lecture 8: protein purification –protein purification
TRANSCRIPT

Lecture 8: Protein purification
– Protein purification

Column chromatography• After the initial fractionation steps we move to column chromatography.• The mixture of substances (proteins) to be fractionated is dissolved in a liquid or
gaseous fluid called the mobile phase.• This solution is passed through a column consisting of a porous solid matrix called
the stationary phase. These are sometimes called resins when used in liquid chromatography.
• The stationary phase has certain physical and chemical characteristics that allow it to interact in various ways with different proteins.
• Common types of chromatographic stationary phases– Ion exchange– Hydrophobic– Gel filtration– Affinity

Ion exchange chromatography• Ion exchange resins contain charged groups.• If these groups are acidic in nature they interact with positively charged proteins and are called cation
exchangers.
• If these groups are basic in nature, they interact with negatively charged molecules and are called anion exchangers.
DEAE celluloseanion exchanger
CH2-CH2 -NH+(CH2CH2)-
--
-
Negatively charged (acidic) protein or enzyme
CM cellulosecation exchanger
CH2-COO-
CH2-COO-
+++
+
Positively charged (basic) protein or enzyme
CH2-CH2 -NH+(CH2CH2)
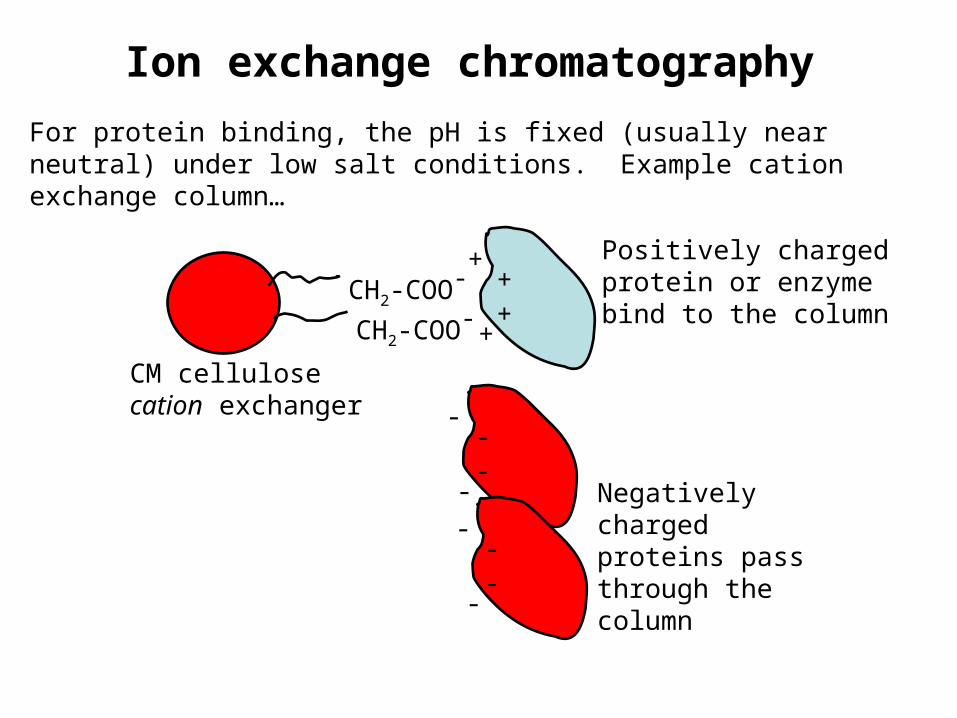
Ion exchange chromatography
CM cellulosecation exchanger
CH2-COO-
CH2-COO-
+++
+
Positively charged protein or enzyme bind to the column
For protein binding, the pH is fixed (usually near neutral) under low salt conditions. Example cation exchange column…
---
-
---
-
Negatively charged proteins pass through the column
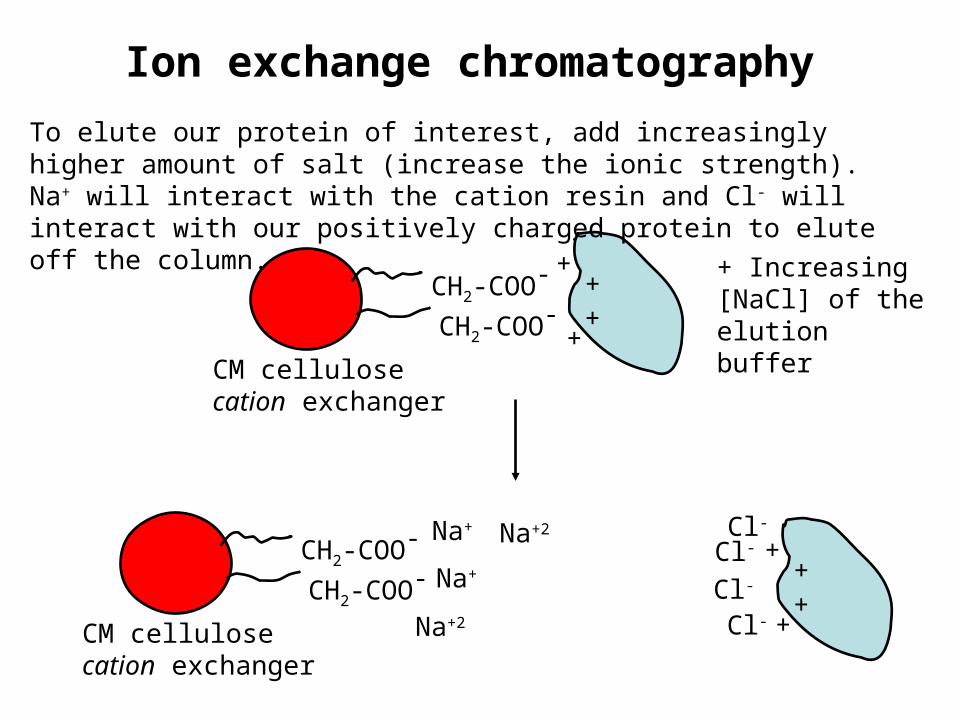
Ion exchange chromatography
CM cellulosecation exchanger
CH2-COO-
CH2-COO-
+++
+
To elute our protein of interest, add increasingly higher amount of salt (increase the ionic strength). Na+ will interact with the cation resin and Cl- will interact with our positively charged protein to elute off the column.
CM cellulosecation exchanger
CH2-COO-
CH2-COO-+
++
+
Na+
Na+Cl-
Na+2
Cl-
Cl-
Cl-
Na+2
+ Increasing [NaCl] of the elution buffer

Ion exchange chromatography• Proteins will bind to an ion exchanger with different affinities.• As the column is washed with buffer, those proteins relatively low affinities
for the ion exchange resin will move through the column faster than the proteins that bind to the column.
• The greater the binding affinity of a protein for the ion exchange column, the more it will be slowed in eluting off the column.
• Proteins can be eluted by changing the elution buffer to one with a higher salt concentration and/or a different pH (stepwise elution or gradient elution).
• Cation exchangers bind to proteins with positive charges.• Anion exchangers bind to proteins with negative charges.
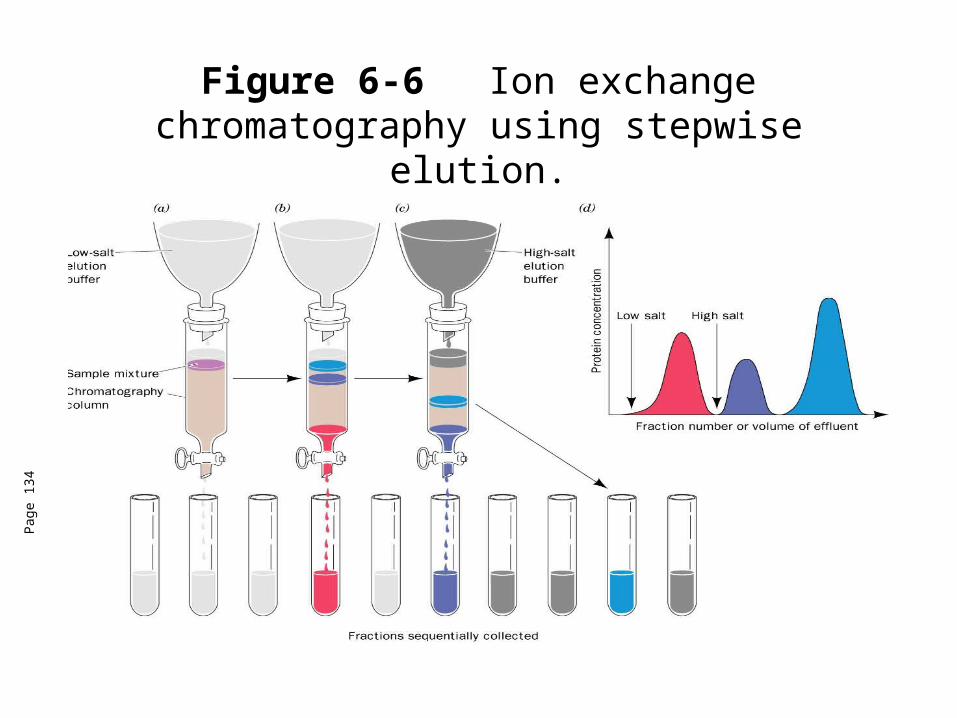
Figure 6-6 Ion exchange chromatography using stepwise elution.
Pag
e 13
4

