mastocytose systémique de l’adulte : aspects ... · ne découlent pas seulement des mutations du...
TRANSCRIPT

Correspondances en Onco-Hématologie - Vol. X - n° 6 - novembre-décembre 20152
d o s s i e r
Hypercytoses singulières
RÉ
SU
MÉ
Su
mm
ar
yLa mastocytose est un groupe hétérogène de pathologies caractérisées par une activation et/ou une prolifération anormales de mastocytes tumoraux. C’est une maladie rare qui atteint l’enfant et l’adulte avec des manifestations cliniques très variées liées, d’une part, à l’activation mastocytaire et, d’autre part, à l’infiltration tissulaire par des mastocytes anormaux. La diversité clinique, formes indolentes et agressives, ainsi que la diversité pronostique ne découlent pas seulement des mutations du gène c-KIT, mais également des anomalies moléculaires additionnelles récemment mises en évidence. Les mesures préventives de la dégranulation mastocytaire associées aux traitements symptomatiques restent la base de traitement des formes indolentes alors les traitements cytoréducteurs sont réservés aux formes avec un handicap fonctionnel et aux formes agressives.
Mots-clés : Mastocytoses – Physiopathologie – KIT
Mastocytosis is a heterogeneous group of disorders characterized by an activation and/or proliferation of abnormal mast cells in diff erent organs. Mastocytosis is a rare disease of children and adults with a broad spectrum of symptoms secondary to mast cell activation and/or mast cell organ infi ltration. The diversity of clinical forms, indolent versus aggressive, as well as the diversity of prognosis could be explained, not only by the abnormalities of KIT gene, but also, by the presence of additionnels mutations. Symtomatic therapy is the main cornerstone for the treatment of ISM, however, cytoreductive therapy are used for the aggressive forms and selected patients with indolent forms and functional handicap.
Keywords : Mastocytosis – Pathophysiology – KIT
Mastocytose systémique de l’adulte : aspects physiopathologiques et manifestations cliniquesSystemic mastocytosis in adults: pathophysiology and clinical manifestationsG. Damaj1, 2, O. Hermine2, 3
1Institut d’hématologie de Basse-Normandie,
CHU de Caen, et université de Caen-Basse-Normandie,
faculté de médecine, Caen.
2Centre de référence national des mastocytoses,
hôpital Necker-Enfants-malades, Paris.
3Service d’hématologie clinique,
et Institut IMAGINE, hôpital Necker-Enfants-
malades, Paris
L e mastocyte est une cellule mononucléée à cytoplasme basophile et noyau rond et excentré, avec des granulations denses hyperchromatiques
fortement marquées par le bleu de toluidine, et riche en médiateurs. Sa surface membranaire est riche en récepteurs, parmi les plus importants, le récepteur à l’IgE et le récepteur du “Stem Cell Factor” (SCF), également appelé CD117 ou c-KIT (1, 2). C’est l’une des cellules les plus précoces de l’immunité innée. Elle joue un rôle important, d’une part, dans la réaction d’hypersensi-bilité immédiate (sécrétion d’histamine, leucotriènes et autres médiateurs vaso-actifs) et d’hyper sensibilité retardée, et, d’autre part, dans la réponse immune innée antibactérienne et antiparasitaire (3). À côté de ce rôle dans l’allergie et la défense immunitaire, le mastocyte
est impliqué dans la pathogénie de plusieurs mala-dies : la polyarthrite rhumatoïde, le lupus, la sclérose en plaques, la fi brose pulmonaire et certaines maladies vasculaires (4). Le mastocyte se situe dans des zones stratégiques à l’intérieur des tissus avec un tropisme périvasculaire et périnerveux. Il privilégie les zones ayant une interface directe avec le milieu extérieur, ce qui lui permet d’exercer ses fonctions et de répondre aux stimuli d’une manière rapide. Les tissus les plus riches en mastocytes sont la peau, les muqueuses et le système nerveux central (SNC) [5].La mastocytose est une maladie considérée comme orpheline et qui est probablement sous-diagnostiquée. L’amélioration récente des connaissances physiopa-thologiques et l’établissement de critères diagnos-

Correspondances en Onco-Hématologie - Vol. X - n° 6 - novembre-décembre 2015 3
Mastocytose systémique de l’adulte : aspects physiopathologiques et manifestations cliniques
tiques − comme le dosage de la tryptase sérique et la recherche de la mutation du gène c-KIT − ainsi que l’organisation des Centres de référence régionaux et nationaux ont suscité un intérêt pour cette pathologie extrêmement variée et ont contribué à l’amélioration des connaissances, permettant probablement un diag-nostic plus précoce.
Défi nition
La mastocytose est une maladie caractérisée par l’accu-mulation et/ou la prolifération de mastocytes anormaux dans un ou plusieurs tissus. Les tissus les plus fréquem-ment atteints sont la peau et la moelle osseuse, suivies du foie, de la rate, du tube digestif et des ganglions.C’est une maladie de l’enfant et de l’adulte. La masto-cytose de l’enfant (qui ne fait pas l’objet de cet article) se présente essentiellement sous une forme cutanée pure, sans atteinte extracutanée. Le pronostic est bon, avec une régression spontanée à l’adolescence et à l’âge adulte (6). La mastocytose de l’adulte, en revanche, se présente sous une forme systémique (MS), avec atteinte d’au moins un organe extracutané. La MS est un groupe hétérogène de maladies de pronostic et d’évolution variables. La plupart des patients présentent une forme indolente (MSI), avec une survie identique à celle de la population générale (7). Les formes avancées sont néanmoins rares, avec une évolution agressive et un pronostic sombre.
