measles virus: early infection, progression and pathogenesis in a
TRANSCRIPT

UNIVERSIDADE DE LISBOA
FACULDADE DE FARMÁCIA
DEPARTAMENTO DE MICROBIOLOGIA
Measles virus: early infection, progression and pathogenesis in a transgenic mouse model
Cláudia Sofia Antunes Ferreira
Tese orientada pelo Prof. Doutor João Gonçalves e
pelo Prof. Doctor Roberto Cattaneo
DOUTORAMENTO EM FARMÁCIA
MICROBIOLOGIA
2010

As opiniões expressas neste trabalho são de exclusiva responsabilidade do
seu autor.
Cláudia Sofia Antunes Ferreira was financially supported by a PhD
scholarship (SFRH / BD / 19785 / 2004) from Programa SFRH, Fundação
para a Ciência e a Tecnologia, Portugal.

To
My Family
For their support, for their understanding and for their love
To
My Grandmother
For teaching me to love life


Preface
v
Preface
The research described in the present thesis was performed under
the supervision of Prof. Roberto Cattaneo and Prof. João Gonçalves.
The studies described in this thesis were performed at the
Department of Molecular Medicine of the Mayo Clinic College of Medicine,
Rochester, USA. The results shown in the third chapter of this thesis were
included in a manuscript already published:
Ferreira CS, Marie Frenzke, Vincent H.J. Leonard, G. Grant Welstead,
Christopher D. Richardson, Roberto Cattaneo. 2010. Measles virus
infection of alveolar macrophages and dendritic cells precedes spread to
lymphatic organs in transgenic mice expressing human signaling
lymphocytic activation molecule (SLAM, CD150). J. Virol. 84:3033-3042.
According with the “Decreto-Lei 388/70”, article 8 point nº 2, the
data presented in this dissertation is the result of the authors work and it is
clearly acknowledge in the text whenever data or reagents produced by
others were used. The author participated in planning and execution of the
experimental procedures such as in results interpretation and manuscript
preparation. The opinions expressed in this publication are from the
exclusive responsibility of the author and have not been previously
submitted for any degree at this or any University.


Acknowledgments
vii
Acknowledgments
First, I want to thank my supervisors Prof. Doutor João Gonçalves
and Prof. Doctor Roberto Cattaneo for their time and effort invested in this
project, for the ideas, scientific input and discussions. Prof. Doutor João
Gonçalves, I for always will thank for supporting all my decisions
concerning this PhD. Prof. Doctor Roberto Cattaneo, I deeply appreciate all
your dedication and commitment.
I also thank Prof. Doutor José Pereira Moniz for receiving me as a
PhD student at the Faculdade de Farmácia da Universidade de Lisboa.
Also I want to acknowledge the Fundação para a Ciência e a
Tecnologia for the funding of my PhD scholarship.
From the Centro de Patologia Molecular/Unidade de Retrovírus e
Infecções Associadas, I would like to thank Ana, Sofia, Andreia, Fred,
Mariana, Iris, Rita and Sylvie for they welcomed me in their lab and shared
their benches with me and taught me a lot.
From the Department of Molecular Medicine, Mayo Clinic College
of Medicine, I specially thank all the members of my laboratory: Sompong,
thank you so much for your infinite kindness and for welcoming me so well
in the lab; Patricia, thank you for teaching me a lot of techniques, for
reading my many attempts of writing science and for being a friend; Jorge,
my desk (almost) and bench colleague, thank you so much for your infinite,
infinite patience and for always helping me and try to solve my science
problems; Vincent, thank you so much for being always a good colleague

Acknowledgments
viii
and for your unmeasurable help in finishing the manuscript; Chanakha,
thank you so much; I wish we had played tennis more often; K.C. thank you
so much for you taught me to look only at the good in people and to accept
challenges with good thoughts, strength and faith. Thank you for your
pleasant company during our Saturday and Sunday lunches at the
Methodist cafeteria; Marie, thank you for your precious help in the lab and
in the animal facility and during all our meetings; Andrew, thank you for
your always good mood and kindness in helping everyone; Guy and
Johanna, thank you.
From the all floor of the Department of Molecular Medicine, I thank
all who helped me to go through these four years. To Richard Vile, Memy
Diaz, Candice Willmon, Ponpimon Wongthida, Feorillo Galivo, Tim Kottke,
Yasuhiro Ikeda, Ruyta Sakuma, Mark Federspiel, a special thank you. I also
thank the former director of the Department of Molecular Medicine,
Stephen Russell.
From the Department of Immunology, Mayo Clinic College of
Medicine, I thank Larry Pease, Hirohito Kita, Richard Vile and Adam Schrum
for valuable scientific ideas and discussions.
From the Transplantation Biology Unit, Mayo Clinic College of
Medicine, I thank Josie Williams, Kim Butters, Karen Lien, Bruce Knudsen,
Brenda Ogle and Gregory Brunn for all their immense support and help.
From the Department of Urology, Mayo Clinic College of Medicine, I
thank Sue Kunts for her precious help with the immuno stainings.

Acknowledgments
ix
From the Mayo Clinic Flow Cytometry and Optical Morphology Core
Facility I thank James Tarara and his team for their technical assistance and
valuable advice.
And I want to thank all my friends from around the world that
became my family during the time spent in Rochester and made my life so
much better: Catarina Cortesão for being so patient and such a good friend
and such a positive company; Susanne Lang, for being unique and for being
always there; Cristina Correia for her infinite patience and nice
conversations during coffee breaks; Sarwa Darwish-Murad for being such a
beautiful person and being there and to all the great people I met and that
I had the pleasure of being friends with: Josef Korinek, Filippo Agnesi,
Clifton Haider, Kostandinos Sideras, Noemi Vidal-Folch, Xavier Frigola-Baro,
Wissam ElRiachy, Justo Sierra Johnson, Angel Gonzalez, Laura Moreno-
Luna, Pedro Geraldes, Carolina Trabuco, Sandra Herrmann, Barry Boilson,
Aisling Carroll, Bolaji Ajao, Chris Van Der Walt, Alain, Jasminka and Dannah
Bernheim, Cristina João and Paulo Nicola and many more…
I could not not thank all the friends I made through my life: Elsa,
Maria, Catarina Noronha, Catarina Loureiro, Orieta, Fernanda, Gabi,
Catarina Cortesão, a special thank you. You mean a lot to me.
I have a special thank to Prof. Doutora Laurentina Pedroso for her
support at the Universidade Lusófona de Humanidades e Tecnologias,
where I’m a collaborator.

Acknowledgments
x
I thank with all my heart to my family, for their abundant support,
understanding and for strength. And I have a very special thank to Tiago,
for his support, for the strength, understanding and love.
.

Resumo
xi
Resumo
O desenvolvimento de novas terapias, de vacinas e a aplicação dos
vírus no tratamento do cancro será melhor sucedido quanto mais
abrangente e aprofundado for o conhecimento da interacção entre o vírus
e o hospedeiro.
Esta tese tem como objectivo global compreender a interacção
entre o vírus do sarampo e o hospedeiro e as consequências dessa
interacção no tropismo e patogénese viral. Em particular, pretende-se
identificar as células alvo iniciais do vírus bem como as células responsáveis
pela disseminação do vírus no hospedeiro. Pretende-se ainda compreender
os mecanismos de atenuação do vírus atenuado do sarampo.
O vírus do sarampo é transmitido através de aerossóis e, apesar da
infecção ter início no aparelho respiratório, as células alvo iniciais do vírus
não são conhecidas. A partir do aparelho respiratório o vírus alcança os
orgãos linfáticos e inicia a replicação nos nódulos linfaticos que drenam o
aparelho respiratório.
Apesar de vários estudos terem sido desenvolvidos, a contribuição
da interacção entre os receptores e o vírus no tropismo viral e progressão
da infecção têm sido restringidos pela falta de um modelo animal
adequado. Os macacos são os únicos primatas não humanos que mostram
susceptibilidade ao vírus do sarampo. No entanto, o seu uso fica muito
limitado devido a questoes éticas e monetárias. O vírus wild type do
sarampo utiliza como receptor celular uma proteína expressa em células

Resumo
xii
activadas do sistema imune denominada de molécula de sinalização de
activação linfocitária (SLAM, CD150). Com o objectivo de desenvolver um
modelo animal para o estudo da infecção pelo vírus do sarampo, ratinhos
transgénicos SLAMGe, ratinhos que expressam o SLAM humano (hSLAM),
foram cruzados com ratinhos que apresentam inactivação do gene que
codifica para o receptor do interferão tipo I, ratinhos Ifnarko
, de forma a
permitir uma replicação do vírus mais eficiente. Os ratinhos resultantes do
cruzamento foram denominados ratinhos Ifnarko
-SLAMGe. São deficientes
na produção de interferão tipo I e expressam hSLAM com um perfil de
expressão idêntico ao dos humanos. Estes ratinhos foram utilizados neste
estudo para caracterizar as células infectadas pelo vírus do sarampo
imediatamente após infecção respiratória bem como as células
responsáveis pela progressão e disseminação do vírus no hospedeiro.
Em primeiro lugar caracterizamos a expressão do hSLAM nos
ratinhos Ifnarko
-SLAMGe. A expressão de hSLAM foi detectada em linfócitos
B e T extraídos do baço e em macrófagos produzidos a partir da medula
óssea, através de citometria de fluxo, após activação destas células ex vivo.
Uma vez que o linfonodo mediastinal foi previamente demonstrado ser
relevante na infecção pelo vírus do sarampo em ratinhos SLAM, estudamos
a expressão de hSLAM em células imunes provenientes do linfonodo
mediastinal de ratinhos Ifnarko
-SLAMGe. Através de citometria de fluxo
demonstramos que os linfócitos B e T não activados provenientes do
linfonodo mediastinal expressavam baixos níveis de hSLAM.
Documentamos ainda que, mais células B do que células T expressavam

Resumo
xiii
hSLAM, como previamente demonstrado em tecidos linfáticos humanos.
Uma vez que SLAM é expresso em células imunes e não em células
epiteliais, as células imunes presentes nas vias respiratórias são candidatas
a células alvo para a replicação do vírus. Desta forma, analizamos a
expressão de hSLAM em células imunes provenientes dos pulmões do
ratinho Ifnarko
-SLAMGe. Cerca de 9% de células B e 3% de células T
expressavam hSLAM nos pulmões do ratinho Ifnarko
-SLAMGe e baixa
percentagem de macrófagos alveolares expressavam hSLAM.
De seguida, estes ratinhos foram inoculados via intranasal (IN) com
o vírus do sarampo wild type. Um vírus derivado da estírpe Ichinoise B e ao
qual foi adicionado o gene que codifica para a expressão da proteína verde
fluorescente, wtMVgreen. Vários orgãos foram recolhidos um, dois e três
dias após a inoculação e analizados para a expressão de proteína
fluorescente. Um dia após inoculação a replicação viral foi detectada nos
pulmões, e, de seguida, em vários orgãos do sistema linfático; inicialmente
no linfonodo que drena o aparelho respiratório, o linfonodo mediatinal, e
mais tarde em outros orgãos linfáticos, nomeadamente nos linfonodos
mandibular, inguinal e mesentérico, no baço e em células mononucleares
do sangue periférico.
Para responder à questão quais as células que sustentam a
replicação do vírus imediatamente após inoculação, os ratinhos foram
inoculados via IN com wtMVgreen; os pulmões e vários orgãos linfáticos
foram recolhidos e as células infectadas foram identificadas pela expressão
de proteína verde fluorescente através de citometria de fluxo.

Resumo
xiv
Os macrófagos alveolares foram identificados como as principais
células que sustêm a replicação do vírus do sarampo imediatamente após
inoculação respiratória. As células dendríticas foram 5 vezes menos
infectadas. Outras populações de células imunes foram infectadas em
menor extensão.
Apesar da expressão de SLAM pelos macrófagos alveolares ser
muito reduzida, 24 horas apos a infecção a expressão de SLAM aumentou
drasticamente; 0.86% de macrófagos alveolares expressava SLAM antes da
infecção, passando 16% a expressarem SLAM após a infecção. Este
resultado sugere que o vírus do sarampo utiliza um receptor celular que é
facilmente induzido nas células alvo, contribuindo assim para uma rápida e
eficiente disseminação no hospedeiro.
Dois dias após a inoculação IN o vírus propagou-se ao sistema
linfático sendo o linfonodo mediastinal o primeiro orgão linfático a ser
infectado e o que suportou título viral mais elevado. Através de citometria
de fluxo identificaram-se os linfócitos B e T como as principais células
infectadas bem como células dendríticas, pese embora em menor grau.
Por último, com o objectivo de compreender os mecanismos de
atenuação da estírpe do vírus usada como vacina do sarampo, foi
caracterizada a infecção e disseminação do vírus do sarampo atenuado,
denominado aqui de MVvac. Ratinhos Ifnarko
-SLAMGe foram inoculados
com MVvac pela via de infecção intraperitoneal (IP) e o título viral foi
determinado em vários orgãos linfáticos (nomeadamente nos linfonodos
mandibular, mediastinal, mesentérico e inguinal e no baço). A replicação

Resumo
xv
do vírus atenuado nos orgãos do ratinho foi muito baixa comparada com a
replicação do wild type. Três dias após inoculação, o orgão que apresentou
um título viral mais elevado foi o linfonodo mediastinal seguido do
linfonodo mesentérico e do baço. A replicação nos linfonodos mandibular e
inguinal ficou aquém do limite de detecção. Apesar de o linfonodo
mediastinal ser o orgão linfático onde o vírus mais eficientemente se
replicou a replicação neste linfonodo pelo MVvac foi cerca de 100 vezes
inferior à replicação pelo vírus wild type. De seguida, as células que
sustentam a replicação deste vírus bem como do wild type foram
identificadas. Um e três dias após a infecção, o linfonodo mediastinal e o
baço foram recolhidos e secções destes orgãos foram analisadas através de
microscopia confocal. Um dia após inoculação, ambos os vírus infectaram
uma população de macrófagos presente na região subcapsular dos
linfonodo e na zona marginal do baço, denominados macrófagos do seio
subcapsular (macrófagos SSC). Após uma primeira interacção com os
macrófagos, foi observada uma disseminação generalizada do vírus do wild
type nos folículos do linfonodo bem como na polpa branca do baço. Em
contraste, quando os ratinhos foram inoculados com um vírus do sarampo
atenuado, apesar de o vírus ser encontrado na região subcaspular do
linfonodo e zona marginal do baço no primeiro dia após infecção,
colocalizando com os macrófagos SSC, esta infecção inicial não foi seguida
de infecção massiva das células linfáticas nestes orgãos. O vírus foi
eliminado, observando-se apenas alguma replicação no linfonodo
mediastinal e baço mas inferior à replicação pelo wild type.

Resumo
xvi
Ainda com o objectivo de esclarecer o mecanismo de atenuação do
vírus do sarampo atenuado caracterizamos a replicação de um virus wild
type com uma substitução de um aminoácido na proteína hemaglutinina.
Esta mutação, asparagina por tirosina, no aminoácido 481 na proteína
hemaglutinina (N481Y), está presente em todas as estirpes atenuadas do
vírus do sarampo mas não no vírus wild type. Esta substituição introduzida
no genoma do vírus wild type permite ao vírus, que apenas usa o SLAM
como receptor celular, passar também a utilizar o receptor CD46. O
receptor CD46 ou cofactor proteico membranar (MCP) é expresso em
todas as células nucleadas do corpo humano. Todas estirpes atenuadas do
vírus do sarampo para além de SLAM utilizam o CD46 como receptor
celular. Para aceitar ou revogar a hipótese de que esta mutação presente
no vírus atenuado pode afectar o fitness do vírus, introduzimos esta
mutação num vírus wild type, denominado aqui wtMVN481Y e
comparamos a replicação in vivo deste vírus com um vírus wild type
original, wtMV. Para tal, ratinhos Ifnarko
-SLAMGe forma inoculados via IP
com wtMVN481Y ou wtMV. Três dias após inoculação, o título do vírus foi
determinado em vários linfonodos e no baço. Demonstramos que esta
mutação não afecta a replicação nem o tropismo do vírus uma vez que não
se verificaram diferenças relevantes na replicação dos vírus nos diferentes
orgãos. De seguida, para compreender se a interacção com o receptor
CD46 para além do SLAM afecta a replicação e disseminação do vírus,
obtivemos ratinhos transgénicos que expressam o CD46 humano (hCD46)
para além do hSLAM, aqui denominados de ratinhos Ifnarko
-CD46Ge-

Resumo
xvii
SLAMGe. Inoculamos ratinhos Ifnarko
-CD46Ge-SLAMGe e ratinhos Ifnarko
-
SLAMGe com o vírus wtMVN481Y e comparamos a replicação e
disseminação deste vírus em vários orgãos linfáticos destes ratinhos. Não
foram encontradas diferenças significativas na replicação e tropismo deste
vírus o que sugere que o uso do receptor CD46 é pouco relevante na
atenuação do vírus do sarampo.
Este estudo demonstra que os macrófagos e as células dendríticas
são as células alvo iniciais do vírus do sarampo e que podem estar
envolvidas na rápida disseminação do vírus para outras células imunes no
organismo. Demonstra-se ainda que, os macrófagos como células alvo
iniciais, aliado à baixa replicação do vírus do sarampo atenuado parecem
desempenhar um papel importante na atenuação deste vírus. O uso do
receptor CD46 parece ser pouco relevante no mecanismo de atenuação do
vírus. Para além disso, a linha de ratinhos transgénicos que expressam o
receptor hSLAM são uma peça chave no estudo das fases iniciais de
infecção do vírus do sarampo.
Palavras Chave: macrófagos alveolares, células dendríticas, vírus do
sarampo, SLAM, CD46, tropismo.


Abstract
xix
Abstract
Measles virus (MV) is one of the most infectious viruses but several
aspects of MV biology and pathogenesis remain poorly understood. The
virus is transmitted by aerosol droplets and initial infection is believed to
occur in the upper airways but the cells that support early infection and
viral spread throughout the body are not well defined. As wild type MV
(wtMV) enters cells through SLAM but not CD46 receptor, we generated a
transgenic mice expressing human SLAM (hSLAM) and used these mice to
identify the cells supporting primary MV infection. We first characterized
hSLAM expression in immune cells of these mice by flow cytometry. We
documented hSLAM expression in resting B and T cells in the mediastinal
lymph node (LN) and in B and T cells and in a small fraction of alveolar
macrophages (AM) collected from the lungs. Next, we inoculated these
animals intranasally and assessed viral infection in the nasal associated
lymphoid tissue, lungs, several LN, spleen and thymus. One day post
inoculation (p.i.), viral replication was documented in the lungs; the AM
and dendritic cells (DC) were the main infected cells. It was observed that
MV infection temporarily enhanced hSLAM expression in AM. From the
lungs, infection spread to all lymphatic organs and in particular to the
mediastinal LN. This LN supported high levels of infection upon IN and
intraperitoneal inoculation. Finally, to understand the mechanisms of MV
vaccine attenuation, we compared the spread of wtMV with that of a
vaccine strain (MVvac) in Ifnarko
-SLAM mice. One day p.i. MV was detected

Abstract
xx
in the subcapsular macrophages in the mediastinal LN. Three days later,
the wtMV spread to hSLAM expressing leukocytes whereas the MVvac was
cleared. As the wtMV does not enter cells through CD46 receptor but the
attenuated strain does, we compared the replication of a wtMV carrying a
mutation in the hemagglutinin protein allowing CD46 usage. CD46 usage
seems to have no relevant effect on MV attenuation. Thus, SLAM
expressing mice allowed characterization of the early steps of MV
infection. The alveolar macrophages and dendritic cells are the key players
in MV infection and propagation.
Keywords: alveolar macrophages, dendritic cells, measles virus, SLAM,
CD46, tropism.

Abbreviations
xxi
AAbbbbrreevviiaattiioonnss
aa amino acid
APC antigen presenting cell
APC-Cy7 allophycocyanin-Cy7
B95a marmoset B-cell line
BM bone marrow
CAT chloramphenicol acetyltransferase
CCPs complement control protein repeats
CD46 membrane cofactor protein
cDNA complementary DNA
CDV canine distemper virus
Cyt cytoplasmic
DAPI 4',6-diamidino-2-phenylindole
DC dendritic cells
DMEM dulbeco’s modified eagle medium
DNA deoxyribonucleic acid
EDTA ethylenediaminetetraacetic acid
ER endoplasmic reticulum
F fusion
FACS flow activated cell sorter
FBS fetal bovine serum
FITC fluorescein isothiocyanate
G guanosine
GFP green fluorescent protein
GM-CSF granulocyte-macrophage colony-stimulating factor
H hemagglutinin
hCD46 human CD46
hSLAM human SLAM
HδR hepatitis delta virus ribozyme
i.e. id est
IC-B ichinoise-B
IFN interferon
IFNAR interferon type I receptor
Ifnarko
interferon type I receptor knock out

Abbreviations
xxii
Ig immunoglobulin
IL interleukin
IN intranasal
IP intraperitoneal
IRF IFN regulatory factor
ISGF3 IFN-stimulated gene factor 3
ISRE IFN-stimulated response element
Kb kilobase
kDa kilo Dalton
KO knock out
L large
LcK lymphocyte protein tyrosine kinase
LN lymph node
LPS lipopolysaccharide
M matrix
M.O.I. multiplicity of Infection
MCP membrane cofactor protein
MDA-5 melanoma differentiation-associated gene 5
mL milliliter
Mm millimeter
mM millimolar
mRNA messenger RNA
MHC major histocompatibility complex
MS marginal sinus
MV measles virus
MW molecular weight
N nucleocapsid
NALT nasal associated lymphoid tissue
Nm nanometer
ºC degree celsius
ORF overlapping reading frame
P phosphoprotein
p.i. post infection
PBMC peripheral blood mononuclear cells
PBS phosphate buffered saline
PCR polymerase chain reaction

Abbreviations
xxiii
PCT C-terminal domain
PE phycoerythrin
PerCP peridin chorophyll protein
PNT N-terminal domain
PRRs pattern-recognition receptors
RdRp RNA dependent RNA polymerase
RIG-I retinoic acid-inducible gene I
RLRs retinoic acid-inducible gene I (RIG-I)-like receptors
RNA ribonucleic acid
RNP ribonucleocapsid
Rpm revolutions per minute
RPMI Roswell Park Memorial Institute
RT room temperature
SAP SLAM-associated protein
SCR short consensus repeats
SCS subcapsular Sinus
SDS-PAGE sodium dedecyl sulfate-polyacrylamide gel electrophoresis
SLAM signaling lymphocytic activation molecule
SSPE subacute sclerosing panencephalitis
STAT signal transducers and activators of transcription
STP serine-threonine-proline
TBS tris-buffered saline
TCID50 tissue culture infectious dose 50%
Th2 T helper 2
TLC thin layer chromatography
TLRs toll-like receptors
TNF-α tumor necrosis factor alpha
Tyr110 tyrosine 110
UV ultra violet
VSV vesicular stomatitis virus
WT wild type
μg microgram
μl microlitre
μm micrometer


Table of Contents
xxv
Table of Contents
Preface
v
Acknowledgments vii
Resumo xi
Abstract xix
Abbreviations xxi
Table of Contents xxv
Chapter 1 General introduction 1
1.1. Taxonomy 3
1.2. History of measles 4
1.3. Clinical features of measles 5
1.4. Basic biology 6
1.4.1. Virion structure and genome 6
1.4.1.1. Nucleocapsid protein 9
1.4.1.2. P, V, and C proteins 10
1.4.1.3. Matrix protein 12
1.4.1.4. Fusion protein 13

Table of Contents
xxvi
1.4.1.5. Hemagglutinin protein 14
1.4.1.6. Polymerase protein 15
1.4.2. Measles virus life cycle 16
1.4.3. Measles virus receptors 19
1.4.3.1. CD46 20
1.4.3.2. SLAM 23
1.4.3.3. Other receptors 24
1.5. Measles virus pathogenesis 27
1.5.1. Measles virus entry and spread 27
1.5.2. Immune response to measles virus infection 28
1.5.2.1. Innate immunity 28
1.5.2.2. Humoral immunity 29
1.5.2.3. Cellular immunity 30
1.5.3. Measles virus-induced immunosuppression 31
1.6. Mouse models for the study of measles pathogenesis: SLAM
transgenic mice
32
Aims 37
Chapter 2 Measles virus infection of transgenic mice expressing
human signaling lymphocytic activation molecule (SLAM, CD150)
39

Table of Contents
xxvii
Abstract 41
Introduction 43
Materials and Methods 45
Mouse strains 45
Mouse infections 45
Generation of a wild type MV expressing the reporter gene
chloramphenicol acetyl transferase
45
Rescue of viruses and preparation of virus stocks 46
Isolation and activation of B and T lymphocytes 47
Isolation and activation of macrophages 48
Flow cytometry 49
Immunohistochemistry 49
CAT assay 49
Results 51
Human SLAM expression in lymphocytes of hSLAM transgenic
mice
51
Human SLAM expression in macrophages of hSLAM transgenic
mice
52
Ifnar deficiency increases susceptibility to wild type MV
replication in hSLAM transgenic mice
54

Table of Contents
xxviii
Spread of MV in Ifnarko
-SLAMGe mice 57
Discussion 60
Chapter 3 Measles virus infection of alveolar macrophages and
dendritic cells precedes spread to lymphatic organs in transgenic
mice expressing human signaling lymphocytic activation molecule
(SLAM, CD150)
63
Abstract 65
Introduction 67
Materials and Methods 70
Mouse strains 70
Mouse infections 71
Viruses and virus stock preparation 72
Blood and organ collection, and preparation of single cell
suspensions
73
Immunohistochemistry and immunofluorescence 74
Flow cytometry 76
Quantification of virus load in lymphatic organs 77
Results 78
Ifnarko
-SLAMGe transgenic mice 78

Table of Contents
xxix
Human SLAM expression in lymphatic tissue of Ifnarko
-SLAMGe
transgenic mice
79
Human SLAM expression in immune cells of the lungs of Ifnarko
-
SLAMGe transgenic mice
80
Early MV replication in the lungs of Ifnarko
-SLAMGe transgenic
mice
82
AM sustain MV replication in the lungs 85
MV infects AM expressing SLAM, and MV infection enhances
SLAM expression
88
Spread in the lymphatic organs 90
Discussion 93
A new small animal model of MV infection 93
Crossing the epithelial barrier: alternatives 95
SLAM expression and virus amplification 97
Chapter 4 Pathways of lymphatic dissemination of measles virus
attenuated and wild type strains in SLAM expressing mice
99
Abstract 101
Introduction 103
Materials and Methods 104
Cells 104

Table of Contents
xxx
Cell infections 104
Mouse strains 105
Mouse infections 105
Site-specific mutagenesis of measles virus H protein 105
Viruses and virus stock preparation 106
Organ collection and preparation of single cell suspensions 106
Flow cytometry 106
Immunofluorescence 106
Quantification of virus load in lymphatic organs 107
Results 107
Low efficiency of replication of MV vaccine strain 107
Early capture of wild type and vaccine strains of MV by
subcapsular macrophages
108
Only wild type MV infection progresses to B and T cell areas 110
Replication of a recombinant wild type MV with a single
mutation allowing different receptor usage
114
Discussion 119
Chapter 5 Main conclusions and future perspectives 123
References 131

General Introduction
1
CHAPTER 1
General introduction


General Introduction
3
1.1. Taxonomy
Measles virus (MV) is a member of the family Paramyxoviridae, genus
Morbillivirus within the Mononegavirales order. The Paramyxoviridae are
enveloped viruses with a single-stranded nonsegmented RNA genome of
negative polarity. This family includes some of the most relevant pathogens
of humans and animals (Table 1).
Family
Subfamily Genus Species
Paramyxoviridae Paramyxovirinae Respirovirus Bovine parainfluenza virus
3
Human parainfluenza virus
1 and 3
Sendai virus
Rubulavirus Human parainfluenza virus
2, 4a and 4b
Parainfluenza virus 5
Mumps virus
Morbillivirus Canine distemper virus
Dolphin morbillivirus
Cetacean morbillivirus
Measles virus
Peste-des-petits-ruminants
virus
Phocine distemper virus
Rinderpest virus
Avulavirus Newcastle disease virus

