la enfermerÍa en ensayos clinicos - fundació … · · 2015-12-09en un tratamiento asistencial...
TRANSCRIPT

LA ENFERMERÍA EN ENSAYOS CLINICOS
Verónica Martínez Fabra Enfermera Coordinadora de Ensayos Clínicos
IRB-HUAV Lleida 18NOVIEMBRE2015

INTRODUCCIÓN • La investigación en salud constituye un instrumento clave
para mejorar la calidad y expectativas de vida de las personas
• Su objetivo es mejorar la prevención, el diagnóstico y el tratamiento de las enfermedades
• En el ámbito farmacéutico, antes de aprobar un nuevo fármaco, debe ensayarse su eficacia, comprobar su seguridad y compararlo con las terapias disponibles

TIPOS DE INVESTIGACIÓN
Industria Farmacéutica
Doctorado
Becas de
diferentes
instituciones
Investigación propia/
Independiente

¿QUÉ ES UN ENSAYO CLÍNICO?

¿QUÉ ES UN ENSAYO CLÍNICO?
Evaluación experimental de un producto, sustancia, medicamento, técnica diagnóstica o terapéutica que,
aplicada en seres humanos, pretende valorar su eficacia y seguridad

“Toda investigación efectuada en seres humanos para determinar o confirmar los efectos clínicos, farmacológicos y/o demás efectos farmacodinámicos, y/o detectar las reacciones adversas, y/o estudiar la absorción, distribución, metabolismo y excreción de uno o varios medicamentos en investigación con el fin de determinar su seguridad y/o eficacia”.

¿Para qué participar en un ensayo clínico?
• Para tener acceso a tratamientos novedosos antes de su comercialización
• Para recibir seguimientos más estrictos y frecuentes durante el estudio, con la realización de más pruebas de seguridad y eficacia que en un tratamiento asistencial
• Para contribuir a la investigación médica

ENSAYOS CLÍNICOS
• DEPENDIENTES industria farmacéutica
• INDEPENDIENTES investigación propia
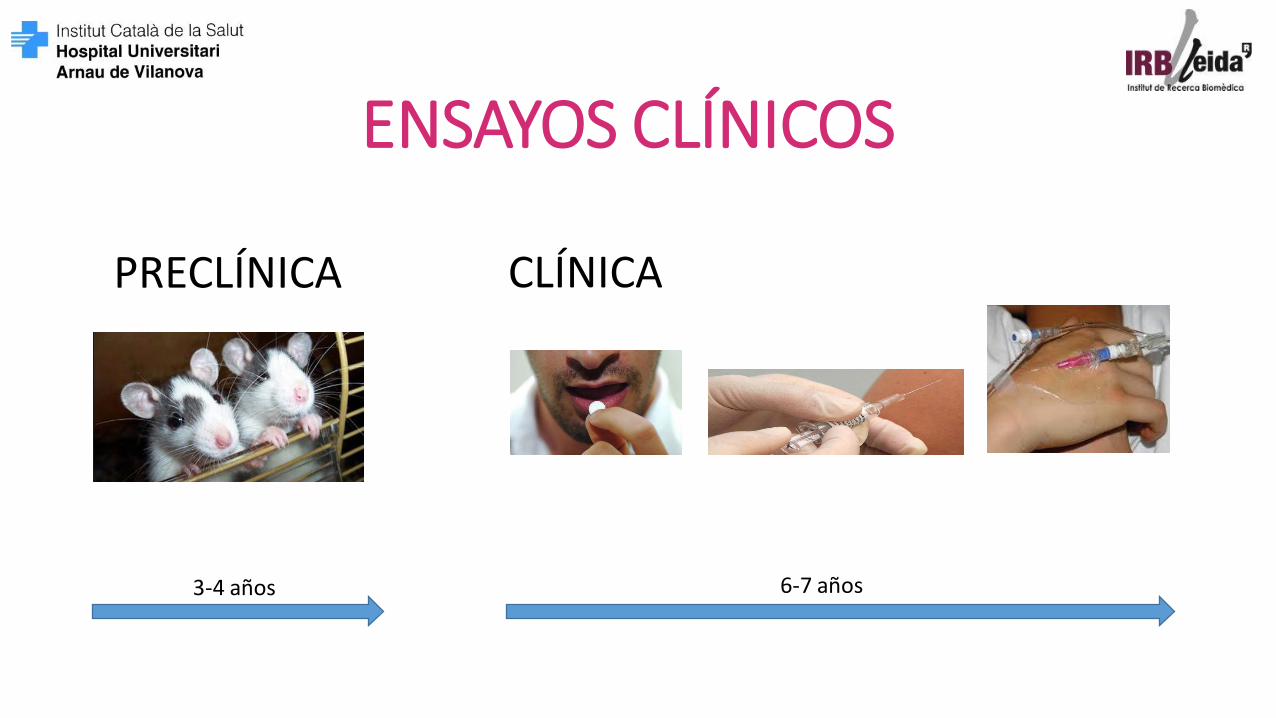
CLÍNICA
ENSAYOS CLÍNICOS
PRECLÍNICA
3-4 años 6-7 años