Anomalies moléculaires et physiopathologiques
Les progéniteurs mastocytaires sont libérés dans la cir-culation à partir de la moelle osseuse. Ils sont identifi és par leurs marqueurs CD34+/CD117+/CD33+/CD13+/FCεRI– (8). Le SCF est le facteur qui joue le rôle le plus important dans la diff érenciation, la migration, l’ad-hésion et la survie des mastocytes. Les progéniteurs mastocytaires migrent vers les tissus et se diff érencient sous l’infl uence du SCF. L’absence totale du SCF, ou de son récepteur c-KIT, entraîne une absence totale de mastocytes. En revanche, l’activation constitutive de c-KIT entraîne une augmentation de la survie, de la prolifération, de la diff érenciation, de la migration et des fonctions des mastocytes (2).Le récepteur du SCF, ou c-KIT, est un récepteur trans-membranaire à activité tyrosine kinase (TK). La fi xa-tion du ligand (le SCF) au niveau de son site de liaison entraîne la dimérisation du récepteur et l’activation de
plusieurs voies de signalisation, les voies de Ras, Raf et ErK ; la voie PI3-kinases/AKT/mTOR et, d’une manière moins importante, la voie de JAK2-STAT1/5. L’activation de c-KIT va également entraîner une activation de la voie de Src kinases (SFK) et des phospholipases (par-ticulièrement la phospholipase C-g) qui jouent un rôle dans l’activation des fonctions des mastocytes (9-11).Les mutations de c-KIT sont des mutations activatrices responsables de l’activation constitutive des voies de signalisation intracellulaires et, par conséquent, d’une diff érenciation, d’une prolifération et d’une survie pro-longée des mastocytes anormaux (12, 13).Les mutations de c-KIT les plus fréquemment rencon-trées sont les mutations du domaine TK au niveau de l’exon 17-18 du gène (D816) [fi gure 1]. Elles sont retrouvées dans plus de 90 % des cas de mastocy-toses indolentes. D’autres mutations, ou mutations additionnelles, ont été récemment décrites. Parmi
Figure 1. Structure normale et principales mutations du récepteur c-KIT en fonction du sous-type de mastocytose systémique et de l’âge d’apparition (d’après [14 avec modifi cations]).
ECD
Site de fixation du ligand
Site de dimérisation
Hélice transmembranaire
Domaine juxtamembranaire
Membrane
Y568544
581
596
Exon 1Exon 2Exon 3
Exon 4-6
Exon 6, 7
Exon 8, 9
Exon 10
Exon 11
Exon 12, 13
Exon 14, 15
Exon 17, 18
Exon 19-21
601630
648671676694
753790
797
810839
938
976
Y568
Y703Y721Y730
Y823Y900
Y937
Cycle catalytique
Cycle d’activation
Queue N-terminale
Domainekinase
Boucle P
Hélice C
Région charnière
KID
Domainekinase
Lobe N-terminal
Lobe C-terminal
Enfants : 40-50 %Régression : 85 %
Adultes et enfantsLeucémies et sarcomes agressifs
Adultes : 85-90 %Formes indolentesAHNMDLeucémies à mastocytes

Correspondances en Onco-Hématologie - Vol. X - n° 6 - novembre-décembre 20154
d o s s i e r
Hypercytoses singulières
les gènes atteints, on note ceux jouant un rôle dans l’épigénétique − TET2 et ASXL1 −, ceux jouant un rôle dans la régulation de l’épissage des ARN − SRSF2 et SF3B1 −, et ceux jouant un rôle dans la transcription − RUNX1 et CBL. Ces mutations sont trouvées de manière intéressante dans les formes avancées des mastocy-toses avec une fréquence plus élevée dans les MS associées à une maladie hématologique clonale non mastocytaire (MS-AHNMD) [15-19].Dans une récente étude portant sur des patients atteints de MS-AHNMD, l’isolement des mastocytes et des cellules myéloïdes du clone non mastocytaire de la moelle a permis de mettre en évidence la présence des mutations de c-KIT, SRSF2 et TET2 dans les masto-cytes anormaux ainsi que dans les cellules myéloïdes associées, témoignant par conséquent de la même origine clonale des 2 types cellulaires (19, 20). Il a aussi été démontré que certaines mutations additionnelles − notamment celles de TET2 et de SRSF2 (mutations fondatrices) − précèdent l’apparition des mutations de c-KIT (mutations “drivers” ou phénotypiques), témoi-gnant ainsi de l’existence d’une hiérarchie mutation-nelle (19, 20). L’hétérogénéité clinique et phénotypique
de la mastocytose peut donc s’expliquer par les ano-malies moléculaires. En eff et, les mutations du gène c-KIT sont les plus fréquentes des MSI, et l’acquisition secondaire de mutations additionnelles est exception-nelle, expliquant la faible incidence de transformation des MSI en formes plus avancées (19, 20). En revanche, dans les formes avancées, les mutations additionnelles, notamment celles de TET2, coopèrent avec les muta-tions de c-KIT et entraînent une transformation de la maladie vers une forme plus agressive (fi gure 2) [20].
Critères diagnostiques et classifi cation
Circonstances de découvertePlusieurs scénarios peuvent constituer des circons-tances de découverte d’une MS. Les plus fréquents sont les suivants.
Lésions cutanées anormalesElles sont découvertes par le patient ou par son médecin traitant. Parmi les lésions les plus fréquemment trouvées, on observe l’urticaire pigmentaire (fi gure 3).
Figure 2. Hétérogénéité moléculaire et évolutive des mastocytoses.A. Formes indolentes des mastocytoses, avec un risque faible d’acquisition d’anomalies additionnelles et de transformation vers une forme agressive.B. Formes avancées de mastocytoses avec une fréquence élevée d’anomalies moléculaires additionnelles et une coopération génique avec c-KIT pour une trans-formation vers une forme agressive.
A. FORMES INDOLENTES
KIT D816VSRSF2
Autres ?
Mastocytose agressive
RareMastocytose indolente
KIT D816V“driver” ou
phénotypique
SMP
SM-LAM, MDS
SM-LMMC
TET2
TET2JAK2
FIP1L1-PDGFRETNK1
TET2SRSF2ASXL1Autres
Événements pré-KIT mutations fondatrices
Mastocytose agressive
MSA MSA
MSA
TET2SRSF3ASXL1Autres
± KIT D816V
± KIT D816V ± KIT D816V
± KIT D816V
B. FORMES AVANCÉES
Correspondances en Onco-Hématologie - Vol. X - n° 6 - novembre-décembre 2015 5
Mastocytose systémique de l’adulte : aspects physiopathologiques et manifestations cliniques
Symptômes d’activation ou de dégranulation mastocytaireLeur intensité est très variable et elle n’est pas corrélée à l’importance de l’infi ltration mastocytaire. Ces mani-festations sont multiples et aspécifi ques. On constate parmi elles :
✓ les bouff ées vasomotrices paroxystiques ou fl ushes : il s’agit d’accès subis d’érythème et de chaleur, souvent prurigineux, généralisés ou limités. Elles sont souvent associées à des palpitations, à des précordialgies, à de l’hypotension, ou même à une syncope pouvant aller jusqu’à l’arrêt cardiaque ;
✓ les réactions anaphylactoïdes avec possibilité de choc anaphylactique sans cause allergique notable. L’urticaire et l’angiœdème sont rarement des signes de découverte de mastocytose ;
✓ le prurit, généralisé ou localisé, qui peut survenir en l’absence de lésions cutanées ;
✓ les manifestations digestives : elles sont non spéci-fi ques, de type crises douloureuses, diarrhées profuses, épigastralgies, nausées et/ou vomissements et bal-lonnements abdominaux sans causes évidentes (21) ;
✓ les manifestations ORL et respiratoires, qui sont de type rhinite, otite, pharyngite à répétition, dyspnée d’allure spastique, hypersécrétion de mucus, toux ;
✓ les troubles neuropsychiatriques atypiques, de type anxiété, dépression, troubles de la mémoire, de l’atten-tion et de la concentration (22, 23) ;
✓ la cystite interstitielle avec pollakurie, qui est un symptôme très fréquent mais peu connu (24).
