maladies neuromusculaires: classification, prise en charge cours des myopathies2012 (1).pdf · dr...
TRANSCRIPT

Maladies neuromusculaires: classification, prise en charge
C O U R S D E S D E M P R , S É M I N AI R E N E R F S E T
M U S C L E S , M AR S 2 0 1 2
D R V I N C E N T T I F F R E AU , M C U - P H , L I L L E


Classification topographique
• Maladies du
neurone moteurs
– SLA
– SMA (amyotrophie
spinale
– polio

Classification topographique
• Nerf périphérique
– NP héréditaires
(CMT)
– NP acquises:
• Toxiques, métabol.
• Dysimmunitaires
• …

Classification topographique
• JNM
– Myasthénie
• Auto-immune
• Congénitale
– Lambert-Eaton

Classification topographique
• Myopathies

epidémiologie
• De la maladie fréquente
– NP diabétique
– NP alcoolo-carentielle
• A la maladie rare (< 1/2000)
– Duchenne/Becker
– Steinert 1/25 000 Europe de l’Ouest
– FSH 5-10/100 000 Europe

étiologie
• Génétique AD, AR, lié à X, mitochondriale
• Inflammatoire, immunitaire (myosites,
PRN chronique, myasthénie)
• Métabolique
• Dégénérative,SLA

Tableaux Clinique • D’asymptomatique (HyperCK isolée) à
gravissime
• Age d’apparition : congénital /infantile /adulte
• Evolution: – Aigue: paralysie périodique des canalopathies, SGB
– Subaiguë: PRNc
– Rapide: SLA
– Chronique
– Par poussée • du fait de l’histoire naturelle: myasthénie, PRNc
• du fait de facteurs de décompensation: infections, rhabdomyolyse
• du fait des traitements: neuropathies dysimmunitaires, myasthénie

Symptomes Musculaire • Déficit moteur: quasiment toutes les MNM (sauf
SNP sensitive pure) – Amyotrophie: NP, myopathies /hypertrophie
musculaire (Becker) – Fatigabilité à l’effort: syndrome myasthénique – Myalgies d’effort – Conséquences orthopédiques:
• pieds creux: CMT, HSP • Scoliose • Rétractions tendineuses
– Bulbaires: dysphagie, dysarthrie, dysphonie – Ophtalmologiques – respiratoires

• Neurosensoriels
– Visuel: Steinert, FSH, mitochondrie
– Auditif: FSH, mitochondrie
• Cognitif: DM1, mitochondrie

• Extra-neurologiques:
– Respiratoire
– Cardiaque (myocardiopathie, troubles du
rythmes et de conduction)
– Orthopédique
– Endocrinien
– Rénal
– Gastro-entérologique
– Asthénie,somnolence diurne excessive

Problèmes spécifiques
• Multidisciplinarité: – Neurologue et électrophysiologiste, neuropédiatre
– MPR
– Anatomopathologiste
– Maladies métaboliques
– Interniste
– Généticien
– Pneumologue, réanimateur
– Cardiologue
– kiné, ergo
– Nutritionniste
– Psychologues
– Coordination

Examens utiles au diagnostic
• ENMG – Détection
– Stimulodétecti
on (vitesses
de conduction,
blocs)
– décrément

• Biologie : CPK, lactates à l’effort,
recherches d’AC spécifiques
• Imagerie musculaire (topographie de
l’atteinte)

La biopsie musculaire • Sous AL • Sur un muscle atteint (mais pas trop)
– Répartition types de fibres
– Dystrophie musculaire
– Inclusions/accumulations
– Cellules de l’inflammation
– Fibrose endomysiale
– Mitochondries
– Immunohistochimie (ac spécifiques)
– Western Blot
– enzymologie
– Structure myofibrillaire au ME

Fibrose Régénération
Nécrose

Explorations respitatoires
• EFR
– CV assis , couché, CVF
– VEMS
– Pressions : PImax, PEmax, SNIF
• Gaz du Sang : (hypercapnie)
• Polysomnographie
– Hypoventilation : hypercapnie
– apnées

Exploration cardiaques
• Echocardiographie : FEVG
• Ventriculographie isotopique (FEVG) et IRM
• ECG, HOLTER
• Electrophysiologie intracardiaque (intervalle HV)
(DM1)

Critères entrant dans la
classification
• Ages d’apparitions; ex: Myopathies congénitales/Dystrophies musculaires congénitales
• Topographie musculaire: myopathies distales, oculopharyngée, FSH
• Mécanisme physiopathologique (inflammatoire : polymyosites)
• Morphologiques (dystrophies musculaires, myopathies myofibrillaires)
• Neurophysiologique : myotonies, canalopathies, CMT

Classification topographique
• Maladies du
neurone moteurs
– SLA
– SMA (amyotrophie
spinale
– polio

L A S L A
L A P O L I O
L’ AM Y O T R O P H I E S P I N AL E
Les pathologies du neurone moteur

Amyotrophies spinales
• Génétique : AR, délétion exon 7 du gène SMN
• 1/6000 naissance (fréquence des
hétérozygotes)
– Types I début dans les 3 premiers mois
– Ibis, n’a pas tenu assis
– II a tenu assis mais n’a pas marché
– III a marché (Kukelberg Welander)
– IV = III forme adulte
• Déficience musculaire progressive proximale
• Atteinte respiratoire

