journal of chemistry vol. no. 45, 11, q in denatured state ... · denatured state of ovalbumin in...
TRANSCRIPT

T m JOURNAL OF BIOLOGICAL CHEMISTRY Q 1994 by The American Society for Biochemistry and Molecular Biology, IN.
Vol. 269, No. 45, Issue ofNovember 11, pp. 28062-28067, 1994 Printed in U.S.A.
Denatured State of Ovalbumin in High Concentrations of Urea as Evaluated by Disulfide Rearrangement Analysis*
(Received for publication, June 20, 1994, and in revised form, August 22, 1994)
Eizo Tatsumi, Nobuyuki Takahashi, and Masaaki Hirosel From the Research Institute for Food Science, Kyoto University, Uji, Kyoto 611, Japan
To investigate the highly denatured state of ovalbu- min (molecular mass of 42.7 kDa, four cysteine sulfhy- dryls and one cystine disulfide) using the disulfide re- arrangement approach, we established the peptide- mapping procedure using a cysteine-labeling technique with a fluorescent dye that allows the quantitative analyses for the disulfide-involved half-cystines. Ovalbumin denatured at a low protein concentration in 8-10 Y urea, in which the protein showed complete un- folding as evaluated by far-UV CD spectra, was analyzed for the disulfide-involved half-cystines using the pep- tide-mapping procedure. Data clearly showed that the number of free sulfhydryls and intrachain disulfides were four and one, respectively, but that all six half- cystines are labeled with the dye. These results strongly suggested that 15 disulfide isomers that are theoreti- cally possible for a molecule having one disulfide and four sulfhydryls are all generated during the denatur- ation. The quantitative data for the ratios of the ob- served labeling values for the six half-cystines, relative to the overall labeling values, were consistent with the view that the distribution of the 15 possible disulfide isomers at equilibrium depends on the number of amino acid residues separating the two half-cystines to the power of -1.9 to -2.0. Essentially, the same non-gaussian chain nature was also observed with kinetic data for sulfhydryl-disulfide exchanges after denaturation of native ovalbumin.
Studies of proteins under strongly denaturing conditions are crucial for understanding the mechanism of protein folding, since highly denatured proteins are generally employed as the starting materials for refolding studies. Several lines of evi- dence supported the conclusion that global protein conforma- tion is highly unfolded by denaturation in high concentrations of guanidine hydrochloride or urea (Aune et al., 1967; Tanford, 1968; Greene and Pace, 19741, although recent microscopic studies have demonstrated in some cases the occurrence of a residual conformation even in the presence of a denaturant (Amir and Haas, 1988; Amir et al., 1992; Bierzynski and Bald- win, 1982; Neri et al., 1992). In a highly denatured state, all fixed internal noncovalent interactions that maintain the na- tive conformation are disrupted, and all parts of the protein molecule are mobile (Tanford, 1970). Consequently, a protein molecule behaves like a random coil or random flight chain, so that all possible conformational isomers exist (Tanford, 1961).
*This work was supported in part by a grant-in-aid for scientific research from the Ministry of Education, Science and Culture of Japan. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$ To whom correspondence should be addressed. Tel.: 0774-32-3111 (ext. 2725); Fax: 0774-33-3004.
For the investigation of conformational distribution, the sulf- hydryl-disulfide rearrangement problem may provide a unique model system. It has been known that sulfhydryl-disulfide re- arrangements take place intramolecularly in denatured states, giving many disulfide isomers with non-native disulfide bonds (Creighton, 1984). If a protein that contains multiple sulfhy- dryls and a disulfide in a molecule is in an ideal random-coil state, every sulfhydryl will be able to freely encounter a disul- fide, thereby all possible disulfide isomers are generated. Such a disulfide isomer corresponds to a subset consisting of a large number of conformational isomers, and an entropically favor- able disulfide isomer that includes the greatest number of con- formational isomers dominates over the distribution. Early sta- tistical calculations have shown that the probability of the occurrence of such disulfide isomers is proportional to n-1.5, where n is the number of amino acid residues separating the two half-cystines that are involved in a disulfide bond (Kauz- mann, 1959; Poland and Scheraga, 1965). Chan and Dill (1990, 1991) have recently pointed out that, unlike such Gaussian distributions, the probability may be more consistent with a dependence upon n-2.4 for random coil polymers with significant excluded volume effects. Few attempts, however, have been made to experimentally investigate the feasibility of the poly- mer chain theories for the highly denatured state of a protein.
As a useful model system, we have been interested in the disulfide rearrangement problem of ovalbumin. The egg white protein consists of a single polypeptide chain of 385 amino acid residues, and contains six half-cystines of SH-1 (Cys-111, SH-2 (Cys-301, SH-3 (Cys-731, SH-4 (Cys-l20), SH-5 (Cys-3671, and SH-6 (Cys-382). In the native state, SH-3 and SH-4 form a disulfide bond, and the other four occur as free cysteine sulf- hydryls (Thompson and Fisher, 1978; Nisbet et aZ., 1981). Our indirect evidence has, however, shown that some non-native disulfide bonds may be produced in this large disulfide protein in a high concentration of urea (Takahashi and Hirose, 1992). In this protein, the six half-cystines are widely allocated from the N-terminal to the C-terminal region, indicating that disul- fide rearrangement reactions will occur in the overall molecule. In the present report, we have established a peptide-mapping procedure that allows the quantitative analysis for the ratios of disulfide-involved half-cystines that are directly related to the distribution of the 15 disulfide isomers. Using this peptide- mapping procedure, sulfhydryl-disulfide exchanges in highly denatured ovalbumin were analyzed kinetically and at the equilibrium state. On the basis of the exchange data, it was investigated whether or not ovalbumin behaves as a Gaussian chain in its highly denatured state.
