methodenentwicklung für die trennung verschiedener
TRANSCRIPT

Universität Kassel
Fachbereich Ökologische Agrarwissenschaften
Fachgebiet Agrartechnik
Prof. Dr. sc. agr. Oliver Hensel
Methodenentwicklung für die Trennung verschiedener Proteinhydrolysate in Peptidfraktionen per Crossflow Diafiltration & sensorische Bewertung der Fraktionen
Dissertation zur Erlangung des akademischen Grades
Doktor der Agrarwissenschaften (Dr. agr.)
Vorgelegt im Fachbereich Ökologische Agrarwissenschaften
der Universität Kassel
Von M. Sc. Markus Stefan Friedrich
aus Öhringen
Witzenhausen, 2019

Die vorliegende Arbeit wurde am 25.01.2019 vom Fachbereich für Ökologische
Agrarwissenschaften, Fachgebiet Agrartechnik der Universität Kassel als Dissertation zur
Erlangung des Grades eines Doktor der Agrarwissenschaften angenommen.
Tag der mündlichen Prüfung: 05.06.2019
Hauptberichter: Prof. Dr. Oliver Hensel
Mitberichter: Prof. Dr. Joachim J. Schmitt
Mündliche Prüfung: Prof. Dr. Oliver Hensel
Prof. Dr. Joachim J. Schmitt
Prof. Dr. Johannes Kahl
Prof. Dr. Elke Pawelzik

Vorwort
Mein besonderer Dank gilt Herrn Prof. Dr. Oliver Hensel für die wissenschaftliche Betreuung
der Arbeit, sowie Herrn Prof. Dr. Joachim J. Schmitt, der die Durchführung dieser Arbeit
ermöglichte.
Bei Frau Prof. Dr. Ingrid Seuß-Baum bedanke ich mich für die hilfreichen Diskussionen zur
Planung und Durchführung der Sensorik-Studie.
Weiter gilt mein Dank Herrn Ralf Schäfer für die organisatorische Unterstützung, sowie
moralischen Zuspruch. Zudem bedanke ich mich bei Frau Hannelore Borck und Herrn
Alexander Maxones für klärende Diskussionen bei der Einarbeitung in die SE-HPLC-Analytik.
Besonderer Dank gilt Herrn Daniel Koch, Herrn Dominik Hieß und Herrn Alexander Buchmüller
für hilfreiche wissenschaftliche Diskussionen, sowie Frau Giulia Kutzner und Frau Carine
Mollon für die Unterstützung bei der Vorbereitung der Sensorik-Termine.
Bei den Studienenden des Fachbereichs Lebensmitteltechnologie der Hochschule Fulda
bedanke ich mich für das rege Interesse und die zuverlässige Teilnahme als Panelisten im
Rahmen der Sensorik-Studie.
Abschließend bedanke ich mich bei meiner Familie, insbesondere meiner Frau Aziza
Schwenke, sowie meinen Eltern Wolfgang & Gerda Friedrich für die jahrelange Unterstützung,
motivierende Worte, Geduld und Zuversicht.
Ich widme diese Arbeit meiner verstorbenen Mutter,
Gerda Friedrich (25.02.1956 – † 21.01.2017)

I
Inhaltsverzeichnis
1. Einleitung, Zielsetzung & Hypothesenformulierung .............................. 1
1.1 Einleitung ..................................................................................................................... 1
1.2 Zielsetzung & Hypothesenformulierung ........................................................................ 2
2. Stand der Forschung ................................................................................ 5
2.1 Filtrations- und Fraktionierungstechnik ......................................................................... 5
2.1.1 Die Crossflow Filtrationstechnik ............................................................................. 6
2.1.2 Fouling und Konzentrationspolarisation ................................................................. 7
2.1.2.1 Die Konzentrationspolarisation.......................................................................... 7
2.1.2.2 Das Fouling ....................................................................................................... 9
2.1.3 Mathematische Charakterisierung von Filtrationsprozessen .................................10
2.1.3.1 Idealisierte Charakterisierung von Filtrationsprozessen - Das Hagen-Poiseulle-
Model ...............................................................................................................10
2.1.3.2 Charakterisierung der Konzentrationspolarisation – Das Gel-Polarisation-Modell
........................................................................................................................11
2.1.3.3 Charakterisierung druckgetriebener Filtrationsprozesse unter Berücksichtigung
prozessspezifischer Widerstände – Das Widerstands-Modell ..........................13
2.1.4 Einfluss von Prozess-Hydrodynamik und physikalisch-chemischen Eigenschaften
von Feed und Membran auf Fouling und Filtrationsprozess ..................................14
2.1.4.1 Fouling aufgrund der Prozess-Hydrodynamik ..................................................15
2.1.4.2 Fouling aufgrund der physikalisch-chemischen Eigenschaften von Feed und
Membran .........................................................................................................17
2.1.4.3 Weitere Verfahren zur Fouling-Kontrolle & Prozessoptimierung .......................23
2.1.5 Die Dialyse ...........................................................................................................26
2.2 Rohstoffcharakterisierung ............................................................................................28
2.2.1 Milchproteine ........................................................................................................28
2.2.1.1 Caseine ...........................................................................................................28
2.2.1.2 Molkenproteine ................................................................................................29
2.2.2 Algen & Algenproteine ..........................................................................................30
2.3 Hydrolyse von Proteinquellen ......................................................................................32
2.3.1 Die enzymatische Hydrolyse ................................................................................34
2.3.1.1 Hydrolyse von Algen & Algenproteinen ............................................................34
2.4 Analytik – Die Size-Exclusion HPLC (SE-HPLC) .........................................................35
2.5 Sensorik – Geschmackswahrnehmung & deren Modulation ........................................37
2.5.1 Geschmacksrezeptoren – Geschmackspapillen, -Knospen & -Sinneszellen .........38

II
2.5.2 Die fünf Grundgeschmacksarten & deren Wahrnehmung .....................................39
2.5.3 Zusammenhänge der Geschmackswahrnehmung & deren Modulation ................43
2.5.3.1 Zusammenhänge der Wahrnehmung von „süß“, „bitter“ und „umami“ ..............43
2.5.3.2 Modulation der Geschmackswahrnehmung .....................................................44
2.5.4 Geschmacksqualitäten von Peptiden ....................................................................47
3. Material & Methoden ............................................................................... 49
3.1 Rohstoffe .....................................................................................................................49
3.1.1 Molkenprotein-Isolat .............................................................................................49
3.1.2 Alge (Chlorella vulgaris) .......................................................................................50
3.2 Crossflow-Diafiltration .................................................................................................51
3.2.1 Die Methodenerstellung ........................................................................................51
3.2.1.1 Das Anlagensetup ............................................................................................51
3.2.1.2 Ablauf der Membrancharakterisierung – Bestimmung des „kritischen Drucks“ .52
3.2.2 Optimierung der Crossflow-Performance & der Trennschärfe ...............................54
3.2.2.1 Einfluss von pH-Wert und Ionenstärke .............................................................54
3.2.2.2 Einfluss von Ultraschall ....................................................................................56
3.2.2.3 Eignung von regenerierten Cellulose Membranen für die Fraktionierung .........58
3.2.2.4 Dialyse zur Abtrennung kleiner Moleküle (< 2 kDa) aus dem Feed ..................59
3.3 Hydrolyse der Alge (Chlorella vulgaris) ........................................................................60
3.3.1.1 Die Methodenerstellung ...................................................................................61
3.3.1.2 Methode zur Hydrolyse der Alge (Chlorella vulgaris) ........................................63
3.3.2 Aufbereitung und Analytik der Hydrolysate ...........................................................63
3.4 Instrumentelle Analytik & Probenaufbereitung .............................................................63
3.4.1 Aufkonzentration der Peptidfraktionen ..................................................................63
3.4.2 Spektralphotometrische Proteingehaltbestimmung ...............................................64
3.4.2.1 Referenzmethodische Bestimmung des Proteingehalts der hydrolysierten Alge
(Chlorella vulgaris) ...........................................................................................65
3.4.3 Bestimmung des Hydrolysegrads der Proteinhydrolysate .....................................65
3.4.4 Analytik der Peptidfraktionen mittels SE-HPLC .....................................................67
3.4.4.1 Methodenerstellung .........................................................................................67
3.4.4.2 Methode zur SE-HPLC Analyse .......................................................................68
3.4.4.3 Probenvorbereitung .........................................................................................68
3.5 Sensorik ......................................................................................................................68
3.5.1 Schulung des Sensorikpanels ..............................................................................68
3.5.1.1 Prüfung auf Geschmacksblindheit (gemäß DIN EN ISO 8586:2014) ................70
3.5.1.2 Prüfung zur Wahrnehmung eines Reizes (gemäß DIN EN ISO 8586:2014) .....70

III
3.5.1.3 Ermittlung der Reiz- und Erkennungsschwelle (gemäß ISO 3972:2011) ..........70
3.5.1.4 Prüfung zur Unterscheidung von Intensitätsstufen eines Reizes (gemäß DIN EN
ISO 8586:2014) ...............................................................................................71
3.5.1.5 Schulung zur Anwendung der Skale ................................................................72
3.5.2 Prüferauswahl ......................................................................................................72
3.5.3 Sensorik der Peptidfraktionen ...............................................................................73
3.6 Statistische Auswertung & Datenverarbeitung der Messergebnisse ............................74
3.6.1 Datenverarbeitung ................................................................................................74
3.6.1.1 Crossflow-Diafiltration ......................................................................................74
3.6.1.2 Ultraschall ........................................................................................................74
3.6.1.3 Eignung von RC-Membranen ...........................................................................76
3.6.1.4 Absolute Proteinmenge von Retentat (mRet) und Permeat (mPer) ......................76
3.6.2 Statistische Auswertung .......................................................................................76
3.6.2.1 Hydrolyse .........................................................................................................76
3.6.2.2 Crossflow-Filtration ..........................................................................................77
3.6.2.3 Ultraschall ........................................................................................................77
3.6.2.4 Dialyse .............................................................................................................78
3.6.2.5 Sensorik-Schulung ...........................................................................................78
3.6.2.6 Prüferauswahl ..................................................................................................78
3.6.2.7 Sensorik ...........................................................................................................78
4. Ergebnisse ............................................................................................... 79
4.1 Analytik .......................................................................................................................79
4.1.1 Size-Exclusion HPLC ...........................................................................................79
4.1.1.1 Tosoh TSKgel G 3000 SWXL (7,8 mm x 30 mm, 5 µm) .....................................80
4.1.1.2 PSS Suprema 1000 Â (8 mm x 300 mm, 10 µm) .............................................82
4.1.1.3 Agiltent Zorbax GF 250 (4,6 mm x 250 mm, 4 µm) ..........................................83
4.1.1.4 Übertragung der Methode auf eine Agilent 1100 Series HPLC.........................85
4.1.2 Spektralphotometrische Proteingehaltbestimmung ...............................................87
4.1.2.1 Molkenprotein-Isolat .........................................................................................87
4.1.2.2 Alge (Chlorella vulgaris) ...................................................................................88
4.2 Hydrolyse ....................................................................................................................89
4.2.1 Alge (Chlorella vulgaris) .......................................................................................89
4.2.1.1 Behandlung mit Rohament® PL ........................................................................93
4.2.1.2 Behandlung mit HCl, pH 3 ................................................................................97
4.2.1.3 Vergleich – Behandlung mit HCl, pH 3 vs. Behandlung mit Rohament® PL .... 100
4.3 Crossflow-Diafiltration & Fraktionierung ..................................................................... 101

IV
4.3.1 Membran- & Anlagen-Charakterisierung ............................................................. 101
4.3.1.1 Molkenprotein-Isolat ....................................................................................... 106
4.3.1.2 Alge (Chlorella vulgaris) ................................................................................. 106
4.3.2 Einfluss des pH-Werts & der Ionenstärke ........................................................... 107
4.3.2.1 Molkenprotein-Isolat ....................................................................................... 108
4.3.2.2 Alge (Chlorella vulgaris) ................................................................................. 122
4.3.3 Einfluss der Ultraschall-Anwendung ................................................................... 138
4.3.4 Eignung von Membranen aus regenerierter Cellulose ........................................ 142
4.3.5 Dialyse zur Abtrennung kleiner Moleküle (< 2 kDa) aus dem Feed ..................... 144
4.4 Sensorik .................................................................................................................... 153
4.4.1 Schulung des Sensorik-Panels ........................................................................... 153
4.4.1.1 Prüfung auf Geschmacksblindheit (gemäß DIN EN ISO 8586:2014) .............. 153
4.4.1.2 Prüfung zur Wahrnehmung eines Reizes (gemäß DIN EN ISO 8586:2014) ... 154
4.4.1.3 Ermittlung der Reiz- und Erkennungsschwelle (gemäß ISO 3972:2011) ........ 160
4.4.1.4 Prüfung zur Unterscheidung von Intensitätsstufen eines Reizes (gemäß DIN EN
ISO 8586:2014) ............................................................................................. 166
4.4.1.5 Schulung zur Anwendung der Skale .............................................................. 169
4.4.2 Prüferauswahl .................................................................................................... 169
4.4.3 Sensorik der Peptidfraktionen ............................................................................. 176
5. Diskussion ............................................................................................. 179
5.1 Analytik ..................................................................................................................... 179
5.2 Hydrolyse .................................................................................................................. 179
5.2.1 Alge (Chlorella vulgaris) ..................................................................................... 179
5.2.1.1 Zusammenfassung der Diskussionspunkte .................................................... 181
5.2.1.2 Zusammenfassung & Darstellung der Ergebnisse.......................................... 181
5.3 Crossflow-Diafiltration & Fraktionierung ..................................................................... 182
5.3.1 Membran- & Anlagencharakterisierung ............................................................... 182
5.3.1.1 Molkenprotein-Isolat & Alge (Chlorella vulgaris) ............................................. 182
5.3.2 Einfluss des pH-Werts & der Ionenstärke ........................................................... 183
5.3.2.1 Molkenprotein-Isolat ....................................................................................... 183
5.3.2.2 Alge (Chlorella vulgaris) ................................................................................. 192
5.3.2.3 Zusammenfassung der Diskussionspunkte .................................................... 201
5.3.2.4 Zusammenfassung & Darstellung der Ergebnisse.......................................... 201
5.3.3 Einfluss der Ultraschall-Anwendung ................................................................... 203
5.3.3.1 Zusammenfassung der Diskussionspunkte .................................................... 206
5.3.3.2 Zusammenfassung & Darstellung der Ergebnisse.......................................... 207

V
5.3.4 Eignung von Membranen aus regenerierter Cellulose ........................................ 207
5.3.4.1 Zusammenfassung der Diskussionspunkte .................................................... 208
5.3.4.2 Zusammenfassung & Darstellung der Ergebnisse.......................................... 209
5.3.5 Dialyse zur Abtrennung kleiner Moleküle (< 2 kDa) aus dem Feed ..................... 209
5.3.5.1 Zusammenfassung der Diskussionspunkte .................................................... 212
5.3.5.2 Zusammenfassung & Darstellung der Ergebnisse.......................................... 212
5.3.6 Einfluss weiterer Faktoren .................................................................................. 212
5.4 Sensorik .................................................................................................................... 217
5.4.1 Schulung des Sensorik-Panels ........................................................................... 217
5.4.1.1 Zusammenfassung der Diskussionspunkte .................................................... 222
5.4.1.2 Zusammenfassung & Darstellung der Ergebnisse.......................................... 223
5.4.2 Prüferauswahl .................................................................................................... 223
5.4.2.1 Zusammenfassung der Diskussionspunkte .................................................... 226
5.4.2.2 Zusammenfassung & Darstellung der Ergebnisse.......................................... 226
5.4.3 Sensorik der Peptidfraktionen ............................................................................. 226
5.4.3.1 Zusammenfassung der Diskussionspunkte .................................................... 233
5.4.3.2 Zusammenfassung & Darstellung der Ergebnisse.......................................... 233
5.5 Evaluation der Ergebnisse ......................................................................................... 234
5.5.1 Evaluation der Ergebnisse in Bezug auf die formulierten Hypothesen ................ 234
5.5.2 Methodische Aspekte ......................................................................................... 237
5.5.2.1 Rohstoffe ....................................................................................................... 237
5.5.2.2 Crossflow-Diafiltration .................................................................................... 238
5.5.2.3 Hydrolyse der Alge Chlorella vulgaris ............................................................. 241
5.5.2.4 Instrumentelle Analytik & Probenaufbereitung ................................................ 241
5.5.2.5 Sensorik ......................................................................................................... 242
5.5.3 Ausblick .............................................................................................................. 245
6. Zusammenfassung ................................................................................ 246
Literaturverzeichnis ..................................................................................... 250
Anhang ......................................................................................................... 294
Crossflow-Fraktionierung - Versuchsdaten ....................................................................... 294
Dialyse - Versuchsdaten .................................................................................................. 318
Hydrolyse - Versuchsdaten .............................................................................................. 319
Sensorik - Versuchsdaten ................................................................................................ 320
Publikationsliste .......................................................................................... 336

VI
Abbildungsverzeichnis
Abbildung 1: Klassifizierung der Trennverfahren (Kofod Nielsen, 2004; modifiziert) .......... 5
Abbildung 2: Gegenüberstellung - Crossflow Filtration und Dead-End Filtration (Bhave,
1997) ............................................................................................................ 6
Abbildung 3: Schematische Darstellung des Konzentrationsprofils an der
Membranoberfläche (Hiersig, 1995) ............................................................. 8
Abbildung 4: Schematische Darstellung der Konzentrationspolarisation (Chen et al., 1997;
modifiziert) .................................................................................................... 8
Abbildung 5: Schematische Darstellung der Foulingmechanismen (Wang & Tarabara,
2008; modifiziert) .......................................................................................... 9
Abbildung 6: Sherwood-Zahl zur Charakterisierung des Stoffübergangs ..........................13
Abbildung 7: Einfluss einer Deckschichtausbildung auf den Flux JF,t in Abhängigkeit des
Transmembrandrucks ∆pTMP für Reinstwasser und ein beladenes Medium
(Irmler, 2001; modifiziert) .............................................................................15
Abbildung 8: Kritische Flux Bereich (Brans et al., 2004) ...................................................16
Abbildung 9: "harte/starke" und "weiche/schwache" Form des kritischen Flux JCF (Bacchin
et al., 2006) .................................................................................................17
Abbildung 10: Flux/Feed-Konzentration Abhängigkeit (links: Harker et al., 2013; modifiziert;
rechts: Cheryan, 1998; modifiziert) ..............................................................19
Abbildung 11: Einfluss der Feed-Konzentration CF auf den Permeatflux JF,t (links: She et al.,
2009; rechts: Tarleton & Wakeman, 1994) ..................................................19
Abbildung 12: Einfluss der Partikelgröße auf die foulinginduzierte Abnahme des Permeatflux
JF,t für niedrige (links) und hohe (rechts) Strömungsgeschwindigkeiten
(Tarleton & Wakeman, 1993) .......................................................................20
Abbildung 13: pH-Abhängigkeit des ζ-Potentials einer Polysulfon-Membran (Melin &
Rautenbach, 2007; modifiziert) ....................................................................22
Abbildung 14: Schematische Darstellung der Porengrößenverteilung einer Membran
(Mulder, 1996) .............................................................................................23
Abbildung 15: BSA-Konzentration und Reinheit von Immunglobulin G (IgG) in Abhängigkeit
der Diavolumina-Anzahl Nd (Venkiteshwaran et al., 2008; modifiziert) .........24
Abbildung 16: Permeatflux JF,t in Abhängigkeit der Ultraschallfrequenz fUS einer 0,5 %igen
(w/w) Milchlösung (Kobayashi et al., 2003) ..................................................25
Abbildung 17: Abhängigkeit des Permeatflux von der Konzentration CF des Feed für die
Crossflow-Filtration von Molke mit und ohne die Verwendung von US
(Muthukumaran, Kentish, Ashokkumar, et al., 2005; modifiziert) .................25
Abbildung 18: Druckabhängige Änderung des Permeatflux bei der Crossflow-Mikrofiltration
von Bäcker-Hefe (Matsumoto et al., 1996) ..................................................26

VII
Abbildung 19: Schematische Darstellung des Dialyseprozess (Ballew, Martinez, Markee, &
Eddleman, 2002; modifiziert) .......................................................................27
Abbildung 20: Schematische Darstellung von Chlorella (links: Linne von Berg, Hoef-Emden,
& Melkonian, 2012, modifiziert) und transmissionselektronenmikroskopische
Aufnahme von Chlorella vulgaris (rechts: Yamamoto, Kurihara, & Kawano,
2005, modifiziert) .........................................................................................31
Abbildung 21: Zusammensetzung verschiedener Algen (Becker, 2007) .............................31
Abbildung 22: Aminosäurenprofil von Chlorella vulgaris (Safi et al., 2014; modifiziert;
basierend auf a Safi et al. 2012; b Faheed & Fattah, 2008 & Shaaban, 2001; c
Naik, Goud, Rout, & Dalai, 2010) ................................................................32
Abbildung 23: Hydrolyse einer Peptidbindung (Pasupuleti, Holmes & Demain, 2010;
basierend auf: Adler-Nissen, 1986) .............................................................33
Abbildung 24: Hydrolysegrad in Abhängigkeit der Zeit für die Hydrolyse von Chlorella mit
Subtilisin (Tchorbanov & Bozhkova, 1988; modifiziert) ................................35
Abbildung 25: Bestimmung von Totzeit t0/-Volumen V0 und Retentionszeit tr/-Volumen Vr
(Siouffi, 2000) ..............................................................................................36
Abbildung 26: Arbeitsweise einer Size-Exclusion Chromatography (Kremer & Bannwarth,
2014; modifiziert) .........................................................................................37
Abbildung 27: Geschmackrezeptoren - Geschmackspapillen, -Knospen und -Sinneszellen
(Becker-Carus & Wendt, 2017; modifiziert) ..................................................38
Abbildung 28: Bevorzugte Lokalisation der Geschmacksqualitäten auf der Zunge (Hatt,
2011; modifiziert) .........................................................................................40
Abbildung 29: Zusammenfassung der Geschmackstransduktion (Silverthorn, 2009;
modifiziert) ...................................................................................................40
Abbildung 30: Signaltransduktion in Geschmackszellen (Hatt, 2006; modifiziert) ...............41
Abbildung 31: Mechanismus der "süß"-, "bitter"- und "umami"-Wahrnehmung (Feher, 2017;
modifiziert) ...................................................................................................43
Abbildung 32: Schematische Darstellung des „Crosstalk“ zwischen "umami"- und "bitter"-
Rezeptoren (Temussi, 2009; modifiziert; basierend auf: Tomchik et al., 2007)
....................................................................................................................44
Abbildung 33: Schematische Darstellung möglicher Wirkmechanismen von Natrium-
Kationen während der „bitter“-Transduktion (Keast et al., 2001) ..................46
Abbildung 34: Schematischer Aufbau der Crossflow-Anlage ..............................................51
Abbildung 35: Schematischer Ablauf der Diafiltration .........................................................55
Abbildung 36: Schematischer Aufbau der Crossflow-Anlage mit Ultraschalleinheit ............57
Abbildung 37: Schematischer Aufbau der Crossflow- Anlage (RC-Membran) ....................59
Abbildung 38: Schematische Übersicht der Dialyseversuche .............................................60

VIII
Abbildung 39: Schematische Übersicht der Hydrolyseversuche .........................................62
Abbildung 40: Ablaufplan der Sensorik-Schulung...............................................................69
Abbildung 41: 7-Punkt Skale zur Bewertung der Prüfproben ..............................................72
Abbildung 42: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des
BioRad Gel-Filtration Standards mittels Säule: Tosoh TSKgel G 3000 SWXL –
Puffer: 0,05 mol/l Natriumphosphatpuffer +0,05 % NaN3, pH 6,7, quantitative
Darstellung ..................................................................................................80
Abbildung 43: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des
BioRad Gel-Filtration Standards mittels Säule: Tosoh TSKgel G 3000 SWXL
– Puffer: 0,1 mol/l Natriumphosphatpuffer + 0,1 mol/l Na2SO4 + 0,05 % NaN3,
pH 6,7, quantitative Darstellung ...................................................................81
Abbildung 44: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des
BioRad Gel-Filtration Standards mittels Säule: Tosoh TSKgel G 3000 SWXL
– Puffer: 0,2 mol/l Natriumphosphatpuffer + 0,2 mol/l Na2SO4 + 0,05 % NaN3,
pH 7, quantitative Darstellung ......................................................................81
Abbildung 45: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des
BioRad Gel-Filtration Standards mittels Säule: PSS Suprema 1000 Â – Puffer:
0,05 mol/l Natriumphosphatpuffer +0,05 % NaN3, pH 6,9, quantitative
Darstellung ..................................................................................................82
Abbildung 46: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des
BioRad Gel-Filtration Standards mittels Säule: PSS Suprema 1000 Â – Puffer:
0,1 mol/l Natriumphosphatpuffer +0,05 % NaN3, pH 7,0, quantitative
Darstellung ..................................................................................................83
Abbildung 47: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des
BioRad Gel-Filtration Standards mittels Säule: PSS Suprema 1000 Â – Puffer:
0,2 mol/l Natriumphosphatpuffer +0,05 % NaN3, pH 7,0, quantitative
Darstellung ..................................................................................................83
Abbildung 48: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des
BioRad Gel-Filtration Standards mittels Säule: Agilent Zorbax GF 250 – Puffer:
0,05 mol/l Natriumphosphatpuffer + 0,05 % NaN3, pH 6,9, quantitative
Darstellung ..................................................................................................84
Abbildung 49: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des
BioRad Gel-Filtration Standards mittels Säule: Agilent Zorbax GF 250 – Puffer:
0,1 mol/l Natriumphosphatpuffer + 0,05 % NaN3, pH 7,0, quantitative
Darstellung ..................................................................................................84
Abbildung 50: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des
BioRad Gel-Filtration Standards mittels Säule: Agilent Zorbax GF 250 – Puffer:

IX
0,2 mol/l Natriumphosphatpuffer + 0,05 % NaN3, pH 7,0, quantitative
Darstellung ..................................................................................................85
Abbildung 51: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des
BioRad Gel-Filtration Standards mittels Säule: Agilent Zorbax GF 250 – Puffer:
0,2 mol/l Natriumphosphatpuffer + 0,05 % NaN3, pH 7,0, quantitative
Darstellung ..................................................................................................86
Abbildung 52: Kalibrationsfunktion für die photometrische Bestimmung des Proteingehlts von
hydrolysiertem Molkenprotein-Isolat bei 280 nm ..........................................87
Abbildung 53: Kalibrationsfunktion zur photometrischen Bestimmung des Proteingehalts von
hydrolysierter Alge (Chlorella vulgaris) bei 280 nm ......................................88
Abbildung 54: Molekülgrößenverteilung – Hydrolysat der Alge Chlorella vulgaris mit Pronase
(0,25 % (112,5 PU); 45 min), qualitative Darstellung ...................................90
Abbildung 55: Molekülgrößenverteilung – Hydrolysat der Alge Chlorella vulgaris mit Pronase
(0,50 % (225 PU); 45 min), qualitative Darstellung ......................................90
Abbildung 56: Molekülgrößenverteilung – Hydrolysat der Alge Chlorella vulgaris mit
Pancreatin (0,25 % (20 USP); 10 min), qualitative Darstellung ....................92
Abbildung 57: Molekülgrößenverteilung – Hydrolysat der Alge Chlorella vulgaris mit Pronase
(0,25 % (112,5 PU); 10 min), qualitative Darstellung ...................................92
Abbildung 58: Hydrolysegrad des Algen-Hydrolysats in Abhängigkeit des Enzyms und der
Dauer der Vorbehandlung mit Rohament® PL .............................................93
Abbildung 59: Molekülgrößenverteilung – Hydrolysate der Alge Chlorella vulgaris mit a)
Pancreatin (0,25 % (20 USP); 45 min; Behandlung: Rohament® PL, 18 h) und
b) Pronase (0,25 % (112,5 PU); 45 min; Behandlung: Rohament® PL, 18 h),
qualitative Darstellung .................................................................................95
Abbildung 60: Molekülgrößenverteilung – Hydrolysate der Alge Chlorella vulgaris mit a)
Pancreatin (0,25 % (20 USP); 45 min; Behandlung: Rohament® PL, 48 h) und
b) Pronase (0,25 % (112,5 PU); 45 min; Behandlung: Rohament® PL, 48 h),
qualitative Darstellung .................................................................................95
Abbildung 61: Molekülgrößenverteilung – Hydrolysate der Alge Chlorella vulgaris mit a)
Pancreatin (0,25 % (20 USP); 45 min; Behandlung: Rohament® PL, 72 h) und
b) Pronase (0,25 % (112,5 PU); 45 min; Behandlung: Rohament® PL, 72 h),
qualitative Darstellung .................................................................................96
Abbildung 62: Hydrolysegrad des Algen-Hydrolysats in Abhängigkeit des Enzyms und der
Dauer der Vorbehandlung mit HCl, pH 3 .....................................................97
Abbildung 63: Molekülgrößenverteilung – Hydrolysate der Alge Chlorella vulgaris mit a)
Pancreatin (0,25 % (20 USP); 45 min; Behandlung: HCl, pH 3, 18 h) und b)

X
Pronase (0,25 % (112,5 PU); 45 min; Behandlung: HCl, pH 3, 18 h), qualitative
Darstellung ..................................................................................................99
Abbildung 64: Molekülgrößenverteilung - Hydrolysate der Alge Chlorella vulgaris mit a)
Pancreatin (0,25 % (20 USP); 45 min; Behandlung: HCl, pH 3, 48 h) und b)
Pronase (0,25 % (112,5 PU); 45 min; Behandlung: HCl, pH 3, 48 h), qualitative
Darstellung ..................................................................................................99
Abbildung 65: Gelelektrophorese der Fraktionen von hydrolysiertem Molkenprotein-Isolat
.................................................................................................................. 102
Abbildung 66: Molekülgrößenverteilung - SE-HPLC-Chromatogramme von fraktioniertem,
hydrolysiertem Molkenprotein-Isolat (Feed-Konzentration 3 % (bezogen auf
den Proteingehalt) a) Fraktion > 50 kDa b) Fraktion < 50 kDa – 30 kDa,
quantitative Darstellung ............................................................................. 104
Abbildung 67: Molekülgrößenverteilung - SE-HPLC-Chromatogramme von fraktioniertem,
hydrolysiertem Molkenprotein-Isolat (Feed-Konzentration 1,5 % (bezogen auf
den Proteingehalt) a) Fraktion > 100 kDa b) Fraktion < 100 kDa, qualitative
Darstellung ................................................................................................ 105
Abbildung 68: Membrancharakterisierung 100 kDa – hydrolysiertes Molkenprotein-Isolat106
Abbildung 69: Membrancharakterisierung 100 kDa – hydrolysierte Alge (Chlorella vulgaris)
.................................................................................................................. 107
Abbildung 70: Permeatflux JFm von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit von
pH-Wert und Ionenstärke .......................................................................... 109
Abbildung 71: Absolute Proteinmenge der Retentat-Fraktionen von hydrolysiertem
Molkenprotein-Isolat in Abhängigkeit von pH-Wert und Ionenstärke .......... 110
Abbildung 72: Absolute Proteinmenge der Permeat-Fraktionen von hydrolysiertem
Molkenprotein-Isolat in Abhängigkeit von pH-Wert und Ionenstärke .......... 111
Abbildung 73: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die
Fraktionierung von hydrolysiertem Molkenprotein-Isolat, pH 4, a) ohne NaCl
b) mit NaCl, qualitative Darstellung ............................................................ 114
Abbildung 74: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die
Fraktionierung von hydrolysiertem Molkenprotein-Isolat, pH 6, a) ohne NaCl
b) mit NaCl, qualitative Darstellung ............................................................ 114
Abbildung 75: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die
Fraktionierung von hydrolysiertem Molkenprotein-Isolat, pH 7, a) ohne NaCl
b) mit NaCl, qualitative Darstellung ............................................................ 115
Abbildung 76: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die
Fraktionierung von hydrolysiertem Molkenprotein-Isolat, pH 8, a) ohne NaCl
b) mit NaCl, qualitative Darstellung ............................................................ 115

XI
Abbildung 77: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die
Fraktionierung von hydrolysiertem Molkenprotein-Isolat, pH 9, a) ohne NaCl
b) mit NaCl, qualitative Darstellung ............................................................ 116
Abbildung 78: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und
Permeatfraktion von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit der
Ionenstärke, pH 6, a) Retentat ohne NaCl b) Permeat ohne NaCl c) Retentat
mit NaCl d) Permeat mit NaCl, qualitative Darstellung ............................... 118
Abbildung 79: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und
Permeatfraktion von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit der
Ionenstärke, pH 7, a) Retentat ohne NaCl b) Permeat ohne NaCl c) Retentat
mit NaCl d) Permeat mit NaCl, qualitative Darstellung ............................... 119
Abbildung 80: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und
Permeatfraktion von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit der
Ionenstärke, pH 8, a) Retentat ohne NaCl b) Permeat ohne NaCl c) Retentat
mit NaCl d) Permeat mit NaCl, qualitative Darstellung ............................... 120
Abbildung 81: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und
Permeatfraktion von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit der
Ionenstärke, pH 9, a) Retentat ohne NaCl b) Permeat ohne NaCl c) Retentat
mit NaCl d) Permeat mit NaCl, qualitative Darstellung ............................... 121
Abbildung 82: Permeatflux von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit von
pH-Wert und Ionenstärke .......................................................................... 123
Abbildung 83: Absolute Proteinmenge der Retentat-Fraktionen von hydrolysierter Alge
(Chlorella vulgaris) in Abhängigkeit von pH-Wert und Ionenstärke ............ 124
Abbildung 84: Absolute Proteinmenge der Permeat-Fraktionen von hydrolysierter Alge
(Chlorella vulgaris) in Abhängigkeit von pH-Wert und Ionenstärke ............ 124
Abbildung 85: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die
Fraktionierung von hydrolysierter Alge (Chlorella vulgaris), pH 3, a) ohne NaCl
b) mit NaCl, qualitative Darstellung ............................................................ 129
Abbildung 86: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die
Fraktionierung von hydrolysierter Alge (Chlorella vulgaris), pH 6, a) ohne NaCl
b) mit NaCl, qualitative Darstellung ............................................................ 129
Abbildung 87: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die
Fraktionierung von hydrolysierter Alge (Chlorella vulgaris), pH 7, a) ohne NaCl
b) mit NaCl, qualitative Darstellung ............................................................ 130
Abbildung 88: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die
Fraktionierung von hydrolysierter Alge (Chlorella vulgaris), pH 8, a) ohne NaCl
b) mit NaCl, qualitative Darstellung ............................................................ 130

