key decision makers characterization and analysis of...
TRANSCRIPT

LIME (UBE)
Master thesis in medical science
with a major in biomedicine
Stockholm, 2009
Key decision makers –
characterization and analysis of
their attitudes toward the
authorization process and market
of biosimilars in Sweden and
Denmark
Authors: Adam Sierakowiak and Tanya Syed
Working paper version 2.0
Authors: Adam Sierakowiak and Tanya Syed
Supervisor: Jessica Norrbom, Lime (UBE)
Co-supervisor: Carl Johan Sundberg
Examinator: Anneka Ehrnst

2
LIME (UBE)
Master thesis in medical science with
a major in biomedicine
Stockholm, 2009
Key decision makers – characterization and analysis of
their attitudes toward the authorization process and
market of biosimilars in Sweden and Denmark
Abstract
A biosimilar is a biopharmaceutical that claims to be similar to a marketed reference product. These medicines have gained relevance in the last few years because of patent expirations of first generation biopharmaceuticals. The price reduction for biosimilars (estimated to 20-30%) will play a crucial role in the saving potential of medical costs, although it will not reach the same levels as that of generic chemical drugs (estimated to 60-80%). Nevertheless, this price differential still represents a great drop of spendings in terms of magnitude. This study examines how the knowledge and attitudes of key decision makers in Sweden and Denmark affect the authorization process, implementation, and markets of biosimilars. Based on this analysis, propositions on marketing strategies for biosimilar manufacturers were formulated. A qualitative, explorative, multicase study was selected and complemented with a literature review. An inductive approach was chosen for the data analysis. Representatives from the European Medicines Agency, the Medicinal Products Agency in Sweden, The Danish Medicines Agency, the European Generics Association, the European Biopharmaceutical Enterprises, Teva Pharmaceutical Industries Ltd, and Swedish medicinal products committees were interviewed. EMA has pioneered in the field of biosimilars in all regulatory aspects by creating a dynamic process for biosimilar applications, independent from political influence. The market of biosimilars looks very promising thanks to patent expiries, an increasing demand of biopharmaceutical therapies, and the need for the R&D of the biotech industry. Marketing strategies of biosimilars should involve proper product design, QSE profiling, and communication to counteract aggressive campaigning from originator companies and a general lack of knowledge on the topic.

3
“Science should drive the issue. Science should pave the road!” -Suzette Kox, 9th of December, 2009

4
Table of Content
Abstract 2
Abbreviations 7
Background 8
What are biosimilars? 8
What is the difference from other generics? 8
Manufacturing process 10
Production 11
Future economics 11
Teva 12
EU | European Medicines Agency 13
Regulatory and legal framework 14
Guidelines 14
Sweden | Läkemedelsverket 15
Denmark | Lægemiddelsstyrelsen 17
Key approval agencies outside the EU 18
USA | Food and Drugs Administration 18
China | Canada | Japan | Korea | India | Iran 18
Key organizations and projects 19
UN | World Health Organization 19
ICH | International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use 20
EGA | European Generics Association 20
EBE | European Biopharmaceuticals Enterprises 20
Research question and study design 21
Problem formulation 21
Research question 22
Aim 23
Delimitations 23
Research design 24
First phase | Literature review 24
Second phase | Multicase study 25
Why a multicase study? 26
Limitations of multicase studies 26
Prior to data collection 27
Process of data collection 27
Data reduction and analysis 27
Third phase | Data reduction and analysis 27

5
Comparison of different methods of analysis 28
How to perform an inductive analysis 29
Evaluation by the researchers 30
Limitations regarding data reduction and analysis 30
Results 32
Authorities in the EU, Sweden, and Denmark 32
Organization and documents 33
Processing 36
Future 40
Additional comments 41
Organizations (EBE and EGA) 42
Additional comments 47
Teva 49
Additional comments 52
Medicinal products committees in Sweden 53
Organization and documents 53
Processing 55
Future 58
Additional comments 60
Analysis and discussion 61
Methods 61
Past 63
The generics of the 1990s 63
EU number one 64
…And then there were biosimilars. 65
Strategies by originator companies 65
Present 67
Chain of communication and trust 67
Lack of knowledge 68
Authorization is European, implementation is national 69
European 70
National 70
Regional/local 70
Clinical 71
Limiting factors and the current strategies of biosimilar companies 71
Future 72
New products 72
Biobetters 72
Insulin 72
Monoclonal antibodies and low-molecular heparin 73
The American approach to biosimilars 73

6
Proposed strategies for biosimilar manufacturers 74
Conclusions 76
Key approval agencies 76
What are the current attitudes and knowledge regarding biosimilars? 76
What do approval agencies expect from biotech companies that wish to manufacture, market, and sell biosimilar medicines in the future? 76
What are the attitudes of national approval agencies toward decisions made by EMA regarding biosimilars? 77 How will the key approval agencies’ attitudes affect the future of the authorization process for biosimilars? 77
Future market strategies of biosimilar medicines 77
What are the main obstacles? 77
What are the potentials? 78
What are the market demands on the biopharmaceutical industry wishing to manufacture, market, and sell biosimilar medicines in the future? 78 Closing words 79
Key approval agencies 79
Future market strategies of biosimilar medicines 79
Future studies 79
Trust between EMA and prescribers in the clinics 79
Monoclonal antibodies 80
Regional regulation and implementation of biosimilars in Denmark 80
Tender issues of biosimilars and expert groups 80
Networks 80
Asia 81
Acknowledgements 82
References 84
Appendix 87
Interview questionnaire for Teva 87
Interview questionnaire for organizations 89
Interview questionnaire for the European Medicines Agency 91
Interview questionnaire for government agencies 93
Interview questionnaire for medicinal products committees 95

7
Abbreviations
BET Bioentrepreneurship Team
CHMP Committee for Medicinal Products for Human Use
DKMA Danish Medicines Agency, Lægemiddelstyrelsen
EBE European Biopharmaceutical Enterprises
EC European Commission
EFTA European Free Trade Association
EGA European Generics Medicines Association
EMA European Medicines Agency, formerly abbreviated EMEA
EPAR European Assessment Report
EU European Union
FDA Food and Drug Administration in the US
FoB Follow-on biological, alternatively follow-on protein product
ICH International Conference on Harmonization of Technical Requirements for Registration of
Pharmaceuticals for Human Use
INN International Nonproprietary Name
I. V. Intravenous
MAA Market Authorization Application
MPA Medical Products Agency in Sweden, Läkemedelsverket
NRA National Regulatory Authority
QSE Quality, safety, and efficacy
R&D Research and Development
SKL Swedish Association of Local Authorities and Regions, Sveriges kommuner och landsting
UBE Unit for Bioentrepreneurship
UN United Nations
US United States
USA United States of America
WHO World Health Organization

8
Background
What are biosimilars? Biosimilars are a type of biologically produced pharmaceuticals. “Biopharmaceuticals are
biological medicinal products that have been developed through biotechnology practices,
including recombinant human technology, gene technology or antibody methods. Biological
medical products that are biologically similar to registered biopharmaceuticals are referred
to as ‘biosimilars’ in Europe and South-East Asia and ‘follow-on biologicals’ (FoB) in the
USA.”[1]
The EMA (European Medicines Agency) has defined biosimilars as a biological medicinal
product that claims to be “similar” to a reference medicinal product. One of the criteria is
that it has been granted a marketing authorization within the region where it is to be used.
Another criterion states that studies of comparability with special regard to quality, safety,
and efficacy have to be conducted. One of the aims of such studies is also to support and
generate evidence substantiating the similarity between the biosimilar and its reference
product.[2]
What is the difference from other generics? To understand the distinction between biosimilar medicines and generic medicines, one
must first comprehend the two different concepts. According to EMA, a generic medicine is:
“a medicine which is similar to a medicine that has already been authorized (the “reference
medicine”). A generic medicine contains the same quantity of active substance(s) as the
reference medicine. Generic and reference medicines are used at the same dose to treat the
same disease, and they are equally safe and effective.”[3]
Further, EMA states that generic medicines can contain other inactive ingredients in addition
to the reference drug, but must then clearly include those substances in the package leaflet
as well as on the package itself.
According to EGA (The European Generic Medicines Association), “generic medicines”, also
called “generics”, must have an active ingredient that is bioequivalent (the mode of action
within the body must be identical) to that of the originator product. Additionally, the generic
product must comply with same standards of quality, safety, and efficacy (QSE) as are
imposed on other pharmaceutical products. [4]

9
An important point to stress is that biosimilars are not generic in the same sense as chemical
drugs (low molecular weight compounds) due to that the production process of the
innovator is impossible to be duplicated by other manufacturers.[1]
Yet another difference is that it is easier to analyze and detect small impurities in a small
molecular compound because the techniques to purify molecules under 500 kDa are much
more specific and sensitive. Biopharmaceuticals are larger; e.g. insulin has a weight of
approximately 5.8 kDa and growth hormone has an approximate weight of 2.2 kDa, whereas
traditional pharmaceuticals (low molecular weight compounds) have a weight of about 0.05-
1 kDa.[5]
The EMA does not define biosimilar drugs as generic due to differences relating to raw
materials or manufacturing processes of the biological medicinal product compared to their
reference biological medicinal product.[6]
The EGA Handbook on Biosimilar Medicines states that since all biopharmaceuticals are
produced from living organisms, the variability of the product is inevitable; even within
batches. Thus, these products require more extensive as well as very expensive development
due to the high demands of registration data.[4]
Figure 1 provides an overview of the similarities and differences between the definitions of
biosimilars and their reference products.
Figure 1. This sketch is a graphical overview on the field of biopharmaceuticals.

10
Manufacturing process The manufacturing process of biosimilar drugs is a challenging process that requires
adequate competence. It is a multifactorial development with many implications that will
exert an effect on the final product.[7]
The different stages of manufacturing of biopharmaceuticals:
1. Selection of appropriate host cell and establishment of a cell bank.
2. Protein production and purification.
3. Analysis.
4. Formulation.
5. Storage and handling.[1]
Problems could arise from the very beginning when it comes to biopharmaceuticals in
general. The selection of a host cell is vital to the whole process, as well as specific to each
manufacturer. Hence, it is of grand importance to the whole process. Every lab will handle
the cells slightly differently and since it is produced in such large scales, one can be sure of
variations in every batch of pharmaceuticals produced.[7]
Another important factor is that highly efficient expression vectors are used to gain a
maximum yield of protein product. Because these are all developed for each company
separately, they are also one of the most protected company secrets. Once again, these
aspect leads to the possibility of different scenarios in protein production.[7]
Purification processing to achieve a high quality product for the market is yet one of the
essential steps in the manufacturing process which also is considered to be sensitive
information for each company; thus, leading to great diversity among the producers.[7]
Preservatives and stabilizing compounds are used to maintain a good “shelf-life” for the
products and are all exerted to extensive immunogenicity examinations, and as seen in the
other cases, a very important and well-protected knowledge in every company
manufacturing biological agents.[7]
Since biosimilars represent a segment within the field of biotechnology, one could see on a
further perspective that they contribute to the development of a novel field of the drug
market. Biotechnology has already contributed greatly with the evolvement of protein
production. A continuation in this field could therefore lead to increased understanding of
proteomics as well as biological drug development that has the potential in saving huge
number of lives.[8]

11
Production
The adverse events that theoretically could take place with biopharmaceuticals are based on
numerous reasons. Some of the important product related factors such as the epitopes of
the biological molecule, glycosylation state, and type of cell utilized in the production line
are of great importance to the drug profile. Host related (patient) factors such as genetic
background determining antibody production and supplementary medical problems that
might have an impact in drug metabolism are vital features when determining possible
adverse event reactions. [5]
Small molecular medicines have a known risk of intervening with the immune defense
system (by triggering B-cell activation), and it is almost certain that biological
pharmaceuticals will exert the same effect. Since biological medicines are different in the
aspect of their physical and chemical nature, most biologically derived drugs induce immune
responses but with almost no clinical relevance. On the other hand, drugs believed to be
identical to human proteins can cause immune responses due to that the immune system
might confuse them with viruses because of their physiological settings; such as aggregation
if not totally dissolved. This can be hard to evaluate in preclinical studies, but one could take
into account that there have been other biological compounds, not derived from human
cells that have not led to increased numbers of adverse events. [5]
Future economics Generic and originator pharmaceuticals are said to be interchangeable. This leads to the
notion that implementation of generic medicines is thought to reduce cost burden on
healthcare systems (since generics are less expensive). The existence of these products also
stimulates the development of novel patentable products by originator companies.[4]
It has so far been shown that biosimilars in Europe contributed with a reduction of €2-3
billion per year in costs for the healthcare system on only five products.[9]
Biologicals represented the segment of the overall prescription drug expenditures that grew
the fastest between 2006 and 2007, by growing over $45 billion in the USA. This accounted
for more than 16% of the total prescription drug expenditures.[10]
Generic chemical drugs have contributed to 60-80% price reductions overall whereas
biosimilars are expected to only reduce prices by 20-30%. In any case, biosimilars are
generally more expensive and, therefore, assumed to yield greater savings due to the higher
prices.[11]
There are many similar features between the introduction of generics medicines to the
market and the introduction of biosimilars.[12]

12
The future economic perspective of the biopharmaceutical sector (biosimilars included)
seems to expand in an almost exponential rate according to this graph:
Figure 2. The expected prognosis for the biopharmaceutical market.
Biologics are predicted to drive future pharmaceutical growth.
Teva Teva Pharmaceutical Industries Ltd. (listed in NASDAQ, Seaq International in London, and the
Frankfurt Stock Exchange) is among the largest generic pharmaceutical companies in the
world, with more than 38’000 employees worldwide.[12, 13]
The company specializes in the development, production, and marketing of generic and
proprietary branded pharmaceuticals as well as active pharmaceutical ingredients. In
Sweden, Teva also supplies medical devices and are involved in parallel import of
pharmaceuticals in order to lower the costs for society.[12]
The North American and Western European markets contributed with more than 80% of
Teva’s total sales (about US$11.1 billion) in 2008. [13] More than 10% of all medicines in the
USA are supplied from Teva. In Sweden the same year, net sales almost reached half a billion
SEK (US$75.8 million).[12]
The company headquarter is situated in Israel and the company runs production facilities in
Israel, North America, Europe, and Latin America.[13]
Teva Sweden (founded in 2004) is situated in Helsingborg and also functions as the
headquarters for the Nordic countries headquarter with 29 employees.[12]

13
EU | European Medicines Agency The European Medicines Agency (EMA) is a decentralized European agency that attempts to
harmonize the functions and activities of existing national medicines regulatory bodies
within the European Union.[14, 15]
According to the EU Regulation (EC) 726/2004, a centralized authorization process is
compulsory for all biotechnical products (such as biosimilars) "...in order to maintain the high
level of scientific evaluation of these medicinal products in the European Union and thus to
preserve the confidence of patients and the medical professions in the evaluation".[16]
In a centralized authorization process, the scientific evaluation is firstly carried out by national medicinal agencies in two separate countries independently. These countries are labeled reporter and co-reporter, respectively.[17, 18]
Afterwards, the evaluations are sent to the EMA for further assessment. In the case of biosimilars, the EMA's assessments on MAAs are carried out by the Committee for Medicinal Products for Human Use (CHMP). The committee, staffed by experts from the NRAs of all EU and EFTA member states, prepares the agency's opinions with regards to medicinal products for human use.[17-19] The applicant company has an opportunity to address any inquiries formulated by the CHMP in the first assessment of the MAA.[20, 21]
If the inquiries are properly addressed, an updated assessment from the EMA/CHMP is sent to all member states and the European Commission (EC).[18, 20, 21] The EC formally casts a decision regarding the market approval of the applicant drug in all EU member states as well as Norway, Iceland, and Lichtenstein.[14, 22]
Biosimilars were officially referred to in the EU legislation in 2003.[18] The EU, unlike many
other regions, has already established an advanced legal framework and a regulatory
pathway for biosimilar products. Guidelines have also been drafted by the CHMP to clarify
the technical aspects of the Union’s policies.[23, 24] See Figure 3 for an overview of the
development process for market approval of a biosimilar drug in the EU.
The first patents for a licensed biopharmaceutical within the EU expired in 2001. As of yet,
five biosimilars have been granted market authorization within the region.[4]
If a better, unified and more efficient authorization process were to develop (for
biosimilars), one could see an increase in the accessibility of biopharmaceuticals for patients
which would be an immense benefit.[25]

14
Regulatory and legal framework
The term, “similar biological medicinal product”, was first introduced year 2003 in the EU
Directive 2003/63/EC (amending 2001/83/EC), but they are commonly known as
“biosimilars”. In this directive, they were defined as a biological medicine with proven
similarity to another biological medicine previously licensed within the EU.[18]
Directive 2003/63/EC also explained that marketing application packages must include
pharmaceutical, chemical, and biological data. Moreover, the package must contain
information on bioequivalence and bioavailability to prove comparability.[18] These features
must be characterized for both the biosimilar and the reference product.[26] Due to the
varying complexity of biopharmaceuticals, supplementary data from non-clinical and clinical
phases (thereby, providing additional evidence of comparability) may be required. The
amount of data will depend on the type and nature of the applicant drug as well as the
specifications in related guidelines.[18, 26, 27] In 2004, Directive 2004/27/EC elaborated on
legislation introduced in prior directives.[6] The legal framework was officially adopted in
October 2005.[27] Therefore, each market authorization application (MAA) is processed on a
case-by-case basis.[24]
Guidelines
Scientific guidelines published by the EMA aim to clarify the amount of data required in most
types of biosimilar application packages.[27] Three types of guidelines (overarching, general
and annex) address key issues within the biosimilar field.
The overarching guideline, “Guideline on Similar Biological Medicinal Products”, came into
effect in October 2005. It defines and outlines the concept of biosimilars as well as provides
applicants with relevant information in their quest for granted biosimilarity.[2, 24]
General guidelines on biotechnology-derived proteins shed light on quality, non-clinical,
clinical, and immunogenicity issues for both biosimilars and reference products that have
altered their manufacturing processes. [24]
Quality “Guideline on Similar Biological Medicinal Products Containing Biotechnology-
Derived Proteins As Active Substance: Quality Issues” came into effect June
2006.[24]
This document specifies quality requirements for biosimilars with regards to the choice of
reference product, manufacturing, comparability, purity, and analytical methods.[28]

