mapping the protein-dna interface and the metal-binding site of the major human...
TRANSCRIPT

doi:10.1006/jmbi.2000.3653 available online at http://www.idealibrary.com on J. Mol. Biol. (2000) 298, 447±459
Mapping the Protein-DNA Interface and theMetal-binding Site of the Major HumanApurinic/Apyrimidinic Endonuclease
Lam H. Nguyen, Daniel Barsky, Jan P. Erzberger andDavid M. Wilson III*
Molecular and StructuralBiology Division, LawrenceLivermore National LaboratoryP.O. Box 808, L-441Livermore, CA 94551, USA
Present address: J. P. Erzberger, DMolecular and Cellular Biology UniBerkeley CA 94720, USA.
Abbreviations used: BER, base exapurinic/apyrimidinic; OH � , hydrotetrahydrofuran; WT, wild-type; D,
E-mail address of the [email protected]
0022-2836/00/030447±13 $35.00/0
Apurinic/apyrimidinic (AP) endonuclease Ape1 is a key enzyme in themammalian base excision repair pathway that corrects AP sites in thegenome. Ape1 cleaves the phosphodiester bond immediately 50 to APsites through a hydrolytic reaction involving a divalent metal co-factor.Here, site-directed mutagenesis, chemical footprinting techniques, andmolecular dynamics simulations were employed to gain insights intohow Ape1 interacts with its metal cation and AP DNA. It was found thatApe1 binds predominantly to the minor groove of AP DNA, and thatresidues R156 and Y128 contribute to protein-DNA complex stability.Furthermore, the Ape1-AP DNA footprint does not change along its reac-tion pathway upon active-site coordination of Mg2� or in the presence ofDNA polymerase beta (polb), an interactive protein partner in AP siterepair. The DNA region immediately 50 to the abasic residue was deter-mined to be in close proximity to the Ape1 metal-binding site. Exper-imental evidence is provided that amino acid residues E96, D70, andD308 of Ape1 are involved in metal coordination. Molecular dynamicssimulations, starting from the active site of the Ape1 crystal structure,suggest that D70 and E96 bind directly to the metal, while D308 coordi-nates the cation through the ®rst hydration shell. These studies de®ne theApe1-AP DNA interface, determine the effect of polb on the Ape1-DNAinteraction, and reveal new insights into the Ape1 active site and overallprotein dynamics.
# 2000 Academic Press
Keywords: AP endonuclease; Ape1; base excision repair; DNA binding;metal coordination
*Corresponding authorIntroduction
In order to maintain genetic integrity, organismshave developed various means to repair damagedDNA. The base excision repair (BER) pathwaytypically involves the removal of a single damagednucleotide or baseless site from the DNA(reviewed by Mol et al., 1999). Apurinic/apyrimi-dinic (AP) sites are formed from spontaneoushydrolysis of the N-glycosyl bond, from attack of
epartment ofversity of California,
cision repair; AP,xyl radical; F,distance.ng author:
bases by free radicals, or by the action of repairenzymes called DNA N-glycosylases whichremove damaged or unconventional bases(reviewed by McCullough et al., 1999). Ape1, themajor mammalian AP endonuclease, is an essentialcomponent of the BER pathway (Xanthoudakiset al., 1996).
Ape1, in the presence of Mg2�, cleaves the phos-phodiester bond immediately 50 to an AP site, gen-erating a 30 OH group and a 50 deoxyribose moiety(reviewed by Demple et al., 1994). The single resi-due gap can then be ®lled by DNA polymerasebeta (polb) (reviewed by Wilson, 1998), andthe remaining 50-deoxyribose phosphate groupexcised by its phosphodiesterase activity(Matsumoto & Kim, 1995). The ®nal nick is sealedby DNA ligase I or an XRCC1-DNA ligase III com-plex (Prasad et al., 1996; Caldecott et al., 1994).
# 2000 Academic Press

448 AP DNA Interactions and Divalent Metal Coordination of Ape1
Although Ape1 and polb do not form a stable pro-tein-protein complex (Dimitriadis et al., 1998), polbdoes bind Ape1-AP DNA binary complexes toform a higher-order ternary complex, and thephosphodiesterase activity of polb is accelerated bythe presence of Ape1 (Bennett et al., 1997). polbalso forms a complex with XRCC1 (Caldecott et al.,1996; Kubota et al., 1996) or mammalian DNAligase I (Prasad et al., 1996). In addition, polbb, theXRCC1 N-terminal domain, and a gapped DNAsubstrate can form a higher-ordered ternary com-plex in vitro (Marintchev et al., 1999). These datasuggest coordination of various enzymatic steps inthe BER pathway. Besides AP endonucleaseactivity, Ape1 acts as a 30-phosphodiesterase,removing lesions resulting from oxidative damageof DNA such as 30-phosphoglycolates (Suh et al.,1997), and has both 30 to 50 exonuclease andRNAseH activities (reviewed by Demple &Harrison, 1994; Rothwell & Hickson, 1997). It isnoteworthy that these activities of Ape1 are rela-tively poor in comparison to those exhibited by itsbacterial counterpart, exonuclease III (ExoIII).
AP endonucleases are classi®ed into two familiesbased on amino acid sequence homology to Escher-ichia coli ExoIII or endonuclease IV (EndoIV)(reviewed by Demple & Harrison, 1994). Ape1 ishomologous to ExoIII. The three-dimensional struc-tures of Ape1, ExoIII, EndoIV, and the EndoIV-APDNA complex have been determined by X-raycrystallography (Mol et al., 1995; Gorman et al.,1997; Hos®eld et al., 1999). EndoIV inserts side-chains into the DNA base stack through the minorgroove, compresses the DNA backbone, bends theDNA 90 �, and promotes double-nucleotide ¯ip-ping to sequester the extrahelical AP site into theenzyme catalytic pocket (Hos®eld et al., 1999). Themolecular details of the Ape1-AP DNA interactionsare not fully known, yet biochemical studies haveshed signi®cant light on its repair reaction.
Based on kinetic and binding studies of Ape1and its mutants, the reaction pathway, with a mini-mal number of complexes, is as follows: Ape1binds speci®cally to AP DNA in the absence ofMg2� to form a stable intermediate complex(Wilson et al., 1997). This complex is thenconverted to a catalytically competent complex inthe presence of Mg2�. Catalysis subsequentlyoccurs, resulting in cleavage of the phosphodiesterbond immediately 50 to the AP site and the for-mation of a protein-product complex. This complexthen dissociates, releasing Ape1 from nicked APDNA (Lucas et al., 1999). Product dissociationappears to be Mg2� concentration-dependent(Masuda et al., 1998b).
Ape1 requires at least four base-pairs 50 andthree base-pairs 30 of an AP site for incision activity(Wilson et al., 1995). For the AP strand, methyl-ation of guanine residues located one or threebase-pairs 50 of the AP site, or ethylation of phos-phate groups two or three positions 30 of the APsite prevented Ape1-AP DNA binary complex for-mation. While no phosphate ethylation interference
was detected for the complementary strand,methylation at two base-pairs 50, or one or threebase-pairs 30 of the AP site impaired Ape1 binding(Wilson et al., 1997). These data provided the®rst information of how Ape1 engages its targetsubstrate.
Here, we re®ne our understanding of the mol-ecular interactions of Ape1 and AP DNA, examinethe effect of polb and Mg2� on the DNA structureof Ape1-AP DNA binary complexes, map the Ape1metal-binding site in terms of proximity to theDNA substrate, and provide direct evidence of theamino acid residues involved in metal coordi-nation.
Results
Ape1 protects six to seven bases on eitherDNA strand around the AP site and thepresence of polbbb does not change the footprint
The purity of the Ape1 proteins used in thiswork is shown in Figure 1(a). To determine howApe1 interacts with AP DNA, we employed thechemical footprinting reagent hydroxyl radical(OH � ). Due to their small size, OH � are usefulprobes for studying DNA contacts at high resol-ution (Dixon et al., 1991). OH � cleave the DNAdirectly by attacking the deoxyribose ring(Hertzberg & Dervan, 1984; Balasubramanian et al.,1998). The double-stranded DNA used in thisstudy was a 26 bp duplex with tetrahydrofuran(F), an abasic site analog (Wilson et al., 1995), nearthe center (Figure 1(b)).