EXPERIMENTAL PROCEDURES Muteria2s”Ovalbumin was purified from fresh egg white by crystal-
lization in an ammonium sulfate solution and recrystallized five times (Sorensen and H~ymp, 1915). From the crystallized ovalbumin, A,- ovalbumin (diphosphorylated form) was purified by ion-exchange chro- matography in the same way as described by Kitabatake et al. (1988),
28062

Denatured State of Ovalbumin in Denaturant
except that their DEAE-cellulose column was replaced by a HPLC' column (HiLoad 26/10 Q Sepharose; Pharmacia Biotech Inc.) connected to a HPLC apparatus (Shimadzu, LC-1OA). Trypsin treated with diphe- nylcarbamyl chloride (type X I ) and chymotrypsin (type 11) were pur- chased from Sigma. Achromobacter protease I (EC 3.4.21.50) was ob- tained from Wako Pure Chemical Industries. Urea was specially prepared reagent grade and other chemicals were guaranteed grade from Nacalai Tesque.
CD Analyses for Denaturation Transition-Ovalbumin was incu- bated at 0.2 mg/ml and 37 "C in TE buffer (50 mM Tris-HC1, 1.0 mM EDTA, pH 8.8) containing different concentrations of urea. CD elliptic- ity at 222 nm was determined at various time intervals with a spec- tropolarimeter (JASCO, 5-700) using a 0.1-cm cuvette.
Polyacrylamide Gel Electrophoresis-The number of intrachain dis- ulfide bonds in ovalbumin was determined by selective two-step alkyl- ation and subsequent PAGE as described previously (Takahashi and Hirose, 1990). Briefly, ovalbumin denatured in TE buffer containing 9 M urea was alkylated as the first step with 50 m iodoacetic acid, precipi- tated with cold acetone, 1 N HC1 (98:2), washed three times with ace- tone, 1 N HC1, H,O (98:2:10), dissolved in TE buffer containing 9 M urea, reduced by 12 mM DTT, and then alkylated by 35 mM iodoacetamide as the second step. The protein was electrophoresed on a polyacrylamide gel using a high pH urea-denaturing buffer and stained with Coomassie Blue.
The protein sample that had been alkylated in the first step with iodoacetic acid was also analyzed by non-reducing SDS-PAGE; the alkylated sample was mixed with a 0.33 volume of 0.2 M Tris-HC1, pH 7.0, containing 4% SDS and 40% glycerol, incubated at 60 "C for 30 min, and electrophoresed on 10% polyacrylamide gel according to the stand- ard method by Laemmli (1970).
Peptide Mapping--As a prerequisite for the determination of half- cystines involved in disulfide bonds, all six half-cystines were labeled with a fluorescent alkylation reagent, IAEDANS, and subjected to the peptide mapping analysis. Ovalbumin was incubated at 0.2 mg/ml in TE buffer containing 5 lll~ DTT and 8 M urea at 37 "C for 30 min. The free sulfhydryls in the disulfide-reduced, denatured protein were la- beled by incubation at 37 "C for 10 min with 15 mM IAEDANS in TE buffer containing 8 M urea. Excess IAEDANS was trapped by incubation with 18 nm DTT at 37 "C for 5 min. To the mixture, 10 volumes of cold acetone, 1 N HC1 (98:2) solution was added, and then proteins were precipitated by centrifugation (3000 x g , 5 min). After the precipitates were washed three times with cold acetone, 1 N HC1, H,O (98:2:10), the labeled ovalbumin was mssolved in TE buffer containing 8 M urea giving a protein concentration of 2 mg/ml, fully denatured by incubation at 30 "C for 60 min, and diluted 4-fold with TE buffer giving 2 M urea and 1.0 mg/ml ovalbumin. The protein was extensively digested in a two- step manner: in the first step with 20 unitsiml trypsin and 1 unitfml chymotrypsin at 30 "C for 20 h; in the second step with an additional 20 units/ml Achromobacter protease I and 1 uniuml chymotrypsin at 30 "C for 20 h. To obtain reproducible digestion, prior to the ovalbumin diges- tion, we assayed the protease activities as described by Walsh and Wilcox (1970) using synthetic substrates: p-toluenesulfonyl-L-arginine methyl ester for trypsin activity, N-benzoyl-L-tyrosine ethyl ester for chymotrypsin activity and p-toluenesulfonyl-L-lysine methyl ester for Achromobacter protease I activity. One unit was defined as an enzyme activity that hydrolyzed 1 pmol of substrate per min at 30 "C.
The protein digestion was terminated by the addition of a 0.1 volume of 5% trifluoroacetic acid solution. The mixture corresponding to 0.25 mg of the original ovalbumin was applied to a reverse phase HPLC column (Cosmosil 5C4-AR-300: ODS, 4.6 x 150 mm) connected to a HPLC apparatus (Shimadzu, LC-4A). Peptides were eluted with an acetonitrile linear gradient (040%) in 0.1% trifluoroacetic acid. Cys- teine peptides detected by AEDANS fluorescence (excitation, 340 nm; emission, 520 nm) were collected. The fractionation was performed six times and identical peaks were pooled. The peaks were further purified by rechromatography using the same column but with a different buffer system (040% acetonitrile gradient in 10 m triethylamine, acetic acid buffer, pH 5.0). Purified cysteine peptides were analyzed for their amino acid compositions with an amino acid analyzer (Hitachi, model 835-30) and for primary sequences with a gas-phase protein sequenator (Applied Biosystems, model 477N120A). For the amino acid analysis, the peptides were hydrolyzed in the gas phase with 6 M hydrochloric
matography; AEDANS-labeled, N-acetyl-N"(5-sulfo-l-naphthyl)ethyl- ' The abbreviations used are: HPLC, high performance liquid chro-
enediamine-labeled; DTT, dithlothreitol; IAEDANS, N-iodoacetyl- N"(5-sulfo-1-naphthy1)ethylenediamine; PAGE, polyacrylamide gel electrophoresis.
