lee-supporting online material - science€¦ · dong-hwan lee, ki-hyeok kwon, chae s. yi* *to whom...
TRANSCRIPT

www.sciencemag.org/cgi/content/full/333/6049/1613/DC1
Supporting Online Material for
Selective Catalytic C–H Alkylation of Alkenes with Alcohols
Dong-Hwan Lee, Ki-Hyeok Kwon, Chae S. Yi*
*To whom correspondence should be addressed. E-mail: [email protected]
Published 16 September 2011, Science 333, 1613 (2011)
DOI: 10.1126/science.1208839
This PDF file includes:
SOM Text Figs. S1 to S3 Tables S1 to S3 Full Reference List

Supporting Online Material
Selective Catalytic C–H Alkylation of Alkenes with Alcohols
Dong-Hwan Lee, Ki-Hyeok Kwon and Chae S. Yi*
Department of Chemistry, Marquette University, Milwaukee, Wisconsin 53201-1881 United States
E-mail: [email protected]; Fax: 1-414-288-7066; Tel: 1-414-288-3536
Table of Contents
1. General information S1 2. General procedure of the catalytic alkylation reaction S1 1H NMR spectrum of the reaction of propene with ethanol in toluene-d8 (Fig. S1) S2 3. Catalyst screening and activity study for the alkylation of indene with 4-methoxybenzyl alcohol S3
Catalyst survey (Table S1) S3 Catalyst activity study (Table S2) S4
4. Deuterium labeling study S4 1H and 2H NMR spectra of the reaction of indene with 2-propanol-d8 (Fig. S2) S5 5. Carbon isotope effect study S5 Carbon isotope effect of 5e (Table S3) S6 6. Hammett study S7
Hammett plot (Fig. S3) S7 7. Characterization data of the products S8 8. 1H and 13C NMR spectra of the products S21

S1
1. General information. All operations were carried out in a nitrogen-filled glove box or by using standard
high vacuum and Schlenk techniques unless otherwise noted. Solvents were freshly distilled over the
appropriate drying reagents. Tetrahydrofuran, benzene, toluene, hexanes and ether were distilled from purple
solutions of sodium and benzophenone immediately prior to use. Dichloromethane (CH2Cl2) and 1,2-
dichloroethane (ClCH2CH2Cl) were dried over calcium hydride. The NMR solvents were dried from activated
molecular sieves (4 Å). All organic substrates were received from Acros, Sigma-Aldrich, TCI and Alfa-Aesar
and were used without further purification. The 1H, 2H, 13C and 31P NMR spectra were recorded on a Varian
300 or 400 MHz FT-NMR spectrometer, and the data are reported as: s = singlet, d = doublet, t = triplet, q =
quartet, m = multiplet, b = broad, app = apparent; coupling constant(s) in Hz; integration. Mass spectra were
recorded from a Agilent 6850 GC-MS spectrometer with a HP-5 (5% phenylmethylpolysiloxane) column (30
m, 0.32 mm, 0.25 µm). The conversion of organic products was measured from a Hewlett-Packard HP 6890 GC
spectrometer. High resolution mass spectra were obtained at the Center of Mass Spectrometry, Washington
University, St. Louis, MO. Optical rotations were measured by using a 1 mL cell with a 1 dm path length on a
Perkin-Elmer 341 polarimeter with a sodium lamp and are reported as follows: [_]TC (c = g/100 mL, solvent).
Elemental analyses were performed at the Midwest Microlab, Indianapolis, IN.
2. General procedure of the catalytic alkylation reaction. In a glove box, complex 1 (3 mg, 0.5 mol %),
an alkene (1.0 mmol) and an alcohol (1.1 mmol) were dissolved in CH2Cl2 (2 mL) in a 25 mL Schlenk tube
(Kontes) equipped with a Teflon stopcock and a magnetic stirring bar. The tube was brought out of the box, and
was stirred for 2-5 h in an oil bath which was preset at 75 °C (C6H5Cl for >80 °C). The reaction tube was taken
out of the oil bath, and was immediately cooled in a dry ice/acetone bath. After the tube was open to air, the
solution was filtered through a short silica gel column (hexanes/ethyl acetate (EtOAc) = 2:1), and the filtrate
was analyzed by GC and GC-MS. Analytically pure product was isolated by a simple column chromatography
on silica gel (Silicycle Inc., 280 to 400 mesh, hexanes/EtOAc = 40:1 to 4:1).
Alkylation of propene with ethanol. In a glove box, complex 1 (2 mg, 0.01 mol %) and ethanol (1.84 g,
40 mmol) and n-dodecane (0.51 g, 3 mmol, internal standard) were dissolved in C6H5Cl (3 mL) in a 100 mL
Fisher-Porter pressure tube (Andrews Glass Co.) equipped with a magnetic stirring bar. The tube was brought
out of the box, and was degassed three times by freeze-pump-thaw cycles. The tube was immersed in a dry
ice/acetone bath, and propene gas (1.3 g, 30 mmol) was condensed into the tube via a vacuum transfer. The
tube was warmed to room temperature and stirred in an oil bath which was preset at 110 °C for 6 h. After the
tube was cooled in a dry ice/ethylene glycol bath (-25 °C) for 10 min, the tube stopcock was slowly open to the
vacuum line and the volatiles were removed in vacuo to remove unreacted propene. After the tube was opened
to air, the residue was filtered through a short silica gel column, and the resulting filtrate was analyzed by both

S2
NMR and GC. GC analysis conditions: helium carrier gas pressure at 7.7 psi, flow rate at 1.0 mL/min, oven
temperature at 30 to 250 °C. Retention time of the products: 2a (2.40 minute) and 3a (1.95 minute). The
combined GC yield of 2a and 3a = 46% (2a:3a = 9:1, E/Z = 20:1). The product mixture 2a and 3a was isolated
by the distillation of product residue (obtained without using n-dodecane) under a reduced pressure (0.01 torr)
at room temperature (40% yield, >95% pure as analyzed by 1H NMR). Analogous procedure was used for the
reaction of 3,3-dimethyl-1-butene (30 mmol) with ethanol (40 mmol) and 1 (0.02 mol %) to obtain a mixture of
products 2b and 3b in 56% yield.
Reaction in a NMR tube. In a glove box, complex 1 (1.5 mg, 0.25 mol %) and ethanol (70 mg, 1.5 mmol)
were dissolved in toluene-d8 (0.4 mL) in a J-Young NMR tube with a Teflon screw cap stopcock (Wilmad).
The tube was brought out of the box, and was degassed three times by freeze-pump-thaw cycles. The tube was
immersed in a dry ice/acetone bath, and propene gas (42 mg, 1.0 mmol) was condensed into the tube via a
vacuum transfer. The tube was immersed in an oil bath at 110 °C for 6 h. Figure S1 shows the 1H NMR
spectrum of the reaction mixture after 6 h (2a:3a = 20:1, E/Z = 25:1). Generally, the reaction rate in a NMR
tube is considerably lower than the preparatory scale reaction as described above.
Figure S1. 1H NMR spectrum of the reaction of propene with ethanol in toluene-d8 after 6 h of heating at
110 °C in a J-Young NMR tube.
Representative procedure for the alkylation of a bioactive olefin compound with 4-methoxybenzyl
alcohol. In a glove box, complex 1 (6 mg, 1.0 mol %), an alkene (1.0 mmol) and 4-methoxybenzyl alcohol (1.2
mmol) were dissolved in C6H5Cl (3 mL) in a 25 mL Schlenk tube equipped with a Teflon stopcock and a

S3
magnetic stirring bar. The tube was brought out of the box, and was stirred for 2-6 h in an oil bath at 90-110 °C.
The tube was taken out of the oil bath, and was immediately cooled in a dry ice/acetone bath. After the tube
was opened to air, the solution was filtered through a short silica gel column (hexanes/EtOAc = 1:1), and the
resulting solution was analyzed by GC and GC-MS. Analytically pure product was isolated by a simple column
chromatography on silica gel (hexanes/EtOAc = 40:1 to 1:1).
3. Catalyst screening and activity study for the alkylation of indene with 4-methoxybenzyl alcohol.
Catalyst screening. In a glove box, indene (116 mg, 1.0 mmol), 4-methoxybenzyl alcohol (152 mg, 1.1
mmol) and a catalyst (1.0 mol %) were dissolved in CH2Cl2 (2 mL) in a 25 mL Schlenk tube equipped with a
Teflon stopcock and a magnetic stirring bar. The tube was brought out of the box, and was stirred for 2 h in an
oil bath which was preset at 75 °C. The reaction tube was taken out of the oil bath, and was immediately cooled
in a dry ice/acetone bath. After filtering through a short silica gel column (hexanes/EtOAc = 1:1), the solution
mixture was analyzed by GC and GC-MS. The results are summarized in Table S1.
Catalyst activity study. In a glove box, indene (116 mg, 1.0 mmol), 4-methoxybenzyl alcohol (152 mg,
1.1 mmol) and 1 (0.5 mmol % to 3.0 mol %) were dissolved in a 2 mL of a solvent in a 25 mL Schlenk tube
equipped with a Teflon stopcock and a magnetic stirring bar. The tube was brought out of the box, and was
stirred in an oil bath which was preset at 20-150 °C for 2 h. The reaction tube was taken out of the oil bath, and
was immediately cooled in a dry ice/acetone bath. After the tube was opened to air, the solution was filtered
through a short silica gel column (hexanes/EtOAc = 1:1), and the filtrate was analyzed by GC and GC-MS. The
results are summarized in Table S2.
Table S1. Catalyst survey for the alkylation of indene with 4-methoxybenzyl alcohol.†
+HO
OCH3
catalyst (1.0 mol %)
CH2Cl2, 75 oC, 2h
OCH3
5f
+HO
OCH3
1 (0.5 mmol % to 3.0 mol %)
conditions
OCH3
5f
entry Catalyst additive time (h) yield (%) 1 [(C6H6)(PCy3)(CO)RuH]BF4 (1) 2 96 2 HBF4·OEt2 2 0 3 [RuH(CO)(PCy3)]4(O)(OH)2 2 2 4 [RuH(CO)(PCy3)]4(O)(OH)2 HBF4·OEt2 2 72 5 RuCl3·3H2O HBF4·OEt2 2 0