Lésions osseusesElles sont de type déminéralisation osseuse, voire ostéoporose, notamment chez l’homme jeune ; plus rarement des lésions lytiques et/ou condensantes, fractures spontanées costales, vertébrales ou des os longs (25-28).
Manifestations hématologiquesElles sont de type cytopénie (anémie, neutropénie, thrombopénie, hyperleucocytose), splénomégalie ou hépatomégalie inexpliquée.
Fatigue et/ou perte de poids inexpliquéesLa fatigue et/ou la perte de poids inexpliquées sont également à prendre en considération.
Diagnostic positifLe diagnostic d’une MS repose sur des critères majeurs et des critères mineurs (tableau I, p. 6) qui ont été défi nis par un groupe d’experts en 2000 et repris dans la classifi cation OMS en 2008 (29, 30).
Le diagnostic d’une mastocytose cutanée est fondé sur la coexistence de lésions cutanées typiques et d’infi ltrats histologiques multifocaux ou diff us de cel-lules mastocytaires. En revanche, le diagnostic d’une MS nécessite, conformément à la classifi cation OMS, la présence du critère majeur et d’un critère mineur ou de 3 critères mineurs si le critère majeur est absent (tableau I, p. 6).La suspicion d’une mastocytose doit déclencher en premier lieu (première étape) un examen cutané minutieux à la recherche de lésions spécifi ques qui témoignent d’une infi ltration de la peau par des masto-cytes anormaux. Ce diagnostic doit être confi rmé (deuxième étape) par une biopsie cutanée (critère majeur), qui sera suivie par la recherche d’une atteinte systémique extracutanée, d’organomégalie et/ou
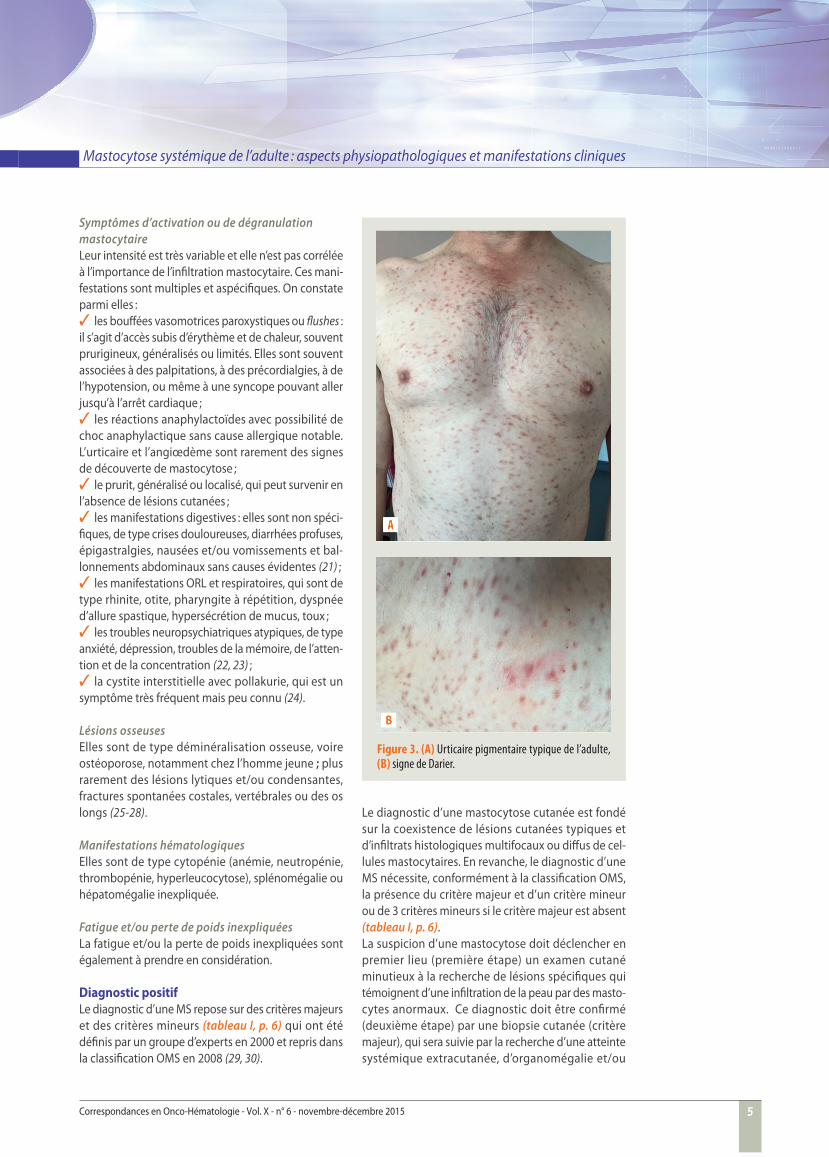
Figure 3. (A) Urticaire pigmentaire typique de l’adulte, (B) signe de Darier.

