la génomique - ens - départment de biologie « génomique évolutive » taatggtaccagttagcagagt…...
TRANSCRIPT

La Génomique���Biologie, informatique, évolution
Hugues Roest Crollius [email protected]
Dyogen Group
L3 – Introduction aux sciences du vivant – 03.12.2013

La « génomique fonctionnelle »

La « génomique évolutive » TAATGGTACCAGTTAGCAGAGT…
CCATGGTTCCCGTAGCCAGAGT…
TAATGGTACCGGTTAACAGAGT…
TTATGGTACCTGTTAACAGAGT…
CGATGGTGCCGGTCGACAGAGC… CTATGGTCCCTGTTATCAGAGC… GTATGGTCCCTGTCGTCAGAGC… CCATGGTTCCCGTAGCCAGAGT…
human baboon mouse dog cat cow pig chicken
human
mouse
rat
dog

Applied Biosystems 3730 (ici au Broad Institute (USA))
1 Mb / jour
1990 2008
La production des données de génomique
2013
Illumina MySeq2500 Capable de re-séquencer 1 génome humain / jour
(40X; 135 Gb)
Séquençage manuel par radioactivité
100 b / jour

La production des données de séquençage
326 millions 686 milliards

1990 91 92 93 94 95 96 97 98 99 2000 01 02 03 04 05 06 07 08 09
Cartographie génétique
Cartographie physique
Human Genome Project (HGP)
Projet Celera
Projet HapMap
Séquençage très haut débit
J.C. Venter
Aujourdhui l’information issue du génome humain et du génome d’espèces modèles nous permet de mieux comprendre certains processus biologiques Bientôt, l’information issue de milliers de génomes humains, intégrée à des données épidémiologiques et de structure de la population, seront la base d’une nouvelle médecine « personnalisée ».
Bactérie Levure Nématode Drosophile Humain Souris Poule Chimpanzee
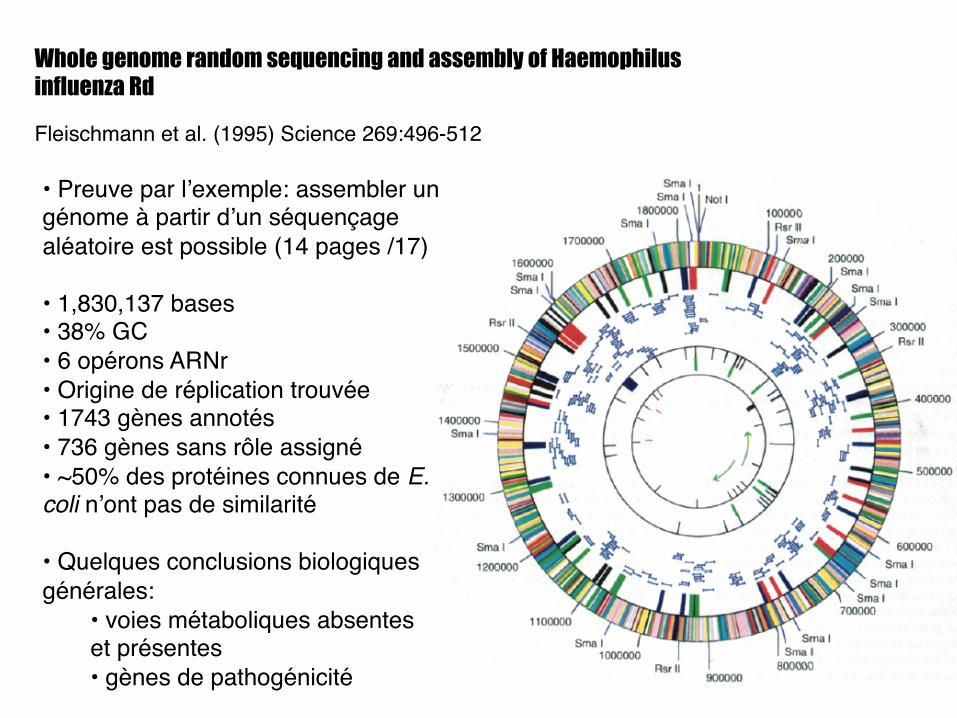
Whole genome random sequencing and assembly of Haemophilus influenza Rd
Fleischmann et al. (1995) Science 269:496-512!
• Preuve par l’exemple: assembler un génome à partir d’un séquençage aléatoire est possible (14 pages /17)!
• 1,830,137 bases!• 38% GC!• 6 opérons ARNr!• Origine de réplication trouvée!• 1743 gènes annotés!• 736 gènes sans rôle assigné!• ~50% des protéines connues de E. coli n’ont pas de similarité !
• Quelques conclusions biologiques générales: !
• voies métaboliques absentes et présentes!• gènes de pathogénicité!