(Standard) (Sample)
28063
Alkylation - by IAM ]
by DTT "-I
t HPLC
teine peptides labeled with a fluorescent dye. Ovalbumin has one FIG. 1. Schematic representation for the determination of cys-
intramolecular disulfide bond and four sulfhydryls. In this figure, the case for protein species with a non-native disulfide bond (FA) consist- ing of SH-5 and SH-6 is shown as an example. Protein sulfhydryls were first quenched in acid-urea solution. In the sample run, the four cys- teine sulfhydryls (SH) were trapped with a high concentration of io- doacetamide (W) yielding alkylated cysteines (SM). The disulfide was then reduced by DTT, and newly generated sulfhydryls were la- beled with a fluorescent alkylation reagent, IAEDANS giving AEDANS- labeled cysteines (SO) . The labeled protein was extensively proteo- Iyzed and fractionated by HPLC. In the standard run, the first alkylation step was skipped, thereby all six half-cystines were allowed to be labeled with the fluorescent dye. Fluorescence peak areas corre- sponding to cysteine peptides were determined, and the relative ratios, R,, (i = 1, 2, . . . , or 6) of disulfide-involved half-cystines were deter- mined using Equation 1. In this example, either R,,, or RSH.6 should be 1.0, whereas R,,, for the other half-cystines should be zero. When the original ovalbumin consists of the 15 possible disulfide isomers, R,,, should distribute in all six half-cystines depending on the distribution of the isomers, but the sum of R,, should be 2.0 regardless of the state of original ovalbumin.
acid containing 0.1% (v/v) phenol for 24 h at 110 "C under vacuum. Quantification of Disulfide-involved Half-cystines-& schematically
shown in Fig. 1, disulfide-involved half-cystines were determined by alkylation with IAEDANS and subsequent peptide mapping analyses. To avoid air oxidation of cysteine sulfhydryls and intermolecular sulf- hydryl-disulfide exchange, ovalbumin denaturation was performed un- der a nitrogen atmosphere and at a low protein concentration. Ovalbu- min was denatured by incuhation at 0.2 mg/ml in TE buffer containing 8-10 M urea for various times at 37 "C. Protein sulfhydryls were quenched in acid by the addition of a 0.54 volume of a urea-HC1 solution giving a final concentration of 0.25 M HC1 and 8 M urea.
For the "sample" (see Fig. l), the acid quenched protein was vigor- ously mixed with 1.6 volumes of 0.3 M Tris-base containing 0.33 M iodoacetamide and 9.5 M urea giving a final pH of 8.8, and alkylation of protein sulfhydryls was performed by incubation at 37 "C for 10 min. The protein was precipitated in cold acetone, 1 N HCl (9821, washed three times with cold acetone, 1 N HC1, H,O (98:2:10), dissolved in TE buffer containing 8 M urea giving a protein concentration of 2 mg/ml, and then fully reduced by incubation with 5 mM DTT at 37 "C for 30 min. The disulfide-reduced ovalbumin was labeled at 37 "C for 10 min

28064 Denatured State of Ovalbumin in Denaturant with 15 mM IAEDANS. For the "standard," the acid-quenched protein was mixed with the same 1.6 volume of 0.3 M "&base containing 9.5 M urea and 5 mM DTT, incubated at 37 "C for 30 min, precipitated in acetone-HC1, dissolved in TE-urea buffer, and labeled with IAEDANS in the same way. For the "blank" (not depicted in Fig. l), the acid-quenched protein was mixed with the same Tris-base containing 5 m~ DTT and urea, incubated at 37 "C for 30 min, alkylated with 0.2 M iodoacetamide, precipitated in acetone-HCI, and then labeled with IAEDANS in the same way.
The mDANS-labeled proteins were precipitated in acetone-HC1, ex- tensively proteolyzed in the two-step way, and analyzed by reverse- phase HPLC in the same way as in the peptide-mapping procedure. In this HPLC analysis, the mixture corresponding to 0.05 mg of original ovalbumin was analyzed, since fluorescence peak areas in the standard run were linear up to 0.25 mg of original ovalbumin. We confirmed that in the blank run, no mDANS-labeled peaks were detected for all six half-cystines. For each of the six half-cystines, SH-i ( i = 1, 2, . . . , or 6), fluorescence peak areas in the sample and standard runs were deter- mined. In the HPLC, either SH-1 (Cys-ll), SH-5 (Cys-367), or SH-6 (Cys-382) was eluted as a single peak, but SH-2 (Cys-30), SH-3 (Cys-73), or SH-4 (Cys-120) was separated into two peaks (see Figs. 2 and 3). Thus, the sums of two peak areas for SH-2, SH-3, and SH-4 were determined. The occurrence of only one intramolecular disulfide was unchanged in either the native or denatured state (see Fig. 41, indicat- ing that two labeled half-cystines existed all the time in an ovalbumin molecule. Thus, the ratio of a disulfide-involved half-cystine (RSH.i, i = 1, 2,. . . , or 6) to the total of 2.0 was determined using the following equation:
6
= 2 X (AsH&&d/X (Asd%d (Eq. 1) '=l
where and B,,, represent the peak areas for AEDANS-labeled half-cystine in the sample and standard runs, respectively, with respect to one of the half-cystines, SH-i.
Fitting the Datu-The theoretical value for the ratio of a labeled half-cystine, R,, was calculated on the basis of the assumption that the distribution of 15 disulfide isomers at equilibrium of the sulfhydryl- disulfide exchanges depends on loop length to a power of -p. To estimate a suitable value for p , we calculated the sums of squares o,f the devia- tions of the observed data, R,,, from the theoretical one, R,,,
6
2 (RSH-I - & - i ) 2 (Eq. 2) I d
at varying p (ranged from 1 to 3 in 0.01 steps) values and searched for a p value giving a minimum deviation (see Fig. 5).