Correspondances en Onco-Hématologie - Vol. X - n° 6 - novembre-décembre 20156
d o s s i e r
Hypercytoses singulières
d’organopathie. L’infi ltration d’un organe peut être responsable de son dysfonctionnement. Ces atteintes symptomatiques sont désignées sous le terme de “signes C”, par opposition à l’infi ltration d’organes avec organomégalie sans organopathie” qui est, elle, désignée par le terme “signes B” (tableau II).En l’absence de lésions cutanées de mastocytose, l’examen histologique d’un autre organe ou tissu est nécessaire (critère majeur de diagnostic). Il faudrait préférer la moelle osseuse, du fait de la fréquence élevée de l’atteinte médullaire et des diffi cultés du diagnostic histologique dans d’autres tissus (9, 29, 33).À côté de l’examen anatomopathologique, le myélo-gramme (mastocytes atypiques fusiformes dans
la grande majorité des cas), avec phénotypage des mastocytes médullaires (expression anormale du CD2 et du CD25), est nécessaire, ainsi que le dosage de la tryptase sérique et la recherche des mutations du gène c-KIT sur la moelle et/ou la peau et/ou d’autres organes infi ltrés par les mastocytes anormaux.L’hémogramme, le taux d’albumine sérique, les enzymes hépatiques sont utiles pour identifi er d’éventuels signes d’agressivité de la maladie (signes C) [tableau II].Enfin, l’ostéodensitométrie à la recherche d’une ostéopénie ou d’une ostéoporose est obligatoire dans le bilan de toute mastocytose de l’adulte pour détecter et prévenir les complications osseuses.
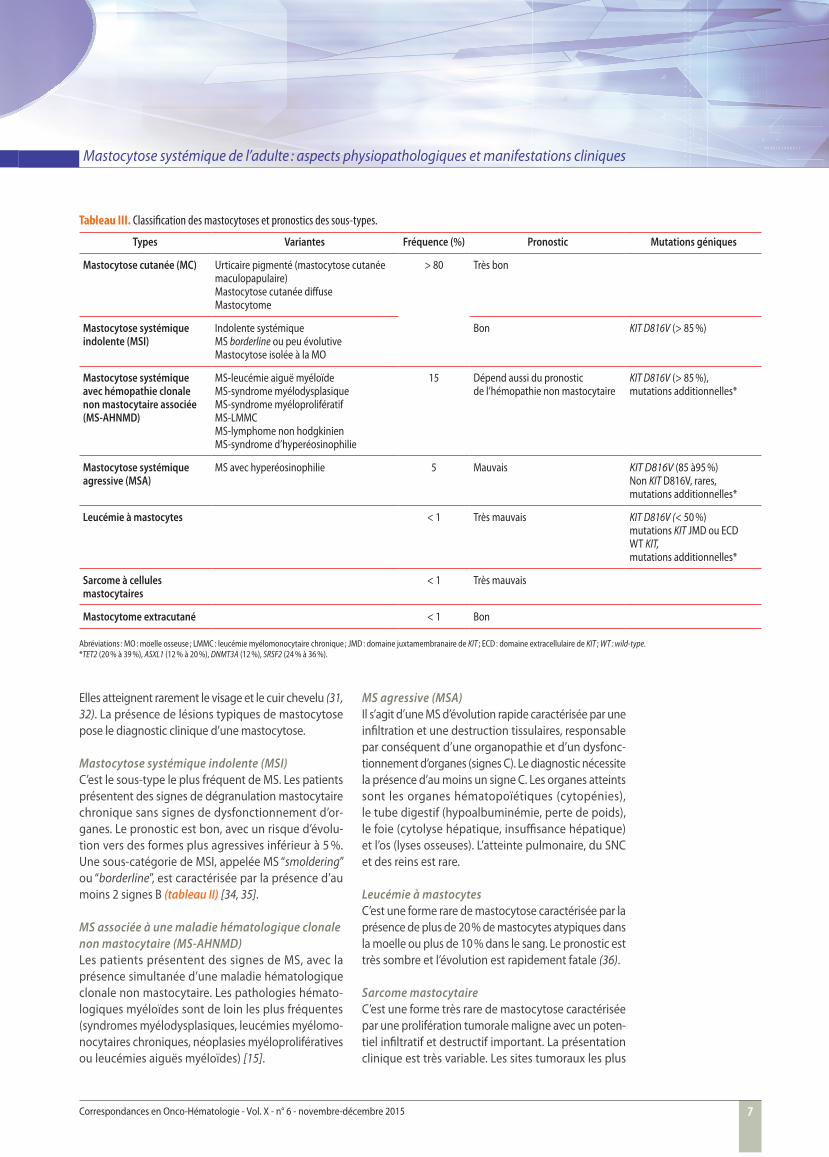
Classifi cationL’ensemble des données issues des évaluations cli-nique, morphologique et biologique permettent de défi nir la catégorie de mastocytose selon la classifi -cation OMS (tableau III) [30].
Mastocytose cutanéeLes lésions cutanées les plus fréquentes sont les lésions maculo-papulaires dans 95 % des cas. Ce sont des lésions fi xes, non labiles. L’urticaire pigmentaire (figure 3A) est la sous-catégorie la plus fréquente. Le signe de Darier est un signe pathognomonique de mastocytose (fi gure 3B).Les lésions se développent habituellement au niveau des cuisses, des aisselles et de la partie inférieure du tronc, et évoluent progressivement vers d’autres sites.
Tableau I. Critères majeurs et critères mineurs du diagnostic de MS*.
Critère majeur Infi ltrat dense multifocal de mastocytes dans la moelle osseuse ou dans un autre organe extracutané, avec plus de 15 mastocytes par agrégat
Critères mineurs 1. Morphologie anormale des mastocytes médullaires ou d’un autre organe extracutané
2. Mutation de c-KIT au codon 816
3. Immunophénotypage des mastocytes médullaires exprimant le CD2 et/ou le CD25
4. Taux de tryptase sérique > 20 ng/ml
*Le diagnostic positif nécessite la présence d’un critère majeur et d’un critère mineur ou de 3 critères mineurs.Le critère majeur est constitué par la présence d’agrégats de plus de 15 mastocytes anormaux au sein de la biopsie ostéo-mé-dullaire ou de tout autre organe atteint en dehors de la peau.Le taux de tryptase sérique doit être > 20 ng/ml en dehors d’une association à une autre hémopathie myéloïde. Un taux de tryptase sérique < 20 ng/ml est présent dans 20 à 30 % des cas de MS.
Tableau II. Signes B et signes C liés à l’infi ltration d’organes par les mastocytes pathologiques.