Life with 6000 genes
• 1er génome eucaryote séquencé!
• 600 chercheurs, 100 laboratoires, le plus grand projet décentralisé de la biologie moléculaire !!!• Seules 43,3 % des protéines ont une fonction connue ou « suggérée »!
• Beaucoup de régions du génome sont dupliquées!
• Tous les gènes d’histones sont présents (dont H1)!
Science (1996) Vol. 274: 546 - 567
(Saccharomyces cerevisae)!

Conséquences politiques!!• Un génome eucaryote complexe peut-être séquencé. !• le projet a révélé l’importance de la bioinformatique (AceDB, GeneFinder)!• Un modèle de projet « ouvert »: accès libre au matériel et aux données!
Résultats scientifiques!!• 19099 gènes, trois fois plus que la levure!• La densité en gène est plus importante près des centromères (sauf sur le X)!• Les éléments répétés sont plus nombreux vers les télomères!• Les extrémités des chromosomes seraient des régions à évolution plus rapide!• 32 % des protéines de C. elegans sont similaires à des protéines humaines, 70% des protéines humaines sont similaires à celles de C. elegans !
Genome Sequence of the Nematode C. elegans: A Platform for Investigating Biology Science (1998) vol. 282: 2012-2018.


Initial sequence of the chimpanzee genome and comparison with the human genome Nature (2005) vol 439:69-87!
• 1,23 % de divergence nucléotidique avec l’espèce humaine sous forme de SNPs, dont 1,06% fixé au cours de l’évolution (ce qui fait ~ 30 millions de bases). !
• 1,5 % de la séquence euchromatique de chaque espèce lui est spécifique (insertions ou délétions; ~45 Mb)!
• 29% des protéines sont identiques entre les 2 espèces, la plupart des autres ne divergent que par 2 acides aminés!
• Les protéines de la réponse immunitaire, de la reproduction et de l’olfaction divergent plus vite que les autres!
• De nombreuses «pépites » sur les gènes spécifiques à l’espèce humaine (éliminé du chimpanzé) ou vice-versa, parfois en liaison avec des maladies humaines. Certaines mutations humaines causant des maladies sont en fait l’allèle sauvage « ancestral » (ex: predisposition au diabète de type 2)!

Le génome Humain
~
Tout un symbole

Un symbole de l’opposition « privé - public »!!• Celera (Craig Venter)!!• Human Genome Project (F. Collins, R. Waterston, J. Sulston, P. Green!!
!Opposition !! !- sur les finalités!! !- l’accès aux données!! !- la stratégie!
Un symbole de la médiatisation de la science!!• Course à la (aux) publication(s)!
• battage médiatique intense!
• Reconnaissance par le monde politique!

La variabilité génétique
« La » séquence du génome humain disponible dans les bases de données représente en réalité un génome fictif: il s’agit d’un assemblage de l’ADN obtenus de plusieurs individus. Cette séquence ne contient pas de variabilité (polymorphisme allélique). Cette séquence est conventionnellement utilisée comme référence.
Mais la population humaine est composée de > 6 milliards d’individus, chacun avec un génome qui lui est unique. En plus des influences de l’environnement, cette variabilité entre individus est l’un des déterminants majeurs de la morphologie, des propriétés physiologique, du comportement, de la santé des individus. Comment se manifeste cette variabilité génétique?