S4
† Reaction conditions: indene (1.0 mmol), 4-methoxybenzyl alcohol (1.1 mmol), catalyst (1.0 mol %), additive (1.0 equivalent to Ru), CH2Cl2 (2 mL), 75 °C. The product yield was determined by GC and GC-MS.
Table S2. Catalyst activity study for the alkylation of indene with 4-methoxybenzyl alcohol.†
† Reaction conditions: indene (1.0 mmol), 4-methoxybenzyl alcohol (1.1 mmol), 1 (0.5 mmol % - 3.0 mol %), solvent (2 mL). The product yield was determined by GC and GC-MS.
4. Deuterium labeling study. In a glove box, indene (0.2 mmol), 2-propanol-d8 (0.6 mmol, 99% D) and 1
(2.5 mg, 2 mol %) were dissolved in toluene-d8 (0.4 mL) in a J-Young NMR tube with a Teflon screw cap
stopcock (Wilmad). The tube was brought out of the box, and the reaction progress was monitored by both 1H
and 2H NMR at 25 °C. After 1 h, the indene substrate was found to contain 21% deuterium on the C(2) position,
1 (2 mol %)
toluene-d8, 25 oCD
D3CC
D3COD+
D
1
2
3
+
D
6 RuCl2(PPh3)3 2 0 7 RuCl2(PPh3)3 HBF4·OEt2 2 trace 8 RuH2(CO)(PPh3)3 2 0 9 RuH2(CO)(PPh3)3 HBF4·OEt2 2 trace 10 [RuCl2(COD)]x HBF4·OEt2 2 0 11 [RuH(CO)(PCy3)2(CH3CN)2]+BF4
- 2 <5 12 [(p-cymene)RuCl2]2 2 0 13 Ru3(CO)12 NH4PF6 2 0 14 CF3SO3H 2 trace 15 BF3·OEt2 2 0 16 Cy3PH+BF4
- 2 trace 17 AlCl3 2 trace 18 FeCl3·H2O 2 0 19 Eu(OTf)3 2 <5
entry catalyst (mol %) solvent temp (°C) time (h) yield (%) 1 1 (1.0) CH2Cl2 75 2 96 2 1 (0.5) CH2Cl2 75 2 95 3 1 (0.1) C6H5Cl 100 2 91 4 1 (0.01) C6H5Cl 130 6 88 (TOF = 9,600/h) 5 1 (0.001) C6H5Cl 150 24 42 6 1 (0.0005) C6H5Cl 150 48 51 (TON = 102,000) 7 1 (0.5) CH2Cl2 50 12 31 8 1 (3.0) CH2Cl2 20 48 11 9 1 (0.5) C6H6 75 3 41 10 1 (0.5) toluene 75 3 58 11 1 (0.5) THF 75 3 73 12 1 (0.5) ClCH2CH2Cl 75 3 74

S5
and a nearly equal amount of indan with 15% deuterium on Cα was also detected, as analyzed by both 1H and 2H NMR. Figure S2 shows the 1H and 2H NMR spectra of the reaction mixture after 1 h of reaction time.
Figure S2. 1H and 2H NMR spectra of the reaction mixture of indene with 2-propanol-d8 at 25 °C.
5. Carbon isotope effect study. In a glove box, indene (1.16 g, 10 mmol), 1-(4-methoxyphenyl)ethanol
(1.67 g, 11 mmol) and complex 1 (60 mg, 1.0 mol %) were dissolved in C6H5Cl (20 mL) into each of three
separate 100 mL Schlenk tubes equipped with a Teflon screw cap stopcock and a magnetic stirring bar. The
tubes were brought out of the box, and were stirred for 10, 15 and 20 minutes, respectively, in an oil bath which
was preset at 75 °C. The product 5e was isolated separately after filtering through a short silica gel column
(hexanes/EtOAc = 20:1), and the product conversion for each solution was analyzed by GC (11, 15 and 18%
+OH
MeO
1 (1.0 mol %)
C6H5Cl, 75 oC
OMe
CH3
5e
2
16
3
7
8
9 10
45

S6
conversion).
The 13C{1H} NMR analysis of the isolated product 5e was performed by following Singleton’s 13C NMR
method (22). The NMR sample was prepared identically by dissolving 100 mg of isolated 5e in CDCl3 (0.5 mL)
in a 5 mm high precision NMR tube. The 13C{1H} NMR spectra were recorded with H-decoupling and 45
degree pulses. A 60 s delay between pulses was also imposed to minimize T1 variations (d1 = 60 s, at = 5.0 s,
np = 245098, nt = 704). The data are summarized in Table S3.
Table S3. Carbon isotope ratio of 5e.
carbon no. recovered (95% conv.)
recovered (11% conv.) 95%/11% change (%)
1 0.997 0.999 0.998 -0.20 2 1.133 1.061 1.068 6.80 3 1.051 1.049 1.002 0.20 4 0.992 0.995 0.997 -0.30 5 1.054 1.060 0.994 -0.60 6 (ref) 1.000 1.000 1.000 0.00 7 1.020 1.016 1.004 0.40 8 1.055 1.054 1.001 0.10 9 1.071 1.073 0.998 -0.20 10 2.232 2.229 1.001 0.10
carbon no. recovered (95% conv.)
recovered (15% conv.) 95%/15% change (%)
1 0.997 0.995 1.002 0.20 2 1.133 1.086 1.043 4.30 3 1.051 1.052 0.999 -0.10 4 0.992 0.994 0.998 -0.20 5 1.054 1.055 0.999 -0.10 6 (ref) 1.000 1.000 1.000 0.00 7 1.020 1.020 1.000 0.00 8 1.055 1.055 1.000 0.00 9 1.071 1.069 1.002 0.20 10 2.232 2.230 1.001 0.10
carbon no. recovered (95% conv.)
recovered (18% conv.) 95%/18% change (%)
1 0.997 0.999 0.998 -0.20 2 1.133 1.094 1.036 3.60 3 1.051 1.051 1.000 0.00 4 0.992 0.991 1.001 0.10 5 1.054 1.056 0.998 -0.20 6 (ref) 1.000 1.000 1.000 0.00

S7
7 1.020 1.018 1.002 0.20 8 1.055 1.055 1.000 0.00 9 1.071 1.068 1.003 0.30 10 2.232 2.229 1.001 0.10
6. Hammett study. In a glove box, complex 1 (3 mg, 0.5 mol %), indene (116 mg, 1.0 mmol) and p-X-
C6H4CH2OH (X = OCH3, CH3, H, F, Cl) (1.1 mmol) were dissolved in C6H5Cl (2 mL) in 4-6 separate 25 mL
Schlenk tubes each equipped with a Teflon screw cap stopcock and a magnetic stirring bar. The tubes were
brought out of the box, and stirred in an oil bath at 80 °C. A small portion of the aliquot was drawn from each
reaction tube in 5-10 minute intervals, and the conversion was determined by GC by measuring the appearance
of the product. The kobs was determined from a first-order plot of -ln([p-X-C6H4CH2OH]t/[p-X-C6H4CH2OH]0)
vs time. Analogous procedure was used to obtain the rate data from the coupling reaction of p-Y-C6H4CH=CH2
(Y = OCH3, CH3, H, F, Cl) (1.0 mmol) with PhCH2OH (1.1 mmol). The Hammett plot of log(kX/kH) versus σp
is shown in Figure S3.
Figure S3. Hammett plot of the alkylation of indene with p-X-C6H4CH2OH (X = OCH3, CH3, H, F, Cl)
(▲), and p-Y-C6H4CH=CH2 (Y = OCH3, CH3, H, F, Cl) with PhCH2OH (■).
+OH
X
1 (0.5 mol%)
C6H5Cl, 80 oC
X = OMe, CH3, H, F, Cl
X
5
Y
+OH 1 (0.5 mol%)
YY = OMe, CH3, H, F, Cl
9C6H5Cl, 80 oC

S8
7. Characterization data of the products.
Table 1, compound 4a. Data for 4a: 1H NMR (400 MHz, CDCl3) δ 5.20 (m, 1H), 2.01-
1.8 (m, 6H), 1.65-1.55 (m, 4H), 0.95 (t, J = 7.2 Hz, 3H) ppm; GC-MS m/z = 110 (M+). The
conversion of the product (>88%) was determined from GC and GC-MS analysis. No other
byproducts were detected by GC. The 1H and 13C NMR spectral data are in good agreement
with the literature data (34).
Table 1, compound 4b. A C6H5Cl (2 mL) solution of complex 1 (3 mg, 0.5
mol %), cyclohexene (1.0 mmol) and 1-hexanol (1.1 mmol) was stirred for 6 h at
90 °C. The product 4b was isolated by a column chromatography on silica gel
(hexanes/EtOAc = 250:1 to 40:1). Yield: 131 mg, 79%. Data for 4b: 1H NMR (400
MHz, CDCl3) δ 5.38 (br, 1H), 1.95 (m, 2H), 1.8-1.6 (m, 4H), 1.43 (m, 2H), 1.4-1.1
(m, 10H), 0.89 (t, J = 7.1 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 139.4, 120.3, 37.7, 30.9, 30.0, 29.2,
27.9, 25.3, 22.9, 22.4, 22.3, 14.1 ppm; GC-MS m/z = 166 (M+); Anal. Calcd for C12H22: C, 86.67; H, 13.33.
Found: C, 86.34; H, 13.18.
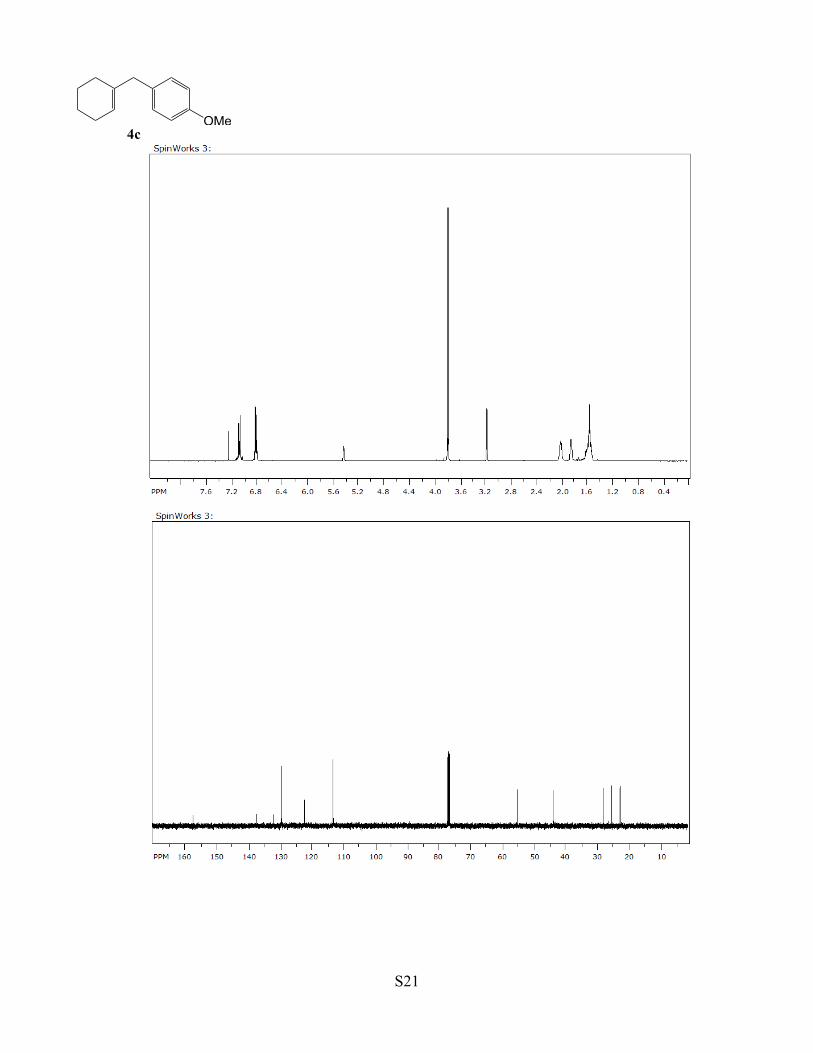
Table 1, compound 4c. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5
mol %), cyclohexene (1.0 mmol) and 4-methoxybenzyl alcohol (1.1 mmol) was
stirred for 4 h at 75 °C. The product 4c was isolated by a column chromatography
on silica gel (hexanes/EtOAc = 250:1 to 4:1). Yield: 181 mg, 90%. Data for 4c: 1H
NMR (400 MHz, CDCl3) δ 7.08 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.6 Hz, 2H), 5.37
(br, 1H), 3.79 (s, 3H), 3.18 (s, 2H), 2.00 (m, 2H), 1.85 (m, 2H), 1.56 (m, 4H) ppm; 13C NMR (100 MHz,
CDCl3) δ 157.7, 137.6, 132.4, 129.8, 122.6, 113.5, 55.2, 43.7, 28.0, 25.3, 22.9, 22.4 ppm; GC-MS m/z = 202
(M+); Anal. Calcd for C14H18O: C, 83.12; H, 8.97. Found: C, 83.43; H, 8.69.
Table 1, compound 4d. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5
mol %), cyclohexene (1.0 mmol) and (±)-1-(4-methoxyphenyl)ethanol (1.1 mmol)
was stirred for 5 h at 75 °C. The product (±)-4d was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 250:1 to 4:1). Yield: 197 mg,
91 %. Data for (±)-4d: 1H NMR (400 MHz, CDCl3) δ 7.04 (d, J = 8.8 Hz, 2H),
6.75 (d, J = 8.8 Hz, 2H), 5.49 (br, 1H), 3.72 (s, 3H), 3.17 (q, J = 7.0 Hz, 1H), 1.97
(m, 2H), 1.59 (m, 2H), 1.46 (m, 4H), 1.24 (d, J = 7.0 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 158.4, 141.5,