Investigación preclínica
3-4 años 5.000-10.000 compuestos (moléculas)
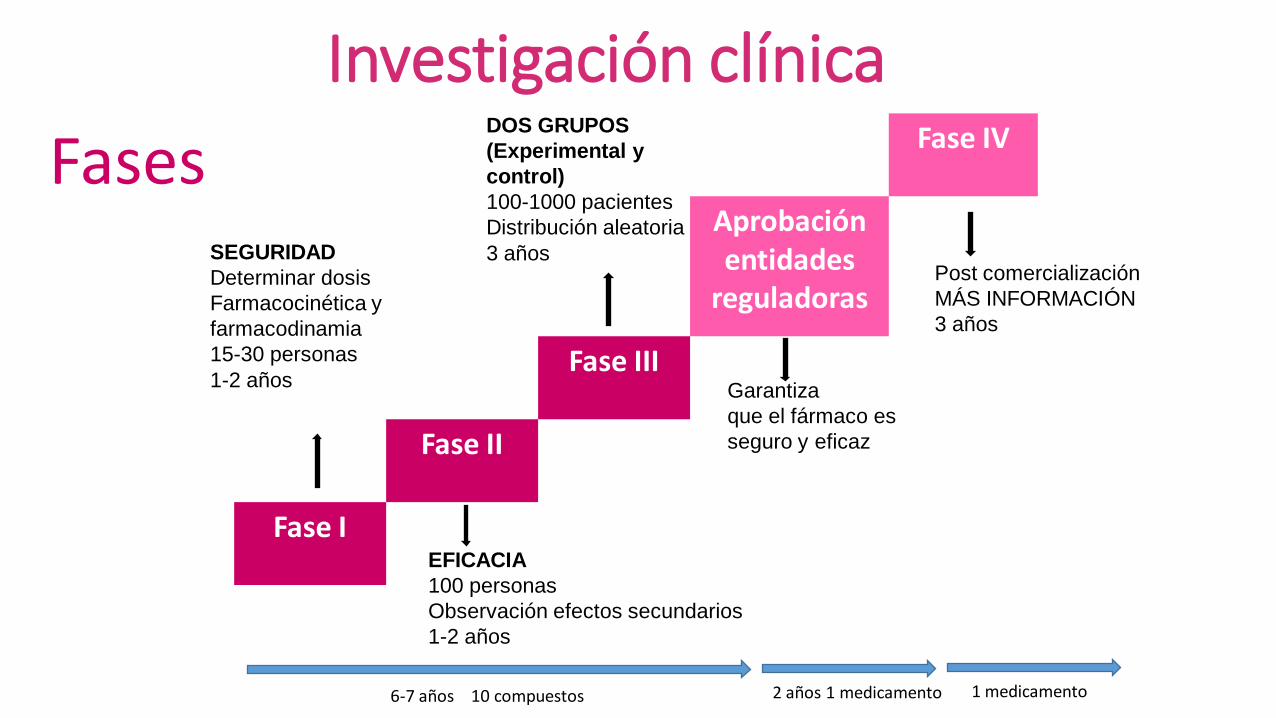
Fase IV
Aprobación entidades
reguladoras
Fase III
Fase II
Fase I
SEGURIDAD
Determinar dosis
Farmacocinética y
farmacodinamia
15-30 personas
1-2 años
EFICACIA
100 personas
Observación efectos secundarios
1-2 años
DOS GRUPOS
(Experimental y
control)
100-1000 pacientes
Distribución aleatoria
3 años
Garantiza
que el fármaco es
seguro y eficaz
Post comercialización
MÁS INFORMACIÓN
3 años
Fases
6-7 años 10 compuestos 2 años 1 medicamento 1 medicamento
Investigación clínica

Ensayo clínico
(Sujeto)
Investigador Principal
Subinvestigadores
CEIC
Study Nurse
Study Coordinator
Data Manager Administrativ@
Técnico Laboratorio
Estadístic@
Monitor
(CRO)
Promotor
Roles participantes en un ensayo clínico