A haplotype map of the human genome Nature (2005) vol 437:1299-1320!
• Nous ignorons encore les causes génétiques de la plupart des maladies humaines: troubles maniaco-depressifs, réponses aux anti-hypertensenseurs, etc…!
• Nous savons que probabement la moitié des facteurs de risques à la racine de ces maux sont d’origine génétique. !
• 1 007 329 SNPs ont été testés dans 269 individus appartenant à 4 groupes:!• population des Yoruba (Ibadan) au Niger!• familles du CEPH (Utah, USA)!• population chinoise (Han) de Beijing!• population japonaise de Tokyo !

A haplotype map of the human genome Nature (2005) vol 437:1299-1320!
Quelques surprises:!!La plupart des variants dans la population sont rares: !
!- 46 % des SNPs ont une fréquence d’allèle minoritaire (FAM) < 0.05!!- 9% ne sont vus que dans un seul individu. !
!La plupart des variants sont largement partagés!
!- 90% des variants observés dans un individu sont des SNPs ! « communs » !
Des confirmations:!!Les échantillons ne sont pas homogènes!
!- la population du Niger est plus riche en SNPs de faible fréquence!!!
Mais nous sommes bien de la même espèce :-)!!- seulement 16 SNPs sur 1 million sont « fixés » dans une population par !
rapport aux autres!!!
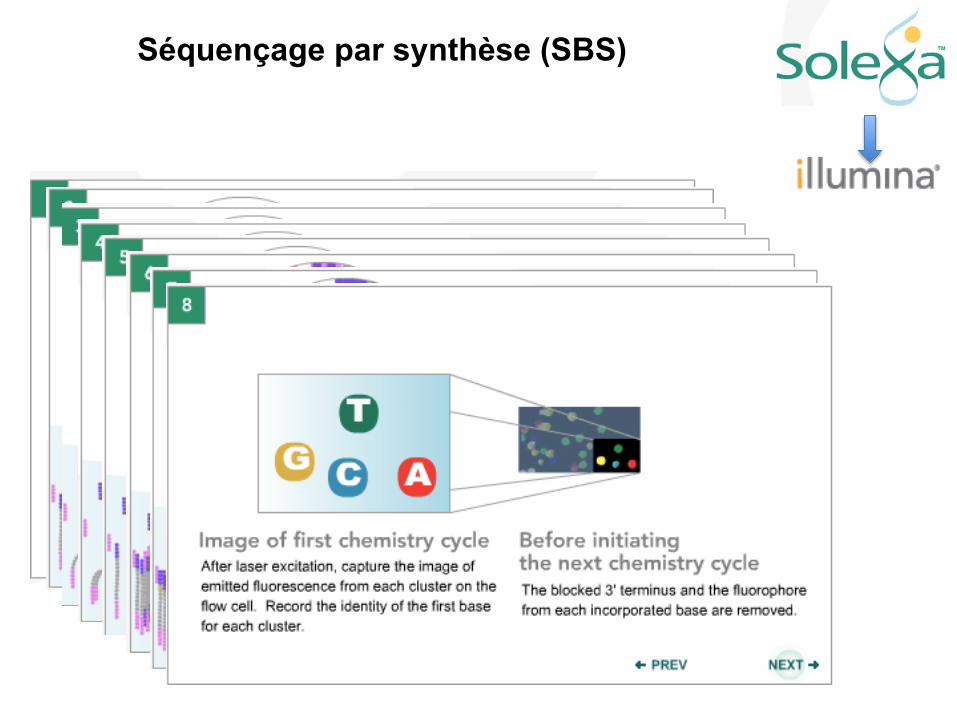
Séquençage par synthèse (SBS)