XII
Abbildung 89: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die
Fraktionierung von hydrolysierter Alge (Chlorella vulgaris), pH 11, a) ohne
NaCl b) mit NaCl, qualitative Darstellung ................................................... 131
Abbildung 90: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und
Permeatfraktion von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit
der Ionenstärke, pH 3, a) Retentat ohne NaCl b) Permeat ohne NaCl c)
Retentat mit NaCl d) Permeat mit NaCl, qualitative Darstellung ................ 133
Abbildung 91: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und
Permeatfraktion von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit
der Ionenstärke, pH 6, a) Retentat ohne NaCl b) Permeat ohne NaCl c)
Retentat mit NaCl d) Permeat mit NaCl, qualitative Darstellung ................ 134
Abbildung 92: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und
Permeatfraktion von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit
der Ionenstärke, pH 7, a) Retentat ohne NaCl b) Permeat ohne NaCl c)
Retentat mit NaCl d) Permeat mit NaCl, qualitative Darstellung ................ 135
Abbildung 93: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und
Permeatfraktion von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit
der Ionenstärke, pH 8, a) Retentat ohne NaCl b) Permeat ohne NaCl c)
Retentat mit NaCl d) Permeat mit NaCl, qualitative Darstellung ................ 136
Abbildung 94: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und
Permeatfraktion von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit
der Ionenstärke, pH 11, a) Retentat ohne NaCl b) Permeat ohne NaCl c)
Retentat mit NaCl d) Permeat mit NaCl, qualitative Darstellung ................ 137
Abbildung 95: Permeatflux JFm von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit einer
Ultraschall-Anwendung .............................................................................. 139
Abbildung 96: Absolute Proteinmenge der Retentat-Fraktionen von hydrolysiertem
Molkenprotein-Isolat in Abhängigkeit einer Ultraschall-Anwendung ........... 139
Abbildung 97: Absolute Proteinmenge der Permeat-Fraktionen von hydrolysiertem
Molkenprotein-Isolat n in Abhängigkeit einer Ultraschall-Anwendung ........ 139
Abbildung 98: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und
Permeatfraktion von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit der
Anwendung von Ultraschall, pH 7, a) Retentat & Permeat ohne US, Wdh. 1 b)
Retentat & Permeat mit US, Wdh. 1 c) Retentat & Permeat ohne US, Wdh. 5
d) Retentat & Permeat mit US, Wdh. 5, qualitative Darstellung ................. 141
Abbildung 99: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und
Permeatfraktion von hydrolysiertem Molkenprotein-Isolat unter Verwendung

XIII
einer RC-Membran, pH 9, a) Retentat, b) Permeat, qualitative Darstellung
.................................................................................................................. 142
Abbildung 100: Ausflockung einer Dialyseprobe (pH 9, 48 h) ............................................. 146
Abbildung 101: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der
Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach
Dialyse in Abhängigkeit des pH-Werts, a) pH 3, 1 %, 24 h, b) pH 5, 1 %, 24 h,
c) pH 6, 1 %, 24 h, d) pH 7,5, 1 %, 24 h, qualitative Darstellung ................ 147
Abbildung 102: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der
Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach
Dialyse in Abhängigkeit des pH-Werts, a) pH 9, 1 %, 24 h, b) pH 11, 1 %, 24
h, qualitative Darstellung ........................................................................... 148
Abbildung 103: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der
Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach
Dialyse in Abhängigkeit des pH-Werts, a) pH 9, 2 %, 24 h, b) pH 11, 2 %, 24
h, qualitative Darstellung ........................................................................... 148
Abbildung 104: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der
Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach
Dialyse in Abhängigkeit des pH-Werts a) pH 9, 2 %, 48 h, b) pH 11, 2 %, 48
h, qualitative Darstellung ........................................................................... 149
Abbildung 105: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der
Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach
Dialyse in Abhängigkeit des pH-Werts, a) pH 9, 4 %, 24 h, b) pH 11, 4 %, 24
h, qualitative Darstellung ........................................................................... 149
Abbildung 106: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der
Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach
Dialyse in Abhängigkeit des pH-Werts, a) pH 3, 1 %, 24 h, b) pH 5, 1 %, 24 h,
c) pH 6, 1 %, 24 h, d) pH 7,5, 1 %, 24 h, quantitative Darstellung ............. 150
Abbildung 107: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der
Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach
Dialyse in Abhängigkeit des pH-Werts, a) pH 9, 1 %, 24 h, b) pH 11, 1 %, 24
h, quantitative Darstellung ......................................................................... 151
Abbildung 108: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der
Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach
Dialyse in Abhängigkeit des pH-Werts, a) pH 9, 2 %, 24 h, b) pH 11, 2 %, 24
h, quantitative Darstellung ......................................................................... 151
Abbildung 109: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der
Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach

XIV
Dialyse in Abhängigkeit des pH-Werts a) pH 9, 2 %, 48 h, b) pH 11, 2 %, 48
h, quantitative Darstellung ......................................................................... 152
Abbildung 110: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der
Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach
Dialyse in Abhängigkeit des pH-Werts, a) pH 9, 4 %, 24 h, b) pH 11, 4 %, 24
h, quantitative Darstellung ......................................................................... 152
Abbildung 111: Grafische Darstellung der a) Prüfersensibilität, b) Reiz- und
Erkennungsschwelle „süß“ ........................................................................ 161
Abbildung 112: Grafische Darstellung der a) Prüfersensibilität, b) Reiz- und
Erkennungsschwelle „sauer“ ..................................................................... 162
Abbildung 113: Grafische Darstellung der a)Prüfersensibilität, b) Reiz- und
Erkennungsschwelle „salzig“ ..................................................................... 163
Abbildung 114: Grafische Darstellung der a) Prüfersensibilität, b) Reiz- und
Erkennungsschwelle „bitter“ ...................................................................... 164
Abbildung 115: Grafische Darstellung der a) Prüfersensibilität, b) Reiz- und
Erkennungsschwelle „umami“ .................................................................... 165
Abbildung 116: Sensorische Bewertung - Fraktionen hydrolysierten Molkenprotein-Isolats
(Retentat > 100 kDa, pH 9; Permeat < 100 kDa, pH 9) .............................. 178
Abbildung 117: Sensorische Bewertung - Fraktionen hydrolysierter Alge (Chlorella vulgaris)
(Retentat > 100 kDa, pH 6; Permeat < 100 kDa, pH 6) .............................. 178

XV
Tabellenverzeichnis
Tabelle 1: Zusammensetzung und Eigenschaften der Milchproteinfraktionen (Ternes,
2008; Topel, 2004; modifiziert) ....................................................................28
Tabelle 2: Zusammensetzung des Molkenprotein-Isolats "Thermax 690" (Glanbia
Nutritionals Ltd., 2012) ................................................................................49
Tabelle 3: Aminosäure-Profil des Molkenprotein-Isolats „Thermax 690“ (Glanbia
Nutritionals Ltd., 2012) ................................................................................50
Tabelle 4: Zusammensetzung des Algen-Pulvers „Chlorella vulgaris Powder“ (Allma,
2014) ...........................................................................................................50
Tabelle 5: Charakterisierte UF- und NF-Membranen des Herstellers Microdyn Nadir
GmbH (Wiesbaden, GER) ...........................................................................52
Tabelle 6: 100 kDa UF-Membranen (Microdyn Nadir GmbH, Wiesbaden, GER) ..........53
Tabelle 7: Übersicht – Nutschenfilter ...........................................................................53
Tabelle 8: Übersicht - pH-Werte je Proteinquelle .........................................................54
Tabelle 9: Pumpenfördermengen und daraus resultierenden
Strömungsgeschwindigkeiten ......................................................................55
Tabelle 10: Übersicht - Parameter der Crossflowfraktionierung .....................................56
Tabelle 11: Herstellerangaben zu den Enzymen Corolase, Pronase und Pancreatin .....61
Tabelle 12: Herstellerangaben zu Rohament® PL ..........................................................62
Tabelle 13: Konzentrationsstufen CF der Kalibrationslösungen ......................................64
Tabelle 14: SEC-Säulen und zugehöriger Anlagenfluss .................................................67
Tabelle 15: Laufmittel zur SE-HPLC Methodenerstellung ..............................................68
Tabelle 16: Rohstoffe zur Herstellung der Schulungsstandards .....................................69
Tabelle 17: Probenkonzentrationen für die Prüfung auf Geschmacksblindheit ...............70
Tabelle 18: Probenkonzentrationen zur Wahrnehmung eines Reizes ............................70
Tabelle 19: Probenkonzentrationen C zur Ermittlung der Reiz-/Erkennungsschwelle.....71
Tabelle 20: Probenkonzentrationen C zur Prüfung zur Unterscheidung von
Intensitätsstufen eines Reizes .....................................................................71
Tabelle 21: Probenkonzentrationen C und Skalenwerte zur Schulung zur Anwendung der
Skale ...........................................................................................................72
Tabelle 22: Probenkonzentrationen C für die sensorische Bewertung der Peptidfraktionen
....................................................................................................................73
Tabelle 23: Zusammensetzung des BioRad Gel-Filtration Standards # 151-1901 (BioRad
Laboratories, Inc., n.d.) ................................................................................79
Tabelle 24: Retentionszeiten tr und prozentuale Standardabweichung (SD) der
Komponenten des BioRad Gel-Filtration Standards ....................................86

XVI
Tabelle 26: Hydrolysegrad des Algen-Hydrolysats in Abhängigkeit des Enzyms und der
Dauer der Vorbehandlung mit Rohament® PL .............................................94
Tabelle 25: Hydrolysegrad des Algen-Hydrolysats in Abhängigkeit des Enzyms und der
Dauer der Vorbehandlung mit HCl, pH 3 .....................................................98
Tabelle 27: Hydrolysegrad des Algen-Hydrolysats in Abhängigkeit des Enzyms und der
Dauer der Vorbehandlung mit HCl, pH 3 und Rohament® PL .................... 100
Tabelle 28: Laufzeit tFm & Permeatflux JFm von hydrolysiertem Molkenprotein-Isolat in
Abhängigkeit von pH-Wert und Ionenstärke .............................................. 108
Tabelle 29: Absolute Proteingehalte von hydrolysiertem Molkenprotein-Isolat in Retentat
und Permeat in Abhängigkeit von pH-Wert und Ionenstärke ..................... 110
Tabelle 30: Wiederfindungsrate und Anteile der Retentat- und Permeat-Fraktion bei der
Fraktionierung von hydrolysiertem Molkenprotein-Isolat ............................ 112
Tabelle 31: Laufzeit tFm & Permeatflux JFm von hydrolysierter Alge (Chlorella vulgaris) in
Abhängigkeit von pH-Wert und Ionenstärke .............................................. 122
Tabelle 32: Absolute Proteingehalte von hydrolysierter Alge (Chlorella vulgaris) in
Retentat und Permeat in Abhängigkeit von pH-Wert und Ionenstärke ....... 123
Tabelle 33: Wiederfindungsrate und Anteile der Retentat- und Permeat-Fraktion bei der
Fraktionierung von hydrolysierter Alge (Chlorella vulgaris) ........................ 126
Tabelle 34: Bestimmtheitsmaß R2 und pH-Mittelwert, -Minima und -Maxima der
Fraktionierung von hydrolysierter Alge (Chlorella vulgaris) ........................ 127
Tabelle 35: Laufzeit tFm, Permeatflux JFm und absolute Proteingehalte der Retentat- und
Permeat-Fraktion von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit der
Anwendung von Ultraschall ....................................................................... 138
Tabelle 36: Wiederfindungsrate und Anteile der Retentat- und Permeat-Fraktion bei der
Fraktionierung von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit der
Anwendung von Ultraschall ....................................................................... 140
Tabelle 37: Laufzeit tFm, Permeatflux JFm und absolute Proteingehalte der Retentat- und
Permeat-Fraktion von hydrolysiertem Molkenprotein-Isolat ....................... 143
Tabelle 38: Absoluter Proteingehalt der Dialyseproben (Retentat) von hydrolysiertem
Molkenprotein-Isolat vor (mvD) und nach (mnD) der Dialyse in Abhängigkeit von
pH-Wert, Konzentration und Dialysedauer ................................................. 144
Tabelle 39: Ergebnisse des Tests auf Geschmacksblindheit (gemäß DIN EN ISO
8586:2014) ................................................................................................ 154
Tabelle 40: Ergebnisse des Test zur Prüfung der Wahrnehmung eines Reizes (gemäß
DIN EN ISO 8586:2014) ............................................................................ 155
Tabelle 41: Ergebnisse des Test zur Prüfung der Wahrnehmung eines Reizes (gemäß
DIN EN ISO 8586:2014) - Fortsetzung ...................................................... 156

XVII
Tabelle 42: Ergebnisse des Test zur Prüfung der Wahrnehmung eines Reizes (gemäß
DIN EN ISO 8586:2014) - Fortsetzung ...................................................... 157
Tabelle 43: Ergebnisse des Test zur Prüfung der Wahrnehmung eines Reizes (gemäß
DIN EN ISO 8586:2014) - Fortsetzung ...................................................... 158
Tabelle 44: Gruppenauswertung des Test zur Prüfung der Wahrnehmung eines Reizes
(gemäß DIN EN ISO 8586:2014) ............................................................... 159
Tabelle 45: Anteil der Prüferpersonen die eine Geschmacksausprägung erkennen –
Verlauf der Schulung - Tag 1 – Tag 3 ........................................................ 159
Tabelle 46: Reiz- und Erkennungsschwelle "süß" ........................................................ 161
Tabelle 47: Reiz- und Erkennungsschwelle "sauer" ..................................................... 162
Tabelle 48: Reiz- und Erkennungsschwelle "salzig" ..................................................... 163
Tabelle 49: Reiz- und Erkennungsschwelle "bitter" ...................................................... 164
Tabelle 50: Reiz- und Erkennungsschwelle "umami" ................................................... 165
Tabelle 51: Diskriminierungsfähigkeit der Prüfer - Friedman-Test ................................ 166
Tabelle 52: Diskriminierungsfähigkeit der Prüfer - Wilcoxon-Test................................. 167
Tabelle 53: Diskriminierungsfähigkeit der Prüfer - Mittlerer Rang ................................. 168
Tabelle 54: Diskriminierungsfähigkeit der Prüfer – Deskriptive Teststatistik der
Rangordnungstests ................................................................................... 168
Tabelle 55: Skalenbewertungen der Prüfer - Deskriptive Statistik der Skalenschulung 169
Tabelle 56: Prüferauswahl - Friedman-Test zur Überprüfung der Diskriminierungsfähigkeit
.................................................................................................................. 171
Tabelle 57: Prüferauswahl - Differenz der Prüfpersonen zum Gruppenmittelwert – „süß“
.................................................................................................................. 172
Tabelle 58: Prüferauswahl - Differenz der Prüfpersonen zum Gruppenmittelwert – „salzig“
.................................................................................................................. 172
Tabelle 59: Prüferauswahl - Differenz der Prüfpersonen zum Gruppenmittelwert – „sauer“
.................................................................................................................. 173
Tabelle 60: Prüferauswahl - Differenz der Prüfpersonen zum Gruppenmittelwert – „bitter“
.................................................................................................................. 174
Tabelle 61: Prüferauswahl - Differenz der Prüfpersonen zum Gruppenmittelwert – „umami“
.................................................................................................................. 174
Tabelle 62: Prüferauswahl - Panelhomogenität "süß" .................................................. 175
Tabelle 63: Prüferauswahl - Panelhomogenität "sauer" ............................................... 175
Tabelle 64: Prüferauswahl - Panelhomogenität "salzig" ............................................... 175
Tabelle 65: Prüferauswahl - Panelhomogenität "bitter" ................................................ 176
Tabelle 66: Prüferauswahl - Panelhomogenität "umami" .............................................. 176

XVIII
Tabelle 67: Sensorische Bewertung – Fraktionen hydrolysierten Molkenprotein-Isolats
(Retentat > 100 kDa, pH 9; Permeat < 100 kDa, pH 9) .............................. 177
Tabelle 68: Sensorische Bewertung - Fraktionen hydrolysierter Alge (Chlorella vulgaris)
(Retentat > 100 kDa, pH 6; Permeat < 100 kDa, pH 6) .............................. 177
Tabelle 70: Konzentrationen der Erkennungsschwelle verschiedener Geschmacksstoffe
(Schiffman et al., 19901; Paulus & Reisch, 19802; Pfaffmann et al., 19713) 219

XIX
Formelverzeichnis
Formel 1: Hagen-Poiseulle Gleichung – Flux durch Membran, ideal ...........................10
Formel 2: Transmembrandruck, ideal ..........................................................................11
Formel 3: Massenbilanz der Konzentrationspolarisation ..............................................11
Formel 4: Permeatflux JP,t bei Konzentrationspolarisation nach dem Gel-Polarisations-
Modell .........................................................................................................12
Formel 5: Stoffüberhangskoeffizient k .........................................................................12
Formel 6: Filtrationswiderstand RT ...............................................................................14
Formel 7: Darcy-Gleichung - Flux durch Membran in Abhängigkeit fluxreduzierender
Parameter ...................................................................................................14
Formel 8: Widerstand durch Fouling ............................................................................14
Formel 9: Degree of Hydrolysis ...................................................................................33
Formel 10: Berechnung von h .......................................................................................67
Formel 11: Rechnerische Angleichung der Prozesslaufzeit in Abhängigkeit des
Permeatvolumen und des Initialflux der Membran mit destilliertem Wasser .74
Formel 12: Rechnerische Angleichung des Permeatvolumens der performanteren
Vivaflow-Einheit ...........................................................................................75
Formel 13: Rechnerische Angleichung des Permeatvolumens der weniger performanten
Vivaflow-Einheit ...........................................................................................75
Formel 14: Rechnerische Angleichung der Prozesszeit in Abhängigkeit des
Permeatvolumens .......................................................................................75
Formel 15: Rechnerische Ermittlung der absoluten Proteinmengen ..............................76
Formel 16: Kalibrationsfunktion zur photometrischen Bestimmung des Proteingehalts von
hydrolysiertem Molkenprotein-Isolat ............................................................88
Formel 17: Kalibrationsfunktion zur photometrischen Bestimmung des Proteingehalts von
hydrolysierter Alge (Chlorella vulgaris) ........................................................88

XX
Abkürzungen
AS Aminosäure
ANOVA Analysis of Variance
AOAC Association of Analytical Communities
BSA Bovine-Serum-Albumin
CA Cellulose Acetat
CFF Crossflow Filtration
CDF Continuous Diafiltration/Kontinuierliche Diafiltration
DDF Discontinuous Diafiltration/Diskontinuierliche Diafiltration
DH Degree of Hydrolysis/Hydrolysegrad
DIN Deutsches Institut für Normung
EH-W extensively hydrolysed whey/extensiv hydrolysiertes
Molkenprotein
EPS extrazellulärer polymerer Substanzen
EOM extracellular organic matter/extrazelluläre organische
Substanzen
EU Europäischen Union
ES Erkennungsschwelle
GPC Gel Permeation Chromatography
H2O Wasser
HCl Salzsäure
HMW high molecular weight/hochmolekular
HIC Hydrophobic-Interaction Chromatography
IBM International Business Machines Corporation
IEP Isoelektrischer Punkt
IgG Immunglobulin G
IUB International Union of Biochemistry

XXI
KCl Kaliumchlorid
kDa Kilo-Dalton
kHz Kilo-Hertz
LMW low molecular weight/niedermolekular
MF Mikrofiltration
MgCl2 Magnesiumchlorid
MSG Monosodium L-Glutamat
MMW middle molecular weight/mittelmolekular
MWCO Molecular Weight Cut-Off
NaCl Natriumchlorid
NaOH Natriumhydroxyd
NaN3 Natriumazid
Na2SO4 Natriumsulfat
n. d. nicht datiert
NF Nanofiltration
NP-HPLC Normal Phase High Pressure Liquid Chromatography
NSF National Sanitation Foundation
OPA O-phthaldialdehyd
PES Polyethersulfon
PH-W partially hydrolysed whey/teilhydrolysiertes Molkenprotein
PS Polysulfon
PSS Polymer Standards Service
RC Regenerierte Celluose
RPS released polysaccharide/freie Polysaccharide
RS Reizschwelle
RP-HPLC Reversed Phase High Pressure Liquid Chromatography

XXII
S Schwefel
SD Standardabweichung
SE-HPLC Size Exclusion High Pressure Liquid Chromatography
TFF Tangentialfluss Filtration
TNBS Trinitro-Benzene-Sulfonic Acid
TOC total organic carbon/Gesamter organischer Kohlenstoff
UF Ultrafiltration
US Ultraschall
VD Verdünnungsfaktor
WFR Wiederfindungsrate
WPC Whey Protein Concentrate/Molkenprotein-Konzentrat
WPH Whey Protein Hydrolysate/Molkenprotein-Hydrolysat
WPI Whey Protein Isolate/Molkenprotein-Isolat

XXIII
Formelzeichen
a Konstante (Charakterisierung der Strömungskanalgeometrie)
α‘ Exponent (Abhängig von Strömungsform)
β‘ Exponent (Abhängig von Strömungsform)
C Konzentration eines Stoffes
CF Konzentration des Feeds
CS Spektralphotometrisch ermittelte Konzentration des Mediums
CS-Ret spektralphotometrisch ermittelte Konzentration des Retentats
CS-Per spektralphotometrisch ermittelte Konzentration des Permeats
CS-vD Spektralphotometrisch ermittelte Konzentration des Feeds vor
Dialyse
CS-nD Spektralphotometrisch ermittelte Konzentration des Retentats
nach Dialyse
CM Konzentration an der Membranoberfläche
δ Dicke der Grenzschicht
D Diffusions-Koeffizient
dH Hydraulischer Durchmesser
ε Porosität der Membranoberfläche
fUS Frequenz des Ultraschalls
h Anzahl hydrolysierter Peptidbindungen
htot Gesamtzahl der Peptidbindungen pro Protein-Äquivalent
JF,t Permeatflux zum Messzeitpunkt t
JFm/ JFmOUS Mittlerer/durchschnittlicher Permeatflux (ohne den Einfluss von
Ultraschall)
JFmUS Mittlerer/durchschnittlicher Permeatflux unter Einfluss von
Ultraschall
JCF Critical Flux/Kritischer (Permeat-)Flux
JD Rückdiffusion von der Membran

XXIV
JK Konvektion zur Membran
JLF Limitierender Permeatflux
JV-W-max Permeatflux von dest. Wasser der performanter Vivaflow-
Einheit
JV-W-min Permeatflux von dest. Wasser der weniger performanten
Vivaflow-Einheit
JV-W-Initial Initialflux von dest. Wasser einer Vivaflow-Einheit
JV-W-W Permeatflux von dest. Wasser einer Vivaflow-Einheit vor Start
eines Wiederholungsversuchs
JV-Wvor-OUS Permeatflux von dest. Wasser einer neuen Membran (die ohne
die Verwendung von Ultraschall betrieben wird)
JV-Wvor-US Permeatflux von dest. Wasser einer neuen Membran (die unter
Verwendung von Ultraschall betrieben wird)
JWnach Permeatflux von dest. Wasser einer gereinigten Membran
JWvor Permeatflux von dest. Wasser einer neuen Membran
k Stoffübergangskoeffizient
mPer absolute Proteinmenge im Permeat
mRet absolute Proteinmenge im Retentat
mvD absolute Proteinmenge vor Dialyse
mnD absolute Proteinmenge im Retentat nach Dialyse
Mr Molmasse
η Dynamische Viskosität
pCP Critical Pressure/Kritischer Druck
pA Ausgangsdruck
pE Eingangsdruck
pF Filtratseitiger Druck
pTMP/∆pTMP Transmembrandruck
pTMP(ideal)/∆pTMP(ideal) Transmembrandruck, ideal

XXV
r Mittlerer Porenradius
RAD Widerstand durch Adsorption
Re Reynolds-Zahl
RF Widerstand durch Fouling
RiF Widerstand durch irreversibles Fouling
RKP Widerstand durch Konzentrationspolarisation
RM Membranwiderstand
RrF Widerstand durch reversibles Fouling
RT Kombinierter Filtrationswiderstand
Sc Schmidt-Zahl
Sh Sherwood-Zahl
∑ Summe
t Prozesslaufzeit
TCF Temperatur des Feeds
TOH Temperaturoptimum des Enzyms der Hydrolyse
tDL Dauer der Dialyse
tE Dauer der Enzymeinwirkung
tEV Dauer der enzymatischen Vorbehandlung
tF Unkorrigierte Prozesslaufzeit des Versuchs
tFm Mittlere/durchschnittliche korrigierte Prozesslaufzeit
tF-korrigiert Korrigierte Prozesslaufzeit des Versuchs
tVF Unkorrigierte Prozesszeit einer Vivaflow-Einheit nach
Performanceanpassung
tVF-korrigiert Korrigierte Prozesszeit einer Vivaflow-Einheit nach
Performanceanpassung
tr Retentionszeit
TSV Dauer der sauren Vorbehandlung (HCl, pH 3)

XXVI
TWH Temperatur des Wasserbads zur Temperierung des
Rotationsverdampfers
TWK Temperatur des Wasserbads zur Kondensation des verdampften
Wassers
ʋ Strömungsgeschwindigkeit
ϑ Kinematische Viskosität
VDia Diavolumen
VPer Volumen des Permeats
VPer,t Volumen des Permeats zum Messzeitpunkt t
VPer-ist Permeat-Volumen des Versuchs
VPer-max Maximale Permeatmenge einer Versuchsreihe
Vr Retentionsvolumen
VRet Volumen des Retentats
VV-Per-korrigert korrigiertes Permeatvolumen
VV-Per-korrigert,t korrigiertes Permeatvolumen zum Messzeitpunkt t
WFRRet Wiederfindungsrate im Retentat
WFRPer Wiederfindungsrate im Permeat
xA Abstand zur Membran
xM Porenlänge/Membrandicke
yExtinktion Spektralphotometrisch ermittelte Extinktion
yEx-vD Extinktion des Feeds vor Dialyse
.yEx-nD Extinktion des Retentats nach Dialyse
yEx-Ret Spektralphotometrisch ermittelte Extinktion des Retentats
yEx-Per Spektralphotometrisch ermittelte Extinktion des Permeats
γM Scherrate an der Membranoberfläche

XXVII
Kurzfassung
Bestimmte Peptidverbindungen besitzen Geschmacksqualitäten, welche sie für einen Einsatz
in Lebensmitteln qualifizieren. Neben einer chemischen Synthese bestimmter Verbindungen
können Peptide auch durch Hydrolyse intakter Proteine gewonnen werden. Zur Trennung und
Aufreinigung stellt die Crossflow-Filtrationstechnik eine skalierbare Alternative zu
chromatographischen Trennverfahren dar.
Im Rahmen dieser Arbeit wurde ein Crossflow-Diafiltrationsverfahren unter Verwendung
hydrophilisierter Polysulfon-Membranen (MWCO: 100 kDa) entwickelt, um hydrolysierte
Protein-Feeds aus hydrolysiertem Molkenprotein-Isolat, sowie hydrolysierter Alge Chlorella
vulgaris zu fraktionieren. Zur Optimierung des Fraktionierungsprozesses wurde der Einfluss
von pH-Wert und Ionenstärke (Anpassung durch Zugabe von 150 mM NaCl) auf Permeatflux,
Wiederfindungsrate, Transmission und die mittels SE-HPLC ermittelten molekulare
Zusammensetzung der Fraktionen beider Proteinquellen bestimmt. Ausgewählte Retentat-
und Permeatfraktionen wurden mittels eines im Zuge der Studie geschulten Sensorik-Panels
hinsichtlich der fünf Grundgeschmacksarten „süß“, „sauer“, „salzig“, „bitter“ und „umami“,
sowie ihrer Eignung als „bitter“-Blocker bewertet. Darüber hinaus wurde die Eignung von
Ultraschall (35 kHz) und die Verwendung alternativer Membranmaterialien zur Verbesserung
des Fraktionierungsprozesses, sowie die Eignung der Dialyse zur Abtrennung kleiner Moleküle
(< 2 kDa) aus dem vorfiltrierten Feed, überprüft.
Bei der Fraktionierung hydrolysierten Molkenprotein-Isolats wirkte sich der pH-Wert nicht
signifikant auf Permeatflux, Wiederfindungsrate und Transmission aus. Die Erhöhung der
Ionenstärke durch Zugabe von 150 mM NaCl resultierte in einem für die pH-Stufen pH 7, pH
8, pH 9 signifikant erhöhten Permeatflux. Die Wiederfindungsrate wurde in diesem Zuge für
die pH-Stufen pH 8, pH 9 jedoch signifikant verringert. Die chromatographisch ermittelte
Molekülgrößenverteilung der Fraktionen offenbarte einen weitestgehend pH-unabhängigen
Rückhalt von Strukturen ≥ 20 kDa im Retentat, bei gleichzeitig hohem Rückhalt von Strukturen
≤ 1,35 kDa, sowie geringe Anteile an Verbindungen ≥ 20 kDa im Permeat. Die Erhöhung der
Ionenstärke wirkte sich positiv auf den Anteil an Verbindungen ≥ 20 kDa im Permeat aus.
Bei der Fraktionierung hydrolysierter Alge Chlorella vulgaris wirkte sich der pH-Wert nicht
signifikant auf Permeatflux und Wiederfindungsrate aus. Die Erhöhung der Ionenstärke führte
für die pH-Stufen pH 7 und pH 11 zu signifikant geringerem Permeatflux. Unabhängig vom pH-
Wert wurden in Retentat und Permeat hauptsächlich Strukturen ≤ 1,35 kDa festgestellt. Die
Erhöhung der Ionenstärke führte für die pH-Stufen pH 3, pH 6 und, in geringem Umfang, pH 8
zu erhöhten Anteilen an Strukturen ≥ 20 kDa im Retentat.

XXVIII
Bei der sensorischen Bewertung der Fraktionen aus hydrolysiertem Molkenprotein-Isolat
wurde im Retentat ein sehr schwacher „süß“-, im Permeat ein schwacher „salzig“-Geschmack
identifiziert. Die Retentatfraktion hydrolysierter Alge Chlorella vulgaris wurden als schwach
„salzig“-, sowie sehr schwach „umami“ schmeckend, die Permeatfraktion als schwach „salzig“,
sowie sehr schwach „sauer“ und „umami“ beschrieben. Die „bitter“-Ausprägung des Koffein-
Standard (0,22 g/l) konnte durch Zugabe von Permeat (1g/l) signifikant reduziert werden.
Der Einsatz von Ultraschall (35 kHz), sowie alternativen Membranmaterialien erwies sich im
Rahmen dieser Studie als nicht für die Optimierung des Fraktionierungsprozess geeignet. Der
Anteil an Molekülen < 2 kDa konnten für Feed-Konzentration zwischen 1 % (w/v) und 4 % (w/v)
durch Dialyse verringert werden.

XXIX
Abstract
Certain peptide compounds have taste qualities which qualify them for the use in foods. In
addition to chemical synthesis of certain compounds, peptides can also be obtained by
hydrolysis of intact proteins. For separation and purification purposes, the crossflow filtration
technology represents a scalable alternative to chromatographic separation processes.
In this work, a crossflow diafiltration process, using hydrophilized polysulfone membranes
(MWCO: 100 kDa) was developed to fractionate hydrolysed protein feeds from hydrolyzed
whey protein isolate, as well as hydrolyzed alga chlorella vulgaris. The influence of pH and
ionic strength (adaptation by addition of 150 mM NaCl) on permeate flux, recovery,
transmission and molecular composition (determined by SE-HPLC) of the fractions of both
protein sources was determined to optimize the fractionation process. Selected retentate and
permeate fractions were evaluated with regard to the five basic tastes "sweet", "sour", "salty",
"bitter" and "umami" and their qualification as "bitter"-blocker, using a trained sensory panel.
In addition, the qualification of ultrasound (35 kHz) and the use of alternate membrane
materials to improve the fractionation process, as well as the qualification of dialysis for the
separation of small molecules (< 2 kDa) from the prefiltered feed was checked.
For hydrolyzed whey protein isolate fractionation, pH did not significantly affect permeate flux,
recovery and transmission. The increase of the ionic strength by addition of 150 mM NaCl
resulted in a significantly increased permeat flux for pH 7, pH 8 and pH 9. However, the
recovery rate was significantly reduced for pH 8 and pH 9. The chromatographically
determined molecular size distribution of the fractions revealed a largely pH-independent
retention of structures ≥ 20 kDa in the retentate, with simultaneously high retention of
structures ≤ 1.35 kDa and low proportion of compounds ≥ 20 kDa in the permeate. The
increase in the ionic strength had a positive effect on the proportion of compounds ≥ 20 kDa
in the permeate.
For hydrolyzed algae chlorella vulgaris fractionation, pH did not significantly affect permeate
flux and recovery. The increase of the the ionic strength led to significantly lower permeate flux
for pH 7 and pH 11. Regardless of the pH, mainly structures ≤ 1.35 kDa were found in the
retentate and permeate. The increase of the ionic strength led to increased proportions of
structures ≥ 20 kDa in the retentate for pH 3, pH 6 and, to a lesser extent, pH 8.
In the sensory evaluation of the fractions of hydrolysed whey protein isolate a very weak
"sweet" taste was noted in the retentate and a weak "salty" taste was identified in the permeate.
The retentate fraction of hydrolyzed algae chlorella vulgaris were described as weak "salty",
as well as very weak "umami", the permeate fraction was described as slightly "salty", as well

XXX
as very weak "sour" and "umami". The "bitter" expression of the caffeine standard (0.22 g/l)
could be significantly reduced by adding permeate (1 g/l).
The use of ultrasound (35 kHz), as well as alternate membrane materials, did not prove
suitable for optimizing the fractionation process in this study. The proportion of molecules
< 2 kDa could be reduced for feed concentration between 1 % (w/v) and 4 % (w/v) by dialysis.