Ion exchange chromatography• Gradient elution can improve the washing of ion exchange columns.• The salt concentration and/or pH is continuously varied as the column is eluted so as
to release sequentially the proteins bound to the column.• The most widely used gradient is the linear gradient where the concentration of
eluant solution varies linearly with the volume of the solution passed.
• The solute concentration, c, is expressed as
c = c2 - (c2 - c1)fc1 = the initial concentration of the solution in the mixing chamber
c2 = the concentration of the reservoir chamberf = the remaining fraction of the combined volumes of the solutions initially present in
both reservoirs.
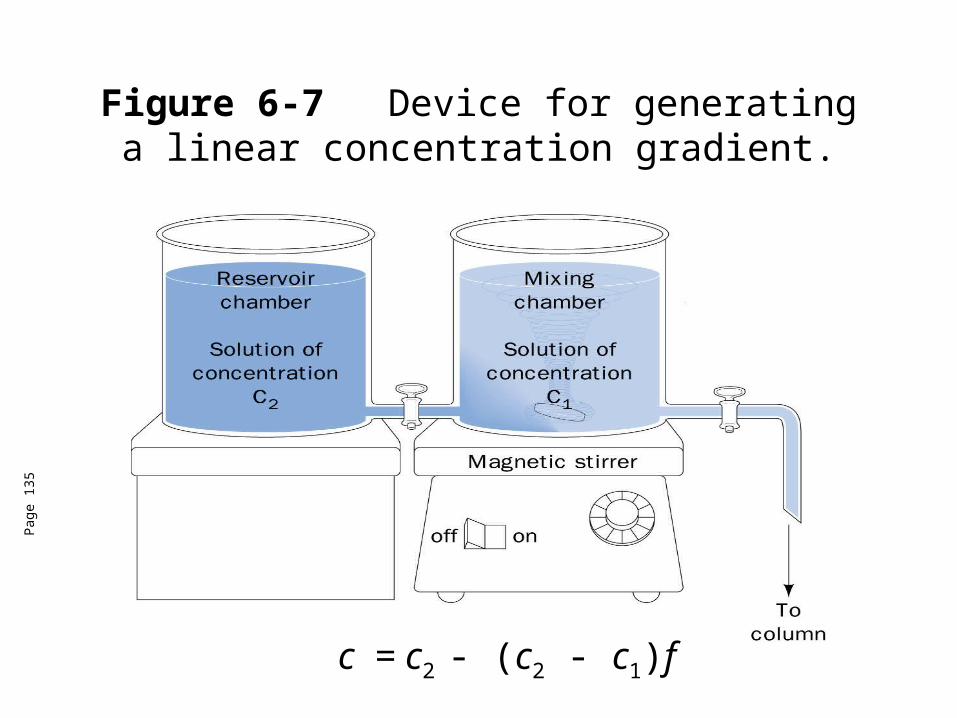
Figure 6-7 Device for generating a linear concentration gradient.
Pag
e 13
5
c = c2 - (c2 - c1)f
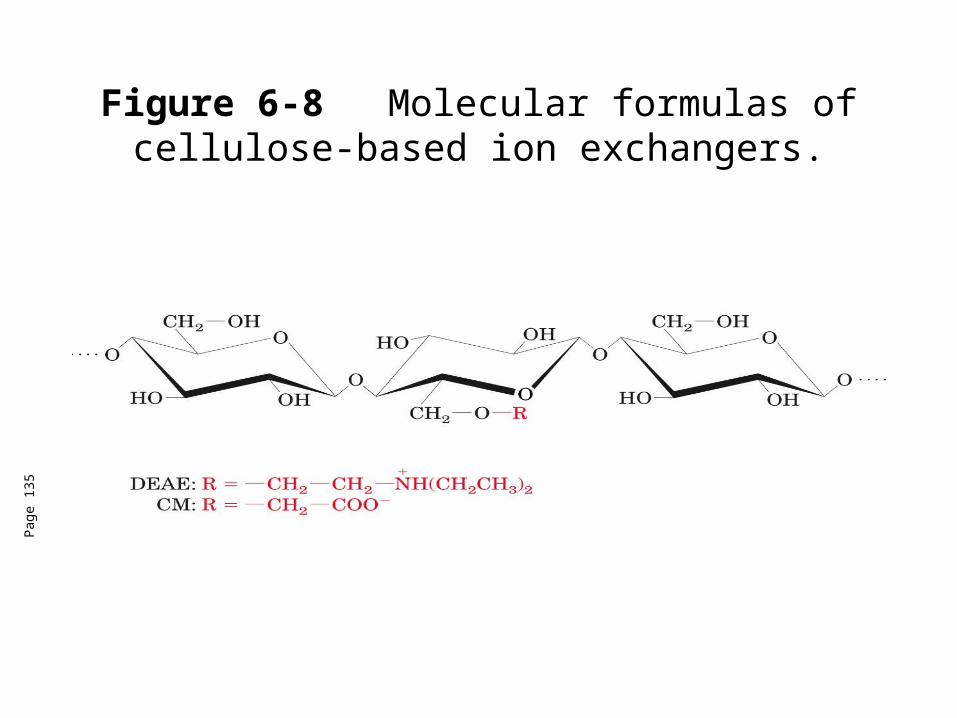
Figure 6-8 Molecular formulas of cellulose-based ion exchangers.
Pag
e 13
5
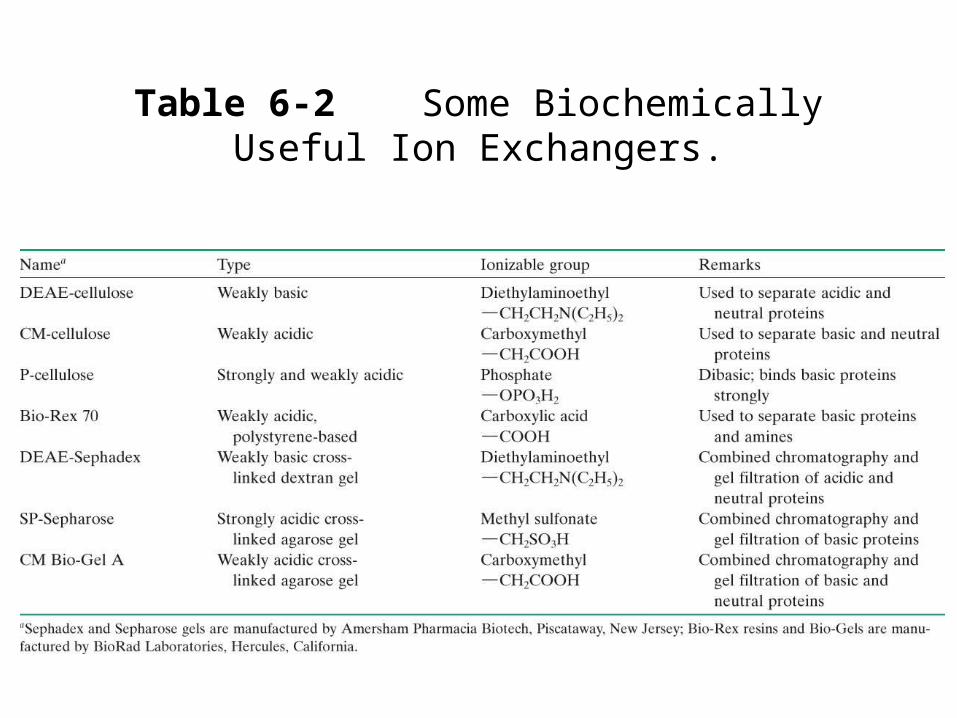
Table 6-2 Some Biochemically Useful Ion Exchangers.