Le séquençage des génomes
Il a fallu créer une nouvelle division dans les bases de données: Short Read Archives (SRA)
4,5 trillions 573 trillions

Le séquençage des génomes
La séquence d’un génome est donc une succession de conDgs organisés en scaffolds. Selon le degré de finiDon, les scaffolds peuvent être ancrés sur une carte généDque, ordonnés et orientés, et les trous de séquence entre les conDgs et scaffolds peuvent être bouchés.
Les génomes eucaryotes séquencé à très haut niveau de qualité (< 1.106 erreurs/base) Saccharomyces cerevisiae Levure de boulanger
Caenorhabdi2s elegans Ver nématode
Drosophila melanogaster Mouche à vinaigre
Arabidopsis thaliana ArabeTe
Homo sapiens Humain
Mus musculus Souris
Danio rerio Poisson zèbre

Le séquençage des génomes
Le « N50 », une mesure devenue classique pour évaluer la conDnuité d’un assemblage. Le N50 est la taille du scaffold (ou conDg) tel que 50% des bases de l’assemblage sont comprises dans des scaffolds de taille supérieures à ceTe taille.
La taille du segment (scaffold) telle que la moitié de la somme des bases de tous les segments (assemblage) soit compris dans des segments de taille supérieure.
N50 Scaffolds de l’assemblage
Trier par taille
50% des bases 50% des bases

Le génome humain en 2013

Un génome à l’état de « brouillon »
Le génome du cheval (Equus caballus) L’assemblage actuel (2013) est la version version EquCab2, obtenu par la technique Whole Genome Shotgun (WGS) avec une couverture de 6.79x en lecture « Sanger ». Une jument appelée "Twilight" fut sélecDonnée pour obtenir le génome référence de l’espèce. Le projet fut coordonné et le génome séquencé par Le Broad InsDtute (USA). La taille N50 des conDgs est de 112.38 kb, et la somme totale des conDgs est de 2.43 Gb. En incluant la taille esDmé des trou entre les conDgs dans les scaffolds, l’assemblage couvre 2.68 Gb.

Un génome à l’état de « brouillon »
Platyfish (Xiphophorus maculatus)
L’assemblage (version XipMac4.4.2) a été produit par The Genome InsDtute, Washington University School of Medicine (USA). Cet assemblage a été réalisé par whole genome shotgun à parDr de séquences produites par la technologie “454” et Illumina, pour une couverture totale du génome de ~19.6X.

Le séquençage du génome humain
Après le séquençage, la première étape de « valorisaDon » de la séquence est d’y idenDfier (annoter) les régions foncDonnelles, principalement les gènes codant les protéines. Chaque génome eucaryote conDent des milliers de gènes. On ne peut pas envisager de faire une « expérience » pour idenDfier chaque gène: il faut recourir à des logiciels pour réaliser une annotaDon automaDque, ou à des ressources génomiques. Annoter les gènes automaDquement est une tâche difficile et un champs encore très « ouvert » de la bioinformaDque. Dans les génomes eucaryotes, les gènes ont des structures extrêmement variables: il difficile d’établir des « règles ».
Les gènes ….

25
Chr. 20 Chr. 21 Chr. 22
Taille chromosome 59,42 Mb 33,54 Mb 33,46 Mb
Gènes connus 335 127 270 Autres 392 98 298
Pseudogènes 168 (18,7%) 59 (20,7%) 134 (19,1%) Densité en gènes 12,2 g./Mb 6,7 g./Mb 17,0 g./Mb
Tailles des gènes
Connus 51,3 kb 57,0 kb 1 ↔ 593 kb Pseudogènes 1,9 kb
Taille des exons
Connus 294 bp 8 ↔ 7600 bp Pseudogènes 499 bp
Nombre d’exons
Connus 10,3 Pseudogènes 1,4
Combien(y(a(t,il(de(gènes(dans(le(génome(humain?(Premières(estimations((année(2000)(((
40000 20000 50000

EsDmaDons du nombre de gènes dans le génome
92 93 94 95 96 97 98 99 00 01 02 03 04 05 06
20 000
40 000
160 000
140 000
120 000
100 000
80 000
60 000
(Antequera and Bird)
(Fields et al.)
(Roest Crollius et al.)
(Lander et al.)
EsDmaDons publiées
(Ewing and Green et al.) (Liang et al.)

