два ε4 алела u2 =0.01; p<0.9); групата без ε4 и групата с два ε4 алела (u2 =0.002; p<1).
ЕУФОРИЯ
Проучването на честотата на „еуфория” при трите групи не установява значима разлика между: групата болни носители на един ε4 алел и групата на болните без ε4 (u2 =2.8; p<0.09); групата на болните носители на един ε4 алел и групата с два ε4 алела (u2 =0.7; p<0.4); групата без ε4 и групата с два ε4 алела u2 =0.2; p<0.7).
АПАТИЯ
Проучването на честотата на „апатия” при трите групи не установява значима разлика между: групата болни носители на един ε4 алел и групата на болните без ε4 (u2 =0.009; p<0.9); групата на болните носители на един ε4 алел и групата с два ε4 алела u2 =0.009; p<0.9); групата без ε4 и групата с два ε4 алела (u2 =0.2; p<0.9).
ДЕЗИНХИБИЦИЯ
Проучването на честотата на „дезинхибиция” при трите групи не установява значима разлика между: групата на болните носители на един ε4 алел и групата на болните без ε4 (u2 =1.2; p<0.3); групата на болните носители на един ε4 алел и групата с два ε4 алела u2 =0.005; p<0.9); групата без ε4 и групата с два ε4 алела u2 =0.5; p<0.5).
РАЗДРАЗНИТЕЛНОСТ
Проучването на честотата на „раздразнителност” при трите групи не показва значима разлика между: групата на болните носители на един ε4 алел и групата на болните без ε4 (u2 =0.7; p<0.4); групата на болните носители на един ε4 алел и групата с два ε4 алела (u2 =0.01; p<0.9); групата без ε4 и групата с два ε4 алела (u2 =0.09; p<0.7).
НЕКОНТРОЛИРАНА ДВИГАТЕЛНА АКТИВНОСТ
Проучването на честотата на „Неконтролирана дви-гателна активност” при трите групи не установява значима разлика между групата: болни носители на един ε4 алел и групата на болни без ε4 u2 =1.2; p<0.3); групата на болните носители на един ε4 алел и групата с два ε4 алела u2 =0.005; p<0.9); групата без ε4 и групата с два ε4 алела u2 =0.5; p<0.5).
ОБСЪЖДАНЕ
Резултатите от нашето проучване показват, че съществува значима разлика между появата и тежест-та на поведенческите нарушения и наличието на ε4 алел. Тази значимост основно се изразява в значимо по-честа-та поява на налудности, възбуда и агресия при групата болни носители на ε4 алел. Не се установява дозов ефект спрямо наличието на ε4 алела. Акцентът на нашето проучване е изясняване ролята на АпоЕ- ε4 върху психи-атричните нарушения при БА.
АпоЕ ε4 алелът е добре известен рисков фактор при пациенти със сърдечно-съдови заболявания в средна въз-раст и за БА в късна възраст (20). Биологичната роля на АпоЕ е комплексна. Някои изследвания показват, че ефектът на ε4 алела е специфична по отношение на различните отдели на главния мозък. Hashimoto и съавт. (2001) показват, че при пациенти носители на ε4 алел обемът на темпоралния дял е по-малък, в сравнение с тези без ε4 алел. По-тежките увреждания на хипокампа и амигдалата са в съответствие с неврохимичните и невропатологичните проучвания. При носителите на ε4 алел холинергичната функция е снижена в хипокампа

Българска Неврология Bulgarian Neurology 57Май 2011
и темпоралния дял, докато честотата на сенилните плаки и неврофибрилерните дегенерации се увеличава.
В противоречие с тези резултати са данните от друго проучване (14), което демонстрира ефект на АпоЕ ε4 върху атрофията на темпоралния дял в съчетание с атрофия на фронталния дял.