Critères BInfi ltration mastocytaire sans dysfonction organique
Critères CDysfonction organique
Défaillance d’organe
Importante infi ltration mastocytaire - Infi ltration > 30 % sur la BOM
et- Taux de tryptase sérique > 200 ng/ml
Atteinte médullaire avec - PNN < 1 000/mm3 et/ou - Hb < 10 g/dl et/ou- Pq < 100 000/mm3
Pancytopénie sévère et progressive : - PNN < 500/mm3 avec infections récurrentes - Nécessité de transfusions - Pq < 20 000/mm3 avec syndrome hémorragique
Dysmyélopoïèse : hypercellularité médullaire avec perte de cellules lipidiques. Signes de myélodysplasie ou de myéloprolifération, mais NFS normale ou discrètement altérée sans aggravation progressive
Atteinte hépatique : hépatomégalie palpable avec ascite, perturbation du bilan hépatique et/hypertension portale
Altération progressive des fonctions hépatiques, avec insuffi sance hépatocellulaire jusqu’au coma
Organomégalie - Hépatomégalie palpable sans ascite ou autre signe
d’altération des fonctions hépatiques et/ou - Adénopathies palpables ou à l’échographie/TDM et/ou - Splénomégalie sans hypersplénisme
Atteinte splénique : splénomégalie palpable avec hypersplénisme
Atteinte du tractus digestif : syndrome de malabsorption avec hypoalbuminémie et perte de poids (> 10 % poids de base)
Correspondances en Onco-Hématologie - Vol. X - n° 6 - novembre-décembre 2015 7
Mastocytose systémique de l’adulte : aspects physiopathologiques et manifestations cliniques
Elles atteignent rarement le visage et le cuir chevelu (31, 32). La présence de lésions typiques de mastocytose pose le diagnostic clinique d’une mastocytose.
Mastocytose systémique indolente (MSI) C’est le sous-type le plus fréquent de MS. Les patients présentent des signes de dégranulation mastocytaire chronique sans signes de dysfonctionnement d’or-ganes. Le pronostic est bon, avec un risque d’évolu-tion vers des formes plus agressives inférieur à 5 %. Une sous-catégorie de MSI, appelée MS “smoldering” ou “borderline”, est caractérisée par la présence d’au moins 2 signes B (tableau II) [34, 35].
MS associée à une maladie hématologique clonale non mastocytaire (MS-AHNMD)Les patients présentent des signes de MS, avec la présence simultanée d’une maladie hématologique clonale non mastocytaire. Les pathologies hémato-logiques myéloïdes sont de loin les plus fréquentes (syndromes myélodysplasiques, leucémies myélomo-nocytaires chroniques, néoplasies myéloprolifératives ou leucémies aiguës myéloïdes) [15].
MS agressive (MSA)Il s’agit d’une MS d’évolution rapide caractérisée par une infi ltration et une destruction tissulaires, responsable par conséquent d’une organopathie et d’un dysfonc-tionnement d’organes (signes C). Le diagnostic nécessite la présence d’au moins un signe C. Les organes atteints sont les organes hématopoïétiques (cytopénies), le tube digestif (hypoalbuminémie, perte de poids), le foie (cytolyse hépatique, insuffi sance hépatique) et l’os (lyses osseuses). L’atteinte pulmonaire, du SNC et des reins est rare.
Leucémie à mastocytesC’est une forme rare de mastocytose caractérisée par la présence de plus de 20 % de mastocytes atypiques dans la moelle ou plus de 10 % dans le sang. Le pronostic est très sombre et l’évolution est rapidement fatale (36).
Sarcome mastocytaireC’est une forme très rare de mastocytose caractérisée par une prolifération tumorale maligne avec un poten-tiel infi ltratif et destructif important. La présentation clinique est très variable. Les sites tumoraux les plus
Tableau III. Classifi cation des mastocytoses et pronostics des sous-types.
Types Variantes Fréquence (%) Pronostic Mutations géniques
Mastocytose cutanée (MC) Urticaire pigmenté (mastocytose cutanée maculopapulaire)Mastocytose cutanée diff useMastocytome
> 80 Très bon
Mastocytose systémique indolente (MSI)
Indolente systémique MS borderline ou peu évolutive Mastocytose isolée à la MO
Bon KIT D816V (> 85 %)
Mastocytose systémique avec hémopathie clonale non mastocytaire associée (MS-AHNMD)
MS-leucémie aiguë myéloïde MS-syndrome myélodysplasique MS-syndrome myéloprolifératifMS-LMMCMS-lymphome non hodgkinienMS-syndrome d’hyperéosinophilie
15 Dépend aussi du pronostic de l’hémopathie non mastocytaire
KIT D816V (> 85 %), mutations additionnelles*
Mastocytose systémique agressive (MSA)
MS avec hyperéosinophilie 5 Mauvais KIT D816V (85 à95 %)Non KIT D816V , rares,mutations additionnelles*
Leucémie à mastocytes < 1 Très mauvais KIT D816V (< 50 %)mutations KIT JMD ou ECDWT KIT,mutations additionnelles*
Sarcome à cellules mastocytaires
< 1 Très mauvais
Mastocytome extracutané < 1 Bon
Abréviations : MO : moelle osseuse ; LMMC : leucémie myélomonocytaire chronique ; JMD : domaine juxtamembranaire de KIT ; ECD : domaine extracellulaire de KIT ; WT : wild-type.*TET2 (20 % à 39 %), ASXL1 (12 % à 20 %), DNMT3A (12 %), SRSF2 (24 % à 36 %).

Correspondances en Onco-Hématologie - Vol. X - n° 6 - novembre-décembre 20158
d o s s i e r
Hypercytoses singulières
fréquents sont l’os et le tube digestif, avec ou sans signes d’activation mastocytaire. Cette forme touche l’enfant et l’adulte, avec un âge médian au diagnostic de 40 ans. L’évolution est agressive, avec une résistance aux trai-tements disponibles et une survie inférieure à 1 an.
Mastocytome extracutanéC’est une tumeur mastocytaire bénigne. La localisation extracutanée est très rare et rapportée seulement sous la forme de cas cliniques (37).
