lingquan deng - divakth.diva-portal.org/smash/get/diva2:445661/fulltext01.pdf · synthesis,...
TRANSCRIPT

Akademisk avhandling som med tillstånd av Kungl Tekniska Högskolan i Stockholm framlägges till offentlig granskning för avläggande av doktorsexamen i kemi med inriktning mot organisk kemi, fredagen den 21 October, kl 13:00 i sal F3, KTH, Lindstedtsvägen 26, Stockholm. Avhandlingen försvaras på engelska. Opponent är Dr. Javier Rojo, Instituto de Investigaciones Químicas, CSIC, Seville, Spain.
Photochemical Surface Functionalization: Synthesis, Nanochemistry and
Glycobiological Studies
Lingquan Deng
邓灵泉
Doctoral Thesis
Stockholm 2011

ISBN 978-91-7501-072-4 ISSN 1654-1081 TRITA-CHE-Report 2011:47 © Lingquan Deng, 2011 E-Print, Stockholm

献给我的父亲、母亲和姐姐
To my family


Lingquan Deng, 2011: “Photochemical Surface Functionalization: Synthesis, Nanochemistry and Glycobiological Studies”, Chemistry/Organic Chemistry, KTH Chemical Science and Engineering, Royal Institute of Technology, SE-100 44 Stockholm, Sweden.
Abstract
This thesis mainly deals with the development of photochemical approaches to immobilize carbohydrates on surfaces for glycobiological studies. These approaches have been incorporated into a number of state-of-the-art nanobio-platforms, including carbohydrate microarrays, surface plasmon resonance (SPR), quartz crystal microbalance (QCM), atomic force microscopy (AFM), and glyconanomaterials. All the surfaces have displayed good binding capabilities and selectivities after functionalization with carbohydrates, and a range of important data have been obtained concerning surface characteristics and carbohydrate-protein interactions, based on the platforms established. Besides, a variety of non-carbohydrate and carbohydrate-based molecules have been synthesized, during which process the mutarotation of 1-glycosyl thiols and the stereocontrol in 1-S-glycosylation reactions have been thoroughly studied. Keywords: Carbohydrates; Glycobiology; Carbohydrate-protein interactions; Perfluorophenylazide (PFPA); Photochemistry; Carbohydrate surface immobilization; Carbohydrate microarrays; Surface plasmon resonance (SPR); Quartz crystal microbalance (QCM); Atomic force microscopy (AFM); Glyconanomaterials; Concanavalin A (Con A); Cyanovirin-N (CV-N); 1-Glycosyl thiols; Mutarotation; 1-S-glycosylation; Stereocontrol.

Abbreviations
Ac Acetyl AFM Atomic force microscopy aq. Aqueous Bn Benzyl BS-II Bandeiraea simplicifolia II BSA Bovine serum albumin Bz Benzoyl CBD Carbohydrate binding domain CD Circular dichroism Con A Concanavalin A CuAAC Cu-catalyzed azide alkyne cycloaddition CV-N Cyanovirin-N Cys Cysteine DCC N,N'-Dicyclohexylcarbodiimide DCM Dichloromethane DIPEA N,N-Diisopropylethylamine DLS Dynamic light scattering DMAP 4-Dimethylaminopyridine DMF Dimethylformamide DMPU N,N'-Dimethylpropyleneurea DMSO Dimethyl sulfoxide DNA Deoxyribonucleic acid EDAC 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide EF Enhancement factor EG Ethylene glycol ELISA Enzyme-linked immunosorbent assay Et Ethyl Gal Galactose GBP Glycan-binding protein Glc Glucose GlcNAc N-Acetylglucosamine GNP Glyconanoparticle HIV Human immunodeficiency virus HSQC Heteronuclear single quantum correlation ISC Intersystem crossing ITC Isothermal titration calorimetry Man Mannose Me Methyl MS Mass spectrometry MW Molecular weight NHS N-Hydroxysuccinimide

NIS N-Iodosuccinimide NMR Nuclear magnetic resonance NOESY Nuclear Overhauser effect spectroscopy OEG Oligoethylene glycol PAAm Polyacrylamide PDT Photodynamic therapy PEG Poly(ethylene glycol) PEO Poly(ethylene oxide) PEOX Poly(2-ethyl-2-oxazoline) PFPA Perfluorophenylazide PNA Peanut agglutinin PP Polypropylene PS Polystyrene PSA Pisum sativum QCM Quartz crystal microbalance QSAR Quantitative structure-activity relationship quant. Quantitative RCA-I Ricinus communis Agglutinin I RNA Ribonucleic acid RT Room temperature SEM Standard error of the mean SFM Scanning force microscopy SPM Scanning probe microscopy SPR Surface plasmon resonance TBA Tetrabutylammonium TEA Triethylamine TEG Triethylene glycol TEM Transmission electron microscopy TfOH Trifluoromethanesulfonic acid THF Tetrahydrofuran TMSOTf Trimethylsilyl trifluoromethanesulfonate Ts 4-Toluenesulfonyl UV Ultraviolet UV-vis Ultraviolet-visible WGA Wheat-germ agglutinin w/w Weight to weight


List of Publications
This thesis is based on the following papers, referred to in the text by their Roman numerals I-VIII:
I. pH-Dependent Mutarotation of 1-Thio-Aldoses in Water. Unexpected Behavior of (2S)-D-Aldopyranoses Rémi Caraballo*, Lingquan Deng*, Luis Amorim, Tore Brinck, and Olof Ramström * These authors contributed equally to this work
J. Org. Chem. 2010, 75, 6115-6121.
II. Stereocontrolled 1-S-Glycosylation and Comparative Binding Studies of Photoprobe-Thiosaccharide Conjugates with Their O-Linked Analogs Lingquan Deng, Xin Wang, Suji Uppalapati, Oscar Norberg, Hai Dong, Adrien Joliton, Mingdi Yan, and Olof Ramström
Submitted for publication
III. Stereoselective Synthesis of Light-Activatable Perfluorophenylazide-Conjugated Carbohydrates for Glycoarray Fabrication and Evaluation of Structural Effects on Protein Binding by SPR Imaging
Lingquan Deng, Oscar Norberg, Suji Uppalapati, Mingdi Yan, and Olof Ramström
Org. Biomol. Chem. 2011, 9, 3188-3198.
IV. Perfluorophenyl Azide Immobilization Chemistry for Single Molecule Force Spectroscopy of the Concanavalin A/Mannose Interaction
Carolin Madwar, William Chu Kwan, Lingquan Deng, Olof Ramström, Rolf Schmidt, Shan Zou, and Louis Cuccia
Langmuir 2010, 26, 16677-16680.
V. Photogenerated Carbohydrate Microarrays to Study Carbohydrate-Protein Interactions Using Surface Plasmon Resonance Imaging
Anuradha Tyagi, Xin Wang, Lingquan Deng, Olof Ramström, and Mingdi Yan
Biosens. Bioelectron. 2010, 26, 344-350.
VI. Multivalent Glyconanoparticles with Enhanced Affinity to the Anti-Viral Lectin Cyanovirin-N

Xin Wang*, Elena Matei*, Lingquan Deng*, Olof Ramström, Angela Gronenborn, and Mingdi Yan * These authors contributed equally to this work
Chem. Comm. 2011, 47, 8620-8622.
VII. Photo-Click Immobilization of Carbohydrates on Polymeric Surfaces - A Quick Method to Functionalize Surfaces for Biomolecular Recognition Studies
Oscar Norberg, Lingquan Deng, Mingdi Yan, and Olof Ramström
Bioconjugate Chem. 2009, 20, 2364–2370.
VIII. Photo-Click Immobilization on Quartz Crystal Microbalance Sensors for Selective Carbohydrate-Protein Interaction Analyses
Oscar Norberg, Lingquan Deng, Teodor Aastrup, Mingdi Yan, and Olof Ramström
Anal. Chem. 2011, 83, 1000-1007.
Papers not included in this thesis:
IX. Control of the Ambident Reactivity of the Nitrite Ion
Hai Dong, Martin Rahm, Lingquan Deng, Tore Brinck, and Olof Ramström Submitted for publication
X. Synthesis of Glyconanomaterials via Photo-Initiated Coupling Chemistry
Xin Wang, Oscar Norberg, Lingquan Deng, Olof Ramström and Mingdi Yan
ACS Symp. Ser. 2011, in press.
XI. A Novel Carbohydrate-Anion Recognition System
Hai Dong, Lingquan Deng, and Olof Ramström
Preliminary manuscript

Table of Contents
Abstract Abbreviations List of publications 1. Introduction ..................................................................................... 1
1.1. Carbohydrates and glycobiology – A general background ..................... 1 1.2. Carbohydrate chemistry ......................................................................... 2
1.2.1. Introduction .................................................................................................. 2 1.2.2. Mutarotation, conformation and anomeric effect ......................................... 3 1.2.3. Glycomimetics ............................................................................................. 5
1.3. Glycobiology .......................................................................................... 6 1.3.1. Biological roles of glycans ........................................................................... 6 1.3.2. Glycans in medicine and biotechnology ...................................................... 6 1.3.3. Glycan-mediated recognition ....................................................................... 7 1.3.4. Multivalency in carbohydrate-protein interactions ....................................... 7
1.4. Methods and tools in glycobiological studies ......................................... 7 1.5. Carbohydrate immobilization ............................................................... 10
1.5.1. Survey of methods .................................................................................... 10 1.5.2. Photochemical immobilization methods .................................................... 13 1.5.3. Three photochemical carbohydrate immobilization approaches ............... 15
1.6. Aims of this thesis ................................................................................ 17 2. Syntheses ..................................................................................... 19
2.1. Synthesis of non-carbohydrate molecules ........................................... 19 2.1.1. Synthesis of linker molecules .................................................................... 19 2.1.2. Synthesis of PFPA derivatives .................................................................. 21
2.2. Synthesis of non-PFPA derivatized glycosides .................................... 23 2.2.1. Synthesis of 1-thioglycosides .................................................................... 23 2.2.2. Synthesis of oligosaccharides ................................................................... 25 2.2.3. Synthesis of carbohydrate-azides ............................................................. 27
2.3. Synthesis of PFPA-conjugated carbohydrates .................................... 28 2.3.1. Synthesis of O-linked PFPA-carbohydrates .............................................. 28 2.3.2. Synthesis of S-linked PFPA-carbohydrates .............................................. 29 2.3.3. Synthesis of oligosaccharide-PFPA conjugates ........................................ 31
3. Mutarotation of 1-thioaldoses and stereocontrol in 1-thioglycosylation (Papers I and II) ........................................................... 35
3.1. Mutarotation of 1-thioaldoses in aqueous media (Paper I) .................. 35 3.1.1. Introduction ................................................................................................ 35 3.1.2. pD-dependence of the mutarotation .......................................................... 35 3.1.3. Reversibility of the mutarotation ................................................................ 38 3.1.4. Proposed mechanisms of the mutarotation of 1-thioaldoses .................... 39 3.1.5. Conclusions ............................................................................................... 39

3.2. Stereocontrol in 1-thioglycosylation (Paper II) ..................................... 40 3.2.1. Introduction ................................................................................................ 40 3.2.2. Occurrence and identification of the anomerization .................................. 40 3.2.3. Controlling the stereochemistry in the synthesis ....................................... 41 3.2.4. Conclusions ............................................................................................... 44
4. Development of three carbohydrate immobilization approaches and their applications (Papers II-VIII) ...................................................... 45
4.1. Carbohydrate-conjugated photoprobes (Papers II-IV) ......................... 46 4.1.1. Photo-induced glycosurface fabrication approach I .................................. 46 4.1.2. SPR imaging analysis................................................................................ 47 4.1.3. Affinity analyses using SPR and ITC ......................................................... 49 4.1.4. AFM single molecular force analysis ......................................................... 51 4.1.5. Conclusions ............................................................................................... 55
4.2. Direct photoconjugation (Papers V and VI) .......................................... 56 4.2.1. Photo-induced glycosurface fabrication approach II ................................. 56 4.2.2. Carbohydrate microarray studies .............................................................. 57 4.2.3. Glyconanoparticle platform for probing CV-N ............................................ 60 4.2.4. Conclusions ............................................................................................... 65
4.3. Photo-click carbohydrate immobilization (Papers VII and VIII) ............ 66 4.3.1. Photo-induced glycosurface fabrication approach III ................................ 66 4.3.2. Protein binding and surface specificity studies by QCM ........................... 67 4.3.3. Conclusions ............................................................................................... 71
5. Concluding remarks and outlooks ................................................ 73 Acknowledgements Appendix References

1
1. Introduction
1.1. Carbohydrates and glycobiology – A general background
Life is an extraordinarily complex phenomenon, and the understanding of living things has been an everlasting quest in human history. In the 1950s to 1970s, the molecular biology revolution initiated the interrogation of living systems at the molecular level. The central dogma in biology, that biological information flows from DNA to RNA and then to proteins, has been established since then. It is thus tempting to assume that the makeup and functions of cells, tissues, organs, physiological systems and intact organisms can be explained merely by these molecules. However, in fact, a few more classes of molecules, including lipids and carbohydrates, are required to fulfill the task and complete the puzzle.
Lipids and carbohydrates take part in some of the major posttranslational modifications of proteins, and thus help to generate a vast variety of biological complexes inherent in the development, growth and function of intact organisms. Due to the modifications, these entities greatly diversify the effects of the relatively small number of genes in a typical genome. Details and functions of lipids are however not discussed in this thesis. Instead, it will focus on carbohydrates and carbohydrate-containing biomolecules, which are ubiquitous in living systems and play essential roles in various biological processes, including modulating or mediating various fundamental processes in cell-cell-, cell-matrix-, and cell-molecule interactions, or in the interactions between different organisms, and acting as regulatory switches within the nucleus and cytoplasm.1-3
The chemistry of carbohydrates has drawn considerable interest since the late 19th century. However, carbohydrates had long been considered mainly as an energy source or as structural materials, and were believed to lack other biological functions. Nevertheless, with the development of new technologies for elucidating the structures and functions of carbohydrates, the word “glycobiology” was first coined in the late 1980s,4 to recognize the new frontier of molecular sciences, which combined the disciplines of carbohydrate chemistry and biochemistry with a modern understanding of the cell biology of carbohydrates, especially that of their conjugates with proteins and lipids.
Glycobiology can be defined as the branch of science concerned with the role of sugars in biological systems.3 In the broadest sense, it is the study of the structure, biosynthesis and biology of saccharides that are widely distributed in nature. This research field has grown very rapidly in recent years. It involves studies in the chemistry of carbohydrates, the enzymology of carbohydrate

2
formation and degradation, the recognition of carbohydrates by specific proteins, the roles of carbohydrates in complex biological systems and their analysis or manipulation by various techniques.3
1.2. Carbohydrate chemistry
1.2.1. Introduction
In nature, carbohydrates are produced in the process of photosynthesis, which converts the light energy of the sun to chemical energy by combining carbon dioxide and water to form carbohydrates and molecular oxygen (Scheme 1.1).
Scheme 1.1 Process of photosynthesis.
Structurally, the smallest carbohydrate would have three carbons, with an aldehyde or keto group and two hydroxyl groups. Carbohydrates with three or more carbons are successively called trioses, tetroses, pentoses, hexoses, heptoses, octoses, and so on. Carbohydrates exist as aldoses, which are polyhydroxyaldehydes, and ketoses, being polyhydroxyketones. Each aldose and ketose further exists as two enantiomers, and the prefixes “D” and “L” have been used to distinguish them. Thus a D- or L-carbohydrate is defined by the fact that the hydroxyl group on the asymmetric carbon atom furthest from the most oxidized carbon atom (aldehydo, keto, or carboxyl group) is positioned right or left in the carbohydrate’s Fischer projection (Figure 1.1, C5-OH), respectively.
Figure 1.1 Open-chain and ring forms of mannose.
A monosaccharide is the simplest form of carbohydrates that cannot be further hydrolyzed. It can exist in either open-chain or ring form. Oligosaccharides are linear or branched chains of monosaccharides attached to one another by glycosidic linkages, and polysaccharides are typically referred to structures composed of repeating oligosaccharide motifs.
A chiral center, termed the anomeric center, at C-1 for aldo carbohydrates or at C-2 for keto carbohydrates is generated in the formation of the carbohydrate ring. From this center, the monosaccharides are attached to other residues by glycosidic linkage, and typically the residues are hydroxy-, amino-, or

3
thio-groups, or interlinked with the carbohydrate ring by a carbon atom, forming O-, N-, S- or C-glycosides, respectively. When the attached residue is a non-carbohydrate structure, it is usually termed as aglycone, and the carbohydrate counterpart as glycone. Depending on the stereochemical relationship between the hydroxyl groups attached to the anomeric carbon and to the highest numbered chiral carbon in the carbohydrate ring, α or β linkages are designated. More specifically, when the groups are cis (bound to the center carbons in the same direction), the anomer is defined as α anomer, while if they are trans (bound to the center carbons in opposite directions), the β configuration is designated. The anomers are diastereomers.
It is important to realize that the two anomers confer very different structural properties and biological functions upon saccharide sequences that are otherwise identical in composition. A good illustration for this in nature is the marked differences between starch and cellulose. Though both are homopolymers of glucose, the former is mainly α1-4 linked and the latter β1-4 linked throughout the molecule. Even for monosaccharide anomers, they can possess distinct biological activities.5 In this case, they are normally of similar physicochemical properties which can hamper the separation of the two stereoisomers.
1.2.2. Mutarotation, conformation and anomeric effect
The anomeric center of all reducing saccharides (those with the reducing ability of the aldehyde or ketone in its terminal monosaccharide component) can undergo an interconversion of isomers, accompanied by a change in optical rotation. This process is known as mutarotation. It can be catalyzed by dilute acid or base. In the process, the monosaccharide ring opens up and then recloses to form a ring with the other anomeric configuration, or form other isomers with different ring sizes (Scheme 1.2). Aldohexoses form six-membered rings through a C-1—O—C-5 ring closure, and five-membered rings via C-1—O—C-4 ring closure, producing pyranoses and furanoses, respectively.

4
Scheme 1.2 Mutarotation of D-mannose.
Pyranoses mainly exist in a chair conformation, but can also exist in for example half-chair, boat and skew conformations. The chair form is the most stable and only the skew conformation has a comparable energy minimum, although still about 20 kJ higher than the chair form. Major conformations for furanoses are the envelope and twist forms. The chair conformation of pyranoses further exists in two isomeric forms, as 1C4 and 4C1. The letter C stands for “chair” and the numbers indicate the relative locations of the C-1 and C-4 carbon atoms, either above or below the reference plane of the chair, which is made up by the other four atoms in the carbohydrate ring (cf. Figure 1.1).
In a molecule with a chair conformation, the equatorially positioned substituent should normally be energetically favored for steric reasons, compared to its axial form. However, the anomerically bound groups (aglycones) in carbohydrates do not completely follow this rule. Thus, the term “anomeric effect” was coined to describe the increased preference for an electronegative substituent, at the anomeric carbon in a carbohydrate, to be in an axial rather than equatorial orientation.6,7 The intramolecular electrostatic interactions of the two dipoles next to the anomeric center have been used to explain the anomeric effect (Figure 1.2). Thus the anomeric configurations arise as a result of the partial neutralization of the two dipoles. However, it is noteworthy that a modern understanding of the anomeric effect includes electrostatic effects, molecular orbital interactions, as well as solvent effects, which all dictate the conformational preferences.

5
Figure 1.2 Dipole interactions and anomeric effect.
1.2.3. Glycomimetics
Due to the difficulties of purifying necessary carbohydrate ligands from natural sources or synthesizing oligosaccharides that occur naturally, the strategy of designing carbohydrate ligands as simplified carbohydrate derivatives, which are often called glycomimetics, emerged and has been constantly developed. Many advantages are associated with the synthesis of glycomimetics, including easier access than the natural analogs, facile modification of the structural properties, and that the glycomimetics can be designed as non-biodegradable compounds, as well as low molecular weight derivatives (drug candidates). It is not guaranteed that glycomimetics will always work as ligands for the target receptors, but it is possible that they display even higher receptor affinities than their naturally-occurring analogs.
When the structure of the receptor under investigation is known, it dramatically helps the rational design of glycomimetics. If the receptor structures, for example those of lectins, are unknown, then systematic derivatization of the ligands is the way to go for identifying the essential structural features for potent receptor binding. With the help of synthetic glycomimetics, carbohydrate-protein interactions can be studied with regard to their structural and functional details. Furthermore, in a therapeutic context, carbohydrate-protein interactions can also be modulated or inhibited by glycomimetics.
Two means of producing glycomimetics, among others, are worth mentioning. The first concerns the strategies to block the anomeric reactivities of the glycosidic linkage, which are based on the substitution of the hemiacetal function with a more stable one. These enable the generation of glycomimetics that are more resistant to glycosidases and to acidic cleavage, providing promising potential therapeutics. To achieve such a goal, one of the two hemiacetal oxygen atoms, or both, should be replaced. Figure 1.3 shows some possible modifications around the anomeric center to generate such glycomimetics.