As shown in Figure 2, Ape1 protects seven baseson the strand containing F and six bases on thecomplementary strand. The F residue is located inthe center of the protected region on the abasicstrand. The guanine base opposite the F residue isstrongly protected, whereas the surrounding basesare less protected by Ape1 from OH � -mediatedcleavage. There is a hypersensitive band within thefootprint that migrates at the same position on thegel as the Ape1 cleavage product. A simpleinterpretation is that this ``hypersensitivity'' is theresult of Ape1 incision due to residual enzymaticactivity even in the presence of EDTA. Consistentwith this interpretation, at the necessarily highlevels of Ape1 used in the footprinting assays,Ape1 incised an amount of labeled AP DNA(�1 %) similar to that present in the hypersensitiveband. In addition, as shown below, such hypersen-sitivity is not observed within the footprint of thecatalytically inactive Ape1 mutant D210N. AD210N mutation reduces incision activity by25,000-fold, without affecting the speci®c DNA-binding activity of Ape1, indicating a critical rolefor this residue in the catalytic reaction (Erzberger& Wilson, 1999).
Using the OH � footprinting approach, we exam-ined whether the presence of polb, which has beenshown to form a ternary complex with Ape1 andAP DNA (Bennett et al., 1997), causes any change

Figure 1. Protein and AP DNA substrate reagentsused in this work. (a) Puri®ed Ape1 mutant proteins.Ape1 proteins (�1.0 mg) used in this work were fractio-nated on an SDS/12 % polyacrylamide gel and stainedwith Coomassie blue dye. Lane 1, wild-type Ape1; lane2, D210N mutant; lane 3, D308A; lane 4, D70R, lane 5,D210N/D308A double mutant; lane 6, E96Q; lane 7,H309S; lane 8, N68A; lane 9, R156Q; lane 10, Y128A,and lane 11, D210N/D70A double mutant. The proteinmolecular mass standards (in kDa) are indicated on theright. (b) The duplex DNA substrate. 26F is 50-AATT-CACCGGTACCFTCTAGAATTCG-30, 26G is the comp-lementary strand where a G is positioned directlyopposite F. F is the tetrahydrofuran residue, a syntheticabasic site analog (Wilson et al., 1995). The arrow indi-cates the phosphodiester linkage incised by Ape1.
Figure 2. Ape1 binds in the minor groove and DNApolymerase b does not cause a change in the Ape1-speci®c footprint. (a) The OH � footprint of Ape1-APDNA complex. Lanes 1 to 7 are samples with labeled26F strand; lanes 8 to 14 are with labeled 26G strand.Lanes 1 and 14 are the no cleavage agent controls.Lanes 2 and 13 are the no protein controls with 10 mMFe(AS)2. Lanes 3 and 12 are the no protein controls with5 mM Fe(AS)2. Lanes 4 to 11 are with 10 mM Fe(AS)2.Lanes 4 and 11 are reactions with 160 nM Ape1; lanes 5and 10, 160 nM Ape1 and 580 nM polb; lanes 6 and 9,160 nM Ape1 and 1.74 mM polb; lanes 7 and 8, 1.74 mMpolb. (b) Summary of the OH � footprint data. The ®lledvertical bars above each base indicate protection fromcleavage by OH � in solution. The height indicates therelative strength of footprint protection as determinedby Phosphorimager scans of three independent exper-iments.
AP DNA Interactions and Divalent Metal Coordination of Ape1 449
in the footprint. As shown in Figure 2, we cannotdetect any difference in the footprint pattern ofApe1-AP DNA complexes in the presence of polb,indicating that polb does not bind elsewhere to theDNA, and that either polb binds directly to Ape1without changing the Ape1-DNA interactions orreplaces exactly some of the Ape1-DNA contacts.Ternary complexes were observed by gel retar-dation assays (Bennett et al., 1997; data not shown).
Ape1 mutants Y128A and R156Q have reducedAP DNA binding activity
The non-speci®c nuclease DNaseI displays struc-tural similarity to the ExoIII family of proteins(Mol et al., 1995). Comparison of the crystal struc-tures of Ape1 and the DNaseI-DNA complex led toa proposed model for the Ape1-AP DNA binarycomplex (Gorman et al., 1997). In this model, Y128and R156 residues of Ape1 are implicated in DNAcontacts. To test this prediction and to better de®nethe DNA-binding interface of Ape1, we con-
structed Y128A and R156Q Ape1 mutants andasked if there was a loss in DNA-binding af®nityby gel retardation assays. As shown in Figure 3,both the Y128A and R156Q mutations resulted in a>100-fold reduced DNA-binding capacity (with acorresponding reduction in speci®c incision activityof �fourfold and �70-fold, respectively), consistentwith the involvement of these residues in AP DNAcomplex stability.
Sites of AP DNA in proximity to themetal-binding site of Ape1
To gain additional information regarding thetopography of Ape1-AP DNA binary complex, wedetermined which bases of the AP DNA substrateare located near the metal-binding site of Ape1 byemploying an Fe2�-cleavage assay (Mustaev et al.,
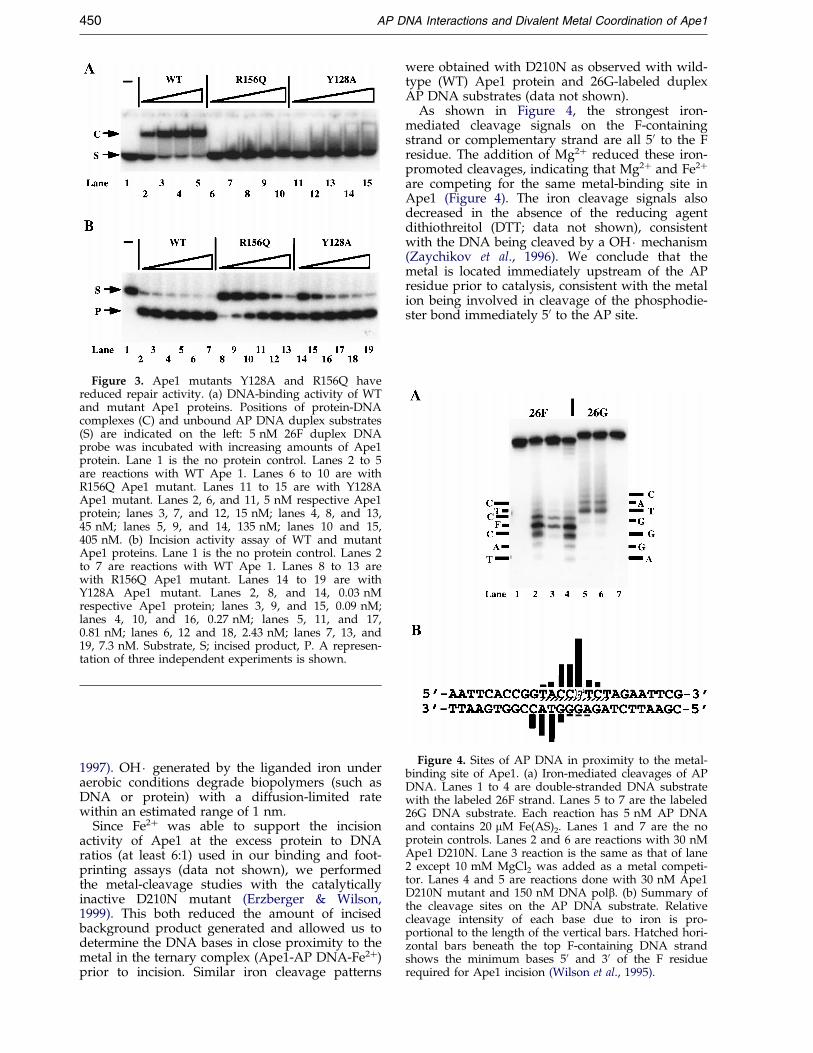
Figure 3. Ape1 mutants Y128A and R156Q havereduced repair activity. (a) DNA-binding activity of WTand mutant Ape1 proteins. Positions of protein-DNAcomplexes (C) and unbound AP DNA duplex substrates(S) are indicated on the left: 5 nM 26F duplex DNAprobe was incubated with increasing amounts of Ape1protein. Lane 1 is the no protein control. Lanes 2 to 5are reactions with WT Ape 1. Lanes 6 to 10 are withR156Q Ape1 mutant. Lanes 11 to 15 are with Y128AApe1 mutant. Lanes 2, 6, and 11, 5 nM respective Ape1protein; lanes 3, 7, and 12, 15 nM; lanes 4, 8, and 13,45 nM; lanes 5, 9, and 14, 135 nM; lanes 10 and 15,405 nM. (b) Incision activity assay of WT and mutantApe1 proteins. Lane 1 is the no protein control. Lanes 2to 7 are reactions with WT Ape 1. Lanes 8 to 13 arewith R156Q Ape1 mutant. Lanes 14 to 19 are withY128A Ape1 mutant. Lanes 2, 8, and 14, 0.03 nMrespective Ape1 protein; lanes 3, 9, and 15, 0.09 nM;lanes 4, 10, and 16, 0.27 nM; lanes 5, 11, and 17,0.81 nM; lanes 6, 12 and 18, 2.43 nM; lanes 7, 13, and19, 7.3 nM. Substrate, S; incised product, P. A represen-tation of three independent experiments is shown.