15
Non-clinical and clinical “Guideline on Similar Biological Medicinal Products Containing
Biotechnology-Derived Proteins as Active Substance: Non-Clinical
and Clinical Issues” came into effect June 2006.[24]
The non-clinical guidelines address the pharmaco-toxicological assessment of biosimilars
while the clinical section covers the requirements for pharmacokinetic, pharmacodynamic,
and efficacy studies. In addition to these sections, the document also states that application
data package must include a pharmacovigilance plan (for the post-marketing phase) to
address immunogenicity and the risk for rare adverse events.[24, 29]
Safety “Guideline on Immunogenicity Assessment of Biotechnology-Derived
Therapeutic Proteins” came into effect April 2008.[24, 30]
This document contains key information on immunogenicity and provides general
recommendations for performing a systematic immunogenicity assessment that satisfies the
requirements for marketing authorization.[30]
Annex guidelines detail specific requirements for marketing authorization approval of human
insulin, growth hormone, G-CSF, erythropoietin, IFN-α, and LMWH. These requirements
include mandatory trial sizes and how to satisfyingly demonstrate equivalence to a reference
product.[24]
Figure 3. Overview of the development process of a biosimilar medicine in the EU.[4]
Sweden | Läkemedelsverket Läkemedelsverket (the Medical Products Agency, MPA) is a national agency of the Swedish
Ministry of Health and Social Affairs and oversees the regulation and surveillance of the
development, manufacturing, and marketing of medicinal products in Sweden. Its main
responsibility is to secure realistic and cost-effective ways in which patients and healthcare
professionals can access satisfactory medicinal products.[31]

16
In Sweden, the term “biosimilar” has been adopted by the MPA to denote the role of
biological drugs that are similar to a reference drug.[32] The MPA has thus far refused
automatic substitution of biosimilars and their reference drugs due to the elevated risk for
immunogenic responses. Instead, physicians must choose the most suitable medicines
depending on the needs of their individual patients. This position was reached after an
internal discussion on the EMA’S drafted guidelines for similarity between
biopharmaceuticals.[32]
Sweden also consists of local government bodies known as landsting (county councils) and regions with specific responsibilities within healthcare and dental care; more precisely, 18 county councils, two regions (Skåne and Västra Götaland), and Gotland's municipality (which holds county council responsibilities).[33]
According to the Medicinal Products Committees Act 1996:1157, each county council must have at least one läkemedelskommitté (medicinal products committee) consisting of medical and pharmaceutical experts. A local set of regulations regarding work processes and activities must also be established by the county council to provide clear guidance to the committee.[34]
The committees provide recommendations to health care professionals on the implementation of medical drugs within the county council. Recommendations must be based on previous scientific evidence and experience.[34]
See Figure 4 for an overview of the regulation of medicines in Sweden.
Figure 4. Overview of the regulation of medicines in Sweden.

17
Denmark | Lægemiddelstyrelsen Lægemiddelstyrelsen (the Danish Medicines Agency, DKMA) is a national agency within the
Danish Ministry of Health and Prevention. The agency enforces Danish legislation to ensure
that safe and effective medicinal products are available and implemented in the country.[35]
The DKMA collects and provides information on medicinal products to the public and
healthcare, with special focus on QSE aspects for all types of medicines (including
biosimilars). Additionally, it manages the pricing and reimbursement of prescribed drugs.[36,
37]
Because current biosimilar products are classified as new medicines only administered in
hospitals, the reimbursement of these medicines are instead handled by Sundhedsstyrelsen
(National Board of Health, SST).[38, 39] SST coordinates the administration of healthcare
service by advising authorities (on national, regional, and municipal levels) and the public on
specific health issues.[38, 39]
The hospitals and its services are also managed by five regions.[39, 40] The regions operate
“…healthcare, hospitals, special education, regional development, environment and
finances…” from a regional perspective.[40, 41] To manage the expenses of hospital
medicines, the regions have created an organization known as Amgros that coordinates
tenders of and hearings on public pharmaceuticals. The organization also purchases all
pharmaceuticals for the public hospitals in Denmark.[42]
See Figure 5 for an overview of the regulation of medicines in Denmark.
Figure 5. Overview of the regulation of medicines in Denmark.

18
Key approval agencies outside the EU
USA | Food and Drug Administration
The Food and Drug Administration (FDA) is an agency of the United States Department of
Health and Human Services. It is responsible for the regulation and surveillance of safety
within such areas as foods, vaccines, and biopharmaceuticals.[43]
In the US, biologically similar medicinal products are called “follow-on biologicals” or
“follow-on protein products” (FoB). The FDA lacks legal authority to approve most of these
drugs because of restrictions in the Hatch-Waxman Act of 1984.[1] The act amended The
Federal Food, Drugs and Cosmetic Act, subsequently introducing an abbreviated pathway for
FDA approval of small molecule drugs to accelerate market entry of generic products.
Therefore, legal pathways for review and approval of smaller, well-characterized proteins
(i.e. human insulin and growth hormone) already exist.[23] On the other hand, the Hatch-
Waxman Act did not amend the Public Health Service Act. Hence, a specific regulatory
process has not been established for larger biological medicines.[24, 44, 45]
China | Canada | Japan | Korea | India | Iran
China already has a regulatory framework in place for biopharmaceuticals and biosimilars.
Complete conformity between the reference product and the biosimilar must be proven
with quantifiable and extremely accurate studies.[23]
Canada is still developing a regulatory system for “subsequent entry biologicals”, which will
be based on existing frameworks for biologics, pharmaceuticals, and generics. Automatic
substitution will not be granted by the Canadian authorities.[23]
Although regulatory pathways exist for generics in Japan, a new framework is being built
exclusively for biosimilars. Biosimilars are called “follow-on biologicals” and must comply
with guidelines set by the ICH Guideline Q5E. The guidelines state that the reference and
follow-on product must be highly similar while quality differences cannot interfere with
safety and efficacy.[23]
Korea has a regulatory pathway for biosimilars but specific definitions and criteria are yet to
be formulated. Full non-clinical and quality data must be submitted for authorization. Clinical
data may be simplified if they confirm comparability. However, extrapolation of clinical
indications is not granted by the Korean NRA.[23]

19
Biosimilars are called “biogeneric products” in India and treated as entirely new drugs.
Therefore, the regulatory pathway is identical to that of innovator drugs. The first biogeneric
product was introduced in 1997 with no major adverse events reported for 10 years.[23]
In Iran, biosimilars are called “biogeneric products” and are reviewed on an individual basis.
The country already has an established criterion for biogeneric products.[23]
Key organizations and projects
UN | World Health Organization
The World Health Organization (WHO) is an agency of the United Nations, specialized in the
assessment and coordination of international public health.[46] The organization officially
implements the term, “similar biotherapeutic products”, when referring to biosimilars.[47]
In 1950, the WHO started the International Nonproprietary Name (INN) program to establish
a global system of assigning a nonproprietary name (commonly referred as an INN) to
biological and biotechnological substances. An INN allows these substances to be recognized
globally by a unique name and facilitates the regulatory process in countries where a
nonproprietary name is required for license application.[23]
In 2007, the agency hosted an informal consultation on the regulatory evaluation of
therapeutic biological medicinal products. Representatives from regulatory agencies,
industry, and academia worldwide convened to discuss the current status of regulatory
pathways and challenges in evaluating quality, safety, and efficacy of biosimilars. Although
global consensus was not reached at the conference, participants highlighted the need for
global harmonization of the regulation and marketing of biosimilars (including assignment of
nonproprietary names).[23]
Drafts on the guidelines of evaluation of similar biotherapeutic products are currently being
processed. The guidelines will address QSE aspects, regulatory issues and the relationship
between biosimilars and their reference products. [47]

20
ICH | International Conference on Harmonization of Technical
Requirements for Registration of Pharmaceuticals for Human Use
The aim of the International Conference on Harmonization of Technical Requirements for
Registration of Pharmaceuticals for Human Use (ICH) is to present recommendations on the
harmonization of technical guidelines and requirements for product registration. The
organization regularly invites representatives from regulatory authorities and the
pharmaceutical industry in the US, EU and Japan.[48]
The ICH has drafted several guidelines regarding “the characterization and quality
assessment as well as the manufacturing process for biotechnological/biological products” to
facilitate a global harmonization of the authorization of biosimilars.
The latest guideline, Guideline Q5E from 2002, is a tripartite document aimed at harmonizing
the quality assessments in comparability tests between biotechnological or biological
products with dissimilar manufacturing processes (as is the case with biosimilars with
regards to their reference products).[49] Guidelines on basic requirements for
bioequivalence (based on the FDA) and the registration of biosimilars (based on the EMA)
are still being drafted.[50]
EGA | European Generic Medicines Association
The European Generic Medicines Association (EGA) is an international non-profit
organization that represents the European generic and biosimilar pharmaceutical industry. It
closely consults European national governments and the EU in issues related to policy-
making in the given industries.[26]
EBE | European Biopharmaceutical Enterprises
European Biopharmaceutical Enterprises (EBE) is a European trade organization that
represents biopharmaceutical originator companies of all sizes in Europe. Situated in
Brussels, it was established in 2000 to provide support for its members in a wide range of
activities. These activities range from regulatory aspects, business strategies to networking
and education.[51]

21
Research question and study design
Problem formulation The life science industry is at the brink of what could be large-scale shifts in its global
markets. As patents for innovator biopharmaceuticals expire in the coming years, competing
manufacturers will have the opportunity to develop, produce, and sell new versions of these
drugs (commonly known as “biosimilars” in Europe). Hence new competitors will have to
penetrate a market that has otherwise been noticeably impermeable due to patent
regulations.
Advocates contend that biosimilars can expedite access to affordable biological medicines
for a wider patient population. Subsequent reductions in pricing can also alleviate spending
on healthcare and medication. These developments not only help increase competition
between rival companies, but can galvanize entrepreneurship and innovation within the
industry.
Although this situation is highly reminiscent of the birth of the generics field in the 1990s,
the emerging biosimilars market presents a new set of ambiguities regarding market
dynamics, legal frameworks, manufacturing, substitution, and patient safety.
Originator companies, for instance, want to maintain their presence in the market and,
subsequently, do not welcome the introduction of biosimilars from competing companies.
Amongst other issues, they argue that batch variations are a key manufacturing issue that
can interfere with the quality and therapeutic effect of biosimilar drugs. Therefore, patients
and healthcare providers will have to weigh in on the safety issues of these drugs; especially
with regards to the risk for immunogenic responses. Moreover, approval agencies are facing
difficulties in establishing and implementing new legal frameworks because of the unique
problems linked to the use of biosimilars (such as naming, labelling, interchangeability, etc).

22
In 2007, Sweden was the first country in the world to grant market approval to Sandoz for
the biosimilar medicine, Omnitrope®[1]* The market approval opened the door for the
introduction of new biosimilar medicines in the Swedish and Danish markets. A continuation
of this development is seen, when one of the world's largest providers of generics, Teva
Pharmaceutical Industries Ltd received a market approval for its biosimilar medicine,
TevaGrastim®, in 2008. Yet there are several questions to be addressed if one seeks to
market biosimilar drugs in Sweden and Denmark:
What are the current attitudes of key decision makers regarding biosimilars?
How do these attitudes affect the approval pathway for biosimilars?
What will the market look like for biosimilars in the coming years?
What must companies do if they wish to market biosimilars successfully in the given
markets?
Research question This paper pursues the following overall research question:
What are the attitudes of key decision makers and how do those attitudes affect the
authorization process and market of biosimilars in Sweden and Denmark?
Characterize and analyze the attitudes of key approval agencies towards biosimilars:
What are the current attitudes and knowledge regarding biosimilars?
What do approval agencies expect from biotech companies that wish to manufacture, market, and sell biosimilar medicines in the future?
What are the attitudes of national approval agencies toward decisions made by EMA regarding biosimilars?
How will the key approval agencies’ attitudes affect the future of the authorization process for biosimilars?
Characterize and analyze the current and future market of biosimilar medicines:
What are the main obstacles?
What are the potentials?
What are the market demands on the biopharmaceutical industry wishing to manufacture, market, and sell biosimilar medicines in the future?
Regulatory approval was first granted by the EMA, but EU members independently choose if they
wish to implement these grants in their own country.

23
Aim The aim of the study is to examine how the knowledge and attitudes of key decision makers
in Sweden and Denmark affect the authorization process, implementation, and markets of
biosimilars. The analysis will be processed to formulate propositions for businesses seeking
to develop, produce, market, and sell biosimilars within Sweden and Denmark. The market
of biosimilars in the given countries will see an expedient growth in the coming years,
making this field of research very relevant for the future of both healthcare and life science
industry.
Delimitations This study will look at the authorization process and markets of biosimilar medicines within
Sweden and Denmark because these two markets are closely interconnected.** The EU,
namely the EMA, will serve as a backdrop due to its intimate interactions with the Swedish
and Danish approval agencies. General information on the global arena of biosimilars will be
illuminated in the Background. However, further elaborations on the developments in other
countries and regions will not be carried out unless they have a direct link to Sweden and
Denmark.
The investigation will identify current knowledge and attitudes of key players within the field
of biosimilars in Sweden and Denmark. It will also explain how their knowledge and attitudes
influence the approval pathways and markets of biosimilars. Thus, the four key groups of
research units are:
Figure 6. List of key research units in the given study.
** Medicon Valley, http://www.mediconvalley.com/

24
Due to time constraints and thanks to their extensive experience and knowledge within the
field of biosimilar and generic drugs, the third key group of research units (Group III) will be
represented by the world’s largest provider of generics, Teva Pharmaceutical Industries Ltd.
Also, clinics and reference companies will not be contacted during the course of this
investigation due to time constraints and, therefore, do not qualify as research objects. They
will be represented by relevant trade organizations. Time constraints also prevent contact
with representatives from the Danish regions or the National board of Health. Instead, a
member from the DKMA with direct knowledge on the national regulatory dynamics and a
representative from Teva’s Danish headquarters will be interviewed. Because of the
extensive material available on monoclonal antibodies, this study will not elaborate on the
scientific aspects nor any other aspect of this matter to any larger extent.
Research design The study was performed according to the BET (BioEntrepreneurship Team) format. In a BET
project, several students collaborate with external companies/organizations and UBE to
process the research question. Two students from the Biomedical Program at Karolinska
Institute participated in the given project.
Figure 6 provides an overview of the research design of the given study.
Figure 7. Overview of the research design.
First phase | Literature review
In the first phase of this study, background information was gathered through a literature
review of several key sources to form a better understanding of biosimilars, the
pharmaceutical market, and the authorization process of drugs. The review mainly involved
articles from the online databases listed in Figure 8.

25
Figure 8. List of online databases accessed during the investigation.
Because of large informational gaps in current literature on the biosimilars field and its
authorization pathways, the method was complemented by the implementation of a
multicase study.
Second phase | Multicase study
In the second phase, a qualitative, explorative multicase study was conducted via semi-
structured interviews with key persons, people with special competence of biosimilars
working for Teva, and organizations within the biopharmaceutical industry. Agencies
involved in the authorization process in Sweden, Denmark, and the EU were also
interviewed for a more extensive investigation of their attitudes and role in the regulation of
biosimilars. Members of advisory committees in Sweden were interviewed for a deeper
understanding of the local regulation and implementation of biosimilars. Table 9 provides an
overview of the interviewee groups in this study.
Table 9. Groups of interviewees in the given study.

26
Why a multicase study?
Properly executed case studies are sometimes mandated as a supplementary research tool
when existing literature alone cannot provide enough data. The approach is known as
triangulation, referring to the use of additional data sources.[52] Because this report aims to
investigate a relatively new and continuously growing field within the biopharmaceutical
industry, case studies can help evaluate areas that have yet to be examined in current
literature.[53]
Due to current consensus which lacks theories in this topic, the approach chosen for this
study involves building theories as opposed to theory-testing.[53]
A case study is an investigational method in which an empirical inquiry is conducted to
explore a phenomenon in its physical setting. Each case is defined as a singular experiment
and not as a sample. They must be selected on the grounds that they will provide
indispensable information and aid in theory formulation; also known as theoretical sampling.
Therefore, the number of cases and their characteristics as well as the execution of each
case study determine the scientific rigor of the entire investigation. This approach can be
contrasted to random sampling which is applied during statistical analysis of an unknown
parameter of a population.[53]
A well-executed single case study can produce a rich description of a specific case. Because
the given report will examine the growing biosimilars market and authorization process in
several countries, multiple case studies (also known as multicase studies) are more suitable
for this investigation. Multicase studies provide a stronger foundation for theory formulation
because the theories are based on a wider range of empirical data.[53]
Limitations of multicase studies
“The human factor is the great strength and the fundamental weakness of qualitative inquiry
and analysis.” [54]
In a quantitative study, empirical data is analyzed according to a positivistic understanding of
research (in which a method is said to be reliable if it yields reproducible results). Qualitative
studies, however, are not aimed at producing exact algorithmic data but general depictions
of complex social issues. These depictions are unique in nature, cannot be replicated, and
necessitate the application of appropriate methods and precautionary measures.
Subsequently, several limitations regarding the different phases of multicase studies must be
taken into consideration when reviewing this paper.[52, 53, 55]

27
Prior to data collection
Interviews must be carefully prepared, executed, and assayed to limit bias and insufficient
data. Such actions include theoretical sampling of cases (and interviewees) and carefully
formulated interviews.[53]
Process of data collection
Confidentiality issues in the life science industry limited our ability to retrieve crucial
information considerably. Also, interviews can only provide situation-dependent depictions
and were complemented by other data sources (such as the aforementioned literature
review). However, the ambition of this investigation is not to develop general time-
independent theories but to understand the complex phenomenon of the growing
biosimilars field as well as shed light on a very important process of authorization which
today has vast differences depending on geographical location.
Data reduction and analysis
See “Third phase | Data reduction and analysis” for more information.
Third phase | Data reduction and analysis
Reduction and analysis of data from the literature review and multicase study were carried
out in close consultation with UBE and Teva. The objective was to characterize and analyze
the attitudes of key decision makers towards the authorization process and market of
biosimilars mainly in Sweden and Denmark. The findings also served as a platform to
formulate propositions for improvement of businesses seeking to produce, market, and sell
biosimilar medicines.
Data collected during the literature review helped build a basic understanding of the core
issues in the research topic as well as compose the textual contents of Theoretical
Framework, Background, and Methods. The sources, mainly retrieved from online databases,
were critically assessed on an individual basis.
A general inductive approach was chosen to analyze the qualitative data collected in the
semi-structured interviews. The purpose hereof was to reduce the amount of qualitative
data in a structured and logical manner as well as draw adequate conclusions in order to
form a platform for further theory development.

