lineare: non lineare: generazione di seconda...
TRANSCRIPT

Dipolo indotto da E Lineare:
Non lineare:
Generazione di seconda armonica:

Raman (vibrazionale)
Una molecola non è ferma:
Consideriamo una frequenza (di vibrazione) ω' :
Dipolo indotto Generalizzazione: ω' ωkn (frequenza di risonanza del sistema)

From Cyrus Farokh Hirjibehedin Ph.D. Thesis
Example

Ripasso Conosciamo le soluzioni dell’equazione di Schrödinger di base:
Rappresentazione nello spazio degli autostati di H0:
Considero una perturbazione:
Riscrivo l’equazione di Schrödinger completa dipendente dal tempo nello spazio degli autostati di H0:
(3.2.1)

Teoria delle perturbazioni dipendente dal tempo:
Ordine 1:
Caso particolare: perturbazione sinusoidale su un autostato
Regola d’oro di Fermi
(3.3.1)
(3.3.2)
(3.3.3)
(3.3.4)

Alcune non-linearità: ordine 2
Consideriamo uno spettro non banale per f(t); per mantenere le cose semplici:
Il doppio integrale sarà quindi dato da una somma di termini del tipo:

Sostituendo il risultato dell’integrale nell’espressione per , considerando solo I termini derivanti dall’espressione nel riquadro , e con considerazioni simili a quelle che portano alla regola d’oro di Fermi (e considerando solo termini non trascurabili…) un contributo al secondo ordine alla probabilità di transizione dal livello n al livello j è legato a:
Assorbimento a 2 fotoni: ω1=ω2=ω
g
n
ω
ω
(3.15.1)

Spettroscopia ottica Spettroscopia a due fotoni
Le frequenze di risonanza e le regole di selezione sono diverse. La probabilità di transizione dipende dal “momento di transizione”
Sono attualmente allo studio marcatori specifici per le tecniche a due fotoni, che sono generalmente molecole più complesse con strutture di risonanza diradicaliche, o strutture dendrimeriche

Spettroscopia ottica
La probabilità di transizione (e quindi il segnale) è più basso a potenze ordinarie, ma ci sono diversi vantaggi Le risonanze sono ad energie più basse di quelle usualmente usate (circa la metà, di solito nell’infrarosso), e quindi c’è minor probabilità di danneggiare il campione
Spettroscopia a due fotoni

Spettroscopia ottica
Le regole di selezione sulla simmetria tra stati iniziali e finali sono diverse di quelle ad un fotone ⇒ è possibile avere informazioni su stati eccitati inaccessibili alla spettroscopia ad un fotone La probabilità di transizione (e quindi l’eventuale segnale di fluorescenza) è quadratica nell’intensità di radiazione incidente⇒ varia molto rapidamente per variazioni spaziali dell’intensità del fascio, creando una selettività locale molto utile nella microscopia di a fluorescenza, come vedremo più avanti. Per tutti questi motivi, le tecniche legate all’assorbimento a due fotoni stanno prendendo piede nell’analisi di sistemi biologici
Spettroscopia a due fotoni

2 photon vs. 1-photon excitation

Alcune non-linearità: ordine 2
Consideriamo uno spettro non banale per f(t); per mantenere le cose semplici:
Il doppio integrale sarà quindi dato da una somma di termini del tipo:

Raman ωnk+ ωkj = ωnk− ωjk= ωnj ω2 = ωL (-) ω1 = ωS (+)
γ
(3.16.1)

Raman non risonante
Per W(x) ~ e Ei x, la probabilità per il Raman diventa circa proporzionale a:
(Questa semplificazione si trova in alcuni testi; ha una certa validità per sistemi con pochi livelli molto ben separati, e lontano dalla risonanza)
20,
24SL EEe
(3.16.1)
(3.17.1)

Is/Ias=[(ν0-νvib)/ (ν0+νvib)]3exp(hνvib/kT)
F
Fluorescence
F
Thermally activated fluorescence

Misure Raman: Set-up

Spettrometro
Risoluzione: dispersione grating; distanza fra pixel; apertura fenditura



Rumore
• Elettronico • Schottky (sqrt(N)) • Raggi cosmici
• Possibili fonti: baseline, background
• No rumore: offset


La spettroscopia Raman
• Prerisonanza selezione modi cromoforo
• Misure in soluzione acquosa
Rayleigh
Stokes Anti-Stokes
Inte
nsit
y
Energy
νvib
Difficoltà: Bassa sezione d’urto rilevante segnale di fondo
laser Ar-Kr
0,00 0,02 0,04 0,06 0,08 0,10 0,12 0,14 0,16 0,18 0,20
Ass
orba
nza
250 300 350 400 450 500 550 600 650 Lunghezza d'onda (nm)
EYQ1 in DEA, pH = 8.3 BFPF in DEA, pH = 8.3

La spettroscopia Raman
• Prerisonanza selezione modi cromoforo
• Misure in soluzione acquosa
Rayleigh
Stokes Anti-Stokes
Inte
nsit
y
Energy
νvib
Difficoltà: Bassa sezione d’urto rilevante segnale di fondo 800 1000 1200 1400 1600 1800
Raman shift (cm-1)
psGFP 250 μM in DEA, pH = 7.95 DEA 20 mM, pH = 7.95 laser (Ludox)

Vibrational spectroscopy
Vibrational spectra: fingerprint of a molecule 1. Presence and/or localization of molecules 2. Conformational changes of a molecule
Ene
rgy
S0
S1 Example of “normal mode” for a BFPF chromophore (691 cm-1 )

stretching symmetrical asymmetrical
scissor twisting
rocking wagging
Vibrational spectroscopy Localized modes

Spettroscopia vibrazionale
ω
Frequenze tipiche di modi intra-molecolari La frequenza dei modi vibrazionali si misura in cm-1 f=cν(cm-1)= 1/λ inverso della lunghezza d’onda EM a freq corrispondente
hν[meV] = 0.124/ λ[cm]=0.124 f [cm-1]
Freq f (cm-1) T(fsec) hν (meV)
C-H,O-H,N-H 3000-3700 10 400
C C, C N 2100-2300 15 250
C=O,C=C,C=N 1500-1600 20 200
C-O,C-C,C-N 1000-1400 30 130
Bending, twisting,…
500-1100 50 60-140
Modi “fingerprint” delocalizzati
<600 >70 <70
La spaziatura dei livelli vibrazionali è tipicamente 1-2 ordini di grandezza inferiore a quella delle transizioni elettroniche ottiche o UV
KT(300K)=25meV ⇒ Solo i modi a frequenza molto bassa (quelli più “delocalizzati) possono essere trattati “classicamente” a temperatura ambiente!

