jaargang epilepsie...periodiek voor professionals epilepsie jaargang nummer 7 maart 200 112...
TRANSCRIPT

Periodiek voor professionals
Epilepsie
Jaargangnum
mer
7maart 2009
1
Periodiek voor professionals
Epilepsie
Jaargangnum
mer
12september 2014
3
| 3
| 5
| 8
| 12
| 15
| 18
| 20
| 22
| 24
Actueel
Het Kinder Epilepsie Centrum SEIN-Heemstede Onno van Nieuwenhuizen
Casuïstiek
Antistoffen tegen glutamaatdecarboxylase bij epilepsieRob Rouhl, Mariëlle Vlooswijk, Sylvia Klinkenberg, Joost Nicolai en Alexander Peeraer
Wetenschappelijk onderzoek
Carbamazepine-afbouw bij volwassen patiënten met het syndroom van Dravet Monique Veendrick, Francesca Snoeijen-Schouwenaars, Petra van Mierlo, Gerard van Erp, Bert Kleine, Jurgen Schelhaas en Francis Tan
Nieuwe genen voor diagnostiek van ernstige epilepsieBobby Koeleman, Marjan van Kempen en Eva Brilstra
Historische wetenswaardigheden
Hoe groot is de schade? Paul Eling
Verantwoorde epilepsiezorg
De nieuwe aanvals- en epilepsieclassificatie is niet beter dan de oude Paul Augustijn
EEG diagnostiek bij epilepsie: is een richtlijn zinvol?Frans Leijten
Proefschriftbesprekingen
Hersennetwerken en neuronale dynamiekJaap Jansen
Agenda

Door: Geert Thoonen, GZ-psycholoog, Onderwijscentrum De Berkenschutse en
John van de Corput, landelijk coördinator Steunpunten, Landelijk Werkverband Onderwijs & Epilepsie.
Casuïstiek
Epilepsie en Leerling Gebonden Financiering: de rol
2 Periodiek voor professionals 12 | nr 3 | 2014 Titel
Nederlandse Liga tegen Epilepsie De vereniging van professionals werkzaam in de epilepsiezorg en op aanverwante terreinen
Inspiratie Netwerk Kennis
De inbreng van de overheid en de medische en
maatschappelijke veranderingen in de epilepsiezorg
vragen aandacht. U wilt op de hoogte blijven en uw vak
goed uitoefenen. Verpleegkundigen, maatschappelijk
werkers, medewerkers uit het onderwijs, (kinder)neuro-
logen, kinderartsen, psychologen, neurochirurgen en
andere professionals binnen de epilepsiezorg hebben de
weg naar de Liga inmiddels gevonden.
Eén van de speerpunten van de Liga is het stimuleren van
en informeren over wetenschappelijk onderzoek naar
epilepsie. De Liga slaat daarbij een brug tussen weten-
schap en praktijk. Speciaal voor dit doel is de Sectie
Wetenschappelijk Onderzoek (SWO) opgericht. Als
Ligalid kunt u zich aansluiten bij de SWO. De SWO levert
een vaste bijdrage aan dit blad. De werkgroep
Multidisciplinaire Psychosociale Hulpverlening inventari-
seert en evalueert het psychosociale hulpverleningsaanbod.
De commissie Epilepsieverpleegkundigen is een platform
dat zich richt op de professionalisering van een relatief
nieuwe beroepsgroep.
Maar het lidmaatschap biedt meer:
- Het vakblad ‘Epilepsie’
- Korting op toegang Nationaal Epilepsie Symposium
- Korting op diverse internationale vakbladen
Bent u beroepsmatig werkzaam in de epilepsiezorg?
Dan zult u de Liga als een inspiratiebron ervaren. Als student
of assistent in opleiding (AIO) bent u ook welkom.
Bel 030 63 440 63 of mail naar [email protected].
Colofon‘Epilepsie’ is een uitgave van de Nederlandse Liga tegen Epilepsie, de Nederlandse afdeling van de International League Against Epilepsy.
Redactie:Pauly Ossenblok, hoofdredacteur Gerrit-Jan de Haan Loretta van ItersonGovert HooglandBen VledderMarian Majoie Odile van Iersel, bladmanager
Redactieraad:Eleonora Aronica, Eva Brilstra, Renée Dabekaussen-Spiering, Paul Eling, Anita Geertsema, Richard Lazeron, Olaf Schijns, Hans Stroink, Ton Tempels, Roland Thijs, Mariëlle Vlooswijk, Rob Voskuyl en Al de Weerd.
Aan dit nummer werkten verder mee:Paul Augustijn, Gerard van Erp, Jaap Jansen, Marjan van Kempen, Bert Kleine, Bobby Koeleman, Sylvia Klinkenberg, Frans Leijten, Petra van Mierlo, Joost Nicolai, Onno van Nieuwenhuizen, Alexander Peeraer, Rob Rouhl, Jurgen Schelhaas, Francesca Snoeijen-Schouwe-naars, Francis Tan en Monique Veendrick.
Projectredactie:Epilepsiefonds, Houten
Lay-out:Duotone grafisch ontwerp, De Bilt
Drukwerk: ZuidamUithof Drukkerijen, Utrecht
‘Epilepsie’ verschijnt vier maal per jaar en wordt toegezonden aan iedereen die lid is van de Nederlandse Liga tegen Epilepsie. Jaarlijks komt er een speciaal nummer uit, dat tevens wordt toegezonden aan neurologen in Nederland en Vlaanderen. Het lidmaatschap kost € 25,– per jaar. Voor studenten en AIO’s is dit € 12,50.
Wilt u reageren op de inhoud van dit blad? Laat dit dan binnen één maand ná verschijning weten aan het redactie-secretariaat. Ingezonden kopij wordt beoordeeld door de kernredactie, die zich het recht voorbehoudt om deze te weigeren of in te korten.
De redactie is niet verantwoordelijk voor de inhoud van bijdragen die onder auteursnaam zijn opgenomen.
Secretariaat:Nederlandse Liga tegen Epilepsie Odile van Iersel Postbus 270, 3990 GB HoutenTelefoon 030 634 40 63E-mail [email protected]
U kunt indien u meer informatie wenst rechtstreeks contact opnemen met de auteur of met het secretariaat.
Niets uit deze uitgave mag zonder vooraf gaande, schriftelijke toestemming van de uitgever worden overgenomen of vermenigvuldigd.
ISSN 1571 - 0408
Van de redactie
Hoe groot was de schade voor HM?
Meteen na de bilaterale mediale tempo-
rele lobectomie die bij HM werd uitge-
voerd, was duidelijk dat de geheugen-
functie ernstig was aangetast. Dergelijke
bilaterale resecties zijn daarna niet meer
uitgevoerd bij patiënten met epilepsie,
wel unilaterale temporaalkwabresecties.
Uitgangspunt bij een dergelijke operatie
is het bereiken van aanvalsvrijheid,
waarbij de diverse functies, zoals ge-
heugen- en gezichtsvermogen zoveel
mogelijk gespaard blijven. Recente
ontwikkelingen in functioneel en struc-
tureel beeldvormend onderzoek kunnen
hieraan bijdragen. Door analyse van
het epileptogene netwerk in relatie met
functionele netwerken, kan mogelijke
schade na operatie beter ingeschat wor-
den. Ook de ontwikkeling van minimaal
invasieve technieken voor diagnostiek
en behandeling beloven veel goeds.
Hiervoor is terecht steeds meer belang-
stelling, ook in ‘Epilepsie, periodiek
voor professionals’.
Pauly Ossenblok

Periodiek voor professionals 12 | nr 3 | 2014 3
Epilepsie behelst voor veel kinderen meer dan alleen de
aanvallen. Frequent zijn er bijkomende cognitieve en
sociaal emotionele ontwikkelingsstoornissen die aan-
leiding zijn voor algemene problemen op school, speci-
fieke leerstoornissen, gedragsproblemen en psychische
stoornissen. De samenhang tussen de epilepsie en bij-
komende problemen is lang niet altijd duidelijk. Al in
een vroege fase wordt daarom naast een goede aanvals-,
epilepsie- en etiologische diagnose, diagnostiek op de
andere bovengenoemde gebieden van belang geacht.
Door de organisatie van het onderzoek in het Kinder
Epilepsie Centrum SEIN-Heemstede wordt het kind
minimaal belast.
Het centrum bundelt de nieuwste kennis op het gebied
van diagnostiek, behandeling en begeleiding van kinde-
ren met epilepsie. Het centrum levert state of the art zorg,
is gemakkelijk bereikbaar en is laagdrempelig voor zorg-
verleners, verwijzers en patiënten. Het draagt zorg voor
vroegtijdige en snelle diagnostiek en daarmee een snelle
start van de juiste behandeling. Het centrum levert met
een groot patiëntencohort de data voor wetenschappelijk
onderzoek en vertaalt de uitkomsten van dit onderzoek
in evidence based zorgpaden. Dit en de inzet op de ver-
schillende terreinen van de epilepsiezorg door een mul-
tidisciplinair team, het nauwgezet volgen van innovaties
en ontwikkelingen in de epilepsiezorg en nationale en
internationale samenwerking (in netwerkverband), geven
het centrum zijn positie als expertisecentrum.
Epilepsie en bijkomende problemenEen goede aanvals- en epilepsiediagnose en zo mogelijk
etiologische diagnose is de basis voor adequate behan-
deling. Naast EEG-onderzoek en goede beeldvorming,
speelt vroegtijdige analyse van genetische factoren/oor-
zaken een steeds grotere rol. Genetische consultatie is
Het Kinder Epilepsie Centrum SEIN-Heemstede, dat nu ruim een jaar bestaat, biedt een op maat gemaakt multi-
disciplinair diagnostische programma dat wordt uitgevoerd binnen 36 uur.
Actueel
Door: Onno van Nieuwenhuizen ([email protected]), emeritus hoogleraar epileptologie, Universitair Medisch
Centrum, Utrecht.
Actueel
Het Kinder Epilepsie Centrum SEIN-HeemstedeMultidisciplinaire diagnostiek bij kinderen met aanvallen
dan ook nadrukkelijk een onderdeel bij de analyses.
Kinderen met epilepsie hebben een aanzienlijke kans
op ontwikkelingsproblemen, (specifieke) leerstoornis-
sen, kinderpsychiatrische stoornissen zoals ADHD, en
emotionele problematiek. Dit is zelfs ook aangetoond
bij kinderen met zogenaamde idiopathische epilepsie,
absence epilepsie op de kinderleeftijd en Rolandische
epilepsie. Gelukkig bezitten de meeste kinderen voldoen-
de capaciteiten waardoor er geen grote manifeste leer- en
of gedragsproblemen ontstaan. Bij een aantal kinderen is
dat niet het geval en treden deze wel op. Daarbij is het dan
lang niet altijd duidelijk wat de oorzaak is. Is er sprake
van onderliggende organische pathologie, komt het door
een intellectueel probleem, door de gebruikte medicatie
of een beperkt copings-mechanisme van kind en gezin.
Tevens doet zich dan het stapelen van problemen voor.
Aangetoond is dat kinderen die medicamenteus moeilijk
instelbaar zijn, vaker specifieke leerstoornissen hebben en
visa versa. Moeilijk instelbaar zijn gaat ook gepaard met
een grotere kans op intellectuele beperking en andersom.
Hetzelfde geldt voor kinderpsychiatrische stoornissen,
zoals ADHD en autisme spectrum stoornissen. Over-
vraging op school en thuis zal leiden tot emotionele
schade bij het kind. Sommige kinderen gaan dus naast
de epilepsie gebukt onder een opeenstapeling van bij-
komende problemen.
Door middel van multidisciplinaire diagnostiek kunnen
al deze facetten worden geanalyseerd en kan een behandel
en begeleidingsvoorstel gemaakt worden dat verder gaat
dan uitsluitend de correcte diagnose van de epilepsie en
medicamenteuze behandeling.
Diagnose in 36 uurHet centrum regelt een laagdrempelige, snelle toegang
voor kinderen met (verdenking van) epilepsie, vooral voor