6
Figure 1.3 Glycomimetics with modifications around the anomeric center.
The other means is the preparation of multivalent glycoconjugates which mimic the polyantennary complex-type carbohydrates found on naturally-occurring molecular constructs or biological surfaces. The design of multivalent neoglycoconjugates with respect to the multivalency principle is of fundamental importance in studying carbohydrate-protein interactions.8 Various scaffolds, including dendrimers, polymers, liposomes and different nano-materials, have been developed to help achieving this goal.
1.3. Glycobiology
1.3.1. Biological roles of glycans
Glycan is a term that refers to any form of carbohydrates, from mono- to polysaccharides, and either free or covalently linked to another molecule.3 Due to the ubiquitous and complex nature of glycans, their biological roles are substantially varied. Glycans thus play various subtle to crucial roles for the development, growth, function and survival of an organism. Furthermore, the diverse roles can be categorized into two parts: one is about the structural and modulatory functions, which involve the glycans themselves or their modulation toward the molecule to which they are attached; the other mainly concerns specific recognition of glycans by glycan-binding proteins.
1.3.2. Glycans in medicine and biotechnology
Numerous biotherapeutic agents contain glycan components. These range from natural products to rationally designed glycomimetics and recombinant glycoconjugates, for example glycoproteins. The glycan components in these agents play important roles for their biological activity and therapeutic efficacy. Heparin, a sulfated glycosaminoglycan, and its derivatives are among the most commonly used drugs all over the world.9,10 Glycoproteins are by now one of the major products in the biotechnology industry, with sales in tens of billions of dollars annually.11 Moreover, a number of human disease states are characterized by changes in the glycosylation processes that could be of potential diagnostic and/or therapeutic significance.3 As a result, carbohydrate chemistry and glycobiology have been gaining increasing attention and interest in modern biotechnology and pharmaceutical industries.

7
1.3.3. Glycan-mediated recognition
As mentioned above, glycans play two major classes of biological roles. Besides their structural and modulatory functions, however, many of their more specific biological roles are revealed via their recognition by glycan-binding proteins (GBPs). Except for glycan-specific enzymes and antibodies, the GBPs can be classified into two major groups: lectins and glycosaminoglycan-binding proteins.3 Lectins are glycan-binding proteins that are highly specific for their carbohydrate moieties. They bind to glycans specifically by fitting them into shallow but relatively well-defined binding pockets. In contrast, glycosaminoglycan-binding proteins interact with sulfated glycosaminoglycans by cation-anion attractive forces. Though this type of binding is based on ionic interactions, it can be specific as well. These groups of proteins can be either endogenous to the organism that synthesizes their cognate glycans or exogenous. In the former case, studies focus on the development and function of a complex multicellular organism, while in the latter, the interactions between microbial proteins that bind to specific glycans on host cells are mostly studied.
1.3.4. Multivalency in carbohydrate-protein interactions
It has been generally recognized that single-site binding affinities in many lectins are relatively low, usually with Kd values in the micromolar range. For those lectins with low affinities, multivalent interactions between multiple carbohydrate-binding domains (CBDs) and multiple glycans are usually required to generate high-affinity binding, which is more relevant in vivo. Thus, multivalent interactions can be defined as specific simultaneous association of multiple ligands present on a molecular construct or biological surface binding in a cooperative way to multiple receptors expressed on a complementary entity.8
1.4. Methods and tools in glycobiological studies
The development of methods for obtaining various glycan ligands and tools for studying carbohydrate-protein interactions has greatly facilitated modern advances in glycobiology. Two major synthetic strategies have been developed for efficient preparation of oligosaccharides and glycoconjugates: enzymatic synthesis and chemical synthesis.12 Programmable one-pot synthesis and automated solid-phase assembly of oligosaccharides are excellent examples in this pursuit.13,14 Tools for studying carbohydrate-protein interactions have also evolved rapidly in the past few decades. Techniques like X-ray crystallography, UV-vis, fluorescence spectroscopy, circular dichroism (CD) and NMR have greatly aided the insight into the molecular basis of structural features and functional roles of various glycans. More recently, a number of

8
other techniques and tools have been incorporated in glycobiological studies, such as microarrays, biosensors, nano-based technologies and bioinformatical tools, to name just a few. More specifically, these techniques and tools include isothermal titration calorimetry (ITC),15 mass spectrometry (MS),16 carbohydrate microarrays,17 surface plasmon resonance (SPR),18,19 quartz crystal microbalance (QCM),20,21 scanning probe microscopy (SPM),22 glyconanotechnologies,23 computational simulations.24 Among these, two major methods are especially advantageous: solution-based ITC and solid-supported binding techniques, the combination of which can result in comprehensive elucidations of binding performances. ITC can directly provide thermodynamic data of molecular interactions, thus reliably conveying important binding information and supporting binding implications. On the other hand, techniques and tools involving solid supports, such as microarrays, biosensor instrumentations and nanoplatform-based techniques, have drawn increasing attention and interest in glycobiology.25
ITC is a physical technique used for determining thermodynamic parameters of interactions in solution. The binding of glycans to proteins can be measured as a change in free energy by the ITC technique.15 It directly measures the binding affinity (Ka), enthalpy changes (ΔH), and binding stoichiometry (n) of the interactions between binding partners in solution, while the Gibbs energy changes (ΔG) and the entropy changes (ΔS) can be derived from the thermodynamic relationships: ΔG = -RTInKa = ΔH-TΔS, where R is the gas constant and T is the absolute temperature. In operation, increasing amounts of glycan are injected to a fixed concentration of protein solution in a microcalorimetry cell by a syringe. The glycan solution is added at intervals and the heat taken up or evolved from the binding is measured. The heat flow spikes are then integrated with respect to time, providing the total heat exchanged per injection. This heat effect relative to the molar ratio [ligand]/[macromolecule] is then plotted. These data can be used to directly define the thermodynamic parameters of binding and calculate the Kd of the carbohydrate-protein interaction of interest.
Carbohydrate microarrays constitute a welcome development for elucidating carbohydrate-protein interactions in a wide variety of contexts, from cell-protein-, cell-cell- to host-pathogen interactions. It is an extension of both enzyme-linked immunosorbent assay (ELISA)-type formats and modern DNA and protein microarray technologies. The main advantages of microarray analysis lie in its capability to screen a number of biological interactions in parallel, its potential to present arrayed ligands that can mimic cell surface display and generate high binding affinities of the interactions, as well as the minimization of the amount of glycan probes needed for interaction studies. On a single slide, up to hundreds or even thousands of spots can be printed at

9
the same time. This microarray technique has received much attention ever since its introduction into the glycoscientific community, and it has revolutionalized the study of carbohydrate-protein interactions.17,26-34
SPR is a technique for measuring the association and dissociation kinetics of analytes with their complementary receptors. When either the analyte or the receptor is immobilized on the SPR sensor chip and associated with the other in a free form, a change in the refractive index of the layer in contact with a gold film will occur. This change then leads to the transduction of an SPR signal measured in resonance units (RU). The interactions are measured in real time, and thus the association and dissociation data can be obtained. Compared to carbohydrate microarrays, which usually rely on various protein labeling formats in a step-wise batch mode, the SPR technique can evaluate biological interactions on surfaces in a label-free manner.18,19,35-40 Further advantages of SPR include the capability to measure low affinities from millimolar to picomolar range, the requirement of small amount of samples and very fast measurements.
QCM is known as a very sensitive mass-measuring technique in both gas phase and liquid environment. It measures a mass per unit area by monitoring the change in frequency of a quartz crystal resonator. The resonance frequency is disturbed by addition or removal of a small mass on the surface of the acoustic resonator. As a mass is deposited on the surface of the crystal, the mass of the crystal increases. Consequently, the frequency of oscillation decreases compared to the initial value. This frequency change can be quantified and correlated to the mass change using the Sauerbrey equation.41 In the gas phase, the technique has been widely used in determining thin-layer thickness and for gas-adsorption studies.42,43 In liquid environment, affinities of molecules to surface-bound binding partners can be effectively measured, and interactions between biomolecules, carbohydrate-protein interactions, for example, have been investigated.20,21,44,45

10
Atomic force microscopy (AFM), also known as scanning force microscopy (SFM), is a type of scanning probe microscopy with very high resolution, which can reach the order of fractions of a nanometer (over 1000 times better than the optical diffraction limit). AFM has been recognized as one of the foremost tools for measuring, imaging and manipulating matters at nanoscale. The AFM uses a cantilever with a sharp tip at its far end for scanning the specimen surface. During scanning, forces between the tip and the sample, when they are brought in close proximity, would result in a deflection of the cantilever according to Hooke’s law. Different forces can be measured in different situations, including mechanical contact force, van der Waals’ forces, chemical bonding, electrostatic forces, solvation forces, etc. Most commonly, a laser spot is used to measure the deflection by reflecting from the top surface of the cantilever into an array of photodiodes. In recent years, single molecule force spectroscopy, based on AFM, has been extensively used to address, detect and measure biomolecular interactions at the single molecule level.22,46-49
Carbohydrate-functionalized nanomaterials, or glyconanomaterials, are a variety of nanomaterials carrying carbohydrate ligands on their surfaces. These functional nanomaterials have emerged as a very promising platform for studies of carbohydrate-mediated recognitions, and for biomedical imaging, diagnostics, therapeutics and drug delivery. Gold-, silver- and magnetic nanoparticles, quantum dots, polymer nanoparticles, liposomes, silica nanoparticles, carbon-based nanomaterials including fullerenes, carbon nanotubes and graphene sheets, and virus-based nanoparticles, have been demonstrated to be great core materials for the build-up of such nanoplatforms. The platforms combine the unique properties of nanometer-scale objects with the capability to present carbohydrate ligands in a multivalent manner, greatly enhancing the binding affinities to their complementary partners and better mimicking relevant biological recognition events.23,50-52 To achieve a good performance of such glyconanomaterials, the proper presentations of carbohydrate ligands on the surfaces, which are closely related to the coupling chemistry, the type and length of the spacer and the ligand density, are critical.
1.5. Carbohydrate immobilization
1.5.1. Survey of methods
Partly because of the fact that solid-surfaces are better simulacra to cell surfaces than solution systems, and in part due to their ability to provide comprehensive hierarchical levels of molecular information,53-55 solid-supported techniques are playing increasingly important roles in studies of various carbohydrate-protein interactions. As a result, the development of

11
methods for immobilizing carbohydrates on surfaces and resins has become an important research topic.
Methods for carbohydrate immobilization fall into two general strategies: the non-covalent and covalent immobilizations. For the former strategy, the carbohydrate ligands are physisorbed to surfaces via non-covalent interactions, including electrostatic- and van der Waals’ interactions, hydrophobic effects, as well as hydrogen bonding. Non-conjugated polysaccharides,34 neoglycoproteins,56 neoglycolipids,26,57-59 carbohydrates with fluorous tags,60,61 ionic glycoconjugates,62,63 and biotinylated carbohydrates,64,65 have for example been developed as glycan-bearing ligands in non-covalent immobilization routes (Figure 1.4). Various surface-based carbohydrate-receptor studies are subsequently feasible as long as sufficient stability of the system can be preserved after incubation with the biological target and the washing steps.
non-covalentimmobilization
O
OH
nitrocellulosenitrocellulose
O
OH
OO
OH
OO
OH
O n
a
b c
d
ef
O
SAu
SSS SS
SAu
SSS SS
O
OH
O
O
OH
O
f luorinated f luorinated
O
OH
O
C8F17
O
OH
O
O
OH
O
streptavidin
streptavidin
b
O
OH
O
b
O
OH
O
Figure 1.4 Different non-covalent immobilization methods. (a) Physisorption of
non-conjugated polysaccharides on nitrocellulose coated surfaces. (b) Attachment of
neoglycoproteins. (c) Monolayer formation of neoglycolipids. (d) Immobilization of
fluorous tag derivatized carbohydrates on fluorinated surfaces. (e) Assembly of ionic
glycoconjugates via layer-by-layer deposition. (f) Attachment of biotinylated
carbohydrates to streptavidin derivatized surfaces.

12
Covalent immobilization methods, in comparison, offer more stable functional surfaces than the non-covalent alternatives. Thus the covalent strategy has drawn greater attention and the majority of surface functionalization methods are based on covalent bond formation. Figures 1.5.1 and 1.5.2 depict a range of different chemical protocols for covalent immobilization of carbohydrates on surfaces. These include:
1) Immobilization of functionalized glycans on surfaces: self-assembly of carbohydrate-alkanethiols on gold surfaces,40,66-68 coupling between maleimide-thiol,69,70 hydrazide-epoxide,71,72 amine-cyanuric chloride,73 amine-N-hydroxysuccinimide (NHS) ester,74-76 and aminooxy-aldehyde functionalities,77 attachment via Diels-Alder,28 Staudinger,78 and copper catalyzed 1,3-dipolar cycloaddition reactions,20,21,57,68 immobilization of neoglycoproteins to aldehyde slides,70,79 and site-specific photo-immobilization of carbohydrates on surfaces.17,18,80
Figure 1.5.1 Different covalent immobilization methods, part 1. (a) Self-assembly of
alkanethiol-functionalized carbohydrates on gold surfaces. (b) Immobilization of
carbohydrates using different coupling chemistries between two typical functionalities. (c)
Attachment of neoglycoproteins to aldehyde surfaces. (d) Immobilization of
photoprobe-derivatized carbohydrates on surfaces.
2) Direct immobilization of non-modified glycans on derivatized surfaces: either through a two-step procedure, e.g., first reacting the free carbohydrates with 2,6-diaminopyridine or N-methylaminooxy-containing bifunctional

13
linkers and then coupling the intermediates to NHS ester-coated surfaces;81,82 or via one-step routes, which are exemplified by immobilizing free carbohydrates on aminooxy- or hydrazide-derivatized surfaces,83-85 and by covalent attachment of unmodified glycans to surfaces derivatized with a layer of photoreactive groups.19,23,86,87
photoprobe
photoprobe
Figure 1.5.2 Different covalent immobilization methods, part 2. (e) Attaching free glycans
via 2,6-diaminopyridine linker on NHS-ester surfaces. (f) Immobilizing free glycans via
N-methylaminooxy linker on NHS-ester surfaces. (g) Direct immobilization of free
carbohydrates on aminooxy- or hydrazide-derivatized surfaces. (h) Direct attachment of
unmodified glycans to surfaces derivatized with a layer of photoreactive groups.
1.5.2. Photochemical immobilization methods
Recently, photochemical immobilization methods to attach carbohydrates on solid supports have shown great potential as a result of their simplicity, robustness and versatility. Photoreactive functional groups like phenylazido groups,17-23,44,88-93 benzoyl groups,87,94 and diazirine groups,80,86 which can be converted under UV irradiation into nitrenes, ketyl radicals and carbenes,

14
respectively, have been used for such photochemical functionalization purposes. Photochemical methods have proven to be advantageous from both chemical and surface engineering points of view. From the chemical point of view, the methods 1) provide a clean and easy means for carbohydrate immobilization in which only photons are required as a reagent, 2) give robust surfaces that ensure reproducible results and withstand storage, 3) are versatile to be applicable to a wide range of target molecules, even those that are normally not reactive to form covalent linkages, 4) are compatible with glycan structures and aqueous media, and 5) are adaptable to both free and derivatized carbohydrate structures. From the surface engineering point of view, the photochemical functionalization methods 1) are versatile to be applicable to a big variety of substrate structures, 2) are easily adaptable to many state-of-the-art biotechnologies, and 3) are controllable by external means, for example, patterning of such surfaces can be achieved by applying a photomask.
Aryl azides are well-known chromophores that can insert into C-H, N-H bonds and react with unsaturated carbon-carbon bonds.95 The photochemistry utilized in our studies is based on a subgroup of aryl azides, the perfluorophenylazides (PFPAs).89,96,97 The photochemistry of phenyl azide is complex (Scheme 1.3). Under UV irradiation or heat, the phenyl azide decomposes and forms a singlet phenyl nitrene, which is highly reactive and can quickly rearrange to a seven-membered ketenimine (Scheme 1.3, pathway 1). The ketenimine can further react with amines to give azepinamines or produce polymer tars in the absence of a nucleophile. The singlet phenyl nitrene can also relax through intersystem crossing (ISC) to generate the triplet phenylnitrene, which preferably happens at low temperature and particularly in the presence of alcohols (pathway 2). The lower energy triplet species can either take part in hydrogen abstraction to produce aniline-type of structures or undergo bi-molecular reactions to yield the corresponding azo-compound. However, these are the two reaction pathways that are not useful for carbohydrate immobilization. To increase the percentage of useful insertion or addition reactions as shown in the third pathway (pathway 3), fluorine substituents have been introduced on the phenyl ring, either in a perfluorinated manner or ortho to the azido group. This modification can raise the energy barrier of the ring-expansion reaction and the lifetime of the fluorinated singlet phenyl nitrene can thus be greatly increased, so that the desired insertion or addition reaction yields are largely enhanced, too.

15
Scheme 1.3 Simplified illustration of phenylazide photochemistry. Modified from
reference 97.
In fact, the PFPA chemistry has been applied to attaching organic molecules on surfaces, by either thermal- or photo-activation, resulting in versatile surface modifications for a wide range of applications.17-23,44,88-93,98-100
1.5.3. Three photochemical carbohydrate immobilization approaches
Three different approaches to photochemically immobilize carbohydrates on surfaces can be envisaged: 1) a carbohydrate-conjugated photoprobe approach, 2) a direct-photoconjugation approach, and 3) a “hybrid” carbohydrate immobilization approach. The first approach (referred to as approach I throughout following text) is achieved by first derivatizing the carbohydrate structures with a photoprobe, and then applying the entire conjugate molecule for surface functionalization in a one-step procedure under UV irradiation (Figure 1.6a). In our studies, PFPA was used as the photoprobe (Papers II-IV).18,22,101 This approach ensures site-specific attachment of carbohydrate ligands on surfaces, but it usually requires more labor-demanding work on the synthesis of each carbohydrate-photoprobe conjugate.

16
Figure 1.6 The three approaches developed to photochemically immobilize
carbohydrates on surfaces. (a) The carbohydrate-conjugated photoprobe approach. (b)
The direct-photoconjugation approach. (c) The “hybrid” carbohydrate immobilization
approach. The letters X, Y, Z and W represent different functional groups.
The direct-photoconjugation approach (approach II) concerns first attaching a bifunctional photoprobe on surfaces and then photochemically inserting the photoprobe into the intended target carbohydrate structures (Figure 1.6b). Bifunctional PFPA-thiols were chosen as model photoreactive molecules in our studies (Papers V and VI).19,102 The advantage of this approach lies in its ability to immobilize un-derivatized glycans directly onto surfaces, which can greatly circumvent the difficulties in chemical derivatization of glycan structures, facilitate the process of surface modification and make the method easily applicable to immobilizing complex oligosaccharides and polysaccharides.
The third approach is a hybrid type of carbohydrate immobilization (approach III), based on a “photo-click” immobilization protocol (Figure 1.6c). Specifically, it refers to the method of first attaching the bifunctional PFPA-alkyne linker on surfaces and then coupling it to different carbohydrate-azide molecules using the copper catalyzed 1,3-dipolar cycloaddition “click” chemistry (Papers VII and VIII).20,21 This approach enables site-specific attachment of carbohydrate ligands on surfaces for subsequent biological interrogations, and considerably reduces the work load in organic synthesis, since the simply modified carbohydrate-azide structures are more easily obtainable compared to the entire carbohydrate-photoprobe conjugate molecules. In addition, a large number of carbohydrate-azide

17
structures are readily accessible from commercial sources and among research groups in the glycoscience community. Thus, this approach would enable the direct use of a wide range of carbohydrate structures.
1.6. Aims of this thesis
The aims of the projects in this thesis are to: 1) Develop different approaches to photochemically functionalize surfaces with carbohydrates for glycobiological studies. Three such approaches have been envisaged and developed for the purpose. In approach I, the main task was to further develop synthetic methodologies for obtaining target carbohydrate-photoprobe structures in a simpler, yet more efficient way. In approach II, the focus has been directed to engineering surfaces and interfaces for direct photoconjugation of free carbohydrates. In approach III, the effort on developing methods to easily and significantly expand the carbohydrate ligand diversity was emphasized. A variety of carbohydrate structures, ranging from monosaccharides to more complex structures, have been designed and synthesized. Both photoprobe-derivatized and unmodified glycan structures, from monosaccharides to trisaccharides, were targeted.
2) Design and develop different surface coatings and surface chemical structures that are adapted to the photoligation process and can minimize non-specific adsorption and enhance signal-to-noise ratio; applying the carbohydrate-photoprobe structures to a variety of surface materials was also considered. Concerning surface coatings, different polymeric materials were mainly targeted and evaluated; while for surface chemical structures, the effects of different structural features, including carbohydrate linkage type, spacer length and spacer structure on subsequent protein binding have been evaluated. Moreover, different glycan structures have been applied to various types of surface materials, such as bare gold surfaces, polymer-coated gold surfaces or glass slides, and carbon-based nanomaterials (data concerning the last type of materials not shown in this thesis).
3) Combine the photochemical immobilization methods with modern nano- and biotechniques to establish a variety of state-of-the-art platforms, and apply such platforms for carbohydrate-protein binding studies. These nano- and biotechniques include the microarray-, SPR-, QCM-, AFM- and nanomaterials-based technologies as mentioned above. Producing functionalized surfaces with good specificity and sensitivity and trying to obtain quantitative analysis of the interactions are basic goals in this part. Moving from “proof-of-concept” studies to probing more biologically relevant systems utilizing the established platforms is also within our research pursuit.