Spettroscopia vibrazionale
La soluzione del problema elettronico (con DFT o altre tecniche) definisce l’insieme delle PES (per stato fondamentale ed eccitati)
R
E
Il moto nucleare in approssimazione adiabatica è guidato dalle PES, che agiscono come energie potenziali, i cui gradienti rispetto alle coordinate nucleari sono le forze
Fin ({R}) = −∇Ri
E n ({R})
E0
E1
E2

Spettroscopia vibrazionale Approx armonica, Stati vibrazionali
Vibrational levels
E i(R) = E i(R0) +∂E i
∂RI
I∑
0
(RI − RI0) +
12
∂ 2E i
∂RI∂RJ
IJ∑
0
(RI − RI0)(RJ − RJ
0) + ...
Sviluppo fino al termine quadratico intorno ai minimi delle PES
Passaggio in coordinate normali {Q} (diagonalizzazione della matrice dinamica)
E i(Q) =12
ω I2
I∑ QI
2
⇒Il sistema viene descritto come un insieme di oscillatori armonici semplici indipendenti
χ i(Q) = φ1i(Q1) ⋅ φ2
i (Q2) ⋅ ...⇒Per ogni stato elettronico (i) la funzione d’onda nucleare totale si disaccopia nelle componenti dovute ai singoli modi
Ogni singolo modo può essere trattato quantisticamente
QI ,ωI
Eυ i , I
i,I = ω I12
+ ν i,I
χ i = φυ i , I
i,I
I∏ = ν i,I
I∏
Funz d’onda nucleare totale:
ν=1 ν=2

Spettroscopia vibrazionale Calcolo di spettri vibrazionali con DFT
Ottimizzazione della struttura Soluzione delle equazioni di Kohn-Sham e ricerca dei minimi delle PES ⇒ Geometria del minimo locale R0
Calcolo della matrice dinamica (=Hessiano della PES nel minimo) Calcolo diretto delle derivate seconde oppure calcolo numerico con il metodo dei piccoli spostamenti dal minimo
Diagonalizzazione ⇒ Autovalori (frequenze di vibrazione) e autostati (modi di vibrazione) Risultato finale: per ogni modo normale, frequenza, pattern di spostamenti (autostato di vibrazione)
∂ 2E i
∂RI∂RJ
0

Spettroscopia vibrazionale
I livelli vibrazionali non sono più equispaziati I modi vibrazionali a diverse frequenze interagiscono tra loro e hanno una larghezza e vita media finita
Anarmonicità e accoppiamento vibronico
E i(Q) =12
ω I2
I∑ QI
2 +13!
∂ 3E i
∂QI∂QJ∂QK
0
QIQJQKI ,J ,K∑
All’ordine cubico
QI ,ωI
R
E INOLTRE, in vicinanza di intersezioni coniche, dove le PES si avvicinano tra loro, l’approssimazione adiabaitica fallisce, e possono verificarsi accoppiamenti elettronici-vibrazionali (vibronici) e conseguenti salti tra le PES

1 1
Electronic transitions
1
L’elemento di matrice di dipolo vale: Fattori di Franck Condon

Spettroscopia vibrazionale
Le spaziature tra I picchi dello spettro di fluorescenza misurano la frequenza dei modi vibrazionali dello stato fondamentale ∆E= hνi
Q
Ei
Ef
Spettro a basse temperature della GFP S65T (picco A soppresso) La struttura vibrazionale dello spettro di assorbimento si vede chiaramente Le spaziature tra i picchi corrispondono alle frequenze dei modi vibrazionali dello stato eccitato: ∆E= hνf
I0-0
hνf
hνi
Non tutti i modi vibrazionali contribuiscono alla struttura del picco di ass/emiss: È necessario che le PES dello stato fondamentale ed eccitato siano spostate l’una rispetto all’altra e/o abbiano frequenze sensibilmente diverse, altrimenti i vibrazionali non fondamentali non vengono eccitati dalla transizione elettronica (modi vibrazionali correlati con l’eccitazione) Le frequenze dei modi siano non troppo piccole (altrimenti i picchi vibrazionali non si distinguono) e non troppo grandi (altrimenti i picchi vibrazionali sono troppo spaziati e non si vedono)⇒ Alta selettività su specifici modi caratteristicamente correlati con l’eccitazione elettronica
Esempio:
∆λ~15nm ∆E~77meV hνf~620cm-1
∆λ~17nm ∆E~87meV hνi~700cm-1

Spettroscopia vibrazionale
R
Ei
Ef
I0-0
hνf
ma anche la transizione principale tra vibrazionali “fondamentali” delle PES di stato fondamentale ed eccitato Ef,0→ Ei,0 ha energia inferiore a quella inversa Ei,0→ Ef,0 (Stokes shift del picco principale). Questo è dovuto al rilassamento “solvente” (o ambiente chimico circostante) che si riaggiusta in seguito all’eccitazione spostando la PES
hνi
Origine dello Stokes shift
Lo spettro di fluorescenza presenta una struttura analoga (speculare), con energia media di transizione inferiore a quella di assorbimento (Stokes shift “medio”)
Ei,0
Ef,0

Spettroscopia vibrazionale Effetto della temperatura
A temperatura ambiente, però la struttura vibrazionale di solito non è distinguibile a causa dell’allargamento termico dei picchi vibrazionali L’innalzamento della temperatura produce uno spostamento effettivo dei picchi, modificando di solito anche lo Stokes shift
GFP-wt (solo assorbimento)
GFP S65T
Rimangono tuttavia di solito visibili i picchi dovuti a strutture conformazionali diverse, come gli stati A e B nella GFP-wt, oppure picchi dovuti a transizioni diverse della stessa struttura, che sono separati da differenze di energie maggiori