Einleitung, Zielsetzung & Hypothesenformulierung
1
1. Einleitung, Zielsetzung & Hypothesenformulierung
1.1 Einleitung
Der Ersatz von Lebensmittelinhaltsstoffen wie Zucker oder Salz durch Stoffe mit
vergleichbaren Eigenschaften hat eine lange Tradition. Die bekanntesten Beispiele hierfür sind
die Zuckerersatzstoffe Saccharin und Aspartam, die 1878 bzw. 1965 entdeckt wurden, oder
das in der Europäischen Union (EU) im Jahre 2011 zugelassene Stevia (Buddrus, 2011;
Verordnung (EU) Nr. 1131/2011, 2011; Pilz, 2010). Ersatzstoffe verfügen, gestützt durch
ausgeklügelte Marketingstrategien, über ein großes Absatzpotential im Lebensmittelsektor
und rücken so immer weiter in den Mittelpunkt der Forschung (Pförtsch & Müller, 2006).
Gerade große Unternehmen sichern potentielle Stoffe gerne frühzeitig durch Patente ab
(Kienle, 2010).
Peptidverbindungen gewannen hierbei schon früh an Bedeutung, wie sich z. B. anhand von
Aspartam belegen lässt (Ternes, 2008; Pförtsch & Müller, 2006). Aber auch bisher kommerziell
nicht genutzte Verbindungen wie Brazzein sind bereits seit einiger Zeit Gegenstand der
Forschung (Temussi, 2002; Assadi-Porter, Aceti, Cheng, & Markley, 2000; Ming & Hellekant,
1994). Neben Süßstoffen und Bitterkomponenten lassen sich aus Peptiden und
Peptidverbindungen auch „umami“ und „sauer“ schmeckende Stoffe realisieren (Temussi,
2012; Kirimura, Shimizu, Kimizuka, Ninomiya, & Katsuya, 1969).
Der Einfluss der Peptidstruktur auf die Geschmacksqualitäten von Peptiden ist sehr komplex.
Bereits Kirimura et al. (1969) führten die verschiedenen Geschmacksausprägungen auf
unterschiedliche chemische Strukturen zurück. „sauer“ schmeckende Peptide sind
beispielsweise reich an Säureresten, bittere Peptide verfügen hingegen über einen hohen
Gehalt an hydrophoben Resten. Der Verlust der Peptid-Sekundärstruktur erhöht den
Bittergeschmack zusätzlich (Ternes, 2008). Mazur et al. (1969) belegten für den Süßstoff
Aspartam einen Einfluss der Peptidkonformation auf die Geschmacksqualität. Nur die L-
Konformation des Stoffes (L-aspartyl-L-phenylalanine methyl ester) verfügt über eine stark
süßende Wirkung. Alle weiteren chiralen Isomere schmecken bitter (Mazur et al., 1969).
Ähnliches scheint auch für andere, sowohl nieder- als auch hochmolekulare Verbindungen zu
gelten (Ternes, 2008). Solms et al. (1965) untersuchten 18 Aminosäuren auf, mit der
Konformation verknüpfte, Änderungen des Geschmacks und dokumentierte z.B. für L-Leucin
einen bitteren Geschmack, wohingegen D-Leucin süß schmeckt. Des Weiteren konnte bei
vorwiegend geschmacklosen Aminosäuren wie dem L-Arginin in der D-Form ein leicht süßer
Geschmack festgestellt werden (Solms et al., 1965).

Einleitung, Zielsetzung & Hypothesenformulierung
2
Neben Aspartam existieren auch noch weitere Aminoverbindungen mit hoher Süßkraft.
Darunter fallen Thaumatin, Monellin, Brazzein, Pentadin, Curculin und Mabinlin. Für die
Industrie sind hiervon insbesondere Thaumatin (hohe Thermostabilität), Brazzein
(kostengünstige Herstellung) und Curculin (Umwandlung von „sauer“ in „süß“) interessant
(Ternes, 2008).
Die Erzeugung und Erforschung von Proteinen deren Peptidfraktionen besondere
Geschmacksqualitäten aufweisen ist einer Vielzahl von Faktoren unterworfen, sodass neue
Stoffe oftmals nur zufällig entdeckt werden (Ley, 2008; Mazur et al., 1969). Neben der zumeist
aufwendigen, chemischen Synthese von Peptiden besteht die Möglichkeit verschiedenste
Peptide durch die Hydrolyse von intakten Proteinen zu erzeugen. Dabei entsteht ein Vielzahl
verschiedener Peptide unterschiedlicher chemischer Struktur (Wong & Whitesides, 1994; Jung
& Beck-Sickinger, 1992; Bodanszky, 1984). Die Fraktionierung dieser Peptide kann, in
Labormaßstab, beispielsweise mittels Gel-Permeation Chromatography (GPC) in Verbindung
mit Reversed Phase High Pressure Liquid Chromatography (RP-HPLC) erfolgen
(Schlichtherle-Cerny & Amadò, 2002). Im Hinblick auf eine großtechnische Anreicherung
stellen Membrantrennverfahren eine vielversprechende Alternative zu den technisch
aufwendigen Chromatographieverfahren dar.
Membrantrennverfahren finden bereits seit vielen Jahren in der Lebensmittelindustrie
Verwendung. Besonders im Bereich der Gewinnung von Molkenprotein-Konzentraten und der
Aufreinigung von Molke gibt es zahlreiche Veröffentlichungen, welche die Fortschritte
dokumentieren (Saxena, Tripathi, Kumar, & Shahi, 2009; Zydney, 1998). Für die
Fraktionierung intakter Proteine sind eine Vielzahl von Einflussfaktoren verifiziert. Durch
gezielte Steuerung von pH, Feed-Konzentration, Ionenstärke oder die Verwendung von
Ultraschall lassen sich Flux und Ausbeute verbessern, sowie unerwünschtes Fouling
reduzieren (Almécija, Ibáñez, Guadix, & Guadix, 2007; de la Casa, Guadix, Ibáñez, & Guadix,
2007; Huisman, Prádanos, & Hernández, 2000; Balakrishnan & Agarwal, 1996; Chai,
Kobayashi, & Fujii, 1998).
1.2 Zielsetzung & Hypothesenformulierung
Ziel des Forschungsvorhabens ist die Entwicklung einer Methode zur Fraktionierung von
hydrolysierten Proteinquellen. Hintergrund des Vorhabens ist die Überprüfung der Nutzung
eines Membrantrennverfahrens für die Gewinnung funktioneller Fraktionen aus
Proteinhydrolysaten. Zur Methodenerstellung werden Molkenprotein-Isolat-Hydrolysate
verwendet. Diese verfügen über eine standardisierte, von produzierenden Unternehmen
analytisch kontrollierte Zusammensetzung, sind kommerziell erhältlich, preiswert in der
Anschaffung und werden darüber hinaus von einer Vielzahl von Herstellern angeboten.

Einleitung, Zielsetzung & Hypothesenformulierung
3
Die bereits hydrolysierten Molkenprotein-Isolate werden unter Verwendung einer Kombination
von Crossflow-Filtrationstechnik und Diafiltration aufgetrennt. Anschließend erfolgt ein
Nachweis des Trennerfolges mittels Size Exclusion High Pressure Liquid Chromatography
(SE-HPLC). Die Trennung basiert hierbei nicht auf der Polarität oder Affinität des
Probenmaterials zur Säule, sondern auf einer Auftrennung der Peptidproben anhand deren
Molekül-Größenverteilung im Probenmedium (Meyer, 1988).
Nach erfolgter Etablierung einer Methode zur Fraktionierung von hydrolysierten
Molkenprotein-Isolaten mittels Crossflow Diafiltration erfolgt die Übertragung der Methode für
die Nutzung der Alge Chlorella vulgaris als Proteinquelle. In diesem Zuge ist die Entwicklung
von geeigneten Hydrolyseverfahren für die Alge Chlorella vulgaris erforderlich.
Die gewonnenen Fraktionen werden abschließend durch ein zu schulendes Panel sensorisch
auf ihre Geschmacksqualitäten überprüft. Besonderes Augenmerk liegt hierbei auf einer den
Bittergeschmack hemmenden Eigenschaft bestimmter Peptidfraktionen.
Im Rahmen der Studie sollen folgende Hypothesen überprüft werden:
Hypothese 1: Proteinhydrolysate lassen sich durch Filtration in Fraktionen auftrennen.
Experimenteller Ansatz: Per Crossflow Diafiltration sollen Peptidfraktionen erzeugt werden.
Die Filtration ist hinsichtlich ihrer Performance durch Herausarbeiten der optimalen
Betriebsbedingungen zu optimieren.
Hypothese 2: Durch Hydrolyse kann aus der Alge Chlorella vulgaris ein für die
Fraktionierung nutzbares Feed gewonnen werden.
Experimenteller Ansatz: Ziel des Hydrolyseprozesses ist die Erzeugung eines Hydrolysats mit
breiter Molekülgrößenverteilung durch Ermittlung der für diesen Zweck optimierten
Hydrolysebedingungen.
Hypothese 3: Die anhand von hydrolysiertem Molkenprotein-Isolat gewonnenen
Kenntnisse hinsichtlich der Fraktionierung lassen sich auf weitere Proteinquellen
übertragen.
Experimenteller Ansatz: Die Erkenntnisse aus der Molkenprotein-Isolat-Fraktionierung werden
auf die Proteinquelle Alge (Chlorella vulgaris) übertragen. Anpassungen werden, falls
notwendig, vorgenommen.

Einleitung, Zielsetzung & Hypothesenformulierung
4
Hypothese 4: Einzelne Peptid-Fraktionen verfügen über unterschiedliche
Geschmacksqualitäten.
Experimenteller Ansatz: Die Peptidfraktionen werden durch ein zu schulendes Sensorik-Panel
auf ihre Geschmacksqualitäten überprüft. Die Schulung erfolgt in Übereistimmung mit den
gängigen ISO-Normen.
Hypothese 5: Eine Peptidfraktion besitzt die Eigenschaft eines Bittergeschmack-
Blockers.
Experimenteller Ansatz: Ein Bitterstandard wird mit einer Probe Bitterstandard + Peptidfraktion
hinsichtlich des Geschmackseindrucks „bitter“ durch ein geschultes Sensorik-Panel
verglichen. Stellt das Panel einen signifikant (p < 0,05) geringeren Bittergeschmack fest,
verfügt die Peptidfraktion über die Funktion eines Bittergeschmack-Blockers.
Hypothese 6: Die Peptidfraktionen lassen sich mit der erarbeiteten Methode in
produktionstauglichem Maßstab herstellen.
Experimenteller Ansatz: Die erarbeitete Methode wird anhand der Ergebnisse hinsichtlich ihrer
Skalierbarkeit bewertet.

Stand der Forschung
5
2. Stand der Forschung
2.1 Filtrations- und Fraktionierungstechnik
Grundlegend lassen sich Separationsverfahren anhand ihrer Trenneigenschaften einteilen. Je
nach Trennbereich erfolgt die Klassifizierung anhand der Porengröße der Membran bzw. dem
Molecular Weight Cut-Off (MWCO) (Dosmar & Pinto, 2007; Kofod Nielsen, 2004; Bhave,
1997). Der MWCO ist definiert als die kleinste Molekülmasse einer Struktur, welche durch die
verwendete Membran zu 90 – 95 % (je nach Membran und Hersteller) zurückgehalten wird
(Melin & Rautenbach, 2007). Eine übersichtliche Klassifizierung der Trennverfahren ist in
Abbildung 1 dargestellt. Im Rahmen dieser Arbeit wurde aufgrund der
Molekülgrößenverteilung der Hydrolysate im Ultra- (UF) bzw. Nanofiltrationsbereich (NF)
gearbeitet.
Abbildung 1: Klassifizierung der Trennverfahren (Kofod Nielsen, 2004; modifiziert)
Membrantrennverfahren im Mikro- (MF), Ultra- und Nanofiltrationsbereich haben sich im Life
Science Bereich als Trennprozess zur Produktaufreinigung und Rückgewinnung etabliert
(Schuchmann & Schuchmann, 2005). Besonderes Augenmerk der Forschung gilt der

Stand der Forschung
6
Verbesserung der Selektivität membranbasierter Trennprozesse (Saxena et al., 2009; van
Reis & Zydney, 2001). Basierend auf den Forschungserfolgen der vergangenen Jahre steigen
die an Membrantrennverfahren gestellten Anforderungen. Neben der Aufkonzentration
bestimmter Proteine spielen Fraktionierungsprozesse komplexer werdender Feed-Medien,
wie beispielsweise Proteinhydrolysaten, zunehmend eine tragende Rolle (Butylina, Luque, &
Nyström, 2006; Wijers, Pouliot, Gauthier, Pouliot, & Nadeau, 1998).
Als unverzichtbarer Baustein des Fortschritts in der Filtrationstechnik kristallisierte sich die
Crossflow Filtration heraus, die gegenüber der Dead-End Filtration eine Vielzahl an Vorteilen
hinsichtlich Effektivität und Effizienz bietet (Melin & Rautenbach, 2007; Kofod Nielsen, 2004;
Irmler, 2001).
2.1.1 Die Crossflow Filtrationstechnik
Bei der Crossflow Filtration (CFF), stellenweise auch Tangentialfluss Filtration (TFF) genannt,
handelt es sich um ein druckgetriebenes Membrantrennverfahren (Bhave, 1997; Aimar,
Meireles, Bacchin, & Sanchez, 1994). Im Gegensatz zur Dead-End Filtration, dargestellt in
Abbildung 2, wird der Feed-Strom tangential zur Membran geführt (Dosmar & Pinto, 2007;
Bhave, 1997).
Abbildung 2: Gegenüberstellung - Crossflow Filtration und Dead-End Filtration (Bhave, 1997)
Es erfolgt eine Auftrennung des Feed-Stroms in zwei Fraktionen; Permeat und Retentat. Als
Permeat bezeichnet man den Anteil des ursprünglichen Feeds, der die Membran passiert
(siehe Abbildung 2). Der über der Membran verbleibende Anteil wird als Retentat bezeichnet
(Melin & Rautenbach, 2007; Cheremisinoff, 1998; Bhave, 1997).

Stand der Forschung
7
Die tangentiale Betriebsweise bietet den Vorteil, dass Partikel auf der Membranoberfläche
durch strömungsbedingte Scherkräfte abgetragen werden können. Unter idealisierten
Bedingungen ist die Filtration folglich nicht weiter von Kompressibilität und Größe der Partikel
anhängig, sondern lediglich von der tangentialen Fließgeschwindigkeit (Aimar et al., 1994).
In der Praxis lässt sich die Vermeidung einer Deckschichtbildung durch Fouling und
Konzentrationspolarisation jedoch nicht realisieren (Melin & Rautenbach, 2007; Aimar et al.,
1994).
2.1.2 Fouling und Konzentrationspolarisation
Die Ausbildung einer Deckschicht auf Membranoberflächen stellt für die Filtrationstechnik ein
ernstzunehmendes Problem dar, da es unter anderem zu Veränderungen der
Membranselektivität und Abnahme des Flüssigkeitstransports durch Membran, auch Fluss
oder Flux genannt, kommen kann (Kofod Nielsen, 2004; Hiersig, 1995; Ousman & Bennasar,
1995). Für membranbasierte Trennaufgaben führen deckschichtinduzierte Veränderungen der
Leistungsfähigkeit zu erhöhten Produktionskosten und geringem Durchsatz (Choi, Zhang,
Dionysiou, Oerther, & Sorial, 2005; Ousman & Bennasar, 1995). Trotz intensiver Forschungen
sind die komplexen Vorgänge der Deckschichtbildung nicht vollständig charakterisiert (Hiersig,
1995).
Grundsätzlich unterteilt man die Deckschichtbildung in den reversiblen Prozess der
Konzentrationspolarisation und den irreversiblen Prozess des Fouling (Chen, Fane, Madaeni,
& Wenten, 1997; Balakrishnan & Agarwal, 1996; Hiersig, 1995). Der Übergang zwischen
beiden Phänomenen ist, trotz abgegrenzter Definitionen, schwer zu bestimmen, sodass einige
Autoren neben dem reversiblen Prozess der Konzentrationspolarisation auch von reversiblen
und irreversiblem Fouling sprechen (Choi et al., 2005; Cheryan, 1998; Chen et al., 1997).
2.1.2.1 Die Konzentrationspolarisation
Unter dem Begriff „Konzentrationspolarisation“ versteht man die Aufkonzentration
zurückgehaltener Moleküle des Feeds auf der Retentatseite der Membran. Es kommt zur
Ausbildung eines Konzentrationsgradienten zwischen Membranoberfläche und Feed. Hierbei
ist die Konzentration gelöster Stoff, wie in Abbildung 3 dargestellt, nahe der Membran am
höchsten (Foley, 2013; Hiersig, 1995; Zydney & Colton, 1986).

Stand der Forschung
8
Abbildung 3: Schematische Darstellung des Konzentrationsprofils an der Membranoberfläche (Hiersig, 1995)
Mit steigendem Transmembranflux steigt, abhängig von der Feed-Konzentration, die
Konzentration gelöster Stoffe an der Membranoberfläche. Die dadurch entstehende
bewegliche Grenzschicht wächst bis zum Erreichen eines stationären Zustands und führt zu
einer Abnahme des Flux und zu einer Verschlechterung des Trennprozesses (Shirazi, Lin, &
Chen, 2010; Toledo, 2004; Hiersig, 1995; Ousman & Bennasar, 1995). Im stationären Zustand
wird von einem Gleichgewicht zwischen einer Konvektion zur Membran mit anschließender
Permeation und einer Rückdiffusion ins Feed ausgegangen (Foley, 2013; Zydney & Colton,
1986). Durch hohe Strömungsgeschwindigkeiten an der Membranoberfläche lässt sich, in
Verbindung mit optimiertem Transmembranflux, eine Verringerung der beweglichen
Grenzschichtdicke durch Scherkräfte erreichen (Toledo, 2004; Chen et al., 1997).
Legende: 𝐷 ×∆𝐶
∆𝑦 = Rückdiffusion, 𝐽 × 𝐶 × (1 − Φ) = Transmission, 𝐽 = Flux, 𝐶 = Feed-Konzentration, 𝛿fl = Dicke
„beweglicher Filterkuchen“, 𝛿st = Dicke „unbeweglicher Filterkuchen“
Abbildung 4: Schematische Darstellung der Konzentrationspolarisation (Chen et al., 1997; modifiziert)

Stand der Forschung
9
Wird das Gleichgewicht zwischen Konvektion zur Membran, Rückdiffusion und
scherkraftbedingtem Abtransport von Molekülen an der Membranoberfläche, dargestellt in
Abbildung 4, verschoben, kommt es zu einer Zunahme der beweglichen Grenzschichtdicke,
deren Verdichtung und, gegebenenfalls, zur Ausbildung einer unbeweglichen Ablagerung auf
der Membran (Toledo, 2004; Chen et al., 1997; Hiersig, 1995). Durch
Konzentrationspolarisation induzierte Veränderungen des Filtrationsprozesses, treten in der
Regel sehr schnell auf, oftmals in weniger als einer Minute nach Filtrationsbeginn (Chudacek
& Fane, 1984). Zur mathematischen Charakterisierung der Konzentrationspolarisation
existieren mehrere Modelle, von denen das „Gel-Polarisation Modell“ in Kapitel 2.1.3.2
vorgestellt wird.
2.1.2.2 Das Fouling
Ablagerungen von gelösten Bestandteilen auf Membranoberflächen oder in den Poren der
Membran bezeichnet man als Fouling (Toledo, 2004; Aimar et al., 1994). Dabei unterschiedet
man nach Wang & Tarabara (2008) und Bowen, Calvo, & Hernández (1995) zwischen den in
Abbildung 5 dargestellten Mechanismen des
„Complete Blocking“, der vollständigen Blockierung von Membranporen durch geloste
Stoffe ohne Partikelüberlagerung,
„Standard Blocking“, der Ablagerung von Partikeln in den Membranporen, welche zu
einer Abnahme des Porendurchmessers proportional zur Partikelgröße des
angelagerten Moleküls führt,
„Intermediate Blocking“, der vollständigen Blockierung einzelner Membranporen
(vergleichbar mit „Complete Blocking“) in Verbindung mit Verblockungseffekten, die
durch eine Auf- und Aneinanderlagerung von Partikeln entsteht und der
„Cake Filtration“, der Ausbildung einer dichten Partikelschicht auf der
Membranoberfläche.
Legende: a) „Complete Blocking“, b) „Standard Blocking“, c) „Intermediate Blocking“, d) „Cake Filtration“
Abbildung 5: Schematische Darstellung der Foulingmechanismen (Wang & Tarabara, 2008; modifiziert)

Stand der Forschung
10
Es ist davon auszugehen, dass die Abnahme des Flux während eines Filtrationsprozesses
nicht nur auf einen Foulingmechanismus zurückzuführen ist, sondern auf einem
Zusammenspiel der verschiedenen Mechanismen beruht. Dabei kommt es zu Beginn zu einer
Verblockung der kleinsten Poren und im Verlauf zu einer Ablagerung von Partikeln in größeren
Poren. Es wird davon ausgegangen, dass sogar Partikel, die ein oder zwei Größenordnungen
unter der Porenweite der Membran liegen, durch Adsorption an den Porenwänden signifikant
zum Foulingprozess beitragen. Weiter kommt es zu Ab- und Überlagerung von Partikeln auf
der Membranoberfläche, die schlussendlich in der Ausbildung eines Filterkuchens resultieren
(Güell & Davis, 1996; Bowen et al., 1995). Diese Prozesse laufen, aufgrund der mehr oder
weniger unregelmäßigen Verteilung der Porengröße, die von Hersteller zu Hersteller variieren
kann, überlagert ab (Balakrishnan & Agarwal, 1996; Bowen et al., 1995). Vereinfacht
unterscheidet man zwischen internem Fouling, darunter fallen „Complete Blocking“ und
„Standard Blocking“ und externem Fouling durch „Intermediate Blocking“ und „Cake Filtration“
(Shirazi et al., 2010; Tracey & Davis, 1994).
Zum besseren Verständnis der Einflüsse von Konzentrationspolarisation und Fouling auf den
Crossflow Prozess wird dieser nachfolgend mathematisch näher charakterisiert (Kapitel 2.1.3).
Im Anschluss wird der Einfluss der Prozess-Hydrodynamik und der physikalisch-chemischen
Eigenschaften von Membran und Feed auf die Deckschichtbildung dargestellt (Kapitel 2.1.4).
2.1.3 Mathematische Charakterisierung von Filtrationsprozessen
2.1.3.1 Idealisierte Charakterisierung von Filtrationsprozessen - Das Hagen-Poiseulle-Model
Der Transport einer Flüssigkeit durch eine Membran, lässt sich für einen idealisierten Fall, in
der Abwesenheit einer gelösten Substanz, mit der Hagen-Poiseulle Gleichung beschreiben
(Dosmar & Pinto, 2007):
𝐽𝐹,𝑡 =휀 × 𝑟4 × ∆𝑝𝑇𝑀𝑃(𝑖𝑑𝑒𝑎𝑙)
8 × 𝜂 × ∆𝑥𝑀
Formel 1: Hagen-Poiseulle Gleichung – Flux durch Membran, ideal
JF,t = Flux durch die Membran zum Zeitpunkt t
ε = Porosität der Membranoberfläche
r = Mittlerer Porenradius
∆pTMP(ideal) = Transmembrandruck, ideal
η = dynamische Viskosität der Flüssigkeit
xM = Länge der Pore/Dicke der Membran

Stand der Forschung
11
Der Transmembrandruck ∆pTMP(ideal) ist dabei definiert als (Irmler, 2001):
∆𝑝𝑇𝑀𝑃(𝑖𝑑𝑒𝑎𝑙) = 𝑝𝐸 + 𝑝𝐴
2 × 𝑝𝐹
Formel 2: Transmembrandruck, ideal
∆pTMP(ideal) = Transmembrandruck, ideal
pE = Eingangsdruck
pA = Ausgangsdruck
pF = filtratseitiger Druck
Im Idealfall, bei gleichbleibender Viskosität η der Flüssigkeit, ist der Flux durch die Membran
JF,t folglich proportional zum Transmembrandruck ∆pTMP(ideal) (Dosmar & Pinto, 2007; Irmler,
2001).
Unter realen Bedingungen manifestiert sich bei zunehmendem Transmembrandruck eine
Abweichung von der linearen Zunahme des Flux. Diese ist auf die Konzentrationspolarisation
und Fouling auf der Membranoberfläche zurückzuführen (Irmler, 2001; Cheryan, 1990).
2.1.3.2 Charakterisierung der Konzentrationspolarisation – Das Gel-Polarisation-Modell
Die Grundlage für die Charakterisierung der Konzentrationspolarisation ist die Massenbilanz
für den Transport von suspendierten Stoffen an der Grenzschicht der Flüssigkeit zur
Membranoberfläche (Shirazi et al., 2010; Toledo, 2004). Im stationären Zustand wird davon
ausgegangen, dass ein Gleichgewicht zwischen Konvektion zur Membran JK und
Rückdiffusion JD in das Feed stattfindet (Foley, 2013; Shirazi et al., 2010; Zydney & Colton,
1986):
𝐽𝐹,𝑡 × 𝐶𝐹 = − 𝐷 ×∆𝐶
∆𝑥𝐴
Formel 3: Massenbilanz der Konzentrationspolarisation
JF,t * CF = Konvektion JK zur Membran mit Konzentration CF des Feeds
D * ∆C/∆x = Rückdiffusion JD von der Membran
D = Diffusion-Koeffizient
xA = Abstand zur Membran

Stand der Forschung
12
Integriert man Formel 3 über die Dicke δ der Grenzschicht ergibt sich für den Permeatflux
durch die Membran (Toledo, 2004; Zydney & Colton, 1986):
𝐽𝐹,𝑡 = 𝐷
𝛿× 𝑙𝑛 (
𝐶𝑀
𝐶𝐹) = 𝑘 × 𝑙𝑛 (
𝐶𝑀
𝐶𝐹)
Formel 4: Permeatflux JP,t bei Konzentrationspolarisation nach dem Gel-Polarisations-Modell
JF,t = Flux durch die Membran zum Zeitpunkt t
D = Diffusion-Koeffizient
δ = Dicke der Grenzschicht
CM = Konzentration an der Membranoberfläche
CF = Konzentration im Feed
k = Stoffübergangskoeffizient
Der Stoffübergangskoeffizient k ist dabei nährungsweise, in Analogie zum Wärmeübergang
an undurchlässigen Oberflächen, definiert als (Shirazi et al., 2010; Toledo, 2004; Zydney &
Colton, 1986):
𝑘 = 0,538 × (𝐷2 × 𝛾𝑀
𝑥𝑀)
13
Formel 5: Stoffüberhangskoeffizient k
D = Diffusion-Koeffizient
γM = Scherrate an der Membranoberfläche
xM = Porenlänge
Für höhere Membranflüsse ist eine Anpassung des Stoffübergangskoeffizienten k notwendig.
Abhängig von der Geometrie der Poren und der Strömungsform existieren hierfür in der
Literatur verschiedene Modelle, die prozessspezifisch angepasst werden müssen (Melin &
Rautenbach, 2007). Soll der Einfluss der Strömungsgeschwindigkeit bei der Bestimmung des
Stoffübergangskoeffizienten miteinbezogen werden, eignet sich dafür die Verwendung der
Sherwood-Zahl. Hierbei wird deutlich, dass eine Steigerung des Permeatflusses in
Abhängigkeit der Re Zahl, sprich idealerweise im turbulenten Bereich, erreicht werden kann
(Shirazi et al., 2010; Melin & Rautenbach, 2007; Toledo, 2004; Matthiasson & Sivik, 1980):

Stand der Forschung
13
𝑆ℎ = 𝑘 × 𝑑ℎ
𝐷= 𝑎 × (
𝑑ℎ × 𝑉
𝜗)
𝑎′
× (𝜗
𝐷)
𝛽′
= 𝑎 × 𝑅𝑒𝛼′ × 𝑆𝑐𝛽′
Abbildung 6: Sherwood-Zahl zur Charakterisierung des Stoffübergangs
Sh = Sherwood Zahl
Re = Reynolds Zahl
Sc = Schmidt Zahl
D = Diffusion-Koeffizient
k = Stoffübergangskoeffizient
dh = hydraulischer Durchmesser
a = Konstante (Charakterisierung der Strömungskanalgeometrie)
V = Strömungsgeschwindigkeit
ϑ = kinematische Viskosität
α‘ = Exponent (Abhängig von Strömungsform)
β‘ = Exponent (Abhängig von Strömungsform)
Grundlage für die Gültigkeit des Gel-Polarisation Modells ist die Annahme eines konstanten
Widerstands der Grenzschicht und der Membran. Des Weiteren wird die Konzentration an der
Membranoberfläche CM als unveränderliche Größe vorausgesetzt (Shirazi et al., 2010). Dies
entspricht allerdings nicht den Realbedingungen, da die Konzentration CM sowohl von der
Konzentration CF des Feeds, als auch der Strömungsgeschwindigkeit abhängig ist (Mulder,
1996). Auch das Diffusionsvermögen von Makromolekülen ist konzentrationsabhängig,
wodurch die Annahme eines konstanten Stoffübergangskoeffizienten k zu Ungenauigkeiten
führt (Shirazi et al., 2010).
2.1.3.3 Charakterisierung druckgetriebener Filtrationsprozesse unter Berücksichtigung
prozessspezifischer Widerstände – Das Widerstands-Modell
Für die Charakterisierung druckgetriebener Filtrationsverfahren hat sich die Verwendung des
Widerstandsmodells etabliert, da Flux-Transmembrandruck abhängige Fouling-Phänomene
mit dem Diffusionsmodell der Gel-Polarisation nicht dargestellt werden können (Melin &
Rautenbach, 2007; Choi et al., 2005; Sablani, Goosen, Al-Belushi, & Wilf, 2001). Hierbei wird
angenommen, dass die Ausbildung einer Deckschicht in einem zusätzlichen, in Reihe zum
Membranwiderstand geschalteten Widerstand, resultiert (Shirazi et al., 2010; Melin &
Rautenbach, 2007; Choi et al., 2005; Mulder, 1996). Der kombinierten Filtrationswiderstand
RT ergibt sich aus der Summe der Einzelwiderstände, bestehend aus dem
Membranwiderstand RM, dem Widerstand durch Fouling RF, dem Widerstand durch
Konzentrationspolarisation RKP und dem Widerstand durch Adsorption RAD (Choi et al., 2005):

Stand der Forschung
14
𝑅𝑇 = 𝑅𝑀 + 𝑅𝐹 + 𝑅𝐾𝑃 + 𝑅𝐴𝐷
Formel 6: Filtrationswiderstand RT
RT = kombinierter Filtrationswiderstand
RM = Membranwiderstand
RF = Widerstand durch Fouling
RKP = Widerstand durch Konzentrationspolarisation
RAD = Widerstand durch Adsorption
Für den Flux JF,t ergibt sich unter Berücksichtigung der Widerstände (Choi et al., 2005; Choo
& Lee, 1996; Ousman & Bennasar, 1995):
𝐽𝐹,𝑡 = ∆𝑝𝑇𝑀𝑃
𝜂 × (𝑅𝑀 + 𝑅𝐹 + 𝑅𝐾𝑃 + 𝑅𝐴𝐷)
Formel 7: Darcy-Gleichung - Flux durch Membran in Abhängigkeit fluxreduzierender Parameter
JF,t = Flux durch die Membran zum Zeitpunkt t
∆pTMP = Transmembrandruck
η = dynamische Viskosität des Permeat
RM = Membranwiderstand
RF = Widerstand durch Fouling
RKP = Widerstand durch Konzentrationspolarisation
RAD = Widerstand durch Adsorption
Der durch Fouling erzeugte Widerstand RF kann wiederum in reversibles und irreversibles
Fouling unterteilt werden (Choi et al., 2005):
𝑅𝐹 = 𝑅𝑟𝐹 + 𝑅𝑖𝐹
Formel 8: Widerstand durch Fouling
RF = Widerstand durch Fouling
RrF = Widerstand durch reversibles Fouling
RiF = Widerstand durch irreversibles Fouling
2.1.4 Einfluss von Prozess-Hydrodynamik und physikalisch-chemischen
Eigenschaften von Feed und Membran auf Fouling und Filtrationsprozess
Das Auftreten von Fouling in Membranfiltrationsprozessen ist durch eine Vielzahl von Faktoren
beinflussbar. Grundlegend kann man zwischen Fouling aufgrund der Hydrodynamik des
Prozesses und Fouling hervorgerufen durch die physikalisch-chemischer Eigenschaften von
Membran und Feed unterscheiden (Huisman et al., 2000).