By searching for the best fitting value of p by kinetic analyses, the time course curves calculated on the basis of a model shown in Fig. 7 were fitted to the observed RSH4 at various denaturation times (see Fig. 6) by using a program written in this laboratory that utilizes a Leven- berg-Marquardt algorithm (Marquardt, 1963) and a Runge-Kutta algo- rithm in combination.
RESULTS AND DISCUSSION Peptide Mapping for Labeled Cysteines--To establish the
peptide mapping for the six half-cystines, the disulfide-reduced ovalbumin was labeled with IAEDANS, extensively digested with proteases, and fractionated by reverse-phase HPLC. As shown in Fig. 2, many fluorescent peaks were detected. Amino acid analyses for 20 major peaks revealed that only nine peaks denoted A to I are cysteine peptides, and that the others are non-peptide substances or some non-cysteine peptides. All nine peaks labeled A to I were further fractionated by rechromatog- raphy on the same HPLC column using a different buffer sys- tem. The purified cysteine peptides were analyzed for their amino acid sequences from the N-terminal to C-terminal with a sequenator. According to the established sequence of ovalbu- min (Nisbet et al., 1981), the nine peptides were assigned as shown in Fig. 3. The data for amino acid analyses were con- sistent with the sequence; in all nine peptides, the occurrence of one carboxymethyl cysteine that was the acid-hydrolysis prod- uct of the AEDANS-labeled cysteine was confirmed (data not shown).
".. ""
"" ""
""" "_.--- _ - - ""
"" "" G H _ "
40 - E
20 = 0 0
Y
1 L 0 20 4(
Time (min)
Ovalbumin was reduced with Dm, labeled with IAEDANS, extensively FIG. 2. Fractionation of AEDANS-labeled cysteine peptides.
digested with proteases, and fractionated by a reverse phase HPLC as described in the text. Nine peaks denoted A-I as detected by AEDANS fluorescence (excitation, 340 nm; emission, 520 nm) were found to be cysteine peptides.
SHl H CFDVF 75.2 11
SH2 G CPlAlM 30
I YCPlAlM 69.2 7.5
73 SH3 E CGTSVNVH 19.4
F CGTSVNVH 41.1
120
SH4 &:: 63.3 4.3
SH5 C CIK 81.9 367
382 sH6 D CVSP 69.7
cysteine peptide peaks denoted A-1 in Fig. 2 were further purified by FIG. 3. Amino acid sequences for nine cysteine peptides. The
rechromatography. Purified cysteine peptides were analyzed for their primary sequences with a gas-phase protein sequenator. The peptide recoveries were determined by amino acid analyses and are shown as molar recoveries from pre-proteolyzed ovalbumin.
It is generally observed that a large peptide is poorly recov- ered on reverse-phase HPLC. This may interfere with repro- ducible peptide quantifications by HPLC. The proteolysis con- ditions employed here that included extensive digestion with the three different proteases in combination generated small peptides consisting of up to eight amino acids with quite high recoveries of at least 60.5% (SH-3: 19.4 + 41.1%) from the original albumin (Fig. 3). We confirmed that the peptide recov- eries are closely correlated (correlation coefficient, 0.930) with the relative fluorescence peak areas for the corresponding cys- teine-peptide. In addition, the ratios of the fluorescence peak areas for the six half-cystines in the standard run were highly reproducible. We, therefore, concluded that the present peptide mapping procedure is suitable for the quantitative analysis for disulfide-involved half-cystines in ovalbumin.
Denaturation Dunsition and the Number of Intrachain Disulfide Bond-Using far-UV CD-analyses, we searched for conditions that induced the fully denatured state in ovalbumin. Ovalbumin was incubated with various concentrations of urea.
Denatured State of Ovalbumin in Denaturant 28065
A) O\ 2,
:7 0 15 30 60 120
Time (min)
B)
kDa) 97.4 -
55.6 - - OVA-SH
39.2 -
0 15 30 60 120 Time (min)
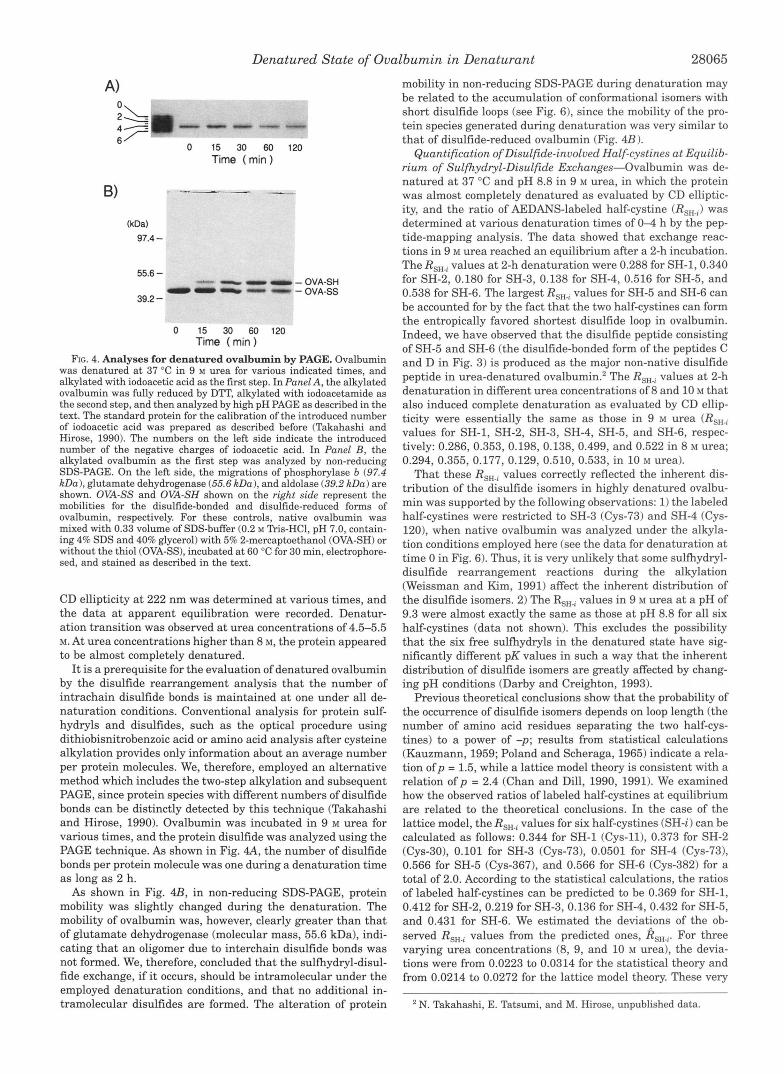