Las Normas de Buenas Prácticas Clínicas

Las Normas de Buenas Prácticas Clínicas
• La guía de Buena Práctica Clínica (BPC) es una norma internacional de calidad ética y científica aplicable al diseño, realización, registro y comunicación de los ensayos clínicos en los que participen seres humanos. El cumplimiento de esta norma proporciona una garantía pública de la protección de los derechos, la seguridad y el bienestar de los sujetos del ensayo de acuerdo con los principios de la Declaración de Helsinki (1964), así como también garantiza la credibilidad de los datos del ensayo clínico

Las Normas de Buenas Prácticas Clínicas
1. Los ensayos clínicos deben realizarse de acuerdo con los principios éticos que tienen su origen en la Declaración de Helsinki, y que sean coherentes con la guía de la BPC y la legislación vigente
2. Antes de iniciar un ensayo, deberán considerarse los riesgos e inconvenientes previsibles en relación con el beneficio esperado, tanto para el sujeto individual del ensayo como para la sociedad. Un ensayo deberá iniciarse y continuarse únicamente en el caso de que los beneficios previstos justifiquen los riesgos

Las Normas de Buenas Prácticas Clínicas
3. Los derechos, la seguridad y el bienestar de los sujetos de un ensayo son las consideraciones más importantes y deberán prevalecer sobre los intereses de la ciencia y de la sociedad
4. La información clínica y no clínica disponible sobre un medicamento en investigación deberá ser suficiente para avalar el ensayo clínico propuesto
5. Los ensayos clínicos deberán estar científicamente justificados y estar descritos en un protocolo claro y detallado

Las Normas de Buenas Prácticas Clínicas
6. El ensayo se deberá realizar de acuerdo con el protocolo que previamente ha recibido un dictamen favorable de un CEIC
7. El cuidado médico que reciben los sujetos y las decisiones médicas tomadas en su nombre serán siempre responsabilidad de un médico cualificado
8. Cada individuo implicado en la realización de un ensayo deberá estar cualificado, por su titulación, formación y experiencia, para realizar sus tareas y responsabilidades respectivas

Las Normas de Buenas Prácticas Clínicas
9. El investigador deberá obtener el consentimiento informado, otorgado de forma libre, de cada sujeto antes de su participación en el ensayo clínico
10. Toda la documentación del ensayo clínico deberá ser registrada, manejada y archivada de forma que permita su comunicación, interpretación y verificación exactas
11. Se deberá proteger la confidencialidad de los registros que pudieran identificar a los sujetos para respetar su privacidad

