ksi ąż ki z podstaw i zastosowa ń modelowania molekularnego
TRANSCRIPT

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ1
Książki z podstaw i zastosowań modelowania molekularnego
1. “Dynamics of protein and nucleic acids”. J. A. McCammon & S. C. Harvey. Cambridge Univ. Press, 1987.
2. “Biological membranes. A molecular perspective from computation and experiment”. Eds. K. M. Merz, Jr. & B. Roux. Birkhäuser, Boston, 1996.
3. “Molecular Modelling: Principles and Applications”. A. R. Leach, Longman, 1996.
4. “Simulating the Physical World, From Quantum Mechanics to
Fluid Dynamics”. Herman J.C. Berendsen. Cambridge Univ. Press, 2007 March

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ2
Tytuły wykładu:
„Zastosowanie modelowania molekularnego w biotechnologii”
BT152, specjalizacja biochemia i biofizyka

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ3
Co to jest modelowanie molekularne?
Modelowanie molekularne służy do tworzenia komputerowych
modeli cząsteczek (bio- lub nie), na których możemy prowadzić
badania
Modelowanie molekularne umożliwia nam zajrzenie w
(pseudo-) świat cząsteczek i jego modyfikowanie
Wyjaśnienie tytułu „Zastosowanie modelowania molekularnego w biotechnologii”

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ4
Zastosowanie metod modelowania molekularnego w biotechnologii
nie polega na użyciu jakiegoś szczególnego wariantu tych metod
(podobnie jak zastosowanie matematyki w biologii), ale na
wskazaniu typowych dla biotechnologii zagadnień, które możemy
rozwiązać metodami modelowania molekularnego. Przed
wskazaniem tych zagadnień należy więc określić jaką problematyką
zajmuje się biotechnologia.
Co to jest biotechnologia?
DEFINICJA: Biotechnologia jest dyscypliną, w której stosuje się
technologię do produkcji i modyfikowania cząsteczek, oraz do
manipulowania żywymi organizmami, aby uzyskać użyteczne
produkty, procesy lub usługi (?).

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ5
Komentarz: Cząsteczkę białka (produkt) o zadanych własnościach
możemy otrzymać na drodze wymiany reszt aminokwasowych
przeprowadzając kontrolowane mutacje (manipulacja na materiale
biologicznym). Przeprowadzenie mutacji punktowej i
„wyprodukowanie” odpowiedniej ilości białka – w celu prowadzenia
dalszych badań – jest czaso- i pracochłonne i wymaga sporych
nakładów finansowych.
Przeprowadzenie analogicznych czynności przy pomocy
komputerowych metod modelowania molekularnego jest znacznie
tańsze i mniej czaso- i pracochłonne. Dlatego zanim zaczniemy
badania eksperymentalne, należy spróbować wygenerować daną
strukturę „w komputerze”. Jeżeli to się uda i gdy otrzymana struktura
posiada pożądane przez nas cechy, dopiero wtedy rozpocząć prace
eksperymentalne.

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ6
DEFINICJA cd: Biotechnologia łączy wiele dziedzin, między
innymi biologię, chemię, techniki komputerowe i informatyczne
(computer science), medycynę, weterynarię, rolnictwo, nauki o
środowisku oraz inżynierię. Korzysta też z szeregu technologii jak:
recombinant DNA technology, gene transfer, embryo manipulation
and transfer, monoclonal antibody production, and bioprocess
engineering.
Jeśli stosujemy biotechnologię na poziomie molekularnym, to
metody modelowania molekularnego będą bardzo przydatne
PRZYKŁADY: Możemy wyznaczyć miejsce i rodzaj wymiany
reszty aminokwasowej,
Możemy wyznaczyć i określić własności miesca wiązania liganda
np. przy projektowaniu leków
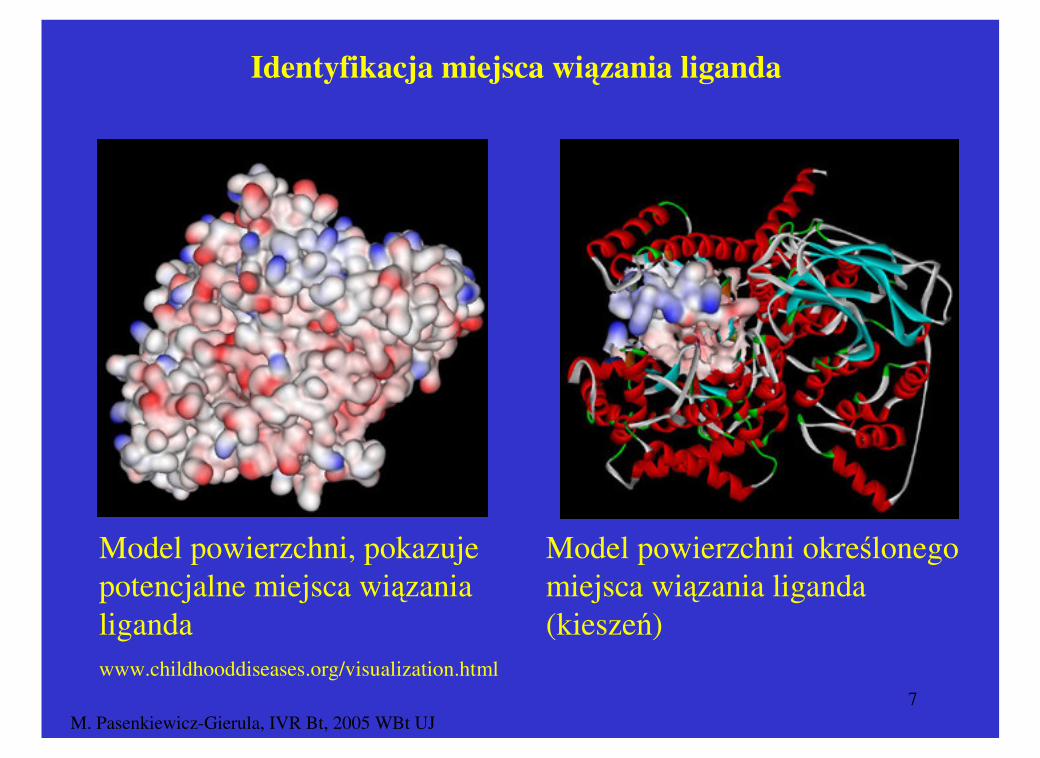
M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ7
Identyfikacja miejsca wiązania liganda
Model powierzchni, pokazuje potencjalne miejsca wiązania liganda
Model powierzchni określonego miejsca wiązania liganda (kieszeń)
www.childhooddiseases.org/visualization.html
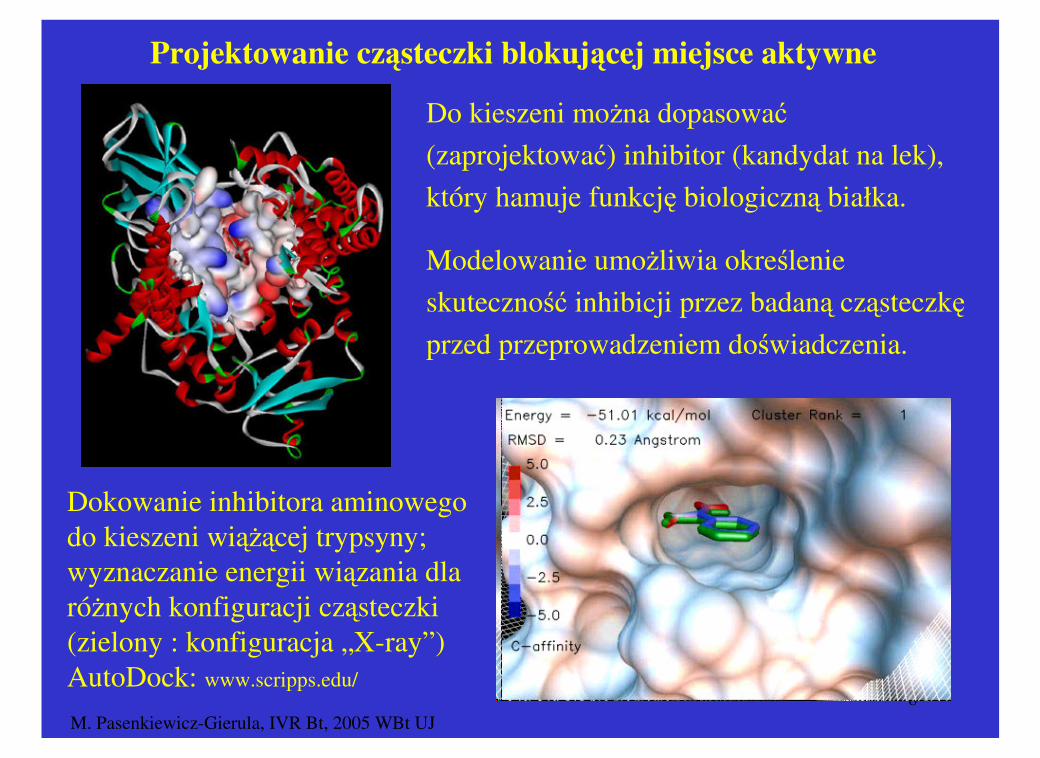
M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ8
Do kieszeni można dopasować
(zaprojektować) inhibitor (kandydat na lek),
który hamuje funkcję biologiczną białka.
Modelowanie umożliwia określenie
skuteczność inhibicji przez badaną cząsteczkę
przed przeprowadzeniem doświadczenia.
Projektowanie cząsteczki blokującej miejsce aktywne
Dokowanie inhibitora aminowego do kieszeni wiążącej trypsyny; wyznaczanie energii wiązania dla różnych konfiguracji cząsteczki (zielony : konfiguracja „X-ray”)AutoDock: www.scripps.edu/

