may 8th, 2012 higher ed & research. molecular dynamics applications overview amber namd gromacs...
TRANSCRIPT

Molecular DynamicsGPU Applications Catalog
May 8th, 2012Higher Ed & Research

Molecular Dynamics Applications Overview
AMBER
NAMD
GROMACS
LAMMPS
Sections Included *
* In fullscreen mode, click on link to view a particular module. Click on NVIDIA logo in each slide to return to this page.

Application Features Supported
GPU Perf Release Status Notes/Benchmarks
AMBERPMEMD Explicit Solvent & GB
Implicit Solvent
89.44 ns/day JAC
NVE on 16X 2090s
ReleasedMulti-GPU, multi-node
AMBER 12 http://ambermd.org/gpus/benchmarks.
htm#Benchmarks
NAMDFull electrostatics
with PME and most simulation features
6.44 ns/days STMV 585X 2050s
Released100M atom capable
Multi-GPU, multi-node
NAMD 2.8.2.9 version April 12
http://biowulf.nih.gov/apps/namd/namd_ bench.html
GROMACSImplicit (5x), Explicit
(2x) Solvent via OpenMM
165 ns/Day DHFR
4X C2075s
4.5 Single GPU released
4.6 Multi-GPU Released
http://biowulf.nih.gov/apps/gromacs-gpu.html
LAMMPS Lennard-Jones, Gay-Berne, Tersoff
3.5-15xReleased.
Multi-GPU, multi-node
1 billion atom on Lincoln: http://lammps.sandia.gov/bench.html# machine
GPU Perf compared against Multi-core x86 CPU socket.GPU Perf benchmarked on GPU supported features
and may be a kernel to kernel perf comparison
Molecular Dynamics (MD) Applications

Application Features Supported GPU Perf Release
Status Notes
Abalone TBDSimulations
4-29X(on 1060
GPU)Released
Single GPU. Agile Molecule, Inc.
ACEMD Written for use on GPUs
160 ns/day Released
Production bio-molecular dynamics (MD) software
specially optimized to run on single and multi-GPUs
DL_POLYTwo-body Forces,
Link-cell Pairs, Ewald SPME forces, Shake
VV
4xV 4.0 Source onlyResults Published
Multi-GPU, multi-node supported
HOOMD-Blue
Written for use on GPUs
2X(32 CPU
cores vs. 2 10XX GPUs)
Released, Version 0.9.2
Single and multi-GPU.
New/Additional MD Applications Ramping
GPU Perf compared against Multi-core x86 CPU socket.GPU Perf benchmarked on GPU supported features
and may be a kernel to kernel perf comparison

GPU Value to Molecular Dynamics
What
Why
How
Study disease & discover drugsPredict drug and protein interactions
Speed of simulations is criticalEnables study of:
Longer timeframes Larger systemsMore simulations
GPUs increase throughput & accelerate simulations
AMBER 11 Application4.6x performance increase with 2 GPUs with only a 54% added cost*
• AMBER 11 Cellulose NPT on 2x E5670 CPUS + 2x Tesla C2090s (per node) vs. 2xcE5670 CPUs (per node)• Cost of CPU node assumed to be $9333. Cost of adding two (2) 2090s to single node is assumed to be $5333
GPU Test DrivePre-configuredApplications
AMBER 11NAMD 2.8
GPU Ready Applications
AbaloneACEMDAMBER
DL_PLOYGAMESS
GROMACSLAMMPS
NAMD

All Key MD Codes are GPU Ready
AMBER, NAMD, GROMACS, LAMMPS
Life and Material Sciences
Great multi-GPU performance
Additional MD GPU codes: Abalone, ACEMD, HOOMD-Blue
Focus: scaling to large numbers of GPUs

AMBER

Outstanding AMBER Results with GPUs

Run AMBER FasterUp to 5x Speed Up With GPUs
DHFR (NVE)23,558 Atoms
GPU+CPU CPU
ns/Day 58.28
ns/Day 14.16
“…with two GPUs we can run a single simulation as fast as on 128 CPUs of a Cray XT3 or on 1024 CPUs of an IBM BlueGene/L machine. We can try things that were undoable before. It still blows my mind.” Axel Kohlmeyer Temple University
1 node 2 nodes 4 nodes 8 nodes NICS Kraken (Athena)0
10
20
30
40
50
60
2.444.55
8.22
13.49
46.01
21.29
31.57
45.58
56.45
Cluster Performance ScalingAMBER 11 JAC Simulation time, ns/day
CPU only CPU + 1x C2050 per node
ns/day
CPU Supercomputer

Cost of 1 Node Performance Speed-up 0.0
1.0
2.0
3.0
4.0
5.0
AMBER 11 on 2X E5670 CPUs (per node)
AMBER 11 on 2X E5670 CPUs + 2X Tesla M2090s (per node)
Re
lati
ve C
ost
an
d P
erf
orm
an
ce B
en
efi
t S
cale
AMBER Make Research More Productive with GPUs
Adding Two 2090 GPUs to a Node Yields a > 4x Performance Increase
Base node configuration:
Dual Xeon X5670s and DualTesla M2090 GPUs per node
318% Higher Performance
54% Additional Expense
No GPU
With GPU