28
The inductive approach provides an opportunity for a researcher to analytically essentialize
and characterize the great amounts of raw data into lesser, more organized and further
developed topics.[56]
One could consider this method as a means of mainly transforming the collected interview-
data to initial brief summaries of texts that will provide the establishment of clear justified
linkage between the data and the research objectives, and thus leading to evidentially based
theory development.[56]
The method described on the previous page is mainly based on these principles[56]:
The evaluation objectives play a critical role in the data analysis, since it provides a
narrowed spectrum of the main topics as well as it maintains the focus. Multiple
reviews of the data and thorough investigations will ensure that the discoveries
made are solely derived from the source data and not outcomes of any predefined
models.
Characterization of categories will provide a firm layout for further classification of
the data collected.
The conductors of the investigation are responsible for evaluating the relevancy of
the categories. The experience along with the judgment of the evaluators plays an
equally important role.
Conclusions drawn from the raw data, by the investigators, might differ.
Assessment of the relevance and adequacy in the results conducted can be carried
out in the same fashion as when reviewing other qualitative analyse methods.
In consideration of the results retrieved in a study strategically using the inductive approach,
one will gain a greater multitude of perspectives, thus preserving the nature of the research
objective by not limiting the variance in the outcome of the data analysis.[56]
Comparison of different methods of analysis
Phenomenology is another analysis method used for summarizing experiences by single
subjects that were selected indiscriminately based on their experiences (see comparison
figure on next page). Due to this specific reasoning, it would not suit a study with an aim to
build theory based on semi structured interviews.[56]
Discourse analysis shines a light on the rhetorical perspectives of a text and emphasizes on
the different perspectives present in such a report, therefore not so useful in this study.[56]

29
Grounded theory, however, is an analytical method closely related to the general inductive
approach. The main difference is that grounded theory keeps another type of coding system,
which in this case could limit the multitude of perspectives that we would like to maintain.
Indeed the grounded theory is an excellent approach when it comes to theory building, yet
there is a risk of failing in keeping a strong relationship with the main objectives through
time. It allows oneself to maybe develop a theory that is less based on the aim of the study
and deeper related to the philosophy behind it.[56]
When discussing different methodologies suitable for this study, it has been agreed upon
choosing the general inductive approach because of the above reasoning. See Table 10 for a
comparison of different qualitative analysis approaches.
[56]
Table 10. Comparison of qualitative analysis approaches.
How to perform an inductive analysis
Initially, one has to collect all the raw data, in this case the interview answers, and carefully
study it, while categorizing different segments of text. The segments will now each be
assigned a category and further be organized in a mesh of relationships.[56]
This could be explained as such (see Table 11 for a graphical representation)[56];
I. Collection of data; writing it down and processing it into a uniform format.
II. Thorough reading and understanding of the data, in such detail so that one obtains a
vast understanding with its implications.

30
III. Category founding, on two levels; the first level to be derived from the study objectives
(this category is superior in a hierarchical relationship to the second category), and a
second level of categories derived from the interview replies.
IV. Organization phase; i.e. sorting out text segments that do not belong to any category
as well as detect segments of text that might fit the description of multiple categories.
V. Further metamorphosis and processing; defining subtitles within the categories and
further reducing the number of categories by fusion of similar statements.
Table 11. Explanation of the coding process using an Inductive Analysis.
Evaluation by the researchers
It has been decided upon an “Independent parallel approach” for evaluation processing. I.e.
when an initial investigator processes all the data into a system of categories, and a second
researcher is then asked (without knowledge of the initial categories) to carry out the same
procedure. The data has then been evaluated from two different examinators and can then
be compared. If there is a major consensus in what the initial categories are then further
development can be proceed as planned. If, however, there is a significant deviation in
points of view, then one has to investigate the reasons behind it.[56]
Limitations regarding data reduction and analysis
Data reduction and analysis from a literature review does not guarantee a realistic reflection
of the given topic even if an extensive and thorough search is conducted. To increase the
scientific rigor of this phase, each data source was carefully considered and critically
analyzed. As previously mentioned, the literature review was complemented by a multicase
study to fill any informational gaps and to address the research question more
accurately.[52]

31
In the final stage of a multicase study, informant and investigator bias can strongly influence
the outcome of data reduction and analysis. A number of methods were enforced to limit
these problems.[52]
Triangulation (in this case, a combination of a literature review and a multicase study) can
reduce investigator bias if performed properly. Each method provides its own set of valuable
information that cannot be retrieved by other means. Comparing and contrasting data from
different sources can, therefore, bring depth and credence to the findings.[52]
Informant bias can also be lessened during the categorization process by allowing two
evaluators to independently assess and categorize the data. Afterwards, overlapping and
differences in their results are discussed, hopefully bringing several viewpoints to this stage
of the process without losing the overall focus.[52]
Interpretations of empirical evidence must be put into a historical and societal setting to
bring contextual relevance to the conclusions. For instance, data as well as the particular
interests of the interviewees were juxtaposed to reduce informant bias. It should be noted
that “tactical answering”, a form of bias, can render crucial information in certain
studies.[52]

32
Results
The semi-structured interviews were simultaneously led by both thesis students but the
collected data was summarized in the following manner:
Table 12. The summaries of the interviews were divided between the two thesis students.
The results hereby presented are divided into the previously described interviewee groups
(see Table 9 in Methods). Considering the amplitude of the answers received, the following
section is a summary of all interviews conducted.
Authorities in the EU, Sweden and Denmark
The following individuals were interviewed for in-depth information on the EMA’s, MPA’s and DKMA’s approach toward biosimilars:
Table 13. List of interviewees from agencies in the given investigation.

33
Organization and documents
What are the areas of responsibilities within the biosimilar field and how are they divided
between units/people?
EMA: John Purves and his working party are responsible for the coordination of biosimilar MAAs in the following sectors:
Table 14. Sectors within the EMA that are involved with biosimilar MAAs.
Reporting countries and their respective teams are assigned by Purves' team to assess all sectors listed above for a given biopharmaceutical MAA. The assessments are, thereafter, sent to the CHMP and reviewed by all member states. Questions are generated during the reviews which must be properly addressed by the applicant company. The procedure is repeated once more and the outcome of the MAA will depend on how the company resolves the questions.
An additional sector is known as Information to Healthcare and the Public.
MPA: There are no specific biosimilar working parties within the MPA. Instead, these products are processed in the following sectors:
Table 15. Sectors within the MPA that are involved with the regulation of biosimilars.
DKMA: Denmark does not view biosimilars as common generics. All biological drugs are firstly approved by a centralized procedure via the EMA. The DKMA can act as a reporter or co-reporter. It can also review assessments through the CHMP. Although biosimilars mainly lie outside the reimbursement responsibilities of the DKMA, they are processed in the following sectors:

34
Table 16. Sectors within the DKMA that are involved with the regulation of biosimilars. *Several departments manage the spending and safety reports on approved biosimilars. Quality issues will
also involve inspectors and testing laboratories.
Seeing that the EMA is the only approval agency within the EU, what does the interviewed
agency feel are the local authorities’ areas of responsibilities with regards to biosimilars?
EMA: The EMA is responsible for the management and coordination of reviews. European Assessment Reports (EPARs) are summaries of approved characteristics reports. NRAs from member states review the EPARs from a cost perspective and must set up proper pharmacovigilance systems. Data from such systems are fed into a common database.
MPA: The EMA is not the formal approval agency for medicines in Europe. Instead, the EMA assists the CHMP in its work procedures, while the EC formally grants MAs. The MPA is one of 25 member states in the CHMP. Thus, it can function as a reporter, co-reporter or a member state that expresses opinions on assessment reports. Additionally, the MPA publishes monographs on how biosimilar post-licensed products should be implemented within Sweden.
DKMA: See response in the previous question!
How well-informed is the agency with regards to the legal framework and guidelines set up
by the EMEA (Please use the Likert scale): 1 Very poor 2 Fair 3 Good 4 Very good 5 Excellent
Please state what information is generally lacking in the documents and how these gaps
affect the agency’s work with biosimilars.
EMA: MAAs are not only assessed case-by-case but also according to experience. The guidelines are organized in relation to a theoretical hierarchical pyramid, in which more detailed guidelines are supported by the extensive contents of the underlying guidelines (See Figure 17). As of yet, guidelines have been drafted in therapeutic areas prior to the market authorization of a biosimilar product in those areas (annex guidelines).

35
Figure 17. The guidelines are drafted according to a hierarchical pyramid.
Figure 18. The relationship between the physicochemical complexity and analytical limitations of a pharmaceutical.
In general, pharmaceuticals exhibit a spectrum of varying physicochemical complexity, which
can be visualized on a graph (see Figure 18). The limitations on the analysis of a
pharmaceutical escalate with increasing complexity of its active substance. The spectrum of
complexity also holds true for the varied complexity of biosimilar products. Insulins are
simple and easier to define, while EPOs and GCSFs are still considered complex and difficult
to characterize with contemporary tools.
Purves and Lönngren do not believe that the documents contain gaps from a procedural or scientific point of view, because the EMA held a series of workshops with interested parties to create proper legislation and guidelines. Guidelines function as an area of neutrality that allows interested parties to reach a common understanding in an otherwise highly polarized field of the medical industry.

36
In the beginning, the implementation of such documents was slightly problematic but those issues have been addressed. There is a need for more guidelines in new therapeutic areas, such as low-molecular heparin and molecular antibodies.
MPA: Jonsson believes that the agency's knowledge on the documents listed above are very good. Greater emphasis is placed on the quality aspects of a biosimilar MAA than its efficacy properties, because efficacy is already well-characterized by the originator company.
The MPA was active in the debate on biosimilarity and the drafting of the guidelines; either via Working Parties or by expressing its opinions on these documents. Guidelines are not binding documents but provide great freedom in the navigation of an investigation. Johnson cannot identify any gaps but explains that the guidelines are meant to be dynamic and easily altered in the wake of new problems.
DKMA: Ersböll believes that the DKMA staff is well-acquainted with the guidelines. He is uncertain about the agency's knowledge on the EMA's legal framework, because the DKMA is primarily comprised of scientific personnel with limited understanding of legal terminology.
Processing
How much difference between innovator drugs and biosimilars is acceptable? Elaborate.
EMA: The legal framework is used as a reference for the definition of biosimilars. Guidelines
are based on the legal framework and address administrative and scientific aspects.
Currently, the tests listed in Table 19 must be executed between the reference product and
the biosimilar equivalent for market approval. Differences detected in the quality module
must be justified clinically and non-clinically. Assessments of the MAAs are product-specific
and company-specific.
Table 19. Comparability and quality must be characterized in the data package of a biosimilar MAA.
MPA: The amino acid sequence, three-dimensional structure and glycosylation patterns
should be identical. Any dissimilarity must be shown to have very little impact on the
product as a whole.
DKMA: Ersböll explains that there should be no meaningful clinical differences. He cannot
provide details but explains that subtle difference can be justified if they do not affect safety
and efficacy. The DKMA consults biological experts in these matters.

37
How many biosimilar drugs have or have not been granted authorization by your agency
upon application? Why? Please exemplify.
EMA: 17 applications were assessed. 13 applications were granted, three applications were
withdrawn by the company while one application was given a negative opinion (due to poor
scientific data).
MPA: It is difficult to state how many biosimilars are currently available on the Swedish
market. Johnson estimates that about 15-20 different products are marketed, although most
of them are duplicates with different names from the same manufacturers.
DKMA: The DKMA officially recognizes all drugs that are authorized by the EMA. Individual
physicians, however, decide if they will opt for an originator product or a biosimilar
equivalent. Amgros, a public body owned by the five Danish regions, is in charge of the
purchase of hospital pharmaceuticals. Once per year, the body organizes tenders and
purchases pharmaceuticals for all public hospitals in Denmark.
Neupogen (Amgen), the first authorized filigrastim, was initially used to diminish
neutropenia in a variety of conditions. Nowadays, it is administered to patients in
preparation for stem cell transplants. In later years, physicians have opted for pegylated***
filigrastims to treat neutropenic patients. Since Neupogen is still the only originator non-
pegylated filigrastim on the market, physicians in Denmark are hesitant about shifting to its
biosimilar equivalents (which are deemed unsafe).
In contrast, several growth hormone (GH) products have existed on the market for several
years prior to the development of biosimilars. Physicians in Denmark are, therefore, less
anxious about opting for a biosimilar growth hormone.
What are the obstacles/problems regarding the authorization/implementation of
biosimilars and how are they dealt with? (Labeling/naming, immunogenicity, etc.)
EMA: Biosimilar MAAs are more complex than that of conventional generics, and their
outcomes largely depend on the competence of the applicant companies.
Obstacles/problems regarding the implementation of biosimilars are not addressed by the
EMA. Other agencies and bodies deal with these issues from other perspectives. WHO, for
instance, has established an INN program for biosimilars. Its application on glycosylated
molecules, however, is still widely debated because such molecules exhibit inherent
heterogeneity within and across manufacturers. The European Commission sent a general
reminder to all member states about the importance of proper pharmacovigilance systems.
More education in medical schools about reporting adverse effects could be advisory, Purves
suggests.
*** Pegylation is the process in which a polyethyline glycol polymer is convalently attached to another molecule, as to camouflage the substance from the host’s immune system; thereby, reducing the risk for immunogenic responses.

38
Table 20. Obstacles/problems of biosimilars
identified by the regulatory authorities.
Table 21. Important issues identified by the
regulatory authorities that should be addressed
by applicant companies.
MPA: No biosimilar product has been
withdrawn from the Swedish market.
DKMA: Many physicians refuse to use
biosimilar filigrastims. As of yet,
Amgros has not purchased any
biosimilar filigrastims. Because of
lobbying from originator companies,
the public body will probably not
change its stance on these products.
There seems to be a clash of
communication between interested
parties.
Aside from comparability and pharmacovigilance, what are the most important issues
biosimilar companies should keep in mind when applying for market approval? Please list
them in descending order of importance.
EMA: Competence and knowledge are the most vital assets for an applicant company. Most
problems can be addressed through proper comparability exercises and pharmacovigilance
plans. Seek advice from the EMA before submitting MAAs. Such interactions can only be
productive if the company willingly and regularly shares its data with the agency, preferably
from an early stage.
MPA: Biosimilar products face the
same sort of complications as any
other biopharmaceutical and these
concerns are addressed through the
centralized procedure. Because
originator companies often change
their manufacturing process once the
authorization is granted, biosimilar
products should not be viewed any
differently.
All companies should follow the guidelines
and secure competent personnel. They
should also adhere to the principles of GMP,
and build product familiarity and
conceptualization within the company.
DKMA: Although the debate is reimbursement-driven in the EU, immunogenicity is still the
key issue. Ersböll suggests that applicant companies must choose the right PK models and

39
experiments. He adds: if the Obama healthcare reform is enforced, the US might have to
look at the saving potential in biosimilars because of increased healthcare costs.
Are there standardized follow-up routines for post-marketing data of biosimilar drugs
(compared to that of innovator drugs)?
EMA: Pay attention to QSE concerns and pharmacovigilance plans.
MPA: There are currently no pharmacovigilance systems specifically designed for biosimilar
drugs. Jonsson personally feels that the need for such systems would indicate a need for a
re-assessment of the entire biosimilar field. A product would never be authorized by the EC
if its QSE properties were deemed unsafe (especially since the originator products are
available on the market). Sweden also has a functional and safe prescription system.
DKMA: There are currently no pharmacovigilance plans but such a system will be set up in
the future; probably separate from that of the reference products. In the long-run,
biosimilars must show benefit ratios (safety) and efficacy.
Does the agency work with external non-government parties to address the issue of
biosimilars? If so, could you describe these interactions?
EMA: The EMA is not involved with cost aspects (which is a national issue) and can,
therefore, steer clear from political issues. The agency seeks advice and expertise from
member states which basically strengthens the European network. Occasionally, universities
are contacted for indispensible insights. Guidelines are drafted in-house by member state
experts before they are sent to the industry for feedback. Although industry responses are
taken into account, the EMA has the final say. The EMA also interacts with ICH, various NRAs
and other types of agencies around the world.
MPA: The MPA interacts with pharmacies, authorities, and medicinal products committees.
It also provides information upon request. There are no regular collaborations with regards
to biosimilars.
DKMA: So far, the DKMA has carried out the evaluations independently. In the future,
physicians might be consulted for the development of biosimilar-specific systems. Such
initiatives might also help the professionals familiarize themselves with the concept of
biosimilars.

40
Table 22. Important issues identified by the
regulatory authorities that should be addressed
by applicant companies.
Future
Excluding the price reduction, which other benefits of biosimilars have been identified by
the agency? Please rank them according to importance in descending order.
EMA: Purves cannot identify any other difference between biosimilar and reference
products but the price reduction. The role of the EMA is to grant marketing licenses to drug
companies.
Lönngren explains that once the patent has expired, competition within the market
increases accessibility to the product. He concludes that a variety of options is favorable
because patients can react differently to available medicines.
MPA: The potential price
reduction is the greatest benefit
of biosimilars. Some biosimilar
products can also increase
product accessibility and
diversity. If only a few options
exist for a given type of
biopharmaceutical, production
disturbances can greatly hamper
access to that drug. As of yet,
originator products and their
biosimilar equivalents are not
considered automatically
interchangeable by the MPA because
more information is needed on
immunogenic risks.
DKMA: Ersböll says that the potential price reduction is the only societal benefit of
biosimilars. Even though these products promote competition, ”...it still boils down to
economy”.
Can the agency foresee changes with regards to the authorization process of biosimilars
within the EU in the next five years? If yes, what should the applicant company consider
when handing in an application in the next five years with regards to these changes?
EMA: Changes in current documents are not expected but additional guidelines will probably
be drafted in the next few years.
MPA: The MPA cannot provide a definite answer. Depending on the progress of biosimilars
in the next few years, demands might be alleviated or broadened.