S9
138.1, 129.4, 128.3, 120.4, 113.3, 55.2, 40.8, 27.0, 25.3, 23.0, 22.6, 19.8 ppm; GC-MS m/z = 216 (M+); Anal.
Calcd for C15H20O: C, 83.28; H, 9.32. Found: C, 83.12; H, 8.97.
Table 1, compound 4e. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5 mol %),
cyclopentene (1.0 mmol) and 4-methylbenzyl alcohol (1.1 mmol) was stirred for 4 h at
75 °C. The product 4e was isolated by a column chromatography on silica gel
(hexanes/EtOAc = 250:1 to 40:1). Yield: 158 mg, 92%). Data for 4e: 1H NMR (400
MHz, CDCl3) δ 7.08 (dd, J = 8.0, 3.2 Hz, 4H), 5.34 (m, 1H), 3.35 (s, 2H), 2.33 (s, 3H), 2.32 (m, 2H), 2.19 (m,
2H), 1.85 (m, 2H) ppm; 13C NMR (100 MHz, CDCl3) δ 144.0, 137.0, 135.3, 128.9, 128.7, 125.2, 37.5, 34.8,
32.4, 23.5, 21.0 ppm; GC-MS m/z = 172 (M+); Anal. Calcd for C13H16: C, 90.64; H, 9.36. Found: C, 90.43; H,
9.41.
Table 1, compound 4f. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5
mol %), cycloheptene (1.0 mmol) and 4-methoxybenzyl alcohol (1.1 mmol) was
stirred for 4 h at 75 °C. The product 4f was isolated by a column chromatography
on silica gel (hexanes/EtOAc = 250:1 to 4:1). Yield: 181 mg, 84%. Data for 4f: 1H NMR (400 MHz, CDCl3) δ 7.02 (d, J = 8.8 Hz, 2H), 6.75 (d, J = 8.8 Hz, 2H),
5.54 (t, J = 6.4 Hz, 1H), 3.72 (s, 3H), 3.15 (s, 2H), 2.03 (q, J = 6.4 Hz, 2H), 1.95
(m, 2H), 1.62 (m, 2H), 1.40 (m, 2H), 1.29 (m, 2H) ppm; 13C NMR (100 MHz, CDCl3) δ 157.7, 143.9, 132.4,
130.0, 127.7, 113.5, 55.2, 45.5, 32.5, 32.4, 28.4, 27.4, 26.8 ppm; GC-MS m/z = 216 (M+); Anal. Calcd for
C15H20O: C, 83.28; H, 9.32; O, 7.40. Found: C, 83.17; H, 9.41.
Table 1, compound 4g. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5
mol %), cyclooctene (1.0 mmol) and 4-methoxybenzyl alcohol (1.1 mmol) was
stirred for 4 h at 75 °C. The product 4g was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 250:1 to 4:1). Yield: 200 mg,
87%. For 4g: 1H NMR (400 MHz, CDCl3) δ 7.02 (d, J = 8.6 Hz, 2H), 6.75 (d, J
= 8.6 Hz, 2H), 5.28 (t, J = 8.0 Hz, 1H), 3.72 (s, 3H), 3.16 (s, 2H), 2.00 (m, 4H), 1.38 (m, 8H) ppm; 13C NMR
(100 MHz, CDCl3) δ 157.8, 140.5, 132.5, 130.0, 129.7, 125.5, 113.5, 55.2, 43.2, 30.0, 28.5, 28.4, 26.5, 26.4,
26.3 ppm; GC-MS m/z = 230 (M+); Anal. Calcd for C16H22O: C, 83.43; H, 9.63. Found: C, 83.11; H, 9.97.

S10
Table 1, compound 5a. A C6H5Cl (2 mL) solution of complex 1 (3 mg, 0.5 mol %),
indene (1.0 mmol) and ethanol (1.1 mmol) was stirred for 5 h at 90 °C. The product 5a
was isolated by a column chromatography on silica gel (hexanes/EtOAc = 250:1 to 40:1).
Yield: 120 mg, 84%. Data for 5a: 1H NMR (400 MHz, CDCl3) δ 7.40-7.05 (m, 4H), 6.48
(m, 1H), 3.28 (s, 2H), 2.47 (q, J = 7.2 Hz, 2H), 1.20 (t, J = 7.2 Hz, 3H), ppm; 13C NMR
(100 MHz, CDCl3) δ 152.6, 146.0, 143.3, 126.1, 125.3, 123.5, 123.4, 120.0, 41.1, 24.8, 14.1 ppm; GC-MS m/z
= 144 (M+). The 1H and 13C NMR spectral data are in good agreement with the literature data (35).
Table 1, compound 5b. A C6H5Cl (2 mL) solution of complex 1 (3 mg, 0.5
mol %), indene (1.0 mmol) and 1-hexanol (1.1 mmol) was stirred for 5 h at
90 °C. The product 5b was isolated by a column chromatography on silica gel
(hexanes/EtOAc = 250:1 to 40:1). Yield: 142 mg, 71%. For 5b: 1H NMR (400
MHz, CDCl3) δ 7.21 (d, J = 7.2 Hz, 1H), 7.13 (d, J = 7.2 Hz, 1H), 7.04 (t, J =
7.2 Hz, 1H), 6.92 (t, J = 7.2 Hz, 1H), 6.40 (s, 1H), 3.21 (s, 2H), 2.45 (t, J = 7.2 Hz, 2H), 1.62-1.53 (m, 2H),
1.40-1.25 (m, 6H), 0.94 (t, J = 7.2 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 149.9, 145.8, 143.2, 127.1,
126.2, 123.4, 123.2, 119.5, 41.2, 32.1, 31.6, 30.5, 29.4, 22.8, 14.0 ppm; GC-MS m/z = 200 (M+); Anal. Calcd
for C15H20: C, 89.94; H, 10.06. Found: C, 89.59; H, 10.37.
Table 1, compound 5c. A C6H5Cl (2 mL) solution of complex 1 (3 mg, 0.5
mol %), indene (1.0 mmol) and 2-ethyl-1-hexanol (1.1 mmol) was stirred for 5
h at 90 °C. The product 5c was isolated by a column chromatography on silica
gel (hexanes/EtOAc = 250:1 to 40:1). Yield: 130 mg, 57%. Data for (±)-5c: 1H
NMR (400 MHz, CDCl3) δ 7.19 (d, J = 7.0 Hz, 1H), 7.12 (d, J = 7.0 Hz, 1H),
7.05 (t, J = 7.2 Hz, 1H), 6.90 (t, J = 7.2 Hz, 1H), 6.35 (s, 1H), 3.19 (s, 2H), 2.0 (dd, J = 7.2, 2.8 Hz, 2H), 1.58
(m, 1H), 1.40-1.20 (m, 8H), 0.87 (m, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 145.7, 145.1, 143.8, 127.1,
126.0, 123.9, 123.0 119.5, 41.9, 39.8, 33.6, 32.4, 28.8, 25.5, 23.1, 14.2, 11.1 ppm; GC-MS m/z = 228 (M+);
Anal. Calcd for C17H24: C, 89.41; H, 10.59. Found: C, 89.43; H, 10.62.
Table 1, compound 5d. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5 mol %),
indene (1.0 mmol) and (R)-PhCH(CH3)OH (1.1 mmol) was stirred for 3 h at 75 °C.
The product 5d was isolated by a column chromatography on silica gel
(hexanes/EtOAc = 250:1 to 10:1). Yield: 189 mg, 86%. Data for (±)-5d: 1H NMR