FIG. 4. Analyses for denatured ovalbumin by PAGE. Ovalbumin was denatured at 37 "C in 9 M urea for various indicated times, and alkylated with iodoacetic acid as the first step. In Panel A, the alkylated ovalbumin was fully reduced by D m , alkylated with iodoacetamide as the second step, and then analyzed by high pH PAGE as described in the text. The standard protein for the calibration of the introduced number of iodoacetic acid was prepared as described before (Takahashi and Hirose, 1990). The numbers on the left side indicate the introduced number of the negative charges of iodoacetic acid. In Panel B, the alkylated ovalbumin as the first step was analyzed by non-reducing SDS-PAGE. On the left side, the migrations of phosphorylase b (97.4 kDa), glutamate dehydrogenase (55.6 kDa), and aldolase (39.2 kDa) are shown. OVA-SS and OVA-SH shown on the right side represent the mobilities for the disulfide-bonded and disulfide-reduced forms of ovalbumin, respectively. For these controls, native ovalbumin was mixed with 0.33 volume of SDS-buffer (0.2 M Tris-HC1, pH 7.0, contain- ing 4% SDS and 40% glycerol) with 5% 2-mercaptoethanol (OVA-SH) or without the thiol (OVA-SS), incubated at 60 "C for 30 min, electrophore- sed, and stained as described in the text.
CD ellipticity at 222 nm was determined at various times, and the data at apparent equilibration were recorded. Denatur- ation transition was observed at urea concentrations of 4.5-5.5 M. At urea concentrations higher than 8 M, the protein appeared to be almost completely denatured.
I t is a prerequisite for the evaluation of denatured ovalbumin by the disulfide rearrangement analysis that the number of intrachain disulfide bonds is maintained at one under all de- naturation conditions. Conventional analysis for protein sulf- hydryls and disulfides, such as the optical procedure using dithiobisnitrobenzoic acid or amino acid analysis after cysteine alkylation provides only information about an average number per protein molecules. We, therefore, employed an alternative method which includes the two-step alkylation and subsequent PAGE, since protein species with different numbers of disulfide bonds can be distinctly detected by this technique (Takahashi and Hirose, 1990). Ovalbumin was incubated in 9 M urea for various times, and the protein disulfide was analyzed using the PAGE technique. As shown in Fig. 4A, the number of disulfide bonds per protein molecule was one during a denaturation time as long as 2 h.
As shown in Fig. 4B, in non-reducing SDS-PAGE, protein mobility was slightly changed during the denaturation. The mobility of ovalbumin was, however, clearly greater than that of glutamate dehydrogenase (molecular mass, 55.6 m a ) , indi- cating that an oligomer due to interchain disulfide bonds was not formed. We, therefore, concluded that the sulfhydryl-disul- fide exchange, if it occurs, should be intramolecular under the employed denaturation conditions, and that no additional in- tramolecular disulfides are formed. The alteration of protein
mobility in non-reducing SDS-PAGE during denaturation may be related to the accumulation of conformational isomers with short disulfide loops (see Fig. 6), since the mobility of the pro- tein species generated during denaturation was very similar to that of disulfide-reduced ovalbumin (Fig. 4B).
Quantification of Disulfide-involved Half-cystines a t Equilib- rium of Sulfiydryl-Disulfide Exchanges-Ovalbumin was de- natured at 37 "C and pH 8.8 in 9 M urea, in which the protein was almost completely denatured as evaluated by CD elliptic- ity, and the ratio of AEDANS-labeled half-cystine (RsH.i) was determined a t various denaturation times of 0-4 h by the pep- tide-mapping analysis. The data showed that exchange reac- tions in 9 M urea reached an equilibrium after a 2-h incubation. The R,,, values at 2-h denaturation were 0.288 for SH-1,0.340 for SH-2, 0.180 for SH-3, 0.138 for SH-4, 0.516 for SH-5, and 0.538 for SH-6. The largest R,,, values for SH-5 and SH-6 can be accounted for by the fact that the two half-cystines can form the entropically favored shortest disulfide loop in ovalbumin. Indeed, we have observed that the disulfide peptide consisting of SH-5 and SH-6 (the disulfide-bonded form of the peptides C and D in Fig. 3) is produced as the major non-native disulfide peptide in urea-denatured ovalbumin.2 The R,,, values at 2-h denaturation in different urea concentrations of 8 and 10 M that also induced complete denaturation as evaluated by CD ellip- ticity were essentially the same as those in 9 M urea (R,,, values for SH-1, SH-2, SH-3, SH-4, SH-5, and SH-6, respec- tively: 0.286, 0.353, 0.198, 0.138, 0.499, and 0.522 in 8 M urea; 0.294, 0.355, 0.177, 0.129, 0.510, 0.533, in 10 M urea).