General Introduction
4
Family
Subfamily Genus Species
Paramyxoviridae
Pneumovirinae Pneumovirus Bovine respiratory syncytial
virus
Human respiratory
syncytial virus
Ovine respiratory syncytial
virus
Pneumonia virus of mice
Henipaviruses Hendra virus
Nipah virus
Table 1. Examples of members of the family Paramyxoviridae. (Adapted
from Lamb, R.A. and Parks, G.D., Fields Virology, 2007).
1.2. History of measles
Measles is postulated to have emerged in the early civilization centers
in the Middle East around 3000 B.C.. Abu Becr is known as the first to
distinguishing measles from smallpox in the 9th
century. Many of the
epidemiological aspects of measles were elucidated by a young Danish
physician during a measles epidemic in 1846 in the Faroe Islands. He
postulated the respiratory route of transmission, the highly contagious
nature of the disease, a 14 days incubation period and the lifelong
immunity present in the older population (135). In 1790, an English
surgeon described measles complications, namely a case of post-measles
encephalomyelitis in a young woman who developed paresis as the rash
started to fade (103). Nineteenth century text books associated measles

General Introduction
5
infection with reactivation of latent tuberculosis and immunosuppression
caused by MV is connoted to von Pirquet, who observed, in 1908,
suppression of the tuberculin test response during measles (197).
1.3. Clinical features of measles
Measles is transmitted by aerosol droplets, and initial infection is
believed to be established in the respiratory tract although primary target
cells are not well defined. From the respiratory tract the virus spreads to
the regional lymph nodes where viral amplification occurs resulting in
viremia. The disease is characterized by an incubation period of 10-14 days,
followed by a 2 to 4 days of prodrome of fever, anorexia, cough and
conjunctivitis (59). During this phase, small white spots, Koplik’s spots, may
become visible in the buccal mucosa and are pathognomonic of measles
(90). The appearance of a maculopapular rash marks the end of the
prodrome and lasts 3 to 4 days. Recovery begins soon after onset of the
rash (59) with the appearance of a marked cellular and humoral immune
response conferring lifelong protective immunity (135). Concomitantly with
activation of the immune system a transient but profound
immunosuppression occurs. This immunosuppression continues for weeks
after apparent recovery and increases the host’s susceptibility to
opportunistic infections which accounts for measles associated deaths.
Several complications are associated with measles, including pneumonia,
encephalitis, otitis media, blindness, and secondary infections by bacteria
and viruses. Interstitial pneumonia caused by MV replication and

General Introduction
6
inflammation in the lower respiratory tract can occur in healthy patients
(58, 68) whereas giant cell pneumonia is seen mostly in
immunocompromised individuals (131). Diarrhea is a common
gastrointestinal complication of measles and frequently is associated with
bacterial infections.
A rare complication of measles is subacute sclerosing
panencephalitis (SSPE). SSPE is caused by a progressive dissemination of
defective virus replication in the central nervous system and occurs in
young individuals several years after infection (91, 122). In about 1 in 1000
cases measles is complicated by a post-infectious encephalitis that usually
occurs within 14 days after the onset of rash and mostly in individuals
under the age of 2 years (83). In contrast to SSPE, little evidence of viral
invasion of the central nervous system is observed and pathological studies
suggest an autoimmune etiology (51). Another measles-induced
neurological complication is measles inclusion body encephalitis or
progressive infectious encephalitis. It occurs 3 to 6 months after the
regular disease and is seen in immunocompromised individuals (115).
1.4. Basic biology
1.4.1. Virion structure and genome
Measles viral particles are pleomorphic or spherical with diameters
ranging from 120 to 300-1000 nm and therefore have variable cargo
volume and tolerate well foreign gene insertion (59, 148). The envelope,
composed of a lipid bilayer derived from the plasma membrane of the host

General Introduction
7
cell, surrounds the virions and contains surface projections composed by
the hemagglutinin (H) and the fusion (F) glycoproteins. The matrix (M) lines
the inner surface of the envelope and associates with the cytoplasmic tails
of the H and F proteins as well as the viral core particle or nucleocapsid.
The viral RNA genome is encapsidated by the nucleoprotein (N) to
form a helical ribonucleocapsid (RNP) that is the substrate for both
transcription and replication. These latter activities are carried out by the
RNA dependent RNA polymerase (RdRp) that is composed of the large (L)
protein and of the phosphoprotein (P). A schematic representation of a MV
particle is shown in Fig. 1 (A).
The 15,894 nucleotide long MV genome contains six genes, coding
for eight proteins in the order 3’-N-P/V/C-M-F-H-L-5’. The P gene uses
overlapping reading frames to code for three proteins, P, V and C. These
genes are flanked by a 3’ extracistronic region, the leader, and a 5’
extracistronic region, the trailer, that are essential for transcription and
replication Fig. 1 (B).

General Introduction
8
Figure 1. (A) Schematic diagram of a polyploid MV particle containing
three genomes. The viral nucleocapsid (N), phosphoprotein (P),
polymerase (large, L), matrix (M), fusion (F), and hemagglutinin (H) proteins
are indicated with different symbols. (B) Diagram of the MV antigenome
(plus strand). The coding regions of MV genes are represented by arrow-
shaped boxes.
M
H (tetramer)
F (trimer)
N
L
P
A
NN P/V/C M H LF
1 15894
B

General Introduction
9
1.4.1.1. Nucleocapsid protein
The nucleocapsid messenger RNA, the first to be transcribed, codes
for the N protein (525 aa), the most abundant viral protein. The N protein
is an RNA-binding protein that encapsidates the full length viral (-) sense
genomic and (+) sense antigenomic RNAs within a helical nucleocapsid
template. The helical nucleocapsid rather than the free genome RNA is the
template for all the RNA synthesis (95). Each N protein coats 6 nucleotides,
the so called “rule of six” i.e. their genome must be of polyhexameric
length to efficiently replicate (20). The binding of N to RNA to form a helical
structure is thought to have several functions, including protection from
nuclease digestion and providing interaction sites for assembly of
nucleocapsids into budding virions (95).
Biochemical and mutational studies have identified two structural
regions in the N protein: N core, a conserved N-terminal domain and a non
conserved N tail, C-terminal. The N core has ~ 400 aa and is essential for
self assembly of N with RNA, RNA binding and for RNA replication. The C-
terminal N tail region is ~ 100 aa and interacts with a C-terminal domain of
P protein. In infected cells, two forms of N exist: a monomeric form
(referred to as N0) and an assembled form (referred to as N
NUC) (55). In its
assembled form, N interacts with P and the polymerase complex (L–P),
which is essential to RNA synthesis by the viral polymerase (18). In its
monomeric form, it is responsible for the encapsidation of the nascent RNA
chain during genome and antigenome replication. During virus assembly, N

General Introduction
10
interacts with the M protein. Finally, it interacts with cellular factors,
including cytoskeleton components.
During the course of infection, polymerase activity changes from
transcription to replication. By analogy with studies made in vesicular
stomatitis virus (VSV) (14), the intracellular concentration of unassembled
N is thought to be a major factor controlling the rates of transcription and
replication from the genome templates.
1.4.1.2. P, V, and C proteins
The P protein, 507 aa, plays multiple roles in both transcription and
replication. It is an essential component of the viral polymerase and binds
to the nucleocapsid tethering the polymerase complex L-P to the
nucleocapsid (NNUC
) template (79, 88). It is a modular protein consisting of
at least 2 domains: an N-terminal domain (PNT) and a C-terminal domain
(PCT). The acidic and highly phosphorylated N-terminal domain includes aa
1 to 230 and is poorly conserved. It is the assembly module required to
chaperone unassembled N protein as a P-N complex during the nascent
chain assembly step of genome replication (73, 173). The C-terminal
domain (aa 231-507) is well conserved; it represents the polymerase
cofactor module and mediates binding of L to the N-RNA template.
In addition to the P protein, the P gene gives rise to two other
polypeptides by means of using overlapping reading frames (ORF) on a
single transcript. The C ORF is accessed by translational choice using an
initiator methionine codon 19 nucleotides downstream from that of P (10).

General Introduction
11
In contrast, the V protein shares the initiator methionine and the amino-
terminal 231 aa of the P protein but has a different C-terminal region. An
extra non-template-directed guanosine (G) residue at position 751 is added
following three Gs by a process known as RNA editing or pseudotemplated
transcription (25). This nucleotide addition results in a shift of the
translational reading frame into an alternative reading frame and hence a
different C-terminal region. The last 276 aa normally encoded by the P
mRNA are replaced with a cyteine-rich domain of 68 aa (25) that has zinc-
binding properties (100).
The N-terminal region of the P protein, as the collinear region of the
V protein, binds to STAT1, with the Tyr110 residue playing a key role in this
interaction (19, 38, 128). The C and V proteins play a role in the regulation
of transcription and replication and are not found in the virions (101, 188).
The V and C proteins have been analyzed for their ability to modulate
either interferon (IFN) type I or its signaling pathways. The C protein is
implicated in inhibition of IFNAR downstream signaling (166), modulation
of the viral polymerase activity, inhibition of cell death and of inflammatory
responses (9, 150, 185). Other reports show that V protein can counteract
the interferon antiviral activity and can inhibit the inflammatory response
in infected peripheral blood monocytic cells of experimentally infected
monkeys (36).

General Introduction
12
1.4.1.3. Matrix protein
The M protein, coded by the third gene, is a basic protein and
underlies the viral lipid bilayer. M is thought to play a crucial role in the
assembly of progeny virus by interacting with the RNP (70, 71). In addition,
M interacts with the cytoplasmic tails of H and F proteins (23, 174) and
drives the apical assembly of MV leading to the release of infectious
particles early after infection (116). Abrogation of M function dramatically
alters the assembly of MV (12, 22). M inactivation or failure to associate
with budding structures or with the viral nucleocapsid is likely to be a
factor responsible for MV persistent infection where release fails to occur.
In SSPE the M protein is either absent (12) or, when present, is not
associated with budding structures in cultivated cells and is not able to
bind to viral nucleocapsid in vitro (27, 70, 71). In addition, the M protein
controls fusion function of the viral envelope glycoproteins (22). A
genetically engineered recombinant MV that lacks M shows increased cell-
to-cell fusion and decreased production of infectious particles (22). More
recently, Tahara et al. (179), identified two substitutions in the M protein
of attenuated MV, P64S and E89K, that, by allowing a strong interaction
with the cytoplasmic tail of H, enhanced the assembly of infections
particles in Vero cells but inhibited MV signaling lymphocytic activation
molecule (SLAM)-dependent cell-cell fusion, reducing viral growth in B
lymphoid cells. By modulating viral growth, M protein may partly account
for the attenuation of MV attenuated strains (178, 179).

General Introduction
13
1.4.1.4. Fusion protein
The F protein mediates viral entry with release of the RNP into the
cytoplasm by fusion between the virion envelope and the host cell plasma
membrane at neutral pH. Later in infection, the F protein, expressed at the
cell surface membrane, can mediate fusion with neighboring cells to form
syncytia (giant multinucleated cells), a cytopathic effect caused by MV that
is an alternative mechanism of viral spread.
The fusion protein, 533 aa, is a type I transmembrane glycoprotein
(151). MV F protein includes an N terminal cleavable signal sequence that
targets the nascent polypeptide chain synthesis to the membrane of the
endoplasmic reticulum (ER). At the C terminus, a hydrophobic stop-transfer
domain anchors the protein in the membrane leaving a short 33 aa
cytoplasmic tail.
F is synthesized as an inactive precursor, F0 and gains its fusogenic
activity by cleavage by the ubiquitous intracellular protease furin, localized
in the trans Golgi network (15, 201). Proteolytic cleavage results in a
disulfide-linked fusion competent F protein that consists in a
transmembrane F1 and a membrane distal subunit F2. F1 contains a stretch
of hydrophobic aa at the N-terminus constituting the fusion peptide that is
inserted into the target membrane when fusion occurs. The F2 subunit
contains all three N-linked glycans that have a role in proteolytic cleavage,
stability and transport to the cell surface (77, 193).
The MV F-protein is classified as a class I fusion protein (94). Class I
fusion proteins mediate membrane fusion by coupling irreversible protein

General Introduction
14
refolding to membrane juxtaposition. This is accomplished by discrete
conformational changes of a metastable F protein structure to a lower
energy structure. The cleavage of F0 into F1-F2 primes the protein for
membrane fusion. Activation of the F-protein results in the insertion of the
fusion peptide located in the F1 subunit into the target membrane. This is
followed by dramatic conformational rearrangements of the F trimer and
results in juxtaposition of the target and donor membranes (205). As the F-
protein goes from a high-energy metastable structure to a low-energy
post-fusion structure, this eventually leads to the formation of a fusion
pore through which the RNP complex enters the cell (95).
1.4.1.5. Hemagglutinin protein
The H protein is the receptor attachment protein and is an
important determinant of cellular tropism. It is a 617 aa type II
transmembrane glycoprotein which is comprised of a N-terminal
cytoplasmic tail of 34 aa followed by a membrane-spanning domain and an
extracellular membrane - proximal stalk region connected to a C-terminal
receptor-binding head domain (3). Receptor-binding residues have been
mapped to this head domain (76, 99, 121, 177, 198). While other
paramyxovirus attachment proteins are globular, MV H protein exhibits a
cube shaped structure. A significant area of the H protein is covered with
N-linked oligosacharides which makes it unavailable for receptor
interaction (67). In contrast to other paramyxovirus attachment proteins, H
only forms dimers (142). The two H protein molecules making up the

General Introduction
15
homodimer are highly tilted with respect to each other. This has further
consequences for receptor specific fusion.
The first step in the fusion process is binding of H to its receptor. On
binding ligand, the H protein undergoes its own conformational change,
which in turn triggers a conformational change in F to release the fusion
peptide. The H protein of the Edmonston strain has five predicted N-linked
glycosylation sites; the first four of these sites are used. Glycosylation is
necessary for proper folding, antigenicity, dimerization, and export of H
from the Golgi (78).
1.4.1.6. Polymerase (large)
The L protein is the least abundant but the largest protein (240
KDa). The L gene codes for the paramyxovirus RdRp which possess all
enzymatic activities necessary to synthesize mRNA, like nucleotide
polymerization, 5’ end mRNA capping and methylation, and
polyadenylation of mRNA (57, 69, 124). L adds poly-A tails to nascent viral
mRNAs cotranscriptionally by stuttering on a stretch of U residues
occurring at the end of each viral gene.
L includes six highly conserved domains that were identified by
sequence comparisons. The exact role of several domains is not clear but
mutations in some of these domains resulted in L proteins that although
were able to transcribe viral mRNA were defective in RNA replication (143,
169).

General Introduction
16
L binding to the viral P protein is mapped to the N-terminal 408 aa
of L (73) and this interaction confers stability to L (65, 72). Within the L-P
complex, the P protein bridges the L polymerase to the nucleocapsid
template. The L protein besides interacting with P can interact with host
cell proteins (172).
1.4.2. Measles virus life cycle
MV contains nonsegmented single-stranded RNA genome of
negative polarity and replication occurs entirely in the cytoplasm. An
overview of the life cycle of MV is depicted in Fig. 2. The H protein contacts
the receptors whereas the F protein mediates fusion. After the viral
membrane fuses with the cellular plasma membrane at the neutral pH, the
helical nucleocapsids are released into the cytoplasm. The uncoating
process takes place by disrupting of the M-N contacts.
Early in virus infection, before the viral translation products
accumulate to high levels, the viral RdRp activity is restricted to the
production of a leader RNA and mRNAs from the incoming virion
nucleocapsid in what is called primary transcription. At later times
following infection, this input RNP is used as a template to produce (+)
sense antigenomes which are in turn used as templates to produce new (-)
sense genomic RNA. After primary transcription and translation, when
abundant progeny genomes have been produced, they can serve as
additional templates in what is called the secondary transcription to
produce much higher levels of viral mRNA transcripts (95). The processive

General Introduction
17
viral polymerase transcribes the N-encapsidated genome RNA into 5’
capped and 3’ polyadenylated mRNAs. Beginning at the 3’ end of the
genome, the polymerase transcribes the genes in a sequential and polar
manner by terminating and reinitiating at each end of the gene junctions.
This sequential “stop-start” mechanism continues across the viral genome
in a 3’ to 5’ direction. Polyadenylation occurs by reiterative copying of four
to seven uridylates in the gene end sequence, followed by release of the
mRNA. Initiation and capping of the downstream mRNA is specific by the
gene-start sequence following the short intergenic region. The RdRp
sometimes fails to reinitiate and this leads to a gradient of mRNA
abundance that decreases according to the distance from the genome
3’end with N mRNA being the most abundant and the L mRNA the least
abundant (26).
Paramyxovirus particles are formed by a budding process. Buds
emerge from the plasma membrane in locations where the viral
components are assembled. Assembly is thought to require coordinated
localization of multiple virus proteins that are preferently incorporated into
nascent viral particles, whereas most of the host proteins are excluded. The
assembly of the envelope is at the cell surface and in polarized epithelial
cells MV egress from the apical surface.

General Introduction
18
Figure 2. Measles virus life cicle. The top of the figure illustrates an
incoming virion which fuses with the cell plasma membrane followed by
release of the helical nucleocapsid in the cytoplasm. Viral mRNAs are
indicated by lines and 5’ mRNA cap is represented by a filled circle whereas
3’ poly A tail by an An. Genome replication carried out by the N-P-L
complex and primary and secondary transcription carried out by P-L
complex are represented in solid lines. Intracellular transport of RNP and M
protein to the plasma membrane and of F and H proteins from the ER to
Golgi to the plasma membrane is indicated with dotted lines. (Adapted
from Lamb, R.A. and Parks, G.D., Fields Virology, 2007).

General Introduction
19
1.4.3. Measles virus receptors
MV was isolated first by Enders and his team in 1954 by inoculating
blood of David Edmonston, a child with measles, onto a primary culture of
human kidney cells (42). However, it was not until 1993 that the first
cellular receptor for MV was identified. Two independent groups reported
that laboratory adapted strains of MV could enter host cells via the
ubiquitous regulator of complement activation membrane cofactor protein
(MCP; CD46) (39, 119). The observation that strains isolated in B95a cells or
human B-cell lines grow inefficiently in cells susceptible to the attenuated
strain, lead to hypothesize that these strains might use other cellular
receptor. Indeed, a few years later, Tatsuo et al. (186) using a functional
expression cloning approach, showed that a cDNA clone could render a
resistant cell line susceptible to a B95a cell-isolated MV. This cDNA was
shown to encode human SLAM or signaling lymphocytic activation
molecule, expressed on several immune cells and first described as
involved in T cell activation (29, 170). Other authors confirmed these
findings (44, 75).
MV is able to infect several tissues and cells that do not express any
of the known receptors, suggesting that additional receptors may still await
identification. Indeed, recent studies showed that human polarized
epithelial cell lines support MV entry, replication, and cytopathic effect
independent of SLAM and CD46 (99, 177).

General Introduction
20
1.4.3.1. CD46
CD46 (MCP) was identified by several research groups in the
eighties as a cell surface molecule with a broad double pattern on SDS-
PAGE and later denominated CD46. Further studies characterized this
glycoprotein belonging to the regulators of complement activation family
with the function to protect host cells from complement mediated attack
(102). CD46 protects the cells from complement mediated damage by
serving as a cofactor for the factor I-mediated cleavage of C3b to C3bi and
C4b to C4c and C4d, preventing the C3b and C4b forming the C3/C5
convertase required for complement regulatory function (102).
CD46 is a cell surface, type I transmembrane glycoprotein of 57-67
kDa encoded by a single gene that is ~ 50 kb long and comprises 14 exons.
The N-terminal region of the CD46 protein consists of four short consensus
repeats (SCR) also named complement control protein repeats (CCPs) (Fig.
3). These SCR are domains of about 60 aa. SCR 1, 2 and 4 are N-
glycosylated; N-glycosylation of SCR 2 is essential for its function as MV
receptor (105). The SCR are followed by one or two serine-threonine-
proline (STP) rich regions, sites of O-glycosylation. Following the STP region
are 12 aa of unknown function, a transmembrane domain and one of two
cytoplasmic tails (CYT-1 or CYT-2).
CD46 is expressed as four predominant isoforms. These isoforms
are the result of the alternative splicing of CD46 gene and differ in the
presence of one (C) or two (BC) STP region and the 16 amino acid (CYT-1)
or 23 amino acid (CYT-2) CYT domains (102). These isoforms are termed

General Introduction
21
STP-BC1, STP-BC2, STP-C1, and STP-C2 and migrate on a SDS-PAGE as two
band pattern with molecular weight (MW) of 62,000-67,000 (STP-BC1 and
STP-BC2 isoforms) and 54,000-60,000 (STP-C1 and STP-C2 isoforms) (102).
All isoforms can serve as receptors for MV (52, 106). Functional studies
mapped the C3b/C4b-regulatory activity on SCR2-4 (2, 80) whereas MV H
protein binding site resides within SCR 1 and 2 (17, 37).
CD46 is expressed on almost all human nucleated cells (165). Three
patterns of expression, differing in the ratio of expression of the four main
isoforms, are recognized in the population: most individuals (65%) express
the STP-BC1/2 forms, 29% express both forms in equal quantities and 6%
express the STP-C1/2 forms (102).
Besides serving as a cellular receptor for MV, CD46 is used as a
receptor and port of entry by other human pathogens. A number of
pathogenic bacteria including Streptococcus pyogenes and Neisseria
meningitides and gonorrhoeae bind to CD46 and different serotypes of
adenovirus and herpesvirus 6 utilize CD46 as cellular receptor (24).

General Introduction
22
Figure 3. Structures of MV receptors SLAM (left) and CD46 (right). SLAM, a
member of the CD2 subset of the immunoglobulin superfamily, regulates
synthesis of T helper 2 cytokines by T cells. The extracellular domain is
composed of a variable (V) and a constant (C2) domains. All MV strains
bind to the V domain of SLAM. CD46, a regulator of complement activation,
has four SCR modules in the extracellular domain. The vaccine strains of
MV interact with SCR1 and 2, whereas SCR 2 and 4 interact with
complement C3b and C4b. APC: antigen presenting cell. (Adapted from
Yanagi, et al., Measles virus: cellular receptors, tropism and pathogenesis.
J. Gen. Virol., 2006).

General Introduction
23
1.4.3.2. SLAM
The lingering question of the relevance of a ubiquitous receptor for
MV pathogenesis was answered when in 2000, the signaling lymphocyte
activation molecule (SLAM; CD150) was identified as the cellular receptor
for clinical isolates of MV (44, 75, 186). Importantly, attenuated strains of
MV can use SLAM as a cellular receptor, in addition to CD46 (45, 132).
SLAM is a 70 kDa membrane glycoprotein that belongs to the CD2
subset of immunoglobulin gene superfamily and has two extracellular
immunoglobulin domains (V and C2) (Fig. 3). The MV H protein interacts
with the N-terminal V domain of SLAM and this domain is necessary and
sufficient to allow MV entry (133). Although mouse SLAM has 60%
sequence identity to human SLAM and similar function, it cannot act as MV
receptor.
SLAM can interact with another SLAM molecule present on a
neighbor cell (Fig. 3) (107). Its cytoplasmic domain contains three tyrosine
residues that are surrounded by SH2 domain-binding sequences. SLAM
associates intracellularly with SH2 domain-containg molecules such as the
SLAM-associated protein (SAP), protein tyrosine phosphatase SHP2 and,
inositol phophatase SHIP (159). Ligation of SLAM on T cells leads to its
binding to SAP, which then recruits and activates FynT, resulting in tyrosine
phosphorylation of SLAM. This triggers the recruitment of multiple players
involved in SLAM signaling (96). This leads to the production of T helper 2
(Th2) cytokines such as IL-4 and IL-13 (43, 104). SLAM regulates the
production of IL-12, tumor necrosis factor-α (TNF-α) and nitric oxide by

General Introduction
24
macrophages (32, 200). In addition, SLAM may induce B cell proliferation
and immunoglobulin (Ig) synthesis (146).
SLAM is expressed on immature thymocytes, on memory T cells,
and is readily induced on B and T cells following activation (7, 29, 170).
According to the same authors, SLAM is not detected on granulocytes,
monocytes and cells from non lymphoid organs (7, 29, 170). However,
SLAM expression on monocytes was readily induced after stimulation with
mitogens or after MV infection (109). Furthermore, SLAM was detected in
murine macrophages following activation with LPS (74). Mature dendritic
cells, but not immature ones express SLAM (13, 92, 125, 144). Distribution
of this receptor overlaps with sensitivity of different cell types to wild type
MV infection and better explains the immunosuppressive characteristics of
the virus.
Besides MV, other morbillivirus like canine distemper virus (CDV)
and rinderpest virus use SLAM (canine and bovine respectively) as their
cellular receptor (187).
1.4.3.3. Other receptors
MV spreads systemically and eventually it infects epithelial tissues
of several organs (108, 157). However these tissues do not express SLAM;
CD46, which is ubiquitously expressed, does not support infection by wild
type MV. This implies that MV may use another receptor (204). The
identification of a putative receptor on epithelial cells still remains elusive
but two groups mapped the residues of the MV attachment protein

General Introduction
25
sustaining epithelial cell receptor mediated cell fusion and showed that the
receptor binding site on the H-protein required to infect epithelial cells
differs from the binding sites for CD46 and SLAM (99, 177). Moreover,
results from these two groups suggest that the epithelial receptor may be a
molecule related with tight junctions and located on the basolateral
surface of the epithelial cells (Fig. 4, top left scheme). Infected immune
cells would infect the epithelial cells through this basolateral molecule and
the virus would initiate replication in epithelial cells. The active release of
infectious viral particles in the airways would explain the high infectious
nature of MV.

General Introduction
26
Immune cellsImmune cells
Figure 4. MV is transmitted by respiratory aerosol droplets containing
MV particles and initial replication takes place in immune cells in the
upper respiratory tract using SLAM receptor (right drawing). Infected
immune cells enter the lymphatic or blood circulation and propagate in
the lymphatic organs throughout the body (bottom drawing). MV
infected immune cells or released viral particles infect epithelia of
several mucosal organs trough the use of a putative epithelial receptor.
Infected epithelial cells release viruses in the airways to achieve
transmission and completing the infectious cycle (left drawing).
(Adapted from Navaratnarajah, C. et al., Measles virus glycoprotein
complex assembly, receptor attachment, and cell entry. Curr. Topics
Microbiol. Immunol., 2009).