Ion exchange chromatography• Ion exchangers can be cellulosic ion exchangers and
gel-type ion exchangers.• Cellulosic ion exchangers most common.• Gel-type ion exchangers can combine with gel filtration
properties and have higher capacity.• Disadvantage-these materials are easily compressed so
eluant flow is low.• There are other materials derived from silica or coated
glass beads that address this problem.
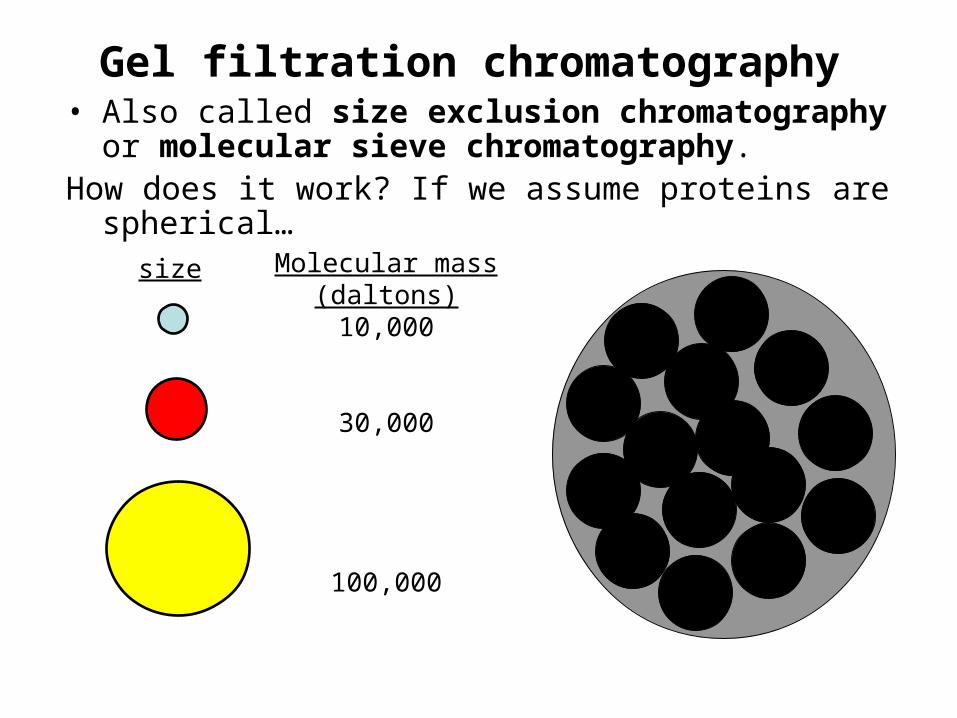
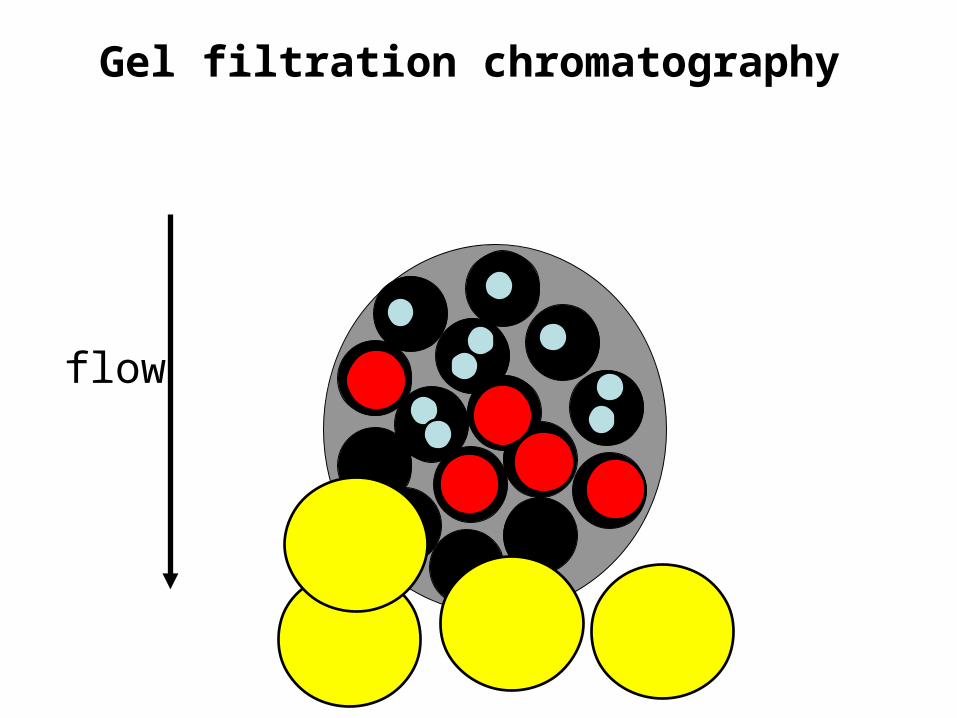
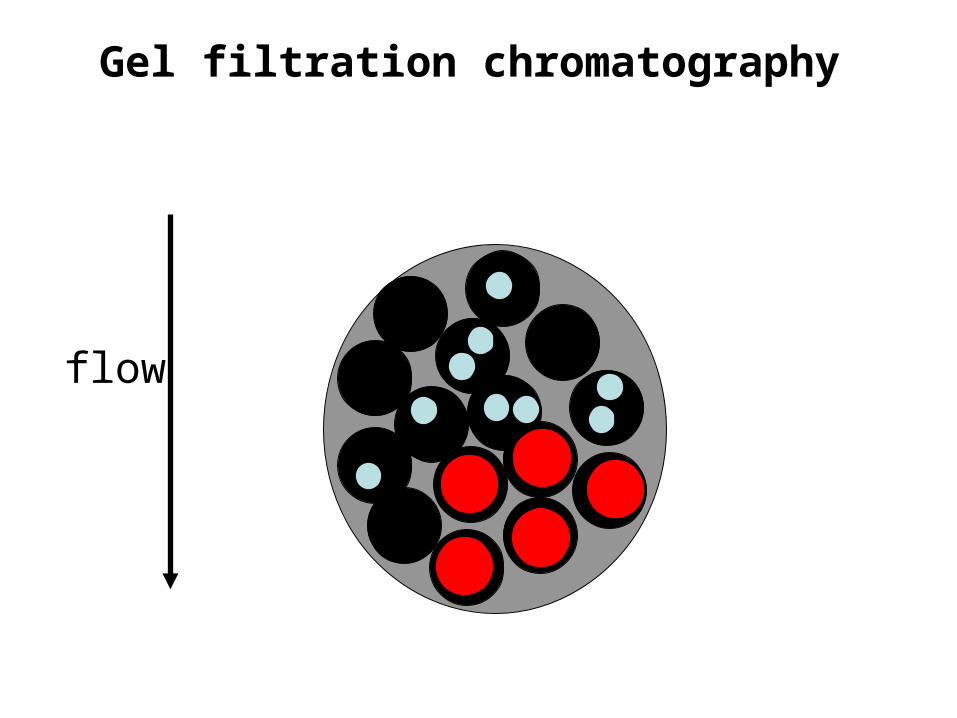
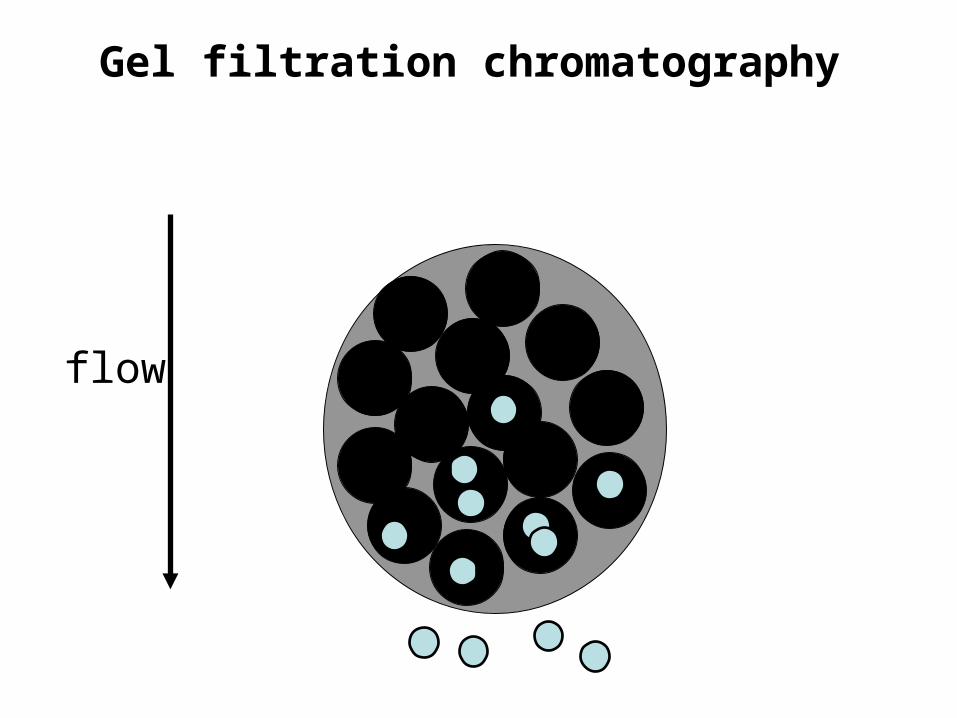
Gel filtration chromatography• Also called size exclusion chromatography or
molecular sieve chromatography.How does it work? If we assume proteins are spherical…
size Molecular mass(daltons)10,000
30,000
100,000
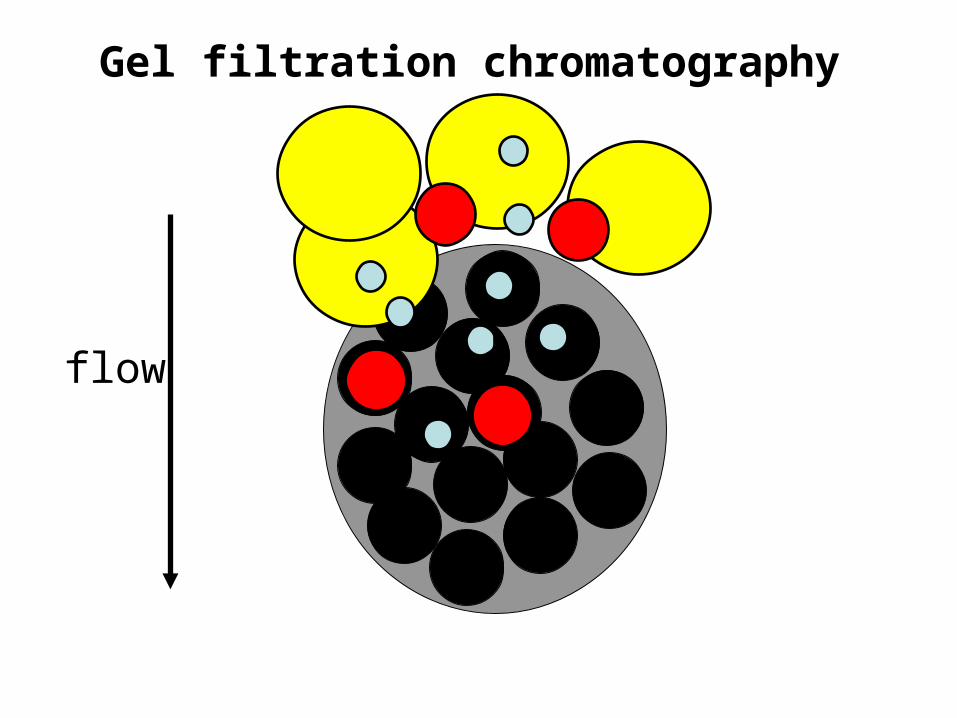
Gel filtration chromatography
flow
Gel filtration chromatography
flow
Gel filtration chromatography
flow

Gel filtration chromatography
flow
Gel filtration chromatography
flow

Gel filtration chromatography• The molecular mass of the smallest molecule unable to penetrate
the pores of the gel is at the exclusion limit.• The exclusion limit is a function of molecular shape, since elongated
molecules are less likely to penetrate a gel pore than other shapes.• Behavior of the molecule on the gel can be quantitatively
characterized.
Total bed volume of the column
Vt = Vx + V0
Vx = volume occupied by gel beads
V0 = volume of solvent space surrounding gel; Typically 35%