18
4) In addition to all the planned aims, solving those unexpected problems identified during the course of research is another continuous effort for us. The studies of mutarotation of 1-thioaldoses and stereocontrol in 1-thioglycosylation reactions, to some extent, belong to this type (Papers I and II).101,103

19
2. Syntheses
In order to develop the photochemical carbohydrate immobilization methodologies and investigate carbohydrate-protein interactions, a variety of chemical structures need to be synthesized. Easy, efficient, and stereoselective syntheses of target molecules are thus highly demanded. To meet the objectives of this thesis, a series of non-carbohydrate structures, including linker molecules and PFPA derivatives were first designed and synthesized. These structures were used either in further synthesis of carbohydrate derivatives or directly for surface modifications and functionalizations.
Secondly, a number of non-PFPA derivatized glycosides were targeted and synthesized. This range of molecules include 1-glycosyl thiols, oligosaccharides and carbohydrate-azides. The original purpose of synthesizing 1-glycosyl thiols was to develop easier, yet more efficient synthetic routes for obtaining desired PFPA-conjugated carbohydrate structures. However, during the process, a conspicuous mutarotation phenomenon of the 1-glycosyl thiols was observed and identified. As a result, more such structures were synthesized and the mutarotation properties of these molecules thoroughly studied. The oligosaccharides and carbohydrate-azide molecules were used in the development of carbohydrate immobilization approaches II and III (see pages 15 and 16 ) for studying protein bindings.
Thirdly, three groups of PFPA-conjugated carbohydrates were stereoselectively synthesized, including O-linked PFPA-carbohydrates, S-linked PFPA-carbohydrates and oligosaccharide-PFPA conjugates. The stereoselective formation of the glycosidic linkage is one of the most challenging aspects in glycoconjugate and oligosaccharide synthesis.104 This remains a challenge for not only O-linked structures but also their S-linked analogs. However, these challenges were addressed to ensure the desired stereochemistry in different target structures. The obtained final structures were then evaluated against the interrogating proteins.
2.1. Synthesis of non-carbohydrate molecules
2.1.1. Synthesis of linker molecules
A number of linker molecules with different lengths were synthesized as spacers to link the carbohydrate and PFPA moieties in the PFPA-carbohydrate structures. The linkers were based on the oligoethylene glycol (OEG) motif, which has been widely adopted for various biotechnological and biomedical applications.105 These were primarily chosen because of their water solubility, stability and their ability to reduce non-specific binding of proteins. The

20
integration of ethylene glycol structures with defined length into other synthetic molecules usually requires a selective funtionalization and prolongation of symmetrical diols or diol derivatives. Thus, an iterative method to produce different bifuntional and monodispersed OEGs with up to 9 units was developed (Scheme 2.1).18
For the shortest linker, triethylene glycol (TEG) derivative 4, the commercially available chloride 1 was chosen as starting material. The TEG azide 2 was prepared quantitatively from the chloride 1 by use of sodium azide in DMF at 90 °C overnight. Tosylation at the other end of azide 2 was achieved in good yield according to an established method.106 SN2 substitution of the tosylate 3 was subsequently achieved by use of sodium iodide in DMF at 50 °C for 5 hours and the desired iodide 4 was obtained in a yield of 82%. To make longer OEG derivatives, OEG diols 5 and 9 were first transformed into OEG ditosylates 6 and 10, respectively. The ditosylates were subsequently prolonged by nucleophilic substitution with the deprotonated azide 2 at one end of the molecule to afford tosylates 7 and 11 in fair yields. The final iodides 8 and 12 were obtained following similar conditions as for linker 4.
Scheme 2.1 Synthesis of iodo-linkers 4, 8, and 12. a) NaN3, DMF, 90 °C, overnight
(quant.); b) TsCl, TEA, DMAP, THF, 0 °C-RT, 12 h (78%); c) NaI, DMF, 50 °C, 5 h (82%);
d) TsCl, TEA, DMAP, THF, 0 °C-RT, overnight (81%); e) 2, NaH, DMF, 1.5 h (59%); f) NaI,
DMF, 50 °C, 5 h (81%); g) TsCl, TEA, DMAP, DCM, 0 °C-RT, 2 h (84%); h) 2, NaH, DMF,
3 h (63%); i) NaI, acetone, reflux, 1 h (86%).
Several other linkers were synthesized to reduce non-specific protein bindings, assist best binding capacity as diluter molecules (molecules that are used to “dilute” the other major functional molecules in the mixture), or correct for the non-specific protein bindings as control molecules in subseuqent surface binding studies. These molecules were used for either the SPR or QCM platform. For the preparation of photoreactive SPR surfaces, non-PFPA-derivatized linkers (see one of the examples in Scheme 2.2) were synthesized as diluter molecules to assist optimal interactions and reduce non-specific binding.19 Again, the commercially available chloride 1 was chosen as starting material. An iodo intermediate was generated by substitution of the chloro functionality in compound 1 using sodium iodide, and thioester 13 was

21
subsequently obtained in the same pot using potassium thioacetate. Hydrolysis of thioester 13 resulted in the desired thiol 14 and disulfide 15 in a combined quantitative yield.
Scheme 2.2 Synthesis of thiol 14 and disulfide 15. a) i. NaI, DMF, 90 °C, 2 h; ii. KSAc,
DMF, 80 °C, 6 h (71%); b) K2CO3, MeOH, 3 h (quant.).
For the QCM platform, a control surface made from azidoethanol 21 (Scheme 2.3) was produced to correct for non-specific protein binding. This was achieved by a three-step procedure: attachment of NHS-derivatized PFPA (NHS-PFPA) ester on the surface, coupling of bifunctional linker 19, and finally introduction of the hydroxyl linker 21 through click chemistry.21 Monofunctionalization of diol 5 produced alkyne derivatized linker 16, followed by tosylation, azido substitution and reduction of the azide group at the other end of the molecule. These yielded the alkyne-functionalized amine linker 19. The azidoethanol 21 was obtained by substitution of the chloro group of chloride 20 using sodium azide.
Scheme 2.3 Synthesis of alkyne-derivatized amine 19 and azide 21. a) KOH, propargyl
bromide, 60 °C, 3 h (36%); b) TsCl, KOH, 0 °C, 2 h (quant.); c) NaN3, TBAI, DMF, 45 °C,
overnight (67%); d) i. PPh3, THF, 30 °C, 19 h; ii. H2O, 30 °C, 25 h (70%); e) NaN3, TBABr,
110 °C, 18 h, 99%.
2.1.2. Synthesis of PFPA derivatives
Besides the non-PFPA derivatized linkers, several PFPA derivatives were also designed and synthesized, either for further incorporation into the PFPA-carbohydrate molecules or for separate use in different surface chemistries. For example, NHS-PFPA ester 25 was produced from commercially available pentafluorophenyl ester 22 (Scheme 2.4), via substitution using sodium azide, hydrolysis, and esterification using carbodiimide chemistry.107 The ester 25 was used for the development of both carbohydrate immobilization approach I in synthesizing PFPA-carbohydrate structures and approach III in stepwise surface derivatization.18,20

22
Scheme 2.4 Synthesis of NHS-PFPA ester 25. a) NaN3, Acetone/H2O (2:1), 90 °C, 2 h
(quant.); b) NaOH aq. (20% w/w):MeOH (1:18), 5 h (97%); c) NHS, EDAC, 35 °C, 25 h
(94%).
PFPA functionalized disulfide linker 27 (Scheme 2.5) was produced by Steglich esterification,108 in which PFPA acid 24 and disulfide diol 26 were reacted in the presence of DCC and catalytic amount of DMAP. The compound was applied for the development of carbohydrate immobilization approaches I and III in double surface photoligation.18,20,22
Scheme 2.5 Synthesis of PFPA disulfide 27. a) DMAP, DCC, DCM, 0 °C-RT, dark, 12 h,
40%.
A longer disulfide linker 29 (Scheme 2.6) was synthesized from starting thiol 28 by iodine promoted oxidation. The resulting disulfide 29 was further reacted with PFPA acid 24 to produce structure 30 by the same method as for its analog 27. Both disulfide 30 and its reduced thiol product 31 could be used for the development of carbohydrate immobilization approach II in goldnanoparticle surface functionalization.102

23
Scheme 2.6 Synthesis of PFPA-disulfide 30 and PFPA-thiol 31. a) I2, EtOH, 0.5 h
(quant.); b) DMAP, EDAC, DCM, 0 °C-RT, dark, 14 h (37%); c) Zn/HCl, EtOH/MeCN (1:1),
1 h (82%).
Alkyne-derivatized PFPA 32 (Scheme 2.7) was produced by reacting the NHS-PFPA ester 25 and amine 19 in a straightforward way. This PFPA derivative was used for further coupling with carbohydrate ligands on solid-support for a stepwise functionalization.21
Scheme 2.7 Synthesis of alkyne-derivatized PFPA 32. a) MeCN, overnight (70%).
2.2. Synthesis of non-PFPA derivatized glycosides
2.2.1. Synthesis of 1-thioglycosides
The initial objective of producing 1-glycosyl thiols was to develop an efficient synthetic route directly using unprotected glycosyl donors for linker conjugation, circumventing the labor-intensive and time-consuming nature of the protection-glycosylation-deprotection routes generally required for synthesizing their O-linked analogs. However, in the process, mutarotation of 1-glycosyl thiols was observed and identified. Thus, a number of 1-glycosyl thiols/thiolates were synthesized to expand the scope of the carbohydrate analogs for a detailed study of the mutarotation process. Moreover, several of these and a few other analogous structures were also used for the original synthetic purpose.

24
Four common 1-glycosyl thiols 33-36 were thus synthesized starting from either their corresponding commercially available free aldopyranoses 33a-36a or peracetylated aldopyranoses 33b-36b (Scheme 2.8). After BF3·Et2O promoted thioacetate substitution and Zemplén deacetylation,45 1-thio-β-D-glucopyranose 33, 1-thio-β-D-galactopyranose 34, 1-thio-α-D-mannopyranose 35 and 1-thio-α-D-altropyranose 36 were accessed in good overall yields.
Scheme 2.8 Synthesis of 1-glycosyl thiolates 33-36. a) I2, Ac2O, 0 °C-RT, 1 h (quant.); b)
BF3·Et2O, HSAc, DCM, 0 °C-RT, 24 h (76-89%); c) NaOMe, MeOH, 2 h (≤ 90%).
Disaccharide thiol derivative, 1-thio-β-D-lactose 40, was also obtained following the same synthetic route (Scheme 2.9).
Scheme 2.9 Synthesis of 1-thio-β-D-lactose 40. a) I2, Ac2O, 0 °C-RT, 1 h (quant.); b)
BF3·Et2O, HSAc, DCM, 0 °C-RT, 24 h (78%); c) NaOMe, MeOH, 2 h (86%).
In synthesizing 1-thio-β-L-fucopyranose 44 (Scheme 2.10), the first two steps were similar, but the final deprotection was achieved using lithium hydroxide.109
O
OH
OH
OH
HO
a O
OAc
OAc
OAc
AcO
O
OAc
SAcOAc
AcO
b O
OH
SHOH
HO
c
41 444342
Scheme 2.10 Synthesis of 1-thio-β-L-fucopyranose 44. a) I2, Ac2O, 0 °C-RT, 1 h (quant.);
b) BF3·Et2O, HSAc, DCM, 0 °C-RT, 24 h (84%); c) i. LiOH, MeOH, H2O, 4 h; ii. H+
exchange resin (92%).
Only few reports on the efficient synthesis of 1-thio-α-L-fucopyranose 47 could be found in the literature. However, the α-thiofucose 47 was still accessible through a straightforward strategy (Scheme 2.11), albeit in relatively low overall yield. Thus, thioacetate 45 was formed prior to the

25
peracetylation, which led to the desired α-anomer 46 in 30% yield over two steps.110 Subsequent deacetylation by lithium hydroxide provided the final product 47.
Scheme 2.11 Synthesis of 1-thio-α-L-fucopyranose 47. a) HSAc, HCl, 0 °C-RT, 5 h; b)
Ac2O, pyridine, DMAP (30% over two steps); c) i. LiOH, MeOH, H2O, 4 h; ii. H+ exchange
resin (90%).
2.2.2. Synthesis of oligosaccharides
To expand the carbohydrate ligand variety used in our studies and extend the study from “proof-of-concept” to more biologically relevant systems, several α1-2-linked mannobiose and mannotriose structures were synthesized. These structures are the outmost moieties from Man9 (Figure 2.1), an oligosaccharide structure on the human immunodeficiency virus (HIV) envelop glycoprotein gp120, which binds anti-HIV proteins like Cyanovirin-N (CV-N) at nanomolar concentration.102,111,112
Figure 2.1 Structure of Man9 N-linked oligosaccharide. The α1-2-linked mannobiose and
mannotriose moieties are shown in the dashed boxes.
A central building block for synthesizing these mannose dimers and trimers is the orthoester 51 (Scheme 2.12). The structure can be conveniently obtained starting from commercially available free D-mannose 35a.113 After peracetylation and bromination, orthoester 49 can be subsequently produced from bromide 48 by lutidine promoted ring-closure. Subsequent deacetylation and benzylation afforded desired building block 51 in good overall yield.

26
Scheme 2.12 Synthesis of orthoester 51. a) HBr in AcOH, DCM, 0 °C-RT, 12 h; b)
2,6-lutidine, MeOH/CHCl3 (1:1), 24 h (72% over two steps); c) K2CO3, MeOH, 2 h; d)
NaH, BnBr, DMF, 0 °C-RT, 24 h (87% over two steps).
Orthoester 51 was then transformed to either glycosyl acceptors 54 and 55 or glycosyl donor 56, both via Lewis acid-promoted/catalyzed ring-opening processes (Scheme 2.13).
Scheme 2.13 Synthesis of glycosyl donor 56 and glycosyl acceptors 54 and 55. a)
BnOH, BF3·Et2O, DCM, 0 °C-RT, 2 h (96% for 52), or TMSOTf, DCM, 0 °C, 1 h (quant. for
53); b) NaOMe, MeOH, H+ exchange resin, 1 h (97% for 54, 92% for 55); c) EtSH, HgBr2,
MeCN, 60 °C, 24h (72%).
The coupling of the respective glycosyl donor and acceptor was achieved using NIS/TfOH-mediated chemistry (Scheme 2.14). The α-linkage in dimannosides 57 and 58 was favored by neighboring group participation from the acetyl group at C-2 in the glycosyl donor 56. Zemplén deacetylation and palladium-catalyzed hydrogenolysis of the dimers 57 and 58 provided the reducing mannobiose 61 and non-reducing mannobioside 62, respectively.

27
Scheme 2.14 Synthesis of mannose dimers 61 and 62. a) NIS, TfOH, DCM, -10 °C, 1 h
(78% for 57, 70% for 58); b) NaOMe, MeOH, H+ exchange resin, 3 h (92% for 59, quant.
for 60); c) H2, Pd/C, MeOH, 72 h (88% for 61, 80% for 62).
However, prior to debenzylation, mannose dimers 59 and 60 can also be further linked to more carbohydrate rings (Scheme 2.15). When glycosyl donor 56 was used again with the NIS/TfOH-mediated coupling, mannose trimers 63 and 64 were successfully obtained. Similarly, two types of deprotections subsequently afforded the reducing carbohydrate 67 and the non-reducing structure 68.
Scheme 2.15 Synthesis of mannose timers 67 and 68. a) 56, NIS, TfOH, DCM, -10 °C, 1
h (79% for 63, 76% for 64); b) NaOMe, MeOH, H+ exchange resin, 3 h (77% for 65, 89%
for 66); c) H2, Pd/C, MeOH, 96 h (94% for 67, 85% for 68).
2.2.3. Synthesis of carbohydrate-azides
A range of carbohydrate-azide molecules were synthesized for the purpose of

28
testing the photo-click carbohydrate immobilization approach. All desired structures were synthesized in few steps in high yields following previously reported procedures. Mannose azide 70 and galactose azide 72 were produced from the corresponding pentaacetates 35b and 34b (Scheme 2.16).26 Structure 76 was prepared from the peracetylated N-acetyl-β-D-glucosamine 73 through the oxazoline intermediate 74, followed by acid-catalyzed ring-opening and linker coupling,114,115 and finally Zemplén deprotection.
Scheme 2.16 Synthesis of carbohydrate-azides 70, 72 and 76. a) 21, BF3·Et2O, DCM,
0 °C-RT, 18 h (83%); b) NaOMe, MeOH, 2.5 h (95%); c) 21, BF3·Et2O, DCM, -40 °C-RT,
24 h (69%); d) NaOMe, MeOH, 2.5 h (97%); e) i. TMSOTf, 50 °C, 0.5 h, ii. TEA, 10 min
(98%); f) 21, H2SO4, DCM, 19 h (23%); g) NaOMe, MeOH, 2.5 h (92%).
2.3. Synthesis of PFPA-conjugated carbohydrates
2.3.1. Synthesis of O-linked PFPA-carbohydrates
One of the most straightforward ways to achieve photochemical immobilization of carbohydrates would be to apply carbohydrate structures derivatized with a PFPA tag on surfaces in a single step. Thus, the aforementioned pentaacetates 33b-35b, 38 and azide linker 2 were utilized as starting materials for the synthesis of such PFPA-carbohydrates (Scheme 2.17). The corresponding carbohydrate-azides were produced with the help of BF3·Et2O chemistry. Subsequent deacetylation and azide reduction yielded the unprotected carbohydrate-amine intermediates, which were further coupled to NHS-PFPA 25 to provide the desired PFPA-carbohydrate products. Three monosaccharide derivatives, PFPA-glucose 77, PFPA-galactose 78, PFPA-mannose 79, and one disaccharide derivative, PFPA-lactose 80, were

29
thus synthesized.
Scheme 2.17 Synthesis of PFPA-carbohydrates 77-80. a) BF3·Et2O, DCM, 0 °C-RT, 24 h
(55-63%); b) NaOMe, MeOH, 1 h (quant.); c) i. H2, Pd/C, MeOH, 1 h, ii. 25, DMF, dark,
2.5 h (58-67%).
In order to evaluate the effect of structural difference of the linkers on subsequent protein binding, a linker bearing a triazole-ring was designed to compare with the linear linker structures (Scheme 2.18). Thus, compound 81 was synthesized based on Cu-catalyzed azide-alkyne cycloaddition, producing the desired triazole-ring in the linker. Subsequently, three steps of reactions including azide substitution, reduction and amination afforded the final PFPA-mannose 83 in good overall yield.
Scheme 2.18 Synthesis of PFPA-carbohydrate 83. a) CuI, DIPEA, MeCN, 48 h (76%); b)
i. NaN3, DMF, 65 °C, 20 h (95%), ii. NaOMe, MeOH, 5 h (80%); c) i. H2, Pd/C, MeOH, 1 h
(92%), ii. 25, MeCN, dark, overnight (69%).
2.3.2. Synthesis of S-linked PFPA-carbohydrates
Taking advantage of the fact that sulfur is less basic and more nucleophilic

30
than oxygen, glycosyl thiols are increasingly used in the synthesis of thiooligosaccharides, thioglycopeptides and thioglycoproteins.116-118 The use of glycosyl thiols was thus adopted in a straightforward, yet more efficient synthetic route, compared to the ones for obtaining O-linked analogs, to produce the S-linked PFPA-carbohydrate structures. Free glycosyl thiolates 33, 35 and 40 were used as donors, and after SN2 reactions with the iodo-linker 4, the corresponding S-linked glycosides 84a-86a were conveniently obtained (Scheme 2.19). Subsequent reduction and PFPA-coupling using the methods mentioned above provided the final S-linked PFPA-carbohydrates 84-86, in total yields up to around 70%. This synthetic pathway proved highly efficient, requiring few steps and minimal purification, potentiating its use for glycobiological studies. However, unexpected anomerization occurred, and efforts to improve the anomeric purities were thus undertaken (see details in the next chapter).
Scheme 2.19 Synthesis of S-linked PFPA-carbohydrates 84-86. a) DMF, 50 °C, 1 h
(82-98%); b) i. H2, Pd/C, MeOH, 1 h (quant.), ii. 25, DMF, dark, 2.5 h (77-86%).
To test the effects of different linker lengths on carbohydrate-protein interactions at surfaces, two additional S-linked PFPA-mannose structures, 89 and 90, were synthesized with hexaethylene glycol- and nonaethylene glycol-based linkers, respectively (Scheme 2.20).