General Introduction
27
1.5. Measles virus pathogenesis
Measles is typically a childhood acute disease characterized by
fever, cough, conjunctivitis and a generalized maculopapular rash. Clinical
symptoms appear after a period of incubation of 10-14 days and this is
accompanied by a transient but profound immunosuppression. Recovery is
accompanied by lifelong protective immunity to re-infection (59).
1.5.1. Measles virus entry and spread
MV is transmitted by aerosol droplets and until recently initial
replication was believed to occur in the epithelia of the respiratory tract
followed by viremia mediated by infected monocytes or macrophages (47,
59). From the respiratory tract the virus spreads to the lymphatic tissues,
where viral amplification occurs and then to other organs like skin,
conjunctiva, kidney, lung, gastrointestinal tract, respiratory and genital
mucosa, and the liver (152).
A classical study of the infection of CDV, a related morbillivirus of
dogs showed that the virus was present in the bronchial LN and tonsils 1
day p.i. and it appeared in the lungs only 3 to 6 days later (4). Moreover, in
a ferret model of CDV pathogenesis (194, 195) primary replication occurred
in the lymphatic tissue of the oral cavity and of the respiratory tract. Viral
replication in epithelial cells, sustaining lung and bladder invasion, was
found only in later stages of infection (194). In addition, experimentally
infection of macaques with a recombinant wild type MV expressing GFP
resulted in predominant infection of SLAM positive lymphocytes and DCs

General Introduction
28
and an occasional infection of epithelial cells at later stages of infection
(34). Based on this, the initial target cells for MV infection are still not well
characterized. Fig. 4 illustrates the proposed model of MV infection and
transmission based on the recent findings.
1.5.2. Immune response to measles virus infection
MV infection induces an efficient immune response leading to viral
clearance and long life immunity against re-infections. It gives rise to a non-
specific activation of the immune system characterized by a spontaneous
proliferation of peripheral blood mononuclear cells (PBMC) and an
upregulation of activation associated cell surface markers. Along with this
immune activation, MV induces a transient but severe immunosuppression
which increases the susceptibility to opportunistic secondary bacterial and
viral infections, mainly in immunocompromised individuals.
Different components of the immune response are elicited by MV
infection. The first immune response to be activated by MV infection is the
innate immune response followed by the development of MV specific
adaptive immune responses.
1.5.2.1. Innate immunity
Innate immunity plays a major role in establishment of adaptive
immune responses. The innate immune system recognizes viral
components through pattern-recognition receptors (PRRs). This recognition
triggers a signal transduction cascade that activates transcriptional

General Introduction
29
responses that culminates in the production of proinflamatory cytokines,
interferons, and TNF-α. Two classes of PRRs have been described in innate
immune cells and involved in the recognition of MV, namely Toll-like
receptors (TLRs) and retinoic acid-inducible gene I (RIG-I)-like receptors
(RLRs). Activation of TLRs and RLRs is important for the production of type I
interferons and cytokines. Binding of type I IFNs to cognate receptors on
the cell surface triggers JAK/STAT signaling pathway leading to a formation
of IFN-stimulated gene factor 3 (ISGF3) complex comprised of STAT1,
STAT2 and IRF9. The ISGF3 complex translocates into the nucleus and
activates the IFN-stimulated response element (ISRE)-mediated gene
transcription gene resulting in synthesis of numerous proteins to establish
the antiviral state.
MV has developed ways to circumvent the host innate immune
response. MV was shown to inhibit signaling for both type I IFN induction
and JAK/STAT signaling pathway. MV is able to shut down IFN-α induction
in response to TLR7 and TLR9 ligands (160) and MV V protein can block
melanoma differentiation-associated gene 5 (MDA-5) mediated IFN
induction pathway (117). Several other reports show that the V, C, and P
proteins are able to block the JAK/STAT signaling pathway (19, 38, 49, 123,
128, 134, 149, 166, 183, 206).
1.5.2.2. Humoral immunity
MV specific neutralizing antibodies play a key role in preventing re-
infection after natural exposure to MV. However, the role of antibodies in

General Introduction
30
viral clearance remains unclear since individuals with deficient antibody
production are still able to resolve the infection, and depletion of B cells in
experimentally infected monkeys does not affect viral clearance (138).
Nevertheless, it was reported that failure to mount an adequate antibody
response to MV infection carries a poor prognosis in children (203).
Antibodies are initially detected at the onset of the rash. During
acute measles IgM are predominant and this is followed by a switch to IgG2
and IgG3, whereas during recovery IgG3 seems to decrease and IgG1 and
IgG4 increase (59). Antibodies are produced to all MV proteins, with
antibodies to the N protein being initially most abundant. Absence of this
antibody is an indicator of seronegativity. The majority of neutralizing
antibodies are specific for the H protein although antibodies to F
contribute to virus neutralization as well (145). MV specific neutralizing
antibodies binding to infected cells alters intracellular virus replication and
this way may contribute to control of infection (50, 54).
1.5.2.3. Cellular immunity
The role of cellular immunity in recovery from MV infection has
been appreciated from humans presenting immune abnormalities. Children
with impaired T cell immunity frequently die of progressive measles while
infection is cleared in individuals with agammaglobulinaemia (138, 140). In
addition, monkeys depleted of CD8+ T cells and challenged with wild type
MV had more severe rash, higher viral load in the blood and a prolonged
time to clear infection (139).

General Introduction
31
The cellular immune response is present at the onset of the rash.
Expansion of cytotoxic CD8 T cells and production of IFN-γ were observed
after MV infection and the establishment of CD8 T cell memory as well (81,
82, 118, 190). In addition, CD4 T responses are activated during MV
infection and the synthesis of cytokines is increased.
Acute measles generates a Th1 response profile, characterized by
IFN-γ and IL-2 production while the recovery phase is characterized by a
skew towards a Th2 response resulting in high levels of IL-4 and IL-10 and
low IFN-γ levels. The cytokine response after MV infection influences
disease outcome. In the acute phase of measles, IFN-γ, neopterin and IL-2
rise whereas at the time of the rash there is an increase in IL-2, in soluble
CD8 and CD4. In the recovery phase, when the rash starts to fade, there is
an elevation in the levels of IL-4, IL-10 and IL-13 persisting for several
weeks (61, 111).
1.5.3. Measles virus-induced immunosuppression
MV infection of immunocompetent host causes a virus-specific
immune response that clears infection and provides lifelong immunity.
However, concomitantly, MV is responsible for a generalized
immunosuppression that dampens immune responses to other pathogens.
Immunosuppression induced by MV is characterized by: abnormal cytokine
production, with an imbalance towards a Th2 response resulting in an
impaired cellular immune response to new antigens (60); suppression of

General Introduction
32
lymphoproliferative responses to mitogens and a marked lymphopenia
(156).
The mechanisms underlying immunosuppression induced by MV
are complex. Apoptosis (46), unidentified soluble mediators produced by
infected cells causing inhibition of proliferation of B and T cells (175, 199),
impaired dendritic cell function, lymphocyte depletion (129, 130) and
interleukin 12 downregulation (6, 87) may account for this immunological
abnormality.
1.6. Mouse models for the study of measles pathogenesis: SLAM
transgenic mice
Studies of MV pathogenesis have been hindered by the lack of
suitable animal models. Humans are the only natural host for MV. Non-
human primates have been successfully infected with MV and can
reproduce many aspects of the disease, however these are scarce and
costly and their use raises ethical questions. Since viral tropism is
determined by the pattern of expression of virus specific cellular receptors,
the discovery of the human MV receptors, CD46 and SLAM, has opened the
possibility of the development of transgenic mice susceptible to MV
infection. Several transgenic mice lines have been developed expressing
one or both receptors. The different outcome obtained with these models
reflects the use of different promoters, composition and integration site of
the transgenic construct.

General Introduction
33
Since the identification of human SLAM as the receptor for both
wild type and attenuated strains of MV several mice lines expressing SLAM
have been developed. Table 2 refers to the most relevant SLAM transgenic
lines and the outcome of infection.
Table 2. Comparison of SLAM transgenic mouse models of MV infection.
(adapted from Sellin C.I. and Horvat B., Current animal models: transgenic
animal models for the study of measles pathogenesis. Curr. Topics
Microbiol. Immunol., 2009).
Model Pattern of
expression
Immunological
status
Clinical signs and
pathology following
infection Lck-CD150
(Hahm et al. 2003)
T
lymphocytes
Immunocompetent Infection of thymocytes
of neonates infected IP
Ex vivo inhibition of T
cell proliferation
CD11c-CD150
(Hahm et al. 2004)
Splenic and
bone
marrow
derived
dendritic
cells
Immunocompetent Infection of 2 to 5%
dendritic cells after i.v.
inoculation
CD150Ge x STAT1-ko
(Welstead et al.
2005)
Human-like
expression
Immunodeficient IN and IP infection of
lymph nodes, spleen
and thymus
Knock-in CD150 x
IFNAR-ko
(Ohno et al. 2007)
Human-like
expression
Immunodeficient IN and IP infection of
spleen and lymph
nodes,
immunosuppression
CD46Ge x CD150Ge
x IFNAR-ko
(Shingai et al. 2005)
Human-like
expression
Immunodeficient Infection of dendritic
cells in lymph nodes

General Introduction
34
The Lck-CD150 transgenic mouse was the first CD150 transgenic
mouse to be generated. Human SLAM was expressed under the control of
Lck promoter and thus restricted to T lymphocytes from thymus, spleen
and blood. Spleen lymphocytes were susceptible to in vitro infection with
MV wild type and vaccine strains and infection correlated with the amount
of SLAM expressed in these cells. In addition, MV infection of T cells
expressing hSLAM inhibited their proliferation and rendered them
unresponsive to mitogen. Intraperitoneal infection of newborns resulted in
infection of SLAM expressing thymocytes (62).
To test the role of infection of DC on MV-induced
immunosuppression a transgenic mouse was generated where hSLAM is
expressed under a CD11c specific promoter; hSLAM expression was
restricted to splenic and bone marrow derived dendritic cells (63). These
mice were also used to study the effect of MV infection on the ability of
dendritic cells to induce IL-12 synthesis via toll-like receptor signaling.
Engagement of TLR-4 on MV infected DCs but not TLR-2, 3, 7 or 9 resulted
in defective IL-12 production. Furthermore, interaction of MV H with
hSLAM, but not the MV V and C proteins, influenced this inhibition. These
results suggested that MV, by altering DC function, renders them
unresponsive to secondary pathogens via TLR4 (64).
Another transgenic mouse was generated expressing the complete
human gene through the use of the human’s gene endogenous promoter
(202). Human SLAM expression profile was equivalent to expression in
humans; expression of SLAM was detected on activated B and T

General Introduction
35
lymphocytes from spleen and on activated bone marrow derived DC.
Intranasal infection of these mice with a wild type MV resulted in transient
infection of the nasal associated lymphoid tissue 6 days after inoculation.
To improve efficiency of MV infection these mice were then bred into a
STAT-1 deficient background. Four to six days upon IN or IP infection,
mRNA of MV was detected on thymus, spleen, and lymph nodes of these
mice. However, these mice did not show clinical signs of disease.
Abnormally number of neutrophils and natural killer cells were shown to
be responsible for the splenomegaly observed after IP infection. Crossing
this parental strain, SLAMGe mice (202), with Ifnarko
-CD46Ge mice (113)
resulted in the strain Ifnarko
-SLAMGe mice used in our studies, further
showed in Chapters 2, 3 and 4.
As the V domain of human SLAM is necessary and sufficient for MV
binding to CD150 receptor (133), Ohno et al (127) used a knock-in
approach to establish a mouse model where the V domain of mouse SLAM
was replaced by that of human SLAM by homologous recombination in
embryonic stem cells. In these mice SLAM had an expected human-like
tissue specificity of expression and retained normal function. Splenocytes
from these mice sustained productive infection when infected in vitro.
However, in vivo infection was limited. To increase efficiency of infection
these mice were crossed on an IFNARko
background. Following IN infection
virus spread to the draining lymph nodes of the respiratory tract and to
other lymphatic tissues throughout the body. In addition, after IP infection,
splenocytes failed to proliferate when stimulated with concanavalin A.

General Introduction
36
Thus, infection of these mice reproduces MV lymphotropism and
immunosuppression as in humans and might be relevant for the study of
MV immunopathology when production of type I interferon is not relevant.

Aims
37
Aims
The aim of this thesis is to study MV receptor-host interactions and
assess the consequences of these interactions for MV spread and
pathogenesis. Towards this we characterized MV infection in a transgenic
mouse expressing the cellular receptor for MV, SLAM (CD150). First, we
characterized the tissue specificity of SLAM expression in these mice and
assessed the spread of wild type MV in lymphoid and non-lymphoid organs
of mice that are expressing human SLAM (Chapter 2). Second, we aimed at
identifying the initial cellular target cells of MV infection after contagion
(Chapter 3) and third, to gain insight on the mechanisms of MV attenuation
we characterized the spread of an attenuated and a wild type strains of MV
in the lymphatic system in the transgenic mice expressing human SLAM
(Chapter 4).


Results Chapter 2
39
CHAPTER 2
Measles virus infection of transgenic mice expressing human
signaling lymphocytic activation molecule (SLAM, CD150)


Results Chapter 2
41
Abstract
We bred a transgenic mouse expressing SLAM with human like
tissue specificity in an interferon type I deficient background (Ifnarko
) to
allow more extensive measles virus (MV) replication. We obtained B cells
from the spleen and T cells from the thymus of these Ifnarko
-SLAMGe mice
and documented an increase in human SLAM (hSLAM) expression after
activation of these cells. Macrophages from the bone marrow of these
mice expressed hSLAM twenty four hours after lipopolysaccharide (LPS)
activation. Furthermore we showed that ex vivo inoculation of
macrophages with a MV expressing green fluorescent protein, wtMVgreen,
resulted in infection in a receptor-dependent manner. To assess the spread
of MV in vivo, we infected these mice intranasally or intraperitoneally with
a virus expressing the reporter protein chloramphenicol acetyltransferase
(CAT), from an additional transcription unit downstream the hemagglutinin
gene, here named wtMVCAT. Three and six days after inoculation, the lungs,
thymus and spleen were collected and the CAT activity was assessed in
homogenates of these tissues. Intranasal (IN) inoculation resulted in viral
replication in the airways. Later the virus spread to the lymphatic system
with involvement of the spleen. MV replication was more efficient in the
spleen and less efficient in the lungs after intraperitoneal (IP) inoculation.
Thus, Ifnarko
-SLAMGe mice seem particularly useful in the study of the
early phases of MV infection and dissemination.


Results Chapter 2
43
Introduction
Despite eradication efforts and the availability of an inexpensive
and safe vaccine, measles is still included in the group of five conditions
that make up to 70% of deaths in children younger than 5 years (41).
As humans are the only natural host for MV, a complete
understanding of many aspects of MV infection and pathogenesis is
hampered by the lack of a suitable animal model. MV is pathogenic only for
non human primates but these are scarce and costly and ethical questions
can be raised. Mice, for their availability and low cost, constitute an
alternative model to study MV pathogenesis. However, MV replication in
mice is inefficient and occurs only after intracranial inoculation of brain-
adapted strains. The lack of human receptors for MV on mice presents a
major obstacle to their use and has motivated the development of
transgenic mice expressing MV receptors with human like tissue specificity
as recently reviewed (164).
Several transgenic mice have been generated in the last decade to
study MV infection and pathogenesis. Initial studies of MV infections were
conducted in transgenic mice expressing human CD46 (MCP, human
membrane cofactor protein). CD46 is the cellular receptor for MV
attenuated strains (39, 119). These studies reported inefficient and short-
lived dissemination of an attenuated strain of MV. To overcome these
limitations CD46 transgenic mice were bred into a type I interferon
receptor knock out (IFNARko
) background (112, 113).

Results Chapter 2
44
CD46 transgenic mice present a major limitation as they only
support infection by the vaccine and laboratory adapted strains of MV; the
identification of SLAM (CD150, signaling lymphocyte activation molecule)
as a receptor for both wild type and laboratory MV strains (75, 186)
opened new perspectives in the generation of transgenic mice to study the
pathogenesis of clinical isolates.
Unlike CD46, which is ubiquitously expressed, SLAM expression is
restricted to immune cells. This protein was originally identified on
activated B and T lymphocytes (29, 170) but is expressed constitutively on
immature thymocytes, memory T cells and certain B cells. Monocytes and
dendritic cells express SLAM only following activation. Importantly, besides
MV, other morbillivirus like canine distemper virus and rinderpest virus use
SLAM (canine and bovine respectively) as their cellular receptor (187).
Although murine immune cells express SLAM, which has 60% sequence
identity to human SLAM and a similar function, it cannot act as MV
receptor (126).
In the next chapter we describe how we generated a mouse line
expressing human SLAM with human-like tissue specificity and defective in
the interferon response. In this chapter we present a detailed
characterization of the expression profile of hSLAM in hSLAMGe transgenic
mice and investigate the susceptibility of these mice to MV infection. We
analyzed hSLAM expression in activated B and T cells, and in bone marrow
derived macrophages. Human SLAM expression increases after activation

Results Chapter 2
45
of these cell populations. Next we characterized the tissues supporting MV
infection upon IN or IP inoculation.
Materials and Methods
Mouse strains. The establishment of a transgenic mouse line
expressing hSLAM is described in Chapter 3.
Mouse infections. All animal experimental procedures were
performed according to protocols previously approved by the Mayo Clinic
Institutional Animal Care and Use Committee. Five to 8-week-old animals
were anesthetized with isoflurane (Novation, IL, USA) and infected IN with
4x105 tissue culture infectious dose 50% (TCID50) MV. Five to 8-week-old
mice were infected IP with 8x106 TCID50 for histological analysis or with
1.5x106 TCID50 for assessment of CAT activity in infected tissues.
Generation of a wild type MV expressing the reporter gene
chloramphenicol acetyltransferase. MVwtIC323 (here abbreviated wtMV)
was obtained from an infectious cDNA derived from the Ichinoise B (IC-B)
wild type strain and cloned in the plasmid p(+)MV323 (provided by Dr. K.
Takeuchi, University of Tsukuba, Tsukuba, Japan) (182). wtMVCAT is a
derivative of MVwtIC323 to which a transcription unit expressing the gene
CAT was added downstream of the hemaglutinin (H) gene. Briefly, the CAT
gene was obtained from the plasmid pcDNA3.1CAT. The restriction sites
MluI and AatII were added to the CAT using the forward primer F

Results Chapter 2
46
5’CGACGCGTATGGAGAAAAAAATCACTGG 3’ and reverse 5’
CGGACGTCTTTACGCCCCGCCCTG 3’ and amplified by a PCR reaction The
CAT was then amplified using the pCR2.1 TOPO, and cloned using the
restriction sites MluI and AatII in the expression plasmid pCG-vac-H. The
segment H-CAT was then obtained from the resulting plasmid, pCG-vac2-H-
CAT, with the restriction sites PacI/SpeI and cloned in the plasmid p(+)
MVvac (named MVvacCAT). The CAT was then obtained with the restriction
sites PvuII/SpeI and cloned in the plasmid pCG-IC323-H. IC323-H-CAT was
then cloned with PacI/SpeI in the full length p(+) MV323 coding for the wt
genome.
Rescue of viruses and preparation of virus stocks. Viruses were
rescued as described by Radecke et al. (147). Briefly, the helper cell line
293-3-46 stably expressing MV-N, MV-P and T7 polymerase was
transfected by calcium phosphate precipitation using ProFection Kit
(Promega, Madison, WI) with two plasmids, one containing the relevant
MV genome and the other coding for the MV polymerase (pEMCLa). Three
days after transfection, the helper cells were overlaid on Vero/hSLAM cells
and the appearance of infectious centers was monitored. Single syncytia
were picked and propagated on Vero/hSLAM cells. To prepare virus stocks,
Vero/hSLAM cells were infected at a multiplicity of infection (M.O.I.) of
0.03 and incubated at 37ºC. Cells were scraped in Opti-MEM
(Gibco/Invitrogen Corp., Grand Island, NY) and viral particles were released
by two freeze-thaw cycles. Titers were determined by TCID50 titration on

Results Chapter 2
47
Vero/hSLAM cells according to the Spearman-Kärber method (86). A UV-
inactivated virus was prepared by incubating a vial containing 50 µl of
wtMVCAT on a UVP CL-1000 UV crosslinker (UVP, Upland, CA) on ice. The
virus was exposed to a UV light of 254 nm at 3 cm from a UV source of 1.85
x105 μJ/cm
2 for 30 minutes.
Isolation and activation of B and T lymphocytes. Spleen and
thymus were collected aseptically and transferred into a 15 ml tube
containing RPMI 1640 (Mediatech Inc., Herndon, VA) with 5% fetal bovine
serum (FBS) (Gibco/Invitrogen Corp.) and 1% Penicillin/Streptomycin
(Mediatech Inc.). To obtain single cell suspensions, spleen and thymus
were transferred to a 70 µm cell strainer (BD Biosciences, Bedford, MA),
smashed with the plunger of a syringe, washed with RPMI 1640 medium
and resuspended in this medium.
T and B cells isolated from the thymus and spleen respectively,
were negatively separated by magnetically labeling cells with MACS
microbeads (MiltenyiBiotec, Bergisch Gladbach, Germany) according with
manufacturer’s recommendations. To activate the T lymphocytes, 96 well
plates were coated over night at 4ºC with CD3 antibody (5 µg/ml) (BD
Pharmingen). T lymphocytes were incubated for 24 hours at 37ºC in anti-
CD3 coated plates and interleukin-2 (IL-2) (100 U/ml). B cells were
activated by incubating B lymphocytes with LPS (Sigma) (20 µg/ml) for 24
hours at 37ºC.

Results Chapter 2
48
Isolation and activation of macrophages. Femurs of 5-8 weeks
Ifnarko
-SLAMGe and C57BL/6 mice were collected and cleaned with the
help of a blade to remove adherent muscle tissue. Intact bones were then
left in 70% ethanol for 5 minutes for disinfection and washed with
phosphate buffered saline (PBS). Both ends were cut with scissors and the
marrow flushed with PBS using a syringe with 0.45 mm diameter needle.
Clusters within the marrow suspension were disintegrated by vigorous
vortexing. After wash with PBS, about 1-1.5x107 leukocytes were obtained
per femur.
At day 0 bone marrow (BM) leukocytes were seeded at 2x106 per
100 mm bacteriological petri dish in 10 ml complete medium (RPMI 1640
(Mediatech Inc.) with 10% heat inactivated and filtered (0.22 µm,
Millipore) FBS (Gibco/Invitrogen Corp.) and 1% Penicillin/Streptomycin
(Mediatech Inc.). Cells were allowed to attach for 2 hours, after which the
unattached were washed off with PBS. The attached cells were cultured
with 10 ml of complete medium containing 200 U/ml of granulocyte-
macrophage colony-stimulating factor (GM-CSF) and allowed to
differentiate to macrophages for 3 to 5 days. Every third day medium was
replaced with fresh medium containing GM-CSF. One day before
harvesting, 500 U/ml of LPS (Sigma) was added to the culture medium. At
day 5, medium was replaced by PBS/0.005% trypsin and the cells were
gentle scraped and collected. Macrophages were analyzed for CD11b (also
named Mac-1), F4/80, and hSLAM expression. C57Bl6 mice served as
negative control.

Results Chapter 2
49
Flow cytometry. Different cell populations were identified with the
following markers: for the identification of CD4+ T cells, anti-CD4-
fluorescein isothiocyanate (FITC) (GK1.5, BD Pharmingen) was used. For the
identification of CD8+ T cells, cells were stained with anti-CD8-FITC
(Lou/Ws1/M, BD Pharmingen). B lymphocytes were identified with anti-
CD45R (B220)-FITC (RA3-6B2, BD Pharmingen). Bone marrow derived
macrophages were identified with the following stainings: anti-Mac-1-
PerCP (Peridinin chlorophyll protein) (M1/70, eBioscience) and anti-F4/80-
APC (BM8, eBioscience).
SLAM expression was detected by incubating cells with PE anti-
human CD150 (A12, BD Pharmingen). Corresponding isotype antibodies
were used as controls for antibody specificity. Acquisition of samples was
performed on a cytometer (FacsCanto) and analyzed using FlowJo software
(Tree Star, Inc., Asland, OR).
Immunohistochemistry. Tissues were collected and prepared as
described in Chapter 3. Briefly, frozen tissue sections 4-6 µm in thickness
were incubated with anti-MV N polyclonal antibody (189), washed,
counterstained and mounted.
CAT assay. Animals were sacrificed and tissues removed and snap
frozen in liquid nitrogen. Frozen tissues were grinded on ice in 500 µl of
Tris-buffered saline (TBS), 1mM EDTA, and antiproteases (Calbiochem).
After grinding, 500 µl of TBS, 1 mM EDTA and 1% Triton x100 were added

Results Chapter 2
50
and the samples were subjected to vortex for 30 seconds and left on ice for
30 minutes. The homogenates were then subjected to two freeze-thaw
cycles and centrifuged at 12,000 rpm at 4ºC for 5 minutes. The supernatant
was recovered and the protein concentration was measured by the
Bradford method. For each tissue, 800 µg of protein in 500 µl volume was
analyzed for CAT activity according to the supplier’s protocol (Molecular
Probes). Briefly, 10 µl of FAST CAT (provided) substrate solution was added
to the tissue extract and incubated at 37ºC for 5 minutes. The mixture was
then incubated at 37ºC for 5 hours with 10 µl of acetyl coA (Sigma). The
reaction was stopped by adding 1 ml of ice-cold ethyl acetate (Sigma) and
the samples were vortexed for about 20 seconds. Samples were
centrifuged at top speed for 3 minutes to separate the liquid phase. The
top 900 µl of ethyl acetate were recovered and transferred to a clean
Eppendorf tube and the solvent allowed evaporating. The residue was
dissolved in 25 µl of ethyl acetate and stored in the dark at – 20ºC until
further analysis. Samples were applied in a thin layer chromatography silica
gel plate. The solvent was allowed to ascend the plate. Samples were air
dried and the results visualized under UV-light.

Results Chapter 2
51
Results
Human SLAM expression in lymphocytes of hSLAM transgenic
mice. To characterize the protein expression profile of hSLAM in the
hSLAM transgenic mice, we purified B and T cells from the spleen and
thymus, respectively, and analyzed hSLAM expression by flow cytometry.
As SLAM expression in resting lymphocytes is expected to be very low (29),
B cells were purified from the spleen and activated for 24 hours with LPS,
stained with a B cell marker and analyzed for hSLAM expression. T cells
were purified from the thymus and activated for 24 hours with CD3
antibody and IL-2.
Figure 1 documents hSLAM expression in one representative
animal. The panels in Figures 1A, B and C document hSLAM expression
levels in activated cells (red line) as compared to non activated cells (blue
line) from Ifnarko
-SLAMGe mice and to non- activated cells from C57BL/6
mice (green line). In non-activated B and CD4 and CD8 T cells hSLAM
expression was very low whereas the expression levels increased after
activation of B cells with LPS (Fig. 1A, red line) and CD4 and CD8T cells with
anti-CD3 and IL-2 (Fig. 1B and C, red line). Thus, resting T and B cells from
Ifnarko
-SLAMGe mice express human SLAM at low levels whereas
expression increases following activation as previously described (29, 153,
170).

Results Chapter 2
52
Figure 1. Analysis of hSLAM expression in activated B and T lymphocytes
from hSLAM transgenic mice. Activated (red lines) and non-activated (blue
lines) B cells were stained with antibodies specific for B cells (B220) (A) and
T cells (CD4) and (CD8) (B and C respectively) and SLAM. Activated cells of
C57BL/6 mice served as negative controls (green lines). Histogram panels, Y
axis: % of Max is a measure of the number of cells normalized to the same
total number in each sample.
Human SLAM expression in macrophages of hSLAM transgenic
mice. Since MV may infect initially macrophages or dendritic cells in the
respiratory tract rather than epithelial cells (34) we characterized hSLAM
expression in BM derived macrophages of Ifnarko
-SLAMGe mice.
To generate macrophages, cells were harvested from the femurs of
transgenic mice. Cells were allowed to attach and cultured with GM-CSF for
3 to 5 days. Half of the cells were further cultured with LPS for activation.
Bone marrow derived macrophages were analyzed for Mac-1, F4/80, and
hSLAM expression.

Results Chapter 2
53
Figures 2 documents hSLAM expression in activated cells (red line)
as compared to non-activated cells (blue line) from Ifnarko
-SLAMGe mice
and non-activated cells from C57BL/6 mice (green line).
A small fraction of non-activated macrophages from Ifnarko
-SLAMGe
mice expresses hSLAM compared with macrophages obtained from
C57BL/6 mice (compare blue line with green line of C57BL/6 negative
control), but a substantial increase in the percentage of cells expressing
SLAM was documented following activation with LPS.
Figure 2. Analysis of hSLAM expression
in activated macrophages cultured
from bone marrow of Ifnarko
-SLAMGe
mice (red lines). Blue lines represent
hSLAM expression in non activated
macrophages. C57BL/6 mice were used
as negative controls (green lines).
To assess whether macrophages from Ifnarko
-SLAMGe mice were
susceptible to MV infection, cells obtained from BM of Ifnarko
-SLAMGe
mice were activated and infected with wtMVgreen at an M.O.I. of 5 and
analyzed by flow cytometry at 48 hours p.i..
As shown in Figure 3, inoculation of activated macrophages from
Ifnarko
-SLAMGe mice resulted in receptor-dependent infection as

Results Chapter 2
54
determined by GFP expression. Compare GFP expression in activated
macrophages from Ifnarko
-SLAMGe mice (red line) with GFP expression in
non-activated macrophages from Ifnarko
-SLAMGe or C57BL/6 mice (blue
and green line respectively).
Figure 3. Analysis of GFP expression in
activated (red line) and in non activated
(blue line) macrophages from Ifnarko
-
SLAMGe mice and C57BL/6 mice (green
line). Cells were inoculated with
wtMVgreen at an M.O.I. of 5 and analyzed
by flow cytometry 48 hours later.
Ifnar deficiency increases susceptibility to wild type MV
replication in hSLAM transgenic mice. Previous studies with mice
expressing hCD46 infected with the vaccine strain of MV, showed that Ifnar
deficiency render mice more susceptible to MV infection (113). This effect
was documented already 2-3 days post-infection. Therefore we bred SLAM
transgenic mice into an Ifnar deficient background. As we plan to use our
mice to characterize even earlier steps of MV infection we sought to
determine whether interferon type I exerts its antiviral effect already one
day after inoculation. Therefore, we inoculated Ifnarko
-SLAMGe and
SLAMGe mice intraperitoneally with wtMV and analyzed MV infection. At

Results Chapter 2
55
one day after inoculation, lymphatic organs (mediastinal LN and spleen)
were harvested and analyzed by immunohistochemistry.
Figure 4A and B shows a representative immunohistochemistry
analysis of mediastinal LN one day after inoculation of SLAMGe (panel A)
and Ifnarko
-SLAMGe mice (panel B). MV N protein was detected in this LN
of Ifnarko
-SLAMGe mice, whereas no virus was detected in the
corresponding LN of SLAMGe mice. In addition, Figure 4B shows that the
virus is localized mainly in the subcapsular sinus of this LN and the
presence of syncytial cells is documented (box in higher magnification on
the low right corner of panel B).
Figure 4C and D shows that MV replicated in the spleen of Ifnarko
-
SLAMGe mice (panel D, arrows), but not in the spleen of SLAMGe mice
(panel C) one day p.i..