Casuïstiek
4 Periodiek voor professionals 12 | nr 3 | 2014 Actueel
Actueel
de maatschappelijk werker en de speltherapeut de
bevindingen in een multidisciplinair beraad. Ook de
verwijzer is hier zo mogelijk bij. De kinderneuroloog/
epileptoloog is case-manager.
7. Tot slot vindt aan het eind van de opname of kort na
de opname een eindgesprek met de kinderneuroloog/
epileptoloog en de patiënt en zijn of haar ouders (en
zo mogelijk) de verwijzer plaats. In dit gesprek krijgt
de patiënt de (voorlopige) diagnose en een voorstel
voor het vervolgtraject.
Eenzelfde concept werd toegepast bij kinderen die in
aanmerking kwamen voor epilepsiechirurgie, in het
Sylvia Tóth Centrum in het Wilhelmina Kinderzieken-
huis van het Universitair Medisch Centrum Utrecht. Het
vraagt een goede organisatie – voorbereiding, tijd van
zorgverleners en planning van onderzoeken – en een
forse inspanning van de kinderen en ouders omdat het
diagnosetraject geconcentreerd is in 36 uur. Maar het le-
vert veel op: een zeer korte doorlooptijd van het diagnos-
tische traject en een diagnose waar een multidisciplinair
team van experts achter staat en die tot stand is gekomen
met de inbreng van de verwijzer(s) en de patiënt en zijn
of haar ouders. Op deze manier staat de patiënt met zijn
of haar zorgvraag echt centraal. En niet het zorgaanbod
van de zorgverlener.
kinderen met complexe epilepsie. En als een kind in het
centrum is, zorgt het centrum met een concept van diagno-
se-in-36-uur dat zo snel mogelijk de diagnose en de daaruit
voortvloeiende behandeling duidelijk is.
Aan het principe van diagnose-in-36-uur wordt als volgt
inhoud gegeven:
1. De huisarts, kinderarts of (kinder)neuroloog verwijst
een kind naar het centrum.
2. Voor een patiënt in het centrum komt, is er een consult
op een polikliniek van SEIN waar de kinderneuroloog,
het diagnosetraject voorbereid. Alle relevante infor-
matie van de patiënt: brief of brieven van verwijzer(s),
uitslagen diagnostische onderzoeken, zoals MRI’s en
EEG’s wordt verzameld. Zo nodig worden de onder-
zoeken voorgelegd aan de klinisch neurofysioloog of
neuroradioloog.
3. Op basis van deze informatie plant de kinderneuroloog
het diagnostische traject.
4. Op de dagen dat er diagnostisch onderzoek wordt
gedaan krijgt de patiënt bij binnenkomst een ‘eigen’
kamer op de EEG-afdeling toegewezen.
5. Vervolgens vinden de noodzakelijke additionele on-
derzoeken – EEG, MRI – plaats, alsmede gesprekken
met andere relevante specialisten zoals bijvoorbeeld
een klinisch geneticus, kinderneuropsycholoog, psy-
choloog, medewerker van de ambulante schoolbege-
leidingsdienst van leerwegondersteunend onderwijs
(LWOO)/’De Waterlelie’, de maatschappelijk werker
of speltherapeut. Voor de onderzoeken beschikt het
centrum over time-slots op de betreffende afdelingen.
De gesprekken en zoveel mogelijk ook de onderzoeken
vinden plaats op de kamer van de patiënt. De patiënt
loopt dus niet de artsen af, maar de artsen komen naar
de patiënt toe.
6. Aan het einde van de 36 uur bespreken de kinderneuro-
loog/epileptoloog, klinisch neurofysioloog, kinderpsy-
choloog, de klinisch geneticus, de neuropsycholoog,
de medewerker van de Ambulante Begeleidingsdienst,
Anton de Louw nieuwe voorzitter Nederlandse Liga tegen Epilepsie Neuroloog dr. Anton de Louw is tijdens de Algemene Ledenvergadering op 15 mei jl. gekozen als nieuwe voor-
zitter van het bestuur van de Nederlandse Liga tegen Epilepsie. Anton de Louw, medisch hoofd epileptologie
van Kempenhaeghe, is benoemd voor een eerste termijn van vier jaar. De Louw: “De Liga is een prachtig plat-
form waarbinnen de professionals werkzaam in de epilepsiezorg verenigd zijn. De in 2011 verkregen status van
werkgroep binnen de Nederlandse Vereniging voor Neurologie heeft het platform een goed fundament gegeven.
Aankomende jaren zal verder worden geïnvesteerd in een coördinerende rol van het Ligabestuur waarbij onder
meer kan worden gedacht aan de ontwikkeling van een Zorgstandaard epilepsie.”
Ervaringen tot nu toeHet Kinder Epilepsie Centrum is in september 2013 – na
een pilotfase – officieel geopend. Sindsdien worden weke-
lijks kinderen in het centrum onderzocht; aanvankelijk één
patiënt per week, vervolgens werd dit uitgebreid tot twee
patiënten. De ervaringen zijn unaniem positief: voor de
patiënt en ouders omdat het snelle diagnostische traject
een lange, vermoeiende reis langs alle disciplines overbodig
maakt; voor het ‘team’ omdat het gemeenschappelijk onder-
zoeken van de patiënt en de open discussie van de verschil-
lende opinies leidt tot verdieping en vermeerdering van
kennis, wat meteen ten goede komt aan de patiënt.

Periodiek voor professionals 12 | nr 3 | 2014 5Casuïstiek
Door: Rob Rouhl ([email protected]), Mariëlle Vlooswijk, Sylvia Klinkenberg, Joost Nicolai, neurologie en kinder-
neurologie, Maastricht Universitair Medisch Centrum en Academisch Centrum voor Epileptologie Kempenhaeghe/
MUMC+, Maastricht en Heeze, Alexander Peeraer, Universiteit Maastricht.
Casuïstiek
Antistoffen tegen glutamaat-decarboxylase bij epilepsieIn deze bijdrage wordt aan de hand van twee casusbeschrijvingen van patiënten met refractaire epilepsie en geheugen-
stoornissen of veranderd gedrag met een positieve antistoftiter de stand van zaken omtrent de kennis over antistoffen
tegen glutamatdecarboxylasein in relatie tot epilepsie besproken.
Casus 1 Patiënt presenteerde zich op zestienjarige leeftijd met
geheugenklachten. Na drie leerjaren vmbo zonder pro-
bleem doorlopen te hebben, doubleerde hij het vierde
leerjaar twee maal. Een jaar nadien kreeg hij epileptische
aanvallen die begonnen met ruiken van een zure geur,
dubbelzien en smakken. Vervolgens slaan met de rechter-
arm op de borst, zijn linkerarm liet hij hangen. Soms ging
een dergelijke aanval over in een algehele verstijving en
schokken. Neurologisch onderzoek en MRI toonden geen
afwijkingen. Het interictale EEG toonde epileptiforme af-
wijkingen rechts temporaal. Behandeling met anti-epilep-
tica gaf onvoldoende aanvalsreductie. Daarom screenden
wij (opnieuw) naar onderliggend lijden (POLG-1 mutatie,
antilichamen passend bij coeliakie en Hashimotoen
antilichamen tegen spanningsafhankelijke kaliumkanalen
(VGKC). Alleen de antistoffen tegen glutamaatdecarboxy-
lase (anti-GAD) waren positief (>2000 IU/ml). Hierop
werd de diagnose anti-GAD auto-immuun encephalitis
gesteld. Vanwege de bijzonder hoge anti-GAD titer werd
direct gestart met plasmaferese.
Een nieuwe MRI-cerebrum toonde nu een mesiotempo-
rale sclerose (figuur 1). Liquoronderzoek toonde geen
leukocyten en de anti-GAD concentratie was 3,3 IU/ml.
Onderhoudsbehandeling met prednison en mycofenolaat
mofetil werd gestart. Na enkele maanden behandeling
waren er nog hooguit twee aura’s per maand. Mycofeno-
laat mofetil werd vervangen door azathioprine, ook de
dosering van de anti-epileptica werd geleidelijk verlaagd.
Anderhalf jaar later namen de bij deze patiënt bekende
aanvallen met hoofdpijn en braken weer in frequentie
toe. Mycofenolaat mofetil werd herstart, in combinatie
met maandelijkse kuren immunoglobulines. Opvallend
genoeg nemen de aanvallen toe in de laatste (vierde) week
voor de gift immunoglobulines. Met driewekelijkse kuren
en anti-epileptica (lamotrigine en clobazam) blijven de
aanvallen weg. Wel houdt hij een hinderlijke down-beat
nystagmus en een fors gestoord geheugen waarbij patiënt
een opschrijfboekje moet gebruiken om dagelijkse dingen
te onthouden. Mogelijke verdere gevolgen zijn onderbre-
king of beperking van onderwijs en begeleid wonen.
Casus 2Een meisje met een blanco voorgeschiedenis, geboren na
een ongecompliceerde zwangerschap en partus debuteert
met epileptische aanvallen op driejarige leeftijd. Vanaf
Figuur 1 De coronale FLAIR-opname toont een hyperintens signaal
van beide hippocampi, links meer dan rechts, bij een nagenoeg
behouden volume (zie pijlen, geduid als hippocampale sclerose).

6 Periodiek voor professionals 12 | nr 3 | 2014 Casuïstiek
Casuïstiek
debuut is de aanvalsfrequentie hoog. Er zijn veelal com-
plex partiële aanvallen, incidenteel secundair gegenerali-
seerde en atone aanvallen. Terugkijkend bleek dat er enige
weken voor het epilepsiedebuut een gedragsverandering
was: zij was onhandelbaar en onrustig. Verder had zij
meer moeite in het richten en behouden van aandacht. Zij
werd opgenomen voor observatie en instellen van behan-
deling. Tijdens de opname vertoonde zij zeer wisselend
gedrag, op momenten was zij agressief, grensoverschrij-
dend en ongeremd. Er werd gestart met risperidon. Op
andere momenten hallucineerde zij. Ook was er een
slaapstoornis. Behoudens de gedragsverandering was het
neurologisch onderzoek niet afwijkend. Beeldvormend
onderzoek was bij herhaling normaal. Initieel toonde het
EEG een diffuus vertraagd beeld, later in de eerste week
ontstonden er multifocale epileptiforme afwijkingen.
Ondanks clobazam, vervolgens levetiracetam en feno-
barbital, ontstond er binnen twee weken na het epilepsie-
debuut een partiële status epilepticus met klinische én
non-convulsieve aanvallen. Na uitgebreid onderzoek
bleken de antistoffen tegen glutmaatdecarboxylase
(GAD) positief (143 IU/ml). De diagnose auto-immuun
encephalitis werd gesteld. Patiëntje werd behandeld met
intraveneuze immuno-globulines gevolgd door steroïden.
In combinatie met carbamazepine en lorazepam werd
aanvalscontrole bereikt. Steroïden werden geleidelijk
afgebouwd en de anti-epileptica werden afgebouwd tot
monotherapie carbamazepine. In de afgelopen negen
maanden heeft zij nog twee aanvallen gehad. Het EEG is
volledig genormaliseerd zowel in waak als in slaap. Er
bestaan nog geringe gedragsproblemen, maar haar neuro-
cognitieve toestand gaat inmiddels weer duidelijk vooruit.
Anti-GADGAD is een intracellulair enzym dat onder andere in neu-
ronen voorkomt en glutamaat omzet in GABA. Anti-GAD
antistoffen worden bij veel verschillende ziekten beschre-
ven, zoals het stiff-person syndroom en cerebellaire ataxie
(Saiz et al., 2008). Antistoffen tegen GAD komen ook
voor (zij het in lage titers) bij patiënten met diabetes
mellitus type 1. De hypothese is dat antistoffen tegen
GAD (met name bij hogere titers) de werking van het
GAD verminderen. Hierdoor wordt glutamaat minder
in GABA omgezet, hetgeen leidt tot een verminderde
GABA-erge synaptische transmissie. Zo zou dit ook
kunnen leiden tot epileptische aanvallen.
Prevalentie bij epilepsieDe prevalentie van een positieve anti-GAD titer varieert
van 3-8 procent (Sokol et al., 2004; Peltola et al., 2000)
bij patiënten met therapieresistente epilepsie tot ongeveer
22 procent bij patiënten met een verdenking op een auto-
immuun encephalitis (epilepsie met geheugenstoornissen
en psychiatrische symptomen) (Quek et al., 2012). Een
positieve antistoftiter komt vaker voor bij vrouwen. Een
positieve anti-GAD titer komt ook voor bij 0,4-1 procent
van de gezonde populatie (Batstra et al., 1999). Een com-
plicerende factor in de literatuur over de prevalentie van
een positieve titer is dat verschillende studies verschil-
lende eenheden gebruiken voor de anti-GAD concen-
tratie en deze met een verschillende nauwkeurigheid
(aantal cijfers achter de komma) weergeven.
Klinisch beeld en neuroradiologieDe eerste case-reports beschreven patiënten met refrac-
taire temporaalkwab-epilepsie en geringe tot ernstige
inprentingsstoornissen, passend bij een limbische
encephalitis (Matà et al., 2008; Malter et al., 2010). Later
bleken ook verhoogde titers voor te komen bij patiënten
met een refractaire temporaalkwabepilepsie zonder dui-
delijke andere klachten. In zeldzamere gevallen hebben
de epileptische aanvallen een extra-temporale origine
(Najjar et al., 2011). Vaak waren er ook afwijkingen te
zien (uni- of bilateraal) in de mesiale temporaalkwab op
MRI (Malter et al., 2010) of, zeer zelden, hyperintense
afwijkingen in de witte stof en cortex met enige contrast-
opname (Najjar et al., 2011).
TherapiePatiënten met een positieve anti-GAD titer reageren
slecht op anti-epileptica. Dit kan ook berusten op een
selectiebias, aangezien juist patiënten die refractair zijn
voor de gebruikelijke behandeling worden gescreend op
de aanwezigheid van anti-GAD. Gezien de (verdenking
op) een actieve auto-immuunrespons worden patiënten
met een positieve anti-GAD titer vaak behandeld met
immunomodulatie, en zeker wanneer het klinisch beeld
met meer diverse problemen (epileptische aanvallen en
geheugenstoornissen) verloopt. De immunomoduleren-
de therapie (corticosteroïden, gammaglobulines, cyclo-
fosfamide of plasmaferese) heeft echter een zeer matig
effect op de aanvalsfrequentie (Matà et al., 2008, Malter
et al., 2010) en op de afname van de antistoftiters (Malter
et al., 2010). Er zijn geen factoren bekend die voorspel-
len welke patiënten wel op immunotherapie reageren.
Vaak is er een recidief wanneer de immuunmodulerende
therapie wordt afgebouwd.
Heeft anti-GAD een direct pathogeen effect?Het is onduidelijk of de anti-GAD antistoffen zelf patho-
geen zijn. Ze zijn gericht tegen een intracellulair eiwit,
maar de intracellulaire ruimte is moeilijk toegankelijk
voor een antistof (Vincent et al., 2011). Daarnaast komen
andere auto-antistoffen ook vaker voor bij patiënten met
anti-GAD antistoffen, zoals anti-TPO en antilichamen
tegen Langerhans-cellen (in de pancreas). Het is daarom