41
DKMA: The DKMA cannot provide a definite answer but the agency feels that clear guidance
on certain products is still lacking. On the other hand, the EMA can still tap into a large bank
of experience since the current system for the market authorization of medicines in Europe
was already in place 25 years ago. Monoclonal antibodies constitute the next big challenge
for the EMA.
What are the views of the agency with regards to the biosimilar debate in the U.S.? How do
these views affect EMEA?
EMA: Purves explains that the EMA was the first agency to set legal framework and
guidelines in place for biosimilar products. In the US, the lack of legislation is largely due to
patent issues. There are some clusters of bilateral discussions, where the EU shares its
experience within the biosimilar field with the US. The American debate, however, has no
influence on the European stance on biosimilars.
MPA: The situation in the US is very different to that of the EU. The US has one federal
agency, one set of law, one country, and one population. There are many member states in
the EU and, subsequently, several sets of laws that must be considered when processing a
European MAA. Therefore, it is logistically easier for the US to make exceptions or create
entirely new pharmaceutical classes. On the other hand, the American debate on FoBs is
lagging, which is probably sensed by the ICH.
DKMA: The American debate has no effect on the Danish market although there are bilateral
talks. Ersböll feels that FoBs should be approved. If so, this change would lead to more
interactions.
Additional comments
EMA: Purves adds that monoclonal antibodies constitute the next area to be processed.
Even though current technology makes it nearly impossible to address such biosimilars
products, the analytical tools might be in place in a few years!
MPA: Patents have only expired for a limited number of biopharmaceuticals in a few
therapeutic areas. Although the implementation of biosimilars varies across the EU, the
knowledge on these medicines is consistent. The MPA does not interfere with the work
procedures of other countries. The ICH is an appropriate forum for such initiatives, but a
global consensus cannot be reached due to the varying challenges around the world.
Biosimilars are mentioned in medical journals published by the MPA.
DKMA: The first originator EPOs were administered subcutaneously. The first biosimilar EPOs
were administered intravenously; thus, they were not biosimilar in terms of administration.
Therefore, physicians were and still are hesitant about using biosimilar EPOs.

42
Table 23. Society benefits of biosimilars identified
by both the originator companies as well as the
biosimilar producers.
Because regular insulin is already inexpensive in developed countries, biosimilar companies
cannot expect much profit and demands in that market. Insulin analogues, however, are
expensive in the West which could be of greater interest to the biosimilar industry.
Organizations (EBE and EGA)
Could you please specify the main society benefits (of biosimilars) identified by the
organization that you represent?
EBE: There would be no biosimilars if there were no originator products. The biosimilars
which have been approved for marketing and placed on the market are beneficial in that it
increases access of biopharmaceuticals to patients. In different markets where biosimilars
have been made available, it has been observed that biosimilars compete with originator
products.
Biosimilars have to compete with innovators as well as “biobetters” which according to us
would qualify as second generation of products that the originator would put on the market.
EGA: Biosimilars support a cost efficient and sustainable healthcare.
Biosimilars introduce competition (both by challenging the originator company as well as
other biosimilar producing
companies) in the market place and
end the market monopoly of the
originator product after its patent
expiry.
The advantages of biosimilar
development is that biosimilar
products are better studied and
characterized than the reference
products (characterized 10 years
earlier or more), using the latest
state of the art technology and quality by
design principles for the development.

43
Table 24. The main obstacles with biosimilars
identified by both the originator companies as well
as the biosimilar producers.
What are the obstacles? (with special regard to the reference companies and with special
regard to competitive generic companies.)
EBE: Apart from Novartis, the innovators have not entered the “biosimilars business”. So the
obstacles to biosimilars development, market authorization, and commercialization are
mainly those faced by generic manufacturers.
The reality is medical practice and the fact that a biosimilar is not identical to its originator
product. In the case of biotechnology medicines, these are complex molecules and a
biosimilar can only be deemed similar to its reference product.
We feel that the substitution from one
originator product to the biosimilar or
even substitution (shift from) one
originator to another originator in the
same class should be carried out under
heavy supervision by the treating
physician, so automatic substitution at
the level of the pharmacy is not
advised.
Another point is that the
interchangeability must be held under
supervision, because you may have a
different clinical response, require a
different dose, induce immunogenic reactions
etc.
EGA: Some interested parties spread
misinformation about biosimilars not at the authority level but in the market place. Fear
mongering started before biosimilars were approved and even before a framework was in
place. They have used different forums, e.g. international organization for patients and the
UK parliament. National authorities within the EU member states are more and more
concerned by this practice and work towards addressing this issue.
It is really noticed when the biosimilar companies are approaching physicians in attempts to
market their products. The companies marketing biosimilars have noticed that the road has
been made slippery and they have to reassure specifically prescribers about the QSE.
Another limiting factor for “number of players” in the biosimilar world is the financial
investment that needs to be done compared to the “small molecule world” where one might
have around 30 different competitors on one small molecule.
The competitive generic companies are not seen as an obstacle, but rather as the nature of
this type of industry.

44
Do you see skepticism among decision makers in authorities – local as well as international
as an obstacle, and if so, what kind of skepticism are they mostly indulged with?
EBE: Once the biosimilar product has gone through the rigorous assessment at the EMA,
there is no need for worrying, since these products have gone through the set of safety,
quality, and efficacy assessment and we can welcome that patients have access to more
alternatives.
There is no skepticism. One just needs to learn by doing and by commercialization.
EGA: There is no known scepticism at authority level during the decision process. QSE
qualities are checked during the approval process like for any other product. Authorities do
not have and cannot have different criteria for the approval of biosimilars.
The data requirements for biosimilar approval in the EU are very high and only products with
a positive benefit/risk balance and which are shown to be comparable at QSE level with the
reference product get a marketing authorization.
The regulatory framework in the EU is delivering and not every application successfully
meets the EU standards.
How many units or people are directly involved with the subject of biosimilars within the
organization?
EBE: It is a small trade association so only one person in addition to me knows the subject
matter.
In addition, there is a biosimilar task force which is a European task force, composed of
experts from around 15 companies (only originator companies) contributing with policy-
experts, regulatory experts, and clinical development experts.
EGA: The member companies that develop biosimilars have their representation within the
association.
Do you have any interaction with other organizations or companies?
EBE: We represent the industry in all official consultations with the European commission,
with the EMA (especially the biosimilars Working Party). Through our member companies,
we interact with member states’ authorities; either in the pricing and re-imbursement of
drugs or the regulatory filings.
EGA: No response.

45
Table 25. The attitude towards the current marketing
of biosimilars identified by both the originator
companies as well as the biosimilar manufacturers.
Which are the current attitudes of the organization towards the marketing process of
biosimilars?
EBE: We welcomed the
sound and robust
regulatory framework for
biosimilars in Europe
which we helped establish,
though there are still a few
issues which remain to be
addressed at a European,
or a member-state level.
These issues were not
properly addressed by the
European regulatory
framework that the EMA
has published. It includes the naming of
biosimilars (the INN-naming), the
substitution and interchangeability,
labeling, and the post-marketing
authorization.
Contact has been made with the WHO, who is responsible for the INN-naming process.
When it comes to substitution and interchangeability, it is a matter for the member states to
act upon and we are pleased to see that after 2 or 3 years of discussion between parties at
the national level, there is a majority of member states now discouraging automatic
substitution in the case of biosimilars.
In the issue of labeling, we were pleased to see that the EMA recognizes that, in the case of
biosimilars, the label has to indicate that it is a biosimilar product.
In post-marketing authorization, there is currently a review of the European legislation on
pharmacovigilance. In that context, we are supporting that there will be better identification
and traceability of all biologics including biosimilars.
Ideally, the physicians should be able to report adverse events by reporting more than just
the INN. They should report INN + brand name + batch number, which would avoid
attribution of adverse events to a product from another manufacturer, e.g. in case of
immunogenicity reaction.

46
EGA: We do not interfere with the marketing process, but try to address and contribute to
the communication on biosimilars. E.g. a handbook was given out in order to inform people
about the subject. Information has been produced in order to inform stakeholders about
biosimilars in an attempt to balance the misinformation provided by the originator
companies. Regulatory, scientific or legislative contacts are also handled by the association
but not physician contacts or anything similar.
Could you please speculate what a future pipeline or “fast-lane” (by EMA) would mean to the
development of biosimilars, do you think that such a pipe-line will exist or do you see the
need of it?
EBE: There is already a European regulatory framework, with a set of guidelines and this is
what we support. If the authorities would suggest a fast track or some kind of simplified
procedure, we would surely provide input and may be very skeptical about a change of the
current system. Legislators might in 5-10 years from now decide about different rules but
that is for the future to tell!
EGA: The EU framework is based on a case-by-case approach. This provides legal certainty as
regards the legal basis for the application and flexibility. Indeed guidelines can be adapted
following experience with biosimilar applications and taking into account technological and
scientific developments.
The timeline of the approval process is the same for both originator products as well as
biosimilars.
The section that actually is abridged in a biosimilar application compared to an originator
application is the clinical trials.
From the quality side of view, one does not only require a quality dossier, but also an
additional comparability dossier, which is more than the requirements placed on the
originator. It is on this basis that the clinical trial package is allowed to be abridged.
What are the views of the organization with regards to the biosimilar debate in the U.S.?
How do these views affect the organization?
EBE: Well, the US for once is learning from Europe and there were various European experts
that attended meetings in Washington D.C. for the past 2-3 years. There has been a very
good transfer of knowledge from regulators and industry players to their counterparts in the
US. It is understandable that the FDA will take some time before they put their regulatory
system in place. It is positive that the FDA will not re-invent the wheel, rather base their
work on what we have developed in Europe which is well implemented.

47
Table 26. The views on FDA and the American
system by both the originator companies and the
biosimilar manufacturers.
EGA: The FDA’s approach is not yet public since there is no legal framework in place in the
US. The ongoing legislative process is very highly influenced by the originator industry’s
millions of dollar lobbying.
The FDA hides behind the fact that
they have no framework and does
not go public with where they are
aiming.
The current version (Dec 2009) of
the legal text in the House and
Senate are unfavourable toward
the biosimilar industry and
consequently hampers access to
affordable high quality
biopharmaceuticals.
The 12-year exclusivity period is not justified
since they have patents. But the EU has a
patent registration that is very different from
the USA thus making the American market
unworkable.
Obama supports a 7-year exclusivity period and this is going to be a big part of his reform.
Current speculations say that the current bill will pass; leading to that biosimilar
development in the USA is impossible.
Additional comments
EGA: We have also been very active in the approval process of the guidelines as well as we
have been involved in all the debates etc.
EBE: It is possible that a biosimilar product in the future may find itself superior. Truly it has
to be bioequivalent, but it may work better for a patient or a subset of the patient
population but in that class, for that indication.
The biosimilars compete with originator drugs, but it does not prevent innovation by the
innovator companies from putting a “next generation biologic” on the market, which would
have much better efficacy and safety than the first generation drug and its biosimilar.
This means that it is going to be tough for biosimilar manufacturers to compete over a
longer period of time because of continuous innovation.

48
Table 27. Additional comments
that were revealed during interview.
So as I have understood, you say that these generics companies or these companies
developing biosimilars seem to stimulate the innovators to develop “biobetters” or second
generation biopharmaceuticals, as you call it?
It is a very good point which is worth exploring. If that statement is confirmed by facts and
confirms the strategy of
some innovators in the
biologics business, we can
only welcome that. There
might have been various
sources pointing out that
biosimilars are actually
fostering innovation
provided that the
framework conditions for
innovation remain there.
Biosimilars are additional
ingredients in the mix,
but they are not the first and only
reason for companies to continue
investing in R&D (research and
development) leading second or
third generation products (so called
“biobetters”).
There were a lot of expectations on biosimilars 4-5 years ago because the analysis and
various consulting firms were predicting an explosion of opportunities. If one looks at the
market now the biosimilars have not been able to penetrate the market as many analysts
were predicting. The manufacturers putting the products on the market are the large
companies like Teva or Sandoz and not small operators since it takes huge investments.
We are supporting that there will be better identification and traceability of all biologics including biosimilars. Ideally, the physicians should be able to report adverse events by reporting more than just the INN. It would be better if they report INN + brand name + batch number, which would avoid attributing adverse events to a product from another manufacturer in case of immunogenicity reactions.
And last but not least the skepticisms I can express are whether biosimilar manufacturers
will be able to compete in the monoclonal antibody business because the regulatory
requirements of biosimilar monoclonal antibodies by EMA are under development. It is
possible that the requirements for the biosimilar monoclones will be as comprehensive as
for stand-alone applications. Why would a company then try to get an abbreviated package,
while they could go on their own and submit for an innovator monoclonal antibody?

49
Teva Some of the questions were removed due to the risk of revealing sensitive information
about the company.
Could you please specify the main society benefits (of biosimilars) identified by Teva?
To make healthcare and especially the treatment of life-threatening and serious diseases
(where biological products are used) more affordable, thus making expensive treatments
available to more patients, broader use, or re-investing the saved money in new
innovative treatments.
It leads to a better and broader
understanding of a biological
product. E.g. sometimes new
indications can be added to the
biosimilar and the side-effects profile
is more developed since the product
has been on the market for a while.
Biosimilars increase the choices of
treatment and leads stimulating a
competitive market.
Another key factor of society benefit is the
development of safe as well as refined
production of biological drugs where the
patent has expired (partially due to
competition).
From the aspects of competition with the originators, one has to build up cost effective
biotechnology meeting the quality requirements and standards of the originators.
Because you have generic versions coming in, this stimulates innovation because the
innovative company cannot continue to sell a product it introduced many years before
indefinitely at high prices. It stimulates a large pipeline of innovative products to replace the
products already on the market. Since that is understood for chemical products, then the
same principle should be applied for biological and biosimilar products.
Another aspect would be to take advantage of ten years of technological progress to come
up with an improved version that may be similar, but in some way even better (a
“biobetter”). But if a product is a “biobetter”, it is certainly not a biosimilar.
Table 28. The society benefits of biosimilars,
identified by Teva.

50
What are the main obstacles?
1) With special regard to the reference companies.
They have been very hostile and aggressive in their communication trying to discourage
doctors from prescribing biosimilars as well as successfully spreading fear to key decision
makers about biosimilars.
Confusion has been spread about
how a biosimilar product is
developed and how its profile
should be presented. I.e. The
originator companies try to make
the biosimilar producing companies
apply all the rules for a new
innovative drug, instead of the
comparability exercise acquired by
the regulatory authorities.
The innovator companies have been
spreading a great deal of misinformation e.g.
that biosimilar products are somehow inferior in quality and may have risks that are not
associated with the risks of a conventional product.
When it comes to the actual implementation of a biosimilar and the comparability, a
biosimilar can only be approved on the basis of being similar, thus one cannot get approval
for a different route of administration. E. g. if the originator product is only allowed for i.v.
(intravenous) administration, one cannot as a biosimilar receive market approval for e.g.
sub-cutaneous administration.
Due to originator companies lobbying activities in the USA, the conditions for biosimilars are
very unfavorable from the aspect of regulatory authorities, thus making it very hard for
biosimilars to reach the American market.
From the business and commercial side, one can note that the originator companies
challenge the safety and quality of the biosimilars but the efficacy is challenge to a much
lesser extent.
In some countries (the main markets of Europe), the innovator companies try to give better
pricing in order to prevent the penetration and spread of biosimilars.
Table 29. The main obstacles of biosimilars,
with special regard to originator companies.

51
Table 30. The main obstacles of biosimilars, with special
regard to other biosimilar producing companies.
2) With special regard to competitive generic companies.
Generic companies (in general) in the Nordic countries lack the infrastructure as well as sales
representatives in this area.
Another aspect is the downward
pressure on pricing, which we
have seen in the conventional
generic area. If you get the same
sort of thing in biosimilars, it
could lead to disillusionment by a
number of companies, because
these are more expensive
products, harder to develop and
riskier products than conventional
generics.
If there is the same reduction in pricing
as in conventional generics, I think
companies will feel that it is not worth
developing biosimilars. Therefore, you will lose the competition and the cost advantages.
That's the danger of excess competition in this field.
To be first in the market gives you a lot of competitive advantages. EMA provides guidelines
that utilize a comparability exercise as a tool. However, they do not state how to carry it out
leading to that a particular biosimilar can be granted market authorization on different
grounds. So one really has to “think outside the box”, how to cleverly apply the guidelines.
The focus of pushing prices is not the main target in the market of biosimilars, rather the
investment in safety, good distribution, and reliable sources play a more critical role.
Development of good relationships with prescribers is more important compared to the
generic market, where good relations with e.g. pharmacies can be crucial, thus competitive
companies have to develop this by learning and by time.
Have you noticed any scepticism among decision makers in authorities?
EMA has a generally very positive attitude, although local authorities are still positive in
Germany and Denmark, but not all local authorities within the EU are that positive. Outside
EMA, the authorities are much more sceptical and one can notice that they really reflect the
concerns and arguments from the originator companies.
The local authorities are perhaps only inquisitive since these are new products, rather than
being sceptic in their view of biosimilars.

52
What would a future pipeline or “fast-lane” (by EMA) for biosimilars mean for Teva, or since
this is not the case, is Teva satisfied with the EMA demands upon application for market
approval of a biosimilar?
(The latter part of this question was modified after the interviews with EMA revealed that there were
no future plans of an even more abbreviated dossier for biosimilars.)
Overall Teva is very happy about what EMA are doing. An even more abbreviated package
might not give any benefits at this point of the introduction of biosimilars. Teva would better
like to have a robust set of data accepted more than the rapidness so that prescribers can
trust the Teva products.
As soon as these types of medicines were called “biosimilars”, everyone got afraid of them
since it was a new phenomenon, even though the products had been on the market for
quite some time.
What are the views of Teva with regards to the biosimilar debate in the U.S. and the FDA
approach to biosimilars? How do these views affect Teva?
It does not change any of the current strategies, attitudes or plans in the local Scandinavian
markets.
For Teva, the US market is very important and also in terms of value. Teva is not happy that
the biosimilar debate in the US is taking too long, and that there are still no finalized
guidelines etc.
The 12 years of data exclusivity period is seen as very problematic.
Since there is no bill on how to treat the biosimilars, the outmost impacted by this is the
American people seeing that the biological treatments are very expensive.
Additional comments
Partnerships have been built between competitive generic companies in order to both
ensure the quality of GMP as well as educate key decision makers about biosimilars.
Since it is hard to estimate the market, it is likewise hard to estimate the capacity of
manufacturing.