Spettroscopia vibrazionale
La misura delle frequenze vibrazionali dallo spettro di assorbimento/emissione ottica è una misura indiretta delle proprietà vibrazionali, difficoltosa (va fatta a bassa temperatura) e dà informazioni su pochi modi Esiste però la possibilità di fare misure dirette delle frequenze vibrazionali di un sistema

Spettroscopia vibrazionale
µFI = ν f ν i µ0(R0) + ν fI
∑ QI ν i∂µ0
∂QI R0
+ ...
Misura diretta degli spettri vibrazionali
Spettroscopia IR = spettroscopia in assorbimento con radiazione IR tale da non eccitare lo stato elettronico (di solito hν ~ 0.05-0.5eV, λ ~ 2000-20000nm)
Sorgente IR accordabile
λ
I0
Campione Rivelatore
I
QI
IR absorption
0 µ(R0) 0
Stati vibrazionali dello stato fondamentale ⇒il termine di ordine 0 è nullo
⇒ ν f ν i = δij
ν f QI ν i =
2ω I
ν i +1δ f ,i+1 + ν iδ f ,i−1( )⇒il termine di ordine 1 è diverso da zero solo per transizioni di singolo livello vibrazionale, cioè per assorbimenti di energia pari a hν! In realtà le transizioni a partire dai vibrazionali eccitati hanno energia leggermente minore, per via dell’anarmonicità, il che introduce rumore. Rumore può essere introdotto anche dall’emissione stimolata da stati vibrazionali eccitati, ma di solito è trascurabile
hν

IR spectroscopy • Absorption spectroscopy; usual range ~300--3000 cm-1 (λ ~ 30--3µm)
• Dispersion usually obtained by Fourier Transform Interferometer
• Best results with water (and CO2) free environment
• Often, differential spectra to highlight changes.
Milano - Rho, 18/11/2010
FTIR

Spettroscopia vibrazionale
µFI = ν fI
∑ QI ν i∂µ0
∂QI R0
Selezione delle frequenze vibrazionali: picchi di assorbimento a ω=ωI
Modulazione dei picchi: l’intensità del picco dipende dalla variazione del dipolo elettrico lungo la coordinata normale
I modi con intensità non nulla si dicono IR-attivi
formaldeide
Esempio: spettro IR della formaldeide n. Totale di modi = (4x3-6)=6

Spettroscopia vibrazionale

Spettroscopia vibrazionale Spettro IR del cromoforo di GFP
stretching double bonds stretching single bonds
bending, scissor delocalized bending-ring def
I modi del cromoforo GFP si possono assegnare con calcoli (DFT): Si calcolano frequenze e autostati dei modi vibrazionali Si confrontano frequenze/ampiezze con quelle sperimentali⇒si assegna il modo L’assegnazione del modo ci dice che tipo di vibrazione ha la molecola
Ad es, i due modi a più alta frequenza (esclusi i modi dell’idrogeno) corrispondono a stretching localizzato di C=O e C=C e la loro frequenza è sensibile all’ambiente chimico circostante (ad es, formazione di legami a idrogeno con O di C=O)
I modi a frequenza <1000cm-1 sono delocalizzati e fortemente caratteristici della molecola e della sua conformazione

Spettroscopia vibrazionale
n. nodi del cromoforo: ~70 di cui circa 30 che coinvolgono l’idrogeno e ~ 40 che coinvolgono anche gli atomi “pesanti”
Nello spettro IR si vedono quasi tutti sono stati assegnati tramite calcoli ⇒ l’assegnazione consente di seguire la variazione di specifici modi per effetto della variazione dell’environment e quindi trarre informazioni strutturali dalla misura dello spettro vibrazionale
Ma la spettroscopia IR è poco selettiva! In una proteina ci sono 3N-6 modi con N~10000!! Risulta impossibile distinguere e classificare i singoli modi tramite semplice spettroscopia IR
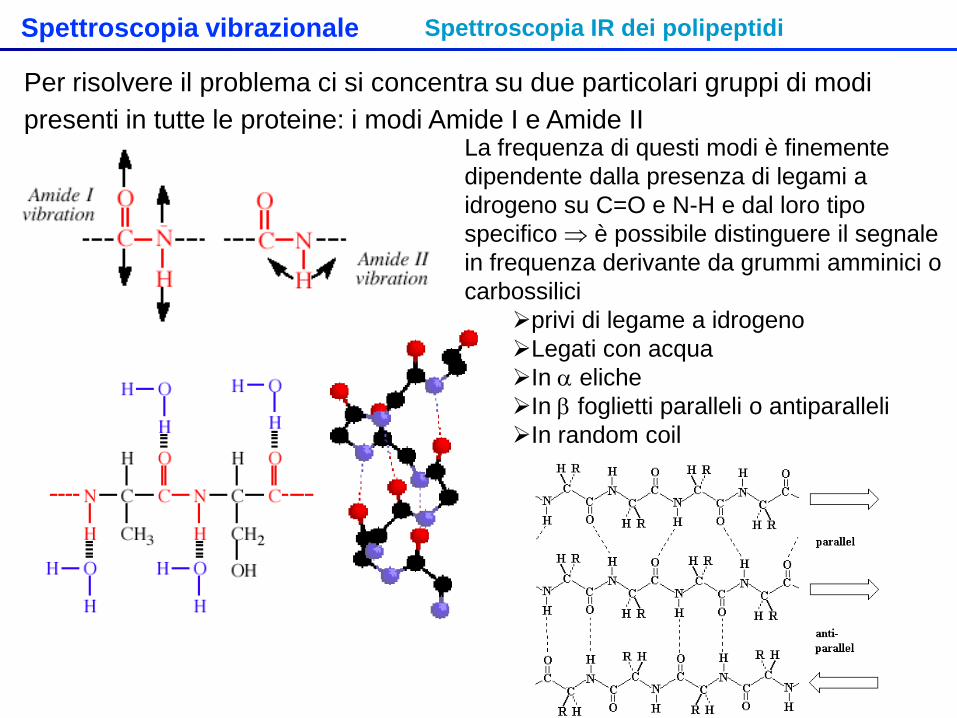
Spettroscopia vibrazionale Spettroscopia IR dei polipeptidi
Per risolvere il problema ci si concentra su due particolari gruppi di modi presenti in tutte le proteine: i modi Amide I e Amide II
La frequenza di questi modi è finemente dipendente dalla presenza di legami a idrogeno su C=O e N-H e dal loro tipo specifico ⇒ è possibile distinguere il segnale in frequenza derivante da grummi amminici o carbossilici
privi di legame a idrogeno Legati con acqua In α eliche In β foglietti paralleli o antiparalleli In random coil