Periodiek voor professionals 12 | nr 3 | 2014 7
Casuïstiek
Casuïstiek
ook mogelijk dat anti-GAD-antistoffen een epifenomeen
bij een ander auto-immuunproces zijn. Bovendien pleit
de prevalentie van anti-GAD in de gezonde populatie
tegen een direct pathogeen effect van anti-GAD.
ConclusieAnti-GAD antistoffen zijn geassocieerd met epilepsie, en
mogelijk meer met refractaire temporaalkwabepilepsie,
zoals bij de patiënt uit de eerste casusbeschrijving (casus
1). De respons op immunomodulerende therapie is wisse-
lend, echter deze lijkt doorgaans teleurstellend. Een oor-
zakelijke rol van anti-GAD antistoffen bij het ontstaan van
epilepsie is nog onopgehelderd. Desondanks verdient het
aanbeveling bij een refractaire epilepsie antistoffen tegen
GAD te bepalen en bij een duidelijk verhoogde titer (>50
IU/ml) een immunomodulerende behandeling te starten,
bij voorkeur in overleg met een centrum met expertise in
behandeling van deze aandoening.
ReferentiesBatstra MR, van Driel A, Petersen JS et al. (1999) Glutamic
acid decarboxylase antibodies in screening for autoim-
mune diabetes: Influence of comorbidity, age, and
sex on specificity and threshold values. Clin Chem.
45:2269-2272.
Malter MP, Helmstaedter C, Urbach H et al. (2010)
Antibodies to glutamic acid decarboxylase define a
form of limbic encephalitis. Ann Neurol. 67:470-478.
Matà S, Muscas GC, Naldi I et al. (2008) Non-
paraneoplastic limbic encephalitis associated with
anti-glutamic acid decarboxylase antibodies. J
Neuroimmunol. 199:155-159.
Najjar S, Pearlman D, Najjar A et al. (2011) Extralimbic
autoimmune encephalitis associated with glutamic
acid decarboxylase antibodies: An underdiagnosed
entity? Epilepsy Behav. 21:306-313.
Peltola J, Kulmala P, Isojarvi J et al. (2000) Auto-
antibodies to glutamic acid decarboxylase in
patients with therapy-resistant epilepsy. Neurology
55:46-50.
Quek AM, Britton JW, McKeon A et al. (2012)
Autoimmune epilepsy: Clinical characteristics and
response to immunotherapy. Arch Neurol. 69:582-593.
Saiz A, Blanco Y, Sabater L et al. (2008) Spectrum of
neurological syndromes associated with glutamic acid
decarboxylase antibodies: Diagnostic clues for this
association. Brain 131:2553-2563.
Sokol DK, McIntyre JA, Wagenknecht DR et al. (2004)
Antiphospholipid and glutamic acid decarboxylase
antibodies in patients with focal epilepsy. Neurology
62:517-518.
Vincent A, Bien CG, Irani SR et al. (2011) Autoantibodies
associated with diseases of the cns: New developments
and future challenges. Lancet Neurol. 10:759-772.
Hebt u al kennisgemaakt met het voorlichtingsmagazine van het Epilepsiefonds: Epilepsie Magazine?
Epilepsie Magazine bevat artikelen over wetenschappelijk onderzoek, ervaringsverhalen over mensen met epilepsie en hun omgeving, medische achtergrond-informatie en epilepsienieuws.
Neem nu een abonnement!Abonnees ontvangen het kwartaalblad voor € 20,– per jaar. Nieuwe abonnees ontvangen het eerste nummer gratis! Als u vragen of opmerkingen hebt, kunt u uiteraard bellen of mailen met Annelies Bakker, hoofdredacteur van Epilepsie Magazine: 030 634 40 63 of [email protected].
Epilepsie Magazine

Casuïstiek
8 Periodiek voor professionals 12 | nr 3 | 2014
Door: Monique Veendrick ([email protected]), Francesca Snoeijen-Schouwenaars, Petra van Mierlo,
Gerard van Erp, Bert Kleine, Jurgen Schelhaas en Francis Tan, Centrum voor Epilepsiewoonzorg, Kempenhaeghe, Heeze.
Wetenschappelijk onderzoek
Wetenschappelijk onderzoek
Carbamazepine-afbouw bij volwassen patiënten met het syndroom van DravetCorrect volgens de literatuur maar pas op in de praktijk
In de literatuur wordt geadviseerd terughoudend te zijn met het gebruik van carbamazepine, lamotrigine en fenytoïne
bij patiënten met het syndroom van Dravet. Bij nieuw gediagnosticeerde patiënten zal deze medicatie dan ook worden
vermeden. Veel volwassen patiënten bij wie op latere leeftijd de diagnose Dravet is gesteld, zijn echter reeds ingesteld
op carbamazepine. Afbouw van deze medicatie blijkt in de praktijk niet altijd goed uit te pakken.
Het syndroom van Dravet is een ernstig epilepsiesyn-
droom dat veroorzaakt wordt door een mutatie in het
SCN1A gen. De aandoening presenteert zich meestal in
het eerste levensjaar met frequent optredende koortscon-
vulsies. Al snel krijgen patiënten ook aanvallen zonder
dat zij koorts hebben. Vanaf het tweede jaar treedt er
een verlies op van vaardigheden, die de patiënten in het
eerste levensjaar normaal hebben ontwikkeld. Epilep-
tische aanvallen die bij het syndroom van Dravet vaak
worden gezien zijn atypische absences, atone aanvallen,
myoclonieën en tonisch-clonische aanvallen. Bij volwas-
sen patiënten is er een afname van de atypische absences
en myoclonieën maar persisteren de tonisch-clonische
aanvallen met een focaal debuut. Daarnaast is er op de
volwassen leeftijd een verminderde mobiliteit op basis
van onder andere cerebellaire stoornissen (Rilstone et
al., 2012). De prognose is slecht met een mortaliteit, ook
in de groep volwassenen, van tussen de 15 en 20 procent
(Genton et al., 2011). De meeste patiënten overlijden aan
de gevolgen van een status epilepticus of door sudden
unexpected death in epilepsy (SUDEP).
Behandeling met anti-epilepticaEr wordt veel onderzoek verricht naar de medicamenteu-
ze behandeling van het syndroom van Dravet. Op basis
van twee randomized controlled trials is er bewijs voor de
effectiviteit van stiripentol. In deze add-on trials werd sti-
ripentol toegevoegd aan de onderhoudsbehandeling met
valproaat en clobazam (STICLO France, Lancet 2000,
STICLO Italy (niet gepubliceerd)). Ook levetiracetam,
topiramaat, valproaat en zonisamide lijken een gunstig
effect te hebben op de aanvalsfrequentie. Daarnaast
wordt geadviseerd om niet te starten met lamotrigine,
fenytoine en carbamazepine. Voor met name lamot-
rigine is dit door Guerrini et al. (1998) zorgvuldig
uitgezocht. Lamotrigine induceerde bij 17 van de 21
patiënten die in deze studie werden onderzocht een
toename van met name tonisch-clonische aanvallen
en myoclonieën. Voor het vermijden van fenytoine en
carbamazepine is minder wetenschappelijk bewijs.
De theoretische achtergrond voor een toename van de
aanvalsfrequentie door lamotrigine, carbamazepine en
fenytoïne wordt gezocht in de mutaties in het SCN1A gen
(Catteral, 2014). Het SCN1A gen codeert voor een deel
van het natriumkanaal. Spanningsafhankelijke kanalen
zijn de basis voor iedere actiepotentiaal, wat de relatie
met prikkelbaarheid en epilepsie goed kan verklaren.
Bij het syndroom van Dravet is er meestal sprake van
loss of function mutaties en zou men dus juist een ver-
minderde excitatie verwachten. Onderzoek bij muizen
met mutaties in het SCN1A gen laten zien dat vooral de
GABA-erge interneuronen een verminderde prikkel-
baarheid vertonen, terwijl de activerende piramidecellen
een normale natriumstroom laten zien. In het muismo-
del wordt de epilepsie dus veroorzaakt door een vermin-
derde functie van de remmende interneuronen. Op basis
van dit mechanisme kunnen de natriumkanaalblokkers
carbamazepine, lamotrigine en fenytoine de inhibitie
verder verminderen. Benzodiazepinen en stiripentol
zouden wel goed werken omdat deze middelen een
GABA versterkende werking hebben. Middelen als
levetiracetam, topiramaat, valproaat en zonisamide

Casuïstiek
Periodiek voor professionals 12 | nr 3 | 2014 9Wetenschappelijk onderzoek
Wetenschappelijk onderzoek
hebben indirecte effecten op GABAerge neuronen en
kunnen op deze manier gunstig uitpakken.
Op basis van bovenstaande overwegingen werd bij een
van onze volwassen Dravet patiënten met polytherapie
en frequent optredende myoclonieën de carbamaze-
pine afgebouwd. Hierop zagen we bij deze patiënt een
afname van de myoclonieën maar ook een toename van,
met name, tonische aanvallen. Deze tonische aanvallen
ontwikkelden zich tot een status epilepticus die werd ge-
compliceerd door een ernstige pneumonie, waaraan hij
uiteindelijk is overleden. Op basis van deze ervaring heb-
ben wij onderzoek gedaan naar het effect van medicatie-
wijzigingen, in het bijzonder carbamazepine-afbouw.
De vraagstelling was, in hoeverre afbouw van carbama-
zepine heeft geleid tot een verslechtering van de aanvals-
controle, en of er sprake is geweest van complicaties. De
reden dat in deze studie uitsluitend wordt gekeken naar
het gebruik van carbamazepine en niet naar fenytoine
en lamotrigine, is omdat juist carbamazepine nog veel-
vuldig wordt gebruikt bij de volwassen patiënten met het
syndroom van Dravet.
Patiënten onderzoekVoor dit onderzoek zijn in Kempenhaeghe de gegevens
van alle volwassen residentiele patiënten verzameld, die
een Dravet syndroom met een SCN1A mutatie hadden.
De volgende parameters zijn specifiek gedocumenteerd:
geslacht, leeftijd, leeftijd debuut epilepsie, type SCN1A
mutatie, ernst van de verstandelijke beperking, het al
dan niet gebruik van carbamazepine, de duur van het ge-
bruik, het effect van het starten of stoppen van de carba-
mazepine en het overlijden van patiënten. De gebruikte
medicatie en de aanvalsregistratie zijn in Kempenhaeghe
digitaal beschikbaar vanaf het begin van de negentiger
jaren. De aanvalsregistratie zoals die door het verpleeg-
kundig personeel wordt bijgehouden werd voor deze
studie wel geraadpleegd, maar de conclusie of er een
verandering is opgetreden na het starten/stoppen van
de carbamazepine is tevens gebaseerd op het (kwalitatief
gedocumenteerde) klinische oordeel van de behandelend
epileptoloog.
ResultatenDe studie populatie betreft negen mannen en zeven
vrouwen (n=16). Van de 16 patiënten zijn er in de loop
van de jaren vier patiënten overleden. Bij één patiënt was
de oorzaak van het overlijden niet gerelateerd aan de
epilepsie. Van de drie andere patiënten die zijn overle-
den zijn er twee overleden aan de complicaties van een
status epilepticus en een patiënt aan een SUDEP. Bij een
van de twee patiënten die is overleden aan de gevolgen
van een status epilepticus (patiënt 9) is er een duidelijke
tijdsrelatie tussen het stoppen van de carbamazepine, het
ontwikkelen van een status epilepticus en het overlijden
van de patiënt. De patiënt die is overleden aan een SUDEP
overleed twee jaar nadat gestopt was met het verder af-
bouwen van de carbamazepine. In totaal hebben 13 van
de 16 patiënten ooit carbamazepine gebruikt. Bij tien
patiënten is geprobeerd carbamazepine af te bouwen
waarbij bij acht de carbamazepine geheel is gestaakt. De
reden van afbouw was bij zeven van de tien patiënten te
achterhalen en varieerde van bijwerkingen, onvoldoende
effect, tot veranderen van medicatie ‘omdat er in de litera-
tuur indicaties zijn dat carbamazepine minder goed werkt
bij patiënten met een SCN1A mutatie’. Bij vier van de tien
patiënten waarbij carbamazepine werd afgebouwd was er
een toename van het aantal tonisch-clonische insulten.
Voor de overige klinische gegevens en de gegevens met
betrekking tot de gevonden mutaties wordt verwezen naar
tabel 1 (pagina 10).
DiscussieHet stoppen dan wel afbouwen van de carbamazepine,
bij de in onze instelling verblijvende Dravet patiënten gaf
aanleiding tot een toename van tonisch-clonische aanval-
len bij vier van de tien patiënten. Bij een patiënt was de
afbouw van carbamazepine in tijd gerelateerd aan zowel
een toename van het aantal convulsieve aanvallen als aan
het overlijden van de patiënt. Onze studie betreft welis-
waar een kleine populatie, maar is unbiased omdat alle bij
ons verblijvende volwassen patiënten met het syndroom
van Dravet geïncludeerd zijn.
Onze retrospectief verkregen data zijn niet in overeen-
stemming met de eveneens retrospectief verzamelde
gegevens uit de literatuur. In 2011 verwijst Dravet zelf
naar drie publicaties waarin een verslechtering van de
aanvalscontrole zou zijn opgetreden na, let wel, het
starten met carbamazepine (Dravet & Guerrine 2011).
In een van deze publicaties (Wakai et al., 1996) was er
bij vier van de zes patiënten sprake van een verslechtering
van de aanvalscontrole en geen effect bij twee patiënten.
De tweede publicatie die door Dravet wordt aangehaald
is de publicatie door Horn (Horn et al., 1986) bij negen
patiënten. Bij twee van de negen was er een toename van
het aantal aanvallen, bij vijf was er sprake van bijwerkin-
gen en bij twee werd een gunstig effect van de carbamaze-
pine gezien. Een vergelijkbaar resultaat werd gezien in de
derde publicatie (Wang et al., 1996), waar tien patiënten
aan deelnamen, maar waarvan in het Engels alleen het
abstract beschikbaar is. Daarnaast zijn er relatief recent
twee grote klinische ‘Dravet’ studies (Brunklaus et al.,
2012; Xu et al., 2013) gepubliceerd waarbij ook gekeken
werd naar het effect van medicatie. In het artikel van
Brunklaus waren van de 241 patiënten 60 die een
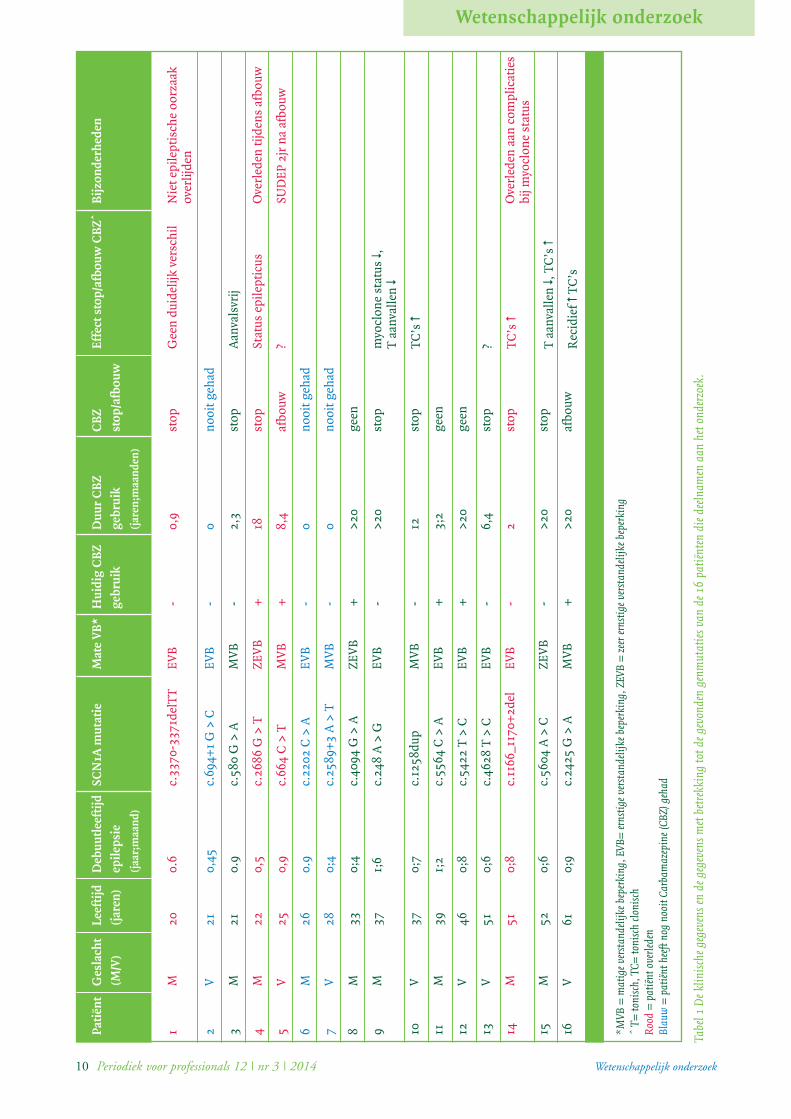
10 Periodiek voor professionals 12 | nr 3 | 2014 Wetenschappelijk onderzoek
Wetenschappelijk onderzoekWetenschappelijk onderzoek
Tabe
l 1 D
e klin
isch
e geg
even
s en
de g
egev
ens m
et b
etre
kkin
g to
t de g
evon
den
genm
utat
ies v
an d
e 16
patië
nten
die
dee
lnam
en a
an h
et o
nder
zoek
.
Pati
ënt
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Ges
lach
t
(M/V
)
M V M M V M V M M V M V V M M V
Leef
tijd
(jar
en)
20 21 21 22 25 26 28 33 37 37 39 46 51 51 52 61
Deb
uutl
eeft
ijd
epile
psie
(jaa
r;m
aan
d)
0.6
0,45
0.9
0,5
0,9
0.9
0;4
0;4
1;6
0;7
1;2
0;8
0;6
0;8
0;6
0;9
SCN
1A m
utat
ie
c.33
70-3
371d
elT
T
c.69
4+1
G >
C
c.58
0 G
> A
c.26
86 G
> T
c.66
4 C
> T
c.22
02 C
> A
c.25
89+
3 A
> T
c.40
94 G
> A
c.24
8 A
> G
c.12
58du
p
c.55
64 C
> A
c.54
22 T
> C
c.46
28 T
> C
c.11
66_1
170+
2del
c.56
04 A
> C
c.24
25 G
> A
Mat
e VB
*
EVB
EVB
MV
B
ZEV
B
MV
B
EVB
MV
B
ZEV
B
EVB
MV
B
EVB
EVB
EVB
EVB
ZEV
B
MV
B
Hui
dig
CB
Z
gebr
uik
- - - + + - - + - - + + - - - +
Duu
r C
BZ
gebr
uik
(jar
en;m
aan
den
)
0,9
0 2,3
18 8,4
0 0 >20
>20
12 3;2
>20
6,4
2 >20
>20
CB
Z
stop
/afb
ouw
stop
nooi
t geh
ad
stop
stop
afbo
uw
nooi
t geh
ad
nooi
t geh
ad
geen
stop
stop
geen
geen
stop
stop
stop
afbo
uw
Effe
ct s
top/
afbo
uw C
BZ^
Gee
n du
idel
ijk v
ersc
hil
Aan
vals
vrij
Stat
us e
pile
ptic
us
? myo
clon
e st
atus
,
T a
anva
llen
TC
’s
? TC
’s
T a
anva
llen
, TC
’s
Rec
idie
f T
C’s
Bijz
onde
rhed
en
Nie
t epi
lept
isch
e oo
rzaa
kov
erlij
den
Ove
rled
en ti
jden
s af
bouw
SUD
EP 2
jr n
a af
bouw
Ove
rled
en a
an c
ompl
icat
ies
bij m
yocl
one
stat
us
*MVB
= m
atig
e ver
stan
delij
ke b
eper
king
, EVB
= er
nstig
e ver
stan
delij
ke b
eper
king
, ZEV
B =
zee
r ern
stig
e ver
stan
delij
ke b
eper
king
^ T=
toni
sch,
TC=
toni
sch
cloni
sch
Roo
d =
pat
iënt
ove
rlede
nBl
auw
= p
atië
nt h
eeft
nog
nooi
t Car
bam
azep
ine (
CBZ)
geh
ad

