john c. stendahl et al- toughening of polymers by self-assembling molecules
TRANSCRIPT

8/3/2019 John C. Stendahl et al- Toughening of Polymers by Self-Assembling Molecules
http://slidepdf.com/reader/full/john-c-stendahl-et-al-toughening-of-polymers-by-self-assembling-molecules 1/4
high-resolution transmission electron microscope operating at 200 kV; andphotoluminescence (PL) spectra recorded on a Renishaw 2000 with an Ar ionlaser at room temperature with an excitation wavelength of 514.5 nm. The SEMimages were taken on a Philips XL-30 instrument.
Received: May 7, 2002Final version: July 25, 2002
±
[1] Z. Y. Pan, X. J. Liu, S. Y. Zhang, G. J. Shen, L. G. Zhang, Z. H. Lu, J. Z.Liu, J. Phys. Chem. B 1997, 101, 9703.
[2] T. Vossmeyer, E. DeIonno, J. R. Heath, Angew. Chem., Int. Ed. Engl.
1997, 36, 1080.[3] P. C. Ohara, J. R. Heath, W. M. Gelbart, Angew. Chem., Int. Ed. Engl.
1997, 36, 1078.[4] T. Yonezawa, S. Onoue, N. Kimizuka, Adv. Mater. 2001, 13, 140.[5] B. A. Korgel, S. Fullam, S. Connolly, D. Fitzmaurice, J. Phys. Chem. B
1998, 102, 8379.[6] R. Maoz, E. Frydman, S. R. Cohen, J. Sagiv, Adv. Mater. 2000, 12, 424.[7] X. M. Liu, H. M. Jaeger, C. M. Sorensen, K. J. Klabunde, J. Phys. Chem.
B 2001, 105, 3353.[8] Z. L. Wang, Z. Dai, S. Sun, Adv. Mater. 2000, 12, 1944.[9] R. P. Andres, J. D. Bielefeld, J. I. Henderson, D. B. Janes, V. R. Kolagun-
ta, C. P. Kubiak, W. J. Mahoney, R. G. Osifchin, Science 1996, 273, 1690.[10] I. Willner, F. Patolsky, J. Wasserman, Angew. Chem., Int. Ed. 2001, 40,
1861.
[11] J. Hu, L.-S. Li, W. Yang, L. Manna, L.-W. Wang, A. P. Alivisatos, Science2001, 292, 2060.
[12] S. J. Park, S. Kim, S. Lee, Z. G. Khim, K. Char, T. Hyeon, J. Am. Chem.
Soc. 2000, 122, 8581.[13] V. F. Puntes, K. M. Krishnan, A. P. Alivisatos, Science 2001, 291, 2115.[14] X. Duan, Y. Huang, Y. Cui, J. Wang, C. M. Lieber, Nature 2001, 409, 66.[15] S. W. Chung, G. Markovich, J. R. Heath, J. Phys. Chem. B 1998, 102, 6685.[16] Y. Cui, C. M. Lieber, Science 2001, 291, 851.[17] S. Kwan, F. Kim, J. Akana, P. Yang, Chem. Commun. 2001, 447.[18] Z. A. Peng, X. Peng, J. Am. Chem. Soc. 2001, 123, 1389.[19] X. Peng, L. Manna, W. Yang, J. Wickham, E. Scher, A. Kadavanich, A. P.
Alivisatos, Nature 2000, 404, 59.[20] S. H. Yu, M. Yoshimura, J. M. C. Moreno, T. Fujiwara, T. Fujino, R. Tera-
nishi, Langmuir 2001, 17 , 1700.[21] L. Manna, E. C. Scher, A. P. Alivisatos, J. Am. Chem. Soc. 2000, 122,
12700.[22] Y. W. Jun, S. M. Lee, N. J. Kang, J. Cheon, J. Am. Chem. Soc. 2001, 123,
5150.[23] L. Vayssieres, N. Beermann, S. E. Lindquist, A. Hagfeldt, Chem. Mater.