53
Medicinal products committees in Sweden
26 medicinal products committees in Sweden were contacted for a possible interview
regarding their approach toward biosimilars. Interviews were conducted with the 15
committees listed in Table 31 below. Due to confidentiality issues, the results from these
interviews are presented without directly linking them to specific committees.
Table 31. List of interviewed medicinal products committees.
Organization and documents
Seeing that medicinal products committees provide recommendations to healthcare
professionals, what does the committee feel is its areas of responsibilities with regards to
biosimilars? How are these areas of responsibilities divided within the committee?
In accordance with the Medicinal Products Committees Act 1996:1157, QSE aspects of basic
medicines are handled by the medicinal products committees. Thus, most committees
mainly evaluate and recommend basic medicines in outpatient or primary healthcare.
Because current biosimilars are specialized products administered in hospitals, a large
number of committees feel that these medicines fall outside the perimeter of their
recommendations responsibility.
Instead, they rely on assessments carried out by so-called expert groups with focus on QSE
and scientific aspects of specialized healthcare. These groups are comprised of staff with
specialized competence in a number of therapeutic areas and an understanding of the local
work procedures and dynamics.
Several committees also handle tenders of both basic and specialized medicines, and
therefore come across biosimilar products. A few county councils are currently employing
biosimilars (growth hormones and EPOs).

54
Some committees believe that biosimilars will gain more relevance in the next few years.
Biosimilar insulins were mentioned as products of great interest because they could
potentially classify as basic medicines.
The responsibility of costs and implementation of specialized medicines are increasingly
shifting from hospitals to their clinics, because their physicians are the actual prescribers
that interact with the patients. Several committees are providing additional support to
clinics as a result of these augmented responsibilities.
How well-informed is the committee with regards to the legal framework and guidelines set
up by the EMEA? 1 Very poor 2 Fair 3 Good 4 Very good 5 Excellent
Please state what information is generally lacking in the documents and how these gaps
affect the committee's work with biosimilars.
Most committees were unfamiliar with the documents and scribed their knowledge as very
poor. In one case, this project initiated a discussion on the topic of biosimilars within the
county council. The committees rely on and trust the competence of their expert groups as
well as assessments carried out by the MPA and the EMA.
Because of the lack of familiarity, a majority would not identify informative gaps in the
documents. Still, several committees feel that the guidelines and legal framework drafted by
the EMA lack informational gaps of greater concern. A large number of committees also
indicated the need for more guidance from the MPA regarding biosimilars.
Figure 32 provides an overview of the responses from the interviewees.
Figure 32. Overview of familiarity among committees toward EMA’s document on biosimilars.

55
Processing
What is the definition of a “biosimilar” according to the committee? Which official
documents are used as a basis for this definition?
Most committees rely on and trust the official definitions on biosimilars drafted by the EMA
and the MPA. Local definitions have not been developed by any committee because it falls
outside their area of responsibility. Nearly all interviewees revealed that very little discussion
had taken place on the topic of biosimilars. Although most committees refrained from trying
to define biosimilars in the interview, some representatives could elaborate on the legal and
scientific facets of the topic.
How much difference between innovator drugs and biosimilars is acceptable? Elaborate.
Several committees rely on and trust the assessments carried out by the EMA and the MPA.
Responses regarding the permitted differences between the reference products and the
biosimilar equivalents varied dramatically in the interviewee pool. Most respondents
explained that subtle differences are allowed but patient safety is always the primary
concern. A few committees stressed that variations in the biosimilar product must match the
degree of variation seen in its reference product.
How many biosimilar drugs have been recommended by the committee upon application?
Most interviewed committees have not encountered biosimilars in their recommendations
of basic medicines. This situation is largely due to the fact that contemporary biosimilars are
specialized drugs and the committees recommend medicines for basic healthcare. Many
committees, however, do evaluate biosimilars during tendering periods. See Figure 33 for an
overview of the responses from the committees.
Figure 33. Overview of the number of biosimilars recommended by the committees.

56
How many biosimilar drugs have not been recommended by the committee upon
application?
One committee has casted a negative opinion on a couple biosimilars upon evaluation. Many
committees, however, have not cast an official decision regarding any biosimilar because it
lies outside their area of responsibility. One representative suspects that the absence of
biosimilar products in major medical journals in Sweden could have an unfavorable effect on
the perception of these medicines. Figure 34 provides an overview of how many biosimilars
have not been recommended by the committee.
Figure 34. Overview of how many biosimilars that have not been recommended by the committees.
What are the obstacles/problems regarding the implementation of biosimilar drugs and
how are they dealt with? (Labeling/naming, immunogenicity, etc)
Many committees highlighted the need for more experience and information regarding
biosimilars, preferably through meetings in addition to written material (See Figure 35).
Such initiatives could counteract what one interviewee described as “pharmaceutical
conservatism” in clinics. One committee rationalized that “fear-mongering campaigns” led
by originator companies might have a negative impact on the potential of the biosimilar
market.
Some respondents fear that the saving potential of biosimilars could lead to tender-technical
problems. For instance, the wish to reduce healthcare spending might force county councils
to frequently switch to cheaper medicines, hampering traceability in case of adverse effects.
As a consequence, several committees requested robust safety profiles and follow-up
routines to ensure patient safety. Other problems included labeling and naming issues as
well as the lack of interchangeability. One representative suspected that the choice of
medicines is a balance between rules and intuition. He explained that the given committee
never recommends biotechnological products that lack a rich bank of phase IV data.
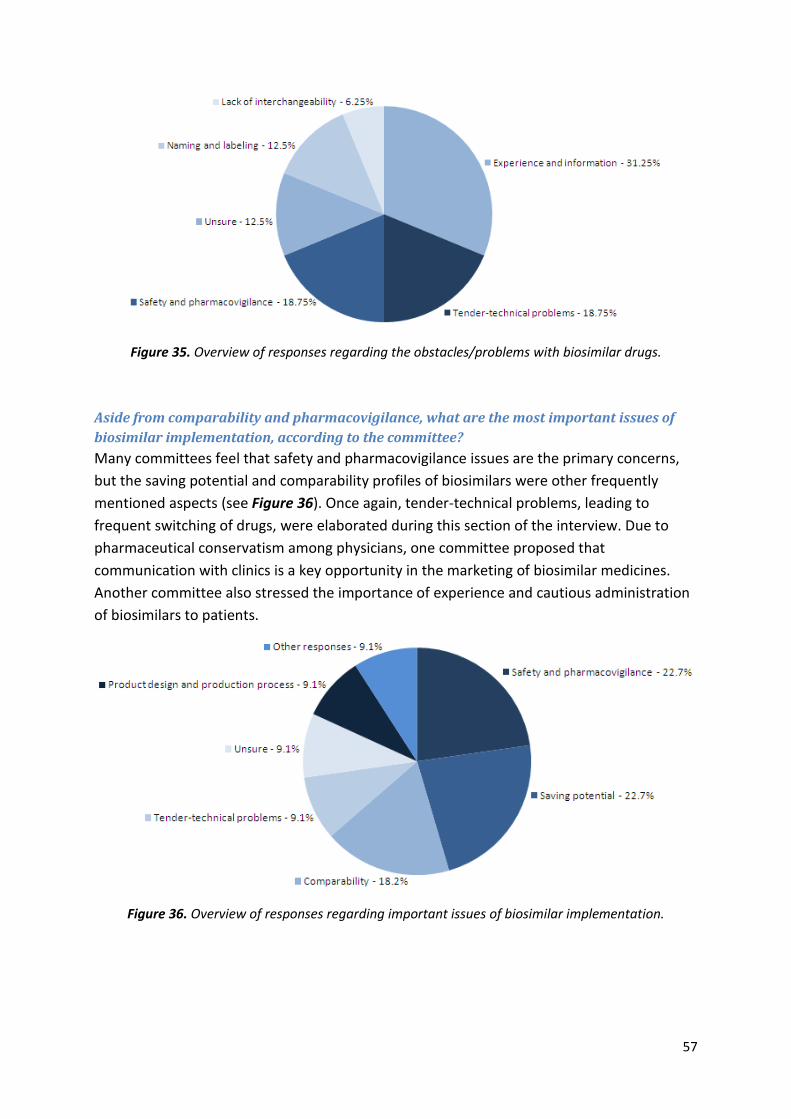
57
Figure 35. Overview of responses regarding the obstacles/problems with biosimilar drugs.
Aside from comparability and pharmacovigilance, what are the most important issues of
biosimilar implementation, according to the committee?
Many committees feel that safety and pharmacovigilance issues are the primary concerns,
but the saving potential and comparability profiles of biosimilars were other frequently
mentioned aspects (see Figure 36). Once again, tender-technical problems, leading to
frequent switching of drugs, were elaborated during this section of the interview. Due to
pharmaceutical conservatism among physicians, one committee proposed that
communication with clinics is a key opportunity in the marketing of biosimilar medicines.
Another committee also stressed the importance of experience and cautious administration
of biosimilars to patients.
Figure 36. Overview of responses regarding important issues of biosimilar implementation.

58
Has the committee recommended standardized follow-up routines for post-marketing data
of biosimilar drugs (compared to that of innovator drugs)?
A large group of committees have not set up standardized follow-up routines for post-
marketing data of biosimilar drugs. However, some committees already coordinate follow-
up routines for pharmaceuticals in general.
A number of committees foresee a growing need for biosimilar-specific follow-up routines.
Such routines could be set up on a national basis after close consultation with the involved
companies. Expert groups and clinics could also play an important role in these
pharmacovigilance systems, they explained. It was noted in one interview that a national
follow-up system for new drugs is already implemented through which biosimilars could
possibly be processed.
Future
Does the committee co-operate with external non-government parties, the national
regulatory authority, or the EMEA to address the issue of biosimilars? If so, could you
describe these interactions?
Most committees are active in the national networks, Läkemedelskommittéernas
ordförandegrupp (LOK) and SKL. Some committees have also established regional networks.
The MPA and TLV continuously interact with the committees in various healthcare issues.
There are currently no specific collaborations regarding biosimilars but a large number of
committees believe that this situation could change in the future. If such collaborations do
take place, expert groups could play a vital role in those interactions.
Excluding the price reduction, which other benefits of biosimilars have been identified by
the committee? Please rank them according to importance in descending order.
Several respondents highlighted the benefit of providing several medical options to different
types of patient populations as well as increased accessibility to niched drugs. Biosimilars are
also believed to promote innovation within the biopharmaceutical industry and drive the
development of currently available products. A large group of committees still feel that the
budgetary aspect is the only societal benefit of biosimilars.
See Figure 37 for an overview of the most frequent responses regarding the societal benefits
of biosimilars.
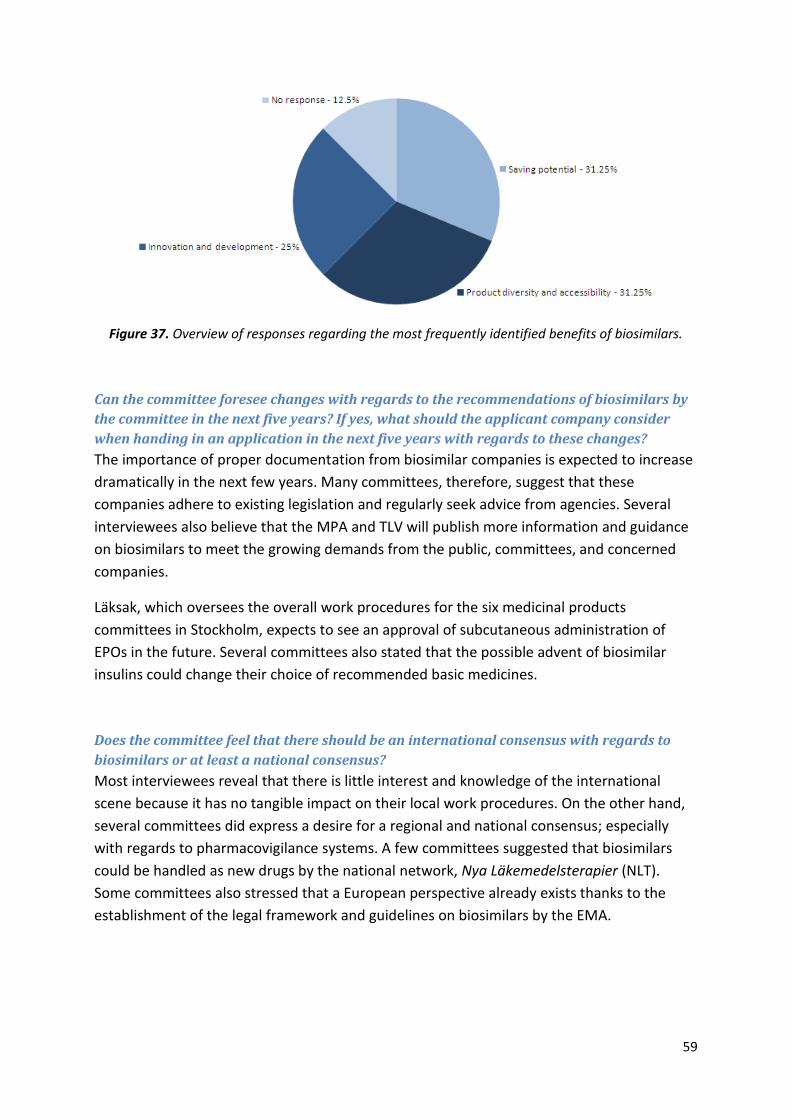
59
Figure 37. Overview of responses regarding the most frequently identified benefits of biosimilars.
Can the committee foresee changes with regards to the recommendations of biosimilars by
the committee in the next five years? If yes, what should the applicant company consider
when handing in an application in the next five years with regards to these changes?
The importance of proper documentation from biosimilar companies is expected to increase
dramatically in the next few years. Many committees, therefore, suggest that these
companies adhere to existing legislation and regularly seek advice from agencies. Several
interviewees also believe that the MPA and TLV will publish more information and guidance
on biosimilars to meet the growing demands from the public, committees, and concerned
companies.
Läksak, which oversees the overall work procedures for the six medicinal products
committees in Stockholm, expects to see an approval of subcutaneous administration of
EPOs in the future. Several committees also stated that the possible advent of biosimilar
insulins could change their choice of recommended basic medicines.
Does the committee feel that there should be an international consensus with regards to
biosimilars or at least a national consensus?
Most interviewees reveal that there is little interest and knowledge of the international
scene because it has no tangible impact on their local work procedures. On the other hand,
several committees did express a desire for a regional and national consensus; especially
with regards to pharmacovigilance systems. A few committees suggested that biosimilars
could be handled as new drugs by the national network, Nya Läkemedelsterapier (NLT).
Some committees also stressed that a European perspective already exists thanks to the
establishment of the legal framework and guidelines on biosimilars by the EMA.

60
Additional comments
Many committees revealed that usability (specifically the product design) has a large impact
on which types of medicines a physician might select. In addition to practical product
designs, physicians tend to opt for medicines from companies that transparently and
regularly provide information to the authorities. A number of respondents feel that
biosimilars should be assessed like any other medicine.
Several committees have experienced what they deemed as “fear-mongering” attempts
from originator companies with regards to the QSE aspects of biosimilars. One committee
initially suspected that this project was sponsored by an originator company.
The Stockholm committees rely on Läksak's list of recommended basic medicines, Kloka
Listan. In addition to the list and a number of joint campaigns, the committees are fairly
autonomous from Läksak.

61
Analysis and discussion
Following the inductive approach, there were no major aberrations, merely some minor
technicalities when it comes to the comparison of categories. Therefore both authors agreed
on common titles and their contents.
Methods In this study, a qualitative approach was chosen prior to a quantitative school of thought.
This was an intentional choice made by both authors in agreement, knowing that certain are
limitations associated with the given methodology.
If we reconnect with the limitations of multicase studies (described in Methods), one can
definitely agree that it was the correct approach to select in order to produce adequate data
in a study of this nature. Although “human factor” intervened with the analysis of the data, it
still seems to be the most suitable way of researching complex social qualitative data.
On the other hand, a single case study could have been chosen (also described in Methods).
Looking from a hindsight perspective, such an approach would not have been sufficient to
cover the multitude of data conducted. I.e. one could probably divide this study into a few
cases which could be examined on a case-by-case basis, and one would get very good results
but loose the variations and the complexity of the aim.
An interview technique called “semi-structured interviews” was chosen for the multicase
study in order to collect data and examine the cases. In this particular method, a major
concern was identified in later stages of study: because the questions were structured
according to the background material, they turned out to be quite biased. On one hand, this
bias was a problem when conducting the actual interviews, since the questions could not be
changed or deleted in correlation to the information that was obtained. On the other hand,
this also limits the investigator bias because it provides a common starting point for all
interviewees the same starting point, which would not be the case of open questions if such
an approach was selected.
A confidentiality issue was a topic that had an impact on the study when trying to investigate
the Teva structure around the biosimilar development. Hints of this aspect were
encountered when consulting the questionnaires under supervision. There was no
structured framework that could foresee to what extent it would impact the study.
Open questions in this study would perhaps be a better choice to in order to develop
profound theories based on cases with entirely different functions and characteristics.