Освен когнитивните нарушения, повечето пациенти с БА страдат от поведенчески разстройства и психоло-гични синдроми, като агресивно поведение, хиперактив-ност, депресия или психози, които са основна причина за приемането на тези болни в социални домове (16). Невропсихиатричните симптоми не са единствено пос-ледица от невродегенеративния процес. Най-вероятно за тяхната поява са намесени и някои нарушения в нев-ротрансмитерните системи.
Важно е да се установи неврохимичната база на пси-хиатричните симптоми при БА с оглед заместващата терапия, която ще доведе до подобрение на поведенчес-ките нарушения, независимо от ефекта върху когни-тивните функции на тези пациенти.
Снижението в холинергичната функция корелира с агресивните прояви. Това доказва двойната роля на холинергичната система върху когнитивните и неког-нитивните симптоми при БА. Предполага се, че не само холинергичния дефицит, но и нарушенията в серотони-нергичната медиация имат значима корелация с когни-тивната и психиатричната картина при БА.
Ролята на серотонинергичната система при висши-те корови функции като памет и заучаване са добре изу-чени. Промените в тази система настъпват дори при нормално стареене. Обсъжда се ролята на серотонин и редукцията на холинергичната трансмисия за прог-ресията на заболяваето и появата на поведенческите разстройства. Промените в серотониновата система показва значима корелация с появата на хиперактивност и психози (6).
Minger et al (2000) потвърждават данните от пре-дишните проучвания, които показват редукция в преси-наптичния маркер на холинергичната система и холина-цетилтлансферазната астивност при БА, в сравнение с допаминергичната система. Поведенческите нарушения са асоциирани със снижение в холинергичната трансми-сия. В това проучване снижението на холинергичната активност в темпоралния дял е свързано с агресивно поведение, докато снижението в префронталния кор-текс довежда до по-честа проява от хиперактивност.
Наличието на ε4 алел може да повиши риска от поява на психотични симптоми (налудности, халюцинации) (11, 21). Особено висок риск съществува за проявите на налудности, най-вероятно поради по-честата им поява сред болни с БА (налудности 29%; Халюцинации 15%). Проучванията със SPECT показват, че появата на налуд-ности при пациенти с БА е свързано с хипоперфузия на темпоралния дял (24). Някои невроизобразяващи изслед-вания показват, че носителите на ε4 алел са с по-голяма атрофия на темпоралния лоб (15).
В заключение, в нашето проучване появата на налуд-ности, възбуда и агресия е значимо по-често при групата болни носители на ε4 алел. Не се установява дозов ефект спрямо наличието на ε4 алел. Тези резултати са в покре-па на връзката между агресивните прояви и налуднос-ти с атрофията на темпоралния дял и холиниргичния дефицит. Нашите резултати подкрепят хипотезата, че АпоЕ-ε4 алела е свързан с по-тежки патологични промени, в резултат на което показваме наличието на специфичен профил на поведенчески разстройства за пациенти носители на ε4 алела.
ЛИТЕРАТУРА1. Трайков, Л. Ранна диагностика при болестта на Алцхаймер и
съдовата деменция. Мозъчносъдови заболявания, 9, 2001, 9-12. 2. Трайков, Л., Раукс, Н., Риго, А-С. Честота на ε4 алела на
Аполипопопротеин Е при пациенти с леки когнитивни нарушения. Българска Неврология, 2006, 6, 2, 88-91.
3. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. Fourth Edition. Washington, DC: American Psychiatric Association, 1994.
4. Ballard, C., Massey, H., Lamb, H., Morris, C. Apolipoprotein E: non-cognitive symptoms and cognitive decline in late-onset Alzheimer’s disease. J Neurol Neurosurg Psychiatr, 1997, 63, 273–274.