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ9
Obecnie, wprowadzenie nowego leku na rynek trwa 10-15 lat
i kosztuje 800 milionów USD
Rational Drug Design (racjonalne projektowanie leków) ma
pomóc w odkrywaniu nowych leków
„szybciej, taniej i bezpieczniej dla pacjenta”
Pierwszy lek wprowadzony dzieki RDD to Relenza (na
grypę)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ10
Zagadnienia omawiane na wykładzie:
1. Podsumowanie metody modelowania molekularnego
2. Analiza wyników symulacji dynamiki molekularnej i weryfikacja
modelu
3. Zastosowanie modelowania molekularnego w badaniach błon
lipidowych
4. Zastosowanie modelowania molekularnego do badania mechanizmu
działania i specyficzności peptydów antybakteryjnych
5. Zastosowanie modelowania molekularnego w badaniach białek
błonowych
6. Zastosowanie modelowania molekularnego w badaniach białek
rozpuszczalnych
7. Analiza konformacyjna polipeptydów

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ11
Wykład I
Modelowanie molekularne – podsumowanie metod
1. Rola modeli w nauce
2. Modele molekularne
a. "poziomy"
b. miejsce modelowania molekularnego w nauce
3. Podstawowe zastosowania modelowania molekularnego
4. Główne metody obliczeniowe modelowania molekularnego
5. Metody wyznaczania struktur makrocząsteczek
6. Białkowa baza danych strukturalnych Protein Data Bank (PDB)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ12
W nauce stosuje się dwie podstawowe grupy modeli:
a. model eksperymentalny (np. hodowla komórek, przepływ
turbulentny itp.)
b. model teoretyczny (matematyczny)
oraz
c. model „numeryczny” (symulacje)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ13
Model układu badawczego pozwala na:
a. wyznaczenie relacji między elementami układu (modeleksperymentalny i teoretyczny); PRZYKŁAD: badanie stabilność struktury drugorzędowej w funkcji temperatury dla danego białka
b. ilościowy opis zachowania układu (model eksperymentalny iteoretyczny); PRZYKŁAD: wyznaczenie temperatury, dla której następuje denaturacja białka, opis procesu denaturacji
c. przewidywanie zachowania układu w nieco innych warunkach lub w innym przedziale czasu (model teoretyczny)– jest to najistotniejsza cecha modelu;
PRZYKŁAD: wyznaczenie związku między strukturą drugorzędową białka, liczbą oddziaływań wewnątrz-cząsteczkowych a termostabilnością białka: UOGÓLNIENIE

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ14
W naukach przyrodniczych oraz innych „ilościowych” naukach
modele są wszechobecne
Bez modeli te nauki byłyby naukami czysto opisowymi
Model jest jednak tylko przybliżeniem i uproszczeniem
rzeczywistości i jest poprawny w warunkach zmieniających się
jedynie w pewnym zakresie
(np. zmiana kinetyki układu z temperaturą – podgrzewanie roztworu
białka)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ15
Wykład I
Modelowanie molekularne – podsumowanie metod
1. Rola modeli w nauce
2. Modele molekularne
a. "poziomy"
b. miejsce modelowania molekularnego w nauce
3. Podstawowe zastosowania modelowania molekularnego
4. Główne metody obliczeniowe modelowania molekularnego
5. Metody wyznaczania struktur makrocząsteczek
6. Białkowa baza danych strukturalnych Protein Data Bank (PDB)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ16
Model molekularny to układ modelowy zbudowany z jednej lub
więcej cząsteczek
„Poziom” modelu określony jest przez podstawowy element modelu
(niepodzielny element układu)
Istnieje ścisła zależność między wielkością układu a szczegółową
informacją o układzie
Strukturę układu molekularnego możemy definiować z różną
„rozdzielczością” (dokładnością)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ17
W klasycznym modelowaniu molekularnym podstawowym
elementem modelu jest ATOM
zarówno z punktu widzenia struktury jak i oddziaływań
Oznacza to, że zaniedbujemy każdy niższy „poziom” tj. nie
uwzględniamy struktury elektronowej atomów tworzących
cząsteczkę

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ18
Np., cząsteczka białka zbudowana jest z reszt aminokwasowych, a
każda reszta zbudowana jest z atomów. W modelu mamy dużą
liczbę atomów, mniejsza liczba aminokwasów i jeszcze mniejszą
cząsteczek białka
ALA
GLU

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ19
W modelu zaniedbujemy fakt, że atomy są zbudowane z elektronów
i jądra atomowego (nie możemy badać reakcje chemicznych)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ20
Miejsce modelowania molekularnego w nauce
Klasyczne modelowanie molekularne jest to zbiór metod
komputerowych, które służą do tworzenia i badania układów
molekularnych na poziomie atomu,

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ21
Ponieważ struktura układu molekularnego jest zdefiniowana na
poziomie atomu, również wszystkie oddziaływania między
cząsteczkami lub grupami w układzie są definiowane poprzez
oddziaływania atom-atom
Międzyatomowe oddziaływania oraz struktura atomowa determinują
konformacje cząsteczki i jej dynamiczne zachowanie (nie ma „sił
zewnętrznych”) (temperatura i ciśnienie)
Metody modelowania molekularnego umożliwiają odtworzenie „w
komputerze” (in silico) zarówno struktury jak i zachowania
cząsteczek rzeczywistych

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ22
Modelowanie molekularne jest szeroko-stosowaną metodą
badawczą w nowoczesnych dyscyplinach naukowych jak biologia
strukturalna, biochemia, farmakologia, a także komercyjnie przy
projektowaniu leków,
Można jej używać jako metody niezależnej, ale najlepiej stosować ją
w powiązaniu z metodami eksperymentalnymi i/lub teoretycznymi –
ta metoda nadal wymaga weryfikacji
(problem nie jest trywialny)
Miejsce modelowania molekularnego w nauce

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ23
Komputerowe modele molekularne nie są modelami
teoretycznymi ponieważ przy ich tworzeniu wprowadzamy
warotści liczbowe otrzymane w badaniach doświadczalnych lub
obliczeniach chemii kwantowej
Modelowanie molekularne jest więc czymś pośrednim między
teorią a empirią