Las Normas de Buenas Prácticas Clínicas
12. Los medicamentos en investigación deberán fabricarse, manejarse y almacenarse de acuerdo con las Normas de Correcta Fabricación (NCF) pertinentes y se deberán utilizar de acuerdo con el protocolo aprobado
13. Se implantarán sistemas con procedimientos que aseguren la calidad de cada aspecto del ensayo
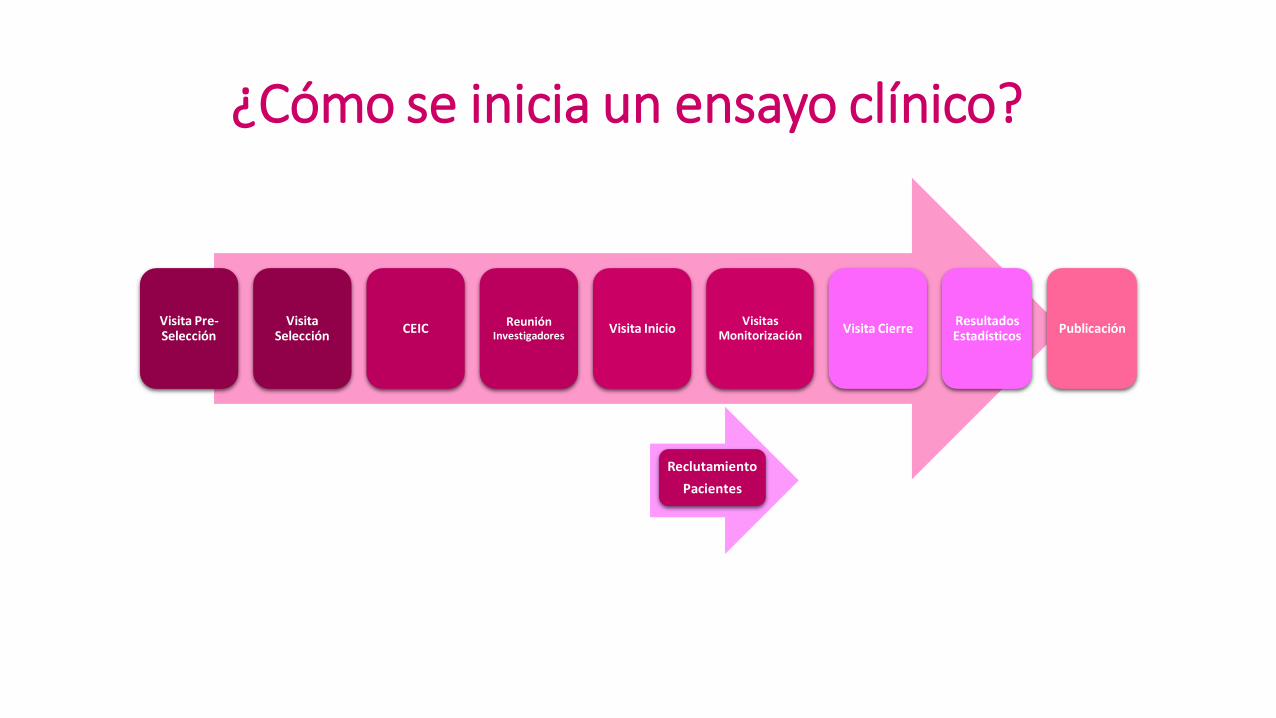
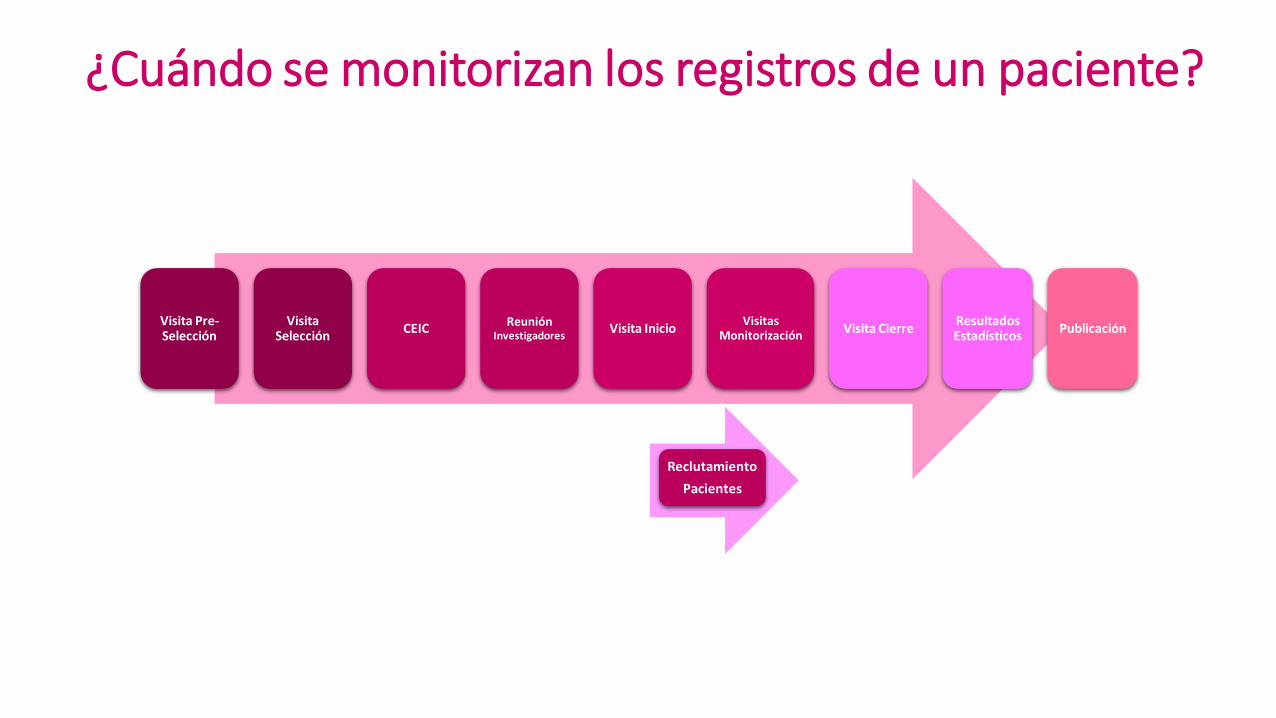
Reclutamiento
Pacientes
Visita Pre-Selección
Visita Selección
CEIC Reunión
Investigadores Visita Inicio
Visitas Monitorización
Visita Cierre Resultados Estadísticos
Publicación
¿Cómo se inicia un ensayo clínico?