Results Chapter 2
56
Figure 4. Identification of MV infection in the mediastinal LN and spleen
of SLAMGe or Ifnarko
-SLAMGe mice one day after IP inoculation. Frozen
sections of mouse mediastinal LN and spleen were stained with an anti-N
antibody and counterstained with hematoxylin. N protein was visualized
with red chromogen.

Results Chapter 2
57
This suggests that interferon type I acts as an important restriction
factor for MV replication in this animal model as early as 24 hours after
infection.
Spread of MV in Ifnarko
-SLAMGe mice. To be in the position of
easily measure the levels of MV-based gene expression in different tissues,
we generated a virus expressing the reporter gene CAT (Materials and
Methods). A map of the plasmid constructed to generate the virus is shown
in Figure 5.
Figure 5. Map of the p(+) MV323 plasmid coding for the wt genome and
location of the inserted CAT gene. Six coding regions of MV genes are
represented by colored boxes. The T7 promoter, hepatitis delta virus
ribozyme (HδR) and selected unique restriction sites are indicated. The CAT
transcription unit was inserted between the PvuII and SpeI restriction sites
downstream the H gene.
Since the respiratory tract is the natural route of MV infection in
humans, mice were inoculated intranasally with 4x105 TCID50 of wtMVCAT.
Lungs, thymus and spleen were harvested and CAT activity was assessed in
the homogenates of these tissues (Fig. 6A). Inoculation with UV-inactivated
virus served as negative control. No virus was detected in the homogenates

Results Chapter 2
58
of tissues of mice infected with UV-inactivated virus (not shown). In
contrast, viral replication was detected three days p.i. in the lungs of all
animals inoculated with live virus and in the thymus of one of six animals.
At day 6 virus was detected in spleen of one of six mice (Fig. 6A).
We also tested an alternative route of inoculation and assessed
viral spread after IP infection. Animals were inoculated with 1.5x106 TCID50
of wtMVCAT and viral replication was documented by the presence of CAT
activity in the homogenates of lungs, thymus and spleen.
As shown in Figure 6B, after IP infection MV replication was
strongest in the spleen, it was detectable in the thymus of all mice at day 6
but not at day 3, and it was detected also in the lungs at both 3 and 6 days
p.i.. This pilot experiment suggests that these mice can serve as a model to
study the initial steps of MV infection and pathogenesis.

Results Chapter 2
59
Figure 6. MV spread in different organs of Ifnarko
-SLAMGe mice. Mice
inoculated intranasally (A) or intraperitoneally (B) with wtMVCAT were
sacrificed at days 3 or 6 p.i., tissues collected and homogenized and tested
for CAT activity. As a positive control Vero/hSLAM cells infected with
wtMVCAT were used (+CAT) and homogenates from organs of a non
infected mice were used as a negative control (-CAT).

Results Chapter 2
60
Discussion
Since the identification of SLAM as the cellular receptor for wild
type MV strains (44, 75, 186) several types of transgenic mice have been
generated with the goal of serving as animals models for MV infection.
These models differ in what concerns to the profile of hSLAM expression.
Some of these animals cannot be used as a model for MV infection because
SLAM expression is restricted to certain cell types (62, 63). In our
transgenic mouse the use of a large fragment of the human genome
including the locus control region resulted in a human-like expression
profile. We documented hSLAM expression in activated B and T
lymphocytes, and in macrophages following their activation. B and T cells
were previously shown to be important targets for MV infection of SLAM
‘’knock-in’’ mice: three days after IP infection up to 1% of B and T cells
were infected by wild type MV (127).
We also generated macrophages from Ifnarko
-SLAMGe transgenic
mice and documented hSLAM expression following activation with LPS.
SLAM expression was reported to be absent in resting monocytes (144,
170). Nevertheless, it could be induced by mitogens and cytokines as well
as with MV infection (109). Another finding was that macrophages
expressing hSLAM were susceptible to MV infection in a receptor-
dependent manner. Consistent with the relevance of these cells as specific
hosts for MV infection and dissemination is the fact that these cells are the
major targets for a vaccine strain of MV in Ifnarko
-CD46Ge mice

Results Chapter 2
61
independent of the route of infection (154). Thus, macrophages may play
an important role in the initial phases of MV infection and dissemination.
We also show that upon IN or IP infection MV replicates in the
airways and in lymphatic tissues of Ifnarko
-SLAMGe mice. One interesting
point is that two of six mice inoculated IP show some infection of the lungs
three days post-inoculation. We do not know whether infection is due to
infiltration of infected immune cells or direct infection of the respiratory
epithelia.
Based on these results we hypothesize that SLAM expressing
macrophages or lymphocytes might constitute the major targets for MV
infection upon respiratory inoculation and may be responsible for
transmission of the virus to the lymphatic tissues. Following amplification
of the virus in the lymphatic tissues infected immune cells may cross the
airway epithelium and promote transmission to a new host. Thus, infection
of these mice provide the ideal opportunity to characterize the early
phases of MV infection and gain insights in the cell types supporting initial
MV infection and dissemination.


Results Chapter 3
63
CHAPTER 3
Measles virus infection of alveolar macrophages and dendritic
cells precedes spread to lymphatic organs in transgenic mice
expressing human signaling lymphocytic activation molecule
(SLAM, CD150)
Claudia S. Antunes Ferreira1, *
, Marie Frenzke1, Vincent H.J. Leonard
1,
G. Grant Welstead2, Christopher D. Richardson
2,3, Roberto Cattaneo
1
1Department of Molecular Medicine, and Virology and Gene Therapy Track,
Mayo Clinic College of Medicine, Rochester, Minnesota 55905, 2Department of Medical Biophysics, University of Toronto, and Ontario
Cancer Institute, Toronto, Ontario, Canada M5G 2M9, 3Department of
Microbiology & Immunology/Pediatrics, Dalhousie University Halifax, Nova
Scotia, Canada B3H 1X5
* Current address: Universidade Lusofona de Humanidades e Tecnologias,
Avenida do Campo Grande, 376, 1749 – 024 Lisboa Portugal
J. Virol. 2010; 83:3033-3042


Results Chapter 3
65
Abstract
Recent studies in primate models suggest that wild type measles
virus (MV) infects immune cells located in the airways before spreading
systemically, but the identity of these cells is unknown. To identify cells
supporting primary MV infection we took advantage of mice expressing the
MV receptor human signaling lymphocyte activation molecule (SLAM,
CD150) with human-like tissue specificity. We infected these mice
intranasally with a wild type MV expressing green fluorescent protein. One,
two or three days after inoculation, nasal-associated lymphoid tissue, the
lungs, several lymph nodes, spleen and thymus were collected and
analyzed by microscopy and flow cytometry, and virus isolation was
attempted. One day after inoculation MV replication was documented only
in the airways, in about 2.5% of alveolar macrophages and 0.5% of
dendritic cells. These cells expressed human SLAM, and it was observed
that MV infection temporarily enhanced SLAM expression. Later, MV
infected other immune cell types including B and T lymphocytes. Virus was
isolated from lymphatic tissue as early as two days post IN inoculation; the
mediastinal lymph node (LN) was an early site of replication, and supported
high levels of infection. Three days after intraperitoneal inoculation 1-8% of
the mediastinal LN cells were infected. Thus, MV infection of alveolar
macrophages and sub-epithelial dendritic cells in the airways precedes

Results Chapter 3
66
infection of lymphocytes in lymphatic organs of mice expressing human
SLAM with human-like tissue specificity.

Results Chapter 3
67
Introduction
Measles virus, a member of the Morbillivirus genus of the
Paramyxoviridae family, causes measles, a highly contagious disease
transmitted by respiratory aerosols that induces a transient but severe
immunosuppression (59, 161). In spite of eradication efforts, MV still
accounts for about 4% of deaths in children aged under five years
worldwide (16, 110), mainly due to opportunistic secondary infections
facilitated by MV-induced immune suppression (59). Experimental analyses
of the mechanisms of pathogenesis, including the characterization of cells
and tissues supporting primary MV infection, is limited by host species
specificity: old world monkeys and humans are the only natural MV hosts.
MV replication has been characterized mainly around the time of
rash appearance, 10-14 days after experimental infection of monkeys (33,
34, 108, 185). Viremia in blood cells peaks at or slightly before rash:
infected B and T lymphocytes, monocytes and dendritic cells (DC) are
detected, while little if any cell-free virus is produced. Infected cells express
the signaling lymphocytic activation molecule (SLAM, CD150), the
lymphatic cell MV receptor (44, 75, 186). More information about the
cellular targets of wild type MV infection in the airways immediately after
contagion is important: recent studies in monkeys have suggested that MV
may replicate initially in immune cells in the airways (34, 99) rather than in
lung epithelial cells as previously postulated (31, 157).

Results Chapter 3
68
Limited availability and high costs of primate experimentation
motivated the development of transgenic rodent models of MV infection.
Studies in the ‘90s were based on mice expressing human membrane
cofactor protein (MCP, CD46), the receptor used only by the attenuated
MV strain (39, 119, 204). These studies indicated that airway macrophages
are infected by the MV vaccine strain in the first days after intranasal (IN)
inoculation, and that blood monocytes and tissue macrophages
disseminate the infection (113, 154). To increase susceptibility to MV
infection, CD46-expressing mice were crossed into an interferon receptor
knock-out (Ifnarko
) background; this did not appear to change the cell type
specificity of viral replication (154).
After human SLAM (hSLAM) was characterized as the immune cell
receptor for wild type and vaccine MV, several mouse strains expressing
this protein were generated, as recently reviewed (164). SLAM is a 70-kDa,
type I transmembrane glycoprotein expressed on immune cells such as
activated T cells, B cells, monocytes/macrophages, and DC (29). It belongs
to the immunoglobulin protein superfamily and has two extracellular
domains named V and C2; V interacts with the MV attachment protein
hemagglutinin (133). SLAM determines Th2 cytokine production such as IL-
4 and it may be involved in the production of IL-12, TNF-α, and nitric oxide
by macrophages (171, 192, 200). In addition, SLAM may induce B cell
proliferation and immunoglobulin synthesis. Importantly, hSLAM-
expressing mice but not CD46-expressing mice can be infected by wild type
MV strains that use SLAM but not CD46 as receptor (120).

Results Chapter 3
69
Initially a model expressing hSLAM under control of the T-cell
specific lck promoter was reported (62). In this model hSLAM expression
was restricted to immature and mature lymphocytes in the spleen, thymus,
and blood; lymphocyte proliferation was observed, but there were no
clinical signs of disease. The second model was a mouse in which the
hSLAM coding sequence was expressed under control of the promoter of
the ubiquitously expressed HMGCoA protein (163). In suckling mice
generalized but not necessarily relevant infections of most of the major
organs were documented. Adult mice were less susceptible to MV
infection: only intracerebral inoculation was productive, and yielded viral
proteins but no infectious virus. The third model consisted of a transgenic
mouse expressing hSLAM in DC from a cDNA under control of the CD11c
promoter (63). MV infections are limited to DC in this model and disrupt
the function of these cells in stimulating adaptive immunity.
An alternative approach to transgenesis seeks human-like tissue
specificity of expression. Towards this, Shingai et al. (168) added a full-
length hSLAM gene to the mouse genome, showed that indeed these mice
express hSLAM with human-like tissue specificity, and crossed them in an
Ifnarko
and human CD46-positive background. In this model CD11c-positive
DC are instrumental to establish MV infections. The fifth mouse strain was
generated by exchanging only the MV binding site on mouse SLAM with
that in the V domain of hSLAM (127). Since human and mice have similar
tissue specificity of SLAM expression, this led to human like expression.
When these mice were crossed in an Ifnarko
background, efficient MV

Results Chapter 3
70
replication suppressing proliferative responses to concanavalin A was
documented.
We previously generated a mouse strain expressing hSLAM with
human-like tissue specificity in a STAT1-deficient background (202) We
showed that hSLAM expression was restricted to B and T lymphocytes and
some monocytes/macrophages, and that it was inducible by LPS, lectins or
anti-CD3 antibodies in immune cells, as in primates. Since the Ifnarko
background allows more efficient MV spread (113, 154) without apparent
effects on the cell type specificity of MV infection in mice (127, 168), we
crossed here these mice in the Ifnarko
background. We then used the
Ifnarko
-SLAMGe mice to identify the cells infected by wild type MV
immediately after IN inoculation. We document efficient early infection of
alveolar macrophages (AM) and DC. We also observed subsequent
infection of all lymphatic organs, and in particular of the mediastinal LN,
upon both IN and intraperitoneal (IP) inoculation.
Materials and Methods
Mouse strains. To establish a transgenic mouse line expressing the
hSLAM gene and deficient in the alpha/beta interferon response, Ifnarko
-
CD46Ge mice (113) were crossed with SLAMGe mice (202). The hSLAM
gene was added to the genome of congenic C57Bl/6 mice (202), but the
Ifnarko
-CD46Ge mice is on a mixed C57Bl/6 and C3H background (113), and
the crossing yields a mixed background. To obtain homozygous Ifnarko
-
SLAMGe mice, the F2 and F3 progeny was screened for inactivation of both

Results Chapter 3
71
copies of the interferon alpha receptor gene (114) for availability of two
copies of the hSLAM gene, and for absence of the CD46 gene.
Since no simple PCR test for the hSLAM genotype was available, the
site of insertion of the hSLAM gene in the mouse genome was
characterized. To accomplish this, primers specific for terminal sequences
of the human genome inserted in the bacterial artificial chromosome used
for transgenesis (202) were synthesized. These primers were used in
combination with primers provided in the GenomeWalker Universal Kit (BD
Pharmingen, San Diego, CA) to identify the insertion site of hSLAM in the
mouse genome, after nucleotide 47,072,810 of chromosome 12, in band
B3 (mouse genome sequence available through www.ensembl.org). The
hSLAM specific analysis was then based on two combined PCR reactions.
SLAM PCR 1 used the primers pairs: 5’–
GTGTCACCTAAATAGCTTGGCGTAATCATG and 5’–
GTTAATATAGACAATGCCCATCTCCAGCAG, amplifying a 1030 base pair band
including part of the hSLAM gene and part of the mouse chromosome 12.
SLAM PCR 2 used the primer pairs 5’-
AGCTTTCTGAATAGGGGTGTTACTTAATGC and 5’-
GTTAATATAGACAATGCCCATCTCCAGCAG, amplifying a 1339 base pair band
of mouse chromosome 12. Mice homozygous for the hSLAM gene had a
positive signal from PCR 1 and no signal from PCR 2.
Mouse infections. All animal experimental procedures were
performed according to protocols previously approved by the Mayo Clinic

Results Chapter 3
72
Institutional Animal Care and Use Committee. Five to 8-week-old animals
were anesthetized with isoflurane (Novation, IL, USA) and infected IN with
8x105 tissue culture infectious dose 50% (TCID50) MV in 100 μl divided in
two doses of 50 μl (25μl in each nare) in Opti-MEM. Ten minutes elapsed
between both inoculations. Five to 8-week-old mice were infected IP with
1.5x106 TCID50 in 1 ml Opti-MEM to measure viral load in lymphatic tissues;
for the experiments of Table 3 the TCID50 was 8x106. IN and IP infections
with ten-fold smaller inocula were productive, but the results were difficult
to quantify.
Viruses and virus stock preparation. MVwtIC323 (here abbreviated
wtMV) was obtained from an infectious cDNA derived from the Ichinoise B
(IC-B) wild type strain and cloned in the plasmid p(+)MV323 (provided by
Dr. K. Takeuchi, University of Tsukuba, Tsukuba, Japan) (182). wtMVgreen is a
derivative of MVwtIC323 to which a transcription unit containing the open
reading frame of enhanced green fluorescence protein (GFP) (40) was
added upstream of the nucleocapsid gene (99). Viruses were amplified on
Vero/hSLAM cells as described by Radecke et al. (147). Cells were scraped
in Opti-MEM (Gibco/Invitrogen Corp., Grand Island, NY) and viral particles
were released by two freeze-thaw cycles. Titers were determined by TCID50
titration on Vero/hSLAM cells according to the Spearman-Kärber method
(86).

Results Chapter 3
73
Blood and organ collection, and preparation of single cell
suspensions. Blood samples obtained by cardiac puncture at terminal
bleeding were collected in a microtainer tube with lithium heparin (Becton
Dickinson, Franklin Lakes, NJ). Peripheral blood mononuclear cells (PBMCs)
were isolated by density gradient centrifugation with Ficoll-Paque Plus (GE
Healthcare, Sweden).
Nasal associated lymphoid tissue (NALT), LN, spleen and lungs were
collected aseptically and transferred into a 15 ml tube containing RPMI
1640 (Mediatech Inc., Herndon, VA) with 5% fetal bovine serum (FBS)
(Gibco/Invitrogen Corp.) and 1% Penicillin/Streptomycin (Mediatech Inc.).
NALT was collected as follows (5): the facial skin was stripped from the
head and the nose tip was cut off including the front teeth. The cheek
muscles and cheek bones were taken off. The soft and the hard palate
were removed and the NALT was excised distal to the emergence of the
palatine nerve. To obtain single cell suspensions, NALT, LN and spleen were
transferred to a 70 µm cell strainer (BD Biosciences, Bedford, MA), mashed
with the plunger of a syringe, washed with RPMI 1640 medium and
resuspended in this medium.
Single cell suspensions from lungs were generated as described in
(158) with some modifications. Briefly, the lungs were aseptically collected
in 10 ml of RPMI 1640 with 5% FBS and 1% Penicillin/Streptomycin,
reduced to small pieces (approximately 1-2 mm2) and digested at 37 ºC for
1 hour in 10 ml phosphate buffered saline (PBS) (Mediatech) containing
300 U/ml collagenase type II (Worthington, Lakewood, NJ) and 0.15 mg/ml

Results Chapter 3
74
DNase (Roche Diagnostics, Germany). After digestion, lung pieces were
minced with the plunger of a syringe and washed in a 70 µm pore size cell
strainer with RPMI 1640. Cells were centrifuged and resuspended in 3 ml of
red cell lysis buffer (ACK lysis buffer, 0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM
EDTA, pH 7.2) and incubated at 37 ºC for 3 minutes. Cell suspensions were
washed once with RPMI 1640 medium in a 70 µm pore size cell strainer,
centrifuged and resuspended in RPMI 1640 medium.
Immunohistochemistry and immunofluorescence. Initial
preparation of tissues was as follows: tissues were collected at different
time points after infection, embedded in Tissue Tek OCT compound (Sakura
Finetek, Torrence, CA) and frozen by immersion in liquid nitrogen-cooled
methylbutane (Sigma-Aldrich Inc., St.Louis, MO). Tissue sections 4-6 µm in
thickness were obtained, fixed in 99.5% histological grade acetone (Sigma-
Aldrich) and stored at -80ºC until further use. Sections were thawed at
room temperature (RT) for 5 minutes and fixed in 2% paraformaldehyde
solution (USB Corporation, Cleveland, OH) for 10 minutes.
For immunohistochemistry staining, frozen sections were incubated
with peroxidase blocking reagent (Dako, Carpinteria, CA) for 30 minutes at
RT, followed by incubation with background reducer solution (Background
Sniper, Biocare Medical, Concord, CA) for 30 minutes at RT, and by
incubation with anti-MV N polyclonal antibody (189) diluted in Background
Sniper solution for 1 hour at RT. Sections were then washed with Bio-Rad
Tris-buffered saline [20 mM tris (hydroxymethyl) aminomethane, 500 mM

Results Chapter 3
75
NaCl] and incubated with rabbit or rodent alkaline phosphatase-polymer
solution (Biocare Medical) for 20 minutes at RT. Alkaline phosphatase was
visualized with red chromogen substrate (Biocare Medical). Sections were
counterstained with hematoxylin (Vector, Burlingame, CA) and mounted
with permanent mounting medium (Vector).
For immunofluorescence stainings, sections were permeabilized for
3 minutes at RT in PBS with 0.1% (v/v) Triton X-100 (Sigma-Aldrich).
Sections were incubated with 5% goat serum blocking solution for 30
minutes at RT. Streptavidine-biotin blocking was performed prior to
incubation of sections with biotinylated anti-MHC class II (I-Ab) antibody,
using a streptavidin-biotin blocking kit (Vector). After blocking, sections
were incubated one hour at RT with antibodies diluted according to
manufacturer’s recommendations in blocking solution. MV N protein was
detected with a rabbit polyclonal anti-MV N antibody (described above) at
a dilution of 1/100 followed by incubation with Alexa Fluor 488-conjugated
anti-rabbit IgG (Invitrogen, Molecular Probes, Eugene, OR). B lymphocytes
were identified with anti-CD45 (B220) (clone RA3-6B2; BD Biosciences
Pharmingen); CD4 and CD8 T cells were identified with anti-CD4 (YTS191.2;
Abcam Inc., Cambridge, MA) and anti-CD8 (YTS169.4; Abcam) respectively;
AMs were characterized with anti-CD2 (LFA-2) (RM2-5, eBioscience, San
Diego, CA); anti-CD11c (223H7; MBL International, Woburn, MA); anti-
F4/80 (Cl:A3-1, Abcam); biotinylated mouse monoclonal anti-MHC class II
(I-Ab) (AF6-120.1, BD Pharmingen) and anti-Mac-1 (M1/70, eBioscience).
Epithelial cells were identified with anti-CD326 (Ep-CAM, Epithelial Cell

Results Chapter 3
76
Adhesion Molecule) (G8.8, BD Biosciences Pharmingen). Secondary
antibodies included Alexa Fluor 594-conjugated anti-rat IgG (Invitrogen).
Sections stained with biotinylated mouse monoclonal anti-MHC class II (I-
Ab) as primary antibody were stained with Alexa 594 conjugated-
streptavidin (Invitrogen) as the secondary antibody. Sections were
mounted in Prolong Gold antifade reagent with DAPI (Invitrogen, Molecular
Probes) and visualized with a Zeiss Axioplan confocal microscope (LSM 510;
Carl Zeiss Inc., Germany).
Flow cytometry. For flow cytometry analysis, whole lungs were
collected without lavage, to allow the inclusion of bronchoalveolar cells. A
single cell suspension was prepared as described above. Cells were counted
using a hemacytometer after diluting with trypan blue dye to exclude dead
cells. Isolated cells were resuspended at a density of 106
cells/50 µl. To
identify specific cell sub-populations a multicolor staining was performed
by incubating cells with appropriate antibodies (1 µg per million cells), for
45 minutes on ice. GFP was assessed to identify MV infected cells. For the
identification of CD4+ T cells, anti-CD4-phycoerythrin (PE) (H129.19, BD
Pharmingen) and anti-CD3-allophycocyanin (APC)-Cy7 (17A2, eBioscience)
antibodies were used. For the identification of CD8+ T cells, cells were
stained with anti-CD8-PE-Cy7 (53-6.7, eBioscience) and anti-CD3-APC-Cy7
(eBioscience) antibodies. B lymphocytes were identified with anti-CD45R
(B220)-APC-Cy7 (RA3-6B2, eBioscience). In order to reduce Fc receptor
binding, cells stained with CD45R (B220) antibody (B cells) were first

Results Chapter 3
77
incubated with CD16/32 antibody (2.4G2, BD Pharmingen) for 5 minutes on
ice. DC (conventional dendritic cells) were identified with anti-MHCII (I-Ab)-
PE (AF6-120.1, BD Pharmingen) and anti-CD11c-APC (HL3, BD Pharmingen).
Macrophages were stained with anti-CD11c-APC (HL3, BD Pharmingen) and
anti-Mac-1-PE-Cy5 (M1/70, eBioscience). AM were identified with the
following staining: anti-Mac-1-PE-Cy5 (M1/70, eBioscience), anti-F4/80-
APC-Cy7 (BM8, eBioscience), anti-CD11c-APC (HL3, BD Pharmingen) and
anti-MHCII (I-Ab)-PE (AF6-120.1, BD Pharmingen). Epithelial cells were
identified with anti-CD326-PE (Ep-CAM) (G8.8, BD Pharmingen). Ifnarko
-
CD46Ge mice were used as negative controls for quantification.
Human SLAM expression was detected by incubating cells with
biotin anti-human CD150 (A12 (7D4), BioLegend, San Diego, CA) followed
by incubation with streptavidin-PerCP (Becton Dickinson
Immunocytometry Systems, San Jose, CA). Ifnarko
-CD46Ge mice were used
as negative controls and mock-infected animals were used as a negative
control for GFP expression. Acquisition of samples was performed on a
cytometer (FacsCanto or LSRII, BD Biosciences) and analyzed using using
FlowJo software (Tree Star Inc., Asland, OR). Statistical analysis was
performed with standard paired t tests using the JMP7 software (SAS, Cary,
NC).
Quantification of virus load in lymphatic organs. Single cell
suspensions from mandibular, mediastinal, mesenteric and inguinal LN and
spleen were obtained as described above. Serial 10-fold dilutions of

Results Chapter 3
78
mononuclear cells were made in DMEM with 5% FBS and 1%
Penicillin/Streptomycin. Four replicates of 101 to 10
5 mononuclear cells
were co-cultured with 5x105 Vero/hSLAM cells in 24-well plates and 10
6
mononuclear cells were co-cultured with 106 Vero/hSLAM cells in a 100
mm dish. Cultures were monitored for cytopathic effects 4 days later. Virus
titers were expressed as TCID50 per 106 cells.
Results
Ifnarko
-SLAMGe transgenic mice. To generate mice susceptible to
wild type MV infection, the hSLAM gene, including all its transcription
control elements, was previously transferred to the mouse genome (202).
In another approach to enhance replication of any exogenous virus in mice
(114), the mouse interferon type I receptor gene was inactivated (Ifnarko
background). For our study we combined these desirable characteristics by
crossing hSLAM-expressing with Ifnarko
mice; to facilitate progeny
screening, we mapped the insertion site of the hSLAM gene in the mouse
genome, as detailed in the materials and methods. The resulting mouse
strain was named Ifnarko
-SLAMGe. It is defective in the interferon type I
response and expresses hSLAM with human-like tissue specificity: for
example, 11-13% CD3 positive T cells of these mice express hSLAM (data
not shown), as compared to 10-20% CD3 positive T cells of humans (29,
170); 28-31% CD19 positive B cells of these mice express hSLAM, as
compared to 20-25% CD20 positive B cells of humans (data not shown).

Results Chapter 3
79
Human SLAM expression in lymphatic tissue of Ifnarko
-SLAMGe
transgenic mice. We document here cell type specificity of hSLAM
expression in the mediastinal LN, previously shown to be important for MV
infection of SLAM “knock-in” mice (127): in these mice MV reaches higher
titers in this LN than in other lymphatic organs. Single cell suspensions
were prepared from the mediastinal LN of five uninfected Ifnarko
-SLAMGe
mice and stained with specific markers to indentify B cells (B220), T cells
(CD3), and macrophages (CD11clow
Mac-1high
). Cells were gated for live
leukocytes and analyzed for hSLAM expression within each cell population.
Figure 1 documents hSLAM expression in one representative
animal: about 5% B cells (Fig. 1A, left panel, compare top left and right
quadrants) and 2% T cells (Fig. 1B, left panel, compare top left and right
quadrants) scored positive; the right panels in Figures 1A and 1B document
hSLAM expression levels in Ifnarko
-SLAMGe mice (red line) as compared to
a negative control (Ifnarko
-CD46Ge mice, blue line). The mean and standard
deviation of SLAM expression in mediastinal LN of five animals were 10 ±
5.9% for B cells and 1.5 ± 0.6% for T cells (data not shown). Thus resting B
and T cells from mediastinal LN of Ifnarko
-SLAMGe mice express hSLAM at
low levels, and more B than T cells express this protein, as in human
lymphatic tissues (7, 30). In CD11clow
Mac-1high
macrophages of this mouse
(Fig. 1C, left panel, bottom right quadrant) hSLAM expression was below
detection level (Fig. 1C, right panel).