Periodiek voor professionals 12 | nr 3 | 2014 11Wetenschappelijk onderzoek
verslechtering lieten zien na het starten van medicatie.
Bij 36 van deze 60 patiënten (60 procent) ging het om
een verslechtering door carbamazepine. In de studie van
Xu et al. (2013) bij 138 patiënten met Dravet, waarvan 40
behandeld met carbamazepine, werd bij 16 (40 procent)
een toename van de aanvalsfrequentie door het gebruik
van carbamazepine gezien. Bij de overige 24 patiënten
had carbamazepine geen effect.
Er zijn een aantal mogelijke verklaringen voor het ver-
schil in uitkomst tussen deze studies en onze resultaten.
In bovenstaande studies gaat het om patiënten waar-
bij gestart wordt met carbamazepine en dus niet over
afbouw van medicatie. Een andere verklaring zou kunnen
zijn dat in de literatuur met name gekeken is naar kinde-
ren terwijl in onze studie uitsluitend volwassen patiënten
zijn geïncludeerd. De kans op en de uitgebreidheid van
cumulatieve secundaire (anoxische) schade die mogelijk
gevoelig is voor carbamazepine is bij de volwassen groep
veel groter. Een argument voor deze laatste overweging
is de hoge frequentie van tonische aanvallen (niet
vermeld in de resultaten) in onze groep patiënten, een
aanvalstype dat bij kinderen met Dravet zelden wordt
gezien. Daarnaast zou een verandering van de leeftijd
ook gepaard kunnen gaan met een verandering van het
expressiepatroon van het SCN1A gen. Belangrijk voor
het huidige concept van aanvallen bij het syndroom van
Dravet is namelijk de selectiviteit van de natriumkanalen.
Er zijn diverse subtypes van natriumkanalen met verschil-
lende regionale expressiepatronen. Het is goed moge-
lijk dat leeftijd, chronische polytherapie of secundaire
neuronale schade het (compenserende) expressiepatroon
van kanalen in een specifiek hersen-gebied verandert en
daarmee de balans tussen inhibitie en excitatie verstoort.
Klinisch zou zich dit kunnen uiten in een verandering van
aanvalstype. Tot slot zijn er ook in de hierboven aange-
haalde studies een aantal patiënten beschreven bij wie de
carbamazepine niet kon worden gestaakt. Dravet schrijft
hierover: ‘In some older patient taking polytherapies, including
carbamazepine, lamotrigine and diphantoin attempt of drug
withdrawal were followed by aggravation of epilepsy’.
Opvallend is het percentage van 25 procent van de
patiënten die zijn overleden in onze studie. Indien echter
rekening wordt gehouden met het kleine aantal patiën-
ten, het feit dat de doodsoorzaak in een van de vier
patiënten niet gerelateerd is aan de epilepsie en het
gegeven dat het hier uitsluitend gaat om zeer ernstig
aangedane Dravet patiënten, dan lijkt het overlijdens-
risico in onze groep vergelijkbaar met die in de literatuur
werd gerapporteerd. Het is in een zo kleine populatie
uiteraard niet gerechtvaardigd om hier stellige uitspraken
over te doen. Deze studie is te klein om een vergelijking
te maken in overlijdensrisico tussen de patiënten die nooit
aan een carbamazepine of een andere natriumblokker zijn
blootgesteld, de patiënten bij wie de carbamazepine is
gestaakt en de groep bij wie de carbamazepine is geconti-
nueerd.
ConclusiesWij concluderen dat op basis van de beschikbare literatuur
bij nieuw gediagnostiseerde patiënten met het syndroom
van Dravet er sprake is van een relatieve contra-indicatie
voor het starten met carbamazepine. Onze gegevens
bieden echter onvoldoende steun voor een advies om de
carbamazepine af te bouwen bij Dravet patiënten die sta-
biel zijn ingesteld op dit medicijn. Ons advies is echter wel
om afbouw van carbamazepine bij volwassen, genetisch
bewezen Dravet patiënten te overwegen, maar de afbouw
geleidelijk uit te voeren en zeer goed te monitoren.
ReferentiesBrunklaus A, Ellis R, Reavey E et al. (2012) Prognostic,
clinical and demographic features in SCN1A muta-
tion-positive Dravet syndrome. Brain 135:2329-2336.
Catterall WA (2014) Sodium channels, inherited epilepsy,
and antiepileptic drugs. Annu Rev Pharmacol Toxicol
54:317-338.
Chiron C, Marchand MC, Tran A et al. (2000) Stiripentol
in severe myoclonic epilepsy in infancy: a randomised
placebo-controlled syndrome-dedicated trial. STICLO
study group. Lancet 356:1638-1642.
Dravet C, Guerrini R (2011) Dravet Syndrome. In: Topics in
Epilepsy series, vol. 3, pp 67-81 Montrouge: John
Libbey Eurotext.
Genton P, Velizarova R, Dravet C (2011) Dravet syndrome:
the long-term outcome. Epilepsia 52 Suppl 2:44-49.
Guerrini R, Dravet C, Genton P et al. (1998b) Lamotrigine
and seizure aggravation in severe myoclonic epilepsy.
Epilepsia 39:508-512.
Horn CS, Ater SB, Hurst DL (1986) Carbamazepine-
exacerbated epilepsy in children and adolescents.
Pediatr Neurol 2:340-345.
Rilstone JJ, Coelho FM, Minassian BA et al. (2012) Dravet
syndrome: seizure control and gait in adults with differ-
ent SCN1A mutations. Epilepsia 53:1421-1428.
Wakai S, Ito N, Sueoka H et al. (1996) Severe myoclonic
epilepsy in infancy and carbamazepine. Eur J Pediatr
155:724.
Wang PJ, Fan PC, Lee WT et al. (1996) Severe myoclonic
epilepsy in infancy: evolution of electroencephalo-
graphic and clinical features. Zhonghua Min Guo Xiao Er
Ke Yi Xue Hui Za Zhi 37:428-432.
Xu X, Zhang Y, Sun H et al. (2013) Early clinical features
and diagnosis of Dravet syndrome in 138 Chinese
patients with SCN1A mutations. Brain Dev. 36(8):676-81.