62
Anyhow this was not the situation for the given investigation, since comparisons between
different cases would have been almost impossible to carry out if all the cases had different
sets of questions. Once more, one can really appreciate the openness, structure-wise, of the
semi-structured questions that were frequently taken advantage of when performing the
interviews. This provides the investigators with another degree of freedom without
compromising the structure of the questionnaires. The relevancy of questions can also be
better adjusted to the cases thanks to the semi-structure and barriers, such as cultural
differences, can be overcome.
The market-oriented literature findings were found to be very one-sided and biased in their
nature, many only reflecting the originator companies views and arguments. The
triangulation basis of the study was therefore a very appropriate choice of analytical
approach to counteract the shortcomings of the biased material. Background information
relating to the governmental agencies and EMA, however, proved to be very balanced.
Triangulation was, in overall, a very good line of work when performing this kind of study.
The inductive school of thought was chosen because it was most suitable tool for analyzing
the data yielded throughout the study. In the coding process, which was carried out
separately and independently (by both authors), very similar categories were pointed out as
being essential.
Another approach, such as grounded theory, which is closely related to the inductive
approach, might have lead to different results and reasoning. This approach might require
another hypothesis rather than an inquisitive formula for the aim. It is possible that
grounded theory, as compared to the general inductive approach, might have made the
study less flexible considering that one would not be allowed to reflect on the aim on a
continuous basis. For example, one could opt for the grounded theory if another study is set
up to focus on a specific part of this study, using a hypothesis based on the findings in the
current investigation.
Phenomenology could be used if the investigator is determined to specifically focus on one
person with a vast set of experiences in the field of biosimilars, e.g. a representative person
from EMA, EBE, or EGA. It would definitely not be advised in a study investigating different
organizations, companies, and authorities.
The people who were approached for a possible interview throughout the study have been
thoroughly investigated to ensure that they are appropriate representatives of their
organizations in the matter of biosimilars. As investigators, one can really appreciate that the
people taking part in the interviews were all of such caliber.

63
When it comes to the market analysis of biosimilars, the fundamental notions are based on
the results from the interviews. This actually diminishes the reconnection with the
theoretical framework in the background of this particular study. An additional limitation is
that the literature sources do not provide market-related information on biosimilars to any
greater extent. The shortage of information in current literature could depend on that this
market is a fairly recent in the field of biopharmaceuticals and under constant development.
Past
The Generics of the 1990s
The 1990s saw the birth of the generics industry for chemical pharmaceuticals. In addition to
increasing product accessibility and diversity, the advent of generics has also contributed to
a price reduction of 60-80% overall.
However, the developments seen in some aspects of the history and regulation of the
generics industry have not been identical to that of biosimilar medicines. Several
respondents from the industry, for instance, highlighted the lack of congruency between the
authorization processes of generics and biosimilars, respectively. They argued that both
scientific factors and their roles as market players have impacted the construction of the two
types of authorization processes, as well as the implementation of these products.
Scientific factors A generic product is a medicinal product that has an identical QSE
profile to its reference product. Because of their small size and
lack of batch variation, chemical drugs are easier to define than
are biopharmaceuticals. Therefore, the interchangeability of
generics has not been as widely debated as that of biosimilars. As
a consequence, Lönngren from the EMA explains, “…the
authorization process for biosimilars is more complicated than for
applications of chemical generics”.
Roles as market players Several respondents highlighted the link between the introduction
of generics into the market to that of biosimilars. One
representative from Teva explained that “…genericisation
stimulates innovation because the innovative company cannot
continue to sell a product it introduced many years before
indefinitely at high prices”. The EBE, however, emphasized that
the same price differential between originator chemical molecules
and generics cannot be expected for biosimilars and their

64
reference products. The debate on the possible coherence
between the development of the generics industry and the growth
of the biosimilar market has certainly been highly polarized and
unresolved between market players.
Despite a number of disparities (economical, political, and scientific), the hope is that
advancements seen in the generics industry in 1990s will also hold true for the progress of
biosimilars. A respondent from Teva rationalized: “One could compare the situation of
biosimilars to the upcoming of generic medicines, when they were first introduced to the
market; i.e. to reduce the burden coming from the high costs of the originator treatments”.
EU number one
The EU was the first region in the world to draft detailed documents that outline the
scientific and technical aspects of biosimilar products. In fact, the birth of the European
biosimilar market has been a joint European effort.
Our interviews revealed that the EMA actively organized workshops that tapped into the
competence of all stakeholders while respecting their individual interests. Representatives
from the CHMP member states, as well as originator and biosimilar companies were invited
to these events. The aim was to create a neutral venue for all parties to convene, allowing
participants to provide feedback although the agency itself had a final say on the finished
results.
The dynamic process produced two sets of pioneering documents: a legal framework for the
definition of biosimilars, and a number of guidelines to provide advice on the practical
approach within Europe. Both sets of documents still exhibit a degree of freedom,
sanctioning the flexible navigation of biosimilar MAAs as well as the implementation of these
products on a national (if not a regional or local) level.
Special attention was paid to the varying complexity of biosimilars (as to reflect the wide
spectrum of complexity seen in the reference biopharmaceuticals) by creating guidelines in a
hierarchical fashion (see Figures 17 and 18). Thanks to this division of information, additional
guidelines can easily be drafted according to future regulatory and market needs and
experiences.
Lönngren from the EMA conceded that an “initial hiccup” with regards to the authorization
process prolonged the outcome of the first biosimilar application. Through experience and
the combined competence of the CHMP member states and market players, these concerns
were quickly addressed. Hence, it has been and will continue to be a learning process for
both the biopharmaceutical industry and the EMA.

65
...And then there were biosimilars.
When the concept of biosimilars first materialized in 2003, it created a focal point around
which the biosimilar-manufacturing companies could rally, unify, and organize its activities.
It also provided an opportunity for originator companies to directly attack the issue. In fact,
the actual minting of the term, similar biological medicinal product, instantly turned the field
into a hostile environment of clashing interests, revealed Lone Juul Darket.
Strategies by originator companies
When reflecting over the data gained in the interviews, it does not take long until it is
possible to clearly see two different approaches. On one hand, there are the originator
companies doing everything that they can in order to protect their market share of the
original biopharmaceutical medicines. On the other hand, the biosimilar manufacturers try
to penetrate the market with their new product and possibly grasp a substantial sector of
the biopharmaceutical market. Because the pharmaceutical business is very competitive to
its nature, and the field of biosimilars is built on the foundation mentioned earlier, it is not
an easy business which could generate capital rapidly.
The development of biosimilars (market-wise) is constantly related to the initial phase of
generic medicines by the people interviewed for this study. If we glance at how the pricing in
generics has developed, one could only speculate that if such development would take place
within the field of biosimilars, it might not even be a business worth investing in.
Supposedly, if the downward pricing on generics would happen to biosimilars, one could
rapidly see a negative market development around the corner and, with that, fewer new
biosimilars reaching the market as the result.
In the main markets of Europe, the originator companies have already lowered their prices
on the products targeted for biosimilar production in order to keep their market segments.
One assumption that can be made is; if biosimilar manufacturers are somewhat intimidated
by the thought that this might lead to harsh downward pricing, they might not be able to
take such great market shares as predicted in advance. Companies would probably then
benefit more if they would formulate other strategies, such as looking eastward toward new
or somewhat unexplored markets as well as targeting many smaller markets in Europe, in
order to gain a substantial market share.

66
Another great problem with the originators other initial counteract towards biosimilars,
which they were very successful with, is the spread of fear to prescribers. The interviews
revealed that International organizations for patients, the UK parliament etc. were exploited
to profoundly intimidate people from biosimilars. It is a great hurdle to cross since a lot of
people have been misinformed about biosimilars. It is much easier to create an opinion than
changing one, so the producers of biosimilar pharmaceuticals have to make many efforts to
counteract current stands.
As it seems, the originator companies constantly question the aspects of quality and safety
with biosimilar medicines. The efficacy is targeted at a much lesser extent. A mere
speculation would be that the strategy might depend on the fact that either they fear legal
retaliation if they were to attack this angle, or they might not want to incinerate their
relationship with the EMA.
A tool used by originators to stir up fear-mongering among prescribers is the attempt to
approach the WHO about the INN program. The originator companies are suggesting that
the naming of a biopharmaceutical should state: INN + brand name + batch number. A
possible theory would be that this would only increase the rift between biosimilars and the
originator products, whereas the manufacturers of biosimilars state that the INN should only
demonstrate the presence of any possible glycosylation patterns.
The EMA, the Swedish authorities, and the Danish authorities seem to be unaffected by such
misinformation, which one indeed can cherish. As the authors of this very thesis, we even
experienced it! Looking backwards at what papers that were used for our background, as
well as when we contacted an advisory committee, the representative was very inquisitive of
our aim with the study since she was to guarantee that we, as students, were not sponsored
by any originator company trying to spread fear to them!
To grasp a scale of how the misinformation leading to fear-mongering is spread can be very
hard. As far as it has been understood, this seems to be a fairly new concept to companies
that manufacture biosimilars as well. In order to gain the expertise, which enables the
biosimilar company to tackle this issue, the company should invest a lot of resources in
building the capacity to go through with such a campaign keeping in mind that it will yield a
very uncertain outcome.

67
Present
Chain of communication and trust
A very interesting relationship that was encountered in this study was the chain of
communication and trust from the EMA all the way to individual prescribers. First of all, one
has to keep in mind that this particular issue has to be investigated further, but the following
is at least some aspects found with this study.
When talking to the EMA, the authority made its stand about biosimilars very clear and
explained their view on the topic in a very informative manner. EMA believes that after the
assessment of a biosimilar product, it should be considered to be indisputable as a
biopharmaceutical medicine that has shown to be on the same level of quality, safety, and
efficacy as its original product. Even the originator companies’ trade association (EBE)
explained that they can only “welcome” the biosimilar if it passed the rigorous and well
developed assessment of EMA and passed. Both the EGA as well as Teva seem pleased with
the current EMA demands on biosimilar drugs (in terms of what they have to present).
The local authorities in Sweden as well as in Denmark are well established members of the
CHMP. Other than that, they work very close with the EMA in many questions and they have
a seemingly very high level of trust for each other. They are also well-informed about the
contents and implications of the EMA documents, although regular collaborations on
biosimilars have yet to take place outside the EMA. The high level of familiarity toward the
documents can be attributed to the regular workshops with member states that were
organized by the EMA.
The medicinal products committees even stated themselves that they look upon one of their
main tasks to basically “channel” information straight from the MPA to prescribers. They do
not intervene in the relationship between the EMA and the MPA but they look at both, and
especially MPA, as the main sources of information. During some interviews, we also noted
that only a few members of the different committees were aware of EMA stands but
everyone stated that they knew what MPA provides. The trust for the local authorities
(MPA) is clear in this case, and they only look upon them as “an extended arm” of EMA, thus
not being totally informed on EMA stands.
When we investigated the chain even further down, we noticed a sudden change of
attitudes in the level of prescribers in the clinics. It is possible to spot hesitations with EMA
decisions, and especially the trust (in case of biosimilars) of the QSE profile and the
assessment executed by EMA. It seems as if there is (conceptualized by one of the
interviewees) pharmaceutical conservatism in Sweden. Could it be that this conservatism is
not unique for the biopharmaceutical industry but a more widespread phenomenon within
the Swedish market in general? The prescribers say that they do adhere to the medicinal

68
Figure 37. The relationships of trust
between the involved parties.
products committees and do trust their judgments in many cases. When it comes to
authority level, a sudden drop in trust was observed, falling even deeper if the focus was
changed from the national authority (MPA) to EMA.
What does this actually mean? One could
argue that since there is a straight downward
chain of trust, where the concept of
governmental or international governmental
trust does not exist. The matter that suddenly
complicates everything is that if one asks the
prescribers to skip one step in the chain (the
most proximate), one sees an immediate
change of attitudes. Since this study was not
designed to answer this particular question,
the issue will not be elaborated any further. It
is, on the other hand, worth keeping in mind
while considering other inquiries of this
study.
Lack of knowledge
When considering another issue experienced by the manufacturers of biosimilars, one
should keep in mind the previously mentioned issue of trust and communication. One of the
most problematic areas to deal with in terms of marketing of a product is the lack of
knowledge by the potential consumer.
Indeed a lot of misinformation about biosimilars has spread, but one of the most recurrent
answers obtained in the interviews (with the medicinal product committees) was that the
topic of biosimilars had not been treated yet. In some cases, our investigation was the first
time the question had been raised in the given county council. It was also noted in the very
beginning of the interview sessions that the concept of biosimilars was unfamiliar to most
committees (when asked in detail). This lack of knowledge could be attributed to the fact
that the topic had not been in any focus since the class of drugs is fairly new and only (so far)
used by specialists and not by general practitioners.
When asked about their views on how the committees are informed about biosimilars, local
authorities stated that it is up to every committee to request more information and that
some information has already been handed out. In general, it did not seem like the
information given had reached a vast majority of committees or they had at least not
grasped it.

69
This leads to somewhat of a dilemma, since it is hard to ask for something that you do not
know exists. Therefore, it might be suggested that the biosimilar producing companies
should try to address this issue with the local authorities to stimulate a better flow of
information.
When building a stable relationship with prescribers, the biosimilar manufacturers have to
take the role of educators in this matter, which is very problematic since they will always be
regarded as “motivated by own interest” and not as sources of reliable information. For
instance, the biosimilar manufacturers (usually generic medicines companies) have in this
particular issue built partnerships (in Sweden and Denmark at least) in order to educate the
prescribers about this topic by e.g. handing out folders of information.
A totally different aspect of “the lack of knowledge” is that prescribers do not, in general,
know what assessments every drug application has to undergo in order to reach the market.
One might say that this leads to mistrust of new products because they are compared to
products that have been on the market for longer periods of time. This is a two-dimensional
problem since prescribers already lack trust for EMA as well as doubt new products in
general. Both of the aspects play a crucial role in this case; first, one does not feel a good
level of trust toward EMA, and secondly, one is not familiar with the EMA assessment.
It is not a coincidence that biosimilars will suffer greatly from this situation since they belong
to both categories; they are new, and people lack knowledge about them in so many
different manners.
Authorization is European, implementation is national
The interview-based multicase study revealed that responsibilities regarding biosimilars are
divided into four levels: European, national, regional/local, and clinical (see Figure 38).
Figure 38. The areas of responsibilities
regarding biosimilars can be divided
according to a European, national,
regional/local and clinical level.

70
European
The authorization process of biosimilars is a European proceeding. Auxiliary aspects of these
products are handled by individual member states, effectively shielding the EMA from
political influence.
All EU and EFTA member states participate in the authorization process; either as a reporter,
co-reporter, or as a member state of the CHMP. The system, based on an older system
known as the concertation procedure, helps authorize medicines that can theoretically be
implemented in all member states.
Although the EMA is not responsible for the running of pharmacovigilance plans, the agency
has taken the proactive initiative of sending general reminders to all member states about
the importance of such systems. Jonsson from the MPA explains that the role of the EMA is
not to formally approve MAAs (which is carried out by the EC), but to assist the CHMP and its
member states in medicinal issues.
National
The implementation of biosimilars is a national issue. Interchangeability and healthcare
spending are, for instance, handled by the NRAs on a national level; responsibilities
commissioned by the EMA and welcomed by individual countries. Sweden and Denmark
have not appointed biosimilar-specific working parties but their NRAs systematically process
these pharmaceuticals in a number of sectors (see Tables 14, 15, and 16). None of the
Swedish and Danish NRAs have recommended automatic substitution of biosimilars
(interchangeability is still permitted) because they are awaiting more pharmacovigilant data.
Regional/local
In Sweden, local medicinal products committees handle the recommendations of basic
medicines. Thus, they rarely come across biosimilar products in this area of their work
procedures. Instead, medical assessments of biosimilar products are conducted by their
expert groups and clinics. On the other hand, several committees did reveal that they
encounter biosimilars when they assess tenders of both basic and specialized medicines. A
number of aspects are assessed during tendering periods for all pharmaceuticals (and not
merely for biosimilars):
Safety and pharmacovigilance Committees expect an abundant amount of information and data on the safety profile of the biosimilar products (both pre- and post-licensing).
Saving potential Although committees consider the saving potential of biosimilar drugs, they still stress that it could never outweigh patient safety.

71
Comparability Comparability must be proven with a large bank of data to counteract perceptions of mistrust regarding biosimilar products.
Practical product design Physicians often base their choice of medicines on the usability of the product; i.e. container design, choice of administration, number and size of injections, etc.
In an effort to streamline their work procedures, Denmark's five regions coordinate the
purchases of pharmaceuticals for the public hospitals through a joint body known as
Amgros. Once per year, hearings are organized, during which companies (including
biosimilar companies) can present their products to the local authorities.
Clinical
In recent years, the responsibility of cost for specialized medicines has shifted to clinics in
Denmark and Sweden. The responsibility has already reached clinical level in Denmark and
an increasing number of Swedish county councils are following suit. The aim is to promote a
more realistic implementation of specialized medicines because the clinics have direct
knowledge of their patients’ medical history and needs. As a result of the added pressure on
clinics, the Swedish committees are providing extra support.
Limiting factors of the biosimilar business and the current strategies of
biosimilar companies
The limiting factor in the field of biosimilars is mainly the economic investment that needs to
be done. It has a range of aspects to it; it is hard to predict the turnover since the market is
very unclear; a biopharmaceutical production line that is more efficient than that of the
competitors must be built; all of the necessary expertise must already be in place etc. As is
demonstrated in previous statements, it is a very risky business to enter and a lot of
investments must be made to create a favorable starting point. These factors limit many
companies of a smaller magnitude, and only large investors with a great financial capacity
can enter this type of business.
The current strategies of biosimilar companies are quite clear to the observer. First and
foremost, the companies need to establish relationships with prescribers, which is a new
phenomenon compared to the generics business where the generic companies must
establish good relationships with pharmacies etc.
Another aspect, which has been mentioned earlier, is that the responsibility of education
about biosimilars is almost solely placed on the biosimilar producing companies. Therefore,
this is a crucial factor that will determine the whole market development.