Épidémiologie et pronostic
ÉpidémiologieLa mastocytose est considérée comme une maladie orpheline. Elle représente 0,3 % de l’ensemble des diagnostics en hématologie et 1,5 % de l’ensemble des maladies myéloprolifératives (30). Sa prévalence et son incidence dans la population générale sont très peu connues du fait de l’absence d’études épidémio-logiques à grande échelle. Dans une récente étude danoise, l’incidence de la maladie dans la population générale a été estimée à 1 cas pour 10 000 personnes par an (38). Dans une deuxième étude, provenant d’une région des Pays-Bas, l’incidence de la maladie, fondée sur un diagnostic histologique et sur une forte suspicion de mastocytose, a été estimée à 13 cas pour 100 000 habitants par an (39). Son incidence dans les Centres de référence est de l’ordre de 1 cas pour 10 000 personnes. C’est une maladie sporadique acquise avec de rares formes familiales (< 10 % des cas de mastocytose [CEREMAST, données non publiées]). L’âge médian de survenue chez l’adulte est de 32 ans. La mastocytose touche préférentiellement les sujets caucasiens, avec une prédominance féminine pour les formes indolentes (61 % des cas) [23].
PronosticLe pronostic est variable et dépend essentiellement du type indolent ou agressif de la maladie. Chez l’adulte, la persistance et/ou la progression sont une évolution typique de la maladie. Néanmoins, de rares cas de régression ou d’amélioration spontanée des lésions cutanées ont été décrits dans des formes indo-lentes (40). La stabilité de la maladie reste l’évolution la plus fréquente des mastocytoses indolentes de l’adulte. Dans ces cas, moins de 5 % des patients présentent une évolution vers des formes plus agressives (41, 42). En eff et, les patients atteints de MSI (mastocytoses cutanées et mastocytoses systémiques indolentes) ont une survie identique à celle de la population géné-
rale de même tranche d’âge. En revanche, les patients atteints de mastocytoses avancées ont une survie inférieure à celle de la population générale allant de 6 mois (leucémies à mastocytes, MCS) à 5 ans pour les MSA (43). En outre, d’autres facteurs prédictifs de la progression de la maladie et d’une diminution de la survie ont été rapportés, à savoir : l’âge supérieur à 60 ans, l’augmentation des phosphatases alcalines et de la β2-microglobuline, ainsi que l’importance de la masse mutationnelle du gène c-KIT et de l’existence d’une pathologie hématologique clonale non masto-cytaire (44-46). Enfin, la présence et le nombre de mutations additionnelles sont des facteurs de mauvais pronostic (15-17, 19). Il est donc important de défi nir des critères pronostiques non seulement pour informer les patients des risques associés à leur maladie, mais aussi pour guider la décision thérapeutique.
Traitements
Le traitement a pour objectifs de limiter les eff ets de la dégranulation mastocytaire et/ou de réduire la proli-fération mastocytaire.
Mesures généralesL’éviction de tous les facteurs repérés par le patient comme déclenchant l’activation mastocytaire est hautement recommandée. Les évictions se font à la carte, individuellement, en fonction des facteurs identifi és par un individu donné. Il est recommandé de disposer en permanence d’adrénaline auto-injec-table (0,15 ml chez l’enfant et 0,3 ml chez l’adulte), surtout chez les patients ayant des antécédents de malaise, de syncope, d’anaphylaxie ou de tachycar-die, et de porter une carte mentionnant la maladie et les coordonnées de leur médecin référent pour la mastocytose. Lorsqu’une imagerie est requise, il faudrait privilégier l’échographie, la radiographie et l’IRM dans la mesure du possible. Néanmoins, si cela s’avère nécessaire, la première injection d’un produit de contraste iodé (PCI) [scanner, coronarographie] peut se faire sous couvert d’une surveillance médicale et avec de l’adrénaline à disposition. Une prémédication par antihistaminiques et corticoïdes oraux la veille et le jour de l’examen n’a pas démontré son effi cacité en cas de mastocytose. En cas de réaction d’intolérance, même minime, la réinjection de PCI est défi nitivement contre-indiquée. La chirurgie sous anesthésie générale est une autre situation à risque et doit être pratiquée sous certaines précautions pour réduire le risque de complications (47).

Correspondances en Onco-Hématologie - Vol. X - n° 6 - novembre-décembre 2015 9
Mastocytose systémique de l’adulte : aspects physiopathologiques et manifestations cliniques
L’auteur déclare ne pas avoir de liens d’intérêts.
Traitement symptomatiqueIl vise la réduction des manifestations inhérentes à la dégranulation mastocytaire.Les antihistaminiques (48), de type antihistaminiques H1, sont utilisés en cas de prurit, de manifestations congestives cutanées. Les antihistaminiques H2 (rani-tidine) sont effi caces pour les manifestations gastro-intestinales en association avec le cromoglycate disodique (ampoules buvables à 100 mg) à la dose de 100 ou 200 mg 4 fois par jour (49).Les antagonistes des récepteurs des leucotriènes de type montélukast ont une activité sur la toux, le prurit et les bouff ées vasomotrices ainsi que sur les symptômes fonctionnels urinaires (50).Les bisphosphonates sont utilisés en cas d’ostéoporose, sous couvert d’une supplémentation adaptée en cal-cium et en vitamine D (25).La photothérapie est conseillée en cas de lésions éten-dues d’urticaire pigmentaire avec préjudice esthétique. Son effi cacité est le plus souvent transitoire (5 à 8 mois).La corticothérapie est très peu recommandée dans les MSI. Elle est par contre utilisée en cas d’urgence vitale à forte dose (dexaméthasone) et dans les formes agressives.
Traitement cytoréducteurLes polychimiothérapies classiques, même à fortes doses, sont ineffi caces.
INFαUtilisé à des doses progressives (1-5 millions d’unités/m2/j, 3 fois/semaine ou 1,5 μg/kg/semaine pour la forme pégylée), il peut induire des réponses partielles, en particulier sur les signes généraux et d’activation mastocytaire (51), mais il est sans eff et signifi catif sur l’infi ltration médullaire. Sa mauvaise tolérance limite son utilisation aux formes avec handicap fonctionnel.
2-chloro-déoxyadénosine (2-CdA)Ce médicament est intéressant dans les formes systé-miques indolentes et borderline avec handicap fonc-
tionnel, et certaines formes agressives. Administré à la posologie de 0,10 à 0,14 mg/kg/j en perfusion courte (2 heures) ou en sous-cutané de préférence pendant 5 jours, à raison de 1 cycle toutes les 4 à 12 semaines pendant 4 à 6 cycles, il entraîne une réponse globale estimée à 72 %, dont 47 % de réponses majeures. Le taux de réponse est supérieur dans les formes indolentes (92 %) versus avancées (50 %), avec une durée médiane de réponse de 3,7 et 2,4 ans res-pectivement (52).