Figure 4. Sites of AP DNA in proximity to the metal-binding site of Ape1. (a) Iron-mediated cleavages of APDNA. Lanes 1 to 4 are double-stranded DNA substratewith the labeled 26F strand. Lanes 5 to 7 are the labeled26G DNA substrate. Each reaction has 5 nM AP DNAand contains 20 mM Fe(AS)2. Lanes 1 and 7 are the noprotein controls. Lanes 2 and 6 are reactions with 30 nMApe1 D210N. Lane 3 reaction is the same as that of lane2 except 10 mM MgCl2 was added as a metal competi-tor. Lanes 4 and 5 are reactions done with 30 nM Ape1D210N mutant and 150 nM DNA polb. (b) Summary ofthe cleavage sites on the AP DNA substrate. Relativecleavage intensity of each base due to iron is pro-portional to the length of the vertical bars. Hatched hori-zontal bars beneath the top F-containing DNA strandshows the minimum bases 50 and 30 of the F residuerequired for Ape1 incision (Wilson et al., 1995).
450 AP DNA Interactions and Divalent Metal Coordination of Ape1
1997). OH � generated by the liganded iron underaerobic conditions degrade biopolymers (such asDNA or protein) with a diffusion-limited ratewithin an estimated range of 1 nm.
Since Fe2� was able to support the incisionactivity of Ape1 at the excess protein to DNAratios (at least 6:1) used in our binding and foot-printing assays (data not shown), we performedthe metal-cleavage studies with the catalyticallyinactive D210N mutant (Erzberger & Wilson,1999). This both reduced the amount of incisedbackground product generated and allowed us todetermine the DNA bases in close proximity to themetal in the ternary complex (Ape1-AP DNA-Fe2�)prior to incision. Similar iron cleavage patterns
were obtained with D210N as observed with wild-type (WT) Ape1 protein and 26G-labeled duplexAP DNA substrates (data not shown).
As shown in Figure 4, the strongest iron-mediated cleavage signals on the F-containingstrand or complementary strand are all 50 to the Fresidue. The addition of Mg2� reduced these iron-promoted cleavages, indicating that Mg2� and Fe2�
are competing for the same metal-binding site inApe1 (Figure 4). The iron cleavage signals alsodecreased in the absence of the reducing agentdithiothreitol (DTT; data not shown), consistentwith the DNA being cleaved by a OH � mechanism(Zaychikov et al., 1996). We conclude that themetal is located immediately upstream of the APresidue prior to catalysis, consistent with the metalion being involved in cleavage of the phosphodie-ster bond immediately 50 to the AP site.

Figure 5. The effect of Ape1 mutations proximal tothe metal-binding site. (a) The effect of mutations ofamino acid residues in proximity to the metal-bindingsite on iron-mediated cleavage of the labeled 26G DNAstrand. Each reaction has 5 nM AP DNA. Lane 1 is theno iron control. Lane 2 is the no protein control. Lane 3is reaction with 30 nM D210N mutant; lane 4, 30 nMD308A; lane 5, 30 nM D70R; lane 6, 30 nM E96Q; lane7, 30 nM N68A; lane 8, 30 nM H309S; and lane 9,90 nM D308A. (b) Mutations D70A and D308A reduceiron-mediated cleavage of both the 26F strand and itscomplement 26G. Lanes 1 to 5 are with labeled 26Fstrand, and lanes 6 to 9 are with labeled 26G comp-lementary strand. Each reaction has 5 nM AP DNA.Lane 1 is the no iron control reaction. All other lanes arereactions with 20 mM Fe(AS)2. Lanes 2 and 6 are the noprotein controls. Lanes 3 and 7 are reactions with 30 nMD210N Ape1 mutant; lanes 4 and 8, 30 nM D210N/D70A double mutants; and lanes 5 and 9, 30 nMD210N/D308A. The arrows on the left and/or right sideindicate the positions of four strongest iron cleavagesignals.
AP DNA Interactions and Divalent Metal Coordination of Ape1 451
Effects of amino acid mutations on themetal-binding function of Ape1
In the crystal structure of Ape1, a samarium ionis located within 6 AÊ of residues E96, D70, D308,H309, and N68 (Gorman et al., 1997), all of whichare capable of coordinating a cationic metal. Toexamine which residues among these affect Ape1metal-binding activity, we determined the iron-mediated cleavage signals of correspondingmutant proteins. We reasoned that if an aminoacid is directly or indirectly involved in metal-binding, then appropriate mutation of this residuewould cause a decrease in the binding of Fe2�,thereby reducing the iron-mediated cleavage signalof the complementary strand of duplex AP DNAsubstrates. We assayed only the complementarystrand because several Ape1 mutants, excludingD210N and H309S, display signi®cant catalyticactivity at the saturating protein concentrationsused in these experiments.
When compared to the D210N Ape1 mutant,H309S and N68A did not cause a decrease in iron-promoted cleavages (Figure 5(a)). Conversely,E96Q, D308A, and D70R, all exhibited a substantialreduction in the iron cleavage signal, with D70Rand D308A showing the most severe reduction.Even though the D308A mutant has been shown toexhibit a modest twofold decrease in AP DNA af®-nity (Erzberger & Wilson, 1999), increasing theD308A protein concentration by threefold does notlead to a corresponding threefold increase in ironcleavage signal. Thus, D308A is at saturating levelsrelative to the DNA substrate in this assay and thedecrease in signal is not due to a decrease in theDNA equilibrium binding constant. E96Q, D70Rand N68A mutants have an AP DNA-binding af®-nity nearly identical with WT (Erzberger & Wilson,1999; data not shown). The speci®c AP endonu-clease activities of E96Q, D70R, and N68A Ape1mutants are reduced �2200, �27, and �600-fold,respectively, relative to that of WT Ape1(Erzberger & Wilson, 1999; data not shown).
We also constructed double mutants containingD210N with D70A or D308A to allow us to assayboth DNA strands for iron-promoted cleavage. Weconstructed the double mutant with D70A insteadof D70R because alanine is a more subtle mutation.Both double mutants showed a severe decrease iniron-speci®c cleavage on both strands of F-contain-ing DNA duplexes (Figure 5(b)), consistent withthe results for the D308A and D70R single mutants(Figure 5(a)). Both double mutants were found tobind F DNA substrates as well as D210N and WTApe1 protein (data not shown).
D70R and D308A Ape1 mutants exhibitreduced activity in low Mg2�
Previous characterization of D70R and D308Amutants revealed only a minor reduction in enzy-matic activity in buffers containing 10 mM MgCl2(Erzberger & Wilson, 1999). The results of the Fe2�-
cleavage studies led us to assess the effect of Mg2�
concentration on the incision activity of thesemutants, as previous studies revealed that an E96Amutant is more sensitive to reduced Mg2� concen-trations relative to WT (Barzilay et al., 1995). Athigh levels of Mg2� (5 mM), D70R and D308AApe1 mutants cleaved AP DNAs nearly as well asWT Ape1 protein. However, at Mg2� concen-trations of 1 mM and less, the activity of the

452 AP DNA Interactions and Divalent Metal Coordination of Ape1
mutants was sharply reduced in comparison toWT protein (Figure 6). This result further supportsa role for D70 and D308 in metal coordination.