12 Periodiek voor professionals 12 | nr 3 | 2014
Door: Bobby Koeleman ([email protected]), Marjan van Kempen en Eva Brilstra, afdeling medische
genetica, Universitair Medisch Centrum Utrecht.
Nieuwe genen voor diagnostiek van ernstige epilepsie
Wetenschappelijk onderzoek
Wetenschappelijk onderzoek
Detectie van epilepsie-gen mutatiesGenetisch onderzoek en diagnostiek van monogene
aandoeningen is in de afgelopen jaren sterk veranderd
door de komst van een nieuwe DNA-techniek, genoemd
Next Generation Sequencing (NGS)1. Met deze techniek
kunnen nu met ongekende snelheid en voor relatief lage
kosten alle bekende humane genen simultaan onderzocht
worden op ziekte veroorzakende mutaties. Hierdoor is
de detectie van mutaties in bekende en nieuwe epilepsie-
genen enorm toegenomen, niet alleen in het wetenschap-
pelijk onderzoek, maar ook in de reguliere DNA-diagnos-
tiek bij patiënten met epilepsie.
De impact van NGS op de reguliere genetische diagnos-
tiek is substantieel. De snelle ontdekking van nieuwe
genen die betrokken zijn bij epilepsie en de vertaling van
deze nieuwe kennis naar nieuwe klinische diagnostiek
vormt een uitdaging voor zowel de laboratoriumspecialist
als de klinisch geneticus. In de meeste nieuwe epilepsie-
genen is slechts bij enkele patiënten een mutatie gevon-
den, waardoor een goede correlatie tussen dragerschap
van de mutatie (genotype) en de daarbij passende klini-
sche kenmerken (fenotype) ontbreekt. Nieuwe richtlijnen
voor interpretatie van NGS data en ethische aspecten
daarvan zijn nog in ontwikkeling.
Hier geven wij een kort overzicht van de belangrijkste
nieuwe genen voor ernstige epilepsie die recent zijn
gevonden met NGS en van de gevolgen van implementatie
van NGS voor de genetische diagnostiek.
Onderzoek naar nieuwe epilepsiegenenDe meest gebruikte toepassing van NGS is Whole Exome
Sequencing (WES), waarbij de eiwit coderende DNA
sequenties van alle genen worden bepaald. Het voordeel
is dat de onderzoeker direct kan zoeken naar mogelijke
pathogene variaties en niet eerst op basis van het klinisch
beeld het ziektegen hoeft te kennen; het nadeel is dat in
ieder individu duizenden varianten in duizenden genen
zichtbaar worden. De uitdaging is om pathogene- en neu-
trale varianten van elkaar te onderscheiden. Kennis van
de bekende epilepsiegenen en statistisch bewijs voor het
ziekte veroorzakende karakter van nieuw geïdentificeerde
epilepsie-genen blijft noodzakelijk. Familieonderzoek,
waarbij getest wordt of binnen een familie een pathogene
mutatie overerft met epilepsie, is de klassieke manier om
nieuwe genen te vinden en NGS heeft ook gen-identificatie
in familiaire epilepsie versneld.
Familiaire epilepsieMet WES zijn nieuwe genen gevonden in families met
epilepsie, waarin al meer dan een decennium onderzoek
was gedaan. Een goed voorbeeld is het KCNT1 gen dat
geïdentificeerd werd in drie families met autosomal domi-
nant nocturnal frontal lobe epilepsy (ADNFLE). Opval-
lend is dat er gelijktijdig een publicatie verscheen die ook
rapporteerde dat mutaties in het KCNT1 gen voorkomen bij
patiënten met malignant migrating partial seizures of infancy.
(Barca et al., 2012; Heron et al., 2012).
Dit soort heterogeniteit van het klinische fenotype bij
patiënten met een mutatie in hetzelfde epilepsie-gen is
typerend voor veel epilepsie-genen, en vormt een uitda-
ging voor de klinisch geneticus. Een ander voorbeeld van
de complexe fenotype-genotype correlaties zijn GRIN2A
mutaties in de epilepsie-afasie spectrum stoornis (EAS).
Een aantal studies lieten zien dat GRIN2A mutaties in
families worden overgeërfd door patiënten met verschil-
Door nieuwe DNA-technieken worden nieuwe genen voor ernstige epilepsie in een hoog tempo geïdentificeerd.
Implementatie van deze nieuwe epilepsie-genen in de DNA-diagnostiek heeft er toe geleid dat bij ten minste twee
keer zoveel patiënten een genetische diagnose gesteld kan worden.
1 NGS is inmiddels in de DNA-diagnostiek geïmplementeerd en selecties van bekende en kandidaatgenen (genpanels) zijn sinds 2013
in gebruik (www.umcutrecht.nl/subsite/genome-diagnostics; www.umcg.nl/NL/Zorg/Professionals/Verwijsgids/Klinische_Genetica_
Polikliniek).

Periodiek voor professionals 12 | nr 3 | 2014 13Wetenschappelijk onderzoek
Wetenschappelijk onderzoek
lende klinische EAS fenotypes, waarbij Landau-Kleffner
syndroom (LKS) en/of continuous spike and wave during sleep
(CSWS) het meeste voorkwam (Lesca et al., 2013; Carvill
et al., 2013a; Lemke et al., 2013).
Sporadische gegeneraliseerde epilepsie Een van de eerste NGS-studies gebruikte een selectie van
19 bekende en 46 kandidaatgenen (genpanel) voor mutatie
detectie in 500 patiënten met epileptische encefalopathie
(Carvill et al.,2013b). Daarbij werden CHD2 en SYNGAP1
mutaties als nieuwe oorzaken van epileptische encefalo-
pathie geïdentificeerd. Daarnaast werd bij 9 procent van
de patiënten mogelijk pathogene mutaties gevonden in
bekende genen en kandidaat-genen, waaronder 36 muta-
ties in STXBP1, CDKL5, SCN1A, SCN2A, PCDH19. Opvallend
is dat de klinische verschijnselen van meerdere patiënten
niet binnen het tot dan bekende fenotypisch spectrum
viel dat is geassocieerd met mutaties in deze genen. Zo
werden SCN1A mutaties gevonden bij drie patiënten met
EAS, terwijl tot voor kort deze mutatie alleen bekend was
bij het Dravet syndroom. Wel hadden twee van deze drie
patiënten febriele convulsies. Ten slotte beschrijft de
studie één patiënt met een SCN8A mutatie. Daarvoor was
slechts één andere patiënt beschreven in de literatuur.
Het Amerikaanse Epi4K consortium (www.epgp.org/
epi4k) gebruikte exome sequencing voor mutatie detectie
in 149 patiënten met infantiele spasmen en 115 patiënten
met Lennox Gastaut (Epi4K Consortium,2013). Er werden
329 mogelijk pathogene de novo mutaties gevonden (muta-
ties gevonden in het DNA van de patiënt maar niet in
de niet aangedane ouders, omdat de mutatie in de kiem-
baan van een van de ouders is ontstaan). Een statistische
analyse toonde dat er significant vaker mutaties werden
gevonden in genen waarin neutrale variaties in gezonde
individuen bijna nooit gevonden worden (mutatie intole-
rante genen). Voor twee nieuwe genen was er statistisch
bewijs- GABRB3 en ALG13 mutaties werden gevonden in
respectievelijk vier en twee patiënten. In een aantal andere
genen die in de literatuur reeds zijn beschreven werd
slechts bij één patiënt een mutatie gevonden, waaronder
CHD2 en SCN8A.
Meer bewijs voor deze twee genen werd geleverd door
het Euroepinomics consortium (www.euroepinomics.
org) (Suls et al., 2013). Allereerst werden er drie CHD2
mutaties in een groep van 159 patiënten met epileptische
encefalopathie gevonden en liet een zebravismodel zien
dat een uitschakeling van het gen een epilepsie fenotype
veroorzaakt2. In 16 patiënten zijn SCN8A mutaties gevon-
den waardoor fenotype-genotype correlatie mogelijk is.
Interessant is dat alle SCN8A mutaties suggereren dat het
gemuteerde eiwit tot expressie komt en een abnormale
hogere activiteit heeft (gain of function).
Ten slotte is een lang verwacht kandidaatgen voor
humane epilepsie gevonden, HCN1. In diermodellen
was al gevonden dat de expressie van Hcn1 verlaagd is
bij epilepsie. Daartegenover staat dat verschillende
anti-epileptica juist de expressie van HCN1 verhogen.
Een recente publicatie beschrijft HCN1 mutaties bij zes
patiënten met epileptische encefalopathie (Nava et al.,
2014). Het fenotype van deze patiënten wordt geken-
merkt door initiële koorts-geïnduceerde aanvallen
tussen de vier en dertien maanden, die zich ontwikkelen
naar voornamelijk absences en focale aanvallen. Daar-
naast hebben de patiënten een verstandelijke beperking
en gedragsstoornissen, en is bij vier patiënten autisme,
en één patiënt ADHD gediagnosticeerd.
Inmiddels is het aantal gepubliceerde studies dat gebruik
maakt van NGS explosief gestegen. Een aantal nieuw
ontdekte epilepsie genen wordt hier niet verder bespro-
ken. Het is duidelijk dat mutaties in een groot aantal
verschillende genen epilepsie kunnen veroorzaken. Om
nieuwe genen te identificeren is samenwerking op inter-
nationaal niveau daarom essentieel om genoeg patiënten
te vinden met mutaties in hetzelfde gen. Zulke grotere
series zijn ook uitermate waardevol om het fenotypische
spectrum van deze genen in kaart te brengen voor verder
onderzoek en voor translatie naar de kliniek.
Genetische diagnostiekIn de genetische diagnostiek bij epilepsie heeft NGS
zijn intrede gedaan in de vorm van genpanelanalyse en
WES. Bij genpanelanalyse worden in één test alle be-
kende ziektegenen voor een specifiek epilepsie-fenotype
onderzocht. Zo zijn er genpanels onder andere voor
epileptische encefalopathie, epilepsie in combinatie
met andere paroxysmale neurologische verschijnselen
en voor lokalisatiegebonden epilepsie. Als de epilepsie
moeilijk is te classificeren kan ook analyse van meerdere,
of alle genpanels worden aangevraagd. Onderzoek van
alle genpanels houdt op dit moment in dat in één test
meer dan 130 epilepsiegenen worden onderzocht. Als
in wetenschappelijk onderzoek nieuwe epilepsiegenen
worden geïdentificeerd, worden de genpanels hiermee
aangevuld. Wanneer met genpanelanalyse geen gene-
tische oorzaak wordt gevonden, kan vervolgonderzoek
in de vorm van WES plaatsvinden. Binnen de diagnos-
tiek vindt dit vooral nog zijn toepassing bij epileptische
encefalopathie.
2 http://videos.videopress.com/BZ0R1BJq/chd2_hd.mp4

14 Periodiek voor professionals 12 | nr 3 | 2014 Wetenschappelijk onderzoek
Wetenschappelijk onderzoek
Waar voorheen op geleide van de klinische verschijn-
selen met Sanger sequencing slechts één of enkele genen
werden onderzocht, worden nu per patiënt grote aantal-
len genen en soms alle genen onderzocht. Hiermee zal
vaker en sneller een genetische oorzaak voor epilepsie
kunnen worden opgespoord. Ook leidt deze diagnostiek
tot nieuwe kennis. Soms worden mutaties gevonden in
genen waarvan we niet verwacht hadden dat deze betrok-
ken zouden zijn, waarmee het spectrum van geassoci-
eerde fenotypes zich verbreedt. Ook komt het voor dat
patiënten mutaties hebben in meerdere genen, en moet
de hypothese van een monogenetische oorzaak worden
verlaten.
Ons onderzoekIn ons instituut hebben we recent de meerwaarde van
NGS DNA-diagnostiek in epilepsie onderzocht in een
serie van 280 patiënten die nog geen DNA-diagnose
hadden, en nu met het epilepsie genpanel onderzocht
zijn. Bij de meeste van deze patiënten was al eerder
DNA-onderzoek verricht van de genen die pasten bij
het fenotype. Met deze ‘oude’ DNA-diagnostiek wordt
bij ongeveer 10 procent van de patiënten een klinisch
relevante mutatie gevonden. De resultaten van de nieuwe
genpanelanalyse in deze groep is dus de extra opbrengst
door toepassing van NGS. Bij 40 procent van de patiënten
werden één of meer mogelijk klinisch relevante mutaties
gevonden. Na evaluatie door de klinisch geneticus en la-
boratoriumspecialist werd bij 8 procent van de patiënten
de mutatie geclassificeerd als (waarschijnlijk) verklarend
voor de epilepsie. Bij nog eens 12 procent kan de mutatie
een verklaring zijn, maar moet lopend onderzoek nog
worden afgerond om een definitieve uitspraak te doen.
NGS lijkt dus de opbrengst van DNA-diagnostiek te ver-
dubbelen. Opvallend is dat het merendeel van de nieuw
gedetecteerde mutaties gevonden is in de recent ontdekte
genen zoals die hierboven beschreven zijn. De verwach-
ting is dus dat de opbrengst zal blijven groeien bij het
voortschrijden van het onderzoek naar de genetische
oorzaken van epilepsie.
Tot slotHet is van belang om patiënten en ouders voorafgaand
aan de test goed te informeren over de mogelijke uit-
komsten van het onderzoek. Vooral dient vermeld te
worden dat vaak varianten worden aangetoond waarvan
de klinische relevantie onduidelijk is, en bij WES is er
daarnaast de mogelijkheid van ‘niet-gezochte bevindin-
gen’. Hierbij gaat het om de aanleg voor een genetische
aandoening die losstaat van de epilepsie, maar die wel
belangrijke implicaties kan hebben voor de gezondheid
van de patiënt en zijn of haar familie. Om varianten met
onduidelijke klinische relevantie zo goed mogelijk te
kunnen interpreteren moet de laboratoriumspecialist
over klinische gegevens van de patiënt kunnen beschik-
ken. Soms is het nodig om na de testuitslag nog aanvul-
lende onderzoeken te verrichten, of ook familieleden
te onderzoeken. De dialoog en samenwerking tussen
(kinder)neuroloog, kinderarts, klinisch geneticus en
laboratoriumspecialist is in het NGS-tijdperk dan ook
van groot belang.
ReferentiesBarcia G, Fleming MR, Deligniere A et al. (2012) De novo
gain-of-function KCNT1 channel mutations cause
malignant migrating partial seizures of infancy. Nat
Genet. 44(11):1255-9.
Carvill GL, Regan BM, Yendle SC et al. (2013a) GRIN2A
mutations cause epilepsy-aphasia spectrum disorders.
Nat Genet. 45(9):1073-6.
Carvill GL, Heavin SB, Yendle SC et al. (2013b) Targeted
resequencing in epileptic encephalopathies identifies
de novo mutations in CHD2 and SYNGAP1. Nat Genet.
45(7):825-30.
Epi4K Consortium; Epilepsy Phenome/Genome Project.
(2013) De novo mutations in epileptic encephalopa-
thies. Nature. 501(7466):217-21.
Heron SE, Smith KR, Bahlo M et al. (2012) Missense
mutations in the sodium-gated potassium channel gene
KCNT1 cause severe autosomal dominant nocturnal
frontal lobe epilepsy. Nat Genet. 44(11):1188-90.
Lemke JR, Lal D, Reinthaler EM et al. (2013) Mutations in
GRIN2A cause idiopathic focal epilepsy with rolandic
spikes. Nat Genet. 45(9):1067-72.
Lesca G, Rudolf G, Bruneau N et al. (2013) GRIN2A
mutations in acquired epileptic aphasia and related
childhood focal epilepsies and encephalopathies
with speech and language dysfunction. Nat Genet.
45(9):1061-6.
Nava C, Dalle C, Rastetter A et al. (2014) EuroEPINOMICS
RES Consortium, Haaf T, LeGuern E, Depienne. De
novo mutations in HCN1 cause early infantile epileptic
encephalopathy. Nat Genet. 46(6):640-5.
Suls A, Jaehn JA, Kecskés A (2013) EuroEPINOMICS RES
Consortium. De novo loss-of-function mutations in
CHD2 cause a fever-sensitive myoclonic epileptic
encephalopathy sharing features with Dravet syndrome.
Am J Hum Genet. 93(5):967-75.