ThalidomideUtilisé à la dose de 50 à 200 mg/j dans les formes indolentes ou agressives en cas d’échec des autres traitements, il permet un bon contrôle des signes d’ac-tivation mastocytaire dans deux tiers des cas et une réduction partielle du syndrome tumoral dans un tiers des cas (53, 54).
Inhibiteurs de tyrosine kinaseLes mastocytoses avec des mutations KIT D816V sont résistantes à l’imatinib mésylate, au nilotinib, au dasati-nib (55) et au masitinib (56). En revanche, les mutations du domaine juxtamembranaire, que l’on observe plus rarement dans les mastocytoses, sont sensibles à l’ima-tinib mésylate. L’imatinib mésylate est effi cace dans les mastocytoses avec hyperéosinophilie lorsqu’il existe un transcrit de fusion FIP1L1-PDGFRα (57).En revanche, la midostaurine, ou PKC412, inhibe effi ca-cement in vitro la prolifération de lignées mastocytaires mutées D816V. Les résultats préliminaires d’un essai de phase II international et de son utilisation en com-passionnel sous couvert d’une autorisation transitoire d’utilisation en France pour les patients souff rant de formes avancées de mastocytose sont extrêmement encourageants (CEREMAST, données non publiées) [58].
Allogreffe de cellules souchesL’allogreff e de cellules souches hématopoïétiques est une option thérapeutique dans les formes agressives, en particulier en cas d’AHNMD (59). ■

Correspondances en Onco-Hématologie - Vol. X - n° 6 - novembre-décembre 201514
d o s s i e r
Hypercytoses singulières
1. Castells M. Mast cell mediators in allergic inflamma-tion and mastocytosis. Immunol Allergy Clin North Am 2006;26(3):465-85.
2. Metcalfe DD. Mast cells and mastocytosis. Blood 2008;112(4):946-56.
3. St John AL, Abraham SN. Innate immunity and its regulation by mast cells. J Immunol 2013;190(9):4458-63.
4. Voehringer D. Protective and pathological roles of mast cells and basophils. Nat Rev Immunol 2013;13(5):362-75.
5. Marshall JS. Mast-cell responses to pathogens. Nat Rev Immunol 2004;4(10):787-99.
6. Méni C, Bruneau J, Georgin-Lavialle S et al. Paediatric mastocytosis: a systematic review of 1747 cases. Br J Dermatol 2015;172(3):642-51.
7. Pardanani A, Lim KH, Lasho TL et al. Prognostically rele-vant breakdown of 123 patients with systemic mastocy-tosis associated with other myeloid malignancies. Blood 2009;114(18):3769-72.
8. Kirshenbaum AS, Goff JP, Semere T et al.. Demonstration that human mast cells arise from a progenitor cell population that is CD34(+), c-kit(+), and expresses aminopeptidase N (CD13). Blood 1999;94(7):2333-42.
9. Kim MS, Radinger M, Gilfi llan AM. The multiple roles of phos-phoinositide 3-kinase in mast cell biology. Trends Immunol 2008;29(10):493-501.
10. Vadlakonda L, Dash A, Pasupuleti M et al. The paradox of Akt-mTOR interactions. Front Oncol 2013;3:165.
11. Lennartsson J, Blume-Jensen P, Hermanson M et al. Phosphorylation of Shc by Src family kinases is necessary for stem cell factor receptor/c-kit mediated activation of the Ras/MAP kinase pathway and c-fos induction. Oncogene 1999;18(40):5546-53.
12. Longley BJ, Tyrrell L, Lu SZ et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet 1996;12(3):312-4.
13. Longley BJ, Metcalfe DD, Tharp M et al. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc Natl Acad Sci USA 1999;96(4):1609-14.
14. Bibi S, Langenfeld F, Jeanningros S et al. Molecular defects in mastocytosis: KIT and beyond KIT. Immunol Allergy Clin North Am 2014;34:239-62.
15. Damaj G, Joris M, Chandesris O et al. ASXL1 but not TET2 mutations adversely impact overall survival of patients suff e-ring systemic mastocytosis with associated clonal hematologic non-mast-cell diseases. PLoS One 2014;9(1):e85362.
16. Schwaab J, Schnittger S, Sotlar K et al. Comprehensive mutational profi ling in advanced systemic mastocytosis. Blood 2013;122(14):2460-6.
17. Soucie E, Hanssens K, Mercher T et al. In aggressive forms of mastocytosis, TET2 loss cooperates with c-KITD816V to transform mast cells. Blood 2012;120:4846-9.
18. Traina F, Visconte V, Jankowska AM et al. Single nucleotide polymorphism array lesions, TET2, DNMT3A, ASXL1 and CBL mutations are present in systemic mastocytosis. PLoS One 2012;7:e43090.
19. Jawhar M, Schwaab J, Schnittger S et al. Molecular pro-fi ling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifi es KIT D816V as a distinct and late event. Leukemia 2015;29:1115-22.
20. Hanssens K, Brenet F, Agopian J et al. SRSF2-p95 hotspot mutation is highly associated with advanced forms of mastocytosis and mutations in epigenetic regulator genes. Haematologica 2014;99:830-5.
21. Sokol H, Georgin-Lavialle S, Canioni D et al. Gastrointestinal manifestations in mastocytosis: a study of 83 patients. J Allergy Clin Immunol 2013;132:866-73.
22. Moura DS, Sultan S, Georgin-Lavialle S et al. Depression in patients with mastocytosis: prevalence, features and eff ects of masitinib therapy. PLoS One 2011;6:e26375.
23. Hermine O, Lortholary O, Leventhal PS et al. Case-control cohort study of patients’ perceptions of disability in mastocy-tosis. PLoS One 2008;3:e2266.
24. Roth TM. Interstitial cystitis in a woman with syste-mic mastocytosis. Int Urogynecol J Pelvic Floor Dysfunct 2007;18:963-5.
25. Barete S, Assous N, de Gennes C et al. Systemic mastocytosis and bone involvement in a cohort of 75 patients. Ann Rheum Dis 2010;69:1838-41.