27
BLAST
Altschul et al. (1990) Basic Local Alignment Search Tool. J. Mol. Biol. 215:403-410
Nombre total de citations : 36103 (en novembre 2013) L’article le plus cité en sciences du vivant

28
Query: SPWTFPS*FLMSSSMKVPSWSRISSPM*GIL*STVSSST SPWTFPS* L+SSS+KV S S SSPM*GIL T SSST Sbjct: SPWTFPS*LLISSSIKVSSSSFTSSPM*GILHKTXSSST
Query: LLFQLFLALSDLKQLRILHTDLKPDNVMLVD--EKELKIKLMDFGLALLTHEAKT--GTI +L Q+ AL LK L ++H DLKP+N+MLVD + ++K++DFG A +H +KT T Sbjct: ILQQVATALKKLKSLGLIHADLKPENIMLVDPVRQPYRVKVIDFGSA--SHVSKTVCSTY
Query: VNALAQYSHNEDEEEEEEHDFKVDKT-DLCDSKKHPE VNAL QY+ ++D+++ ++ + + +K DL D + E Sbjct: VNALGQYNDDDDDDDGDDPEEREEKQKDLEDHRDDKE
Query: RYKELTEQQMPGALPPECTPNMDGPHARSVRREQSLHSFHTLFCRRCFKYDRFLH +YKELTEQQ+PGALPPECTPN+DGP+A+SV+REQSLHSFHTLFCRRCFKYD FLH Sbjct: KYKELTEQQLPGALPPECTPNIDGPNAKSVQREQSLHSFHTLFCRRCFKYDCFLH

29
BLAST
A T T G C G T A T G C A G C G T A G C A A T T G C G A T A C!
T T A C G C G A T G T A G A C A G C G T A G C A A T G T T G C A!
Match exact
Query
Subject
“mot” de taille W = 11 bases

30
A T T G C G T A T G C A G C G T A G C A A T T G C G A T A C!
T T A C G C G A T G T A G A C A G C G T A G C A A T G T T G C A!
Blast:
Query
Subject
T A T G C A G C G T A G C A A T!
Matrice de score NUC.4.4
A T G C N!A 5 -4 -4 -4 -2!T -4 5 -4 -4 -2!G -4 -4 5 -4 -2!C -4 -4 -4 5 -2!N -2 -2 -2 -2 -1!
+5-4-4+5!
- 8 < X!
X = seuil maximal de mismatch autorisé = 21 par défaut
W

31
Mot “W” = 3 a. a.
(Seuil “X”)
(Seuil “T”)
L E C N Q L I P I A H K T C P E G K N L
H K T!H L T!H V T!H Y T!Y K T!N K T!
L K C H N T Q L P F I Y K T C P E G K N
Extension
Automate
TBLASTX, BLASTP, BLASTX