That these RSHi values correctly reflected the inherent dis- tribution of the disulfide isomers in highly denatured ovalbu- min was supported by the following observations: 1) the labeled half-cystines were restricted to SH-3 (Cys-73) and SH-4 (Cys- 120), when native ovalbumin was analyzed under the alkyla- tion conditions employed here (see the data for denaturation a t time 0 in Fig. 6). Thus, it is very unlikely that some sulfhydryl- disulfide rearrangement reactions during the alkylation (Weissman and Kim, 1991) affect the inherent distribution of the disulfide isomers. 2) The values in 9 M urea at a pH of 9.3 were almost exactly the same as those a t pH 8.8 for all six half-cystines (data not shown). This excludes the possibility that the six free sulfhydryls in the denatured state have sig- nificantly different pK values in such a way that the inherent distribution of disulfide isomers are greatly affected by chang- ing pH conditions (Darby and Creighton, 1993).
Previous theoretical conclusions show that the probability of the occurrence of disulfide isomers depends on loop length (the number of amino acid residues separating the two half-cys- tines) to a power of -p; results from statistical calculations (Kauzmann, 1959; Poland and Scheraga, 1965) indicate a rela- tion ofp = 1.5, while a lattice model theory is consistent with a relation o fp = 2.4 (Chan and Dill, 1990, 1991). We examined how the observed ratios of labeled half-cystines at equilibrium are related to the theoretical conclusions. In the case of the lattice model, the RsH.i values for six half-cystines (SH-i) can be calculated as follows: 0.344 for SH-1 (Cys-ll), 0.373 for SH-2 (Cys-30), 0.101 for SH-3 (Cys-73), 0.0501 for SH-4 (Cys-731, 0.566 for SH-5 (Cys-367), and 0.566 for SH-6 (Cys-382) for a total of 2.0. According to the statistical calculations, the ratios of labeled half-cystines can be predicted to be 0.369 for SH-1, 0.412 for SH-2, 0.219 for SH-3, 0.136 for SH-4, 0.432 for SH-5, and 0.431 for SH-6. We estimated the deviations of the ob- served RSH.i values from the predicted ones, RsH.i. For three varying urea concentrations (8, 9, and 10 M urea), the devia- tions were from 0.0223 to 0.0314 for the statistical theory and from 0.0214 to 0.0272 for the lattice model theory. These very
N. Takahashi, E. Tatsumi, and M. Hirose, unpublished data.
28066 Denatured State of Ovalbumin in Denaturant
0 ' ' I , 1
1 1.5 2 2.5 3 p value
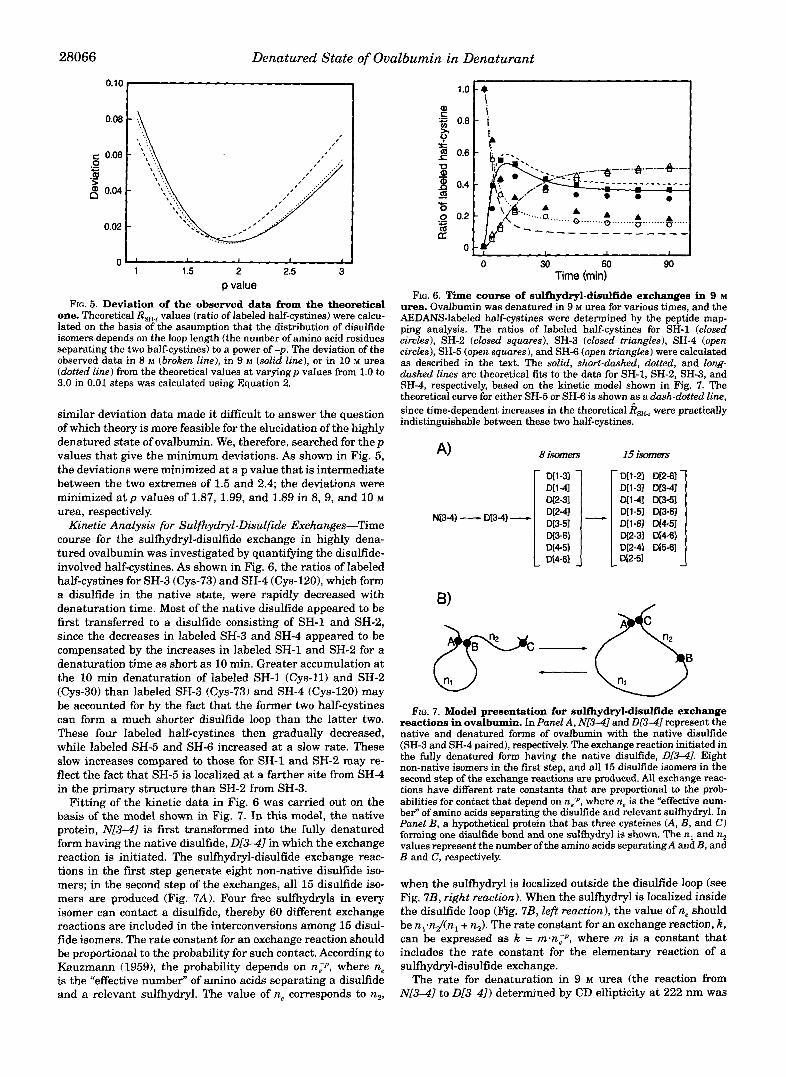
FIG. 5. Deviation of the observed data from the theoretical one. Theoretical R,, values (ratio of labeled half-cystines) were calcu- lated on the basis of the assumption that the distribution of disulfide isomers depends on the loop length (the number of amino acid residues separating the two half-cystines) to a power of -p. The deviation of the observed data in 8 M (broken line), in 9 M (solid line), or in 10 M urea (dotted line) from the theoretical values at varyingp values from 1.0 to 3.0 in 0.01 steps was calculated using Equation 2.
similar deviation data made it difficult to answer the question of which theory is more feasible for the elucidation of the highly denatured state of ovalbumin. We, therefore, searched for thep values that give the minimum deviations. As shown in Fig. 5, the deviations were minimized at a p value that is intermediate between the two extremes of 1.5 and 2.4; the deviations were minimized at p values of 1.87, 1.99, and 1.89 in 8, 9, and 10 M urea, respectively.