M YAS T H É N I E AU T O - I M M U N E
AU T R E S
Les pathologies de la jonction neuro-musculaire

Classification topographique
• JNM
– Myasthénie
• Auto-immune
• Congénitale
– Lambert-Eaton

MG: physio-pathologie 1971-1973: microscopie électronique (Engel)
Moins de RAch
Fente
inter-syn
élargie
Replis
post-syn
éffacés

MG: physio-pathologie
• Anti-RAch:
IgG se fixant près du site liaison Ach (MIR sur partie extra-C de α) des RAch nicotiniques
* Perte de RAch fonctionnels par 3 méca:
- Blocage sites liaison Ach
- Down-regulation (accélération dégradation)
- Lyse mbne post-syn via complément
* Moins de RAch fonctionnels, fatigabilité

MG: clinique
• S’observe à tout âge
• 2 pics chez F: 15-30 ans et > 60 ans
1 pic chez H: > 60 ans
Nette prédo féminine avant 40 ans puis
moindre
• 5-10 % < 15 ans
≠ myasthénie néonatale et ≠ congénitales


MG: clinique
• F. classique (QS):
ROT normaux, ML normal (pupille+++
≠ botulisme)
Pas de systématisation tronc/radicul
prédominance céphalique
50 % début oculaire
Fluctuation+++ en intensité et topographie

MG: paraclinique (EMG)
Myasthénie: décrément > 10 % (A5/A1 ou A4/A1)
Cupule typique, sans retour à 100 % ( vs électrode qui roule)
Baisse relative d’A est maxi entre 1 et 2
A la + basse vers 4

MG: paraclinique (Ac)
• Anti-RAch
Très spécifiques mais
MG séronégatives
= 50 % des MG oculaires
15 % des MG généralisées
NB: refaire à M6-M12 si anti-RAch- au début

MG: paraclinique (Ac)
• MG « séronégatives »
prudence pour dg+
Groupe hétérogène (clinique et
physiopatho)
Origine auto-immune mais Ag ≠ RAch

MG: paraclinique (Ac)
• 20-50 % anti-MuSK+ (Muscle specific
kinase, rôle assemblage initial RAch)
Prédo oculo-bulbaire
+ sévères
Pas de thymome associé, moins anomalies
thymiques
Thymectomie non ou peu efficace

Syndromes myasthéniques congénitaux (SMC)

SMC: généralités
• = maladies génétiques de la transmission
neuromusculaire
• Prévalence 1/ 500 000 en Europe
• Travaux de Engel depuis 25 ans :
(déficit en Achesterase puis déficit pré et
postsynaptique)
cf revue Engel et al. Muscle & nerve Jan
2003

SMC: généralités
• Nbeuses mutations
≈ récessives, entraînent perte de fonction
-SMC présynaptiques (8%)
-SMC synaptiques (16 %)
-SMC post-synaptiques (76 %)+++

S Y N D R O M E P A R A N É O P L A S I Q U E
A C A N T I H U
B l o c p r é s y n a p t i q u e
A t t e i n t e d u s y s t è m e n e r v e u x a u t o n o m e
Syndrome de Lambert-Eaton

Classification des
myopathies
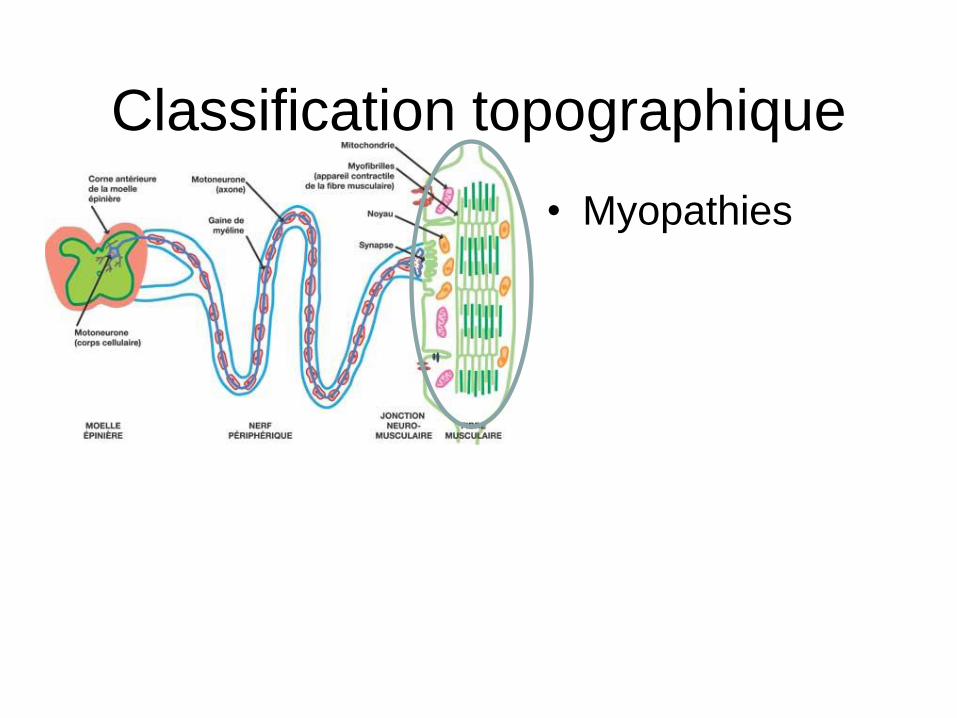
Classification topographique
• Myopathies