31
Scheme 2.20 Synthesis of S-linked PFPA-carbohydrates 89 and 90. a) DMF, 50 °C, 1 h
(84% for 87 and 88); b) i. H2, Pd/C, MeOH, 1 h (quant.), ii. 25, DMF, dark, 2.5 h (81% for
89, 82% for 90).
2.3.3. Synthesis of oligosaccharide-PFPA conjugates
To expand the carbohydrate ligand diversity and investigate more biologically relevant systems, α1-2-linked mannobiose- and mannotriose-PFPA molecules 96 and 102 were synthesized (Scheme 2.21 and 2.22). The azide linker 2 was allowed to react with orthoester 51 directly to yield the carbohydrate-azide 91, which was then deacetylated and further allowed to react with the glycosyl donor 56, leading to azide derivatized dimannoside 93. Compound 93 was deprotected in a similar manner as for its linker-free analogs (57 and 58), described previously. The resulting amine 95 was finally allowed to react with NHS-PFPA ester 25 through amidation, yielding the desired compound 96.

32
Scheme 2.21 Synthesis of PFPA-mannobiose 96. a) TMSOTf, DCM, 0 °C, 2.5 h (72%);
b) NaOMe, MeOH, H+ exchange resin, 1 h (quant.); c) 56, NIS, TfOH, DCM, -10 °C, 1 h
(63%); d) NaOMe, MeOH, H+ exchange resin, 2 h (93%); e) H2, Pd/C, MeOH, 4 d; f) 25,
DMF, dark, overnight.
A similar protocol to obtain the trimer derivative 102 from the dimer 94 was utilized as for its linker-free analog 67 and 68 (Scheme 2.22). However, in the case of 102, when the glycosyl donor 56 was allowed to react with acceptor 94, a mixture of α- and β-anomers was produced that turned out to be difficult to purify. To circumvent this problem, a different glycosyl donor 98 was synthesized and allowed to react with the acceptor 94.119 The desired α-product 99 was then obtained in 58% yield. Deprotection and amidation then afforded the final mannotriose-PFPA structure 102.

33
Scheme 2.22 Synthesis of mannotriose 102. a) NaOMe, MeOH, 2 h; b) BzCl, pyridine,
DCM, 6 h (89% over two steps); c) 98, NIS, TfOH, DCM, -15 °C, 1 h (58%); d) NaOMe,
MeOH, H+ exchange resin, 2 h (92%); e) H2, Pd/C, MeOH, 5 d; f) 25, DMF, dark,
overnight.

34

35
3. Mutarotation of 1-thioaldoses and stereocontrol in 1-thioglycosylation (Papers I
and II)
3.1. Mutarotation of 1-thioaldoses in aqueous media (Paper I)
3.1.1. Introduction
The mutarotation of natural reducing O-saccharides has been known for a long time.120,121 The composition of the mutarotation products is likely governed by a complex combination of factors including steric hindrance, stereoelectronic- and solvent effects.122 As a result, even after over 160 years since its discovery, the mutarotation of carbohydrates still remains a relatively unpredictable/uncontrollable phenomenon.
Certain aldopyranose analogs, in which the endocyclic oxygen and/or the glycosidic oxygen atom were/was replaced by a heteroatom (cf. Figure 1.3), also undergo mutarotation.120,123,124 For the sulfur analogs, however, previous studies on mutarotation have mainly been confined to structures with an endocyclic sulfur at the C-5 position.125 Mutarotation of 1-thioaldoses, on the other hand, have not been thoroughly investigated. In fact, free glycosyl thiols/thiolates were generally considered relatively stable, partly explained by the poor orbital overlap between the anomeric carbon and the sulfur atom, which could possibly block the ring-opening and subsequent mutarotation. In other words, it was relatively unclear under what conditions 1-thioaldoses would effectively mutarotate. The importance of these molecules in synthesis and in biological contexts, and the quest for fundamental understanding of chemical processes led us to investigate and clarify whether 1-thioaldoses undergo mutaroation. The pH-dependent mutarotation of 1-thioaldoses and their equilibrium anomeric ratios in aqueous media were thus studied.
3.1.2. pD-dependence of the mutarotation
Stereochemically pure 1-thioaldoses, 1-thio-β-D-glucopyranose 33, 1-thio-β-D-galactopyranose 34, 1-thio-α-D-mannopyranose 35, and 1-thio-α-L-fucopyranose 47, were initially synthesized and tested under acidic (pD 4), neutral (pD 7) and basic (pD 9) conditions. The influences of the different pD’s on the final anomeric compositions were thus demonstrated. The mutarotation processes of the four 1-thioaldoses were conveniently followed by 1H-NMR spectroscopy (Figure 3.1). Under acidic or neutral conditions, mutarotation for all the thiocarbohydrates tested readily occurred and the process was highly conspicuous.

36
SNa
OHOHO
OHHO
35
SNaOHO
HOOH
HO
33 47
SNa
OHO
HOOH
OH
34
Starting 1-thioaldoses
O
OHOH
OH
SH
Figure 3.1 1H-NMR kinetic studies of 1-thioaldose mutarotation. Fractions of β-anomers
formed from starting 1-thioaldoses 33, 34, 35 and 47 under acidic condition (pD 4).
More specifically, from Figure 3.1, it can be seen that gluco- (33), galacto- (34), and fuco- (47) derivatives yielded anomeric mixtures with β-anomer ratios of 74.1%, 78.1% and 74.3%, respectively, under acidic conditions (pD 4). These values are slightly larger compared to the β-anomer ratios of their corresponding hydroxy-aldoses under the same conditions, but still following the same trend in favoring one of the anomers. In the mannose case, for the hydroxy-mannose analog, 66.6% α-anomer was obtained at pD 4; however, for the thio-mannose 35, the opposite anomer was preferentially yielded (66.2% β-anomer in the final mixture) under the same experimental conditions.
Under basic conditions, the 1-thioaldoses showed different mutarotation behaviors and α-/β- compositions. The mutarotation of thiol analogs 33, 34 and 47 was almost completely restricted at pD 9 (> 95%, 94%, and < 5% of β-anomer, respectively). To confirm these trends, 1-thio-β-L-fucopyranose 44 was synthesized, which was then tested and compared to its α-analog 47. As can be seen from Figure 3.2, compound 44 barely mutarotated at pD 9, the same as its α-analog 47. However, when 44 was subjected to acidic or neutral conditions, a similar anomeric composition (80% of β-anomer) was formed as for the α-anomer 47. Thus, the pD-dependence of the mutarotation for 1-thioaldoses was clearly demonstrated.

37
OHO
OHOH
SH
47
Starting 1-thioaldoses
OHO
OHOH
SH
44
Figure 3.2 1H-NMR kinetic studies of 1-thioaldose mutarotation. Fractions of β-anomers
formed from starting 1-thioaldoses 44 and 47 under basic condition (pD 9).
The 1-thio-α-D-mannopyranose 35 showed slightly different mutarotation behavior (Figure 3.3). Nevertheless, under basic conditions, the compound still proved to be more stable compared to under acidic or neutral conditions, yielding only 24% of β-anomer in the final mixture at pD 9.
SNa
OHOHO
OHHO
35
Starting 1-thioaldose
Figure 3.3 1H-NMR kinetic studies of 1-thioaldose mutarotation. Fractions of β-anomers
formed from starting 1-thioaldose 35 under acidic, neutral and basic conditions (pD 4,
pD 7, pD 9).
These results pointed to the unusual importance of the axial hydroxyl group at the C-2 position of the carbohydrate ring in determining the mutarotation behavior. It was further hypothesized that when 1-thioaldoses pocessed a

38
(2S)-configuration, they would tend to display significant β-anomer preference under acidic/neutral conditions, yet α-preference under basic conditions.
To test this hypothesis, 1-thio-α-D-altropyranose 36 was prepared, and the mutarotation behaviors of this compound were tested under acidic, neutral and basic conditions. The results proved consistent with the predictions. In this case, complete conversion of the starting α-anomer to β-anomer occurred within 5 minutes at pD 4 and pD 7, whereas no mutarotation was observed within the same time span at pD 9.
3.1.3. Reversibility of the mutarotation
The mutarotation of reducing O-saccharides is known to be a reversible process. Thus the property was also probed for two of the 1-thioaldoses, both 1-thio-α-D-mannopyranose 35 and 1-thio-α-D-altropyranose 36.
SNa
OHOHO
OHHO
35
Starting 1-thioaldose
Figure 3.4 1H-NMR kinetic studies of 1-thioaldose mutarotation. pD switches for
1-thiomannose 35.
In the thiomannose case, the anomeric mixture compositions changed from favoring the β-anomer to the α-anomer in response to the pD switch from acidic to basic conditions, or vice versa (Figure 3.4). These processes were carried out over several cycles, and the mutarotation rates and anomeric equilibrium ratios proved to be consistent. However, in the thioaltrose case, the possibility of multiple pD switches was excluded due to a concomitant conformational change of the structure 36 (Scheme 3.1). Nevertheless, the reversibility of the thioaltrose analog was still demonstrated.

39
Scheme 3.1 Proposed 1-thio-altropyranose mutarotation processes.
3.1.4. Proposed mechanisms of the mutarotation of 1-thioaldoses
It was hypothesized that the mutarotation of 1-thioaldoses could follow two mechanistic pathways (Scheme 3.2). One is likely to go through an exo-ring-opening process similar to the 1-hydroxy- or 1-aminoaldoses.124,126 The other pathway would involve the formation of the oxocarbenium ion intermediate (109). The generation of trace amount of thiofuranoses during the kinetic experiments pointed to the acyclic intermediate formation during mutarotation. On the other hand, the slow formation of aldoses (110) as well as the characteristic smell of H2S indicated the involvement of the oxocarbenium ion pathway.
Scheme 3.2 Proposed mutarotation mechanistic pathways.
3.1.5. Conclusions
The mutarotation of 1-thioaldopyranoses proved to occur readily in aqueous media. A strong pD-dependence was demonstrated for the process; comparable mutarotation rates were shown at lower and neutral pD, whereas at higher pD the process proceeded slower or was almost blocked. The (2S)-D-thioaldopyranoses tested showed an interesting tendency to

40
preferentially form the β-anomer under acidic or neutral conditions but α-anomer under basic conditions. The reversibility of the mutarotation process for these stuctures upon pD-switching was also demonstrated.
3.2. Stereocontrol in 1-thioglycosylation (Paper II)
3.2.1. Introduction
Thiosaccharides and S-linked glycoconjugates are highly valuable substrate analogs for studies in glycobiology. Considerable effort has been made towards the synthesis and biological evaluations of such glycomimetics.116-118,127-130 The use and importance of 1-thioaldoses for 1-thioglycosylation reactions were documented as early as in the 1960’s.131 Recently, this type of molecules have been further applied to numerous novel systems including biohybrid polymer systems,132,133 dynamic combinatorial chemistry,45,109,134,135 glycoclusters based on self-organizable scaffolds,8,136,137 glycoporphyrin-assisted photodynamic therapy (PDT),8,138 and nanotechnology.52,139,140
Though the value of thioaldoses and thioglycosides has been increasingly appreciated, anomerization during the 1-thioglycosylation processes, especially when using unprotected 1-glycosyl thiols as donors, is rarely reported in the literature. In fact, it has been generally regarded that glycosyl thiolates retain their anomeric configuration in alkylation reactions.141 However, anomerization of glycosyl thioethers promoted by Lewis or protic acid has been more widely realized and studied.142-144 In the following paragraphs, the process of identifying mutarotation of glycosyl thiols and the optimization of conditions to control the stereochemistry of the product in 1-thioglycosylation reactions will be discussed and presented.
3.2.2. Occurrence and identification of the anomerization
To synthesize the PFPA-derivatized, S-linked carbohydrates 84-86, 89 and 90, a general route was initially designed, and the synthesis of mannose derivative 85 was first implemented (Scheme 3.3). Unprotected mannose thiolate 35 was directly alkylated with a tosylated TEG linker 3 carrying an azide functionality. Methanol was first used as the reaction solvent due to solubility considerations. After reaction under ambient conditions, however, a mixture of products was obtained after purification. Reducing (0 °C) or increasing (40-60 °C) the temperature, did not prevent the mixture formation.

41
SNa
OHOHO
OHHO
TsOO
ON3
SO
ON3
OHOHO
OHHO
SO
ONH
O F
F
F
F N3
OHOHO
OHHO
+
35 3 85a
85
a
b, c
Scheme 3.3 Initial route for the synthesis of S-linked PFPA-mannose 85. a) MeOH, RT,
72 h, 52% (α/β = 5/1); b) H2, Pd/C, MeOH, 1.5 h, quant.; c) 25, DMF, dark, 2 h, 84%
(α/β = 5/1).
Although to some extent unexpected, a combination of NMR analyses identified the formation of the β-anomer in the product mixture. This observation was corroborated by a value of the homonuclear three-bond coupling constant (J1,2) of 0.63 Hz,145 and the presence of a multiplet resonating at δ 3.41 ppm, which was assigned to mannose H-5 (Figure 3.5).146 In addition, NOESY-NMR of the anomer displayed a strong nuclear Overhauser effect between the H-1, H-3 and H-5 protons, clearly suggesting a β configuration at the anomeric center. Finally, a 2D heteronuclear single quantum correlation (HSQC) experiment indicated the stereochemistry of the obtained structures. Thus, that anomerization was taking place under the present conditions could be confirmed.
Figure 3.5 1H-NMR spectrum of α- and β-anomers of compound 85a in D2O.
3.2.3. Controlling the stereochemistry in the synthesis
In order to obtain the desired final products with specific stereochemistry, a number of factors were investigated including reaction time, temperature, solvent, electrophile and presence of base. Evaluations of the first three conditions in synthesizing mannose-azide 85a are summerized in Table 3.1.

42
Four different polar aprotic solvents, DMSO, DMF, N,N’-dimethylpropyleneurea (DMPU) and MeCN, were tested. DMSO turned out to be the best solvent in controlling the stereochemisry of the product; it promoted the fastest coupling rates and yielded only desired α-product at all different temperatures tested. Compared to DMSO, DMF gave relatively less good results, however, in this case, under the conditions of 3 h of reaction at 50 °C, full conversion of the reaction and fairly high stereoselectivity (α/β = 11/1) were achievable. Considering practical reasons, DMF was chosen as reaction solvent, and further optimization of the reaction conditions were conducted. DMPU and MeCN proved not suitable for the reaction.
Table 3.1 Time, temperature and solvents evaluated a
Entry Solvent Temp.(°C) Time (h) b Product (α/β ratio, yield)
1 DMSO 20 8 only α c
2 DMSO 50 0.5 only α c
3 DMSO 100 0.08 only α c
4 DMF 0 No reaction d -
5 DMF 20 50 α/β: 7/1, 58% e
6 DMF 50 3 α/β: 11/1, 64% e
7 DMF 100 0.5 α/β: 4/1, 36% e
8 DMPU 0 24 side products
9 DMPU 20 16 side products
10 DMPU 50 1.5 side products a In all entries, 1.2 equiv. of glycosyl thiolate 35 and 1 equiv. of linker azide 3 were used. b Time to reach full conversion. c Determined by NMR, yields not determined. d No reaction after 24 h, no mutarotation observed. e Isolated yields, based on 50 mg of glycosyl thiolate scale reactions, α/β ratio determined by NMR.
The effect of the electrophile on the stereocontrol of the reaction was subsequently tested. Thus, the iodo linker 4 instead of the tosyl linker 3 was used as the electrophile. Compared to the originally used tosyl moiety, the iodo functionality was expected to result in an increase in reaction rate of the SN2 displacement, to trap the starting 1-α-mannose thiolate 35 and produce the corresponding α-glycoside in high anomeric purity. This was supported by the results: when the iodo linker was used with the thiolate at 50 °C in DMF, the reaction reached completion within 1 hour, and only α-product was produced with an isolated yield of 87% (Figure 3.6). In contrast, when the iodo-linker was reacted with 1-mannose thiolate in MeOH at 50 °C, a mixture of anomeric

43
products was again formed.
Figure 3.6 1H-NMR spectrum of stereo-controlled compound 85a in D2O.
From the presentation in the first part of this chapter, it is clear that mutarotation of 1-glycosyl thiols is pH-dependent, where the process is generally impaired under basic conditions. As a consequence, the effect of added base was further tested. In this case, the tosyl electrophile 3, which originally led to a mixture of anomeric products at 50 °C in DMF, was subjected to glycosylation using 1-α-mannose thiolate 35. The results showed that the added base, triethylamine (TEA), helped to increase the anomeric purity at 50 °C in DMF. However, at higher temperature, 100 °C, the basic environment was not able to efficiently block the mutarotation of the 1-thiomannose.
Moreover, the origin of the anomeric product mixture was clarified. Anomerization of 1-thioglycosides has been proposed to be able to occur, albeit under Lewis acidic conditions.147 This could as well lead to the final anomeric mixture. However, experiments were carried out to exclude the possibility of product anomerization in the present case. Thus, leaving the glycoside product in the reaction vessel after reaction completion for prolonged times did not result in increased anomerization. Likewise, reactions using the purified α-mannose thioether 85a, in the presence of NaOTs (≥1 equivalent) in DMF at 50 °C for up to one week, yielded no anomerization products. This supported the mechanistic origin to be mutarotation of the 1-glycosyl thiols, resulting in the formation of final anomeric product mixture.
Following the the optimized conditions for obtaining the anomerically pure 1-thioglycoside 85a, several other S-linked PFPA-carbohydrate conjugates were synthesized, including monosaccharide derivative 84, disaccharide derivative 86, as well as two analogs to PFPA-thiomannose 85, the

44
hexaethyleneglycol- and nonaethyleneglycol linker bearing molecules 89 and 90, respectively (cf. Scheme 2.19 and 2.20).
3.2.4. Conclusions
The occurrence and circumvention of anomerization during 1-S-glycosylation reactions were demonstrated. Highly efficient and stereocontrolled synthesis of a number of photoprobe-thiosaccharide conjugates was achieved. Mutarotation of glycosyl thiols proved to be the origin of the anomeric mixtures formed. Kinetic effects were used to circumvent anomerization. This highly efficient and stereocontrolled protocol, in combination with a one-step method to convert free O-saccharides to their corresponding S-analogs,127 will provide a promising route to various S-linked glycoconjugates for glycobiological studies.