PROTOCOLO DE ENSAYO CLÍNICO
• Código: CLOROTIC
• Título: Estudio aleatorizado, doble ciego, multicéntrico, para evaluar la eficacia y la seguridad del tratamiento diurético combinado (diurético de asa e hidroCLOROTiazida) comparado con diurético de asa y placebo en pacientes con Insuficiencia Cardiaca descompensada.
• Fase: III
• Investigador Principal: Dr. José Luís Morales
• Sponsor: Sociedad Española de Medicina Interna (SEMI)
• Fármaco en investigación: Hidroclorotiazida vs. placebo
• Dosis: Cl. de creatinina > 50ml/min: ½ comprimido de HCTZ (25mg)
Cl. de creatinina 20-50ml/min: 1 comprimido de HCTZ (50mg)
Cl. de creatinina < 20ml/min: 2 comprimidos de HCTZ (100mg)
• Pauta: X-0-0 5 días consecutivos (x= ½ ó 1 ó 2)

¿Cómo se seleccionan a los pacientes candidatos a participar en el ensayo clínico?

CRITERIOS DE INCLUSIÓN (TODOS SÍ)
1. Pacientes hombres o mujeres con edad igual o superior a 18 años capaces de entender la Hoja de Información al Paciente (HIP) y otorgar su Consentimiento Informado (CI). En caso contrario se prevé que pueda otorgar dicho consentimiento un familiar o tutor legal competente para ello
2. Pacientes con IC crónica previa (de cualquier etiología e independientemente de del grado de disfunción ventricular) según los criterios de la Sociedad Europea de Cardiología que ingresan por descompensación definida como presencia de al menos un síntoma (disnea, ortopnea o edema) y un signo (crepitantes en auscultación, tercer ruido cardiaco, edema periférico, ascitis, congestión vascular pulmonar en la radiografía de tórax)

CRITERIOS DE INCLUSIÓN (TODOS SÍ)
3. Los pacientes deben estar tomando diurético de asa por vía oral de forma crónica (como mínimo un mes)
4. Las dosis previas de diurético de asa por vía oral deben ser las siguientes: entre 80mg y 160mg de furosemida por vía oral y dosis equivalentes para los otros diuréticos de asa (20mg de torasemida y 1mg de bumetanida se consideran equivalentes a 40mg de furosemida)

CRITERIOS DE INCLUSIÓN (TODOS SÍ)
5. Los pacientes deberán otorgar su consentimiento informado por escrito para participar en el estudio y antes de que se realice ninguna de las evaluaciones relacionadas con el estudio

CRITERIOS DE EXCLUSIÓN (TODOS NO)
1. Hipersensibilidad a tiacidas o a alguno de sus excipientes
2. Niveles plasmáticos de BNP < 100 pg/ml o de NT-pBNP< 300 pg/ml
3. Pacientes previamente diagnosticados de estenosis aortica severa
4. Pacientes con descompensación edematosa de etiología distinta a la IC: IC derecha secundaria a hipertensión pulmonar y/o tromboembolismo pulmonar, cirrosis hepática, síndrome nefrotico, etc.

CRITERIOS DE EXCLUSIÓN (TODOS NO)
5. Pacientes con hipoK+ en el momento de la selección: será excluido
cualquier valor de hipoK+ igual o inferior a 2.5 mEq/L
6. Pacientes con hiponatremia en el momento de la selección: será excluido cualquier valor de hipoNa+ que sea sintomática o cuando el sodio (Na+) sérico sea inferior o igual a 125mEq/L
7. Pacientes con síndrome coronario agudo en el momento del ingreso
8. Pacientes en situación de shock cardiogénico en el momento del ingreso

CRITERIOS DE EXCLUSIÓN (TODOS NO)
9. Pacientes que requieran ingreso en la Unidad de Cuidados Intensivos /Unidad Coronaria
10. Pacientes que requieran tratamiento con drogas vasoactivas (dopamina, dobutamina, noradrenalina) previo a la randomizacion. Se permite que los pacientes reciban tratamiento con drogas vasoactivas posteriormente a la randomización (siempre que se requiera a criterio del medico responsable) y este no será un motivo por si mismo suficiente para la retirada del paciente del estudio

CRITERIOS DE EXCLUSIÓN (TODOS NO)
11. Pacientes que requieran terapias de sustitución renal: hemodiálisis,
hemodiafiltración, ultrafiltración, etc.
12. Pacientes con expectativa de vida inferior a 6 meses en el momento del ingreso no atribuible a la propia insuficiencia cardiaca
13. Pacientes con insuficiencia cardiaca en fase terminal
14. Pacientes bajo tratamiento con HCTZ u otro diurético tiacídico durante el mes previo al ingreso

CRITERIOS DE EXCLUSIÓN (TODOS NO)
15. Pacientes bajo tratamiento con antialdosterónicos iniciados en los 30 días previos al ingreso. Se permite que el paciente esté tomando antialdosterónicos si los toma de forma estable desde hace mas de un mes
16. Mujeres embarazadas o en periodo de lactancia
17. Insuficiencia hepática
18. Enfermedad de Addison
19. Cetoacidosis diabética
20. Antecedentes o indicios de toxicomanía o alcoholismo en los ultimos 12
meses