2001, 13, 233.[24] Z. Zhang, G. Ramanath, P. M. Ajavan, D. Goldberg, Y. Bando, Adv. Ma-
ter. 2001, 13, 197.[25] A. Hatzor, P. S. Weiss, Science 2001, 291, 1019.[26] Z. L. Wang, R. P. Gao, J. L. Gole, J. D. Stout, Adv. Mater. 2000, 12, 1938.[27] M. Li, H. Schnablegger, S. Mann, Nature 1999, 402, 393.[28] J. K. N. Mbindyo, B. D. Reiss, B. R. Martin, C. D. Keating, M. J. Natan,
T. E. Mallouk, Adv. Mater. 2001, 13, 249.[29] Y. Jun, Y. Jung, J. Cheon, J. Am. Chem. Soc. 2002, 124, 615.[30] M. Ciampolini, C. Mengozzi, O. Orioli, J. Chem. Soc., Dalton Trans. 1975,
2051.[31] Y. Li, D. Xu, Q. Zhang, D. Chen, F. Huang, Y. Xu, G. Guo, Z. Gu, Chem.
Mater. 1999, 11, 3433.[32] J. Yang, J. H. Zeng, S. H. Yu, L. Yang, G. E. Zhou, Y. T. Qian, Chem. Ma-
ter. 2000, 12, 3259.[33] J. J. Ramsden, S. E. Webber, M. Grätzel, J. Phys. Chem. 1985, 89, 2740.
[34] G. Q. Xu, B. Liu, S. J. Xu, C. H. Chew, S. J. Chua, L. M. Gana, J. Phys.
Chem. Solids 2000, 61, 829.
Toughening of Polymers by Self-AssemblingMolecules**
By John C. Stendahl , Leiming Li, Eugene R. Zubarev,
Yau-Ru Chen, and Samuel I. Stupp*
The toughening of brittle polymers has been a significant
challenge in materials science over the past several dec-
ades.[1±4] We report here that small quantities of self assem-
bling molecules known as dendron rod±coils (DRCs)[5] impart
higher orientational order in drawn polystyrene (PS) and sig-
nificantly improve impact strength. When added to styrene
monomer in amounts as low as 0.1 wt.-%, DRCs sponta-
neously assemble into gel-forming networks. The self-as-
sembled gels are birefringent and contain supramolecular rib-
bons that are approximately 10 nm wide, 2 nm thick, and up
to 10 lm long.[6] After thermal polymerization of the gels, the
resulting polymer is also birefringent and reveals enhanced
chain orientation relative to pure PS when samples aremechanically drawn under identical conditions.
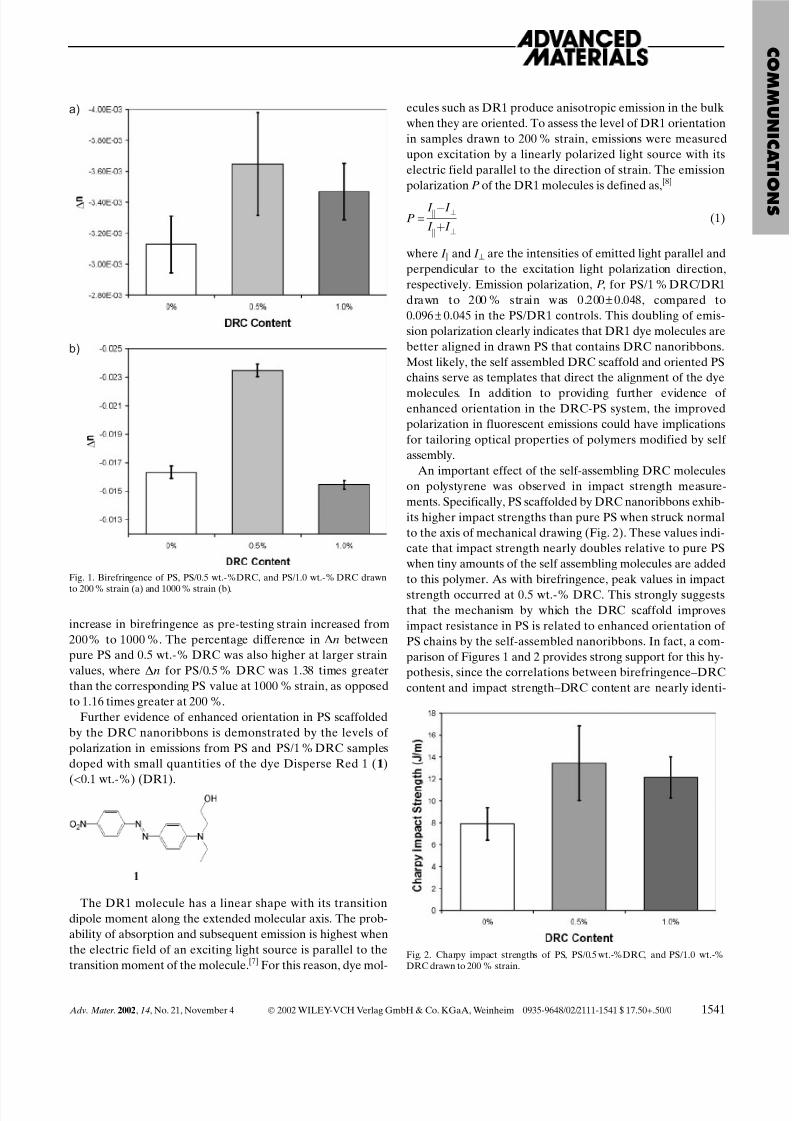
The increased orientation in mechanically drawn PS speci-
mens scaffolded by DRC is revealed by their greater birefrin-
gence relative to pure PS samples (Fig. 1). Interestingly, the
birefringence reaches a maximum for 200 % and 1000 %
strain when the concentration of DRC molecules is approxi-
mately 0.5 wt.-%. The fact that the magnitude of birefrin-