Gel filtration chromatography• Elution volume (Ve) is the volume of a solvent required to elute a
given solute from the column after it has first contacted the gel.
• Relative elution volume (Ve/V0) is the behavior of a particular solute on a given gel that is independent of the size of the column.
• This effectually means that molecules with molecular masses ranging below the exclusion limit of a gel will elute from a gel in the order of their molecular masses with the largest eluting first.
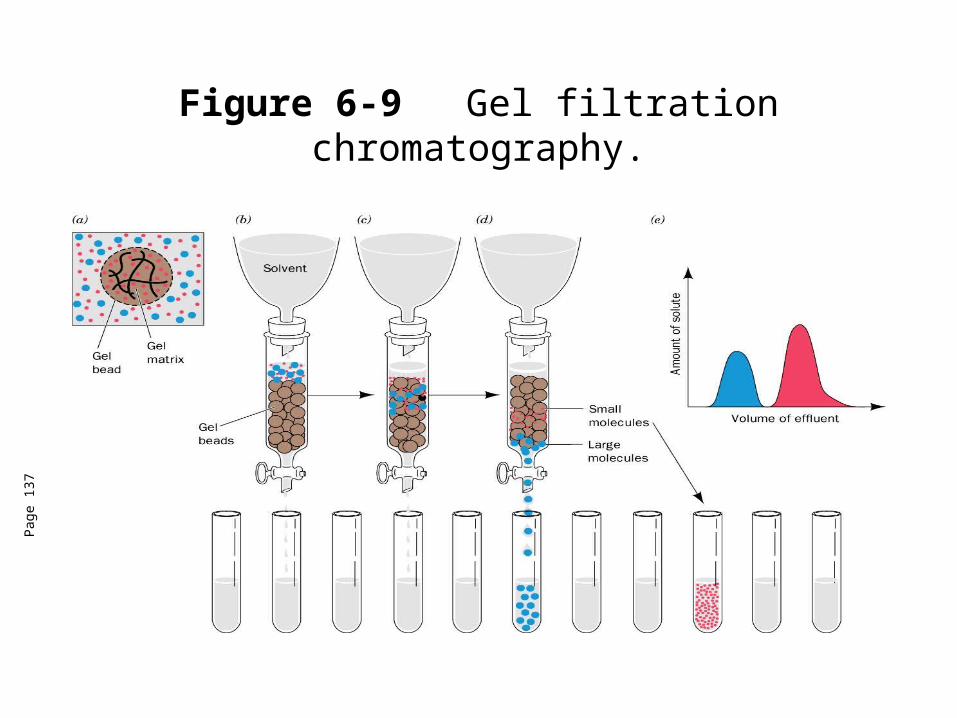
Figure 6-9 Gel filtration chromatography.
Pag
e 13
7
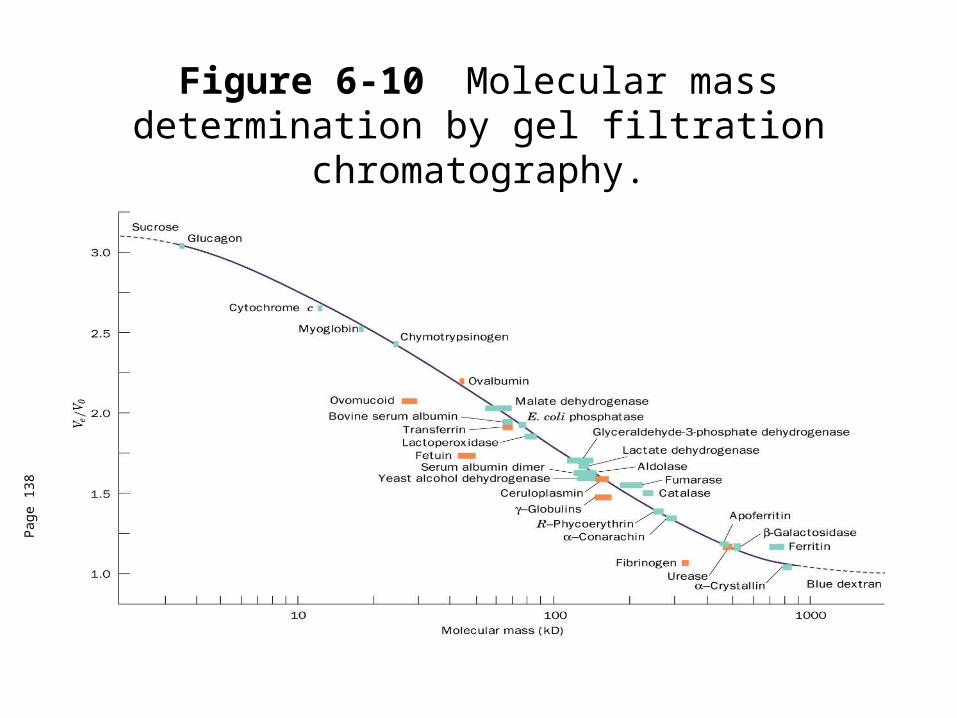
Figure 6-10 Molecular mass determination by gel filtration chromatography.
Pag
e 13
8
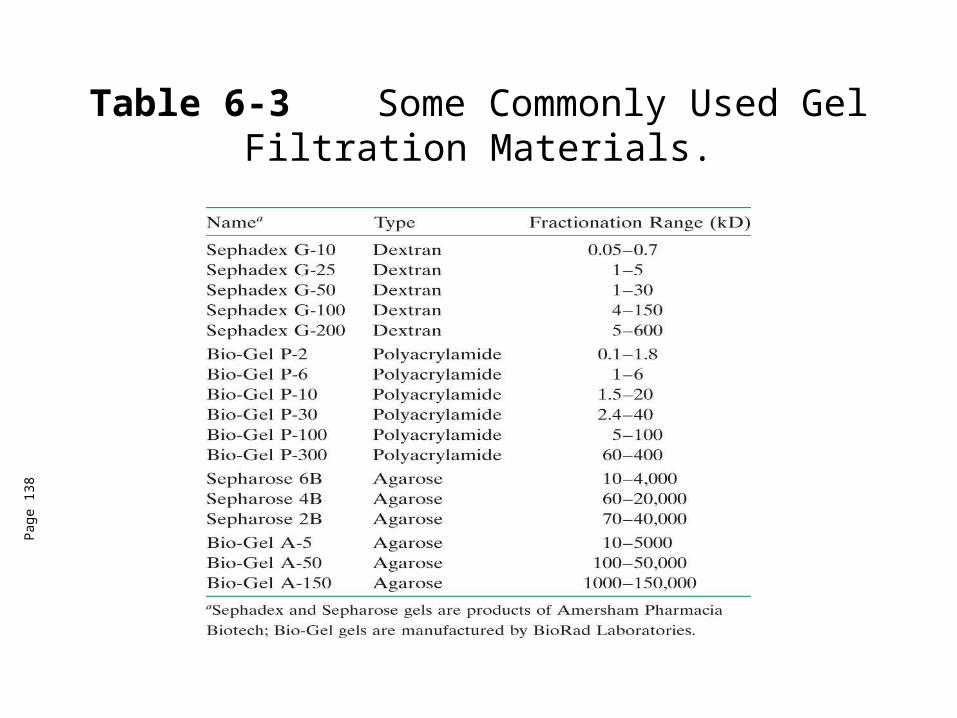
Table 6-3 Some Commonly Used Gel Filtration Materials.
Pag
e 13
8