Kinetic Analysis for Sulfhydryl-Disulfide Exchanges-Time course for the sulfhydryl-disulfide exchange in highly dena- tured ovalbumin was investigated by quantifying the disulfide- involved half-cystines. As shown in Fig. 6, the ratios of labeled half-cystines for SH-3 (Cys-73) and SH-4 (Cys-1201, which form a disulfide in the native state, were rapidly decreased with denaturation time. Most of the native disulfide appeared to be first transferred to a disulfide consisting of SH-1 and SH-2, since the decreases in labeled SH-3 and SH-4 appeared to be compensated by the increases in labeled SH-1 and SH-2 for a denaturation time as short as 10 min. Greater accumulation at the 10 min denaturation of labeled SH-1 (Cys-11) and SH-2 (Cys-30) than labeled SH-3 (Cys-73) and SH-4 (Cys-120) may be accounted for by the fact that the former two half-cystines can form a much shorter disulfide loop than the latter two. These four labeled half-cystines then gradually decreased, while labeled SH-5 and SH-6 increased at a slow rate. These slow increases compared to those for SH-1 and SH-2 may re- flect the fact that SH-5 is localized at a farther site from SH-4 in the primary structure than SH-2 from SH-3.
Fitting of the kinetic data in Fig. 6 was carried out on the basis of the model shown in Fig. 7. In this model, the native protein, N [ 3 4 ] is first transformed into the fully denatured form having the native disulfide, D[341 in which the exchange reaction is initiated. The sulfhydryl-disulfide exchange reac- tions in the first step generate eight non-native disulfide iso- mers; in the second step of the exchanges, all 15 disulfide iso- mers are produced (Fig. 7A). Four free sulfhydryls in every isomer can contact a disulfide, thereby 60 different exchange reactions are included in the interconversions among 15 disul- fide isomers. The rate constant for an exchange reaction should be proportional to the probability for such contact. According to Kauzmann (19591, the probability depends on nip, where n, is the "effective number" of amino acids separating a disulfide and a relevant sulfhydryl. The value of ne corresponds to n2,
0 30 60 90 Time (mid
FIG. 6. Time course of sulthydryl-disulfide exchanges in 9 M urea. Ovalbumin was denatured in 9 M urea for various times, and the AEDANS-labeled half-cystines were determined by the peptide map- ping analysis. The ratios of labeled half-cystines for SH-1 (closed circles), SH-2 (closed squares), SH-3 (closed triangles), SH-4 (open circles), SH-5 (open squares), and SH-6 (open triangles) were calculated as described in the text. The solid, short-dashed, dotted, and long- dashed lines are theoretical fits to the data for SH-1, SH-2, SH-3, and
theoretical curve for either SH-5 or SH-6 is shown as a dash-dotted line, SH-4, respectively, based on the kinetic model shown in Fig. 7. The
indistinguishable between these two half-cystines. since time-dependent increases in the theoretical R,,, were practically
N13-41- D[3-41- I D[3-51 1 - 1 D W I D[1-51 D[3-61
D[3-61 DE-31 M-61 DL1 -61 D[4-51
FIG. 7. Model presentation for sulfhydryl-disulfide exchange reactions in ovalbumin. In Panel A, Nr3-41 and D ( 3 4 represent the native and denatured forms of ovalbumin with the native disulfide (SH-3 and SH-4 paired), respectively. The exchange reaction initiated in the fully denatured form having the native disulfide, D[341. Eight non-native isomers in the first step, and all 15 disulfide isomers in the second step of the exchange reactions are produced. All exchange reac- tions have different rate constants that are proportional to the prob- abilities for contact that depend on n,", where n, is the "effective num-
Panel B, a hypothetical protein that has three cysteines (A, B, and C) ber" of amino acids separating the disulfide and relevant sulfhydryl. In
forming one disulfide bond and one sulfhydryl is shown. The n, and nz values represent the number ofthe amino acids separatingA and B, and B and C, respectively.
when the sulfhydryl is localized outside the disulfide loop (see Fig. 7B, right reaction). When the sulfhydryl is localized inside the disulfide loop (Fig. 7B, left reaction), the value of n, should be nl.n2/cnl + n2). The rate constant for an exchange reaction, k , can be expressed as k = rn.n,-P, where m is a constant that includes the rate constant for the elementary reaction of a sulfhydryl-disulfide exchange.
The rate for denaturation in 9 M urea (the reaction from N[34] to D [ 3 4 ] ) determined by CD ellipticity at 222 nm was

Denatured State of Ovalbumin in Denaturant 28067
quite rapid with a first-order rate constant of 0.264 min". Us- ing this rate for the reaction from NL3-41 to 01341, the ob- served data for labeled half-cystines in Fig. 6 were fitted to the model and m and p were allowed to vary. The best fit curves shown in Fig. 6 gave K = 820 n;1.92. The value for p (1.92) was again an intermediate between the two extremes of 1.5 and 2.4, and was almost exactly the same as the data obtained for the equilibrium analysis in 9 M urea. In a previous report, Darby and Creighton (1993) have pointed out that the rates for dis- ulfide formation in bovine pancreatic trypsin inhibitor in the presence of urea depends on n. (the number of amino acids separating two half-cystines) more suitably to a power of -2.4 than to -1.5. Our best-fit calculation for their data, however, are consistent with 1.84 for the p value. The intermediate val- ues for p may be therefore common to the urea-denatured states of ovalbumin and bovine pancreatic trypsin inhibitor.