5. Cummings, J.L., Mega, M., Gray, K., Rosenberg-Thompson, S., Carusi, D.A., Gornbein, J. The Neuropsychiatric Inventory: com-prehensive assessment of psychopathology in dementia. Neurology, 1994 , 44, 2308–2314. Dringenberg, H.C., Zalan, R.M. Serotonin-dependent maintenance of spatial performance and electroencepha-lography activation after cholinergic blockade: effects of serotoner-gic receptor antagonists. Brain Res., 1-2, 1999, 837, 242-253.
6. Farrer, L.A., Cupples, L.A., Haines, J.L., Hyman, B., Kukull, W.A., Mayeux, R., Myers, R.H., Pericak-Vance, M.A., Risch, N., van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA, 1997, 278, 1349-1356.
7. Geroldi, C., Pihlajamaki, M., Laakso, M.P., DeCarli, C., Beltramello, A., Bianchetti, A., Soininen, H., Trabucchi, M., Frisoni, G.B. APOE-epsilon4 is associated with less frontal and more medial temporal lobe atrophy in AD. Neurology, 8, 1999, 53, 1825-1832.
8. Goate, A., Chartier-Harlin, M.C., Mullan, M., Brown, J., Crawford, F., Fidani, L., Giuffra, L., Haynes, A., Irving, N., James, L. Segregation of a missense mutation in the amyloid precursor pro-tein gene with familial Alzheimer’s disease. Nature, 349, 1991, 6311, 704-706.
9. Gomez-Isla, T., West, H.L., Rebeck, G.W., Harr, S.D., Growdon, J.H., Locascio, J.J., Perls, T.T., Lipsitz, L.A., Hyman, B.T. Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer’s disease. Ann Neurol., 1, 1996, 39, 62-70.
10. Harwood, D.G., Barker, W.W., Ownby, R.L., St George-Hyslop, P., Duara, R. Apolipoprotein-E (APO-E) genotype and symptoms of psychosis in Alzheimer’s disease. Am J Geriatr Psychiatry, 2, 1999, 7, 119-123.
11. Hashimoto, M., Yasuda, M., Tanimukai, S., Matsui, M., Hirono, N., Kazui, H., Mori, E. Apolipoprotein E epsilon 4 and the pattern of regional brain atrophy in Alzheimer’s disease. Neurology, 8, 2001, 57, 1461-1416.
12. Hixson, J.E., Powers, P.K. Restriction isotyping of human apoli-poprotein A-IV: rapid typing of known isoforms and detection of a new isoform that deletes a conserved repeat. J Lipid Res, 1991, 32, 1529–1535.
13. Johnson, J.K., McCleary, R., Oshita, M.H., Cotman, C.W. Initiation and propagation stages of beta-amyloid are associated with distinc-tive apolipoprotein E, age, and gender profiles. Brain Res., 1-2, 1998, 798, 18-24.
14. Lehtovirta, M., Laakso, M.P., Soininen, H., Helisalmi, S., Mannermaa, A., Helkala, E.L., Partanen, K., Ryynanen, M., Vainio, P., Hartikainen, P., et al. Volumes of hippocampus, amygdala and frontal lobe in Alzheimer patients with different apolipoprotein E genotypes. Neuroscience, 1, 1995, 67, 65-72.
15. Levy, M.L., Cummings, J.L., Fairbanks, L.A., Sultzer, D.L., Small, G.W. Apolipoprotein E genotype and noncognitive symptoms in Alzheimer’s disease. Biol Psychiatr, 1999, 45, 422–425.
16. Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D.M., Oshima, J., Pettingell, W.H., Yu, C.E., Jondro, P.D., Schmidt, S.D., Wang, K. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science, 269, 1995, 5226, 973-977.
17. McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D., Stadlan, E. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of the Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology, 1984, 34, 939-944.
18. Minger, S.L., Esiri, M.M., McDonald, B., Keene, J., Carter, J., Hope, T., Francis, P.T. Cholinergic deficits contribute to behavioral disturbance in patients with dementia. Neurology, 10, 2000, 55, 1460-1467.