Molecular dynamics simulations show flexibleloop regions and suggest a potentialmechanism for metal coordination
To gain new insights into the overall Ape1 pro-tein dynamics, active-site residue interactions, andmetal-binding, all-atom molecular dynamics simu-lations were performed. Two simulations of theWT human Ape1 protein, F-sim and E-sim (whichdiffer methodologically in that they employ eithera force-shifted cutoff or a particle mesh Ewaldsummation to calculate the electrostatic inter-actions, respectively), were executed. Both 500 pssimulations were started from the crystal structurereported by Gorman et al. (1997), except that theSm3� cation was replaced with Mg2� as describedin Materials and Methods. The average deviationof backbone heavy atoms (non-hydrogen) for theentire protein molecule converged within 100 ps,yielding an overall RMSD of 1.07(�0.05) AÊ and1.11(�0.04) AÊ over the last 400 ps for E-sim and F-sim, respectively. This result suggests that the pro-tein maintains structural integrity and similardynamics throughout both simulations. The RMSDvaluesfor all heavy atoms (backbone and side-chains)are slightly larger at 1.48(�0.05) AÊ (E-sim) and1.61(�0.05) AÊ (F-sim), as would be expected whenfactoring in typical side-chain movements. Predic-tably, the highest motion occurs primarily on theprotein surface (Figure 7). Most notably, threeregions (one that includes residue R177, one thatincludes N229, and one that includes residueM270), which appear to be involved in AP site rec-
Figure 6. The AP endonuclease activity of D70R andD308A mutants is hypersensitive to low Mg2� concen-trations: 10 nM labeled AP DNA and 0.34 nM (0.13 ng)(Hang et al., 1997) wild-type, D70R, or D308A Ape1were incubated together at 37 �C for ten minutes. Lane 1is the no protein control (ÿ). Lanes 2 to 5 are reactionswith wild-type Ape1; lanes 6 to 9, D70R Ape1 mutant;and lanes 10 to 13, D308A Ape1 mutant. Concentrationof MgCl2 in mM (0.1, 0.5, 1, or 5) is indicated aboveeach lane. Substrate, S; incised product, P.
ognition (Gorman et al., 1997; Cal et al., 1998),show the most dramatic side-chain movements. Infact, the backbone of the M270-containing region isthe most ¯exible section of the entire protein back-bone, showing twice the average RMS ¯uctuations.We suggest that these ¯exible loop regions mayundergo conformational shifts during speci®cDNA binding.
Relevant to the biochemical studies here, weexamined in detail the active-site catalytic residuessurrounding the metal cation. Our analysis is pre-sented for the E-sim only, since it showed slightlysmaller deviations from the crystal structure thanthe F-sim. The duration of binding to or molecularcoordination of an ion is expressed as the prob-ability (Pdist) that the bond exists at any given time,based on a maximum distance criterion denotedby dist. Such bonds can also be expressed bythe average distance (D) between the two atoms(closest non-hydrogen atoms) throughout the simu-lation. A carbonyl oxygen atom of E96 binds Mg2�
at D � 1.83(�0.05) AÊ with P2.4 AÊ � 1.0 (Figure 8),while the other carbonyl oxygen atom of E96 ishydrogen bonded to Y171 (D � 3.06(�0.61) AÊ withP3.2 AÊ � 0.69). The Mg2� is also coordinated by D70(D � 1.83(�0.04) AÊ , P2.4 AÊ � 1.0) and four active-sitewater molecules (all with D � 2.0(�0.1) AÊ ,P2.4 AÊ � 1.0). Thus, Mg2� is bound tightly by boththe carboxylate residues of D70 and E96 and ashell of four water molecules. This bindingoccurred after rapid rearrangements of the watermolecules and amino acid residues in the crystalstructure, yet is so tight that it cannot be said tohave sampled all possible arrangements. D308does not bind to the ion directly, but rather the®rst hydration shell of Mg2� (relative to Mg2�,D � 4.3(�0.4) AÊ , P5 AÊ � 0.92) as shown in Figure 8.This binding is considerably less tight than direction coordination, and one noteworthy difference ofthe F-sim is the ability of D308 to ``¯ip-out'' intothe solvent (D � 5.8(�1.7) AÊ , P5 AÊ � 0.6 for the F-sim). While N68 more consistently coordinates the®rst solvation shell of Mg2� (D � 4.2(�0.2) AÊ ,P5 AÊ � 1.0) than D308, this is likely a result of N68simply being more rigidly positioned within theactive-site, since the biochemical studies do notsupport a major role for this residue in metalcoordination. D210 rarely reaches the ®rst sol-vation shell of Mg2� (D � 6.6(�0.2) AÊ , P5 AÊ � 0.0),consistent with the biochemical data indicating noprominent role for D210 in metal coordination.
No dramatic DNA conformational change wasdetected in the Ape1-DNA binary complexupon the addition of Mg2�
To analyze whether there is any change in DNAsubstrate conformation upon metal coordination,but prior to phosphate hydrolysis, we determinedthe OH � footprint of the catalytically inactive Ape1mutant, D210N, with and without a divalent metalion. We reasoned that Ape1 forms the ®rst Ape1-AP DNA complex in the presence of EDTA, and

Figure 7. The solvent-accessible surface of the non-hydrogen atoms of Ape1 (stereo image), depicting atompositions averaged over a 500 ps molecular dynamics simulation (E-sim). The color ordering, blue, green, yellow andrust, indicates the degree of motional ¯uctuations (RMSF) from largest to smallest. The magnesium ion within theactive site is colored red. The three ¯exible loop regions harboring residue R177 or N229 (to the right) or encompass-ing residues Y269, M270, M271 and N272 (to the left) are shown. These Figures were created within VMD(Humphrey et al., 1996), using SURF (Varshney et al., 1994) to calculate the surfaces.
AP DNA Interactions and Divalent Metal Coordination of Ape1 453
that D210N would form a catalytically competentcomplex in terms of metal coordination (Figure 5).As shown in Figure 9, the Ape1 D210N footprintdoes not change upon the addition of Mg2�. Fur-thermore, the D210N footprint in the presence ofMg2� is indistinguishable from the footprint of WTApe1 (Figure 2; with the exception of the hypersen-sitivity, which likely represents residual cleavageactivity seen with the WT protein). We concludethat there is no major DNA structural changewhen the Ape1-AP DNA complex is converted to aMg2�-coordinating ternary complex.
Figure 8. In the same orientation as Figure 7 is shown alation (left) or captured after 25 ps of dynamics (right). The Fwithin the crystal structure without hydrogen atoms (Gormathe carbonyl oxygen atoms of D70 and E96 coordinate the mthe simulation (right). All electrostatic interactions of less thalines.
Discussion
Ape1 interactions with AP DNA
It has been shown that the abasic deoxyribosesugar ring or an extrahelical base are not essentialcomponents in AP site recognition by Ape1(Wilson et al., 1995; Erzberger et al., 1998; Erzberger& Wilson, 1999). The molecular dynamics simu-lations performed here reveal that Ape1 possessesthree ¯exible loop domains (Figure 7) that areimportant for speci®c binding to AP site-containing
detailed view of the Ape1 active site, prior to the simu-igure to the left indicates the positions of Ape1 residues
n et al., 1997). Four water oxygen atoms (navy blue) andagnesium ion (green sphere) uninterruptedly throughoutn 3 AÊ (left) and 2.4 AÊ (right) are shown as broken black

Figure 9. No major DNA conformational change isdetected upon the coordination of Mg2�. Lanes 1 to 5are with labeled 26F strand; lanes 6 to 10 are withlabeled 26G strand. Lanes 1 and 10 are the no cleavageagent controls. Each reaction contains 4 nM AP DNAduplex. Lanes 2 and 9 are the no protein controls with10 mM Fe(AS)2. Lanes 3 and 8 are the no protein con-trols with 5 mM Fe(AS)2. Lanes 4 to 7 are with 10 mMFe(AS)2. Lanes 4 and 7 are reactions with 160 nMD210N Ape1 with 5 mM EDTA; lanes 5 and 6, 160 nMApe1 and 5 mM MgCl2 instead of 5 mM EDTA. Com-plexes of AP DNA-Ape1-Mg2� were formed prior to the30 second treatment with the footprint reagents. Thevertical bars on the left and right side of the Figure indi-cate the DNA region protected from cleavage by OH �generated by the Fe(EDTA) complex.
454 AP DNA Interactions and Divalent Metal Coordination of Ape1
DNA substrates (Gorman et al., 1997; Cal et al.,1998).