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ24
Wykład I
Modelowanie molekularne – podsumowanie metod
1. Rola modeli w nauce
2. Modele molekularne
a. "poziomy"
b. miejsce modelowania molekularnego w nauce
3. Podstawowe zastosowania modelowania molekularnego
4. Główne metody obliczeniowe modelowania molekularnego
5. Metody wyznaczania struktur makrocząsteczek
6. Białkowa baza danych strukturalnych Protein Data Bank (PDB)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ25
1. Wizualizacja cząsteczki w różnej reprezentacji (konieczna jest
znajomość struktury)
2. „Wspomagana” modyfikacja struktury kowalencyjnej cząsteczki
(strukturę zmodyfikowaną należy poddać procesowi
optymalizacji)
3. Budowa układu wielocząsteczkowego (strukturę układu należy
poddać procesowi optymalizacji)
4. Generowanie ruchów molekularnych – obserwacja zachowania
dynamicznego cząsteczek oraz oddziaływań wewnątrz- i
międzycząsteczkowych
Podstawowe zastosowania modelowania molekularnego

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ26
Stick model
C – atom węgla, szaryH – atom wodoru, białyO – atom tlenu, czerwonyN – atom azotu niebieski
Ball and stick model
CPK (Corey, Pauling, Koltun) model
Wizualizacja cząsteczki w różnych reprezentacjach
www.childhooddiseases.org/visualization.html
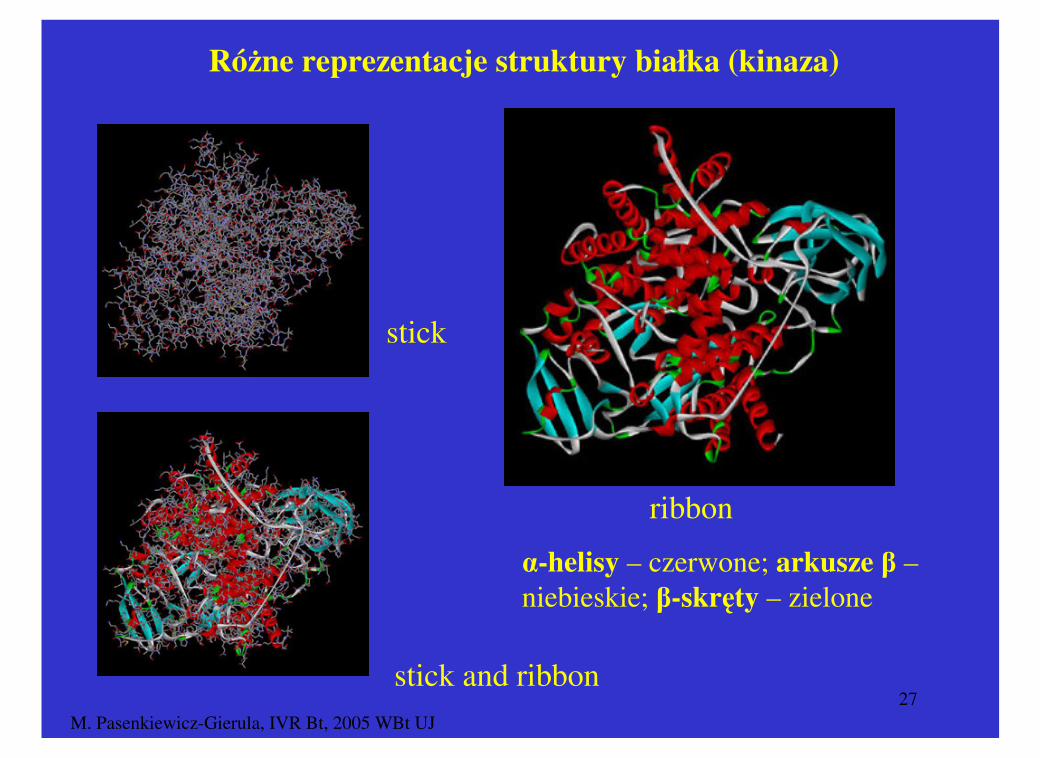
M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ27
Różne reprezentacje struktury białka (kinaza)
stick
stick and ribbon
α-helisy – czerwone; arkusze β –niebieskie; β-skręty – zielone
ribbon

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ28
Wizualizacja cząsteczki jest możliwa jeśli znana jest jej
struktura przestrzenna
nie jest natomiast konieczna znajomość oddziaływań
między-atomowych

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ29
Struktura cząsteczki:
1. Informacja o tym jak atomy tworzące cząsteczkę są ze
sobą powiązane – struktura kowalencyjna
2. Informacja o tym jak atomy tworzące cząsteczkę są
względem siebie położone – struktura przestrzenna

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ30
Struktura przestrzenna
cząsteczki może ulec zmianie
np. w wyniku zmian
konformacyjnych, oddziaływania
z inną cząsteczką, podniesienia
temperatury, zmiany
rozpuszczalnika itp.
Struktura cząsteczki
W klasycznym modelowaniu molekularnym struktura
kowalencyjna danej cząsteczki jest niezmienna (rozdzielczość
atomowa)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ31
Modyfikacja struktury i budowa cząsteczki
W komputerze możemy zmodyfikować strukturę lub zbudować
dowolną, „nierzeczywistą” cząsteczkę jako obiekt „czysto graficzny”
Zbudowaną „nierzeczywistą” strukturę (niepoprawne długości
wiązań, nieoptymalna względna orientacja łańcuchów bocznych itp.)
możemy przeprowadzić „rzeczywistą” jedynie gdy poprawnie
zdefiniowane są:
struktura kowalencyjna zbudowanej cząsteczki
oddziaływania międzyatomowe
oraz
gdy zastosujemy określone metody obliczeniowe
PRZYKŁADY: ćwiczenia w poprzednim roku

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ32
Od momentu zastosowania metod obliczeniowych
przeprowadzających strukturę „nierzeczywistą” w
„rzeczywistą”, struktura kowalencyjna cząsteczki nie może
ulec zmianie
Te metody mogą jedynie zoptymalizować stukturę
przestrzenna cząsteczki
Modyfikacja struktury i budowa cząsteczki

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ33
Modyfikacja struktury białka – „ręczna” wymiana reszt
aminokwasowych
α1-antytrypsynaGlu342� Lys
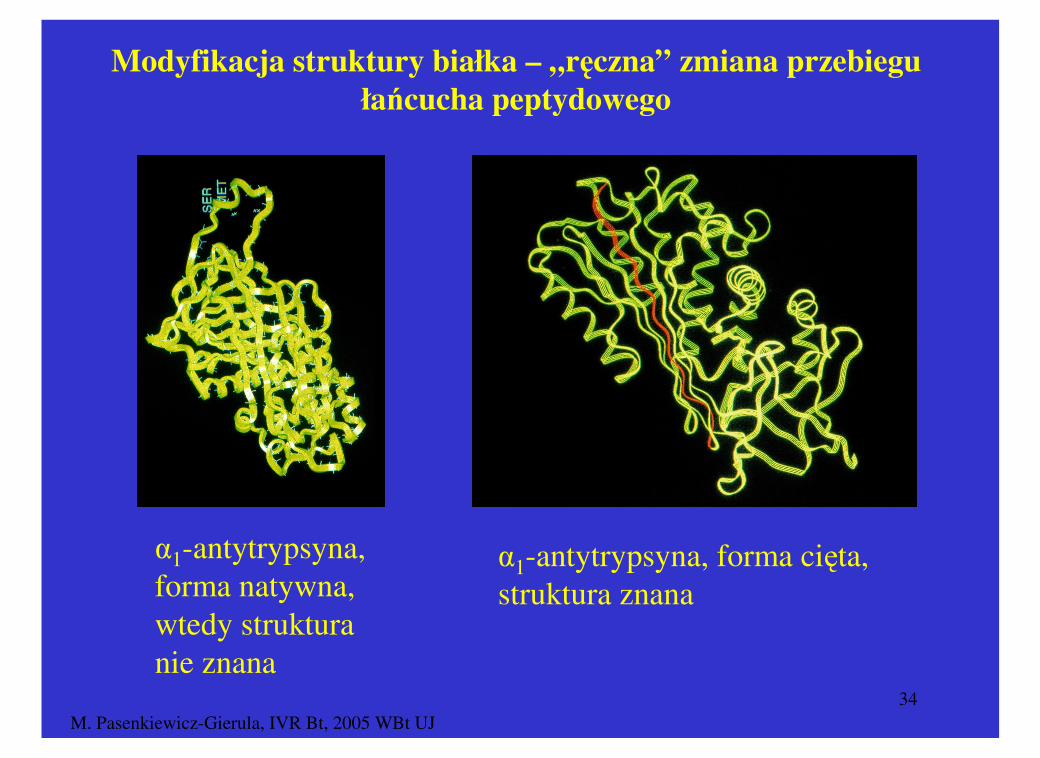
M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ34
Modyfikacja struktury białka – „ręczna” zmiana przebiegu
łańcucha peptydowego
α1-antytrypsyna, forma natywna, wtedy struktura nie znana
α1-antytrypsyna, forma cięta, struktura znana