Stand der Forschung
15
2.1.4.1 Fouling aufgrund der Prozess-Hydrodynamik
Transmembrandruck ∆pTMP/Permeatflux JF,t/JFm:
Für Reinstmedien steigt der Permeatflux JFm, in Übereinstimmung mit den theoretischen
Annahmen, proportional zum Transmembrandruck ∆pTMP. Bei beladenen Medien zeigt sich,
abhängig von Feed-Konzentration und Strömungsgeschwindigkeit, eine Abweichung vom
linearen Zusammenhang des Permeatflux JF,t bei steigendem Transmembrandruck ∆pTMP,
dargestellt in Abbildung 7 (Kofod Nielsen, 2004; Irmler, 2001; Lojkine, Field, & Howell, 1992).
Abbildung 7: Einfluss einer Deckschichtausbildung auf den Flux JF,t in Abhängigkeit des Transmembrandrucks ∆pTMP für Reinstwasser und ein beladenes Medium (Irmler, 2001; modifiziert)
Die Abnahme des Permeatflux JFm ist hierbei im Anfangsstadium auf die Ausbildung einer
Konzentrationspolarisation an der Membranoberfläche, und im weiteren Verlauf durch die
Ausbildung einer irreversiblen Foulingschicht auf der Membran zurückzuführen (Taddei,
Daufin, Aimar, & Sanchez, 1989). Die Ausbildung von Fouling kann, gemäß der „Critical-Flux“-
Theorie von Howell (1995) und Field, Wu, Howell, & Gupta (1995), durch nicht-überschreiten
eines kritischen Flux JCF verhindert werden. Als kritischen Flux JCF bezeichnet man den
Permeatflux JF, bei dessen Überschreiten es zur einer Abweichung vom linearen
Transmembrandruck-Flux Zusammenhang bzw. zur Ausbildung einer wahrnehmbaren
Fouling-Schicht kommt (Bacchin, Aimar, & Field, 2006; Field et al., 1995; Howell, 1995). Eine
weitere Möglichkeit der Prozessführung ist die Verwendung eines konstanten
Transmembrandrucks ∆pTMP. Unterhalb eines kritischen Drucks pCP entspricht der kritische
Flux JCF weitestgehend dem limitierend Flux JLF. Unter limitierendem Flux JLF versteht man den
Flux, für den eine Steigerung des Transmembrandrucks ∆pTMP nicht länger in einem Anstieg
des Permeatflux resultiert (Vyas, Bennett, & Marshall, 2000; Defrance & Jaffrin, 1999).
Betrachtet man die in Abbildung 8 dargestellte Transmembrandruck-Flux-Abhängigkeit näher,
Transmembrandruck ∆pTMP [bar]
Flu
x J
F,t [
l/m
2h]

Stand der Forschung
16
ist eine Einteilung in drei Bereiche möglich (Brans, Schroën, van der Sman, & Boom, 2004;
Field et al., 1995).
Abbildung 8: Kritische Flux Bereiche (Brans et al., 2004, modifiziert)
Bereich I ist gekennzeichnet durch einen linearen Anstieg des Transmembrandruck/Flux-
Verhältnisses bis zum Erreichen des kritischen Flux JCF/kritischen Druck pCP (Brans et al.,
2004). Eine weitere Unterteilung in eine „harte/starken“ und „weiche/schwachen“ Form,
dargestellt in Abbildung 9, ist möglich. Bei der „harten/starken“ Form steigt das Flux-Druck-
Verhältnis analog zum Reinstwasserflux bis JCF bzw. pCP erreicht wird. Bei der
„weichen/schwachen“ Form wird ebenfalls von einem linearen Anstieg bis zum Erreichen von
JCF bzw. pCP ausgegangen, jedoch weicht das Flux/Druck-Verhältnis aufgrund einer
unverzüglich auftretenden, fouling-/konzentrationspolarisation-induzierten Fluxminderung
bereits zu Beginn vom Reinstwasserflux ab (Bacchin et al., 2006; Brans et al., 2004; Field et
al., 1995).
In Bereich II ist der limitierende Flux erreicht. Durch die konzentrationspolarisation-induzierte
Ausbildung einer stationären Grenzschicht kommt es zur Stagnation des Permeatflux und
somit zu einem Übergang vom druckabhängigen Bereich in den deckschichtabhängigen
Bereich des Filtrationsprozesses. Bei weiterem Anstieg des Transmembrandrucks ∆pTMP,
charakterisiert als Bereich III, bildet sich Fouling auf der Membranoberfläche, welches zur
einer drastischen Reduzierung des Permeatflux und der Selektivität der Membran, unter
anderem durch eine druckabhängige Kompression des Filterkuchens, führt (Brans et al., 2004;
Field et al., 1995). Entscheidend für eine Crossflow Filtration ist das Operieren unterhalb des
deckschichtkontrollierten Bereichs durch Bestimmung des kritischen Flux JCF oder kritischen
Druck pCP der Anlagenkonzeption.

Stand der Forschung
17
Abbildung 9: "harte/starke" und "weiche/schwache" Form des kritischen Flux JCF (Bacchin et al., 2006)
Strömungsgeschwindigkeit ʋ:
Ein weiterer Parameter zur Kontrolle des Fouling-Prozesses und der
Konzentrationspolarisation ist die Strömungsgeschwindigkeit ʋ des Crossflow-Prozesses
(Melin & Rautenbach, 2007; Atra, Vatai, Bekassy-Molnar, & Balint, 2005; Punidadas & Rizvi,
1998). Abhängig von der Strömungsgeschwindigkeit ʋ kommt es zur Ausbildung von
Scherkräften auf der Membranoberfläche, durch die ein Abtragen der Deckschicht möglich ist.
Der Permeatflux ist umso höher, je höher die Scherkräfte sind, da die Rückdiffusion von
Partikelen in das Feed verbessert wird (Melin & Rautenbach, 2007; Wang & Song, 1999).
Insbesondere der Rücktransport größerer Partikel kann, bei gesteigerter
Strömungsgeschwindigkeit ʋ in Verbindung mit dem Transmembrandruck ∆pTMP, allerdings zu
einer Verminderung der Deckschichtporosität führen, wodurch der Deckschichtwiderstand
ansteigt (Fane, Beatson, & Li, 2000; Lojkine et al., 1992). Darüber hinaus ist bei hohen
Strömungsgeschwindigkeiten eine Denaturierung der Proteine möglich, wie Meireles, Aimar,
& Sanchez (1991) für Bovin-Serum-Albumin (BSA) zeigten.
2.1.4.2 Fouling aufgrund der physikalisch-chemischen Eigenschaften von Feed und
Membran
pH-Wert & Ionenstärke:
Der Einfluss des pH-Wertes auf den Fouling-Prozess ist seit langem intensiver Gegenstand
der Forschung (Wang & Tang, 2011; She, Tang, Wang, & Zhang, 2009; Almécija et al., 2007;
Persson, Jönsson, & Zacchi, 2003; Chan & Chen, 2001; Huisman et al., 2000; Herrero,
Prádanos, Calvo, Tejerina, & Hernández, 1997; Fane, Fell, & Suki, 1983). Kernaussage vieler
Studien ist, dass starkes Fouling in Verbindung mit niedrigem Permeatflux grundsätzlich in der

Stand der Forschung
18
Nähe des isoelektrischen Punkts (IEP) auftritt (Wang & Tang, 2011; She et al., 2009; Huisman
et al., 2000; McDonogh, Bauser, Stroh, & Chmiel, 1990; Fane et al., 1983). Sowohl über- als
auch unterhalb des IEP ist eine vergleichsweise reduzierte Auswirkung von Fouling auf den
Permeatflux JF zu beobachten (Wang & Tang, 2011; She et al., 2009; Palecek, Mochizuki, &
Zydney, 1993). Grund für verstärktes Membranfouling im Bereich des IEP sind vor allem
Veränderungen des Ladungszustandes von Proteinen und Peptiden. Diese führen durch
verstärkte hydrophobe Interaktion zur Adsorption von Molekülen auf der Membran (Ricq,
Narçon, Reggiani, & Pagetti, 1999; Martin-Orue, Bouhallab, & Garem, 1998; Nyström,
Pihlajamäki, & Ehsani, 1994). Des Weiteren kann es durch die am IEP reduzierte
elektrostatische Abstoßung zur aggregationsbedingtem Fouling kommen (Wang & Tang,
2011; She et al., 2009; Kelly & Zydney, 1995). Der pH-Wert beeinflusst, in Verbindung mit der
Partikelgrößenverteilung, auch die Struktur der Fouling-Schicht (Huisman et al., 2000; Vyas et
al., 2000). Huisman et al. (2000) charakterisierten anhand von rasterkraftmikroskopischen
Aufnahmen die Struktur von BSA-Foulingschichten. Eine besonders dichte, unregelmäßige
Foulingschicht manifestierte sich am IEP, wohingegen niedrige pH-Werte zur Ausbildung einer
lockeren Deckschicht führten. In Verbindung mit dem pH-Wert spielt die Ionenstärke des
Feeds eine wichtige Rolle bei der Entstehung und Strukturbildung der Deckschicht. Eine
allgemeingültige Aussage ist hinsichtlich widersprüchlicher Forschungsergebnisse jedoch
nicht uneingeschränkt möglich. In vielen Fällen wird von einer Zunahme des Fouling-Effekts
bei steigender Ionenstärke berichtet (Teng, Lin, Wu, & Juang, 2006; Tarabara, Koyuncu, &
Wiesner, 2004; Palecek & Zydney, 1994; Heinemann, Howell, & Bryan, 1988; Fane et al.,
1983). Vereinzelt berichten Autoren auch von gegenteiligen Effekten (Salgın, 2007; She et al.,
2009; Chan & Chen, 2001). Aussagen bezüglich des Einflusses der Ionenstärke müssen
folglich einer ganzheitlichen Betrachtung, in Verbindung mit weiteren Prozessparametern,
unterzogen werden. Zudem spielt die Zusammensetzung des Feeds, die Art der Membran und
das Trennziel eine wichtige Rolle. Bei der Filtration von BSA resultierten erhöhte Ionenstärken
in Verbindung mit einem pH-Wert in der Nähe des IEP beispielsweise in einer verbesserten
Transmission der BSA-Moleküle (de la Casa et al., 2007; Persson et al., 2003). Für komplexere
Feed-Medien hat sich zur Verbesserung der Membran-Selektivität eine pH-Führung am IEP
der niedermolekularen Feed-Bestandteile und möglichst weit entfernt vom IEP der
höhermolekularen Bestandteile bewährt (Zydney, 1998; Saksena & Zydney, 1994; Nakatsuka
& Michaels, 1992). Hierbei wird der Einfluss der Ionenstärke auf die elektrochemische
Doppelschicht eines geladenen Proteins ausgenutzt. Mit steigender Ionenstärke nimmt deren
Dicke ab, was in einer Reduktion des hydrodynamischen Volumens des Moleküls resultiert.
Bei genauer Kenntnis des Feeds lässt sich das hydrodynamische Volumen bestimmter
Proteine folglich durch Anpassung der Ionenstärke steuern (Teng et al., 2006; Zydney, 1998).

Stand der Forschung
19
Feed-Konzentration:
Eine höhere Feed-Konzentration CF resultiert, abhängig von der Strömungsgeschwindigkeit ʋ,
wie in Abbildung 10 dargestellt, in einem niedrigeren Permeatflux (Harker, Backhurst, &
Richardson, 2013; Wakeman & Tarleton, 2005; Cheryan, 1998).
Abbildung 10: Flux/Feed-Konzentration Abhängigkeit (links: Harker et al., 2013; modifiziert; rechts: Cheryan, 1998; modifiziert)
Stellt man den Permeatflux für verschiedenen Feed-Konzentrationen CF in Abhängigkeit der
Zeit dar (Abbildung 11), ähnelt sich der stationäre Flux in vielen Fällen. Abhängig von den
Eigenschaften und der Zusammensetzung des Feeds kann sich der stationäre Flux aber auch
deutlich unterscheiden (She et al., 2009; Wakeman & Tarleton, 2005; Tarleton & Wakeman,
1994).
Abbildung 11: Einfluss der Feed-Konzentration CF auf den Permeatflux JF,t (links: She et al., 2009; rechts: Tarleton & Wakeman, 1994)

Stand der Forschung
20
Die Zeit bis zum Erreichen des stationären Zustands ist dabei abhängig von der Partikelgröße
der Moleküle im Feed. Besonders bei kleinen Partikeln stellt sich schnell ein stationärer Flux
ein (Wakeman & Tarleton, 2005; Tarleton & Wakeman, 1994).
Partikelgröße & -Verteilung:
Größe, Form und Verteilung der Partikel eines Feeds beeinflussen die Eigenschaften der
Fouling-Schicht bei der Crossflow Filtration. Eine Zunahme der Partikelgröße resultiert dabei,
dargestellt in Abbildung 12, in einer Zunahme des Permeatflux (Park, Lee, Lee, & Hong, 2008;
Vyas et al., 2000; Tarleton & Wakeman, 1993).
Abbildung 12: Einfluss der Partikelgröße auf die foulinginduzierte Abnahme des Permeatflux JF,t für niedrige (links) und hohe (rechts) Strömungsgeschwindigkeiten (Tarleton & Wakeman, 1993)
Dies lässt sich durch die Art der ausgebildeten Deckschicht erklären. Kleine Partikel führen
verstärkt zu internem Fouling, sodass ein strömungsbedingter Rücktransport der Partikel und
somit ein Abtragen der Deckschicht nicht möglich ist. Die Dicke der darüber ausgebildeten
externen Fouling-Schicht entspricht in den meisten Fällen der größerer Partikel, kann diese
aber auch überschreiten. Eine Erhöhung der Strömungsgeschwindigkeit oder des
Transmembrandruck kann, abhängig von der Strömungsform, in der Ausbildung dichterer
Deckschichten mit geringerer Durchlässigkeit und höherem Filterkuchenwiderstand führen
(Park et al., 2008; Lawler & Kweon, 2004; Vyas et al., 2000; Tarleton & Wakeman, 1993). Laut
Tarleton & Wakeman (1993) wird die Ausbildung der Fouling-Schicht auch bei
unterschiedlicher Partikelgrößenverteilung hauptsächlich von kleinen Partikeln bestimmt.
Dabei spielt auch die Form der Partikel eine Rolle. Je geringer die Kugelhaftigkeit von Partikeln
ist, desto höher ist die Durchlässigkeit der Foulingschicht, sofern es sich nicht um
plättchenförmige Partikel handelt (Foust, Wenzel, Clump, Maus, & Andersen, 2008; Dullien,
1992).

Stand der Forschung
21
Temperatur:
Gezielte Steuerung der Prozesstemperatur TCF kann zu einem erhöhten Permeatflux führen.
Der Einfluss erhöhter Temperatur TCF auf Membranfouling bedarf aufgrund widersprüchlicher
Studienergebnisse jedoch einer differenzierten Betrachtung. Atra et al., (2005) (1),
Samuelsson, Dejmek, Trägårdh, & Paulsson (1997) (2), Vetier, Bennasar, & de la Fuente
(1988) (3) sowie Hwang, Chang, & Chen (1996) (4) berichten bei der Mikrofiltration von Milch
(Rohmilch (1) und entrahmte Milch (2)), Molke (3) und für ein Modellsystem mit Polystyrene
Latex Partikeln (4) von verbesserten Permeatflüssen zwischen 40 °C (Polystyrene Latex
Partikel) und 55 °C (Rohmilch). Vetier et al. (1988) führen dies auf eine Verringerung der
Deckschichtdicke aufgrund gesteigerter Viskosität des Feeds zurück. Mo, Tay, & Ng (2008)
und van Boxtel, Otten & van der Linden (1991) stellten bei Umkehrosmoseprozessen von BSA
und im Rahmen der Käseherstellung anfallender Molke eine Abnahme des Permeatflux bei
steigenden Temperaturen fest. Mo et al. (2008) erklären dies anhand struktureller
Veränderungen der BSA-Moleküle bei steigender Temperatur, die zu Verminderung der
elektrostatischen Abstoßung zwischen Molekülen und Membran führen. Protein-Membran und
Protein-Protein Interaktionen resultieren in Folge pH-abhängig in Deckschichtbildung und
Aggregation von BSA-Molekülen. Eine temperaturabhängige Verringerung der Viskosität führt
also nicht zwangsweise zur Vermeidung von Fouling. Temperatur-, pH- und Ionenstärke-
bedingte Veränderungen der Proteinstruktur- und Ladung müssen bei der Auswahl der
Prozesstemperatur miteinbezogen werden.
Membranen:
Morphologie und hydrophobe/hydrophile Eigenschaften beeinflussen neben den
Trenneigenschaften einer Membran auch deren Neigung zu Fouling (Melin & Rautenbach,
2007). Hydrophobe Membranen wird allgemein eine gesteigerte Fouling-Anfälligkeit
zugeschrieben. Abhängig vom Grad der Hydrophobizität von Membran und Molekül kommt es
zur Ausbildung von hydrophoben Wechselwirkungen oder Anlagerungseffekten aufgrund von
van-der-Walls-Kräften die, besonders in der Startphase eines Filtrationsprozesses, in der
verstärkten Ausbildung von Konzentrationspolarisation und Fouling resultieren. Hydrophobe
Membranen neigen darüber hinaus zur Ausbildung von Filterkuchen mit höherem Widerstand,
was in erhöhtem Rückhalt gelöster Substanzen resultiert (Maximous, Nakhla, & Wan, 2009;
Melin & Rautenbach, 2007; Kofod Nielsen, 2004; Huisman et al., 2000; Matthiasson, 1983).
Einige Studien wiedersprechen mittlerweile der lange geltenden Annahme und führen die
unterschiedlichen Erkenntnisse auf mit der Hydrophobizität verknüpfte Änderungen der
Membranmorphologie zurück, die eine Deutung des Hydrophobizität/Fouling
Zusammenhangs erschweren (Choo & Lee, 2000; Nomura, Fujii, & Suzuki, 1997). Entgegen
der Hydrophobizität stehen elektrostatische Wechselwirkungen zwischen Membran und Feed.

Stand der Forschung
22
Abbildung 13: pH-Abhängigkeit des ζ-Potentials einer Polysulfon-Membran (Melin & Rautenbach, 2007; modifiziert)
Die Oberflächenspannung einer Membran wird dabei anhand des ζ-Potentials beschrieben.
Das ζ-Potential einer Membran ist, aufgrund von Ionisationsvorgängen, abhängig vom pH-
Wert des Feeds, dargestellt in Abbildung 13. Ein steigender Absolutwert des ζ-Potentials steht
in Verbindung mit einer Abnahme der Hydrophobizität der Membran (Melin & Rautenbach,
2007; Huisman et al., 2000). Die Abnahme des Permeatflux ist abhängig von der Porosität der
Membran. Eine steigende Porosität führt dabei, bis zum Erreichen eines Grenzwertes, zu
einem höheren Permeatflux durch Reduktion des Fouling. Dies lässt sich unter anderem auf
die Entstehung einer lockereren Deckschicht mit steigender Porosität der Membran
zurückführen. Wird eine Grenzporosität überschritten, fallen Fouling-Effekte, durch effektivere
Verblockung der Poren, dafür umso schwerwiegender aus. In Verbindung mit der
Porengrößenverteilung beeinflusst die Porosität der Membran die Art des auftretenden
Foulingphänomens (internes Fouling/externes Fouling) (Melin & Rautenbach, 2007; Kofod
Nielsen, 2004; Choo & Lee, 2000; Ho & Zydney, 1999; Riedl, Girard, & Lencki, 1998). Wichtig
für das Verständnis von Membrantrennprozessen ist darüber hinaus die Tatsache, dass der
MWCO einer Membran (z. B. 100 kDa) nicht bedeutet, dass die Porengeometrie aller Poren
identisch ist. Der MWCO definiert vielmehr die kleinste Molekülmasse einer Struktur, welche
durch die verwendete Membran zu 90 – 95 % zurückgehalten wird (Melin & Rautenbach,
2007). Die Membranporung genügt also einer bestimmten Porengrößenverteilung, dargestellt
in Abbildung 14, die einen annährend vollständigen Rückhalt von Molekülen ermöglicht, die
oberhalb der nominalen Trenngrenze liegen (Mulder, 1996).

Stand der Forschung
23
Abbildung 14: Schematische Darstellung der Porengrößenverteilung einer Membran (Mulder, 1996)
2.1.4.3 Weitere Verfahren zur Fouling-Kontrolle & Prozessoptimierung
Diafiltration:
Unter dem Begriff Diafiltration versteht man die kontinuierliche (Continuous Diafiltration; CDF)
oder diskontinuierliche (Discontinuous Diafiltration; DDF) Zugabe von Lösungsmittel im
Rahmen eines Filtrationsprozesses (Wakeman & Tarleton, 2005; Irmler, 2001). Als ein
Diavolumen (VDia) wird dabei die Menge an Lösungsmittel definiert, die der Menge des Feeds
zu Prozessbeginn entspricht. Bei der diskontinuierlichen Diafiltration erfolgt eine schrittweise
Verdünnung des Feeds zu bestimmten Prozesszeiten. Nach Zugabe eines oder mehrerer
Diavolumina wird das Feed solange verarbeitet, bis ein festgelegtes Minimalvolumen des
Feeds erreicht ist. Je nach Trennziel kann zu diesem Zeitpunkt die Zugabe weiterer
Diavolumina erfolgen. Bei kontinuierlicher Fahrweise wird das Lösungsmittel in Abhängigkeit
des Permeatflux über die Laufzeit des Prozesses zugeführt (Wakeman & Tarleton, 2005).
Das Verfahren der Diafiltration eignet sich in der Crossflow-Filtration zur Verbesserung der
Trennschärfe bei der Trennung von hoch- und niedermolekularen Substanzen (Irmler, 2001).
Unter Verwendung des Diafiltrationsverfahren lässt sich, wie in Abbildung 15 dargestellt,
abhängig von der Häufigkeit der Zugabe eines Diavolumens, eine erhöhte Reinheit der
Retentatfraktion bei verbesserter Auswaschung permeabler, niedermolekularer Bestandteile
erreichen (Baldasso, Barros, & Tessaro, 2011; Venkiteshwaran, Heider, Teysseyre, & Belfort,
2008; Wakeman & Tarleton, 2005; van Eijndhoven, Saksena, & Zydney, 1995). Dies liegt in
einer Verhinderung des Viskositätsanstiegs des Feeds durch Aufkonzentration von Feed-
Partikeln im Retentat (Irmler, 2001).

Stand der Forschung
24
Abbildung 15: BSA-Konzentration und Reinheit von Immunglobulin G (IgG) in Abhängigkeit der Diavolumina-Anzahl Nd (Venkiteshwaran et al., 2008; modifiziert)
Ultraschall:
Die Verwendung von Ultraschall (US) hat sich bei der Optimierung von Filtrationsprozessen,
besonders bei Crossflow-Systemen, als vielversprechend erwiesen. Mehrere Studien
berichten von deutlichen Steigerungen des Permeatflux unter Ultraschalleinfluss
(Muthukumaran, Kentish, Ashokkumar, & Stevens, 2005; Kobayashi, Chai, & Fujii, 1999; Chai
et al., 1998; Wakeman & Tarleton, 1991). Die fluxsteigernde Wirkung des Ultraschalls wird
dabei auf eine Reduktion von Konzentrationspolarisation und eine Abschwächung von Fouling
durch Aufbrechen der Deckschicht zurückgeführt (Kobayashi et al., 1999; Lamminen, Walker,
& Weavers, 2004; Muthukumaran et al., 2004). Dies kann, gemäß Muthukumaran, Kentish,
Ashokkumar, & Stevens (2005), anhand von vier ultraschall-induzierten Effekten erklärt
werden:
Verminderung der Filterkuchendichte und Reduktion von internem Fouling durch
Agglomeration von feinen Partikeln
Verbesserung der Suspensionsfähigkeit und Auswaschung von Partikeln durch
mechanische Schwingungsenergie
Abtragen von Partikeln durch das Einwirken akustischer Gleichströmung
Durch Kavitation verursachte Reinigung der Membranoberfläche
Die Steigerung des Permeatflux JF,t ist, wie in Abbildung 16 dargestellt, unter anderem
abhängig von der verwendeten Frequenz fUS. Niedrigere US-Frequenzen (28 – 50 kHz)
erwiesen sich als effektiv (Kyllönen, Pirkonen, & Nyström, 2005; Muthukumaran, Kentish,
Ashokkumar, et al., 2005; Lamminen et al., 2004; Kobayashi et al., 2003).

Stand der Forschung
25
Abbildung 16: Permeatflux JF,t in Abhängigkeit der Ultraschallfrequenz fUS einer 0,5 %igen (w/w) Milchlösung (Kobayashi et al., 2003)
Die Wirksamkeit der US-Anwendung kann dabei nicht losgelöst von weiteren
Prozessparametern betrachtet werden. US-Anwendung bei Filtrationsprozessen erlaubt
höhere Feed-Konzentrationen ohne Abnahme des Permeatflux. Bei Annäherung an eine
Grenzkonzentration, dargestellt in Abbildung 17, nimmt der positive Effekt des US jedoch ab
(Muthukumaran, Kentish, Ashokkumar, et al., 2005; Wakeman & Tarleton, 1991).
Abbildung 17: Abhängigkeit des Permeatflux von der Konzentration CF des Feed für die Crossflow-Filtration von Molke mit und ohne die Verwendung von US (Muthukumaran, Kentish, Ashokkumar, et al., 2005; modifiziert)

Stand der Forschung
26
Hierbei spielt auch die Viskosität und Partikelgrößenzusammensetzung des Feed eine Rolle.
Besonders bei kleinen Partikeln wird von erhöhter Partikelbewegung ausgegangen, die einer
Deckschichtausbildung entgegenwirkt oder durch Agglomeration von feinen Partikeln in einer
Verminderung des Deckschichtwiderstands resultiert (Muthukumaran, Kentish, Ashokkumar,
et al., 2005; Wakeman & Tarleton, 1991). Steigt die Viskosität und die Oberflächenspannung
des Feeds jedoch an, wird besonders das Auftreten kavitations-basierender Effekte durch
erschwerte Ausbildung der Kavitation reduziert (Wakeman & Tarleton, 1991; Mason & Lorimer,
1988). Auch bei Ansteigen des Transmembrandrucks ∆pTMP, dargestellt in Abbildung 18,
verringert sich der fluxsteigernde Effekt des US, abhängig von den Membraneigenschaften,
teilweise deutlich. Die Drucksteigerung führt zu einer Verdichtung des Filterkuchens und somit
zu erhöhtem Wiederstand. Darüber hinaus kommt es bei steigendem Druck zu
Unregelmäßigkeiten im US-Feld, die dafür sorgen, dass US-induzierte Effekte nicht mehr
gleichmäßig auf der kompletten Membranfläche wirken (Kyllönen et al., 2005; Matsumoto,
Miwa, Nakao, & Kimura, 1996).
Abbildung 18: Druckabhängige Änderung des Permeatflux bei der Crossflow-Mikrofiltration von Bäcker-Hefe (Matsumoto et al., 1996)
2.1.5 Die Dialyse
Die Trennung im Dialyse-Prozess erfolgt, im Gegensatz zu druckgetriebenen Ultra- und
Nanofiltrationsverfahren, durch einen Konzentrationsgradienten zwischen zwei
membrangetrennten Phasen (Baynes & Dominiczak, 2014; de Castro, Capote, & Ávila, 2008).
In der Praxis werden Dialyse-Prozesse, neben medizinischen Anwendungen, zur Abtrennung
niedermolekularen Substanzen wie beispielsweise Salzen eingesetzt (Baynes & Dominiczak,
2014; Luo, Wu, Xu, & Wu, 2011; Pingoud & Urbanke, 1997). Des Weiteren ist eine Nutzung
zur Aufkonzentration von Makromolekülen möglich (Dennison, 2003). Eine Dialyse lässt sich

Stand der Forschung
27
im Labormaßstab, dargestellt in Abbildung 19, mit einfachen Mitteln durchführen (Ballew,
Martinez, Markee, & Eddleman, 2002).
Abbildung 19: Schematische Darstellung des Dialyseprozess (Ballew, Martinez, Markee, & Eddleman, 2002; modifiziert)
Abhängig vom MWCO passieren kleine Partikel die semipermeable Membran im Bestreben
eines Konzentrationsausgleichs zwischen Probe und Puffer, während große Moleküle
zurückgehalten werden (Baynes & Dominiczak, 2014). Durch mehrfachen Austausch des
Puffers kann die Konzentration kleiner Partikel deutlich abgesenkt werden (Koolman & Röhm,
2003). Zur Verbesserung des Diffusionsvorgangs ist ein Rühren des Puffers, sowie ein
möglichst großes Membranoberflächen-Probenvolumen-Verhältnis, von Vorteil (Dennison,
2003). Eine erhöhte Diffusionsrate lässt sich bei erhöhten Temperaturen und niedrigen
Viskositäten erreichen (Ballew et al., 2002). Unter Berücksichtigung der Proteinstabilität
werden Dialyseprozess aber bevorzugt bei niedrigen Temperaturen oder bei Raumtemperatur
durchgeführt (Dennison, 2003; Ballew et al., 2002). Die Performance der Dialyse ist darüber
hinaus, vergleichbar mit druckgetriebenen Trennverfahren, abhängig von den Feed-
Charakteristika, Membraneigenschaften und Konzentrationspolarisations-Effekten (Dennison,
2003; Ballew et al., 2002). Besondere Wichtigkeit obliegt dabei Membrancharakteristika wie
Dicke und Oberflächeneigenschaften (Porosität, Dimensionierung) der Membran (de Castro
et al., 2008).

Stand der Forschung
28
2.2 Rohstoffcharakterisierung
2.2.1 Milchproteine
Die Proteine der Milch sind seit vielen Jahren Gegenstand intensiver Forschungen (Hartmann
& Meisel, 2007; Meisel, 1997). Der Schwerpunkt liegt hierbei, neben der Erforschung
bioaktiver Substanzen, auf dem funktionellen Potential der Milchproteine für die
Lebensmittelindustrie (Foegeding, Davis, Doucet, & McGuffey, 2002; Meisel, 1997). Der
Gesamtproteingehalt der Milch beträgt 2,8 – 3,6 %. Der Großteil davon, etwa 98 %, entfällt auf
die beiden Proteingruppen Casein und Molkenprotein. Die restlichen 2 % verteilen sich auf
Hüllproteine und Enzyme (Töpel, 2004). Tabelle 1 gibt einen Überblick über die prozentuale
Zusammensetzung der beiden Proteinfraktionen, sowie weiteren Eigenschaften.
Tabelle 1: Zusammensetzung und Eigenschaften der Milchproteinfraktionen (Ternes, 2008; Topel, 2004; modifiziert)
Protein
%-Anteil
der
Fraktion
%-Anteil in
der
Fraktion
Anzahl der
Aminosäuren
Mr
(kDa) IEP
Casein: 80
αS1-Caseine 35 199 23,6 4,9
αS2-Caseine 10 207 25,3 5,3
β-Caseine 30 209 24,0 5,2
χ-Caseine 12 169 19,0 5,4
Sonstige 13
Molkenproteine: 20
β-Lactoglobulin 55 162 18,0 5,1
α-Lactoglobulin 25 123 14,2 4,2
Immunoglobuline 8 - > 145,0 5,5 – 8,3
Rinderserumalbumin 7 582 66,3 5,0
Lactoferrin 5 700 82,0 8,0
Transferrin - -
Legende: Mr, Molmasse. IEP, Isoelektrischer Punkt.
2.2.1.1 Caseine
Die Proteinfraktion der Caseine bildet mit einem Anteil von ca. 80 % die quantitative
Hauptkomponente der Milchproteine (Töpel, 2004). Im Gegensatz zu den Molkenproteinen
verfügen sie nicht über eine globuläre Struktur und können, begründet durch ihren hohen
Prolingehalt, nur bedingt Sekundärstrukturen ausbilden (Töpel, 2004; Graham, Malcolm, &
McKenzie, 1984; Slattery, 1976; Herskovits, 1966).