Spettroscopia vibrazionale
Amide I C=O Amide I N-H Amide II
α-helix 1650 || 1652 ⊥
3290-3300 1516 II 1546 ⊥
β-sheet parallelo
1685 || 1632 ⊥
3280-3300 1530
β-sheet antiparallelo
1645 || 1630 ⊥
1530 II 1550 ⊥
Random coil 1656 1535 Non legati 1680-1700 ~3400 ~1520
Il modo AmideI vengono indeboliti (frequenze diminuite) dai legami a idrogeno in maniera struttura specifica Il modo AmideII viene generalmente rafforzato (frequenza aumentata) dai legami a idrogeno, sempre in maniera struttura specifica Nelle eliche e nei foglietti si verifica il fenomeno del dicroismo (frequenze e intensità differenziate a seconda del’orientazione della polarizzazione della luce incidente rispetto all’asse dell’elica o al piano del foglietto) ⇒ Si possono ricavare informazioni sulla presenza, abbondanza e orientazione di strutture secondarie

Spettroscopia vibrazionale
In ogni caso i dati non forniscono misure dirette della struttura: sono necessari modelli per interpretare gli spostamenti e le intensità delle bande, oppure specifiche tecniche che introducono conoscenza a priori sul sistema Difference IR: Si determina la natura di una variazione strutturale del sistema dalla variazione del suo spettro IR (differenza dello spettro IR dei due stati)
Spettri IR differenziali
GFP
Ad esempio: gli spettri differenza (stato A - stato B) delle GFP mostrano dei picchi sia nelle bande Amide, sia tra i modi caratteristici del cromoforo, che indicano la deptrotonazione (comparsa del picco 1149, legato al legame singolo C-O del fenolo) NB: di solito si fanno gli spettri diff-IR usando acqua deuterata come solvente perché le bande di assorbimento dell’acqua normale si sovrappongono e creano rumore

Raman (vibrazionale)
Una molecola non è ferma:
Consideriamo una frequenza (di vibrazione) ω' :
Dipolo indotto Generalizzazione: ω' ωkn (frequenza di risonanza del sistema)
( ) ( )00
RRR R
−∂∂
↔′ αωα

Spettroscopia vibrazionale
La polarizzabilità è la funzione di risposta che misura la variazione di dipolo al campo elettrico considerato come perturbazione, e può essere calcolata nell’ambito della teoria delle perturbazioni in risposta lineare Si usano le seguenti approssimazioni/assunzioni Approssimaziona adiabatica Approssimazione armonica Frequenza del fascio incidente sufficientemente piccola da NON eccitare lo stato elettronico
Livelli elettronici Livelli vibrazionali
Anche se stati elettronici iniziali e finali sono gli stessi, la polarizzabillità, come altre funzioni di risposta, coinvolge una somma su stati intermedi, elettronici e vibrazionali NB: αβ sono indici cartesiani, la polarizzabilità è un tensore
I = 0 ν i
I = 0 ν f
R = r ν n
Spettroscopia Raman

Spettroscopia vibrazionale
Oltre che dalla frequenza ω la polarizzabilità dipende anche dalla configurazione nucleare R; usando l’approssimazione armonica e passando in coordinate normali si può scrivere lo sviluppo:
Il primo termine sopravvive solo quando anche gli stati vibrazionali iniziale e finale sono gli stessi (a causa della loro ortonormalità) La conservazione dell’energia implica che le frequenze della radiazione incidente e diffusa siano le stesse, dal momento che stato finale e iniziale del sistema dopo il passaggio della radiazione sono gli stessi Diffusione elastica della luce (Rayleigh)
QI
Ee,i hν hν
Diffusione ELASTICA
α0 è la polarizzabilità statica, e sarebbe l’unico contributo se la radiazione incidente non inducesse lo spostamento degli atomi
NB: la somma coinvolge stati elettronici eccitati

Spettroscopia vibrazionale
Ee,i
Raman Anti-Stokes hνI
hν hν’’
hνI
La diffusione Raman può essere vista come un’interazione inelastica di un fotone con il mezzo: il fotone cede o assorbe energia al sistema generando o assorbendo un singolo “fonone” (quanto di vibrazione della molecola)
Diffusione inelastica Raman
Considerando che in approssimazione armonica vale
il secondo termine risulta non nullo solo se lo stato vibrazionale finale differisce dall’iniziale per un singolo livello vibrazionale ⇒ si deve avere hν-hν’=±hνI
Raman Stokes
L’ampiezza di diffusione coinvolge la derivata della polarizzabilità rispetto allo spostamento lungo la coordinata normale Questa contiene la somma sugli stati elettronici eccitati.

Spettroscopia vibrazionale
Tipico spettro Raman
Lo spettro della radiazione diffusa presenta un picco a frequenza ω=ω’, ∆ω=ω’-ω=0 (Rayleigh) e picchi a frequenze ω’-ω= ωI (i.e. un picco per ogni modo)
Le intensità dei picchi dipendono dal (quadrato della) derivata della polarizzabilità rispetto alla coordinata normale: vengono selezionati i modi per effetto dei quali più facilmente il sistema si polarizza (Raman-attivi)
Generalmente I modi Stokes sono più forti degli anti-Stokes perché i livelli vibrazionali inferiori sono più popolati
La polarizzabilità è un tensore e quindi l’intensità della radiazione diffusa dipende dall’angolo di raccolta della radiazione