While the structure of AP DNA has been shownto deviate only slightly from unmodi®ed DNA,NMR and computational studies have found kink-ing, melting, and structural ¯uctuations around theabasic site, indicating a unique ¯exibility local tothe AP lesion (Coppel et al., 1997; Ayadi et al.,1999; Barsky et al., 2000). Similar increased ¯exi-bility is also observed with gapped DNA (Rollet al., 1998), an oligo substrate that is bound byApe1 (Masuda et al., 1998a). However, nicked
DNA, which retains an unkinked B-DNA confor-mation with only slight distortions at the lesionsite, is not bound by Ape1. Thus, Ape1 may beprobing, using its unique adjustable loop domains,for speci®c DNA structures and/or increased DNAbackbone ¯exibility.
Since AP DNA is thermodynamically less stablethan normal B-DNA (Gelfand et al., 1998), Ape1may induce speci®c DNA distortions that permitbinding to AP DNA. In fact, previous footprintingstudies revealed that Ape1 promotes hypersensitiv-ity to the cleavage agent Cu-1,10-phenanthroline atthe abasic residue (Wilson et al., 1997). However,the hypersensitive band in the footprinting studiespresented here (Figure 2) appears to be the resultof Ape1 incision, even in the presence of EDTA.Furthermore, when using potassium permanganate(KMnO4) as a probe (Sasse-Dwight, 1991), we didnot observe any Ape1-enhanced reactivity of thy-mine in AP DNA substrates, even when thyminewas placed in the complementary strand immedi-ately across from the AP site (L.N. and D.M.W.III,unpublished observations); KMnO4 strongly oxi-dizes unpaired or distorted pyrimidines in DNA,reacting much more strongly with thymine thancytosine residues (Sasse-Dwight, 1991). These datasuggest that Ape1 does not cause a large meltingof duplex DNA upon binding, yet does not excludethe possibility that a more subtle DNA confor-mational change occurs. The ability of Ape1 to pro-mote cleavage in EDTA at acyclic AP site analogs,which display increased backbone ¯exibility, andnot cyclic AP structures, suggests that Ape1 alone(i.e. in the absence of Mg2�) can induce structuralrearrangements in DNA necessary for hydrolysis(Erzberger & Wilson, 1999).
The groove in which Ape1 is located can beidenti®ed by analysis of the DNA protection pat-tern (Dixon et al., 1991). If the protected region oneach strand is offset by two to three base-pairs inthe 30 direction, the protein-DNA interaction takesplace in the minor groove. In contrast, if bindingoccurs in the major groove, then the protectedregions on each strand are offset in the 50 direction.The results of the footprinting experiments pre-sented here are consistent with Ape1 binding pre-dominantly in the minor groove of AP DNA(Figure 10). The other major AP endonuclease,E. coli EndoIV, also binds to the minor groove ofAP DNA (Hos®eld et al., 1999), suggesting that thismode of complex formation may be a commonmechanism used by such repair endonucleases.Thus, damage-speci®c proteins may recognizeunique structural conformations or deformities inthe minor groove of damaged DNA, in a mannersimilar to the way in which architectural proteinsrecognize unique minor groove structural distor-tions of sequence-speci®c DNA (reviewed byBewley et al., 1998).
Although Ape1 binds primarily in the minorgroove, the protein also contacts AP DNA throughmajor groove interactions as determined by pre-vious methylation interference studies (Wilson

Figure 10. The Ape1-DNA interface. Hydroxyl radical footprinting studies revealed that Ape1 protects an area ofthe AP DNA (left) of about seven bases on the abasic strand (blue) and six bases on the complementary strand(orange), with the F residue (gold) located in the center of the protected region. The arrow indicates the site of Ape1incision. It is noteworthy that previous ethylation interference studies (Wilson et al., 1997) indicate phosphate contactstwo or three positions 30 of the abasic site on the F-containing strand. In addition, methylation interference exper-iments revealed base contacts both in the minor and major grooves (not shown). Site-directed mutagenesis hasrevealed a role for speci®c amino acid residues of Ape1 in AP DNA complex stability (right), unveiling the DNA-binding groove of Ape1 (see the text for discussion). The ¯exible loop domains (harboring R177, N229 and M270) areindicated for orientation. The magnesium ion is indicated by a red sphere.
AP DNA Interactions and Divalent Metal Coordination of Ape1 455
et al., 1997). Perhaps major groove interactions aremost critical during the initial stages of AP DNAcomplex formation (i.e. during recognition, prior tothe formation of the speci®c binary complex), assigni®cant major groove contacts were notobserved with the pre-formed Ape1-AP DNA com-plexes here. Alternatively, certain methylationsmay have prevented Ape1-DNA complex for-mation indirectly by either modifying neighboringbase-protein contacts or by causing a more generalconformational change in the DNA substrate thatinterferes with Ape1 association (Yang & Carey,1995).
Ape1-polbbb coordination
No obvious difference in the Ape1 protein foot-print was detected upon the addition of polb, aprotein that forms a ternary complex with Ape1and AP DNA (Bennett et al., 1997). Thus, polb doesnot appear to alter Ape1-speci®c interactions withAP DNA or to interact with DNA directly uponbinding the Ape1-AP DNA binary complex,although polb interactions with DNA ends (whereOH � footprinting resolution is poor) cannot beexcluded. We propose that transient associationof polb with this complex is mediated throughprotein-protein interactions that are dependent onconformational changes in Ape1 that occur upon
binding AP DNA, since polb-Ape1 binary com-plexes have not been observed in the absence ofDNA (Bennett et al., 1997; Dimitriadis et al., 1998).Whether speci®c substrate conformational changesoccur during the repair reaction of Ape1 thatpromote subsequent polb DNA binding and its2-deoxyribose-5-phosphate lyase activity (Bennettet al., 1997; Xu et al., 1998) will need to bedetermined.
The Ape1 repair steps
Upon location of the AP site, the ®rst stableApe1-AP DNA binary complex is formed (Wilsonet al., 1997). Based on the crystal structure of theDNaseI-DNA complex (Lahm & Suck, 1991;Weston et al., 1992), structurally equivalent resi-dues in Ape1, Y128 and R156, were predicted tointeract with DNA and mediate binary complexassembly (Gorman et al., 1997). When these tworesidues were mutated, there was a decrease inboth protein-DNA complex stability and AP endo-nuclease activity, consistent with the predictionthat these residues are involved in AP DNA-bind-ing. N212, F266, W267, H309 and Y171 have alsobeen shown to affect Ape1-AP DNA complex stab-ility (Rothwell & Hickson, 1996; Erzberger et al.,1998; Erzberger & Wilson, 1999; Masuda et al.,1998a). The combination of the footprinting and

456 AP DNA Interactions and Divalent Metal Coordination of Ape1
site-directed mutagenesis studies provides anemerging picture of the Ape1-AP DNA interface(summarized in Figure 10).
The ®rst speci®c binary complex is then con-verted to the catalytically competent complex inthe presence of Mg2� (Lucas et al., 1999). Usingthe high-resolution OH � footprinting technique, nomajor rearrangements of the DNA structurewere detected upon the coordination of Mg2�,suggesting that there are no large conformationalchanges in DNA upon metal-binding. The lack ofmajor conformational changes in the DNA uponmetal coordination is not unprecedented. Structur-al and biochemical studies of the restriction endo-nuclease EcoRI indicate that there is no majorconformational change induced in DNA upon theaddition of Mg2� to the protein-DNA complex(reviewed by Jen-Jacobson, 1997). However, thereare structural rearrangements required for catalysisin the immediate vicinity of the scissile phospho-diester bond (reviewed by Jen-Jacobson, 1997), asseems to be the case for Ape1 (Erzberger & Wilson,1999).
Protein conformational changes are also likelynecessary after Mg2� binding for steric comple-mentarity and ef®cient catalysis to occur. ResidueD308 is likely to be necessary for such tertiarystructural changes in Ape1, since with the D308Amutant, a conformational shift involving metal-binding became an experimentally observable,rate-limiting step in the kinetic pathway (Lucaset al., 1999). The observation that D308 is capableof rotating out of the active-site (in F-sim only)may provide evidence that this residue is involvedin recruiting or releasing the metal cation to orfrom (or correctly orienting the metal within) thecatalytic pocket of Ape1.