S11
(400 MHz, CDCl3) δ 7.30 (m, 4H), 7.24 (m, 4H), 7.10 (td, J = 7.2, 1.0 Hz, 1H), 6.60 (s, 1H), 3.92 (q, J = 7.2 Hz,
1H), 3.23 (s, 2H), 1.60 (d, J = 7.2 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 154.4, 145.7, 145.2, 143.4,
128.4, 127.5, 126.3, 126.2, 123.9, 123.5, 120.4, 41.7, 40.1, 21.2 ppm; GC-MS m/z = 220 (M+); Anal. Calcd for
C17H16: C, 92.68; H, 7.32. Found: C, 92.18; H, 7.49.
Table 1, compound 5e. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5
mol %), indene (1.0 mmol) and (±)-1-(4-methoxyphenyl)ethanol (1.1 mmol)
was stirred for 3 h at 75 °C. The product 5e was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 40:1 to 4:1). Yield: 242 mg,
97%. Data for (±)-5e: 1H NMR (400 MHz, CDCl3) δ 7.32 (t, J = 6.0 Hz, 2H),
7.23 (t, J = 7.6 Hz, 1H), 7.16 (d, J = 8.6 Hz, 2H), 7.10 (t, J = 7.6 Hz, 1H),
6.85 (d, J = 8.6 Hz, 2H), 6.67 (s, 1H), 3.88 (q, J = 7.6 Hz, 1H), 3.80 (s, 3H), 3.23 (s, 2H), 1.58 (d, J = 7.2 Hz,
3H) ppm; 13C NMR (100 MHz, CDCl3) δ 158.0, 154.9, 145.2, 143.4, 137.9, 128.3, 126.3, 125.8, 123.9, 123.5,
120.3, 113.8, 55.3, 40.9, 40.1, 21.3 ppm; GC-MS m/z = 250 (M+); Anal. Calcd for C18H18O: C, 86.36; H, 7.25.
Found: C, 86.17; H, 7.45.
Table 1, compound 5f. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5
mol %), indene (1.0 mmol) and 4-methoxybenzyl alcohol (1.1 mmol) was
stirred for 2 h at 75 °C. The product 5f was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 40:1 to 4:1). Yield: 224 mg,
95%. Data for 5f: 1H NMR (400 MHz, CDCl3) δ 7.24 (d, J = 7.2 Hz, 1H),
7.17 (d, J = 7.2 Hz, 1H), 7.11 (t, J = 7.2 Hz, 1H), 7.05 (d, J = 8.6 Hz, 2H),
7.00 (t, J = 7.2 Hz, 1H), 6.75 (d, J = 8.6 Hz, 2H), 6.39 (s, 1H), 3.69 (s, 3H), 3.65 (s, 2H), 3.16 (s, 2H) ppm; 13C
NMR (100 MHz, CDCl3) δ 158.1, 149.9, 145.4, 143.5, 132.1, 129.8, 127.6, 126.3, 123.9, 123.5, 120.5, 113.9,
55.3, 40.8, 37.1 ppm; GC-MS m/z = 236 (M+); Anal. Calcd for C17H16O: C, 86.40; H, 6.82. Found: C, 86.42; H,
6.93.
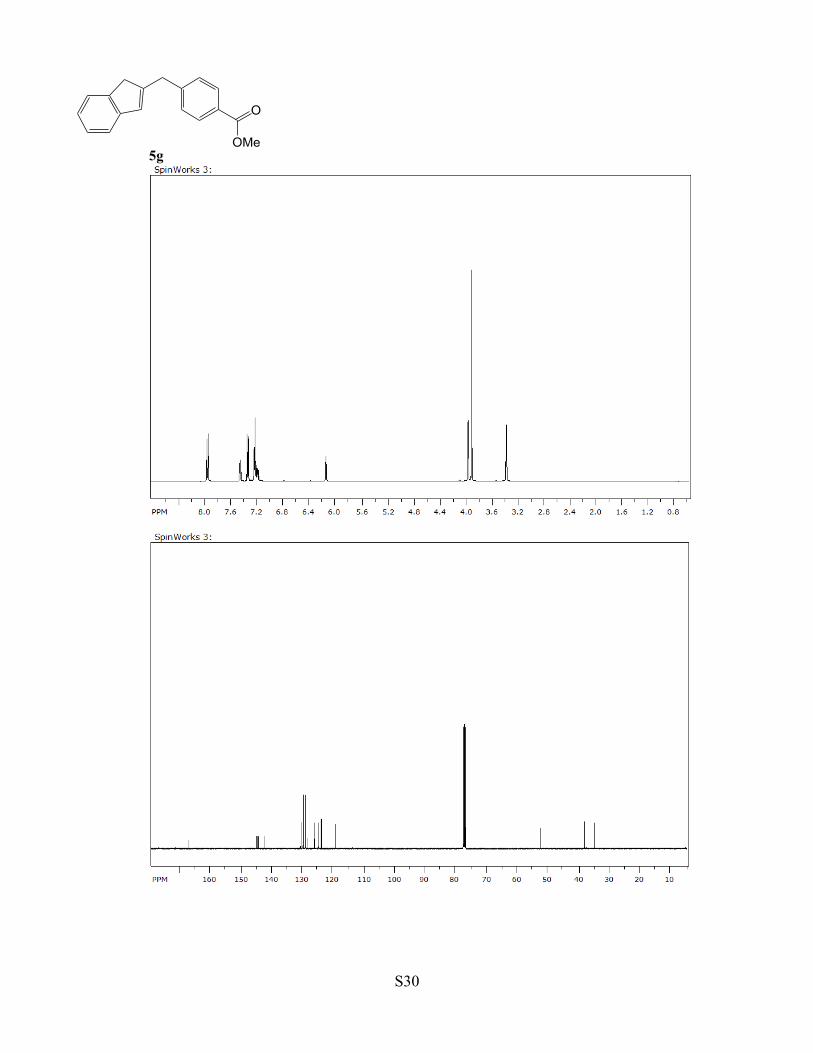
Table 1, compound 5g. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5
mol %), indene (1.0 mmol) and methyl-4-(hydroxymethyl)benzoate (1.1
mmol) was stirred for 3 h at 75 °C. The product 5g was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 40:1 to 1:1). Yield: 240 mg,
91%. Data for 5g: 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.0 Hz, 2H),

S12
7.47 (d, J = 7.2 Hz, 1H), 7.35 (d, J = 8.0 Hz, 2H), 7.26-7.17 (m, 3H), 6.15 (s, 1H), 3.96 (s, 2H), 3.90 (s, 3H),
3.36 (s, 2H) ppm; 13C NMR (100 MHz, CDCl3) δ 167.1, 144.9, 144.8, 144.5, 142.5, 130.5, 129.8, 128.9, 128.2,
126.1, 124.8, 123.8, 119.3, 52.0, 37.9, 34.5 ppm; GC-MS m/z = 264 (M+); Anal. Calcd for C18H16O2: C, 81.79;
H, 6.10. Found: C, 81.62; H, 6.49.
Table 1, compound 6a. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5
mol %), an N-methylindole (1.0 mmol) and 1-hexanol (1.1 mmol) was stirred
for 3 h at 75 °C. The product 6a was isolated by a column chromatography on
silica gel (hexanes/EtOAc = 40:1 to 4:1). Yield: 206 mg, 96%. Data for 6a: 1H
NMR (400 MHz, CDCl3) δ 7.65 (d, J = 8.1 Hz, 1H), 7.32 (d, J = 8.1 Hz, 1H),
7.26 (t, J = 8.1 Hz, 1H), 7.15 (t, J = 7.2 Hz, 1H), 6.86 (s, 1H), 3.77 (s, 3H),
2.78 (t, J = 7.8 Hz, 2H), 1.75 (p, J = 7.8 Hz, 2H), 1.50-1.34 (m, 6H), 0.95 (t, J = 7.0 Hz, 3H) ppm; 13C NMR
(100 MHz, CDCl3) δ 137.0, 128.0, 125.9, 121.4, 119.1, 118.4, 115.7, 109.1, 32.6, 31.9, 30.5, 29.4, 25.1, 22.8,
14.2 ppm; GC-MS m/z = 215 (M+); Anal. Calcd for C15H21N: C, 83.67; H, 9.83; N, 6.50. Found: C, 83.48; H,
9.87; N, 6.59.
Table 1, compound 6b. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5 mol %), an
N-methylindole (1.0 mmol) and 2-methyl-1-propanol (1.1 mmol) was stirred for 3 h at
75 °C. The product 6b was isolated by a column chromatography on silica gel
(hexanes/EtOAc = 40:1 to 4:1). Yield: 178 mg, 95%. Data for 6b: 1H NMR (400 MHz,
CDCl3) δ 7.50 (d, J = 7.8 Hz, 1H), 7.19 (d, J = 8.1 Hz, 1H), 7.12 (t, J = 8.0 Hz, 1H),
7.01 (t, J = 7.8 Hz, 1H), 6.72 (s, 1H), 3.65 (s, 3H), 2.52 (d, J = 7.0 Hz, 2H), 1.88 (m,
1H), 0.87 (d, J = 6.6 Hz, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 136.9, 128.4, 126.8, 121.3, 119.3, 118.4,
114.4, 109.1, 34.5, 32.6, 29.4, 22.8 ppm; GC-MS m/z = 187 (M+); Anal. Calcd for C13H17N: C, 83.37; H, 9.15;
N, 7.48. Found: C, 83.42; H, 9.43; N, 7.11.
Table 1, compound 6c. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5
mol %), an N-methylindole (1.0 mmol) and 4-methoxybenzyl alcohol (1.1
mmol) was stirred for 2 h at 75 °C. The product 6c was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 40:1 to 4:1). Yield: 241 mg,
96%. Data for 6c: 1H NMR (400 MHz, CDCl3) δ 7.25 (d, J = 7.8 Hz, 1H),
7.02 (d, J = 7.0 Hz, 1H), 6.92 (m, 3H), 6.80 (d, J = 7.2 Hz, 1H), 6.56 (d, J =