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ35
Strukturę formy natywnej α1-antytrypsyna otrzymano poprzez modyfikację znanej struktury formy ciętej
oraz zastosowanie metod optymalizacyjnych
Modyfikacja struktury

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ36
Metody komputerowe można wykorzystać do wyznaczania
struktury przestrzennej białka w oparciu o podobieństwo jego
sekwencji aminokwasowej z białkiem o znanej strukturze
przestrzennej. Ta grupa metod bioinformatycznych i modelowania
molekularnego nosi nazwę
„Homology Modelling”
lub
„Similarity Modelling”
Budowa cząsteczki

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ37
Budowa cząsteczki – Similarity (Homology) Modelling
Bazuje na obserwacji, że „podobne sekwencje dają podobne struktur przestrzennych” (?)
Znana struktura służy jako wzorzec przy modelowaniu nieznanej (ale prawdopodobnie podobnej) struktury
Po raz pierwszy zastosowana w latach 70-tych XX wieku przez Toma Blundella
Professor Sir Tom L. Blundell, University ofCambridge
Structural Biology and Bioinformatics
Biochemical, X-ray and computational analysis of multiprotein complexes

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ38
„Homology medelling” stosujemy w przypadkach gdy nie można,
ze względów technicznych czy metodycznych, wyznaczyć
struktury przestrzennej białka metodą dyfrakcji rentgenowskiej
czy wielowymiarowego NMR
Np. dla dużej rodziny receptorów sprzężonych z białkiem G
(GPCR), znana struktura rodopsyny służy za wzorzec
Budowa cząsteczki – Similarity (Homology) Modelling

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ39
Similarity (Homology) Modelling – postępowanie
1. Wyszukanie białek o podobnej sekwencji do „naszego” białka w bazie PDB – co najmniejn 30% podobieństwa;
2. Porównanie (alignment) sekwencji „naszego” białka z podobnymi sekwencjami;
3. Identyfikacja strukturalnie zachowanych obszarów (structurally
concerved regions, SCR);
4. Identyfikacja strukturalnie zmiennych obszarów (structurally
variable regions, SVR);
5. Przepisanie współrzędnych atomów łańcucha obszaru SCR
6. Dofitowanie współrzędnych atomów pętli (Biblioteki)
7. Dodanie łańcuchów bocznych (Biblioteki)
8. Udokładnienie struktury metodami mechaniki molekularnej
9. Weryfikacja struktury

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ40
Generowanie ruchów – obserwacja zachowania dynamicznego
cząsteczek oraz oddziaływań wewnątrz- i międzycząsteczkowych
Użyteczność metod modelowania w biologii wynika przede wszystkim z faktu, że wiązania wodorowe i mostki solne, które są bardzo ważnymi oddziaływaniami w biocząsteczkach, można opisać klasycznie. Metodami modelowania molekularnego możemy więc badać ich tworzenie się i rozpadanie

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ41
Generowanie dynamicznego zachowania cząsteczek oraz
obserwacja oddziaływań wewnątrz- i międzycząsteczkowych
są możliwe jedynie wtedy, gdy w układzie molekularnym
zostaną zdefiniowane oddziaływania międzyatomowe oraz
zastosowane pewne metody obliczeniowe

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ42
Wykład I
Modelowanie molekularne – podsumowanie metod
1. Rola modeli w nauce
2. Modele molekularne
a. "poziomy"
b. miejsce modelowania molekularnego w nauce
3. Podstawowe zastosowania modelowania molekularnego
4. Główne metody obliczeniowe modelowania molekularnego
5. Metody wyznaczania struktur makrocząsteczek
6. Białkowa baza danych strukturalnych Protein Data Bank (PDB)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ43
Główne metody obliczeniowe modelowania molekularnego
służące do tworzenia realistycznych układów molekularnych
1. Mechanika Molekularna (Molecular Mechanics MM)
2. Symulacja Dynamiki Molekularnej (Molecular Dynamics, MD, Simulation)
Obie metody wymagają zdefiniowania oddziaływań między
atomami oraz zdefiniowania struktury kowalencyjnej a także
początkowej struktury przestrzennej cząsteczek tworzących układ
molekularny

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ44
Idea metody mechaniki molekularnej opiera się na związku
między stabilnością układ a odpowiadającą mu energią potencjalną
Przyjmujemy, że cząsteczka ma optymalną (stabilną) strukturę
gdy funkcja opisująca jej energię osiągnęła minimum
(GLOBALNE MINIMUM ENERGETYCZNE)
Mechanika Molekularna

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ45
Zastosowania Mechaniki Molekularnej:
Wyznaczanie stabilnej struktury cząsteczki lub układu cząsteczek
poprzez minimalizację energii potencjalnej
Energia potencjalna jednoznacznie zależy od aktualnej struktury
przestrzennej cząsteczek oraz zdefiowanych oddziaływań
międzyatomowych
UWAGA na ograniczenia metody!!!
Wyznaczenie optymalnej struktury jest możliwe jedynie dla małych
cząsteczek (ćwiczenia)
Najczęściej mechanika molekularna umożliwia znalezienie
„lokalnie stabilnej” struktury (problem lokalnych minimów)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ46
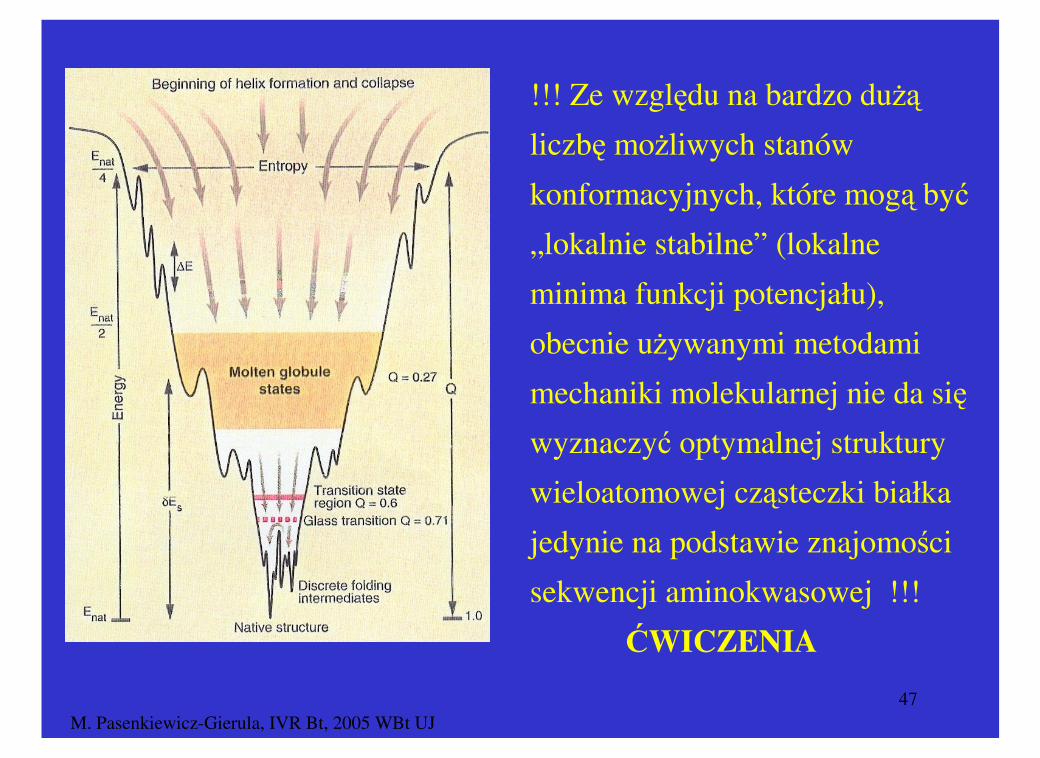
Peptyd zbudowany z N reszt aminokwasowych o znanej sekwencji:
Liczba konformacji: ≅ 2N (N = 20, l. konf. = 1 048 578)
Białko zbudowane z N reszt aminokwasowych o znanej sekwencji:
Liczba konformacji ≅ 1.4N (N = 50, l. konf. = 20 248 916)
Liczba Konformacji
M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ47
!!! Ze względu na bardzo dużą
liczbę możliwych stanów
konformacyjnych, które mogą być
„lokalnie stabilne” (lokalne
minima funkcji potencjału),
obecnie używanymi metodami
mechaniki molekularnej nie da się
wyznaczyć optymalnej struktury
wieloatomowej cząsteczki białka
jedynie na podstawie znajomości
sekwencji aminokwasowej !!!
ĆWICZENIA