Maladies de la fibre musculaire – Dystrophies musculaires
• Dystrophinopathies = DMD, BMD • Dystrophies des ceintures LGMD • FSHD • Dystrophies myotoniques DM1 (Steinert), DM2 • DMC (mérosine, ColVI, FKRP) • Emery Dreifuss et laminopathies • Dystrophie musculaire oculopharyngée
– Collagénopathies (Col VI) Bethlem – Myopathies congénitales (Cores, batonnets, minicores,
myotubulaires) – Canalopathies (paralysie périodiques, myotonies congénitales) – Myopathies distales – Myopathies myofibrillaires – Myopathies métaboliques
• Glycogénose • Anomalies de la béta-oxydation • Cytopathies mitochondriales
– Dermato et polymyosites

Dystrophies musculaires
• Dystrophinopathies = DMD, BMD
• Dystrophies des ceintures (LGMD)
• FSHD = dystrophie facioscapulohumérale
• Dystrophies myotoniques (DM1 =Steinert, DM2)
• DMC = sdystrophies musculaires congénitales
• Emery Dreifuss et laminopathies
• Dystrophie musculaire oculopharyngée (OPMD)


Fibrose Régénération
Nécrose

Dystrophies musculaires
type Duchenne (DMD)
• Transmission liée à l ’X (1garçon /3500)
• Début première décennie (avant 5 ans)
• Déficit musculaire progressif symétrique, CPK++
• Sélectivité de l ’atteinte musculaire:
– quadriceps, psoas, moyen fessier, jambier ant., biceps et triceps
brachial
– grand fessiers, deltoïdes, avant-bras, triceps suraux
(hypertrophie)
• Cardiomyopathie
• Insuffisance respiratoire
• Déficit cognitif: 1/3 des DMD



Dystrophies musculaires
type Becker: les particularités
• Expression phénotypique variable: DMD-like ou
formes très modérées
• Formes type intolérance à l ’effort
• Formes cardiaques pures
Les conductrices:
– asymptomatiques
– intolérance à l ’effort
– présentation type “dystrophies des ceintures”
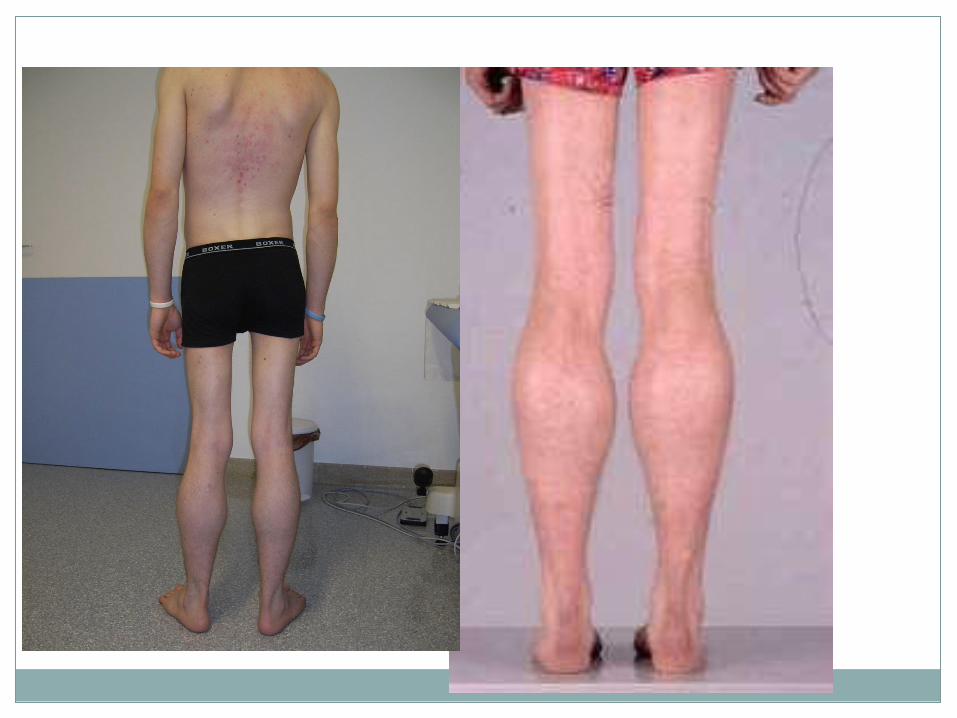

Dystrophies progressives des ceintures
(LGMD)
• Hétérogénéité clinique et génétique +++ • Classification complexe • Progrès considérable depuis la découverte du gène de la
dystrophine (1895) • Le complexe glycoprotéique sarcolemmique (Autosom.
Récessives) – Sarcoglycanes – Dysferline – -dystroglycan
• Les protéines cytoplasmiques (Autosom. Récessives) – Calpaïne
• Les protéines de l’enveloppe nucléaire – Emerine (liée à l ’X) – Lamine A/C (Autosom. Dominant)


Myopathie facio-scapulo-
humérale (FSH)
• Décrite en 1885 par Landouzy et Déjerine
• Prévalence: 1/20 000
• Transmission autosomique dominante
• Age de début: très variable de 4 à 30 ans
• Signes cliniques: déficit et amyotrophie
– ASYMETRIQUE
– des muscles faciaux
– des fixateurs de l’épaule
– des muscles pectoraux
– des extenseurs et des releveurs des orteils