Stand der Forschung
29
Caseine sind, abhängig vom pH-Wert des Umgebungsmilieus, sehr hitzetolerant und bleiben
bis zu einer Temperatur von 120 – 140 °C stabil (Anema & Klostermeyer, 1997; Ternes, 2008;
Töpel, 2004). Durch Zusatz von Natriumchlorid (NaCl), Natriumcitrat (C6H5Na3O7) oder
Natriumphosphat (Na3PO4) und den damit verbundenen pH-Wert-Veränderungen kann
beispielsweise die Caseinstabilität bei 95 °C für Erhitzungsdauern von bis zu 30 min erhöht
werden (Le Ray et al., 1998). Bei einem pH von 4,6 fallen Caseine aus und bilden, unter
Labeinwirkung und in Gegenwart von Calcium-Ionen (Ca2+), ein Gel (Ternes, 2008; Töpel,
2004). Caseine liegen in nativem Zustand überwiegend in Form von Caseinmicellen vor. Als
Caseinmicellen bezeichnet man einen Komplex aus Caseinmolekülen der Caseinfraktionen
αS1, αS2, β und κ sowie Wasser (Töpel, 2004). Den Aufbau der Caseinmicellen betreffend
existieren verschiedene Modelle, von denen das Casein-Submicellen-Modell am weitesten
verbreitet ist (Holt, de Kruif, Tuinier, & Timmins, 2003; Horne, 1998; Ternes, 2008; Töpel,
2004). Die Eigenschaften der Caseine werden lebensmitteltechnologisch besonders bei der
Herstellung von Käse- und Sauermilchprodukten wie Joghurt oder Kefir ausgenutzt. Durch
Säurefällung, Behandlung unter Hochdruck oder das Einwirken von proteolytischen Enzymen
kommt es zur Koagulation der Caseinmicellen und, abhängig von Umgebungsmilieu und
mechanischen Einflüssen, zur Gelbildung (Ternes, 2008; Töpel, 2004).
2.2.1.2 Molkenproteine
Molkenproteine galten lange Zeit als unerwünschtes Nebenprodukt der Käseherstellung. Ihre
Bedeutung als bioaktive Substanz und ihr technologischer Nutzen wurden erst
verhältnismäßig spät, durch Einführung neuer gesetzlicher Regulationen zur Entsorgung und
technologischen Fortschritt offenkundig und Gegenstand intensiver Forschung. Zuvor lag der
Fokus verstärkt auf der Erforschung und Nutzung der Caseinfraktion (Smithers, 2008; Töpel,
2004). Molkenproteine bilden mit 20 % den quantitativ kleineren Anteil am
Gesamtproteingehalt der Milchproteine (Töpel, 2004). Der größte Anteil wird von β-
Lactoglobulin (55 %) und α-Lactoglubulin (25 %) abgedeckt (Ternes, 2008). Abhängig von der
Wahl der Literaturquelle existieren geringfügig abweichende Konzentrationsangaben. Im
Gegensatz zu den Caseinen liegen Molkenproteine in nativer Form als globuläre Strukturen
vor und sind somit vergleichsweise hitzeempfindlicher. Ihre Denaturierungstemperatur liegt, je
nach Beschaffenheit (z.B. pH-Wert, Vorhandensein von Ca2+-Ionen) des
Umgebungsmediums, zwischen 60 °C und 72 °C (Ternes, 2008; Töpel, 2004).
Spricht man von Molkenproteinen, ist eine Unterscheidung zwischen
Molkenproteinkonzentrat, -Isolat oder -Hydrolysat notwendig (DeMan, 1999). Die Unterteilung
in Konzentrat, Isolat und Hydrolysat erfolgt aufgrund unterschiedlicher Herstellungsprozesse.
Molkenproteinkonzentrate (WPC – Whey Protein Concentrate) werden unter Verwendung
mehrstufiger Ultrafiltrationsanlagen gewonnen (DeMan, 1999; Mehrens, 2004). Hohe

Stand der Forschung
30
Proteingehalte von bis zu 80 % erreicht man durch Auswaschen niedermolekulare
Bestandteile mittels Diafiltration. Molkenproteinisolate (WPI – Whey Protein Isolate) können
einen Proteingehalt von über 90 % aufweisen (Mehrens, 2004). Hierzu wird zusätzlich zur
Ultrafiltration eine Mikrofiltration vorgeschaltet oder eine Kombination aus Mikrofiltration und
anschließender Aufarbeitung durch Ionenaustauscher verwendet (DeMan, 1999; Mehrens,
2004). Bedingt durch die feinere Filtration werden Fettbestandteile, Mikroorganismen und
große Proteinaggregate abgetrennt (Mehrens, 2004). Die Herstellung von
Molkenproteinhydrolysaten (WPH – Whey Protein Hydrolysate) findet bevorzugt unter
Verwendung von Proteasen statt (DeMan, 1999). Das Ergebnis der Hydrolyse ist abhängig
von der Spezifität der verwendeten Enzyme und deren Einwirkzeit. Nach erfolgter Hydrolyse
wird die erhaltene Lösung filtriert und getrocknet (DeMan, 1999; Lahl & Braun, 1994).
Abhängig vom Hydrolysegrad werden hydrolysierte Molkenproteine in „partially hydrolysed
whey proteins“ (PH-W) und „extensively hydrolysed whey proteins“ (EH-W) eingeteilt (Schütte
& Paschke, 2009).
2.2.2 Algen & Algenproteine
Mit dem Begriff „Alge“ klassifiziert man eine Vielzahl überwiegend im Wasser lebender
Organismen (Rogers, 2011; Hoek, Mann, & Jahns, 1995). Die Systematik der Algen ist
aufgrund laufender Forschung ständiger Veränderung unterworfen und umfasst
quellenabhängig Pro- und Eukaryoten (Rogers, 2011; Hoek et al., 1995; Harlin & Darley,
1988). Eine Einteilung in einzellige Mikroalgen und komplexere Makroalgen erleichtert eine
Übersicht. Basierend auf ihrer Pigmentierung werden Makroalgen in Braun- (Phaeophyceae),
Rot- (Rhodophyceae) und Grünalgen (Chlorophyceae) eingeteilt. Mikroalgen sind in
Diatomeen (Bacillariophyta), Dinoflagellaten (Dinophyceae) und prokaryotische Blaualgen
(Cyanbateria) unterteilt (Rogers, 2011; Garson, 1989). Mikroalgen rückten in den letzten
Jahren besonders als Lieferant für Biomasse zur Kraftstoffproduktion in den Fokus der
Forschung (Demirbas & Demirbas, 2010; Mata, Martins, & Caetano, 2010). Mit der
Entdeckung aus Algenprotein gewonnener, bioaktiver Peptide stieg das Interesse an Makro-
und Mikroalgen auch abseits der Kraftstoffproduktion (Samarakoon & Jeon, 2012; Harnedy &
FitzGerald, 2011). Ein hoher Proteinanteil macht Mikroalgen auch als Lebensmittel interessant
(Mata et al., 2010; Becker, 2007; Bewicke & Potter, 1984).
Neben Dunaliella und Spirulina gilt Chlorella als Mikroalgen-Gattung mit großem Potential als
Lebensmittelzusatz (Mata et al., 2010; Bewicke & Potter, 1984). Die den Grünalgen
zugehörige, kugelförmig bis elliptische Chlorella vulgaris, dargestellt in Abbildung 20, verfügt
über eine Zellwand mit hoher Stabilität (Renneberg & Berkling, 2012). Die Zellwand der
meisten Pflanzen besteht zu 15 – 30 % aus Cellulose, zu ca. 30 % aus Pektin, sowie zu 20 –

Stand der Forschung
31
25 % aus Hemicellulose (Chen, 2014). Cellulose, Pektin und Hemicellulose sind auch in
Mikroalge maßgebende Bestandteile der Zellwand (Bastos, 2018; Domozych, 2016).
a) ganze Zelle; b) hochauflösende Vergrößerung der Zellwand
Abbildung 20: Schematische Darstellung von Chlorella (links: Linne von Berg, Hoef-Emden, & Melkonian, 2012, modifiziert) und transmissionselektronenmikroskopische Aufnahme von Chlorella vulgaris (rechts: Yamamoto, Kurihara, & Kawano, 2005, modifiziert)
Die durchschnittliche Dicke einer Chlorella vulgaris Zellwand liegt bei 17 – 20 nm (Yamamoto
et al., 2005). Der Proteingehalt von Chlorella, wird, je nach Art und Quelle, mit 51 – 58 %
angegeben (Safi, Zebib, Merah, Pontalier, & Vaca-Garcia, 2014; Morris, Almarales, Carrillo, &
Bermúdez, 2008; Becker, 2007). Eine von Übersicht der Zusammensetzung verschiedener
Algen ist beispielhaft in Abbildung 21 dargestellt (Becker, 2007).
Abbildung 21: Zusammensetzung verschiedener Algen (Becker, 2007)
Hydrolysate von Chlorella können aufgrund ihres hohen Anteils an Glutaminsäure, dargestellt
in Abbildung 22, einen fleischig-herzhaften (umami) Geschmack aufweisen (Yamaguchi, 1997;
Tchorbanov, Bozhkova, Dilov, & Litchev, 1986). Die Aminosäuren-Zusammensetzung von
Chlorella vulgaris kann, je nach Analysemethode, Umrechnungsfaktor und Quelle jedoch
schwanken (Safi et al., 2014).
Zellwand
Zellkern
Chloroplast

Stand der Forschung
32
Abbildung 22: Aminosäurenprofil von Chlorella vulgaris (Safi et al., 2014; modifiziert; basierend auf a Safi et al. 2012; b Faheed & Fattah, 2008 & Shaaban, 2001; c Naik, Goud, Rout, & Dalai, 2010)
Die technologischen Eigenschaften von Chlorella im Lebensmittelbereich sind trotz großem
Potential bis jetzt wenig erforscht (Ursu et al., 2014). Ursu et al. (2014) charakterisierten die
aus Chlorella vulgaris gewonnenen Proteine mit Hilfe einer Natriumdodecylsulfat-
Polyacrylamidgelelektrophorese (SDS-PAGE). Abhängig von pH-Wert während der Zell-Lysis
identifizierten sie Proteine im Bereich von 25 – 60 kDa mit Schwerpunkt bei 30 kDa (pH 7)
bzw. 60 kDa (pH 12). Der IEP lag für einen Großteil der Proteine im Bereich 4,0 – 5,5, sowie
6,0 – 8,0. Morris et al. (2008) stellten bei der Gel-Filtration von Chlorella vulgaris sechs
Proteinfraktionen im Bereich von 12 – 120 kDa fest. Die aus Chlorella vulgaris gewonnen
Proteine verfügen über eine Emulgierfähigkeit und emulsionsstabilisierende Eigenschaften,
die mit kommerziellen Produkten wie Natrium-Caseinat vergleichbar sind (Ursu et al., 2014).
2.3 Hydrolyse von Proteinquellen
In der Lebensmittelverarbeitung werden Proteine aufgrund ihrer funktionellen Eigenschaften
geschätzt. Ihre stabilisierenden, texturbildenden und wasserbindenden Eigenschaften können
in Produktionsabläufen jedoch durch thermische-mechanische Beanspruchung, Änderungen
des pH-Werts oder der Ionenstärke beeinträchtigt oder aufgehoben werden (Zayas, 1997). Die
Hydrolyse von Proteinen bietet eine gangbare Möglichkeit die Funktionalität des
Ursprungsproteins bei verbesserter Stabilität beizubehalten oder sogar zu steigern (Nielsen,
1997).
Unter dem Begriff Hydrolyse versteht man die Spaltung einer Peptid-Bindung durch eine
Reaktion mit Wasser (H2O), dargestellt in Abbildung 23 (Tavano, 2013; Watson et al., 2010).

Stand der Forschung
33
Das Produkt dieses Prozesses wird als Hydrolysat bezeichnet (Pasupuleti, Holmes & Demain,
2010).
Abbildung 23: Hydrolyse einer Peptidbindung (Pasupuleti, Holmes & Demain, 2010; basierend auf: Adler-Nissen, 1986)
Wichtiger Kontrollparameter bei der Herstellung von Proteinhydrolysaten ist der Hydrolysegrad
(DH - Degree of Hydrolysis). Der DH, in Formel 9 dargestellt, erlaubt eine Aussage über die
prozentuale Anzahl gespaltener Peptidbindungen im Medium.
𝐷𝐻 = ℎ
ℎ𝑡𝑜𝑡 × 100 %
Formel 9: Degree of Hydrolysis
h = Anzahl hydrolysierter Peptidbindungen
htot = Gesamtzahl der Peptidbindungen pro Protein-Äquivalent
htot, die Gesamtzahl der Peptidbindungen pro Protein-Äquivalent ist hierbei abhängig von der
Aminosäurenzusammensetzung des ursprünglichen Proteins (Nielsen, Petersen, &
Dambmann, 2001).
Für die Bestimmung des DH gibt es eine Vielzahl an Methoden von denen die pH-stat, O-
phthaldialdehyd- (OPA) und Trinitro-Benzene-Sulfonic Acid (TNBS) Methode am weitesten
verbreitet sind (Spellman, McEvoy, O’Cuinn, & FitzGerald, 2003; Nielsen et al., 2001; Church,
Porter, Catignani, & Swaisgood, 1985; Adler-Nissen, 1979; Fields, 1971; Jacobsen, Léonis,
Linderstrøm-Lang, & Ottesen, 1957). Die Ermittlung des DH basiert für OPA- und TNBS-
Methode auf einer Reaktion zwischen spezifischen Aminosäuren und einer Reagenz (O-
Phthaldialdehyd bzw. Trinitro-Benzene-Sulfonic Acid). Hierbei entsteht ein Komplex der
spektralphotometrisch bei 340 nm detektiert werden kann (Nielsen et al., 2001; Adler-Nissen,
1979). Die pH-stat-Technik nutzt die Freisetzung von Protonen bei einer Hydrolyse im
neutralen oder alkalischen Bereich, die zu einem Absinken des pH-Wert führt. Durch Titration
einer Base wird der pH-Wert konstant gehalten. Die Bestimmung des DH erfolgt rechnerisch
unter Verwendung der zum Konstanthalten des pH-Wertes benötigten Menge an Base (Adler-
Nissen, 1986).

Stand der Forschung
34
Die Hydrolyse von Proteinen kann chemisch, im sauren oder alkalischen Bereich, oder
enzymatisch erfolgen (Tavano, 2013; Pasupuleti, Holmes & Demain, 2010).
2.3.1 Die enzymatische Hydrolyse
In der Lebensmittelverarbeitung werden aufgrund erhöhter Spezifität und Wirtschaftlichkeit,
sowie einer reduzierter Menge an Nebenprodukten, bevorzugt enzymatische Verfahren
eingesetzt (van Oort, 2009). Die grundlegende Charakterisierung von Enzymen erfolgt anhand
der katalysierten Reaktion. Die von der International Union of Biochemistry (IUB) initialisierte
Nomenklatur umfasst sechs Enzymgruppen (van Oort, 2009). Zur Hydrolyse von Proteinen
werden Proteasen aus der Gruppe der Hydrolyasen (EC 3) verwendet (Tavano, 2013; van
Oort, 2009; Clemente, 2000). Proteolytische Enzyme lassen sich anhand ihrer Wirkweise
weiter in Endo-und Exopeptidasen unterteilen (Tavano, 2013; Clemente, 2000).
Exopeptidasen trennen Aminosäuren systematisch am Anfang oder Ende einer Sequenz ab,
wohingegen Endopeptidasen verschieden große Peptidfragmente erzeugen (Clemente,
2000). Die Aktivität der Enzyme ist dabei abhängig von Umgebungsbedingungen. Temperatur,
pH, Substrat- und Enzymkonzentration müssen prozess- und produktspezifisch angepasst
werden (van Oort, 2009).
2.3.1.1 Hydrolyse von Algen & Algenproteinen
Aufgrund ihres hohen Proteingehalts sowie antioxidativer, antibakterieller und antiviraler
Eigenschaften besitzen Mikroalgen Potential für Anwendungen in der Lebensmittel- und
Pharmawirtschaft (Plaza, Cifuentes, & Ibáñez, 2008; Suzuki et al., 2004). Um funktionelle
Komponenten von Mikroalgen zu nutzen, ist das Aufbrechen der oftmals stabilen Zellwände
notwendig (Morris et al., 2008). Dies ist beispielsweise durch das Einwirken von Druck,
organischen Lösungsmitteln oder enzymatisch möglich (Tchorbanov & Bozhkova, 1988). Der
Erfolg der nachfolgenden Hydrolyse des Algenproteins ist abhängig vom verwendeten Enzym
und der Enzymkonzentration. Während eine Hydrolyse mit Pepsin nur zu niedrigen
Hydrolysegraden führt, erwiesen sich Subtilisin, Papin, Pronase und Pancreatin als geeignete
Wahl zur Hydrolyse von Proteinen aus Mikroalgen der Gattung Chlorella und Scenedesmus
(Morris et al., 2008; Tchorbanov & Bozhkova, 1988; Tchorbanov et al., 1986; Barashkov,
Trubachev, & Gitel’zon, 1979). Bei der Hydrolyse von Chlorella mit Pancreatin bzw. Subtilisin
erreichten Morris et al. (2008) und Tchorbanov & Bozhkova (1988) eine DH von 20 – 22 %,
dargestellt in Abbildung 24.

Stand der Forschung
35
Abbildung 24: Hydrolysegrad in Abhängigkeit der Zeit für die Hydrolyse von Chlorella mit Subtilisin (Tchorbanov & Bozhkova, 1988; modifiziert)
Eine vierstündige Hydrolyse von Chlorella vulgaris mit Pancreatin führt zum Abbau aller
Peptide > 28 KDa. Die entstehenden Hauptpeptidfraktionen liegen zwischen 2 – 5 kDa (Morris
et al., 2008).
2.4 Analytik – Die Size-Exclusion HPLC (SE-HPLC)
Die High Pressure Liquid Chromatography (HPLC) hat sich, bedingt durch den technischen
Fortschritt der vergangenen Jahrzehnte, zur leistungsfähigsten Methode der Flüssigkeits-
Chromatografie entwickelt (Gritter, Bobbitt, & Schwarting, 1987). Die Trennung einer Probe
basiert dabei, vereinfacht dargestellt, auf einer Interaktion des durch eine mobile Phase
(Puffer, Lösungsmittel, Eluent) transportierten Probenmaterials mit einer stationären Phase
(Trennsäule). Abhängig von der Beschaffenheit der Trennsäule wird zwischen adsorptiven und
nicht-adsorptiven Wechselwirkungen unterschieden (Bradley & Desai, 2000). Trennverfahren
wie Normal-Phase (NP)-, Reversed-Phase (RP)- und Hydrophobic-Interaction
Chromatography (HIC) basieren auf adsorptiven Interaktionen, wohingegen bei der Trennung
in SE-HPLC-Prozessen die unterschiedlichen Molekülgrößen einzelner Probenbestandteile
ausschlaggeben sind (Kremer & Bannwarth, 2014; Meyer, 1988). Nicht-adsorptive
Trennprozesse werden in der Regel isokratisch, sprich ohne Veränderung der
Zusammensetzung des Eluenten durchgeführt, während bei adsorptiven Prozessen häufig
Gradienten zum Einsatz kommen. Unter einer Gradientenfahrweise versteht man die
schrittweise Veränderung der Zusammensetzung des Eluenten während des Trennprozesses.
Dies resultiert in einer Veränderung der Interaktion zwischen Probenmolekül und stationärer
Phase, sowie Probemolekül und mobiler Phase und beeinflusst somit dessen
Elutionszeitpunkt (Bradley & Desai, 2000; Amersham Pharmacia Biotech, 1999; Pharmacia
LBK Biotechnology, 1991; Meyer, 1988).

Stand der Forschung
36
Die Zeit die zwischen Aufbringen einer Substanz und der Detektion des Peakmaximums
vergeht, bezeichnet man als Retentionszeit tr, dargestellt in Abbildung 25. Mit der
Retentionszeit tr korrespondiert das Retentionsvolumen Vr, definiert als das Eluenten-
Volumen, das von Aufbringen bis Detektion des Peakmaximums benötigt wird (Siouffi, 2000;
Galensa, Bahadir, Engelhardt, & Böhm, 1995).
Abbildung 25: Bestimmung von Totzeit t0/-Volumen V0 und Retentionszeit tr/-Volumen Vr (Siouffi, 2000)
Als Totzeit t0 bezeichnet man die Zeitspanne, die eine Substanz, die keine Wechselwirkung
mit der stationären Phase erfährt, von Injektion bis Peakmaximum des ersten Peaks benötigt
(Kromidas, 2008; Galensa et al., 1995). Das Totvolumen V0 ist das durch Kapillaren, Fittings
und weitere Bauteile des HPLC-Systems verursachte Volumen zwischen Injektor und
Detektor, ausgenommen der Trennsäule (Kromidas, 2008). Mit Kenntnis von tr und t0 lässt sich
die Nettoretentionszeit ts als Differenz beider Größen bestimmen (Kromidas & Kuss, 2008).
Die SE-HPLC gilt als leistungsfähiges Verfahren zur Größencharakterisierung einer
Probensubstanz und findet dadurch begründet, unter anderem, Anwendung in der Protein-
und Peptid-Charakterisierung (Hong, Koza, & Bouvier, 2012). Als stationäre Phase werden
Trennsäulen mit einer Füllung aus chemisch modifizierten, porösen Silica-Partikeln verwendet
(Hong et al., 2012; Tayyab, Qamar, & Islam, 1991). Beim SE-HPLC Verfahren lassen sich
anhand der Retentionszeiten einzelner Proben-Moleküle Rückschlüsse auf deren
Molekülgröße ziehen. Dazu ist eine Kalibrierung der jeweiligen Säule mit einer bekannten

Stand der Forschung
37
Standardsubstanzen notwendig (Kostanski, Keller, & Hamielec, 2004; Tayyab et al., 1991;
Meyer, 1988). Große Moleküle passieren die Säule dabei am schnellsten, dargestellt in
Abbildung 26, da sie nicht in die Porenstruktur des Silica-Gels eindringen, wohingegen kleine
Teilchen eine längere Wegstrecke durch die Poren zurücklegen und später eluieren (Hong et
al., 2012; Tayyab et al., 1991).
Abbildung 26: Arbeitsweise einer Size-Exclusion Chromatography (Kremer & Bannwarth, 2014; modifiziert)
Die Trennung erfolgt dabei, im Idealfall, lediglich anhand des hydrodynamischen Radius der
Moleküle (Tieke, 2014; Yau & Kirkland, 1981). In der Praxis lassen sich Wechselwirkungen
zwischen Probe und stationärer Phase jedoch nicht vollkommen ausschließen, sodass es zu
Adsorption oder Degradation von Molekülen kommen kann (Hong et al., 2012; O’Callaghan,
Donnelly, Slattery, & Mulvihill, 1995; Kopaciewicz & Regnier, 1982; Yau & Kirkland, 1981).
Diese Effekte gilt es experimentell im Rahmen der Methodenerstellung, beispielsweise durch
die Anpassung von Ionenstärke und pH-Wert des Puffers, zu reduzieren (Arakawa, Ejima, Li,
& Philo, 2010; Golovchenko, Kataeva, & Akimenko, 1992; Kopaciewicz & Regnier, 1982).
2.5 Sensorik – Geschmackswahrnehmung & deren Modulation
Der Geschmack eines Stoffes gibt Auskunft über dessen Genusstauglichkeit (Frings & Müller,
2013). Ein Geschmackseindruck wird auf der Zunge, aber auch im Gaumen und Rachenraum
durch Geschmacksrezeptoren wahrgenommen. Die objektive Einordnung des
Geschmackseindrucks erfolgt durch Interpretation der Reize im Gehirn (Rehner & Daniel,
2010).
Um geschmacksmodulatorische Eigenschaften von Peptidverbindungen verstehen und
bewerten zu können, sind nachfolgend grundlegende Begrifflichkeiten und Abläufe der

Stand der Forschung
38
Wahrnehmung gustatorischer Reize dargestellt. Darüber hinaus werden mögliche
Mechanismen zur Modulation der Geschmackswahrnehmung erläutert.
2.5.1 Geschmacksrezeptoren – Geschmackspapillen, -Knospen & -
Sinneszellen
Die Geschmackswahrnehmung erfolgt durch Geschmackssinneszellen, dargestellt in
Abbildung 27 (c). Diese sind, in Gruppen von 40 - 60, in Geschmacksknospen gebündelt (c),
die wiederum in Zungenpapillen lokalisiert sind (b) (Becker-Carus & Wendt, 2017; Rehner &
Daniel, 2010). Auch Teile der Mundhöhle tragen zur Geschmackswahrnehmung bei.
Vereinzelte Geschmacksknospen befinden sich am weichen Gaumen, der hinteren
Rachenwand, sowie dem Kehldeckel (Rehner & Daniel, 2010).
Abbildung 27: Geschmackrezeptoren - Geschmackspapillen, -Knospen und -Sinneszellen (Becker-Carus & Wendt, 2017; modifiziert)
Die Zungenpapillen werden unter anderem in Abhängigkeit von Form, Größe, Anzahl, sowie
der Anzahl der Geschmacksknospen charakterisiert (Rehner & Daniel, 2010; Büsing &
Heinzeller, 2001). Im Bereich des Zungenrund liegen 7 – 12 unverhornte Wallpapillen (Papillae
vallatae), die jeweils bis zu 200 Geschmacksknospen enthalten (Rehner & Daniel, 2010; Hatt,

Stand der Forschung
39
2006; Büsing & Heinzeller, 2001). Am hinteren Zungenrand sitzen 15 – 30 Blätterpapillen
(Papillae foliatae), die über bis zu 50 Geschmacksknospen verfügen. Über die ganze Zunge
verteilt sind darüber hinaus 150 – 400 unverhornte Pilzpapillen (Papillae fungiformes)
vorzufinden, die jeweils nur 3 – 4 Geschmacksknospen enthalten (Hatt, 2006). Die verhornten
Fadenpapillen (Papillae filiformes) dienen der Tastempfindung (Schulz, Hundeiker, & Kreusch,
2016; Hatt, 2006). Insgesamt verfügt der Mensch über ca. 2000 – 4000 Geschmacksknospen
(Schmidt, Lang, & Heckmann, 2007; Hatt, 2006; Birbaumer & Schmidt, 1991).
Der Aufbau einer Geschmacksknospe ist in Abbildung 27 (c) dargestellt. Gelangen
wasserlösliche Reizstoffe, beispielweise aus der Nahrung, in den Pore/Porus genannten,
flüssigkeitsgefüllten Raum einer Geschmacksknospe, diffundieren diese in Richtung der
fingerförmigen Membranfortsätze (Mikrovilli) der Geschmackssinneszellen (Hatt, 2006;
Birbaumer & Schmidt, 1991). Von dort aus wird der Geschmacksreiz durch afferente
Nervenfasern ins Gehirn weitergeleitet (Birbaumer & Schmidt, 1991). Dort findet die
hedonische Bewertung des Geschmackseindrucks statt (Frings & Müller, 2013).
Überschneidungen von Sinneseindrücken während des Verzehrs können die individuelle
Einordung des Reizes beeinflussen (Rehner & Daniel, 2010). Darüber hinaus kann sich die
Wahrnehmung eines Geschmacksreizes von Person zu Person, beispielsweise aufgrund
unterschiedlicher Anzahl an Geschmacksknospen, unterscheiden (Oehlenschläger &
Schneider-Häder, 2012).
2.5.2 Die fünf Grundgeschmacksarten & deren Wahrnehmung
Klassischerweise wird zwischen den vier Grundgeschmacksarten „süß“, „sauer“, „salzig“ und
„bitter“ unterschieden (Becker-Carus & Wendt, 2017; Rehner & Daniel, 2010; Bujas, Szabo,
Ajdukovć, & Mayer, 1989). Darüber hinaus ist mittlerweile auch „umami“ als fünfte
Grundgeschmacksart anerkannt (Rehner & Daniel, 2010; Birbaumer & Schmidt, 1991). Neben
den Grundgeschmacksarten existieren die Nebengeschmacksqualitäten alkalisch (seifig) und
metallisch (Birbaumer & Schmidt, 1991).
Die Wahrnehmung der einzelnen Grundgeschmackseindrücke ist, mit geringfügigen
Unterschieden (max. 10 %), über die gesamte Zungenfläche moglich. „bitter“ wird jedoch
vorwiegend im hinteren Bereich der Zunge wahrgenommen (Becker-Carus & Wendt, 2017;
Hatt, 2006). Darüber hinaus gilt die Zungenmitte als geschmacksunempfindlich (Becker-Carus
& Wendt, 2017). Die bevorzugte Lokalisation der Geschmacksqualitäten auf der Zunge ist in
Abbildung 28 dargestellt.

Stand der Forschung
40
Abbildung 28: Bevorzugte Lokalisation der Geschmacksqualitäten auf der Zunge (Hatt, 2011; modifiziert)
Grundlegend gehen einer Geschmackswahrnehmung zwei Prozesse voraus; Eine lokale
Depolarisation/Hyperpolarisation der Zellmembran der Geschmackssinneszellen, sowie die
darauf folgende neuronale Codierung des Reizes (Rehner & Daniel, 2010).
Die (Signal-) Transduktion, sprich, die Umsetzung eines chemischen (Geschmacks-) Reizes
in eine elektrische Antwort der Sinneszelle, die in der Depolarisation/Hyperpolarisation
resultiert, erfolgt dabei für „bitter“ und „süß“ rezeptorvermittelt, während „sauer“- und „salzig“-
Empfindungen durch eine direkte Interaktion des ionisierten Geschmacksstoffes mit
Ionenkanälen zustande kommen (Rehner & Daniel, 2010; Hatt, 2006).
Auf die Reizverarbeitung folgt die Reizweiterleitung. Das zuvor erzeugte Sensorpotential der
Geschmackssinneszelle resultiert in einer Transmitterfreisetzung. Diese führt wiederum zu
einer Erregung der afferenten Nervenfasern und der Weiterleitung des Reizes in Form eines
Aktionspotentials (Birbaumer & Schmidt, 1991). Abbildung 29 und Abbildung 30 zeigen die
zugrundeliegenden Vorgänge.
Abbildung 29: Zusammenfassung der Geschmackstransduktion (Silverthorn, 2009; modifiziert)

Stand der Forschung
41
Abbildung 30: Signaltransduktion in Geschmackszellen (Hatt, 2006; modifiziert)
Eine „sauer“-Wahrnehmung wird durch Wasserstoffionen (H+-Ionen) dissoziierter Säuren
ausgelöst. Die Intensität des Geschmacks nimmt dabei mit steigender H+-Ionen-Konzentration
zu, sodass stark/vollständig dissoziierte Säuren, mit wenigen Ausnahmen, eine stärkere
Geschmacksempfindung auslösen (Hatt, 2006). In der Zellmembran der
Geschmacksinneszellen sind zwei Kanalproteine für die “sauer“-Wahrnehmung zuständig;
Amiloridsensitive Natriumionen-Kanäle (Na+-Ionen Kanäle), sowie hyperpolarisationsaktivierte
Kationenkanäle (Hatt, 2011). Die Kanalproteine sind im Ruhezustand Kalium-Ionen (K+-Ionen)
und in geringem Maße auch für Na+-Ionen permeabel (Hatt, 2006). In Anwesenheit von H+-
Ionen wird der Ausstrom von K+-Ionen blockiert. Dies resultiert in der Ausbildung eines
positiven Membranpotentials, sowie in Folge, der Depolarisation der Zelle (Hatt, 2011). Durch
die Zelldepolarisation öffnen sich Calcium-Kanäle (Ca2+-Kanäle), wodurch ein Calcium-Signal
erzeugt wird, das letztendlich zur Transmitterfreisetzung führt (Silverthorn, 2009).
Für milde Säuren, wie Apfel-, Essig- oder Milchsäure wird ein anderer Mechanismus vermutet.
Hierbei wird davon ausgegangen, dass die wenig dissoziierten Säuren die Zellmembran der
Geschmackssinneszellen passieren und erst im Zellinneren weiter dissoziieren und in Folge
die Depolarisation der Zelle auslösen (Frings & Müller, 2013).
„salzig“ schmeckende Stoffe dissoziieren im Mundraum in Kationen und Anionen. Beide
Moleküle tragen zur Geschmackswahrnehmung bei (Hatt, 2006). Die in der Zellmembran der

Stand der Forschung
42
Geschmackssinneszelle verankerten Ionenkanäle sind unspezifisch für Kationen permeabel.
Eine Erhöhung der Kationen außerhalb der Zelle führt zu einem Kationeneinstrom und in Folge
zur Depolarisation (Frings & Müller, 2013; Hatt, 2006). Vergleichbar mit „sauer“ führt diese
Depolarisation zur Öffnung von Ca2+-Kanälen und gipfelt in der Transmitterfreisetzung
(Silverthorn, 2009). Amiloridsensitive Na+-Kanäle können durch Amilorid blockiert werden
(Hatt, 2006).
Die „bitter“-Wahrnehmung ist wesentlich komplexer als es im Falle von „sauer“- oder „salzig“-
Reizen der Fall ist. Aufgrund der Vielfältigkeit von Bitterstoffen verfügt der Mensch über ca. 25
verschiedene Bitterrezeptoren (Frings & Müller, 2013). Bindet ein Bitterstoff an einen Rezeptor
wird eine mehrstufige Signalkaskade ausgelöst. Hierbei sind verschiedene G-Protein-
gekoppelte Mechanismen bekannt, die sich „second messenger“ Botenstoffen bedienen. Eine
Möglichkeit ist der Abbau von cyclischem Adenosinmonophosphat (cAMP) durch
Phosphodiesterase (PDE) zu nicht-cyclischen 5‘-AMP. Der Abfall der cAMP-Konzentration
führt in Folge zur Öffnung der Ca2+-Kanäle und somit zu einer Depolarisation der Zelle (Rehner
& Daniel, 2010; Aidley, 1998).
Die Zelldepolarisation ist ebenfalls durch eine Phospholipase C (PLC) vermittelte Abspaltung
des „second messenger“ Botenstoffs Inositoltrisphosphat (IP3) aus der Plasmamembran
möglich (Frings & Müller, 2013; Aidley, 1998). Gelangt IP3 zu den intrazellulären Ca2+-
Speichern, werden Ca2+-Ionen freigesetzt. Die Erhöhung des zellinternen Ca2+-Spiegels führt
zur Öffnung von Ionenkanälen. Der daraus resultierende Einstrom von Na+-Ionen führt
letztendlich zur Depolarisation der Zelle und zur Transmitterfreisetzung (Feher, 2017; Boron &
Boulpaep, 2016; Frings & Müller, 2013).
Die „süß“-Wahrnehmung erfolgt ebenfalls G-Protein-vermittelt. Durch natürliche oder
synthetische Zucker ausgelöste Reize werden dabei in Form unterschiedlicher
Signalkaskaden verarbeitet (Rehner & Daniel, 2010; Birbaumer & Schmidt, 1991). Bindet ein
natürlicher Zucker an einen Rezeptor, wird Adenosintriphosphat (ATP) durch das Enzym
Adenylat-Cyclase (AC) in den „second messenger“ cAMP umgewandelt. cAMP aktiviert
darauffolgend die Proteinkinase C. Diese überträgt ihren Phosphat-Rest auf K+-Kanalproteine
und führt dadurch zu einer Blockade des K+-Ionenstroms aus der Zelle. Die Folge ist die
Depolarisation der Zelle (Rehner & Daniel, 2010; Birbaumer & Schmidt, 1991).
Im Falle von synthetischem Zucker wird anstelle der AC eine PLC aktiviert. Die PLC-Aktivität
führt zur Ausbildung der Botenstoffe IP3 und Diacylglycerol (DAG). DAG regt die Proteinkinase
C wiederum zur Übertragung des Phosphat-Rests auf K+-Kanalproteine an und führt, analog
des vorausgehend beschriebenen Prozesses, zur Depolarisation der Zelle (Rehner & Daniel,
2010). Darüber hinaus wird durch IP3 eine Erhöhung des zellinternen Ca2+-Spiegels, mit darauf
folgender Öffnung von Ionenkanälen, getriggert. Dieser Vorgang trägt zur Depolarisation bei
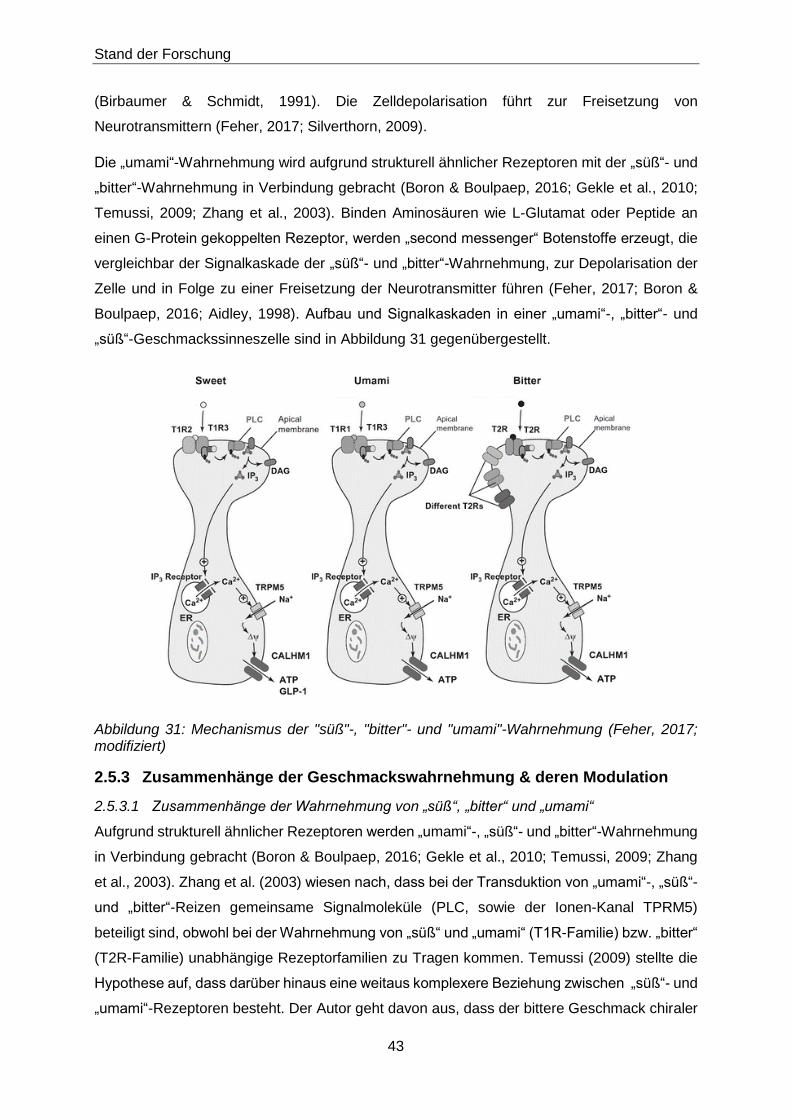
Stand der Forschung
43
(Birbaumer & Schmidt, 1991). Die Zelldepolarisation führt zur Freisetzung von
Neurotransmittern (Feher, 2017; Silverthorn, 2009).
Die „umami“-Wahrnehmung wird aufgrund strukturell ähnlicher Rezeptoren mit der „süß“- und
„bitter“-Wahrnehmung in Verbindung gebracht (Boron & Boulpaep, 2016; Gekle et al., 2010;
Temussi, 2009; Zhang et al., 2003). Binden Aminosäuren wie L-Glutamat oder Peptide an
einen G-Protein gekoppelten Rezeptor, werden „second messenger“ Botenstoffe erzeugt, die
vergleichbar der Signalkaskade der „süß“- und „bitter“-Wahrnehmung, zur Depolarisation der
Zelle und in Folge zu einer Freisetzung der Neurotransmitter führen (Feher, 2017; Boron &
Boulpaep, 2016; Aidley, 1998). Aufbau und Signalkaskaden in einer „umami“-, „bitter“- und
„süß“-Geschmackssinneszelle sind in Abbildung 31 gegenübergestellt.
Abbildung 31: Mechanismus der "süß"-, "bitter"- und "umami"-Wahrnehmung (Feher, 2017; modifiziert)
2.5.3 Zusammenhänge der Geschmackswahrnehmung & deren Modulation
2.5.3.1 Zusammenhänge der Wahrnehmung von „süß“, „bitter“ und „umami“
Aufgrund strukturell ähnlicher Rezeptoren werden „umami“-, „süß“- und „bitter“-Wahrnehmung
in Verbindung gebracht (Boron & Boulpaep, 2016; Gekle et al., 2010; Temussi, 2009; Zhang
et al., 2003). Zhang et al. (2003) wiesen nach, dass bei der Transduktion von „umami“-, „süß“-
und „bitter“-Reizen gemeinsame Signalmoleküle (PLC, sowie der Ionen-Kanal TPRM5)
beteiligt sind, obwohl bei der Wahrnehmung von „süß“ und „umami“ (T1R-Familie) bzw. „bitter“
(T2R-Familie) unabhängige Rezeptorfamilien zu Tragen kommen. Temussi (2009) stellte die
Hypothese auf, dass darüber hinaus eine weitaus komplexere Beziehung zwischen „süß“- und
„umami“-Rezeptoren besteht. Der Autor geht davon aus, dass der bittere Geschmack chiraler