32
A R N D C Q E G H I L K M F P S T W Y V B Z X * A 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0 -2 -1 0 -4 R -1 5 0 -2 -3 1 0 -2 0 -3 -2 2 -1 -3 -2 -1 -1 -3 -2 -3 -1 0 -1 -4 N -2 0 6 1 -3 0 0 0 1 -3 -3 0 -2 -3 -2 1 0 -4 -2 -3 3 0 -1 -4 D -2 -2 1 6 -3 0 2 -1 -1 -3 -4 -1 -3 -3 -1 0 -1 -4 -3 -3 4 1 -1 -4 C 0 -3 -3 -3 9 -3 -4 -3 -3 -1 -1 -3 -1 -2 -3 -1 -1 -2 -2 -1 -3 -3 -2 -4 Q -1 1 0 0 -3 5 2 -2 0 -3 -2 1 0 -3 -1 0 -1 -2 -1 -2 0 3 -1 -4 E -1 0 0 2 -4 2 5 -2 0 -3 -3 1 -2 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4 G 0 -2 0 -1 -3 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 -1 -2 -1 -4 H -2 0 1 -1 -3 0 0 -2 8 -3 -3 -1 -2 -1 -2 -1 -2 -2 2 -3 0 0 -1 -4 I -1 -3 -3 -3 -1 -3 -3 -4 -3 4 2 -3 1 0 -3 -2 -1 -3 -1 3 -3 -3 -1 -4 L -1 -2 -3 -4 -1 -2 -3 -4 -3 2 4 -2 2 0 -3 -2 -1 -2 -1 1 -4 -3 -1 -4 K -1 2 0 -1 -3 1 1 -2 -1 -3 -2 5 -1 -3 -1 0 -1 -3 -2 -2 0 1 -1 -4 M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 -3 -1 -1 -4 F -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6 -4 -2 -2 1 3 -1 -3 -3 -1 -4 P -1 -2 -2 -1 -3 -1 -1 -2 -2 -3 -3 -1 -2 -4 7 -1 -1 -4 -3 -2 -2 -1 -2 -4 S 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4 1 -3 -2 -2 0 0 0 -4 T 0 -1 0 -1 -1 -1 -1 -2 -2 -1 -1 -1 -1 -2 -1 1 5 -2 -2 0 -1 -1 0 -4 W -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11 2 -3 -4 -3 -2 -4 Y -2 -2 -2 -3 -2 -1 -2 -3 2 -1 -1 -2 -1 3 -3 -2 -2 2 7 -1 -3 -2 -1 -4 V 0 -3 -3 -3 -1 -2 -2 -3 -3 3 1 -2 1 -1 -2 -2 0 -3 -1 4 -3 -2 -1 -4 B -2 -1 3 4 -3 0 1 -1 0 -3 -4 0 -3 -3 -2 0 -1 -4 -3 -3 4 1 -1 -4 Z -1 0 0 1 -3 3 4 -2 0 -3 -3 1 -1 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4 X 0 -1 -1 -1 -2 -1 -1 -1 -1 -1 -1 -1 -1 -1 -2 0 0 -2 -1 -1 -1 -1 -1 -4 * -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 1
Matrice de score BLOSUM62

33
Levure 6000 Drosophile 13600 Humain 25000
Nematode 19000
Arabidopsis 25000
Nombre de gènes dans les génomes eucaryotes

34
EVOLUTION MOLECULAIRE
Quelques principes

MutaDon
Dérive généDque
SélecDon posiDve
SélecDon négaDve
FixaDon 100%
Avantageuse Neutre Délétère
DispariDon 0%
Fréquence Intermédiaire
0-‐100%

Evolution moléculaire
Les fréquences des variations au sein d’une population fluctuent au cours du temps.
P
0
1
Générations (temps)
Pour estimer les fréquences dans une population, il faut échantillonner de nombreux individus
Les variations AVANTAGEUSES sont sélectionnées et augmentent en fréquence Les variations DELETERES sont éliminées et diminuent en fréquence
Les variations NEUTRES fluctuent de manière aléatoire

La sélection naturelle

Cys Ser Arg Cys Lys Gly His Cys Arg Ala Arg!TGT TCG AGA TGT AAG GGC CAT TGT CGA GCA AGA!!!!Cys Leu Arg Cys Lys Arg His Cys Arg Ala Lys!TGT TTG AGA TGT AAA CGC CAT TGT AGA GCT AAA!!!!
Observé Attendu neutre Substitutions synonymes 3 Substitutions non-synonymes 3 ~3 X 4 = 12 è 75% des
mutations sont délétères

dS: taux de substitution synonyme (Ks) dN: taux de substitution non-synonymes (Ka) ω = dN / dS ω ~ 1 è ω << 1 è evolution sous sélection négative ω >> 1 è evolution sous sélection positive