Spettroscopia vibrazionale
IR L’intensità del picco a ωI dipende da (∂µ/∂QI) Raman L’intensità del picco a ωI dipende da (∂α/∂QI) ⇒Le due tecniche danno risultati diversi, e spesso “complementari”: dipendentemente dalla simmetria della molecola, di solito i modi IR attivi non sono Raman attivi e viceversa
Nel caso del cromoforo GFP in soluzione, solo circa 1/3 del totale dei modi sono Raman attivi In particolare nella zona 500-900cm-1 ci sono i modi delocalizzati caratteristici della molecola
Selettività dei processi Raman Spettri vibrazionali del cromoforo GFP neutro
IR
Raman

Spettroscopia vibrazionale
In generale, anche se la tecnica Raman è più selettiva rispetto all’IR, in una proteina rimangono pur sempre qualche migliaio di modi diversi Raman-attivi che in linea di principio contribuiscono tutti quanti allo spettro Tuttavia, in certi casi è possibile rendere la tecnica più specificamente selettiva Nella formula della polarizzabilità la frequenza della radiazione incidente compare al denominatore
Negli esperimenti Raman usuali ω è nell’infrarosso (condizioni “fuori risonanza”) e quindi si può trascurare nel calcolo, risultando in un valore della polarizzabilità praticamente indipendente dalla frequenza Se l’esperimento viene fatto con ω nel vicino infrarosso/visibile, e l’ampiezza di scattering dipenderà in modo più complesso dalla frequenza; questo complica i calcoli e quindi anche il confronto esperimento-teoria
( )
0~ EEr −<ω

Spettroscopia vibrazionale
In condizioni di “risonanza”, cioè quando l’energia della radiazione incidente è uguale o molto vicina all’energia della transizione elettronica hν~E1-E0, solo un termine della somma sugli stati eccitati (r=1) contribuisce alla polarizzabilità
Ee,i Resonance Raman scattering
Raman risonante
In queste condizioni si può ulteriormente assumere che la dipendenza principale di α da Q provenga dalla variazione della PES (si trascura la variazione di µ); usando anche il fatto che si parte dal minimo della PES dello stato fondamentale si ha infine
I'∝ (ω −ω I )4
ω I
∂E1
∂QI
2
⇒

Spettroscopia vibrazionale
Ee,i Resonance Raman scattering
Interpretazione “euristica”: Poiché l’energia hν è sufficiente ad eccitare elettronicamente il sistema, la transizione non è più “virtuale”, ma reale
I'∝ (ω −ω I )4
ω I
∂E1
∂QI
0
2
Successivamente il sistema torna sullo stato elettronico fondamentale, con le velocità acquisite; l’ampiezza di vibrazione a frequenza ωI sarà proporzionale a queste velocità, e quindi a ⇒ Saranno maggiormente amplificati i modi con maggiore valore di
Sullo stato eccitato, il sistema si trova fuori equilibrio e acquista una velocità che in prima approssimazione è proporzionale alle forze che sente; riferito a un singolo modo normale, la forza è precisamente il gradiente della PES di stato eccitato rispetto alla coordinata Q calcolata nel minimo della PES fondamentale,
∂E1 /∂QI
∂E1 /∂QI

Spettroscopia vibrazionale
La DFT è in grado di calcolare molto bene gli spettri Raman
⇒rende possibile l’identificazione e assegnazione delle bande vibrazionali a specifici modi di vibrazione
Ma come si usano queste informazioni?
Come si possono utilizzare i dati vibrazionali per acquisire informazioni strutturali?

Spettroscopia vibrazionale
La frequenza dei modi vibrazionali localizzati su specifici legami (ad es C=O, C=C, C=N) è linearmente correlata con la lunghezza del legame ⇒ La misura Raman da un’informazione diretta sulla struttura del cromoforo
La frequenza di C=O (IR attivo) e il C=C (Raman attivo) sono correlati con lo stato di protonazione del cromoforo (distinguono anioni da neutri), mentre C=N è correlato con il “colore” ⇒ ulteriori informazioni sulla proprietà strutturali/ottiche
Correlazione frequenze-struttura

Spettroscopia vibrazionale
Glu222 Ser205
Excitation Energy (eV)
quinonoid
benzenoid
Chr
omop
hore
geo
met
ry (B
LA)
mod
e fre
quen
cy (c
m-1
)
C=O
C=C
La relazione lineare tra le frequenze dei modi C=C (C=O) e la struttura esiste anche all’interno delle proteine, e implica una correlazione tra frequenze e “colore”: ωC=O e ωC=C sono linearmente correlate alla BLA La BLA è correlata all’energia di eccitazione ⇒ Frequenze ed energie di eccitazione sono correlate!

Spettroscopia vibrazionale
Neutral-like (benzenoid)
anionic-like (quinonoid)
La correlazione lineare tra frequenza del modo C=C ed energia di assorbimento è stata esperimentalmente misurata, e c’è accordo con la teoria ⇒ Dalla misura delle frequenza del modo C=C si può risalire alle proprietà elettroniche e a quelle strutturali (interazioni tra cromoforo e amminoacidi nel sito attivo)
teoria
esperimento

Spettroscopia vibrazionale
Relazione lineare tra frequenze dei modi localizzati (~1500-1800cm-1) e lunghezze dei legami corrispondenti Frequenze (e quindi lunghezze di legame) finemente dipendenti dall’environment chimico ⇒ gli spettri Raman (o IR) potrebbero essere usati, al pari degli spettri NMR, per porre vincoli sulla struttura ⇒Determinazione indiretta della struttura! Di recente sta prendendo piede la spettroscopia IR 2D, dove, similmente a quello che si fa con l’NMR, si misurano mappe di correlazione tra coppie di modi, che sono molto più selettive e informative degli spettri unidimensionali
Problema: questa tecnica è facilmente utilizzabile solo per i modi localizzati
Mappa 2D dei modi amide per diverse strutture secondarie

Conformational changes in fluorescent proteins studied by Raman
Raman scattering: from excitations to
conformational properties
ON OFF
switching
switching
514
kT 416
N
N O
H O N N
O O
Fluorescent proteins
Results: synthetic cromo-phores and folded proteins
Conclusions

BFPF EYQ1
IN ON OFF
switching
switching
photoactivation
† bleaching
Photophysics of Fluorescent Proteins