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ48
Symulacja Dynamiki Molekularnej
Symulacja dynamiki molekularnej służy do generowania ruchów w
układzie molekularnym, tj. dynamicznego zachowania układu,
poprzez rozwiązanie równania ruchu dla każdego atomu w układzie
molekularnym. Aby prowadzić symulacje, oprócz zdefiniowania
oddziaływań międzyatomowych musimy w układzie molekularnym
zdefiniować jeszcze równanie ruchu
Rozwiązując numerycznie równanie ruchu (Newtona) dla każdego
atomu co krok czasowy ∆t, otrzymujemy trajektorię układu, tj.,
zbiór położeń atomów w funkcji czasu.

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ49
Makroskopowe zachowanie układu jest jednoznacznie
zdeterminowane poprzez zdefiniowane oddziaływania
międzyatomowe oraz strukturę cząsteczek tworzących
układ molekularny, tj. wynika jednoznacznie z opisu
mikroskopowego

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ50
Wykład I
Modelowanie molekularne – podsumowanie metod
1. Rola modeli w nauce
2. Modele molekularne
a. "poziomy"
b. miejsce modelowania molekularnego w nauce
3. Podstawowe zastosowania modelowania molekularnego
4. Główne metody obliczeniowe modelowania molekularnego
5. Metody wyznaczania struktur makrocząsteczek
6. Białkowa baza danych strukturalnych Protein Data Bank (PDB)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ51
Ponieważ na razie brak sukcesu w wyznaczaniu struktur
przestrzennych makrocząsteczek z „praw podstawowych”
(kolejne etapy procesu zwijania białka nie są znane, nie
można więc stworzyć algorytmu opisującego ten proces)
musimy wyznaczać je eksperymentalnie
CASP6 – grudzień 2004

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ52
Doświadczalne metody wyznaczania struktury makrocząsteczki
z rozdzielczością atomową
Metody wyznaczania struktury kowalencyjnej makrocząsteczek:
analiza sekwencyjna lub wysoko-rozdzielcza dyfrakcja
Metody wyznaczania struktury przestrzennej makrocząsteczek:

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ53
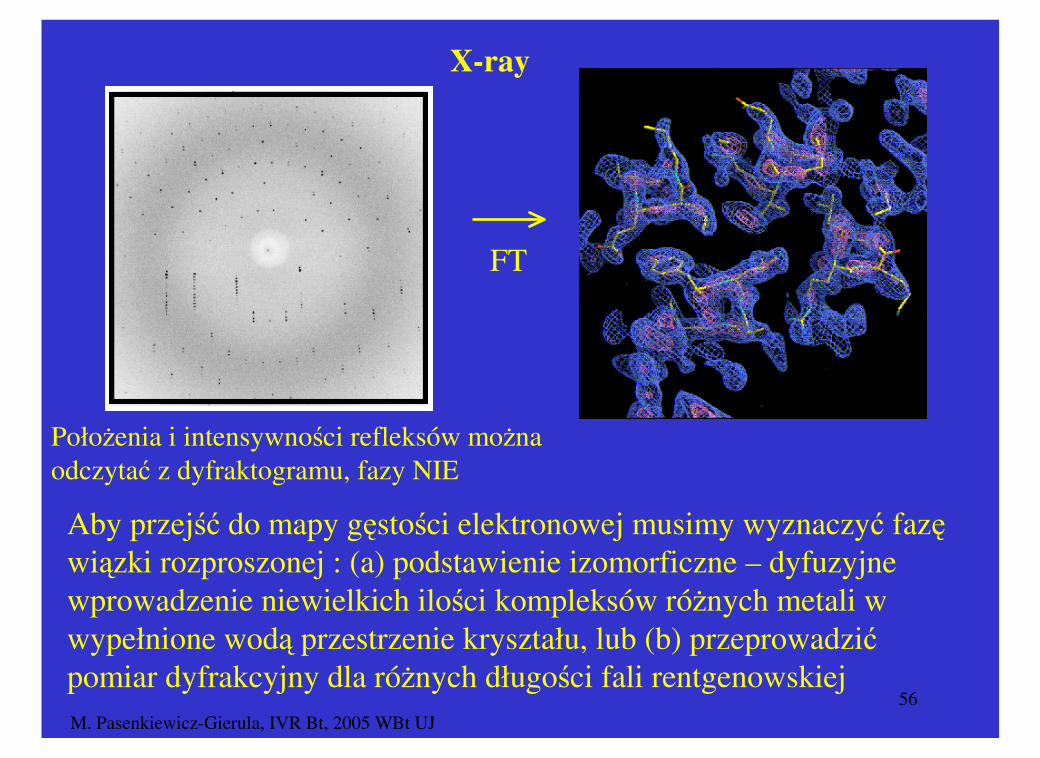
a. Dyfrakcja promieni rentgenowskich (X-ray diffraction)
Obserwacja dyfrakcji promieni rentgena na elektronach atomów
cząsteczek, które tworzą kryształ (obraz dyfrakcyjny, zbiór
refleksów).
Doświadczalne metody wyznaczania struktury przestrzennej
makrocząsteczki z rozdzielczością atomową

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ54
b. Magnetyczny Rezonans Jądrowy (1D-, 2D-, 3D- ... NMR),
Jądrowy Efekt Overhausera (Nuclear Overhauser Effect, NOE)
Homojądrowy (Homonuclear) NOE (1H, I = ½)
Heterojądrowy (Heteronuclear) NOE (15N, I = ½; 13C, I = ½; 31P, I = ½)
Rejestracja przesunięcia chemicznego (chemical shift) linii
rezonansowych atomów ze spinem jądrowym I ≠ 0.
Doświadczalne metody wyznaczania struktury przestrzennej
makrocząsteczki z rozdzielczością atomową

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ55
Porównanie metod dyfrakcji rentgenowskiej i magnetycznego rezonansu jądrowego w aspekcie badań struktury makrocząsteczek
X-ray NMRwielkość badanych nieograniczona ~50 kDcząsteczek
ilość materiału ~ 0.1 µmol ~ 1 µmol
próbka monokryształ roztwór
stężenie próbki ~ 15 mM ~ 1 mM
zjawisko dyfrakcja promieni magnetyczny rezonansX na elektronach jądrowy
obserwacja natężenie refleksów NOE, stałe sprzężenia
fitowanie mapy gęstości lokalnych odległościmodelu do elektronowej międzyatomowych
Brak bezpośredniego „pomiaru” położeń atomów w cząsteczce
M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ56
FT
Położenia i intensywności refleksów można odczytać z dyfraktogramu, fazy NIE
Aby przejść do mapy gęstości elektronowej musimy wyznaczyć fazę wiązki rozproszonej : (a) podstawienie izomorficzne – dyfuzyjne wprowadzenie niewielkich ilości kompleksów różnych metali w wypełnione wodą przestrzenie kryształu, lub (b) przeprowadzić pomiar dyfrakcyjny dla różnych długości fali rentgenowskiej
X-ray

