mathematics and computation behind blast and fasta xuhua xia [email protected]
TRANSCRIPT

Mathematics and computation behind BLAST and FASTA
Xuhua Xia
http://dambe.bio.uottawa.ca

Slide 3
Why string matching?• Biological significance
– "We have discovered a new gene that has major effect on liver cancer…"
– "We have discovered a new cancer-arresting function of a previously reported reported gene …"
• Early applications: Sequence similarity between an oncogene (genes in viruses that cause a cancer-like transformation of the infected cells), v-sis, and the platelet-derived growth factor (PDGF)
• M. D. Waterfield et al. 1983. Nature 304:35-39• R. F. Doolittle et al. 1983. Science 221:275-227
• Fast computational methods in string matching– FASTA– BLAST– Local pair-wise alignment by dynamic programming

Slide 4
FASTA• A commonly used family of alignment and search
tools
• Generally considered to be more sensitive than BLAST.
• Illustration with two fictitious sequences used in the Contig Assembly lecture:Seq1: ACCGCGATGACGAATASeq2: GAATACGACTGACGATGGA
Seq1: ACCGCGATGACGAATASeq2: GAATACGACTGACGATGGA

Slide 5
String Match in FASTA1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Query A C C G C G A T G A C G A A T A Move N Move NTarget G A A T A C G A C T G A C G A T G G A -1 3 1 6
-2 5 2 7A C G T -3 1 3 31 2 4 8 -4 3 4 37 3 6 15 -5 7 5 610 5 9 -6 1 6 313 11 12 -7 1 7 314 -8 4 8 516 -9 1 9 2
-10 1 10 21 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 -11 5 11 3G A A T A C G A C T G A C G A T G G A -12 1 12 2-3 1 2 -4 4 4 3 7 7 2 7 11 11 10 14 8 13 14 18 -13 1 13 1-5 -5 -4 -11 -2 3 1 1 6 -5 5 5 10 8 8 1 11 12 12 -14 1 14 2-8 -8 -7 -5 1 -2 -2 4 2 2 8 5 5 8 9 9 -15 0 15 0-11 -11 -10 -8 -5 -5 -5 -2 -1 -1 2 2 2 5 6 6 16 0
-12 -11 -9 -6 -2 1 5 17 0-14 -13 -11 -8 -4 -1 3 18 1
Left Right
Left and Right: -n means moving the query left by n sites and n means moving the query right by n sites.

Slide 6
Alternative Matched Strings
Query: ACCGCGATGACGAATATarget:GAATACGACTGACGATGGA
From lecture on contig assembly:
Query: ACCGCGATGACGAATATarget: GAATACGACTGACGATGGA
From FASTA algorithm:
Query: ACCGCGATGACGAATATarget: GAATACGACTGACGATGGA
Query: ACCGCGATGACGAATATarget: GAATACGACTGACGATGGA
Which one is best based on YOUR judgment?
Move N Move N-1 3 1 6-2 5 2 7-3 1 3 3-4 3 4 3-5 7 5 6-6 1 6 3-7 1 7 3-8 4 8 5-9 1 9 2-10 1 10 2-11 5 11 3-12 1 12 2-13 1 13 1-14 1 14 2-15 0 15 0
16 017 018 1
Forw. Back
Best
2nd best
One of the three 3rd best

Slide 7
Word length of 21 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Query A C C G C G A T G A C G A A T A Move N Move NTarget G A A T A C G A C T G A C G A T G G A -1 1 1 3
-2 2 2 5AA AC AG AT CA CC CG CT GA GC GG GT TA TC TG TT -3 0 3 113 1 7 2 3 6 4 15 8 -4 1 4 1
10 14 5 9 -5 4 5 211 12 -6 0 6 1
-7 0 7 1-8 1 8 4-9 0 9 1-10 0 10 1
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 -11 4 11 1GA AA AT TA AC CG GA AC CT TG GA AC CG GA AT TG GG GA -12 0 12 1-5 -11 -4 -11 4 3 1 7 2 5 11 10 8 8 8 12 -13 0 13 0-8 -11 -5 1 -2 -2 2 2 8 5 1 9 -14 0 14 0-11 -5 -5 -1 2 2 6 15 0
16 017 0
Left Right
Query: ACCGCGATGACGAATATarget: GAATACGACTGACGATGGA
Query: ACCGCGATGACGAATATarget: GAATACGACTGACGATGGA Best
One of the three 2nd best

Slide 8
BLAST• Adapted from Crane & Raymer 2003
• Motivation: matching short sequences are faster than matching longer ones
• Input sequence: AILVPTVIGCTVPT
• Algorithm:– Break the query sequence into words:
AILV, ILVP, LVPT, VPTV, PTVI, TVIG, VIGC, IGCT, GCTV, CTVP, TVPT
– Discard common words (i.e., words made entirely of common amino acids)– Search for matches against database sequences, assess significance and
decide whether to discard to continue with extension using dynamic programming: AILVPTVIGCTVPTMVQGWALYDFLKCRAILVPTVIACTCVAMLALYDFLKC
• Critical decision: Discard or continue?
• The E-value as an answer.