0.13
0.12
0.11
0.10
0.09
0.08
0.07
0.06
0.05
0.04
0.03
0.02
0.01
0.00
600 550 500 450 400 350 300
T=0' (totally relaxed)
T=15', 20 mW @ 514 nm
T=1', 40 mW @ 416 nm
T=+60' relaxation after react
Wavelength (nm)
Abs
orba
nce
EYQ1 pH 8.5
X
Photochromic Fluorescent Proteins
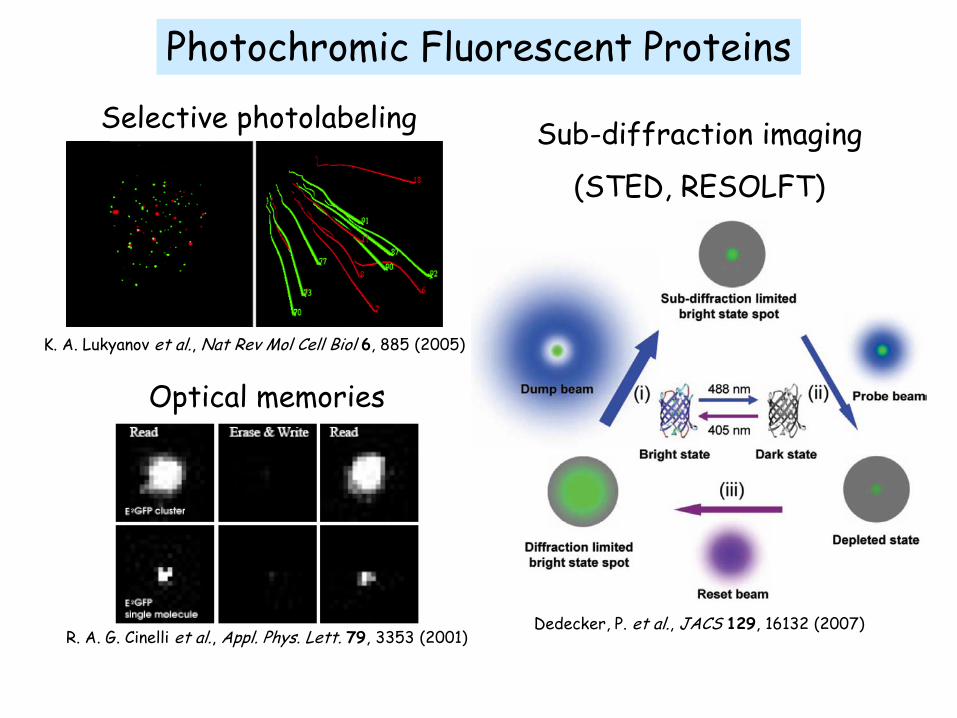
Photochromic Fluorescent Proteins
K. A. Lukyanov et al., Nat Rev Mol Cell Biol 6, 885 (2005)
Selective photolabeling
R. A. G. Cinelli et al., Appl. Phys. Lett. 79, 3353 (2001)
Optical memories
Dedecker, P. et al., JACS 129, 16132 (2007)
Sub-diffraction imaging
(STED, RESOLFT)

NEUTRAL ANION
Reversible photophysics/chemistry
CIS TRANS
Photochromism Mechanisms

BFPF EYQ1
250 300 350 400 450 500 550 6000,00
0,02
0,04
0,06
0,08
0,10
0,12
0,14
0,16
Asso
rban
za
Lunghezza d'onda (nm)
psGFP in DEA, pH = 8.3 psGFP, pH = 8.3,
dopo 60 min irraggiamento
400 410 4200,0000,0020,0040,0060,0080,010
240 270 300 330 360 390 420 450
0,00
0,02
0,04
0,06
0,08
0,10
0,12
0,14
Asso
rban
za
Lunghezza d'onda (nm)
BFPF in DEA, pH = 8.3 BFPF in DEA,
dopo 1 h irraggiamento
EYQ1 EYQ1

600 800 1000 1200 1400 1600 1800
600 800 1000 1200 1400 1600 1800
Inten
sity
(a.u
.)
cBFPFExperiments
Theory
Energy (cm-1)
Raman on Chromophores: cBFPF

600 800 1000 1200 1400 1600 1800
600 800 1000 1200 1400 1600 1800
Inten
sity
(a.u
.)
cBFPFExperiments
Theory
Energy (cm-1)
Raman on Chromophores: cBFPF
350nm

(a)
(b)
600 800 1000 1200 1400 1600 1800
600 800 1000 1200 1400 1600 1800
Inten
sity
(a.u
.)
cBFPF
cBFPFExperiments
tBFPF
(LB-B3LYP pre-resonance)
Theory
Energy (cm-1)
tBFPF
(c)
(d)
(GB-B3LYB pre-resonance)
Raman on Chromophores: cBFPF

1000 1500
1000 1500
1000 1500
1000 1500
(b)
Inten
sity
(a.u
.)
cCFP
cCFPExperiments
tCFP
(d)
(LB-B3LYP pre-resonance)
Theory
Energy (cm-1)
tCFP
Inten
sity
(a.u
.)
cGFP
cGFPExperiments
tGFP
(a)
(c)
(LB-B3LYP pre-resonance)
Theory
Energy (cm-1)
tGFP(GB-B3LYB pre-resonance) (GB-B3LYB pre-resonance)
Raman on Chromophores: cGFP and cCFP

Raman on Chromophores: Conclusions
•Similar results on chromophores of BFPF, GFP and CFP
•Synthetic chromophores are
photochromic
•Identification of peaks in the Raman spectra of native and photoconverted
forms
•Photoconverted form is a trans isomer

Raman on Proteins: Preresonance Conditions
250 300 350 400 450 500 550 600 650
laser Ar-Kr A
bsor
banc
e
Wavelength (nm)
EYQ1 in DEA, pH = 8.3 BFPF in DEA, pH = 8.3
Ar laser
Ar-Kr laser
1000 1200 1400 16001000 1200 1400 1600
Inte
nsity
(a. u
.)
Raman shift (cm-1)
Inte
nsity
(a. u
.)
Raman shift (cm-1)
BFPF ~0.2mM pH=8.3
* * * # %
CH 3 N N
O
CH 3
cBFPF
*
cGFP ~1mM in H2O pH=9.9
Protein: EYQ1 0.3mM pH=8
CH 3 O
N N
O
CH 3