Stand der Forschung
44
Isomere süßer Moleküle von „umami“-Rezeptoren wahrgenommen werden kann, somit folglich
kein separater Rezeptor für die „bitter“-Wahrnehmung chiraler „süß“-Isomere existiert.
Temussi (2009) stützt seine Hypothese auf die Studienergebnisse von Tomchik, Berg, Kim,
Chaudhari, & Roper (2007). Die Autoren arbeiten heraus, dass die meisten präsynaptischen
Zellen (83 %) Signale von zwei oder mehreren Geschmackssinneszellen empfangen. Eine
durch chirale „süß“-Isomere ausgeloste „bitter“-Empfindung kann folglich per „Crosstalk“,
dargestellt in Abbildung 32, auch von „umami“-Rezeptoren erfasst und weitergegeben werden
(Temussi, 2009).
Abbildung 32: Schematische Darstellung des „Crosstalk“ zwischen "umami"- und "bitter"-Rezeptoren (Temussi, 2009; modifiziert; basierend auf: Tomchik et al., 2007)
Weitere Autoren berichteten von nicht-rezeptorspezifischen Geschmackswahrnehmung.
Sowohl Caicedo, Kim, & Roper (2002) als auch Gilbertson, Boughter, Zhang, & Smith (2001)
arbeiten heraus, dass 38 – 73 % der Geschmackssinneszellen auf mehr als eine
Geschmacksausprägung reagieren. Ein einheitliches Meinungsbild hinsichtlich dieser These
existiert jedoch nicht (Scott, 2004). Zhang et al. (2003) wiederlegten die Ergebnisse der zuvor
genannten Autoren in „in vivo“ Knockout-Versuchen mit modifizierten Mäusezellen durch
selektive Modulation der PLC-Funktionalität. Auch Zhao et al. (2003) wiesen durch gezielte
Modulation des T1R2-Rezeptors nach, dass ein bestimmter Rezeptortyp dezidierte
Verhaltensweisen auslöst und die weitergeleitete Geschmacksempfindung abhängig von der
Art des Rezeptors ist.
2.5.3.2 Modulation der Geschmackswahrnehmung
Lebensmittel können bedingt durch Herstellungsprozesse oder das Zusammenwirken von
Inhaltsstoffen unerwünschte Off-Flavours, darunter fallen beispielsweise „bitter“ und
„adstringierend“, aufweisen. Um eine breite Akzeptanz des Produktes zu erreichen, ist in vielen

Stand der Forschung
45
Fällen eine Maskierung des Geschmackseindrucks oder eine Modulation der
Geschmackswahrnehmung erforderlich (Nadathur & Carolan, 2016; Dürrschmid, 2009). Um
unerwünschte oder Fehlgeschmäcker zu reduzieren, existieren verschiedene Methoden
(Nadathur & Carolan, 2016; Dürrschmid, 2009; Ley, 2008). Darunter fallen nach Ley (2008) …
die Entfernung schlecht schmeckender Komponenten,
die Ausbildung physischer Barrieren (Verkapselung, Coating),
der Einsatz von Radikalfängern oder komplexbildenden Stoffen,
die Verwendung stark süßer, salziger, saurer oder fruchtiger Geschmacksstoffe,
kongruenter und maskierender Aromen,
sowie Modulationen auf molekularer Ebene.
Auf molekularer Ebene sind, gemäß Ley (2008), mehrere Möglichkeiten zur Unterdrückung
der „bitter“-Transduktion denkbar. Darunter fallen …
das Einwirken von Molekülen die eine Komplexbildung bewirken, Bitterstoffe zersetzen
oder deren Transport zum Rezeptor einschränken,
als Gegenspieler der T2R-Rezeptor Bindungsstellen wirken, sowie
Moleküle die die T2R-Rezeptorbindungsstellen verändern,
Moleküle die weitere Proteine der Transduktionskette modulieren,
Moleküle die die Funktion des TRPM5-Ionenkanals oder die
Ausschüttung, Bindung und Wiederaufnahme von Neurotransmittern beeinflussen.
In den folgenden Abschnitten wird entsprechend der Zielsetzung der Studie die Modulation
der Bitterwahrnehmung näher beleuchtet. Hierbei wird schwerpunktmäßig auf eine Modulation
auf molekularer Ebene eingegangen.
Für die „bitter“-Wahrnehmung sind Rezeptoren der T2R-Familie verantwortlich (Mueller et al.,
2005). Ein gangbarer Weg zur Reduktion des „bitter“-Geschmacks auf molekularer Ebene
stellt die Hemmung der Bindung von Bitterstoffen mit den relevanten T2R-Rezeptoren dar
(Nadathur & Carolan, 2016). Slack et al. (2010) identifizierten GIV3727 (4-(2,2,3-
trimethylcyclopentyl)-Buttersäure) als Gegenspieler bestimmter Bitterrezeptoren,
insbesondere des hTAS2R31-Rezeptors. Den Autoren gelang es sowohl in „in vitro“
Versuchen, als auch in humansensorischen Panels, die bittere Geschmackskomponente von
Acesulfam-K und Saccharin in wässrigen Lösungen signifikant zu reduzieren. Neben GIV3727
scheint auch Probenecid (4-(Dipropylsulfamoyl)benzoesäure) mit bestimmten T2R-
Rezeptoren zu interagieren. Greene et al. (2011) berichten, neben einer Unterbindung des
Rezeptorsignals „in vitro“, von einer signifikanten Reduktion der Bitterwahrnehmung einer 10
mM Salicin-Lösung nach Benetzen der Mundraumes mit einer 10 mM Probenecid-Lösung. Die

Stand der Forschung
46
Autoren führen dies auf eine Interaktion von Probenecid mit dem hTAS2R16-Rezeptor zurück
und gehen dabei von einem allosterischen Mechanismus aus.
Keast, Breslin, & Beauchamp (2001) verwendeten Natriumsalze und den „bitter“-Geschmack
von verschiedenen Modellösungen zu reduzieren. Die Reduktion der Geschmackintensität ist
dabei abhängig von der Bittersubstanz. Während sich Natriumsalze zur Reduktion der „bitter“-
Wahrnehmung von Kaliumchlorid, Harnstoff und Amilorid eignen, konnte die von Koffein und
Chinin-Hydrochlorid-Dihydrat ausgelöste Geschmacksempfindung nur geringfügig reduziert
werden. Im Gegensatz zu GIV3727 kann der Wirkmechanismus der Natriumsalze nicht alleinig
auf das Gegenspieler-Prinzip zurückgeführt werden.
Abbildung 33: Schematische Darstellung möglicher Wirkmechanismen von Natrium-Kationen während der „bitter“-Transduktion (Keast et al., 2001)
Keast et al. (2001) postulieren verschiedene Wirkmechanismen, dargestellt in Abbildung 33.
Ein möglicher Mechanismus ist die Ausbildung eines ladungsbedingten Abschirmungseffekts
im Bereich des Rezeptors (1). Dieser vermindert potentiell die Rezeptor-Interaktion der „bitter“-
Substanz. Darüber hinaus ist eine Modulation der am Transduktionsprozess beteiligten Ionen-
Kanäle verstellbar (2). Weiter ist eine durch Na+-Ionen ausgelöste Stabilisierung der
Zellmembran denkbar, die Bitterstoffen den Zugang zu in der Membran eingebetteten
Rezeptorproteinen erschwert (3). Auch eine Interaktion der Na+-Ionen mit „second-
messenger“-Botenstoffen kann zu einer Modulation der Geschmackswahrnehmung führen (4)
(Keast et al., 2001).
Bryant et al. (2007) gelang es, die Aktivität des am Transduktionsprozess beteiligten Ionen-
Kanals TRPM5 in HEK293 Zellsystemen auf 40 % zu reduzieren. Die Autoren verwendeten

Stand der Forschung
47
dafür geringe Konzentrationen von Hydrazin-Derivaten. Die Ausprägung der Wirkung von
Hydrazin-Derivaten wurde von Bryant et al. (2007) nicht dokumentiert. Somit ist nicht bekannt,
ob die Substanz zur selektiven Abschwächung eines Geschmacksreizes in der Lage ist, oder
die Geschmackswahrnehmung generell einschränkt (Ley, 2008).
Nicht bei allen Substanzen mit geschmacksmodulatorischen Eigenschaften sind die
Wirkmechanismen geklärt. Zhang, Xia, & Peterson (2014) identifizierten „bitter“-blockenden
Eigenschaften bei einem Maillard-Reaktionsprodukt des flavan-3-ol−spiro-C-Glycosids ohne
dessen Einfluss auf die Geschmackstransduktion zu betrachten. Hofmann et al. (2007)
unterbanden den „bitter“-Geschmack einer wässrigen L-Phenlyalanin-Lösung durch den
Zusatz verschiedener Pyridinium Betaine (Glypyridaine, β-Alapyridaine, Gabapyridaine).
Pyridinium Betaine wirken nicht nur als „bitter“-Blocker sondern verstärken auch den „süß“-,
„salzig“- und „umami“-Geschmack (Hofmann et al., 2007). Neodiosmin vermindert ebenfalls
die „bitter“-Wahrnehmung, wie Guadagni, Horowitz, Gentili, & Maier (1979) bei der
humansensorischen Bewertung von Koffein- und Chinin-Sulfat-Lösungen feststellten. Die
Verwendung von Neodiosmin wird jedoch unter anderem durch eine begrenzte Erhältlichkeit
eingeschränkt (Ley, 2008). Weitere Verbindungen, die ein quantitative Veränderung der
Bitterwahrnehmung bewirken, sind Adenosin-5‘-Monophosphat und Chlorhexidin
(Dürrschmid, 2009).
Generell ist die Modulation des „bitter“-Geschmacks aufgrund der Vielzahl der am
Transduktionsprozess beteiligten T2R-Rezeptoren eine Herausforderung. Bis heute gibt es
kein allumfassende Lösung, sodass zur Reduktion der „bitter“-Wahrnehmung zumeist
Kombinationen aus Verkapselung, dem Zusatz von kongruenten und maskierenden Aromen
oder weiteren Methoden Anwendung finden (Ley, 2008).
2.5.4 Geschmacksqualitäten von Peptiden
Peptidverbindungen gewannen als Ersatz für Lebensmittelinhaltsstoffe bereits früh an
Bedeutung, wie sich anhand des Beispiels Aspartam (als Zuckerersatz) belegen lässt (Ternes,
2008; Pförtsch & Müller, 2006). Die zufällige Entdeckung von Aspartam bildete die Grundlage
für weitere Forschungen hinsichtlich potentieller Geschmackseigenschaften von
Peptidverbindungen. In Anlehnung an die Struktur von Aspartam synthetisierten Polinelli et al.
(1992) und Toniolo et al. (1993) Süßstoffe mit vergleichbarer oder höherer Süßkraft und
gesteigerter Stabilität. In den vergangenen Jahren rückten weitere Verbindungen mit hoher
Süßkraft wie Thaumatin, Monellin, Brazzein, Pentadin, Curculin und Mabinlin in den Fokus der
Forschung (Ternes, 2008). Das aus 54 Aminosäuren bestehende Brazzein trug, wie auch
Monellin und Thaumatin dazu bei, die Annahme zu wiederlegen, dass Verbindungen mit einer
molaren Masse > 2,5 kDa generell nicht über Geschmackseigenschaften verfügen (Ming &
Hellekant, 1994).

Stand der Forschung
48
Neben Süßstoffen lassen sich durch Peptidverbindungen auch „umami“ schmeckende
Komponenten realisieren. Tamura et al. (1989), Ohyama, Ishibashi, Tamura, Nishizaki, & Okai
(1988), sowie Fujimaki, Arai, Yamashita, Kato, & Noguchi (1973) berichteten von
mehrgliedrigen Aminosäureverbindungen, die einen „umami“-Geschmackseindruck auslösen.
Die Existenz „umami“-schmeckender Peptidstrukturen wird jedoch angezweifelt. Bei der
sensorischen Bewertung einer Auswahl von 16 Di- und Tri-Peptiden der zuvor genannten
Autoren, konnten van den Oord & van Wassenaar (1997) bei keiner der untersuchten
Komponenten eine Geschmacksqualität ermitteln, die Monosodium L-Glutamat (MSG) gleicht.
Die Autoren führen dies, unter anderem, auf unzureichende Reinheit der in den
Referenzstudien verwendeten Peptidproben zurück.
Peptide mit einem hohen Gehalt an hydrophoben Resten werden als „bitter“ wahrgenommen
(Matoba & Hata, 1972; Kirimura et al., 1969). Ney (1971) erarbeitete, basierend auf den
Erkenntnissen von Tanford (1962), den Q-Wert zur Vorhersage der Bitterkeit von
Peptidverbindungen. Dieser erlaubt die Bestimmung der mittleren Hydrophobität eines
Peptides. Der Autor postulierte, dass Verbindungen mit Q > 1400 „bitter“ und Verbindungen
mit Q < 1300 nicht „bitter“ schmecken. Guigoz & Solms (1976) bestätigten diese These bei der
Auswertung von diversen Studienergebnissen zu 61 natürlichen und 145 synthetisch
gewonnenen Peptiden, identifizierten allerdings 3 Verbindungen mit Q < 1300 die ebenfalls
einen „bitter“-Geschmack aufwiesen. Darüber hinaus ist der Q-Wert nicht für
Peptidverbindungen geeignet deren Struktur Glycin-Reste aufweist (Guigoz & Solms, 1976).
Cho, Unklesbay, Hsieh, & Clarke (2004) stellten bei der Untersuchung von aus Soja-
Hydrolysat gewonnenen Peptidverbindungen ebenfalls keinen Zusammenhang zwischen Q-
Wert und „bitter“-Geschmack fest.
Die strukturellen Eingeschalten, sowie die Konformität einer Peptidstruktur, ist grundsätzlich
maßgeblich entscheidend für deren Geschmackseigenschaften (Temussi, 2011).
Säurerestreiche Peptide losen beispielweise eine „sauer“-Empfindung aus (Kirimura et al.,
1969). Hydrochloride einiger Peptidverbindungen werden mitunter als „salzig“ empfunden
(Belitz, Grosch, & Schieberle, 2008). Darüber hinaus führt die L- und D-Konformität einer
Struktur zu unterschiedlichen Geschmacksempfindungen, wie Solms et al. (1965) für 18
Aminosäuren herausarbeiteten. Auch der bekannte Süßstoff Aspartam verfügt nur in seiner L-
Konformität über eine süßende Wirkung. Alle weiteren chiralen Isomere schmecken „bitter“
(Mazur et al., 1969). Die Komplexität und Vielzahl der Einflussfaktoren führt jedoch dazu, dass
geschmacksaktive Peptide häufig nur durch Zufall entdeckt werden (Ley, 2008; Mazur et al.,
1969).

Material & Methoden
49
3. Material & Methoden
Im Fokus der Versuchsreihe standen die Entwicklung einer Methode zur Fraktionierung von
Proteinhydrolysaten mittels Crossflow Diafiltration, sowie die sensorische Charakterisierung
von aus Proteinhydrolysaten gewonnenen Peptidfraktionen. Die Methodenerstellung erfolgte
unter Verwendung von kommerziell erhältlichem, hydrolysiertem Molkenprotein-Isolat (Kapitel
3.2.1). Nach Etablierung der Modellmethode wurden rohstoffspezifische Anpassungen (Feed-
pH, Ionenstärke) experimentell ermittelt (Kapitel 3.2.2). Für hydrolysiertes Molkenprotein-Isolat
wurde darüber hinaus der Einfluss einer Fraktionierung unter Anwendung von Ultraschall,
sowie die Verwendung alternativer Membranen, untersucht (Kapitel 3.2.2.2 und Kapitel
3.2.2.3). Um die Möglichkeit der Abtrennung kleiner Moleküle (< 2 kDa) aus dem vorfiltrierten
Feed (Nullprobe) zu untersuchen, wurden Dialyseversuche durchgeführt (Kapitel 3.2.2.4).
Für die Fraktionierung der Alge Chlorella vulgaris war die Entwicklung eines geeigneten
Hydrolyseverfahrens notwendig (Kapitel 3.3). Die Proteingehalte der verwendeten Hydrolysate
wurden spektralphotometrisch (280 nm) ermittelt (Kapitel 3.4.2). Die Bestimmung des
Hydrolysegrads basiert auf der OPA-Methode von Nielsen, Petersen, & Dambmann (2001)
(Kapitel 3.4.3). Die Analyse der Peptidgrößenverteilung der Fraktionen erfolgte mittel SE-
HPLC unter Zuhilfenahme eines Standards (Kapitel 3.4.4)
3.1 Rohstoffe
3.1.1 Molkenprotein-Isolat
Im Rahmen dieser Studie wurde das hydrolysierte Molkenprotein-Isolat „Thermax 690“ (Lot.
02923210) (Glanbia Nutritionals Ltd., IRL), gewonnen aus Süßmolke, verwendet. Der
Hydrolysegrad des Pulvers liegt bei 8 – 12 %. Der Proteingehalt „dry basis“ ist mit 90 %, der
pH-Wert mit 7,2 – 7,8 angegeben (Glanbia Nutritionals Ltd., 2012). In Tabelle 2 sind weitere
Analysedaten des Molkenprotein-Isolats aufgeführt. In Tabelle 3 ist das Aminosäure-Profil
dargestellt.
Tabelle 2: Zusammensetzung des Molkenprotein-Isolats "Thermax 690" (Glanbia Nutritionals Ltd., 2012)
Bezeichnung Gehalt (%)
Proteingehalt „dry basis“ > 90
Feuchte < 5
Fett < 1
Mineralstoffe < 7
Lactose < 1,5

Material & Methoden
50
Tabelle 3: Aminosäure-Profil des Molkenprotein-Isolats „Thermax 690“ (Glanbia Nutritionals Ltd., 2012)
Bezeichnung Gehalt (%)
Asparaginsäure 10,7
Threonin 6,6
Serin 4,3
Glutaminsäure 16,7
Glycin 1,6
Alanin 4,4
Valin 4,9
Isoleucin 6,3
Leucin 9,6
Tyrosin 2,8
Phenylalanin 2,7
Histidin 1,6
Lysin 9,8
Arginin 1,9
Prolin 5,7
Cystein 2,4
Methionin 1,9
Tryptophan 2,0
3.1.2 Alge (Chlorella vulgaris)
Neben dem hydrolysiertem Molkenprotein-Isolat „Thermax 690“ (Glanbia Nutritionals Ltd., IRL)
fand die Alge „Chlorella vulgaris Powder“ (Lot. 201410215) (Allma, Lissabon, PRT) bei den
nachfolgend dargestellten Untersuchungen Anwendung. Der Proteingehalt des
sprühgetrockneten Algenpulvers liegt bei 52 % (Allma, 2014). Weitere Analysedaten sind in
Tabelle 4 dargestellt.
Tabelle 4: Zusammensetzung des Algen-Pulvers „Chlorella vulgaris Powder“ (Allma, 2014)
Bezeichnung Gehalt (%)
Proteingehalt 52,0
Feuchte 6,0
Fett 9,6
Asche 6,8
Kohlenhydrate 12,0
Ballaststoffe 13,6

Material & Methoden
51
3.2 Crossflow-Diafiltration
3.2.1 Die Methodenerstellung
Zur Fraktionierung der hydrolysierten Proteinquellen (Molkenprotein-Isolat, Alge (Chlorella
vulgaris)) wurde ein Crossflow-Anlagensetup konzipiert und entsprechend der Zielsetzung
dieser Studie optimiert. Die hierzu durchgeführten Versuche sind nachfolgend dargestellt.
3.2.1.1 Das Anlagensetup
45 °C
CrossflowAnlage
Feed/Retenat
pTMP (bar)
Wasserbad (beheizt)
TCF (°C)
Waage
Permeat
Datenlogger
pH
Abbildung 34: Schematischer Aufbau der Crossflow-Anlage
Das zur Fraktionierung verwendete Anlagesetup (Abbildung 34) bestand aus einer Polyacryl-
Crossflow Einheit (OPTISEP CL, Schleicher & Schuell BioScience GmbH, Dassel, GER) mit
integriertem Druckmesser, geeignet für die Verwendung von Flat-Sheet Membranen. Die
Regulierung des Transmembrandrucks pTMP erfolgte über ein Ventil an der Ausflussseite der
Crossflow Einheit. Für die Förderung der Feed-Lösung wurde eine Schlauchpumpe (CD 1500,
Heraeus Instruments, Hanau, GER) samt geeigneter Schläuche (Pharmed BPT, Ø 9,5 mm,
NSF 51, R-Biopharm AG, Darmstadt, GER) verwendet. Die Temperierung des Feeds erfolgte
mit Hilfe eines Wasserbads (Ecoline RE 206, Lauda Dr. R. Wobser GmbH & Co.KG, Lauda-
Königshofen, GER). Der pH-Wert des Retentats wurde mittels eines pH-Meters (FE 20,
Mettler-Toledo AG, Schwerzenbach, AUT) gemessen. Zur Kontrolle der Temperatur wurde ein
Thermometer (Testo 110, Testo AG, Lenkirch, GER) herangezogen. Die Permeatmenge
wurde gravimetrisch bestimmt (TP 3002, Denver Instruments, Bohemia, USA) und durch einen

Material & Methoden
52
Datenlogger (YDP03-0CE, Sartorius AG, Göttingen, GER) dokumentiert (nur für die Filtration
der Alge Chlorella vulgaris).
3.2.1.2 Ablauf der Membrancharakterisierung – Bestimmung des „kritischen Drucks“
Entscheidend für optimale Selektivität bei hohem Flux ist das Operieren unterhalb eines
deckschichtkontrollierten Bereichs (Pearce, 2007). Da jeder Membrantyp über verschiedene
prozessspezifische Eigenschaften verfügt, wurde der kritische Druck pCP der verwendeten
Membranen im Rahmen der Methodenerstellung ermittelt.
Vor Beginn der Messungen wurde der Permeatflux JWvor von deionisiertem, auf 45 ± 0,2 °C
temperiertem Wasser bei steigendem Druck bestimmt. Dazu erfolgte zur Erzeugung eines
stationären Zustandes ein 15 minütiges Rundführen des Feeds. Im Anschluss wurde nach
weiteren 5 Minuten Laufzeit Permeat über einen Zeitraum von 5 Minuten per Messzylinder
aufgefangen und der Mittelwert JWvor für den jeweiligen pTMP bestimmt.
Zur Charakterisierung der Membranen wurde der Permeatflux JF,t des Feeds bei steigendem
Transmembrandruck pTMP dokumentiert. pTMP wurde, in Intervallen von ∆pTMP = 0,2 bar,
schrittweise von 0 bar bis max. 3 bar (je nach Membran) angehoben. Nach jeder Druckstufe
erfolgte eine Reinigung der Membran und eine Überprüfung des Reinigungserfolgs durch
Vergleich des Wasserflux JWvor mit dem nach der Reinigung bestimmten Flux JWnach. Der
kritische Druck pCP wurde durch Auftragen des Permeatflux JF über ∆pTMP grafisch bestimmt.
Tabelle 5: Charakterisierte UF- und NF-Membranen des Herstellers Microdyn Nadir GmbH (Wiesbaden, GER)
Bezeichnung Trenngrenze (kDa) Material Trennbereich
UH 050 P 50 Polyethersulfon UF
UH 030 P 30 Polyethersulfon UF
UH 020 P 20 Polyethersulfon UF
UH 010 P 10 Polyethersulfon UF
UH 005 P 5 Polyethersulfon UF
UH 004 P 4 Polyethersulfon UF
NP 010 1 Polyethersulfon NF
NP 030 0,5 Polyethersulfon NF
Die initiale Charakterisierung verschiedener Membranen (Trenngrenze 0,5 kDa – 50 kDa;
siehe Tabelle 5) des Herstellers Microdyn Nadir GmbH (Wiesbaden, GER) wurde mittels des
gleichen Anlagensetups unter Verwendung hydrolysierten Molkenprotein-Isolats (Thermax
690, Glanbia Nutritionals Ltd., IRL) bei einer Feed-Konzentration 3 % (w/v) (bezogen auf den
Proteingehalt), pH 8 durchgeführt. Das Permeat der ersten Fraktionierung (Trenngrenze: 50
kDa, Fraktion < 50 kDa) diente dabei als Feed für den jeweils folgenden Trennprozess.

Material & Methoden
53
Die Charakterisierung der in dieser Studie verwendeten 100 kDa Membran (Microdyn Nadir
GmbH, Wiesbaden, GER) (Tabelle 6) erfolgte, abgesehen von der Feed-Konzentration (0,15
% (w/v)), analog der beschriebenen Vorgehensweise (siehe Kapitel 4.3.1).
Tabelle 6: 100 kDa UF-Membranen (Microdyn Nadir GmbH, Wiesbaden, GER)
Bezeichnung Trenngrenze (kDa) Material Trennbereich
US 100 P 100 Polysulfon
(hydrophiliert) UF
Die Prozessparameter des Feeds (Temperatur TCF, pH, CF) wurden gemäß der Prämisse der
Vermeidung/Reduzierung von Fouling, Konzentrationspolarisation sowie Bakterienwachstum
basierend auf Erkenntnissen von Pelegrine & Gomes (2012), Nigam, Bansal, & Chen (2008),
Butylina et al. (2006) und Britten, Giroux, & Gaudin (1994) gewählt. Als Rohstoff für das Feed
wurde ein hydrolysiertes Molkenprotein-Isolat (Thermax 690, Glanbia Nutritionals Ltd., IRL)
verwendet. Für optimale Löslichkeit des hydrolysierten Molkenprotein-Isolats (Thermax 690,
Glanbia Nutritionals Ltd., IRL) in 500 ml deionisiertem Wasser, bei gleichzeitig minimiertem
Bakterienwachstum über die Prozesszeit, wurde das Feed (0,15 % (w/v) (bezogen auf den
Proteingehalt)) auf 45 ± 0,2 °C temperiert und mittels Natriumhydroxyd (NaOH) auf pH 8 ±
0,01 eingestellt.
Tabelle 7: Übersicht – Nutschenfilter
Bezeichnung Trenngrenze (µm) Hersteller Material
Polypropylen/Polyethylen
Tiefenfilter 10
Pall Corporation (Port
Washington, USA)
Polypropylen/
Polyethylen
GLA 5000 PVC 5 Pall Corporation (Port
Washington, USA) Polyvinylchlorid
Typ A/E Glass-Fiber
Filter 1
Pall Corporation (Port
Washington, USA)
Borsilikat
Glasfaser
Supor 800 PES 0,80 Pall Corporation (Port
Washington, USA) Polyethersulfon
Cellulose Nitrate Filter 0,45 Sartorius AG
(Göttingen, GER) Cellulose Nitrat
Cellulose Nitrate Filter 0,20 Sartorius AG
(Göttingen, GER) Cellulose Nitrat
Um große Moleküle abzutrennen wurde das Feed vor Prozessbeginn via Nutsche (Sartorius
AG, Göttingen, GER) aufgereinigt. Die verwendeten Filter sind in Tabelle 7 aufgeführt. Das
vorfiltrierte Feed wird als Nullprobe bezeichnet.

Material & Methoden
54
3.2.2 Optimierung der Crossflow-Performance & der Trennschärfe
3.2.2.1 Einfluss von pH-Wert und Ionenstärke
Da die Trennschärfe eines membranbasierten Trennverfahrens hauptsächlich auf gezielter
Auswahl von Feed-pH-Wert und Ionenstärke basiert (Huisman et al., 2000; Zydney, 1998),
wurden für jede Proteinquelle eine umfassende pH-Versuchsreihe durchgeführt. Die Auswahl
der pH-Stufen, dargestellt in Tabelle 8, orientierte sich, sofern vorhanden, an aktueller
Fachliteratur (Wang et al., 2006; Almécija et al., 2007). Neben der pH-Reihe wurde der Einfluss
einer 150 mM Natriumchlorid (NaCl) Zugabe überprüft (Zydney, 1998).
Die Untersuchungen erfolgten neben hydrolysiertem Molkenprotein-Isolat (Thermax 690,
Glanbia Nutritionals Ltd., IRL) für die hydrolysierte Alge Chlorella vulgaris (Allma, Lissabon,
PRT).
Tabelle 8: Übersicht - pH-Werte je Proteinquelle
pH/Proteinquelle Molkenprotein-Isolat Alge (Chlorella vulgaris)
3 X
4 X
6 X X
7 X X
8 X X
9 X
11 X
Zur Fraktionierung wurde eine hydrophilierte Polysulfon-Membran mit einer Trenngrenze von
100 kDa (Microdyn Nadir GmbH, Wiesbaden, GER) (Tabelle 6) verwendet. Für die Alge
(Chlorella vulgaris) war zuvor die Entwicklung eines geeigneten Hydrolyseverfahrens
notwendig (Kapitel 3.3). Die Auswahl der Trenngrenze orientierte sich an der per SE-HPLC
ermittelten Peptidgrößenverteilung des Feeds.
Um einem starken Konzentrationsanstieg im Retentat vorzubeugen, wurde das Verfahren der
diskontinuierlichen Diafiltration, vergleichbar Baldasso, Barros, & Tessaro (2011), eingesetzt.
Abbildung 35 zeigt schematisch den Ablauf der Lösungsmittelzugabe. Dem Feed wurde zu
Beginn des Prozesses ein Diavolumen VDia deionisiertes Wasser zugeführt. Sobald eine dem
Diavolumen VDia entsprechende Permeatmenge VPer aufgefangen wurde, erfolgte die Zugabe
des nächsten Diavoluminas. Der pH-Wert des Feeds wurde nach Zugabe eines Diavoluminas
VDia auf den vor Zugabe ermittelten Wert eingestellt. Eine Fraktionierung endete, sobald vier
Volumenanteile (4 x VDia) Permeat erzeugt wurden.