FSH: autres signes cliniques
• Surdité de perception
• Telangiectasies et microanévrysmes rétiniens
• Troubles du rythme supra-ventriculaire
• Insuffisance respiratoire restrictive
– ventilation assistée exceptionnelle

Diagnostic de la FSH
CPK < 5x normale
Biologie moléculaire : liaison avec
une anomalie en 4q35
Région D4Z4 : 12 à 100 répétitions
FSH : moins de 10 répétitions
Infiltrats inflammatoires
périvasculaires
Atteinte dystrophique





Dystrophie myotonique de Steinert
= DM1
Myotonie: lenteur à la décontraction musculaire
D’action et/ou de percussion
Liée à une hyperexcitabilité des fibres
musculaires
Décharge myotonique en EMG:
Non spécifique du Steinert

La DM1 : Une maladie des ARN
• Héréditaire (AD)
• Expansion de triplets (CTG)n dans le gène
DMPK (19q13.3) en région non codante
(n>50)
• Rétension nucléaire des CUGexp-RNAs
(foci)

• Les Focis retiennent des facteurs d’épissages
alternatif MBNL1 et CUG-BP
• Anomalies d’épissage de Pre-mRNA
– CLCN1 (canal Chlore)
– BIN1 (formation des TubulesT)
• Relation (CTG)n, toxicité, sévérité clinique
DM1
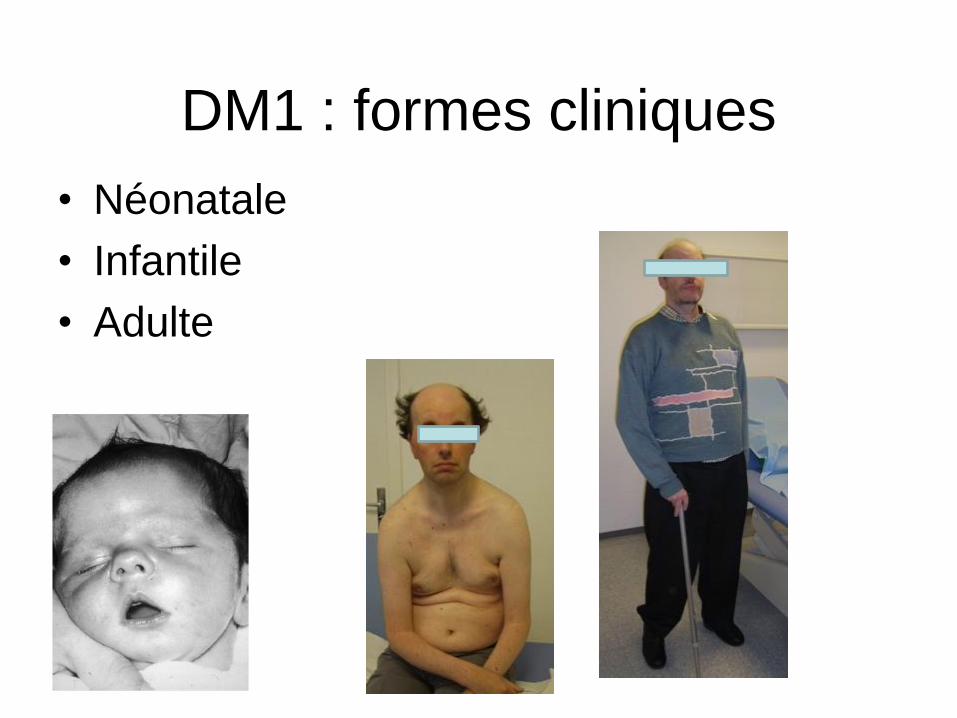
DM1 : formes cliniques
• Néonatale
• Infantile
• Adulte

Manifestations cliniques
• Déficience musculaire : distale, faciale et axiale
• Myotonie
• Cardiopathie (TDC)
• Déficience respiratoire
• Troubles digestifs
• Troubles de déglutition
• Cataracte précoce (rétinopathies)
• Troubles endocriniens (diabète, hypofertilité)
• Troubles cognitifs (retard scolaire, troubles perception des émotions, démence)

DM1 infantile
• Sous diagnostiqué
• Atteinte faciale
• Myotonie souvent marquée (mains)
• Dysarthrie
• Pb de scolarisation, QI faible
• Conduction cardiaque

DM1 adulte
• Parfois présentations atypiques:
– Trb du rythme cardiaque
– Insuffisance respiratoire
– Somnolence diurne excessive
– Trb du transit
– Naissance d’un enfant avec DM1 congénital
• Handicap sévère 50-60 ans





Dystrophie myotonique proximale =
PROMM=DM2
• AD, gène ZNF9
• Déficience proximale et axiale
• Douleurs musculaires
• Moins de signes extramusculaires

Maladies de la fibre musculaire
– Dystrophies musculaires • Dystrophinopathies = DMD, BMD • Dystrophies des ceintures LGMD • FSHD • Dystrophies myotoniques DM1 (Steinert), DM2 • DMC (mérosine, ColVI, FKRP) • Emery Dreifuss et laminopathies • Dystrophie musculaire oculopharyngée
– Collagénopathies (Col VI) Bethlem – Myopathies congénitales (Cores, batonnets, minicores,
myotubulaires) – Canalopathies (paralysie périodiques, myotonies congénitales) – Myopathies distales – Myopathies myofibrillaires – Myopathies métaboliques
• Glycogénose • Anomalies de la béta-oxydation • Cytopathies mitochondriales
– Dermato et polymyosites

Maladies de la fibre musculaire – Dystrophies musculaires
• Dystrophinopathies = DMD, BMD • Dystrophies des ceintures LGMD • FSHD • Dystrophies myotoniques DM1 (Steinert), DM2 • DMC (mérosine, ColVI, FKRP) • Emery Dreifuss et laminopathies
– Collagénopathies (Col VI) Bethlem – Myopathies congénitales (Cores, batonnets, minicores,
myotubulaires) – Canalopathies (paralysie périodiques, myotonies congénitales) – Myopathies distales – Myopathies myofibrillaires – Myopathie oculopharyngée – Myopathies métaboliques
• Glycogénose • Anomalies de la béta-oxydation • Cytopathies mitochondriales
– Dermato et polymyosites

DMC
• Début congénital
• Muscles dystrophiques
• Possibles atteintes du SNC
• Atteinte du tissus de soutien



Maladies de la fibre musculaire – Dystrophies musculaires
• Dystrophinopathies = DMD, BMD • Dystrophies des ceintures LGMD • FSHD • Dystrophies myotoniques DM1 (Steinert), DM2 • DMC (mérosine, ColVI, FKRP) • Emery Dreifuss et laminopathies • Myopathie oculopharyngée
– Collagénopathies (Col VI) Bethlem – Myopathies congénitales (Cores, batonnets, minicores,
myotubulaires) – Canalopathies (paralysie périodiques, myotonies congénitales) – Myopathies distales – Myopathies myofibrillaires – Myopathies métaboliques
• Glycogénose • Anomalies de la béta-oxydation • Cytopathies mitochondriales
– Dermato et polymyosites

Emery Dreifuss
• XR (emerine) + formes AR (lamine A/C)
• Flexums
• Cardiopathie (tdc)

Maladies de la fibre musculaire – Dystrophies musculaires
• Dystrophinopathies = DMD, BMD • Dystrophies des ceintures LGMD • FSHD • Dystrophies myotoniques DM1 (Steinert), DM2 • DMC (mérosine, ColVI, FKRP) • Emery Dreifuss et laminopathies • Myopathie oculopharyngée
– Collagénopathies (Col VI) Bethlem – Myopathies congénitales (Cores, batonnets, minicores,
myotubulaires) – Canalopathies (paralysie périodiques, myotonies congénitales) – Myopathies distales – Myopathies myofibrillaires – Myopathies métaboliques
• Glycogénose • Anomalies de la béta-oxydation • Cytopathies mitochondriales
– Dermato et polymyosites
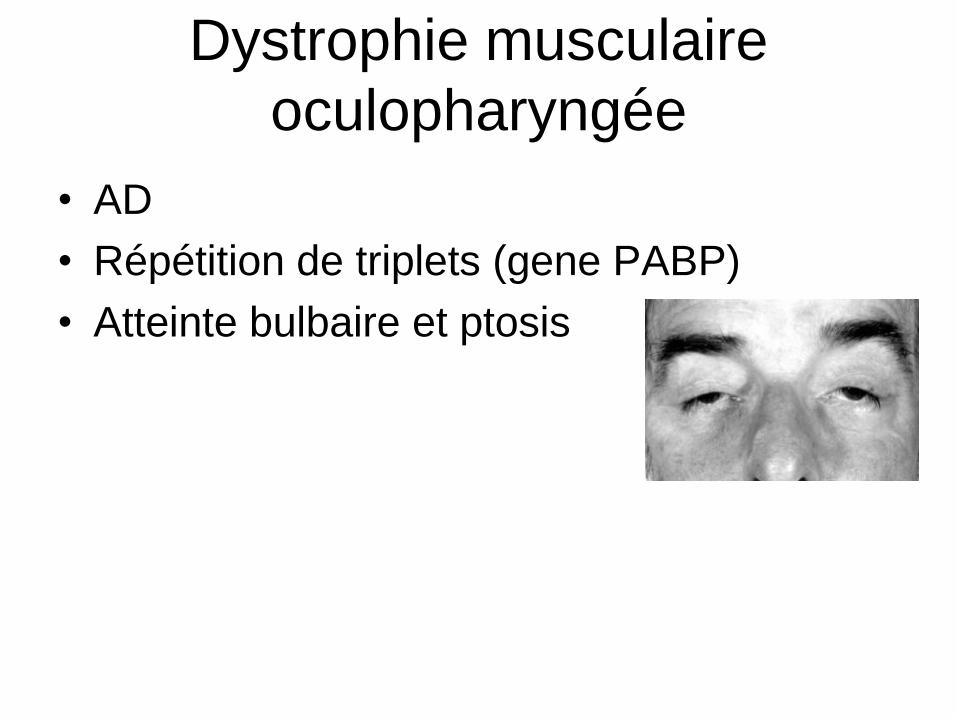
Dystrophie musculaire
oculopharyngée
• AD
• Répétition de triplets (gene PABP)
• Atteinte bulbaire et ptosis