gence actually decreases as DRC content increases from 0.5
to 1.0 wt.-% for both strain values suggests that the enhanced
birefringence in DRC samples is primarily due to enhanced
alignment of PS chains, and not to the intrinsic birefringence
of DRC nanoribbons. At 0.5 wt.-% DRC, the dispersed DRC
nanoribbons apparently aid alignment of the PS chains duringmechanical stretching. However, as DRC concentration in-
creases, it is likely that greater phase separation occurs
between DRC nanophases and PS, and the aligning effects of
the DRC are reduced. As expected, higher degrees of drawing
resulted in greater chain orientation, which is revealed by an
1540 Ó 2002 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim 0935-9648/02/2111-1540 $ 17.50+.50/0 Adv. Mater. 2002, 14, No. 21, November 4
______________________
±
[*] Prof. S. I. StuppDepartments of Materials Science and EngineeringChemistry, and Medicine, Northwestern UniversityEvanston, IL (USA)E-mail: [email protected]
J. C. Stendahl, Dr. L. Li
Department of Materials Science and EngineeringNorthwestern UniversityEvanston, IL (USA)
Dr. E. R. ZubarevDepartment of Chemistry, Northwestern UniversityEvanston, IL (USA)
Y.-R. ChenDepartment of Materials Science and Engineering, University of IllinoisUrbana, IL (USA)
[**] This work made use of the Electron Probe Instrumentation Center(EPIC) at Northwestern University and was supported by United StatesDepartment of Energy grant DE-FG02-00ER45810/A001, United StatesArmy Research Office grant DAAG55-97-1-0126, and United States AirForce Office of Scientific Research grant F49620-00-1-283/P0002. Theauthors are thankful for useful discussions with Prof. Edward Kramer of the University of California at Santa Barbara.
8/3/2019 John C. Stendahl et al- Toughening of Polymers by Self-Assembling Molecules
http://slidepdf.com/reader/full/john-c-stendahl-et-al-toughening-of-polymers-by-self-assembling-molecules 2/4
increase in birefringence as pre-testing strain increased from
200% to 1000 %. The percentage difference in Dn between
pure PS and 0.5 wt.-% DRC was also higher at larger strain
values, where Dn for PS/0.5 % DRC was 1.38 times greater
than the corresponding PS value at 1000 % strain, as opposed
to 1.16 times greater at 200 %.