Gel filtration chromatography• Elution volume (Ve) is the volume of a solvent required to elute a
given solute from the column after it has first contacted the gel.
• Relative elution volume (Ve/V0) is the behavior of a particular solute on a given gel that is independent of the size of the column.
• This effectually means that molecules with molecular masses ranging below the exclusion limit of a gel will elute from a gel in the order of their molecular masses with the largest eluting first.
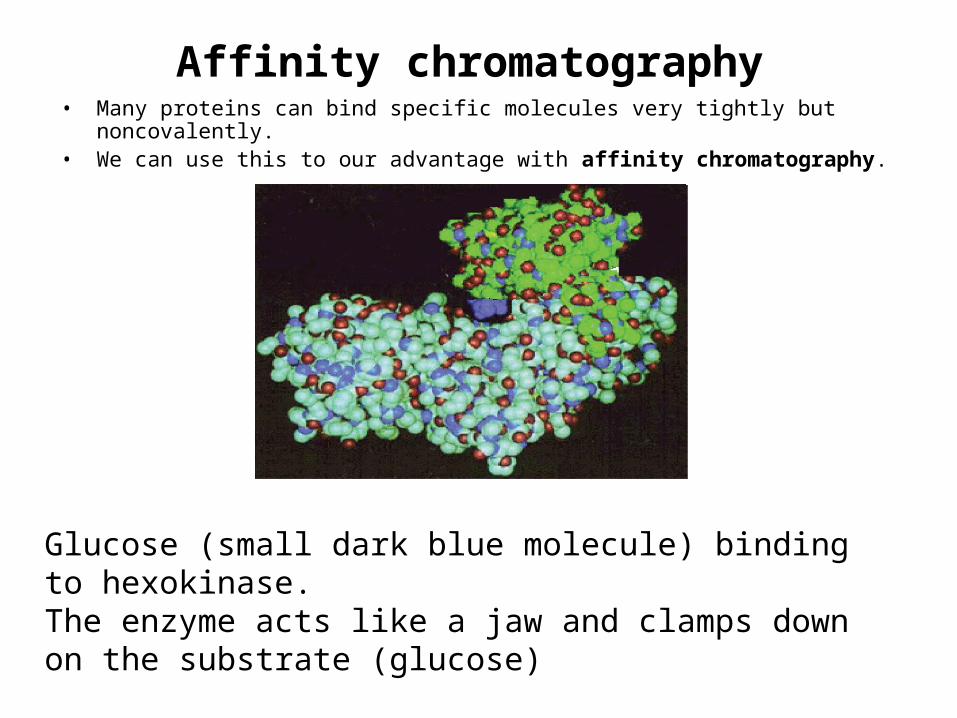
Affinity chromatography• Many proteins can bind specific molecules very tightly but noncovalently. • We can use this to our advantage with affinity chromatography.
Glucose (small dark blue molecule) binding to hexokinase. The enzyme acts like a jaw and clamps down on the substrate (glucose)
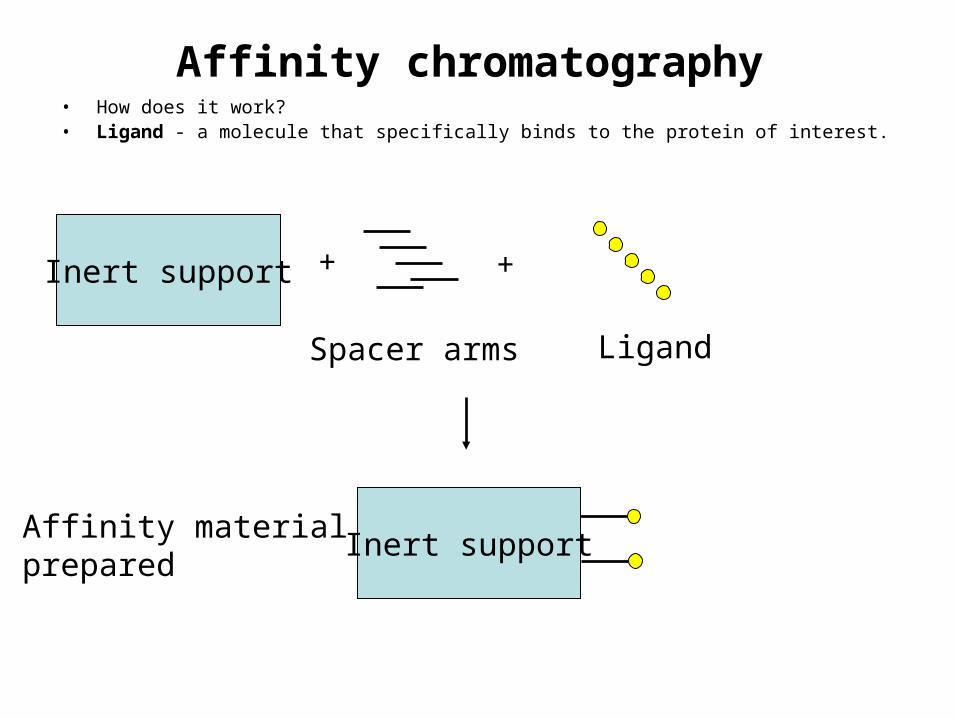
Affinity chromatography• How does it work?• Ligand - a molecule that specifically binds to the protein of interest.
Inert support
Spacer arms
+ +
Ligand
Inert supportAffinity materialprepared
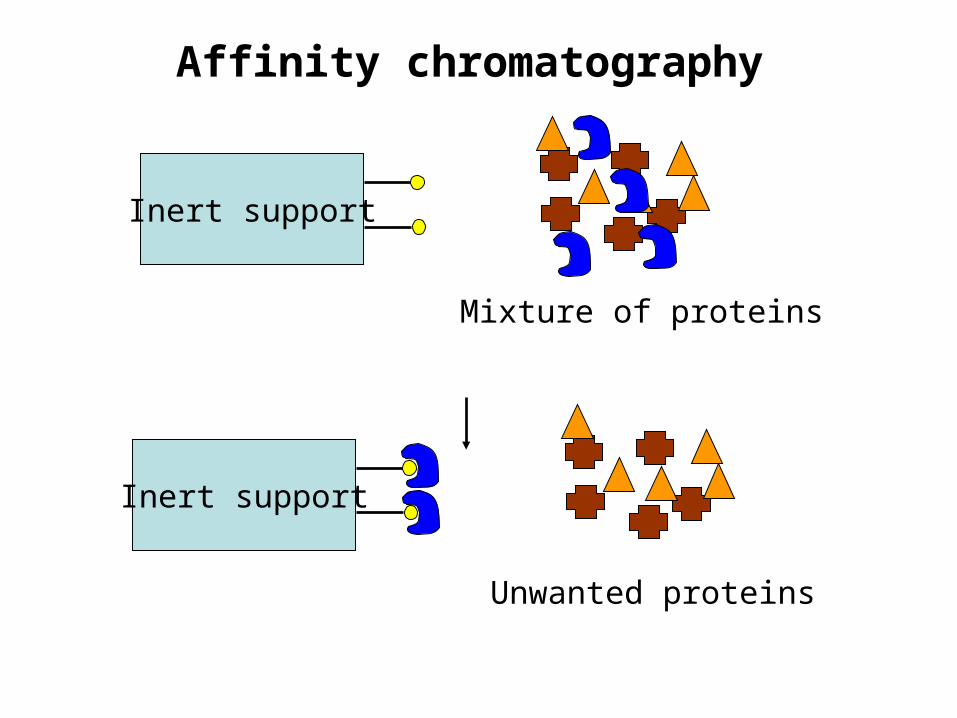
Affinity chromatography
Inert support
Mixture of proteins
Inert support
Unwanted proteins
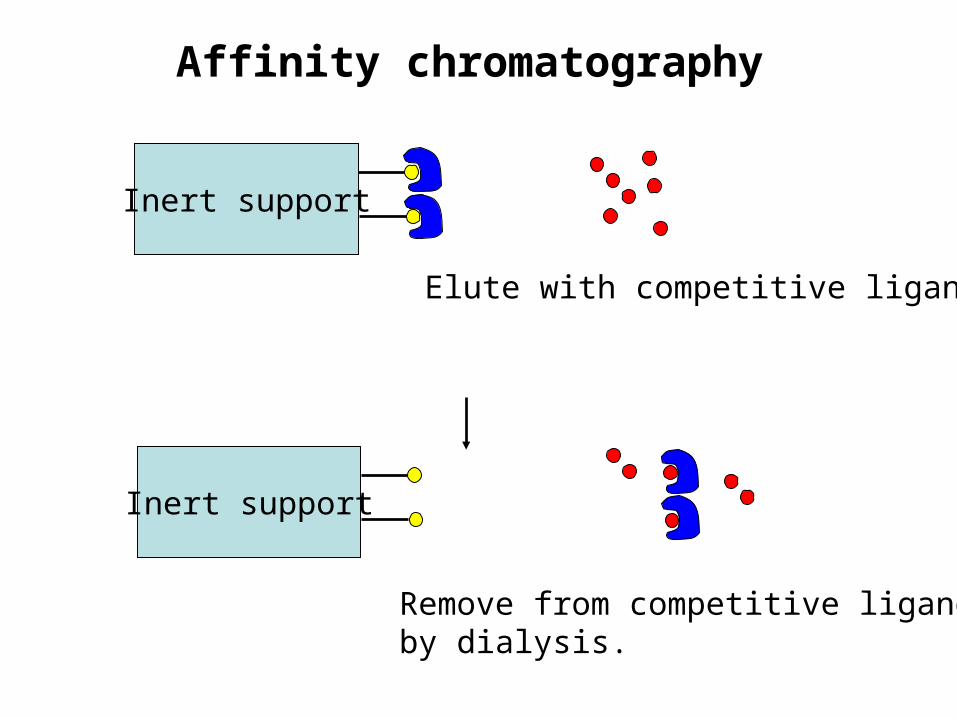
Affinity chromatography
Inert support
Remove from competitive ligandby dialysis.
Inert support
Elute with competitive ligand.

Affinity chromatography• To remove the protein of interest from the column, you
can elute with a solution of a compound with higher affinity than the ligand (competitive)
• You can change the pH, ionic strength and/or temperature so that the protein-ligand complex is no longer stable.

Immunoaffinity chromatography
• Monoclonal antibodies can be attached to the column material.• The column only binds the protein against which the antibody has
been raised.• 10,000-fold purification in a single step!• Disadvantges
– Difficult to produce monoclonal antibodies (expensive $$!)– Harsh conditions to elute the bound protein

Other chromatographic methods
• Adsorption chromatography - nonpolar molecules physically adosrbed on the surface of an insoluble substance (alumina, diatomeceous earth, silica gel, etc.) through Van der Waals forces.
• Molecules eluted from the column by organic solvents (chloroform, hexane, ethyl ether).
• Based on the partition of polar column material and nonpolar solvent.• Not used often with proteins.• Hydroxyapatite chromatography - gels of crystalline hydroxyapatite
(an insoluble form of calcium phosphate) adsorb proteins.• Separation occurs with a gradient elution of the column with
phosphate buffer.

Other chromatographic methods• Paper chromatography - separation of small polar molecules. Mostly used to separate amino acids,
oligopeptides. Historically the first chromatography but not really used today. However, principles of its use are useful to know.
• Rates of migration of the substances are determined by relative solubilities in the polar stationary phase (paper) and the nonpolar mobile phase
• A given solute is distributed between the mobile and stationary phases according to its partition coefficient
Kp =concentration in stationary phase
concentration in mobile phase
Molecules are separated according to their polarities, with nonpolar molecules moving faster than polar molecules

Other chromatographic methods• After the solvent has migrated an appropriate distance, the chromatogram is removed from the solvent
and dried. If not colored, the separated materials can be detected by radioactivity, fluroescence, etc.• The migration rate of the substance is expressed by the following ratio:
• Each substance has a characteristic Rf value for a given solvent and paper type.
• Reverse-phase chromatography (RPC)- separates nonpolar substances including denatured proteins.
• Form of liquid-liquid partition chromatography in which the polar character of the phases is reversed relative to paper chromatography. Stationary phase is nonpolar and the mobile phase is a more polar liquid. Used to separate lipids but can also be used for proteins.
• Solvent must be highly nonpolar usually high concentration of organic solvent (acetonitrile) so it denatures proteins so that the hydrophobic cores can interact with the matrix.
Rf =Distance traveled by substance
Distance traveled by solvent front

Other chromatographic methods• Hydrophobic interaction chromatography (HIC)- the stationary phase is hydrophillic
(agarose gel) with substituted hydrophobic groups.• Interactions with column are relatively weak and can be used for the separation of
native proteins (not denatured), so proteins are separated based on surface hydrophobicity.
• High performance liquid chromatography (HPLC)- may be based on adsorption, ion exchange, size exclusion, HIC or RPC but is improved because of the noncompressible matrix.
• Can be made of silica and withstand very high pressures (up to 5000 psi) so flow rates can be very high.
• Advantages of HPLC– High resolution– Fast– High sensitivity– Can be easily automated