Slide 9
Basic stats in string matching• Given PA, PC, PG, PT in a target (database) sequence, the
probability of a query sequence, say, ATTGCC, having a perfect match of the target sequence is:
prob = PAPTPT PGPCPC = PA (PC)2 PG (PT)2
• Let M be the target sequence length and N be the query sequence length, the “matching operation” can be performed (M – N +1) times, e.g., Query: ATGTarget CGATTGCCCG
• The probability distribution of the number of matches follows (approximately) a binomial distribution with p = prob and n = (M – N +1)

Slide 10
Basic stats in string matching• Probability of having a sequence match: p• Probability of having no match: q = 1-p• Binomial distribution:
• When np > 50, the binomial distribution can be approximated by the normal distribution with the mean = np and variance = npq
• When np < 1 and n is very large, binomial distribution can be approximated by the Poisson distribution with mean and variance equal to np (i.e., = 2 = np).
1! !( ) ... ...
( 1)!1! ( )! !n n n n x x nn n
p q p p q p q qn n x x
2
2
( )
21( )
2
x
P x e
( )!
xeP x
x

Slide 11
From Binomial to Poisson
1! ! !( ) ... ... ...
( 1)!1! ( )! ! ( )! !n n n n x x x n x nn n n
p q p p q p q p q qn n x x n x x
1
( )
( 1)
!( )
( )! !
!( )
( )! !
(0)
n
n
n x x
x n x
n
P n p
P n np q
nP n x p q
n x x
nP x p q
n x x
P q
!
( )! !
( 1)( 2)...( 1)
!(
!
( )
)(
! ! ! !
)! !
(1 )
x n x
nx x
x
n
npx x x
x p px
nx
n
nP x p q
n x x
qp q
p
q
pp
p
e
n
n x x
n n n n x
x
n n npe e
xe
x x x

Slide 12
Matching two sequences without gap• Assuming equal nucleotide frequencies, the probability of a
nucleotide site in the query sequence matching a site in the target sequence is p = 0.25.
• The probability of finding an exact match of L letters is a = pL = 0.25L = 2-2L = 2-S, where S is called the bit score in BLAST.
• M: query length; N: target length, e.g., M = 8, N = 5, L = 3AACGGTTCCGGTT
• A sequence of length L can move at (M – L +1) distinct sites along the query and (N – L +1) distinct sites along the target.
• m = (M-L+1) and n = (N-L+1) are called effective lengths of the two sequences.
• The expected number of matches with length L is mn2-S, which is called E-value in ungapped BLAST.
• S is calculated differently in the gapped BLAST

Slide 13
Blast Output (Nuc. Seq.)BLASTN 2.2.4 [Aug-26-2002]...Query= Seq1 38 Database: MgCDS 480 sequences; 526,317 total letters Score ESequences producing significant alignments: (bits) ValueMG001 1095 bases 34 7e-004 Score = 34.2 bits (17), Expect = 7e-004 Identities = 35/40 (87%), Gaps = 2/40 (5%)
Query: 1 atgaataacg--attatttccaacgacaaaacaaaaccac 38 |||||||||| ||||||||||| |||||| ||||||||Sbjct: 1 atgaataacgttattatttccaataacaaaataaaaccac 40
Lambda K H 1.37 0.711 1.31 Matrix: blastn matrix:1 -3Gap Penalties: Existence: 5, Extension: 2…effective length of query: 26effective length of database: 520,557
Matches: 35*1 = 35Mismatches: 3*(-3) = -9Gap Open: 1*5 = 5Gap extension: 2*2 =4R = 35 - 9 - 5 - 4 = 17S = [λR – ln(K)]/ln(2) =[1.37*17-ln(0.711)]/ln(2) = 34E = mn2-S = 26 * 520557 * 2-34 = 7.878E-04x p(x)0 0.9992652171 0.0007345132 0.0000002703 0.000000000
( )( )
!
E xe Ep x
x
Typically one would count only 1 GE here.
Constant gap penalty vs affine function penalty