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ57
2D COSY NMR
2D NOESY NMR
sprzężenie między 1H związanymi kowalencyjnie poprzez 1 lub 2 inne atomy(sprzężenie poprzez wiązanie)
sprzężenia między spinami jądrowymi atomów znajdujących się blisko w przestrzeni (r < 5 Å) inależących do różnych resztaminokwasowych (daleko w sekwencji; sprzężenie przez przestrzeń)
NMR

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ58
Wielowymiarowy NMR pozwala jedynie na wyznaczenie odległości
(górny zakres) między obserwowanymi w eksperymencie atomami
WIĘZY

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ59
Zbiór odległości należy przetworzyć na zbiór współrzędnych
kartezjańskich wszystkich atomów w cząsteczce
Distance
geometry,symulowane wyżarzanie
Wszystkie wyznaczone struktury spełniają warunki więzów

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ60
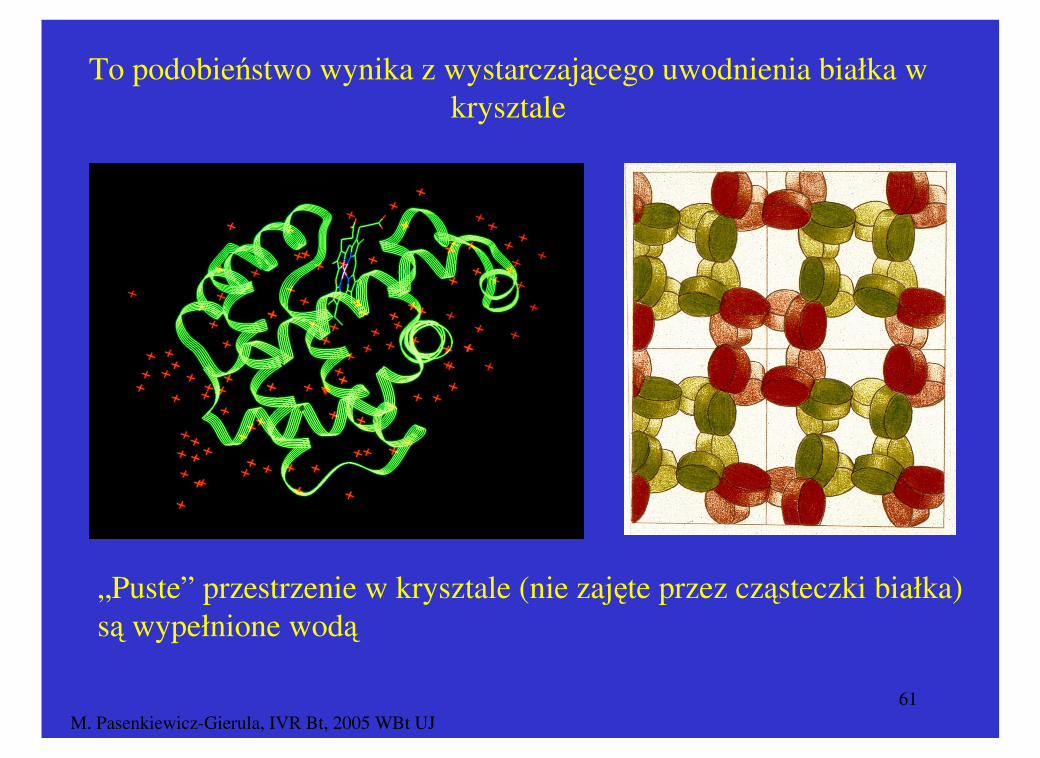
Struktury przestrzenne białka wyznaczone metodą dyfrakcji promieni rentgenowskich oraz wielowymiarowego NMR są bardzo podobne
NMR – roztwór białka X-ray – kryształ białka
M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ61
To podobieństwo wynika z wystarczającego uwodnienia białka w krysztale
„Puste” przestrzenie w krysztale (nie zajęte przez cząsteczki białka) są wypełnione wodą

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ62
Wykład I
Modelowanie molekularne – podsumowanie metod
1. Rola modeli w nauce
2. Modele molekularne
a. "poziomy"
b. miejsce modelowania molekularnego w nauce
3. Podstawowe zastosowania modelowania molekularnego
4. Główne metody obliczeniowe modelowania molekularnego
5. Metody wyznaczania struktur makrocząsteczek
6. Białkowa baza danych strukturalnych Protein Data Bank (PDB)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ63
Obecnie PDB zawiera jedynie struktury wyznaczone metodami eksperymentalnymi
The Protein Data Bank – PDB (Bank Danych Białkowych)http://www.rcsb.org/pdb

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ64
PDB Holdings List: 18-Oct-2005
Molecule Type
Proteins,
Peptides,
and Viruses
Protein/
Nucleic Acid
Complexes
Nucleic
Acids
Carbohyd
ratestotal
E
x
p
.
T
e
c
h
.
X-ray
Diffraction
and other26235(87%)
1239(91%)
851(56%)
11 28336
NMR4033
(13%)118
(9%)66344%
2 4816
Total 30268 1357 1514 13 33152

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ65
Wykład I
Modelowanie molekularne – podsumowanie metod
1. Rola modeli w nauce
2. Modele molekularne
a. "poziomy"
b. miejsce modelowania molekularnego w nauce
3. Podstawowe zastosowania modelowania molekularnego
4. Główne metody obliczeniowe modelowania molekularnego
5. Metody wyznaczania struktur makrocząsteczek
6. Białkowa baza danych strukturalnych Protein Data Bank (PDB)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ66
Wykład I
Modelowanie molekularne – podsumowanie metod
1. Oddziaływania międzyatomowe, funkcja potencjału
2. Generator ruchu, rozwiązanie równania Newtona

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ67
Wybór oddziaływań międzyatomowych zależy od nas!
(struktura nie jest dowolna)
Dzielimy je na:
oddziaływania wiążące
ioddziaływania niewiążące

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ68
Oddziaływania wiążące są odpowiedzialne za zachowanie „poprawnej” struktury kowalencyjnej cząsteczek tworzących układ, tj. zachowanie równowagowych długości wiązań, kątów walencyjnych i torsyjnych

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ69
Do modelowania makrocząsteczek lub układów wielocząsteczkowych zazwyczaj definiuje się trzy proste oddziaływania wiążące:
1. Oddziaływanie harmoniczne zachowujące równowagową długość
wiązań kowalencyjnych oraz umożliwiające drgania wibracyjne
wiązań wokół wartości równowagowych
( )2
0bbKb
b−∑
http://www.biochemistry.bham.ac.uk/osmart/course/