Dialysis• Dialysis-a process that separates molecules according to size
through the use of semipermeable membranes containing pores of less than macromolecular dimensions.
• Pores in the membrane allow solvents, salts and small metabolites to diffuse across but block larger molecules.
• Cellophane (cellulose acetate) most commonly used dialysis material.
• Usually used to change the solvent in which the protein is dissolved in.
• Can also be used to concentrate a protein solution by placement in a polymeric dessicant (PEG) which cannot go through the membrane but absorbs water through the membrane.
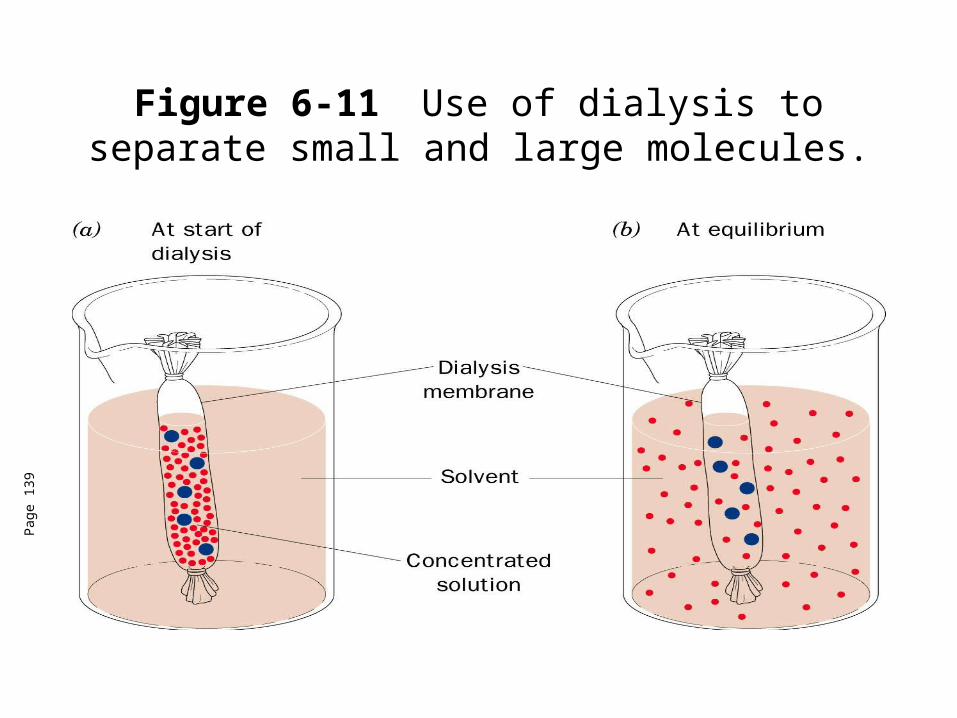
Figure 6-11 Use of dialysis to separate small and large molecules.
Pag
e 13
9
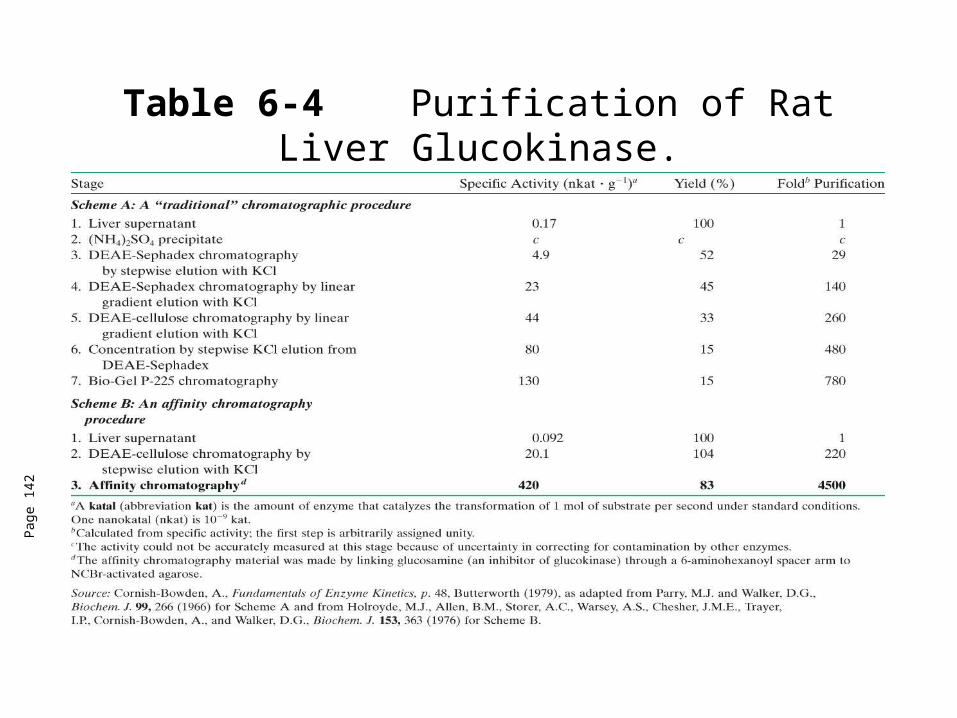
Table 6-4 Purification of Rat Liver Glucokinase.
Pag
e 14
2

Electrophoresis• The migration of ions in an electric field to separate molecules.• Many forms of electrophoresis-we will focus on polyacrylamide gel electrophoresis (PAGE).• PAGE techniques are often used determine the purity of proteins.
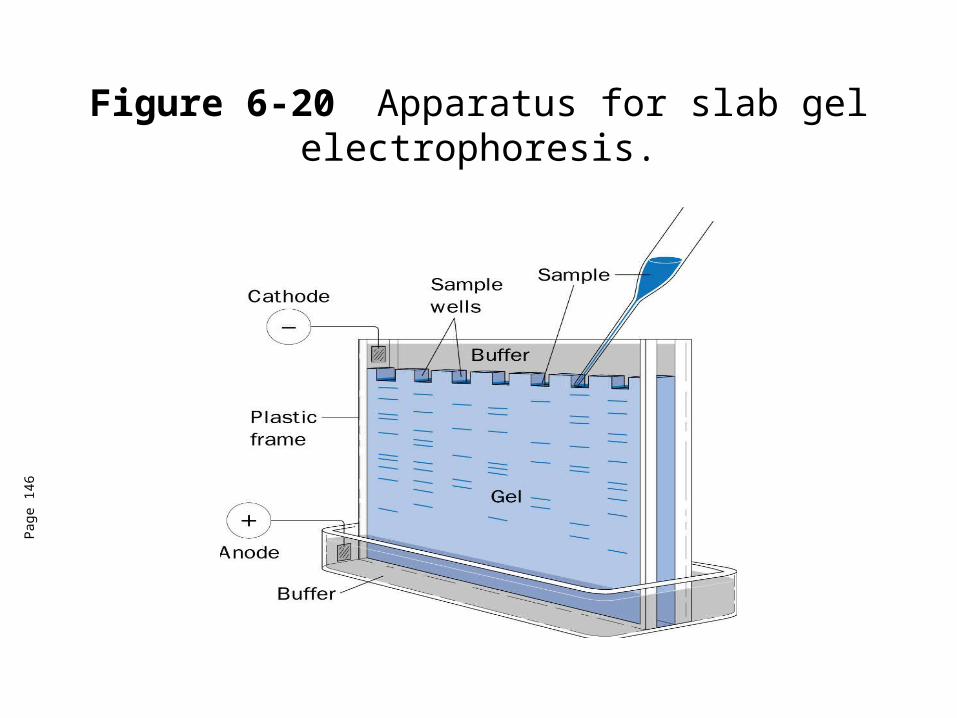
Figure 6-20 Apparatus for slab gel electrophoresis.
Pag
e 14
6
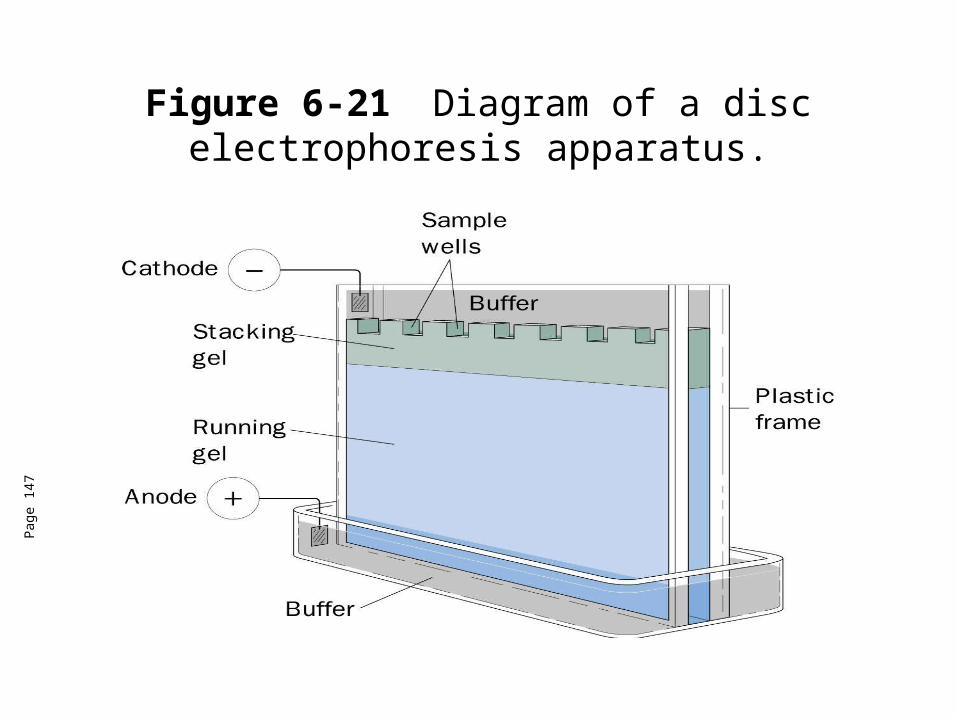
Figure 6-21 Diagram of a disc electrophoresis apparatus.
Pag
e 14
7

Sodium dodecyl sulfate (SDS-PAGE)
• Native protein is unfolded by heating in the presence of -mercaptoethanol and SDS.
• SDS binds to the protein so that it stays in solution and denatures.
• Large polypeptides bind more SDS than small polypeptides, so proteins end up with negative charge in relation to their size.
• Thus, we can separate the proteins based on their mass.
Native protein
Heat+
Reductant+
SDS
Denatured protein with bound SDS
N
C
--
-
---
- -
--
--
- -
-
- -
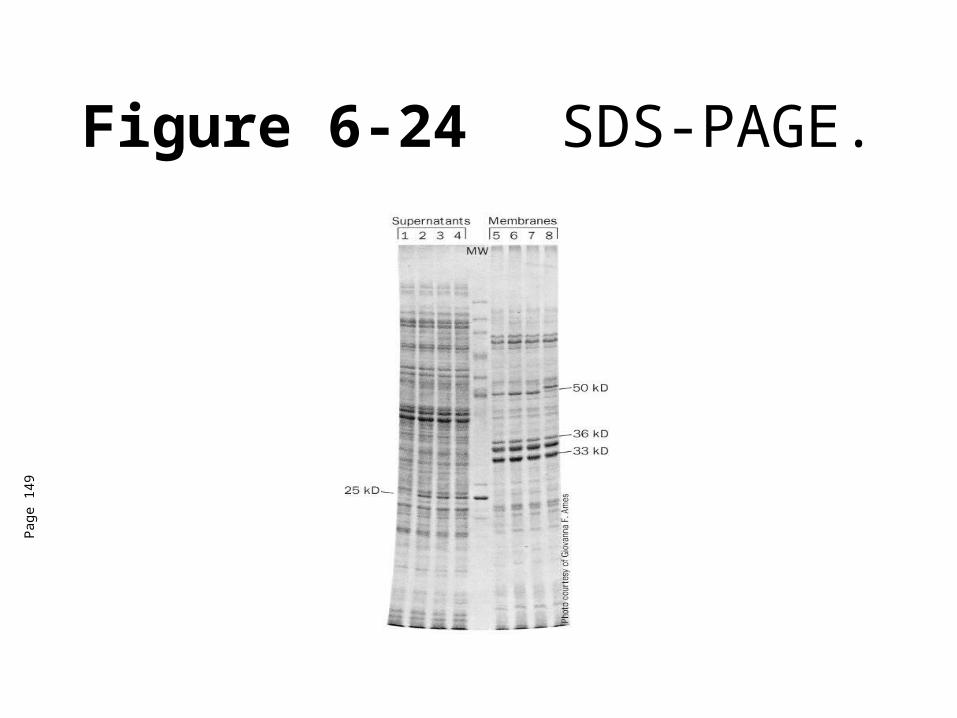
Figure 6-24 SDS-PAGE.
Pag
e 14
9
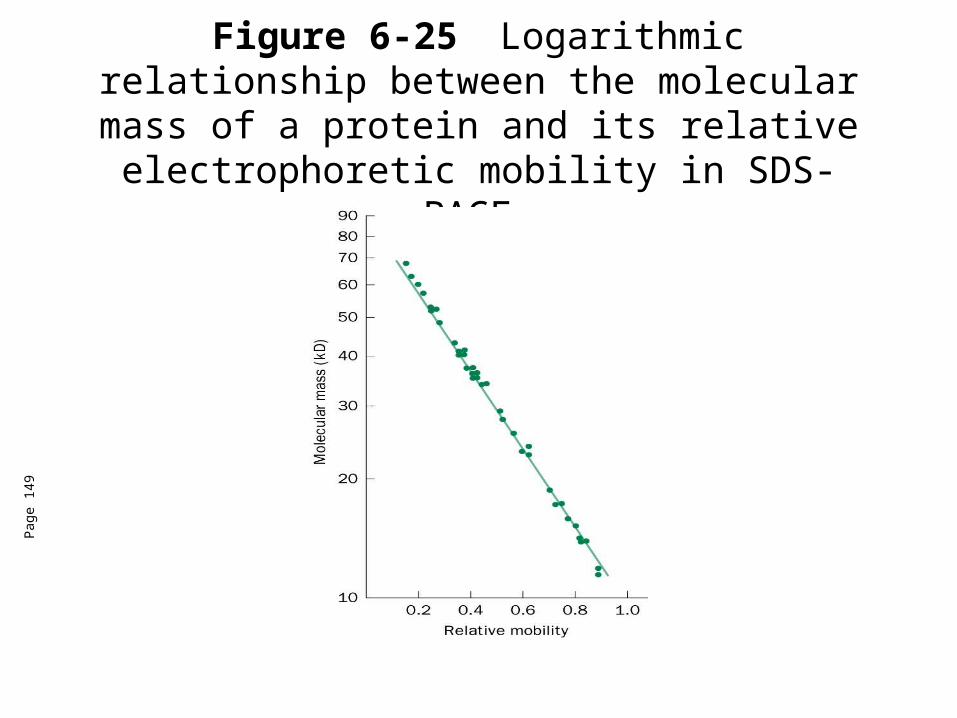
Figure 6-25 Logarithmic relationship between the molecular mass of a protein and its relative electrophoretic mobility in SDS-PAGE.
Pag
e 14
9