S13
8.6 Hz, 2H), 6.45 (s, 1H), 3.78 (s, 2H), 3.50 (s, 3H), 3.43 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 157.8,
137.2, 133.5, 129.5, 127.8, 127.0, 121.6, 119.3, 118.8, 114.8, 113.8, 109.2, 55.3, 32.6, 30.7 ppm; GC-MS m/z =
251 (M+); Anal. Calcd for C17H17NO: C, 81.24; H, 6.82; N, 5.57. Found: C, 81.40; H, 6.52; N, 5.66.
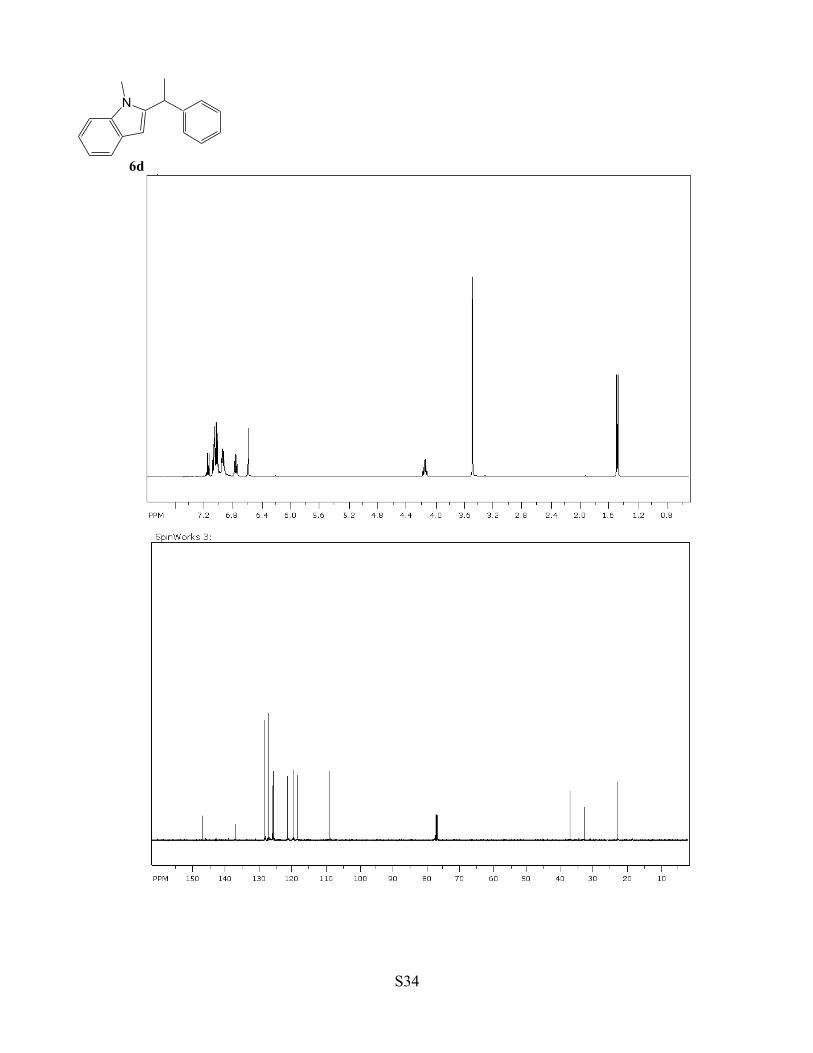
Table 1, compound 6d. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5 mol %),
an N-methylindole (1.0 mmol) and (±)-PhCH(CH3)OH (1.1 mmol) was stirred for 2
h at 75 °C. The product 6d was isolated by a column chromatography on silica gel
(hexanes/EtOAc = 40:1 to 4:1). Yield: 221 mg, 94%. Data for (±)-6d: 1H NMR (400
MHz, CDCl3) δ 7.15 (d, J = 7.8 Hz, 1H), 7.10-7.00 (m, 5H), 6.95 (m, 2H), 6.77 (t, J
= 7.8 Hz, 1H), 6.60 (s, 1H), 4.14 (q, J = 7.2 Hz, 1H), 3.49 (s, 3H), 1.47 (d, J = 7.2
Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 147.0, 137.4, 128.4, 127.5, 126.0, 125.9, 121.6, 119.8, 118.7,
109.2, 36.9, 32.7, 22.6 ppm; GC-MS m/z = 235 (M+); Anal. Calcd for C17H17N: C, 86.77; H, 7.28; N, 5.95.
Found: C, 86.43; H, 7.21; N, 5.99.
Table 1, compound 7a. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5
mol %), benzopyrene (1.0 mmol) and 4-methoxybenzyl alcohol (1.1 mmol)
was stirred for 2 h at 75 °C. The product 7a was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 40:1 to 4:1). Yield: 226 mg,
95%. Data for 7a: 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 7.6 Hz, 1H),
7.32 (d, J = 7.6 Hz, 1H), 7.17-7.05 (m, 4H), 6.78 (d, J = 8.6 Hz, 2H), 6.25 (s, 1H), 3.96 (s, 2H), 3.70 (s, 3H)
ppm; 13C NMR (100 MHz, CDCl3) δ 158.5, 158.3, 154.9, 129.9, 129.3, 123.4, 122.5, 120.4, 114.0, 110.9, 103.1,
55.3, 34.1 ppm; GC-MS m/z = 238 (M+); Anal. Calcd for C16H14O2: C, 80.65; H, 5.92. Found: C, 80.33; H, 5.82.
Table 1, compound 7b. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5
mol %), benzopyrene (1.0 mmol) and (±)-1-(4-methylphenyl)ethanol (1.1 mmol)
was stirred for 2 h at 75 °C. The product 7b was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 40:1 to 4:1). Yield: 212 mg,
91%. For (±)-7b: 1H NMR (400 MHz, CDCl3) δ 7.50 (m, 1H), 7.42 (m, 1H),
7.27-7.05 (m, 6H), 6.45 (s, 1H), 4.24 (q, J = 7.2 Hz, 1H), 2.36 (s, 3H), 1.70 (d, J
= 7.2 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 162.4, 154.8, 140.4, 136.4, 129.3, 128.6, 127.3, 123.3,
122.4, 120.4, 111.0, 101.9, 39.3, 21.1, 20.4 ppm; GC-MS m/z = 236 (M+); Anal. Calcd for C17H16O: C, 86.40;
H, 6.82. Found: C, 86.31; H, 6.96.

S14
Table 1, compound 8. A CH2Cl2 (2 mL) solution of complex 1 (3 mg, 0.5
mol %), indoline (1.0 mmol) and 4-methoxybenzyl alcohol (1.1 mmol) was
stirred for 3 h at 75 °C. The product 8 was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 40:1 to 4:1). Yield: 216 mg,
91%. Data for 8: 1H NMR (400 MHz, CDCl3) δ 7.67 (br, 1H), 7.46 (d, J = 7.4
Hz, 1H), 7.16 (d, J = 7.6 Hz, 1H), 7.09 (d, J = 8.6 Hz, 2H), 7.00 (m, 2H), 6.78 (d, J = 8.6 Hz, 2H), 6.23 (s, 1H),
4.00 (s, 2H), 3.72 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 158.4, 138.3, 136.2, 129.8, 128.6, 121.2, 119.9,
119.7, 114.1, 110.4, 100.8, 55.3, 33.8 ppm; GC-MS m/z = 237 (M+); Anal. Calcd for C16H15NO: C, 80.98; H,
6.37; N, 5.90. Found: C, 80.83; H, 6.55; N, 5.92.
Table 1, compound 9a. A C6H5Cl (2 mL) solution of complex 1 (3 mg,
0.5 mol %), 4-methylstyrene (1.0 mmol) and 1-hexanol (1.1 mmol) was
stirred for 5 h at 110 °C. The product 9a was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 250:1 to 20:1). Yield:
170 mg, 84 %, (E/Z = 24:1). Data for 9a: 1H NMR (400 MHz, CDCl3) δ 7.19 (q, J = 8.4 Hz, 4H), 6.36 (d, J =
15.8 Hz, 1H), 5.60 (dt, J = 15.8, 7.0 Hz, 1H), 2.33 (s, 3H), 2.30 (m, 2H), 1.44 (m, 2H), 1.38-1.20 (m, 6H), 0.87
(t, J = 6.6 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 136.3, 135.3, 132.79, 129.05, 129.0, 128.8, 31.8, 30.1
ppm; GC-MS m/z = 202 (M+); Anal. Calcd for C15H22: C, 89.04; H, 10.96. Found: C, 89.41; H, 10.84.
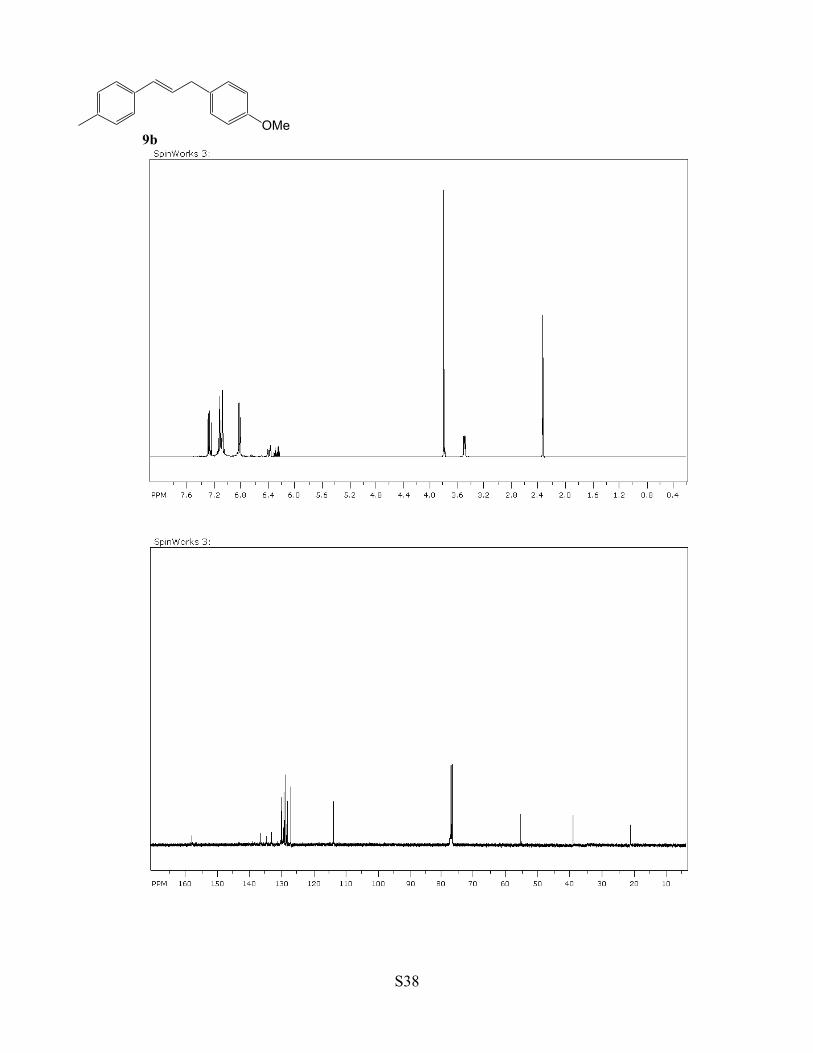
Table 1, compound 9b. A C6H5Cl (2 mL) solution of complex 1 (3 mg,
0.5 mol %), 4-methylstyrene (1.0 mmol) and 4-methoxybenzyl alcohol
(1.1 mmol) was stirred for 5 h at 90 °C. The product 9b was isolated by a
column chromatography on silica gel (hexanes/EtOAc = 40:1 to 4:1).
Yield: 217 mg, 91% (E/Z = 22:1). Data for 9b: 1H NMR (400 MHz,
CDCl3) δ 7.29 (d, J = 8.6 Hz, 2H), 7.19-7.10 (m, 4H), 6.83 (d, J = 8.6 Hz, 2H), 6.40 (d, J = 15.6 Hz, 1H), 6.25
(m, 1H), 3.80 (s, 3H), 3.48 (d, J = 6.8 Hz, 2H), 2.32 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 158.2, 136.9,
134.9, 132.5, 130.7, 129.7, 129.3, 128.7, 126.1, 114.0, 55.4, 38.6, 21.3 ppm; GC-MS m/z = 238 (M+). The 1H
and 13C NMR spectral data are in good agreement with the literature data (36).
Table 1, compound 9c. A C6H5Cl (2 mL) solution of complex 1 (3 mg, 0.5 mol %), 4-chlorostyrene (1.0
mmol) and 1-hexanol (1.1 mmol) was stirred for 8 h at 110 °C. The product 9c was isolated by a column