Further evidence of enhanced orientation in PS scaffolded
by the DRC nanoribbons is demonstrated by the levels of
polarization in emissions from PS and PS/1 % DRC samples
doped with small quantities of the dye Disperse Red 1 ( 1)
(<0.1 wt.-%) (DR1).
1
The DR1 molecule has a linear shape with its transition
dipole moment along the extended molecular axis. The prob-
ability of absorption and subsequent emission is highest when
the electric field of an exciting light source is parallel to the
transition moment of the molecule.[7] For this reason, dye mol-
ecules such as DR1 produce anisotropic emission in the bulk
when they are oriented. To assess the level of DR1 orientation
in samples drawn to 200 % strain, emissions were measured
upon excitation by a linearly polarized light source with its
electric field parallel to the direction of strain. The emission
polarization P of the DR1 molecules is defined as,[8]
P = I
iÀ I
c
I i I
c
(1)
where I i and I ̂ are the intensities of emitted light parallel and
perpendicular to the excitation light polarization direction,
respectively. Emission polarization, P , for PS/1 % DRC/DR1
drawn to 200 % strain was 0.200± 0.048, compared to
0.096± 0.045 in the PS/DR1 controls. This doubling of emis-
sion polarization clearly indicates that DR1 dye molecules are
better aligned in drawn PS that contains DRC nanoribbons.
Most likely, the self assembled DRC scaffold and oriented PS
chains serve as templates that direct the alignment of the dye
molecules. In addition to providing further evidence of enhanced orientation in the DRC-PS system, the improved
polarization in fluorescent emissions could have implications
for tailoring optical properties of polymers modified by self
assembly.
An important effect of the self-assembling DRC molecules
on polystyrene was observed in impact strength measure-
ments. Specifically, PS scaffolded by DRC nanoribbons exhib-
its higher impact strengths than pure PS when struck normal
to the axis of mechanical drawing (Fig. 2). These values indi-
cate that impact strength nearly doubles relative to pure PS
when tiny amounts of the self assembling molecules are added
to this polymer. As with birefringence, peak values in impact
strength occurred at 0.5 wt.-% DRC. This strongly suggests
that the mechanism by which the DRC scaffold improves
impact resistance in PS is related to enhanced orientation of
PS chains by the self-assembled nanoribbons. In fact, a com-
parison of Figures 1 and 2 provides strong support for this hy-
pothesis, since the correlations between birefringence±DRC
content and impact strength±DRC content are nearly identi-
Adv. Mater. 2002, 14, No. 21, November 4 Ó 2002 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim 0935-9648/02/2111-1541 $ 17.50+.50/0 1541
a)
b)
Fig. 1. Birefringence of PS, PS/0.5 wt.-%DRC, and PS/1.0 wt.-% DRC drawn
to 200 % strain (a) and 1000 % strain (b).
Fig. 2. Charpy impact strengths of PS, PS/0.5 wt.-%DRC, and PS/1.0 wt.-%DRC drawn to 200 % strain.

8/3/2019 John C. Stendahl et al- Toughening of Polymers by Self-Assembling Molecules
http://slidepdf.com/reader/full/john-c-stendahl-et-al-toughening-of-polymers-by-self-assembling-molecules 3/4
cal. This result is not surprising, since enhancement of impact
strength in polymers as a result of orientation is well docu-
mented in the literature.[9,10] In unoriented, isotropic poly-
mers, crack propagation requires the breaking of a combina-
tion of relatively weak interchain bonds as well as stronger
covalent bonds. In oriented systems, the propagation of a
crack normal to the axis of orientation requires severing a sig-nificantly higher fraction of covalent bonds. Since a larger per-
centage of high strength bonds must be broken, fracture ener-
gies in oriented systems can be significantly higher.