45
4. Development of three carbohydrate immobilization approaches and their
applications (Papers II-VIII)
Multivalent binding is prevalent in carbohydrate-protein interactions.148 It has been well established that multivalency can offer numerous benefits regarding enhanced receptor selectivity and affinity, and may induce particular organization on the cell surface to control signal transduction pathways within cells, which is not achievable with monovalent interactions.149 To mimic the multivalent interactions, a number of solid-support based techniques have emerged and since played increasingly important roles in glycobiological studies. A key step and prerequisite to these studies, however, is the fabrication of carbohydrate functionalized surfaces. Thus, the development of methods to immobilize carbohydrates on surfaces has become an important research topic. Meanwhile, it is also highly desirable to optimize both factors involved in multivalency and the intrinsic properties of individual ligands to obtain high specificity and increased affinity.
One of the ultimate applicable goals of studying carbohydrate-protein interactions is to generate useful information for the development of diagnostic tools and therapeutics. In this pursuit, a number of methods and tools have been involved.8 To develop potent carbohydrate-based drugs, the following steps can be undertaken: first, the presence of a carbohydrate binding protein, assumingly on a bacterial or viral surface, needs to be confirmed. This can be achieved by assessing the bacterial/viral genomes, proteomic analysis, and searching for carbohydrate binding protein homology through bioinformatics. Then, the isolated proteins can be labeled with fluorogenic probes and subjected to carbohydrate microarrays to identify the best glycan lead. Afterwards, the lead candidates can be validated using a range of other techniques such as ELISA, ITC, SPR, QCM, AFM, and X-ray crystallography. Next, a panel of simpler structures, compared to the original lead glycans, are usually evaluated, mainly for the purpose of easing potential manufacturing. Further lead optimizations can be achieved by classical quantitative structure-activity relationship (QSAR) methods. The resulting glycomimetics can be subsequently incorporated into multivalent scaffolds to obtain even higher binding affinities.
Our efforts in developing robust and versatile carbohydrate immobilization approaches using photochemistry will be presented in this chapter. Moreover, the incorporation of these approaches in different nano- and biotechniques to build useful and general platforms for studying carbohydrate-protein

46
interactions will be elaborated. Such techniques include carbohydrate microarrays, ITC, SPR, QCM, AFM, and carbohydrate functionalized goldnanoparticles. These techniques are among the most common tools that have been extensively applied to the development of novel diagnostics and therapeutics, as illustrated in the paragraph above. Optimization of the platforms to achieve good selectivities and the example of the more biologically relevant system will be emphasized.
4.1. Carbohydrate-conjugated photoprobes (Papers II-IV)
In this section, the photo-initiated glycosurface fabrication using carbohydrate-conjugated photoprobes will be introduced. This carbohydrate immobilization approach was subsequently incorporated in SPR and AFM platforms to study carbohydrate-protein interactions. In the SPR case, the effects of different structural features among the PFPA-carbohydrate ligands on protein binding were evaluated. In the AFM study, single molecular forces in the model mannose-Con A interaction system were measured, yielding fundamental data like the unbinding/rupture forces and kinetics. Furthermore, some of the solid supported SPR binding data were compared with the solution-based ITC results. The ITC results proved consistent with the SPR data, thus demonstrating the reliability of this solid-support platform for studying protein binding.
4.1.1. Photo-induced glycosurface fabrication approach I
The first approach of photochemical fabrication of glycan-functionalized surfaces was realized using carbohydrate-conjugated photoprobes by a double surface photoligation methodology (Figure 4.1). Either aminated- or gold-coated- glass substrates could be utilized by treating with NHS-PFPA ester 25 or PFPA-disulfide 27, respectively. The resulting photoprobe-derivatized surfaces were subsequently covered with a polymer film followed by UV irradiation. As a result, the polymer was covalently immobilized on the surfaces by the fast and efficient C-H insertion reaction of the photogenerated perfluorophenyl nitrene from PFPA. The matrix then served as the substrate for the second round of photoligation to introduce the different PFPA-derivatized carbohydrates. The resulting glycosurfaces were thus prepared for subsequent protein binding studies.

47
F F
F
N3
F
O O
NO O
25
OS
SO
F F
F
N3
F
O
F
F
F
N3
F
O
27
Figure 4.1 Immobilization of carbohydrates on surfaces using carbohydrate-conjugated
photoprobes by double surface photoligation.
In the fabrication process, a range of protein-resistant polymers, including poly(2-ethyl-2-oxazoline) (PEOX), poly(ethylene glycol) (PEG) and poly(ethylene oxide) (PEO), were evaluated. Of these, PEOX provided a more stable performance in the SPR setup, and PEG was selected for the AFM study.
4.1.2. SPR imaging analysis
To evaluate the effects of chemical and structural differences from individual ligands on protein binding, five mannose-derivatized photoprobes, 79, 83, 85, 89 and 90, were applied on the SPR sensor surface (Figure 4.2). These structures were varied with respect to anomeric attachment, S- and O-linked carbohydrates, as well as linker structure and length. Structure 79 was composed of a mannose head group linked by an O-glycosyl bond to a TEG-based linker, which was further coupled to the PFPA moiety. In contrast, structure 85 was linked through an S-glycosyl bond while all the other features were the same as for compound 79. Compounds 85, 89 and 90 were all S-linked glycoconjugates, however differing in linker lengths composed of three to nine ethylene glycol (EG) units. Stucture 83 was designed to possess a triazole ring in the linker part, with a linker length comparable to that of

48
compound 89. In addition, a galactose-derivatized PFPA structure (78) was used as a reference compound in the study.
Figure 4.2 SPR responses of six different PFPA-carbohydrates (10 mM) toward Con A
(10 μM) interrogation. Each data point (± SEM) represents the average response of six
replicates for each compound. Δ%R in the Y-axis means the percentage of change in
reflectivity.
Con A,150 the plant lectin originally extracted from the jack bean, Canavalia ensiformis, was used as a model lectin to probe the carbohydrate-protein interactions. The binding data are shown in Figure 4.2. First of all, the results clearly show that Con A selectively binds to its cognate mannose ligands (compounds 79, 83, 85, 89 and 90), but not to the control compound (galactose-derivative 78) on the sensor. This matches the established specificity of the lectin, thus demonstrating good lectin selectivity and minimum nonspecific binding for the photogenerated glycosurface.
The effects of structural variations among the mannose-derivatives on lectin binding were also evidenced. As can be seen from the figure, the performances of the O- and S-linked mannoside analogs, 79 and 85, respectively, upon Con A interrogation were comparable. Similar binding capacities were obtained, with that for the O-linked analog 79 being slightly higher. The result correlated well with the literature reported solid-support ELISA comparison of S- and O-linked glycoconjugates.151 The two molecules possessed only one

49
atom difference (the glycosyl S- and O-atom, respectively), thus providing a minimally varied pair of ligands for assessing the reliability and sensitivity of the photogenerated glycosurfaces. To confirm the result, the solid supported SPR binding experiments were subjected to a range of different lectin concentrations for further tests and were also cross-checked with the solution based ITC data, which will be discussed below.
The effects of other structural variations, i.e., the different linker lengths and linker structures, on protein binding were also addressed. Three different linker lengths (3, 6 and 9 EG units) and two different linker structures (linear and triazole-containing) were evaluated for protein binding on the same sensor. Based on previous studies, linkers shorter than 3 EG units in the PFPA-carbohydrate conjugates showed reduced binding efficiency. Only linkers longer than the TEG moiety were targeted in the present study. Structures 85, 89, and 90 possessed linear linkers with three, six and nine ethylene glycol units, respectively. Compound 83 had an equivalent chain length to 89 however containing a triazole-ring within the linker. As shown in Figure 4.2, comparing compounds 85, 89 and 90, the lectin binding capacity decreases as the spacer length increases over the tested range in the system used. This observation is, however, to some extent expected, likely due to a combination of potential entanglement and increased intramolecular photoinsertion during UV irradiation. With respect to the linker structural difference, the triazole-ring-bearing compound 83 shows slightly higher binding capacity compared to its linear analog 89, when applied to lectin interrogation. This agrees with previous studies, indicating that biomolecules retain their biological activities after incorporation of the triazole rings.152-154
4.1.3. Affinity analyses using SPR and ITC
As mentioned above, further SPR experiments were carried out with varying concentrations of interrogating Con A solution toward the O- and S-linked mannose-PFPA conjugates. The carbohydrate concentration was thus fixed at 10 mM, and three different concentrations of Con A solution, 100 nM, 1 μM and 10 μM, were employed for the lectin binding studies (Figure 4.3). Consistent and reproducible results were obtained with good signal-to-noise ratios at all concentrations tested, showing that the S-linked PFPA-mannose 85 bound the lectin slightly weaker than its O-linked analog 79.

50
Figure 4.3 SPR responses of O- and S-linked PFPA-carbohydrates 79 and 85,
respectively, toward Con A interrogation. Each data point (± SEM) represents the average
response of six replicates for each compound.
ITC analyses were subsequently conducted to support the results. ITC is a well-established method which can provide accurate and quantitative thermodynamic information of various binding events. Binding affinity data can be conveniently derived from the measurement, so it was chosen for cross-checking the surface carbohydrate-protein interactions on the SPR sensor chips. The resulting titration curves of O- and S-linked PFPA-mannose towards Con A are displayed in Figure 4.4. The obtained thermodynamic data for the binding are presented in Table 4.1. Compared to the binding affinity of methyl α-D-mannopyranoside towards Con A (Ka = 7.0 × 103 M-1),155 the Ka values of both compounds 79 and 85 against Con A fall in the same range, with that of O-linked conjugate 79 being slightly higher than that of the S-linked 85. This result is in good agreement with the results obtained from the solid-supported SPR measurements, thus demonstrating the reliability of the comparison of lectin bindings between the S- and O-linked carbohydrate conjugates on the SPR platform, built upon the photochemical carbohydrate immobilization approach I.

51
Figure 4.4 Calorimetric titration of Con A (80 µM) with carbohydrate derivatives 79 (a)
and 85 (b) (5 mM) at 25 °C: raw data obtained from 20 automatic injections, each of 2 µL.
The integrated curves show experimental points (square dots) and the best fit (solid line).
Table 4.1 Thermodynamic binding parameters of S- and O-linked PFPA-conjugated
carbohydrates with lectins at 25 °C
Comp. Lectin Ka M-1 × 10-3
-ΔG -ΔH -TΔS kcal/mol
79 Con A a 6.50 ± 1.04 5.2 ± 0.7 13.5 ± 1.0 8.3 ± 1.3 85 Con A a 4.06 ± 0.65 4.9 ± 0.4 12.8 ± 1.1 7.9 ± 0.9
a Average of six experiments.
4.1.4. AFM single molecular force analysis
In the AFM instrumentation setup, not only carbohydrate-functionalized surfaces are needed, but also AFM tips modified with proteins are prerequisite. Thus, AFM tips functionalized with Con A using a bifunctional PEG, NHS-PEG-NHS, were produced (Figure 4.5). The PEG moiety was used to suppress non-specific interactions. Meanwhile, excess amount of a monofunctional PEG, NHS-PEG-OMe, was applied on the tip to limit the number of proteins attached and increase the possibility of obtaining single molecular interactions.156

52
O
O N
O
O
PEGO
ON
O
O
NHS-PEG-NHS
OMePEGO
ON
O
O
NHS-PEG-OMe
Figure 4.5 AFM tip functionalization. Tip functionalization with Con A via amide formation
between the NHS-ester and the terminal amino group or surface-exposed lysine residues
of Con A.
Mannose-PFPA conjugate 79 was used for probing the interactions, and it is noteworthy that all the force measurements were carried out in pH<7 environment, where Con A would exist as a dimer rather than a tetramer,157 so that measurement of multiple Con A/mannose interactions could be reduced.
The interaction force between single mannose/Con A pairs was obtained by force-distance measurements, that is, after the AFM tip approaches the surface and interactions between Con A and mannose are established, the tip is subsequently retracted to measure the unbinding forces required to detach Con A from mannose. In most cases, the recognition events could not be observed since the probability of detecting single molecular interactions with minimally functionalized AFM tips was very low. However, when it occurred, force curves displaying rupture events could be obtained (Figure 4.6). The shape of the nonlinear repture peak reflects the elastic properties of the tethering PEG polymer, whereas the height of the rupture peak is determined by the strength of the Con A/mannose interaction at a particular tip velocity. The curves were fitted with a simplified extended freely-jointed chain model (e-FJCPEG) to determine whether the unbinding events were resulted from the specific binding partners.158

53
Figure 4.6 Typical retraction force curves for the unbinding of a Con A to a
PFPA-mannose-functionalized surface. The loading rate was 7120 pN/sec (tip velocity
400 nm/s, cantilever spring constant 17.8 pN/nm) with a relative trigger of 100 pN toward
the surface and a dwell time of 1 s. The fits to a simplified extended freely jointed chain
model (e-FJCPEG) are shown as solid lines. The Kuhn lengths determined from the fits
are: 868, 708, and 649 pm from top to bottom.
Control experiments were also performed to confirm the results. Several ways of eliminating or blocking the Con A/mannose interactions were designed, including the use of galactose-functionalized surfaces that do not specifically bind Con A, using monofunctionalized PEG spacer on the AFM tip that do not link to Con A, or adding free mannose to the buffer to saturate the Con A binding sites in advance. These control experiments resulted in the absence of specific interactions (<0.3%), thus reinforcing the reliability of the aforementioned force measurements.
The random insertion points along the PEG polymer chains by the PFPA photochemistry however led to the fact that the rupture distance can not be used as a discriminating factor in analyzing the force curves. Nevertheless, the PEG Kuhn lengths could be used to serve the analysis in this context.159 Thus, curves with PEG Kuhn lengths between 600 and 900 pm at each tip velocity were chosen for further analysis (Figure 4.7a). The distribution of unbinding forces of the chosen curves was then analyzed with a Gaussian fit, yielding the most probable unbinding force for the Con A/mannose interaction (Figure 4.7b). The value is typically reported with its corresponding loading rate, which is the product of the tip velocity and the spring constant.46 In the present study, for mannose surfaces on aminated glass, an average unbinding force of 70-80 pN for loading rates between 8000 and 40000 pN/s was obtained, while for mannose surfaces on gold-coated glass, an unbinding force of 57 ± 20 pN

54
at 6960 pN/s was acquired. The results are in agreement with previous reported unbinding forces in related mannose/Con A studies.
Figure 4.7 Histograms of single molecular force spectroscopy events for unbinding of
Con A from mannose at a loading rate of 8140 pN/sec (tip velocity 400 nm/s, cantilever
spring constant 20.35 pN/nm). (a) Distribution frequency of Kuhn lengths (n = 80); the
highlighted curves between 600 and 900 pm were selected for further analysis. (b)
Distribution frequency of unbinding forces (n = 39); a Gaussian fit was used to determine
the most probable unbinding force to be 69.5 ± 2.7 pN.
Furthermore, measuring the rupture forces of biomolecular interactions at variable probe loading velocities (dynamic force spectoscopy) allowed exploring the kinetics of the mannose/Con A interaction.160,161 In our experiments, the loading rates were varied from 8140 to 40700 pN/s to probe the binding kinetics. A single linear plot in the Con A/mannose dynamic force spectroscopy could be obtained (Figure 4.8). From the plot, the dissociation rate constant (koff) and the distance between the binding complex and the transition state (xβ) were determined to be 0.16 s-1 and 5.3 Å, respectively. Both the rupture forces and the kinetics obtained in our study are in good agreement with previously reported results.162,163
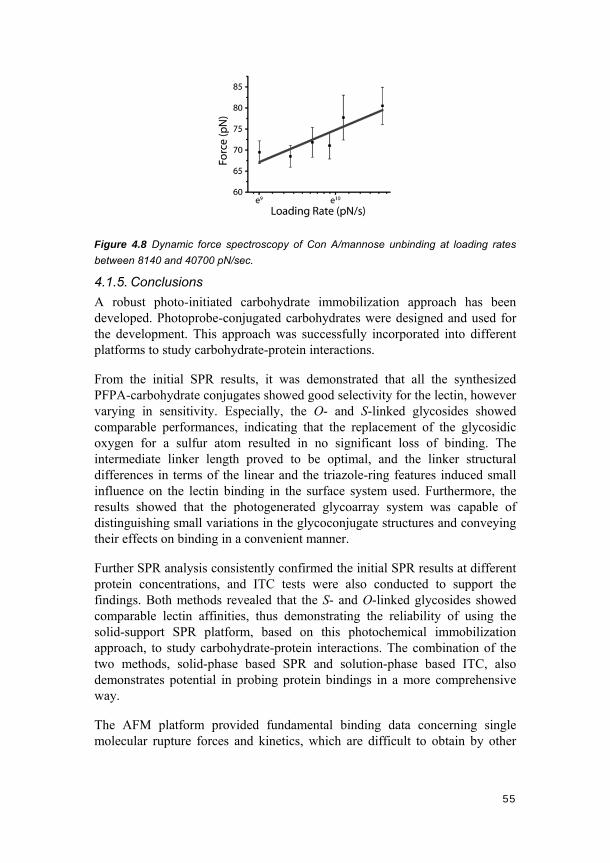
55
Figure 4.8 Dynamic force spectroscopy of Con A/mannose unbinding at loading rates
between 8140 and 40700 pN/sec.
4.1.5. Conclusions
A robust photo-initiated carbohydrate immobilization approach has been developed. Photoprobe-conjugated carbohydrates were designed and used for the development. This approach was successfully incorporated into different platforms to study carbohydrate-protein interactions.
From the initial SPR results, it was demonstrated that all the synthesized PFPA-carbohydrate conjugates showed good selectivity for the lectin, however varying in sensitivity. Especially, the O- and S-linked glycosides showed comparable performances, indicating that the replacement of the glycosidic oxygen for a sulfur atom resulted in no significant loss of binding. The intermediate linker length proved to be optimal, and the linker structural differences in terms of the linear and the triazole-ring features induced small influence on the lectin binding in the surface system used. Furthermore, the results showed that the photogenerated glycoarray system was capable of distinguishing small variations in the glycoconjugate structures and conveying their effects on binding in a convenient manner.
Further SPR analysis consistently confirmed the initial SPR results at different protein concentrations, and ITC tests were also conducted to support the findings. Both methods revealed that the S- and O-linked glycosides showed comparable lectin affinities, thus demonstrating the reliability of using the solid-support SPR platform, based on this photochemical immobilization approach, to study carbohydrate-protein interactions. The combination of the two methods, solid-phase based SPR and solution-phase based ITC, also demonstrates potential in probing protein bindings in a more comprehensive way.
The AFM platform provided fundamental binding data concerning single molecular rupture forces and kinetics, which are difficult to obtain by other

56
means.
4.2. Direct photoconjugation (Papers V and VI)
In this part, the direct photoconjugation approach to immobilize carbohydrates on surfaces will be introduced. As discussed in several earlier chapters, current preparation approaches to carbohydrate-functionalized surfaces are generally based on derivatized carbohydrate structures. However, the direct conjugation of underivatized carbohydrates to surfaces remains a challenging task. In this area, available methods mainly target the reducing end of the glycan chains, but these methods usually induce ring opening of the terminal saccharide unit, which can affect the binding properties of smaller saccharide structures. Another often associated disadvantage with these methods is the requirement of relatively harsh conditions. In constrast, the PFPA photochemistry provides a facile and efficient conjugation method to immobilize underivatized carbohydrates on surfaces.19,102,164
The direct photoconjugation approach was applied to generate carbohydrate arrays on flat sensor substrates, and SPR was used to read out the binding events in real time and at high throughput. To our delight, the carbohydrate ligands immobilized on surfaces in this manner well retained their biological activities, and the ligand presentation and surface composition had strong influences on subsequent binding. These aspects will be discussed below in details.
As already mentioned, multivalent display of carbohydrate ligands has proven to be of high potential in addressing challenges in probing various carbohydrate-involved recognition events. Scaffolds coupled with high density of glycans have thus been developed to mimic the glycan clustering effects on cell surfaces, enhancing the affinity to carbohydrate-binding entities.54,165,166 In this context, glyconanoparticles (GNPs) became our desired target to exercise the direct photoconjugation approach. The anti-HIV lectin, CV-N, and its mutants, were probed in terms of binding sites and affinities. The idea of preparing “multivalent glycomimetics” by grafting simple glycans on a multivalent scaffold, instead of synthesizing complex glycan structures to obtain high affinities, was tested.
4.2.1. Photo-induced glycosurface fabrication approach II
A key step of the direct photoconjugation approach is the introduction of PFPA photoprobes to surfaces. Towards this end, the PFPA moiety can be derivatized with different terminal groups, including thiols/disulfides, phosphates or silanes, and these structures can subsequently be used to functionalize different surfaces based on gold-, iron oxide- and silica

57
substrates, respectively. Following the PFPA-modification of surfaces, a solution of underivatized carbohydrates is applied to the surfaces and the attachment of underivatized carbohydrates can be achieved by fast UV irradiation.
In the carbohydrate array case, gold-coated optical glass slides were first treated with PFPA-derivatized thiols, and solutions of each carbohydrate were subsequently printed onto the SPR sensors using a robotic printer (Figure 4.9a). Surfaces with different PFPA photoprobes, diluter molecules and ligand densities were tested, and various underivatized carbohydrates including mono- (Man, Glc, Gal), di- (Manα1-2Man) and trisaccharides (Manα1-3[Manα1-6]Man) were immobilized. In the glyconanoparticle case, uniform, ~22 nm gold nanoparticles were synthesized and in situ functionalized with a PFPA-disulfide (Figure 4.9b), following a previously established procedure.91 Di- (Manα1-2Man) and trisaccharides (Manα1-2Manα1-2Man) were then photochemically conjugated to the PFPA-functionalized gold nanoparticle surfaces by C-H insertion reactions as described earlier.
Figure 4.9 The direct photoconjugation of free carbohydrates on: (a) flat SPR gold
surfaces, and (b) gold nanoparticle surfaces.
4.2.2. Carbohydrate microarray studies
Initially, the quest for an optimal PFPA photoprobe was pursued. Thus, a number of thiol-functionalized PFPAs with varying lengths of spacers, PFPA-C2-SH, PFPA-C6-SH, PFPA-C11-SH and PFPA-MUTEG (Figure 4.10), were used to derivatize the SPR sensor surfaces. After exposure to each of

58
these thiols in ethanol (4 mM) for 3 h, the surfaces were printed with free mannose (Man), glucose (Glc) and galactose (Gal) solutions as quadruplets. Subsequent UV irradiation and washing provided the functionalized SPR surfaces for protein interrogation. Con A was used again as a model protein, and the results showed that the SPR responses increased with the length of the spacer (Figure 4.10). PFPA-MUTEG gave the highest signal and was therefore chosen as the photo-coupling probe in subsequent studies.
O
O
F
N3 F
F
F O
SH
49O
SH
F
N3 F
F
F O
O
F
N3 F
F
F O
SH
O
F
N3F
F
FO
SH
PFPA-C2-SH PFPA-C6-SH PFPA-C11-SH PFPA-MUTEG
Figure 4.10 SPR responses of Con A with Man, Glc and Gal immobilized on SPR
sensors functionalized with four different PFPA-thiol linkers. Each data point was the
average of three SPR sensors with four replicates for each ligand. Error bars represent
the standard derivation of 12 data sets.
Subsequently, a few diluter molecules were evaluated with respect to their ability to suppress non-specific interactions. Mercapto-1-hexanol (HS(CH2)6OH), 1-decanethiol (HS(CH2)9CH3), MUTEG and MDEG (Figure 4.11) were chosen and applied to the SPR sensor surfaces, respectively, to form monolayers. The resulting surfaces were then exposed to Con A and the SPR responses were recorded. From the results (Figure 4.11), it can be seen that surfaces covered by HS(CH2)6OH or HS(CH2)9CH3 gave large non-specific adsorption signals, while the responses from MUTEG and MDEG surfaces showed to be much lower. The results are consistent with the general finding that ethylene glycol units reduce non-specific protein adsorption. MDEG proved to provide minimal background adsorption, and was thus chosen as the diluter molecule to be mixed with PFPA-MUTEG in different molar ratios to treat the SPR sensors.