Material & Methoden
55
Feed H2O H2O H2O H2O
Filtration
Permeat Permeat Permeat Permeat
Retentat
+ + +
Abbildung 35: Schematischer Ablauf der Diafiltration
Die Feed-Konzentration wurde, basierend auf den Erkenntnissen der Pilot-Versuche (siehe
Kapitel 4.3.1), auf 0,15 % (w/v) festgelegt. Für optimale Löslichkeit des hydrolysierten
Molkenprotein-Isolats, bzw. der hydrolysierten Alge (Chlorella vulgaris) in 500 ml deionisiertem
Wasser, wurde das Feed auf 45 ± 0,2 °C temperiert. Der jeweilige pH-Wert des Versuchs
(siehe Tabelle 8) wurde mittels Natriumhydroxyd (NaOH) bzw. Salzsäure (HCl) eingestellt.
Neben der pH-Reihe wurde der Einfluss einer 150 mM NaCl-Zugabe überprüft. Hierzu wurden
dieselben pH-Stufen verwendet.
Um große Moleküle abzutrennen, wurde das Feed vor Prozessbeginn via Nutsche (Sartorius
AG, Göttingen, GER) aufgereinigt. Die verwendeten Filter sind in Tabelle 7 aufgeführt. Das
vorfiltrierte Feed wird als „Nullprobe“ bezeichnet.
Die im Rahmen dieser Studie verwendete Schlauchpumpe (CD 1500, Heraeus Instruments,
Hanau, GER) verfügt über eine variable Einstellung der Fördermenge Q. In Tabelle 9 sind die
mit verschiedenen Fördermengen korrelierenden Strömungsgeschwindigkeiten ʋ dargestellt.
Tabelle 9: Pumpenfördermengen und daraus resultierenden Strömungsgeschwindigkeiten
Fördermenge Q (ml/min) Strömungsgeschwindigkeit ʋ (m/s)
1000 0,24
2000 0,47
4000 0,94
6000 1,41
8000 1,88

Material & Methoden
56
Während der Pilot-Versuche wurde der Einfluss der Strömungsgeschwindigkeit ʋ auf die
Performance der Anlagen-Konzeption überprüft. Bei Strömungsgeschwindigkeiten ʋ zwischen
0,24 – 0,47 m/s (Fördermenge: 1000 ml/min – 2000 ml/min) konnten während des Anfahrens
des Systems entstandene Luftblasen nicht vollständig aus der Filtrationseinheit transportiert
werden. Das Forderverhalten erschien „ungleichmäßig“, sowie „schubartig“. Ein Anheben der
Strömungsgeschwindigkeit ʋ über 0,94 m/s (Fördermenge: 4000 ml/min) war im Rahmen
dieser Studie für die gegebene Anlagenkonzeption ebenfalls nicht über längere Zeiträume
möglich. In Abhängigkeit vom Transmembrandruck ΔpTMP führten höhere
Strömungsgeschwindigkeiten ʋ zu mechanisch bedingtem, unrundem Pumpenlauf und
darüber hinaus zum Aufblähen und schlussendlich Platzen der Schläuche. Schläuche mit
höherer Wandstärke/Stabilität konnten auf der verwendeten Schlauchpumpe nicht verwendet
werden, da Rotor und Stator aufgrund der geringeren Elastizität nicht zur Kompression des
Schlauchmaterials imstande waren und somit der notwendige Ansaugunterdruck nicht erzeugt
werden konnte.
Die für die Versuche in dieser Studie angewendeten Parameter pTMP und Fördermenge Q der
Pumpe sind in Tabelle 10 dargestellt.
Tabelle 10: Übersicht - Parameter der Crossflowfraktionierung
Proteinquelle Trenn-
grenze (kDa)
Transmembrandruck
pTMP (bar)
Pumpenfördermenge Q
(ml/min)
Molkenprotein-
Isolat 100 0,5 4000
Alge (Chlorella
vulgaris) 100 0,5 4000
Während des Fraktionierungsprozess wurden die Prozesszeit tF, die Permeat- und
Retentatmengen VPer bzw. VRet, sowie der pH-Verlauf (nur für die hydrolysierte Alge (Chlorella
vulgaris)) (Messintervall: Minute 1 – 10, minütlich; Minute 10 – Ende, alle 5 Minuten) während
des Prozesses dokumentiert. Nach Abschluss des Fraktionierungsprozesses erfolgte die
Bestimmung des Proteingehalts CS-Ret/Per (Kapitel 3.4.2), die Aufkonzentration der Fraktionen
(Kapitel 3.4.1), sowie die Probenvorbereitung für die SE-HPLC Analyse (Kapitel 3.4.4.3).
Alle Messungen erfolgten in Dreifachbestimmung.
3.2.2.2 Einfluss von Ultraschall
Für intakte Molkenproteine resultiert eine Fraktionierung unter Ultraschallanwendung (50 kHz)
in einer signifikanten Steigerung des Permeatflux und somit in einer Verminderung der
Prozesszeit durch Beeinflussung der Deckschichtstabilität (Muthukumaran, Kentish, Stevens,

Material & Methoden
57
Ashokkumar, & Mawson, 2007; Muthukumaran, Kentish, Lalchandani, et al., 2005;
Muthukumaran, Kentish, Ashokkumar, et al., 2005). Der Einfluss von Ultraschall auf die
Trennschärfe der Fraktionierung wurde bis dato wenig beachtet.
In dieser Studie sollte der Einfluss von Ultraschall auf den Prozessparameter Permeatflux JFm,
sowie auf die Trennschärfe der Fraktionierung für hydrolysierte Molkenprotein-Isolate
untersucht werden. Der Versuchsaufbau ist in Abbildung 36 dargestellt. Abweichend zu den
Hauptversuchen wurden für diese Versuchsreihe zwei mit Polyethersulfon (PES) Membranen
ausgestattete Crossflow-Filtrationskammern (Vivaflow 50, Sartorius AG, Göttingen, GER) mit
einer Trenngrenze von 100 kDa verwendet. Filtrationskammer 1 wurde in einem beheizbaren
Wasserbad (RM6 S, Lauda Dr. R. Wobser GmbH & Co.KG, Lauda-Königshofen, GER)
platziert und auf 48 ± 0,2 °C temperiert. Filtrationskammer 2 wurde in ein Ultraschallwasserbad
mit einer Frequenz von 35 kHz (Super RKS 10 H, Bandelin Sonorex, Berlin, GER) eingebracht.
Die Temperierung auf 48 ± 0,2 °C erfolgte in diesem Fall durch den Erwärmungseffekt des
Ultraschalls. Die Temperatur der Wasserbäder wurde mit Hilfe von Thermometern (Testo 110,
Testo AG, Lenkirch, GER) überprüft.
48 °C
Feed/Retenat
Wasserbad (beheizt)
Waage
PermeatSartorius Crossflow Kammer
Sartorius Crossflow Kammer
Ultraschall-Wasserbad (beheizt)
Feed/Retenat
48 °CWaage
Permeat
pH pH
Abbildung 36: Schematischer Aufbau der Crossflow-Anlage mit Ultraschalleinheit
Je 100 ml eines 0,30 %igen (w/v), vorfiltrierten Feeds (Tabelle 7) aus hydrolysiertem
Molkenprotein-Isolat (Thermax 690, Glanbia Nutritionals Ltd., IRL) wurden mit je zwei
Diavolumina VDia deionisiertem Wasser verdünnt und auf pH 7 eingestellt. Die Zugabe von
insgesamt zwei weiteren Diavolumina VDia erfolgte, sobald jeweils eine dem Diavolumen
entsprechende Menge Permeat VPer aufgefangen wurde. Der pH-Wert des Feeds wurde nach
Zugabe eines Diavoluminas VDia auf den vor Zugabe ermittelten Wert eingestellt. Die

Material & Methoden
58
Permeatmengen VPerUS und VPerOUS wurden gravimetrisch (LSP 3102, VWR International
GmbH, Darmstadt, GER) in einem Intervall von zwei Minuten bestimmt. Der pH-Wert des
Feeds wurde alle 5 Minuten unter Verwendung eines pH-Meters (FE 20, Mettler- Toledo AG,
Schwerzenbach, AUT) gemessen. Das Versuchssetup wurde simultan mit einer
Schlauchpumpe (Minipuls 2, Gilson Inc., Middelton, USA) bei 25 min-1 betrieben. Vor
Versuchsstart wurden beide Filtrationskammern für 15 min mit deionisiertem Wasser gespült.
Im Anschluss wurde nach weiteren 5 Minuten Laufzeit Permeat über einen Zeitraum von 5
Minuten per Messzylinder aufgefangen und der Flux JV-Wvor-US bzw. JV-Wvor-OUS beider
Filtrationskammern bestimmt. Eine Fraktionierung endete sobald vier Volumenanteile (4 x VDia)
Permeat erzeugt wurden.
Nach Abschluss des Fraktionierungsprozesses erfolgte die Bestimmung des Proteingehalts
CS-Ret/Per (Kapitel 3.4.2), die Aufkonzentration der Fraktionen (Kapitel 3.4.1), sowie
anschließend die Probenvorbereitung für die SE-HPLC Analyse (Kapitel 3.4.4.3).
Die Filtrationskammern wurden nach jedem Versuchsdurchgang einer Reinigung unterzogen.
Hierzu wurden die Kammern jeweils 15 min mit deionisiertem Wasser, 30 min mit 0,075 %
NaOH-Lösung und abschließend abermals 15 min mit deionisiertem Wasser gespült. Der
Permeatflux JV-Wvor-US bzw. JV-Wvor-OUS der Kammern wurde vor jedem Versuchsstart überprüft.
Alle Messungen erfolgten in Sechsfachbestimmung.
3.2.2.3 Eignung von regenerierten Cellulose Membranen für die Fraktionierung
Neben synthetischen Polyethersulfon Membranen finden auch Filtermedien aus regenerierter
Cellulose (RC) Anwendung in der Proteinaufreinigung (Walter et al., 2012). In einem
Pilotversuch wurde, unter Verwendung der zuvor ermittelten optimalen Bedingungen für das
Feed, ermittelt, ob die Nutzung von RC-Membranen in einer Verbesserung der Trennleistung
resultiert.
Der Versuchsaufbau ist in Abbildung 37 dargestellt. Abweichend zu den Hauptversuchen
wurde für diese Versuchsreihe eine mit Membranen aus regenerierter Cellulose ausgestattete
Crossflow-Filtrationskammer (Vivaflow 50, Sartorius AG, Göttingen, GER) mit einer
Trenngrenze von 100 kDa genutzt. Als Feed wurden 100 ml einer vorfiltrierten (Tabelle 7),
0,15 %igen (w/v) Molkenprotein-Isolat-Lösung, pH 9 (Thermax 690, Glanbia Nutritionals Ltd.,
IRL) verwendet. Das Feed wurde mit Hilfe eines beheizbaren Wasserbads (RM6 S, Lauda Dr.
R. Wobser GmbH & Co.KG, Lauda-Königshofen, GER) auf 45 ± 0,2 °C temperiert.

Material & Methoden
59
Wasserbad (beheizt)
45 °C
45 °C
Feed/Retenat
Wasserbad (beheizt)
Sartorius Crossflow Kammer
Waage
Permeat
pH
Abbildung 37: Schematischer Aufbau der Crossflow- Anlage (RC-Membran)
Zugabe der Diavolumina, Dokumentation und Aufnahme der Messwerte, Betriebsparameter
und Art der Geräte, sowie Reinigungs- und Aufarbeitungsprocedere, wurden von den
Versuchen zum Einfluss von Ultraschall (Kapitel 3.2.2.2) übernommen.
Alle Messungen erfolgten in Dreifachbestimmung.
3.2.2.4 Dialyse zur Abtrennung kleiner Moleküle (< 2 kDa) aus dem Feed
Die Trennleistung von membranbasierten Trennverfahren kann durch die Ausbildung von
Konzentrationspolarisation, Membran-Protein-Interaktionen und damit in Verbindung
stehendem Fouling beeinträchtigt werden (Huisman et al., 2000; Aimar et al., 1994; Marshall,
Munro, & Trägårdh, 1993). Durch Bildung eines Filterkuchens auf der Membranoberfläche
oder durch Adsorption in den Poren der Membran wird die nominale Trenngrenze
herabgesetzt, sodass auch niedermolekulare Partikel zurückgehalten werden können (Wang
& Tarabara, 2008; Marshall et al., 1993).
Im Rahmen des Versuchs wurde überprüft, ob niedermolekulare Bestandteile (< 2 kDa) aus
einem zur Fraktionierung vorgesehenen, vorfiltrierten Feed per Dialyse abgetrennt werden
können. Hintergrund des Versuchs war die Überprüfung der Eignung der Dialyse zur
Abtrennung kleiner Moleküle (< 2 kDa) aus der zur Fraktionierung vorgesehenen Nullprobe.
Als Feed wurde eine Molkenprotein-Isolat-Lösung (Thermax 690, Glanbia Nutritionals Ltd.,
IRL) (1 % (w/v), 2 % (w/v), 4 % (w/v); bezogen auf den Proteingehalt) verwendet.

Material & Methoden
60
Hochmolekulare Bestandteile wurden per Vorfiltration des Feeds abgetrennt (Tabelle 7).
Neben der Feed-Konzentration wurde der Einfluss von Initial-pH-Wert (3, 5, 6, 7,5, 9, 11) und
Dialysezeit (24 h, 48 h) auf die Trennschärfe ermittelt. Unter Initial-pH wird der pH-Wert der
Dialyseprobe verstanden, der zu Beginn des Dialyseprozess, sowie bei Wechsel der
Dialyselösung eingestellt wurde. Als Dialyselösung wurde destilliertes Wasser verwendet. Die
Dialyselosung „destilliertes Wasser“ wurde mit dem Hintergrund gewählt, das Feed frei von
den zur Herstellung von Dialysepuffern üblichen Salzen, Aminen und ähnlichen Zusätzen mit
Pufferwirkung zu halten, die eine Aufreinigung des Retentats vor einem nachfolgenden
Fraktionierungsprozess notwendig machen würden. Ein Wechsel der Dialyselösung, sowie die
Justierung des pH-Werts der Dialyseprobe auf den Initial-pH, erfolgten jeweils nach 2, 4 und
6 h (24 h Versuch) sowie nach 2, 4, 6, 24, 26 und 28 h (48 h Versuch). Eine schematische
Übersicht der Versuche ist in Abbildung 38 dargestellt.
Molkenprotein-Isolat
4 % (w/v)2 % (w/v)1 % (w/v)
pH 6 pH 7,5 pH 9 pH 11pH 5pH 3 pH 9 pH11 pH 9 pH 11
24 h 48 h
pH 9 pH 11
4 % (w/v)
24 h 24 h
Abbildung 38: Schematische Übersicht der Dialyseversuche
23 ml Feed wurden in einen nach Herstellerangaben vorbereiteten Dialyseschlauch mit einer
nominalen Trenngrenze von 2 kDa (Spectra/Por 6, Ø 24 mm, Spectrum Labs Inc., Rancho
Dominguez, CA) eingebracht. Der verschlossene Dialyseschlauch wurde in 2,3 l Dialyselösung
überführt und unter Rühren (IKAMAG RCT Basic, IKA Labortechnik GmbH & Co. KG, Staufen,
GER) bei Raumtemperatur dialysiert. Nach Abschluss der Dialyse wurde der Proteingehalt
CS-Ret des Retentats photometrisch (Kapitel 3.4.2) erfasst. Die Analyse der
Peptidgrößenverteilung wurde mittels SE-HPLC (Kapitel 3.4.4) durchgeführt.
Alle Messungen erfolgten in Dreifachbestimmung.
3.3 Hydrolyse der Alge (Chlorella vulgaris)
Für die Alge (Chlorella vulgaris) war die Entwicklung eines eigenes Hydrolyseverfahrens
notwendig. Ziel war die Erzeugung eines Feeds mit einer möglichst breiten

Material & Methoden
61
Molekülgrößenverteilung unter Verwendung von in der Lebensmittelindustrie genutzten
Enzymen.
Im Rahmen der Hydrolyseversuche für die Alge Chlorella vulgaris wurden die Enzyme
Corolase 2 TS (AB Enzymes GmbH, Darmstadt, GER), Pronase - Streptomyces griseus
(Merck KGaA, Darmstadt, GER) sowie Pancreatin - Porcine pancreas (Sigma Aldrich GmbH,
Steinheim, GER) auf ihre Eignung für die Hydrolyse überprüft. Von den Herstellern
angegebene Temperatur- und pH-Optima der Enzyme sind in Tabelle 11 dargestellt.
Tabelle 11: Herstellerangaben zu den Enzymen Corolase, Pronase und Pancreatin
Enzym pH-Optimum Temp.-Optimum TOH (°C) Enzymaktivität
Corolase 2 TS 6,5 65 1100 UHb/g
Pronase 7,5 40 45000 PU/g
Pancreatin 7,5 40 8000 USP/g
3.3.1.1 Die Methodenerstellung
Je 200 ml destilliertes Wasser wurden mit Hilfe eines beheizbaren Magnetrührers (IKAMAG
RCT Basic, IKA Labortechnik GmbH & Co. KG, Staufen, GER) auf das Temperatur-Optimum
TOH erwärmt. Anschließend wurden 2,5 % (w/v) Alge (Chlorella vulgaris Powder, Allma,
Lissabon, PRT) (bezogen auf den Proteingehalt) im temperierten Wasser suspendiert und der
pH-Wert auf 7 (Corolase) bzw. 8 (Pancreatin/Pronase) eingestellt.
Der Hydrolyseprozess startete mit Zugabe des jeweiligen Enzyms in den in Abbildung 39
dargestellten Konzentrationen.
Je Enzym (Corolase, Pronase, Pancreatin) erfolgte zuerst eine Hydrolyse der Algen für 45 min
unter Verwendung von 0,125 % Pronase (56,25 PU), 0,25 % Pronase (112,5 PU), sowie 0,5
% Pronase (225 PU), 0,125 % Pancreatin (10 USP), 0,25 % Pancreatin (20 USP), sowie 0,5
% Pancreatin (40 USP) und 0,1 % Corolase (2,75 UHb), 1,5 % Corolase (41,25 UHb), sowie 3,0
% Corolase (82,5 UHb).
Darüber hinaus wurden für die Enzym Pronase und Pancreatin Versuche mit einer
Vorbehandlung im sauren Bereich durchgeführt. Hierzu wurde die Algensuspension (2,5 %
(w/v), bezogen auf den Proteingehalt) mit HCl auf pH 3 eingestellt und für 18 h, sowie 48 h im
Wasserbad (Ecoline RE 206, Lauda Dr. R. Wobser GmbH & Co.KG, Lauda-Königshofen,
GER) bei 45 ± 0,2 °C inkubiert. Anschließend erfolgte die Anhebung des pH auf pH 8 ± 0,04.
Das inkubierte Feed wurde darauf folgend für 45 min bei Optimal-Temperatur unter
Verwendung von 0,25 % Pronase (112,5 PU) bzw. 0,25 % Pancreatin (20 USP) hydrolysiert.

Material & Methoden
62
Pancreatin Pronase
0,25 %0,125 % /
Hydrolyse: 45 min0,5 % /
Hydrolyse: 45 min0,125 % /
Hydrolyse: 45 min0,5 % /
Hydrolyse: 45 min0,25 %
Vorinkubation: 0,02 %
Rohament PL
Vorinkubation:HCl, pH 3
18 h 48 h
72 h
48 h18 h
Hydrolyse: 45 min
Hydrolyse:135 min
Vorinkubation: 0,02 %
Rohament PL
Vorinkubation:HCl, pH 3
18 h 48 h
72 h
48 h18 h
0,25 %
Hydrolyse: 45 min Hydrolyse: 45 min Hydrolyse: 45 min Hydrolyse: 45 min
Hydrolyse: 45 min
Hydrolyse:135 min
Corolase
1,5 % / Hydrolyse: 45 min
0,1 % / Hydrolyse: 45 min
3 % /Hydrolyse: 45min
Legende: Einfachversuch
Abbildung 39: Schematische Übersicht der Hydrolyseversuche
Zusätzlich zur Vorbehandlung im sauren Bereich wurde der Einfluss eines proteolytischen
Enzymes (Pektinase, Rohament® PL, AB Enzymes GmbH, Darmstadt, GER) untersucht. Das
vom Hersteller angegebene Temperatur- und pH-Optima der Pektinase ist in Tabelle 12
dargestellt.
Tabelle 12: Herstellerangaben zu Rohament® PL
Enzym pH-Optimum Temp.-Optimum TOH (°C) Enzymaktivität
Rohament® PL 4 - 5 - 28000 PGU/mg
Der auf pH 4,5 ± 0,05 eingestellten Algensuspension (2,5 % (w/v), bezogen auf den
Proteingehalt) wurden 0,02 % Rohament® PL (14000 PGU) zugegeben. Im Anschluss erfolgte
eine Inkubation für 18 h, 48 h, sowie 72 h bei 45 ± 0,2 °C im Wasserbad (Ecoline RE 206,

Material & Methoden
63
Lauda Dr. R. Wobser GmbH & Co.KG, Lauda-Königshofen, GER). Rohament® PL wurde durch
eine fünfminütige Erhitzung auf 100 °C inaktiviert. Nach der proteolytischen Vorbehandlung
wurde der pH-Wert auf 8 ± 0,05 eingestellt und die Algensuspension für 45 min unter
Verwendung von 0,25 % Pronase (112,5 PU) bzw. 0,25 % Pancreatin (20 USP) hydrolysiert.
Der Hydrolyseprozess endete jeweils mit einer Inaktivierung der Enzyme durch eine
zehnminütige Erhitzung auf 100 °C.
pH-Wert und Temperatur wurden jede Minute (Minute 1 – 5) bzw. in einem Fünf-Minuten-
Intervall (Minute 5 – Ende) dokumentiert. Bei einem Abfall des pH-Wertes unter 6,2 (Corolase)
bzw. 7,2 (Pronase/Pancreatin) erfolgte eine Anhebung auf 6,5 (Corolase) bzw. 7,5 (Pronase/
Pancreatin).
Sofern nicht anders gekennzeichnet, erfolgten alle Messungen in Dreifachbestimmung.
3.3.1.2 Methode zur Hydrolyse der Alge (Chlorella vulgaris)
Zur Herstellung des Hydrolysats für die Crossflow Diafiltration wurden, basierend auf den
Ergebnissen der Methodenerstellung, 500 ml einer 2,5 %igen (w/v) Algensuspension
(Chlorella vulgaris Powder, Allma, Lissabon, PRT) (bezogen auf den Proteingehalt) mit HCl
auf pH 3 eingestellt und 48 h im Wasserbad bei 45 ± 0,2 °C inkubiert. Anschließend erfolgte
die Anhebung des pHs auf 8 ± 0,06 und die Temperierung des Feeds auf TOH 40 ± 1 °C. Der
Hydrolyseprozess wurde durch Zugabe von 0,25 % Pronase (112,5 PU) (bezogen auf den
Proteingehalt) gestartet und nach 45 min durch eine Inaktivierung der Enzyme bei 100 °C
beendet.
3.3.2 Aufbereitung und Analytik der Hydrolysate
Im Anschluss an die Hydrolyse wurde das Medium für 30 min bei 3000 min-1 zentrifugiert
(Megafuge 1.0 R, Thermo Fisher Scientific, Waltham, USA). Der Hydrolysegrad des
Überstandes wurde mittels OPA-Methode (Kapitel 3.4.3), die
Molekülgrößenzusammensetzung mittels SE-HPLC (Kapitel 3.4.4) ermittelt. Bis zur
Verwendung wurde das Hydrolysat bei -18 °C gelagert.
3.4 Instrumentelle Analytik & Probenaufbereitung
3.4.1 Aufkonzentration der Peptidfraktionen
Die während des Fraktionierungsprozesses gewonnen Retentate und Permeate wurden für
die weitere Verwendung mit Hilfe eines Rotationsverdampfers-Setups, bestehend aus einem
Rotationsverdampfer mit Wasserbad, temperiert auf TWH 75 ± 5 °C (Rotavapor R-124 + B-
490, Büchi Labortechnik GmbH, Essen, GER), einer Vakuumpumpe (MZ2C, Vaccumbrand
GmbH, Wertheim, GER) zu Erzeugung des Unterdrucks (pU 0,3 ± 0,02 bar), sowie einem
Wasserbad (TWK 8 ± 2 °C) zur Kondensation des Wasserdampfs (Ecoline RE 212, Lauda Dr.

Material & Methoden
64
R. Wobser GmbH & Co.KG, Lauda-Köngishofen, GER) auf ein Volumen von ca. 100 ml
aufkonzentriert und im Anschluss bei -18 °C gelagert.
3.4.2 Spektralphotometrische Proteingehaltbestimmung
Gemäß des Lambert-Beer-Gesetzes besteht bei konstanter Schichtdicke des durchstrahlten
Mediums ein proportionaler Zusammenhang zwischen der Extinktion eines
monochromatischen Lichtstrahls und der molaren Konzentration einer gelösten Substanz, die
eine Bestimmung der molaren Konzentration bei Vorhandensein einer Kalibrationsfunktion
erlaubt (Gressner & Arndt, 2007). Die Bestimmung des Proteingehalts der Fraktionen erfolgte
spektralphotometrisch bei einer Wellenlänge von 280 nm (Aitken & Learmonth, 2009).
Zur Erstellung der Kalibrationsfunktionen wurden Stammlösungen (3 g/l Molkenprotein-Isolat
(bezogen auf den Proteingehalt); 1,6 g/l Alge (Chlorella vulgaris) (bezogen auf den
Proteingehalt)) aus hydrolysiertem Molkenprotein-Isolat und hydrolysierter Alge (Chlorella
vulgaris) hergestellt und in Anlehnung an Whitaker & Granum (1980) mit 20 mM Natrium-
Phosphat Puffer schrittweise verdünnt, um verschiedene Konzentrationsstufen CF abzubilden.
Für die hydrolysierte Alge Chlorella vulgaris war zuvor eine referenzmethodische Bestimmung
des Proteingehalts notwendig (siehe Kapitel 3.4.2.1). Die verwendeten Konzentrationsstufen
sind in Tabelle 13 dargestellt.
Tabelle 13: Konzentrationsstufen CF der Kalibrationslösungen
Probe Molkenprotein-Isolat (g/l) Alge (Chlorella vulgaris) (g/l)
1 0,00 0,00
2 0,30 0,20
3 0,60 0,40
4 0,90 0,60
5 1,20 0,80
6 1,50 1,00
7 1,80 1,20
8 2,10 1,40
9 2,40 1,60
10 2,70
11 3,00
Die Konzentration der Molkenprotein-Isolat-Stammlösung wurde unter Berücksichtigung des
Proteingehalts des verwendeten Rohstoffs (Thermax 690, Glanbia Nutritionals Ltd., IRL)
ermittelt und ist somit als absolut zu betrachten. Der Proteingehalt der hydrolysierten Alge
(Chlorella vulgaris) wurde referenzmethodisch per Kjeldahl-Stickstoffbestimmung ermittelten
(Kapitel 3.4.2.1).

Material & Methoden
65
Das Puffermedium wurde für hydrolysiertes Molkenprotein-Isolat in Anlehnung an Britten et al.
(1994) auf pH 8 und für die hydrolysierte Alge Chlorella vulgaris in Anlehnung an Hadjoudja,
Deluchat, & Baudu (2010) auf pH 6,5 eingestellt. Die Messungen erfolgten mit Hilfe eines
Spektralphotometers (Ultraspec III, Pharmacia LKB Biochrom Ltd., Cambridge, UK). Durch
Korrelieren der gemessenen Extinktionen mit den Konzentrationen der Kalibrationslösungen
konnten Kalibrationsfunktionen ermittelt werden.
Alle Messungen erfolgten in Dreifachbestimmung.
3.4.2.1 Referenzmethodische Bestimmung des Proteingehalts der hydrolysierten Alge
(Chlorella vulgaris)
Zur Erstellung der Spektralphotometerkalibrationen ist Kenntnis bezüglich der Proteingehalte
der verwendeten Kalibrationsstandards notwendig. Der Proteingehalte organischer Stoffe
kann per Kjeldahl-Stickstoffbestimmung ermittelt werden (Nielsen, 2010; Kjeldahl, 1883).
Die Bestimmung des Proteingehalts der hydrolysierten Alge (Chlorella vulgaris) wurde extern
an das Labor KWALIS Qualitätsforschung (Fulda GmbH, Fulda, GER) vergeben.
Alle Messungen erfolgten in Dreifachbestimmung.
3.4.3 Bestimmung des Hydrolysegrads der Proteinhydrolysate
Die Bestimmung des Hydrolysegrads der Algenhydrolysate (Chlorella vulgaris) erfolgte gemäß
der OPA-Methode von Nielsen, Petersen, & Dambmann (2001).
Die OPA-Methode ist eine analytische Schnellmethode zur Bestimmung des Hydrolysegrads,
die auf der Reaktion primärer Aminogruppen mit o- Phthaldialdehyd beruht (Nielsen et al.,
2001). Die Herstellung der benötigten Reagenzien, sowie die Durchführung der Bestimmung,
sind nachfolgend beschrieben.
Die OPA-Reagenz, sowie ein Serinstandard, wurden vor jeder Anwendung frisch hergestellt.
OPA-Reagenz:
Zur Herstellung der OPA-Reagenz wurden 7,62 g di-Natriumtetraborat Decahydrat (di-
Natriumtetraborat Decahydrat 99,0 - 103,0 % AnalaR NORMAPUR®, VWR International
GmbH, Darmstadt, GER) und 200 mg Natriumdodecylsulfat (SDS) (Natriumdodecylsulfat ≥
98,0 %, VWR International GmbH, Darmstadt, GER) in 150 ml destilliertem Wasser vollständig
gelöst. 160 mg o-Phthaldialdehyd (Phthaldialdehyd ≥ 97 %, Sigma Aldrich GmbH, Steinheim,
GER) wurden in 4 ml Ethanol gelöst und in die bestehende Lösung überführt. Abschließend
wurden der Lösung 176 mg Dithiothreitol (Dithiothreitol ≥ 98 %, Alfa Aesar GmbH & Co. KG,
Karlsruhe, GER) zugefügt und die Lösung mit destilliertem Wasser auf 200 ml aufgefüllt.

Material & Methoden
66
Serin-Standard:
Die Herstellung des Serinstandards erfolgte durch Zugabe von 50 mg Serin (L(+)-Serin, Merck
KGaA, Darmstadt, GER) zu 500 ml destilliertem Wasser.
Die Bestimmung des Hydrolysegrads mittels OPA-Methode erfolgte rechnerisch unter
Verwendung spektralphotometrischer Extinktions-Messwerte von Blindproben, Serinstandard,
sowie der eigentlichen Probe.
Für die Messungen wurden Blindprobe, Serinstandard, sowie die eigentliche Probe gemäß
nachfolgendem Schema in Küvetten (Brand GmbH & Co. KG, Wertheim, GER) pipettiert.
Blindprobe:
3 ml OPA-Reagenz + 400 µl destilliertes Wasser
Serinstandard:
3 ml OPA-Reagenz + 400 µl Serinstandard
Probe:
3 ml OPA-Reagenz + 400 µl Probe
Verdünnung hydrolysierte Alge (Chlorella vulgaris): 1:20
Nach dem Pipettieren beider Komponenten erfolgte ein sofortiges Mischen für 10 sec. Nach 2
min Standzeit wurden die Proben bei 340 nm spektralphotometrisch analysiert (Ultraspec III,
Pharmacia LKB Biochrom Ltd., Cambridge, UK). Die gemessene Extinktion dient als
Berechnungsgrundlage für die Bestimmung des Hydrolysegrads.
Die Extinktion von Blindprobe und Serinstandard wurde je Hydrolysatprobe vierfach, die
Extinktion der zu untersuchenden Probe dreifach bestimmt.
Der Hydrolysegrad wurde gemäß Formel 9 (Seite 33) ermittelt. Hierzu ist vorab die
Berechnung von h (Anzahl hydrolysierter Peptidbindungen), sowie die Kenntnis von htot
(Gesamtzahl der Peptidbindungen pro Protein-Äquivalent) notwendig.
h wurde mit Hilfe der ermittelten mittleren Extinktionen von Blindprobe, Serinstandard, sowie
der zu untersuchenden Probe gemäß Formel 10, berechnet (Nielsen et al., 2001). Die
rohstoffspezifischen Konstanten α (1,00), β (0,40), sowie htot (8,3) entsprechend den
Referenzwerten von Nielsen et al. (2001), sowie Adler-Nissen (1986).