NAMD

Run NAMD FasterUp to 7x Speed Up With GPUs
ApoA-192,224 Atoms
GPU+CPU CPU
ns/Day 2.94
ns/Day 0.51
STMV1,066,628 Atoms
1 2 4 8 12 160
0.5
1
1.5
2
2.5
3
3.5 NAMD 2.8 Benchmark
GPU+CPU
CPU only
# of Compute Nodes
Ns/Day
Test Platform: 1 Node, Dual Tesla M2090 GPU (6GB), Dual Intel 4-core Xeon (2.4 GHz), NAMD 2.8, CUDA 4.0, ECC On.Visit www.nvidia.com/simcluster for more information on speed up results, configuration and test models.
NAMD 2.8 B1 + unreleaesd patch, STMV BenchmarkA Node is Dual-Socket, Quad-core x5650 with 2 Tesla M2070 GPUsPerformance numbers for 2 M2070 8 cores (GPU+CPU) vs. 8 cores
(CPU)

Cost of 1 Node Performance Speed-up 0.0
1.0
2.0
3.0
4.0 NAMD 2.8 on 2X E5670 CPUs (per node)NAMD 2.8 on 2X E5670 CPUs + 2X Tesla C2070s (per node)
Re
lati
ve C
ost
an
d P
erf
orm
an
ce B
en
efi
t S
cale
Make Research More Productive with GPUs
Get up to a 250% Performance Increase(STMV – 1,066628 atoms)
No GPU
With GPU
250% Higher
54% Additional Expense

GROMACS

GROMACS Partnership Overview
Erik Lindahl, David van der Spoel, Berk Hess are head authors and project leaders. Szilárd Páll is a key GPU developer.
2010: single GPU support (OpenMM library in GROMACS 4.5)
NVIDIA Dev Tech resources allocated to GROMACS code
2012: GROMACS 4.6 will support multi-GPU nodes as well as GPU clusters

GROMACS 4.6 Release Features
Multi-GPU support - GPU acceleration is one of main focus: majority of features will be accelerated in 4.6 in a transparent fashion
PME simulations get special attention, and most of the effort will go into making these algorithms well load-balanced
Reaction-Field and Cut-Off simulations also run accelerated
List of non-supported GPU accelerated features will be quite short
GROMACS Multi-GPU Expected in April 2012

GROMACS 4.6 Alpha Release Absolute Performance
Absolute performance of GROMACS running CUDA- and SSE-accelerated non-bonded kernels with PME on 3-12 CPU cores and 1-4 GPUs. Simulations with cubic and truncated dodecahedron cells, pressure coupling, as well as virtual interaction sites enabling 5 fs are shown
Benchmark systems: RNAse in water with 24040 atoms in cubic and 16816 atoms in truncated dodecahedron box
Settings: electrostatics cut-off auto-tuned >0.9 nm, LJ cut-off 0.9 nm, 2 fs and 5 fs (with vsites) time steps
Hardware: workstation with 2x Intel Xeon X5650 (6C), 4x NVIDIA Tesla C2075

GROMACS 4.6 Alpha Release Strong Scaling
Strong scaling of GPU-accelerated GROMACS with PME and reaction-field on:
Up to 40 cluster nodes with 80 GPUs
Benchmark system: water box with 1.5M particles
Settings: electrostatics cut-off auto-tuned >0.9 nm for PME and 0.9 nm for reaction-field, LJ cut-off 0.9 nm, 2 fs time steps
Hardware: Bullx cluster nodes with 2x Intel Xeon E5649 (6C), 2x NVIDIA Tesla M2090, 2x QDR Infiniband 40 Gb/s

GROMACS 4.6 Alpha Release PME Weak Scaling
Weak scaling of the GPU-accelerated GROMACS with reaction-field and PME on:
3-12 CPU cores and 1-4 GPUs. The gradient background indicates the range of system
Sizes which fall beyond the typical single-node production size.
Benchmark systems: water boxes size ranging from 1.5k to 3M particles.
Settings: electrostatics cut-off auto-tuned >0.9 nm for PME and 0.9 nm for reaction-field, LJ cut-off 0.9 nm, 2 fs time steps.
Hardware: workstation with 2x Intel Xeon X5650 (6C), 4x NVIDIA Tesla C2075.