59
HO
O
SH
49 O
SH
HO
SH SH
MDEGMUTEGCH3(CH2)9SHHO(CH2)6SH
O
HO
Figure 4.11 SPR responses of Con A with different thiol-linker derivatized and bare gold
surfaces. Each data point was the average of three SPR sensors with four replicates for
each ligand. Error bars represent the standard derivation of 12 data sets.
Various ratios of photoprobe molecule PFPA-MUTEG versus diluter molecule MDEG, from neat PFPA-MUTEG to a mixture containing 90% MDEG (Figure 4.12), were attached to the surfaces, and five underivatized carbohydrates including Man, Glc, Gal, Manα1-2Man and Manα1-3[Manα1-6]Man were subsequently printed and covalently attached on the photoreactive sensor surfaces, in quadruplets in a 5 × 4 patterned array (Figure 4.12). Con A binding results showed that the SPR responses generally decreased with decreasing ratio of PFPA-MUTEG in the bifunctional linker mixture, except when the surfaces were treated with the 90:10 [PFPA-MUTEG]/[MDEG] mixture. The results also revealed the rank of binding affinities of the applied carbohydrate ligands against Con A. It can be seen from the figure that trisaccharide Manα1-3[Manα1-6]Man displayed the highest affinity, followed by disaccharide Manα1-2Man. The binding of Glc was weaker than that of Man, while Gal gave minimal binding. This order of affinities and selectivities agreed with that of the corresponding free ligands in solution,167 thus demonstrating that the surface-attached ligands well retained their recognition abilities on the platform used.
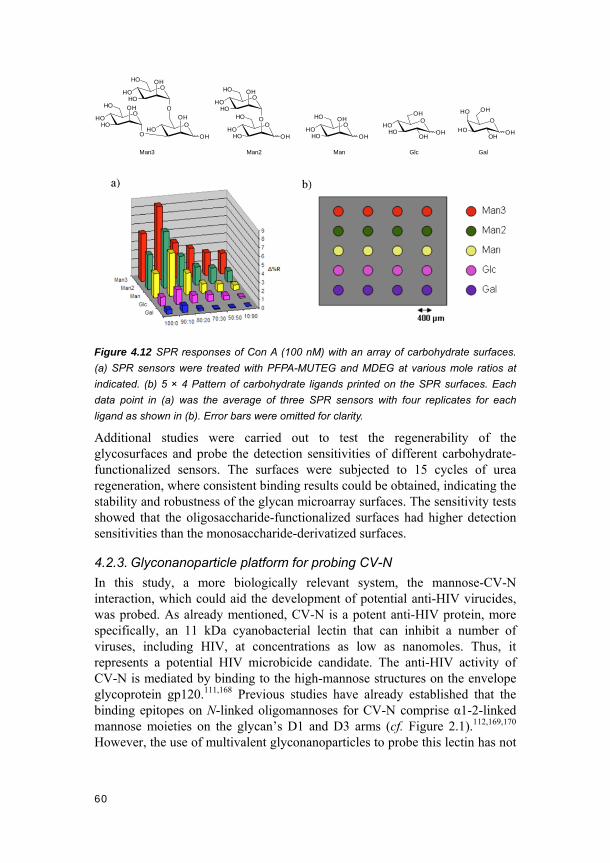
60
OHO O
HOHO
OHO OH
HOHO
OH
OOH
HOOH
OHO OH
HOHO
O
OHO OH
HOHO
O
OHO OH
HOHO
OH
OOH
OHHO
HOOH
O
OH
OHHO
HO
OH
Man3 Man2 Man Glc Gal
Figure 4.12 SPR responses of Con A (100 nM) with an array of carbohydrate surfaces.
(a) SPR sensors were treated with PFPA-MUTEG and MDEG at various mole ratios at
indicated. (b) 5 × 4 Pattern of carbohydrate ligands printed on the SPR surfaces. Each
data point in (a) was the average of three SPR sensors with four replicates for each
ligand as shown in (b). Error bars were omitted for clarity.
Additional studies were carried out to test the regenerability of the glycosurfaces and probe the detection sensitivities of different carbohydrate-functionalized sensors. The surfaces were subjected to 15 cycles of urea regeneration, where consistent binding results could be obtained, indicating the stability and robustness of the glycan microarray surfaces. The sensitivity tests showed that the oligosaccharide-functionalized surfaces had higher detection sensitivities than the monosaccharide-derivatized surfaces.
4.2.3. Glyconanoparticle platform for probing CV-N
In this study, a more biologically relevant system, the mannose-CV-N interaction, which could aid the development of potential anti-HIV virucides, was probed. As already mentioned, CV-N is a potent anti-HIV protein, more specifically, an 11 kDa cyanobacterial lectin that can inhibit a number of viruses, including HIV, at concentrations as low as nanomoles. Thus, it represents a potential HIV microbicide candidate. The anti-HIV activity of CV-N is mediated by binding to the high-mannose structures on the envelope glycoprotein gp120.111,168 Previous studies have already established that the binding epitopes on N-linked oligomannoses for CV-N comprise α1-2-linked mannose moieties on the glycan’s D1 and D3 arms (cf. Figure 2.1).112,169,170 However, the use of multivalent glyconanoparticles to probe this lectin has not

61
yet been reported.
Low-mannose structures were thus attached onto gold nanoparticles by the direct photoconjugation approach to study the glycan binding specificities and affinities for CV-N. In order to quantitatively probe the affinity enhancement introduced by the GNPs, a fluorescence competition assay was developed to determine the apparent dissociation constants of GNP binding to CV-N (KD). The results obtained from this assay were compared with the Kd values of monomeric glycan binding to CV-N measured by ITC. The information gained from the study was envisaged to help the development of new diagnostic and therapeutic tools directed at combating viral infections.
Two CV-N variants were used in this study: CVNQ50C and CVNMutDB. CVNQ50C is essentially a more stable wild-type variant, comprising two separate glycan binding sites, one on Domain A and the other on Domain B.112,170 α1-2-Linked mannotriose (Man3) moieties preferentially bind to Domain A while α1-2-linked mannobiose (Man2) structures show preference for Domain B.170,171 The Cys substitution at position 50 was introduced to allow for specific fluorescence labeling of the CV-N without interfering with glycan bindings (referred to as Cy5-labeled CV-Ns throughout text below). In contrast, the CVNMutDB has its Domain B completely eliminated, keeping only the binding site on Domain A active.
After the carbohydrate functionalization of the gold nanoparticles, the surface ligand density was determined by a colorimetric assay using anthrone/sulfuric acid.91 The mannobiose and mannotriose functionalized gold nanoparticles (GNP-M2 and GNP-M3) (Figure 4.13) showed to present 1516 ± 232 Man2- and 1037 ± 148 Man3-entities per particle, respectively.

62
Figure 4.13 Synthesis of the α1-2-linked mannobiose and mannotriose functionalized
gold nanoparticles (GNP-M2 and GNP-M3).
With these two sets of binding partners prepared, a series of interaction studies were subsequently implemented. It can be expected that when CVNQ50C is used to interact with the GNPs, crosslinking between the two partners will occur leading to formation of complexes. However, no or less crosslinking should be possible between the GNPs and CVNMutDB. Indeed, when GNP-M3 was treated with CVNQ50C, a red shift from 529 nm to 542 nm in the surface plasmon resonance band of gold nanoparticles could be observed (Figure 4.14b), indicating particle size growth.172 Transmission electron microscopy (TEM) tests confirmed this indication by showing the presence of clusters of aggregated particles (Figure 4.14d). Similar tests were also conducted for CVNMutDB, but the results revealed neither SPR shift nor particle aggregations (Figure 4.14a,c). Dynamic light scattering (DLS) measurements of CVNQ50C-and CVNMutDB-treated GNP-M3 particles yielded average particle sizes of 38.3 ± 4.6 nm and 25.9 ± 3.5 nm, respectively (Figure 4.14e), further confirming the above conclusions. These results are in good agreement with previous structural studies on the CV-N mutants.173

63
Figure 4.14 UV-vis spectra of GNP-M3 before (solid line) and after (dotted line) treatment
with: (a) CVNmutDB, and (b) CVNQ50C. TEM micrographs of GNP-M3 treated with: (c)
CVNmutDB, and (d) CVNQ50C. (e) DLS measurements of CVNMutDB- (middle) and
CVNQ50C- (right) treated GNP-M3 particles.
Next, the binding affinities of the GNPs with the CV-N variants were quantitatively evaluated using a fluorescence-based competition assay.23 Free ligand competitors, Man2 and Man3, were incubated with a fixed concentration of Cy5-labeled CVNQ50C, in the presence of varying concentrations of GNP-M2 and GNP-M3, respectively. After incubation, the solution was centrifuged to remove all GNP-CVN complexes, and the fluorescence intensity of the resulting supernatant was measured. The difference in fluorescence intensity of Cy5-CVNQ50C before and after incubation corresponds to the amount of the bound (and removed) CVNQ50C. Concentration response curves for GNP-M2 and GNP-M3 thus permit to determine the IC50 values (Figure 4.15a). However, to determine the binding constants of GNPs with CVNQ50C, it is as well necessary to obtain the Kd values of the monomeric ligands, Man2 and Man3, with CVNQ50C

64
(Figure 4.15b,c). These values were conveniently determined by ITC and the dissociation constants, Kd1 for Domain A and Kd2 for Domain B (Figure 4.15d), were calculated based on a two-site binding model.
Figure 4.15 Fluorescence binding assay studies. (a) Concentration response curves of
GNP-M2 (right) and GNP-M3 (left). (b) Schematic representation of a binding scenario.
(c) Modified Cheng-Prusoff equation based on a competitive two-site binding model,
where [M] is the concentration of the free ligand for the glycan-binding sites on Domains
A and B of CVNQ50C, respectively. The data were fitted using the maximum bound
fractions, fBmax1 and fBmax2, corresponding to the two binding sites, and dissociation
constants KD1 and KD2 as adjustable parameters. (d) ITC measurement of bindings
between free Man2 (left) / Man3 (right) and CVNQ50C.
These data, together with the IC50 values, were then used to calculate the

65
apparent dissociation constants, KD1 and KD2, of GNPs for the two binding sites on CVNQ50C, Domain A and B, respectively. The data are summarized in Table 4.2. From the results, it can be seen that both GNP-M2 and GNP-M3 exhibit affinity enhancements of several orders of magnitude compared to their free monomeric glycan ligands, when binding to CVNQ50C. Considering the enhancement of affinity per ligand, an increase of up to several hundred times is still present for the gold nanoparticle-bound glycans (Table 4.2, enhancement factor (EF) values in parentheses). Interestingly, for both GNP-M2 and GNP-M3, the affinity enhancement is more pronounced for their better binding domain. That is, when Man2 exhibits a higher affinity for the binding site on Domain B, its multivalent analog GNP-M2 induces the affinity enhancement by 176 folds for Domain B versus 8.2 folds for Domain A. In contrast, for GNP-M3, the opposite was observed that the EF is higher for the Domain A site (309) than for Domain B (3.6).
Table 4.2 Affinities for Man2/3 (Kd) and GNP-M2/3 (KD) binding to CVNQ50C. Numbers in
parentheses correspond to EF (= Kdi/( KDi × number of ligands on GNP))
Ligand Kd1 or KD1 (Domain A) Kd2 or KD2 (Domain B)
Man2 700 ± 50 µM 64 ± 4 µM GNP-M2 56.4 ± 7 nM (8.2) 0.24 ± 0.1 nM (176)
Man3 3.4 ± 0.2 µM 43 ± 2 µM GNP-M3 0.011 ± 0.007 nM (309) 11.8 ± 2.3 nM (3.6)
4.2.4. Conclusions
A new approach, the PFPA-based direct photoconjugation approach, to immobilize carbohydrates on surfaces has been developed. This new approach was applied to fabricate carbohydrate arrays that could be analyzed directly by SPR. Using this approach, underivatized carbohydrates were covalently attached to the SPR sensors, leading to robust and stable surfaces that could be used repeatedly. Equally importantly, the surface bound glycan ligands retained their affinities and selectivities toward their binding proteins. Interaction studies also showed that the affinity was highly dependent on the ligand density.
Furthermore, two mannose structures, Man2 and Man3, were successfully grafted onto gold nanoparticles using the same approach. The resulting GNPs were used to probe two CV-N variants, the single binding-site protein CVNMutDB and the two binding-site protein CVNQ50C. The bindings occurred in a predictable manner, where crosslinked complexes and aggregates were not observed for CVNMutDB but were seen for CVNQ50C. Moreover, the GNPs exhibited significantly higher affinity towards the CV-N lectins, compared to

66
the free glycan ligands. Substantial affinity enhancements per carbohydrate ligand attached were also recorded. These data showed that gold nanoparticles indeed serve as efficient multivalent scaffolds to enhance the apparent affinities and to provide glycan clustering effects. The enhanced affinities compare well with those of natural or synthetic high-mannose structures as well as other types of synthetic multivalent ligands. Therefore, the idea of grafting simple glycans on gold nanoparticle scaffolds to prepare multivalent glycomimetics, instead of synthesizing complex glycan structures, to obtain high affinities, was also demonstrated. These approaches and ideas hold strong promise in the development of new and effective diagnostic tools and therapeutics.
4.3. Photo-click carbohydrate immobilization (Papers VII and VIII)
In this section, the design of carbohydrate immobilization approach III will be described. This approach is a “hybrid” of PFPA photo-immobilization and Cu-catalyzed azide alkyne cycloaddition (CuAAC, one of the best known “click” reactions) methods. The photo-click approach combines the advantages of approaches I and II described above. It introduces site-specific carbohydrate attachment, reduces the synthetic burden, as well as extends this approach to a large range of readily accessible carbohydrate-azide structures. The photo-click concept was designed to be realized in two experimental procedures, which were further compared with immobilization approach I of using the entire PFPA-carbohydrate entity. Moreover, one of the photo-click procedures was elaborated with various polymer coatings, multiple carbohydrate structures and interrogated by a number of different lectins.
The immobilization approach was tested on the QCM platform. Comparisons of bindings on the constructed surfaces by the three different experimental procedures, and additional evaluations of different surface performances using one of the photo-click procedures will be discussed.
4.3.1. Photo-induced glycosurface fabrication approach III
Initially, two procedures of the photo-click approach were designed to immobilize carbohydrates on the QCM chip surfaces. They were further compared to approach I. All of the three procedures should theoretically result in comparable final surfaces (Figure 4.16). The earlier described double-surface photoligation method was used here to generate the polymer surfaces and subsequent glycosurfaces, except for certain polystyrene (PS) surfaces which were obtained from a commercial source. More specifically, one of the photo-click procedures consisted of two steps (Figure 4.16, procedure B): firstly, alkyne-derivatized PFPA 32 was attached to the surface

67
by UV irradiation. The alkyne surface was then coupled to the carbohydrate-azide 70 using the CuAAC chemistry. The other procedure, however, comprised three steps (Procedure C), and the alkyne surface in this case was produced in two separate steps: first, the polymeric surface was derivatized by the NHS-PFPA ester 25. The resulting surface was subsequently immersed in a solution of alkyne-derivatized amine linker 19. Amidation reactions on the surface afforded the same alkyne-functionalized surface as in procedure B, which was then subjected to the carbohydrate-azide 70. In comparison, carbohydrate immobilization approach I (Procedure A) was also utilized, in which the mannose-derivatized photoprobe 83 was immobilized on the surface in a single photoligation step under UV light.
OHO OH
HOHO
O
70N3
72
O
OH
OHHO
HO
ON3
OOH
NHAcHO
HO ON3
76
HON3
21
Figure 4.16 Two photo-click procedures (B and C), in comparison with photochemical
approach I (procedure A), to immobilize carbohydrates on surfaces.
Subsequently, procedure C was utilized to further probe the capability of this photo-click approach in generating a general platform to study carbohydrate-protein interactions. Thus, a number of additional polymers, including polyacrylamide (PAAm), PEG, PEOX, and polypropylene (PP), were used to coat the QCM sensor surfaces in the first photoligation step (cf. Figure 4.1). Three different carbohydrate-azide structures, 70, 72 and 76, were used in the third step of procedure C (cf. Figure 4.16). The polymeric surfaces were also functionalized with 2-azidoethanol 21 to produce control surfaces.
4.3.2. Protein binding and surface specificity studies by QCM
The biorecognition properties of the surfaces were evaluated using the QCM flow-through instrumentation, with repeated injections of lectins specific for the carbohydrate ligands used in the studies. The QCM technique allows for
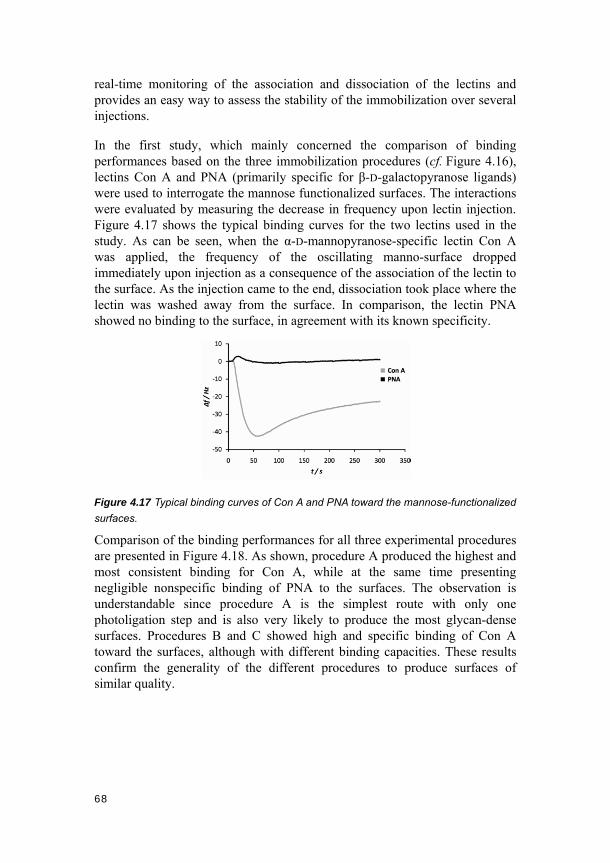
68
real-time monitoring of the association and dissociation of the lectins and provides an easy way to assess the stability of the immobilization over several injections.
In the first study, which mainly concerned the comparison of binding performances based on the three immobilization procedures (cf. Figure 4.16), lectins Con A and PNA (primarily specific for β-D-galactopyranose ligands) were used to interrogate the mannose functionalized surfaces. The interactions were evaluated by measuring the decrease in frequency upon lectin injection. Figure 4.17 shows the typical binding curves for the two lectins used in the study. As can be seen, when the α-D-mannopyranose-specific lectin Con A was applied, the frequency of the oscillating manno-surface dropped immediately upon injection as a consequence of the association of the lectin to the surface. As the injection came to the end, dissociation took place where the lectin was washed away from the surface. In comparison, the lectin PNA showed no binding to the surface, in agreement with its known specificity.
Figure 4.17 Typical binding curves of Con A and PNA toward the mannose-functionalized
surfaces.
Comparison of the binding performances for all three experimental procedures are presented in Figure 4.18. As shown, procedure A produced the highest and most consistent binding for Con A, while at the same time presenting negligible nonspecific binding of PNA to the surfaces. The observation is understandable since procedure A is the simplest route with only one photoligation step and is also very likely to produce the most glycan-dense surfaces. Procedures B and C showed high and specific binding of Con A toward the surfaces, although with different binding capacities. These results confirm the generality of the different procedures to produce surfaces of similar quality.