1200 1600Raman shift (cm-1)
600 800 1000 1200 1400 1600 1800
Inten
sity
(arb
. uni
ts)
Raman shift (cm-1)
CH 3 N N
O
CH 3
cBFPF (Chromo-
phore)
(Protein) BFPF ~0.2mM pH=8.3
* * * # %
Raman on Proteins: BFPF

1200 1600Raman shift (cm-1)
600 800 1000 1200 1400 1600 1800
Inten
sity
(arb
. uni
ts)
Raman shift (cm-1)
CH 3 N N
O
CH 3
CH 3 N
N O CH
3
cBFPF
tBFPF
(Chromo-phore)
(Protein)
BFPF Photoconverted
BFPF ~0.2mM pH=8.3
* * * # %
Raman on Proteins: BFPF

600 800 1000 1200 1400 1600 1800
Inten
sity
(arb
. uni
ts)
Raman shift (cm-1)
CH 3
O N
N
O
CH 3
*
cGFP ~1mM in H2O @ pH=9.9
Protein: EqGFP ~0.3mM pH=8
Raman on Proteins: EYQ1

600 800 1000 1200 1400 1600 1800
Inten
sity
(arb
. uni
ts)
Raman shift (cm-1)
H O
CH 3 N
N O CH
3
CH 3
O N
N
O
CH 3
*
cGFP ~1mM in H2O @ pH=9.9 tGFP ~2.5mM in acetonitrile
Protein: EYQ1 ~0.3mM pH=8 Photoconverted form (@ 514nm ~50mW ~30min)
Raman on Proteins: EYQ1

1200 1500 1600Raman shift (cm-1)
600 800 1000 1200 1400 1600 1800
Inten
sity
(arb
. uni
ts)
Raman shift (cm-1)
H O CH 3 N
N
O
CH 3
H O
CH 3 N
N O CH 3 cGFP
tGFP
(Chromophore)
(Protein)
Chromophore in acetonitrile
Photoconverted protein @ pH=8
EYQ1 ~0.1mM pH=4.7
Raman on Proteins: EYQ1

1200 1500 1600Raman shift (cm-1)
(Chromophore)
(Protein)
Raman on Proteins: EYQ1
trans
cis
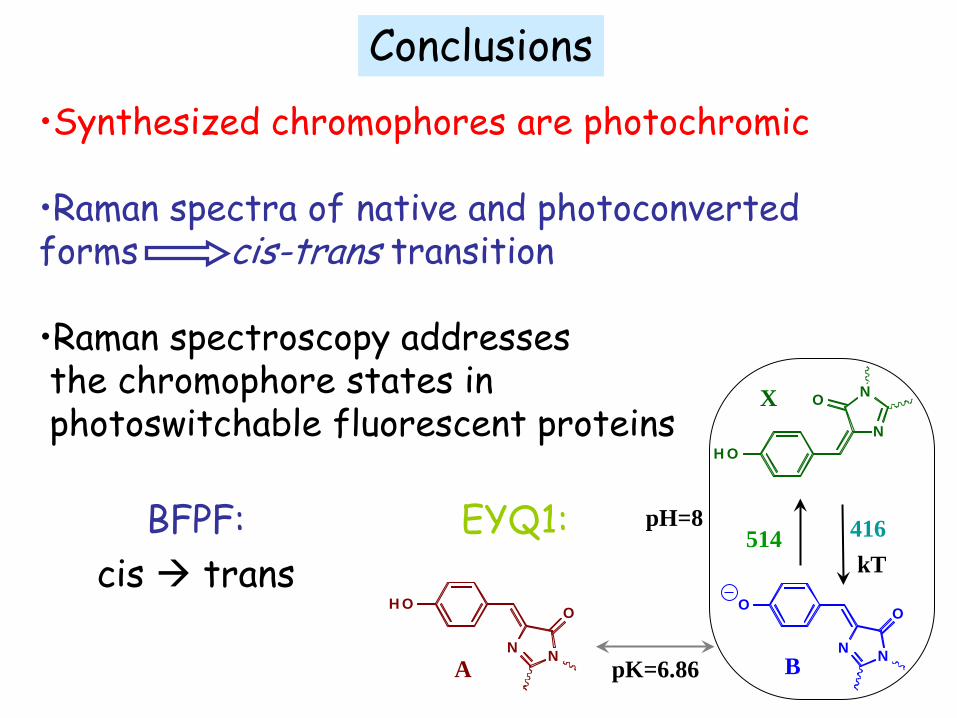
BFPF: cis trans
•Synthesized chromophores are photochromic
•Raman spectra of native and photoconverted forms cis-trans transition
•Raman spectroscopy addresses the chromophore states in photoswitchable fluorescent proteins
514 kT
416
X N
N O
H O
A B N N
O O
N N
O H O
pK=6.86
pH=8 EYQ1:
Conclusions
Spettroscopia vibrazionale
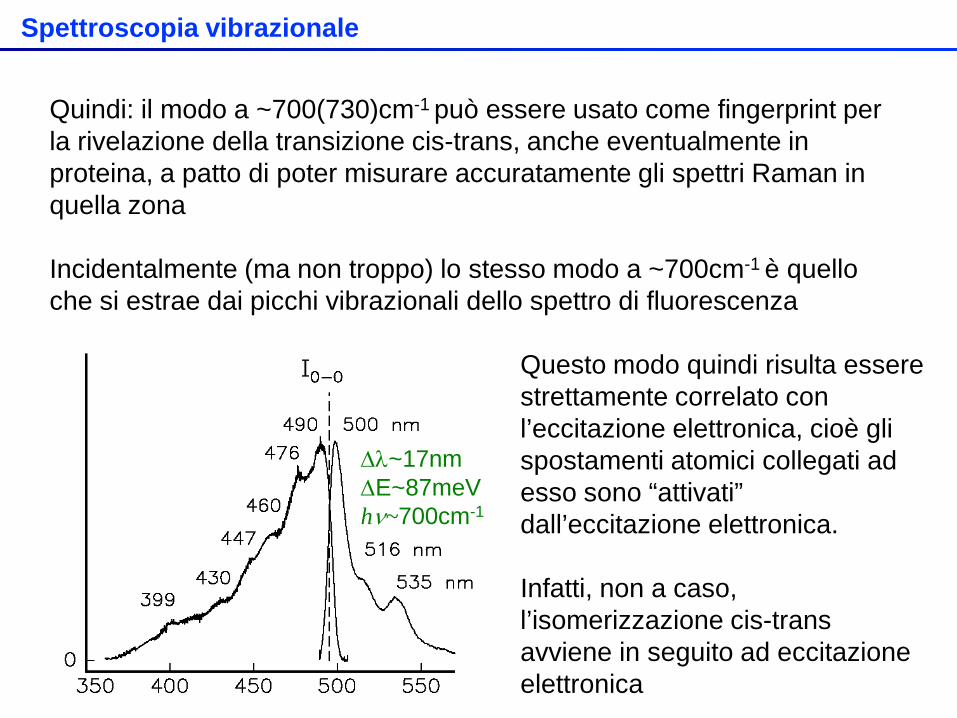
Quindi: il modo a ~700(730)cm-1 può essere usato come fingerprint per la rivelazione della transizione cis-trans, anche eventualmente in proteina, a patto di poter misurare accuratamente gli spettri Raman in quella zona Incidentalmente (ma non troppo) lo stesso modo a ~700cm-1 è quello che si estrae dai picchi vibrazionali dello spettro di fluorescenza
∆λ~17nm ∆E~87meV hν~700cm-1
Questo modo quindi risulta essere strettamente correlato con l’eccitazione elettronica, cioè gli spostamenti atomici collegati ad esso sono “attivati” dall’eccitazione elettronica. Infatti, non a caso, l’isomerizzazione cis-trans avviene in seguito ad eccitazione elettronica