Slide 14
E-Value in BLAST• The e-value is the expected number of random
matches that is equally good or better than the reported match. It can be a number near zero or much larger than 1.
• It is NOT the probability of finding the reported match.
• Only when the e-value is extremely small can it be interpreted as the probability of finding 1 match that is as good as the reported one (see next slide).
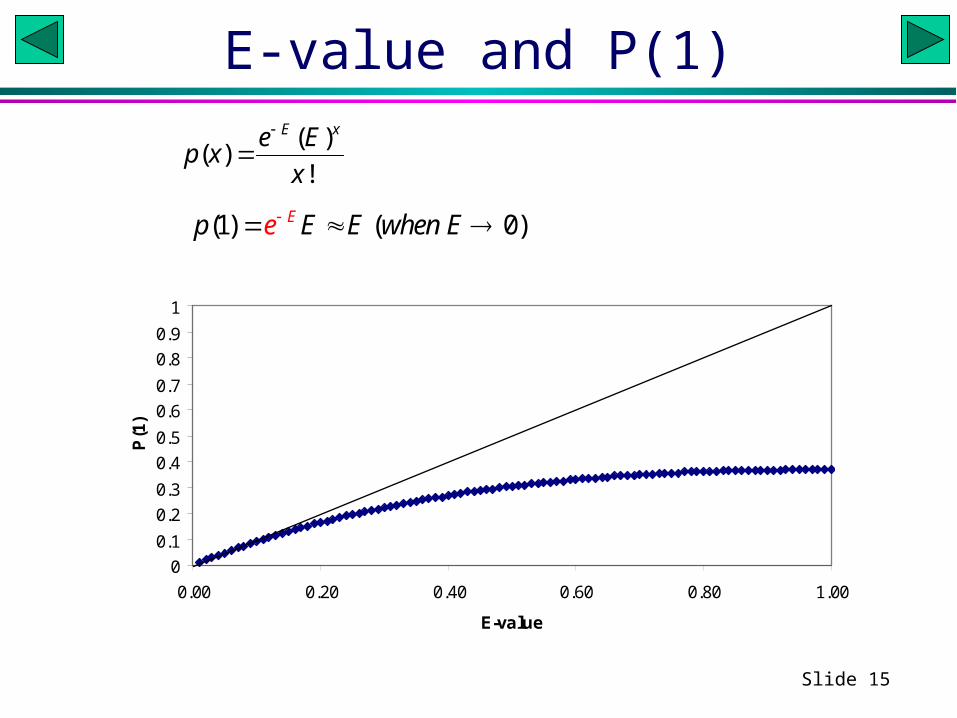
Slide 15
E-value and P(1)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
0.00 0.20 0.40 0.60 0.80 1.00
E-value
P(1)( )
( )!
E xe Ep x
x
(1) ( 0)Ep E E when Ee

Slide 16
BLAST ProgramsProgram Database Query Typical Uses
BLASTN/MEGABLAST
Nucleotide Nucleotide MEGABLAST has longer word size than BLASTN
BLASTP Protein Protein Query a protein/peptide against a protein database.
BLASTX Protein Nucleotide Translate a nuc sequence into a “protein” in six frames and search against a protein database
TBLASTN Nucleotide Protein Unannotated nuc sequences (e.g., ESTs) are translated in six frames against which the query protein is searched
TBLASTX Nucleotide Nucleotide 6-frame translation of both query and database
PHI-BLAST Protein Protein Pattern-hit iterated BLAST
PSI-BLAST Protein Protein Position-specific iterated BLAST
RPS-BLAST Protein Protein Reverse PSI-BLAST

Slide 17
Comparison: BLAST and FASTA• BLAST starts with exact string matching, while
FASTA starts with inexact string matching (or exact string matching with a shorter words). BLAST is faster than FASTA.
• For the examples given, both BLAST and FASTA will find the same best match, i.e., shifting the query sequence by 2 sites to the right.
• Both perform dynamic programming for extending the match after the initial match.

Optional: BLAST Parameters• Lambda and Karlin-Altschul (K) parameters are important
because they directly affect the computation of E value.
• Both and K depend on – nucleotide (or aminon acid) frequencies– match-mismatch matrix
• All BLAST implementations generally assume that nucleotide (or amino acid) sequences have roughly equal frequencies.
• For nucleotide (or amino acid) sequences with strongly biased frequencies, BLAST E value obtained with the assumption can be quite misleading, i.e., one should use appropriate and K.