Results Chapter 3
80
Figure 1. Analysis of
hSLAM expression in
leukocytes from the
mediastinal LN of Ifnarko
-
SLAMGe mice. Cells were
stained with antibodies
specific for B cells (B220+)
(A), T cells (CD3+) (B) or
macrophages
(CD11clow
Mac-1high
) (C).
Ifnarko
-CD46Ge mice were
used as negative controls
(right panels, blue lines).
Right panels, Y axis: % of
Max is a measure of the
number of cells normalized
to the same total number
in each sample.
Human SLAM expression in immune cells of the lungs of Ifnarko
-
SLAMGe transgenic mice. Since primary wtMV replication occurs mainly if
not exclusively in immune cells, rather than in epithelial cells (99), SLAM-
expressing immune cells in the airways or in sub-epithelial tissue are
candidate hosts for virus replication. We thus characterized hSLAM
expression in single cell suspensions obtained from lungs of uninfected
Ifnarko
-SLAMGe mice, including B cells (B220+), T cells (CD3
+) and alveolar

Results Chapter 3
81
macrophages (AM) defined as CD11chigh
MHCIIlow
Mac-1low
F4/80high
cells (53,
93).
Figure 2. Analysis of SLAM
expression in leukocytes from
the lungs of Ifnarko
-SLAMGe
mice. For (A) and (B),
lymphocytes were gated and
sorted with antibodies against
B cells (B220+) or T cells
(CD3+), respectively. For AM
(C), total live cells were
stained with three antibodies
and sorted for the
combination
CD11chigh
MHCIIlow
Mac-1low
.
Ifnarko
-CD46Ge mice were
used as negative controls.
Histogram panels, Y axis: % of
Max is a measure of the
number of cells normalized to
the same total number in each
sample. (C) y axis: FSC,
forward scatter.

Results Chapter 3
82
Figure 2 documents hSLAM expression in these cells: about 5% of B
cells (Fig. 2A, left panel, compare top left and right quadrants) and 4% of T
cells (Fig. 2B, left panel, compare top left and right quadrants) of one
animal scored positive; the right panels in Figures 2A and 2B document
hSLAM expression levels in Ifnarko
-SLAMGe mice (red line) as compared to
a negative control (Ifnarko
-CD46Ge mice, blue line). The mean and standard
deviation of SLAM expression in lung tissue of five animals were 8.7 ± 1.9%
for B cells and 3.3 ± 0.7% for T cells (data not shown). A small fraction of
AM also express SLAM (Fig. 2C, lower left panel, compare red line of
Ifnarko
-SLAMGe mice with blue line of Ifnarko
-CD46Ge negative control
mice).
Early MV replication in the lungs of Ifnarko
-SLAMGe transgenic
mice. We then sought to identify the tissue and cell types sustaining wtMV
replication immediately after IN inoculation of Ifnarko
-SLAMGe mice.
Candidate host tissues/cell types include NALT and immune cells in the
airway lumen or sub-epithelial airway tissue. Mice were inoculated with
wtMVgreen, a GFP-expressing virus; since wtMV interacts with SLAM but not
CD46, Ifnarko
mice expressing CD46 but not SLAM served as negative
control. One to seven days after IN inoculation, as indicated in Table 1,
NALT and the lungs were harvested; in addition, mandibular and
mediastinal LN, thymus, spleen and PBMC were collected. Cells from these
organs were co-cultured with Vero/hSLAM cells and the appearance of
syncytia was monitored over 4 days.

Results Chapter 3
83
As shown in Table 1 MV was isolated from the lungs of every
Ifnarko
-SLAMGe mouse at every time point. Virus was also isolated from the
lungs of all three Ifnarko
-CD46Ge mice sacrificed 1 day p.i., but not at later
time points (data not shown) suggesting that some of the virus re-isolated
at day 1 may be the inoculum. At 2 days p.i., virus was recovered from the
mediastinal LN in 2 out of 3 animals. At 3 and 7 days p.i., virus was isolated
from the mediastinal LN and the spleen of most mice, and from the
mandibular LN, thymus and PBMC of some mice. However, no virus was
isolated from the NALT of any animal. These observations suggest that MV
replicates initially in the lungs of Ifnarko
-SLAMGe mice, and then spreads to
lymphatic organs through infected cells circulating in the lymphatics rather
than in the blood.

Results Chapter 3
84
TABLE 1. MV re-isolation from organs of Ifnarko
-SLAMGe mice after IN
infection
Organs Nº. positive/total nº.
Day 1a Day 2 Day 3 Day 7
Lungs 5/5 3/3 3/3 3/3
NALT 0/5 0/3 0/3 0/3
Mandibular LN 0/5 0/3 1/3 2/3
Mediastinal LN 0/5 2/3 3/3 3/3
Thymus 0/5 NDb 0/3 2/3
Spleen 0/5 0/3 3/3 2/3
PBMC 0/5 0/3 1/3 1/3
a day post-infection.
b ND, not done.
To assess whether the inoculum accounts for a significant fraction
of the signal detected after inoculation, we infected Ifnarko
-SLAMGe mice
IN with UV-inactivated MVgreen. Mice were sacrificed one, two or three days
after inoculation and whole lungs examined under a fluorescence
microscope; no fluorescence was detected in mice infected with UV-
inactivated virus. On the other hand, the pattern of GFP expression was
quite homogeneous; in four of four Ifnarko
-SLAMGe mice inoculated with
live virus green fluorescence was observed in all the lobes of the lung, and
reached into the lower respiratory airways (data not shown). As an
additional control, we infected Ifnarko
-CD46Ge mice with MVgreen but again

Results Chapter 3
85
no fluorescence was detected in the lungs of these animals (data not
shown). Since the UV-inactivated inoculum is not detectable by analysis of
GFP fluorescence, we conclude that one day post-inoculation significant
MV replication accounts for strong GFP expression.
AM sustain MV replication in the lungs. We then assessed by
immunohistochemistry (Fig. 3A and B) and immunofluorescence (Fig. 3C to
H) which cell types sustain MV replication in the airways. Ifnarko
-SLAMGe
mice were infected IN with wtMVgreen, lungs collected one day post-
inoculation, frozen, and stained with an anti-N antibody (red colour). Figure
3, panels A and B, shows one representative immunohistochemistry
analysis: MV N protein was most often detected in cells located on the
luminal side of respiratory bronchioles, a location typical for AM.
To assess the identity of infected cells and differentiate between
AM and other immune cells including DCs, we stained lung sections with an
anti-N antibody (green colour) and different cellular markers: the immune
cells markers CD11c, F4/80, CD2 (also named LFA-2), Mac-1 (also named
CD11b) and MHCII, and the epithelial cell marker EpCAM (53, 93). None of
the infected cells were EpCAM positive (Fig. 3G), suggesting that epithelial
cells are not infected. Infected cells were often CD11c, F4/80, and CD2
positive (Fig. 3 C-E), but very rarely Mac-1 or MHCII positive (Fig. 3F and
Fig. 3H). Even if none of the above markers is entirely specific for one type
of immune cells, altogether these results suggest that AM may sustain
primary MV infection in the lungs.

Results Chapter 3
86
Figure 3. Characterization of wtMV-infected cells in the lungs one day
after mouse inoculation. (A and B) Frozen sections of mouse lung stained
with an anti-N antibody and counterstained with hematoxylin. N protein
was visualized with red chromogen. Arrows indicate positive cells, which
localize in the airway lumen. (C to H) Identification of the cell types
accumulating MV N by confocal micrographs analysis of lung frozen
sections. N protein was visualized with Alexa Fluor 488 (green). The specific
cell markers indicated on the top right of each panel were visualized with
Alexa Fluor 594 (red). Nuclei were conterstained in blue with DAPI. Scale
bars represent 10 µm.
To quantify the level of infection of AM and other immune cell
types mice inoculated IN with wtMVgreen were sacrificed 1 or 3 days later.
Lungs were collected, tissue cells were dissociated, and GFP expression
analyzed using flow cytometry. The analysis presented in Table 2 indicates
that AM (sorted as CD11chigh
MHCIIlow
Mac-1low
F4/80high
cells) were infected
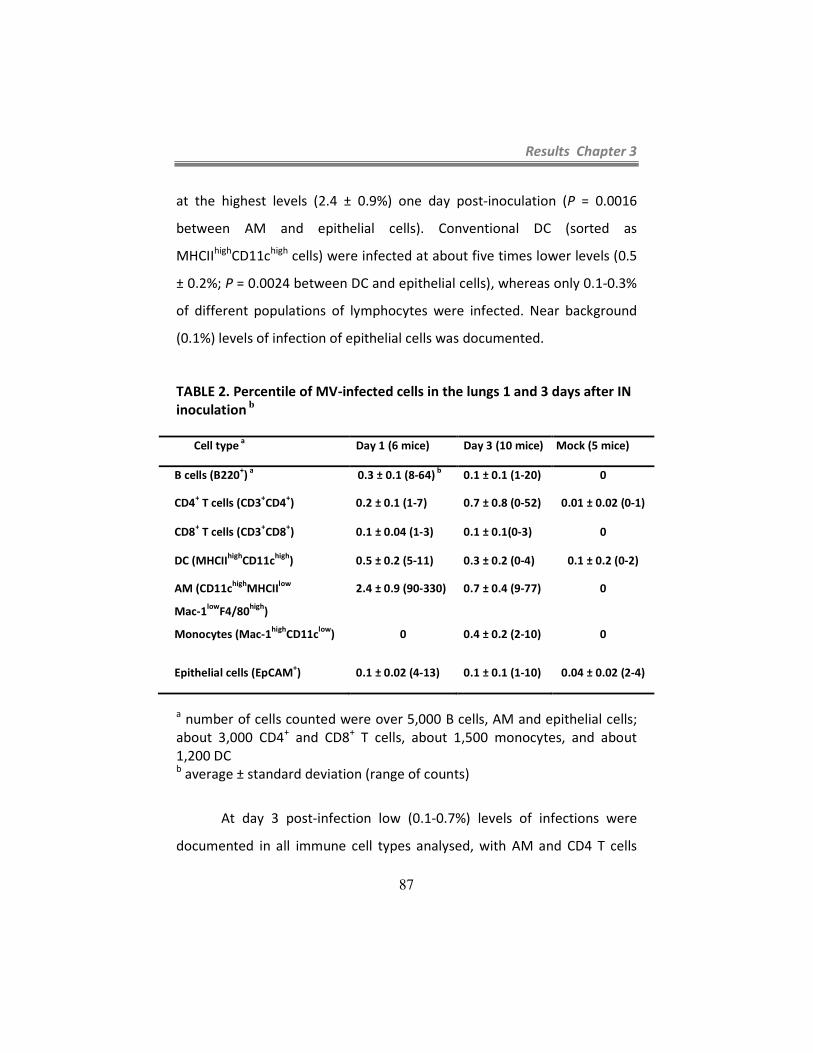
Results Chapter 3
87
at the highest levels (2.4 ± 0.9%) one day post-inoculation (P = 0.0016
between AM and epithelial cells). Conventional DC (sorted as
MHCIIhigh
CD11chigh
cells) were infected at about five times lower levels (0.5
± 0.2%; P = 0.0024 between DC and epithelial cells), whereas only 0.1-0.3%
of different populations of lymphocytes were infected. Near background
(0.1%) levels of infection of epithelial cells was documented.
TABLE 2. Percentile of MV-infected cells in the lungs 1 and 3 days after IN
inoculation b
Cell type a
Day 1 (6 mice) Day 3 (10 mice) Mock (5 mice)
B cells (B220+)
a 0.3 ± 0.1 (8-64)
b 0.1 ± 0.1 (1-20) 0
CD4+ T cells (CD3
+CD4
+) 0.2 ± 0.1 (1-7) 0.7 ± 0.8 (0-52) 0.01 ± 0.02 (0-1)
CD8+ T cells (CD3
+CD8
+) 0.1 ± 0.04 (1-3) 0.1 ± 0.1(0-3) 0
DC (MHCIIhigh
CD11chigh
) 0.5 ± 0.2 (5-11) 0.3 ± 0.2 (0-4) 0.1 ± 0.2 (0-2)
AM (CD11chigh
MHCIIlow
Mac-1low
F4/80high
)
Monocytes (Mac-1high
CD11clow
)
2.4 ± 0.9 (90-330)
0
0.7 ± 0.4 (9-77)
0.4 ± 0.2 (2-10)
0
0
Epithelial cells (EpCAM+) 0.1 ± 0.02 (4-13) 0.1 ± 0.1 (1-10) 0.04 ± 0.02 (2-4)
a number of cells counted were over 5,000 B cells, AM and epithelial cells;
about 3,000 CD4+ and CD8
+ T cells, about 1,500 monocytes, and about
1,200 DC b average ± standard deviation (range of counts)
At day 3 post-infection low (0.1-0.7%) levels of infections were
documented in all immune cell types analysed, with AM and CD4 T cells

Results Chapter 3
88
being infected at the highest levels; monocytes that are available in
unperfused lungs were infected at similar levels as other immune cell types
(0.4%). Epithelial cell infection remained near background (0.1%). These
data confirm that AM are the main initial target cells for MV infection, and
also indicate that MV replication in immune cells located in the mouse
airways is limited and short-lived. We also detected cellular infiltrates in
the lungs 3 days p.i., which was due mainly to neutrophils (data not
shown).
MV infects AM expressing SLAM, and MV infection enhances
SLAM expression. To assess whether hSLAM expression correlates with MV
infection, we inoculated Ifnarko
-SLAMGe mice IN with MVgreen and analyzed
hSLAM expression on AM collected one day after inoculation. AM from
mock-infected Ifnarko
-SLAMGe mice, and from infected Ifnarko
-CD46Ge
mice, served as controls. AM were sorted as CD11chigh
(Fig. 4, left panels),
MHCIIlow
(Fig. 4, center-left panels), Mac-1low
(Fig. 4, center-right panels)
cells.
Figure 4A (right panel, right quadrants) shows that background
(0.12%) SLAM expression was detected in AM from control Ifnarko
-CD46Ge
mice. On the other hand, 0.86% AMs of Ifnarko
-SLAMGe mice mock-
infected with the post-nuclear fraction of uninfected Vero/hSLAM cells
expressed SLAM (Fig. 4B, right panel, right quadrants). Remarkably, about
16% (1.44% GFP-positive and 14.8% GFP-negative) AMs of infected Ifnarko
-

Results Chapter 3
89
SLAMGe mice expressed SLAM (Fig. 4C, right panel), which may reflect AM
activation.
D

Results Chapter 3
90
Figure 4. Analysis of hSLAM expression in AM before and after MV
infection. AM from non-infected Ifnarko
-CD46Ge mice (A), mock-infected
Ifnarko
-SLAMGe (B) or MV-infected Ifnarko
-SLAMGe mice (C). Cells were
gated for CD11chigh
(left panels), MHCIIlow
(center left panels), and Mac-1low
(center right panels) and analyzed for infection (GFP) versus SLAM
expression (right panels). Effect of MV infection on hSLAM expression in AM
(D). X axis: FSC, forward scatter. Ifnarko
-CD46Ge mice were used as negative
controls to set the quadrants.
Importantly, about one in 11 SLAM-expressing AM were infected
(Fig. 4C, compare numbers in top and bottom right panels), whereas one in
28 SLAM-negative AM were infected (Fig. 4C, compare numbers in top and
bottom left panels). Analysis of AM from additional four animals confirmed
2-3 times preferential infection of SLAM-expressing cells (data not shown).
These data confirm that infection of AMs in a SLAM-dependent manner is a
crucial step in the early stages of MV infection. Enhanced hSLAM
expression on infected and non-infected AM is short-lived: 3 days after
infection, hSLAM expression returned to levels slightly above the values
found on AM from mock infected mice (data not shown).
Spread in the lymphatic organs. We further characterized MV
spread by quantifying viral replication in different lymphatic organs of
Ifnarko
-SLAMGe mice 3 days after IN inoculation. As shown in Figure 5A, the
mediastinal LN was infected with high efficiency in two mice and at lower
levels in other three animals, whereas the other lymphatic tissues
(mandibular, mesenteric, and inguinal LN, and spleen) were infected at low
or undetectable levels. Thus the mediastinal LN that drains the respiratory

Results Chapter 3
91
tract appears to be a prominent site of MV replication after IN infection of
Ifnarko
-SLAMGe mice, as well as knock-in hSLAM expressing mice (127).
Figure 5. MV infection
levels in lymphatic organs.
Data were obtained three
days after IN inoculation (A),
three days after IP
inoculation (B), or six days
after IP inoculation (C). Each
dot represents a single
mouse. Group average is
represented by a horizontal
bar and detection limit by a
dotted line. Man:
mandibular LN; Med:
mediastinal LN; Mes:
mesenteric LN; Ing: inguinal
LN; Spl: spleen.

Results Chapter 3
92
We then asked whether this effect is due to the IN route of
infection or to a stronger susceptibility of cells in the mediastinal LN to MV
infection. We inoculated mice IP and compared replication levels in a LN
located in the peritoneum (mesenteric), one in the thoracic cavity
(mediastinal), and two control lymph nodes (mandibular and inguinal).
Three days later lymphatic tissues were harvested and the viral load
measured. Again, a high MV titer was documented in the mediastinal LN
(Fig. 5B): in different mice 1-8% of the cells of this LN were infected 3 days
p.i. The average levels of infected cells in the mandibular, mesenteric and
inguinal LN, and the spleen, were 10-100 times lower, but the differences
between different LN were not statistically significant. Six days p.i. (Fig. 5C)
0.01-0.1% of the cells were infected in all lymph nodes and in the spleen.
Finally, we sought to identify which cells support MV replication in
the mediastinal LN. A group of 6 Ifnarko
-SLAMGe mice was inoculated IP,
mediastinal LN collected three days p.i., single cell suspensions obtained
and GFP expression analyzed by flow cytometry. As shown in Table 3,
lymphocytes were infected at the highest levels: 2 ± 0.7% of B cells (B220+),
1.1 ± 0.3% of CD4+ T cells (CD3
+CD4
+), and 2 ± 0.4% of CD8
+ T cells
(CD3+CD8
+). DC (MHCII
highCD11c
high) were infected at similar levels (1.4 ±
0.4% infection) but macrophages (CD11clow
Mac-1high
MHCIIhigh
) were
infected at lower levels (0.6 ± 0.1%). We note that DC and macrophages
account only for a small fraction of cells in the mediastinal LN, and thus
that lymphocytes are by far the most abundant cell type infected.

Results Chapter 3
93
TABLE 3. Percentile of infected cells in the mediastinal LN after IP
inoculation a
Cell type Day 3 Mock
B cells (B220+) 2 ± 0.7 (362-851) <0.01 (0-1)
CD4+ T cells (CD3
+CD4
+) 1.1 ± 0.3 (159-332) <0.01 (0-1)
CD8+ T cells (CD3
+CD8
+) 2 ± 0.4 (188-313) 0.02 ± 0.01 (1-5)
DC (MHCIIhigh
CD11chigh
Mac-1low
) 1.4 ± 0.4 (18-54) 0.08 (0-2)
Macrophages (CD11clow
Mac-1high
) 0.6 ± 0.1 (10-22) 0.01 (0-2)
a average ± standard deviation (range of counts); group size was 6 animals
(both groups)
Discussion
A new small animal model of MV infection. Small animal models
for measles do not exactly mimic the human disease but allow one to focus
on particular aspects of infection. We focused our study on the question of
which cells are infected immediately after respiratory inoculation, and
documented that hSLAM-expressing AM, as well as DC, are the initial
targets in the lungs. These cells may ferry the infection through the
epithelial barrier. Similarly, recent work with wild type and selectively
receptor-blind MVs (34, 99), and with a selectively receptor-blind canine
distemper virus (196) brought evidence for a model of morbillivirus
dissemination postulating that these viruses take advantage of SLAM-
expressing immune cells situated in the lumen of the respiratory tract of
natural hosts to cross the epithelial barrier after contagion.

Results Chapter 3
94
Our improved mouse model has an immune deficient background:
we generated hSLAM transgenic mice whose innate immune system is
compromised by the deletion of the interferon alpha receptor. As
expected, the Ifnarko
background allowed more extensive MV infections
than the previously used STAT1-defective background (202). Minimal MV
replication in hSLAM-expressing mice that are immune-competent did not
allow a sensitive study of the cell type specificity of infection, and we do
not know whether prevention of the interferon type I response induces a
different cell type specificity of infection. However, we know this was not
the case for other mouse strains expressing CD46 or hSLAM in an Ifnarko
background mice (127, 168), and we do not have indications of the
contrary for our mouse strain.
Most characteristics of the Ifnarko
and the STAT1-defective strain
were similar: for example, equivalent levels of SLAM expression were
observed in different cell types. Moreover, in the STAT1-defective mice we
reported enlarged lymph nodes and splenomegaly due to immune cell
infiltration (202); similarly, six days post-inoculation we observed enlarged
lymph nodes in the Ifnarko
mice, and the weight of their spleens was on
average double than normal (119.8 ± 23.4 mg versus 54.8 ± 6.2 mg in
uninfected mice, average and standard deviation in groups of five mice).
The cell-type specificity of expression of hSLAM suggests that a wild
type MV might infect certain target lymphocytes,
monocytes/macrophages, and DC, in transgenic mice. Indeed we
monitored and quantified MV entry in these cell types, and documented

Results Chapter 3
95
predominant interactions with AM and DC one day after IN inoculation. To
our knowledge, this report is the first direct analysis of the events that
support primary MV replication and early viral spread in an animal model.
Later, infection progresses to lymphatic tissues, with an early (2 days p.i.)
involvement of the mediastinal LN. Lymphocytes sustain the bulk of MV
infection not only in the mediastinal LN (Table 3) but also in other LN and
lymphatic organs (data not shown). Thus, our model reproduces the
lymphatic phases of the infections of MV and other morbilliviruses in their
natural hosts (108, 194).
Crossing the epithelial barrier: alternatives. One day after IN
inoculation with 106 TCID50, MV replication was documented in about 2.5%
of AM and 0.5% of DC of hSLAM-expressing mice. MV replication was
below detection levels in Ifnarko
mice not expressing hSLAM, indicating that
scavenging of infectious virus by Ifnarko
AM rarely if at all results in
productive infection. Since there are about 2 million AM in a mouse (191)
about 5x104 AM were productively infected and expressed GFP. Even if
only 1 in 1000 infected AM crosses the lung epithelium, about 50 of these
cells would have the opportunity to transmit infectivity to the lymphatic
organs. AM are implicated in the translocation of particles from the lungs
to the draining lymph nodes (66), a process that MV may exploit to cross
the epithelial barrier: infected AM might migrate to the draining lymph
nodes and spread infection to B and T lymphocytes. The fact that
replication of a MV vaccine strain in CD46-expressing transgenic mice is

Results Chapter 3
96
most prominent in macrophages after inoculation by three different routes
(154) is consistent with these cells sustaining the immediate early phase of
MV infection. Moreover, in Ifnarko
-CD46Ge mice in vivo depletion of AM
resulted in strong local DC infiltration, virus infection of infiltrating DCs and
macrophages, and ultimately higher virus titer in lymphatic tissues (154).
Thus, while protecting other immune cells from infection, AM also support
MV replication in the lungs, and are likely to be one of the pathways
exploited by MV to cross the epithelial barrier.
Another potential ferry for MV are DC present in the sub-epithelial
tissues of the airways. We have observed that there are about 10 times
less DC than AM in the lungs of mice, and estimate that about 0.5% of
2x105
DC, or about 1000 DC, are infected. It has been reported that DC
infected with influenza virus rapidly migrate from the respiratory tract to
the draining LN (56), beginning 2 hours after infection (98). Analogously, DC
productively infected with MV might cross the epithelial barrier and rapidly
disseminate virus in the lymphatic organs including the mediastinal LN.
Alternatively, the virus would attach to DC through DC-SIGN but not
replicate, and DC would trans-infect T lymphocytes (35). DC that carry low
levels of MV may be detected inefficiently by flow cytometry, resulting in
an under-estimation of the efficiency of infection. Finally, it is also possible
that cell-free virus exploits breaks in the epithelial barrier to reach immune
organs through the lymphatics, but the efficiency of AM and DC infection is
consistent with either cell type being more important than epithelium
leakage as primary mean for MV to cross the epithelial barrier.

Results Chapter 3
97
SLAM expression and virus amplification. Ohno et al. (127)
observed that the mediastinal LN is preferentially infected in mice
expressing a human-mouse SLAM hybrid, and several lines of evidence in
our study suggest that a similar phenomenon occurs in our mice expressing
hSLAM. To assess whether this is simply explained by higher receptor
expression, we measured hSLAM expression levels in this and the
mesenteric, mandibular and inguinal LN of uninfected animals, but failed to
document significant differences (data not shown). Thus the cause for
preferential MV spread in the mediastinal LN remains to be determined.
In transgenic mice (Fig. 4) as in human tonsillar tissue (30), MV
infects predominantly, but not exclusively, immune cells expressing SLAM.
We do not know if the apparently SLAM-independent entry really occurs: it
is possible that hSLAM expression is undetectable with antibodies but still
sufficient to facilitate viral entry.
After IN inoculation with MV, hSLAM expression is enhanced in
infected and non-infected AM. Similarly, induction of SLAM expression
after MV infection was previously reported ex vivo in monocytes (11, 109).
The mechanisms activating SLAM expression in both systems are unknown
and could be indirect. Nevertheless, since SLAM is a low affinity self-ligand
glycoprotein (107), increase of its expression in few target cells may cause
a feed-back loop of cell activation. This feed-back loop may in turn sustain
extremely rapid spread of Morbillivirus infections in natural hosts (194).
The new mouse model developed in this study allowed
characterization of the immediate early phases of wild type MV infection in

Results Chapter 3
98
the respiratory airways of an interferon-defective host, and gave
quantitative insights in the cell types supporting primary MV replication in
the lungs of this host. Future studies will address the relative importance of
AM and DC for supporting primary MV infection and ferrying virus through
the epithelial barrier in mice, and the question of the levels of infection of
these cells in interferon-competent hosts.

Results Chapter 4
99
CHAPTER 4
Pathways of lymphatic dissemination of measles virus
attenuated and wild type strains in SLAM expressing mice


Results Chapter 4
101
Abstract
MV vaccine has outstanding safety and efficacy records but the
mechanisms underlying its attenuation are poorly understood. We
characterized the spread of a vaccine strain and compared with that of a
wild type. We also characterized the host cells supporting early infection
and dissemination in the lymphatics. We infected Ifnarko
-SLAMGe mice
intraperitoneally with a MV vaccine strain (MVvac) or with a wild type
strain (wtMV). To identify which cells sustained the wild type MV and the
attenuated strain infection, several lymph nodes (LN) and spleen were
collected one or three days after intraperitoneal (IP) inoculation and
analyzed by confocal microscopy. One day after inoculation MV was
detected in a macrophage population in the subcapsular sinus (SCS) of the
mediastinal LN and marginal sinus (MS) of the spleen (here named SCS
macrophages). Later, three days post infection, the wtMV efficiently spread
to SLAM expressing leukocytes in the mediastinal LN and spleen whereas
the vaccine strain was cleared. The spread of a wild type virus strain
carrying a mutation in the H protein (N481Y) allowing CD46 binding in
addition to SLAM did not alter viral tropism or replication efficiency. Thus,
vaccine attenuation may be caused by suboptimal replication and early
clearance by the host innate immune system.