19. Poirier, J., Delisle, M.C., Quirion, R., Aubert, I., Farlow, M., Lahiri, D., Hui, S., Bertrand, P., Nalbantoglu, J., Gilfix, B.M., Gauthier, S. Apolipoprotein E4 allele as a predictor of cholinergic deficits and treatment outcome in Alzheimer disease. Proc Natl Acad Sci U S A., 26, 1995, 92, 12260-12264.

Българска Неврология Bulgarian Neurology 58Май 2011
20. Scarmeas, N., Brandt, J., Albert, M., Devanand, D.P., Marder, K., Bell, K., Ciappa, A., Tycko, B., Stern, Y. Association between the APOE genotype and psychopathologic symptoms in Alzheimer’s disease. Neurology, 8, 2002, 58, 1182-1188.
21. Schmechel, D.E., Saunders, A.M., Strittmatter, W.J., Crain, B.J., Hulette, C.M., Joo, S.H., Pericak-Vance, M.A., Goldgaber, D., Roses, A.D. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A., 20, 1993, 90, 9649-9653.
22. Sherrington, R., Rogaev, E.I., Liang, Y., Rogaeva, E.A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K., Tsuda, T., Mar, L., Foncin, J.F., Bruni, A.C., Montesi, M.P., Sorbi, S., Rainero, I., Pinessi, L., Nee, L., Chumakov, I., Pollen, D., Brookes, A., Sansosu, P., Polinsky, R.J., Wasco, W., Da Silva, H.A.R., Haines, J.L., Pericak-Vance, M.A., Tanzi, R.E., Roses, A.D., Fraser, P.E., Rommens, J.M., St George-Hyslop, P. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature, 375, 1995, 6534, 754-760.
23. Starkstein, S.E., Vazquez, S., Petracca, G., Sabe, L., Migliorelli, R., Teson, A., Leiguarda, R. A SPECT study of delusions in Alzheimer’s disease. Neurology, 11, 1994, 44, 2055-2059.
24. Wilson, R.S., Schneider, J.A., Barnes, L.L., Beckett, L.A., Aggarwal, N.T., Cochran, E.J., Berry-Kravis, E., Bach, J., Fox, J.H., Evans, D.A., Bennett, D.A. The apolipoprotein E epsilon 4 allele and decline in different cognitive systems during a 6-year period. Arch Neurol., 7, 2002, 59, 1154-1160.
Адрес за кореспонденция: д-р Шима Мехрабиан УМБАЛ “Александровска”, Клиника по неврология, бул. “Георги Софийски” 1, София 1431тел.: 0887428676e-mail: [email protected]

������������������������������
�����������
����������
��������������������
��������������������������������
��������������
��������������� �!"#�
����������������������
• ������������������������������������������
�����������
�����������
• �$%&' �(''��#)'#)*+�
,"! �+ �-"(#)*),"! � "&"," '#)*
• �.+�,"! �%�&)*�+�)/)'- )*)
+�)/�%�&)*,'."-�(�-� '0!)-)"
• �.+�,"! �+'-)1,')/ �,)1,'��%('
• �$%&' �(''��#)'#)*+�"+)%"+�)*
• �.+�&%'!���%)")��%('
• �$%&' �(�,'21,�. 23"�-!�
4."5",#))6
• �$%&' �(�. 23"�-!�+�(%),)1,'
,"! �7)0)�%�&)*

• '��#)'#)*4.!)&'-"%,),' 28",)*6
• '��#)'#)*+�5,�3"�-!",'�(%" �0'
• 7�,.'#)*6+ ".+'0!',"�-5�0$1,)
),�2%-)6
• �$%&' �(''��#)'#)*+�
,"! ���,�& '7)*)5�0$1,'
/"5�.),'5)('
• �. 23",)"9,"! �%�&)*�+ �7�.:
�'8���3),�!4
• . 23"�-!�+ �-)!"+)%"+�)*
• '��#)'#)*+�(%),)1,'"5&)
"!�() ',)+�-",#)'%)
�������������������������
�������������
• ��������������������
• �������������������
• ��������������������
���������������
• ���������������������
������������������
��������������������������
����
�� !"#$%&'($�!�)"*+$#!,*-*.$�$#*"&-*&�$�/// 01234565789:7 0;5<
�;�<����������=
���������������������������������
������������������������
����������������
������������������� ��� ����� ���� �� �
!!!����� �

Българска Неврология Bulgarian Neurology 61Май 2011
На 24 януари 2011 г. след кратко боледуване си отиде от живота видният български учен, невролог – неврове-гетолог проф. д-р Венцеслав Боснев, д. м., д. м. н.