CONCLUSIONS Unlike conventional methods for protein analysis, the disul-
fide-rearrangement approach employed in the present study was found to provide unique information about the denatured state of a protein; some information about the distribution of conformational isomers by an equilibrium analysis and about the dynamic chain nature by a kinetic approach. Both equilib- rium and kinetic data for highly denatured ovalbumin revealed that all cysteine sulfhydryls in this large protein can freely encounter a disulfide bond. In addition, the greatest ratios of labeled half-cystines were detected with SH-5 and SH-6, which can form the entropically favored shortest disulfide loop. We, therefore, qualitatively conclude that ovalbumin essentially be- haves as a random coil polymoer in high concentrations of urea.
More quantitatively, the previous theoretical conclusions demonstrate that the probability of the occurrence of disulfide isomers depends on loop length to a power of -p; the p value is 1.5 for an ideal Gaussian chain (Kauzmann, 1959; Poland and Scheraga, 1965), but it is estimated to be 2.4 for a self-avoiding chain with significant exclusion volume (Chan and Dill, 1990, 1991). The p value is a highly important factor for the predic- tion of the effect of a disulfide bond on the conformational entropy in a protein; previous estimations have been all based on the Gaussian behavior for disulfide proteins (Lin et al., 1984; Ueda et al., 1985; Goto et al., 1987; Pace et al,, 1988). Both the equilibrium and kinetic data from urea-denatured ovalbumin were, however, consistent with a p value from 1.9 to 2.0. Highly denatured ovalbumin, therefore, appears to behave as an in-
termediate between the two extremes of the ideal Gaussian chain and the self-avoiding chain. The present experimental data, however, are restricted to the protein species of ovalbu- min and the denaturing conditions in high concentrations of urea. Also, the previous theoretical models (Kauzmann, 1959; Poland and Scheraga, 1965; Chan and Dill, 1990,1991) depend on the calculation using a model polymer consisting of homol- ogous monomers. Extensive examinations of a variety of disul- fide proteins under various denaturing conditions and intro- duction of a theoretical model for a protein-like heteropolymer would help better understand the highly denatured states of proteins.
Acknowledgments-We are grateful to Dr. Yoshisuke Tsunashima (Institute for Chemical Research, Kyoto University) and Professor Tokuji Ikeda (Faculty ofAgriculture, Kyoto University) for their helpful suggestions and discussions.
REFERENCES
Amir, D., Krausz, S., and Haas, E. (1992) Proteins Struct. Funct. Genet. 13, 162- Amir, D., and Haas, E. (1988) Biochemistly 27,8889-8893
Aune, K C., Salahuddin, A,, Zarlengo, M. H., and Tanford, C. (1967) J. Biol. Chem.
Bierzynski, A., and Baldwin, R. L. (1982) J. Mol. Biol. 162, 173-186 Chan, H. S., and Dill, K A. (1990) J. Chem. Phys. 92,3118-3135 Chan, H. S., and Dill, K. A. (1991)Annu. Rev. Biophys. Biophys. Chem. 20,447-490 Creighton, T. E. (1984) Methods Enzymol. 107, 305-329 Darby, N. J., and Creighton, T. E. (1993) J. Mol. B i d . 232, 873-896 Goto, Y., Tsunenaga, M., Kawata, Y., and Hamaguchi, K. (1987) J. Biochem. (7bkyo)
Greene, Jr., R. F., and Pace, C . N. (1974) J. Biol. Chem. 249,5388-5393 Kauzmann, W. (1959) in Sulfur in Proteins (Benesch, R., Benesch, R. E., and Boyer,
Kitabatake, N., Ishida, A,, and Doi, E. (1988) Agric. Biol. Chem. 52,967-973 Laemmli, U. K. (1970) Nature 227,680-685 Lin, S. H., Konishi, Y., Denton, M. E., and Scheraga, H. A. (1984) Biochemistry 23,
Marquardt, D. W. (1963) J. SOC. Ind. Appl. Math. 11,431-441 Neri, D, Billeter, M., Wider, G., and Wuthrich, K. (1992) Science 267, 1559-1563 Nisbet, A. D., Saundry, R. H., Moir, A. J. G., Fothergill, L. A,, and Fothergill, J. E.
Pace C. N., Grimsley, G. R., Thomson, J. A,, and Barnett, B. J. (1988) J. Biol. Chem.
Poland, D. C., and Scheraga, H. A. (1965) Biopolymers 3,379-399 Smensen, S. P. L., and H e p p , M. (1915) C . R. Puu. Lab. Carlsberg 12, 12-67 Takahashi, N., and Hirose, M. (1990) Anal. Biochem. 188,359365
Tanford, C. (1961) Physical Chemistry ofMacromolecuZes, John Wiley & Sons, New Takahashi, N., and Hirose, M. (1992) J. B i d . Chem. 267, 11565-11572
Tanford, C . (1968) Adu. Protein Chem. 23, 121-282 Tanford, C. (1970) Adu. Protein Chem. 24, 1-95 Thompson, E. 0. P., and Fisher, W. K. (1978) Aust. J. B i d . Sci. 31,433-442 Ueda, T., Yamada, H., Hirata, M., and Imoto, T. (1985) Biochemistry 24,63166322 Walsh, K. A., and Wilcox, P. E. (1970) Methods EnzymoZ. 19, 31-63 Weissman, J. S., and Kim, P. S. (1991) Science 263, 1386-1393
173
242,4486-6189
101,319329
P. D. eds) pp. 95-105, Academic Press, New York
5504-5512
(1981) Eur: J . Biochem. 115,335-345
263,11820-11825
York