A Fe2� cleavage technique (Mustaev et al., 1997)was employed to map the sugars of the AP DNAsubstrate proximal to the metal-binding site ofApe1 and to identify residues of Ape1 that areinvolved in metal coordination. The sugars on bothDNA strands located immediately upstream of theAP site were found to be cleaved, with the stron-gest signal at the sugar immediately 50 to the APresidue. These data are consistent with the metalbeing involved in the hydrolysis of the phospho-diester bond 50 to the lesion. We found that H309Sor N68A mutations in Ape1 did not cause adecrease in iron cleavage signals, indicating nomajor or a less signi®cant involvement for theseresidues in metal coordination. However, mutatingD70, E96, and D308 caused a substantial decreasein the intensity of the iron cleavage signals. Fur-thermore, as observed with the E96A mutant(Barzilay et al., 1995), D70R and D308A exhibitedhypersensitivity to low Mg2� concentrations incomparison to WT Ape1. These results are consist-ent with other studies that have suggested a rolefor E96 and D308 in metal-binding (Barzilay et al.,1995; Masuda et al., 1998b; Lucas et al., 1999). Ourresults are the ®rst to implicate D70 in metalcoordination, and may suggest that Ape1 main-
tains a unique metal-binding site (which may playa role in substrate speci®city) as this aspartate resi-due is not conserved in ExoIII or DNaseI (see thediscussion by Erzberger & Wilson, 1999).
Molecular dynamics simulations provided avisualization of WT Ape1 active-site dynamics andsuggest that E96 and D70 bind directly to themetal ion, while D308 interacts with the metal ionindirectly through the ®rst hydration shell. Fromthe results presented here, it seems likely that thesethree residues (and not N68, D210, or H309), incooperation with three active-site water molecules(Figure 8), form the primary pre-incision metal-binding site for orienting the target scissile phos-phate. Additional roles for these amino acids or forthe metal ion in nucleophile generation or leavinggroup stabilization, and the number of metalswithin the active-site, cannot be discerned fromour studies here. It is noteworthy that the muta-genic pattern of D210, where a histidine substi-tution has a less dramatic negative effect than analanine mutation on Ape1 catalytic activity(Erzberger & Wilson, 1999), is consistent with arole for this residue in stabilizing the negativelycharged, post-incision leaving group, eitherthrough proton donation or metal chelation.Further studies are clearly needed, and the exactrole of the metal cation in the dissociation step(Masuda et al., 1998b) will need to be determined.The data presented here extend our current work-ing knowledge of how Ape1 engages DNA, com-municates with polb during BER, and coordinatesits catalytically critical divalent metal co-factor.
During the review of this paper, a report describ-ing three crystal structures of Ape1 and DNA waspublished (Mol et al., 2000). This study providesdirect evidence that the ¯exible loop regionsdescribed here are central to AP site-speci®c bind-ing and that Ape1 may be a structure-speci®cnuclease that detects and productively binds DNAthat adopts a kinked conformation and can presenta ¯ipped-out AP site to a selective recognitionpocket.
Material and Methods
Buffers and reagents
Reagents were purchased from Sigma unless other-wise indicated. Restriction enzymes were purchasedfrom New England Biolabs. Radio-labeled nucleotideswere from Amersham. Spectrophotometric-grade glycer-ol was obtained from Fisher. All pH values were deter-mined at 21 �C. Oligos were purchased from OperonBiotechnologies. Oligos were further puri®ed by dena-turing 20 % polyacrylamide gel electrophoresis prior touse (Sambrook et al., 1989).
Plasmid constructions
Site-directed mutagenesis was performed as described(Erzberger & Wilson, 1999). To generate a D210N/D308A double mutant Ape1 expression construct, plas-mids containing an APE1 gene with either D210N or

AP DNA Interactions and Divalent Metal Coordination of Ape1 457
D308A mutation (Erzberger & Wilson, 1999) weredigested with PstI. Appropriate restriction fragmentswere then puri®ed by agarose gel electrophoresis andligated. To construct an APE1 D210N/D70A gene, plas-mids containing a D210N or D70A mutation (Erzberger& Wilson, 1999) were digested with AlwNI, and appro-priate DNA fragments were gel-puri®ed and ligated.Plasmid constructs were sequenced as described byWilson et al. (1998).
Purification of recombinant proteins
All Ape1 proteins were overexpressed in bacteria andpuri®ed as described (Erzberger & Wilson, 1999), excepta gel-®ltration chromatography step was added for theD210N double mutants (see above). Brie¯y, followingion-exchange chromatographies (Erzberger & Wilson,1999), Ape1 protein fractions were pooled, concentratedby 80 % (w/v) ammonium sulfate, and then fractionatedon a BioRad BioSil SEC 125-5 gel ®ltration column(7.8 mm � 300 mm) in 50 mM NaHepes (pH 7.5), 5 %glycerol (w/v), 0.1 mM EDTA, and 0.1 mM DTT. The¯ow rate was 0.25 ml/minute. Size markers used forcalibration are thyroglobulin (670 kDa), gamma globulin(158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa),and vitamin B-12 (1.35 kDa). Proteins were detected byultraviolet absorbance at 280 nm. Ape1 proteins weredialyzed overnight against 50 mM Tris-HCl (pH 7.9),50 mM KCl, 20 % glycerol, 1 mM PMSF and 0.1 mMDTT, and stored at ÿ70 �C. Proteins were more than95 % pure as determined by Coomassie blue staining ofSDS/polyacrylamide gels (Figure 1(a)). All mutants hadsimilar chromatographic pro®les, suggesting structuralintegrity has been maintained.
For polb puri®cation, protein extracts fromBL21(lDE3) bacteria harboring pT7Polb (a generous giftfrom Dr Stuart Linn, University of California, Berkeley)were generated from IPTG-induced 2 l culture asdescribed above for Ape1, except that the resuspensionbuffer was 50 mM Tris (pH 8.0), 0.1 mM EDTA, 1 mMDTT, 5 % glycerol (buffer PC) containing 400 mM KCl.In addition, PMSF was added to a ®nal concentration of1 mM prior to sonication. After centrifugation, clari®edextracts were applied to a Q20 column pre-equilibratedwith buffer PC containing 400 mM KCl. Flowthroughmaterial was collected, diluted with an equal volume ofbuffer PC, and applied to an S10 column pre-equili-brated with buffer PC containing 200 mM KCl. Proteinwas eluted with a linear gradient of KCl from 200 mMto 800 mM. Polb protein was detected by SDS-PAGEand Coomassie blue staining (>95 % pure).
Incision and binding assays
Nuclease and gel retardation assays were done asdescribed by Erzberger & Wilson (1999).
Hydroxyl radical footprinting protection assay
Either oligo 26F or 26G (Figure 1(b)) was labeled atthe 50-end and annealed to a molar equivalent ofunlabelled complementary strand (Erzberger & Wilson,1999). Double-stranded DNA was then puri®ed througha G25 desalting spin column (Pharmacia) according tothe instruction of the manufacturer, and stored atÿ70 �C. Binary Ape1-DNA complexes were formed byincubating on ice, 0.1 pmol of the labeled DNA fragmentwith at least 3 pmol of various Ape1 proteins in 50 ml of
50 mM KHepes (pH 7.5), 1 mM DTT, 100 mg/ml BSA,50 mM KCl, and 0.01 % (v/v) Triton X-100 for 20 min-utes with either 5 mM EDTA or 5 mM MgCl2. The reac-tions were then ``footprinted'' by adding 4 ml of 50 mMsodium ascorbate, 4 ml of 3.2 % H2O2, and 4 ml of125 mM Fe(EDTA)ÿ2 at 4 �C for 30 seconds (Dixon et al.,1991). The reactions were stopped with 100 ml of 60 mMthiourea. The samples were extracted with an equalvolume of phenol/chloroform/isoanyl alcohol (25:24:1,by vol.), and precipitated with 0.1 vol. of 10 M LiCl,10 mg of glycogen and 300 ml of 100 % (v/v) ethanol. Thesamples were resuspended in 7 ml of 50 % formamide,heated at 80 �C for ®ve minutes, and analyzed on a 20 %(w/v) polyacrylamide, 7 M urea denaturing gel. Visual-ization of the labeled substrate was achieved using aMolecular Dynamics (Sunnyvale, CA) STORM 860 Phos-phorimager and quantitative analysis was performedusing Molecular Dynamics ImageQuant v1.11 software.The locations of protected bases were mapped relative tovarious chemical sequencing ladders which were gener-ated as described by Kassavetis et al. (1989).