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ70
Copyright: Sebastian Szytuła
M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ71
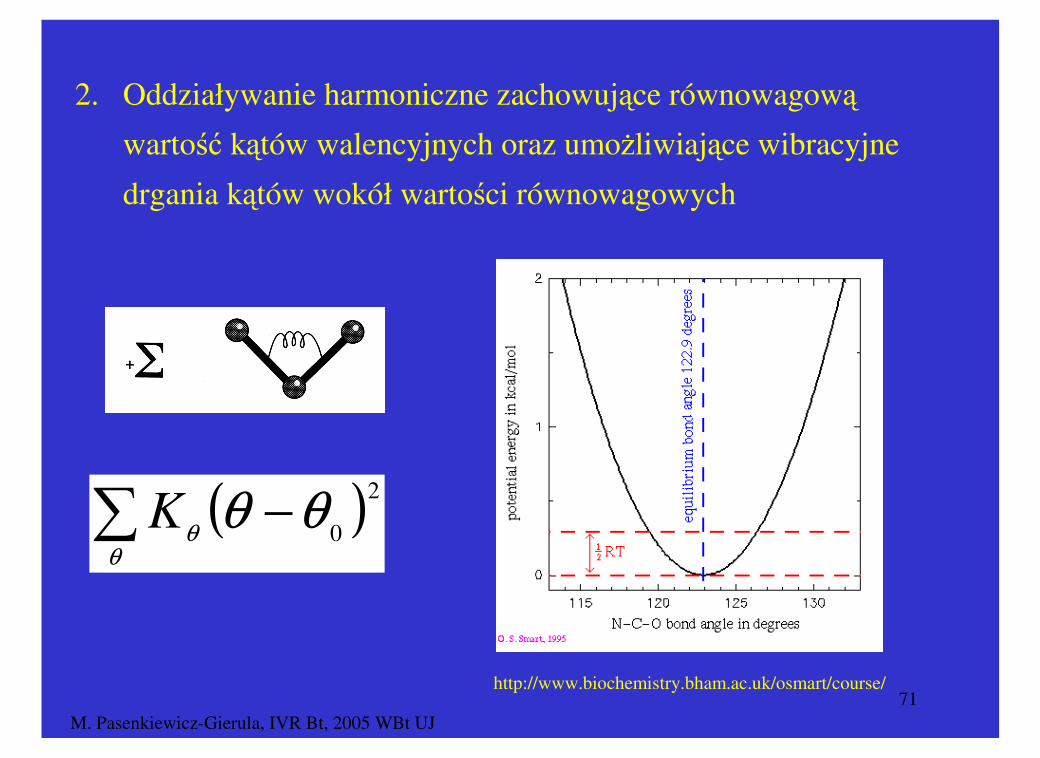
2. Oddziaływanie harmoniczne zachowujące równowagową
wartość kątów walencyjnych oraz umożliwiające wibracyjne
drgania kątów wokół wartości równowagowych
( )2
0θθθ
θ−∑K
http://www.biochemistry.bham.ac.uk/osmart/course/

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ72
3. Oddziaływanie periodyczne zachowujące stabilne konformacje
kątów torsyjnych oraz umożliwiający przejścia konformacyjne
[ ]∑ −+ϕ
ϕϕ 0cos(12
nV
n
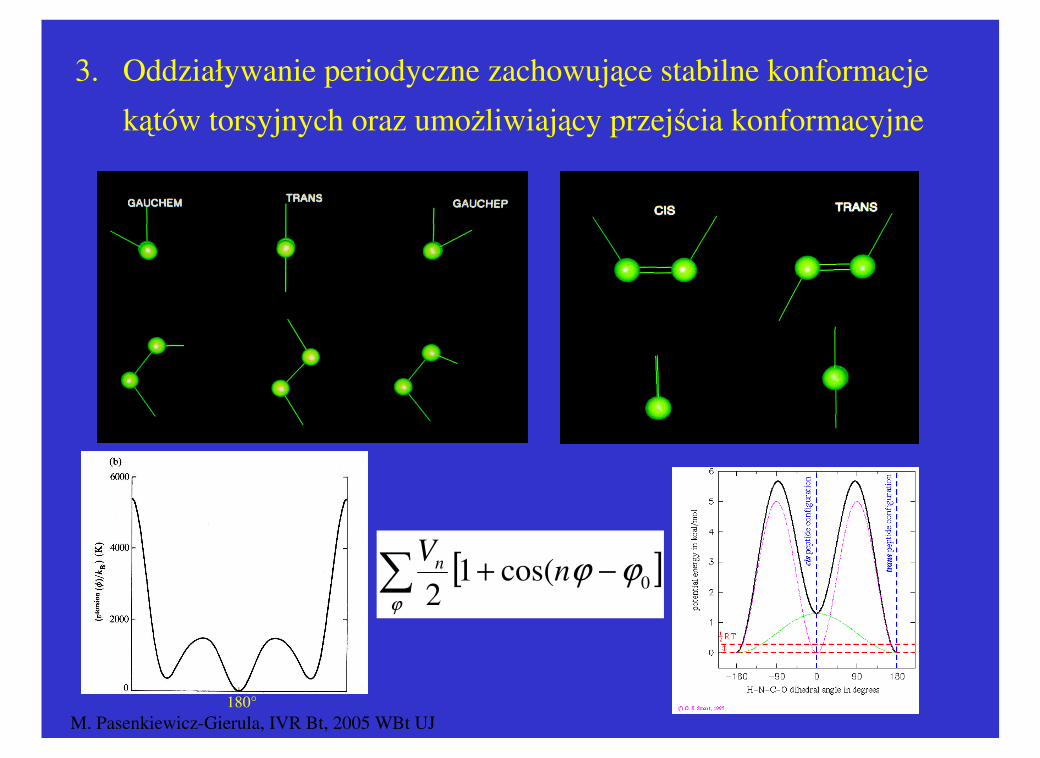
M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ73
3. Oddziaływanie periodyczne zachowujące stabilne konformacje
kątów torsyjnych oraz umożliwiający przejścia konformacyjne
[ ]∑ −+ϕ
ϕϕ 0cos(12
nVn
180°

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ74
3. Oddziaływanie periodyczne zachowujące stabilne konformacje
kątów torsyjnych oraz umożliwiający przejścia konformacyjne
http://www.missouri.edu/~chemrg/

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ75
3. Oddziaływanie periodyczne zachowujące stabilne konformacje
kątów torsyjnych oraz umożliwiający przejścia konformacyjne

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ76
Oddziaływania niewiążące muszą poprawnie odtwarzać:
wiązania wodorowe
efekt hydrofobowy

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ77
Cząstka polarna Cząstka niepolarna
Wiązania wodorowe i efekt hydrofobowy

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ78
Oddziaływania niewiążące muszą poprawnie odtwarzać:
oddziaływania między grupami polarnymi – mostki solne (α1-antytrypsyna)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ79
Oddziaływania niewiążące muszą poprawnie odtwarzać:
oddziaływania między grupami niepolarnymi(lipoproteina)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ80
Wiązania wodorowe, efekt hydrofobowy, oddziaływania między
grupami polarnymi oraz oddziaływania między grupami
niepolarnymi mają zasadniczy wpływ na
strukturę przestrzenną
cząsteczek tworzących układ molekularny oraz determinują
oddziaływania wewnątrz- i międzycząsteczkowe
M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ81
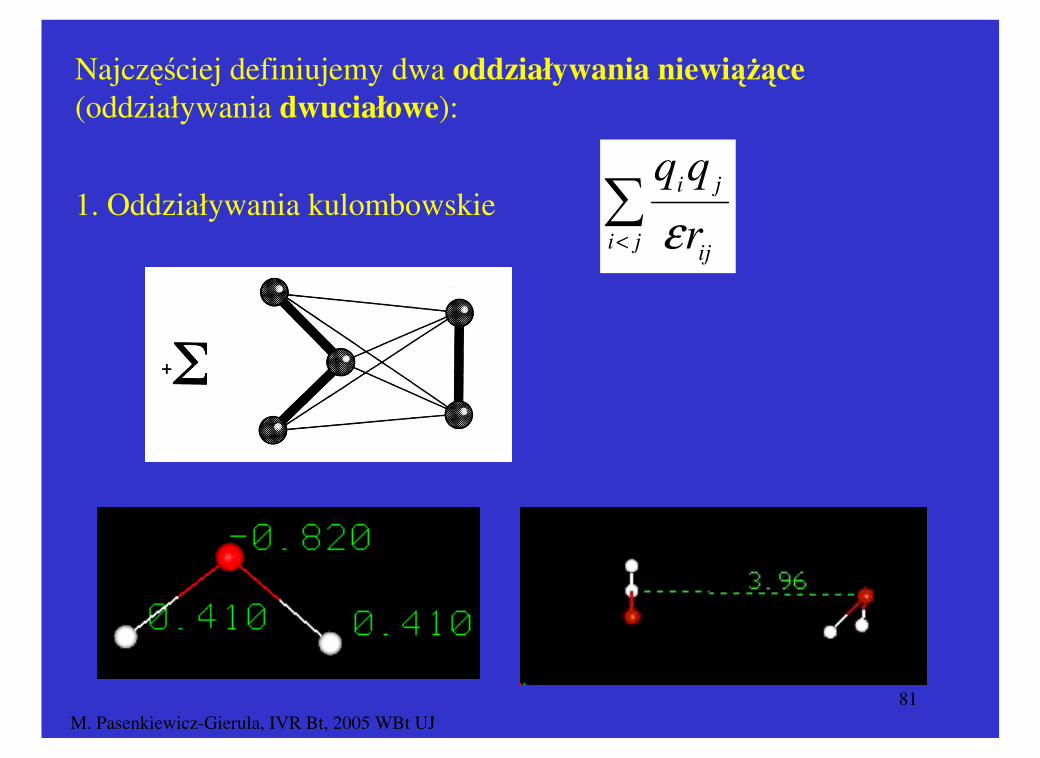
Najczęściej definiujemy dwa oddziaływania niewiążące
(oddziaływania dwuciałowe):
∑< ji
ij
ji
r
ε1. Oddziaływania kulombowskie