72
Obviously, the main aspect is the pricing of biosimilars which has to be lower than products
that already exist on the market. Equally important in Sweden and Denmark at least, is to
make great deals with the regional county councils and in that sense offer better prices.
Future
New products
A range of biopharmaceuticals were highlighted as potential biosimilars in the conducted
interviews.
Biobetters
Augmented competition in the biopharmaceutical industry by the advent of new market
players (such as biosimilar-producing companies) is generally believed to drive innovation
and development, possibly even generating the birth of so-called biobetters. Because these
products are second or third generation biopharmaceuticals with an improved QSE profile,
biobetters could potentially seize market shares previously secured by their predecessors.
However, as rationalized by one of the respondents: a biobetter can never be a biosimilar
because it is better. Likewise, a biosimilar can never be better because it is similar. Thus, the
biobetter market could be an area of interest to both generic and originator companies. One
respondent, however, suggested that the advent of biobetters greatly limits the longevity of
the biosimilar market, because of the improved QSE properties in these products in
comparison to biosimilars and first generation biopharmaceuticals.
Insulin
Insulin was presented as an interesting product by many respondents. In fact, several
representatives from the medicinal products committees highlighted the potential of
biosimilar insulins (which would be classified as basic medicines). If such products are
available in the future, they could greatly affect the recommendations and choice of insulin
medicines, perhaps even allow for an increased familiarity toward biosimilars in general.

73
One interviewee did identify variations in a potential biosimilar market for insulin medicines.
Because the pricing of insulin products in developed countries already are greatly reduced,
creating biosimilar equivalents for insulin analogues (which are priced much higher in the
West) could be an area of interest to generics companies. In developing countries, however,
where regular insulin is expensive, both regular insulin and insulin analogues could function
as potential biosimilar products.
Monoclonal antibodies and low-molecular heparin
Because monoclonal antibodies were excluded in the scope of this study (see Delimitations),
a theoretical framework for the topic was not introduced in the Background. Several
respondents did reveal that monoclonal antibodies present the next great challenge for the
EMA. With the purpose of drafting guidelines, a workshop was organized in the summer of
2009 and additional evaluations are expected in the near future. Guidelines might also be
drafted for low-molecular heparin. The outcome of these initiatives remains to be seen and
largely depends on experience and the development of analytical tools.
The American approach to biosimilars
The cultural and political parameters in the US present an entirely different environment in
which biopharmaceutical companies must operate their enterprises. As a reflection of the
federal nature of this market, the US is comprised of one set of laws, one population, and
one political system. From a logistical perspective, overcoming hurdles in the American
market is less problematic than marketing a pharmaceutical in the multifarious European
market.
Due to restrictions in current legislation, however, the FDA lacks legal authority to approve
most follow-on-biologicals (See USA | Food and Drug Administration in Background). Several
attempts in igniting a debate on FoBs have been fruitless, and many interviewees suspect
that the lack of momentum is largely due to massive lobbying from originator companies as
well as strict patent laws. The American debate on biosimilars is, in effect, lagging.
Close collaborations and dialogue have transpired between Europe (its regulators and
industry players) and the US, where the European counterparts share their experience
within the biosimilar field. So far, these interactions have not translated into tangible
activities in the American debate. Many respondents expect a lengthy and cautious debate
in the US before the FDA establishes a regulatory system for FoBs. One respondent
explained: “It is positive that the FDA will not re-invent the wheel, rather base their work on
what we have developed in Europe.”

74
Nevertheless, the American debate, or lack thereof, has no influence on Europe. The EMA
seems to be highly confident about its own system. One interviewee explained:
“We do not interfere with the intrigues that take place in the US. The events in the US do not
affect us at all, especially since we already have biosimilars and we have casted a political
decision to implement these products. There is an interest from the American side on a
political and regulatory level.”
Proposed strategies for biosimilar manufacturers
In order to tackle a few of the problems faced by the manufacturers of biosimilars,
partnerships have been built. For instance, in the production line, companies form
partnerships in order to gain full competence. As stated before, in the field of
communication and information, the Swedish and the Danish branch of Teva
Pharmaceuticals has created informative brochures together with other companies
producing biosimilar medicines.
When it comes to future strategies of how to develop biosimilars, a sometimes disregarded
matter is actually the product design. Often this concern can be vital to clinicians while one
product can be totally misfit for the use it is intended when it comes to product handling in
terms of physical features. One way of overcoming this problem might be to conduct a
market analysis before producing e.g. the actual container.
A hypothetical future approach to somewhat compensate for the lack of information among
the prescribers about the EMA could be to explain EMA’s demands and exemplify questions
that were answered during the assessment of the biosimilar in question.
In order to meet the fear and misinformation that biosimilars have been exposed to, one
should notify authorities of the problems in order for them to spread the correct information
within the given system, instead of tackling it all alone since there is a risk of disregard due
to company bias.
In the interview with the European Biopharmaceutical Enterprises, a few arguments about
why biosimilars would not be a successful business to develop were mentioned. It was
argued that a few years ago, many business analysts said that it would be a good business to
enter and that today there are still not so many involved parties. It was also stated that the
originator companies will continuously develop new versions (second generation or third
generation) of the same biopharmaceutical which is meant to be superior to the latter, thus
making biosimilars a discouraged choice of treatment.

75
It might be that only a few major companies possess the capacity of developing biosimilars
these days, but that could solely depend on time. It is possible, since this is the start-up
phase of the biosimilar industry when only a handful patents have expired, only a few major
companies have had the economical means to promote such a development.
Seeing that partnerships are being built within this field of biopharmaceuticals, more and
more companies are getting involved which would contradict a negative market
development. The current framework set by EMA is also quite robust, prolonging the time it
takes for an applicant company to obtain market authority acceptance. This framework was
not in place at the time when the analysts conducted their assessments, and it might have
been assumed that a similar system of approving generics was to be applied.
More and more patents will expire in the next few years and therefore, one has to take in to
consideration that some of those might present very lucrative business opportunities in the
future, even more than the ones already expired.
The continuous market development of the originator companies might be an aspect of
consideration, but since the new products will be more expensive than the biosimilars of the
first generation biopharmaceuticals, many might continue prescribing them. Equally
important is the aspect of continuous development of biosimilars that will only expand over
the next few years according to preliminary prognosis.

76
Conclusions
Key approval agencies
What are the current attitudes and knowledge regarding biosimilars?
The knowledge within the approval agencies in Sweden and Denmark is generally very good
and not influenced by market interests. In addition, the attitudes are very objective in this
matter, not reflecting any industrial interests to the point that it does not inflict on patient
safety. This also holds true for the EMA.
Since the European market pioneered in this area of the life science industry, high demands
were placed on the EMA to develop a functional, yet dynamic, system of approval. EMA
successfully established a robust system for biosimilar applications, as a result of a joint
effort that involved all of its member states and industry players. Therefore, the structure is
highly lauded by all involved parties.
This does not seem to be the case for the FDA, nor any companies seeking to sell biosimilars
in the American market. A different study design could possibly examine this issue more
thoroughly.
What do approval agencies expect from biotech companies that wish to
manufacture, market, and sell biosimilar medicines in the future?
The keywords are: competence, knowledge, documentation, and communication. Close
interactions with relevant authorities are, therefore, highly advised, even at an early stage.
Manufacturers are currently creating relationships in order to gain competence and
knowledge and meet the criteria of the regulatory demands.
The agencies are presently content with the material provided by applicant companies. Only
future developments can tell if more detailed dossiers will be requested.

77
What are the attitudes of national approval agencies toward decisions made
by EMA regarding biosimilars?
They take an active part in the decision-making on the EU level and they do not feel run over
by any overlying authority. Instead, they see themselves very much a part of the CHMP and,
thus, the EMA.
Every member state can provide feedback on any subject matter within the organization;
therefore, demonstrate the values they represent in an international environment.
As of yet, decisions made by the EMA in the question of biosimilars have been welcomed by
all of its member states, including Sweden and Denmark.
How will the key approval agencies’ attitudes affect the future of the
authorization process for biosimilars?
No changes are expected in current guidelines, but additional documents might be drafted
according to market needs and experiences.
Thanks to the infrastructure, the EMA is shielded from political influence regarding
biosimilars, and can independently compose new material. Since NRAs are aware of fear-
mongering campaigns led by originator companies, they can more easily validate
information.
Future market strategies of biosimilar medicines
What are the main obstacles?
Hostile and aggressive marketing initiatives from the originator companies have, so far, had
a negative impact on the prospect of the biosimilar industry. This behavior has led to fear-
mongering among prescribers along with confusion of the QSE profile of biosimilars. The
originators companies have also lowered the pricing of their own products to sustain a
substantial market share over a longer period of time.
A lack of knowledge within different sectors of society is a great hurdle for every biosimilar
manufacturer due to the already unfavorable market conditions created by the reference
companies.

78
The conceptualization of biosimilar products are strongly orchestrated by the preconditions
and nature of the reference products, which limits the area of usability of an applicant
product. It is much easier to create an opinion than changing one that already exists.
What are the potentials?
So far, only a few patents have expired in a limited number of therapeutic areas. Many
biosimilars are already on the market, but the expiration of additional patents will boost the
market developments. The current market is mainly focused on specialized medicines, but
new biosimilars are expected within the areas of insulin, monoclonal antibodies, and low-
molecular heparin.
Another important aspect of the biosimilar market is the possible advent of biobetters.
These products might see itself superior to its predecessors in certain indications or specific
patient populations.
Biosimilars drive innovation and development within biopharmaceutical industry, leading to
new inventions from both originator and biosimilar manufacturers.
What are the market demands on the biopharmaceutical industry wishing
to manufacture, market, and sell biosimilar medicines in the future?
The market demands can be divided into five categories:
Product design – the biosimilar product must meet the requirements and expectations of the
clinicians, providing an easy, useable, and functional design.
Saving potential – a biosimilar manufacturer should offer a more affordable healthcare, thus
making expensive treatments available to a larger group of patients, broader use, or re-
investing the saved money in new innovative treatments.
QSE – companies seeking to produce biosimilar medicines should be able to provide
demonstrative data in order to reassure the clinicians on the prospects of QSE.
Documentation – more (pre-licensing and post-licensing) documentation is expected with
time in order to meet the growing demands of the healthcare and public. Experience within
the field of biosimilars may lead to other documentation demands in the future.
Communication – companies must invest in proper expertise with the purpose of
successfully reaching and correctly informing the different stakeholders on the benefits of
biosimilars.
The only way to develop the sector is to approach the biosimilar field with an open-
mindedness and willingness, thus “…learning by doing”.

79
Closing words
Key approval agencies
The EMA has pioneered in the field of biosimilars, securing knowledge and competence from
industry players and the CHMP member states. Its infrastructure successfully protected the
agency from political influence. Thus, the EMA could draft documents that are highly
appreciated by both the industry and all member states. The dynamic process still allows for
the creation of additional guidelines according to future needs and experience. When
interacting with approval agencies, biosimilar companies should adhere to the following
keywords: competence, knowledge, documentation, and communication.
Future market strategies of biosimilar medicines When it comes to the marketing of biosimilars, one of the main obstacles that manufacturers have to
overcome is a general lack of knowledge in this topic. Due to aggressive marketing by originator
companies that has led to fear-mongering among clinicians is another very important obstacle.
However, the preliminary market prognosis looks very promising thanks to an increasing demand of
biopharmaceutical therapies and the expiration of patents in more therapeutic areas within the field
of biotechnology. The current presence of biosimilars is also thought to drive innovation and
development of biological medicines.
Marketing strategies of biosimilars should not only focus on product design and proper QSE profiling,
but equally important is a communicative approach to relevant stakeholders.
Future studies Because the biosimilar field is a relatively new topic within life science, current literature
cannot alone address all of its aspects. Although a supplementary aim of this study was to fill
some of these gaps, several key areas were identified as appropriate subjects for future
studies:
Trust between EMA and prescribers in the clinics
An analysis of trust issues between the EMA and prescribers in the clinics, and why these
interactions have appeared, can shed some light on the lack of trust among prescribers
toward the European agency with regards to biosimilars and how it can be addressed.

80
Monoclonal antibodies
Although monoclonal antibodies were initially excluded from the scope of this study, the
future of these products as biosimilars was identified as the next great challenge for the
EMA and the market. The growing market share of monoclones holds a potential economic
benefit for healthcare and industry players.
Analyzing the progression of biosimilar monoclones would not only provide knowledge on
how the EMA creates new legislation and guidelines, but also how all stakeholders interact
with the European agency to tackle new issues (especially with regards to biosimilar
products).
Regional regulation and implementation of biosimilars in Denmark
Due to time constraints and other technical limitations, the investigation could not analyze
the regulation and implementation of biosimilars on a regional and local level in Denmark
and other European countries. A representative from the Danish headquarters for Teva
revealed that the regional and local dynamics in Sweden can still be applied to those of
Denmark. However, thorough investigation is advised in order to obtain a realistic view in
this subject matter.
Tender issues of biosimilars and expert groups
Current biosimilars are administered in specialized healthcare such as clinics. Although most
committees do not encounter biosimilars in their recommendations work procedures, they
occasionally assess these products during tendering periods. Thus, more information on the
dynamics of tendering periods could provide knowledge on how to strategically market
specialized biosimilars on a regional and local level.
Likewise, the work procedures for expert groups (that are coupled to the Swedish medicinal
products committees) and how they assess biopharmaceuticals (such as biosimilars) could
facilitate future marketing strategies for specialized biosimilar products.
Networks
Several networks, such as LOK, SKL, and NLT, were suggested as appropriate forums for the
marketing of biosimilars and other new drugs on a regional and local level. An analysis of
their work procedures and how these correlate to the biosimilar industry is yet another
interesting area of investigation for marketing purposes.

81
Asia
This region is yet another somewhat unexplored market with huge potential for Western-
based companies. India already has a system in place but processes these products as
entirely new drugs, while other countries have yet to develop such an infrastructure. Special
focus could be placed on how to apply the structure and procedures from the EMA to
equivalent agencies in Asia.

82
Acknowledgements
We, the authors, would like to express our heartfelt gratitude to everyone who has contributed to our master thesis, both from within the university and outside of the academic realm.
First and foremost, we would like to thank our primary supervisor Jessica Norrbom, PhD, at LIME (UBE) for her continuous support and mentorship. Her valuable insights and encouragement have inspired and guided us throughout the progress of this investigation.
Moreover, we want to acknowledge the guidance from our assistant supervisor, Carl Johan Sundberg, MD PhD, Associate Professor at LIME (UBE).
We would also like to thank Fredrik Lindqvist at Teva Sweden AB for his creative inputs as well as internal contacts within Teva Pharmaceuticals Ltd.
We deeply appreciate the enlightening and rewarding interview with Thomas Lönngren, the Executive Director at the EMA, on the EMA's approach to biosimilars.
Also, we would like to express our gratitude to John Purves, Head of Quality of Medicines at the EMA, for a rewarding interview on the EMA's experience with biosimilar products.
We would like to thank Ann Jonsson, Senior Expert at the MPA, for her indispensible information on the Swedish regulatory system for biosimilars.
Likewise, we want to express our appreciation to Jens Ersbøll, the Chief Medical Officer at the DKMA and CHMP, for his illustrative testimonial on the Danish regulatory approach to biosimilars.
Additionally, we want to thank Per Helboe, Senior Director of Licensing Division at the DKMA, for providing information on the overall regulation of medicines in Denmark.
We would like to thank Suzette Kox, Senior Director Scientific Affairs at the EGA, for an informative interview and for providing us with supplementary material.
Furthermore, we wish to thank Emmanuel Chanterlot, Executive Director at the EBE, for an insightful disclosure on the originators' perspective on biosimilars.
During our study, we established contact with many creative and interesting people within Teva, to whom we would like to show our appreciation:
Lone Juul Darket, General Manager in TEVA Denmark A/S
Sandy Eisen, MB BChir, Chief Medical Officer at Teva Pharmaceuticals Europe

83
Udo Müller, MD, PhD, Global Medical Director BioGenerics Global Specialty Pharmaceutical Group, Teva Pharmaceutical Industries Ltd, Israel and Teva Deutschland GmbH
Bram van Dijck, MBA, Head of Hospital and Specialty Business Europe, TEVA Pharmaceuticals Europe BV 83
Last but not least, we want to extend our gratitude to all of the representatives from the Swedish medicinal products committees that we had initially contacted:
Blekinge – Mats Josefsson
Dalarna – Elisabeth Kallin
Gävleborg – Stefan Back
Kalmar – Ellen Vinge
Kronoberg – Stephan Quittenbaum
Norrbotten – Anders Bergström
Stockholm (LÄKSAK) – Peter Bárány
Stockholm (Södra LK) – Eva Andersson Karlsson
Sörmland – Marie Portström
Värmland – Gunilla Welander
Västerbotten – Håkan Larsson
Västernorrland – Leif Rentzhog
Västmanland – Carina Westberg
Örebro – Leif Kronberg
Östergötland – Mikael Svensson

84
References
1. Roger, S.D., Biosimilars: how similar or dissimilar are they? Nephrology (Carlton), 2006. 11(4): p. 341-6.
2. EMA, GUIDELINE ON SIMILAR BIOLOGICAL MEDICINAL PRODUCTS, in COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP). 2005: London, UK. p. 1-7.
3. EMA, Questions and Answers on generic medicines. 2007: London, UK. p. 1. 4. EGA, EGA HANDBOOK ON BIOSIMILAR MEDICINES, B. Dr. Clark, Dr. Eisen, S., Huinink, L., et
al., Editor. 2008, Palgrave MacMillan: Brussels, Belgium. 5. Genazzani, A.A., et al., Biosimilar drugs : concerns and opportunities. BioDrugs, 2007. 21(6):
p. 351-6. 6. EMA, DIRECTIVE 2004/27/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 31
March 2004. Official Journal of the European Union, 2004. 1: p. 35:§15. 7. Wurm, F.M., Manufacturing of biopharmaceuticals and implications for biosimilars. Kidney
Blood Press Res, 2007. 30 Suppl 1: p. 6-8. 8. Johnson, P.E., Implications of biosimilars for the future. Am J Health Syst Pharm, 2008. 65(14
Suppl 6): p. S16-22. 9. Oldham, T., EGA Position Paper on Biosimilar Drugs. 2006, EGA: Brussels. p. 1-6. 10. Hoffman, J.M., et al., Projecting future drug expenditures--2009. Am J Health Syst Pharm,
2009. 66(3): p. 237-57. 11. Blackstone, E.A. and J.P. Fuhr Jr, Why Biosimilars Will Have a Smaller Competitive Edge Than
Generics. Journal of Health Care Finance, 2008. 35(2): p. 84-95. 12. Lindqvist, F., Personal Interview with representative from Teva Sweden AB, A. Sierakowiak,
Editor. 2009: Stockholm. 13. Desheh, E., The 2008 Annual Report of Teva Pharmacuetical Indusries Ltd. 2008, Financial:
Petach Tikva. p. 1-345. 14. EMA. Human Medicines. 2009 2009-10-13 [cited 2009 2009-11-18]; Available from:
http://www.emea.europa.eu/index/indexh1.htm. 15. EMA, European Medicines Agency. 2009: London. p. 1-2. 16. EMA, REGULATION (EC) No 72&2004 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL
of 31 March 2004, in Official Journal of the European Union. 2004, European Parliament and European Council. p. 1:§33.
17. EMA. Committee for Medicinal Products for Human Use (CHMP) Rules of Procedure. EMEA/45110/2007 2007 [cited 2009 20091219]; Available from: http://www.ema.europa.eu/pdfs/human/regaffair/4511007en.pdf.
18. EuropeanCommission, COMMISSION DIRECTIVE 2003/63/EC. 2003, Official Journal of the European Union: Brussels. p. 46-94.
19. EMA. Committee for Medicinal Products for Human Use (CHMP). 2008 20081107 [cited 2009 20091219]; Available from: http://www.ema.europa.eu/htms/general/contacts/CHMP/CHMP.html.
20. Pignatti, F., H. Boone, and I. Moulon, Overview of the European regulatory approval system. J Ambul Care Manage, 2004. 27(2): p. 89-97.
21. Singer, E., [Registration of new medicinal products in Europe]. Onkologie, 2008. 31 Suppl 2: p. 64-6.