S15
chromatography on silica gel (hexanes/EtOAc = 250:1 to 20:1). Yield:
173 mg, 78% (E/Z = 25:1). Data for 9c: 1H NMR (400 MHz,
CDCl3) δ 7.26 (s, 4H), 6.32 (dt, J = 15.6, 1.2 Hz, 1H), 6.21 (dt, J =
15.6, 6.8 Hz, 1H), 2.20 (q, J = 6.8 Hz, 2H), 1.50 (m, 2H), 1.40-1.20
(m, 6H), 0.90 (t, J = 6.8 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ
136.5, 132.2, 132.0, 128.5, 128.4, 127.1, 33.0, 31.7, 29.2, 28.9, 22.6, 14.1 ppm; GC-MS m/z = 222 (M+). The 1H and 13C NMR spectral data are in good agreement with the literature data (37).
Table 1, compound 9d. A C6H5Cl (2 mL) solution of complex 1 (3 mg,
0.5 mol %), 4-chlorostyrene (1.0 mmol) and 4-methoxybenzyl alcohol
(1.1 mmol) was stirred for 6 h at 90 °C. The product 9d was isolated by
a column chromatography on silica gel (hexanes/EtOAc = 250:1 to
20:1). Yield: 214 mg, 83% (E/Z = 23:1). Data for 9d: 1H NMR (400
MHz, CDCl3) δ 7.35-7.20 (m, 4H), 7.18 (d, J = 8.6 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 6.41 (d, J = 15.9 Hz, 1H),
6.38-6.31 (m, 1H), 3.83 (s, 3H), 3.53 (d, J = 6.0 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3) δ 158.1, 136.0,
132.6, 131.8, 130.4, 129.6, 129.5, 128.6, 127.3, 113.9, 55.3, 38.4 ppm; GC-MS m/z = 258 (M+). The 1H and 13C
NMR spectral data are in good agreement with the literature data (36).
Table 1, compounds 11 and 12. A C6H5Cl (2 mL) solution of complex 1 (3
mg, 0.5 mol %), allybenzene (1.0 mmol) and 1-hexanol (1.1 mmol) was
stirred for 5 h at 90 °C. The product was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 250:1 to 4:1). Combined
yield of 11 and 12: 214 mg, 83% (11:12 = 5:1). Data for 11: 1H NMR (400
MHz, CDCl3) δ 7.28-7.05 (m, 7H), 6.76 (d, J = 8.6 Hz, 2H), 6.34 (s, 1H), 3.72 (s, 3H), 3.41 (s, 2H), 1.71 (s, 3H)
ppm; 13C NMR (100 MHz, CDCl3) δ 157.9, 140.0, 138.5, 130.0, 129.9, 128.9, 128.8, 128.3, 128.0, 126.4, 126.2,
113.7, 55.3, 47.1, 17.6 ppm; GC-MS m/z = 238 (M+); GC-MS m/z = 238 (M+); Anal. Calcd for C17H18O: C,
85.67; H, 7.61. Found: C, 85.42; H, 7.49.
Data for 12: 1H NMR (400 MHz, CDCl3) δ 7.38-7.16 (m, 5H), 7.09 (d, J = 8.6 Hz,
2H), 6.85 (d, J = 8.6 Hz, 2H), 5.48 (q, J = 6.8 Hz, 1H), 3.82 (s, 3H), 3.24 (s, 2H),
1.56 (d, J = 6.8 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 158.0, 140.4, 138.5,

S16
130.0, 129.7, 128.7, 128.6, 128.1, 128.0, 126.4, 124.8, 114.2, 55.1, 43.2, 12.8 ppm; GC-MS m/z = 238 (M+);
Anal. Calcd for C17H18O: C, 85.67; H, 7.61. Found: C, 85.48; H, 7.53.
Table 1, compound 13. A C6H5Cl (2 mL) solution of complex 1 (3 mg, 0.5
mol %), vinylcyclohexane (1.0 mmol) and 4-methoxybenzyl alcohol (1.1 mmol)
was stirred for 5 h at 90 °C. The product 13 was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 250:1 to 20:1). Yield: 205 mg,
89%. Data for 13: 1H NMR (400 MHz, CDCl3) δ 7.00 (d, J = 8.6 Hz, 2H), 6.73 (d,
J = 8.6 Hz, 2H), 3.71 (s, 3H), 3.22 (s, 2H), 2.08 (q, J = 7.6 Hz, 2H), 1.96 (m, 2H),
1.76 (m, 2H), 1.48 (m, 4H), 0.92 (t, J = 7.6 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 157.6, 133.5, 133.1,
129.8, 129.2, 128.2, 113.5, 55.2, 37.6, 29.3, 28.9, 26.4, 23.4, 23.3, 13.2 ppm; GC-MS m/z = 230 (M+); Anal.
Calcd for C16H22O: C, 83.43; H, 9.63. Found: C, 83.55; H, 9.38.
Table 1, compound 14. A C6H5Cl (8 mL) solution of complex 1 (6 mg, 0.5 mol %) and
geraniol (2.0 mmol) was stirred for 8 h at 110 °C. The product 14 was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 250:1 to 20:1). Yield: 190 mg, 71%. Data
for 14: 1H NMR (400 MHz, CDCl3) δ 7.20-7.10 (m, 4H), 2.95 (m, 1H), 2.34 (s, 3H) 1.30
(d, J = 6.8 Hz, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 145.9, 135.2, 129.0, 126.3, 33.8,
24.2, 21.0 ppm. The 1H and 13C NMR spectral data are in good agreement with the literature data (38).
Table 1, compound 15. A C6H5Cl (2 mL) solution of complex 1 (3 mg, 0.5
mol %), indene (1.0 mmol) and (R)-2-phenyl-1-propanol (1.1 mmol) was
stirred for 3 h at 90 °C. The product 15 was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 250:1 to 10:1). Yield: 204 mg,
87%. Data for (R)-15: 1H NMR (400 MHz, CDCl3) δ 7.28-7.15 (m, 8H), 7.10
(m, 1H), 6.35 (s, 1H), 3.21 (s, 2H), 2.95 (m, 1H), 2.48 (dd, J = 10.0, 6.4 Hz, 1H), 2.38 (dd, J = 10.0, 6.4 Hz,
1H), 1.38 (d, J = 7.0 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 154.3, 143.9, 143.4, 140.6, 128.4, 128.3,
127.4, 126.6, 125.8, 124.9, 114.5, 45.8, 40.7, 37.4, 18.4 ppm; GC-MS m/z = 234 (M+); Anal. Calcd for C18H18:
C, 92.26; H, 7.74. Found: C, 91.94; H, 7.39; [α]23D +23.2° (c = 0.5, CH2Cl2).