Periodiek voor professionals 12 | nr 3 | 2014 15
Door: Paul Eling ([email protected]), Donders Center for Brain, Cogition and Behaviour, Radboud Universiteit
Nijmegen.
Hoe groot is de schade?Over de rapportage van de mediaal-temporale lobectomie bij HM
Historische wetenswaardigheden
Historische wetenswaardigheden
HM overleed in 2008 en volgens afspraak kwamen zijn hersenen beschikbaar voor pathologisch-anatomisch onder-
zoek. Recent verscheen het verslag van een eerste studie over de omvang van de beschadiging van de hippocampus,
die werd aangericht door de mediaal-temporale lobectomie bij HM. De bevindingen worden vergeleken met eerdere
beschrijvingen.
Op 2 december 2008 overleed HM. Vanaf zijn tiende jaar
leed hij aan epileptische aanvallen. Omdat de ernst van de
aanvallen de kwaliteit van leven te zeer verstoorden stelde
de neurochirurg William Bleecher Scoville (1906-1984) in
1953 voor om in te grijpen; HM was toen 27. De ingreep
had dramatische gevolgen: HM’s geheugen was hierdoor
ernstig aangetast. HM bleef een vriendelijke persoon,
die altijd bereid was aan onderzoek mee te werken en hij
heeft ook zijn lichaam ter beschikking gesteld voor weten-
schappelijk onderzoek. De Amerikaanse neuropsychologe
Suzanne Corkin begeleidde HM in de vele decennia dat hij
werd onderzocht. Zij schreef een prachtige biografie over
HM (Corkin, 2013), waarin ook gedetailleerd wordt inge-
gaan op de talloze studies over de diverse aspecten van het
geheugen van HM.
Wat er aan vooraf gingScoville had ruime ervaring met lobotomie bij mensen
met psychiatrische aandoeningen, vooral bij psychoses.
Gebleken was dat een volledige lobotomie tot een te grote
afstomping van de persoonlijkheid leidde en daarom
onderzocht Scoville al vanaf 1949 het effect van een frac-
tionele lobotomie. Undercutting, aan beide zijden, van het
orbitale oppervlak zou een ideale oplossing bieden voor
deze patiënten (Scoville et al., 1951). Met deze techniek
werd getracht de gyrus anterior cingulatum en de pos-
terieure orbitale cortex te isoleren. In 1954 presenteerde
Scoville een overzicht van resultaten bij zo’n 230 patiën-
ten met deze undercutting techniek en met wat hij noemt
mediaal- temporale lobectomie. Deze lobectomie had slechts
geringe gevolgen op het mentaal functioneren. Echter,
bij twee patiënten zou hij bilateraal een groter deel van de
mediale temporaalkwab weggenomen hebben, over een
lengte van 8-9 cm in plaats van de meer gebruikelijke 5 cm
en bij hen was een ernstige geheugenstoornis opgetreden
voor recent memory. HM werd bekend; de andere patiënt
was D.C., een psychotische man, van wie later niets meer
is vernomen.
Bilaterale mediale temporele lobectomieScoville (1954) presenteerde die gegevens onder de titel:
The limbic lobe in man, en verwees naar het ‘monumentale’
werk van Paul McLean. McLean had in 1952 voorgesteld
om ondermeer de orbitale frontaalschors en het voorste
deel van de temporaalkwab aan het circuit van Papez toe te
voegen en vormde zo een uitgebreider limbisch systeem,
betrokken bij emotie. Scoville stelde toen ook de vraag of
een bilaterale mediale temporele lobectomie een effectieve
behandeling van epilepsie zou kunnen zijn, omdat die
vooral zou ontstaan in de uncus. Hij had die operatie toen
al bij HM uitgevoerd!
Bij de ingreep bij HM boorde Scoville twee gaatjes met een
doorsnee van 3,8 cm net boven de ogen en verwijderde
langs die weg delen van de mediale temporaalkwab. Hij
wilde een symmetrische resectie verrichten van de mediale
temporaalkwab, van het midden van de punt van de tempo-
raalkwab tot zo’n 8 cm verder. Met een dergelijke ingreep
zou hij de uncus, amygdala en het hippocampale complex
weghalen, inclusief de parahippocampale gyrus. Hij had
na de operatie een verslag gemaakt met tekeningen van het
weefsel dat hij had weggehaald. Tijdens de operatie was
met behulp van acute elektrocorticografie geen afwijking
vastgesteld en ook in het weggehaalde weefsel werden
geen tekenen van pathologie gevonden.
De techniek van de mediale temporele lobectomie kwam
dus voort uit de psychochirurgie en werd bij HM gebruikt
om ondermeer het epilepsiegevoelige gebied, de uncus
en de hippocampus, te verwijderen. Er zijn nog minstens
twee andere patiënten bij wie een soortelijke ingreep was
uitgevoerd en die er ook een ernstige geheugenstoornis

16 Periodiek voor professionals 12 | nr 3 | 2014 Historische wetenswaardigheden
Historische wetenswaardigheden
aan overhielden. Dit waren patiënten van Penfield (Milner
& Penfield, 1955). Penfield meende dat de geheugenstoor-
nis bij deze patiënten was ontstaan doordat er weliswaar
een unilaterale resectie had plaatsgevonden maar dat de
contralaterale temporaalkwab al was aangetast, hetgeen
later postmortem onderzoek bevestigde (Penfield & Ma-
thieson, 1974).
‘Imaging’: omvang van de resectieIn 1984 beschreef Corkin (Corkin, 1984) de langetermijn
gevolgen van de ingreep, vooral de neuropsychologische
effecten. Op basis van de CT-scan die was gemaakt kon
over de omvang van de resectie niets gezegd worden.
Daarom werden in mei 1992 en augustus 1993 MRI-scans
gemaakt. Het vergde heel wat detectivewerk om na te gaan
of HM met de clips, waarmee tijdens de ingreep in 1953
bloedvaten waren afgeklemd, de scanner in kon (Corkin et
al., 1997). De resectie bleek minder uitgebreid dan Scoville
had gedacht: 5,4 cm links en 5,1 cm rechts en omvatte:
de schors van de mediale temporale pool, het grootste
deel van het amygdala complex en de entorhinale schors,
en ongeveer de helft van het rostro-caudale deel van het
intraventriculaire deel van de hippocampus formatie
(gyrus dentatus, hippocampus en subiculum). Delen van
de ventrale perirhinale schors waren gespaard gebleven.
Corkin et al. (1997) meenden dat het resterende deel van
de hippocampus nauwelijks meer verbindingen had en
daardoor niet meer functioneel zou zijn.
In de periode van 2002 tot 2004 werden gedurende
vier sessies MRI-scans gemaakt (Salat et al., 2006). Op
de leeftijd van HM zou men een hippocampus volume
verwachten van 3.3 ± 0,4 cm3. maar bij HM bleek er links
0,65 en rechts 0,88 cm3 over te zijn. Op die leeftijd zou
men een amygdala verwachten van ongeveer 1,7 cm3,
maar bij HM was links nog 0,2 en rechts 0,3 cm3 over. De
resterende delen van de hippocampus waren sedert 1993
verder geatrofiëerd. Men zag enige aan de leeftijd gerela-
teerde veranderingen, maar opvallend waren de witte stof
afwijkingen vooral in de frontale en parietale gebieden,
die van recente datum moeten zijn geweest en pasten bij
zijn medische conditie.
Post mortemNa zijn dood zijn de hersenen van HM zo snel mogelijk uit
de schedel verwijderd en ingevroren. De gang van zaken en
het vervoer waren tot in detail gepland en staan uitvoerig
beschreven in Corkin (2013). Jacopo Annese stond aan het
hoofd van de hele ‘operatie’ en publiceerde de eerste resul-
taten van de postmortemanalyse (Annese et al., 2014). De
lengte van de resectie bleek links 54,5 mm te zijn en rechts
44 mm. De lengte van het gespaarde deel van de hippo-
campus was 23,6 mm links en 24,3 mm rechts. Bij de
3D-reconstructie van de hersenen bleek dat de hippocampus
wat steiler omhoog liep dan gewoonlijk. Nu het volume
van het gespaarde deel van de hippocampus kon worden
gemeten, bleek dat het intacte deel zelfs nog iets groter was.
Opvallend was dat neuropathologische kenmerken die men
kan verwachten als gevolg van epilepsie niet gevonden wer-
den in het resterende deel van de hippocampus. Ook opval-
lend was de integriteit van neuronale cellen in het CA4 deel
van het resterende deel van de hippocampus. De vraag is nu
welke functionele betekenis dit heeft gehad, gezien de afwe-
zigheid van verbindingen met de rest van de hippocampus.
VergelijkingScoville merkte in 1954 in een voetnoot op dat bij twee
patiënten (D.C.en HM) beiderzijds ongeveer 8 à 9 cm was
weggehaald met een ernstig geheugenverlies tot gevolg. Hij
schreef dat het gehele complex met de hippocampale gyrus
weggehaald was, terwijl hij in 1957 schreef dat hij het voor-
ste tweederde deel van het complex zou hebben weggehaald.
Het valt op dat hij nauwelijks aangaf waarom hij een groter
gebied weghaalde. Het enige argument leek te zijn dat de
uncus en het hippocampale complex epileptogeen konden
zijn. Dat de ingreep zo dramatisch uitpakte leidde ook niet
tot een discussie over deze andere aanpak. Wel beweerde
Scoville dat bij een kleinere laesie ook geheugenproblemen
konden optreden en dat er geen duidelijke correlatie was
tussen omvang van de laesie en ernst van het geheugen-
probleem.
Corkin et al. (1997) constateerden dat de lengte van het deel
van de punt van de temporale pool tot het caudale einde van
de hippocampale formatie bij HM ongeveer 7 cm bedroeg
en dat als Scoville inderdaad 8 cm had weggehaald, hij ook
een deel van de calcarine schors zou hebben weggehaald.
De schattingen van de laesie van Corkin et al. (1997) worden
min of meer bevestigd door de latere studies. Hoewel dus
nog een deel van de hippocampus was blijven zitten na de
operatie, kan nauwelijks meer worden vastgesteld wat de be-
tekenis hiervan is. De functionele status van het resterende
weefsel is onduidelijk. Wel lijkt het cognitieve beeld bij HM
opvallend stabiel over de jaren, met alleen in de laatste paar
jaar een lichte achteruitgang. Vanuit die gegevens zijn er
geen aanwijzingen dat dat weefsel aanvankelijk functioneel
actief was en na verloop van tijd zijn functie verloor.
Tot slotHet valt op dat Scoville in de regel de ingreep bilateraal uit-
voerde zonder expliciet aan te geven waarom dat van belang
was. Bij hem ging het meestal om psychiatrische patiënten.
Penfield voerde ook unilaterale resecties uit. In een enkel ge-
val pakte het toch dramatisch uit omdat de andere hemisfeer
al aangetast was. Penfield vond dit een cruciaal punt en ging

Periodiek voor professionals 12 | nr 3 | 2014 17Historische wetenswaardigheden
Historische wetenswaardigheden
daarom in 1974 uitvoerig in op dit punt en hij en Scoville
waren het er over eens dat de geheugenstoornis alleen op-
trad bij bilaterale resecties. Penfield gaf twee verklaringen
voor de rol van de hippocampus bij het geheugen: daar
zou de recording of the stream of consciousness plaatsvinden, of
die registratie zou elders in de hersenen plaats vinden en
vanuit de hippocampus geactiveerd worden. Aanvankelijk
geloofde hij de eerste maar na verloop van tijd hechtte hij
meer geloof aan de tweede verklaring. Maar een overtui-
gende discussie over het cruciale verschil tussen een unila-
terale en een bilaterale ingreep leverde dit ook niet op.
ReferentieAnnese JS, Schenker-Ahmed NM, Bartsch H et al. (2014)
Postmortem examination of patient H.M.’s brain based
on histological sectioning and digital 3D reconstruc-
tion. Nature Communications, 5:3122, 1-9.
Corkin S. (1984) Lasting consequences of bilateral medial
temporal lobectomy: Clinical course and experimental
findings in H.M. Seminars in Neurology, (4):249-259.
Corkin S., Amaral DG, Gonzalez RG, Johnson KA,
Hyman BT (1997) H.M.’s medial temporal lobe lesion:
Findings from magnetic resonance imaging. Journal of
Neuroscience, 17:3964–3979.
Corkin S (2013) Permanent tegenwoordige tijd. Het onver-
getelijke leven van de man die zijn geheugen verloor.
Amsterdam: Prometheus.
Milner B & Penfield W (1955) The effect of hippocampal
lesions on recent memory. Transactions of the American
Neurological Association, 80:42– 48.
Penfield W & Mathieson GM (1974) Memory: autopsy
findings and comments on the role of hippocampus in
experiential recall. Archives of Neurology, 31:145–154.
Salat DH, van der Kouwe AJW, Tuch DS et al. (2006)
Neuroimaging H.M.: A 10-Year Follow-Up Examination.
Hippocampus, 16:936–945.
Scoville WB & Milner B (1957) Loss of recent memory after
bilateral hippocampal lesions. Journal of Neurology,
Neurosurgery and Psychiatry, 20:11–21.
Scoville WB (1954) The limbic lobe in man. Journal of
Neurosurgery, 11:64–66.
Scoville WB, Wilk EK & Pepe AJ (1951) Selective cortical
undercutting. Results in New Method of Fractional
Lobotomy. American Journal of Psychiatry, 107:730-738.
Subsidies voor epilepsieonderzoek in 2016Het Epilepsiefonds wil wetenschappelijk onderzoek stimuleren en stelt daarom subsidies beschikbaar voor onderzoeksprojecten over epilepsie en de behandeling/bestrijding daarvan.
Uitgangspunt is dat de instelling waarbij de onderzoeker werkt een belangrijke inbreng heeft in het te subsidiëren onderzoek. De subsidie heeft in principe een aanvullend karakter. Belangrijke beoordelingscriteria zijn kwaliteit van het onderzoek en klinische en maatschappelijk relevantie. Projecten mogen in principe de duur van vier jaar niet overschrijden. De subsidie bedraagt maximaal € 203.000,–. Bij een combinatie van een specialistenopleiding en wetenschappelijk onderzoek behoort een langere duur van het project, binnen eenzelfde budget, tot de moge-lijkheden.
Het is tot en met 15 januari 2015 mogelijk om subsidieaanvragen in te dienen voor onderzoeken die in 2016 beginnen.
In maart 2015 wordt uit de ontvangen subsidieaanvragen de eerste selectie gemaakt door de Wetenschappelijke Adviesraad van het Epilepsiefonds. In juni 2015 wordt een besloten hoorzitting gehouden. Subsidieaanvragers die door de eerste selectieronde heen zijn, worden voor deze hoorzitting uitgenodigd om hun onderzoeksvoorstel nader toe te lichten aan de Wetenschappelijke Adviesraad.
Een aanvraag indienen?Subsidieaanvraagformulieren en meer informatie kunt u vinden op www.epilepsiefonds.nl, kijk bij ‘Wat doen wij?’ en vervolgens bij ‘Wetenschappelijk onderzoek’.
EpilepsiefondsSecretariaat Wetenschappelijke AdviesraadPostbus 270, 3990 GB HOUTENTelefoon: 030 634 40 63E-mail: [email protected]