Spettroscopia vibrazionale Tipo di tecnica Proprietà
elettroniche Proprietà strutturali Risoluzione
Proprietà vibrazionali e dinamiche
Raggi X Diffrazione
Mappa di densità elettronica
Posizioni degli atomi pesanti; ~Å
Neutroni Diffrazione Posizioni atomiche; ~Å
NMR Misura dello spostamento chimico
Posizioni atomiche, ottenute adattando modelli ai vincoli ricavati dai dati sperimentali; pochi Å
Strutture alternative dovute alle fluttuazioni termiche o piccole transizioni strutturali
Microscopia (crio-)elettronica
Mappa di densità elettronica
Posizione dei Cαo dei P; ~10Å
Strutture alternative dovute a transizioni strutturali
Spettroscopia ottica-UV Assorbimento o Fluorescenza
Spettro dei livelli energetici elettronici
Indicazioni indirette, derivanti dal confronto con la teoria
Indicazioni su variazioni strutturali, dal confronto con la teoria A bassa temperatura, frequenze dei modi vibrazionali correlati con l’eccitazione
Spettroscopia IR Assorbimento
Indicazioni indirette, derivanti dal confronto con la teoria
Spettro vibrazionale dello stato fondamentale
Spettroscopia Raman Diffusione inelastica di radiazione EM
Indicazioni indirette, derivanti dal confronto con la teoria
Spettro vibrazionale dello stato fondamentale Indicazioni su variazioni strutturali dal confronto con la teoria

Spettroscopia vibrazionale
Teoria-modelli
r-X neutroni CrioEM NMR Spettr ottica-
UV
IR Raman Struttura
Variazioni strutturali Dinamica
Livelli elettronici
Struttura vibrazionale
Sinergie tra tecniche

Spettroscopia vibrazionale
SERS cromoforo
Surface Enhanced Resonance Raman (SERS)
SERS proteina
La luce incidente eccita plasmoni di superficie, della sferetta, la cui frequenza si aggira intorno a 500 cm-1, a seconda delle dimensioni Questi creano un campo elettromagnetico locale che è anche più di 3 ordini di grandezza maggiore di quello esterno, e che interagisce con le vibrazioni della molecola Questo risulta in un sensibile aumento del campo effettivo, e quindi del segnale Raman, per frequenze vicine a questi valori ⇒ Amplificazione del segnale proprio nella regione di interesse
Le bande a bassa frequenza (300-700 cm-1), che sono anche le più caratterizzanti di una struttura, hanno solitamente intensità Raman molto bassa È possibile amplificarle selettivamente con una tecnica che prevede che le molecole (cromofori o proteine) siano legati (o adsorbiti) a sferette metalliche in sospensione acquosa
Raman cromoforo

Risonanze plasmoniche La tecnica: dal Raman al SERS
J.Fe
ldm
ann
et a
l, N
ano
Lett
. 200
7,7
(9) 2
753-
2757
W. Rechberger et al. Opt. Comm. 220 (2003)


30 100 nm

SER(R)S
SEM images of deposited silver sol
100nm
20nm
1µm
200nm
SERS (Surface Enhanced Raman Spectroscopy): Surface plasmon enhance electric field; chemical effects
Singola molecola in hot spot

S. Nie, S. R. Emory, Science 275, 1102 (1997)
SERS

Oltre al fattore elettrico, l’aumento del segnale Raman è portato anche, in minor parte, da un “fattore chimico”, in cui si considera una transizione mediata dall’accoppiamento con il gas elettronico nel vicino metallo: oltre allo scambio di fotoni, si considera nel singolo processo di diffusione anche un possibile scambio di elettroni.

( ) ( )( ) o
3
o
o Er2εωεεωεωP
+−
∝
. r
d
Au or Ag nanoparticle
ε(ω) – frequency complex dielectric constant
SERS EFFECT: METALLIC NANOPARTICLES
If Re[ε(ω)] = -2εo {real part of ε(ω)} and the imaginary part of ε(ω) is small, the dipole moment (hence the local electric field) will be significantly enhanced.
( )
+−
+=γωω
εωεi
fo 2
Higher enhancement close to tips or between nanoparticles in aggregrates
( )3
drr ωP~
+∝
frequency dependent dipole moment of a spherical nanoparticle
Eo – incident electric field from laser beam
“perfect” metals (free electrons)
Behaviour of “induced” electric field: