manual de laboratorio de qumica orgnica 2006
DESCRIPTION
TRANSCRIPT

UNIVERSIDAD DE CARABOBO
Facultad Experimental de Ciencias y
Tecnología
Departamento de Química
Guía de Laboratorio de
Química Orgánica
Elaborado por: Prof. Douglas J Escalante Ayala
Valencia, Noviembre del 2006

ii

INDICE
iii
INDICE
PRACTICA # 1: TECNICAS EXPERIMETALES ....................................................................................... 1
PUNTO DE FUSIÓN SIMPLE Y MIXTO ..................................................................................................... 1
ANTECEDENTES .............................................................................................................................................. 1
TEORIA ......................................................................................................................................................... 1
Punto de fusión .......................................................................................................................................... 1
Punto de fusión de compuesto impuros; diagrama de presión de vapor-temperatura .............................. 2
Punto de fusión de mezcla de compuestos ................................................................................................. 3
Punto de fusión de compuestos desconocidos ........................................................................................... 4
Métodos, aparatos y equipos para determinar el punto de fusión ............................................................ 4
CUESTIONARIO ............................................................................................................................................... 4
SOLUBILIDAD EN DISOLVENTES ORGÁNICOS Y CRISTALIZACIÓN SIMPLE ........................... 6
ANTECEDENTES ............................................................................................................................................... 6
TEORIA ............................................................................................................................................................ 6
Solubilidad ................................................................................................................................................ 6
TEORÍA DE LA CRISTALIZACIÓN ...................................................................................................................... 8
SELECCIÓN DE DISOLVENTE ............................................................................................................................ 9
TÉCNICA DE CRISTALIZACIÓN ....................................................................................................................... 10
CRISTALIZACIÓN ........................................................................................................................................... 13
CUESTIONARIO ............................................................................................................................................. 14
PUNTO DE EBULLICIÓN, DESTILACIÓN SIMPLE Y FRACCIONADA ........................................... 16
ANTECEDENTES ............................................................................................................................................. 16
TEORIA ......................................................................................................................................................... 16
Punto de ebullición, relación estructura-punto de ebullición ................................................................. 16
Destilación simple ................................................................................................................................... 17
Destilación fraccionada .......................................................................................................................... 18
Diagrama de fase .................................................................................................................................... 19
Eficiencia de la columna ......................................................................................................................... 20
CUESTIONARIO ............................................................................................................................................. 21
EXTRACCIÓN CON DISOLVENTES ORGÁNICOS Y DISOLVENTES ACTIVOS .......................... 23
ANTECEDENTES ............................................................................................................................................ 23
TEORIA ......................................................................................................................................................... 23
Extracción con disolventes orgánicos ..................................................................................................... 23

INDICE
iv
El embudo de separación ........................................................................................................................ 24
Emulsiones ............................................................................................................................................... 26
Agente desecante químico ....................................................................................................................... 26
Extracción selectiva con disolventes activos ........................................................................................... 28
CUESTIONARIO .............................................................................................................................................. 29
SEPARACIÓN Y PURIFICACIÓN DE UNA MEZCLA DE TRES COMPONENTES ......................... 31
OBJETIVOS .................................................................................................................................................... 31
PARTE EXPERIMENTAL .................................................................................................................................. 31
MATERIAL .............................................................................................................................................. 31
SUSTANCIAS .......................................................................................................................................... 31
PROCEDIMIENTO ..................................................................................................................................... 31
Tratamiento de residuos .......................................................................................................................... 32
BIBLIOGRAFIA ............................................................................................................................................... 32
PRACTICA # 2: EL PETRÓLEO Y SUS DERIVADOS ............................................................................ 35
OBJETIVO ................................................................................................................................................... 35
INTRODUCCIÓN. ...................................................................................................................................... 35
TEORIA ....................................................................................................................................................... 36
Destilación simple ................................................................................................................................... 36
Destilación fraccionada .......................................................................................................................... 37
Destilación a presión reducida ................................................................................................................ 39
MATERIAL ................................................................................................................................................. 40
SUSTANCIAS ............................................................................................................................................. 40
PROCEDIMIENTO ..................................................................................................................................... 40
CUESTIONARIO ........................................................................................................................................ 41
PRACTICA # 3: CROMATOGRAFÍA DE CAPA FINA Y CROMATOGRAFIA DE COLUMNA ..... 43
OBJETIVO ...................................................................................................................................................... 43
ANTECEDENTES ....................................................................................................................................... 43
TEORIA ....................................................................................................................................................... 43
Separación sólido-líquido........................................................................................................................ 44
Secuencia de elución por compuesto ....................................................................................................... 45
Parámetros que afectan la separación .................................................................................................... 46
Cromatografía en capa fina .................................................................................................................... 46
Revelador ................................................................................................................................................. 47
Frente de referencia (Rf) ......................................................................................................................... 48
MATERIALES ................................................................................................................................................. 49
MATERIALES ADICIONALES ........................................................................................................................... 49

INDICE
v
SUSTANCIAS ................................................................................................................................................. 49
PROCEDIMIENTO ........................................................................................................................................... 50
Preparación de las placas ....................................................................................................................... 50
Preparación de las cámaras de desarrollo ............................................................................................. 51
Obtención de los valores de Rf a las muestras patrón ............................................................................ 52
Identificación de los compuestos presentes en la muestra problema ...................................................... 52
Extracción de los colorantes del tomate ................................................................................................. 52
CUESTIONARIO ........................................................................................................................................ 53
SEPARACIÓN DE UNA MEZCLA MEDIANTE CROMATOGRAFÍA DE COLUMNA .................... 54
OBJETIVO ...................................................................................................................................................... 54
ANTECEDENTES ............................................................................................................................................ 54
TEORÍA ......................................................................................................................................................... 54
PARTE EXPERIMENTAL ................................................................................................................................. 56
Materiales por equipo de 2 alumnos ....................................................................................................... 56
Sustancias ................................................................................................................................................ 56
PROCEDIMIENTO ........................................................................................................................................... 56
Determinación de las condiciones de separación de los componentes del extracto de los colorantes del
tomate. ..................................................................................................................................................... 56
Empaquetamiento de la columna ............................................................................................................ 56
Carga de la muestra ................................................................................................................................ 58
Elución de la columna ............................................................................................................................. 58
Comparación de la separación realizada, mediante cromatografía en capa fina .................................. 59
CUESTIONARIO ........................................................................................................................................ 61
PRÁCTICA # 4: SINTESIS DEL CICLOHEXENO ................................................................................... 63
OBJETIVOS ................................................................................................................................................ 63
ANTECEDENTES....................................................................................................................................... 63
REACCIÓN A EFECTUAR Y ESTEQUIMETRIA .................................................................................... 63
MECANISMO ............................................................................................................................................. 63
PARTE EXPERIMENTAL .................................................................................................................................. 64
MATERIAL .............................................................................................................................................. 64
SUSTANCIAS .......................................................................................................................................... 65
INFORMACIÓN ...................................................................................................................................... 65
PROCEDIMIENTO ..................................................................................................................................... 65
Método A. Por destilación fraccionada. .................................................................................................. 65
Método B. Por reflujo directo. ................................................................................................................ 67
PRUEBAS DE INSATURACIÓN. ............................................................................................................ 68

INDICE
vi
REACCIÓN CON BR2/CCL4. ............................................................................................................................ 68
REACCIÓN CON KMNO4. ............................................................................................................................... 68
NOTAS ..................................................................................................................................................... 68
DIAGRAMA ECOLOGICO ................................................................................................................................. 69
CUESTIONARIO ........................................................................................................................................ 70
BIBLIOGRAFÍA .......................................................................................................................................... 70
ESPECTROS DE I.R. ........................................................................................................................................ 71
PRÁCTICA # 5A: OBTENCION DE ACIDO BENZOICO Y ALCOHOL BENZILICO ...................... 73
OBJETIVOS .................................................................................................................................................... 73
REACCIÓN ..................................................................................................................................................... 73
MECANISMO ................................................................................................................................................. 73
PARTE EXPERIMENTAL............................................................................................................................ 74
MATERIAL ................................................................................................................................................. 74
SUSTANCIAS ............................................................................................................................................. 75
PROCEDIMIENTO........................................................................................................................................ 75
ESPECTROS DE I.R. ........................................................................................................................................ 76
BIBLIOGRAFÍA ............................................................................................................................................. 76
PRÁCTICA # 5B: OBTENCIÓN DE ACIDO BENZOICO A PARTIR DE ACETOFENONA ............. 77
OBJETIVOS ................................................................................................................................................ 77
ANTECEDENTES ....................................................................................................................................... 77
REACCIÓN A EFECTUAR Y ESTEQUIMETRIA .................................................................................... 77
MECANISMO ............................................................................................................................................. 78
PARTE EXPERIMENTAL .......................................................................................................................... 81
MATERIAL .............................................................................................................................................. 81
SUSTANCIAS .......................................................................................................................................... 81
PROCEDIMIENTO ..................................................................................................................................... 81
DIAGRAMA ECOLÓGICO ........................................................................................................................ 82
CUESTIONARIO .............................................................................................................................................. 83
ESPECTROS DE I.R. ........................................................................................................................................ 83
BIBLIOGRAFÍA .......................................................................................................................................... 84
PRÁCTICA # 6: SISTESIS DEL BENZOATO DE METILO ................................................................... 85
OBJETIVOS ................................................................................................................................................ 85
Objetivo general: ..................................................................................................................................... 85

INDICE
vii
Objetivos Específicos: ............................................................................................................................. 85
ANTECEDENTES ............................................................................................................................................. 85
REACCIÓN A EFECTUAR Y ESTEQUIMETRIA................................................................................................ 86
MECANISMO .................................................................................................................................................. 86
PARTE EXPERIMENTAL .................................................................................................................................. 88
MATERIAL .............................................................................................................................................. 88
SUSTANCIAS .......................................................................................................................................... 88
PROCEDIMIENTO ..................................................................................................................................... 88
PRECAUCIONES ESPECIALES. ...................................................................................................................... 89
DIAGRAMA ECOLÓGICO ................................................................................................................................ 91
CUESTIONARIO ............................................................................................................................................. 92
ESPECTROS DE I.R. ....................................................................................................................................... 92
BIBLIOGRAFÍA ......................................................................................................................................... 94
PRÁCTICA # 7: OBTENCIÓN DE UNA ESENCIA NATURAL ............................................................. 95
OBJETIVOS ................................................................................................................................................ 95
Objetivo general:..................................................................................................................................... 95
ANTECEDENTES ............................................................................................................................................. 95
INTRODUCCIÓN ............................................................................................................................................. 95
PARTE EXPERIMENTAL .................................................................................................................................. 96
MATERIAL .............................................................................................................................................. 96
SUSTANCIAS .......................................................................................................................................... 97
PROCEDIMIENTO ..................................................................................................................................... 97
Aceites de clavo o de la pimienta inglesa ................................................................................................ 97
Preparación de un derivado .................................................................................................................... 98
Aceite de cinamomo ................................................................................................................................ 98
Preparación del derivado ........................................................................................................................ 99
PRÁCTICA # 8A: REACCION DE GRIGNARD. SINTESIS DEL ACIDO VALERICO O SINTESIS
DEL 4-METIL-3-HEPTANOL ...................................................................................................................... 81
OBJETIVOS ................................................................................................................................................ 81
ANTECEDENTES....................................................................................................................................... 81
REACCIONES A EFECTUAR Y ESTEQUIMETRIA ........................................................................................... 81
Síntesis de ácido valerico ........................................................................................................................ 81
Sintesis del 4-metil-3-heptanol ................................................................................................................ 82
MECANISMO .................................................................................................................................................. 82
PARTE EXPERIMENTAL .................................................................................................................................. 82
MATERIAL .............................................................................................................................................. 82

INDICE
viii
SUSTANCIAS .......................................................................................................................................... 83
PROCEDIMIENTO ..................................................................................................................................... 83
Acido Valerico ......................................................................................................................................... 83
4-metil-3-heptanol ................................................................................................................................... 84
PRÁCTICA # 8B: REACCION DE GRIGNARD PREPARACION DE TRIFENIL CARBINOL ........ 82
OBJETIVOS ................................................................................................................................................ 82
ANTECEDENTES ............................................................................................................................................. 82
REACCIÓN A EFECTUAR Y ESTEQUIMETRIA ................................................................................................ 82
MECANISMO .................................................................................................................................................. 83
FORMACIÓN DE BROMURO DE FENIL MAGNESIO: ........................................................................................... 83
FORMACIÓN DEL TRIFENIL CARBINOL ........................................................................................................... 85
PARTE EXPERIMENTAL .................................................................................................................................. 87
MATERIAL .............................................................................................................................................. 87
SUSTANCIAS .......................................................................................................................................... 87
PROCEDIMIENTO ..................................................................................................................................... 87
Bromobenceno ......................................................................................................................................... 88
Bromuro de fenil magnesio...................................................................................................................... 89
Adición de benzofenona ........................................................................................................................... 90
Adición de benzoato de etilo .................................................................................................................... 90
DIAGRAMA ECOLÓGICO .......................................................................................................................... 91
REACCIÓN DE GRIGNARD .............................................................................................................................. 91
OBTENCIÓN DE TRIFENIL CARBINOL .............................................................................................................. 91
REACCIÓN DE GRIGNARD .............................................................................................................................. 92
OBTENCIÓN DE TRIFENIL CARBINOL .............................................................................................................. 92
CUESTIONARIO ........................................................................................................................................... 93
BIBLIOGRAFÍA ............................................................................................................................................. 93
PRÁCTICA # 9: CONDENSACION DE CLAISEN-SCHMIDT. SÍNTESIS DE DIBENZALACETONA
.......................................................................................................................................................................... 95
OBJETIVOS ................................................................................................................................................ 95
ANTECEDENTES ............................................................................................................................................. 95
REACCIÓN A EFECTUAR Y ESTEQUIOMETRIA ............................................................................................. 95
MECANISMO DE LA REACCIÓN ....................................................................................................................... 96
PARTE EXPERIMENTAL ................................................................................................................................ 100
MATERIAL ............................................................................................................................................ 100
SUSTANCIAS ........................................................................................................................................ 100

INDICE
ix
PROCEDIMIENTO ................................................................................................................................... 100
Datos para la cromatografía en capa fina (c.c.f) .................................................................................. 100
CUESTIONARIO ...................................................................................................................................... 101
BIBLIOGRAFÍA ............................................................................................................................................ 101
DIAGRAMA ECOLÓGICO ............................................................................................................................... 102
ESPECTROS DE I.R. ..................................................................................................................................... 103
PRÁCTICA # 10: SALES DE DIAZONIO. OBTENCIÓN DEL ACIDO SULFANÍLICO, NARANJA
DE METILO Y NARANJA II ..................................................................................................................... 105
OBJETIVOS .............................................................................................................................................. 105
ANTECEDENTES..................................................................................................................................... 105
REACCIÓN A EFECTUAR Y ESTEQUIOMETRIA ............................................................................... 106
MECANISMO DE LA REACCIÓN .......................................................................................................... 108
Acido sulfanílico .................................................................................................................................... 108
Sal de diazonio ...................................................................................................................................... 110
PARTE EXPERIMENTAL ................................................................................................................................ 112
MATERIAL ............................................................................................................................................ 112
SUSTANCIAS ........................................................................................................................................ 113
PROCEDIMIENTO ................................................................................................................................... 113
Acido sulfanílico .................................................................................................................................... 113
Anaranjado de metilo ............................................................................................................................ 113
Naranja II .............................................................................................................................................. 114
DIAGRAMA ECOLÓGICO .............................................................................................................................. 115
Preparación del ácido sulfanílico ......................................................................................................... 115
Obtención de anaranjado de metilo ...................................................................................................... 116
Obtención de naranja II ........................................................................................................................ 117
CUESTIONARIO ...................................................................................................................................... 118
BIBLIOGRAFÍA ............................................................................................................................................ 119
ESPECTROS DE I.R. ..................................................................................................................................... 120
PRÁCTICA # 11: SINTESIS DE CICLOHEXANONA POR OXIDACION DE CICLOHEXANOL
CON HIPOCLORITO DE SODIO ............................................................................................................. 121
OBJETIVOS .............................................................................................................................................. 121
ANTECEDENTES ........................................................................................................................................... 121
REACCIÓN A EFECTUAR Y ESTEQUIOMETRIA ........................................................................................... 121
MECANISMO DE LA REACCIÓN ..................................................................................................................... 122
PARTE EXPERIMENTAL ................................................................................................................................ 123
MATERIAL ............................................................................................................................................ 123

INDICE
x
SUSTANCIAS ........................................................................................................................................ 123
PROCEDIMIENTO ................................................................................................................................... 123
Pruebas de identificación ...................................................................................................................... 124
DIAGRAMA ECOLÓGICO .............................................................................................................................. 125
CUESTIONARIO ...................................................................................................................................... 126
BIBLIOGRAFÍA ............................................................................................................................................. 127
ESPECTROS DE I.R. ...................................................................................................................................... 127
PRACTICA 12: SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA: NITRACIÓN DE LA ANILINA
........................................................................................................................................................................ 129
INTRODUCCIÓN ........................................................................................................................................... 129
REACTIVOS ................................................................................................................................................. 130
1. PREPARACIÓN DE LA ACETANILIDA (2) .................................................................................................... 130
2. PREPARACIÓN DE LA P-NITROACETANILIDA (3) ....................................................................................... 130
3. DESPROTECCIÓN DEL GRUPO NH2: PREPARACIÓN DE P-NITROANILINA (4) .......................... 130
PROCEDIMIENTO ......................................................................................................................................... 130
1. Preparación de la acetanilida (2) ..................................................................................................... 130
2.Preparación de la p-nitroacetanilida (3) ........................................................................................... 131
3. Desprotección del grupo NH2: Preparación de p-nitroanilina (4) .................................................... 131
CUESTIONES ................................................................................................................................................ 131
ESPECTROS DE I.R. ...................................................................................................................................... 132

Practica # 1 Técnicas Experimentales
1
PRACTICA # 1: TECNICAS EXPERIMETALES
PUNTO DE FUSIÓN SIMPLE Y MIXTO
ANTECEDENTES
Punto de fusión como constante física.
Factores estructurales que determinan la fusión de un sólido.
Factores experimentales que determinan la fusión de un sólido.
Influencia de las impurezas en el punto de fusión.
Métodos, aparatos y equipos para determinar el punto de fusión.
Mezclas eutécticas.
Punto de fusión mixto.
TEORIA
Punto de fusión
El punto de fusión de un sólido cristalino es la temperatura a la cual, la fase sólida y
líquida se encuentra en equilibrio bajo la presión de una atmósfera.
Cuando una pequeña cantidad de una sustancia sólida se calienta lentamente, se lee
la temperatura a la cual se observa la primera gota de líquido en la muestra sólida, ésta es la
temperatura mínima del intervalo de fusión de dicha sustancia. La temperatura a la que, la
muestra finalmente resulta líquida, es la temperatura máxima del intervalo de fusión. Para
sustancias puras, el intervalo entre esos dos puntos es muy corto (máximo 1 grado), un
intervalo mayor indica que el producto está impuro.
El punto de fusión es una propiedad física característica de cada sustancia; es una
constante muy sensible a la presencia de impurezas, entre más pura sea una sustancia
mayor será su punto de fusión y menor será el intervalo de fusión. Un compuesto orgánico
impuro presenta no solo un amplio intervalo de fusión sino que su punto de fusión se abate
en fusión de la cantidad de impurezas presentes.
Por esta razón el punto de fusión es un índice de pureza usado para un compuesto
orgánico sólido.

Practica # 1 Técnicas Experimentales
2
Punto de fusión de compuesto impuros; diagrama de presión de
vapor-temperatura
La razón por la que el punto de fusión de una sustancia pura y cristalina es constante
e instantáneo puede verse mediante el diagrama presión de vapor-temperatura.
En la figura podemos observar que la curva AB corresponde a la presión de vapor,
determinada experimentalmente, de un sólido X puro a las temperaturas comprendidas entre
TA a TB; BC muestra la presión de vapor del líquido puro X a las temperaturas
comprendidas entre TA a TC. A la temperatura TB, las presiones de vapor de las fases sólidas
y líquidas de la sustancia pura X son iguales a PB; o lo que es lo mismo, las curvas de
presión de vapor para la fase sólida y líquida se cortan en el punto B. La temperatura TB es
por lo tanto el punto de fusión de X, es decir la temperatura en la que la fase sólida y
líquida de X existe en equilibrio, una en presencia de la otra a la presión de una atmósfera.
Se debe evaluar el efecto que una pequeña cantidad de una impureza Y (que es
soluble en el líquido X) ejerce sobre el punto de fusión de un producto. En el preciso
momento en que el último cristal de X ha fundido, toda la impureza Y se habrá disuelto en
X. El punto de fusión sigue siendo la temperatura a la cual el líquido y el sólido de X
existen en equilibrio a la presión de 1 atmósfera; la presión de vapor que hay que considerar
aquí no es la total de la solución líquida (debido a X y Y), sino solamente la presión de
vapor parcial debida al líquido X. Como la presencia de esta impureza disuelta disminuirá
la presión de vapor debida al líquido X en el intervalo total de temperaturas de la solución
líquida (curva B1C1), está claro que la presión de vapor del sólido X será igual a la debida al
líquido X en la solución a la temperatura TB1, que es más baja que la del punto de fusión TB
de la sustancia pura.

Practica # 1 Técnicas Experimentales
3
En otras palabras. En presencia de esta cantidad de impurezas Y, la temperatura a la
que funden las últimas trazas de X disminuye hasta TB1, y el efecto de la impureza Y es
precisamente bajar el punto de fusión de X, lo mismo que cuando al hielo se le agrega sal o
alcohol, se baja su punto de fusión por debajo de los 0 °C.
Punto de fusión de mezcla de compuestos
El diagrama que muestra el comportamiento de una mezcla de dos componentes en
fusión (A + B), lo podemos observar a continuación.
Este diagrama se construye determinando los puntos de fusión de mezclas
preparadas a diferentes proporciones de un producto B en un producto A.
Esta gráfica al mismo tiempo indica que el punto de congelación (paso de líquido a
sólido), de un producto puro se bate por la presencia de una impureza.
Si la sustancia A pura tiene menor punto de fusión que B pura, se observa que el
punto de fusión disminuye hasta un punto mínimo llamado punto eutéctico, en el cual,
además, el intervalo de fusión se hace mínimo, pudiéndose pensar que fuera pura, bajo ese
criterio.
No todas las mezclas binarias tienen un punto eutéctico y en cambio otras mezclas
tienen varios puntos eutécticos.
Cuando una mezcla de 80% de A y 20% de B (en moles), se calienta, antes de
hacerse miscible la primera gota de líquido la mezcla se reblandece debido a que B se
empieza a disolver en el producto A fundido, cuando se alcanza la máxima solubilidad de

Practica # 1 Técnicas Experimentales
4
B en A, se observa la primera gota, punto inicial de la fusión; luego más cantidad de B
funde hasta que todo es un líquido, máxima temperatura del punto de fusión. El punto
mínimo de fusión de cualquier mezcla siempre será el punto eutéctico (Tc), sin embargo no
siempre es fácil de observar esta temperatura y la aparición de la primera gota generalmente
ocurre arriba de Tc.
Punto de fusión de compuestos desconocidos
En la identificación de un compuesto desconocido es sumamente útil recurrir a la
determinación de un punto de fusión mixto. Se prepara una mezcla (1:1) del compuesto
desconocido y el compuesto puro que se supone es idéntico (compuesto de referencia), los
cuales tienen un punto de fusión muy similar o igual. A continuación se determinan los
puntos de fusión del compuesto desconocido, del compuesto de referencia y de la mezcla
de ambos, al mismo tiempo y en el mismo aparato. Si los tres se funden simultáneamente,
el compuesto desconocido y el de referencia son idénticos. Si la mezcla tiene el punto de
fusión más bajo, los dos compuestos son diferentes.
Métodos, aparatos y equipos para determinar el punto de fusión
Existen varios tipos de aparatos para determinar el punto de fusión.
Uno de stos aparatos utiliza un tubo de Thiele y tubos capilares de vidrio para
contener la muestra, los cuales se colocan en un baño de calentamiento. Otros aparatos
cuentan con planchas de calentamiento en la que se coloca la muestra entre dos
cubreobjetos, ambos aparatos cuentan con un termómetro y en algunos casos con un
reóstato que regula la velocidad de calentamiento.
Los termómetros para la realización del punto de fusión deben ser calibrados,
mediante la determinación del punto de fusión de cinco o seis compuestos puros que
abarquen un amplio rango de temperatura del termómetro.
CUESTIONARIO
1. Explique si mezclas de sustancias diferentes, pero con punto de fusión igual, funden a
temperatura igual o diferente a las de as sustancias originales.
2. Explique por qué una sustancia impura presenta un abatimiento en su punto de fusión.
3. ¿Cuáles son los parámetros que influyen en el valor del punto de fusión?
4. Si tiene los siguientes isómeros; explique la diferencia en los puntos de fusión de éstos.

Practica # 1 Técnicas Experimentales
5
C C
C C
H H
OH OH
O O
C C
C
CH
HOH
OH
O
O
Acido maléicop.f. 140-142 °C
Acido fumáricop.f. 299-300 °C (sublima)
5. ¿Qué parámetro experimental deben controlarse en la determinación del punto de fusión
por el método de Thiele?

Practica # 1 Técnicas Experimentales
6
SOLUBILIDAD EN DISOLVENTES ORGÁNICOS Y CRISTALIZACIÓN SIMPLE
ANTECEDENTES
Solubilidad.
Disolventes utilizados en cristalización. Sus propiedades y características.
Pruebas de solubilidad. Factores que determinan la solubilidad.
Elección de un disolvente para una cristalización.
Factores que influyen en la cristalización.
Formas de inducir la cristalización.
Diversas técnicas de filtración.
Etapas del proceso de cristalización como método de purificación de sólidos.
TEORIA
Solubilidad
Por lo general, los compuestos orgánicos sólidos se pueden purificar por
cristalización. La técnica general incluye la disolución del sólido que va a ser cristalizado,
en un disolvente o mezcla de disolvente calientes a punto de ebullición y, posteriormente, al
enfriar la solución, cristaliza o precipita como sólido amorfo. A este fenómeno se le llama
cristalización si el crecimiento del cristal es relativamente lento y selectivo o también se le
conoce como precipitación si el proceso es muy rápido y no selectivo.
La cristalización es un proceso de equilibrio de las moléculas en una red cristalina
con las moléculas en solución, y de este proceso resulta un sólido puro.
Inicialmente se forma un pequeño núcleo cristalino y a partir de él van creciendo las
capas cristalinas en un proceso reversible y selectivo. Las moléculas no adecuadas a la red
cristalina, posiblemente regresen a la solución y las moléculas adecuadas son retenidas en
ella.
En cualquier cristalización, un enfriamiento de la solución demasiado rápido o
demasiado lento debe evitarse. Se debe evitar enfriar muy rápido, así como añadir
rápidamente un disolvente en el cual no es soluble el producto, ambos son errores que
conducen a una mala purificación.

Practica # 1 Técnicas Experimentales
7
La cristalización está fundamentada en las relaciones de solubilidad soluto—
disolvente, por lo tanto, el sólido debe presentar un comportamiento de solubilidad
adecuado. La solubilidad de los compuestos orgánicos es una función de las polaridades del
disolvente y en soluto.
Cuando se disuelve un sólido o un líquido, las unidades estructurales (iones o
moléculas) se separan y el espacio entre ellas es ocupado por moléculas de disolvente.
Para que exista la disolución, debe suministrarse energía para vencer las fuerzas
intermoleculares o interiónicas; esta energía proviene de las interacciones electrostáticas
soluto—disolvente.
En moléculas polares, estas interacciones electrostáticas son dipolo—dipolo, puente
de hidrógeno (fenómenos de solvatación), mientras que en moléculas no polares las
interacciones son del tipo de las fuerzas de van der Waals.
Existe una regla empírica qu establece “lo semejante disuelve a lo semejante”. Si el
soluto es muy polar, lo disolverá un disolvente muy polar; si es no polar, un disolvente no
polar lo disolverá. Los compuestos que tienen grupos funcionales capaces de formar
puentes de hidrógeno (por ejemplo, -OH, -NH2, -COOH, -CONH2) son:
Estos compuestos son más solubles en disolventes hidroxílicos como el agua o
metanol que en hidrocarburos como benceno o hexano. Si el grupo funcional no predomina
en la molécula, la solubilidad cambia. Por ejemplo, el alcohol dodecilico, CH3-(CH2)10-
CH2-OH, es insoluble en agua, su cadena de doce átomos de carbono causa que sea más
parecido a un hidrocarburo que a un alcohol. Otra molécula interesante es el p-terbutil

Practica # 1 Técnicas Experimentales
8
fenol, la cual por la presencia del grupo –OH es soluble en agua pero también es soluble en
hexano por la parte alquilarílica de la molécula.
La estructura de la red cristalina también afecta la solubilidad. Por ejemplo, entre
isómeros, aquel de punto de fusión mayor, presentara menor solubilidad que los otros con
respecto a determinado disolvente.
TEORÍA DE LA CRISTALIZACIÓN
La cristalización de un sólido depende de la diferencia de solubilidad de éste en un
disolvente a temperatura ambiente y a temperatura de ebullición. También es importante
considerar la solubilidad de las impurezas, cuando la solubilidad de ellas es igual en el
disolvente frió como en el disolvente caliente, no se logra purificar el sólido por
cristalización.
Un sólido puede ser purificado por cristalización cuando la solubilidad del sólido y
las impurezas son diferentes y las impurezas representan una pequeña proporción con
relación al peso total del sólido; al enfriar la solución el sólido cristaliza y las impurezas no.
Lo anterior se puede entender con el siguiente diagrama 1 que considera un sólido
“A” y su impureza “B”. A la solución remanente después de la cristalización se le llama
líquido madre.

Practica # 1 Técnicas Experimentales
9
SELECCIÓN DE DISOLVENTE
La selección del disolvente para cristalización se realiza mediante pruebas de
solubilidad del sólido por purificar con una variedad de disolventes. El sólido debe reunir
ciertas condiciones de solubilidad con respecto al disolvente.
El sólido debe ser ligeramente soluble o insoluble a temperatura ambiente y
totalmente soluble a la temperatura de ebullición del disolvente; el disolvente que reúna
estas características será el “ideal” para cristalizar el sólido.
La gráfica de solubilidad ante la temperatura nos muestra el comportamiento de un
disolvente ideal respecto a otros disolventes.
A: disolvente ideal en el que el sólido es poco soluble a temperatura ambiente y
muy soluble a ebullición.

Practica # 1 Técnicas Experimentales
10
B: disolvente en el que el sólido es poco soluble.
C: disolvente en el que el sólido es muy soluble.
Al realizar las pruebas de solubilidad es importante tener la relación correcta
sólido/disolvente, por esta razón se utiliza 0.1 g de sólido por 3 mL de disolvente. Otras
características que debe reunir el disolvente ideal son las siguientes:
No debe reaccionar con el sólido.
Tener punto de ebullición inferior al punto de fusión del sólido por purificar; debe
evitarse utilizar disolventes de punto de ebullición más alto que el punto de fusión del
sólido por cristalizar, para evitar que éste se funda en vez de disolverse.
Debe presentar volatilidad moderada (bajo p. eb.) para eliminar con cierta facilidad
de los cristales.
Experimentalmente, las pruebas se realizan en una serie de tubos de ensayos limpios
y secos, en donde se coloca la cantidad de sólido y el disolvente en la relación adecuada.
Las pruebas de solubilidad se realizan con diferentes disolventes para obtener una
información completa acerca de la solubilidad del sólido.
Si el compuesto es conocido, se investiga su solubilidad en libros de constantes
físicas para elegir el disolvente adecuado para la purificación.
TÉCNICA DE CRISTALIZACIÓN
Disolución de sólidos. La disolución de sólidos se realiza de preferencia en
matraces Erlenmeyer en lugar de vasos o cualquier otro recipiente, con el objeto de
proteger la solución del polvo y evitar una evaporación excesiva de disolvente.
El sólido se pulveriza antes de disolverlo; la pérdida de sólido es el líquido madre se
evita con la saturación adecuada de la solución, la cual al enfriar, transformará con alto
rendimiento el sólido en cristales.
Antes de iniciar el calentamiento de un disolvente es necesario agregar un pequeño
fragmento de material poroso (carborundum, tezontle, plato poroso, etc.) para evitar el
sobrecalentamiento del mismo.
El disolvente se calienta a punto d ebullición, se agrega la cantidad necesaria al
sólido para disolverlo y la mezcla se calienta nuevamente hasta ebullición. Si el sólido no
se ha disuelto, se adiciona otro pequeño volumen de disolvente caliente y nuevamente se
lleva a ebullición hasta lograr la disolución total del sólido en la mínima cantidad del
disolvente.

Practica # 1 Técnicas Experimentales
11
Filtración. Es común que el sólido impuro presente partículas de impurezas
insolubles, polvo, fibras de papel y restos de otros materiales; las cuales se pueden
pretender disolver sin lograrlo y aumentar sin necesidad, el volumen del disolvente usado.
Para eliminar estas impurezas, se utiliza la filtración por gravedad en caliente en el embudo
de vidrio de tallo corto. Es conveniente antes de filtrar, precalentar el embudo y el matraz
donde se va a recibir el filtrado, para evitar la cristalización del sólido en el tallo del
embudo.
El papel filtro debe utilizarse estriado o en forma de cono para aumentar la
velocidad de filtración. La solución caliente se filtra por gravedad para eliminar las
impurezas insolubles y restos de carbón activado. Si los cristales empiezan a formarse en el
filtro durante la filtración por gravedad, se adiciona una cantidad mínima de disolvente
caliente para disolverlos.

Practica # 1 Técnicas Experimentales
12

Practica # 1 Técnicas Experimentales
13
CRISTALIZACIÓN
La solución filtrada se deja reposar a temperatura ambiente durante 5 minutos y
posteriormente se enfría en un baño de hielo. Si al enfriar la solución no cristaliza, debe
inducirse la cristalización empleando diferentes técnicas:
a) Siembra de la solución con cristales puros de la misma sustancia que se está
purificando y que sirven como núcleos de cristalización.
b) Raspar las paredes del matraz con una varilla o espátula. Este movimiento debe
ser vertical y firme para producir vibraciones de alta frecuencia que induzcan la
cristalización. También es probable que la inducción de la cristalización se deba
a la introducción de pequeños cristales formados por evaporación del disolvente
en las paredes del matraz y que sirve como núcleo de cristalización.
c) Si no se logra la cristalización utilizando los incisos a y b, puede deberse al
exceso de disolvente; si es el caso, es necesario evaporar un poco y volver a
enfriar, repitiendo lo indicado en los incisos.
Separación de cristales. La filtración a vacío se utiliza para separar los cristales del
líquido madre. El material necesario para esta operación es el siguiente: embudo Büchner,
adaptador de hule, matraz Kitasato, tubo de hule para vacío, pinzas, soporte y papel filtro
circular a la medida para que cubra los orificios del embudo Büchner.

Practica # 1 Técnicas Experimentales
14
CUESTIONARIO
1. ¿Qué información se obtiene de un sólido al que se le realizan pruebas de solubilidad
con disolventes orgánicos de polaridad conocida?
2. Un sólido que es soluble en frió, ¿podrá recristalizarse del disolvente en que se
encuentra? Fundamente su respuesta.
3. Un sólido que es insoluble en caliente, ¿podrá recristalizarse del disolvente en que se
encuentra? Fundamente su respuesta.
4. ¿Cuáles son las principales características que debe presentar un sólido para
recristalizarlo de su disolvente ideal?
5. En qué parte del proceso de recristalización es eliminada cada una de las impurezas
solubles e insolubles?
6. ¿Qué condiciones se deben controlar en el enfriamiento de la solución para tener
cristales de máxima pureza?
7. Prediga cuales de los siguientes compuestos son más solubles en benceno, justifique su
respuesta:
8. a) Acido oxálico b) etilendiamina c) malonato de etilo
9. De la siguiente lista de compuestos orgánicos (izquierda) y disolventes (derecha), elija
el disolvente en el cual cada sólido sea semejante para ser soluble (no necesariamente
disolvente ideal).
a)
CH3 CH2 CH2 OH4
CH3 CH2 C
O
O-Na
+
b)
c)
d)
OH2
CH3 CH2 OH
éter de petróleo
a)
b)
c)

Practica # 1 Técnicas Experimentales
15
10. ¿Qué se hace con los sólidos contenidos en las aguas madres?
11. ¿Cuál es el objetivo de determinar el punto de fusión antes y después de cristalizar un
sólido?
12. ¿Por qué razón no se debe desechar por el drenaje, disolventes orgánicos como: hexano,
acetato de etilo, acetona, etanol y metanol?

Practica # 1 Técnicas Experimentales
16
PUNTO DE EBULLICIÓN, DESTILACIÓN SIMPLE Y FRACCIONADA
ANTECEDENTES
Relación entre las propiedades físicas y la estructura molecular de los alcanos y los
alcoholes.
Propiedades físicas de alcanos y alcoholes.
Presión de vapor. Punto de ebullición.
Destilación simple y fraccionada. Características y diferencias.
Diagrama de composición vapor-líquido.
Concepto de plato teórico y número de platos teóricos en una columna de destilación
fraccionada.
Eficiencia de la columna de destilación fraccionada. Factores que intervienen en una
destilación.
Ley de Raoult.
TEORIA
Punto de ebullición, relación estructura-punto de ebullición
El punto de ebullición de un líquido es la temperatura a la cual su presión de vapor
es igual a la presión externa.
Para que una sustancia alcance su punto de ebullición, es necesario suministrar la
energía necesaria para que pase del estado líquido al estado de vapor.
En general, los factores que determinan el punto de ebullición son: el peso
molecular, la forma lineal o ramificada de las moléculas, su polaridad y la asociación
intermolecular.
En el caso de los alcoholes se ha observado que a medida que aumenta el número de
carbono, de la cadena lineal, el punto de ebullición también aumenta; sin embargo, éste
disminuye al aumentar las ramificaciones moleculares.
Entre los hidrocarburos, los factores que determinan puntos de ebullición suelen ser
principalmente el peso molecular y la forma, lo que es de esperar de moléculas que se
mantienen unidas esencialmente por fuerzas de van der Waals.

Practica # 1 Técnicas Experimentales
17
Dentro de una familia es de esperar que cuanto más grande sea una molécula y, por
lo mismo, su superficie, más intensa son las fuerzas intermoleculares. El punto de
ebullición aumenta conforme crece el número de carbonos; el punto de ebullición sube
porque las fuerzas intermoleculares se intensifican a medida que aumenta el tamaño
molecular.
Un isómero ramificado tiene un punto de ebullición más bajo que uno de cadena
recta. Al aumentar las ramificaciones en una molécula, la forma de ésta tiende a
aproximarse a la de una esfera, con lo que disminuye sus superficie; esto se traduce en un
debilitamiento de las fuerzas intermoleculares.
Los alcoholes también exhiben un aumento del punto de ebullición, con un número
creciente de átomos de carbono y una disminución del mismo, con una mayor ramificación.
Sus puntos de ebullición anormalmente elevados se deben a que son líquidos
asociados y por lo mismo requieren mayor energía para romper los puentes de hidrógeno
que mantienen unidas a las moléculas.
Destilación simple
La destilación es un proceso que consiste en la evaporación de un líquido,
condensación del vapor y colección del condensado.
Cuando una sustancia líquida se contamina con pequeñas cantidades de impurezas,
éstas pueden eliminarse por algún método de destilación, Se dice entonces que se efectúa
una purificación.
Cuando dos o más sustancias líquidas se encuentran formando mezclas en
proporción relativamente parecida, se dice que la destilación puede usarse para la
separación de componentes.

Practica # 1 Técnicas Experimentales
18
En cada caso, deberá elegirse la técnica de destilación más acorde con las
características de la muestra.
Existen cuatro métodos de destilación: destilación simple, destilación fraccionada,
destilación a presión reducida y destilación por arrastre con vapor.
La relación entre la presión y la temperatura de ebullición de un líquido está
determinada por su comportamiento presión de vapor-temperatura.
Si un líquido contiene una impureza volátil, la separación de ambos se realiza
generalmente por destilación. Cualquier par de sustancias que no tenga presiones de vapor
idénticas se pueden separar por destilación.
Como regla general, se puede indicar que una mezcla de dos componentes con una
diferencia de por lo menos 80 °C puede separarse por una destilación sencilla. Sustancias
cuyos puntos de ebullición difieren por menos de 80 °C se separan por destilación
fraccionada. La destilación simple se utiliza para la purificación de compuestos orgánicos
líquidos.
Las mezclas, cuyos componentes no presentan grandes diferencias en sus puntos de
ebullición, se separan por destilación fraccionada; esta técnica se utiliza continuamente en
la industria y laboratorios.
Destilación fraccionada
La destilación fraccionada debe emplearse para separar y purificar mezclas de
sustancias de puntos de ebullición cercanos. Actualmente se dispone de equipos de
destilación fraccionada muy eficaces para lograrlo.
Si una mezcla formada por benceno (p. eb.: 80 °C) – tolueno (p. eb.: 110 °C) cuyos
componentes difieren 30 °C en sus puntos de ebullición, se separan por destilación simple,
y al trazar una gráfica de punto de ebullición ante volumen de destilado podemos concluir
al observarla, que se trata de una mezcla de dos componentes de punto de ebullición
similar, por lo mismo, no se observa una separación definida con este método de
destilación.
Con base en la gráfica, se observa que no hay constancia en el punto de ebullición
porque éste aumenta continuamente.
Esta misma mezcla se puede separar eficazmente utilizando la técnica de destilación
fraccionada, al emplear una columna de fraccionamiento que proporciona una gran
superficie para el intercambio de calor, en las condiciones de equilibrio, entre el vapor
ascendente y el condensado descendente.

Practica # 1 Técnicas Experimentales
19
Al trazar una gráfica de los puntos de ebullición ante volumen de condensado, se
observa una buena separación donde hay definición de las fracciones separadas.
Diagrama de fase
El diagrama de fase para una destilación fraccionada de un sistema ideal de dos
componentes (A y B), se utiliza para explicar el funcionamiento de una columna de
fraccionamiento. El diagrama relaciona la composición del líquido en ebullición con su
vapor como una función de la temperatura.

Practica # 1 Técnicas Experimentales
20
Diagrama de fase de la destilación fraccionada de un sistema ideal de dos
componentes
Las líneas horizontales (L1 V1, L2 V2, L3 V3, etc.) representan la evaporación en el
ciclo evaporación↔condensación e indica la composición del vapor en equilibrio con el
líquido. Por ejemplo, a 63 °C un líquido con una composición de 50% de A (L3 sobre el
diagrama) puede producir vapor con la siguiente composición: 80% A (V3 en el diagrama)
en el equilibrio. El vapor es más rico en el componente de menor punto de ebullición (A)
con relación al líquido original.
Las líneas verticales (V1 L2, V2 L3 etc.) representan la condensación en el ciclo
evaporación↔condensación. Por ejemplo, el vapor en V3 se condensa para dar un líquido
(L4) con la siguiente composición: 80% de A. Este proceso de evaporación↔condensación
se presenta a lo largo de la columna de fraccionamiento, como se muestra en la figura.
Eficiencia de la columna
La eficiencia de la columna se da con relación a los platos teóricos. Una columna
puede tener un plato teórico si el primer destilado tiene la composición localizada en L2
(20% A) iniciando con un líquido de composición L1 (5% A) como se observa en el
diagrama. Esto corresponde a una destilación simple en el ciclo evaporación condensación.
Una columna puede tener dos platos teóricos si el destilado tiene la composición L3
(50% A) partiendo de un líquido de composición L1 (5% A).
Proceso de evaporación↔condensación en una columna de fraccionamiento
Los dos platos teóricos de la columna efectuarán dos destilaciones simples
correspondiendo a las líneas L1V1L2 y L2V2L3 como se observa en el diagrama.

Practica # 1 Técnicas Experimentales
21
Para separar A de B se necesitan cinco platos teóricos, o sea, se efectuaron a través
de ella cinco destilaciones simple.
CUESTIONARIO
1. De acuerdo as su formula estructural ordene los siguientes isómeros del pentano
y hexano según su punto de ebullición creciente:
CH3 C CH3
CH3
CH3
CH3 CH2 CH2 CH2 CH3
CH3 CH CH2 CH3
CH3
CH3 CH CH CH3
CH3
CH3
CH3 C CH2 CH3
CH3
CH3
CH3 CH CH2 CH2 CH3
CH3
a)
b)
c)
d)
e)
f)
2. ¿Qué establece la Ley de Raoult? Conteste en palabras y matemáticamente.

Practica # 1 Técnicas Experimentales
22
3. Tabla de presión de vapor para el benceno y tolueno a diferentes temperaturas:
Temperatura mm.Hg Temperatura mm.Hg
Benceno 30 °C 120 Tolueno 30 °C 37
40 180 40 60
50 270 50 95
60 390 60 140
70 550 70 200
80 760 80 290
90 1010 90 405
100 1340 100 560
111 711
4. ¿Cuál es la fracción molar de cada componente si 39 g de benceno (C6H6) se
disuelven en 46 g de tolueno (C7H8)’
5. Considere a esta mezcla ideal, de acuerdo a la Ley de Raoult. ¿Cuál es la presión
de vapor parcial del benceno en esta mezcla a 50 °C?
6. ¿Cuál es la composición del vapor en el punto de ebullición (50 °C)?
7. ¿Qué finalidad tiene conectar el agua a contracorriente en el refrigerante?
8. ¿Qué criterio se sigue para separar diferentes fracciones durante una destilación?
Explique.
9. ¿En qué caso se recomienda utilizar la destilación simple y en cuáles la
destilación fraccionada?.

Practica # 1 Técnicas Experimentales
23
EXTRACCIÓN CON DISOLVENTES ORGÁNICOS Y DISOLVENTES ACTIVOS
ANTECEDENTES
Coeficiente de reparto o de distribución.
Métodos de extracción: simple, múltiple y selectiva.
Disolventes orgánicos y activos empleados para la técnica de extracción. Sus
características físicas y químicas.
Diseño de diagramas de separación de mezclas: acido, base, neutro.
Reacciones ácido-base ocurridas al extraer compuestos con disolventes activos.
Agentes desecantes.
TEORIA
Extracción con disolventes orgánicos
La extracción es la transferencia de un soluto de un disolvente a otro. El soluto se
extrae por un proceso de distribución.
Cuando una solución (soluto A en disolvente 1) se agita con un segundo disolvente
(disolvente 2) con el cual es inmiscible, el soluto se distribuye entre las dos fases hasta
lograr una situación de equilibrio. Al separarse las dos capas de los disolvente inmiscibles
se determina la concentración del soluto en cada capa, la relación de las concentraciones en
cada capa fase es una constante. Esta constante, es llamada coeficiente de distribución (o
partición), K, la cual es definida por:
1
2
C
CK
Conde C1 y C2 son las concentraciones en equilibrio, en gramos/litro, del soluto en
el disolvente 1 y en el disolvente 2 a una temperatura determinada.
Esta relación es independiente de la concentración total y de los volúmenes de los
disolventes. El coeficiente de distribución tiene un valor constante para cada soluto y es
dependiente de la naturaleza del disolvente utilizado en cada caso.
Con base en el coeficiente de distribución, no todo el soluto se transfiere al
disolvente 2 en una sola extracción a menos que el valor de K sea muy alto. Generalmente

Practica # 1 Técnicas Experimentales
24
se requieren varias extracciones para eliminar el soluto del disolvente 1. Los disolventes
orgánicos utilizados en extracción deben tener baja solubilidad en agua, alta capacidad de
solvatación hacia la sustancia que se va a extraer y bajo punto de ebullición para facilitar su
eliminación posterior. La extracción tiene amplia aplicación en la Química Orgánica. Se
utiliza para extraer productos y eliminar impurezas de las mezclas de reacción, se emplea
también para extraer productos de tejidos animales o de plantas. Para extraer un soluto de
una solución es más efectivo realizar varias extracciones empleando volúmenes pequeños
(extracción múltiple), que realizar una sola extracción (extracción simple) mediante el
empleo de un volumen grande de disolvente.
Matemáticamente esto se comprueba con la siguiente expresión:
sK
KWW
v
n
Vo
n
Wn = g de soluto remanentes en la fase acuosa después de n extracciones
Wo = g de soluto en la fase acuosa
V = volumen de fase acuosa
S = volumen del disolvente de extracción
K = coeficiente de partición
El embudo de separación
El embudo de separación es la pieza que se utiliza en la extracción. La forma
correcta de agitar el embudo se muestra en la Figura 1. El tapón y la llave, que deben estar
bien ajustados, que se lubrican con una grasa adecuada antes de usarlos. El embudo debe
agitarse moderadamente y purgarlo (aliviar presión) con frecuencia, para evitar la presión
en el interior (Figura 1 y 2).

Practica # 1 Técnicas Experimentales
25
Figura 1: Agitación del embudo
Después de agitar el embudo se debe reposar para la formación de una interfase que
da la pauta para la separación de las fases. (Figura 3) En la identificación de fases es
importante conocer la densidad de los disolventes orgánicos para ubicar su posición en el
embudo con respecto a la fase acuosa. El número de extracciones en cada caso depende del
coeficiente de distribución del disolvente.
Figura 2: Purga del embudo

Practica # 1 Técnicas Experimentales
26
Figura 3: Separación de fases
Emulsiones
Con frecuencia se forman emulsiones durante el proceso de extracción. Estas
pueden romperse mediante:
Un movimiento de giro suave al líquido del embudo.
Agitación vigorosa de la capa emulsionada.
Agitación de la fase acuosa con solución saturada de cloruro de sodio.
Centrifugación.
Agente desecante químico
Un desecante debe reunir ciertas condiciones:
No reaccionar con la sustancia que se va a secar.
Ser eficaz, o sea, tener alto poder desecante; esto es, eliminar el agua
completamente o casi completamente cuando se alcanza el equilibrio.

Practica # 1 Técnicas Experimentales
27
Tener gran capacidad de desecación, es decir, eliminar una gran cantidad de
agua por unidad de peso de desecante.
Secar rápidamente (alcanzar rápido el equilibrio)
Separarse fácilmente de la sustancia una vez seca.
Los desecantes químicos se pueden dividir en dos grupos:
a) Aquellos que reaccionan químicamente con el agua en un proceso no reversible,
lo cual da lugar a un nuevo compuesto libre de agua.
b) Los que se combinan reversiblemente con el agua para formar un hidrato.
En la tabla 1 se presenta una serie de agentes desecantes con sus principales
características y que son eliminados por filtración.
Tabla 1. Agentes desecantes
Acidez Hidratación Capacidad*
Eficiencia**
Velocidad***
Uso
Sulfato de
Magnesio
Neutro MgSO4.7H2O Alta Media Rápida General
Sulfato de
Sodio
Neutro Na2SO4.7H2O
Na2SO4.10H2O
Alta Baja Media General
Cloruro de
Calcio
Neutro CaCl2.2H2O
CaCl2.6H2O
Baja Alta Rápido Hidrocarburos
Haluros
Sulfato de
Calcio
Neutro CaSO4.1/2H2O
CaSO4.2H2O
Baja Alta Rápida General
Carbonato
de Potasio
Básico K2CO3.1/2H2O
K2CO3. 2H2O
Media Media Media Aminas, ésteres,
bases, cetonas
Hidróxido
de Potasio
Básico Rápida General
* Cantidad de agua eliminada por peso de agente secante
** Se refiere a la cantidad de H2O en solución en equilibrio con el agente secante
*** Se refiere a la velocidad de secado

Practica # 1 Técnicas Experimentales
28
Extracción selectiva con disolventes activos
La extracción selectiva se emplea para separar mezclas de compuestos orgánicos,
en función de la acidez, de la basicidad o de la neutralidad de éstos. Para realizar estas
separaciones, es necesario utilizar disolventes activos, éstos pueden ser ácidos o básicos.
Los disolventes activos ácidos que más se utilizan son HCl y H2SO4 en solución acuosa
al 5 o 10%.
Los disolventes activos básicos pueden ser fuertes como NaOH, KOH o moderados
como NAHCO3 y Na2CO3 en solución acuosa al 5 y 10%.
Las extracciones selectivas se basan en una reacción ácido-base entre el producto a
separar y el disolvente activo ademado. Los compuestos iónicos son más solubles en agua
que los compuestos covalentes y éstos más solubles en disolventes orgánicos que aquellos.
Los compuestos básicos como aminas, se extraen con disolventes activos ácidos
(HCl al 5 o 10%); la reacción que ocurre es la siguiente:
R NH2 + HCl R NH3
+Cl
-
Unión covalente Unión iónica
Menos polar Más polar
Solubles en disolventes Soluble en agua
Orgánicos
Los ácidos carboxílicos reaccionan con disolventes activos básicos como NaOH,
Na2CO3 o NAHCO3 al 5 o 10%,
Las reacciones ácido-base que se efectúan son las siguientes:
+CR OH
O
NaOH CR O-
O
Na+
+ OH2
Unión covalente Unión iónica
Menos polar Más polar
Solubles en disolventes Soluble en agua
Orgánicos

Practica # 1 Técnicas Experimentales
29
Los fenoles son compuestos menos ácidos que los ácidos carboxílicos. Por esta
característica, reaccionan únicamente con bases fuertes como NaOH al 5 o 10%, lo anterior
constituye la base para separar ácidos de fenoles.
OH
+ NaOH
O-
Na+
+ OH2
FenolFenóxido de sodioSal soluble en agua
La extracción selectiva también se utiliza para eliminar impurezas ácidas o básicas a
un producto aislado de una mezcla de reacción. Los compuestos que no tienen
características ácidas o básicas los consideramos neutros. Los compuestos extraídos pueden
recuperarse por neutralización del disolvente activo para extraer.
Si un compuesto está disuelto en fase básica éste puede recuperarse acidificando la
solución. Por el contrario, si el compuesto está disuelto en fase acuosa ácida, puede
recuperarse al agregar una base y modificar el pH hasta lo básico. Los compuestos neutros
disueltos en un disolvente orgánico, se recuperar al eliminar el disolvente por destilación.
CUESTIONARIO
1. ¿Qué es un disolvente activo? Cite cinco ejemplos.
2. ¿En qué casos debe utilizarse la extracción múltiple?
3. ¿En qué caso debe utilizarse la extracción selectiva?
4. ¿Por qué el compuesto neutro debe obtenerse por destilación del disolvente en el que se
encuentra y no por cristalización en dicho disolvente?
5. Diga cuál de los siguientes sistemas de disolventes son factibles para la extracción. De
acuerdo a su densidad, ¿en qué fase quedarian ubicados los disolventes?
a) Hexano-agua b) Tolueno-agua c) Ac. Acetico-agua d) Ac. Clorhídrico-agua
6. Diseñe un diagrama de separación para una mezcla de m-nitroanilina, b-naftol y p-
dicloro-benceno.

Practica # 1 Técnicas Experimentales
30
7. De acuerdo con un criterio de calidad, especificidad y costo ¿qué tipo de hidróxido de
sodio recomendaría para un proceso de extracción de la fase orgánica?
a) Sosa en escamas b) Sosa al 50% (sol. Acuosa)
c) Hidróxido de sodio grado R.A. d) Sosa cáustica grado Rayón

Practica # 1 Técnicas Experimentales
31
SEPARACIÓN Y PURIFICACIÓN DE UNA MEZCLA DE TRES COMPONENTES
OBJETIVOS
Conocer la técnica de extracción como método de separación y purificación de
sustancias integrantes de una mezcla.
Elegir los disolventes adecuados para un proceso de extracción.
Realizar diferentes tipos de extracción: simple, múltiple y selectiva. Aplicarlos a
problemas específicos.
Conocer el proceso de destilación simple.
PARTE EXPERIMENTAL
MATERIAL
Embudo de separación con tapón 1 Embudo de vidrio 1
Probeta de 25 ml 1 Matraz pera Quickfit 1
Vaso de precipitado de 250 ml 1 T de destilación 1
Vaso de precipitado de 150 ml 2 Refrigerente de agua con mangueraa 1
Matraz Kitasato con manguera 1 Colector 1
Espátula 1 Pinzas de 3 dedos con nuez 2
Agitador de vidrio 1 Recipiente de peltre 1
Embudo Büchner 1 Manta de calentamiento 1
Matraces Erlenmeyer de 125 ml 3 Tubos de ensayo 8
Matraces Erlenmeyer de 50 ml 4 Pipetas graduadas de 10 ml 2
SUSTANCIAS
Se entregara a cada grupo dos muestras problemas, las cuales pueden ser líquidas o
sólidas.
PROCEDIMIENTO
El estudiante deberá realizarle un análisis de la muestra y tomando en cuenta el
estado físico en el cual se presente la muestra, para luego tomara la decisión de que tipo de
técnica según la teoría estudiada debe aplicar para la separación y purificación de los
componentes presentes en la muestra.
Deberán realizar un esquema de separación y purificación, dependiendo del caso a
estudiar, y proponer que hacer con los desechos generados.

Practica # 1 Técnicas Experimentales
32
Una vez separados y purificados los compuestos el estudiante determinara los
grupos funciónales o grupo funcional principal presente en los compuestos puros, además
deberá determinarle las diferentes propiedades físicas, dependiendo del estado de la
muestra.
Todos los análisis realizados a la muestra se deberán documentar, tanto en tablas
como por escrito.
Tratamiento de residuos
Los residuos de este experimento son los siguientes.
a) Residuos ácidos: deben neutralizarse y desecharse en el embase dispuesto para
los mismos. Si presentan sólidos, se filtran previamente y se empacan para su
posterior tratamiento.
b) Residuos básicos: el tratamiento es igual que para los residuos ácidos.
c) Residuos Etéreos: recuperar l disolvente por destilación para uso posterior; las
colas de la destilación se almacenan para su posterior tratamiento.
d) Residuos de Na2SO4: lavar con etanol y secar en la estufa para su uso posterior.
BIBLIOGRAFIA
1. Brewster, R.Q., Vanderwerf, C.A. y McEwen, W.E., Curso práctico de química
orgánica, 3ª. Ed., Alambra, S.A., Madrid, España, 1979.
2. Fessender, R.J. y Fessender, J.S., Techniques and Experiments for Organic
Chemistry, Willard Grant Press, Boston, EU, 1983.
3. Mayo, D.W., y Pike, R.M., Microscale Organic Laboratory, John Wiley and
Sons, Nueva York, EU, 1986.
4. Moore, J.A. y Dalrymple, D.L., Experimental Methods in Organic Chemistry,
2a. ed., W.B. Saunders, Philadelphia, EU, 1976.
5. Pavia, D.L., Lampman, G.M. y Kriz, G.S., Introduction to Organic Laboratory
Techniques, Saunders College Publishing, 1988.
6. Perry, Chemical Engineer’s Handbook, McGraw-Hill, EU.
7. Roberts, R.M., Gilbert, J.C., Rodewald, L.B., Wingrove, A.S., Modern
Experimental Organic Chemistry, 3ª. Ed., Holt, Reinehart and Winston, Nueva
York, EU, 1979.

Practica # 1 Técnicas Experimentales
33
8. Vogel, A.I., A Textbook of Practical Organic Chemistry, 3a. ed., Longman,
Londres, Inglaterra, 1962.
9. Zubrick, J.W., Techniques and Experiments for Organic Chemistry, Survival
Manual, John Wiley and Sons, 1984.
10. Zubrick, J.W., The Organic Chemistry Laboratory, John Wiley and Sons, EU,
1988.

Practica # 1 Técnicas Experimentales
34

Practica # 2 El Petróleo y sus Derivados
35
PRACTICA # 2: EL PETRÓLEO Y SUS DERIVADOS
OBJETIVO
Que el alumno entre en contacto con el petróleo crudo, sus orígenes, sus
principales componentes, los métodos usados para sus preparaciones y su
aprovechamiento.
Conocer los procesos de destilación simple y fraccionada, sus
características y factores que en ellas intervienen.
Aplicar la técnica de destilación simple y fraccionada en la separación de
los componentes de un crudo.
Aplicar la técnica de destilación a presión reducida en la purificación de
líquidos.
INTRODUCCIÓN.
El petróleo crudo, al igual que el gas natural, se extrae del subsuelo, su origen
es igual que el gas. El petróleo natural no presenta utilidad comercial así como se
encuentra, y por ello es necesario someterlo a un proceso de refinación para obtener
una gran cantidad de compuestos útiles para la industria química. La refinación del
petróleo se hace por medio de una destilación. La refinación del petróleo consiste en
calentarlo progresivamente para que los hidrocarburos comiencen a evaporarse y a
separarse de los demás componentes. Industrialmente la refinación del petróleo se
realiza en una columna de fraccionamiento en la cual se separan las distintas
fracciones de hidrocarburos.
Las fracciones más volátiles o ligeras están constituidas por los compuestos
gaseosos y líquidos ligeros; estas fracciones salen de la columna por la parte superior.
Las fracciones más pequeñas se destilan por la parte inferior de la columna. Este
proceso se conoce como destilación primaria y es el primer paso de la refinación del
petróleo.
“Para nosotros el petróleo es esencial y sin el nos quedaríamos inútiles a
menos de que hubiera un nuevo y abundante combustible.”
El petróleo es esencialmente una mezcla de hidrocarburos que resultan de la
descomposición de materias orgánicas de origen animal y vegetal. Los derivados del
petróleo pueden tener varias propiedades, algunas iguales y otras diferentes. Por

Practica # 2 El Petróleo y sus Derivados
36
ejemplo, algunos derivados reaccionan al calor muy fácilmente, mientras que otros se
necesita de mucho calor para llegar a su combustión. El derivado que más conocemos
es el plástico y que lo usamos en un gran porcentaje de las cosas y útiles que
necesitamos en la vida diaria.
Las reservas de petróleo que quedan están por agotarse, si se siguen usando
tan desmesuradamente. Y nos quedaríamos sin el principal combustible de nuestra
era.
Pero ahora ya se piensa en usar un nuevo combustible para ahorrar el petróleo
y además, causa menos contaminación.
La forma de obtener el petróleo es la destilación fraccionada de la cual se
sacan todos sus derivados y por supuesto los hidrocarburos, alcanos, alquenos y
alquinos. Usualmente, un yacimiento contiene: una capa gaseosa conocida como gas
natural, una capa de hidrocarburos líquidos que es petróleo y otra de agua salada. La
presencia de agua salada indica que la materia orgánica que dio lugar a la formación
del petróleo fue primeramente depositada en el fondo de los mares.
Debido a que el interés económico de las diferentes fracciones obtenidas es
variable, se desarrollaron procesos para incrementar la producción de las fracciones
de mayor consumo tales como gasolina y keroseno.
El cracking es un proceso durante el cual los hidrocarburos de peso molecular
alto son transformados en compuestos más pequeños como hidrógeno, el etileno y los
componentes de las gasolinas. Actualmente, esta operación se realiza con mayor
eficiencia en presencia de catalizadores para evitar temperaturas elevadas.
TEORIA
Destilación simple
La destilación es un proceso que consiste en la evaporación de un líquido,
condensación del vapor y colección del condensado.
Cuando una sustancia líquida se contamina con pequeñas cantidades de
impurezas, éstas pueden eliminarse por algún método de destilación, Se dice entonces
que se efectúa una purificación.
Cuando dos o más sustancias líquidas se encuentran formando mezclas en
proporción relativamente parecida, se dice que la destilación puede usarse para la
separación de componentes.

Practica # 2 El Petróleo y sus Derivados
37
En cada caso, deberá elegirse la técnica de destilación más acorde con las
características de la muestra.
Existen cuatro métodos de destilación: destilación simple, destilación
fraccionada, destilación a presión reducida y destilación por arrastre con vapor.
La relación entre la presión y la temperatura de ebullición de un líquido está
determinada por su comportamiento presión de vapor-temperatura.
Si un líquido contiene una impureza volátil, la separación de ambos se realiza
generalmente por destilación. Cualquier par de sustancias que no tenga presiones de
vapor idénticas se pueden separar por destilación.
Como regla general, se puede indicar que una mezcla de dos componentes con
una diferencia de por lo menos 80 °C puede separarse por una destilación sencilla.
Sustancias cuyos puntos de ebullición difieren por menos de 80 °C se separan por
destilación fraccionada. La destilación simple se utiliza para la purificación de
compuestos orgánicos líquidos.
Las mezclas, cuyos componentes no presentan grandes diferencias en sus
puntos de ebullición, se separan por destilación fraccionada; esta técnica se utiliza
continuamente en la industria y laboratorios.
Destilación fraccionada
La destilación fraccionada debe emplearse para separar y purificar mezclas de
sustancias de puntos de ebullición cercanos. Actualmente se dispone de equipos de
destilación fraccionada muy eficaces para lograrlo.
Si una mezcla formada por benceno (p. eb.: 80 °C) – tolueno (p. eb.: 110 °C)
cuyos componentes difieren 30 °C en sus puntos de ebullición, se separan por

Practica # 2 El Petróleo y sus Derivados
38
destilación simple, y al trazar una gráfica de punto de ebullición ante volumen de
destilado podemos concluir al observarla, que se trata de una mezcla de dos
componentes de punto de ebullición similar, por lo mismo, no se observa una
separación definida con este método de destilación.
Con base en la gráfica, se observa que no hay constancia en el punto de
ebullición porque éste aumenta continuamente.
Esta misma mezcla se puede separar eficazmente utilizando la técnica de
destilación fraccionada, al emplear una columna de fraccionamiento que proporciona
una gran superficie para el intercambio de calor, en las condiciones de equilibrio,
entre el vapor ascendente y el condensado descendente.
Al trazar una gráfica de los puntos de ebullición ante volumen de condensado,
se observa una buena separación donde hay definición de las fracciones separadas.
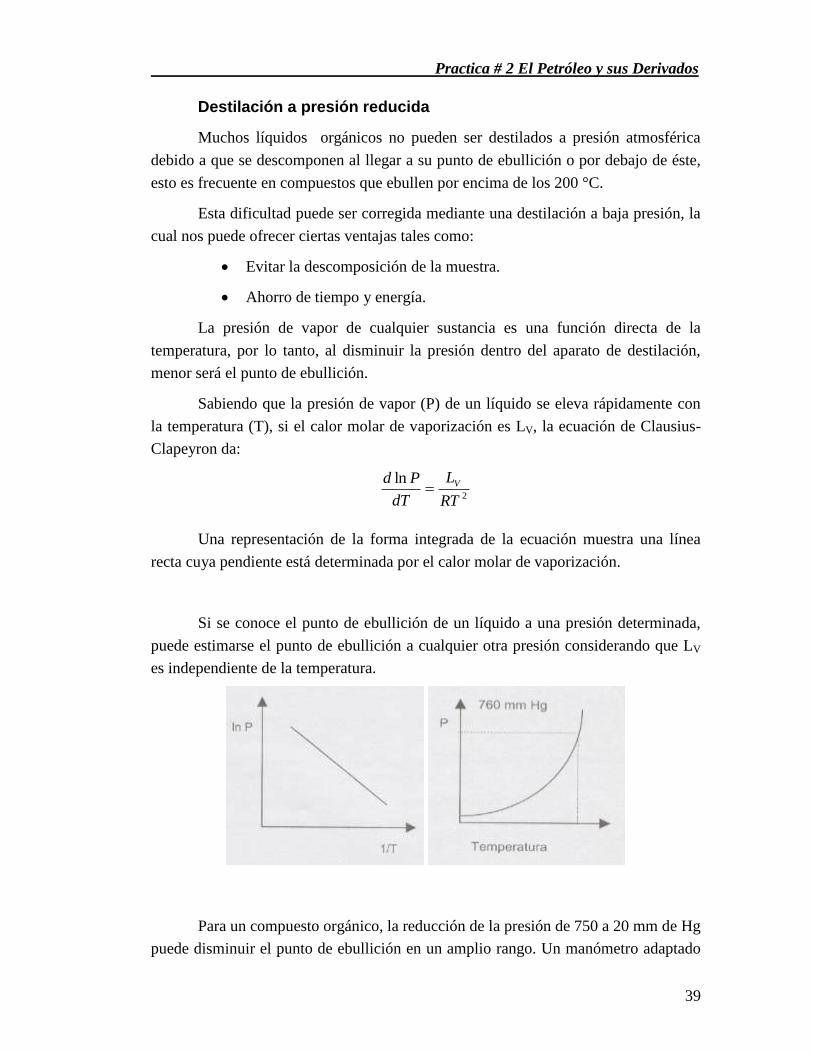
Practica # 2 El Petróleo y sus Derivados
39
Destilación a presión reducida
Muchos líquidos orgánicos no pueden ser destilados a presión atmosférica
debido a que se descomponen al llegar a su punto de ebullición o por debajo de éste,
esto es frecuente en compuestos que ebullen por encima de los 200 °C.
Esta dificultad puede ser corregida mediante una destilación a baja presión, la
cual nos puede ofrecer ciertas ventajas tales como:
Evitar la descomposición de la muestra.
Ahorro de tiempo y energía.
La presión de vapor de cualquier sustancia es una función directa de la
temperatura, por lo tanto, al disminuir la presión dentro del aparato de destilación,
menor será el punto de ebullición.
Sabiendo que la presión de vapor (P) de un líquido se eleva rápidamente con
la temperatura (T), si el calor molar de vaporización es LV, la ecuación de Clausius-
Clapeyron da:
2
ln
RT
L
dT
Pd V
Una representación de la forma integrada de la ecuación muestra una línea
recta cuya pendiente está determinada por el calor molar de vaporización.
Si se conoce el punto de ebullición de un líquido a una presión determinada,
puede estimarse el punto de ebullición a cualquier otra presión considerando que LV
es independiente de la temperatura.
Para un compuesto orgánico, la reducción de la presión de 750 a 20 mm de Hg
puede disminuir el punto de ebullición en un amplio rango. Un manómetro adaptado

Practica # 2 El Petróleo y sus Derivados
40
al equipo de destilación puede ayudarnos a conocer la medida real de la presión. Otra
medida para conocer la presión a la cual se está llevando a cabo la destilación es
mediante el uso de un nomograma (este método en menos preciso).
MATERIAL
1 Matraz de destilación de 250 ml
1 Refrigerante
2 Soportes universales
4 Matraces Erlenmeyer
1 Termómetro de 0 a 200 °C
1 Pinzas para matraz
1 Pinza para refrigerante
1 Anillo y tela de asbesto
1 Mechero bunsen
SUSTANCIAS
100 ml de Petróleo crudo.
PROCEDIMIENTO
1. Monte el equipo para la destilación como lo muestra la figura.
2. Coloque 100 ml de petróleo crudo en un matraz de destilación.
3. Procede a calentar el matraz lenta y uniformemente (destilación simple).
4. Cuando la destilación empiece, recoja por separado en los matraces Erlenmeyer,
las diferentes fracciones que observe, anotando las temperaturas registradas en el
momento de colectar las primeras gotas de destilado. Puede que solo obtenga una
fracción que destile en un rango amplio.
5. Monte un equipo para destilación al vacío, y proceda a destilar el residuo que
obtuvo de la destilación simple, y recoja las fracciones que sean posible. Al igual
que en el punto anterior puede que obtenga un solo corte con un amplio rango de
temperatura.
6. Tome la fracción obtenida en la primera destilación y sométala a una destilación
fraccionada, recoja por separado en los matraces Erlenmeyer, las diferentes

Practica # 2 El Petróleo y sus Derivados
41
fracciones que observe, anotando las temperaturas registradas en el momento de
colectar las primeras gotas de destilado. Guarde las fracciones en ferascos
herméticos.
7. Tome la fracción obtenida en la segunda destilación y sométala a una destilación
fraccionada al vacio, recoja por separado en los matraces Erlenmeyer, las
diferentes fracciones que observe, anotando las temperaturas registradas en el
momento de colectar las primeras gotas de destilado. Guarde las fracciones en
ferascos herméticos.
8. Observe y analice las características de las muestras obtenidas y con ayuda de las
temperaturas a las cuales obtuvo las muestras, trate de encontrar un nombre para
cada fracción destilada.
NOTA: Es importante contar con la bibliografía referente a los derivados del
petróleo.
CUESTIONARIO
1. Explique brevemente la teoría sobre el origen del petróleo:
2. ¿A qué se debe que la composición del petróleo no sea uniforme en todas
3. las partes de la Tierra?
4. ¿Cuáles son los productos que se pueden obtener con la destilación del Explica
como se realiza la refinación del petróleo y que importancia tiene realizarla:
5. Explica que importancia tiene actualmente el petróleo en Venezuela
6. Anota el nombre de 20 productos que sean derivados del petróleo y que estos sean
de uso cotidiano.
Con los datos obtenidos en su experimento, complete la siguiente tabla:
Fracción Temperatura, °C No, de átomos de
carbono
Observaciones
1
2
3
4

Practica # 2 El Petróleo y sus Derivados
42
5
6
7
8
9
10
Residuo
COMPLETE LA SIGUIENTE TABLA:
Fracción Temperatura de
destilación, °C
Número de átomos de
carbono
Gas natural
Nafta ligera o éter de
petróleo
Gasolina
Benceno
Keroseno
Gas oil
Aceites lubricantes
Grasas
Minerales
Parafinas
Asfaltos

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
43
PRACTICA # 3: CROMATOGRAFÍA DE CAPA FINA Y
CROMATOGRAFIA DE COLUMNA
OBJETIVO
Conocer y aplicar la técnica de cromatografía en capa fina c.c.f., sus
características y los factores que en ella intervienen.
Deducir, a través del r.f., la relación que existe entre la polaridad de las sustancias
que se analizan y la de los eluyentes utilizados.
Aplicar la técnica de c.c.f. como criterio de pureza e identificación de las
sustancias.
ANTECEDENTES
Concepto de cromatografía. Clasificación de la cromatografía. Cromatografía de
adsorción y cromatografía de partición. Ejemplos.
Fenómenos de reparto o partición. Coeficiente de reparto. Factor de retención.
Concepto de r.f.
Eluyentes, adsorbentes y reveladores más comunes para cromatografía en capa
fina.
Factores que influyen en una separación por cromatografía de capa fina.
TEORIA
Un método de separación de mezcla que la química orgánica tiene disponible
es la cromatografía. La cromatografía se define como la separación de una mezcla de
dos o más compuestos por distribución entre dos fases, una de las cuales es
estacionaria y la otra una fase móvil. Varios tipos de cromatografía son posibles,
dependiendo de la naturaleza de las dos fases involucradas: sólido-líquido (capa fina,
papel o columna), líquido-líquido y gas-líquido (fase vapor).
Básicamente, el método depende de la adsorción, éste es un fenómeno de
superficie en donde interaccionan electrostáticamente los solutos, de acuerdo a su
polaridad la interacción es fuerte o débil.

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
44
Separación sólido-líquido
Esta es una técnica de separación por partición sólido-líquido. La fase sólida,
fase estacionaria, corresponde al adsorbente que debe ser un material que no se
disuelva en la fase líquida; los materiales más comunes utilizados son:
Celulosa
Silicato de magnesio
Sulfato de calcio
Acido silícico
Gel de sílice
Alúmina
Oxido de magnesio
Carbón activado
Estos adsorbentes son usados en finos granos, usualmente de un tamaño de
malla de 100 a 200 para capa fina y de 200 a 400 para columna.
La acción del adsorbente se basa en la atracción intermolecular tal como
atracción dipolo-dipolo o de enlaces de hidrógeno. Diferentes tipos de adsorbentes
atraen diferentes tipos de moléculas. Un adsorbente altamente polar atrae moléculas
fuertemente polares, pero no presenta gran atracción por moléculas no polares, como
por hidrocarburos.
El eluyente corresponde a la fase móvil de la cromatografía, siendo por lo
general, disolventes o mezcla de éstos, dependiendo de la muestra a separar. Dentro
de los eluyentes más utilizados podemos mencionar:
Eter de petróleo
Ciclohexano
Tetracloruro de carbono
Benceno
Cloroformo
Cloruro de metileno
Dietiléter
Aumento de la fuerza de
interacción de enlace
hacia compuestos polares
Incrementa la polaridad y
poder de disolvente hacia
grupos funcionales polares

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
45
Acetato de etilo
Acetona
Etanol
Metanol
Agua
Acido acético.
Cuando se desea llevar a cabo la separación de una mezcla de compuestos, lo
más recomendable es iniciar la elusión con un disolvente no polar; eluyendo
posteriormente, con disolventes medianamente polares, hasta finalizar con los más
polares, separando al inicio los compuestos menos polares de la mezcla a separar y
terminar con los compuestos más polare.
Sin embargo, el peso molecular también es un factor determinante en el orden
de elución.
Un compuesto no polar de alto peso molecular se desplaza más lento que un
compuesto no polar de bajo peso molecular.
Secuencia de elución por compuesto
Hidrocarburos
Olefinas
Eteres
Halogenuros de alquilo
Aromáticos
Cetonas
Aldehidos
Esteres
Alcoholes
Aminas
Acidos y bases fuertes
Elución rápida con
disolventes no polares
Elución lenta, necesita un
disolvente polar

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
46
Parámetros que afectan la separación
La versatilidad de la cromatografía depende de muchos factores que deben ser
considerados:
Elección del adsorbente adecuado.
Polaridad del disolvente o disolventes seleccionados.
Velocidad de elución.
Cromatografía en capa fina
La cromatografía en capa fina es una técnica para la separación rápida y el
análisis cualitativo de pequeñas cantidades de muestra.
Este tipo de cromatografía se fundamenta en la separación por partición
sólido-líquido, fase móvil (eluyente) y fase sólida (adsovente), llevándose a cabo la
elución en forma ascendente (por capilaridad).
El adsorbente se encuentra adherido a una superficie lisa formando una capa
delgada de aproximadamente 100 m de espesor, en la cual una pequeña cantidad de
mezcla (2-20 g) es adsorbida para su separación.
La muestra a separar se aplica con una micropipeta cerca de la base de la
placa, como se muestra en la figura.
Aplicar la muestra mediante un capilar
Una vez aplicada la muestra, la placa se introduce en una cámara de elución,
la cual contiene el disolvente seleccionado para llevar a cabo la separación en una
cantidad tal que no rebase el punto de aplicación de la muestra (a 5mm de la base de
la placa).
Durante el proceso de separación se dice que la muestra está eluyendo.

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
47
Cámara de solución
Cuando la muestra ha terminado de eluir (el disolvente ha recorrido la placa
hasta 1 cm antes del borde superior), la placa se retira de la cámara de elución y se
deja secar, procediendo a revelar laplaca.
Dentro de las principales aplicaciones de la cromatografía en capa fina,
podemos mencionar que es una técnica eficiente para:
Establecer si dos compuestos son idénticos.
Determinar el número de componentes de una mezcla.
Determinar el disolvente adecuado para una separación cromatográfica en
columna.
Monitorear la separación de una mezcla por cromatografía en columna.
Seguir el proceso de una reacción.
Como criterio de pureza.
Revelador
Una vez que se llevó a cabo una separación por cromatografía en capa fina
para identificar cada uno de los compuestos, se requiere revelar las placas.
El método más utilizado de revelado es el uso del yodo, éste forma complejos
coloreados que van de café a amarillos, llevándose a cabo en una cámara de revelado,

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
48
en la cual están contenidos los cristales de yodo; la placa una vez eluida, se deja secar
y se introduce en esta cámara, hasta que aparecen las manchas definidas, donde se
encuentra cada compuesto separado.
El segundo método más utilizado es el revelado con luz ultravioleta,
aprovechando que muchos compuestos presentan fluorescencia bajo luz UV. Esto se
puede lograr también, utilizando un adsorbente con un indicador (sulfuro de cadmio o
zinc) fluorescente, el cual, después de eluir una muestra y dejar secar, se observa bajo
luz UV.
Existen otros métodos de revelado, disponibles principalmente a base de
reactivos químicos, sin embargo, no son muy utilizados, ya que este tipo de reactivos
son muy específicos para ciertos grupos funcionales.
Frente de referencia (Rf)
La relación de la distancia que recorre la sustancia en la placa y la distancia
que recorre el eluyente a partir del punto de aplicación se le conoce como “Frente de
Referencia” Rf, y se expresa como una fracción decimal:
eluyente el recorre que distancia
sustancia la recorre que tanciadisRf
Este valor se puede usar para seleccionar el disolvente o la mezcla de
disolventes ideales para la separación.

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
49
El valor del frente de referencia es de gran utilidad cuando se desea
seleccionar el eluyente adecuado en una separación de compuestos, por lo que
podemos decir que para los siguientes valores de rf la información que obtendremos
es:
Si el rf < 0,5 el eluyente es de muy baja polaridad para la separación.
Si el rf = 0,5 el eluyente es el ideal para la separación.
Si el rf > 0,5 el eluyente es de muy alta polaridad para la separación.
MATERIALES
Placa para cromatografía (portaobjetos) 6 Frascos cámara 5
Matraces Erlenmeyer de 10 ml o viales 5 Agitador de vidrio 1
Pipetas graduadas de 10 ml 2 Capilares 3
Probeta de 25 ml 1 Espátula 1
MATERIALES ADICIONALES
Envase resistente cerrada
Lámpara de ultravioleta
Aspersor
SUSTANCIAS
Muestra Problema
Muestra Patrón
Solvente A
Solvente B
Se le entregara a cada grupo una muestra problema la cual consta de dos
compuestos que los estudiantes deberán resolver por c.c.f., mediante el uso de los
solventes A y B. Además deberán realizar la extracción de los colorantes de la pasta

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
50
de tomate, que luego serán resueltos por c.c.f., y posterior mente separados por
cromatografía de columna.
PROCEDIMIENTO
Preparación de las placas
Se prepara en un frasco de boca ancha, una suspensión de 35 g de sílice gel en
75 ml de solución A aproximadamente. Para ello se coloca el disolvente y luego se
añade la sílice lentamente, agitando continuamente con una varilla de vidrio, hasta
lograr la consistencia apropiada, que es algo espesa y bien homogénea. Luego se
introducen en esta suspensión dos portaobjetos superpuestos, previamente lavados
con acetona y bien secos, tratando de cubrir la máxima superficie posible, según se
muestra en la figura.
Se secan enseguida, con movimiento regular, para que el recubrimiento se
deposite uniformemente. Se separan las placas y si el resultado es satisfactorio se
dejan secar al aire, limpiando cuidadosamente los bordes con el dedo. Si se desea
mayor activación del adsorbente, puede colocarse en la estufa a 100 ºC por unos
minutos. Cada estudiante debe preparar seis placas como mínimo.
Por otra parte, se preparan capilares finos usando tubos para medidas de
fusión, que sirven para aplicar las muestras sobre las placas. Para ello es necesario
que la muestra esté disuelta en un disolvente de cierta volatilidad, se sumerge la punta
del capilar en la solución y se toca suavemente la superficie de la placa con el capilar,
una vez cargado de manera de transferir una pequeña gota, levantando enseguida el
capilar. Esto se ilustra en la siguiente figura.

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
51
Al evaporarse el disolvente, la muestra queda fija sobre el adsorbente. Se
puede aplicar varias muestras en una misma placa, dependiendo del tamaño de ésta
ultima. Las aplicaciones se realizan a 0,5 cm aproximadamente del borde de la placa.
Preparación de las cámaras de desarrollo
Se cubren las tres cuartas partes de la superficie inferior del recipiente a
utilizar, con una banda de papel de filtro, la cual se impregna del disolvente elegido,
cubriendo además el fondo del recipiente con el mismo disolvente. Se tapa para que
el ambiente interior se sature de vapores. Existen cámaras de vidrio especialmente
diseñadas para tal fin, equipadas con tapas esmeriladas. En su defecto usarse, para
placas pequeñas, un beaker de 400 ml o un frasco de boca ancha.
Posteriormente se coloca con cuidado, la placa con la muestra que se desea
analizar, como se ilustra en la figura, de manera que el borde inferior del adsorbente
entre en contacto con el disolvente, pero sin llegar a sumergir la zona donde se aplicó
la muestra.

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
52
El disolvente sube a través de la placa por capilaridad, produciéndose el
desarrollo del cromatograma.
Obtención de los valores de Rf a las muestras patrón
Se colocan en una misma placa, soluciones al 2% de las muestras patrón. Al
depositar la gota y evaporar el disolvente, queda fijo en el adsorbente el soluto
correspondiente. La placa se introduce en la cámara preparada con el solvente A, se
tapa y se desarrolla el cromatograma. Cuando el frente del disolvente llegue a la parte
superior de la placa, se saca esta de la cámara y se deja secar, colocándola
posteriormente en una cámara de vapores de yodo para revelar el cromatograma, ya
que los compuestos utilizados son incoloros. Se marca el lugar al cual llegó cada
muestra, pues este revelado no es permanente, se mide la distancia recorrida por cada
soluto, desde el punto de aplicación hasta el centro de la mancha y la distancia
recorrida por el disolvente. Con estos valores se calcula el Rf de cada sustancia en el
solvente A. De igual forma se procede hacer con el solvente B. Al utilizar estos dos
solventes se puede observar como varia el valor del Rf al aumentar la polaridad del
disolvente. Comente al respecto en las discusiones del informe.
Identificación de los compuestos presentes en la muestra
problema
Se realiza una experiencia similar a la anterior pero en lugar de utilizar la
muestra patrón se toma la muestra problema. En esta experiencia se utilizan ambos
solventes probados anteriormente. Al final de la experiencia cuando se realiza el
revelado de la muestra problema se comparan los valores de Rf obtenidos en cada
solvente, con los valores obtenidos para las muestras patrón, y se identifican los
compuestos presentes en la muestra problema.
Extracción de los colorantes del tomate
En un erlenmeyer de 125 ml se colocan 5 gr de pasta de tomate y se le
agregan 10 ml de etanol al 95% y se somete a calentamiento suave con agitación
continua durante 10 minutos, Luego se filtra por succión, tomando la precaución de
pasar todo el sólido al embudo. El sólido debe presionarse con la parte plana de la
espátula, para tratar de eliminar el resto de líquido. El filtrado se guarda y el sólido se
regresa al elenmeyer, añadiendo 10 ml del diclorometano y se coloca a reflujo por 10
minutos. Posteriormente se filtra por succión de nuevo, uniendo este filtrado al
anterior. Se repite la extracción con otra porción del diclorometano, se unen los
filtrados y se satura con cloruro de sodio.

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
53
Se coloca el filtrado en un embudo de separación y se añaden porciones de
solución saturada de cloruro de sodio, para favorecer la separación de las capas. Se
agita y se deja en reposo para que ambas fases se separen nuevamente y se desaloja el
embudo. La capa orgánica se seca con sulfato de magnesio anhidro y se procede a
realizar la destilación sencilla para eliminar los disolventes, teniendo cuidado de no
destilar a sequedad. Luego se agrega al residuo de la destilación, una porción del
solvente B y se destila. El extracto obtenido en el balón de destilación se transvasa
con hexano a un recipiente pequeño con tapa y se guarda para el próximo periodo de
laboratorio.
CUESTIONARIO
1. ¿Por qué la c.c.f. es un criterio parcial y no total de identificación?
2. ¿Cuál será el resultado de los siguientes errores en cromatografía en capa fina?
i. Aplicación de soluciones muy concentradas.
ii. Utilizar eluyentes de alta polaridad
iii. Emplear gran cantidad de eluyente en la cámara de cromatografía.
3. Una serie de colorante es separada por cromatografía en capa fina. Calcular los
valores de Rf de cada colorante de acuerdo a la siguiente tabla.
Colorante Distancia recorrida
por el colorante (mm)
Distancia recorrida
por el eluyente (mm)
Rojo de metilo 56.0 70
Rodamina B 38.0 65
Rojo congo 0.5 72
4. ¿Qué tratamiento es necesario darle a las sílice gel para reutilizarla?

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
54
SEPARACIÓN DE UNA MEZCLA MEDIANTE CROMATOGRAFÍA DE
COLUMNA
OBJETIVO
Conocer y aplicar la cromatografía en columna como técnica de
separación de mezcla de sustancias, sus características y los factores
que en ella intervienen.
Controlar con cromatografía en capa fina, el proceso de separación de
mezclas por cromatografía en columna.
ANTECEDENTES
Cromatografía en columna y capa fina. Características y aplicaciones.
Cromatografía de adsorción y de partición o reparto.
Selección de eluyentes para separar mezclas de compuestos por
cromatografía en columna.
Técnicas de separación cromatográfica por elución, adsorción y
desplazamiento.
Eluyentes y adsorbentes para cromatografía en columna y capa fina.
Factores que influyen en la separación por cromatografía.
TEORÍA
La cromatografía de columna es una técnica basada en la adsorción y
solubilidad. Es una técnica de partición sólido-líquido, puesto que existe un equilibrio
dinámico entre la adsorción y desorción (elución).
Adsorción ↔ desorción
Existen diferentes clases de interacciones intermoleculares que originan que
los compuestos orgánicos se unan a la superficie del adsorbente con mayor o menor
fuerza dependiendo de la naturaleza polar o no polar de los compuestos.
Los compuestos no polares interaccionan débilmente con el adsorbente
mediante fuerzas de van der Waals, mientras que las interacciones más fuertes se
manifiestan en compuestos polares que van desde fuerzas del tipo dipolo-dipolo hasta
interacciones más directas como: formación de sales, enlaces de coordinación o
puentes de hidrógeno.

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
55
La fase móvil y la fase estacionaria, son las mismas que para el caso de
cromatografía en capa fina, variando para el caso del adsorbente, el tamaño de
partícula.
La columna de cromatografía esta construida por un tubo de vidrio en
posición vertical en el cual, por la parte baja, se colectarán las diferentes fracciones.
En el fondo del tubo se introduce un pequeño trozo de algodón que cumple la función
de un filtro que impide que el gel de sílice salga. Una vez colocado el algodón, se
coloca una delgada capa de arena, la cual no permite que el algodón se mueva de su
posición. Posteriormente, por la parte superior, se introduce el adsorbente, el cual ha
sido previamente suspendido en el disolvente seleccionado (vía húmeda) para la
separación con el objetivo de eliminar las burbujas que se encuentran en las partículas
de adsorbente y posteriormente el eluyente seleccionado.
La cantidad requerida de fase estacionaria se determina de acuerdo a la
cantidad de muestra a separar. Una regla común es usar 30 a 100 veces el peso de la
muestra a separar de adsorbente. Las dimensiones de la columna se diseñan según las
características de la muestra a separar, siendo por lo general una relación de 10:1 de
largo de la columna por el radio de ésta.
Una vez que se ha preparado la columna, la muestra a separar se disuelve en la
mínima cantidad de disolvente y se aplica por la parte superior de la columna,
llevándose a cabo la elución por gravedad, colectando por la parte baja de la columna,
las diferentes fracciones separadas. El grado de elución de los diferentes compuestos
separados depende del grado de polaridad de cada uno de ellos.
Es muy importante señalar que se debe cuidar que no se seque la columna. La
velocidad del flujo debe permitir el tiempo necesario para que exista un equilibrio
entre las dos fases.
La identificación de los compuestos separados por cromatografía en columna
se realiza por cromatografía en capa fina.

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
56
PARTE EXPERIMENTAL
Materiales por equipo de 2 alumnos
Frascos viales de 15 ml 10 Placas para cromatografía 8
Vasos de precipitado de 150 ml 2 Pinzas 3 dedos con nuez 1
Espátula 1 Columna para cromatografía 1
Frasco para cromatografía con tapa 3 Varilla de vidrio 1
Capilares 5 Embudo de vidrio 1
Pipeta de 125 ml 1 Probeta graduada de 25 ml 1
Agitador de vidrio 1
Sustancias
Sílice gel para columna
Sílice gel para cromatografía en capa fina
Extracto de los colorantes del Tomate
PROCEDIMIENTO
Determinación de las condiciones de separación de los
componentes del extracto de los colorantes del tomate.
Mediante un procedimiento similar a los realizados la semana anterior (c.c.f),
se analiza la solución del extracto de colorantes del tomate, obtenidos la semana
anterior, para determinar las condiciones de separación. Se utilizan placas de sílice
gel y hexano, solvente A y solvente B y/o mezcla de ellos como disolventes de
desarrollo. Con los resultados obtenidos se planifica la separación de -caroteno y
licopeno mediante cromatografía en columna.
Empaquetamiento de la columna
Existen dos formas de empaquetamiento. El empaquetamiento húmedo en el
cual se añade una mezcla del eluyente y del soporte o el empaquetamiento seco que

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
57
consiste en añadir primero el soporte y a continuación el eluyente. Aquí emplearemos
el empaquetamiento húmedo.
Pesar 30 gramos de gel de sílice y depositarlos en un vaso de precipitados. (Es
importante manipular el gel de sílice lo máximo posible en la campana pues es
perjudicial respirarlo).
Añadir sobre el gel de sílice poco a poco sobre el disolvente seleccionado
removiendo con una varilla de vidrio hasta que quede una suspensión del gel en el
líquido. Procurar que no se formen burbujas en el interior de esta suspensión, en este
caso remover hasta que desaparezcan. También hay que tener en cuenta que en el
momento de añadir el eluyente la mezcla se calentará al liberarse calor de
solvatación.
Antes de introducir esta mezcla en la columna y mientras enfría se introduce
un poco de algodón ó lana de vidrio y se ajusta en el estrechamiento interior de la
llave, no apretándolo demasiado.
Fijar la columna verticalmente con dos pinzas a un soporte universal.
Ayudados por un embudo de sólidos, añadir una parte de la suspensión
soporte/eluyente (es conveniente remover esta suspensión justo antes de verterla en la
columna) Con el vaso de precipitados de la mezcla debajo de la llave, abrir ésta y
dejar pasar el eluyente golpeando suavemente con una varilla provista de goma para
que se liberen las burbujas de aire. (Es importante que no quede aire en el soporte
pues esto dificultaría la separación de los productos, tampoco debe pasar ningún
sólido a través de la llave, en este caso se debe empezar de nuevo con el algodón más
ajustado).
Esperar hasta que baje el nivel del eluyente y que el soporte compacte en el
fondo de la columna. Cuando el eluyente esté un centímetro por encima del nivel del
soporte, cerrar la llave y añadir más suspensión soporte/eluyente a la columna.
Repetir el mismo proceso hasta que tengamos todo el soporte en la columna, arrastrar
con un poco de eluyente el gel de sílice que queda por las paredes de la columna.
Dejar siempre el nivel del eluyente por encima del soporte, evitando que este
se seque.
La superficie del adsorbente debe ser completamente horizontal, pues de lo
contrario la eficiencia de la separación disminuye, como se visualiza en la figura.

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
58
Carga de la muestra
Disolver disolver 1.5 g de la mezcla de colorantes del tomate en la mínima
cantidad posible de eluyente. (aprox. 1.5 ml).
Abrir la llave de la columna hasta que el nivel del eluyente alcance casi el
nivel del soporte y cerrar la llave.
Añadir la disolución de la muestra sobre la superficie del soporte con la ayuda
de una pipeta larga dejando correr la mezcla por las paredes interiores de la columna.
Abrir de nuevo la llave hasta alcanzar casi el nivel del soporte.
Enjuagar el matraz con una pequeña cantidad de eluyente (aprox. 1.5 ml) e
introducir la disolución de nuevo en la columna.
Rellenar la columna con unos 150 ml de eluyente.
Elución de la columna
Se debe dejar eluir la columna despacio, gota a gota
Una vez que toda la muestra está dentro del adsorbente, se agrega suficiente
disolvente y se sigue desarrollando el cromatograma. Poco a poco se van separando
los componentes, que se visualizan por ser muy coloreados. Se eluyen las diferentes
fracciones y se recogen en erlenmeyers, a medida que se va variando la polaridad de

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
59
la fase móvil, de acuerdo a las condiciones previamente determinadas en capa fina. El
procedimiento se esquematiza en la siguiente figura
Comparación de la separación realizada, mediante cromatografía
en capa fina
Las fracciones recolectadas de la columna se concentran un poco, calentando
suavemente en una plancha.

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
60
Por otra parte, se preparan placas de sílice gel, de acuerdo a la técnica ya
conocida, así como cámaras de desarrollo con los disolventes usados en la elución de
los colorantes.
Se aplican muestras de cada fracción y se desarrollan en la cámara
correspondiente, para comprobar si la separación ha sido efectiva.

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
61
CUESTIONARIO
1. Si una mezcla de antraceno y naftaleno se separan por cromatografía sobre
alúmina, ¿Cuál de los dos hidrocarburos eluirá primero cuál al ultimo?, ¿Cuál
sería más polar?
2. Un colorante desconocido, se piensa que puede ser azul de metileno, ¿Cómo se
podría comprobar esta suposición usando un procedimiento asado en una técnica
cromatográfica?
3. ¿Qué debe hacerse para encontrar el eluyente adecuado para una sustancia en una
cromatografía en columna?
4. La recuperación cuantitativa del producto principal de la práctica, sería más
completa si se recogieran fracciones mayores o menores de 10 ml, ¿por qué?
5. Indique alguna de las aplicaciones de la cromatografía de adsorción en columna y
capa fina.
6. Cual es la diferencia entre la cromatografía en capa fina y la cromatografía en
papel.

Practica # 3 Cromatografía de capa fina y Cromatografía de columna
62

Practica # 4 Síntesis del Ciclohexeno
63
PRÁCTICA # 4: SINTESIS DEL CICLOHEXENO
OBJETIVOS
Preparar ciclohexeno por deshidratación catalítica de ciclohexanol.
Comprender la influencia de factores experimentales que modifican una reacción
reversible.
ANTECEDENTES
Propiedades físicas, químicas y toxicidad de reactivos y productos.
Deshidratación catalítica de alcoholes para obtener alquenos. Mecanismo de reacción.
Influencia de las condiciones experimentales en la reversibilidad de una reacción.
Reacciones de adición a dobles ligaduras.
REACCIÓN A EFECTUAR Y ESTEQUIMETRIA
OH
+ H2SO4 + H2O
Ciclohexano Acido sulfúrico Ciclohexeno
PM 100.13 98.08 82.15
Peso (g) 9.48 0.9 7.77
Moles 9.46 x 10-2
9.0 x 10-3
9.46 x 10-2
MECANISMO
El presente caso es un ejemplo de una eliminación monomolecular E1. Es
importante observar que el paso lento de la reacción es la formación del carbocatión
secundario; para que esto ocurra se debe protonar previamente el grupo –OH para que el
grupo saliente sea la molécula de agua que es estable.
El alcohol en presencia de un ácido fuerte efectúa una reacción ácido-base entre uno
de los pares de electrónicos en los orbitales sp3 del oxígeno del alcohol (1) y el ácido
sulfúrico, dando lugar a la formación del alcohol protonado (2). La formación de este

Practica # 4 Síntesis del Ciclohexeno
64
intermediario trae como consecuencia un aumento en la polarización del enlace C-OH2+
hasta romperse, generando así el carbocatión secundario, el cual puede sufrir 2 tipos de
reacciones: el ataque nucleofílico o la eliminación.
En el primer caso no esta favorecida la reacción ya que los nucleófilos presentes son
sulfato ácido HSO4-, el cual es un nucleófilo débil más agua; sin embargo, es un paso lento
ya que la concentración disminuye por estar en equilibrio con el ácido sulfúrico para formar
el hidronio. La segunda reacción es la eliminación de uno de los hidrógenos al
carbocatión. Aunque existen cuatro hidrógenos vecinos al carbocatión, se facilitará la
eliminación de uno de los hidrógenos axiales, ya que habrá una interpenetración orbital más
efectiva entre los orbitales que forman el enlace C-H y el orbital p vacío. Debido a que se
encuentran cuasi-coplanares (3), mientras que el hidrógeno ecuatorial se encuentra cuasi-
ortogonal, impidiendo una interpenetración orbital.
La pérdida de uno de los hidrógenos axiales tiene como consecuencia la formación
de un doble enlace, teniendo así el ciclohexeno (4).
OH
+ H2SO4O
+H
H+ HSO4
-
(1) (2)
O+
H
H
(2)
-H2O
H
H
H-H
+
(3) (4)
PARTE EXPERIMENTAL
MATERIAL
Agitador de vidrio 1 Probeta graduada de 25 mL 1
Anillo metálico 1 Pipeta graduada de 5 mL 1
Colector 1 Refrigerante con manguera 1
Columna Vigreaux 1 Resistencia eléctrica 1
Embudo de separación con tapa 1 Tela de alambre con asbesto 1
Matraz Erlenmeyer 50 mL 2 Termómetro -10 a 400 |C 1
Matraz balón 25 mL 1 Matraz pera de una boca 1
Mechero con manguera 1 Pinza de tres dedos con nuez 2
T de destinación 1 Porta termómetro 1
Vaso de precipitados 250 mL 1 Tubos de ensayo 2

Practica # 4 Síntesis del Ciclohexeno
65
SUSTANCIAS
Cantidad Calidad
Ciclohexano 10.0 mL Q.P
Tetracloruro de carbono 5.0 mL Q.P
Acido sulfúrico concentrado 0.5 mL Q.P
Permanganato de potasio 0.1 g Q.P
Bicarbonato de sodio 2.0 g Q.P
Bromo 0.1 g Q.P
Sulfato de sodio anhidro 2.0 g Q.P
Cloruro de sodio 2.0 g Q.P
INFORMACIÓN
La reacción para obtener ciclohexeno a partir de ciclohexanol es reversible.
La reversibilidad de una reacción se puede evitar:
Si se elimina el producto del medio de reacción a medida que ésta sucede.
Si se aumenta la concentración de uno o varios de los reactivos.
Si se aumenta o disminuye la temperatura en el sentido que se favorezca la reacción
directa, etc.
Por lo tanto, las condiciones experimentales en las que se efectúa una reacción
determinan los resultados de ésta, en cuanto a calidad y cantidad del producto obtenido.
PROCEDIMIENTO
Preparar el ciclohexeno a partir de ciclohexanol por dos procedimientos diferentes
(Método y Método B) y comparar los resultados obtenidos en cuanto a calidad y cantidad
del producto, con el fin de determinar que método es más eficiente.
Luego se comprobará a través de reacciones específicas de identificación, la
presencia de dobles enlaces C=C en el ciclohexeno obtenido (pruebas de insaturación).
Método A. Por destilación fraccionada.
Monte un equipo de destilación fraccionada (Nota 1).
En el matraz pera de una boca de 50 ml coloque 10 ml de ciclohexanol, agregue
gota a gota y agitando 0.5 ml de ácido sulfúrico concentrado, agregue cuerpos de ebullición
y adapte el resto del equipo. Posteriormente vierta en la trampa 25 ml de la solución de
permanganato de potasio.

Practica # 4 Síntesis del Ciclohexeno
66
Emplee un baño de aire y caliente moderadamente el vaso de precipitado con el
mechero, a través de la tela de asbesto. Reciba el destilado en el matraz de bola y colecte
todo lo que destile entre 80-85 ºC enfriando con un baño de hielo.
Suspenda el calentamiento cuando solo quede un pequeño residuo en el matraz o
bien empiecen a aparecer vapores blancos de SO2. (Nota 2)
Sature el destilado con cloruro de sodio y decántelo en el embudo de separación,
lávelo 3 veces con una solución de bicarbonato de sodio al 5% empleando porciones de 5
ml cada vez. Coloque, la fase orgánica en un vaso de precipitados y séquela con sulfato de
sodio anhídro. Esta fase orgánica debe ser ciclohexeno, el cual deberá purificar por
destilación simple, empleando un baño de aire (Nota 3).
Colecte la fracción que destila a la temperatura de ebullición del ciclohexeno (Nota
4).
La cabeza y la cola de la destilación pueden utilizarse para hacer las pruebas de
insaturación, que se indican al final de este procedimiento.
Mida el volumen obtenido y entréguelo al profesor. Calcule el rendimiento de la
reacción.
1
2
3
45 6
7
8
9
1 10
2
3
45 6
7
8
9
11

Practica # 4 Síntesis del Ciclohexeno
67
Método B. Por reflujo directo.
La realización de este método tiene por objetivo establecer una comparación con el
anterior en cuanto a los resultados que se obtengan. Por esta razón, sólo un alumno pondrá
en práctica este método en tanto los demás deberán tomar en cuenta este resultado para
hacer la comparación respectiva
Monte un equipo de reflujo directo. En el matraz pera de una boca coloque 10 ml de
ciclohexanol, agregue gota a gota y agitando 0.5 ml de ácido sulfúrico concentrado,
agregue cuerpos de ebullición y adapte el resto del equipo.
Caliente el sistema con el mechero a través de la tela de alambre con asbesto,
empleando un baño de aire, durante 45 minutos.
Luego déjelo enfriar un poco y vierta la mezcla de reacción en una suspensión de 2
g de bicarbonato de sodio en 10 ml de agua. Separe entonces la fase orgánica, lávela con 3
porciones de 5 ml de una solución saturada de bicarbonato de sodio y séquela con sulfato
de sodio anhidro. Purifique por destilación simple, empleando un baño de aire, el
ciclohexeno obtenido.
Mida el volumen obtenido y entréguelo al profesor. Calcule el rendimiento de la
reacción.
1
2
3
45 6
7
8
9
1 10
2
3
45 6
7
8
9
11

Practica # 4 Síntesis del Ciclohexeno
68
PRUEBAS DE INSATURACIÓN.
REACCIÓN CON BR2/CCL4.
En un tubo de ensayo coloque 1 ml de solución de bromo en tetracloruro de
carbono, agregue 1 ml de ciclohexeno y agite.
Observe e interprete los resultados. Escriba la reacción que se lleva a cabo.
REACCIÓN CON KMNO4.
En un tubo de ensayo coloque 1 ml de solución de permanganato de potasio (acidule
a pH 2 ó 3), agregue 1 ml de ciclohexeno y agite. Observe e interprete los resultados.
Escriba la reacción que se lleva a cabo.
Resuma en el siguiente cuadro los datos experimentales de los dos métodos de
obtención del ciclohexeno.
Método Condiciones
experimentales
Temperatura
de destilación
Volumen (mL)
del destilado
% de la
reacción
A
B
NOTAS
1. Para aumentar el gradiente de temperatura en la columna cúbrala exteriormente con
fibra de vidrio.
2. Enfrié muy bien el aparato antes de desmontar y coloque el matraz de bola con su tapón
en un baño de hielo.
3. Tenga cuidado de utilizar el material bien limpio y seco.
4. El punto de ebullición del ciclohexeno es de 83-84o C. a 760 mm Hg .

Practica # 4 Síntesis del Ciclohexeno
69
DIAGRAMA ECOLOGICO
D1: separe las fases, mande a incineración la fase orgánica, utilice fase acuosa para
neutralizar D2.
D2: neutralice con D1 y cheque pH.
D3: lave con EtOH y seque para uso posterior.
Destilado
7) Destilar. H2SO4 conc.
Residuo
Na2SO4
Mat. Org.
Degradada
Ciclohexeno
Agua
D1
5) Saturar con NaCl.
6) Lavar con NaHCO3 (5%)
NaCl
NaHCO3
Ciclohexeno
Impuro, húmedo
D2 3) Sevar con Na2SO4.
4) Decantar ó filtrar
Residuo Filtrado
Fase orgánica
Fase acuosa
Ciclohexeno
Impuro
Na2SO4
D3
2) Destilar.
Destilado Residuo
Residuo
Orgánico
D4
Ciclohexeno
puro
1) Tomar muestra
KMnO4 Br2
Bromo
Ciclohexano
MnO2
diol
D5 D6
Ciclohexanol

Practica # 4 Síntesis del Ciclohexeno
70
D4: mande a incineración.
D5: mande a incineración.
D6: filtre el MnO2, etiquetero y confínelo. Cheque pH al líquido y deseche por el
drenaje.
CUESTIONARIO
1. Con base en los resultados obtenidos, ¿cuál de los dos métodos es el más eficiente para
obtener ciclohexeno? Explique.
2. ¿Qué es una reacción reversible?
3. ¿Qué es una reacción irreversible?
4. ¿Qué es una reacción en equilibrio?
5. ¿Cuáles fueron los principales factores experimentales que se controlaron en esta
práctica.
6. ¿Qué debe hacer con los residuos de la reacción depositados en la pera o matraz antes
de desecharlos por el drenaje?
7. ¿Cuál es la toxicidad de los productos que se forman al realizar las pruebas de
insaturación
8. Asigne las bandas principales presentes en los espectros de I.R. a los grupos funcionales
de reactivos y productos.
BIBLIOGRAFÍA
1. Brewster R.Q., Vander Werf C.A. y Mc. Ewen W.E. Curso Práctico de Química
Orgánica, 2a. Edición Alambra Madrid, (1979).
2. Vogel, A.I. A textbook of Practical Organic Chemistry. 5a. Edición , Longmans
Scientifical and Technical, NY, (1989).
3. Pavia D.L., Lampman G.M. y Kriz G.S. Introduction to Organic Laboratory
Techniques.
4. W.B. Saunders Co. Philadelphia, (1976).
5. Lehman, J.W. Operational Organic Chemistry 3er edition Prentice Hall New Jersey,
USA (1999).

Practica # 4 Síntesis del Ciclohexeno
71
6. Mohring, J.R., Hammond, C.N., Morril, T.C., Neckers, D.C. Experimental Organic
Chemistry W.H. Freeman and Company New York,USA (1997).
ESPECTROS DE I.R.
a) Ciclohexanol
b)Ciclohexeno

Practica # 4 Síntesis del Ciclohexeno
72

Practica # 5A Obtención de Acido Benzoico y Alcohol Benzilico
73
PRÁCTICA # 5A: OBTENCION DE ACIDO BENZOICO Y
ALCOHOL BENZILICO
OBJETIVOS
Estudiar la reacción de Cannizzaro
Síntesis de ácido benzoico a partir de benzaldehido
REACCIÓN
C
O
H
+ KOH
COOK CH2 OH
+2
Benzaldehido Hidróxido de
Potasio
Acido
Benzoico
Alcohol Benzilico
PM 106.05 56.10 122.13 108.14
Peso (g) 3.12 (3.0 ml)
(d = 1.04 g/ml)
3.0 3.59 3.18
Moles 29.42 x 10-3
53.47 X 10-3
29.42 x 10-3
29.42 x 10-3
MECANISMO
La adición de bases diluidas a aldehídos o cetonas conduce a la formación de
β- hidroxialdehídos o β-hidroxicetonas a través de una condensación aldólica. Si el
aldehído o la cetona no poseen hidrógenos α, la condensación aldólica no tienen lugar.
Sin embargo, los aldehídos que no poseen hidrógenos α experimentan
autooxidaciónreducción en presencia de álcalis concentrados para dar una mezcla
equimolecular del alcohol y de la sal del correspondiente ácido. Por ejemplo, benzaldehído
produce alcohol bencílico y benzoato potásico en presencia de hidróxido de potasio.

Practica # 5A Obtención de Acido Benzoico y Alcohol Benzilico
74
El mecanismo del proceso de auto-oxidación-reducción descrito anteriormente
implica la transferencia de ion hidruro desde el intermedio de reacción, resultante del
ataque de OH- al carbonilo, hasta una segunda molécula de aldehído.
R C O
H
+ OH-
R C O-
H
OH
R C O-
H
OH
R C O
H
+
R C O
OH
R C O-
H
H
+
R COOH
+
R CH2 O-
R CH2 OH+R COO-
El formaldehído experimenta reacción de Cannizzaro, al igual que los aldehídos
alifáticos (como trimetilacetaldehído) que no contienen hidrógenos en α.
El ácido benzoico es un sólido blanco, cristalino (p.f. 122oC) ligeramente soluble en
agua a temperatura ambiente (0.2 g/100 g a 20oC, 2.2 g/100 g a 75oC). El alcohol bencílico
es un líquido incoloro (p.e. 205oC) ligeramente soluble en agua.
PARTE EXPERIMENTAL
MATERIAL
Matraz de fondo redondo de 50 o 100 mL 1 Refrigerante con manguera 1
Embudo de succión 1 Embudo de decantación 1
Vaso de precipitados 50 ó 100 mL 1 Matraz Erlenmeyer 100 mL 1
Matraz Erlenmeyer 250 mL 1 Termómetro -10 a 400 |C 1
Embudo de vidrio 1 Manta calefactora 1
Balanza 1 Plato poroso 1

Practica # 5A Obtención de Acido Benzoico y Alcohol Benzilico
75
SUSTANCIAS
Cantidad Calidad
Hidroxido de potasio 3.0 g Q.P
Benzaldehido 3.0 mL Q.P
Eter dietílico 16.0 mL Q.P
Sulfato de magnesio 2.0 g Q.P
Acido clorhídrico 8.0 mL Q.P
PROCEDIMIENTO
Disolver 3.0 g del KOH sólido (corrosivo) en 3.0 ml de agua desionizada
(destilada) suavemente revolviendo la mezcla en un beaker de 50 o 100 ml. Enfriar la
mezcla a temperatura ambiente. Colocar 3.0 mL del benzaldehido (toxico/inflamable) (el
goteo deben ser marcados) en un tubo de prueba (o un sistema micro de destilación)
proporcionado y agregar el KOH concentrado que usted acaba de preparar. Tapar el tubo
con corcho por seguridad y agitar el tubo vigorosamente hasta que se forma una emulsión.
Colocar el tubo de prueba parado hasta el próximo período del laboratorio. (o, por lo
menos, día 4). La otra posibilidad es reflujar el sistema a 80 ºC por 1.5 h.
Después del reflujo, agregar cerca de 2.0 mL de agua a la mezcla, tapar y agitar. Si
todo el sólido no se ha disuelto decante la solución en un beaker limpio y agregar más agua.
Repetir la agitación. Si hay sólidos, romperlos con una barra de agitación y agregar el agua
y después agitar la mezcla. Repetir el procedimiento hasta todos o la mayoría del sólido se
halla disuelto. agregar la solución en un embudo de separación, y extraer la solución 2
veces con las porciones de 8 ml del éter dietílico. Combinar los extractos de éter en un
beaker. Si un sólido se separa de la solución cuando se agrega el éter, agregar agua en
cantidades pequeñas hasta que el sólido se redisuelva, y después proceder con la extracción.
Estar seguro de evitar la formación de una emulsión agitando la mezcla suavemente al
principio, y más vigoroso si se observa la persistencia de la emulsión. Se seca la solución
etérea usando Mg2SO4 anhidro. Una alícuota de la solución del éter seca se analiza usando
CROMATOGRAFÍA de GAS para obtener las cantidades relativas de alcohol bencílico a
partir de benzaldehido. El resto de la solución de éter se destila hasta 100 ºC. Comprobar la
cantidad destilada y la fracción restante. Pesar las cantidades.
La solución acuosa restante después de la extracción con éter se vierta en una
mezcla de 8 ml de HCl concentrado (corrosivo), 5 agua y 3 cubos del hielo. Agitar
vigorosamente la mezcla durante la adición. Enfriar la mezcla resultante en un baño del
hielo, y recoger el ácido benzoico sólido por filtración al vacío usando un embudo buchner
de 5,5 cm. Secar el sólido con papel de filtro. Determinar su punto de fusión cuando este

Practica # 5A Obtención de Acido Benzoico y Alcohol Benzilico
76
seco. Después de tomar el punto de fusión guardar del ácido benzoico para la práctica
siguiente.
ESPECTROS DE I.R.
Benzaldehido
BIBLIOGRAFÍA
1. DUPONT DURST H. and GOKEL GEORGE W: Experimental Organic
Chemistry.
2. BREWTER. “Curso practico de Química Orgánica” 2ª Edición. 1970, pp. 19
3. SOLOMONS.T. “Química Orgánica” 2ª Edición. México pp. 940-942.
4. Vogel, A.I., Elementary Practical Organic Chemistry, Part I, Small Scale
Preparation, 2ª. Ed., Longman Londres, Inglaterra, 1970, pp. 263-265.

Practica # 5B Obtención de Acido Benzoico a partir de Acetofenona
77
PRÁCTICA # 5B: OBTENCIÓN DE ACIDO BENZOICO A
PARTIR DE ACETOFENONA
OBJETIVOS
Efectuar la oxidación de una metil-cetona mediante hipoclorito de sodio comercial.
Ilustrar un método de obtención de ácidos carboxílicos.
Preparar ácido benzoico mediante la reacción del haloformo.
Efectuar una aplicación de la reacción del haloformo.
ANTECEDENTES
Reacciones de sustitución electrofílica en carbonos () a grupos carbonilo.
Formación de carbaniones, estabilidad de los mismos.
Oxidación de metil-cetonas.
Reacciones de adición-eliminación.
Métodos de obtención de ácidos carboxílicos.
Reacción de haloformo.
Utilidad de la reacción del haloformo.
Características químicas de los ácidos carboxílicos.
Solubilidad de los ácidos carboxílicos y de sus sales.
REACCIÓN A EFECTUAR Y ESTEQUIMETRIA
CCH3
O
(1)
+ NaOCl2) HCl
CO
O
H
+ HCCl3
(9)
Acetofenona
(1)
Hipoclorito de sodio Acido Benzoico
(9)
PM 120 74 122
Peso (g) 2.06 Variable 2.09
Moles 1.7 x 10-2
Variable 1.7 x 10-2

Practica # 5B Obtención de Acido Benzoico a partir de Acetofenona
78
MECANISMO
La metil-cetona reacciona con halógenos en presencia de bases, realizando
halogenaciones múltiples en el carbono metilo.
La primera reacción se da entre el hipoclorito de sodio y la acetofenona (1)
metilcetona, para que a través de un equilibrio ácido-base se formen el carbanión (2) y el
ácido hipocloroso. El carbanión de la metilcetona esta en resonancia con el enolato
correspondiente, ya que los átomos de hidrógeno en la posición al grupo son ácidos (pKa
≈ 19) debido precisamente a que la base conjugada es estable por dicha resonancia.
CC
O
H
H
HClO
-Na
+
+
CC
-
O
H
H
Na+
CC
O-
H
H
Na+
(1) (2)
El intermediario (2) reacciona a través del carbanión, el cual actúa como nucleófilo
sobre el ácido hipocloroso, que reacciona como electrófilo, para formar el intermediario
(3), que es la -cloroacetofenona, regenerándose el hidróxido de sodio.
CC
-
O
H
H
Na+ + Cl O H
CC
O
H
H
Cl + Na+
OH-
(2) (3)
La siguiente reacción se da entre el hipoclorito de sodio y la -cloroacetofenona (3),
para que a través de un equilibrio ácido-base se forme el carbanión (4), el cual se encuentra
estabilizado por resonancia formándose un enolato de sodio y el ácido hipocloroso. Como
el átomo de cloro es muy electronegativo (EN = 3, escala de Pauling), la acidez de los
hidrógenos en posición a al grupo carbonilo en la -cloroacetofenona (3) son más ácidos
que los de la propia acetofenona (1), favoreciéndose esta reacción ácido-base.
CC
O
Cl
H
H
(3)
Na++ ClO
-
CC
-
O
Cl
HC
C
O-
Cl
H
(4)
Na+
Na+
ClOH

Practica # 5B Obtención de Acido Benzoico a partir de Acetofenona
79
El intermediario (4) reacciona a través del carbanión, el cual actúa como nucleófilo
sobre el ácido hipocloroso, que reacciona como electrófilo, para formar el intermediario
(5), la -dicloroacetofenona, y regenerándose el hidróxido de sodio.
CC
-
O
Cl
H
Na+
(4)
+ Cl O H
CC
O
Cl
H
Cl
(5)
+ Na+
OH-
Como en el intermediario (5) todavía hay un átomo de hidrógeno en la posición al
grupo carbonilo, se vuelve a dar otra reacción ácido-base entre la -dicloroacetofenona
(5) y el hipoclorito de sodio, para que se forme el carbanión (6), el cual se encuentra en
resonancia con el enolato correspondiente y con el ácido hipocloroso. No obstante el
impedimento estérico que presenta la -dicloroacetofenona, la presencia de los dos
átomos de cloro, muy electronegativos (EN = 3, escala de Pauling), la acidez de los
hidrógenos en posición al grupo carbonilo del intermediario (5) son más ácidos que los
de la -dicloroacetofenona y los de la acetofenona (1), favoreciendo esta reacción ácido-
base.
CC
O
Cl
Cl
H
(5)
Na++ ClO
-
CC
-
O
Cl
Cl
Na+
CC
O-
Cl
Cl
(6)
+ ClOH
Y nuevamente el intermediario (6) reacciona a través del carbanión, el cual actúa
como nucleófilo sobre el ácido hipocloroso, que reacciona como electrófilo, para formar el
intermediario (7), que es la -tricloroacetofenona, regenerándose el hidróxido de sodio.
CC
-
O
Cl
Cl
Na+
Cl O H+
(6)
CC
O
Cl
Cl
Cl
(7)
+ Na+
OH-

Practica # 5B Obtención de Acido Benzoico a partir de Acetofenona
80
Como dentro de la estructura del intermediario (7) ya no hay más protones en la
posición al grupo carbonilo, pero hay base presente en el medio de acuerdo a las
reacciones analizadas hasta el momento, el anión hidróxido actúa ahora como un
nucleófilo, y se adiciona sobre el grupo carbonilo de la a,a,a-tricloroacetofenona (7), el cual
actúa como un electrófilo, para formar el intermediario (8). Dentro de la estructura del
intermediario (8), el átomo de carbono del grupo carbonilo cambio de una hibridación sp2
con ángulo de valencia 120º a una sp3 con ángulo de valencia de 109º37’. Para regenerar la
hibridación original del grupo carbonilo en la que se minimiza el impedimento estérico,
ocurre una reacción de eliminación, rompiéndose el enlace C-C, y saliendo el carbanión
(10), el cual es muy estable por la presencia de los tres átomos de cloro. Al quedar el átomo
de carbono deficiente de electrones, el átomo de oxígeno aporta sus electrones para
regenerar el grupo carbonilo con su hibridación sp2, dando lugar al ácido benzoico (9). En
una ultima reacción ácido-base, reaccionan el ácido benzoico (9) y el carbanión (10), para
formar el benzoato de sodio (11) y el cloroformo (12).
CC
O
Cl
Cl
Cl
(7)
+ Na+
OH-
CC
O-
Cl
Cl
Cl
O
H
Na+
(8)
C
O
OH
+ Na+
C-
Cl
Cl
Cl
(9)
CH
Cl
Cl
Cl
+
C
O
O-
(10)
Na+
Al tratar la mezcla de reacción con ácido clorhídrico, el benzoato de sodio (10),
soluble en agua, da lugar al producto final, el ácido benzoico (9), insoluble en la misma y a
cloruro de sodio.

Practica # 5B Obtención de Acido Benzoico a partir de Acetofenona
81
C
O
O-
(10)
Na+
+ ClHOH2
C
O
OH
Na+
+ Cl-
(9)
PARTE EXPERIMENTAL
MATERIAL
Matraz Erlenmeyer de 250 mL con
tapón
1 Embudo de vidrio, tallo corto 1
Agitador de vidrio 1 Recipiente de peltre 1
Anillo metálica 1 Baños de agua eléctrica 1
Tela de alambre con asbesto 1 Baño de temperatura constante 1
Mechero con manguera 1 Matraz de boca esmerilada 125 mL 1
Embudo Büchner con empaque de hule 1 Refrigerante de agua con
mangueras
1
Matraz Kitasato con manguera 1 Colector 1
Probeta de 25 mL 1 T de destilación 1
Vaso de precipitado de 250 mL 1 Tapón esmerilado 1
Espátula 1 Pinzas de tres dedos con nuez 1
SUSTANCIAS
Cantidad Calidad
Solución de hipoclorito de sodio comercial 80.0 mL Cloros
Acetofenona 2.0 mL R.A.
Acetona 1.0 mL R.A.
Acido clorhídrico (1:1) 200 mL Q.P.
Papel pH
PROCEDIMIENTO
Coloque 80 mL de hipoclorito de sodio comercial (solución al 6%) en un matraz
Erlenmeyer, agregue 2 mL de acetofenona y agite vigorosamente durante 50 min.
Pasado este tiempo coloque el matraz en un baño de agua caliente y destile el
cloroformo producido. Agregue 1 ml de acetona o bisulfito de sodio para eliminar el
hipoclorito de sodio que no haya reaccionado y vuelva a colocar el matraz en el baño de
agua otros 10 minutos.
De ser necesario, decolore la solución con carbón activado y filtre.

Practica # 5B Obtención de Acido Benzoico a partir de Acetofenona
82
Acidule la solución en caliente hasta pH = 1 con ácido clorhídrico (1:1), debida a
que la acetofenona que no ha reaccionado es insoluble en agua, se separa en forma de
aceite, el cual se puede eliminar por decantación.
Enfrié a temperatura ambiente y coloque en baño de hielo hasta completar la
cristalización. Filtre el sólido obtenido y purifique el producto por recristalización de agua
caliente, si queda aún acetofenona decante nuevamente antes de enfriar la solución. Calcule
el rendimiento y determine el punto de fusión.
DIAGRAMA ECOLÓGICO
D1: Filtre los sólidos presentes. Neutralice la solución y elimine por el drenaje.
Precausiones: acetofenona LD50 = 810 mg/kg: tóxico e irritante por inhalación,
ingestión o adsorción en la piel
Acido benzoico LD50 = 2538 mg/kg: produce por ingestión problemas
gastrointestinales y alergías.

Practica # 5B Obtención de Acido Benzoico a partir de Acetofenona
83
Disposición (ambos): disuelva en un disolvente inflamable e incinere en la forma
correcta.
Productos de descomposición: monóxido y bióxido de carbono.
CUESTIONARIO
1. Explique qué tipo de reacciones se llevan a cabo.
2. Escriba el mecanismo de la reacción efectuada.
3. ¿Para qué utiliza la acetona o el bisulfito de sodio en la obtención del ácido benzoico?
4. ¿Cómo se obtiene el ácido benzoico a nivel industrial?
5. ¿Qué tratamiento deberá darle a los efluentes líquidos antes de desecharlos en el
drenaje?
ESPECTROS DE I.R.
a) Acetofenona
b) Acido benzoico

Practica # 5B Obtención de Acido Benzoico a partir de Acetofenona
84
BIBLIOGRAFÍA
1. Allinger, N.L., et al., Organic Chemistry, Worth Pub., NUeva York, EU, 1973.
2. March, J., Advanced Organic Chemistry, Wiley Interciencie, 3a. ed., Nueva York, EU,
1985, pp. 567, 788, 789.
3. Robertson, G. Ross, Jacobs, T.L., Truce, W.E., Laboratory Practice of Organic
Chemistry, 5a. ed., McMillan, Nueva York, Eu, 1974, pp. 325.
4. Solomons, T.W.G., Química Orgánica, Limus, México, 1982, pp. 759-762.
5. Streitwieser, A. y Heathcoock, C.H., Introduction to Organic Chemistry, Collier
McMillan Internacional, Eu, 1976, pp. 367.

Practica # 6 Síntesis del Benzoato de Metilo
85
PRÁCTICA # 6: SISTESIS DEL BENZOATO DE METILO
OBJETIVOS
Objetivo general:
Obtención de un éster (benzoato de metilo) a partir de un ácido (ácido benzoico) y
de un alcohol (alcohol metílico) según el método de esterificación de Fischer.
Objetivos Específicos:
Obtener Benzoato de metilo a partir del ácido Benzoico y metanol anhidro.
Separación y purificación del producto por medio de destilación a presión
atmosférica.
Determinar el porcentaje de rendimiento del producto obtenido.
Medir el índice de refracción al producto.
Realizar prueba especificas para identificación de esteres.
ANTECEDENTES
Reacciones de adición nucleofílica sobre el grupo carbonilo.
Reacciones de adición-eliminación en grupos carbonilo.
¿Qué son los reactivos nucleofílicos? Orden de nucleofilicidad.
¿Qué son los grupos salientes?, ¿Cuáles son los más usados?, ¿Cuál es el
orden de facilidad de salida de los mismos?.
Catálisis ácida en las reacciones de adición nucleofílica.
Reacciones en equilibrio; cómo desplazar un equilibrio.
Principales derivados de los ácidos carboxílicos.
¿Qué derivados de los ácidos carboxílicos se pueden transformar en otros
derivados?, ¿Cómo se llevan a cabo dichas transformaciones?
¿Cómo se puede eliminar el agua en una reacción de equilibrio?
Diferentes rutas sintéticas para obtener los derivados de los ácidos
carboxílicos.

Practica # 6 Síntesis del Benzoato de Metilo
86
REACCIÓN A EFECTUAR Y ESTEQUIMETRIA
OH
O
+ CH3 OHH2SO4
O
O
CH3
+ H2O
Acido benzoico Metanol Acido sulfúrico Benzoato de metilo
PM 122 32 98 136
Peso (g) 3 5.51 1.84 3.346
Moles 2.46 x 10-2
17.22 x 10-2
1.88 x 10-2
2.46 x 10-2
MECANISMO
Como la diferencia de electrones en el átomo de carbono del grupo carbonilo del
ácido (1) no es muy alta, o sea su electrofilia es baja, en consecuencia su reactividad es
baja. Por esta razón se emplea un catalizador ácido, para incrementar la electrofilia de dicho
grupo funcional. Dado que el ácido mineral utilizado (ácido sulfúrico) es más fuerte que el
ácido (1), se lleva a cabo una reacción de ácido-base, para formar el intermediario (2) y el
anión bisulfito. Como se puede ver, el intermediario (2) implica la formación de un grupo
carbonilo protonado, el cual se encuentra en resonancia con un carbocatión,
incrementándose así su electrofilia.
+O
O
H
:
:S OO
O
O
H HO
O+
H
HC
+
O
O
H
H
+ S OO-
O
O
H
(1) (2)
Así, una vez que se forma el carbocatión (2), el cual puede actuar como un
electrófilo, éste reacciona con un nucleófilo débil como el metanol, para formar un enlace
C-O y dar lugar al intermediario (3). A través de un equilibrio ácido-base se puede protonar
cualquiera de los dos grupos hidroxilos presentes en la estructura del intermediario (3), para
formar así el intermediario (4), el cual tiene dentro de su estructura agua como grupo
saliente.

Practica # 6 Síntesis del Benzoato de Metilo
87
C+
O
O
H
H
(2)
CH3OH+S OO
-
O
O
H
S OO
O
O
H H
O
O
H
H
HO
+
CH3
(3)
O
O+
H
H
O
CH3
H
(4)
+ S OO-
O
O
HS OO
O
O
H H +
Dentro del intermediario (4), cuando se rompe heterolíticamente el enlace C-O, sale
agua como grupo saliente y se forma un carbocatión el cual es estabilizado por el grupo
hidroxilo, para formar así el intermediario (5), el que es en realidad el benzoato de metilo
protonado. El éster protonado (5) se encuentra en un equilibrio ácido-base con el agua, para
formar el producto final, el benzoato de metilo (6).
O
O+
H
H
O
CH3
H
(4)
O
O+
CH3
H
O
H
H+ O
O
CH3O
+H
H
H
+
(5) (6)
Como se puede observar en el mecanismo de reacción para explicar la formación
del éster, todas las reacciones son reversibles y se establece un equilibrio, que puede
desplazarse hacia el ácido y el alcohol, en este caso las dos materias primas, lo cual sería en
realidad la reacción de hidrólisis ácida de los ésteres; o bien desplazarse hacia la formación
del éster (6).
Una manera de evitar el equilibrio o, más bien, de desplazarlo es eliminar el agua
del medio de reacción (formando un azeótropo o bien por el uso de mallas moleculares).
Otra estrategia es la de desplazar el equilibrio hacia la derecha poniendo el alcohol en
exceso, como es el caso del experimento que aquí se va a efectuar.

Practica # 6 Síntesis del Benzoato de Metilo
88
PARTE EXPERIMENTAL
MATERIAL
Matraz de fondo redondo de 50 mL 1 Anillo de hierro 1
Refrigerante para agua con mangueras 1 Probeta de 25 Ml 1
T de destilación 1 Pipeta de 10 mL 1
Colector 3 Vasos de precipitados 150 mL 2
Pinzas de tres dedos con nuez 1 Espátula 1
Embudo de tallo corto 1 Matraz Erlenmeyer de 50 mL 1
Agitador de vidrio 1 Recipiente de peltre 1
Baño con resistencia electrico 1 Embudo de separación con tapón 1
Vidrio de reloj 1 Termómetro 1
SUSTANCIAS
Cantidad Calidad
Acido benzoico 3.0 g Q.P.
Metanol absoluto 7.0 mL Q.P.
Acido sulfúrico (H2SO4) 1.0 mL Q.P.
Eter etílico 30.0 mL Q.P.
Solución de bicarbonato de sodio al 10% 30.0 mL Técnico
Sulfato de sodio anhidro 10.0 g Q.P.
PROCEDIMIENTO
Coloque en el matraz esmerilado
3 g de ácido benzoico y 7 mL de
metanol. Enfríe la mezcla sobre hielo, y
adicione lenta y cuidadosamente, por las
paredes del matraz, un mL de ácido
sulfúrico (H2SO4) y agite. Adicione
piedras de ebullición, coloque el
refrigerante de agua en posición de
reflujo y caliente hasta tener un reflujo
vigoroso, manteniendo éste durante una
hora (Ver figura).
Pasado este tiempo, enfríe la
solución y verifique que el pH sea ácido,
lave nuevamente con agua hasta pH
neutro, y decante a un embudo de
separación que contenga 10 mL de agua,
verifique que la llave del embudo selle bien y
que la temperatura de la mezcla de reacción
sea baja (+ 10 °C), ya que, al entrar en
contacto con el agua, la mezcla se puede
calentar.
Extraiga dos veces la mezcla de
reacción con 10 mL de éter etílico cada una.
Agite, separe las fases y elimine la fase
acuosa. Lave la fase etérea con 5 mL de agua
y posteriormente con 5 mL de solución de
bicarbonato de sodio al 10 %. Deseche los
lavados y trasvase la fase orgánica a un
Erlenmeyer de 50 mL.

Practica # 6 Síntesis del Benzoato de Metilo
89
1
2
3
45 6
7
8
9
1 10
2
3
45 6
7
8
9
11
Séquela con sulfato de sodio
anhidro, deje reposar de 3 a 5 minutos y
decántela a un matraz esmerilado seco.
Lave el sulfato de sodio con 5 mL más
de éter y reúnalo con la fase orgánica
anterior.
Destile el disolvente a baño
María, y cuando éste haya sido
totalmente eliminado detenga la destilación
8el punto de ebullición del benzoato de metilo
es de 199 °C).
Enfríe el sistema y observe el producto
obtenido. Si no es completamente
transparente, adicione más sulfato de sodio
anhidro hasta que seque, deje reposar 3 a 5
minutos y decante a una probeta.
1
2
3
45 6
7
8
9
1 10
2
3
45 6
7
8
9
11
Mida el volumen del benzoato de
metilo sintetizado. Calcule rendimiento. Si se
requiere, el producto obtenido se puede
purificar por destilación al vacío.
PRECAUCIONES ESPECIALES.
Añadir el H2SO4 al disolvente y no al
revés.
Una vez agregado el ácido sulfúrico
homogeneizar bien la mezcla para evitar que
el ácido queme los reactivos por sobre
concentración local.

Practica # 6 Síntesis del Benzoato de Metilo
90
Manejar el H2SO4 con guantes y
en vitrina.
Tras lavar con NaOH comprobar
que la fase acuosa es neutra o básica, lo
que indica que no quedan restos de
ácido.
Añadir al matraz plato poroso
tanto al iniciar el reflujo como en la
destilación.
Al destilar no dejar que se seque
el matraz, porque podría romperse.
1
2
3
45 6
7
8
9
1 10
2
3
45 6
7
8
9
11

Practica # 6 Síntesis del Benzoato de Metilo
91
DIAGRAMA ECOLÓGICO

Practica # 6 Síntesis del Benzoato de Metilo
92
D1, D2, D3: pueden contener materia prima o producto, en este caso se recupera el
producto. Absorba la solución con carbón activado y confínelo para incineración.
Neutralice la solución deseche.
CUESTIONARIO
1. ¿Podria utilizarse ácido sulfúrico diluido?
2. Si se desea eliminar agua de la reacción para desplazar el equilibrio hacia el éster,
¿Cómo modificaria la tecnica utilizada?
3. ¿Daria buenos resultados usar ácido benzoico en exceso en vez de metanol?
4. ¿Para que extrae la fase etérea con solución de bicarbonato de sodio?
5. ¿Por qué luego de extraer con bicarbonato de sodio se debe lavar con agua hasta pH
neutro?
6. ¿Por qué se debe secar muy bien el benzoato de metilo antes de destilarlo?
7. ¿Como se deben tratar los desechos de D1 a D3?
ESPECTROS DE I.R.
a) Acido Benzoico

Practica # 6 Síntesis del Benzoato de Metilo
93
b) Metanol
c) Benzoato de metilo

Practica # 6 Síntesis del Benzoato de Metilo
94
BIBLIOGRAFÍA
1. Kremlin, R.J.W. y Still, R.H., Named and Miscellaneous Reactions in Practical Organic
Chemistry, 2ª. Ed., Heinemann Educational Book, Londres, Inglaterra, 1967.
2. Giral y Rojahn, Productos Quimicos y Farmaceuticos, Vol. II., Atlante Mexico, 1966,
pp. 112-113.
3. March, J. Advanced Organic Chemistry Reactions, Mechanisms and Structure, 4ª. Ed.,
John Wileey and Sons, Nueva York, EU, 1992, pp. 401-402, 300-393, 418-424.
4. Morrison, Robert T. y Boyd, Robert N., Qu÷imica Org÷anica, 5a. ed., Addison=Wesley
Interamericana, Wilmington, Delawere, EU. 1990, pp. 842-870.
5. Pavia, Donald L., Lampman, Gary M., y Kriz, George S., Introduction to Organic
Laboratory Tecniques, a Contemporary Approach, Saunders College Publishing,
Philadelphia, EU. 1988, pp. 33-44.
6. BREWTER. “Curso practico de Química Orgánica” 2ª Edición. 1970, pp. 19
7. SOLOMONS.T. “Química Orgánica” 2ª Edición. México pp. 940-942.
8. Vogel, A.I., Elementary Practical Organic Chemistry, Part I, Small Scale Preparation,
2ª. Ed., Longman Londres, Inglaterra, 1970, pp. 263-265.

Practica # 7 Obtención de una esencia natural
95
PRÁCTICA # 7: OBTENCIÓN DE UNA ESENCIA NATURAL
OBJETIVOS
Objetivo general:
Aislamiento de un producto natural
Destilación por arrastre con vapor
Formación de derivado
ANTECEDENTES
Destilación por arrastre con vapor.
Terpenos, biosíntesis, características, clasificación, nomenclatura, aislamiento y
purificación.
Fenilpropanos biosíntesis, características, clasificación, nomenclatura, aislamiento y
purificación.
Obtención de esencias naturales y preparación de sus derivados.
INTRODUCCIÓN
Cualquier persona que ha recorrido a través de un bosque del pino o de cedro, o
cualquier persona que ama las flores y especies, sabe que muchas plantas y árboles tienen
distintamente olores agradables. Las esencias o los aromas de plantas son debido a los
aceites volátiles o esenciales, muchos de los cuales se han valorado desde la antigüedad por
sus olores característicos (Ejemplo, frankincense y mirra). Una lista de los aceites
esenciales comercialmente más importantes se recoge en unos 200. La pimienta inglesa, la
almendra, el anís, la albahaca, la bahía, la alcaravea, la canela, el clavo, el comino, el
eneldo, el eucalipto, el ajo, el jazmín, el enebro, la naranja, la hierbabuena, la rosa, el la
sasafrás, el sándalo, la menta verde, el tomillo, la violeta, y wintergreen son algunos
ejemplos de familias de tales aceites esenciales valiosos. Los aceites esenciales se utilizan
principalmente por sus olores y sabores agradables en perfumes, incienso, olores, especia, y
como sustancias aromáticas en alimentos. Algunos también se valoran por su acción anti-
bacteriana y antifungicida. Algunos se utilizan en medicamentos (ejmp., alcanfor y
eucalipto) y otros como repelentes de insecto (citronela). El aceite de Chaulmoogra
representa uno de los pocos agentes curativos sabidos para la lepra. La trementina se utiliza
como un solvente para muchos productos de pintura.

Practica # 7 Obtención de una esencia natural
96
Los componentes del aceite esencial se encuentran a menudo en las glándulas o los
espacios intercelulares en el tejido fino de las plantas. Pueden encontrarse en todas las
partes de la planta, pero se concentran a menudo en las hojas o las flores. Muchos de los
componentes de aceites esenciales son vapores volátiles y se pueden aislar con la
destilación por arrastre con vapor. Otros métodos de aislar los aceites esenciales incluyen la
extracción con solvente y métodos de presión (Prensado). Como es bien sabido los ésteres
son con frecuencia responsables de olores y de sabores característicos de frutas y de flores.
Además de los ésteres, los aceites esenciales se pueden componer de mezclas complejas de
hidrocarburos, de alcoholes, y de compuestos carbonilos. Estos componentes pertenecen
generalmente a uno de los dos grupos de productos naturales llamados los terpenos o los
fenilpropanoides.
Los principales componentes de los aceites esenciales del clavo, de la canela, del
comino, y de la pimienta inglesa son aromáticos y volátiles con vapor. En este experimento
aislaremos el componente principal del aceite esencial derivado de estas especias por la
destilación de vapor. En la opción de su instructor, la conversión de estos aceites en un
derivado sólido puede también ser requerida.
Un derivado es un compuesto nuevo a el cual el compuesto original d es convertido
fácilmente por una reacción simple. El compuesto original puede ser difícil de purificar o
difícil de caracterizar exactamente por sus características físicas puesto que es un líquido o
un aceite. Un derivado es un compuesto cristalino sólido del punto de fusión definido. El
derivado es fácil de caracterizar o de identificar. Su identidad, una vez que esté establecida,
sirve pues de evidencia circunstancial para ayudar a identificar el material del cual fue
obtenido.
La técnica de la destilación por arrastre con vapor permite la separación de
componentes volátiles del material permanente sin la necesidad de elevar la temperatura de
la destilación sobre 100º. La destilación por arrastre con vapor proporciona los medios del
aislamiento de los aceites esenciales sin el riesgo de descomponerlos térmicamente.
PARTE EXPERIMENTAL
MATERIAL
Matraz de fondo redondo de 50 mL 1 Anillo de hierro 1
Refrigerante para agua con mangueras 1 Probeta de 25 Ml 1
T de destilación 1 Pipeta de 10 mL 1
Colector 3 Vasos de precipitados 150 mL 2
Pinzas de tres dedos con nuez 1 Espátula 1
Embudo de tallo corto 1 Matraz Erlenmeyer de 50 mL 1
Agitador de vidrio 1 Recipiente de peltre 1

Practica # 7 Obtención de una esencia natural
97
Baño con resistencia electrico 1 Embudo de separación con tapón 1
Vidrio de reloj 1 Termómetro 1
SUSTANCIAS
Cantidad Calidad
Materia Prima 200 g
Agua 500 ml Destilada
Cloruro de Metileno 30 ml R:A
PROCEDIMIENTO
Aceites de clavo o de la pimienta inglesa
El aceite del clavo (del caryophyllata de Eugenia) y el aceite de la pimienta inglesa
(de Pimenta Officianalis) son ricos en el eugenol (4-allyl-2-methoxyphenol). La
Caryophyllene está presente en cantidades pequeñas junto con otros terpenos. El eugenol
(punto de ebullición 250º) es un fenol o un compuesto aromático del oxidrilo.
En este experimento le pedirán aislar el eugenol y caracterizarlo por medio de la
espectroscopia infrarroja. También tendrá que obtener su derivado por medio de la reacción
con el cloruro del benzoilo.
Ensamblar un aparato para destilación con arrastre por vapor usando un balón de
fondo redondo de 500 ml con tres bocas. El recipiente de recolección puede ser una fiola de
125 ml. La fuente de calor será una llama, usando un mechero Bunsen. Coloque 15 g de la
pimienta inglesa o 10 g del clavo de olor en el balón y agrega 150 ml de agua. Comenzar a
calentar lentamente el líquido en el balón, para asegurar una destilación constante. Durante
la destilación continuar agregando agua del embudo de adición (de separación) en una
velocidad que apenas mantenga el nivel original del líquido en el balón de destilación.
Continuar la destilación por arrastre con vapor hasta que cerca de 100 ml de destilación se
han recogido.
Vaciar el agua del embudo de adición y colocar el destilado en él. Extraer el
destilado con dos porciones de 10 ml del cloruro de metileno. Separar las capas y desechar
la fase acuosa. Si la capa del cloruro de metileno se separa cuidadosamente, no necesitará
ser secada, aunque puede secarse con una cantidad pequeña de sulfato de sodio anhidro.
Decantar la solución de cualquier elemento deshidratador y evaporar la mayoría del
solvente en un baño de vapor en la campana. Transferir el líquido restante a un tubo de
prueba previamente pesado. Concentrar el contenido del tubo de prueba por calentamiento
teniendo cuidado en un baño de vapor hasta que se tenga un residuo aceitoso. Secar el
exterior del tubo de prueba y pesarlo. Calcular el porcentaje en peso recuperado del aceite
basado en la cantidad original de pimienta inglesa o de clavos usados.

Practica # 7 Obtención de una esencia natural
98
Determinar el espectro infrarrojo. El informes deben incluir los espectros que fueron
registrados. Las absorciones principales en los espectros deben ser interpretadas.
Preparación de un derivado
Coloque cerca de 0.2 ml del eugenol en un tubo de prueba pequeño y agrega 1 ml de
agua. Agregar unas gotas de una solución 1 M de hidróxido del sodio al tubo de prueba
hasta que el aceite apenas se disuelva. La solución final puede estar nublada, pero no debe
haber gotitas grandes del aceite evidentes. Agregar cuidadosamente 0.1 ml (4 o 5 gotas) de
cloruro del benzoilo. Un exceso del cloruro del benzoilo debe ser evitado puesto que hará la
cristalización del producto final imposible de alcanzar. Calentar la mezcla en el baño de
vapor por 5 o 10 minutos. Enfriar la mezcla, y raspar el interior del tubo de prueba con una
barrilla de vidrio hasta que solidifica la mezcla. Si no solidifica, enfriar la mezcla en un
baño del hielo. Si todavía no solidifica el aceite, decantar la capa acuosa, agregar algunas
gotas del metanol, y continuar raspando las paredes del tubo y enfriando el aceite. Recoger
el sólido en un embudo de Hirsch y lavarlo cuidadosamente con un volumen pequeño de
agua fría. Recristalizar el sólido en una mínima cantidad de metanol a ebullición. Si un
aceite se separa de la solución, recalentar la solución y enfriarla lentamente mientras que se
raspan la sparedes del tubo y sembrar algunos cristales. Recoger los cristales en un embudo
de Hirsch. Secar los cristales y medir su punto de fusión. El punto de fusión del benzoato
del eugenol es 70º. Entregar los cristales en un frasco etiquetado al instructor.
Aceite de cinamomo
El componente principal del aceite de canela (cinnamomum zeylanicum) es el
cinamaldehido (trans-3-phenylpropenal). El cinnamaldehyde tiene una punta que hierve de
252º.
En este experimento le pedirán aislar el cinnamaldehyde y caracterizarlo por medio
de la espectroscopia de la resonancia magnética infrarroja y nuclear. Usted puede también
ser pedido convertir el cinnamaldehyde a su derivado del semicarbazone por la reacción
con clorhidrato del semicarbazide.
Ensamblar un aparato para la destilación de vapor usando un flash del fondo del
roaund del cuello del threee de 500 ml. El frasco de la colección puede ser un frasco de
125 ml erlenmeyer. El lugar 15 g de la corteza de cinamomo de tierra en el frasco y agrega
100 ml de la agua caliente. Admitir el vapor de la línea del vapor para proporcionar a un
índice lento, pero constante de la destilación. Continuar la destilación de vapor hasta que
cerca de 100 ml de destilado se han recogido.
Colocar el destilado en un embudo y un ectract separatory él con dos porciones de
10 ml del cloruro de metileno. Separar las capas y desechar la fase acuosa. Si la capa del

Practica # 7 Obtención de una esencia natural
99
cloruro de metileno se separa cuidadosamente, no necesitará ser secada, aunque el secarse
con una cantidad pequeña de sulfato de sodio anhidro puede ser recomendable. Decantar la
solución de cualquier elemento deshidratador y evaporar la mayoría del solvente en un
baño de vapor en la capilla. Transferir el líquido restante a un tubo de prueba previamente
pesado. Concentrar el contenido del tubo por la calefacción cuidadosa adicional en un baño
de vapor hasta que sigue habiendo nada pero un residuo aceitoso. Secar el exterior del tubo
de prueba y pesarlo. Calcular la recuperación de los por ciento del peso del basado en la
cantidad original de corteza de cinamomo usada.
Determinar el espectro infrarrojo. El informe debe incluir los espectros fue
registrado, y una interpretación del pico principal en los espectros.
Preparación del derivado
Disolver 0.20 g del clorhidrato del semicarbazide y 0.30 g del acetato anhidro del
sodio, que servirá como almacenador intermediaro, en 2 ml de agua. A esta mezcla agregar
3 ml de etanol absoluto. Agregar esta solución a la gasolina de canela y calentar la mezcla
en el baño de vapor por cinco minutos. Refrescar la mezcla y permitir que el semicarbazone
del cinnamaldehyde se cristalice. Recoger los cristales en un embudo de Hirsch y
recristalizarlos del metanol. Permitir que los cristales sequen y determinen su punto de
fusión. El punto de fusión del semicarbazone del cinnamaldehyde es 215º. Submite el
derivado en un frasco etiquetado al instructor.

Practica # 7 Obtención de una esencia natural
100

Practica # 8ª: Reacción de Grignard. Sintesis del Acido Valerico o del 4-metil-3-heptanol
81
PRÁCTICA # 8A: REACCION DE GRIGNARD. SINTESIS
DEL ACIDO VALERICO O SINTESIS DEL 4-METIL-3-
HEPTANOL
OBJETIVOS
Ilustrar en el laboratorio la reacción de Grignard como un ejemplo de la reacción
de adición nucleofílica a compuestos carbonílicos.
Carbonatación de un reactivo de Grignard para la obtención de un acido
Realizar procedimiento típico de adición de carbaniones a aldehídos en medio
acido, para obtención de alcoholes secundarios.
ANTECEDENTES
Adición nucleofílica, mecanismo general.
Reacción de Grignard. Propiedades de los compuestos organometálicos y
adición a cetonas.
Obtención de ácidos carboxílicos. Propiedades y normas de seguridad en el
manejo de todas y cada una de las sustancias involucradas (reactivos y
productos).
Obtención de alcoholes. Propiedades y normas de seguridad en el manejo de
todas y cada una de las sustancias involucradas (reactivos y productos).
Análisis, reacciones y fundamento químico de la técnica.
REACCIONES A EFECTUAR Y ESTEQUIMETRIA
Síntesis de ácido valerico
CH3
CH2
CH2
CH2
Br+ Mg
CH3
CH2
CH2
CH2
MgBrCO2
H3O+
CH3
CH2
CH2
CH2
COOH
1)
2)

Practica # 8ª: Reacción de Grignard. Sintesis del Acido Valerico o del 4-metil-3-heptanol
82
Bromobutano Magnesio Dióxido de Carbono Acido Valerico
PM 137,018 g/mol 24,305 g/mol 44 g/mol 102,132 g/mol
Peso (g) 14,04 g 2,4 g 100 g Realizar Calculo
Moles 0,1025 mol 0,1 mol 2,2727 mol Realizar Calculo
Sintesis del 4-metil-3-heptanol
1)
2)CH3
CH2
CH2
CH
CH3
Br
+ Mgeter
CH3
CH2
CH2
CH
CH3
MgBr
H3O+
CH3 CH2 CHO
CH3
CH2
CH2
CH
CH3
CH
CH2CH3
OH
2-bromopentano Magnesio Propanal 4-metil-3-heptanol
PM 151,054 g/mol 24,305 g/mol 58 g/mol 130,228 g/mol
Peso (g) 28,06 g 7,3 g 11,6 g Realizar Calculo
Moles 0,1858 mol 0,3042 mol 0,2 mol Realizar Calculo
MECANISMO
Actividad para los estudiantes. “Realizar el mecanismo”
PARTE EXPERIMENTAL
MATERIAL
Agitador 1 Espátula 1
Probeta graduada 25 ml 1 Recipiente para baño 1
Embudo Búchner 1 Vaso de precipitado 150 ml 2
Matraz Kitasato de 250 ml con
manguera
1 Refrigerante para agua (Quickfit) con
manguera
1
Termómetro -10 a 400 °C 1 Baño para calentamiento 1
Embudo de filtración rápida 1 Pinzas de tres dedos con nuez 2
Matraz pera de 2 bocas 1 Matraces Erlenmeyer 125 ml 2
Colector 1 Pipeta de 5 ml (graduada) 1
Embudo de separación Quickfit 1 Vaso de precipitado de 400 ml 1
T de destilación Quickfit 1 Tapón esmerilado (Quickfit) 1
Trampa de humedad (Quickfit) 2

Practica # 8ª: Reacción de Grignard. Sintesis del Acido Valerico o del 4-metil-3-heptanol
83
SUSTANCIAS
Cantidad Calidad
Bromobutano 11,0 mL Q.P.
Magnesio en virutas 2,4 g Q.P.
Eter 100 mL Q.P.
Dioxido de Carbono
(Hielo sco)
100 g
2-bronopentano 23,0 mL Q.P.
Magnesio en virutas 7,3 g Q.P.
Propanal 11,6 g Q.P.
Eter 100 mL
PROCEDIMIENTO
Acido Valerico
Montar un aparato que consiste en un balón de destilación de fondo redondo de
250 ml, una cabeza claisen, un embudo de adición con tapón, y un condensador de reflujo
con el tubo de secado en el tope.
Pesar 2.4 g (0.100 mol) de cinta de magnesio y colocarlo en el balón de fondo
redondo de 250 ml. Flamear del aparato. Poner un tubo de secado encima del condensador
como el aparato de enfriamiento y conectar el agua al condensador de reflujo. Agregar 100
ml de éter para cubrir el sólido y agregar el n-butíl bromuro (11 ml, 0.1 mol) en el embudo
de adición. Adicionar aproximadamente 1 ml del haluro. La reacción comenzará
inmediatamente. La adición del n-butil bromuro se debe realizar de tal forma que el reflujo
sea uniforme y se mantenga apacible. Después de que se haya agregado todo el haluro,
mantener el reflujo de la mezcla de reacción por 10 minutos (baño de vapor) para convertir
todo el magnesio en n – butilmagnesio bromuro.
Enfriar la mezcla de reacción a temperatura ambiente. Desmontar el aparato y
verter la solución en un beaker de 600 ml que contenga el hielo seco machacado 100 g.
Revolver vigoroso la mezcla con una espátula o una barra de cristal pesada de modo que
haya buen contacto entre el hielo seco y la solución del éter. Cuando la adición sea
completa, permitir que el hielo seco sublime. Hidrolizar la pasta restante lentamente
agregando 100 ml de ácido sulfúrico acuoso al 10% frío. Una vez más revolver vigoroso
con una espátula o una barra de cristal pesada para asegurar un buen mezclado y terminar la
hidrólisis. Transferir la mezcla que resulta a un embudo de separación y lavar cualquier
material restante en el beaker, usando una cantidad pequeña de éter en caso de ser
necesario. Tapar el embudo de separación, agitar, y después separar la capa acuosa. Retirar
la capa del éter y volver a colocar la capa acuosa en el embudo de separación. Extraer la

Practica # 8ª: Reacción de Grignard. Sintesis del Acido Valerico o del 4-metil-3-heptanol
84
fase acuosa tres veces con porciones de 50 ml de éter. Combinar todas las capas de éter y
extraerlas (lavarlas) con 50 ml de agua destilada fría. Extraer cuidadosamente la capa de
éter con tres porciones de 25-ml de hidróxido de sodio acuoso al 10%. Agregar la primera
porción de hidróxido del sodio al embudo y agitar suavemente. Sostener el embudo con la
una mano y sostener el tapón y la otra mano que controla la llave de paso. Se calentara un
poco la solución y esto puede ocasionar que un poco de éter se vaporizarse. Dar vuelta al
embudo para colocar su fondo hacia arriba y lejos de usted y abrir la llave de paso para
librar con seguridad la presión. Repetir este procedimiento dos veces más; después
combinar todas las soluciones acuosas y comprobar el pH. (debe ser fuertemente básico.)
Lavar la solución con dos porciones 25 ml de éter y después acidificar
cuidadosamente con HCl hasta que el papel de tornasol azul se torne inmediatamente rojo
brillante o el papel del hydrion demuestra un pH menos de 1. Permitir que la solución se
refresque a la temperatura ambiente y extraer con cuatro porciones de 25 ml de
diclorometano. Combinar las capas del diclorometano y lavar con una solución medio
saturada de 25 ml del cloruro de sodio, preparada diluyendo 25 ml de solución satura de
cloruro de sodio con un volumen igual de agua. Secar las soluciones del diclorometano
sobre sulfato de sodio. Filtrar por gravedad para quitar el elemento deshidratante y después
evaporar el diclorometano en un baño de vapor. Transferir el aceite residual a un aparato de
destilación simple. Destilar el ácido valeriánico con una llama libre y recoger el producto
que destila entre 170 y 180 ºC. El ácido valeriánico es un líquido agua-blanco, odorífero.
La producción de ácido valeriánico puro es 5 a 6 g (cerca de 50%). El olor de este
compuesto es fuerte, así que todas las operaciones se deben realizar en la campana. Algunas
personas encuentra el olor ofensivo y así que sería bien lavarse después del experimento
con un jabón relativamente básico.
4-metil-3-heptanol
Montar un aparatos para la reacción de Grignard colocando una cabeza Claisen en
un balón de fondo redondo de 250 ml. Ajustar la cabeza Claisen a un condensador de
reflujo y un embudo de adición. Cargar el balón con 7.3 g (0.30 mol) de magnesio y la
flamear el aparato. Tapar el embudo de adición con un tapón de cristal ligeramente
engrasado y colocar un tubo de secado en un adaptador ligeramente engrasado del
termómetro en la tapa del condensador de reflujo. Comenzar el flujo agua en el
condensador solamente después que el flameado seque completamente. Cuando el aparato
se ha enfriado totalmente, cubrir el magnesio en el balón de fondo redondo con 100 ml de
éter anhidro agregados cuidadosamente con ayuda del embudo de adición sobre la cabeza
de Claisen. Colocar 28 g de 2-bromopentano en el embudo de adición y después agregar 2 a
3 ml de él a la mezcla del eter-magnesio. La reacción debe comenzar rápidamente.

Practica # 8ª: Reacción de Grignard. Sintesis del Acido Valerico o del 4-metil-3-heptanol
85
Continuar la adición de 2-bromopentano en tal manera que el reflujo sea apacible pero se
mantenga constante. Cuando se ha agregado todo el 2-bromopentano, continuar el reflujo
con un baño de vapor por 15 minutos adicionales.
Una vez que la formación de Grignard sea completa, transferir 11.6 g (0.2 mol, 14.5
ml) del propianaldehido recientemente destilado al embudo de adición y llenar un cilindro
graduado con 20 ml éter. Agregar esta cantidad éter al embudo de adición. Agregar la
solución del propianaldehido del embudo de adición en tal forma que el reflujo sea apacible
pero se mantenga constante. Reflejar la mezcla por 30 minutos, permitir que se enfrie
brevemente, y después verterla en un beaker de 500 ml que contiene una mezcla 100 g de
hielo picado y 100 ml de ácido sulfúrico acuoso al 10%. Todas las sales deben disolver. La
mezcla se debe revolver continuamente con una barra de cristal pesada o una espátula
durante la adición. Después de la disolución de la mayoría de la sal, decantar
cuidadosamente la mezcla en el embudo de separación (un tapón de lana de vidrio se puede
utilizar en el embudo para quitar exceso de magnesio) y separar las capas. Quitar y
desechar la capa más baja. Lavar la capa de éter (que contiene el producto) con dos
porciones de 25 ml de una solución acuosas al 5% de hidróxido de sodio y dos porciones de
25 ml una solución acuosa saturada de cloruro de sodio. Secar la solución de éter que
resulta sobre sulfato de sodio y después filtrar en un erlenmeyer 250 ml para quitar el
sulfato de sodio. Colocar el erlenmeyer en un baño de vapor para evaporar el éter.
Transferir el aceite que resulta con la cantidad más pequeña del éter posible a un aparato de
destilación simple y destilar con una llama libre.
Recoger el material que destila entre 150 y 165 ºC. El producto (aproximadamente
12 g) debe ser un líquido agua-blanco.

Practica # 8ª: Reacción de Grignard. Sintesis del Acido Valerico o del 4-metil-3-heptanol
86

Practica # 8B: Reacción de Grignard Preparación de trifenilcarbinol
82
PRÁCTICA # 8B: REACCION DE GRIGNARD
PREPARACION DE TRIFENIL CARBINOL
OBJETIVOS
Ilustrar en el laboratorio la reacción de Grignard como un ejemplo de la reacción de
adición nucleofílica a compuestos carbonílicos.
Realizar procedimiento típico de adición de carbaniones a cetonas o ésteres en medio
anhidro, para obtención de alcoholes terciarios.
ANTECEDENTES
Adición nucleofílica, mecanismo general.
Reacción de Grignard. Propiedades de los compuestos organometálicos y adición a
cetonas.
Obtención de alcoholes. Propiedades y normas de seguridad en el manejo de todas y
cada una de las sustancias involucradas (reactivos y productos.
Análisis, reacciones y fundamento químico de la técnica.
REACCIÓN A EFECTUAR Y ESTEQUIMETRIA
Br
+Br2
C5H5N
+ HBr

Practica # 8B: Reacción de Grignard Preparación de trifenilcarbinol
83
Br
+ Mg
I2
THF
(cat)
MgBr
MgBr
+
O
1) THF
2) H3O+
C
OH
Benceno Bromo Bromobenceno
PM 78.11 159.81 157.02
Peso (g) 5.0 10.5 10.05
Moles 6.4 x 10-2
6.6 x 10-2
6.4 x 10-2
Bromobenceno Magnesio Bromuro de fenil mg
PM 157.02 24.31 181.33
Peso (g) 1.93 0.3 2.17
Moles 1.2x 10-2
1.2 x 10-2
1.2 x 10-2
Bromuro de fenil mg Benzofenona Trifenil carbinol
PM 181.33 182.22 260.34
Peso (g) 2.17 1.2 1.17
Moles 1.2 x 10-2
6.5 x 10-3
6.5 x 10-3
MECANISMO
Incluir el mecanismo de la sustitución nucleofílica de bromo sobre benceno.
FORMACIÓN DE BROMURO DE FENIL MAGNESIO:
La formación del bromuro de fenil magnesio (8) consiste en cuatro etapas:

Practica # 8B: Reacción de Grignard Preparación de trifenilcarbinol
84
a) La reacción del bromobenceno (1) con el magnesio (2), da lugar a un radical aniónico
(3) y al radical catiónico (4).
b) Esta etapa consiste en la ruptura hemolítica del enlace carbono-bromo, del
correspondiente radical aniónico de bromobenceno (3), esta ruptura da lugar a la
formación del radical fenilo (5) y al anión bromuro (6).
c) La siguiente etapa es la reacción iónica entre el radical catiónico del magnesio (4) con
el anión bromuro (6), formando un radical de bromuro de magnesio (7).
d) Esta última etapa consiste en la unión del radical fenilo (5) y el radical bromuro de
magnesio (7), formado de esta manera el enlace organometálico C-Mg y obteniéndose
así el bromuro de fenil magnesio.
Br
+ Mg-
+
(1) (2) (3) (4)
(3)
+ Br-
Br-.
Br-.
Mg+.
(5) (6)
(4)
Mg+. + Br
-
(6)
.MgBr
.
(7)
(5)
.
+.
MgBr
(8)
MgBr
(7)

Practica # 8B: Reacción de Grignard Preparación de trifenilcarbinol
85
FORMACIÓN DEL TRIFENIL CARBINOL
La adición nucleofílica (1,2), al carbonilo de la acetona (9) por el bromuro de finil
magnesio (8), se inicia con la formación del complejo (10), el cual se genera por la
interacción de uno de los pares electrónicos que se encuentran en el orbital sp2 del oxígeno
con el magnesio del organometálico (8), después el complejo (10) reacciona con otra
molécula de bromuro de fenil magnesio (8) formando un intermediario de 6 miembros (11),
en donde destaca la interacción del orbital entre el fenilo y el carbono carbonílico, dando
lugar a la formación del nuevo enlace sigma (Csp3-Csp
2 Ar) presente en la sal (12) ésta
reacciona con el bromuro de magnesio (13) generando un intermediario de cuatro centros
(14) el cual en equilibrio con la sal (15) y el compuesto organometálico (8) se destruye en
medio ácido para formar el alcohol (16) y el hidrocarburo correspondiente del
organometálico.

Practica # 8B: Reacción de Grignard Preparación de trifenilcarbinol
86
(8)
MgBr
(9)
O + O Mg
Br
(10)
+
(8)
MgBr
O Mg
Br
MgBr
(11)(12)
O-
Mg+
+ MgBr2 (13)
(14)
O+
Mg
BrMg-
Br
O-
MgBr
+MgBr+
(15) (8)
OH
(16)

Practica # 8B: Reacción de Grignard Preparación de trifenilcarbinol
87
PARTE EXPERIMENTAL
MATERIAL
Agitador 1 Espátula 1
Probeta graduada 25 ml 1 Recipiente para baño 1
Embudo Búchner 1 Vaso de precipitado 150 ml 2
Matraz Kitasato de 250 ml con
manguera
1 Refrigerante para agua (Quickfit) con
manguera
1
Termómetro -10 a 400 °C 1 Baño para calentamiento 1
Embudo de filtración rápida 1 Pinzas de tres dedos con nuez 2
Matraz pera de 2 bocas 1 Matraces Erlenmeyer 125 ml 2
Colector 1 Pipeta de 5 ml (graduada) 1
Embudo de separación Quickfit 1 Vaso de precipitado de 400 ml 1
T de destilación Quickfit 1 Tapón esmerilado (Quickfit) 1
Trampa de humedad (Quickfit) 2
SUSTANCIAS
Cantidad Calidad
Benceno 5.0 g Q.P.
Bromo 10.5 g Q.P.
Piridina 0.5 ml Q.P.
Magnesio en virutas 0.7 g Q.P.
Tetrahidrofurano anhidro 18 ml Q.P.
Acido sulfúrico concentrado 1.4 ml Q.P.
Yodo 0.1 g Q.P.
Cloruro de calcio anhidro 30 g Q.P.
Bromobenceno 2.9 ml Q.P.
Benzofenona 1.6 g Q.P.
Cloruro de sodio 15 g Técnico
Sulfato de sodio anhidro 4.0 g Q.P.
Eter etílico 40 ml Q.P.
Cloruro de amonio (sol. 10%) 20 ml
Hexano 20 ml Técnico
Benzoato de etilo 1 ml Q.P.
PROCEDIMIENTO
Secar todo el material perfectamente y preparar dos trampas de humedad con
cloruro de calcio anhidro.

Practica # 8B: Reacción de Grignard Preparación de trifenilcarbinol
88
Bromobenceno
Colocar 50 g (57 ml) de benceno seco. y 0.5 ml de piridina seca (1) (secar sobre
pelotillas de hidróxido del potasio) en un balón de fondo redondo de 500 ml.. Colocar el
condensador de reflujo al balón y un embudo invertido (apenas sumergido en un poco de
agua en un beaker) al tope del condensador.. Sumergir parcialmente el balón en un baño de
agua fría, soportándolo en un trípode y una rejilla. Agregar cuidadosamente 125 g (40 ml)
de bromo a través de un condensador e inmediatamente insertar el dispositivo de absorción
en el extremo superior del condensador. Ocurrirá una vigorosa reacción tan pronto el
bromuro de hidrógeno es absorbido por el agua un el beaker; cuando la reacción disminuya,
calentar el baño a 25-30 °C por 1 hora. Finalmente elevar la temperatura del baño a 65-70
°C por 45 minutos más o hasta que todo el bromo halla desaparecido (no deben haber
vapores rojo visible) y la evolución del bromuro de hidrógeno en el beaker halla también
cesado.
Dos métodos están disponibles para aislar el bromobenceno puro.
Método 1. Arreglar el balón que contiene la mezcla de reacción para una destilación
de vapor como se muestra en la Fig. 11,40,1. Proceder con la destilación de vapor hasta que
los cristales del p-dibromobenceno aparezcan en el condensador. Cambiar el recipiente y
continuar con la destilación hasta que todo el p-dibromobenceno halla pasado a través; de
tiempo en tiempo hacer circular el agua del condensador de modo que los cristales se
derritan y se recolecten en el recipiente. Colectar el residuo en un frasco. Transferir el
primer destilado a un embudo de separación, lavarlo con un poca de agua, y secar la capa
baja con un poco sulfato de magnesio anhidro o cloruro de calcio anhidro: filtrar. Destilar
lentamente mediante una destilación flask; utilizar una rejilla de alambre o un baño del aire.
(la fig. II, 5, 3) Recoge la fracción que destila entre 150-170°; colocar el residuo (R),
mientras que sigue caliente, en un pequeño beaker o una vasija de porcelana para el
aislamiento del p-dibromobenzene. Redestilar la fracción del punto de ebullición. 150-170°
y recolectar el bromobenceno en 154-157°. La producción de bromobenceno es de 60 g.
Para aislar el p-dibromobernzene puro, filtrar la segunda porción de la destilación de
vapor a través de un embudo Büchner pequeño con succión; presionar los cristales y
secarlos tanto como sea posible. Combinar estos cristales con el residuo (R) y
recristalizarlos en alcohol etílico caliente (para los detalles experimentales, ver la sección
IV, 12) con la adición de 1-2 g de carbón activado decolorar la solución; utilizar cerca de 4
ml de alcohol (alcohol desnaturalizado) para cada gramo de material. Filtrar la solución en
caliente a través de un papel de filtro estriado, enfriar en hielo, y filtrar los cristales por
succión. La producción del p-dibromobenceno, de punto de fusión. 89° es cerca de 12 g.

Practica # 8B: Reacción de Grignard Preparación de trifenilcarbinol
89
Método 2. Transferir el producto de color oscuro de la reacción a un embudo de
separación y agitar sucesivamente con agua, con suficiente solución del hidróxido del sodio
al 5-10%, asegurarse de que los lavados sean alcalinos con papel tornasol, y finalmente con
agua. Secarse con el sulfato de magnesio o el cloruro de calcio anhidro. Filtrar a través de
un papel de filtro estriado en un pequeño balón de destilación y destilar lentamente.
Recoger el bromobencene en el rango de 150-170°; agregar el residuo mientras que aún
esta caliente en una pequeña vasija de porcelana. Redestilar el líquido del punto de
ebullición. 150-170° y recogen el bromobenceno en 154-157°; la producción de
bromobenceno es cerca de 60 g.
Aislar el p-dibromobencene puro del residuo en la vasija de porcelana mediante el
procedimiento descrito en el método 1. La producción del p-dibromobenceno esta cerca de
10 g.
Bromuro de fenil magnesio
En el matraz pera de dos bocas coloque 0.3 g (0.125 at. G) de virutas de magnesio y
un cristal de yodo, agregue 1 ml de tetrahidrofurano y adapte el embudo de separación con
la llave cerrada en la boca inclinada; en la otra boca del matraz coloque un refrigerante de
agua en posición de reflujo y en la parte superior de éste una trampa con cloruro de calcio
anhidro.
En el embudo de separación añada 1.3 ml (0.0121 moles) de bromobenceno, de
preferencia recién destilado, tape el embudo con la otra trampa de cloruro de calcio y deje
gotear al matraz alrededor de 0,2 ml de bromobenceno, sin agitar, para tener una
concentración local alta, una vez que la reacción se inicie adicione 7 ml de THF seco en el
embudo de adición (para diluir el bromobenceno restante), agitando constantemente,
agregue el resto de esta solución gota a gota de tal forma de tener una ebullición suave y
continua. Si la reacción no se inicia puede inducirse mediante:
a) Adición de otro cristal de yodo.
b) Calentamiento suave agitando continuamente, con un baño de agua a 50 °C. Si
el color de yodo desaparece y la ebullición continua una vez retirado el baño, la
reacción se ha iniciado.
c) Adición de otro cristal de yodo, frotándolo sobre el magnesio contra la pared del
matraz, agitándolo, con movimiento circular (sin romper el matraz), mediante
una varilla seca.
d) Adición de unas gotas de una reacción ya iniciada.
e) Adición de gotas de dibromometano.

Practica # 8B: Reacción de Grignard Preparación de trifenilcarbinol
90
No permita que ésta se suspenda. Si la ebullición disminuye sensiblemente, caliente
la mezcla de reacción suavemente con el baño de agua a 50 °C.
La reacción termina cuando todo el magnesio se ha desintegrado y la solución
adquiere una turbidez de color café.
Si es necesario, caliente a reflujo durante 10 minutos.
Adición de benzofenona
En el embudo de separación coloque 1.2 g (0.007 moles) debenzofenona y disuelva
en 6 ml de THF seco, adicione lentamente esta solución al reactivo de Grignard, agitando y
enfriando sólo lo necesario para mantener una ebullición suave. Terminando la adición
caliente a reflujo durante 20 minutos.
Vierta la mezcla de reacción en un vaso de precipitado de 150 ml con 10 g de hielo.
Lave el matraz de pera con 5 ml de éter y reúnalo con la mezcla anterior.
Para lograr la hidrólisis completa agregue una solución de ácido sulfúrico al 5%,
hasta disolver el sólido. Transfiera la mezcla a un embudo de separación, apartando la fase
orgánica (THF, éter y producto). La fase acuosa se somete a una extracción múltiple con
éter etílico (2 x 7ml). Reúna los extractos orgánicos, en el embudo de separación, y
enseguida lave con 20 ml de solución de cloruro de amonio al 10% y después con una
solución saturada de cloruro de sodio. Seque la fase orgánica con sulfato de sodio anhidro,
filtre y destile el éter-THF casi a sequedad. El residuo se cristaliza vertiéndolo en 15 ml de
hexano. Filtre el sólido formado y determine punto de fusión y rendimiento.
Adición de benzoato de etilo
En el embudo de separación coloque 0.6 ml (0.004 moles) de benzoato de etilo y 5
ml de THF seco, adicione lentamente y con agitación esta solución al reactivo de Grignard.
Terminada la adición caliente a reflujo durante 20 minutos.
Continué con el procedimiento descrito en la sección anterior.


Practica # 8B: Reacción de Grignard Preparación de trifenilcarbinol
91
DIAGRAMA ECOLÓGICO
REACCIÓN DE GRIGNARD
OBTENCIÓN DE TRIFENIL CARBINOL
1ª parte

Practica # 8B: Reacción de Grignard Preparación de trifenilcarbinol
92
REACCIÓN DE GRIGNARD
OBTENCIÓN DE TRIFENIL CARBINOL
2ª parte
D1: neutralice y deseche por el drenaje.
D2,D3: recupere el disolvente por destilación, envíe las colas a incineración.

Practica # 8B: Reacción de Grignard Preparación de trifenilcarbinol
93
CUESTIONARIO
1. Describa un método para eliminar el agua del éter etílico y tetrahidrofurano.
2. ¿Cuál es la evidencia que muestra que la reacción de Grignard ha terminado?
3. ¿Qué productos se formarán, si la benzofenona o el benzoato de etilo son adicionados al
reactivo de Grignard con trazas de agua?
4. ¿Cómo detiene la reacción?
5. ¿Por qué es necesario acidular?
6. ¿Las soluciones utilizadas en los lavados son inocuas?
BIBLIOGRAFÍA
1. Brewter, R.Q. y Vanderwerf, C.A., “Curso practico de Química Orgánica” 2ª. Ed.,
Alambra, España, 1970, pp. 236.
2. Kremlin, R.J.W. y Still, R.H:, Named and Miscellaneous Reactions in Practical Organic
Chemistry, Heinemann, Londres, Inglaterra, 1967, pp. 66.
3. Morrison, R.T. y Boyd, R.N., Quimica orgánica, Fondo Educativo Interamericano,
México, 1976.

Practica # 8 Reacción de Grignard. Síntesis del Acido Valérico ó 4-metil-2-heptanol.
94

Practica # 9 Síntesis de Dibenzalacetona
95
PRÁCTICA # 9: CONDENSACION DE CLAISEN-SCHMIDT.
SÍNTESIS DE DIBENZALACETONA
OBJETIVOS
Ilustra una reacción de adición nucleofílica-eliminación.
Efectuar una condensación aldólica cruzada.
Realizar reacciones de formación de enlace C-C.
Obtener un producto de uso comercial.
ANTECEDENTES
Reacciones de adición nucleofílica a grupos carbonilo.
Reacciones de condensación aldólica.
Reacciones de condensación aldólica cruzada.
Ejemplos de la reacción de Claisen-Schmidt.
Usos de la dibenzalacetona.
Deshidratación de aldoles en medio básico,
Conjugación de grupos carbonilo con dobles enlaces.
Reactividad de grupos carbonilo , -insaturados.
REACCIÓN A EFECTUAR Y ESTEQUIOMETRIA
C
O
H
+NaOH
H2O/EtOH
C
H
CC
H
CC
O
H HCH3
C
CH3
O
2
(1) (2) (12)
2+ OH2
Benzaldehido Acetona Dibenzalacetona
PM 106 58 234
Peso (g) 3.12 0.79 3.44 (teórico)
Moles 2.9 x 10-2
1.3 x 10-2
1.4 x 10-2

Practica # 9 Síntesis de Dibenzalacetona
96
MECANISMO DE LA REACCIÓN
La reacción que se lleva a cabo es una condensación aldólica cruzada, debido a que
se efectúa con dos compuestos carbonilos diferentes (aldehído y cetona). Esta reacción no
siempre es factible, puesto que puede resultar una mezcla de cuatro productos si ambos
tienen hidrógenos en
Se puede obtener un sólo producto si se siguen las siguientes condiciones:
a) Uno de los reactivos no debe tener hidrógeno , de modo que no pueda
autocondensarse.
b) Mezclar este reactivo con el catalizador.
c) Adicionar al final el compuesto carbonilo con hidrógeno para que haya una
concentración muy baja de compuesto carbonilito iónizable.
La síntesis de dibenzalacetona involucra la condensación de dos moléculas de
benzaldehido con una de acetona, catalizada con hidróxido de sodio.
El mecanismo ocurre gracias a la base presente en el medio de reacción (sosa), cuyo
ión hidróxido ataca al compuesto carbonilo con hidrógenos , la acetona (2), extrayendo un
hidrógeno formando el carbocatión estabilizado por resonancia como el ión enolato (3) y
agua.
CH3
C
CH2
O
H
NaOH+ CH3
C
C-
O
H
H
Na+
CH3
C
C
O-
H
H
Na+
(2) (3)
+ OH2
Posteriormente el anión enolato (3), a través del carbanión, ataca nucleofílicamente
al grupo carbonilo de una molécula de benzaldehido (1), formando un ión alcóxido (4), el
cual es una base fuerte que reacciona con el agua para producir la -hidroxicetona (5) y
regenerando el ión hidróxido.

Practica # 9 Síntesis de Dibenzalacetona
97
Na+
(3)
CH3
C
C-
O
H
H
C
O
H
+
(1)
C
O-
CH3
C
C
O
HH
H
Na+
H OH
OH2
(4)
C
O
CH3
C
C
O
HH
H
H
NaOH+
(5)
La -hidroxicetona (5) reacciona con el ión hidróxido para formar el enolato (6),
que es la base conjugada del compuesto (5).
C
O
CH3
C
C
O
HH
H
H
NaOH +
(5)
H OH +C
O
CH3
C
C-
O
H
H
H
Na+
C
O
CH3
C
C
O-
H
H
HNa
+
(6)
El enolato (6), al pasar al tautómero ceto, elimina con facilidad el ión hidróxido para
formar la monobenzalacetona (7). Esta reacción de eliminación, se ve favorecida gracias a
que la doble ligadura que se genera está conjugada con el grupo carbonilo y el anillo de
benceno.

Practica # 9 Síntesis de Dibenzalacetona
98
C
O
CH3
C
C
O-
H
H
HNa
+
(6)
C
H
CH3
C
C
O
H
(7)
NaOH+
La monobenzalacetona (7) reacciona nuevamente con el ión hidróxido para que al
perder otro hidrógeno de carbono en la posición al grupo carbonilo se genere el carbanión
estabilizado por resonancia como el enolato (8).
C
H
C
C
C
O
H
H
H
HNaOH +H OH +
C
H
C-
C
C
O
H
H
H
(7)
Na+
C
H
C
C
C
O-
H
H
H
(8)
Na+
El enolato (8) reacciona a través del carbanión con la segunda molécula de
benzaldehido (1) para generar el alcóxido (9).
C
O
H +
C
H
C-
C
C
O
H
H
H
Na+
(8)(1)
C
O-
HC
H
C
C
C
O
HH
H
Na+
(9)
El alcóxido (9) toma un protón del agua, al sr una base más fuerte que el ión
hidróxido, para generar la -hidroxicetona (10) y regenerar el ión hidróxido, para que a
través de otra reacción ácido-base, se forme la base conjugada (11) y agua.

Practica # 9 Síntesis de Dibenzalacetona
99
C
O-
HC
H
C
C
C
O
HH
H
Na+
(9)
H OH
OH-
Na+
OH-
+C
OH
C
H
C
C
C
O
HH
H
H
(10)
H OH +
C
OH
C
H
C-
C
C
O
HH
H
Na+
(11)
La base conjugada (11) es estable porque se encuentra en resonancia con el enolato
y al pasar del tautómero enolato al tautómero ceto, se lleva a cabo la eliminación del ión
hidróxido, para formar la dibenzalacetona (12).
C
OH
C
H
C-
C
C
O
HH
H
Na+
C
OH
C
H
C
C
C
O-
HH
H Na+
Na+
OH-
+
C
H
C
H
C
C
C
O
HH
(11)
(12)
El isómero que se obtiene de la dibenzalacetona (12) es el trans-trans, por ser el
menos impedido estéricamente y por lo tanto el más estable. El color que presenta la
dibenzalacetona (12) es el resultado de un largo sistema conjugado el cual puede absorber
en la parte visible del espectro electromagnético (Imax. 330 nnm; = 34, 300)

Practica # 9 Síntesis de Dibenzalacetona
100
PARTE EXPERIMENTAL
MATERIAL
Vasos de precipitados de 250 mL 1 Vidrio de reloj 1
Matraz Erlenmeyer de 125 mL 2 Espátula 1
Termómetro de -10 a 400 °C 1 Agitador de vidrio 1
Probeta de 25 ml 1 Baño con resistencia eléctrica 1
Pipeta de 10 mL 1 Recipiente de peltre 1
Matraz Kitasato con manguera 1 Embudo Büchner con empaque de hule 1
Pinzas de tres dedos con nuez 1 Frasco para cromatografía 1
Potaobjetos para cromatgrafía 2 Embudo de vidrio tallo largo 1
SUSTANCIAS
Cantidad Calidad
Benzaldehido 3.0 mL Destilado
Acetona 1.0 mL R.A.
Hidróxido de sodio 2.25 g R,A.
Etanol 70 mL Q.P
Acetato de etilo 10 mL Q.P.
Gel de sílice para c.c.f. 2.0 g R.A.
Yodo 0.01 g R.A.
PROCEDIMIENTO
En un matraz Erlenmeyer de 125 mL coloque 2.25 g de hidróxido de sodio, 25 mL
de agua y 20 mL de etanol. En seguida agregue poco a poco y con agitación 3 mL de
benzaldehido y 1 mL de acetona. Continue agitando la mezcla de reacción durante 15 min
más, mantenga la temperatura de la mezcla de reacción a 20-25 °C; use (de ser necesario)
un baño de agua fría o tibia según el caso.
Filtre el precipitado, lave con agua fría hasta que las aguas de lavado tengan pH = 7.
Seque y purifique el producto por recristalización de etanol. El producto obtenido bao estas
condiciones tiene una pureza entre 96 y 98% determine por cromatografía de gases.
Si al recristalizar la solución, ésta se torna de color rojo-naranja, es debido a que el
pH es ligeramente alcalino, por lo que debe agregar ácido clorhídrico (1:1), hasta pH = 7.
Pese el producto, calcule el rendimiento y determine el punto de fusión. Si se desea
comprobar la pureza realice unas c.c.f.
Datos para la cromatografía en capa fina (c.c.f)
Suspensión: gel de sílice al 35% CHCl3/MeOH (3:1) o en acetato de etilo.
Disolvente: acetona o acetato de etilo.

Practica # 9 Síntesis de Dibenzalacetona
101
Eluente: hexano-acetato de etilo 3.
Revelador: I2 o luz UV.
CUESTIONARIO
1. Explique por que debe adicionar primero el benzaldehido y después la acetona a la
mezcla de reacción.
2. Diga por que obtiene un solo producto y no una mezcla de productos en este
experimento.
3. Indique por que se pierde fácilmente agua (reacción de eliminación) en medio alcalino
en el producto de adición, para obtener un producto -insaturado.
4. Diga por que para recristalizar el producto obtenido, la solución no debe ser alcalina.
5. ¿Por qué se utilizan 0.029 moles de benzaldehido y sólo 0.013 moles de acetona?
6. La dibenzalacetona puede existir como diferentes isómeros geométricos. ¿Cuáles son?,
¿Cuál se obtiene preferentemente?, ¿Cómo lo analizaría so obtuviera una mezcla?
7. Diga como afecta, en la reacción, la presencia de ácido benzoico en el benzaldehido.
8. ¿Qué característica tienen los hidrógenos unidos a un carbono al carbonilo?
9. Explique qué tratamiento se debería dar a los efluentes líquidos de la reacción, para
poder desecharlos en el drenaje.
10. ¿Cuál sería la temperatura máxima a la que se puede hacer esta reacción?, ¿Por qué?
BIBLIOGRAFÍA
1. Conrad, C.R. y Dolliver, M.A., Organic Síntesis, Coll. 2, 1943, pp. 167.
2. Cremlyn, R.J. y Still, R.H., Named and Miscellaneous Reactions in Practical Organic
Chemistry, Heinemann, Londres, Inglaterra, 1976.
3. Dimwiddie, J.G. Jr., White, H. M., Day, W.J., J. Org. Chem. 27, 1962, pp. 327-328.
4. Morrison, R.T. y Boyd, R.N., Química orgánica, Fondo Educativo Interamericano,
México, 1985, pp. 868.
5. Solomons, T.W.G., Química orgánica, Limusa, México, 1979 , pp. 736.
6. Vogel, A.I., Elementary Practical Organic Chemistry, Pat I, Small Scale Preparations,
2ª. Ed., Longman, Londres, Inglaterra, 1967, pp. 327.

Practica # 9 Síntesis de Dibenzalacetona
102
DIAGRAMA ECOLÓGICO
D1: Recuperar el etanol por destilación. La dibenzalacetona pura se puede emplear
como sustancia problema de cristalización para otros cursos.
D2: haga ensayos para aldehídos. Si resulta negativo, recupere el etanol por
destilación y neutralice el residuo desechándolo posteriormente por el drenaje. Si el
resultado es positivo, agregue más NaOH y repita el proceso para obtener más
dibenzalacetona. Agregue más acetona si es necesario.
Precauciones: benzaldehido. LD50 1 300 mg/kg. Peligroso si se inhala o se absorbe
por la piel cuasando irritación.
Disposición: disuelva en un solvente combustible para ser incinerado en un horno
adecuado.
Productos de descomposición: humos tóxicos de monóxido y bióxido de carbono.

Practica # 9 Síntesis de Dibenzalacetona
103
ESPECTROS DE I.R.
a) Benzaldehido
b) acetona

Practica # 9 Síntesis de Dibenzalacetona
104
c) Dibenzalacetona

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
105
PRÁCTICA # 10: SALES DE DIAZONIO. OBTENCIÓN DEL
ACIDO SULFANÍLICO, NARANJA DE METILO Y
NARANJA II
OBJETIVOS
Ilustrar la fulfonación de una amina primaria.
Efectuar la preparación de un intermediario usado en la obtención de varios compuestos
colorantes.
Ilustrar en el laboratorio las reacciones de acoplamiento (copulación) de las sales de
diazonio.
Obtener colorantes azoicos, a partir de las reacciones de diazotación y acoplamiento de
la sal de diazonio del ácido sulfanílico.
Aplicar las propiedades químicas de los compuestos obtenidos, para la tinción de
diferentes fibras.
ANTECEDENTES
Sustitución electrofílica aromática (SEA).
Comportamiento de las aminas aromáticas en las reacciones de SEA.
Reacciones de sulfonación.
Propiedades de los compuestos que son sales internas (zwitterion)
Formación de las sales de diazonio
Estabilidad de las sales de diazonio aromáticas
Sales de diazonio como agente electrofílicos.
Condiciones de reacción de sustituciones electrofílicas de las sales de diazonio con
fenol y con aminas.
Efecto del pH en las reacciones de acoplamiento (copulación) de las sales de diazonio
con aminas y fenoles.
Características estructurales de los colorantes.
Métodos para aplicar colorantes a los diferentes tipos de fibras.

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
106
Toxicidad de reactivos y productos.
REACCIÓN A EFECTUAR Y ESTEQUIOMETRIA
NH2
H2SO4NH3
+S OH
O
O
O-
H2SO4
NH S OH
O
O
ácido fenilsulfámico
OH2
H2SO41)
2)
NH2 S O
O
O
H NH3
+S O
-
O
O
ácido sulfanílico
H2SO4 Anilina Acido sulfanílico
PM 98 93 173
Peso
(g)
14.72 g (8ml)
(d = 1.84)
2.044 g (2ml)
(d = 1.022)
3.80 g teóricos
Moles 1.5 x 10-1
2.20 x 10-2
2.20 x 10-2
teórico

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
107
NH2 SO3H
NaNO2
ClH
N+
SO3HNCl-
N SO3HNCl- +
N
CH3
CH3
N
CH3
CH3
N SO3HN
NaOH
N
CH3
CH3
N SN
O
O
O-
Na+
N,N-
dimetilanilina
Acido
sulfanílico
Anaranjado de
metilo
Beta-naftol Naranja II
PM 121.18 173.09 327.34 144.17 350.33
Peso
(g)
0.57 (0.6 ml)
(d = 0.956)
1.0 1.539 t
teórico
0.8 1.96
Teórico
Moles 4.7 x 10-3
5.7 x 10-3
4.7 x 10-3
teórico
5.5 x 10-3
5.5 x 10-3
teórico

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
108
MECANISMO DE LA REACCIÓN
Acido sulfanílico
El mecanismo procede con una reacción ácido-base, seguida de una deshidratación
de la sal, formándose de esta manera el ácido fenilsulfámico. Posteriormente, éste sufre una
sulfdonación a través de una reacción de sustitución electrofílica aromática:
NH2
H2SO4NH3
+S OH
O
O
O-
S OH
O
O
OH
S O+
O
O
OH H
H
+NH2N
+
H
H
S
O-
O
O
O+
H
H
H
N+
H
H
S
O
O
OH O HH+
N H
SO O
OH
O+
HH
H
+

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
109
N H
SO O
OH
O+
HH
H
+
S O+
O
O
O H
H
Hadición
N+
H
S
O
O
O
S
H
O-
OO
O+
HH
H
H O+
HH
H
+N
H
S
O
O
O+
H
HSOH
O
O
+ O
H
H
eliminación
En seguida se lleva a cabo la hidrólisis del grupo ácido sulfámico, para dar como
resultado la formación del ácido fulfanílico.
N
H
S
O
O+
O+
H
HSOH
O
O
H
+ O
H
H
N
H
S
O
O+ O H
SOH
O
O
H H
O H
N+
H
S
O
OO H
SOH
O
O
H
O H
H
S
O+
OO
O
HH
H
+
NH2 S O H
O
O
NH3
+S O
-
O
O

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
110
Sal de diazonio
El mecanismo de reacción para la formación de la sal de diazonio del ácido
sulfanílico, se lleva a cabo a través del ataque del grupo amino sobre el electrófilo
nitrosonio formando al reaccionar 2 moléculas de ácido nitroso entre si.
N OO-
Na+
+ ClH N OOH + Na+Cl
-
N OOH2
(a)
(b)
+ O HH
+ O HHN OONO
N OO-
NH O+ :
N OO-
NH O+ :
Posteriormente, se propone existe un equilibrio tautomérico y la protonación de uno
de los isómeros, el cual, al perder agua, conduce a la formación de la sal de diazonio
correspondiente.

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
111
N+
S
H
H
H
O
O-
O
NH O+
:
H+
H+
+
-
N
S
H
H
O
OH
O
:
N+
S
H
H
O
OH
O
N O
H+
:
H+
+
H+
-
N+
S
H
O
OH
O
N O H N
S
H
O
OH
O
N O+H
.. ..H
+
N
S O
OH
O
N O+
H
H
..
H+
+
H+
-
N+
S O
OH
O
N
O
H
H
:
+
N
S O
OH
O
NH
..
.. .. +
Esta sal, mediante una reacción de sustitución electrofílica aromática con la
N.N-dimetil-anilina conlleva a la formación del anaranjado de metilo.

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
112
N
CH3
CH3
..+ N S
O
OH
O
NH..+
adición
N S
O
OH
O
NN+
CH3
CH3
H
eliminación
N S
O
OH
O
NN
CH3
CH3
+H+
PARTE EXPERIMENTAL
MATERIAL
Matraz bola de 2 bocas (Quickfit) 1 Embudo de vidrio 1
Portatermómetro (Quickfit) 1 Espátula 1
Agitador de vidrio 1 Termómetro de -10 a 250 °C 1
Vidrio de reloj 1 Baño de agua electrico 1
Probeta de 25 ml 1 Matraz Kitazato 250 ml con manguera
para vacio
1
Embudo Büchner con alargadera 1 Pipeta graduada de 2 ml 2
Vaso de precipitado de 125 ml 2 Pipeta graduada de 5 ml 1
Matraz Erlenmeyer de 125 ml 2 Pipeta graduada de 10 ml 2
Mechero con manguera 1 Tela de alambre con placa de asbesto 1
Recipiente de peltre R.A. 1 Anillo metálico 1

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
113
SUSTANCIAS
Cantidad Calidad
Anilina 2 ml R.A.
H2SO4 concentrado 8 ml Q.P.
Carbón activado 0.2 g
Carbonato de sodio anhidro 0.4 g Q.P.
Nitrito de sodio 0.4 g Q.P.
Acido clorhídrico 2.5 ml Q.P
N,N-dimetilanilina 0.6 ml Q.P
Hidróxido de sodio 1 g
Cloruro de sodio ind (sal gruesa 20.0 g
Cloruro de sodio 4 g R.A.
Etanol de 96 (alcohol etílico de 96) 2 ml
-naftol 0.8 g Q.P
Acido sulfanílico 1.0 g R.A.
PROCEDIMIENTO
Acido sulfanílico
Coloque en el matraz de bola de dos bocas 2 ml de anilina y agregue lenta y
cautelosamente 8 ml de ácido sulfúrico concentrado (haga esta operación en la campana,
empleando un baño de agua fría para ayudar a disipar el calor generado por la reacción).
Adapte el termómetro en la boca lateral del matraz (cuidando que el bulbo quede sumergido
en la mezcla de reacción) y en la otra boca, el refrigerante de agua. Caliente la mezcla de
reacción a 170 – 180 ºC empleando baño eléctrico (la temperatura debe ser controlada muy
cuidadosamente). Deje enfriar la mezcla de reacción hasta 50 – 60 ºC y vierta lentamente y
con agitación sobre 50 g de una mezcla de hielo-agua. (Si el contenido del matraz bola
fuese muy viscoso, caliente a baño de María hasta que la mezcla se haga fluida y fácil de
verterse sobre hielo.) Separe el sólido formado por filtración al vacío y recristalice en agua
empleando carbón activado. Seque el producto y calcule el rendimiento.
Anaranjado de metilo
En un vaso de precipitado de 125 ml, coloque 1 g de ácido sulfanílico, 0.6 ml de
dimetilanilina y 0.5 ml de ácido clorhídrico concentrado; agregue 5 ml de agua y enfríe la
mezcla hasta tener una temperatura de 0-5 °C.
En otro vaso de precipitado de 125 ml, prepare una solución de 0.35 g de nitrito de
sodio disuelto en 2.5 ml de agua. Manteniendo la temperatura debajo de 5 °C agregue gota
a gota y con agitació constante la solución de nitrito de sodio a la solución de la amina y el
ácido sulfanílico, preparada anteriormente, Una vez terminada la adición agite la mezcla

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
114
hasta que tome la temperatura ambiente. La mezcla adquiere una coloración rojo vino
oscuro.
Agregue gota a gota y agitando, una solución de sosa al 10% hasta tener un pH =
10. Caliente la mezcla de reacción con agitación constante, retire el recipiente en el
momento en que se inicia la ebullición. Enfríe en hielo. Si fuese necesario, induzca la
cristalización y filtre. Lave con agua helada y seque el producto en el desecador o en la
estufa para posteriormente pesarlo. El naranja de metilo precipita como sal sódica.
Naranja II
Reacción de diazotación
Coloque 0.4 g de carbonato de sodio en un vaso de precipitado de 125 ml y adicione
10 ml de agua. Agregue 1 g de ácido sulfanílico hasta disolución total.
Coloque la solución en baño de hielo-sal. Adicione a la mezcla de reacción 10 g de
hielo picado y 4 ml de solución de nitrito de sodio al 10% y 4 ml de ácido clorhídrico al 20
v/v. Al cabo de unos minutos se forma la sal de diazonio.
Reacción de acoplamiento
Disuelva en un matraz Erlenmeyer 0.8 g b-naftol en 4 ml de hidróxido de sodio al
10% calentando si es necesario. Enseguida enfríe en baño de hielo-sal hasta 0-5 °C. Una
vez fría la solución, adiciones la sal de diazonio, manteniendo la mezcla de reacción en
baño de hielo-sal y con agitación constante.
Terminada la adición, dejé reposar la mezcla a temperatura ambiente durante 15-20
minutos.
Agregue 4 g de cloruro de sodio, calentar casi a ebullición hasta disolución
completa y enfríe en baño de hielo hasta la cristalización de la mayor parte del producto.
Filtre al vacío, lave con 2 ml de etanol frío, seque y pese el producto obtenido. Mida el
punto de fusión.
Pruebas de tinción
En un matraz o vaso pequeño, coloque 10 ml de solución al 1% del colorante,
agregue cortes pequeños de diferentes telas: algodón, lana o seda (preferentemente blanco).
Hierva durante 5 minutos, y posteriormente lave los cortes de tela con agua. Observe y
anote sus resultados.

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
115
DIAGRAMA ECOLÓGICO
Preparación del ácido sulfanílico
D1, D2: filtre el sólido y confine. Verifique el pH de la solución, neutralice, adsorba
en carbón activado; el carbón se manda a incineración y la solución se desecha por el
drenaje.

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
116
Obtención de anaranjado de metilo
D1,D2: lleve a sequedad. Almacene el sólido para su posterior uso, incineración o
confinamiento.

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
117
Obtención de naranja II
D1: éste líquido contiene apreciables cantidades de producto, por lo cual debe
eaporarse y guardarse el sólido para su posterior uso o su incineración.
D2: absorba e líquido con carbón activado, después de lo cual podrá desecharse por
el drenaje. El carbón activado se manda a incinerar.

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
118
CUESTIONARIO
1. Escriba un mecanismo alternativo de reacción para la formación del ácido sulfámico
a partir de anilina y ácido sulfúrico.
2. Escriba un mecanismo de reacción alternativo que explique la sulfonación del ácido
bencensulfámico.
3. Explique con reacciones qué sucede al verter la mezcla de reacción sobre hielo-
agua.
4. Explique por qué el grupo sulfamida favorece a la monosustitución en el anillo,
mientras que el grupo amino favorece la polisustitución en reacciones de SEA.
5. ¿Por qué el ácido sulfanílico es soluble en agua caliente pero insoluble en éter o
benceno?
6. Dibuje las estructuras del ácido sulfanílico en una solución ácida, básica y neutra.
7. ¿Cómo deben tratarse los residuos que contienen ácido sulfúrico antes de
desecharse por el drenaje?
8. Explique ¿cuál es la razón por la que las sales de diazonio aromáticas son
relativamente estables?
9. ¿Cómo evita que se descompongan las sales de diazonio?
10. ¿Qué diferencia se requiere en el pH de la mezcla de reacción para que el
acoplamiento de las sales de diazonio sea óptimo con fenol y con aminas?
11. ¿Qué es un colorante y cómo imparte color a una tela?
12. ¿Cómo reacciona el ácido sulfanílico con el carbonato de sodio?
13. ¿Qué productos se tienen en la mezcla de reacción en cada caso?
14. ¿Cómo debe tratar los residuos acuosos antes de eliminarlos en el drenaje?

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
119
BIBLIOGRAFÍA
1. Brewter, R.Q. y Vanderwerf, C.A., “Curso practico de Química Orgánica” 2ª. Ed.,
Alambra, España, 1970, pp. 27-A.
2. Bruse, Paula Yurkanis, Organic Chemistry, Prentice-Hall, EU, 1995, pp. 607.
3. Carey, Francis A. y Sundberg, Richard J., Advanced Organic Chemistry, Part B,
Reactions and Synthesis, Plenum Press, EU, 1990, pp. 504-516.
4. Fieser, Louis F. y Williamson, Kenneth, Organic Experiments, D.C. Heath and
company, EU, 1992, pp. 394, 396, 529, 531.
5. Morrison, R.T. y Boyd, R.N., Química orgánica, Fondo Educativo Interamericano,
México, 1985, pp. 864, 866, 868-873..
6. Roberts, J.D. y Caserio, M.C., Basic Principles of Organic Chemistry, W.A.
Benjamín, California, EU, 1964, pp. 892-895 y 1048-1053.
7. Vogel, A.I., Elementary Practical Organic Chemistry, Pat I, Small Scale
Preparations, 2ª. Ed., Longman, Londres, Inglaterra, 1967, pp. 293.

Practica # 10 Sales de diazoni. Obtención del ácido sulfanílico, anaranjado de
metilo y naranja II
120
ESPECTROS DE I.R.
Acido Sulfanilico

Practica # 11 Síntesis de Ciclohexanona por oxidación de Ciclohexanol
121
PRÁCTICA # 11: SINTESIS DE CICLOHEXANONA POR
OXIDACION DE CICLOHEXANOL CON HIPOCLORITO
DE SODIO
OBJETIVOS
Obtener una acetona a partir de un alcohol secundario
Utilizar como agente oxidante, blanqueador de uso doméstico.
Identificar la cetona obtenida mediante la formación de un derivado de punto de fusión
conocido.
ANTECEDENTES
Propiedades químicas de alcoholes.
Agentes oxidantes empleados comúnmente. Ventajas y desventajas.
Reacciones de identificación y caracterización de alcoholes, aldehídos y cetonas.
Toxicidad de reactivos y productos.
REACCIÓN A EFECTUAR Y ESTEQUIOMETRIA
OH
NaOCl
AcOH
O
+ AcOH + NaCla)
b)
O
+ NH2 N
H
O2N
NO2
H+
N
NH
NO2
NO2

Practica # 11 Síntesis de Ciclohexanona por oxidación de Ciclohexanol
122
Ciclohexanol Ciclohexanona
PM 100.16 98.15
Peso (g) 4.74 g 5.0 ml d = 0.948 4.64 g teóricos
Moles 4.73 x 10-2
4.73 x 10-2
teóricos
MECANISMO DE LA REACCIÓN
El primer paso de la reacción consiste en la protonación del grupo hidroxilo,
seguida del ataque del ión hipoclorito. (Esto conduce a la salida de una molécula de agua).
OH
AcOH
O+
H
H
+ AcO:
Na+ClO
-
OCl
+ AcO: Na+ + OH2
Posteriormente, el protón gem del carbono que soporta al grupo hipoclorito, es
extraído, desplazandose entonces el par electrónico de este enlace hacia el oxígeno, con la
consecuente formación de un nuevo enlace C-O, y la salida del ión cloruro.
OCl
H
AcO: Na+
+
reacciónredox
AcO-H +
O
Na+
+ Cl-
La segunada parte de la reacción consiste en la identificación del producto obtenido
en forma de su 2,4-dinitrofenmilhidrazona.

Practica # 11 Síntesis de Ciclohexanona por oxidación de Ciclohexanol
123
PARTE EXPERIMENTAL
MATERIAL
Matraz Erlenmeyer de 250 mL 1 Tapón de corcho para matraz 1
Matraz Erlenmeyer de 125 mL 1 Erlenmeyer de 250 mL 1
Matraz Erlenmeyer de 50 mL 1 Tapón de hule horadado para matraz
Erlenmeyer de 250 mL
1
Embudo de separación
(Quickfit) copn tapón
1 Termómetro de -10 a 400 ºC 1
Pipeta graduada de 10 mL 1 Tela de alambre con placa de asbesto 1
Pinzas de 3 dedos con nuez 2 Agitador de vidrio 1
Colector 1 Espátula 1
Termopozo 1 Mechero con manguera 1
Refrigerante de agua con
manguera
1 Vaso de precipitado de 250 mL 1
T de destilación 1 Probeta de 25 mL 1
Tubos de ensayo 3 Anillo metálico 1
Recipiente de peltrwe 1 Agitador magnético con barra 1
Pipeta de 5 mL 1 Vidrio de reloj 1
SUSTANCIAS
Cantidad Calidad
Ciclohexanol 5.0 mL R.A.
Hipoclorito de sodio (Sol. Ac. Al 11%) 50.0 mL
Solución acuosa 6M de hidróxido de sodio 20 mL
Anhídrido crómico 1.0 g R.A.
Reactivo de 2,4-dinitrofenilhidracina 0.5 mL
Acido sulfúrico conc. 1.0 mL R.A.
Acido acético glacial 3.0 mL Q.P
Solución saturada de bisulfito de sodio 8.0 mL
Cloruro de sodio sólido 5.0 g
Sulfato de sodio anhidro 5.0 g
Acido sulfùrico Conc. 1.0 mL R.A.
Papel yodo-yodurato 2 tiras
PROCEDIMIENTO
En un matraz Erlenmeyer de 250 mL coloque 5 mL de ciclohexanol y 3 mL de
ácido acético glacial. Adapte un embudo de separación y deje gotear 40 mL de solución de
hipoclorito de sodio al 11%. Esta adición se realizará en un mínimo de 30 minutos, con
agitación constante. La temperatura de la mezcla de reacción deberá mantenerse entre 40-
50 ºC. Terminada la adición, la solución deberá tener un color amarillo-verdoso y dará
prueba positiva con papel yoduro-almidón. (Si termina la adición, y el color de la mezcla

Practica # 11 Síntesis de Ciclohexanona por oxidación de Ciclohexanol
124
no es amarillo-verdoso, adicione de 2 mL en 2 mL más de solución de hipoclorito de sodio
hasta alcanzar esta coloración, verificando que de prueba positiva con papel yoduro-
almidón.) Agregue 5 mL de exceso de solución de hipoclorito de sodio a la reacción, tape el
matraz Erlenmeyer con un tapón de corcho y agite durante 30 minutos más a temperatura
ambiente.
Posteriormente, manteniendo la agitación, adiciones 2 mL de solución saturada de
bisulfito de sodio hasta decolorar la mezcla de reacción y/o hasta reacción negativa a la
prueba de papel yoduro-almidón. Agregue entonces lentamente, y agitando, 10-15 mL de
solución de hidróxido de sodio 6 M(24% acuosa) hasta pH = básico (verifique con papel
indicador de pH). Destile la mezcla de reacción colectando los primeros 30 mL de
destilado.
Adiciones 5 g de cloruro de sodio sólido y agite por 10 minutos. Decante la mezcla
de reacción en un embudo de separación y separe las dos fases. Seque la fase orgánica cxon
sulfato de sodio anhidro y con gotas de este producto haga las siguientes pruebas.
Pruebas de identificación
a) Identificación de la ciclohexanona formada. Coloque en un tubo de ensayo 5 gotas de
reactivo de 2,4-dinitrofenilhidracina, agregue 2 gotas de la ciclohexanona formada,
deje enfriar en baño de hielo y raspe las paredes del tubo hasta formación del sólido.
b) Identificación de presencia de materia prima. Coloque en un tubo de ensayo 1 mL de
reactivo de Jones y 1 gota de ciclohexanona obtenida. El cambio de reactivo a verde
indica prueba positiva para alcoholes.
Preparación del reactivo de Jones:
Pese 1 g de anhídrido crómico, agregue 1 mL de ácido sulfúrico concentrado y
diluya hasta 3 mL con agua.

Practica # 11 Síntesis de Ciclohexanona por oxidación de Ciclohexanol
125
DIAGRAMA ECOLÓGICO
Obtención de ciclohexanona

Practica # 11 Síntesis de Ciclohexanona por oxidación de Ciclohexanol
126
D1: Residuo neutro: verificar pH y desechar por el drenaje.
D2, D3: Ambos residuos se desechan por el drenaje.
D4: Agregue agua, ajuste el pH a 7; filtre la solución; el sólido se confina para su
posterior disposición y el líquido se desecha por el drenaje.
D5: Filtre la solución: el sólido se confina para enviarse a incineración, la solución
se absorbe sobre carbón activado: el carbón se manda a incinerar y la solución se desecha
por el drenaje.
D6: Tratamiento para cromo: lleve la solución a pH 3. Adiciones lentamente y con
agitación una solución saturada de bisulfito de sodio (agregue un exceso de 50%), el Cr VI
será reducido a cromo III que da un color verde a la solución. Precipite el cromo III
agregando sosa diluida calentar a ebullición si es necesario para lograr la precipitación
cuantitativa del Cr III. Si la solución tiene un color rojizo, puede ser materia orgánica
degradada, decolórese con carbón activado. El carbón se manda a incineración.
CUESTIONARIO
1. ¿Cómo sabe que existe un exceso de hipoclorito de sodio en la reacción?
2. Escriba la reacción que ocurre al identificar el hipoclorito de sodio en el seno de la
reacción.
3. ¿Cómo asegura que al finalizar la reacción ya no existe oxidante en el seno de la
reacción?
4. Escriba la reacción que le permite saber que ya no hay oxidante en el seno de la
reacción.
5. ¿Cómo identificó el producto obtenido? Escriba la reacción de identificación.
6. ¿Cómo supo que todo el alcohol había sido oxidado? Escriba la reacción que se lo
indicó.
7. Mecanismo de la reacción.
8. ¿Todos los alcoholes se pueden oxidar con hipoclorito de sodio?
9. Explique en qué consiste la reacción de haloformo. ¿En qué condiciones ocurre?.
¿es este experimento una reacción de haloformo?
10. ¿Puede usted desechar en el drenaje los residuos de la mezcla de reacción?, ¿por
qué?
11. ¿Qué tratamiento debe dar a los residuos para poder descartarlos?

Practica # 11 Síntesis de Ciclohexanona por oxidación de Ciclohexanol
127
BIBLIOGRAFÍA
1. Fessenden, R.J., y Fessenden, J.S., Techniques and Experiments for Organic
Chemistry, Willard Grant Press, Boston, EU, 1938, pp. 188-193.
2. Fieser, Louis F. y Williamson, Kenneth, Organic Experiments., D.C. Heath and
Company, EU, 1992, pp. 261.
3. Mohring, Nienhius, et al., J. Chem., Ed., EU, 62, pp. 519, 1985.
4. Pernkins, R.A. y Chau, Felix, J. Chem., Ed., EU, 59, pp. 981, 1982.
5. Solomons, T.W. Graham, Fundamentals of Organic Chemistry, John Wiley and
Sons, EU, 1994, pp. 435, 532.
6. Streitwieser, Andrew y Clayton H., Heath Cock, Introduction to Organic Chemistry,
McMillan Publishing, Republica de Singapur, 1992, pp. 228.
7. Zuczek, N.M. y Furth P.S., J. Chem., Ed., EU, 58, pp. 824, 1981.
ESPECTROS DE I.R.
a) Ciclohexanol

Practica # 11 Síntesis de Ciclohexanona por oxidación de Ciclohexanol
128
b) Ciclohexanona

Practica # 12 Nitración de la Anilina
129
PRACTICA 12: SUSTITUCIÓN ELECTROFÍLICA
AROMÁTICA: NITRACIÓN DE LA ANILINA
INTRODUCCIÓN
La reacción de nitración del anillo aromático es uno de los recursos más
utilizados para introducir un nuevo grupo funcional en el mismo. Se trata de un
proceso de sustitución electrofílica aromática y, como tal, su regioquímica depende
de los sustituyentes en el anillo aromático.
En nuestro caso el anillo aromático de la anilina (1) está muy activado (rico en
densidad electrónica) debido a la capacidad que tiene el par de electrones del
nitrógeno para deslocalizarse a través del sistema pi ( efecto +R).
Por esta razón la anilina experimenta reacciones de sustitución electrofílica
con mucha facilidad pudiendo dar lugar a productos de polisustitución en las
posiciones orto y para, por lo que no se puede llevar a cabo la reacción de nitración
de la anilina sin un paso de protección del grupo amino. Además, debido al carácter
básico de este grupo tendría lugar una reacción ácido-base con los reactivos que se
utilizan en la nitración.
Transformando la anilina en una amida (ej: acetanilida, 2) el grupo amino
queda protegido. Así se evita que tengan lugar reacciones de tipo ácido-base y se
disminuye la reactividad del sistema. La razón es que en las amidas hay una
estructura resonante resultante de la deslocalización del par de electrones del
nitrógeno en el doble enlace C=O de la amida, haciendo al par de electrones menos
accesible al anillo aromático. Una ventaja adicional de la formación de amidas es que
el mayor tamaño del grupo amida favorece la sustitución en la posición 4 frente a la
2, por estar esta última más impedida.
NH2
Ac2O
NHCOCH3
HNO3/H2SO4
(1) (2) (3)
NH2
NO2
NHCOCH3
NO2
H+
(4)

Practica # 12 Nitración de la Anilina
130
REACTIVOS
1. PREPARACIÓN DE LA ACETANILIDA (2)
Anilina ( d= 1,022) 2,5 mL
Anhídrido acético 3 mL
Ácido acético glacial 6 mL
2. PREPARACIÓN DE LA P-NITROACETANILIDA (3)
Acetanilida
H2SO4(concentrado)
HNO3 ac. Concentrado (70%, d = 1,42)
Etanol
3. DESPROTECCIÓN DEL GRUPO NH2: PREPARACIÓN DE
P-NITROANILINA (4)
p-Nitroacetanilida 1,5 g
Ácido clorhídrico 5M 25 ml
Disolución acuosa de hidróxido sódico al 25 %
Advertencia: La anilina es muy tóxica, por lo que es conveniente utilizarla
siempre en la campana. Los ácidos concentrados producen importantes quemaduras.
Debe evitarse en todo momento el contacto con la piel y la ropa.
PROCEDIMIENTO
1. Preparación de la acetanilida (2)
En un matraz provisto de imán se introduce el ácido acético. A continuación
se adicionan la anilina y el anhídrido acético y se mantiene la mezcla de reacción
agitando a temperatura ambiente durante unos 10 minutos (reacción exotérmica).
Transcurrido este tiempo el matraz estará ya frío y la mezcla de reacción se diluye
con 50-100 mL de agua. El producto se separa mediante extracción y es de pureza
suficiente para ser utilizado en el apartado 2 aunque se debe de reservar una pequeña
porción para recristalizarla en etanol y anotar el punto de fusión del producto
recristalizado.

Practica # 12 Nitración de la Anilina
131
2.Preparación de la p-nitroacetanilida (3)
Pesar 2,5 g de acetanilida en un matraz de 50 mL provisto de imán. Se coloca
el matraz en un baño de hielo y mientras se agita se añaden 5 mL de ácido sulfúrico
concentrado en porciones de 1 mL y durante un tiempo aproximado de un minuto.
Paralelamente se prepara la mezcla sulfonítrica en un vaso de precipitados de
100 mL que deberá estar en baño de hielo. Con cuidado se mezcla 2 mL de ácido
sulfúrico concentrado y 2,7 mL de nítrico concentrado.
¡¡¡MUCHÍSIMA PRECAUCIÓN EN EL MANEJO DE LOS ÁCIDOS
CONCENTRADOS!!!
La mezcla sulfonítrica preparada se añade gota a gota (utilizar pipeta Pasteur)
sobre la disolución de acetanilida sometida a agitación mecánica y rodeada del baño
de hielo. El tiempo de adición debe de ser de aproximadamente 20_25 min. ( Si la
adición es más rápida o la temperatura sube por encima de 10 ºC pueden tener lugar
polinitraciones indeseadas). Una vez concluida la adición se retira el baño de hielo y
se mantiene la agitación otros 10 min.
A continuación se vierte el contenido del matraz sobre un vaso de precipitados
que contenga una cuarta parte de hielo, se agita con una varilla de vidrio hasta que el
hielo se funde y se forma un precipitado granular amarillento. El producto de
reacción se aísla mediante extracción, se purifica por recristalización en etanol y se
determina el punto de fusión del producto recristalizado.
3. Desprotección del grupo NH2: Preparación de p-nitroanilina (4)
En un matraz de 100 Ml se coloca la p-nitroacetanilida y se adiciona el HCl
5M (25 mL). Se coloca un refrigerante en posición de reflujo y se calienta la mezcla a
ebullición hasta que todo el material se disuelve y mantiene el calentamiento de la
mezcla durante otros 10 min. a partir de ese momento. Se enfría la disolución y se
adiciona cuidadosamente la disolución acuosa de hidróxido sódico al 25% hasta pH
alcalino (comprobar utilizando papel indicados). El producto se separa mediante
extracción y se recristaliza con una mezcla de etanol y agua. Tras el aislamiento del
producto recristalizado se determina el punto de fusión y el rendimiento.
CUESTIONES
1) Formular las formas resonantes para los distintos tipos de ataque elctrófilico en la
anilina.

Practica # 12 Nitración de la Anilina
132
2) En soluciones fuertemente ácidas la anilina es muy poco reactiva frente al ataque
electrofílico y se observa un aumento en la proporción del producto sustituido en
meta. Explíquese el por qué.
3) Proponer una ruta sintética para la obtención del isómero de posición 2-
nitroanilina (onitroanilina)
4) Calcular el rendimiento del producto bruto obtenido, así como el del proceso de
recistalización para cada uno de los pasos. ¿Cuál será el rendimiento global de la
secuencia sintética?
ESPECTROS DE I.R.
Anilina

Practica # 12 Nitración de la Anilina
133
Acetanilida
p-nitroacetanilida

Practica # 12 Nitración de la Anilina
134
p-nitroanilina