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ82
Najczęściej definiujemy dwa oddziaływania niewiążące
(oddziaływania dwuciałowe):
∑<
−
jiijij
r
r
r
r6
*12
*
2ε2. Oddziaływania van der Waalsa(model Lennarda-Jonesa „6-12”)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ83
Funkcja potencjału
Funkcja potencjału układu molekularnego to suma wszystkich
oddziaływań (wiążących i niewiążących) jakie zdefiniowaliśmy w
układzie molekularnym

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ84
Oddziaływania występujące w funkcji potencjału są zachowawcze,
tj. zależą jedynie od
chwilowych położeń atomów
To jest podstawą związku struktury cząsteczki z jej energią!!
Funkcja potencjału

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ85
Oddziaływania wiążące występują tylko między kolejnymi
dwoma, trzema i czterema atomami połączonymi wiązaniami
kowalencyjnymi
Funkcja potencjału
Człony oddziaływań wiążących są równe zero dla idealnej
konformacji cząsteczki lub dodatnie (niestabilna konformacja)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ86
Oddziaływania niewiążące występują między wszytkimi
parami atomów nie oddziałujących poprzez oddziaływania
wiążące
Człony oddziaływań niewiążących mogą być dodatnie
(oddziaływania odpychające) lub ujemne (oddziaływania
przyciągające)
Funkcja potencjału

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ87
UWAGA PRAKTYCZNA
Zero funkcji jest jednak zdefiniowane arbitralnie, dlatego
porównywanie energii układu molekularnego obliczonej dla
różnych funkcji potencjału nie ma sensu. Możemy jedynie
porównywać energie różnych konformacji cząsteczki obliczone
przy użyciu tej samej funkcji potencjału

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ88
Funkcja potencjału
Parametry funkcji potencjału:
Wyznaczamy albo empirycznie albo w obliczeniach kwantowo-mechanicznych, najczęściej dla określonego typu układu (gazowy, uwodniony itp.)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ89
Jak możemy wykorzystać zdefiniowaną funkcję potencjału?
Do optymalizacji struktury układu molekularnego (Mechanika Molekularna – Molecular Mechanics)
Do generowania dynamicznego zachowania układu (Symulacja Dynamiki Molekularnej – Molecular Dynamics Simulation).

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ90
Wykład I
Modelowanie molekularne – podsumowanie metod
1. Oddziaływania międzyatomowe, funkcja potencjału
2. Generator ruchu, rozwiązanie równania Newtona

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ91
Ruchy atomów i grup atomowych w cząsteczkach
W nie-zerowej temperaturze cząsteczki podlegają ruchom (na różnych
poziomach strukturalnych)

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ92
Ruchy atomów i grup atomowych w cząsteczkach
ruchy atomów → ruchy grup → zmiany konformacyjne → fluktuacje strukturalne (objętościowe)
Związek między strukturą, dynamiką i funkcją makrocząsteczek: Biologia Strukturalna

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ93
W metodzie symulacji dynamiki molekularnej do zdefiniowanego
układu molekularnego wprowadzamy równanie ruchu
To równanie generuje ruchy molekularne w układzie, tj.
dynamiczne zachowanie układu w pewnej skali czasowej
(generuje ewolucję czasową ukadu)
W dynamice molekularnej równaniem ruchu jest równanie
Newtona opisujące drugą zasadę dynamiki

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ94
Równanie Newtona
F ai
ti i
tm( ) ( )=
F ri
ti
grad V t( ) ( ( ))= −
gradx y z
=
∂∂
∂∂
∂∂
i j k, ,
Fr
r
rr
it
V tm
t
tm t
ii
ii i
( )( ( )) ( )
&& ( )= − = =∂
∂
∂
∂
2
2

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ95
Funkcja potencjału

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ96
Równanie Newtona rozwiązujemy numerycznie
Stosujemy algorytm leapfrog
ttv tr ttr
t tv tv
t
m
tFtt
∆++=∆+
∆+−=+∆
∆∆
)()()(
)()(
2
)(22
Jest to algorytm iteracyjny, tj. znając prędkości i położenia w czasie
t możemy obliczyć te wielkości w czasie ∆t późniejszym

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ97
Schemat numerycznego wyznaczania położeń (i prędkości) atomów w metodzie symulacji dynamiki molekularnej i mechaniki molekularnej

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ98
Rozwiązując numerycznie równanie Newtona dla każdego atomu co
krok czasowy ∆t, otrzymujemy trajektorię układu, tj., zbiór położeń
atomów w funkcji czasu
Otrzymujemy też zbiór prędkości atomów

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ99
6*6*2 LAYER made of POPC A, built of 6 residues; united atom
31949 0.9466000E+03
23.6689567 64.7803866 41.0944437 24.2084846 64.1276521 42.3503141
23.3005781 64.3412346 43.4894241 21.9890438 63.5834207 43.3622669
21.0339638 63.8854831 44.5671746 19.7936690 63.2203522 44.1329786
18.5972271 63.2837801 45.1084069 18.9041160 62.4152601 46.3808788
17.8836478 62.6568132 47.4873951 18.1484763 62.4949013 48.7911322
19.4947570 61.9605219 49.3301369 19.8400878 62.4858792 50.7388486
18.7550477 62.4269931 51.7337683 19.1719719 63.0012332 53.0614203
18.1688185 62.8697911 54.0943868 18.5051257 63.7985092 55.2943021
17.6161882 63.5432961 56.5497843 18.0276000 64.4250195 57.7689137
17.8397159 65.6610175 57.6811261 19.0840960 63.8578614 58.4151114
19.8536217 64.8544128 59.0933408 20.1222681 64.3236762 60.4909903
18.9117605 64.0633365 61.1380845 18.9059599 63.1863742 62.4971722
20.1417722 63.3965449 63.2436460 17.6672145 63.7205446 63.1361581
18.7986175 61.5997432 62.2252364 17.9526703 61.1540734 61.1771877
18.0700027 59.6519669 60.8471474 17.6959929 59.2458115 59.4893770
18.0945944 57.8648251 59.1086975 16.2484431 59.2799871 59.4928062
18.1389489 60.0887036 58.3423607 21.1512760 65.2581816 58.4208670
Trajektoria układu (zbiór położeń atomów w funkcji czasu)
M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ100

M. Pasenkiewicz-Gierula, IVR Bt, 2005 WBt UJ101
Zagadnienia omawiane na wykładzie:
1. Podsumowanie metody modelowania molekularnego
2. Analiza wyników symulacji dynamiki molekularnej i weryfikacja
modelu
3. Zastosowanie modelowania molekularnego w badaniach błon
lipidowych
4. Zastosowanie modelowania molekularnego do badania mechanizmu
działania i specyficzności peptydów antybakteryjnych
5. Zastosowanie modelowania molekularnego w badaniach białek
błonowych
6. Zastosowanie modelowania molekularnego w badaniach białek
rozpuszczalnych
7. Analiza konformacyjna polipeptydów