Permanganate reactivity was done as described bySasse-Dwight (1991) except the reactions were performedon ice instead of at 37 �C.
Metal site proximity cleavage assay
Binary Ape1-DNA complex was formed as describedabove except without EDTA in a 20 ml volume. The clea-vage reactions were initiated by adding 20 mM ferrousammonium sulfate (Fe(AS)2) and the reactions were con-tinued for ten or 20 minutes at 37 �C. The reactions wereextracted with phenol, precipitated with ethanol, frac-tionated on a 7 M urea/20 % (w/v) denaturing poly-acrylamide gel, and analyzed as above.
Computer simulations
Molecular dynamics simulations of Ape1 were per-formed beginning with the protein structure derivedfrom crystallography (Gorman et al., 1997; RCSB PDB#1BIX). Ape1 was simulated under constant pressureand temperature (NPT) by the strong coupling methodof Nose and Hoover, as implemented in CHARMM26(Brooks et al., 1983; Feller et al., 1995) version 26b2(Department of Chemistry, Harvard University). The pri-mary (cubic) cell of this periodic system measures about60 AÊ on a side and contains 21,571 atoms, including5734 water molecules, only 208 of which were present inthe original crystal structure. The image cells contributeanother 22,000 atoms to the simulation. Finite, 12 AÊ
force-shifted cutoffs (F-sim) or Particle-Mesh Ewald sum-mation (E-sim) were used to handle the electrostaticinteractions. We have employed each of these electro-static approximations in two otherwise identical simu-lations, denoted F-sim and E-sim. The simulationsrequired about six weeks for each 500 ps simulation on asingle DEC Alpha workstation.
Protonation states of the titratable residues have beenchosen to be consistent with pH 7.5, where the protein ismost active (Kane & Linn, 1981). To more closely modelin vivo conditions, the samarium ion present in theactive-site of the Ape1 crystal was replaced with mag-nesium, the preferred metal cofactor of the humanenzyme (Barzilay et al., 1995). The three other samariumions from the crystal structure were discarded to reduceprotein self-interaction. The charged residues and Mg2�
gave rise to a net charge of �3 for which three chloride

458 AP DNA Interactions and Divalent Metal Coordination of Ape1
ions were added to the solvent, initially 15 AÊ away fromthe protein, to keep the system neutral (especiallyimportant in studies employing Ewald summations).A sulfur-sulfur distance of 3.5 AÊ was judged too far for adisul®de bond (C93-C208).
Acknowledgments
This work was carried out under the auspices of theUS Department of Energy by Lawrence LivermoreNational Laboratory under contract number W-7405-ENG-48 and supported by an NIH grant (CA79056) toDMWIII. We thank Ms Tina Xi and Dr Harvey Mohren-weiser for sequencing support, and Dr Ian McConnellfor critical inputs.
References
Ayadi, L., Jourdan, M., Coulombeau, C., Garcia, J. &Lavery, R. (1999). Experimental and theoretical stu-dies of the conformational perturbations induced byan abasic site. J. Biomol. Struct. Dynam. 17, 245-257.
Balasubramanian, B., Pogozelski, W. K. & Tullius, T. D.(1998). DNA strand breaking by the hydroxyl rad-ical is governed by the accessible surface areas ofthe hydrogen atoms of the DNA backbone. Proc.Natl Acad. Sci. USA, 95, 9738-9743.
Barsky, D., Foloppe, N., Ahmadia, S., Wilson, D. M., III& MacKerell, A. D., Jr (2000). New insights into thestructure of abasic DNA from molecular dynamicssimulations. Nucl. Acids Res. In the press.
Barzilay, G., Mol, C. D., Robson, C. N., Walker, L. J.,Cunningham, R. P., Tainer, J. A. & Hickson, I. D.(1995). Identi®cation of critical active-site residuesin the multifunctional human DNA repair enzymeHAP1. Nature Struct. Biol. 2, 561-568.
Bennett, R. A., Wilson, D. M., III, Wong, D. & Demple,B. (1997). Interaction of human apurinic endonu-clease and DNA polymerase beta in the base exci-sion repair pathway. Proc. Natl Acad. Sci. USA, 94,7166-7169.
Bewley, C. A., Gronenborn, A. M. & Clore, G. M. (1998).Minor groove-binding architectural proteins: struc-ture, function, and DNA recognition. Annu. Rev.Biophys. Biomol. Struct, 27, 105-131.
Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States,D. J., Swaminathan, S. & Karplus, M. (1983).CHARMM: a program for macromolecular energy,minimization, and dynamics calculations. J. Comput.Chem. 4, 187-217.
Cal, S., Tan, K. L., McGregor, A. & Connolly, B. A.(1998). Conversion of bovine pancreatic DNase I toa repair endonuclease with a high selectivity forabasic sites. EMBO J. 17, 7128-7138.
Caldecott, K. W., McKeown, C. K., Tucker, J. D.,Ljungquist, S. & Thompson, L. H. (1994). An inter-action between the mammalian DNA repair proteinXRCC1 and DNA ligase III. Mol. Cell. Biol. 14, 68-76.
Caldecott, K. W., Aoufouchi, S., Johnson, P. & Shall, S.(1996). XRCC1 polypeptide interacts with DNApolymerase beta and possibly poly (ADP-ribose)polymerase, and DNA ligase III is a novel molecu-lar ``nick-sensor'' in vitro. Nucl. Acids Res. 24, 4387-4394.
Coppel, Y., Berthet, N., Coulombeau, C., Coulombeau,C., Garcia, J. & Lhomme, J. (1997). Solution confor-mation of an abasic DNA undecamer duplexd(CGCACXCACGC) � d(GCGTGTGTGCG): theunpaired thymine stacks inside the helix. Biochemis-try, 36, 4817-4830.
Demple, B. & Harrison, L. (1994). Repair of oxidativedamage to DNA: enzymology and biology. Annu.Rev. Biochem. 63, 915-948.
Dimitriadis, E. K., Prasad, R., Vaske, M. K., Chen, L.,Tomkinson, A. E., Lewis, M. S. & Wilson, S. H.(1998). Thermodynamics of human DNA ligase I tri-merization and association with DNA polymeraseb. J. Biol. Chem. 273, 20540-20550.
Dixon, W. J., Hayes, J. J., Levin, J. R., Weidner, M. F.,Dombroski, B. A. & Tullius, T. D. (1991). Hydroxylradical footprinting. Methods Enzymol. 208, 380-413.
Erzberger, J. P. & Wilson, D. M., III. (1999). The role ofMg2� and speci®c amino acid residues in the cata-lytic reaction of the major human abasic endo-nuclease: new insights from EDTA-resistant incisionof acyclic abasic site analogs and site-directedmutagenesis. J. Mol. Biol. 290, 447-457.
Erzberger, J. P., Barsky, D., Scharer, O. D., Colvin, M. E.& Wilson, D. M., III (1998). Elements in abasic siterecognition by the major human and Escherichia coliapurinic/apyrimidinic endonucleases. Nucl. AcidsRes. 26, 2771-2778.
Feller, S. E., Zhang, Y. H., Pastor, R. W. & Brooks, B. R.(1995). Constant pressure molecular dynamics simu-lation-the langevin piston method. J. Chem. Phys.103, 4613-4621.
Gelfand, C. A., Plum, G. E., Grollman, A. P., Johnson, F.& Breslauer, K. J. (1998). Thermodynamic conse-quences of an abasic lesion in duplex DNA arestrongly dependent on base sequence. Biochemistry,37, 7321-7327.
Gorman, M. A., Morera, S., Rothwell, D. G., de LaFortelle, E., Mol, C. D., Tainer, J. A., Hickson, I. D.& Freemont, P. S. (1997). The crystal structure ofthe human DNA repair endonuclease HAP1suggests the recognition of extra-helical deoxyriboseat DNA abasic sites. EMBO J. 16, 6548-6558.
Hang, B., Rothwell, D. G., Sagi, J., Hickson, I. D. &Singer, B. (1997). Evidence for a common active-sitefor cleavage of an AP site and the benzene-derivedexocyclic adduct, 3,N4-benzetheno-dC, in the majorhuman AP endonuclease. Biochemistry, 36, 15411-15418.