Maladies de la fibre musculaire – Dystrophies musculaires
• Dystrophinopathies = DMD, BMD
• Dystrophies des ceintures LGMD
• FSHD
• Dystrophies myotoniques DM1 (Steinert), DM2
• DMC (mérosine, ColVI, FKRP)
• Emery Dreifuss et laminopathies
– Collagénopathies (Col VI) Bethlem
– Myopathies congénitales (Cores, batonnets, minicores, myotubulaires)
– Canalopathies (paralysie périodiques, myotonies congénitales)
– Myopathies distales
– Myopathies myofibrillaires
– Myopathies métaboliques • Glycogénose
• Anomalies de la béta-oxydation
• Cytopathies mitochondriales
– Dermato et polymyosites

Bethlem (Collagénopathies =
Col IV))

Maladies de la fibre musculaire – Dystrophies musculaires
• Dystrophinopathies = DMD, BMD • Dystrophies des ceintures LGMD • FSHD • Dystrophies myotoniques DM1 (Steinert), DM2 • DMC (mérosine, ColVI, FKRP) • Emery Dreifuss et laminopathies
– Collagénopathies (Col VI) Bethlem – Myopathies congénitales (Cores, batonnets, minicores,
myotubulaires) – Canalopathies (paralysie périodiques, myotonies congénitales) – Myopathies distales – Myopathies myofibrillaires – Myopathie oculopharyngée – Myopathies métaboliques
• Glycogénose • Anomalies de la béta-oxydation • Cytopathies mitochondriales
– Dermato et polymyosites

MYOPATHIES
CONGENITALES
- atteinte primitive de la fibre musculaire
- hétérogénéïté clinique et génétique
- début précoce (0 - 1 an)
- peu ou pas évolutive (histologiquement...) ; CPK normales
- identification précise par microscopie électronique
Clinique commune :
• hypotonie précoce ;
• tr. succion-déglutition ; mb graciles ; visage atone
• déficit proximal
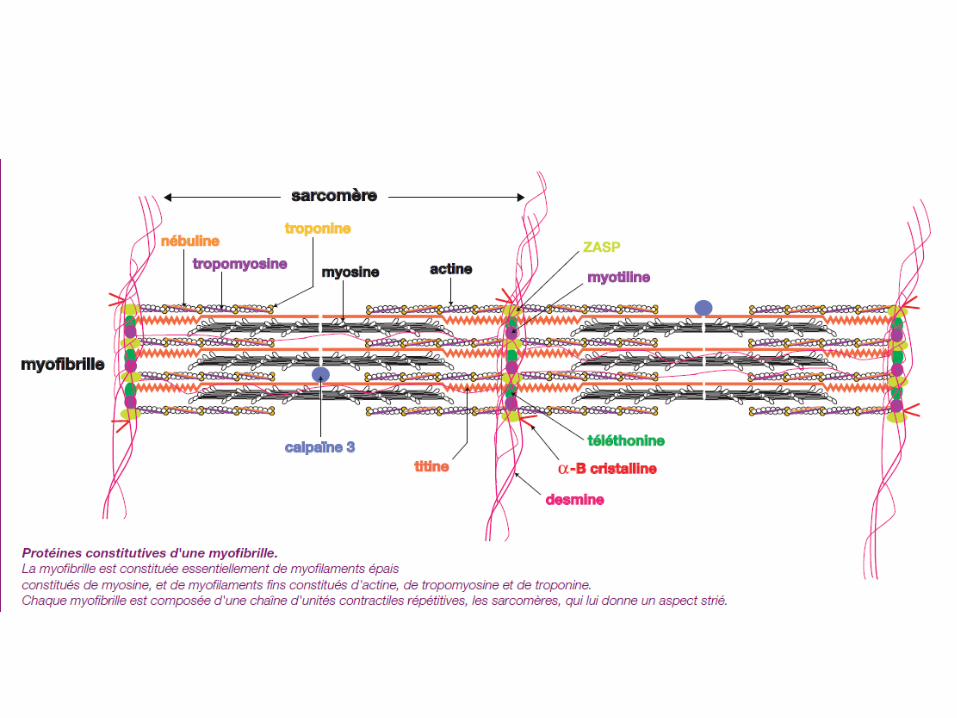

Epidémiologie des myopathies
congénitales Maladies très rares, d’origine génétique
250 enfants souffrant d’hypotonie néonatale, < 2 ans : 14%
180 cas
bâtonnets : 20 % (Acta1,NEB, MYH7, TPM3…)
cores centraux : 16% (Ryr1…)
centronucléaire : 14% (dyn2…)
multiminicore : 10% (sepn1…)
disproportion de fibres / de taille : 21 % (TPM3)
autres (6) : 19%
Transmission : AD, AR, RX


M. congénitale: multiminicores

Maladies de la fibre musculaire – Dystrophies musculaires
• Dystrophinopathies = DMD, BMD • Dystrophies des ceintures LGMD • FSHD • Dystrophies myotoniques DM1 (Steinert), DM2 • DMC (mérosine, ColVI, FKRP) • Emery Dreifuss et laminopathies
– Collagénopathies (Col VI) Bethlem – Myopathies congénitales (Cores, batonnets, minicores,
myotubulaires) – Canalopathies (paralysie périodiques, myotonies
congénitales) – Myopathies distales – Myopathies myofibrillaires – Myopathies métaboliques
• Glycogénose • Anomalies de la béta-oxydation • Cytopathies mitochondriales
– Dermato et polymyosites

Canalopathies
• Myotonies congénitales
• Paramyotonies congénitales
• Paralysie périodique hypokaliémiante
• Paralysie périodique hyperkaliémiante
• Mutations des canaux SCN, CACN,
CLCN, KCN