S17
Fig. 2, compound 16. A C6H5Cl (3 mL) solution of complex 1
(6 mg, 1.0 mol %), cholesterol (1.0 mmol) and 4-methoxybenzyl
alcohol (1.2 mmol) was stirred for 6 h at 90 °C. The product 16
was isolated by a column chromatography on silica gel
(hexanes/EtOAc = 40:1 to 4:1). Yield: 288 mg, 57% (rsm = 108
mg, 28%). For (-)-16: 1H NMR (400 MHz, CDCl3) δ 7.04 (d, J =
8.6 Hz, 2H), 6.81 (d, J = 8.6 Hz, 2H), 3.72 (s, 3H), 3.51 (m, 1H),
3.39 (d, J = 15.1 Hz, 1H), 3.19 (d, J = 15.1 Hz, 1H), 2.30-1.00
(m, 29H), 0.99 (s, 3H), 0.90 (d, J = 6.4 Hz, 3H), 0.90 (dd, J = 6.4,
2.0 Hz, 6H), 0.67 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 158.2, 139.7, 133.1, 130.7, 129.6, 114.1, 72.1,
56.7, 56.1, 55.4, 50.1, 42.3, 42.2, 39.7, 39.6, 37.3, 37.0, 36.5, 36.2, 35.8, 31.9, 31.8, 28.2, 28.0, 24.3, 23.9, 22.8,
22.5, 21.1, 19.4, 18.7, 11.8 ppm; HRMS (ESI-TOF) m/z = 506 (M+); Anal. Calcd for C35H54O2: C, 82.95; H,
10.74. Found: C, 82.87; H, 10.63; [α]23D -45.1° (c = 0.5, CH2Cl2).
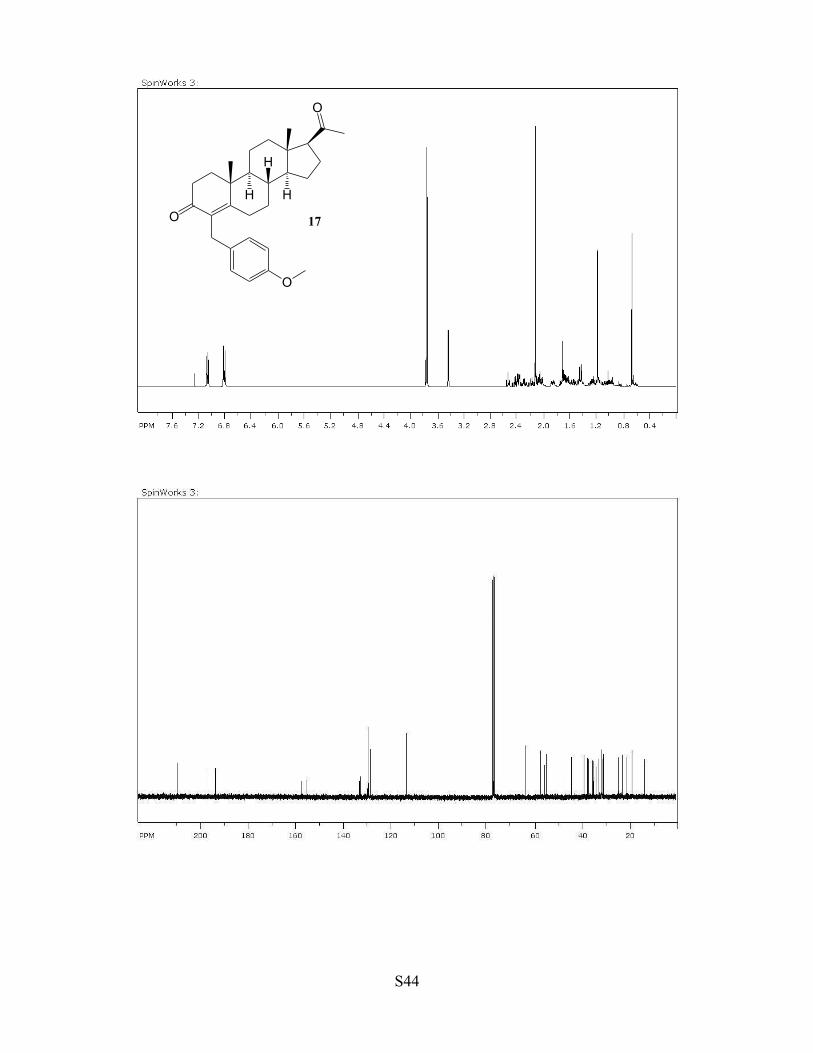
Fig. 2, compound 17. A C6H5Cl (3 mL) solution of complex 1 (6 mg, 1.0
mol %), progesterone (1.0 mmol) and 4-methoxybenzyl alcohol (1.2 mmol)
was stirred for 6 h at 90 °C. The product 17 was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 40:1 to 1:1). Yield: 352 mg,
81% (rsm = 35 mg, 11%). Data for (+)-17: 1H NMR (400 MHz, CDCl3) δ
7.05 (d, J = 8.6 Hz, 2H), 6.77 (d, J = 8.6 Hz, 2H), 3.74 (s, 3H), 3.42 (s, 2H),
2.55 (t, J = 9.1 Hz, 1H), 2.40-0.8 (m, 19H), 2.13 (s, 3H), 1.18 (s, 3H), 0.66 (s,
3H) ppm; 13C NMR (100 MHz, CDCl3) δ 209.2, 186.3, 158.3, 133.1, 132.9,
129.8, 129.0, 113.8, 63.5, 57.8, 55.9, 55.2, 43.9, 39.9, 38.6, 38.4, 35.9, 35.5,
33.9, 32.5, 31.9, 31.5, 24.3, 22.8, 21.0, 18.9, 13.4 ppm; HRMS (ESI-TOF) m/z = 434 (M+); Anal. Calcd for
C29H38O3: C, 80.14; H, 8.81. Found: C, 80.50; H, 8.39; [α]23D +208.3° (c = 0.5, CH2Cl2).
Fig. 2, compound 18. A C6H5Cl (3 mL) solution of complex 1 (6 mg, 1.0 mol %),
N-methoxycarbonyl-L-tryptophan (1.0 mmol) and 4-methoxybenzyl alcohol (1.2
mmol) was stirred for 4 h at 90 °C. The product 18 was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 40:1 to 1:2). Yield: 333 mg, 84%
(rsm = 19 mg, 7%). Data for (-)-18: 1H NMR (400 MHz, CDCl3) δ 7.78 (br, 1H),
7.48 (d, J = 6.3 Hz, 1H), 7.19 (d, J = 6.3 Hz, 1H), 7.09 (m, 4H), 6.84 (d, J = 8.8

S18
Hz, 2H), 5.26 (d, J = 8.0 Hz, 1H), 4.67 (m, 1H), 4.01 (s, 2H), 3.79 (s, 3H), 3.65 (s, 3H), 3.63 (s, 3H), 3.33 (d, J
= 5.8 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.7, 158.5, 156.4, 135.6, 135.4, 130.1, 129.8, 128.6,
121.6, 119.6, 118.1, 114.2, 110.6, 106.2, 55.3, 54.7, 52.5, 52.3, 31.9, 27.2 ppm; HRMS (ESI-TOF) m/z = 396
(M+); Anal. Calcd for C22H24N2O5: C, 66.65; H, 6.10; N, 7.07. Found: C, 66.58; H, 6.43; N, 7.31; [α]23D -11.3°
(c = 0.5, CH2Cl2).
Fig. 2, compound 19. A C6H5Cl (3 mL) solution of complex 1
(6 mg, 1.0 mol %), (-)-strychnine (1.0 mmol) and 4-
methoxybenzyl alcohol (1.2 mmol) was stirred for 5 h at 90 °C.
The product 19 was isolated by a column chromatography on
silica gel (hexanes/EtOAc = 20:1 to 1:2). Yield: 331 mg, 73%
(rsm = 40 mg, 12%). Data for (-)-19: 1H NMR (400 MHz,
CDCl3) δ 8.02 (d, J = 8.0 Hz, 1H), 7.32 (d, J = 8.8 Hz, 2H), 7.17
(d, J = 8.0 Hz, 1H), 7.08 (d, J = 7.4 Hz, 1H), 7.01 (d, J = 7.4 Hz,
1H), 6.87 (d, J = 8.8 Hz, 2H), 4.21 (dd, J = 8.4, 3.3 Hz, 1H), 4.04 (m, 2H), 3.86 (m, 1H), 3.78 (s, 3H), 3.74 (d, J
= 10.4 Hz, 1H), 3.61 (d, J = 15.1 Hz, 1H), 3.20 (s, 2H), 3.12-2.95 (m, 3H), 2.80 (m, 1H), 2.62 (m, 2H), 2.25 (dt,
J = 14.4, 4.3 Hz, 1H), 1.80 (m, 2H), 1.36 (d, J = 14.4 Hz, 1H), 1.19 (m, 1H) ppm; 13C NMR (100 MHz,
CDCl3) δ 168.6, 158.7, 140.0, 138.3, 131.1, 129.8, 129.5, 128.5, 127.7, 127.1, 124.1, 122.2, 113.9, 78.2, 67.8,
60.1, 60.0, 55.3, 52.6, 51.8, 50.3, 48.1, 42.8, 42.4, 40.3, 32.8, 26.8 ppm; HRMS (ESI-TOF) m/z = 454 (M+);
Anal. Calcd for C29H30N2O3: C, 76.63; H, 6.65; N, 6.16. Found: C, 76.76; H, 6.69; N, 6.42; [α]23D -124.6° (c =
0.5, CH2Cl2).
Fig. 2, compound 20. A C6H5Cl (3 mL) solution of complex 1 (6 mg, 1.0
mol %), quinine (1.0 mmol) and 4-methoxybenzyl alcohol (1.2 mmol) was
stirred for 6 h at 110 °C. The product 20 was isolated by a column
chromatography on silica gel (hexanes/EtOAc = 20:1 to 1:2). Yield: 259 mg, 58%
(rsm = 78 mg, 24%). Data for (-)-20: 1H NMR (400 MHz, CDCl3) δ 8.66 (d, J =
4.5 Hz, 1H), 7.99 (d, J = 9.1 Hz, 1H), 7.84 (d, J = 2.8 Hz, 1H), 7.46 (d, J = 8.8
Hz, 2H), 7.36 (dd, J = 9.1, 2.8 Hz, 1H), 7.28 (d, J = 4.5 Hz, 1H), 6.90 (d, J = 8.8
Hz, 2H), 6.22 (br, 1H), 5.45 (d, J = 3.1 Hz, 1H), 4.02 (s, 3H), 3.84 (s, 3H), 3.68
(m, 2H), 3.56 (m, 1H), 3.36 (m, 3H), 2.61 (m, 2H), 2.21 (m, 1H), 2.15 (m, 1H),
1.71 (m, 2H), 1.48 (d, J = 7.0 Hz, 3H), 1.32 (m, 2H) ppm; 13C NMR (100 MHz,

S19
CDCl3) δ 158.6, 157.6, 148.7, 147.1, 143.7, 132.1, 131.0, 129.6, 126.5, 121.2, 118.5, 113.9, 104.3, 72.6, 60.1,
55.6, 55.1, 52.6, 43.2, 41.1, 39.9, 30.2, 28.8, 28.6, 21.4, 12.6 ppm; HRMS (ESI-TOF) m/z = 446 (M+); Anal.
Calcd for C28H34N2O3: C, 75.31; H, 7.67; N, 6.27. Found: C, 75.44; H, 7.53; N, 6.65; [α]23D -140.9° (c = 0.5,
CH2Cl2).
Fig. 2, compound 21. A C6H5Cl (3 mL) solution of complex 1 (3 mg, 1.0
mol %), (-)-sinomenine (0.5 mmol) and 4-methoxybenzyl alcohol (0.65
mmol) was stirred for 6 h at 90 °C. The product 21 was isolated by a
column chromatography on silica gel (hexanes/EtOAc = 20:1 to 1:2).
Yield: 139 mg, 62% (rsm = 78 mg, 21%). Data for (-)-21: 1H NMR (400
MHz, CDCl3) δ 7.34 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 6.76 (d,
J = 1.36 Hz, 1H), 6.70 (d, J = 1.36 Hz, 1H), 6.27 (dd, J = 9.6, 3.2 Hz, 1H),
5.74 (dd, J = 9.6, 3.1 Hz, 1H), 4.61 (d, J = 11.4 Hz, 1H), 4.35 (d, J = 11.4
Hz, 1H), 4.02 (d, J = 15.1 Hz, 1H), 3.87 (s, 3H), 3.80 (s, 3H), 3.60 (m,
1H), 3.43 (m, 1H), 3.35 (s, 3H), 2.56 (m, 1H), 2.51 (d, J = 15.1, 1.6 Hz, 1H), 2.40 (s, 3H), 2.01 (m, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 196.3, 158.4, 148.6, 143.1, 142.7, 132.9, 129.8, 129.5, 127.2, 121.9, 117.1,
114.3, 111.9, 109.0, 58.2, 56.4, 59.9, 55.1, 48.9, 45.1, 38.3, 32.8, 31.4, 23.1 ppm; HRMS (ESI-TOF) m/z = 447
(M+); Anal. Calcd for C27H29NO5: C, 72.46; H, 6.53; N, 3.13. Found: C, 72.38; H, 6.51; N, 3.09; [α]23D -58.7°
(c = 0.3, CH2Cl2).