69
Figure 4.18 Binding results for the three different procedures (normalized for the
molecular weight of the lectins).
In the second study, procedure C was chosen and a few more polymers, carbohydrates and lectins were used for the versatility evaluation of the photo-click approach and the QCM platform. The carbohydrates and lectins were chosen based on their binding specificities and previous experiences of surface binding analyses. Con A and PSA are complementary lectins for α-D-mannose residues, whereas WGA and BS-II bind selectively to N-acetyl-β-D-glucosamine and RCA-I to β-D-galactose. To minimize nonspecific binding, BSA was used to block all non-covered surface areas by several injections in the beginning of the experiment. Several studies have shown that blocking with BSA in the initial phase of the experiment lasts throughout the whole experiment, thus obviating the need for BSA in the running buffer.21,45
The dissociation and association of the lectins to the surfaces could again be clearly observed, with small levels of nonspecific binding found for some of the lectins. These nonspecific interactions are hypothesized to be due to, e.g., surface structure and composition, blocking coverage by BSA, and pI values of the lectins, etc. However, these can be corrected for by a control surface, using the non-carbohydrate linker molecule 2-azidoethanol (21). Thus, a corrected average binding value was obtained for each lectin on each surface with good reproducibility over time (Figure 4.19). As shown, high binding specificities were found for all lectins evaluated, and the expected trends were followed. Con A and PSA displayed binding to the mannose surfaces, WGA and BS-II to the N-acetylglucosamine surfaces, and RCA-I to the galactose surfaces. The difference in binding capacities between the different lectins mainly resulted from parameters like the lectin molecular weight and the affinity for the carbohydrate ligands used in the study.

70
Figure 4.19 Corrected lectin binding to PEG-coated surfaces. Each data represents an
average of binding values from two separate injections of protein, subtracted with the
averaged binding from the 2-azidoethanol control surface.
The lectin binding profiles to all glycan-functionalized QCM sensor surfaces are depicted in Figure 4.20. From the protein point of view, the Con A interactions proved to be the most consistent in the study, with more than 40 Hz corrected binding to all mannose-functionalized surfaces and with relatively insignificant nonspecific binding. PSA followed the same binding pattern as for Con A, though with lower capacity for the selective mannose interactions. The other lectins showed certain variations in binding among the different polymeric surfaces. For example, relatively low bindings of WGA and BS-II to the GlcNAc/PAAm surface can be seen while high bindings to GlcNAc/PEG are observed. RCA-I also showed different capacities of binding to the galactose-functionalized surface, although to a lower extent compared with WGA and BS-II binding to the GlcNAc-surfaces.
Figure 4.20 Corrected binding values of lectins to the functionalized surfaces: (a) PEG.
(b) PEOX. (c) PAAm. (d) PS. (e) PP.

71
The different binding features can also be viewed from the polymeric surface point of view. Comparison of the differences in sensitivity showed that the PS surfaces in general displayed negative bindings to the mismatched carbohydrate-functionalized surfaces, for example, RCA-I to Man and PSA to GlcNAc. Whereas the PP surfaces displayed a resulting positive binding, e.g., PSA to GlcNAc and Con A to Gal. The hydrophilic polymer surfaces (PAAm, PEG, and PEOX) generally showed a more coherent binding than the hydrophobic polymer surfaces (PS and PP), demonstrated by the more coherent corrected lower binding values to the mismatched surfaces.
4.3.3. Conclusions
The photo-click approach has been successfully developed to immobilize carbohydrates on surfaces in a fast, straightforward and site-specific way. The approach makes use of the highly efficient PFPA photoligation, in combination with the CuAAC chemistry. QCM instrumentation was used as the platform to incorporate the immobilization approach and to evaluate subsequent protein binding performances of the glycan-surfaces. High specificities and capacities were recorded for all surfaces, and all the surfaces can also be applied with a variety of polymeric substrates.
In the first study on this platform, three different experimental procedures were adapted for the glycosurface production, all resulting in functional surfaces with good quality, and the two photo-click procedures can be easily expanded with a larger range of carbohydrate-azide structures, as well as other azide-containing molecules.
In the second study on the platform, the versatility of the approach and the platform was further probed by diversifying the polymeric coating materials, the carbohydrate ligands and the complementary interrogating lectins. The predicted protein binding selectivities were observed for all the glyco-surfaces developed, thus demonstrating the potential of this technology. PEG-coated sensors proved to give very good binding performances; other formats also showed high quality. The photochemically fabricated carbohydrate surfaces are inherently robust, requiring only low-cost starting materials and ambient conditions. In general, these features point to a convergent fabrication approach that constitutes a versatile platform for sensing and other types of glycobiological studies.

72

73
5. Concluding remarks and outlooks
This thesis has mainly been directed to developing new, straightforward and robust photochemical approaches to immobilize carbohydrates on surfaces for glycobiological studies. Three different approaches, namely the carbohydrate-conjugated photoprobe approach, the direct photoconjugation approach and the “hybrid” carbohydrate immobilization approach, have been designed and successfully applied into a wide range of nanobio-platforms. These platforms comprise some of the most common tools used in modern glycobiological field, including carbohydrate microarrays, biosensors like SPR and QCM, AFM and glyconanomaterials. The demonstrated compatibility with a large number of applications, the robust and versatile coupling chemistry, and the controllable and adaptable surface features involved in the development, have endowed great potential for the photochemical carbohydrate immobilization approaches presented.
All the glycan-functionalized surfaces have displayed good selectivities toward protein binding. Moreover, the surfaces have been used to evaluate the effects of structural variations in surface chemical components on subsequent binding (SPR platform), to compare with solution-phase based ITC measurement of carbohydrate-protein interactions, and to probe fundamental binding parameters like the single molecular rupture forces and kinetics (AFM platform). The surfaces can also be adapted to fabricating carbohydrate microarrays using common present-day arrayers, enabling evaluation of multiple glycan-protein bindings in parallel. Carbohydrate functionalized surfaces based on gold nanoparticles have also been produced, leading to quantitative analysis of their interactions toward the anti-HIV lectin CV-N. The robustness of the surfaces has been demonstrated by the reproducible binding results on the surfaces and their ability to withstand storage. The versatility of the surfaces was demonstrated by their compatibility with a wide range of polymeric coatings, glycan ligand structures and interrogating proteins (QCM platform).
To make the above studies possible, synthetic methods have been developed and carbohydrate structures synthesized. For example, a highly efficient and stereocontrolled protocol, using unprotected thiocarbohydrates, to obtain various S-linked glycoconjugates was developed. Furthermore, a long-standing question, the mutarotation of 1-thioaldopyranoses, has been proven to occur readily in aqueous media. A strong pH-dependence was demonstrated for the process.
Looking forward, a series of research projects can be further pursued based on the current thesis:

74
In order to more efficiently use the carbohydrate microarray platform for applications in glycobiology, projects on extending the arrays with an appropriate number of carbohydrate structures can be carried out. The carbohydrate structures may be obtained either from synthetic means based on our own experience and effort, or from commercial sources, consortia and other research groups in glycobiology and glycochemistry.
Evaluation of additional methods is desired to expand the photochemical carbohydrate immobilization approaches. For example, a site-specific immobilization method to attach free glycans can be invisaged by using hydroxylamine- or hydrazide-terminated PFPA-linker molecules. In this context, studies to determine and elucidate the point of attachment of the free carbohydrate rings by the direct photoconjugation approach (approach II) have been initiated.
More detailed information about the glycan surfaces is valuable, which can be obtained by various surface characterization tools and methods. For example, to determine the immobilization efficiency of various PFPA-carbohydrates with different linker lengths, respective PFPA-linkers derivatized by certain fluorophores can be synthesized and used for the purpose.
Quantitative analyses of carbohydrate-protein interactions using our SPR, QCM and microarray platforms are also highly desirable. Some attempts in this direction are currently ongoing.
Moreover, the production of higher-density carbohydrate arrays is another desired research aim, the combination of microarray technologies with nanomaterials scaffolds will aid us in achieving such goals. For the applications of the established glycan surfaces, current studies have been mainly focused on different carbohydrate-binding protein analyses, however, much more can be explored. These include for example high-throughput ligand screening by the microarray platform and feasibility study of bacterial or toxin analysis.
Last but not least, preparing carbohydrate-functionalized polymer nanoparticles by use of the established photochemical approaches for various diagnostic and therapeutic applications, such as targeted drug delivery and release, is also of great interest, considering a number of unique properties of polymeric materials and their compatibility in biological systems.

75
Acknowledgements
Herein I would like to take the opportunity to express my deepest gratitude and heartfelt thanks to all the people who have guided, supported and helped me in various ways during the past four years.
First and foremost, I sincerely thank my supervisor, Professor Olof Ramström, for accepting me as a PhD student in the group, for guiding me all the way along, for offering me various support and opportunities, for all kinds of advices, for strong trust in me from day one and for your forever open office door – amazing availability and patience whenever I want to discuss with you. Olof, it has truly been a fantastic experience and great pleasure working with you!
A very sincere thanks also goes to Professor Mingdi Yan, thank you for your recommendation in the first place, for your everlasting guidance and support, and for the fruitful and pleasant collaborations in the last four years.
I also feel grateful to:
Professor Tore Brinck, Professor Louis Cuccia and Professor Angela Gronenborn for nice collaborations.
My other co-authors: Professor Hai Dong, Oscar Norberg, Dr. Rémi Caraballo, Dr. Luis Amorim, Xin Wang, Suji Uppalapati, Dr. Anuradha Tyagi, Carolin Madwar, William Chu Kwan, Dr. Rolf Schmidt, Dr. Shan Zou, Dr. Teodor Aastrup, Dr. Elena Matei, Dr. Martin Rahm, Adrien Joliton, Dr. Delphine Gallois-Montbrun and Dr. Manishkumar Shimpi for pleasant team work in different projects.
Professor Torbjörn Norin for taking time and effort to preview my thesis and provide constructive criticisms and valuable comments. Dr. Rémi Caraballo, Dr. Morakot Sakulsombat and all the current Ramström group members for helping with proof-reading and comment on the thesis.
My colleagues and friends in the Ramström group. A special thanks to Hai for helping me with all sorts of matters when I just started in the group and arrived in the country. Other former members including Zhichao, Rikard, Gunnar, Pornrapee (Jom), Rémi, Marcus and Morakot, current members Oscar, Yan, Lei, Manish, Niranjan, Juan, Na, Yang, Sheng, Fredrik, Javier, and my three Erasmus students Adrien, Katharina and Philipp during the years – thank you all for being good friends, for creating pleasant time in the lab and for spreading a nice atmosphere throughout the group.

76
Professor Christina Moberg, Professor Torbjörn Norin, Professor Licheng Sun, Professor Peter Somfai, Professor Anna-Karin Borg Karlson and Dr. Zoltán Szabó for interesting graduate courses.
Dr. Ulla Jacobsson and Dr. Zoltán Szabó for support with the NMR instrumentation and other matters.
Lena Skowron, Henry Challis and Ilona Mozsi for all kinds of help.
All former and present coworkers in the Chemistry/Organic Chemistry Department for a friendly and pleasant working atmosphere.
The Swedish Research Council, the Royal Institute of Technology and the China Scholarship Council for financial support.
The Aulin-Erdtman foundation and Knut och Alice Wallenbergs Stiftelse for travel grants.
I would also like to warmly thank some of my non-research-related friends: Lin Cao, Xiaoling Cao, Hairong Deng, Junmei Duan, Jing Fu, Hailong Li, Minyue Li, Xiaogai Li, Han-Hsuan Lin, Zhonghai Lu, Zhanyu Ma, Yuxin Pei, Jian Qin, Zhongwei Si, Yongbo Tang, Tianling Wei, Juan Wu, Sihong Wu, Ailing Xu, Chunlin Xu, Ting Yang, Yongliang Yang, Peiqun Ye, Liang Zhang, Qian Zhang, Qinglin Zhang, Chelsea, Mandy, Markus, Victor…. Thank you guys for making my days in Stockholm this colorful and memorable.
I thank all the good friends and teachers in China for everlasting care and support as well.
A special thanks to Aunt Zhilan, for so much care, help and support in the past four years. You have made my life here really much easier and more comfortable. Thank Xinxin for being cute and nice.
Elaine, thank you for the many years of love, and for being patient, considerate and supportive.
My parents and sister in China, my gratitude becomes nonverbal when I think of you. Thank you, thank you and thank you!
最衷心的感谢父母这么多年来对我最无私的爱、培育、鼓励和支持!也谢谢姐姐一直以来的关心和爱。

77
Appendix
The following is a description of my contribution to publications I to VIII, as requested by KTH. Paper I: I contributed to the formation of the research problems (major contribution to the initiation of the project), and shared the experimental work and writing. Paper II: I contributed to the formation of the research problems, performed the majority of the experimental work and wrote the manuscript. Paper III: I contributed to the formation of the research problems, performed the majority of the experimental work and wrote the manuscript. Paper IV: I contributed to the formation of the research problems, and performed part of the experimental work. Paper V: I contributed to the formation of the research problems, and performed part of the experimental work. Paper VI: I contributed to the formation of the research problems, and shared the experimental work and writing. Paper VII: I contributed to the formation of the research problems, and performed part of the experimental work. Paper VIII: I contributed to the formation of the research problems, and performed part of the experimental work.

78

79
References
(1) Varki, A. Glycobiology 1993, 3, 97-130. (2) Dwek, R. A. Chem. Rev. 1996, 96, 683-720. (3) Varki, A.; Cummings, R. D.; Esko, J. D.; Freeze, H. H.; Stanley, P.;
Bertozzi, C. R.; Hart, G. W.; Etzler, M. E. Essentials of Glycobiology, Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press, 2009.
(4) Rademacher, T. W.; Parekh, R. B.; Dwek, R. A. Annu. Rev. Biochem. 1988, 57, 785-838.
(5) Kalyuzhin, O.; Zemlyakov, A.; Kalina, N.; Mulik, E.; Kuzovlev, F.; Makarova, O. Bull. Exp. Biol. Med. 2008, 145, 623-625.
(6) Edward, J. T. Chem. Ind. 1955, 1102-1104. (7) Lemieux, R. U. Pure Appl. Chem. 1971, 25, 527-548. (8) Chabre, Y. M.; Roy, R. Adv. Carbohydr. Chem. Biochem. 2010, 63,
165-393. (9) Coombe, D. R.; Kett, W. C. Cell. Mol. Life Sci. 2005, 62, 410-424. (10) Lever, R.; Page, C. P. Nat. Rev. Drug Discov. 2002, 1, 140-148. (11) The Future of Biotech. The 2010 Guide to Emerging Markets and
Technology, BioWorld Today Publishers, 2010. (12) Bertozzi, C. R.; Kiessling, L. L. Science 2001, 291, 2357-2364. (13) Sears, P.; Wong, C.-H. Science 2001, 291, 2344-2350. (14) Seeberger, P. H.; Werz, D. B. Nature 2007, 446, 1046-1051. (15) Dam, T. K.; Brewer, C. F. Chem. Rev. 2002, 102, 387-430. (16) Mischnick, P.; Niedner, W.; Adden, R. Macromol. Symp. 2005, 223,
67-77. (17) Pei, Z.; Yu, H.; Theurer, M.; Waldén, A.; Nilsson, P.; Yan, M.;
Ramström, O. ChemBioChem 2007, 8, 166-168. (18) Deng, L.; Norberg, O.; Uppalapati, S.; Yan, M.; Ramström, O. Org.
Biomol. Chem. 2011, 9, 3188-3198. (19) Tyagi, A.; Wang, X.; Deng, L.; Ramström, O.; Yan, M. Biosens.
Bioelectron. 2010, 26, 344-350. (20) Norberg, O.; Deng, L.; Aastrup, T.; Yan, M.; Ramström, O. Anal.
Chem. 2011, 83, 1000-1007. (21) Norberg, O.; Deng, L.; Yan, M.; Ramström, O. Bioconjugate Chem.
2009, 20, 2364-2370. (22) Madwar, C.; Chu Kwan, W.; Deng, L.; Ramström, O.; Schmidt, R.;
Zou, S.; Cuccia, L. A. Langmuir 2010, 26, 16677-16680. (23) Wang, X.; Ramström, O.; Yan, M. Anal. Chem. 2010, 82, 9082-9089. (24) Jansson, J. L. M.; Maliniak, A.; Widmalm, G. ACS Symp. Ser. 2006,
930, 20-39.

80
(25) Larsen, K.; Thygesen, M. B.; Guillaumie, F.; Willats, W. G. T.; Jensen, K. J. Carbohydr. Res. 2006, 341, 1209-1234.
(26) Fazio, F.; Bryan, M. C.; Blixt, O.; Paulson, J. C.; Wong, C.-H. J. Am. Chem. Soc. 2002, 124, 14397-14402.
(27) Feizi, T.; Fazio, F.; Chai, W.; Wong, C.-H. Curr. Opin. Struct. Biol. 2003, 13, 637-645.
(28) Houseman, B. T.; Mrksich, M. Chem. Biol. 2002, 9, 443-454. (29) Laurent, N.; Voglmeir, J.; Flitsch, S. L. Chem. Commun. 2008,
4400-4412. (30) Liu, Y.; Feizi, T. In: Glycoscience. Fraser-Reid, B., Tatsuta K., Thiem
J. (Eds), Springer-Verlag Berlin Heidelberg, 2008, p. 2121. (31) Love, K. R.; Seeberger, P. H. Angew. Chem. Int. Ed. 2002, 41,
3583-3586. (32) Park, S.; Lee, M.-R.; Shin, I. Chem. Commun. 2008, 4389-4399. (33) Song, E.-H.; Pohl, N. L. B. Curr. Opin. Chem. Biol. 2009, 13,
626-632. (34) Wang, D.; Liu, S.; Trummer, B. J.; Deng, C.; Wang, A. Nat.
Biotechnol. 2002, 20, 275-281. (35) D'Agata, R.; Grasso, G.; Iacono, G.; Spoto, G.; Vecchio, G. Org.
Biomol. Chem. 2006, 4, 610-612. (36) Karamanska, R.; Clarke, J.; Blixt, O.; MacRae, J.; Zhang, J.; Crocker,
P.; Laurent, N.; Wright, A.; Flitsch, S.; Russell, D.; Field, R. Glycoconjugate J. 2008, 25, 69-74.
(37) Linman, M. J.; Yu, H.; Chen, X.; Cheng, Q. ACS Appl. Mater. Interfaces 2009, 1, 1755-1762.
(38) Liu, W.; Chen, Y.; Yan, M. Analyst 2008, 133, 1268-1273. (39) Mercey, E.; Sadir, R.; Maillart, E.; Roget, A.; Baleux, F.;
Lortat-Jacob, H.; Livache, T. Anal. Chem. 2008, 80, 3476-3482. (40) Smith, E. A.; Thomas, W. D.; Kiessling, L. L.; Corn, R. M. J. Am.
Chem. Soc. 2003, 125, 6140-6148. (41) Sauerbrey, G. Z. Phys. 1959, 155, 206-222. (42) Grate, J. W.; Martin, S. J.; White, R. M. Anal. Chem. 1993, 65,
940A-948A. (43) Guilbaut, G. C. In: Methods and Phenomena. Their Applications in
Science and Technology, Lu C., Czanderna, A. W. (Eds), Elsevier, New York, 1984, p. 251.
(44) Pei, Y.; Yu, H.; Pei, Z.; Theurer, M.; Ammer, C.; Andre, S.; Gabius, H.-J.; Yan, M.; Ramström, O. Anal. Chem. 2007, 79, 6897-6902.
(45) Pei, Z.; Larsson, R.; Aastrup, T.; Anderson, H.; Lehn, J.-M.; Ramström, O. Biosens. Bioelectron. 2006, 22, 42-48.
(46) Bizzarri, A. R.; Cannistraro, S. Chem. Soc. Rev. 2010, 39, 734-749. (47) Neuman, K. C.; Nagy, A. Nat. Methods 2008, 5, 491-505.