CRITERIOS DE EXCLUSIÓN (TODOS NO)
21. Cualquier condición quirúrgica o medica que, en opinión del Investigador, pueda poner al paciente en una situación de mayor riesgo debido a su participación en el estudio, o que es probable que impida que el paciente cumpla los requisitos del estudio o que finalice el estudio
22. Antecedentes de incumplimiento de los regímenes médicos o que no este dispuesto/a a cumplir el protocolo del estudio
23. Cualquier condición que, en opinión del Investigador, pueda poner en peligro la evaluación de la eficacia o seguridad
24. Falta de aceptación por parte del paciente para participar en el estudio
25. Haber participado en otros ensayos clínicos durante los 3 meses previos

DISEÑO DEL ESTUDIO
Ensayo clínico: CLOROTIC Servicio: Medicina Interna

CALENDARIO DE VISITAS Y
EVALUACIONES Ensayo clínico: CLOROTIC Servicio: Medicina Interna

CIRCUITO PACIENTE ENSAYO CLÍNICO

Consentimiento Informado
Identifica los 7 Errores

VISITA MÉDICA DOCUMENTO FUENTE: HISTORIA CLÍNICA Paciente candidato a participar en el ensayo clínico CLOROTIC: Estudio aleatorizado, doble ciego, multicéntrico, para evaluar la eficacia y la seguridad del tratamiento diurético combinado (diurético de asa e hidroCLOROTiacida) comparado con diurético de asa y placebo en pacientes con Insuficiencia Cardiaca descompensada.
Cumple todos los criterios de inclusión y ninguno de los criterios de exclusión. Se le informa, se responden sus dudas y acepta participar. Firma el CI y se le da copia. Realización de la VISITA 1, según protocolo. […]
Señor JOSÉ
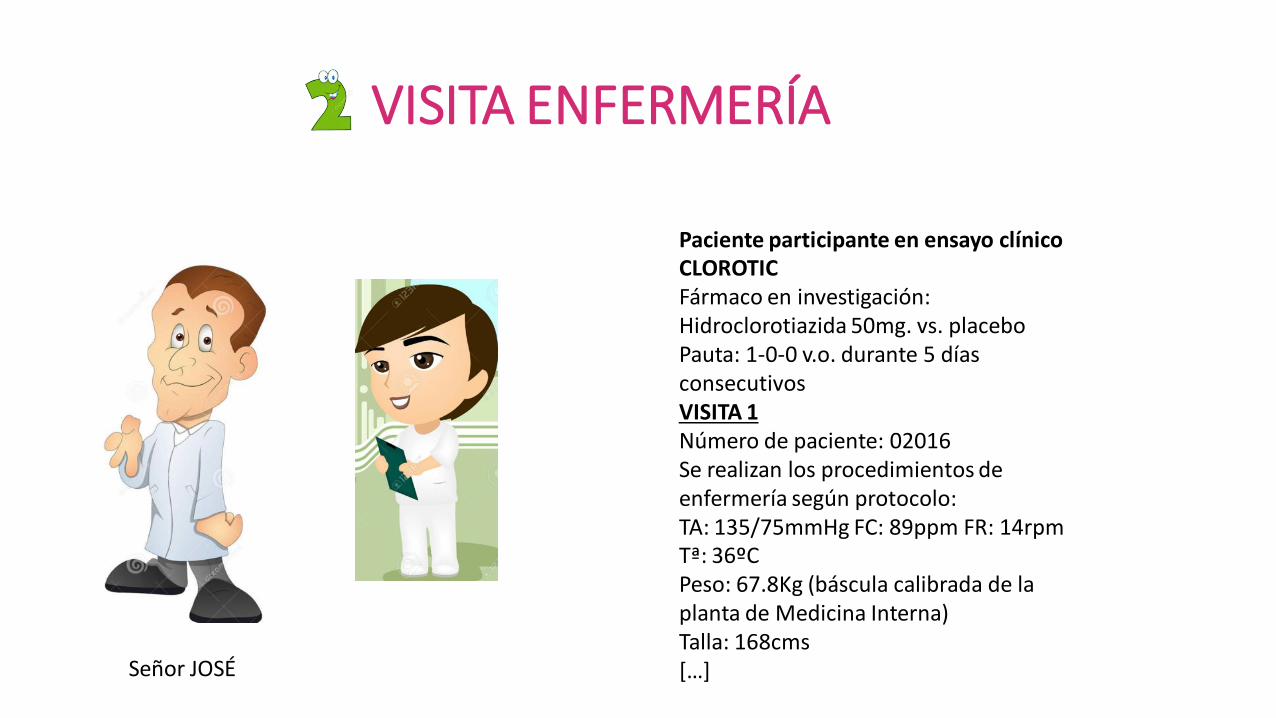
VISITA ENFERMERÍA
Paciente participante en ensayo clínico CLOROTIC Fármaco en investigación: Hidroclorotiazida 50mg. vs. placebo Pauta: 1-0-0 v.o. durante 5 días consecutivos VISITA 1 Número de paciente: 02016 Se realizan los procedimientos de enfermería según protocolo: TA: 135/75mmHg FC: 89ppm FR: 14rpm Tª: 36ºC Peso: 67.8Kg (báscula calibrada de la planta de Medicina Interna) Talla: 168cms […]
Señor JOSÉ