S20
8. 1H and 13C NMR spectra of the products.
4b
S21
OMe 4c

S22
OMe 4d

S23
4e

S24
OMe 4f

S25
OMe 4g

S26
5c

S27
5d

S28
OMe 5e

S29
OMe 5f
S30
OMe
O
5g

S31
N
6a

S32
N
6b

S33
N
OMe
6c
S34
N
6d

S35
O
OMe
7a

S36
O
7b

S37
HN
OMe
8
S38
OMe 9b

S39
Cl OMe 9d

S40
OMe 11

S41
OMe
13

S42
(R)
(R)-15

S43
HO
OMe
16H
H
H
S44
17O
O
O
HH
H

S45
18
NH
OMe
OMe
O
HNOMe
O

S46
N
O
H
N
OOMe
H
H
19

S47
N
MeO
HO N
MeO
HH
(R)(R)
20

S48
21
MeO
HO
O
OMe
N
H
OMe

References and Notes 1. G. A. Olah, A. Molnár, Hydrocarbon Chemistry (Wiley, New York, 2003).
2. R. F. Heck, Palladium-catalyzed reactions of organic halides with olefins. Acc. Chem. Res. 12, 146 (1979). doi:10.1021/ar50136a006
3. J.-Q. Yu, Z. Shi, Eds., C–H Activation (Springer, New York, 2010).
4. S. Murai et al., Efficient catalytic addition of aromatic carbon-hydrogen bonds to olefins. Nature 366, 529 (1993). doi:10.1038/366529a0
5. D. A. Colby, R. G. Bergman, J. A. Ellman, Rhodium-catalyzed C-C bond formation via heteroatom-directed C-H bond activation. Chem. Rev. 110, 624 (2010). doi:10.1021/cr900005n Medline
6. C. S. Yeung, V. M. Dong, Catalytic dehydrogenative cross-coupling: Forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds. Chem. Rev. 111, 1215 (2011). doi:10.1021/cr100280d Medline
7. C.-J. Li, Cross-dehydrogenative coupling (CDC): Exploring C-C bond formations beyond functional group transformations. Acc. Chem. Res. 42, 335 (2009). doi:10.1021/ar800164n Medline
8. D. R. Stuart, K. Fagnou, The catalytic cross-coupling of unactivated arenes. Science 316, 1172 (2007). doi:10.1126/science.1141956 Medline
9. D.-H. Wang, K. M. Engle, B.-F. Shi, J.-Q. Yu, Ligand-enabled reactivity and selectivity in a synthetically versatile aryl C-H olefination. Science 327, 315 (2010); 10.1126/science.1182512. doi:10.1126/science.1182512 Medline
10. M. B. Smith, J. March, March’s Advanced Organic Chemistry (Wiley, New York, 2001).
11. C. S. Yi, D. W. Lee, Regioselective intermolecular coupling reaction of arylketones and alkenes involving C–H bond activation catalyzed by an in situ formed cationic ruthenium hydride complex. Organometallics 28, 4266 (2009). doi:10.1021/om900416k Medline
12. C. S. Yi, D. W. Lee, Intermolecular dehydrative coupling reaction of aryl ketones with cyclic alkenes catalyzed by a well-defined cationic ruthenium-hydride complex: A novel ketone olefination method via vinyl C−H bond activation. Organometallics 29, 1883 (2010). doi:10.1021/om100051h Medline
13. G. E. Dobereiner, R. H. Crabtree, Dehydrogenation as a substrate-activating strategy in homogeneous transition-metal catalysis. Chem. Rev. 110, 681 (2010). doi:10.1021/cr900202j Medline
14. D. V. McGrath, R. H. Grubbs, The mechanism of aqueous ruthenium(II)-catalyzed olefin isomerization. Organometallics 13, 224 (1994). doi:10.1021/om00013a035
15. F. C. Courchay, J. C. Sworen, I. Ghiviriga, K. A. Abboud, K. B. Wagener, Understanding structural isomerization during ruthenium-catalyzed olefin metathesis: A deuterium labeling study. Organometallics 25, 6074 (2006). doi:10.1021/om0509894
16. D. W. Lee, C. S. Yi, Chain-selective and regioselective ethylene and styrene dimerization reactions catalyzed by a well-defined cationic ruthenium hydride complex: New insights

on the styrene dimerization mechanism. Organometallics 29, 3413 (2010). doi:10.1021/om100468q Medline
17. M. Karplus, Contact electron-spin coupling of nuclear magnetic moments. J. Chem. Phys. 30, 11 (1959). doi:10.1063/1.1729860
18. The regioselective deuterium exchange on the C-2 carbon of indene is most consistent with the vinyl C−H activation mechanism. The selective deuterium incorporation on the C-1 and C-3 carbons of indan can readily be explained from regioselective insertion of indene to Ru−D, followed by reductive elimination with the hydride formed from the vinyl C−H activation of another indene molecule. Due to steric hindrance from the arene hydrogen, metalation and insertion of the Ru atom are expected to be favored at the C-2 carbon of indene. Previously, we also observed the formation free benzene molecule in the reactions catalyzed by 1 (12, 16).
19. R. M. Roberts, A. A. Khalaf, Friedel-Crafts Alkylation Chemistry: A Century of Discovery (Dekker, New York, 1984).
20. G. A. Olah, R. Krishnamurti, G. K. S. Prakash, in Comprehensive Organic Synthesis, B. M. Trost, I. Fleming, Eds. (Pergamon, New York, 1991), vol. 3, chap. 1.8.
21. Z.-Q. Liu et al., Iron-catalyzed stereospecific olefin synthesis by direct coupling of alcohols and alkenes with alcohols. Org. Lett. 13, 2208 (2011). doi:10.1021/ol200372y Medline
22. D. E. Frantz, D. A. Singleton, J. P. Snyder, 13C kinetic isotope effects for the addition of lithium dibutylcuprate to cyclohexenone. Reductive elimination is rate-determining. J. Am. Chem. Soc. 119, 3383 (1997). doi:10.1021/ja9636348
23. R. Gao, C. S. Yi, Regioselective formation of α-vinylpyrroles from the ruthenium-catalyzed coupling reaction of pyrroles and terminal alkynes involving C-H bond activation. J. Org. Chem. 75, 3144 (2010). doi:10.1021/jo100269y Medline
24. F. Kakiuchi, S. Murai, Catalytic C-H/olefin coupling. Acc. Chem. Res. 35, 826 (2002). doi:10.1021/ar960318p Medline
25. Transition metal–vinyl complexes have been proposed to be a key intermediate in C–H bond activation chemistry. For representative examples, see (32, 33).
26. The formation of a free benzene molecule (2%) is detected after heating the reaction mixture at 70°C for 15 min using 5 mol % of 1. Previously, we observed similar results in the reactions catalyzed by 1 (12, 16).
27. Y. Feng et al., Evidence for the net addition of arene C-H bonds across a Ru(II)-hydroxide bond. J. Am. Chem. Soc. 127, 14174 (2005). doi:10.1021/ja054101e Medline
28. T. Matsumoto, Y. Nakaya, N. Itakura, K. Tatsumi, A functional hydrogenase model: reversible interconversion of H2 and H2O by a hydroxo/sulfido-bridged dinuclear ruthenium-germanium complex. J. Am. Chem. Soc. 130, 2458 (2008). doi:10.1021/ja711325f Medline
29. D. Rondon, B. Chaudret, X. D. He, D. Labroue, Carbon-hydrogen, carbon-oxygen, and carbon-carbon bond activation by an electrophilic ruthenium complex. J. Am. Chem. Soc. 113, 5671 (1991). doi:10.1021/ja00015a022

30. D. B. Grotjahn, H. C. Lo, Ruthenium alkoxycarbene complexes from an acetal function by C–O bond cleavage and alcohol elimination. Organometallics 15, 2860 (1996). doi:10.1021/om9603171
31. S. Ueno, E. Mizushima, N. Chatani, F. Kakiuchi, Direct observation of the oxidative addition of the aryl carbon-oxygen bond to a ruthenium complex and consideration of the relative reactivity between aryl carbon-oxygen and aryl carbon-hydrogen bonds. J. Am. Chem. Soc. 128, 16516 (2006). doi:10.1021/ja067612p Medline
32. P. O. Stoutland, R. G. Bergman, Insertion of iridium into the carbon-hydrogen bonds of alkenes: the π-complex cannot be an intermediate. J. Am. Chem. Soc. 107, 4581 (1985). doi:10.1021/ja00301a052
33. K. B. Renkema, Y. V. Kissin, A. S. Goldman, Mechanism of alkane transfer-dehydrogenation catalyzed by a pincer-ligated iridium complex. J. Am. Chem. Soc. 125, 7770 (2003). doi:10.1021/ja0289200 Medline
34. J. Barluenga, M. Yus, J. M. Concellon, P. Bernad, Synthesis of olefins from α-chlorocarbonyl compounds. J. Chem. Res. Synopses 1980, 41 (1980).
35. N. E. Grimmer, N. J. Coville, C. B. de Koning, J. M. Smith, L. M. Cook, Zirconium bis-indenyl compounds. Synthesis and x-ray crystallography study of 1- and 2-substituted bis(R-indenyl)zirconium dichloride metallocenes. J. Organomet. Chem. 616, 112 (2000). doi:10.1016/S0022-328X(00)00570-2
36. H. Narahashi, I. Shimizu, A. Yamamoto, Synthesis of benzylpalladium complexes through C–O bond cleavage of benzylic carboxylates: Development of a novel palladium-catalyzed benzylation of olefins. J. Organomet. Chem. 693, 283 (2008). doi:10.1016/j.jorganchem.2007.10.051
37. Y. Nakao, H. Imanaka, J. Chen, A. Yada, T. Hiyama, Synthesis and cross-coupling reaction of alkenyl[(2-hydroxymethyl)phenyl]dimethylsilanes. J. Organomet. Chem. 692, 585 (2007). doi:10.1016/j.jorganchem.2006.04.046
38. G. A. Olah, O. Farooq, S. M. F. Farnia, J. A. Olah, Friedel-Crafts chemistry. 11. Boron, aluminum, and gallium tris(trifluoromethanesulfonate)(triflate): Effective new Friedel-Crafts catalysts. J. Am. Chem. Soc. 110, 2560 (1988). doi:10.1021/ja00216a032