81
(48) Handbook of Molecular Force Spectroscopy. Noy, A. (Ed.), Springer: New York, 2008.
(49) Zlatanova, J.; Lindsay, S. M.; Leuba, S. H. Prog. Biophys. Mol. Biol. 2000, 74, 37-61.
(50) de la Fuente, J. M.; Barrientos, A. G.; Rojas, T. C.; Rojo, J.; Cañada, J.; Fernández, A.; Penadés, S. Angew. Chem. Int. Ed. 2001, 40, 2257-2261.
(51) El-Boubbou, K.; Huang, X. Curr. Med. Chem. 2011, 18, 2060-2078. (52) Marradi, M.; Martín-Lomas, M.; Penadés, S. Adv. Carbohydr. Chem.
Biochem. 2010, 64, 211-290. (53) Cohen, M.; Varki, A. OMICS 2010, 14, 455-464. (54) Lee, Y. C.; Lee, R. T. Acc. Chem. Res. 1995, 28, 321-327. (55) Lundquist, J. J.; Toone, E. J. Chem. Rev. 2002, 102, 555-578. (56) Willats, W. G. T.; Rasmussen, S. E.; Kristensen, T.; Mikkelsen, J. D.;
Knox, J. P. Proteomics 2002, 2, 1666-1671. (57) Bryan, M. C.; Fazio, F.; Lee, H.-K.; Huang, C.-Y.; Chang, A.; Best,
M. D.; Calarese, D. A.; Blixt, O.; Paulson, J. C.; Burton, D.; Wilson, I. A.; Wong, C.-H. J. Am. Chem. Soc. 2004, 126, 8640-8641.
(58) Feizi, T.; Stoll, M. S.; Yuen, C.-T.; Chai, W.; Lawson, A. M. Meth. Enzymol. 1994, 230, 484-519.
(59) Mann, D. A.; Kanai, M.; Maly, D. J.; Kiessling, L. L. J. Am. Chem. Soc. 1998, 120, 10575-10582.
(60) Ko, K.-S.; Jaipuri, F. A.; Pohl, N. L. J. Am. Chem. Soc. 2005, 127, 13162-13163.
(61) Pohl, N. L. Angew. Chem. Int. Ed. 2008, 47, 3868-3870. (62) Uzawa, H.; Ito, H.; Izumi, M.; Tokuhisa, H.; Taguchi, K.; Minoura, N.
Tetrahedron 2005, 61, 5895-5905. (63) Uzawa, H.; Ito, H.; Neri, P.; Mori, H.; Nishida, Y. ChemBioChem
2007, 8, 2117-2124. (64) Linman, M. J.; Taylor, J. D.; Yu, H.; Chen, X.; Cheng, Q. Anal. Chem.
2008, 80, 4007-4013. (65) Shao, M.-C. Anal. Biochem. 1992, 205, 77-82. (66) Houseman, B.; Mrksich, M. Top. Curr. Chem. 2002, 218, 1-44. (67) Qian, X.; Metallo, S. J.; Choi, I. S.; Wu, H.; Liang, M. N.; Whitesides,
G. M. Anal. Chem. 2002, 74, 1805-1810. (68) Zhang, Y.; Luo, S.; Tang, Y.; Yu, L.; Hou, K.-Y.; Cheng, J.-P.; Zeng,
X.; Wang, P. G. Anal. Chem. 2006, 78, 2001-2008. (69) Park, S.; Shin, I. Angew. Chem. Int. Ed. 2002, 41, 3180-3182. (70) Ratner, D. M.; Adams, E. W.; Su, J.; O'Keefe, B. R.; Mrksich, M.;
Seeberger, P. H. ChemBioChem 2004, 5, 379-383. (71) Lee, M.-R.; Shin, I. Angew. Chem. Int. Ed. 2005, 44, 2881-2884. (72) Park, S.; Lee, M.-R.; Shin, I. Nat. Protocols 2007, 2, 2747-2758.

82
(73) Schwarz, M.; Spector, L.; Gargir, A.; Shtevi, A.; Gortler, M.; Altstock, R. T.; Dukler, A. A.; Dotan, N. Glycobiology 2003, 13, 749-754.
(74) Blixt, O.; Head, S.; Mondala, T.; Scanlan, C.; Huflejt, M. E.; Alvarez, R.; Bryan, M. C.; Fazio, F.; Calarese, D.; Stevens, J.; Razi, N.; Stevens, D. J.; Skehel, J. J.; van Die, I.; Burton, D. R.; Wilson, I. A.; Cummings, R.; Bovin, N.; Wong, C.-H.; Paulson, J. C. Proc. Nat. Acad. Sci. 2004, 101, 17033-17038.
(75) Disney, M. D.; Seeberger, P. H. Chem. Biol. 2004, 11, 1701-1707. (76) Liang, P.-H.; Wang, S.-K.; Wong, C.-H. J. Am. Chem. Soc. 2007, 129,
11177-11184. (77) de Paz, J. L.; Spillmann, D.; Seeberger, P. H. Chem. Commun. 2006,
3116-3118. (78) Köhn, M.; Wacker, R.; Peters, C.; Schröder, H.; Soulère, L.;
Breinbauer, R.; Niemeyer, C. M.; Waldmann, H. Angew. Chem. Int. Ed. 2003, 42, 5830-5834.
(79) Adams, E. W.; Ratner, D. M.; Bokesch, H. R.; McMahon, J. B.; O'Keefe, B. R.; Seeberger, P. H. Chem. Biol. 2004, 11, 875-881.
(80) Chevolot, Y.; Bucher, O.; Léonard, D.; Mathieu, H. J.; Sigrist, H. Bioconjugate Chem. 1999, 10, 169-175.
(81) Bohorov, O.; Andersson-Sand, H.; Hoffmann, J.; Blixt, O. Glycobiology 2006, 16, 21C-27C.
(82) Xia, B.; Kawar, Z. S.; Ju, T.; Alvarez, R. A.; Sachdev, G. P.; Cummings, R. D. Nat. Methods 2005, 2, 845-850.
(83) Lee, M.-R.; Shin, I. Org. Lett. 2005, 7, 4269-4272. (84) Zhi, Z.-L.; Powell, A. K.; Turnbull, J. E. Anal. Chem. 2006, 78,
4786-4793. (85) Zhou, X.; Zhou, J. Biosens. Bioelectron. 2006, 21, 1451-1458. (86) Angeloni, S.; Ridet, J. L.; Kusy, N.; Gao, H.; Crevoisier, F.;
Guinchard, S.; Kochhar, S.; Sigrist, H.; Sprenger, N. Glycobiology 2005, 15, 31-41.
(87) Carroll, G. T.; Wang, D.; Turro, N. J.; Koberstein, J. T. Langmuir 2006, 22, 2899-2905.
(88) Liu, L.-H.; Dietsch, H.; Schurtenberger, P.; Yan, M. Bioconjugate Chem. 2009, 20, 1349-1355.
(89) Liu, L.-H.; Yan, M. Acc. Chem. Res. 2010, 43, 1434-1443. (90) Wang, H.; Zhang, Y.; Yuan, X.; Chen, Y.; Yan, M. Bioconjugate
Chem. 2011, 22, 26-32. (91) Wang, X.; Ramström, O.; Yan, M. J. Mater. Chem. 2009, 19,
8944-8949. (92) Wang, X.; Ramström, O.; Yan, M. Chem. Commun. 2011, 47,
4261-4263. (93) Wang, X.; Ramström, O.; Yan, M. Adv. Mater. 2010, 22, 1946-1953.

83
(94) Carroll, G.; Wang, D.; Turro, N.; Koberstein, J. Glycoconjugate J. 2008, 25, 5-10.
(95) Bayley, H.; Scroven, J. V. In: Azides and Nitrenes. Scriven, E. F. (Ed.), 1984, p. 434.
(96) Platz, M. S. Acc. Chem. Res. 1995, 28, 487-492. (97) Yan, M. Chem. Eur. J. 2007, 13, 4138-4144. (98) Liu, L.; Yan, M. Angew. Chem. Int. Ed. 2006, 45, 6207-6210. (99) Liu, L.-H.; Lerner, M. M.; Yan, M. Nano Lett. 2010, 10, 3754-3756. (100) Yan, M.; Cai, S. X.; Wybourne, M. N.; Keana, J. F. W. J. Am. Chem.
Soc. 1993, 115, 814-816. (101) Deng, L.; Wang, X.; Uppalapati, S.; Norberg, O.; Dong, H.; Joliton,
A.; Yan, M.; Ramström, O. Submitted 2011. (102) Wang, X.; Matei, E.; Deng, L.; Ramstrom, O.; Gronenborn, A. M.;
Yan, M. Chem. Commun. 2011, 47, 8620-8622. (103) Caraballo, R.; Deng, L.; Amorim, L.; Brinck, T.; Ramström, O. J.
Org. Chem. 2010, 75, 6115-6121. (104) Demchenko, A. V. In: Handbook of Chemical Glycosylation:
Advances in Stereoselectivity and Therapeutic Relevance. Demchenko, A. V. (Ed.), 2008, pp 1-27.
(105) Harris, J. M. In: Poly(ethylene glycol) Chemistry: Biotechnical and Biomedical Applications; Harris, J. M. (Ed.), 1992, pp 1-14.
(106) Lee, Y.; Koo, H.; Jin, G.-W.; Mo, H.; Cho, M. Y.; Park, J.-Y.; Choi, J. S.; Park, J. S. Biomacromolecules 2005, 6, 24-26.
(107) Keana, J. F. W.; Cai, S. X. J. Org. Chem. 1990, 55, 3640-3647. (108) Neises, B.; Steglich, W. Angew. Chem. Int.Ed. 1978, 17, 522-524. (109) André, S.; Pei, Z.; Siebert, H.-C.; Ramström, O.; Gabius, H.-J.
Bioorg. Med. Chem. 2006, 14, 6314-6326. (110) Hashimoto, H.; Shimada, K.; Horito, S. Tetrahedron: Asymmetry
1994, 5, 2351-2366. (111) Bewley, C. A.; Gustafson, K. R.; Boyd, M. R.; Covell, D. G.; Bax, A.;
Clore, G. M.; Gronenborn, A. M. Nat. Struct. Mol. Biol. 1998, 5, 571-578.
(112) Bewley, C. A.; Otero-Quintero, S. J. Am. Chem. Soc. 2001, 123, 3892-3902.
(113) Beignet, J.; Tiernan, J.; Woo, C. H.; Kariuki, B. M.; Cox, L. R. J. Org. Chem. 2004, 69, 6341-6356.
(114) Eklind, K.; Gustafsson, R.; Tidén, A. K.; Norberg, T.; Åberg, P. M. J. Carbohydr. Chem. 1996, 15, 1161-1178.
(115) Nakabayashi, S.; Warren, C. D.; Jeanloz, R. W. Carbohydr. Res. 1986, 150, c7-c10.
(116) Driguez, H. Top. Curr. Chem. 1997, 187, 85-116. (117) Gamblin, D. P.; Scanlan, E. M.; Davis, B. G. Chem. Rev. 2009, 109,
131-163.

84
(118) Pachamuthu, K.; Schmidt, R. R. Chem. Rev. 2006, 106, 160-187. (119) Zhang, Y.-M.; Mallet, J.-M.; Sinay, P. Carbohydr. Res. 1992, 236,
73-88. (120) Isbell, H. S.; Pigman, W.; Melville L. Wolfrom, R. S. T.; Derek, H.
Adv. Carbohydr. Chem. Biochem. 1970, 24, 13-65. (121) Pigman, W.; Isbell, H. S.; Melville, L. W.; Tipson, R. S. Adv.
Carbohydr. Chem. Biochem. 1968, 23, 11-57. (122) Juaristi, E.; Cuevas, G. Tetrahedron 1992, 48, 5019-5087. (123) Davis, B. G.; Fairbanks, A. J. Carbohydrate Chemistry, Oxford
University Press, Oxford, 2002. (124) Sinnott, M. L. Carbohydrate Chemistry and Biochemistry: Structure
and Mechanism, Royal Society of Chemistry, 2007. (125) Grimshaw, C. E.; Whistler, R. L.; Cleland, W. W. J. Am. Chem. Soc.
1979, 101, 1521-1532. (126) Smiataczowa, K.; Kosmalski, J.; Nowacki, A.; Czaja, M.; Warnke, Z.
Carbohydr. Res. 2004, 339, 1439-1445. (127) Bernardes, G. J. L.; Gamblin, D. P.; Davis, B. G. Angew. Chem. Int.
Ed. 2006, 45, 4007-4011. (128) Bundle, D. R.; Rich, J. R.; Jacques, S.; Yu, H. N.; Nitz, M.; Ling,
C.-C. Angew. Chem. Int. Ed. 2005, 44, 7725-7729. (129) Marcaurelle, L. A.; Bertozzi, C. R. Chem. Eur. J. 1999, 5, 1384-1390. (130) Thayer, D. A.; Yu, H. N.; Galan, M. C.; Wong, C.-H. Angew. Chem.
Int. Ed. 2005, 44, 4596-4599. (131) Horton, D.; Hutson, D. H.; Melville, L. W.; Tipson, R. S. Adv.
Carbohydr. Chem. 1963, 18, 123-199. (132) Chen, Y.; Chen, G.; Stenzel, M. H. Macromolecules 2010, 43,
8109-8114. (133) van Dongen, S. F. M.; de Hoog, H.-P. M.; Peters, R. J. R. W.; Nallani,
M.; Nolte, R. J. M.; van Hest, J. C. M. Chem. Rev. 2009, 109, 6212-6274.
(134) Caraballo, R.; Dong, H.; Ribeiro, J. P.; Jiménez-Barbero, J.; Ramström, O. Angew. Chem. Int. Ed. 2010, 49, 589-593.
(135) Caraballo, R.; Sakulsombat, M.; Ramström, O. Chem. Commun. 2010, 46, 8469-8471.
(136) Dondoni, A.; Marra, A. Chem. Rev. 2010, 110, 4949-4977. (137) Nelson, A.; Belitsky, J. M.; Vidal, S. b.; Joiner, C. S.; Baum, L. G.;
Stoddart, J. F. J. Am. Chem. Soc. 2004, 126, 11914-11922. (138) Hirohara, S.; Obata, M.; Alitomo, H.; Sharyo, K.; Ando, T.; Yano, S.;
Tanihara, M. Bioconjugate Chem. 2009, 20, 944-952. (139) Fernandez, J.; Mellet, C.; Defaye, J. J. Incl. Phenom. Macrocyclic
Chem. 2006, 56, 149-159. (140) Yu, M.; Yang, Y.; Han, R.; Zheng, Q.; Wang, L.; Hong, Y.; Li, Z.; Sha,
Y. Langmuir 2010, 26, 8534-8539.

85
(141) Josse, S.; Le Gal, J.; Pipelier, M.; Cléophax, J.; Olesker, A.; Pradère, J.-P.; Dubreuil, D. Tetrahedron Lett. 2002, 43, 237-239.
(142) Boons, G.-J.; Stauch, T. Synlett 1996, 1996, 906,908. (143) Connolly, D. T.; Roseman, S.; Lee, Y. C. Carbohydr. Res. 1980, 87,
227-239. (144) Kartha, K. P. R.; Cura, P.; Aloui, M.; Readman, S. K.; Rutherford, T.
J.; Field, R. A. Tetrahedron: Asymmetry 2000, 11, 581-593. (145) Yu, H. N.; Ling, C.-C.; Bundle, D. R. J. Chem. Soc., Perkin Trans. 1
2001, 832-837. (146) Crich, D.; Li, H. J. Org. Chem. 2000, 65, 801-805. (147) Encinas, L.; Chiara, J. L. Eur. J. Org. Chem. 2009, 2009, 2163-2173. (148) Kiessling, L. L.; Young, T.; Grubber, T. D.; Mortell, K. H. In:
Glycoscience. Fraser-Reid, B., Tatsuta K., Thiem J. (Eds), Springer-Verlag Berlin Heidelberg, 2008, p. 2484.
(149) Choi, S. K. Synthetic multivalent molecules: concepts and biomedical applications, Wiley, New York, 2004.
(150) Sumner, J. B. J. Biol. Chem. 1919, 37, 137-142. (151) Nilsson, U.; Johansson, R.; Magnusson, G. Chem. Eur. J. 1996, 2,
295-302. (152) Best, M. D. Biochemistry 2009, 48, 6571-6584. (153) Devaraj, N.; Collman, J. QSAR Comb. Sci. 2007, 26, 1253-1260. (154) Hanson, S.; Greenberg, W.; Wong, C. H. QSAR Comb. Sci. 2007, 26,
1243-1252. (155) Schwarz, F. P.; Puri, K. D.; Bhat, R. G.; Surolia, A. J. Biol. Chem.
1993, 268, 7668-7677. (156) Hinterdorfer, P.; Dufrene, Y. F. Nat. Methods 2006, 3, 347-355. (157) Ratto, T. V.; Langry, K. C.; Rudd, R. E.; Balhorn, R. L.; Allen, M. J.;
McElfresh, M. W. Biophys. J. 2004, 86, 2430-2437. (158) Guo, S.; Ray, C.; Kirkpatrick, A.; Lad, N.; Akhremitchev, B. B.
Biophys. J. 2008, 95, 3964-3976. (159) Oesterhelt, F.; Rief, M.; Gaub, H. E. New J. Phys. 1999, 1, 6.1 - 6.11. (160) Bell, G. I. Science 1978, 200, 618-627. (161) Evans, E.; Ritchie, K. Biophys. J. 1997, 72, 1541-1555. (162) Krieg, M.; Helenius, J.; Heisenberg, C.-P.; Muller, D. J. Angew. Chem.
Int. Ed. 2008, 47, 9775-9777. (163) Ratto, T. V.; Rudd, R. E.; Langry, K. C.; Balhorn, R. L.; McElfresh,
M. W. Langmuir 2006, 22, 1749-1757. (164) Wang, X.; Norberg, O.; Deng, L.; Ramström, O.; Yan, M. ACS Symp.
Ser. 2011, in press. (165) Huskens, J. Curr. Opin. Chem. Biol. 2006, 10, 537-543. (166) Mammen, M.; Choi, S. K.; Whitesides, G. M. Angew. Chem. Int. Ed.
1998, 37, 2754-2794.

86
(167) Mandal, D. K.; Kishore, N.; Brewer, C. F. Biochemistry 1994, 33, 1149-1156.
(168) O'Keefe, B. R.; Shenoy, S. R.; Xie, D.; Zhang, W.; Muschik, J. M.; Currens, M. J.; Chaiken, I.; Boyd, M. R. Mol. Pharmacol. 2000, 58, 982-992.
(169) Barrientos, L. G.; Gronenborn, A. M. Mini-Rev. Med. Chem. 2005, 5, 21-31.
(170) Shenoy, S. R.; Barrientos, L. G.; Ratner, D. M.; O'Keefe, B. R.; Seeberger, P. H.; Gronenborn, A. M.; Boyd, M. R. Chem. Biol. 2002, 9, 1109-1118.
(171) Bewley, C. A.; Kiyonaka, S.; Hamachi, I. J. Mol. Biol. 2002, 322, 881-889.
(172) Daniel, M.-C.; Astruc, D. Chem. Rev. 2004, 104, 293-346. (173) Matei, E.; Furey, W.; Gronenborn, A. M. Structure 2008, 16,
1183-1194.