Fréquence des valeurs de ω pour 835 paires de gènes orthologues rat-souris (les valeurs indiquées en abscisse sont la moyenne de la classe)
Hurst DL (2002) TIGS 18:486-487

Génomique Comparative
L’alignement multiple entre génome est un outil fondamental pour identifier des régions conservées au cours de l’évolution (par sélection négative)
UCSC Genome Browser : http://genome.ucsc.edu/ Une région de 100 pb sur Xq26:

Tous les mammifères possèdent à peu près le même nombre de gènes, et partagent les mêmes grandes fonctions de la vie
- reproduction - développement - système nerveux central - système digestif - système musculaire - ….
On estime que les gènes présents dans le génome de la souris ou du chien peuvent être informatifs pour identifier les gènes humains (ou vice-versa) simplement par alignement de séquence. Généralisation: Toutes les informations importantes contenues dans le génome (codage des protéines et autres…) sont susceptibles d’êtres partagées entre espèces différentes et donc d’être découvertes par alignement de séquences.
Génomique ComparaDve: Annoter les Gènes

Génomique Comparative (5)
Les séquences fonctionnelles les mieux connues dans le génome humain sont les exons des gènes codant les protéines. On peut les comparer par paires, mais les comparer toutes ensemble est plus informatif, à l’aide d’un alignement multiple
Les exons codant sont particulièrement ben conservés, à travers l’ensemble des vertébrés (sélection négative). Les régions « UTRs » évoluent plus vite. Les introns ne montrent pas de conservation particulière (évolution neutre) Les espèces trop proches de l’homme sont peu informatives (ex: Macaque)

Migration, adaptation et selection naturelle
Les variations génétiques qui confèrent un avantage pour une meilleure adaptation seront sélectionnés

Mutation avantageuse
Different types de sélection naturelle
Mutation neutre Mutation délétère mutation “balancée”
SELECTION POSITIVE
Ex. G6PD, CD40 protection contre la malaria en Afrique
SELECTION BALANCEE
Ex. MHC worldwide, HbS en Afrique (malaria)
SELECTION PURIFICATRICE
Ex. Beaucoup de gènes humain

La cas de la lactase
La plupart des adultes ne peuvent métaboliser le lactose, sucre principal du lait, car la fonction de l’enzyme lactase-phlorizin hydrolase diminue après le sevrage. Mais certaines population, principalement celles descendantes de population ayant pratiqué la domestication du bétail, maintiennent cette possibilité à l’âge adulte. Fréquences de la « persistance de la lactase »
> 90% chez les suédois et les danois ~ 50% chez les français et les espagnols 5% - 20% chez les africains de l’ouest « non-pastoraux » 1 % chez les chinois
Mais 90% chez les Tutsis, Fulani, … populations africaines « pastorales ». Certains SNPs ont été retrouvés dans les introns d’un gènes voisin de la lactase, et sont associé au phénotype « persistance de la lactase »

Distribution du phénotype « persistance de la lactase » dans le monde
La cas de la lactase

La cas de la lactase
Intron 13
Danois et Suédois
Europe du sud S. A. Tishkoff et al., Convergent adaptation of human lactase persistence in Africa and Europe. Nature genetics 39, 31 (2007).

La cas de la lactase
Danois et Suédois
Europe du sud
Afrique
S. A. Tishkoff et al., Convergent adaptation of human lactase persistence in Africa and Europe. Nature genetics 39, 31 (2007).

La cas de la lactase
Conclusions: Les mutations de la lactase sont un cas classique d’évolution convergente:
le même phénotype est sélectionné de manière indépendante dans des populations différentes, mais pas par le biais du même génotype.
Les mutations favorables sont dans les introns d’un gènes voisin du gène dont la protéine confère l’avantage Les mutations augmentent la production de lactase au cours de la vie adulte (modification de l’expression du gène)