In addition to enhancing molecular orientation in PS, it is
possible that the DRC scaffold provides an impact-absorbing
skeleton that dissipates strain energy. Based on models of
ribbon formation, it is estimated that energy approximately
equivalent to one C±C bond in the PS backbone is absorbed
for each DRC nanoribbon that is severed as the material
deforms.[5] Most importantly, because the DRC scaffold self
assembles through non-covalent bonds, it has the potential to
impart a self-healing quality to the deforming material.Unlike covalent bonds in the PS matrix, the hydrogen bonds
and p±p stacking interactions between DRC monomers have
the potential to reform after breaking under mechanical
stress. The mobility of severed ribbons in the PS matrix may
be high enough for reconnection upon localized heating
caused by deformation.
A very important difference between the effect of DRC in
dissipating strain energy and that of polybutadiene (PB) in
high impact strength PS±PB blends and copolymers is the vol-
ume of the modifying phase. Most high impact strength blends
and copolymers have elastomer contents that are five to thirty
times greater[1] than the content of DRC nanophases de-
scribed here. Moreover, the minority phases in most highimpact PS blends are present as separate, spherical domains
because of immiscibility, and render the material opaque. In
contrast, DRC supramolecular structures do not extensively
phase separate from PS upon polymerization and the modi-
fied material remains transparent. Our group has demonstrat-
ed earlier that the nanoribbons remain dispersed throughout
the bulk polymer in small bundles of less than ten and are
sometimes present individually.[6] The model described here
for dispersing self assembled nanoribbons in bulk polymers
could be useful for altering electrical or optical properties.
This could be accomplished through chemical modification
and tailoring of the self assembling molecules.
The extremely large interfacial area between the DRCnanoribbons and bulk PS suggests a mechanically interlocking
network that may provide multiple sites for crack deflection.
Because interactions at the PS/DRC interface can be ex-
pected to be weaker than the covalent bonds in PS and intra-
ribbon bonds in DRC, cracks extending through PS may shift
their orientation when encountering less resistant paths along
the matrix/scaffold interface. With such a large contact surface
provided by the DRC nanophase, the potential for deflecting
cracks and absorbing strain energy at the PS/DRC interface
may be significantly higher than in conventional polymer
blends. Most importantly, preliminary data indicate DRC
molecules are able to increase impact strength without com-
promising tensile strength. In traditional high impact blends,
elastomer additions usually decrease tensile strength by 20±
40 % below that of the PS homopolymer.[11]
Analysis of the fracture surfaces of impact specimens sug-
gests that failure in 0.5 and 1.0 wt.-% DRC Charpy samples
occurred by different mechanisms than in pure PS. To thenaked eye, the translucent, marbled surfaces of DRC samples
are in stark contrast to the shiny, transparent surfaces of pure
PS (Fig. 3). The whitening in the DRC samples suggests that
crazes and defects are smaller in size and greater in quantity
than in pure PS. The extensive formation of defects at subcri-
tical stresses is a potentially important mechanism for stress
dissipation that could result from structural and/or orienta-
tional effects of the DRC scaffold. Kramer and Farrar have
observed that crazes formed in PS when the tensile axis is par-
allel to the direction of molecular orientation are smaller and
more numerous than those formed when the tensile axis is
normal to the molecular orientation.[12] In a similar manner, it
is reasonable that smaller and more numerous defects form inthe more highly oriented PS/DRC polymers.
Optical and scanning electron micrographs (SEMs) confirm
that PS/DRC impact fracture surfaces are significantly more
textured than the glassy, faceted PS surfaces (Fig. 4). At high
magnification, there are especially noticeable differences
between the rounded and sometimes fibrous surface features
of DRC samples, and the clean, angled features of pure PS
samples. The glassy fracture surfaces in PS suggest failure by a
brittle mechanism,[10] where critical crack extension was initi-
ated prior to significant deformation by the growth of small
numbers of large, planar, easily propagating defects. On the
other hand, the rough, irregular relief and localized whitening
in DRC samples suggest their failures involved large numbersof small defects on different planes. Unlike pure PS, critical
cracks in DRC specimens did not proceed on single planes
and were apparently forced to change orientation numerous
times to accommodate out-of-plane subcritical defects that
formed ahead of crack tips. The fibrils present on some DRC
fracture surfaces are features frequently associated with
highly oriented systems and suggest that defect formation was
accompanied by plastic deformation prior to fracture.[10] The
increased surface area of DRC fracture faces is consistent
with the Charpy data indicating that greater energy was nec-
essary for crack propagation in DRC samples.[10] Although it
1542 Ó 2002 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim 0935-9648/02/2111-1542 $ 17.50+.50/0 Adv. Mater. 2002, 14, No. 21, November 4
0% DRC 0.5% DRC 1.0% DRC
Fig. 3. Macroscopic image of PS, PS/0.5 wt.-%DRC, and PS/1.0 wt.-%DRCimpact specimen fracture surfaces.