GROMACS 4.6 Alpha Release Rxn-Field Weak Scaling Weak scaling of the GPU-accelerated
GROMACS with reaction-field and PME on:
3-12 CPU cores and 1-4 GPUs. The gradient background indicates the range of system
sizes which fall beyond the typical single-node production size
Benchmark systems: water boxes size ranging from 1.5k to 3M particles
Settings: electrostatics cut-off auto-tuned >0.9 nm for PME and 0.9 nm for reaction-field, LJ cut-off 0.9 nm, 2 fs time steps
Hardware: workstation with 2x Intel Xeon X5650 (6C), 4x NVIDIA Tesla C2075

GROMACS 4.6 Alpha Release Weak Scaling
Weak scaling of the CUDA non-bonded force kernel on GeForce and Tesla GPUs. Perfect weak scaling, challenges for strong scaling
Benchmark systems: water boxes size ranging from 1.5k to 3M particles
Settings: electrostatics & LJ cut-off 1.0 nm, 2 fs time steps
Hardware: workstation with 2x Intel Xeon X5650 (6C) CPUs, 4x NVIDIA Tesla C2075

LAMMPS

LAMMPS Released GPU Features and Future Plans
* Courtesy of Michael Brown at ORNL and Paul Crozier at Sandia Labs
LAMMPS August 2009First GPU accelerated support
LAMMPS Aug. 22, 2011Selected accelerated Non-bonded short‐range potentials (SP, MP, DP support)
Lennard-Jones (several variants with &without coulombic interactions)MorseBuckinghamCHARMMTabulated Course grain SDKAnisotropic Gay-BernRE-squared“Hybrid” combinations (GPU accel & no GPU accel)
Particle-Particle Particle-Mesh (SP or DP)Neighbor list builds
Longer Term*Improve performance on smaller particle counts
Neighbor List is the problem
Improve long-range performanceMPI/Poisson Solve is the problem
Additional pair potential support (including expensive advanced force fields) – See “Tremendous Opportunity for GPUs” slide*Performance improvements focused to specific science problems
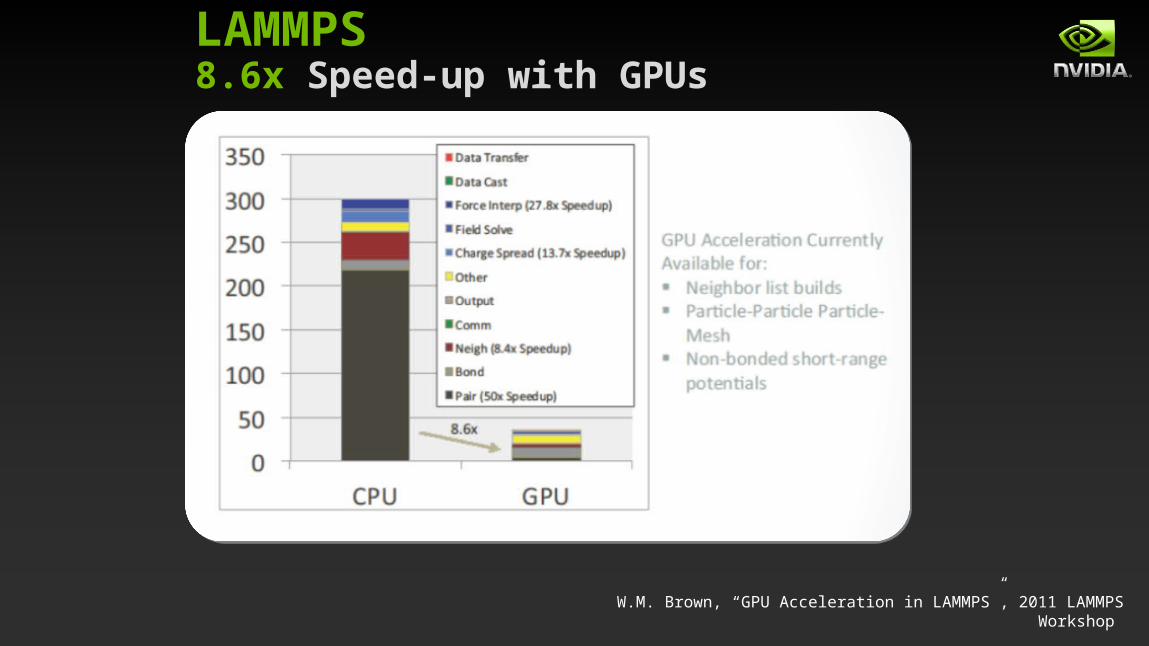
W.M. Brown, “GPU Acceleration in LAMMPS”, 2011 LAMMPS Workshop
LAMMPS8.6x Speed-up with GPUs

LAMMPS4x Faster on Billion Atoms
Test Platform: NCSA Lincoln Cluster with S1070 1U GPU servers attached CPU-only Cluster- Cray XT5
Billion Atom Lennard-Jones Benchmark
Series1
29 Seconds
103 Seconds
288 GPUs + CPUs 1920 x86 CPUs

4X-15X Speedups
Gay-BerneRE-Squared
From August 2011 LAMMPS Workshop
Courtesy of W. Michael Brown, ORNL
LAMMPS

LAMMPS Conclusions
Runs both with individual multi-GPU node, as well as GPU clusters
Outstanding raw performance! Performance is 3x-40X higher than equivalent CPU code
Impressive linear strong scaling
Good weak scaling, scales to a billion particles
Tremendous opportunity to GPU accelerate other force fields

END