Results Chapter 4
103
Introduction
Measles is preventable by a safe and effective vaccine. A
vaccination program led to a significant reduction in the number of MV
related deaths; since 2000 to 2008 the number of deaths related to MV
decreased 78%, from 733.000 in 2000 to 164.000 in 2008 (1). However, in
countries with poor health infrastructure measles remains a major cause of
human mortality and morbidity.
The current available measles vaccines are derived from the original
Edmonston strain, isolated from a patient by Enders and Peebles in 1954
(42). The live attenuated vaccines in use were obtained by passaging of the
original strain in several cell lines, including human kidney and amnion cells
and chicken embryo fibroblasts (59). The vaccine strain used here was
obtained from an infectious cDNA derived from a vial of the Moraten strain
(38). The Moraten is one of several attenuated strains derived from the
original Edmonston clinical isolate and is identical to the Schwartz vaccine
strain (137).
We recently bred a mouse line expressing hSLAM in an interferon
type I deficient background and documented that alveolar macrophages
and dendritic cells support early MV replication in the respiratory tract. In
addition, we observed early infection of all lymphatic organs, and in
particular of the mediastinal LN, upon IP and intranasal (IN) inoculation.
Based on this, we sought to compare the viral tropism and spread of a wild
type strain with that of a vaccine strain in the lymphatic tissues. In

Results Chapter 4
104
addition, we characterized the spread of a wild type strain carrying a single
mutation in the H protein, an asparagine to tyrosine substitution at aa 481
(N481Y), allowing CD46 binding in addition to SLAM. CD46 is the receptor
used only by the attenuated MV strain (39, 119, 204).
One day after IP inoculation we documented that MV colocalized
with SCS and MS macrophages in the mediastinal LN and spleen,
respectively. Later, wild type MV efficiently infected SLAM expressing
leukocytes in the mediastinal LN. In contrast, the vaccine strain was cleared
after an initial infection of SCS or MS macrophages. Thus, early clearance
may account for the MV vaccine attenuation.
Materials and Methods
Cells. B95a (marmoset B-cell line kindly provided by Dr. Gerlier) and
Vero cells were maintained in DMEM and 10% fetal bovine serum (FBS).
Vero/hSLAM cells (Vero cells expressing human SLAM, kindly provided by
Dr. Y. Yanagi) were maintained in DMEM supplemented with 10% FBS and
0.5 mg of G418/ml. The rescue helper cell line 293-3-46 (147) was grown in
DMEM with 10% FBS and 1.2 mg of G418/ml.
Cell infections. B95a (5x105 cells/well), Vero and Vero/hSLAM
(2.5x105 cells/well) were cultured on 12 well plate and infected with wtMV,
MVvac, MVvacY481N, and wtMVN481Y at an M.O.I. of 0.1 in 0.5 ml of Opti-
MEM. Two hours later the opti-MEM was replaced by DMEM

Results Chapter 4
105
supplemented with 10% FBS. Syncytia formation was photographed 24 to
48 hours post infection (p.i.) under a phase-contrast microscopy.
Mouse strains. Generation of Ifnarko
-SLAMGe mice was described in
Chapter 3. To establish a transgenic mouse line expressing the hSLAM and
hCD46 gene and deficient in the alpha/beta interferon response, Ifnarko
-
CD46Ge mice (113) were crossed with SLAMGe mice (202). To obtain
Ifnarko
-CD46Ge-SLAMGe mice, the F2 and F3 progeny was screened for
inactivation of both copies of the interferon alpha receptor gene as
previously described (114) and for the availability of two copies of the
hSLAM gene and hCD46 gene.
Mouse infections. All animal experimental procedures were
performed according to protocols previously approved by the Mayo Clinic
Institutional Animal Care and Use Committee. Five to 8-week-old animals
were infected IP with 1.5x106 tissue culture infectious dose 50% (TCID50)
MV to measure viral load in lymphatic tissues, or with 8x106 TCID50 for
detection of infected cells by flow cytometry or histology.
Site-specific mutagenesis of measles virus H protein. The specific
mutation (asparagine to tyrosine substitution at amino acid 481 (N481Y) or
the reverse (Y481N) was introduced into the wild type measles and vaccine
strain virus H genes with the Quickchange site-directed mutagenesis kit
(Stratagene) on pCG-IC323-H or pCG-vac-H respectively. The mutated H,

Results Chapter 4
106
HN481Y or HY481N, was cloned in the full length p(+)MV323 or
pB(+)MVvac with PacI/SpeI. The full construct was named wtMVN481Y or
MVvacY481N.
Viruses and virus stock preparation. wtMV, wtMVCAT and MVvacCAT
were previously described in Chapter 2 and wtMVgreen in Chapter 3.
MVvac (38), MVvacY481N and wtMVN481Y were generated from full-
length cDNAs as descrived by Radecke et al. (147). Cells were scraped in
Opti-MEM (Gibco/Invitrogen Corp.) and viral particles were released by
two freeze-thaw cycles. Titers were determined by TCID50 titration on
Vero/hSLAM cells according to the Spearman-Kärber method (86).
Organ collection and preparation of single cell suspensions. Organ
collection and preparation of single cell suspensions were as described in
Chapter 3.
Flow cytometry. As previously described in Chapter 3, SLAM
expression was detected by incubating cells with PE anti-human CD150
(A12, BD Pharmingen). Acquisition of samples was performed on a
cytometer (FacsCanto) and analyzed using FlowJo software (Tree Star, Inc.,
Asland, OR).
Immunofluorescence. Frozen sections, obtained as previously
explained in Chapters 2 and 3, were incubated one hour at RT with

Results Chapter 4
107
antibodies diluted according to manufacturer’s recommendations in
blocking solution. MV N protein was detected with a rabbit polyclonal anti-
MV N antibody (described in Chapters 2 and 3) at a dilution of 1/100
followed by incubation with Alexa Fluor 488-conjugated anti-rabbit IgG
(Invitrogen, Molecular Probes, Eugene, OR). Subcapsular macrophages
were identified with rat anti-mouse sialoadhesin (CD169) (clone 3D6.112;
Abcam Inc., Cambridge, MA). Sections were mounted in Prolong Gold
antifade reagent with DAPI (Invitrogen, Molecular Probes) and visualized
with a Zeiss Axioplan confocal microscope (LSM 510; Carl Zeiss Inc.,
Germany).
Quantification of virus load in lymphatic organs. Quantification of
virus load in lymphatic organs was performed as described in Chapter 3.
Results
Low efficiency of replication of MV vaccine strain. As shown in
Chapters 2 and 3, infection by the wild type MV upon IN or IP inoculation
resulted in spread to several lymphatic organs, with preferential replication
in the mediastinal LN.
We then asked whether infection of an attenuated strain of MV,
MVvac, reproduces the tropism and efficiency of infection as that of a wild
type strain. To quantify and characterize the spread of an attenuated strain
of MV, Ifnarko
-SLAMGe mice were inoculated IP with MVvac; three days
later the lymph nodes and spleen were harvested and the viral load

Results Chapter 4
108
measured. As shown in Figure 1, panel A, MVvac infected the mediastinal
and mesenteric LNs and spleen about 100 times less efficiently as
compared to wtMV (panel B). The infection levels of mandibular and
inguinal LN by the attenuated strain were undetectable (panel A). Although
infection of the lymphoid tissues was generally low, the mediastinal LN
supported the highest level of infection. Thus, in this animal model, the
vaccine strain replication is very inefficient compared to wild type.
Figure 1. MVvac (A) and wtMV (B) infection levels in lymphatic organs
three days after IP inoculation. Each dot represents a single mouse. Group
average is represented by a horizontal bar and detection limit by a dotted
line. Man: mandibular LN; Med: mediastinal LN; Mes: mesenteric LN; Ing:
inguinal LN; Spl: spleen. (Panel B is from Chapter 3, Fig. 5B).
Early capture of wild type and vaccine strains of MV by
subcapsular macrophages. We next sought to identify which cells support
early infection by the wtMV and MVvac strains in the lymphatic tissues and
A
B

Results Chapter 4
109
whether the tropism of these two strains differs. To characterize the initial
targets of MV infection in the lymphoid tissues, Ifnarko
-SLAMGe mice were
inoculated IP with wtMVgreen or MVvac and were sacrificed one or three
days after infection. The mediastinal LN and spleen were collected and
frozen sections were obtained and stained with anti-N antibody to
detected viral protein. As shown in Figures 2 and 3, panels A and C, one day
p.i. wtMV and MVvac viruses were localized mainly in the SCS of the
mediastinal LN and to a lesser extent in the MS of the spleen (green
colour).
The subcapsular sinus of LN is an area rich in macrophages (28, 48)
and, recently, VSV was shown to colocalize with a macrophage marker,
CD169 in the subcapsular sinus of LN early after injection into the footpad
(84). This macrophage population, SCS macrophages, is characterized as
CD169+ Mac-1
+ MHCII
+ and CD11c
low (84). To assess whether MV replicates
in SCS macrophages, Ifnarko
-SLAMGe mice were infected IP with wtMV or
MVvac. Frozen sections of LN and MS of spleen were double stained with
anti-CD169 (red) and anti-MVN (green) antibodies. Figures 2 and 3 (A and
C) shows that one day p.i. MV wild type and vaccine strains colocalized
mainly with SCS macrophages in the mediastinal LN and spleen.

Results Chapter 4
110
Only wild type MV infection progresses to B and T cell areas. We
next infected Ifnarko
-SLAMGe mice IP with wtMV or MVvac, and assessed
progression of infection in the mediastinal LN and spleen. Three days p.i.
these organs were harvested, frozen sections obtained and stained with
anti-MV N and anti-CD169 antibodies. Three days after inoculation the
colocalization of wild type or vaccine strains with the SCS macrophages was
strongly reduced (Figs. 2 and 3, B and D). However, MV wild type replicated
extensively and spread to the B and T cell areas of the LN (below the SCS)
and spleen (white pulp area represented by a white dotted circle) (Figure.
2, panels B and D), whereas the vaccine strain did not (Figure 3, panels B
and D). This suggests that only wild type can escape capture by SCS and MS
macrophages in the LN and spleen and continue replicating; replication of
the vaccine strain is impaired after capture by SCS macrophages.

Results Chapter 4
111
Figure 2. Characterization of wtMV-infected cells in the lymphatic organs
one and three days after IP inoculation of Ifnarko
-SLAMGe mice.
Identification of the cell types accumulating MV N, by confocal micrographs
analysis. Frozen sections of mouse mediastinal LN (A and B) or spleen (C
and D) stained with an anti-N and anti-CD169 antibodies. N protein was
visualized with Alexa Fluor 488 (green). The macrophage marker, CD169,
was visualized with Alexa Fluor 594 (red). Nuclei were counterstained in
blue with DAPI. Scale bars represent 200 µm. Scale bar for insert in panel A
represent 10 µm.

Results Chapter 4
112
Figure 3. Characterization of MVvac-infected cells in the lymphatic organs
one and three days after IP inoculation of Ifnarko
-SLAMGe mice.
Identification of the cell types accumulating MV N, by confocal micrographs
analysis. Frozen sections of mouse mediastinal LN (A and B) or spleen (C
and D) stained with an anti-N antibody. N protein was visualized with Alexa
Fluor 488 (green). The macrophage marker, CD169, was visualized with
Alexa Fluor 594 (red). Nuclei were counterstained in blue with DAPI. Scale
bars represent 200 µm (A to C) and 50 µm (D).

Results Chapter 4
113
Next we sought to assess whether infection in the mediastinal LN
correlates with SLAM expression. A group of 6 Ifnarko
-SLAMGe mice were
infected IP with wtMVgreen, three days later the mediastinal LN was
collected and leukocytes were analyzed for GFP versus SLAM expression by
flow cytometry. Leukocytes from mock-infected Ifnarko
-SLAMGe mice and
from non infected Ifnarko
-CD46Ge mice served as controls. Figure 4A (right
quadrants) shows that background (0.81%; 0.75% plus 0.06%) hSLAM
expression was detected in leukocytes from control Ifnarko
-CD46Ge mice
whereas 11.61% (11.57% plus 0.04%) and 13.25% (11.54% plus 1.71%) of
leucocytes from mock-infected Ifnarko
-SLAMGe mice and infected Ifnarko
-
SLAMGe mice respectively expressed hSLAM (Figs. 4B and C, right
quadrants). Importantly, about one in 8 SLAM-expressing leukocytes were
infected (8.3 ± 2.7; average ± standard deviation) (Fig. 4C, compare
numbers in top and bottom right panels), whereas one in 96 SLAM-
negative leucocytes were infected (94.5 ± 32.65, average ± standard
deviation) (Fig. 4C, compare numbers in top and bottom left panels). Thus,
these results confirm the relevance of infection of SLAM expressing
leukocytes in MV propagation.
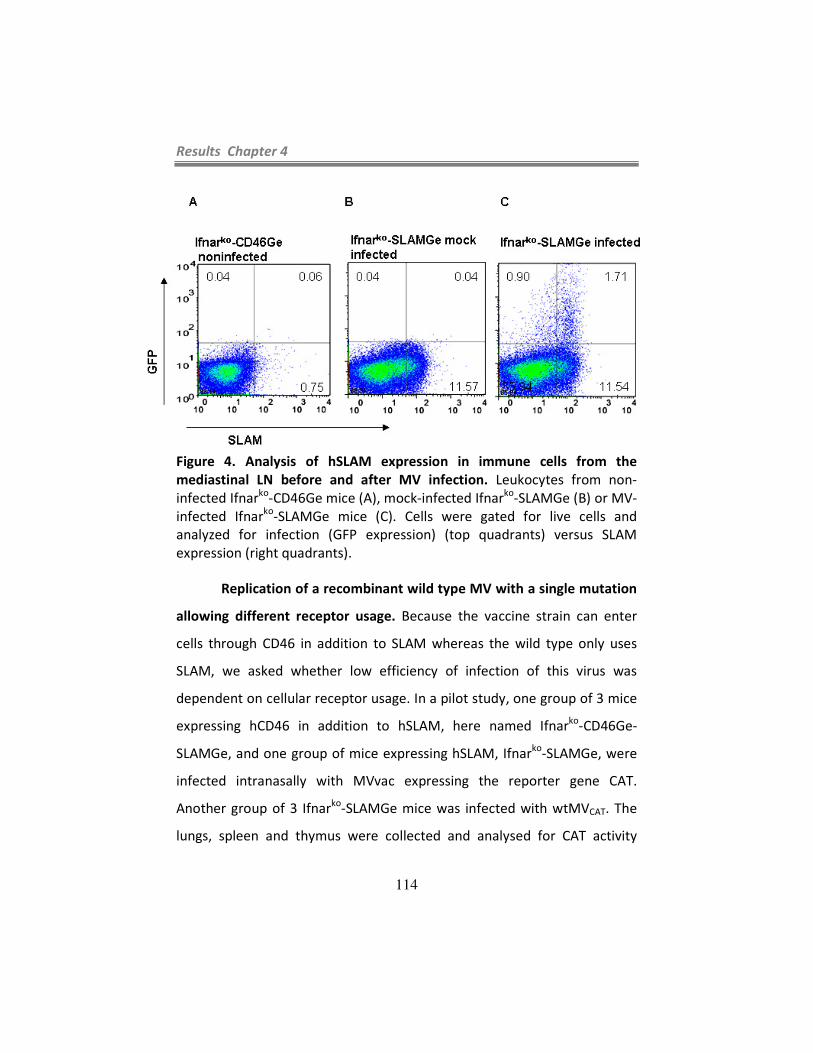
Results Chapter 4
114
Figure 4. Analysis of hSLAM expression in immune cells from the
mediastinal LN before and after MV infection. Leukocytes from non-
infected Ifnarko
-CD46Ge mice (A), mock-infected Ifnarko
-SLAMGe (B) or MV-
infected Ifnarko
-SLAMGe mice (C). Cells were gated for live cells and
analyzed for infection (GFP expression) (top quadrants) versus SLAM
expression (right quadrants).
Replication of a recombinant wild type MV with a single mutation
allowing different receptor usage. Because the vaccine strain can enter
cells through CD46 in addition to SLAM whereas the wild type only uses
SLAM, we asked whether low efficiency of infection of this virus was
dependent on cellular receptor usage. In a pilot study, one group of 3 mice
expressing hCD46 in addition to hSLAM, here named Ifnarko
-CD46Ge-
SLAMGe, and one group of mice expressing hSLAM, Ifnarko
-SLAMGe, were
infected intranasally with MVvac expressing the reporter gene CAT.
Another group of 3 Ifnarko
-SLAMGe mice was infected with wtMVCAT. The
lungs, spleen and thymus were collected and analysed for CAT activity

Results Chapter 4
115
three days after inoculation. Upon IN infection, the vaccine strain was
detected at low levels only in the thymus of one Ifnarko
-CD46Ge-SLAMGe
animal and in the lungs of one Ifnarko
-SLAMGe animal; in contrast, the wild
type virus was detected in the lungs of all Ifnarko
-SLAMGe mice, in the
spleen of two out of three Ifnarko
-SLAMGe animals and in the thymus of
one Ifnarko
-SLAMGe animal (data not shown). Thus, vaccine strain infection
is very inefficient and this inefficiency seems to be independent of receptor
usage.
Adaptation to growth in certain cell lines results in the selection of
certain mutations in MV genomes (136, 137). One frequent mutation is the
substitution of an asparagine to tyrosine at amino acid 481 (N481Y) in the
H protein. This mutation is absent in the wild type strains but common to
all vaccine strains and is linked to the ability of the virus to use an
alternative receptor for entry into cells, CD46 (76, 97). CD46, a
complement regulatory protein, is expressed in all nucleated cells and the
usage of this receptor in addition to SLAM may play a role in vaccine
attenuation. To confirm or deny whether this mutation and adaptation in
receptor specificity alters replication efficiency of MV, we characterized the
replication of a wild type MV with an asparagine to tyrosine substitution in
the hemagglutinin protein allowing binding to CD46.
We generated a wild type virus with an asparagine to tyrosine
substitution and a vaccine virus with a tyrosine to asparagine substitution
in the hemagglutinin protein, wtMVN481Y and MVvacY481N, respectively.
We first documented receptor usage by wtMV, wtMVN481Y, MVvac and

Results Chapter 4
116
MVvacY481N, using several cell lines expressing CD46 and/or SLAM. Figure
5A (top row) shows that all viruses (wtMV, wtMVN481Y, MVvac and
MVvacY481N) induce syncytia on B95a cells, expressing SLAM but not
CD46, whereas only wtMVN481Y and MVvac can fuse Vero cells, expressing
only CD46 (second row). However, MVvac induced syncytia in Vero cells
more efficiently than wtMVN481Y virus. Substitution of a tyrosine to
asparagine in MVvac abolishes CD46 usage and this virus no longer fuses
Vero cells (second row). All viruses can infect Vero cells expressing SLAM
(third row). Thus, the N481Y mutation allows the wild type MV to infect
cells expressing CD46 without compromising its ability to use SLAM
receptor.
wtMV and wtMVN481Y grew to similarly high titers in vitro in
Vero/hSLAM cells (not shown). To analyse whether wtMVN481Y replicates
as efficiently as the wtMV in vivo, we infected Ifnarko
-SLAMGe mice IP with
wtMVN481Y or wtMV; three days after inoculation the viral replication was
measured in several lymph nodes (mandibular, mediastinal, mesenteric,
and inguinal LN) and spleen. Fig. 5 B and C graphs, shows that wtMV and
wtMVN481Y moderately infected the mandibular, mesenteric and inguinal
LNs whereas the mediastinal LN and the spleen were infected with higher
efficiency. The wtMV replicated slightly more efficiently in the mediastinal
LN than the wtMVN481Y whereas the wtMVN481Y infected in average 10
times more the spleen compared to wtMV. But overall, both viruses
replicated efficiently in Ifnarko
-SLAMGe mice.
Results Chapter 4
117
Next, we sought to analyse whether the ability to infect cells
through CD46 in addition to SLAM would affect viral replication. Towards
this, we inoculated a mouse strain expressing hCD46 in addition to hSLAM,
and bred in an interferon type I deficient background, named Ifnarko
-
CD46Ge-SLAMGe mice. Infection of Ifnarko
-SLAMGe mice served as a
control. A group of 6 Ifnarko
-CD46Ge-SLAMGe mice and a group of 6 Ifnarko
-
SLAMGe mice were infected with wtMVN481Y and several lymphatic
organs (lymph nodes and spleen) were collected and the viral load was
assessed 3 days after infection. As shown in Figs. 6A and B, wtMVN481Y
replicated equally well in Ifnarko
-SLAMGe mice and in Ifnarko
-CD46Ge-
SLAMGe mice. Thus, N481Y mutation and CD46 usage does not seem to
affect replication of the wild type strain of MV, at least in this animal
model.
A

Results Chapter 4
118
B C
Ifnar
ko-SLAMGe mice
Figure 5. Characterization of a recombinant MV containing an H protein
with an asparagine to tyrosine substitution at position 481 (N481Y
mutation), wtMVN481Y. (A) Differential receptor usage by MV carrying a
mutation in the hemagglutinin protein. Different cell types expressing
different receptors (indicated at the left of each row) were infected by
several viruses (indicated at the top of each column) at an MOI of 0.1.
Syncytia formation was photographed 24 to 48 hours later under phase-
contrast microscopy.
(B) Replication of wtMV and (C) wtMVN481Y in several lymphatic organs of
Ifnarko
-SLAMGe mice. Titers were measured 3 days after IP inoculation.
Each dot represents a single mouse. Group average is represented by a
horizontal bar and detection limit by a dotted line. Man: mandibular LN;
Med: mediastinal LN; Mes: mesenteric LN; Ing: inguinal LN; Spl: spleen.
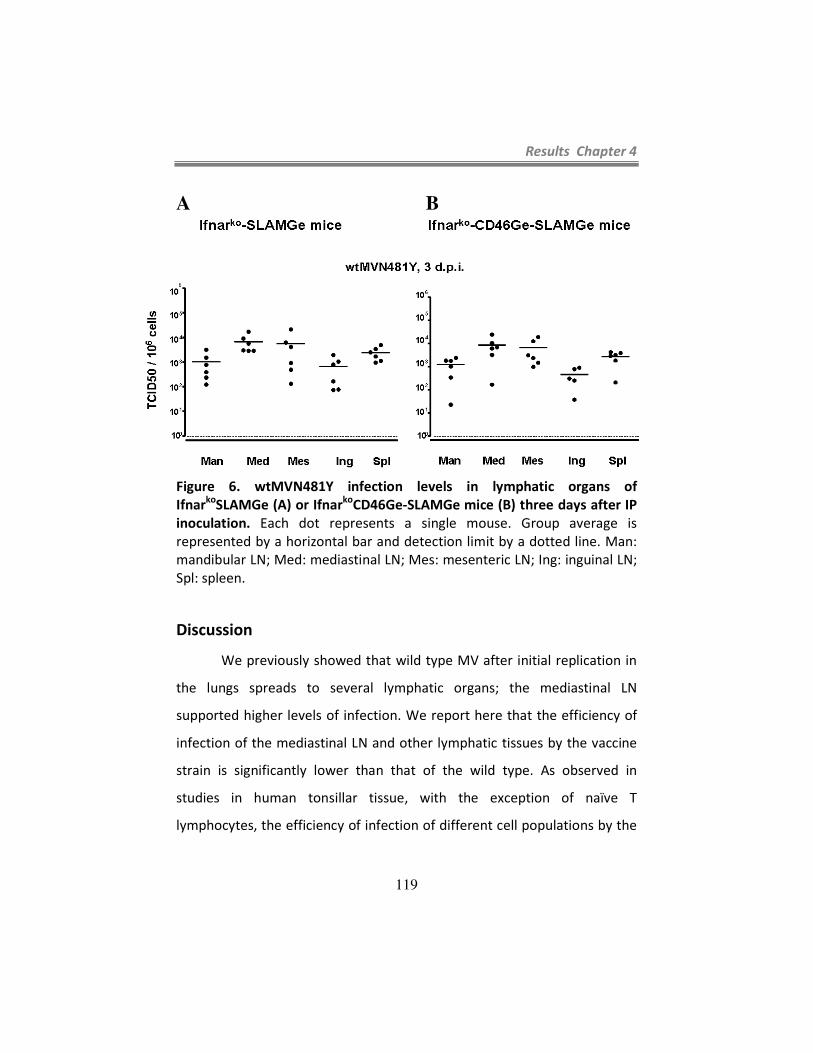
Results Chapter 4
119
A B
Figure 6. wtMVN481Y infection levels in lymphatic organs of
Ifnarko
SLAMGe (A) or Ifnarko
CD46Ge-SLAMGe mice (B) three days after IP
inoculation. Each dot represents a single mouse. Group average is
represented by a horizontal bar and detection limit by a dotted line. Man:
mandibular LN; Med: mediastinal LN; Mes: mesenteric LN; Ing: inguinal LN;
Spl: spleen.
Discussion
We previously showed that wild type MV after initial replication in
the lungs spreads to several lymphatic organs; the mediastinal LN
supported higher levels of infection. We report here that the efficiency of
infection of the mediastinal LN and other lymphatic tissues by the vaccine
strain is significantly lower than that of the wild type. As observed in
studies in human tonsillar tissue, with the exception of naïve T
lymphocytes, the efficiency of infection of different cell populations by the

Results Chapter 4
120
vaccine strain was significantly lower than that of the wild type (30). We
also show that wild type infection of SCS macrophages is followed by
extensive replication in SLAM expressing leukocytes. In contrast, after an
initial interaction with SCS macrophages the vaccine strain of MV does not
propagates as efficiently as the wild type. Thus, initial encounter with
macrophages in the secondary lymphatic organs might be fatal for the
vaccine strain; whereas the wild type overcomes this barrier and
disseminates to lymphocytes.
Subcapsular sinus macrophages are resident macrophages lining
the sinuses of the secondary lymphoid organs involved in filtering
pathogens out of the lymph or blood. These macrophages are poorly
endocytic and have low degradative capacity which enables them to
present intact antigen on their cell surface (84). In fact, electron
microscopy studies of captured VSV reported virions on the cell surface and
also in vacuoles of SCS macrophages (84, 141). It has been reported that B
cells, located immediately beneath the subcapsular sinus in the secondary
lymphoid organs, are able to capture antigens from SCS macrophages (21,
84, 141). Analogously, SCS macrophages infected with MV might present
infectious viral particles to B lymphocytes; a process that MV may exploit
to secure efficient infection and spread. Moreover, following capture of
MV SCS macrophages themselves might become infected and therefore
constitute an early viral reservoir. This associated to the low degradative
capacity of these cells may make them a safe sanctuary for the wild type
MV.

Results Chapter 4
121
We next asked whether the N481Y mutation selected during
adaptation to certain cultivated cells, would affect the tropism or
replication efficiency of MV. We showed that the mutated strain as well as
the parental strain replicated efficiently in the lymphatic organs of Ifnarko
-
SLAMGe mice, suggesting that the mutation did not affect replication
ability. We further showed that wtMVN481Y replicated as efficiently as
wtMV in Ifnarko
-SLAMGe and Ifnarko
-CD46Ge-SLAMGe mice. Thus,
adaptation to entry through CD46 does not strongly alter the tropism of
the virus in vivo. Additional mutations acquired by attenuated strains such
as S546G, N390I, N416D, T446S, T484N and E492G seem to increase the
viral binding affinity for CD46 (155, 162, 167, 176). Thus, it is conceivable
that a wild type virus with all these mutations may enter cells preferably
through CD46 instead of SLAM with a possible implication in viral tropism
and attenuation.
On the other hand, besides mutations in the H protein, cell culture
adaptation of MV has generated several amino acid substitutions in P, C, V
and M proteins of the virus (8, 180, 181, 184). These genetic changes might
affect viral replication, maturation or interaction with host proteins and
may have a cumulative effect on attenuation. Whatever the mechanism or
mechanisms underlying MV vaccine attenuation are, escape from
macrophage infection appears to be a key aspect of attenuation.