¿Cómo se asigna el tratamiento al paciente?
• Sistemas electrónicos interactivos
IWRS Interactive Web Response System
o
IVRS Interactive Voice Response System

¿Cómo se asigna el tratamiento al paciente?
• Listado de aleatorización en papel (Custodiado por la farmacéutica)

VISITA FARMACIA

VISITA FARMACIA
Señor JOSÉ

ENFERMERA COORDINADORA DE ENSAYOS CLÍNICOS

1.Coordinadora
Organizar equipo investigador y sus funciones
Organizar material: kits de laboratorio, electrocardiógrafo…
Programación de visitas de los pacientes, según protocolo
Asistir a la reunión de investigadores

2.Study Nurse
Procedimientos de enfermería, según protocolo:
• TA, FC, FR, Tª, analítica de sangre, analítica de orina, ECG…
• Acontecimientos Adversos (AA) y Acontecimientos Adversos Graves (AAG)
• Cambios en la medicación concomitante
• Escribir cursos clínicos de cada una de las visitas en la historia clínica del paciente (documento fuente)

¿Cómo registrar un AA o AAG en la historia clínica?
• En un ensayo clínico es importante registrar y notificar todos los eventos adversos reflejando:
1. Grado de severidad (leve, moderada, grave)
2. Relación con el fármaco del estudio (sospechada/no sospechada)
3. Duración (fecha de inicio y fin del evento o si continúa en la visita final)
4. Medidas que se toman para resolver el AE/SAE • (Fármaco, medidas físicas, dosis, pauta, inicio, fin)
5. Si constituye un evento adverso grave ¿SAE?

Acontecimiento Adverso Grave (AAG) Serios Adverse Event (SAE)
• Cualquier Acontecimiento Adverso o reacción adversa que a cualquier dosis:
Produzca la muerte del paciente Amenace la vida del sujeto Requiere hospitalización o la prolongación de ésta. Produzca discapacidad/incapacidad persistente o significativa Constituye una anomalía o malformación congénita.
Panace@. Vol. IX, n.º 28. Segundo semestre, 2008
Real Decreto 223/2004, Art. 2 letra q) Real Decreto 1344/2007, art. 2 letra d)

Notificación SAE: 24 horas
FAX 93 260 70XX
HOJA DE ENVIO DE FAX NOTIFICACIÓN DE AAG
(ACONTECIMIENTO ADVERSO GRAVE)
PROTOCOLO BICHO
PROMOTOR: DR. Dolores Curados
Servicio de Enfermedades Infecciosas del Hospital Univestiari de Bellvitge
REMITENTE DEL FAX Hospital Arnau Vilanova de Lleida Investigador Principal Dr. Shoenenberger
PARA Ucicec IDIBELL
TIPO DE AAG: Inicial Seguimiento
PERSONA QUE ENVÍA EL AAG
FECHA
………. / .......... / ……….
NÚMERO DE FAX REMITENTE
NÚMERO DE FAX 93 260 70XX
NÚMERO DE TEL REMITENTE
Nº PÁGINAS (portada incluída)…………….
Asunto: AAG PROTOCOLO BICHO Nº DE PACIENTE: .................
Comentarios
• Notificar al promotor del ensayo clínico dentro de las 24 horas tras su conocimiento.

3.Técnico de laboratorio

4.Data Manager
Registrar los datos recogidos en las visitas en un Cuaderno de Recogida de Datos electrónico (CRDe) o en un cuaderno en papel.

Cuaderno de Recogida de Datos electrónico (CRDe)

Query
• Incongruencia entre datos entrados en la base de datos y el documento fuente (Historia Clínica)/realidad.
• Ejemplo:
Paciente de 65 años
Peso: 6.78kg
TA: 135/75mmHg
Peso: 67.8Kg

¿Y AHORA QUÉ?