18 Periodiek voor professionals 12 | nr 3 | 2014
In 2010 is door de Commission on Classification and Termino-
logy van de International League Against Epilepsy (ILAE) een
nieuw classificatiesysteem voorgesteld ter vervanging van
de aanvalsclassificatie uit 1981 en de epilepsieclassificatie
uit 1989 (Berg et al., 2010). Voor een overzicht van de in-
deling wordt verwezen naar de Epilepsie richtlijn van de
Nederlandse Vereniging voor Neurologie (NVN) (http://
epilepsie.neurologie.nl). De commissie van de ILAE geeft
aan dat de nieuwe classificatie een dynamisch classificatie
is die kan worden aangepast bij nieuwe inzichten.
Indeling van aanvallenAanvallen worden ingedeeld in drie groepen: focaal,
gegeneraliseerd en onbekend. Focaal wil zeggen dat de
aanvalsactiviteit beperkt blijft tot een netwerk dat één
hemisfeer betreft (figuur 1). Dit kan zeer lokaal zijn of
meer uitgebreid binnen één hemisfeer. Gegeneraliseerde
aanvallen spelen zich af in een netwerk dat verdeeld is
over twee hemisferen. Epileptic spasms, hier vertaald door
salaamkrampen, vallen als enige type aanval in de groep
‘onbekend’. Focale aanvallen worden beschreven op basis
van de verschijnselen zoals subjectieve sensorische of psy-
chische gevoelens (voorheen aura genoemd), autonome
en motorische symptomen en veranderd bewustzijn. Bij de
nieuwe classificatie vervallen termen als eenvoudig partieel
en complex partieel. Een focale aanval kan overgaan in
een gegeneraliseerd tonisch-clonisch insult en wordt dan
een ‘focale aanval overgaand in een bilaterale convulsieve
aanval’ genoemd in plaats van een secundair gegenera-
liseerd insult, zoals in het oude classificatiesysteem. De
In deze bijdrage worden de door de ‘International League Against Epilepsy’ voorgestelde epilepsieclassificatie syste-
matiek en de bezwaren hiertegen besproken. Wetenschappelijk onderzoek van Nederlandse origine steunt het stand-
punt van de Epilepsie richtlijn commissie van de Nederlandse Vereniging voor Neurologie dat de nieuwe classificatie
niet veel beter is dan de oude en daarom vooralsnog niet wordt overgenomen.
De nieuwe aanvals- en epilepsie-classificatie is niet beter dan de oude
Door: Paul Augustijn ([email protected]), neurologie, Stichting Epilepsie Instellingen Nederland, Heemstede.
Verantwoorde epilepsiezorg
Verantwoorde epilepsiezorg
Figuur 1 Schematische weergave van het concept ‘ focale aanval’ in het nieuwe epilepsieclassificatie systeem.
*Onstaan in en beperkt tot één hemisfeer
Focale aanval* Onbekend
D.W.Z. onvoldoende bewijs voor focaal
of gegeneraliseerd karakter
- Salaamkrampen
- Andere
Gekenmerkt door een of meer verschijnselen:
- subjectieve sensorische/psychische
- motorisch
- autonoom
- veranderd/verminderd bewustzijn
Overgaand in bilaterale convulsieve aanval

Casuïstiek
Periodiek voor professionals 12 | nr 3 | 2014 19Verantwoorde epilepsiezorg
en ‘farmaca-responsief ’ (vertaling van de auteur).
De plaats van de nieuwe classificatie Er is vanuit diverse invalshoeken en diverse landen kritiek
geuit op de nieuwe classificatie. De Nederlands Epilepsie
richtlijn commissie adviseert het gebruik van de nieuwe
classificatie (nog) niet. Dit in navolging van onder andere de
Engelse (NICE) Epilepsie richtlijn commissie. De bezwaren
betreffen vooral de indeling van de epilepsie (-syndromen)
en van de etiologie, Epilepsiesyndromen kunnen makkelijk
overlappen met de overige epilepsie. Een voorbeeld hiervan
is een patiënt met een CSWS- (continuous spike and wave during
sleep) syndroom bij heterotopieën. De epilepsie kan geclas-
sificeerd worden als ‘epileptische encefalopathie met conti-
nue piekgolven tijdens slaap’, een epilepsiesyndroom van de
kinderleeftijd, maar ook als ‘epilepsie die wordt toegeschre-
ven aan malformaties van de corticale ontwikkeling’. Ook de
etiologie indeling is in deze onduidelijk: heeft de epilepsie
bij deze patiënt een genetische oorzaak of een structureel/
metabole oorzaak? Steeds vaker worden patiënten met epi-
lepsie ten gevolge van een (chronische) niet-paraneoplasti-
sche limbische encefalitis, bijvoorbeeld gepaard gaand met
anti VGKC antistoffen, gediagnosticeerd. Er is al voorgesteld
om naast de categorieën genetisch, structureel/metabool en
onbekend als vierde de categorie ‘auto-immuun’ in te voe-
ren. Ook is er kritiek op de indeling van de epilepsiesyndro-
men. Deze indeling is niet veel verschillend van de vorige,
er zijn geen in- en exclusie criteria geformuleerd en ze zijn
niet aangepast aan nieuwe inzichten. Zo worden nog de
diagnoses ‘syndroom van Dravet’ en ‘febrile seizures plus’
gebruikt, die overlappen en waarbij het al dan niet aanwezig
zijn van mutaties niet als criterium wordt gebruikt.
indeling van de gegeneraliseerde aanvallen is wat beperkt
in vergelijking met het oude systeem, vooral wat betreft de
verschillende types absences. De facto bestaat gegenerali-
seerde epilepsie niet meer.
Indeling van epilepsieEpilepsie wordt ingedeeld in elektroklinische syndromen
en vier andere groepen van epilepsie. De syndromen wor-
den ingedeeld op basis van de gangbare debuutleeftijd
(neonataal, baby-peuter, kinder-, adolescentie-volwassen
en variabele leeftijd) en weerspiegelen niet de etiologie.
De niet-syndromale epilepsieën bestaan uit:
1. Epilepsie die wordt toegeschreven en veroorzaakt door
structurele of metabole oorzaken (bijvoobeeld malfor-
maties van de corticale ontwikkeling zoals heterotopie).
2. Syndromen met een karakteristieke constellatie
(bijvoorbeeld mesiale temporaalkwab epilepsie met
hippocampus sclerose).
3. Epilepsie door onbekende oorzaak.
4. Een groep die situaties beschrijft waarbij epileptische
aanvallen optreden zonder dat er sprake is van epilep-
sie, zoals bijvoorbeeld bij koortsstuipen.
De termen gegeneraliseerde epilepsie en lokalisatiegebon-
den epilepsie komen dus te vervallen in deze classificatie.
De etiologie van niet-syndromale epilepsie bestaat in de
nieuwe indeling uit drie categorieën: genetisch, structu-
reel/metabool en onbekend. Daarmee komen de termen
Idiopathisch, symptomatisch en cryptogeen (vermoedelijk
symptomatisch) te vervallen. De commissie stelt voor om
ook termen als benigne, maligne of catastrofaal niet meer
te gebruiken. Daarvoor in de plaats komen ‘zelflimiterend’
Verantwoorde epilepsiezorg
Figuur 2 Schematische weergave van de indeling van gegeneraliseerde aanvallen in het nieuwe epilepsieclassificatie systeem.
*Onstaan in en snel uitbreidend binnen een bilateraal verdeeld netwerk
Tonisch-clonisch
typisch
Absence
Absences met speciale
verschijnselen
- Myoclonische absences
- Ooglid myoclonus
Clonisch Tonisch
atypisch
Atoon Myoclonus
- myoclonus
- myocloon-atoon
- myocloon-tonisch
Gegeneraliseerde aanvallen*

20 Periodiek voor professionals 12 | nr 3 | 2014 Verantwoorde epilepsiezorg
Verantwoorde epilepsiezorg
Vergelijking classificatiesystemenBeide classificatiesystemen, het oude uit 1989 en het
nieuwe, zijn vergeleken op bruikbaarheid door Van
Campen et al. (2013). De vergelijking vond plaats door de
interobserver agreement van beide systemen te berekenen op
basis van een groep van 80 kinderen met 165 aanvallen.
In het kort samengevat concludeerde men dat het oude
en nieuwe systeem vergelijkbaar zijn, de nieuwe etiologie
indeling leidde tot veel overeenstemming, maar niet meer
dan de oude, er was een betere overeenstemming bij de
nieuwe epilepsiesyndroom classificatie dan bij de oude
maar epilepsie blijft frequent niet te classificeren. Bij de
indeling van aanvallen was er onvoldoende overeenstem-
ming met betrekking tot de diagnose epileptic spasms.
Tot slotIn conclusie: er zijn op dit moment onvoldoende
argumenten om de nieuw voorgestelde classificatie
te propageren.
ReferentiesBerg AT, Berkovic SF, Brodie MB, et al. (2010) Revised
terminology and concepts for organization of seizures
and epilepsies: Report of the ILAE Commission on
Classification and Terminology, Epilepsia, 51(4):676–685.
Van Campen JS, Jansen FE, Brouwer OF et al. (2013)
Interobserver agreement of the old and the newly
proposed ILAE epilepsy classification in children.
Epilepsia 54(4):726–732.
Door: Frans Leijten, ([email protected]), neurologie, Brain Center Rudolf Magnus, Universitair Medisch
Centrum Utrecht.
EEG diagnostiek bij epilepsie: is een richtlijn zinvol?Het zal geen neuroloog zijn ontgaan dat er een nieuwe Richtlijn Epilepsie is. De lezer van de richtlijn zal opvallen
dat de wetenschappelijke onderbouwing van de meeste aanbevelingen matig is. Heeft het zin om deze richtlijn te
raadplegen over zoiets eenvoudigs als diagnostiek, terwijl het aanvragen van een EEG en MRI bij verdenking op
epilepsie voor de hand ligt? Zelfs de classificatie van epilepsie is hetzelfde gebleven in deze richtlijn.
De nieuwe richtlijn voor epilepsie (http://epilepsie.neuro-
logie.nl/cmssite/index.php) is praktisch en vernieuwend
– door haar toegankelijkheid op internet en doordat zij
halfjaarlijks wordt vernieuwd wanneer er nieuwe weten-
schappelijke informatie binnenkomt, vooral als deze aan
strenge evidence based criteria voldoet. Als het om EEG
diagnostiek gaat, is de richtlijn vooral een verzameling
wijsheden uit de praktijk, bekend onder de naam expert
opinion. Maar eigenlijk is dat zo gek nog niet. Het leren
van het lezen van het EEG is gebaseerd op ervaringen in
de praktijk en leerboeken, zoals het nieuwe Nederlandse
Leerboek Klinische Neurofysiologie (Zwarts et al., 2014).
De richtlijn is als het om EEG gaat vooral een verzameling
uitspraken die de nadruk leggen op handelingen die in de
praktijk het vaakst misgaan.
Valkuilen bij de beoordeling van een EEGIn het Wilhelmina Kinderziekenhuis met het speerpunt
kinderepilepsiechirurgie worden per jaar enkele hon-
derden second opinions gedaan, waar de EEG’s worden
herbeoordeeld die elders zijn gemaakt. Het is interes-
sant om te zien waar verschillen in zitten, hoe het komt
dat deskundigen verschillend oordelen, en of de nieuwe
richtlijn daarbij helpt. Een paar voorbeelden:
1. Interpretatie van fysiologische EEG verschijnselen
als epileptiform. Er zijn enkele bekende valkuilen
zoals reeksen scherpe golven temporaal bij volwas-
senen (rhythmic temporal bursts of theta, wicket spikes
of psychomotor variants, zoals deze worden genoemd)
en K-complexen die soms piekgolfcomplexen lijken.
Overinterpretatie leidt tot een onterechte diagnose
van temporale of gegeneraliseerde epilepsie.
2. Overwaardering van artefacten. Vaak is al eerder in
dezelfde registratie een probleem te zien op hetzelfde
kanaal, maar dan lijkt het kanaal zich spontaan te
herstellen en verderop zijn pieken te zien.