Figure 6-23 Detection of proteins by immunoblotting.
Pag
e 14
8

Isoelectric focusing• For looking at proteins without charge, proteins can be treated with
6M urea (denatures but unlike SDS does not put charges on a protein).
• Thus, a mixture of proteins can be electrophoresed through a solution having a a stable pH gradient in from the anode to the cathode and a each protein will migrate to the position in the pH gradient according to its isoelectric point. This is called isoelectric focusing.
• Ampholytes (amphoteric electrolytes)-low molecular mass (600-900D) ooligomers with aliphatic amino and carboxylic acid groups with a range of isoelectric points. Ampholytes help maintain the pH gradiennt in the presence of high voltage.
• Can also use gels with immobilized pH gradients -made of acrylamide derivatives that are covalently linked to ampholytes. Used with a gradient maker to ensure continuously varied mixture when the gel is made.
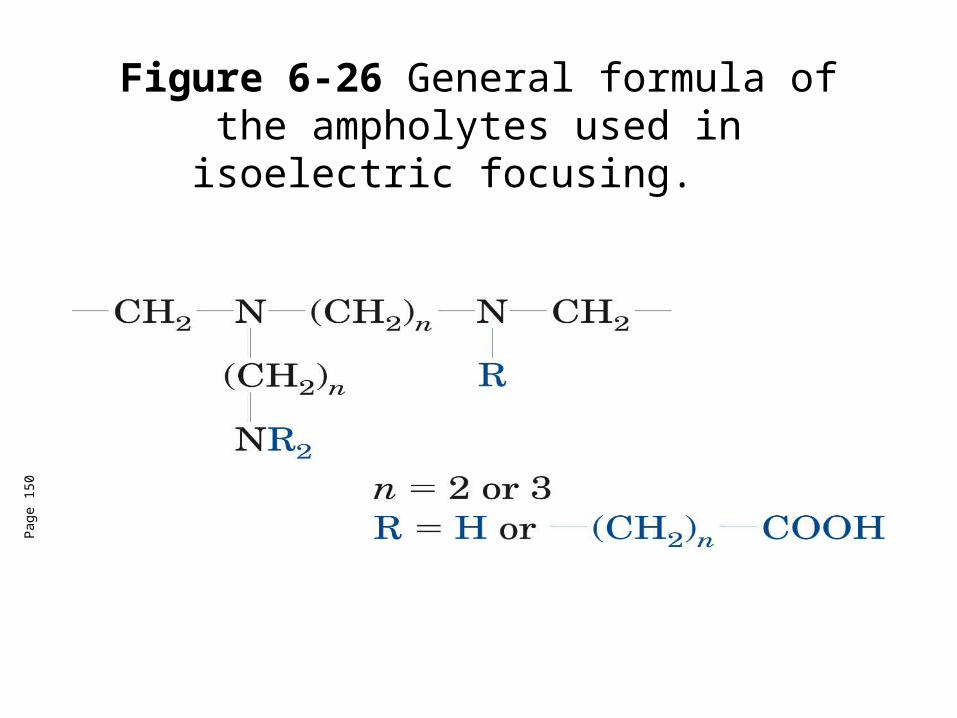
Figure 6-26 General formula of the ampholytes used in isoelectric focusing.
Pag
e 15
0

Isoelectric focusing• 2D-gel electrophoresis is an invalubale tool for proteomics.• Proteome (like genome) is the total number of all proteins expressed
by a cell or organism, but with an emphasis on their quantitation, localization, modifications, interactions, and activities, as well as their identification.
• Individual protein bands froma stained gel can be cut out of a gel, destained, and and the protein can be eluted from the gel fragment for identification and characterization using mass spec.
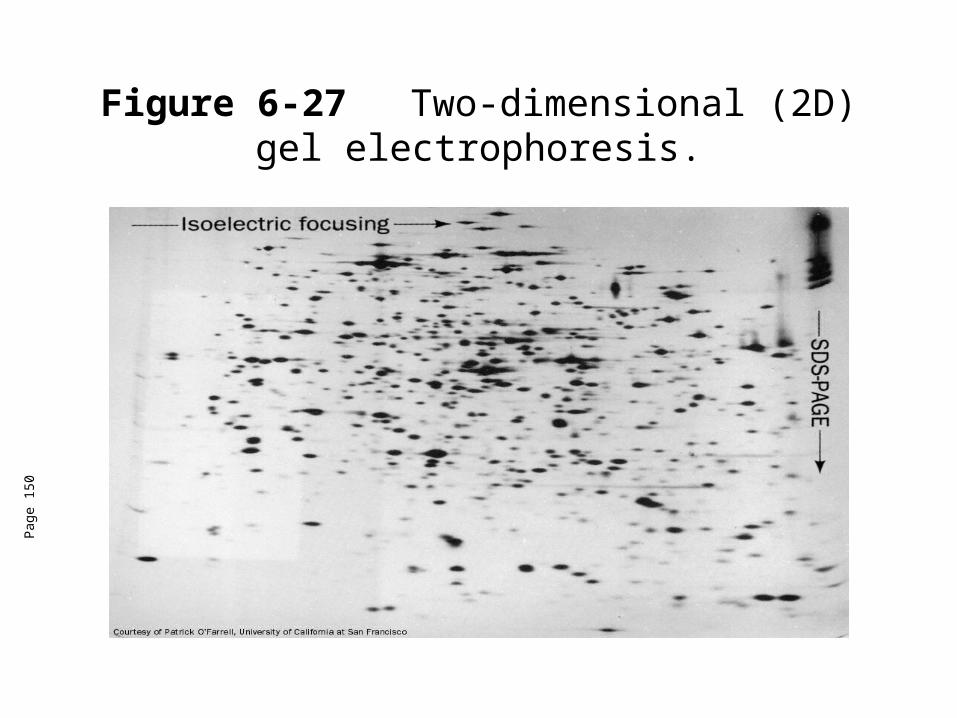
Figure 6-27 Two-dimensional (2D) gel electrophoresis.
Pag
e 15
0
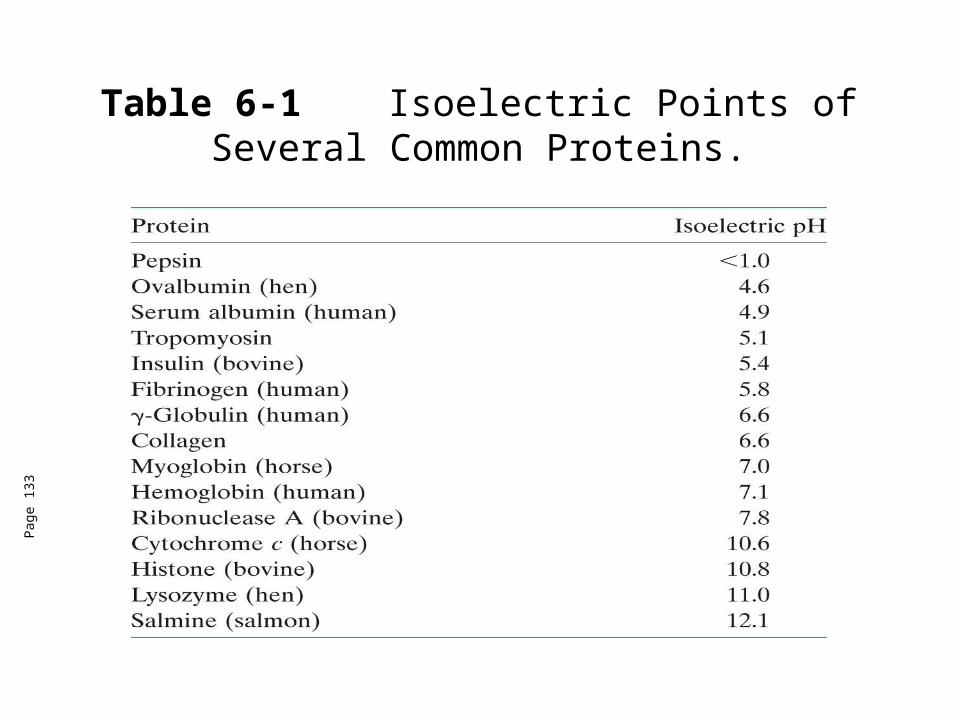
Table 6-1 Isoelectric Points of Several Common Proteins.
Pag
e 13
3