Проф. Боснев завършва Медицинския факултет в София. Професионалното му развитие започва от лекар на село, преминава през основател и ръководител на Лабораторията по клинична невровегетология към Клиниката по неврология в Александровска болница през 1978 г. и ръководител на Клиниката по професионални заболявания от пренапрежение на нервната и мускулно-скелетната система в Университеска болница “Св. Иван Рилски”. Цялото му практическо и научно израстване е свързано с Медицинския университет – София. Той беше дългогодишен национален консултант по професи-онални заболявания, председател на ЦДЕК, член на спе-циализирани научни съвети на Висшата атестационна комисия.
Проф. Боснев е популярен сред колегите и възпита-ниците си със своите професионални интереси върху проблемите на невровегетологията и граничните й състояния, болестите на ръката, професионалните болести на нервната и мускулно-скелетната система, паркинсонизма, болката и невралната терапия в меди-цината. Известни са неговите монографии “Рамо-ръка синдром”, ”Невровегетативна патология на ръката”, “Клинична невровегетология и гранични проблеми”, учас-тия и редакторство на “Болести на ръката”, “Болест на Паркинсон”. Автор е на над 260 научни труда, от които 6 монографии и 28 колективни монографии по дос-тойнства оценени с множество български и междуна-родни отличия и награди за научна и творческа дейност от американски, европейски, руски, български научни общества и редакционни колегии. Научен редактор е на списание Scripta periodica, Acta Medica bulgarica, член на редколегии на редица медицински журнали. Визитиращ професор и консултант е в Японски университет в
Китакюшу, гост-професор в Американската Академия по термодиагностика и в Американското научно дру-жество по микроциркулация.
Проф. Боснев отдаде много сили и време за общес-твена дейност. През 1997 г. основа Атланто-Евро-Средиземноморската Академия на Медицинските Науки (АЕС АМН) и стана неин президент. Той беше замес-тник-председател на Фондацията по паркинсонизъм в България, член на Научния съвет на Европейската асо-циация по паркинсонова болест, на Научния европейски съвет по неврална терапия, на Европейското общество по микроциркулация, на Българската национална акаде-мия по медицина, дългогодишен редовен член на Съюза на ученита в България. Организира и ръководи редица науч-ни прояви – конгреси, конференции и симпозиуми често с мултидисциплинарен характер, адресирани към невроло-зи, психиатри, кардиолози, ендокринолози, специалисти с близки дисциплини и лекари по обща медицина, научно-изследователски национални и международни проекти.
Със своя рядък дар за човешко общуване и организаци-онен ентусиазъм в науката създаде и сплоти около себе си редица съмишленици и колеги, сред които видни про-фесори и учени от България и чужбина. Тези свои усилия отдаде за популяризиране на българската медицинска наука и практика, за съдействие и връзки между учени-те в областта на медицината, медико-биологичните и граничните им науки.
Със своите високо човешки и професионални качест-ва проф. Боснев, любим на своите ученици и последова-тели, дълбоко уважаван от колегите и пациентите си, ще остане вечно в паметта им.
Доц. Д-р Зл. Стойнева, дм
IN MEMORIAM