Main Conclusions and Future Perspectives
123
CHAPTER 5
Main conclusions and future perspectives


Main Conclusions and Future Perspectives
125
This thesis aimed at a better understanding of the interaction
between MV and the cellular receptor SLAM and the consequences for MV
spread and pathogenesis. Towards this we characterized the spread of MV
in a transgenic mouse expressing human SLAM, the cellular receptor for
wild type and attenuated strains of MV.
These studies provided valuable insights about MV initial target
cells upon respiratory infection as well as the development of infection in
the lymphatic system. Moreover we bred a transgenic mice expressing
hSLAM into an IFNAR deficient background and we used these mice to
show that alveolar macrophages and dendritic cells are the major target
cells immediately after intranasal infection. Infection in the airways was
short lived and early the virus spread to the lymphatic tissues. The
mediastinal LN was the LN where the virus reached the highest titre. In the
lymphatic tissues we documented an early involvement of MV with a
specific population of macrophages, SCS macrophages, localized in the
subcapsular sinus of the mediastinal LN and marginal sinus of spleen. Later
the virus spread to the follicular areas and extensively replicated in SLAM
expressing immune cells. In contrast, MV vaccine strain seems to be
impaired by this population of macrophages; after initial infection of SCS
macrophages the vaccine strain showed low levels of replication. Thus,
macrophages seem to play a dual role: they might serve as an early
reservoir of MV and seed infection to other lymphatic cells but they also

Main Conclusions and Future Perspectives
126
protect the host from uncontrolled viral dissemination as documented in
the case of the attenuated strains of MV.
Briefly the main conclusions are presented here as follows:
In Chapter 2
• We showed that SLAM transgenic mice expressed hSLAM
with human like tissue specificity; hSLAM expression in B
and T lymphocytes and in macrophages is dependent on the
activation status of the cells. Resting B and CD4 and CD8 T
and macrophages expressed hSLAM at low levels whereas
activation increased hSLAM expression;
• We documented that macrophages from Ifnarko
-SLAMGe
mice support MV infection ex vivo in a receptor-dependent
manner;
• We showed that Ifnarko
-SLAMGe mice are susceptible to MV
infection after IP or IN inoculation; several lymphatic tissues
and non lymphatic tissues supported replication of MV.
Thus, these mice are suitable to assess the relevance of receptor
usage for MV infection and should give insights into the early steps of MV

Main Conclusions and Future Perspectives
127
infection and into the mechanism of MV pathogenesis including
immunosuppression.
In Chapter 3
We took advantage of mice expressing SLAM with human like tissue
specificity, Ifnarko
-SLAMGe mice, to characterize the early steps of MV
infection. Moreover, we identified the initial target cells after respiratory
infection.
• We identified alveolar macrophages and dendritic cells as
the cells that sustain MV replication immediately after
respiratory infection; these cells expressed hSLAM;
• We documented that MV infection of alveolar macrophages
temporarily enhanced SLAM expression in these cells. Thus,
MV infects cells that easily up-regulate its receptor,
improving spread and infection efficacy;
• We showed that one day after initial replication in the
alveolar macrophages the virus disseminated to the
lymphatic system with early involvement of the mediastinal
LN. Furthermore, the mediastinal LN supported the highest
titre after IN or IP inoculation. In the mediastinal LN MV
infected immune cells such as B and T lymphocytes.

Main Conclusions and Future Perspectives
128
In Chapter 4
The mechanisms underlying MV vaccine attenuation are not well
understood. We compared the spread of an attenuated strain of MV with
that of a wild type in an animal model expressing hSLAM.
• We showed that MVvac replication was very inefficient in
the lymphatic tissues of Ifnarko
-SLAMGe mice. Infection of
mediastinal LN and spleen by the vaccine strain was 100
times less efficient as that of a wild type strain. Infection of
other lymph nodes was under detectable levels;
• We documented by confocal microscopy that at one day p.i.
both, the vaccine and the wild type viruses were retained in
the subcapsular sinus of mediastinal LN and in the marginal
sinus of the spleen; the virus colocalized with a specific
population of macrophages, the subcapsular sinus
macrophages. Subcapsular macrophages are typically
present in the SCS and MS of lymph nodes and spleen
respectively and play an important protective role by
sampling pathogens in the lymphatic organs;
• We showed that infection of SCS macrophages by the wild
type is preceded by massive infection of SLAM expressing

Main Conclusions and Future Perspectives
129
lymphoid cells in the lymphoid tissues whereas the
replication of the vaccine strain is disallowed after
interaction with the macrophages; 3 days after IP infection
the vaccine strain was almost cleared from the mediastinal
LN and spleen;
• We showed that adaptation to CD46 usage by the wild type
MV, in addition to SLAM, has little influence on MV virulence
and does not alter viral tropism.
Three characteristics of the mouse model used in these studies,
Ifnarko
-SLAMGe mouse, allowed the study of the early phases of MV
infection. First, infection of these animals resulted in extensive viral
replication due to an interferon defective response without affecting the
specificity of MV replication (127, 154). Second, the expression of the
lymphatic MV receptor, SLAM, in these animals allowed the
characterization of the early stages of MV infection and lymphatic
dissemination. And third, this mouse does not express the basolateral
epithelial cell receptor necessary for MV to leave the host (99) but allowed
characterization of the early stages of infection and spread of the virus in
the lymphatic tissues. This animal model gave insights in the cellular
targets supporting primary MV replication. Future studies are necessary to
understand whether the conclusion that the AM and DC are the early and

Main Conclusions and Future Perspectives
130
primary target cells can be extended to infection of the natural hosts.
Further studies into the mechanism(s) of how the virus crosses the
epithelial barrier and achieves dissemination throughout the body are
necessary.
It is interesting that replication of MV is detected in the
macrophages by two different routes of infection. AMs have been
implicated in the transport of particles and pathogens from the lungs to the
draining LN (66, 89), whereas SCS macrophages were shown to guide
captured virions across the SCS of the lymph nodes for efficient
presentation and activation of B cells (84, 85). MV may exploit these
characteristics to achieve dissemination throughout the host permissive
cells. Further research will be required to determine how the virus infected
macrophages achieves dissemination to other immune cells. The fact that
after interaction with the macrophages the wild type MV propagates
efficiently whereas the vaccine does not is puzzling. The low efficiency of
replication may disable the vaccine strain to counteract the innate immune
response and thus plays a role in viral attenuation.
Understanding the interactions of MV with macrophages and
dendritic cells may open new avenues for the development of more
efficient vaccines and novel therapeutic approaches.

References
131
References
1. 2009. Global measles mortality, 2000-2008. MMWR Morb Mortal
Wkly Rep 58:1321-1326.
2. Adams, E. M., M. C. Brown, M. Nunge, M. Krych, and J. P.
Atkinson. 1991. Contribution of the repeating domains of
membrane cofactor protein (CD46) of the complement system to
ligand binding and cofactor activity. J Immunol 147:3005-3011.
3. Alkhatib, G., and D. J. Briedis. 1986. The predicted primary
structure of the measles virus hemagglutinin. Virology 150:479-490.
4. Appel, M. J. 1969. Pathogenesis of canine distemper. Am J Vet Res
30:1167-1182.
5. Asanuma, H., A. H. Thompson, T. Iwasaki, Y. Sato, Y. Inaba, C.
Aizawa, T. Kurata, and S. Tamura. 1997. Isolation and
characterization of mouse nasal-associated lymphoid tissue. J
Immunol Methods 202:123-131.
6. Atabani, S. F., A. A. Byrnes, A. Jaye, I. M. Kidd, A. F.
Magnusen, H. Whittle, and C. L. Karp. 2001. Natural measles
causes prolonged suppression of interleukin-12 production. J Infect
Dis 184:1-9.
7. Aversa, G., C. C. Chang, J. M. Carballido, B. G. Cocks, and J. E.
de Vries. 1997. Engagement of the signaling lymphocytic activation
molecule (SLAM) on activated T cells results in IL-2-independent,
cyclosporin A-sensitive T cell proliferation and IFN-gamma
production. J Immunol 158:4036-4044.
8. Bankamp, B., G. Hodge, M. B. McChesney, W. J. Bellini, and P.
A. Rota. 2008. Genetic changes that affect the virulence of measles
virus in a rhesus macaque model. Virology 373:39-50.
9. Bankamp, B., J. Wilson, W. J. Bellini, and P. A. Rota. 2005.
Identification of naturally occurring amino acid variations that affect
the ability of the measles virus C protein to regulate genome
replication and transcription. Virology 336:120-129.
10. Bellini, W. J., G. Englund, S. Rozenblatt, H. Arnheiter, and C.
D. Richardson. 1985. Measles virus P gene codes for two proteins. J
Virol 53:908-919.

References
132
11. Bieback, K., E. Lien, I. M. Klagge, E. Avota, J. Schneider-
Schaulies, W. P. Duprex, H. Wagner, C. J. Kirschning, V. Ter Meulen, and S. Schneider-Schaulies. 2002. Hemagglutinin protein
of wild-type measles virus activates toll-like receptor 2 signaling. J
Virol 76:8729-8736.
12. Billeter, M. A., R. Cattaneo, P. Spielhofer, K. Kaelin, M. Huber,
A. Schmid, K. Baczko, and V. ter Meulen. 1994. Generation and
properties of measles virus mutations typically associated with
subacute sclerosing panencephalitis. Annals of the New York
Academy of Sciences 724:367-377.
13. Bleharski, J. R., K. R. Niazi, P. A. Sieling, G. Cheng, and R. L.
Modlin. 2001. Signaling lymphocytic activation molecule is
expressed on CD40 ligand-activated dendritic cells and directly
augments production of inflammatory cytokines. J Immunol
167:3174-3181.
14. Blumberg, B. M., M. Leppert, and D. Kolakofsky. 1981.
Interaction of VSV leader RNA and nucleocapsid protein may
control VSV genome replication. Cell 23:837-845.
15. Bolt, G., and I. R. Pedersen. 1998. The role of subtilisin-like
proprotein convertases for cleavage of the measles virus fusion
glycoprotein in different cell types. Virology 252:387-398.
16. Bryce, J., C. Boschi-Pinto, K. Shibuya, and R. E. Black. 2005.
WHO estimates of the causes of death in children. Lancet 365:1147-
1152.
17. Buchholz, C. J., D. Koller, P. Devaux, C. Mumenthaler, J.
Schneider-Schaulies, W. Braun, D. Gerlier, and R. Cattaneo. 1997. Mapping of the primary binding site of measles virus to its
receptor CD46. J Biol Chem 272:22072-22079.
18. Buchholz, C. J., C. Retzler, H. E. Homann, and W. J. Neubert.
1994. The carboxy-terminal domain of Sendai virus nucleocapsid
protein is involved in complex formation between phosphoprotein
and nucleocapsid-like particles. Virology 204:770-776.
19. Caignard, G., M. Guerbois, J. L. Labernardiere, Y. Jacob, L. M.
Jones, F. Wild, F. Tangy, and P. O. Vidalain. 2007. Measles virus
V protein blocks Jak1-mediated phosphorylation of STAT1 to
escape IFN-alpha/beta signaling. Virology 368:351-362.

References
133
20. Calain, P., and L. Roux. 1993. The rule of six, a basic feature for
efficient replication of Sendai virus defective interfering RNA. J
Virol 67:4822-4830.
21. Carrasco, Y. R., and F. D. Batista. 2007. B cells acquire
particulate antigen in a macrophage-rich area at the boundary
between the follicle and the subcapsular sinus of the lymph node.
Immunity 27:160-171.
22. Cathomen, T., B. Mrkic, D. Spehner, R. Drillien, R. Naef, J.
Pavlovic, A. Aguzzi, M. A. Billeter, and R. Cattaneo. 1998. A
matrix-less measles virus is infectious and elicits extensive cell
fusion: consequences for propagation in the brain. EMBO J
17:3899-3908.
23. Cathomen, T., H. Y. Naim, and R. Cattaneo. 1998. Measles
viruses with altered envelope protein cytoplasmic tails gain cell
fusion competence. J Virol 72:1224-1234.
24. Cattaneo, R. 2004. Four viruses, two bacteria and one receptor:
membrane cofactor protein (CD46) as a pathogen’s magnet. J Virol
78:4385-4388.
25. Cattaneo, R., K. Kaelin, K. Baczko, and M. A. Billeter. 1989.
Measles virus editing provides an additional cysteine-rich protein.
Cell 56:759-764.
26. Cattaneo, R., G. Rebmann, A. Schmid, K. Baczko, V. ter
Meulen, and M. A. Billeter. 1987. Altered transcription of a
defective measles virus genome derived from a diseased human
brain. EMBO J 6:681-688.
27. Cattaneo, R., A. Schmid, D. Eschle, K. Baczko, V. ter Meulen,
and M. A. Billeter. 1988. Biased hypermutation and other genetic
changes in defective measles viruses in human brain infections. Cell
55:255-265.
28. Clark, S. L., Jr. 1962. The reticulum of lymph nodes in mice
studied with the electron microscope. Am J Anat 110:217-257.
29. Cocks, B. G., C. C. Chang, J. M. Carballido, H. Yssel, J. E. de
Vries, and G. Aversa. 1995. A novel receptor involved in T-cell
activation. Nature 376:260-263.
30. Condack, C., J. C. Grivel, P. Devaux, L. Margolis, and R.
Cattaneo. 2007. Measles virus vaccine attenuation: suboptimal
infection of lymphatic tissue and tropism alteration. J Infect Dis
196:541-549.

References
134
31. Cherry, J. 2003. Measles Virus, p. 2283-2304. In C. J. Buck, G.
Demmler, and S. Kaplan (ed.), Textbook of Pediatric Infectious
Diseases. Elsevier Health Sciences.
32. Davidson, D., X. Shi, S. Zhang, H. Wang, M. Nemer, N. Ono, S.
Ohno, Y. Yanagi, and A. Veillette. 2004. Genetic evidence linking
SAP, the X-linked lymphoproliferative gene product, to Src-related
kinase FynT in T(H)2 cytokine regulation. Immunity 21:707-717.
33. de Swart, R. L. 2009. Measles studies in the macaque model. Curr
Top Microbiol Immunol 330:55-72.
34. de Swart, R. L., M. Ludlow, L. de Witte, Y. Yanagi, G. van
Amerongen, S. McQuaid, S. Yuksel, T. B. H. Geijtenbeek, W. P. Duprex, and A. D. M. E. Osterhaus. 2007. Predominant infection
of CD150+ lymphocytes and dendritic cells during measles virus
infection of macaques. PLoS Pathogens 3:e178.
35. de Witte, L., R. D. de Vries, M. van der Vlist, S. Yuksel, M.
Litjens, R. L. de Swart, and T. B. H. Geijtenbeek. 2008. DC-
SIGN and CD150 have distinct roles in transmission of measles
virus from dendritic cells to T-lymphocytes. PLoS Pathogens
4:e1000049.
36. Devaux, P., G. Hodge, M. B. McChesney, and R. Cattaneo. 2008.
Attenuation of V- or C-defective measles viruses: infection control
by the inflammatory and interferon responses of rhesus monkeys. J
Virol 82:5359-5367.
37. Devaux, P., B. Loveland, D. Christiansen, J. Milland, and D.
Gerlier. 1996. Interactions between the ectodomains of
haemagglutinin and CD46 as a primary step in measles virus entry. J
Gen Virol 77:1477-1481.
38. Devaux, P., V. von Messling, W. Songsungthong, C. Springfeld,
and R. Cattaneo. 2007. Tyrosine 110 in the measles virus
phosphoprotein is required to block STAT1 phosphorylation.
Virology 360:72-83.
39. Dorig, R. E., A. Marcil, A. Chopra, and C. D. Richardson. 1993.
The human CD46 molecule is a receptor for measles virus
(Edmonston strain). Cell 75:295-305.

References
135
40. Duprex, W. P., I. Duffy, S. McQuaid, L. Hamill, S. L. Cosby, M.
A. Billeter, J. Schneider-Schaulies, V. ter Meulen, and B. K. Rima. 1999. The H gene of rodent brain-adapted measles virus
confers neurovirulence to the Edmonston vaccine strain. J Virol
73:6916-6922.
41. Elliman, D., and H. Bedford. 2007. Achieving the goal for global
measles mortality. Lancet 369:165-166.
42. Enders, J. F., and T. C. Peebles. 1954. Propagation in tissue
cultures of cytopathogenic agents from patients with measles. Proc
Natl Acad Sci U S A 86:277-286.
43. Engel, P., M. J. Eck, and C. Terhorst. 2003. The SAP and SLAM
families in immune responses and X-linked lymphoproliferative
disease. Nat Rev Immunol 3:813-821.
44. Erlenhoefer, C., W. J. Wurzer, S. Loffler, S. Schneider-
Schaulies, V. ter Meulen, and J. Schneider-Schaulies. 2001.
CD150 (SLAM) is a receptor for measles virus but is not involved in
viral contact-mediated proliferation inhibition. J Virol 75:4499-
4505.
45. Erlenhofer, C., W. P. Duprex, B. K. Rima, V. ter Meulen, and J.
Schneider-Schaulies. 2002. Analysis of receptor (CD46, CD150)
usage by measles virus. J Gen Virol 83:1431-1436.
46. Esolen, L. M., S. W. Park, J. M. Hardwick, and D. E. Griffin.
1995. Apoptosis as a cause of death in measles virus-infected cells. J
Virol 69:3955-3958.
47. Esolen, L. M., B. J. Ward, T. R. Moench, and D. E. Griffin. 1993.
Infection of monocytes during measles. J Infect Dis 168:47-52.
48. Farr, A. G., Y. Cho, and P. P. De Bruyn. 1980. The structure of
the sinus wall of the lymph node relative to its endocytic properties
and transmural cell passage. Am J Anat 157:265-284.
49. Fontana, J. M., B. Bankamp, W. J. Bellini, and P. A. Rota. 2008.
Regulation of interferon signaling by the C and V proteins from
attenuated and wild-type strains of measles virus. Virology 374:71-
81.
50. Fujinami, R. S., and M. B. Oldstone. 1979. Antiviral antibody
reacting on the plasma membrane alters measles virus expression
inside the cell. Nature 279:529-530.

References
136
51. Gendelman, H. E., J. S. Wolinsky, R. T. Johnson, N. J.
Pressman, G. H. Pezeshkpour, and G. F. Boisset. 1984. Measles
encephalomyelitis: lack of evidence of viral invasion of the central
nervous system and quantitative study of the nature of
demyelination. Ann Neurol 15:353-360.
52. Gerlier, D., B. Loveland, G. Varior-Krishnan, B. Thorley, I. F.
McKenzie, and C. Rabourdin-Combe. 1994. Measles virus
receptor properties are shared by several CD46 isoforms differing in
extracellular regions and cytoplasmic tails. J Gen Virol 75:2163-
2171.
53. GeurtsvanKessel, C. H., M. A. Willart, L. S. van Rijt, F.
Muskens, M. Kool, C. Baas, K. Thielemans, C. Bennett, B. E.
Clausen, H. C. Hoogsteden, A. D. Osterhaus, G. F. Rimmelzwaan, and B. N. Lambrecht. 2008. Clearance of influenza
virus from the lung depends on migratory langerin+CD11b- but not
plasmacytoid dendritic cells. J Exp Med 205:1621-1634.
54. Goldman, M. B., T. A. O'Bryan, D. J. Buckthal, L. M. Tetor, and
J. N. Goldman. 1995. Suppression of measles virus expression by
noncytolytic antibody in an immortalized macrophage cell line. J
Virol 69:734-740.
55. Gombart, A. F., A. Hirano, and T. C. Wong. 1993.
Conformational maturation of measles virus nucleocapsid protein. J
Virol 67:4133-4141.
56. Grayson, M. H., and M. J. Holtzman. 2007. Emerging role of
dendritic cells in respiratory viral infection. J Mol Med 85:1057-
1068.
57. Grdzelishvili, V. Z., S. Smallwood, D. Tower, R. L. Hall, D. M.
Hunt, and S. A. Moyer. 2005. A single amino acid change in the L-
polymerase protein of vesicular stomatitis virus completely abolishes
viral mRNA cap methylation. J Virol 79:7327-7337.
58. Gremillion, D. H., and G. E. Crawford. 1981. Measles pneumonia
in young adults. An analysis of 106 cases. Am J Med 71:539-542.
59. Griffin, D. E. 2007. Measles Virus, p. 1551-1585. In D. M. Knipe
and P. M. Howley (ed.), Fields' Virology, vol. 1. Lippincott
Williams and Wilkins, Philadelphia.
60. Griffin, D. E., and B. J. Ward. 1993. Differential CD4 T cell
activation in measles. J Infect Dis 168:275-281.

References
137
61. Grivel, J. C., M. Garcia, W. J. Moss, and L. B. Margolis. 2005.
Inhibition of HIV-1 replication in human lymphoid tissues ex vivo
by measles virus. J Infect Dis 192:71-78.
62. Hahm, B., N. Arbour, D. Naniche, D. Homann, M. Manchester,
and M. B. Oldstone. 2003. Measles virus infects and suppresses
proliferation of T lymphocytes from transgenic mice bearing human
signaling lymphocytic activation molecule. J Virol 77:3505-3515.
63. Hahm, B., N. Arbour, and M. B. Oldstone. 2004. Measles virus
interacts with human SLAM receptor on dendritic cells to cause
immunosuppression. Virology 323:292-302.
64. Hahm, B., J. H. Cho, and M. B. Oldstone. 2007. Measles virus-
dendritic cell interaction via SLAM inhibits innate immunity:
selective signaling through TLR4 but not other TLRs mediates
suppression of IL-12 synthesis. Virology 358:251-257.
65. Hamaguchi, M., T. Yoshida, K. Nishikawa, H. Naruse, and Y.
Nagai. 1983. Transcriptive complex of Newcastle disease virus. I.
Both L and P proteins are required to constitute an active complex.
Virology 128:105-117.
66. Harmsen, A. G., B. A. Muggenburg, M. B. Snipes, and D. E.
Bice. 1985. The role of macrophages in particle translocation from
lungs to lymph nodes. Science 230:1277-1280.
67. Hashiguchi, T., and e. al. 2007. Crystal structure of measles virus
hemagglutinin provides insight into effective vaccines. Proc Natl
Acad Sci U.S.A. 104:19535-19540.
68. Henneman, P. L., D. M. Birnbaumer, and C. B. Cairns. 1995.
Measles pneumonitis. Ann Emerg Med 26:278-282.
69. Hercyk, N., S. M. Horikami, and S. A. Moyer. 1988. The vesicular
stomatitis virus L protein possesses the mRNA methyltransferase
activities. Virology 163:222-225.
70. Hirano, A., M. Ayata, A. H. Wang, and T. C. Wong. 1993.
Functional analysis of matrix proteins expressed from cloned genes
of measles virus variants that cause subacute sclerosing
panencephalitis reveals a common defect in nucleocapsid binding. J
Virol 67:1848-1853.
71. Hirano, A., A. H. Wang, A. F. Gombart, and T. C. Wong. 1992.
The matrix proteins of neurovirulent subacute sclerosing
panencephalitis virus and its acute measles virus progenitor are
functionally different. Proc Natl Acad Sci U S A 89:8745-8749.

References
138
72. Horikami, S. M., J. Curran, D. Kolakofsky, and S. A. Moyer.
1992. Complexes of Sendai virus NP-P and P-L proteins are required
for defective interfering particle genome replication in vitro. J Virol
66:4901-4908.
73. Horikami, S. M., S. Smallwood, B. Bankamp, and S. A. Moyer.
1994. An amino-proximal domain of the L protein binds to the P
protein in the measles virus RNA polymerase complex. Virology
205:540-545.
74. Howie, D., S. Okamoto, S. Rietdijk, K. Clarke, N. Wang, C.
Gullo, J. P. Bruggeman, S. Manning, A. J. Coyle, E. Greenfield, V. Kuchroo, and C. Terhorst. 2002. The role of SAP in murine
CD150 (SLAM)-mediated T-cell proliferation and interferon gamma
production. Blood 100:2899-2907.
75. Hsu, E. C., C. Iorio, F. Sarangi, A. A. Khine, and C. D.
Richardson. 2001. CDw150(SLAM) is a receptor for a
lymphotropic strain of measles virus and may account for the
immunosuppressive properties of this virus. Virology 279:9-21.
76. Hsu, E. C., F. Sarangi, C. Iorio, M. S. Sidhu, S. A. Udem, D. L.
Dillehay, W. Xu, P. A. Rota, W. J. Bellini, and C. D. Richardson. 1998. A single amino acid change in the hemagglutinin protein of
measles virus determines its ability to bind CD46 and reveals
another receptor on marmoset B cells. J Virol 72:2905-2916.
77. Hu, A., T. Cathomen, R. Cattaneo, and E. Norrby. 1995.
Influence of N-linked oligosaccharide chains on the processing, cell
surface expression and function of the measles virus fusion protein. J
Gen Virol 76:705-710.
78. Hu, A., R. Cattaneo, S. Schwartz, and E. Norrby. 1994. Role of
N-linked oligosaccharide chains in the processing and antigenicity of
measles virus haemagglutinin protein. J Gen Virol 75:1043-1052.
79. Huber, M., R. Cattaneo, P. Spielhofer, C. Orvell, E. Norrby, M.
Messerli, J. C. Perriard, and M. A. Billeter. 1991. Measles virus
phosphoprotein retains the nucleocapsid protein in the cytoplasm.
Virology 185:299-308.
80. Iwata, K., T. Seya, S. Ueda, H. Ariga, and S. Nagasawa. 1994.
Modulation of complement regulatory function and measles virus
receptor function by the serine-threonine-rich domains of membrane
cofactor protein (CD46). Biochem J 304:169-175.

References
139
81. Jaye, A., A. F. Magnusen, A. D. Sadiq, T. Corrah, and H. C.
Whittle. 1998. Ex vivo analysis of cytotoxic T lymphocytes to
measles antigens during infection and after vaccination in Gambian
children. J Clin Invest 102:1969-1977.
82. Jaye, A., A. F. Magnusen, and H. C. Whittle. 1998. Human
leukocyte antigen class I- and class II-restricted cytotoxic T
lymphocyte responses to measles antigens in immune adults. J Infect
Dis 177:1282-1289.
83. Johnson, R. T., D. E. Griffin, R. L. Hirsch, J. S. Wolinsky, S.
Roedenbeck, I. Lindo de Soriano, and A. Vaisberg. 1984. Measles
encephalomyelitis--clinical and immunologic studies. N Engl J Med
310:137-141.
84. Junt, T., E. A. Moseman, M. Iannacone, S. Massberg, P. A.
Lang, M. Boes, K. Fink, S. E. Henrickson, D. M. Shayakhmetov,
N. C. Di Paolo, N. van Rooijen, T. R. Mempel, S. P. Whelan, and
U. H. von Andrian. 2007. Subcapsular sinus macrophages in lymph
nodes clear lymph-borne viruses and present them to antiviral B
cells. Nature 450:110-114.
85. Junt, T., E. Scandella, and B. Ludewig. 2008. Form follows
function: lymphoid tissue microarchitecture in antimicrobial immune
defence. Nat Rev Immunol 8:764-775.
86. Kärber, G. 1931. Beitrag zur kollektiven Behandlung
pharmakologischer Reihenversuche. Arch. Exp. Pathol. Pharmakol.
162:480-483.
87. Karp, C. L., M. Wysocka, L. M. Wahl, J. M. Ahearn, P. J.
Cuomo, B. Sherry, G. Trinchieri, and D. E. Griffin. 1996.
Mechanism of suppression of cell-mediated immunity by measles
virus. Science 273:228-231.
88. Kingston, R. L., W. A. Baase, and L. S. Gay. 2004.
Characterization of nucleocapsid binding by the measles virus and
mumps virus phosphoproteins. J Virol 78:8630-8640.
89. Kirby, A. C., M. C. Coles, and P. M. Kaye. 2009. Alveolar
macrophages transport pathogens to lung draining lymph nodes. J
Immunol 183:1983-1989.
90. Koplik, H. 1962. The diagnosis of the invasion of measles from a
study of the exanthema as it appears on the buccal mucous
membrane. Arch Pediatr 79:162-165.

References
140
91. Kristensson, K., and E. Norrby. 1986. Persistence of RNA viruses
in the central nervous system. Annu Rev Microbiol 40:159-184.
92. Kruse, M., E. Meinl, G. Henning, C. Kuhnt, S. Berchtold, T.
Berger, G. Schuler, and A. Steinkasserer. 2001. Signaling
lymphocytic activation molecule is expressed on mature CD83+
dendritic cells and is up-regulated by IL-1 beta. J Immunol
167:1989-1995.
93. Kumagai, Y., O. Takeuchi, H. Kato, H. Kumar, K. Matsui, E.
Morii, K. Aozasa, T. Kawai, and S. Akira. 2007. Alveolar
macrophages are the primary interferon-alpha producer in
pulmonary infection with RNA viruses. Immunity 27:240-252.
94. Lamb, R. A. 1993. Paramyxovirus fusion: a hypothesis for changes.
Virology 197:1-11.
95. Lamb, R. A., and G. D. Parks. 2007. Paramyxoviridae: the viruses
and their replication, p. 1449-1496. In D. M. Knipe and P. M.
Howley (ed.), Fields Virology, Fifth ed. Lippincott, Williams and
Wilkins, Philadelphia.
96. Latour, S., G. Gish, C. D. Helgason, R. K. Humphries, T.
Pawson, and A. Veillette. 2001. Regulation of SLAM-mediated
signal transduction by SAP, the X-linked lymphoproliferative gene
product. Nat Immunol 2:681-690.
97. Lecouturier, V., J. Fayolle, M. Caballero, J. Carabana, M. L.
Celma, R. Fernandez-Munoz, T. F. Wild, and R. Buckland. 1996. Identification of two amino acids in the hemagglutinin
glycoprotein of measles virus (MV) that govern hemadsorption,
HeLa cell fusion, and CD46 downregulation: phenotypic markers
that differentiate vaccine and wild-type MV strains. J Virol 70:4200-
4204.
98. Legge, K. L., and T. J. Braciale. 2003. Accelerated migration of
respiratory dendritic cells to the regional lymph nodes is limited to
the early phase of pulmonary infection. Immunity 18:265-277.
99. Leonard, V. H. L., P. L. Sinn, G. Hodge, T. Miest, P. Devaux, N.
Oezguen, W. Braun, P. B. McCray, M. B. McChesney, and R. Cattaneo. 2008. Measles virus blind to its epithelial cell receptor
remains virulent in rhesus monkeys but cannot cross the airway
epithelium and is not shed. J Clin Invest 118:2448-2458.
100. Liston, P., and D. J. Briedis. 1994. Measles virus V protein binds
zinc. Virology 198:399-404.