Lambda () and K
4 4
1 1
1ijsi j
i j
p p e
4 42 2 3 3
1 1
4 0.25 12 0.25 0.25 0.75 1ijsi j
i j
p p e e e e e
BLAST output includes lambda () and K. Mathematically, is defined as follows:
where pi, pj are nucleotide frequencies (i,j = A, C, G, or T), and sij is the match (when i = j) or
mismatch (when i j) score. In nucleotide BLAST by default, we have sii = 1 and sij = -3. In the
simplest case with equal nucleotide frequencies, i.e., when p i = 0.25, the equation above is reduced to
See the updated Chapter 1 and BLASTParameter.xlsm on how to compute K.
20 20
1 1
1ijsi j
i j
p p e
(for amino acid sequences)
Now insert different values to the equation above to find which balances the equation (not the trivial solution of = 0)

Finding I: equal , (1, -3)
Slide 20
A G C T
0.25 0.25 0.25 0.25
A 0.25 0.0625 0.0625 0.0625 0.0625
G 0.25 0.0625 0.0625 0.0625 0.0625
C 0.25 0.0625 0.0625 0.0625 0.0625
T 0.25 0.0625 0.0625 0.0625 0.0625
Match-MismatchA 1 -3 -3 -3
G -3 1 -3 -3
C -3 -3 1 -3
T -3 -3 -3 1
Lambda 1
0.169893 0.003112 0.003112 0.003112
0.003112 0.169893 0.003112 0.003112
0.003112 0.003112 0.169893 0.003112
0.003112 0.003112 0.003112 0.169893 0.716911
Double-click it, copy to EXCEL and find by using solver.

Finding II: Different , (1, -3)
A G C T
0.1 0.4 0.4 0.1
A 0.1 0.01 0.04 0.04 0.01
G 0.4 0.04 0.16 0.16 0.04
C 0.4 0.04 0.16 0.16 0.04
T 0.1 0.01 0.04 0.04 0.01
Match-MismatchA 1 -3 -3 -3
G -3 1 -3 -3
C -3 -3 1 -3
T -3 -3 -3 1
Lambda 1
0.027183 0.001991 0.001991 0.000498
0.001991 0.434925 0.007966 0.001991
0.001991 0.007966 0.434925 0.001991
0.000498 0.001991 0.001991 0.027183 0.957075

Finding III: Different , s/v A G C T0.1 0.4 0.4 0.1
A 0.1 0.01 0.04 0.04 0.01G 0.4 0.04 0.16 0.16 0.04C 0.4 0.04 0.16 0.16 0.04T 0.1 0.01 0.04 0.04 0.01 1
Match-MismatchA 1 -1 -3 -3G -1 1 -3 -3C -3 -3 1 -1T -3 -3 -1 1
Lambda 0.98990.02691 0.014865 0.002053 0.0005130.014865 0.430554 0.008211 0.0020530.002053 0.008211 0.430554 0.0148650.000513 0.002053 0.014865 0.02691 1.000046

Finding K: equal , (1, -3)
Slide 23
A G C T
0.25 0.25 0.25 0.25
A 0.25 0.0625 0.0625 0.0625 0.0625
G 0.25 0.0625 0.0625 0.0625 0.0625
C 0.25 0.0625 0.0625 0.0625 0.0625
T 0.25 0.0625 0.0625 0.0625 0.0625
Match-MismatchA 1 -3 -3 -3
G -3 1 -3 -3
C -3 -3 1 -3
T -3 -3 -3 1
Lambda 1
0.169893 0.003112 0.003112 0.003112
0.003112 0.169893 0.003112 0.003112
0.003112 0.003112 0.169893 0.003112
0.003112 0.003112 0.003112 0.169893 0.716911
Double-click it, copy to EXCEL and find by using solver.

Slide 24
Bioinformatics research workflowAccumulation of nucleotide and amino acid sequences:
UUCUCAACCAACCAUAAAGAUAU
UUCUCUACAAACCACAAAGACAU
UUCUCAACCAACCAUAAAGAUAU
UUCUCAACCAACCACAAAGACAU
UUCUCCACGAACCACAAAGAUAU
UUCUCUACAAACCACAAAGAUAU
UUCUCAACCAACCACAAAGACAU
UUCUCUACUAACCACAAAGACAU
Storage and annotation of the sequences
1. Structural annotation with homology search and de novo gene prediction
2. Functional annotation with gene ontologies
Species-specific gene dictionaries, e.g., yeastgenome.org
1. Comparative genomics (the origin of new genes, new features and new species)
2. Phylogenetics (cladogenic process, dating of speciation and gene duplication events)
3. Phylogeny-based inference.
Mutation
Selection
Adaptation
1. Gene/Protein families (e.g., Pfam)
2. Cluster of orthologous genes (e.g., COG)
3. Supermatrix of gene presence/absence
4. Genome-based pair-wise distance distributions
Functional genomicsSystems biologyDigital cells