Maladies de la fibre musculaire – Dystrophies musculaires
• Dystrophinopathies = DMD, BMD • Dystrophies des ceintures LGMD • FSHD • Dystrophies myotoniques DM1 (Steinert), DM2 • DMC (mérosine, ColVI, FKRP) • Emery Dreifuss et laminopathies
– Collagénopathies (Col VI) Bethlem – Myopathies congénitales (Cores, batonnets, minicores,
myotubulaires) – Canalopathies (paralysie périodiques, myotonies
congénitales) – Myopathies distales – Myopathies myofibrillaires – Myopathies métaboliques
• Glycogénose • Anomalies de la béta-oxydation • Cytopathies mitochondriales
– Dermato et polymyosites
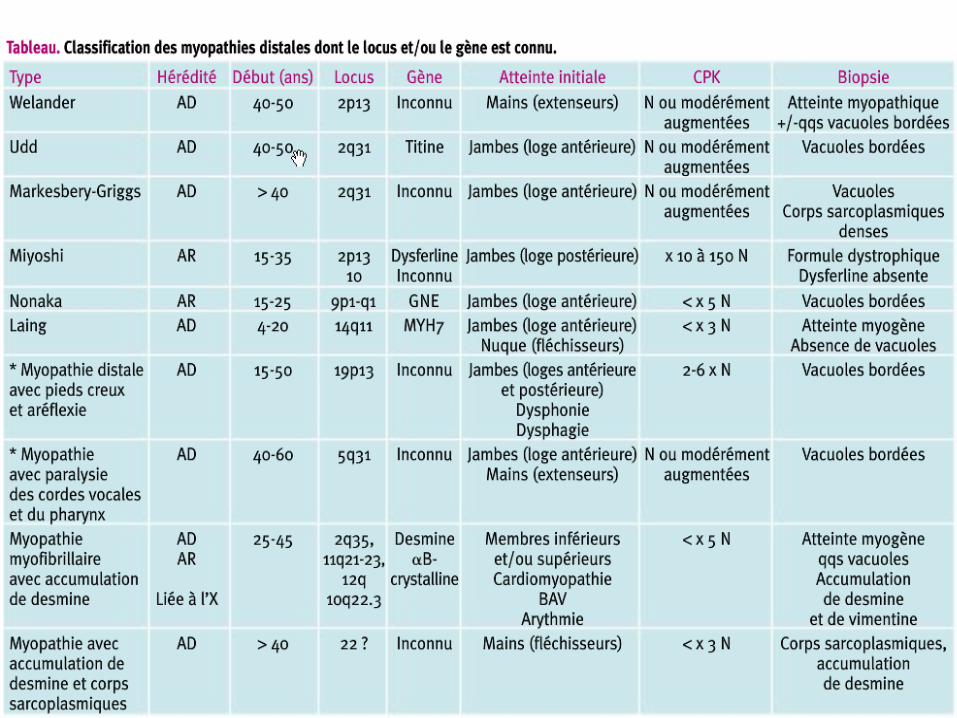
Myopathies distales

Maladies de la fibre musculaire – Dystrophies musculaires
• Dystrophinopathies = DMD, BMD • Dystrophies des ceintures LGMD • FSHD • Dystrophies myotoniques DM1 (Steinert), DM2 • DMC (mérosine, ColVI, FKRP) • Emery Dreifuss et laminopathies
– Collagénopathies (Col VI) Bethlem – Myopathies congénitales (Cores, batonnets, minicores,
myotubulaires) – Canalopathies (paralysie périodiques, myotonies
congénitales) – Myopathies distales – Myopathies myofibrillaires – Myopathies métaboliques
• Glycogénose • Anomalies de la béta-oxydation • Cytopathies mitochondriales
– Dermato et polymyosites
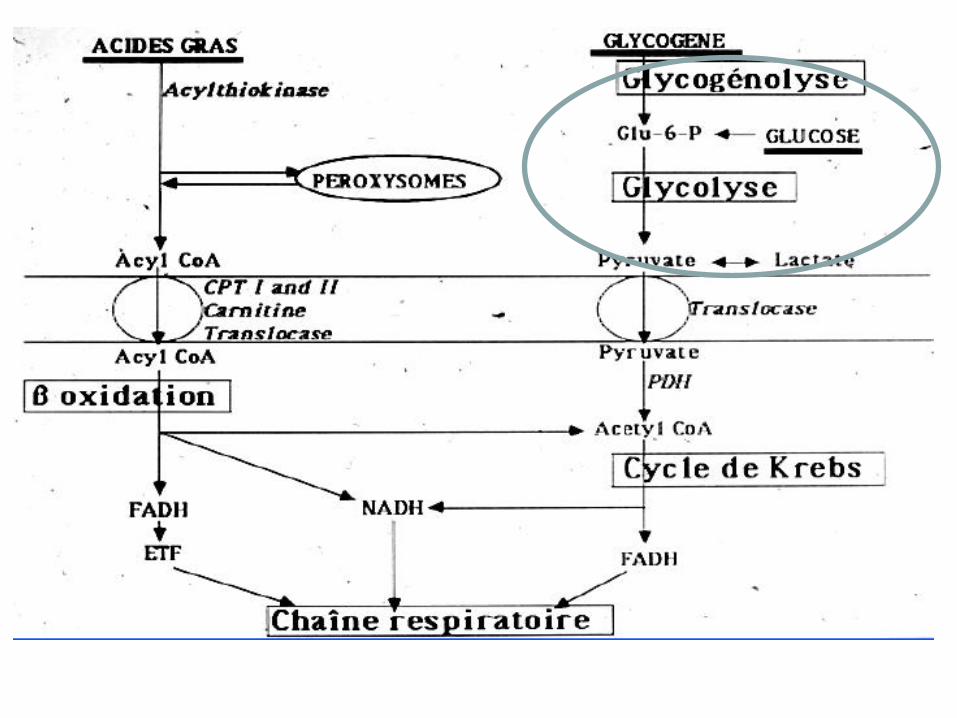