Ensayo clínico
(Sujeto)
Investigador Principal
Subinvestigadores
CEIC
Study Nurse
Study Coordinator
Data Manager Administrativ@
Técnico Laboratorio
Estadístic@
Monitor
(CRO)
Promotor
Roles participantes en un ensayo clínico
Reclutamiento
Pacientes
Visita Pre-Selección
Visita Selección
CEIC Reunión
Investigadores Visita Inicio
Visitas Monitorización
Visita Cierre Resultados Estadísticos
Publicación
¿Cuándo se monitorizan los registros de un paciente?

Monitorización
Acto de vigilar el progreso de un ensayo clínico y de asegurar que es realizado, registrado y comunicado de acuerdo con el protocolo, los procedimientos normalizados de trabajo (PNT), la buena práctica clínica (BPC) y los requisitos reguladores aplicables
OBJETIVOS Velar por la seguridad de los pacientes y que se respeten su derechos éticos Comprobar que los datos recogidos durante el estudio son verificables (no se los ha inventado nadie),
exactos (no hay error al transcribirlos al cuaderno de recogida de datos) y completos Asegurar que el ensayo se lleva a cabo según lo establecido en el Protocolo, según las Normas de Buena
Práctica Clínica y la legislación española (Real Decreto 223/2004)

Monitor
El monitor NO debe formar parte del equipo investigador

LO QUÉ NO ESTÁ ESCRITO, NO EXISTE
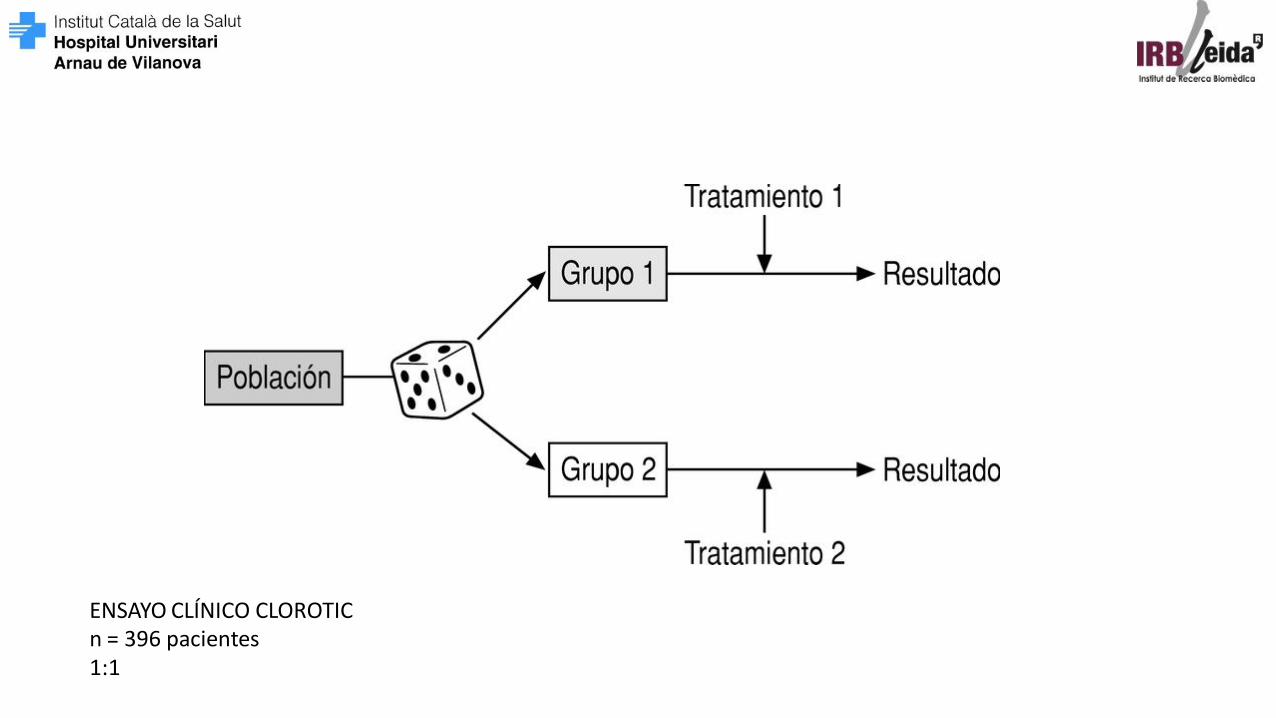
ENSAYO CLÍNICO CLOROTIC n = 396 pacientes 1:1

COMERCIALIZACIÓN DEL FÁRMACO… O NO…

¡En medicina no hay grandes avances sin los ENSAYOS CLÍNICOS!
¡No sería posible sin los PACIENTES que participan y sin los PROFESIONALES
SANITARIOS que se dedican!