References
141
101. Liston, P., C. DiFlumeri, and D. J. Briedis. 1995. Protein
interactions entered into by the measles virus P, V, and C proteins.
Virus Res 38:241-259.
102. Liszewski, M. K., T. W. Post, and J. P. Atkinson. 1991.
Membrane cofactor protein (MCP or CD46): newest member of the
regulators of complement activation gene cluster. Annu Rev
Immunol 9:431-455.
103. Lucas, J. 1790. An account of uncommon symptoms succeeding the
measles; with additional remarks on the infection of measles and
smallpox. London Med J 11:325-331.
104. Ma, C. S., K. E. Nichols, and S. G. Tangye. 2007. Regulation of
cellular and humoral immune responses by the SLAM and SAP
families of molecules. Annu Rev Immunol 25:337-379.
105. Maisner, A., and G. Herrler. 1995. Membrane cofactor protein
with different types of N-glycans can serve as measles virus
receptor. Virology 210:479-481.
106. Manchester, M., M. K. Liszewski, J. P. Atkinson, and M. B.
Oldstone. 1994. Multiple isoforms of CD46 (membrane cofactor
protein) serve as receptors for measles virus. Proc Natl Acad Sci U S
A 91:2161-2165.
107. Mavaddat, N., D. W. Mason, P. D. Atkinson, E. J. Evans, R. J.
Gilbert, D. I. Stuart, J. A. Fennelly, A. N. Barclay, S. J. Davis, and M. H. Brown. 2000. Signaling lymphocytic activation molecule
(CDw150) is homophilic but self-associates with very low affinity. J
Biol Chem 275:28100-28109.
108. McChesney, M. B., C. J. Miller, P. A. Rota, Y. D. Zhu, L.
Antipa, N. W. Lerche, R. Ahmed, and W. J. Bellini. 1997.
Experimental Measles. I. Pathogenesis in the normal and the
immunized host. Virology 233:74-84.
109. Minagawa, H., K. Tanaka, N. Ono, H. Tatsuo, and Y. Yanagi.
2001. Induction of the measles virus receptor SLAM (CD150) on
monocytes. J Gen Virol 82:2913-2917.
110. Moss, W. J. 2009. Measles control and the prospect of eradication.
Curr Top Microbiol Immunol 330:173-189.
111. Moss, W. J., J. J. Ryon, M. Monze, and D. E. Griffin. 2002.
Differential regulation of interleukin (IL)-4, IL-5, and IL-10 during
measles in Zambian children. J Infect Dis 186:879-887.

References
142
112. Mrkic, B., B. Odermatt, M. A. Klein, M. A. Billeter, J. Pavlovic,
and R. Cattaneo. 2000. Lymphatic dissemination and comparative
pathology of recombinant measles viruses in genetically modified
mice. J Virol 74:1364-1372.
113. Mrkic, B., J. Pavlovic, T. Rulicke, P. Volpe, C. J. Buchholz, D.
Hourcade, J. P. Atkinson, A. Aguzzi, and R. Cattaneo. 1998.
Measles virus spread and pathogenesis in genetically modified mice.
J Virol 72:7420-7427.
114. Müller, U., U. Steinhoff, L. F. Reis, S. Hemmi, J. Pavlovic, R. M.
Zinkernagel, and M. Aguet. 1994. Functional role of type I and
type II interferons in antiviral defense. Science 264:1918-1921.
115. Mustafa, M. M., S. D. Weitman, N. J. Winick, W. J. Bellini, C. F.
Timmons, and J. D. Siegel. 1993. Subacute measles encephalitis in
the young immunocompromised host: report of two cases diagnosed
by polymerase chain reaction and treated with ribavirin and review
of the literature. Clin Infect Dis 16:654-660.
116. Naim, H. Y., E. Ehler, and M. A. Billeter. 2000. Measles virus
matrix protein specifies apical virus release and glycoprotein sorting
in epithelial cells. EMBO J 19:3576-3585.
117. Nakatsu, Y., M. Takeda, S. Ohno, Y. Shirogane, M. Iwasaki, and
Y. Yanagi. 2008. Measles virus circumvents the host interferon
response by different actions of the C and V proteins. J Virol
82:8296-8306.
118. Nanan, R., C. Carstens, and H. W. Kreth. 1995. Demonstration of
virus-specific CD8+ memory T cells in measles-seropositive
individuals by in vitro peptide stimulation. Clin Exp Immunol
102:40-45.
119. Naniche, D., G. Varior-Krishnan, F. Cervoni, T. F. Wild, B.
Rossi, C. Rabourdin-Combe, and D. Gerlier. 1993. Human
membrane cofactor protein (CD46) acts as a cellular receptor for
measles virus. J Virol 67:6025-6032.
120. Navaratnarajah, C. K., V. H. Leonard, and R. Cattaneo. 2009.
Measles virus glycoprotein complex assembly, receptor attachment,
and cell entry. Curr Top Microbiol Immunol 329:59-76.

References
143
121. Navaratnarajah, C. K., S. Vongpunsawad, N. Oezguen, T.
Stehle, W. Braun, T. Hashiguchi, K. Maenaka, Y. Yanagi, and R. Cattaneo. 2008. Dynamic interaction of the measles virus
hemagglutinin with its receptor signaling lymphocytic activation
molecule (SLAM, CD150). J Biol Chem 283:11763-11771.
122. Norrby, E., and K. Kristensson. 1997. Measles virus in the brain.
Brain Res Bull 44:213-220.
123. Caignard, G., M. Bourai, Y. Jacob, F. Tangy, and P. O. Vidalain.
2009. Inhibition of IFN-alpha/beta signaling by two discrete peptides
within measles virus V protein that specifically bind STAT1 and
STAT2. Virology 383:112-120.
124. Ogino, T., M. Kobayashi, M. Iwama, and K. Mizumoto. 2005.
Sendai virus RNA-dependent RNA polymerase L protein catalyzes
cap methylation of virus-specific mRNA. J Biol Chem 280:4429-
4435.
125. Ohgimoto, S., K. Ohgimoto, S. Niewiesk, I. M. Klagge, J.
Pfeuffer, I. C. Johnston, J. Schneider-Schaulies, A. Weidmann, V. ter Meulen, and S. Schneider-Schaulies. 2001. The
haemagglutinin protein is an important determinant of measles virus
tropism for dendritic cells in vitro. J Gen Virol 82:1835-1844.
126. Ohno, S., S. Fumio, N. Ono, and Y. Yanagi. 2003. Histidine at
position 61 and its adjacent amino acid residues are critical for the
ability of SLAM (CD150) to act as a cellular receptor for measles
virus. J Gen Virol 84:2381-2388.
127. Ohno, S., N. Ono, F. Seki, M. Takeda, S. Kura, T. Tsuzuki, and
Y. Yanagi. 2007. Measles virus infection of SLAM (CD150)
knockin mice reproduces tropism and immunosuppression in human
infection. J Virol 81:1650-1659.
128. Ohno, S., N. Ono, M. Takeda, K. Takeuchi, and Y. Yanagi. 2004.
Dissection of measles virus V protein in relation to its ability to
block alpha/beta interferon signal transduction. J Gen Virol 85:2991-
2999.
129. Okada, H., F. Kobune, T. A. Sato, T. Kohama, Y. Takeuchi, T.
Abe, N. Takayama, T. Tsuchiya, and M. Tashiro. 2000. Extensive
lymphopenia due to apoptosis of uninfected lymphocytes in acute
measles patients. Arch Virol 145:905-920.

References
144
130. Okada, H., T. A. Sato, A. Katayama, K. Higuchi, K. Shichijo, T.
Tsuchiya, N. Takayama, Y. Takeuchi, T. Abe, N. Okabe, and M. Tashiro. 2001. Comparative analysis of host responses related to
immunosuppression between measles patients and vaccine recipients
with live attenuated measles vaccines. Arch Virol 146:859-874.
131. Okamura, A., O. Itakura, M. Yoshioka, M. Kubota, H. Kikuta,
and K. Kobayashi. 2001. Unusual presentation of measles giant cell
pneumonia in a patient with acquired immunodeficiency syndrome.
Clin Infect Dis 32:E57-58.
132. Ono, N., H. Tatsuo, Y. Hidaka, T. Aoki, H. Minagawa, and Y.
Yanagi. 2001. Measles viruses on throat swabs from measles
patients use signaling lymphocytic activation molecule (CDw150)
but not CD46 as a cellular receptor. J Virol 75:4399-4401.
133. Ono, N., H. Tatsuo, K. Tanaka, H. Minagawa, and Y. Yanagi.
2001. V domain of human SLAM (CDw150) is essential for its
function as a measles virus receptor. J Virol 75:1594-1600.
134. Palosaari, H., J. P. Parisien, J. J. Rodriguez, C. M. Ulane, and C.
M. Horvath. 2003. STAT protein interference and suppression of
cytokine signal transduction by measles virus V protein. J Virol
77:7635-7644.
135. Panum, P. 1938. Observations made during the epidemic of measles
on the Faroe Islands in the year of 1846. Medical Classics 3:829-
886.
136. Parks, C. L., R. A. Lerch, P. Walpita, H. P. Wang, M. S. Sidhu,
and S. A. Udem. 2001. Analysis of the noncoding regions of
measles virus strains in the Edmonston vaccine lineage. J Virol
75:921-933.
137. Parks, C. L., R. A. Lerch, P. Walpita, H. P. Wang, M. S. Sidhu,
and S. A. Udem. 2001. Comparison of predicted amino acid
sequences of measles virus strains in the Edmonston vaccine lineage.
J Virol 75:910-920.
138. Permar, S. R., S. A. Klumpp, K. G. Mansfield, A. A. Carville, D.
A. Gorgone, M. A. Lifton, J. E. Schmitz, K. A. Reimann, F. P. Polack, D. E. Griffin, and N. L. Letvin. 2004. Limited contribution
of humoral immunity to the clearance of measles viremia in rhesus
monkeys. J Infect Dis 190:998-1005.

References
145
139. Permar, S. R., S. A. Klumpp, K. G. Mansfield, W. K. Kim, D. A.
Gorgone, M. A. Lifton, K. C. Williams, J. E. Schmitz, K. A.
Reimann, M. K. Axthelm, F. P. Polack, D. E. Griffin, and N. L. Letvin. 2003. Role of CD8(+) lymphocytes in control and clearance
of measles virus infection of rhesus monkeys. J Virol 77:4396-4400.
140. Permar, S. R., W. J. Moss, J. J. Ryon, M. Monze, F. Cutts, T. C.
Quinn, and D. E. Griffin. 2001. Prolonged measles virus shedding
in human immunodeficiency virus-infected children, detected by
reverse transcriptase-polymerase chain reaction. J Infect Dis
183:532-538.
141. Phan, T. G., I. Grigorova, T. Okada, and J. G. Cyster. 2007.
Subcapsular encounter and complement-dependent transport of
immune complexes by lymph node B cells. Nat Immunol 8:992-
1000.
142. Plemper, R., A. Hammond, and R. Cattaneo. 2000.
Characterization of a region of measles virus hemagglutinin
sufficient for its dimerization. J Virol 74:6485-6493.
143. Poch, O., B. M. Blumberg, L. Bougueleret, and N. Tordo. 1990.
Sequence comparison of five polymerases (L proteins) of
unsegmented negative-strand RNA viruses: theoretical assignment
of functional domains. J Gen Virol 71:1153-1162.
144. Polacino, P. S., L. M. Pinchuk, S. P. Sidorenko, and E. A. Clark.
1996. Immunodeficiency virus cDNA synthesis in resting T
lymphocytes is regulated by T cell activation signals and dendritic
cells. J Med Primatol 25:201-209.
145. Polack, F. P., S. H. Lee, S. Permar, E. Manyara, H. G. Nousari,
Y. Jeng, F. Mustafa, A. Valsamakis, R. J. Adams, H. L. Robinson, and D. E. Griffin. 2000. Successful DNA immunization
against measles: Neutralizing antibody against either the
hemagglutinin or fusion glycoprotein protects rhesus macaques
without evidence of atypical measles. Nat Med 6:776-781.
146. Punnonen, J., B. G. Cocks, J. M. Carballido, B. Bennett, D.
Peterson, G. Aversa, and J. E. de Vries. 1997. Soluble and
membrane-bound forms of signaling lymphocytic activation
molecule (SLAM) induce proliferation and Ig synthesis by activated
human B lymphocytes. J Exp Med 185:993-1004.

References
146
147. Radecke, F., P. Spielhofer, H. Schneider, K. Kaelin, M. Huber,
C. Dotsch, G. Christiansen, and M. A. Billeter. 1995. Rescue of
measles viruses from cloned DNA. EMBO J 14:5773-5784.
148. Rager, M., S. Vongpunsawad, W. P. Duprex, and R. Cattaneo.
2002. Polyploid measles virus with hexameric genome length.
EMBO J 21:2364-2372.
149. Ramachandran, A., J. P. Parisien, and C. M. Horvath. 2008.
STAT2 is a primary target for measles virus V protein-medicated
IFNα/β signaling inhibition. J Virol 82:8330-8338.
150. Reutter, G. L., C. Cortese-Grogan, J. Wilson, and S. A. Moyer.
2001. Mutations in the measles virus C protein that up regulate viral
RNA synthesis. Virology 285:100-109.
151. Richardson, C., D. Hull, P. Greer, K. Hasel, A. Berkovich, G.
Englund, W. Bellini, B. Rima, and R. Lazzarini. 1986. The
nucleotide sequence of the mRNA encoding the fusion protein of
measles virus (Edmonston strain): a comparison of fusion proteins
from several different paramyxoviruses. Virology 155:508-523.
152. Rima, B. K., and W. P. Duprex. 2006. Morbilliviruses and human
disease. J Pathol 208:199-214.
153. Romero, X., D. Benitez, S. March, R. Vilella, M. Miralpeix, and
P. Engel. 2004. Differential expression of SAP and EAT-2-binding
leukocyte cell-surface molecules CD84, CD150 (SLAM), CD229
(Ly9) and CD244 (2B4). Tissue Antigens 64:132-144.
154. Roscic-Mrkic, B., R. A. Schwendener, B. Odermatt, A. Zuniga,
J. Pavlovic, M. A. Billeter, and R. Cattaneo. 2001. Roles of
macrophages in measles virus infection of genetically modified
mice. J Virol 75:3343-3351.
155. Rota, J. S., Z. D. Wang, P. A. Rota, and W. J. Bellini. 1994.
Comparison of sequences of the H, F, and N coding genes of
measles virus vaccine strains. Virus Res 31:317-330.
156. Ryon, J. J., W. J. Moss, M. Monze, and D. E. Griffin. 2002.
Functional and phenotypic changes in circulating lymphocytes from
hospitalized Zambian children with measles. Clin Diagn Lab
Immunol 9:994-1003.
157. Sakaguchi, M., Y. Yoshikawa, K. Yamanouchi, T. Sata, K.
Nagashima, and K. Takeda. 1986. Growth of measles virus in
epithelial and lymphoid tissues of cynomolgus monkeys. Microbiol
Immunol 30:1067-1073.

References
147
158. Sauer, K. A., P. Scholtes, R. Karwot, and S. Finotto. 2006.
Isolation of CD4+ T cells from murine lungs: a method to analyze
ongoing immune responses in the lung. Nat Protoc 1:2870-2875.
159. Sayos, J., C. Wu, M. Morra, N. Wang, X. Zhang, D. Allen, S. van
Schaik, L. Notarangelo, R. Geha, M. G. Roncarolo, H. Oettgen,
J. E. De Vries, G. Aversa, and C. Terhorst. 1998. The X-linked
lymphoproliferative-disease gene product SAP regulates signals
induced through the co-receptor SLAM. Nature 395:462-469.
160. Schlender, J., V. Hornung, S. Finke, M. Gunthner-Biller, S.
Marozin, K. Brzozka, S. Moghim, S. Endres, G. Hartmann, and K. K. Conzelmann. 2005. Inhibition of toll-like receptor 7- and 9-
mediated alpha/beta interferon production in human plasmacytoid
dendritic cells by respiratory syncytial virus and measles virus. J
Virol 79:5507-5515.
161. Schneider-Schaulies, S., and J. Schneider-Schaulies. 2009.
Measles virus-induced immunosuppression. Curr Top Microbiol
Immunol 330:243-269.
162. Schneider, U., V. von Messling, P. Devaux, and R. Cattaneo.
2002. Efficiency of measles virus entry and dissemination through
different receptors. J Virol 76:7460-7467.
163. Sellin, C. I., N. Davoust, V. Guillaume, D. Baas, M. F. Belin, R.
Buckland, T. F. Wild, and B. Horvat. 2006. High pathogenicity of
wild-type measles virus infection in CD150 (SLAM) transgenic
mice. J Virol 80:6420-6429.
164. Sellin, C. I., and B. Horvat. 2009. Current animal models:
transgenic animal models for the study of measles pathogenesis.
Curr Top Microbiol Immunol 330:111-127.
165. Seya T, B. L., Bora NS, Kumar V, Cui W, Atkinson JP. 1988.
Distribution of membrane cofactor protein of complement on human
peripheral blood cells. An altered form is found on granulocytes. Eur
J Immunol 18:1289-1294.
166. Shaffer, J. A., W. J. Bellini, and P. A. Rota. 2003. The C protein
of measles virus inhibits the type I interferon response. Virology
315:389-397.

References
148
167. Shibahara, K., H. Hotta, Y. Katayama, and M. Homma. 1994.
Increased binding activity of measles virus to monkey red blood
cells after long-term passage in Vero cell cultures. J Gen Virol
75:3511-3516.
168. Shingai, M., N. Inoue, T. Okuno, M. Okabe, T. Akazawa, Y.
Miyamoto, M. Ayata, K. Honda, M. Kurita-Taniguchi, M.
Matsumoto, H. Ogura, T. Taniguchi, and T. Seya. 2005. Wild-
type measles virus infection in human CD46/CD150-transgenic
mice: CD11c-positive dendritic cells establish systemic viral
infection. J Immunol 175:3252-3261.
169. Sidhu, M. S., J. P. Menonna, S. D. Cook, P. C. Dowling, and S.
A. Udem. 1993. Canine distemper virus L gene: sequence and
comparison with related viruses. Virology 193:50-65.
170. Sidorenko, S. P., and E. A. Clark. 1993. Characterization of a cell
surface glycoprotein IPO-3, expressed on activated human B and T
lymphocytes. J Immunol 151:4614-4624.
171. Sidorenko, S. P., and E. A. Clark. 2003. The dual-function CD150
receptor subfamily: the viral attraction. Nat Immunol 4:19-24.
172. Sleeman, K., and M. D. Baron. 2005. The polymerase (L) protein
of rinderpest virus interacts with the host cell protein striatin.
Virology 332:225-234.
173. Spehner, D., R. Drillien, and P. M. Howley. 1997. The assembly
of the measles virus nucleoprotein into nucleocapsid-like particles is
modulated by the phosphoprotein. Virology 232:260-268.
174. Spielhofer, P., T. Bachi, T. Fehr, G. Christiansen, R. Cattaneo,
K. Kaelin, M. A. Billeter, and H. Y. Naim. 1998. Chimeric
measles viruses with a foreign envelope. J Virol 72:2150-2159.
175. Sun, X., J. B. Burns, J. M. Howell, and R. S. Fujinami. 1998.
Suppression of antigen-specific T cell proliferation by measles virus
infection: role of a soluble factor in suppression. Virology 246:24-
33.
176. Tahara, M., M. Takeda, F. Seki, T. Hashiguchi, and Y. Yanagi.
2007. Multiple amino acid substitutions in hemagglutinin are
necessary for wild-type measles virus to acquire the ability to use
receptor CD46 efficiently. J Virol 81:2564-2572.

References
149
177. Tahara, M., M. Takeda, Y. Shirogane, T. Hashiguchi, S. Ohno,
and Y. Tanagi. 2008. Mealses virus infects both polarized epithelial
and immune cells by using distrinctive receptor-binding sites on its
hemagglutinin. J Virol 82:4630-4637.
178. Tahara, M., M. Takeda, and Y. Yanagi. 2007. Altered interaction
of the matrix protein with the cytoplasmic tail of hemagglutinin
modulates measles virus growth by affecting virus assembly and
cell-cell fusion. J Virol 81:6827-6836.
179. Tahara, M., M. Takeda, and Y. Yanagi. 2005. Contributions of
matrix and large protein genes of the measles virus edmonston strain
to growth in cultured cells as revealed by recombinant viruses. J
Virol 79:15218-15225.
180. Takeda, M., A. Kato, F. Kobune, H. Sakata, Y. Li, T. Shioda, Y.
Sakai, M. Asakawa, and Y. Nagai. 1998. Measles virus attenuation
associated with transcriptional impediment and a few amino acid
changes in the polymerase and accessory proteins. J Virol 72:8690-
8696.
181. Takeda, M., T. Sakaguchi, Y. Li, F. Kobune, A. Kato, and Y.
Nagai. 1999. The genome nucleotide sequence of a contemporary
wild strain of measles virus and its comparison with the classical
Edmonston strain genome. Virology 256:340-350.
182. Takeda, M., K. Takeuchi, N. Miyajima, F. Kobune, Y. Ami, N.
Nagata, Y. Suzaki, Y. Nagai, and M. Tashiro. 2000. Recovery of
pathogenic measles virus from cloned cDNA. J Virol 74:6643-6647.
183. Takeuchi, K., S. I. Kadota, M. Takeda, N. Miyajima, and K.
Nagata. 2003. Measles virus V protein blocks interferon (IFN)-
alpha/beta but not IFN-gamma signaling by inhibiting STAT1 and
STAT2 phosphorylation. FEBS Lett 545:177-182.
184. Takeuchi, K., N. Miyajima, F. Kobune, and M. Tashiro. 2000.
Comparative nucleotide sequence analyses of the entire genomes of
B95a cell-isolated and vero cell-isolated measles viruses from the
same patient. Virus Genes 20:253-257.
185. Takeuchi, K., M. Takeda, N. Miyajima, Y. Ami, N. Nagata, Y.
Suzaki, J. Shahnewaz, S. Kadota, and K. Nagata. 2005. Stringent
requirement for the C protein of wild-type measles virus for growth
both in vitro and in macaques. J Virol 79:7838-7844.

References
150
186. Tatsuo, H., N. Ono, K. Tanaka, and Y. Yanagi. 2000. SLAM
(CDw150) is a cellular receptor for measles virus. Nature 406:893-
897.
187. Tatsuo, H., N. Ono, and Y. Yanagi. 2001. Morbilliviruses use
signaling lymphocyte activation molecules (CD150) as cellular
receptors. J Virol 75:5842-5850.
188. Tober, C., M. Seufert, H. Schneider, M. A. Billeter, I. C.
Johnston, S. Niewiesk, V. ter Meulen, and S. Schneider-Schaulies. 1998. Expression of measles virus V protein is associated
with pathogenicity and control of viral RNA synthesis. J Virol
72:8124-8132.
189. Toth, A. M., P. Devaux, R. Cattaneo, and C. E. Samuel. 2009.
Protein kinase PKR mediates the apoptosis induction and growth
restriction phenotypes of C protein-deficient measles virus. J Virol
83:961-968.
190. van Binnendijk, R. S., M. C. Poelen, K. C. Kuijpers, A. D.
Osterhaus, and F. G. Uytdehaag. 1990. The predominance of
CD8+ T cells after infection with measles virus suggests a role for
CD8+ class I MHC-restricted cytotoxic T lymphocytes (CTL) in
recovery from measles. Clonal analyses of human CD8+ class I
MHC-restricted CTL. J Immunol 144:2394-2399.
191. van oud Alblas, A. B., and R. van Furth. 1979. Origin, kinetics,
and characterization of pulmonary macrophages in the normal steady
state. J. Exp.Med. 149:1504-1518.
192. Veillette, A. 2006. Immune regulation by SLAM family receptors
and SAP-related adaptors. Nat Rev Immunol 6:56-66.
193. von Messling, V., and R. Cattaneo. 2003. N-linked glycans with
similar location in the fusion protein head modulate paramyxovirus
fusion. J Virol 77:10202-10212.
194. von Messling, V., D. Milosevic, and R. Cattaneo. 2004. Tropism
illuminated: lymphocyte-based pathways blazed by lethal
morbillivirus through the host immune system. Proc Natl Acad Sci U
S A 101:14216-14221.
195. von Messling, V., C. Springfeld, P. Devaux, and R. Cattaneo.
2003. A ferret model of canine distemper virus virulence and
immunosuppression. J Virol 77:12579-12591.

References
151
196. von Messling, V., N. Svitek, and R. Cattaneo. 2006. Receptor
(SLAM, CD150) recognition and the V protein sustain swift
lymphocyte-based invasion of mucosal tissue and lymphatic organs
by a morbillivirus. J Virol 80:6084-6092.
197. von Pirquet, C. 1908. Das Verhalten der kutanen
Tuberkulinreaktion waehrend der Masern. Dtsch. Med. Wochenschr.
30:1297-1300.
198. Vongpunsawad, S., N. Oezgun, W. Braun, and R. Cattaneo.
2004. Selectively receptor-blind measles viruses: identification of
residues necessary for SLAM- or CD46-induced fusion and their
localization on a new hemagglutinin structural model. J Virol
78:302-313.
199. Wang, M., J. E. Libbey, I. Tsunoda, and R. S. Fujinami. 2003.
Modulation of immune system function by measles virus infection.
II. Infection of B cells leads to the production of a soluble factor that
arrests uninfected B cells in G0/G1. Viral Immunol 16:45-55.
200. Wang, N., A. Satoskar, W. Faubion, D. Howie, S. Okamoto, S.
Feske, C. Gullo, K. Clarke, M. R. Sosa, A. H. Sharpe, and C. Terhorst. 2004. The cell surface receptor SLAM controls T cell and
macrophage functions. J Exp Med 199:1255-1264.
201. Watanabe, M., A. Hirano, S. Stenglein, J. Nelson, G. Thomas,
and T. C. Wong. 1995. Engineered serine protease inhibitor
prevents furin-catalyzed activation of the fusion glycoprotein and
production of infectious measles virus. J Virol 69:3206-3210.
202. Welstead, G. G., C. Iorio, R. Draker, J. Bayani, J. Squire, S.
Vongpunsawad, R. Cattaneo, and C. D. Richardson. 2005.
Measles virus replication in lymphatic cells and organs of CD150
(SLAM) transgenic mice. Proc Natl Acad Sci U S A 102:16415-
16420.
203. Wesley, A. G., H. M. Coovadia, and P. Kiepiela. 1982. Further
predictive indices of clinical severity of measles. S Afr Med J
61:663-665.
204. Yanagi, Y., M. Takeda, and S. Ohno. 2006. Measles virus: cellular
receptors, tropism and pathogenesis. J Gen Virol 87:2767-2779.
205. Yin, H. S., X. Wen, R. G. Paterson, R. A. Lamb, and T. S.
Jardetzky. 2006. Structure of the parainfluenza virus 5 F protein in
its metastable, prefusion conformation. Nature 439:38-44.

References
152
206. Yokota, S., H. Saito, T. Kubota, N. Yokosawa, K. Amano, and N.
Fujii. 2003. Measles virus suppresses interferon-alpha signaling
pathway: suppression of Jak1 phosphorylation and association of
viral accessory proteins, C and V, with interferon-alpha receptor
complex. Virology 306:135-146.