8/3/2019 John C. Stendahl et al- Toughening of Polymers by Self-Assembling Molecules
http://slidepdf.com/reader/full/john-c-stendahl-et-al-toughening-of-polymers-by-self-assembling-molecules 4/4
is difficult to draw specific conclusions about the mechanism
by which DRC modifies PS fracture mechanics, the features
of DRC/PS samples are compatible with observations of
greater molecular orientation, and suggest the scaffoldformed by self assembling molecules may increase viscous
flow, multiplanar subcritical defect formation, and crack
deflection.
Experimental
Styrene (Aldrich) inhibited with 10±15 ppm 4-tert -butylcatechol was addedto glass vials containing premeasured amounts of DRC. Sealed vials were ultra-sonicated for two minutes and then heated via heat gun with gentle shaking.When DRC had gone into solution, samples were heated for an additional min-ute to promote supramolecular assembly. After 10 min of ambient cooling toallow for dissipation of excess heat, vials were transferred to a 100 C oven forpolymerization, where a nitrogen atmosphere was maintained to prevent oxida-
tion. After 72 h, caps were removed and vials containing solid polymer werereturned to the oven under vacuum to promote evaporation of residual mono-mer. One day after removal of caps, vials were broken and solid polymer cylin-ders were returned to the oven under vacuum where they were heated for anadditional 24 h before cooling to room temperature.
Portions of all samples were dissolved in tetrahydrofuran and injected into aWaters 2690 gel-permeation chromatography (GPC) unit for molecular weightanalysis. Fractions were monitored by a Waters 2410 refractive index detectorand calibrated to PS standards. The following molecular weight averages wereobtained for each group of polymers: PS: M w = 543000 ± 84000, PDI = 2.7 ±0.2;PS/0.5 % DRC: M w = 561000 ± 78000, PDI = 2.6± 0.2; PS/1% DRC: M w =513000 ± 87 000, PDI = 2.7 ± 0.2.
To prepare Charpy samples, solid polystyrene cylinders were machined intotensile specimens with gauge dimensions 0.25² 0.25² 0.4² (1 inch~ 2.5 cm) bya Bridgeport milling machine connected to a South Western Industries Proto-Track MX2 numerical controller. To promote molecular orientation withoutcracking, specimens were drawn in uniaxial tension at 4.0 mmmin±1 with an
MTS Sintech testing machine equipped with a heating chamber set at 108C(slightly higher than T g for PS). At 200 % strain, drawing was stopped and sam-ples were quenched with a water mist and removed from the chamber to pre-serve orientation. Gauge sections of drawn tensile samples were machined intoCharpy impact specimens with dimensions 3.15 3.15 17.5 mm (±0.05 mm)and 1 mm deep notches aligned normal to the direction of drawing.
Birefringence measurements were made with a Cary 500 UV-vis spectropho-tometer on gauge sections of tensile specimens drawn to 200 % and 1000%
strain at 108 C. Crossed plastic polarizers (Edmund) were placed on either sideof samples so light could not be transmitted through optically isotropic materi-al. Samples were oriented with their axes of drawing at 45 to the polarizer/ana-lyzer pair. The spectrometer was operated in double beam mode while percenttransmission was recorded from 400 to 700 nm. Using the method described byDevon and Rogers [13], transmission minima were numbered by order of appearance from 400 nm to 700 nm, and plotted as the ordinate against 1/ k toprovide a linearly fitted curve with slope 1/(LDn), where L denotes samplethickness and Dn denotes birefringence.