85
22. Läkemedelsverket. Approval of new medicines. 2006 2006-02-05 [cited 2009 2009-11-18]; Available from: http://www.lakemedelsverket.se/english/overview/About-MPA/Activities/Medicines/Approval-of-new-medicines/.
23. Joung, J., et al., WHO informal consultation on regulatory evaluation of therapeutic biological medicinal products held at WHO Headquarters, Geneva, 19-20 April 2007. Biologicals, 2008. 36(4): p. 269-76.
24. Schellekens, H., Biosimilar therapeutics-what do we need to consider? NDT Plus, 2009. 2(Suppl_1): p. i27-i36.
25. Gottlieb, S., Biosimilars: policy, clinical, and regulatory considerations. Am J Health Syst Pharm, 2008. 65(14 Suppl 6): p. S2-8.
26. EGA. About the EGA - Intro. 2004 2004 [cited 2009 2009-11-18]; Available from: http://www.egagenerics.com/ega-intro.htm.
27. Rossignol, N., How the European Union Reviews and Approves 'Follow-on Biologics' or Biosimilar product. 2007, EGA: Washington D.C. p. 1-8.
28. EMA, Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins As Active Substance: Quality Issues, CHMP, Editor. 2006: London. p. 1-8.
29. EMA, Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins As Active Substance: Non-Clinical and Clinical Issues, CHMP, Editor. 2006: London. p. 1-8.
30. EMA, Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins, CHMP, Editor. 2007: London. p. 1-18.
31. Läkemedelsverket. About the Medical Products Agency. 2009 2007-03-06 [cited 2009 2009-11-16]; Available from: http://www.lakemedelsverket.se/english/overview/About-MPA/
32. Läkemedelsverket. "Biosimilars" bedöms inte vara utbytbara. 2007 2007-03-15 [cited; 1]. Available from: http://www.lakemedelsverket.se/malgrupp/Halso---sjukvard/Artikelsamlingar/Lista/Lakemedelsformanerna-och-utbytbarhet/Biosimilars-bedoms-inte-vara-utbytbara/.
33. SKL. Läkemedelskomitéerna. 2009 2009-10-21 [cited 2009 2009-11-25]; Available from: http://www.skl.se/artikel.asp?C=1104&A=1702.
34. Socialdepartimentet, Lag (1996:1157) om Läkemedelskomitéer, in 1996:1157, Socialdepartementet, Editor. 1996: Sweden. p. 1.
35. DKMA. The Danish Medicines Agency. 2009 2009-11-12 [cited; 1]. Available from: http://www.dkma.dk/1024/visUKLSArtikelBred.asp?artikelID=744.
36. DKMA. Medicinal Products. 2010 2010-01-08 [cited 2010 2010-01-14]; Available from: http://www.dkma.dk/1024/visUKLSArtikel.asp?artikelID=732.
37. DKMA. Reimbursement. 2010 2010-01-08 [cited 2010 2010-01-14]; Available from: http://www.dkma.dk/1024/visUKLSArtikel.asp?artikelID=1469.
38. Sundhetsstyrelsen. The Danish National Board of Health. 2010 [cited 2010 2010-01-14]; Available from: http://www.sst.dk/English.aspx.
39. Sundhetsstyrelsen. Sundhetsstyrelsens mål og opgaver. 2010 [cited 2010 2010-01-14]; Available from: http://www.sst.dk/Om%20styrelsen/Maal_og_opgaver.aspx.
40. DanskeRegioner. Danish Regions. 2010 2009-12-10 [cited 2010 2010-01-14]; Available from: http://www.regioner.dk/Danish%20Regions.aspx.
41. DanskeRegioner. Regionernes opgaver. 2009 2009-02-23 [cited 2010 2010-01-14]; Available from: http://www.regioner.dk/Om%20Regionerne/Regionernes%20opgaver.aspx.
42. AMGROS. Amgros I/S. 2009 [cited 2010 2010-01-14]; Available from: http://www.amgros.dk/Sider/AboutAmgros.aspx.
43. FDA, What FDA Regulates. 2009: Silver Spring, MD. p. 1. 44. Barkoff, A.F. (2008) Patent Litigation Under a Future Biosimilar Act. Snippets Volume, 1-3 45. Noonan, K.E. (2008) Follow-on Biologic Drugs and Patent Law: A Potential Disconnect?
Snippets Volume, 2, 9-10

86
46. WHO. About WHO. 2009 2009 [cited 2009 2009-11-18]; International Organization]. Available from: http://www.who.int/about/en/.
47. WHO. Guidelines on evaluation of similar biotherapeutic products (SBPs) Proposed guidelines. WHO/DRAFT/12 June 2009 2009 [cited 2009 20091219]; Available from: http://www.who.int/biologicals/publications/trs/areas/biological_therapeutics/Guidelines_Biotherapeutic_products_FINAL_HK_IK_12_June.pdf.
48. ICH. Welcome to the official web site for ICH. 2009 [cited 2009 2009-11-18]; Available from: http://www.ich.org/cache/compo/276-254-1.html.
49. ICH, Q5E: Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process. 2002. p. 3.
50. ICH. Topics under Harmonisation. 2009 [cited 2009 2009-11-18]; Available from: http://www.ich.org/cache/html/4870-272-1.html.
51. EBE. Overview. 2009 [cited 2009 20091219]; Available from: http://www.ebe-biopharma.org/index.php?option=com_content&task=view&id=1&Itemid=49.
52. Diefenbach, T., Are case studies more than sophisticated storytelling?: Methodological problems of qualitative empirical research mainly based on semi-structured interviews. Quality & Quantity, 2009. 43(6): p. 875-894.
53. Eisenhardt, K.M. and M.E. Graebner, THEORY BUILDING FROM CASES: OPPORTUNITIES AND CHALLENGES. Academy of Management Journal, 2007. 50(1): p. 25-32.
54. Patton, M., Qualitative evaluation and research methods (2nd ed.). 1990. 55. Karami, A., J. Rowley, and F. Analoui, Research and Knowledge Building in Management
Studies: An Analysis of Methodological Preferences. International Journal of Management, 2006. 23(1): p. 43-52.
56. Thomas, D.R., A General Inductive Approach for Analyzing Qualitative Evaluation Data. American Journal of Evaluation, 2006. 27(2): p. 237-246.

87
Appendix
The questionnaires for the four different interviewee groups (See Table 13) are provided below.
Interview questionnaire for Teva
General Information What is your name?
Where are you located?
What are your personal areas of responsibility within the organization?
What is your personal background? (Education, industry experience, authorities, etc)?
Do you have any areas of responsibility outside the organization? If so, could you describe these?
Questions Could you please specify the main society benefits (of biosimilars) identified by Teva?
o Health – Risk benefit? o Economical perspectives (other than the above) o Competition? o Future development?
What are the obstacles?
o What are the main obstacles?
With special regard to the reference companies.
With special regard to competitive generic companies.
o Scepticism among decision makers in authorities (please rank):
Quality issues – GMP? (E.g. purification, shelf life etc.)
Side effects?
The attitude towards production changes?
o Other manufacturing problems?
How many units or people are directly involved with the subject of biosimilars within Teva?
o How are the areas of responsibility divided between these units/people? o Where is it situated (geographically speaking)?

88
Clinical aspects?
Manufacturing aspects?
Other divisions?
o Interaction with other organizations or companies? o Future?
Which are the current attitudes of Teva towards the marketing process of biosimilars?
o Differences between countries/regions? o Phase III trials? o Company – Physician contact? o Company – Patient contact? o Company – Local authority contact? o Company – Commercial? o Company – Pharmacy?
What would a future pipeline or “fast-lane” (by EMEA) for biosimilars mean for Teva?
What are the views of the company with regards to the biosimilar debate in the U.S.? How do these views affect Teva?
o What are the views of Teva (as an international company) with regards to the FDA's approach to biosimilars?

89
Interview questionnaire for organizations
General Information What is your name?
Where are you located?
What are your personal areas of responsibility within the organization?
What is your personal background? (Education, industry experience, authorities, etc)?
Do you have any areas of responsibility outside the organization? If so, could you describe these?
Questions Could you please specify the main society benefits (of biosimilars) identified by the
organization that you represent?
o Health – Risk benefit? o Economical perspectives (other than the above) o Competition? o Future development?
What are the obstacles?
o What are the main obstacles?
With special regard to the reference companies.
With special regard to competitive generic companies.
o Skepticism among decision makers in authorities – local as well as international (please rank):
Quality issues – GMP? (E.g. purification, shelf life etc.)
Side effects?
The attitude towards production changes?
o Other manufacturing problems?
How many units or people are directly involved with the subject of biosimilars within the organization?
o How are the areas of responsibility divided between these units/people? o Where is it situated (geographically speaking)?
Clinical aspects?
Manufacturing aspects?
Other divisions?

90
o Interaction with other organizations or companies? o Future?
Which are the current attitudes of the organization towards the marketing process of biosimilars?
o Organization – Company contact? o Differences between countries/regions? o Phase III trials? o Organization – Physician contact? o Organization – Patient contact? o Organization – Local / International authority contact? o Organization – Commercial?
Could you please speculate what a future pipeline or “fast-lane” (by EMEA) would mean to the development of biosimilars?
What are the views of the organization with regards to the biosimilar debate in the U.S.? How do these views affect the organization?
What are the views of the organization (as a multinational organization) with regards to the FDA's approach to biosimilars?

91
Interview questionnaire for the European Medicines Agency
General background of the agency
What is the location of the agency?
What are the overall areas of responsibility of the agency?
General background of the interviewee
What are your personal areas of responsibility within the agency?
What is your personal background (education, industry experience, etc)?
Do you have any areas of responsibility outside the agency? If so, could you describe these?
Organization and documents
What are the areas of responsibilities within the biosimilar field and how are they divided between units/people? (Steps and their objectives, durations)
Seeing that the EMEA is the only approval agency within the EU, what does the EMEA feel are the local authorities’ areas of responsibilities with regards to biosimilars?
Please state what information is generally lacking in these documents (listed below) and how these gaps affect the agency's work with biosimilars.
o 2001/83/EC o 2003/63/EC o 2004/27/EC o Overarching guidelines o General guidelines:
o Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Quality Issues
o Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Non-Clinical and Clinical Issues
o Guideline on Immunogenicity Assessment of Biotechnology-Derived Therapeutic Proteins
o Annex guidelines

92
Processing How much difference between innovator drugs and biosimilars is acceptable?
Elaborate.
How many biosimilar drugs have not been granted authorization by your agency upon application? Why? Please exemplify.
What are the obstacles/problems regarding the authorization of biosimilar applications and how are they dealt with? (Labeling/naming, immunogenicity, etc.)
Aside from comparability and pharmacovigilance, what are the most important issues biosimilar companies should keep in mind when applying for market approval? Please list them in descending order of importance.
Are there standardized follow-up routines for post-marketing data of biosimilar drugs (compared to that of innovator drugs)?
o No: Future aspects? o Yes: Please specify.
o Does the agency work with external non-government parties to address the issue of
biosimilars? If so, could you describe these interactions? (Why do these interactions take place, how often do they take place, etc
The Future Excluding the price reduction, which other benefits of biosimilars have been identified
by the agency? Please rank them according to importance in descending order.
Can the agency foresee changes with regards to the authorization process of biosimilars within the EU in the next five years?
o If yes, what should the applicant company consider when handing in an application in the next five years with regards to these changes?
What are the views of the agency with regards to the biosimilar debate in the U.S.? How do these views affect EMEA?

93
Interview questionnaire for government agencies
General background of the government agency
What is the name and location of the government agency?
What are the overall areas of responsibility of the government agency?
General background of the interviewee
What are your personal areas of responsibility within the government agency?
What is your personal background (education, industry experience, etc)?
Do you have any areas of responsibility outside the government agency? If so, could you describe these? Organization and documents
What are the areas of responsibilities within the biosimilar field and how are they divided between units/people? (Steps and their objectives, durations)
Seeing that the EMEA is the only approval agency within the EU, what does the government agency feel is its areas of responsibilities with regards to biosimilars?
How well-informed is the government agency with regards to the legal framework and guidelines set up by the EMEA (please use the Likert scale):
Likert scale: 1 Very poor 2 Fair 3 Good 4 Very good 5 Excellent
o 2001/83/EC o 2003/63/EC o 2004/27/EC o Overarching guidelines o General guidelines:
o Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Quality Issues
o Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Non-Clinical and Clinical Issues
o Guideline on Immunogenicity Assessment of Biotechnology-Derived Therapeutic Proteins
o Annex guidelines
Please state what information is generally lacking in these documents and how these gaps affect the government agency's work with biosimilars.

94
Processing What is the definition of a 'biosimilar' according to the government agency? Which
official documents are used as a basis for this definition?
How much difference between innovator drugs and biosimilars is acceptable? Elaborate.
How many biosimilar drugs have been granted authorization by your agency upon application?
How many biosimilar drugs have not been granted authorization by your agency upon application? Why? Please exemplify.
What are the obstacles/problems regarding the authorization of biosimilar applications and how are they dealt with? (Labeling/naming, immunogenicity, etc.)
Aside from comparability and pharmacovigilance, what are the most important issues biosimilar companies should keep in mind when applying for market approval? Please list them in descending order of importance.
Are there standardized follow-up routines for post-marketing data of biosimilar drugs (compared to that of innovator drugs)?
o No: Future aspects? o Yes: Please specify.
Does the government agency work with external non-government parties or the EMEA to address the issue of biosimilars? If so, could you describe these interactions? (Why do these interactions take place, how often do they take place, etc)
The Future
Excluding the price reduction, what other benefits of the introduction of biosimilars into the market have been identified by the government agency? Please rank them according to importance in descending order.
Can the government agency foresee changes with regards to the authorization process of biosimilars within the government agency in the next five years? (i.e. changes or re-interpretations of the legal framework or guidelines)
o If yes, what should the applicant company consider when handing in an
application in the next five years with regards to these changes?
What are the views of the government agency with regards to the biosimilar debate in the U.S.? How do these views affect the work within the given government agency?
o What are the views of the government agency with regards to the FDA's
approach to biosimilars?
Does the government agency feel that there should be a global consensus with regards to biosimilars or at least within the EU??

95
Interview questionnaire for medicinal products committees
General background about the committee What is the location of the committee?
What are the overall areas of responsibility of the committee?
General background of the interviewee What are your personal areas of responsibility within the committee?
What is your personal background (education, industry experience, etc)?
Do you have any areas of responsibility outside the committee? If so, could you describe these?
Organization and documents
Seeing that medicinal products committees provide recommendations to healthcare professionals, what does the committee feel is its areas of responsibilities with regards to biosimilars? How are these areas of responsibilities divided within the committee?
How well-informed is the committee with regards to the legal framework and guidelines set up by the EMEA (Please use the Likert scale):
Likert scale: 1 Very poor 2 Fair 3 Good 4 Very good 5 Excellent
o 2001/83/EC o 2003/63/EC o 2004/27/EC o Overarching guidelines o General guidelines:
o Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Quality Issues
o Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Non-Clinical and Clinical Issues
o Guideline on Immunogenicity Assessment of Biotechnology-Derived Therapeutic Proteins
o Annex guidelines
Please state what information is generally lacking in these documents and how these gaps affect the committee's work with biosimilars.

96
Processing What is the definition of a 'biosimilar' according to the committee? Which official
documents are used as a basis for this definition?
How much difference between innovator drugs and biosimilars is acceptable? Elaborate.
How many biosimilar drugs have been recommended by the committee upon application?
o If yes, which ones?
How many biosimilar drugs have not been recommended by the committee upon application? Why? Please exemplify.
What are the obstacles/problems regarding the implementation of biosimilar drugs and how are they dealt with? (Labeling/naming, immunogenicity, etc)
Aside from comparability and pharmacovigilance, what are the most important issues of biosimilar implementation, according to the committee? Please list them in descending order of importance.
Has the committee recommended standardized follow-up routines for post-marketing data of biosimilar drugs (compared to that of innovator drugs)?
o No: Future aspects? o Yes: Please specify.
Does the committee co-operate with external non-government parties, the national regulatory authority, or the EMEA to address the issue of biosimilars? If so, could you describe these interactions? (Why do these interactions take place, how often do they take place, etc)
The Future Excluding the price reduction, which other benefits of biosimilars have been identified
by the committee? Please rank them according to importance in descending order.
Can the committee foresee changes with regards to the recommendations of biosimilars by the committee in the next five years? (i.e. changes or re-interpretations of the legal framework or guidelines)
o If yes, what should the applicant company consider when handing in an
application in the next five years with regards to these changes?
Does the committee feel that there should be an international consensus with regards to biosimilars or at least a national consensus?