26. Guillaume N, Desoutter J, Chandesris O et al. Bone compli-cations of mastocytosis: a link between clinical and biological characteristics. Am J Med 2013;126:75 e71-77.
27. Rossini M, Zanotti R, Bonadonna P et al. Bone mineral density, bone turnover markers and fractures in patients with indolent systemic mastocytosis. Bone 2011;49:880-5.
28. Rossini M, Zanotti R, Viapiana O et al. Bone involvement and osteoporosis in mastocytosis. Immunol Allergy Clin North Am 2014;34:383-96.
29. Valent P, Horny HP, Escribano L et al. Diagnostic criteria and classifi cation of mastocytosis: a consensus proposal. Leuk Res 2001;25:603-25.
30. Horny AC, Metcalfe DD et al. Mastocytosis. In: Swerdlow , Campo E, Lee Harris N. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. In: Press I, ed. WHO Classifi cation of Tumours of Haematopoietic and Lymphoid Tissues. Lyon; 2008:53-63.
31. Valent P, Akin C, Arock M et al. Defi nitions, criteria and global classifi cation of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal. Int Arch Allergy Immunol 2011;157:215-25.
32. Hartmann K, Escribano L, Grattan C et al. Cutaneous mani-festations in patients with mastocytosis: Consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol 2015 (Epub ahead of print).
33. Akin C, Jaff e ES, Raff eld M et al. An immunohistochemical study of the bone marrow lesions of systemic mastocytosis: expression of stem cell factor by lesional mast cells. Am J Clin Pathol 2002;118:242-7.
34. Jordan JH, Fritsche-Polanz R, Sperr WR et al. A case of ’smouldering’ mastocytosis with high mast cell burden, mono-clonal myeloid cells, and C-KIT mutation Asp-816-Val. Leuk Res 2001;25:627-34.
35. Valent P, Akin C, Sperr WR et al. Smouldering mastocytosis: a novel subtype of systemic mastocytosis with slow progression. Int Arch Allergy Immunol 2002;127:137-9.
36. Georgin-Lavialle S, Lhermitte L, Dubreuil P et al. Mast cell leukemia. Blood 2013;121:1285-95.
37. Castells MC. Extracutaneous mastocytoma. J Allergy Clin Immunol 2006;117:1513-5.
38. Cohen SS, Skovbo S, Vestergaard H et al. Epidemiology of sys-temic mastocytosis in Denmark. Br J Haematol 2014;166:521-8.
39. Van Doormaal JJ, Arends S, Brunekreeft KL et al. Prevalence of indolent systemic mastocytosis in a Dutch region. J Allergy Clin Immunol 2013;131:1429-31.
40. Brockow K, Scott LM, Worobec AS et al. Regression of urti-caria pigmentosa in adult patients with systemic mastocytosis: correlation with clinical patterns of disease. Arch Dermatol 2002;138:785-90.
41. Pardanani A, Baek JY, Li CY et al. Systemic mast cell disease without associated hematologic disorder: a combined retros-pective and prospective study. Mayo Clin Proc 2002;77:1169-75.
42. Travis WD, Li CY, Bergstralh EJ et al. Systemic mast cell disease. Analysis of 58 cases and literature review. Medicine (Baltimore) 1988;67:345-68.
43. Lim KH, Teff eri A, Lasho TL et al. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood 2009;113:5727-36.
44. Teodosio C, Garcia-Montero AC, Jara-Acevedo M et al. An immature immunophenotype of bone marrow mast cells predicts for multilineage D816V KIT mutation in systemic mastocytosis. Leukemia 2012;26(5):951-8.
45. Escribano L, Alvarez-Twose I, Sanchez-Munoz L et al. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol 2009;124:514-21.
46. Erben P, Schwaab J, Metzgeroth G et al. The KIT D816V expressed allele burden for diagnosis and disease monitoring of systemic mastocytosis. Ann Hematol 2014;93:81-8.
47. Dewachter P, Mouton-Faivre C, Cazalaa JB et al. Mastocytosis and anaesthesia. Ann Fr Anesth Reanim 2009;28:61-73.
48. Crawhall JC, Wilkinson RD. Systemic mastocytosis: management of an unusual case with histamine (H1 and H2) antagonists and cyclooxygenase inhibition. Clin Invest Med 1987;10:1-4.
49. Alexander RR. Disodium cromoglycate in the treatment of systemic mastocytosis involving only bone. Acta Haematol 1985;74:108-10.
50. Bouchelouche K, Nordling J, Hald T et al. Treatment of interstitial cystitis with montelukast, a leukotriene D(4) receptor antagonist. Urology 2001;57:118.
51. Casassus P, Caillat-Vigneron N, Martin A et al. Treatment of adult systemic mastocytosis with interferon-alpha: results of a multicentre phase II trial on 20 patients. Br J Haematol 2002;119:1090-7.
52. Barete S, Lortholary O, Damaj G et al. Long-term effi cacy and safety of cladribine (2-CdA) in adult patients with masto-cytosis. Blood 2015;126:1009-16.
53. Damaj G, Bernit E, Ghez D et al. Thalidomide in advanced mastocytosis. Br J Haematol 2008;141:249-53.
54. Gruson B, Lortholary O, Canioni D et al. Thalidomide in systemic mastocytosis: results from an open-label, multicentre, phase II study. Br J Haematol 2013;161:434-42.
55. Purtill D, Cooney J, Sinniah R et al. Dasatinib therapy for sys-temic mastocytosis: four cases. Eur J Haematol 2008;80:456-8.
56. Paul C, Sans B, Suarez F et al. Masitinib for the treatment of systemic and cutaneous mastocytosis with handicap: a phase 2a study. Am J Hematol 2010;85:921-5.
57. Droogendijk HJ, Kluin-Nelemans HJ, van Doormaal JJ et al. Imatinib mesylate in the treatment of systemic mastocytosis: a phase II trial. Cancer 2006;107:345-51.
58. Gotlib J, Kluin-Nelemans H, George T, Akin C et al. Midostaurin (PKC412) demonstrates a high rate of durable responses in patients with advanced systemic mastocytosis: results from the fully accrued global phase 2 CPKC412D2201 trial. ASH 2014; abstr. 636.
59. Ustun C, Reiter A, Scott BL et al. Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J Clin Oncol 2014;32:3264-74.
R é f é r e n c e s