Periodiek voor professionals 12 | nr 3 | 2014 21Verantwoorde epilepsiezorg
Verantwoorde epilepsiezorg
3. Niet goed kijken naar video. Bij piekgolven in de cen-
trale windingen zijn focale myoclonieën te verwachten.
Op basis van de lokalisatie van de pieken is zelfs te
voorspellen of dit in gelaat (T8C4), hand (C4), arm of
been (C4Cz), of voet (CzPz) zal zijn en aan welke zijde.
Als er sprake is van een schokje na zo’n piek, is deze
piekgolf geen interictaal verschijnsel, maar ictaal.
4. Focale of gegeneraliseerde epilepsie. Schijnbaar gege-
neraliseerde ontladingen kunnen consequent focaal
beginnen en dat kan zichtbaar worden door de tijds-
schaal aan te passen. Bij zuigelingen kunnen de eerste
EEGs nog focale verschijnselen vertonen die in latere
EEGs niet meer herkenbaar zijn.
5. Syndroomherkenning. Moeilijk bij zeldzame kinder-
epilepsieën zoals bijvoorbeeld myoclonisch-astatische
epilepsie.
Herbeoordeling van een EEG blijkt minstens zoveel op te
leveren als het herhalen ervan. Dat geldt niet alleen voor
EEG, maar ook voor MRI en PET. De meeropbrengst is in
al deze gevallen 10-15 procent als het om de vraagstelling
epilepsiechirurgie gaat. De redenen hiervoor laten zich
raden. Bij revisie moet de vraagstelling scherp worden
geformuleerd, en dat levert een betere werkhypothese op.
Dit leidt tot een hogere sensitiviteit. Bij patiënten die we
willen reviseren is de a priori kans groter dat er iets mis
is. Het gevoel dat je bij deze patiënt zeker iets niet wilt
missen, is een uitstekende reden om nog eens te (laten)
kijken. De ironie wil dat ook wanneer je zelf de herbeoor-
deling doet, er een meeropbrengst is!
Wat zegt de richtlijn?Staan bovenstaande vijf valkuilen bij interpretatie van een
EEG ook in de nieuwe richtlijn? Nee. De richtlijn is geen
leerboek. Wat er wel in staat is dat:
1. Je alleen een EEG moet doen bij reële verdenking op
epilepsie (en niet bijvoorbeeld als syncope ver boven-
aan staat in de differentiaal diagnostiek). Het adagium
‘baat het niet, dan schaadt het niet’ geldt niet in de
geneeskunde.
2. De sensitiviteit en specificiteit van het EEG bij de
diagnose epilepsie erg variëren in de literatuur. Het kan
heel veel uitmaken wie het EEG beoordeelt. De richtlijn
geeft als advies om bij twijfel over een EEG een herbe-
oordeling van het EEG te overwegen.
De eenvoud van deze adviezen doet bedrieglijk aan. Maar
net als aforismen zou je er motto’s van kunnen maken en
ze overpeinzen met de routine van alledag in het achter-
hoofd.
De richtlijnencommissie geeft ook adviezen op basis van
recente publicaties die interessant lijken voor toepassing
in de dagelijkse praktijk. Ik geef twee voorbeelden: het
geven van melatonine voorafgaand aan een EEG bij een
kind, zodat slaapdeprivatie niet nodig is. Dit advies komt
uit de Engelse NICE richtlijnen. Wilt u meer weten,
dan vindt u de gerelateerde publicatie in de Richtlijn
Epilepsie of op de website van NICE (http://pathways.
nice.org.uk/pathways/epilepsy). Een ander advies geldt
voor kinderen of jong volwassenen met een eerste (voor
epilepsie verdachte) aanval: maak het EEG binnen 24 uur
na deze aanval. De opbrengst blijkt veel hoger te zijn
dan een EEG twee weken later. De richtlijn bevat zo
voor iedereen wetenswaardigheden en is het lezen en
herlezen waard.
StandaardisatieNaast de richtlijn is er een Europees initiatief om te
komen tot uniformering van EEG verslagen, SCORE
geheten van de Holberg groep (https://www.holbergeeg.
com). Dit gaat verder dan gelijke woordkeuze. Wanneer
we overal dezelfde terminologie en opbouw van het
EEG verslag hanteren, wordt het interpreteren van EEG
verslagen voor iedereen gemakkelijker. Er is een kleine
groep binnen de Nederlandse Vereniging voor Neuro-
logie bezig met het voor Nederland geschikt maken van
dit systeem. Het moet uiteindelijk geïmplementeerd
kunnen worden op alle EEG software pakketten. Voorts
is er het E-pilepsy project dat met subsidie van de Euro-
pese Gemeenschap een internetforum heeft opgericht
(http://www.e-pilepsy.eu/about/partners). Het doel is
om allerlei postprocessing op EEGs (en andere vormen van
neuroimaging bij epilepsie) mogelijk te maken. Vele centra
in Europa met speciale expertise brengen hun kennis
daarin samen.
Tot slotZo verandert het EEG mee met de tijdgeest. Het zal niet
lang duren of het wordt eenvoudig om via het internet
iemand op afstand ‘even mee te laten kijken’ naar een
twijfelachtig EEG verschijnsel. Het aanleggen van grote,
multinationale EEG databases gegroepeerd naar ver-
schijnsel of naar ziektebeeld is begonnen. Dit zal ons
alert maken op nieuwe EEG patronen en hun betekenis,
de EEG evolutie zichtbaar maken van bepaalde ziekte-
beelden, of de basis vormen van nieuwe fenotypering.
En het wetenschappelijk onderzoek met modellering
van EEG of netwerkanalyse zal op grote schaal kunnen
worden toegepast en voor iedereen bereikbaar worden.
Als we het EEG beter begrijpen, zal dit ons inzicht in
epilepsie vergroten.
ReferentiesZwarts M, Van Dijk G, Van Putten M et al. (2014)
Leerboek Klinische Neurofysiologie. Bohn, Stafleu
en van Loghum, Houten.

Casuïstiek
22 Periodiek voor professionals 12 | nr 3 | 2014 Proefschriftbesprekingen
Proefschriftbesprekingen
Rolandische epilepsie (RE), één van de meest voorko-
mende kinderepilepsiën, wordt gekarakteriseerd door
centro-temporale ontladingen in het EEG en relatief milde
aanvallen, die vaak ’s nachts plaatsvinden. RE is een idio-
pathische aandoening en gaat vaak gepaard met cogni-
tieve (taal) problemen die mogelijk ernstiger zijn dan de
aanvallen zelf vanwege de impact op het dagelijks leven.
RE dankt zijn naam aan het feit dat de epileptische haard
zich in het sensor-motorische gedeelte van de hersenen
bevindt, ook wel Rolandische cortex genaamd. Frappant is
dat, hoewel de epileptische haard buiten de taalgebieden
ligt, kinderen met RE toch vaak taalproblemen ondervin-
den. Het is momenteel onbekend hoe de epileptiforme
activiteit vanuit de rolandische strip niet alleen spraak
(een motor functie) maar ook andere aspecten van taal,
zoals lezen en verbaal geheugen kan aantasten. Vanwege
de idiopathische aard van RE levert standaard klinische
beeldvorming geen afwijkingen op. In dit promotieon-
derzoek is met behulp van nieuwere, geavanceerde beeld-
vormende technieken gekeken naar de onderliggende
cerebrale afwijkingen van RE.
Afwijkende hersennetwerkenPatronen van fluctuaties in neuronale activiteit werden
bestudeerd middels functionele MRI. Het blijkt dat de
functionele connectiviteit bij RE verminderd is tussen de
rolandische cortex (epileptisch focus) en de traditionele
taalgebieden, met name het gebied van Broca. Bovendien
is deze afname in functionele connectiviteit gecorreleerd
met lagere taalprestaties in RE. Een ander inzicht verkre-
gen middels functionele MRI is dat de rolandische cortex
en belangrijke taalgebieden geïntegreerd zijn, ook bij
1 Promotores van dit onderzoek waren prof. dr. A.P. Aldenkamp en prof. dr. ir. W.H. Backes. Co-promotor: dr. J.F.A. Jansen.
Door: Jaap Jansen ([email protected]), radiologie, Maastricht Universitair Medisch Centrum.
Hersennetwerken en neuronale dynamiekVooruitgang in beeldvorming van focale epilepsie
Op 4 april 2014 promoveerde René Besseling op het proefschrift ‘Brain wiring and neuronal dynamics, advances in
MR imaging of focal epilepsy1’ aan de Universiteit Maastricht. Zijn proefschrift beschrijft de toepassing van geavan-
ceerde MRI-technieken bij kinderen met Rolandische epilepsie om afwijkende hersenverbindingen op te sporen die
een rol spelen bij de leerproblemen (met name taal) die de kinderen ondervinden.
gezonde controlekinderen. Middels analyse van onafhan-
kelijke componenten (independent component analysis;
ICA) werd aangetoond dat deze gebieden in rust deel
uitmaken van hetzelfde spatio-temporele netwerk. Bij RE
is deze integratie aangetast (figuur 1 , pagina 23), hetgeen
een model biedt om rolandische afwijkingen te linken
aan de taalproblemen.
Naast het vaststellen van functionele correlaten voor taal-
problematiek bij RE, is ook gekeken naar afwijkingen in
witte stof connectiviteit middels diffusie-gewogen MRI.
Zowel bij controles als patiënten met RE werd gevonden
dat de rolandische cortex sterk verbonden is middels
witte stofbanen met belangrijke taalgebieden. Echter bij
RE werden connectiviteitsafwijkingen gevonden, met
name afnames in fractionele anisotropie, duidend op
een verminderde witte stof integriteit. Ook de afname in
structurele connectiviteit was geassocieerd met een af-
name in taalprestatie in RE. Deze bevindingen duiden op
structurele afwijkingen van witte stofbanen die relevant
zijn voor taal.
Verder werd ook gekeken naar de koppeling van structu-
rele en functionele connectiviteit. Bij normale hersen-ont-
wikkeling neemt deze structurele en functionele koppeling
toe als gevolg van een gemeenschappelijk maturatieproces
van netwerkoptimalisatie. De infrastructuur van het brein
wordt als het ware afgestemd op het informatieverkeer en
vice versa. Een afname in deze koppeling werd gevonden
voor RE, met name bij de jongere patiënten, wat duidt op
een ontwikkelingsachterstand in de structurele en functio-
nele koppeling voor kinderen met RE.

Periodiek voor professionals 12 | nr 3 | 2014 23Proefschriftbesprekingen
Proefschriftbesprekingen
Tot slotSamenvattend geldt dat RE gepaard gaat met afwijkingen
in functionele en structurele connectiviteit die de epilep-
tische haard in de rolandische strip verbinden met een
verstoord taalnetwerk. Daarnaast lijkt de functionele en
structurele opbouw van het brein de propagatie van rol-
andische pathologie naar het taalnetwerk te faciliteren,
hetgeen een verklaring vormt voor de specifieke taal-
problemen in RE. Met de vondst van zowel afwijkingen in
Figuur 1 Functionele netwerken bestudeerd middels functionele MRI. Sensomotorisch (ofwel Rolandisch) netwerk (groen), en het taal-
netwerk (oranje). Ter hoogte van Broca (groen/oranje) zijn beide netwerken geïntegreerd; deze integratie is aangedaan in Rolandische
epilepsie. De rode gestippelde pijl geeft aan hoe mogelijk de epileptiforme activiteit (rode flits in Rolandische strip) deze verstoring in
Broca veroorzaakt.
functionele en structurele koppelingen tussen hersennet-
werken, is er een biologisch aanknopingspunt gevonden
voor de taalachterstand van kinderen met RE. Voor de
beschreven geavanceerde MRI-technieken zou in de toe-
komst wellicht een rol weggelegd kunnen zijn in de klini-
sche praktijk, bijvoorbeeld voor de prognose van het ver-
loop van de epilepsie van de patiënt of voor het inschatten
of een behandeling gericht op de taalproblematiek wel of
niet succesvol zal zijn.
Lees het actuele overzicht van congressen over epilepsie.
Kijk voor meer informatie op www.epilepsieliga.nl.

De productie van dit blad is mogelijk gemaakt door financiële ondersteuning van:
Agenda
9 - 12 september 2014Audigenic epilepsy: from animal models to the clinic –
first international conference
Locatie: Salamanca, Spanje
Informatie: http://eventum.usal.es
12 - 13 september 2014Forum on Epilepsy at the EPNS Research Meeting
Locatie: Boekarest, Roemenië
Informatie: [email protected], [email protected]
17 september 20143rd Halifax International Epilepsy Conference and
Retreat 2014
Locatie: Halifax, Canada
Informatie: www.aesnet.org, [email protected]
17 - 20 september 20148th Latin American Congress on Epilepsy
Locatie: Buenos Aires, Argentinië
Informatie: www.epilepsycongress.org
5 - 10 oktober 20148th Migrating Course on Epilepsy
Locatie: Dubrovnik, Kroatie
Informatie: [email protected]
5 - 11 oktober 201423rd Annual International Epilepsy Symposia
Locatie: Clevelend, Ohio, USA
Informatie: http://www.clevelandclinicmeded.com
7 - 10 oktober 2014Dietary therapies for Epilepsy and other Neurological
Disorders – 4th global symposium
Locatie: Liverpool, Engeland
Informatie: http://site.matthewsfriends.org/
17 - 19 oktober 2014Canadian League Against Epilepsy (CLAE)
Biennial Meeting
Locatie: London, Ontario, Canada
Informatie: http://claegroup.org/
5 - 9 december 201468th American Epilepsy Society Annual Meeting
Locatie: Seattle, USA
Informatie: www.aesnet.org/
12 - 16 januari 20155th Course on Epilepsy Surgery (EPODES)
Locatie: Brno, Brno, Tsjechië
Informatie: www.ta-service.cz/epodes2015
3 - 8 februari 2015American Clinical Neurophysiology Society 2015
Annual Meeting and Courses
Locatie: Houston, Texas
Informatie: http://www.acns.org
25 - 26 februari 201560th Scientific Meeting
Locatie: Chicago Downtown Marriot, USA
Informatie: http://www.aes-tmj.org/
9 - 11 april 2015London-Innsburck Colloquium on Status Epilepticus and
Acute Seizures
Locatie: London, UK
Informatie: http://www.statusepilepticus2015.eu
1 augustus - 4 september 2015 XIII Workshop on Neurobiology of Epilepsy (WONOEP)
Locatie: Heybeliada Island, Turkey
Informatie: [email protected]
4 - 5 september 20152nd International Epilepsy Symposium
Locatie: Bielefeld-Bethel, Germany
Informatie: [email protected]
6 - 10 oktober 20156th Eilat International Educational Course on the
Pharmacological Treatment of Epilepsy (6th Eilat Edu)
Locatie: Jerusalem, Israël
Informatie: http://www.eilatedu2015.com