Summary of techniques for protein purification
• Cell lysis techniques - osmolysis, mechanical disruption-high speed blender, homogenizer, French press, sonication
• Salting out and salting in
• Chromatography– Ion exchange– Size exclusion– Affinity – others
• Dialysis
• Electrophoresis– SDS PAGE
• Isoelectric focusing

CrystallizationP
age
133
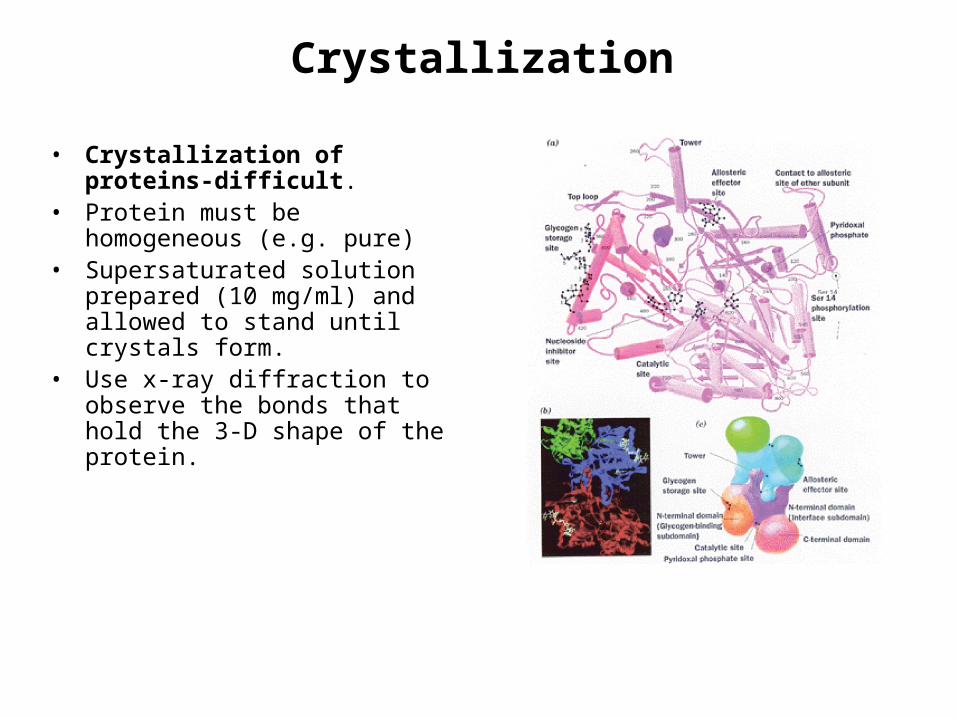
Crystallization
• Crystallization of proteins-difficult.
• Protein must be homogeneous (e.g. pure)
• Supersaturated solution prepared (10 mg/ml) and allowed to stand until crystals form.
• Use x-ray diffraction to observe the bonds that hold the 3-D shape of the protein.

3-D structure of proteins
X-ray sourceSingle crystal of protein
Diffraction pattern
Computational recombination of scattered x-rays
Electron density map
Structural model
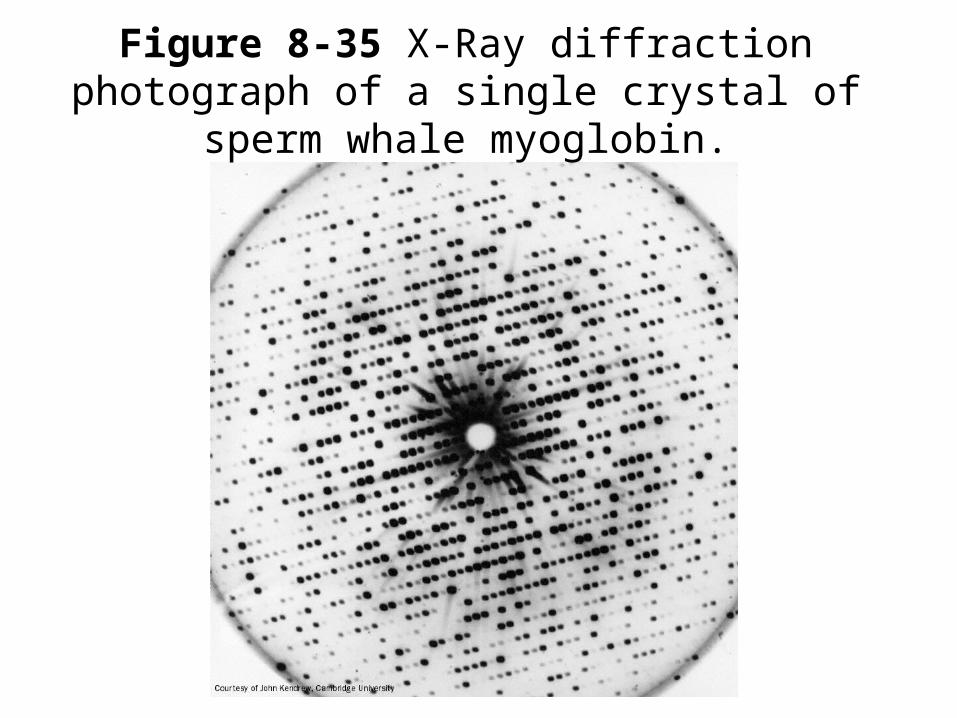
Figure 8-35 X-Ray diffraction photograph of a single crystal of sperm whale myoglobin.
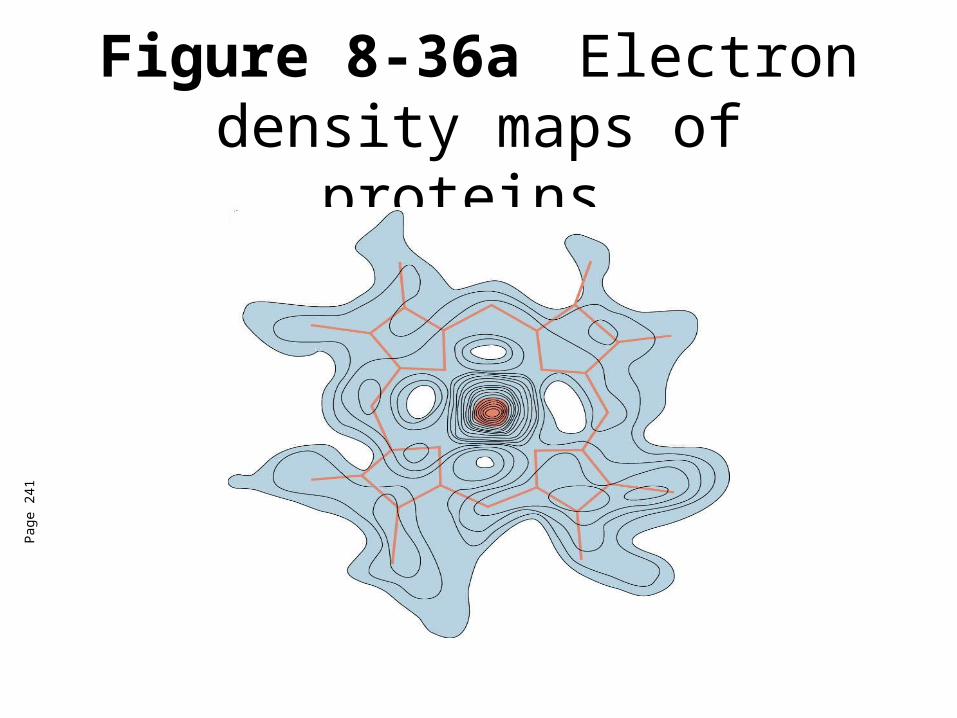
Figure 8-36a Electron density maps of proteins.
Pag
e 24
1
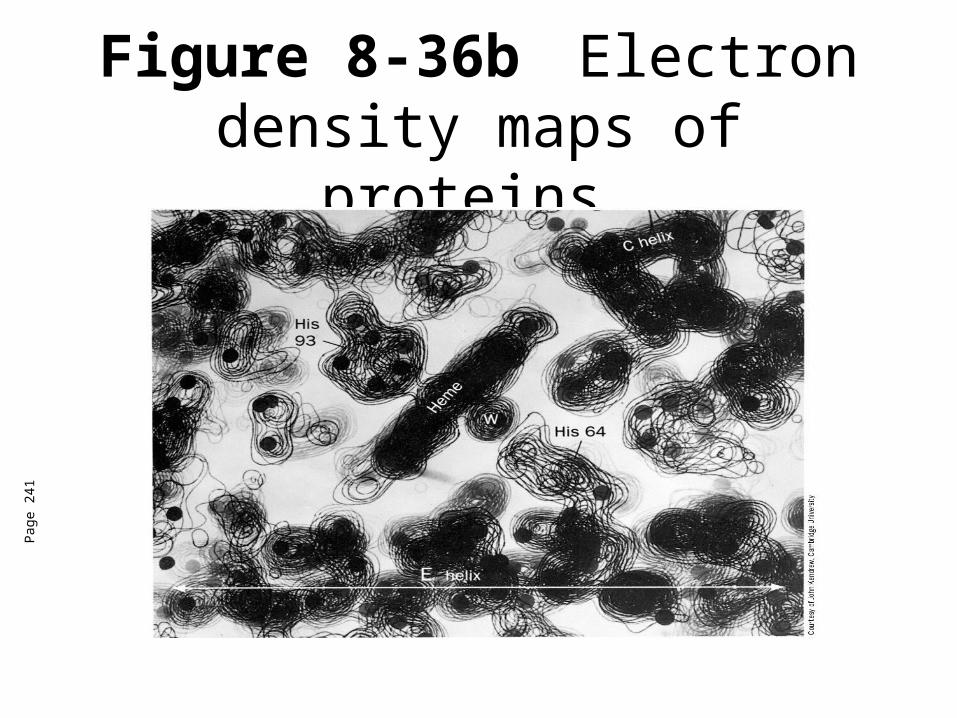
Figure 8-36b Electron density maps of proteins.
Pag
e 24
1

Figure 8-36c Electron density maps of proteins.
Pag
e 24
1