Glycogénoses • Déficit enzymatique touchant la glycolyse
ou la glycogénolyse
– Maladie de Mc Ardle ou Glycogénose de type
V • Intolérance à l’effort
• Élévation des CPK
• Urines foncées
• Douleurs musculaires d’effort, second souffle
• Epreuve d’effort : pas d’élévation des lactates
• Diagnostic en biopsie : surcharge en glycogène, absence de
Myophosphorylase
• Maladies génétique : mutation du gène de la phosphorylase

Glycogénoses
– Glycogénose de type VIII déficit en
Phosphorylase kinase
• Mêmes symptômes
• Possible élévation des lactates d’effort
• Pas de surcharge en glycogène
• Déficit enzymatique sur la biopsie
– Déficit de la glycolyse
• Maladie de Tarui : glycogénose de type VII,
df »ficite en Phosphofructokinase
• Déficit en PGK, PGM, LDH
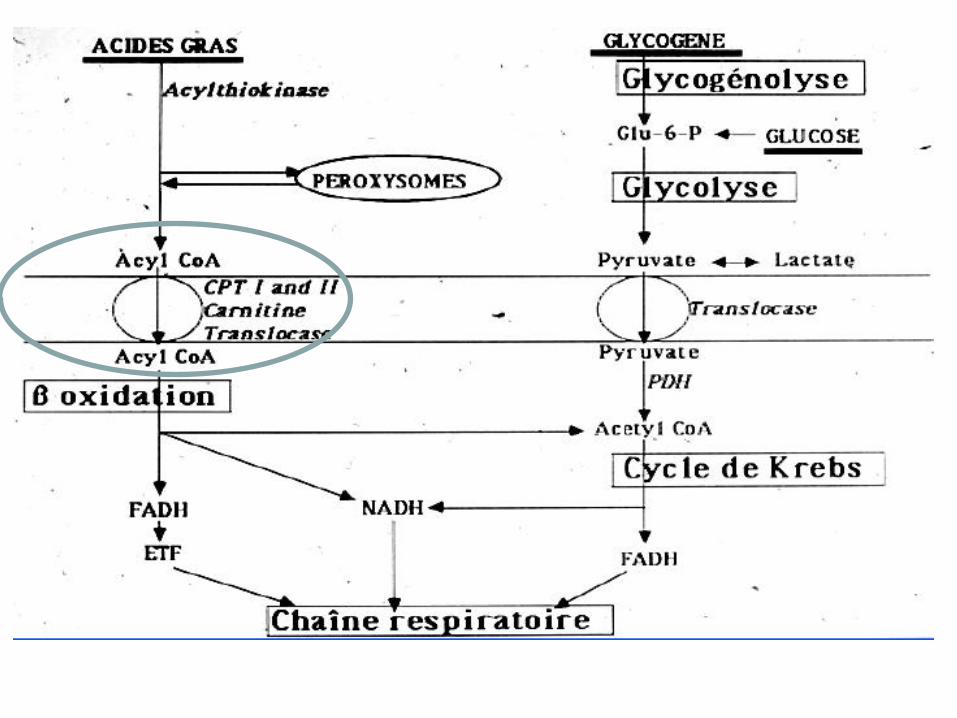

Lipidoses
• Déficit en CPTII – La plus fréquente après la maladie de Mc Ardle
– Pas d’intolérance à l’effort mais Rhabdomyolyse en cas de syndrome infectieux, effort soutenu, jeune
– CPK normales au repos
– La CPT fait entrer les AG dans la mitochondrie
– EE normale
– Biopsie Normale ou surcharge lipidique
– Diagnostic par dosage de l’enzyme sur lymphocyte ou fibroblaste
– AR, 4 mutations connues
– TT régime riche en AG à chaine moyenne


Lipidoses
• Déficit de la Béta Oxydation
– Déficit en Acyl dédhydrogénase à très longue chaine
(VLCAD)
– Rhabdomyolyses d’effort
– Diagnostique :
• chromatographie des acides organiques urinaires
• Spectroscopie de masse des acylcarnitines sanguins
• Etude de l’oxydation des acides gras sur fibroblastes
• Déficit en MAD
– Lactates normaux, hyperammoniémie
– Pathologique?


Cytopathies mitochondriales
• Associé à d’autres atteintes (neuropathie, atteinte centrale, ophtalmo)
• Déficit enzymatique dans la Chaine respiratoire mitochondriale
• Déficit en complexe I, III , IV, coQ
• Étude enzymologique de la chaine respiratoire mitochondriale
• Biopsie : RRF
• Mutations de l’ADN mitochondrial
• Epreuve d’effort : hyperlactatémie
• Traitement : CoQ, riboflavine (vit B12)