Optical emission specimens were prepared by adding DR1 dye (Aldrich) toDRC±styrene solutions before gelation. Gels containing DRC and DR1 werepolymerized and drawn to 200 % strain at 108 C. Samples were excited with alinearly polarized, Q-switched Nd:YAG laser (355 nm, 10 Hz, 35 ps) incidentnormal to their surfaces. Axes of drawing were parallel to the laser polarizationdirection (i.e., electric field). Forward emissions were collected and recorded byCCD. A polarizer was used to selectively filter parallel ( I i) and perpendicular( I ̂ ) emission components.
SEM samples were mounted on aluminum stubs with double-sided carbon tapeand coated in 6 nm gold. Images were collected with a Hitachi 4500 SEM. Errorbars in Figures 1 and 2 represent t distribution confidence intervals at 90 %.
Received: April 8, 2002Final version: July 25, 2002
±
[1] D. Mathur, E. B. Nauman, J. Appl. Polym. Sci. 1999, 72, 1151.[2] H. R. Brown, A. S. Argon, R. E. Cohen, O. S. Gebizlioglu, E. J. Kramer,
Macromolecules 1989, 22, 1004.[3] R. G. Cheatham, A. G. H. Dietz, Mod. Plast . 1951, September , 113.[4] C. C. Chau, W. R. Follette, Adv. Mater . 2000, 12, 1859.[5] E. R. Zubarev, M. U. Pralle, E. D. Sone, S. I. Stupp, J. Am. Chem. Soc.
2001, 123, 4105.[6] E. R. Zubarev, M. U. Pralle, E. D. Sone, S. I. Stupp, Adv. Mater . 2002, 14,
198.[7] M. A. Bopp, A. J. Meixner, G. Tarrach, I. Zschokke-Granacher, L. No-
votny, Chem. Phys. Lett . 1996, 263, 721.[8] J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum Press,New York 1999.
[9] J. Scheirs, Compositional and Failure Analysis of Polymers, John Wileyand Sons, New York 2000.
[10] I. Wolock, S. B. Newman, in Fracture Processes in Polymeric Solids: Phe-
nomena and Theory (Ed: B. Rosen), Interscience, New York, 1964.[11] M. Yokouchi, A. Yokota, Y. Kobayashi, J. Appl. Polym. Sci. 1982, 27 ,
3007.[12] N. R. Farrar, E. J. Kramer, Polymer 1981, 22, 691.[13] M. J. Devon, M. G. Rogers, J. Mater. Sci. 1991, 26, 335.
Eggshell Membrane Templating of HierarchicallyOrdered Macroporous Networks Composedof TiO2 Tubes**
By Dong Yang, Limin Qi,* and Jiming Ma
Recently, much effort has been devoted to the synthesis of
hierarchically ordered macroporous materials with bimodal
porosity because they combine the accessible diffusion path-
Adv. Mater. 2002, 14, No. 21, November 4 Ó 2002 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim 0935-9648/02/2111-1543 $ 17.50+.50/0 1543
a) b)
c)
e) f)
d)
Fig. 4. a) Optical micrograph of PS impact specimen fracture surface. b) SEMof PS impact specimen fracture surface. c) Optical micrograph of PS/0.5 wt-%DRC impact specimen fracture surface. d) SEM of PS/0.5 wt.-% DRC impactspecimen fracture surface. e) Optical micrograph of PS/1.0 wt.-% DRC impactspecimen fracture surface. f) SEM of PS/1.0 wt.-% DRC impact specimen frac-ture surface.
±
[*] Prof. L. Qi, Dr. D. Yang, Prof. J. MaCollege of Chemistry, Peking UniversityBeijing 100871 (China)E-mail: [email protected]
[**] This work was supported by NSFC (20003001), the Special Fund of MOE,China (200020), and the Doctoral Program Foundation of MOE, China.