Hertzberg, R. P. & Dervan, P. B. (1984). Cleavage ofDNA with methidiumpropyl-EDTA-iron(II): reac-tion conditions and product analyses. Biochemistry,23, 3934-3945.
Hos®eld, D. J., Guan, Y., Haas, B. J., Cunningham, R. P.& Tainer, J. A. (1999). Structure of the DNA repairenzyme endonuclease IV and its DNA complex:double-nucleotide ¯ipping at abasic sites and three-metal-ion catalysis. Cell, 98, 397-408.
Humphrey, W. F., Dalke, A. & Schulten, K. (1996).{VMD}-visual molecular dynamics. J. Mol. Graph.14, 33-38.
Jen-Jacobson, L. (1997). Protein-DNA recognition com-plexes: conservation of structure and bindingenergy in the transition state. Biopolymers. 44, 153-180.
Kane, C. M. & Linn, S. (1981). Puri®cation and charac-terization of an apurinic/apyrimidinic endonucleasefrom HeLa cells. J. Biol. Chem. 256, 3405-3414.

AP DNA Interactions and Divalent Metal Coordination of Ape1 459
Kassavetis, G. A., Riggs, D. L., Negri, R., Nguyen, L. H.& Geiduschek, E. P. (1989). Transcription factor IIIBgenerates extended DNA interactions in RNA poly-merase III transcription complexes on tRNA genes.Mol. Cell. Biol. 6, 2551-2566.
Kubota, Y., Nash, R. A., Klungland, A., Schar, P.,Barnes, D. E. & Lindahl, T. (1996). Reconstitution ofDNA base excision-repair with puri®ed human pro-teins: interaction between DNA polymerase betaand the XRCC1 protein. EMBO J. 15, 6662-6670.
Lahm, A. & Suck, D. (1991). DNase I-induced DNAconformation. 2. A structure of a DNase I-octamercomplex. J. Mol. Biol. 222, 645-667.
Lucas, J. A., Masuda, Y., Bennett, R. A., Strauss, N. S. &Strauss, P. R. (1999). Single-turnover analysis ofmutant human apurinic/apyrimidinic endonu-clease. Biochemistry, 38, 4958-4964.
Marintchev, A., Mullen, M. A., Maciejewski, M. W., Pan,B., Gryk, M. R. & Mullen, G. P. (1999). Solutionstructure of the single-strand break repair proteinXRCC1 N-terminal domain. Nature Struct. Biol. 6,884-893.
Masuda, Y., Bennett, R. A. & Demple, B. (1998a).Dynamics of the interaction of human apurinicendonuclease (Ape1) with its substrate and product.J. Biol. Chem. 273, 30352-30359.
Masuda, Y., Bennett, R. A. & Demple, B. (1998b). Rapiddissociation of human apurinic endonuclease(Ape1) from incised DNA induced by magnesium.J. Biol. Chem. 273, 30360-30365.
Matsumoto, Y. & Kim, K. (1995). Excision of deoxyri-bose phosphate residues by DNA polymerase betaduring DNA repair. Science, 269, 699-702.
McCullough, A. K., Dodson, M. L. & Lloyd, R. S. (1999).Initiation of base excision repair: glycosylase mech-anism and structures. Annu. Rev. Biochem. 68, 255-265.
Mol, C. D., Kuo, C. F., Thayer, M. M., Cunningham,R. P. & Tainer, J. A. (1995). Structure and functionof the multifunctional DNA-repair enzyme exonu-clease III. Nature, 374, 381-386.
Mol, C. D., Parikh, S. S., Putnam, C. D., Lo, T. P. &Tainer, J. A. (1999). DNA repair mechanisms for therecognition and removal of damaged DNA bases.Annu. Rev. Biophys. Biomol. Struct. 28, 101-128.
Mol, C. D., Izumi, T., Mitra, S. & Tainer, J. A. (2000).DNA-bound structures and mutants reval abasicDNA binding by APE1 DNA repair and coordi-nation. Nature, 403, 451-456.
Mustaev, A., Kozlov, M., Markovtsov, V., Zaychikov, E.,Denissova, L. & Goldfarb, A. (1997). Modularorganization of the catalytic center of RNA poly-merase. Proc. Natl Acad. Sci. USA, 94, 6641-6645.
Prasad, R., Singhal, R. K., Srivastava, D. K., Molina, J. T.,Tomkinson, A. E. & Wilson, S. H. (1996). Speci®cinteraction of DNA polymerase beta and DNAligase I in a multiprotein base excision repair com-plex from bovine testis. J. Biol. Chem. 271, 16000-16007.
Roll, C., Ketterle, C., Faibis, V., Fazakerley, G. V. &Boulard, Y. (1998). Conformations of nicked andgapped DNA structures by NMR and molecular
dynamic simulations in water. Biochemistry, 37,4059-4070.
Rothwell, D. G. & Hickson, I. D. (1996). Asparagine 212is essential for abasic site recognition by the humanDNA repair endonuclease HAP1. Nucl. Acids Res.24, 4217-4221.
Rothwell, D. & Hickson, I. (1997). Repair of apurinic/apyrimidinic (AP) sites in DNA by AP endonu-cleases. In Base Excision Repair of DNA Damage(Hickson, I., ed.), pp. 67-80, Landes Bioscience,Austin, USA.
Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989). Molecu-lar Cloning: A Laboratory Manual (Nolan, C., ed.),2nd edit., Cold Spring Harbor Laboratory Press,Cold Spring Harbor, NY.
Sasse-Dwight, S. & Gralla, J. D. (1991). Footprintingprotein-DNA complexes in vivo. Methods Enzymol.208, 146-168.
Suh, D., Wilson, D. M., III & Povirk, L. F. (1997).30-phosphodiesterase activity of human apurinic/apyrimidinic endonuclease at DNA double-strandbreak ends. Nucl. Acids Res. 25, 2495-2500.
Varshney, A., Brooks, F. P. & Wright, W. V. (1994).Linearly scalable computation of smooth molecularsurfaces (IEEE comp). Graph. Applicat. 14, 19-25.
Weston, S. A., Lahm, A. & Suck, D. (1992). X-ray struc-ture of the DNase I-d(GGTATACC)2 complex at2.3 AÊ resolution. J. Mol. Biol. 226, 1237-1256.
Wilson, D. M., III, Takeshita, M., Grollman, A. P. &Demple, B. (1995). Incision activity of human apuri-nic endonuclease (Ape) at abasic site analogs inDNA. J. Biol. Chem. 270, 16002-16007.
Wilson, D. M., III, Takeshita, M. & Demple, B. (1997).Abasic site binding by the human apurinic endonu-clease, Ape, and determination of the DNA contactsites. Nucl. Acids Res. 25, 933-939.
Wilson, D. M., III, Carney, J. P., Coleman, M. A.,Adamson, A. W., Christensen, M. & Lamerdin, J. E.(1998). Hex1: a new human Rad2 nuclease familymember with homology to yeast exonuclease 1.Nucl. Acids Res. 26, 3762-3768.
Wilson, S. H. (1998). Mammalian base excision repairand DNA polymerase beta. Mutat. Res. 407, 203-215.
Xanthoudakis, S., Smeyne, R. J., Wallace, J. D. & Curran,T. (1996). The redox/DNA repair protein, Ref-1, isessential for early embryonic development in mice.Proc. Natl Acad. Sci. USA, 93, 8919-8923.
Xu, Y. J., Kim, E. Y. & Demple, B. (1998). Excision ofC-40-oxidized deoxyribose lesions from double-stranded DNA by human apurinic/apyrimidinicendonuclease (Ape1 protein) and DNA polymerasebeta. J. Biol. Chem. 273, 28837-28844.
Yang, J. & Carey, J. (1995). Footprint phenotypes: struc-tural models of DNA-binding proteins from chemi-cal modi®cation analysis of DNA. Methods Enzymol.259, 452-468.
Zaychikov, E., Martin, E., Denissova, L., Kozlov, M.,Markovtsov, V., Kashlev, M., Heumann, H.,Nikiforov, V., Goldfarb, A. & Mustaev, A. (1996).Mapping of catalytic residues in the RNA polymer-ase active center. Science, 273, 107-109.
Edited by J. M. Miller
(Received 21 December 1999; received in revised form 15 February 2000; accepted 24 February 2000)