Material & Methoden
67
ℎ =((𝑦𝐸𝑥.−𝑃𝑟𝑜𝑏𝑒 − 𝑦𝐸𝑥.−𝐵𝑙𝑖𝑛𝑑/𝑦𝐸𝑥.−𝑆𝑒𝑟𝑖𝑛 − 𝑦𝐸𝑥.−𝐵𝑙𝑖𝑛𝑑 ∗ 0,9516 ∗ 0,1 ∗ 100/𝑋 ∗ 𝑃) − 𝛽)
𝛼
Formel 10: Berechnung von h
h = Anzahl hydrolysierter Peptidbindungen
α = Konstante (1,00)
β = Konstante (0,40)
yEx.-Probe = mittlere Extinktion der Probe
yEx.-Blind = mittlere Extinktion der Blindprobe
yEx.-Serin = mittlere Extinktion des Serinstandard
X = Einwaage der Probe
P = Proteingehalt der Probe
3.4.4 Analytik der Peptidfraktionen mittels SE-HPLC
Die Analyse der Peptidgrößenverteilung erfolgte mittels SE-HPLC (Meyer, 1988). Die
Chromatogramme der Fraktionen wurden zur Größenbestimmung der enthaltenen
Molekülstrukturen mit einem Gelfiltrations-Standard verglichen.
3.4.4.1 Methodenerstellung
Um die Performance der SE-HPLC hinsichtlich Auflösung und Analysegeschwindigkeit zu
optimieren, wurden verschiedene Kombinationen aus Säule und Laufmittel unter Verwendung
eines Gelfiltrationsstandards (Gel Filtration Standard Catalog # 151-1901, BioRad
Laboratories Inc., Hercules, USA) auf einer HPLC Anlage (880-PU und UV-2077 Plus, Jasco
Labor- und Datentechnik GmbH, Gross-Umstadt, GER) überprüft. Später erfolgte aufgrund
eines Anlagenwechsels (Agilent 1100 Series, Vacuum Degasser, Binary Pump, UV/VIS-
Detector, Manual Injector, Agilent Technologies Corporation, Santa Clara, USA) die
Übertragung der Methode. Die verwendeten Säulen samt Säulenparameter und
Flussgeschwindigkeit sind in Tabelle 14, die Laufmittel in Tabelle 15 dargestellt. Die Auswahl
der überprüften Laufmittel erfolgte aufgrund von Herstellerangaben und Literatur (Webb,
Naeem, & Schmidt, 2002; Uversky & Ptitsyn, 1994; Singh, Donovan, & MacRitchie, 1990).
Tabelle 14: SEC-Säulen und zugehöriger Anlagenfluss
Säule Säulenparameter Fluss (ml/min)
Agilent Zorbax GF 250 4,6 mm x 250 mm, 4 µm 0,500
PSS Suprema 1000 Â 8 mm x 300 mm, 10 µm 1,000
Tosoh TSKgel G 3000 SWXL 7,8 mm x 30 mm, 5 µm 1,000

Material & Methoden
68
Tabelle 15: Laufmittel zur SE-HPLC Methodenerstellung
Laufmittel Molarität (mol/l) pH-Wert
Natriumphosphatpuffer + 0,05 % NaN3 0,05 6,9
Natriumphosphatpuffer + 0,05 M Na2SO4 + 0,05 % NaN3 0,05 6,9
Natriumphosphatpuffer + 0,05 % NaN3 0,10 6,7 / 7,0
Natriumphosphatpuffer + 0,10 M Na2SO4 + 0,05 % NaN3 0,10 6,7 / 7,0
Natriumphosphatpuffer + 0,05 % NaN3 0,20 6,7 / 7,0
Natriumphosphatpuffer + 0,20 M Na2SO4 + 0,05 % NaN3 0,20 6,7 / 7,0
3.4.4.2 Methode zur SE-HPLC Analyse
Zur Analyse der Peptidfraktionen wurde, basierend auf den Ergebnissen der
Methodenerstellung, ein 0,2 M Natriumphosphatpuffer (+ 0,05 M Natriumazid (NaN3)), pH 7 in
Kombination mit einer Agilent Zorbax GF 250 (4,6 mm x 250 mm, 4 µm) (Agilent Technologies
Corporation, Santa Clara, USA) betrieben bei einem Fluss von 0,500 ml/min gewählt. Die
Aufzeichnung der Chromatogramme erfolgte im UV/VIS-Bereich bei einer Wellenlänge von
280 nm. Es wurde jeweils 100 µl Probe aufgegeben.
3.4.4.3 Probenvorbereitung
Feed, Retentat und Permeat wurden mit deionisiertem Wasser auf einen Proteingehalt von 1
mg/ml verdünnt und mittels Spritzenfilter (0,2 µm, Chromafil Xtra PES 20/25, Macherey-Nagel
GmbH & Co. KG, Düren, GER) filtriert.
3.5 Sensorik
Die Produkte des Fraktionierungsprozesses wurden sensorisch auf ihre
Geschmacksqualitäten geprüft. Hierzu erfolgte eine Prüferschulung auf die fünf
Grundgeschmacksarten „süß“, „sauer“, „salzig“, „bitter“ und „umami“.
3.5.1 Schulung des Sensorikpanels
24 Teilnehmer (8 männlich, 16 weiblich) im Alter von 19 – 29 (MW: 22,75 ± 2,44) Jahren
wurden für die Sensorikschulung ausgewählt. Die Teilnehmer wurden gebeten einen
Fragenbogen auszufüllen um Informationen über Alter, Rauchgewohnheiten, Allergien und
Sensorikerfahrung zu erhalten. Die Studie wurde von der Ethik-Kommission der Hochschule
Fulda genehmigt.
Die Schulung erfolgte an sieben einstündigen, in wöchentlichem Rhythmus stattfindenden
Terminen (siehe Abbildung 40), in Anlehnung an die DIN EN ISO 8586:2014 und die ISO
3972:2011 unter Verwendung von Standards der fünf Grundgeschmacksarten. Die Teilnehmer
wurden in zwei Gruppe à 12 Teilnehmern aufgeteilt. Die Schulung der Gruppen erfolgte an
aufeinanderfolgenden Tagen. Eine Übersicht der verwendeten Rohstoffe ist in Tabelle 16

Material & Methoden
69
dargestellt. Alle Proben wurden mit dreistelligen Zahlencodes codiert. Jeder Prüfer erhielt eine
identische Anordnung der Proben. Zur Neutralisation wurde entionisiertes Wasser verwendet.
Schulung und sensorische Bewertung fanden im klimatisierten (T = 23,1 ± 1 °C) Technikum
der Hochschule Fulda statt.
Sensorik-Schulung
Prüfung auf Geschmacks-
blindheit
Prüfung zur Wahrnehmung
eines Reizes
Ermittlung der Reiz- und
Erkennungs-schwelle
Wdhlg. 1
Wdhlg. 1
Whdlg. 2
Wdhlg. 2
Wdhlg. 3
Wdhlg. 3
Schulung zur Anwendung
der Skale
Prüfung zur Unterscheidung von Intensitäts-
stufen
Wdhlg. 1
Wdhlg. 2
Wdhlg. 3
Wdhlg. 1
Wdhlg. 2
Wdhlg. 3
Termin 1
Termin 2
Termin 3
Termin 4
Termin 5
Termin 6
Termin 7
Abbildung 40: Ablaufplan der Sensorik-Schulung
Tabelle 16: Rohstoffe zur Herstellung der Schulungsstandards
Grundgeschmack Rohstoff Bezugsquelle
süß Saccharose, reinst Merck KGaA, Darmstadt,
GER
sauer Citronensäure Monohydrat,
reinst Bernd Kraft GmbH,
Düssburg, GER
salzig Natriumchlorid, reinst Bernd Kraft GmbH,
Düssburg, GER
bitter Koffein (wasserfrei), reinst AppliChem GmbH, Darmstadt, GER
umami Natrium-L(+)-glutamat
Monohydrat Sigma Aldrich Corporation,
St. Louis, USA gemäß DIN EN ISO 8586:2014

Material & Methoden
70
3.5.1.1 Prüfung auf Geschmacksblindheit (gemäß DIN EN ISO 8586:2014)
Zu Beginn des Tests wurde den Teilnehmern die Möglichkeit gegeben sich gemäß ISO
6658:2005 mit den verschiedenen Grundgeschmacksarten vertraut zu machen. Hierzu wurde
je eine überschwellige Probe pro Grundgeschmackart gereicht. Im Anschluss wurden die
Teilnehmer gebeten die Grundgeschmacksart von zehn Proben (3x „süß“, 1x „sauer“, 1x
„salzig“, 3x „bitter“, 2x „umami“) zu erkennen. Die verwendeten Konzentrationen der Proben
sind in Tabelle 17 dargestellt.
Die Prüfung erfolgte in Einfachbestimmung.
Tabelle 17: Probenkonzentrationen für die Prüfung auf Geschmacksblindheit
Grundgeschmack Rohstoff Konzentration C (g/l)
süß Saccharose, reinst 10,0
sauer Citronensäure Monohydrat,
reinst 0,3
salzig Natriumchlorid, reinst 2,0 bitter Koffein (wasserfrei), reinst 0,3
umami Natrium-L(+)-glutamat
Monohydrat 0,6
gemäß DIN EN ISO 8586:2014
3.5.1.2 Prüfung zur Wahrnehmung eines Reizes (gemäß DIN EN ISO 8586:2014)
Um zu überprüfen, ob die Prüfpersonen den Geschmacksreiz einer überschwelligen Probe
feststellen können, wurde ein Dreieckstest nach DIN EN ISO 4120:2007 durchgeführt. Den
Prüfern wurde für jede Grundgeschmacksart ein Dreieck zur Bewertung gereicht. Die
abweichende Probe sollte ermittelt und die Art der Abweichung angegeben werden. Die
Reihung und Zusammenstellung (AAB, ABA, ABB, BAB, BBA) wurde je Dreieck variiert. Die
verwendeten Konzentrationen der Proben sind in Tabelle 18 dargestellt.
Die Prüfung erfolgte in Dreifachbestimmung.
Tabelle 18: Probenkonzentrationen zur Wahrnehmung eines Reizes
Grundgeschmack Rohstoff Konzentration C (g/l)
süß Saccharose, reinst 7,2
sauer Citronensäure Monohydrat,
reinst 0,2
salzig Natriumchlorid, reinst 1,4 bitter Koffein (wasserfrei), reinst 0,2
umami Natrium-L(+)-glutamat
Monohydrat 0,3
in Anlehnung an DIN EN ISO 8586:2014
3.5.1.3 Ermittlung der Reiz- und Erkennungsschwelle (gemäß ISO 3972:2011)
Um die Reiz- und Erkennungsschwellen der Prüferpersonen zu ermitteln wurde eine einfache
beschreibende Prüfung nach DIN 10964:2014 durchgeführt. Den Prüfern wurden je

Material & Methoden
71
Grundgeschmacksart acht Lösungen einer Geschmacksart, in steigender Konzentration,
gereicht. Als Blindproben dienten zwei Wasserproben. Die Prüfer wurden gebeten anzugeben,
bei welcher Probe ein Geschmackseindruck (Reizschwelle) wahrgenommen, der
Probengeschmack erkannt (Erkennungsschwelle) oder eine Konzentrationsunterschied
(Unterschiedsschwelle) festgestellt wurde. Die verwendeten Konzentrationen der Proben sind
in Tabelle 19 dargestellt.
Die Prüfung erfolgte in Dreifachbestimmung.
Tabelle 19: Probenkonzentrationen C zur Ermittlung der Reiz-/Erkennungsschwelle
Verdünnung/Grundgeschmack süß (g/l) sauer
(g/l)
salzig
(g/l)
bitter
(g/l)
umami
(g/l)
D1 12,00 0,60 2,00 0,27 1,00
D2 7,20 0,48 1,40 0,22 0,70
D3 4,32 0,38 0,98 0,17 0,49
D4 2,59 0,31 0,69 0,14 0,34
D5 1,56 0,25 0,48 0,11 0,24
D6 0,94 0,20 0,34 0,09 0,17
D7 0,55 0,16 0,24 0,07 0,12
D8 0,34 0,13 0,16 0,05 0,08
gemäß ISO 3972:2011
3.5.1.4 Prüfung zur Unterscheidung von Intensitätsstufen eines Reizes (gemäß DIN EN ISO
8586:2014)
Um festzustellen ob die Prüfer Proben mit unterschiedlichen Ausprägungen eines Reizes
unterscheiden können, wurde eine Rangordnungsprüfung gemäß DIN ISO 8587:2010
durchgeführt. Je Grundgeschmacksart erhielten die Prüfer vier Proben die nach steigender
Intensität geordnet werden sollten. Die verwendeten Konzentrationen der Proben sind in
Tabelle 20 dargestellt.
Tabelle 20: Probenkonzentrationen C zur Prüfung zur Unterscheidung von Intensitätsstufen eines Reizes
Verdünnung/Grundgeschmack süß (g/l) sauer
(g/l)
salzig
(g/l)
bitter
(g/l)
umami
(g/l)
D1 12,00 0,60 2,00 0,27 1,00
D3 4,32 0,38 0,98 0,17 0,49
D5 1,56 0,25 0,48 0,11 0,24
D7 0,55 0,16 0,24 0,07 0,12
In Anlehnung an ISO 3972:2011 und DIN EN ISO 8586:2014

Material & Methoden
72
Die Prüfung erfolgte in Dreifachbestimmung.
3.5.1.5 Schulung zur Anwendung der Skale
Um die Geschmackswahrnehmungen zu quantifizieren wurden die Prüfer auf die Nutzung
einer ordinalen 7-Punkt Skale (siehe Abbildung 41) zur Intensitätsmessung trainiert. Die
Schulung erfolgte in Anlehnung an das Kapitel „Ausbildung eines deskriptiven Panels -
Schulungsphase: Training von Intensitätsmessungen“ von Rummel (2002).
schwach mittel stark
Nicht
wahrnehmbar
Sehr
schwach Schwach
Schwach bis
mittel Mittel
Mittel bis
Stark Stark Sehr stark
0 1 2 3 4 5 6 7
Abbildung 41: 7-Punkt Skale zur Bewertung der Prüfproben
Die Prüfer erhielten je Grundgeschmacksart drei Proben unterschiedlicher Konzentration.
Zuerst wurden die Prüfer gebeten die Proben hinsichtlich ihrer Intensität den Kategorien
„schwach“, „mittel“ und „stark“ zuzuordnen. Im nächsten Schritt sollten die Prüfer auf einer 7-
Punkt Skale angeben, wie ausgeprägt sie den jeweiligen Geschmackseindruck empfunden
haben. Die Ergebnisse wurden zusammengefasst und der Gruppe am Ende eines Termins
vorgestellt. Darüber hinaus wurden den Prüfern die Skalenwerte mitgeteilt, die von der
Versuchsleitung für die einzelnen Proben vergeben wurde. Die verwendeten Konzentrationen,
sowie die korrespondierenden Skalenwerte der Proben, sind in Tabelle 21 dargestellt.
Die Schulung erfolgte in Dreifachbestimmung.
Tabelle 21: Probenkonzentrationen C und Skalenwerte zur Schulung zur Anwendung der Skale
Grundgeschmack/Intensität
(Skalenwert) schwach (2) mittel (4) stark (6)
süß 4,32 g/l 7,20 g/l 12,00 g/l
sauer 0,20 g/l 0,38 g/l 0,60 g/l
salzig 0,48 g/l 0,98 g/l 1,40 g/l
bitter 0,07 g/l 0,14 g/l 0,27 g/l
umami 0,12 g/l 0,34 g/l 0,70 g/l
In Anlehnung an ISO 3972:2011
3.5.2 Prüferauswahl
Die Bewertung und Auswahl der Prüfpersonen erfolgte in Anlehnung an die DIN EN ISO
8586:2014. Aufgrund des ordinalen Skalenniveaus wurden verteilungsunabhängige,
nichtparametrische Testverfahren zur Auswertung herangezogen.

Material & Methoden
73
Basierend auf den Ergebnissen der Schulung zur Anwendung der Skale (Kapitel 3.5.1.5)
wurde die Diskriminierungsfähigkeit je Prüfperson für jeden Grundgeschmack mittels
Friedman-Tests überprüft. Prüfer mit nicht signifikanter Diskriminierungsfähigkeit wurden für
den betreffenden Grundgeschmack aus dem jeweiligen Panel ausgeschlossen. Im Anschluss
erfolgte ein Vergleich der Punktevergabe jeder Einzelperson in Bezug auf den
Gruppenmittelwert mit dem reduzierten Panel. Eine Abweichung > 0,6 Punkten je
Konzentrationsstufe oder mehr als eine Abweichung > 0,5 Punkten führten zum Ausschluss
der betroffenen Prüferperson.
Die Homogenität des jeweiligen resultierenden Panels wurde je Grundgeschmack mittels
Kruskal-Wallis H Tests überprüft. Bei unzureichender Homogenität der Merkmalsausprägung
(niedrige, mittlere, hohe Intensität) eines Grundgeschmacks erfolgte eine Überprüfung der
Panelzusammenstellung unter Verwendung von Mann-Whitney U Tests. Signifikante
Unterschiede zwischen den Prüfpersonen resultierten im Ausschluss der Prüfperson mit vom
Rangmittel abweichender Rangsumme.
Die Ermittlung der Teststatistiken basierten auf einem Signifikanzniveau von α = 0,05.
3.5.3 Sensorik der Peptidfraktionen
Im Rahmen der sensorischen Beurteilung der Fraktionen wurden je Proteinquelle vier Proben
zur Bewertung herangezogen. Neben der jeweiligen Permeat- und Retentatfraktion wurde ein
Bitterstandard und eine Mischung aus Bitterstandard und Permeat gereicht um zu überprüfen,
ob die Geschmacksausprägung „bitter“ durch Zusatz einer Permeatfraktion reduziert werden
kann. Die verwendeten Konzentrationen sind in Tabelle 22 dargestellt.
Tabelle 22: Probenkonzentrationen C für die sensorische Bewertung der Peptidfraktionen
Probe Probengrundstoff Konzentration C (g/l)
Retentat Molkenprotein-Isolat 1,0
Permeat Molkenprotein-Isolat 1,0
Bitterstandard Koffein 0,22
Bitterstandard + Permeat Koffein / Molkenprotein-Isolat 0,22 / 1,0
Retentat Alge (Chlorella vulgaris) 1,0
Permeat Alge (Chlorella vulgaris) 1,0
Bitterstandard Koffein 0,22
Bitterstandard + Permeat Koffein / Alge (Chlorella
vulgaris) 0,22 / 1,0

Material & Methoden
74
Die Prüfer wurden gebeten die Proben hinsichtlich der Grundgeschmacksarten „süß“, „sauer“,
„salzig“, „bitter“ und „umami“ auf einer 7-Punkt-Skale zu bewerten. Art und Probengrundstoff
der Probe wurden den Prüfern nicht mitgeteilt.
Die untersuchten Fraktionen von hydrolysiertem Molkenprotein-Isolat wurden bei pH 9 die
Fraktionen hydrolysierter Alge (Chlorella vulgaris) bei pH 6 gewonnen. Auf eine Untersuchung
der korrespondierenden Proben, die unter Zusatz von NaCl gewonnen wurden, wurde
aufgrund des starken „salzig“ Geschmacks verzichtet.
Die Prüfung erfolgte in Dreifachbestimmung.
3.6 Statistische Auswertung & Datenverarbeitung der Messergebnisse
3.6.1 Datenverarbeitung
3.6.1.1 Crossflow-Diafiltration
Zur statistischen Bewertung der Ergebnisse war zuvor eine rechnerische Angleichung der
Daten erforderlich um Unterschiede aufgrund von Schwankungen der Permeatmengen und
des Initialflux der Membranen mit destilliertem Wasser auszugleichen. Diese erfolgte durch
Normierung der Permeatmenge und der Verwendung eines gemittelten Initialflux.
𝑡𝐹−𝑘𝑜𝑟𝑟𝑖𝑔𝑖𝑒𝑟𝑡 =𝑉𝑃𝑒𝑟−𝑚𝑎𝑥
𝑉𝑃𝑒𝑟−𝑖𝑠𝑡×
∑ 𝐽𝑖𝑛𝑖=1𝑛
𝐽𝑊𝑣𝑜𝑟 × 𝑡𝐹
Formel 11: Rechnerische Angleichung der Prozesslaufzeit in Abhängigkeit des Permeatvolumen und des Initialflux der Membran mit destilliertem Wasser
tF-korrigiert = korrigierte Prozesslaufzeit
tF = unkorrigierte Prozesslaufzeit des Versuchs
VPer-max = maximale Permeatmenge einer Versuchsreihe
VPer-ist = Permeatmenge des Versuchs
JWvor = Permeatflux der Membran für destilliertes Wasser des Versuchs
∑ Ji/n = Mittelwert des Permeatflux für destilliertes Wasser
n = Anzahl der Versuche
3.6.1.2 Ultraschall
Anders als bei der Crossflow-Diafiltration der Hauptversuche, wurden die Vivaflow-Einheiten
für die Wiederholungsversuche nach einem Reinigungsprozess wiederverwendet. Um die
geringfügigen, anhand des Flux mit destilliertem Wasser ermittelten Unterschiede zwischen
ursprünglicher Performance der Vivaflow-Einheit und der Ist-Performance des jeweiligen
Wiederholungsversuchs, sowie Unterschiede der Initial-Performance beider Vivaflow-
Einheiten auszugleichen, erfolgte eine rechnerische Angleichung. Die draus resultierenden,
veränderten Probenvolumina VV-Per-korrigiert,t wurden anhand Formel 12 (für die performanter

Material & Methoden
75
Vivaflow-Einheit) und Formel 13 (für die weniger performante Vivaflow-Einheit) berechnet. Ein
Versuch wurde bei dem Erreichen einer Permeatmenge VV-Per-korrigiert von ca. 400 ml als
beendet erklärt. Die korrespondierende korrigierte Zeit tVF wurde als Prozesszeit nach
Performance-Anpassung angenommen.
𝑉𝑉−𝑃𝑒𝑟−𝑘𝑜𝑟𝑟𝑖𝑔𝑖𝑒𝑟𝑡,𝑡 = 𝑉𝑃𝑒𝑟,𝑡 × 𝐽𝑉−𝑊−𝐼𝑛𝑖𝑡𝑖𝑎𝑙
𝐽𝑉−𝑊−𝑊
Formel 12: Rechnerische Angleichung des Permeatvolumens der performanteren Vivaflow-Einheit
VPer,t = Permeatvolumen zum Messzeitpunkt t
VV-Per-korrigiert,t = korrigiertes Permeatvolumen zum Messzeitpunkt t
JV-W-Initial = Initialflux einer Vivaflow-Einheit mit destilliertem Wasser
JV-W-W = Permeatflux einer Vivaflow-Einheit mit destilliertem Wasser vor Start eines Wiederholungsversuchs
𝑉𝑉−𝑃𝑒𝑟−𝑘𝑜𝑟𝑟𝑖𝑔𝑖𝑒𝑟𝑡,𝑡 = 𝑉𝑃𝑒𝑟,𝑡 × 𝐽𝑉−𝑊−𝐼𝑛𝑖𝑡𝑖𝑎𝑙
𝐽𝑉−𝑊−𝑊 ×
𝐽𝑉−𝑊−𝑚𝑎𝑥
𝐽𝑉−𝑊−𝑚𝑖𝑛
Formel 13: Rechnerische Angleichung des Permeatvolumens der weniger performanten Vivaflow-Einheit
VPer,t = Permeatvolumen zum Messzeitpunkt t
VV-Per-korrigiert,t = korrigiertes Permeatvolumen zum Messzeitpunkt t
JV-W-max = Permeatflux von destilliertem Wasser der performanter Vivaflow-Einheit
JV-W-min = Permeatflux von destilliertem Wasser der weniger performanten Vivaflow-Einheit
JV-W-Initial = Initialflux einer Vivaflow-Einheit mit destilliertem Wasser
JV-W-W = Permeatflux einer Vivaflow-Einheit mit destilliertem Wasser vor Start eines Wiederholungsversuchs
JV-W-max entspricht dabei dem Initialflux der Vivaflow-Einheit die für die Filtration mit Ultraschall,
JV-W-min dem Initialflux der Vivaflow-Einheit die für die Filtration ohne Ultraschall verwendet
wurde.
Aufgrund von Schwankungen der Permeatmenge erfolgte eine rechnerische Angleichung der
Prozesslaufzeit.
𝑡𝑉𝐹−𝑘𝑜𝑟𝑟𝑖𝑔𝑖𝑒𝑟𝑡 =𝑉𝑃𝑒𝑟−𝑚𝑎𝑥
𝑉𝑃𝑒𝑟−𝑖𝑠𝑡× 𝑡𝑉𝐹
Formel 14: Rechnerische Angleichung der Prozesszeit in Abhängigkeit des Permeatvolumens
tVF-korrigiert = korrigierte Prozesslaufzeit einer Vivaflow-Einheit
tVF = unkorrigierte Prozesslaufzeit einer Vivaflow-Einheit nach Performanceanpassung
VPer-max = maximale Permeatmenge einer Versuchsreihe
VPer-ist = Permeatmenge des Versuchs

Material & Methoden
76
3.6.1.3 Eignung von RC-Membranen
Die Versuche zur Eignung der RC-Membranen für die Fraktionierung wurden, analog zu den
Ultraschall-Versuchen, unter Verwendung einer Vivaflow-Kammer durchgeführt. Die
rechnerische Korrektur der Messdaten erfolgte gemäß Formel 12 und Formel 14.
3.6.1.4 Absolute Proteinmenge von Retentat (mRet) und Permeat (mPer)
Die Bestimmung der absoluten Proteinmengen in Retentat mRet und Permeat mPer erfolgte
rechnerisch durch die Multiplikation des spektralphotometrisch ermittelten Proteingehalts von
Retentat/Permeat CS-Ret/Per mit dem Retentat-/Permeatvolumen VRet/VPer, dargestellt in Formel
15.
𝑚𝑅𝑒𝑡/𝑃𝑒𝑟 = 𝐶𝑆−𝑅𝑒𝑡/𝑃𝑒𝑟 × 𝑉𝑅𝑒𝑡/𝑃𝑒𝑟
Formel 15: Rechnerische Ermittlung der absoluten Proteinmengen
mRet/Per = Absolute Proteinmenge im Retentat/Permeat
CS-Ret/Per = spektralphotometrisch ermittelte Konzentration von Retentat/Permeat
VRet/Per = Volumen des Retentats/Permeats
3.6.2 Statistische Auswertung
Zur statistischen Auswertung der Daten wurden IBM SPSS Statistics 20 (International
Business Machines Corporation (IBM), Armonk, USA) verwendet.
3.6.2.1 Hydrolyse
Zur Bestimmung des Einflusses von gewähltem Enzym und Vorbehandlung (unabhängige
Variable) der Hydrolysate auf den Hydrolysegrad der Proben (abhängige Variable) wurden die
unabhängigen Stichproben mittels einfaktorieller univariater Varianzanalyse (ANOVA)
ausgewertet. Überschritt der F-Wert des Tests den kritischen F-Wert, Fkritisch, wurde anstelle
der Nullhypothese H0 (die Gruppenmittelwerte sind identisch) die Alternativhypothese H1 (die
Gruppenmittelwerte sind nicht identisch) akzeptiert (Janssen & Laatz, 2007). Im Falle einer
signifikanten ANOVA erfolgten Post-hoc Tests zur Identifizierung signifikant unterschiedlicher
Vergleichspaare.
Voraussetzung für die Verwendung der ANOVA sind mindestens intervallskallierte Daten,
Unabhängigkeit der Stichproben, Normalverteilung der Kriteriumsvariablen sowie
Varianzhomogenität der Gruppe (Eckstein, 2016; Janssen & Laatz, 2007).
Die Normalverteilung der Variablen wurde mittels Shapiro-Wilk Test überprüft, da dieser bei
Stichprobenumfängen n < 50 über eine gute Teststärke verfügt (Janssen & Laatz, 2007). Die
Varianzhomogenität der Gruppe wurde mittels Levene-Test bestimmt.

Material & Methoden
77
Basierend auf den Verteilungseigenschaften der Stichproben erfolgte der Post-hoc
Mehrfachvergleich für die hydrolysierte Alge (Chlorella vulgaris) unter Verwendung des
Bonferroni-Tests.
Die Ermittlung des F-Werts und des kritischen F-Werts basierte auf einem Signifikanzniveau
von α = 0,05 (Rudolf & Kuhlisch, 2008).
3.6.2.2 Crossflow-Filtration
Zur Bestimmung des Einflusses des pH-Werts (unabhängige Variable) auf die Prozesszeit t
der Filtration, dem korrelierenden Flux JF, sowie der absoluten Proteinmenge in
Permeat/Retentat und der Wiederfindungsrate (abhängige Variable), wurden für die beiden
Versuchsgruppen (NaCl-Zusatz, kein NaCl-Zusatz) jeweils einfaktorielle univariate
Varianzanalysen durchgeführt.
Analog zu Kapitel 3.6.2.1 erfolgte eine Überprüfung der Hypothesen der Normalverteilung und
Varianzhomogenität. Basierend auf den Verteilungseigenschaften der Stichproben wurde der
Post-hoc Mehrfachvergleich unter Verwendung des Tukey-HSD-Tests, sowie in Einzelfällen
des Games-Howell-Tests durchgeführt.
Der Einfluss der Änderung der Ionenstärke durch Zugabe von NaCl (unabhängige Variable)
auf die Prozesszeit t der Filtration, dem korrelierenden Flux JF, sowie der absoluten
Proteinmenge in Permeat/Retentat und der Wiederfindungsrate (abhängige Variable) wurde
für jede pH-Stufe und Variable mittels T-Test für unabhängige Stichproben bestimmt. Es
erfolgte eine Überprüfung der Hypothesen der Normalverteilung und Varianzhomogenität. Die
Nichterfüllung einer der beiden Hypothesen resultierte in der Verwendung des
verteilungsunabhängigen, nichtparametrischen Mann-Whitney U Tests (Eckstein, 2016).
Die Ermittlung des F-Werts und des kritischen F-Werts basierte auf einem Signifikanzniveau
von α = 0,05 (Rudolf & Kuhlisch, 2008).
3.6.2.3 Ultraschall
Zur Bestimmung des Einflusses des Ultraschalls (unabhängige Variable) auf die Prozesszeit t
der Filtration, dem korrelierenden Flux JF, sowie der absoluten Proteinmenge in
Permeat/Retentat (abhängige Variablen) wurden jeweils T-Tests für unabhängige Stichproben
durchgeführt. Es erfolgte eine Überprüfung der Hypothesen der Normalverteilung und
Varianzhomogenität. Die Nichterfüllung einer der beiden Hypothesen resultierte in der
Verwendung des verteilungsunabhängigen, nichtparametrischen Mann-Whitney U Tests.
Die Ermittlung der Teststatistiken basierten auf einem Signifikanzniveau von α = 0,05.

Material & Methoden
78
3.6.2.4 Dialyse
Zur Bestimmung des Einflusses des pH-Werts (unabhängige Variable) auf den Proteingehalt
der Proben vor und nach der Dialyse (abhängige Variablen) wurden je Konzentration und
Prozessdauer einfaktorielle univariate Varianzanalysen oder, im Falle von zwei
Vergleichspaaren, T-Tests für unabhängige Stichproben durchgeführt.
Analog zu Kapitel 3.6.2.1 erfolgte eine Überprüfung der Hypothesen der Normalverteilung und
Varianzhomogenität. Basierend auf den Verteilungseigenschaften der Stichproben wurde der
Post-hoc Mehrfachvergleich unter Verwendung des Tukey-HSD-Tests durchgeführt.
Der Einfluss unterschiedlicher Prozesszeiten (unabhängige Variable) auf den Proteingehalt
der Proben vor und nach der Dialyse (abhängige Variablen) wurde je pH-Stufe mittels T-Test
für unabhängige Stichproben bestimmt. Zur Bestimmung des Einflusses der Dialyse
(unabhängige Variable) auf den Proteingehalt der Proben (abhängige Variable) wurden je
Konzentration, Prozessdauer und pH-Stufe T-Tests für abhängige Stichproben durchgeführt.
Es erfolgte eine Überprüfung der Hypothesen der Normalverteilung und Varianzhomogenität.
Die Ermittlung des F-Werts und des kritischen F-Werts basierte auf einem Signifikanzniveau
von α = 0,05 (Rudolf & Kuhlisch, 2008).
3.6.2.5 Sensorik-Schulung
Zur Bestimmung der Diskriminierungsfähigkeit zwischen Proben unterschiedlicher
Konzentrationen erfolgte eine Auswertung der Daten der „Prüfung zur Unterscheidung von
Intensitätsstufen eines Reizes“ mittels Friedman-Test. Im Falle signifikanter Ergebnisse
wurden Wilcoxon-Vorzeichen-Rangtests zur Ermittlung signifikanter Paardifferenzen
durchgeführt (Quadt, Schönberger, & Schwarz, 2009; Lawless & Heymann, 1999).
Die Ermittlung der Teststatistiken basierten auf einem Signifikanzniveau von α = 0,05.
3.6.2.6 Prüferauswahl
Die für die Prüferauswahl angewendeten statistischen Verfahren wurden bereits vorab in
Kapitel 3.5.2 (Seite 72) dargestellt.
Die Ermittlung der Teststatistiken basierten auf einem Signifikanzniveau von α = 0,05.
3.6.2.7 Sensorik
Zur Bestimmung des Einfluss der Zugabe der Permeatfraktion zum gereichten Bitter-Standard
(unabhängige Variable) auf die Bitterwahrnehmung (abhängige Variable) der Probe, wurden
je Proteinquelle verteilungsunabhängige, nichtparametrische Mann-Whitney U Tests
durchgeführt.
Die Ermittlung der Teststatistiken basierten auf einem Signifikanzniveau von α = 0,05.
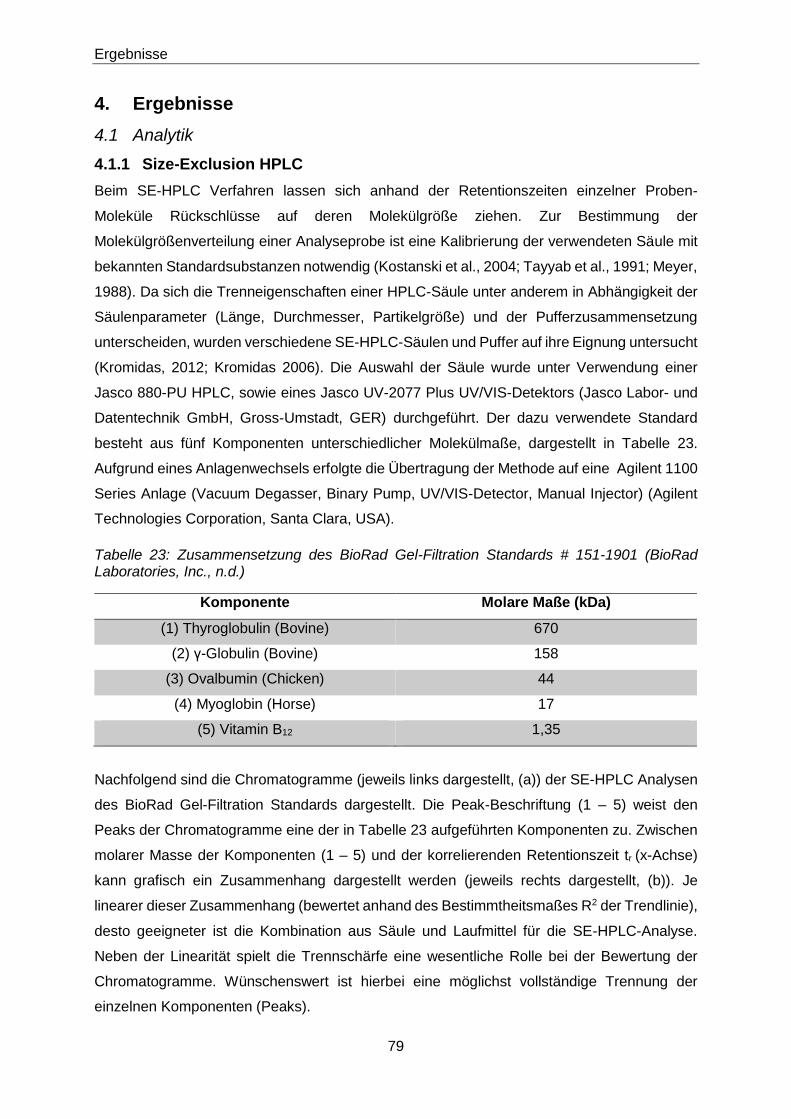
Ergebnisse
79
4. Ergebnisse
4.1 Analytik
4.1.1 Size-Exclusion HPLC
Beim SE-HPLC Verfahren lassen sich anhand der Retentionszeiten einzelner Proben-
Moleküle Rückschlüsse auf deren Molekülgröße ziehen. Zur Bestimmung der
Molekülgrößenverteilung einer Analyseprobe ist eine Kalibrierung der verwendeten Säule mit
bekannten Standardsubstanzen notwendig (Kostanski et al., 2004; Tayyab et al., 1991; Meyer,
1988). Da sich die Trenneigenschaften einer HPLC-Säule unter anderem in Abhängigkeit der
Säulenparameter (Länge, Durchmesser, Partikelgröße) und der Pufferzusammensetzung
unterscheiden, wurden verschiedene SE-HPLC-Säulen und Puffer auf ihre Eignung untersucht
(Kromidas, 2012; Kromidas 2006). Die Auswahl der Säule wurde unter Verwendung einer
Jasco 880-PU HPLC, sowie eines Jasco UV-2077 Plus UV/VIS-Detektors (Jasco Labor- und
Datentechnik GmbH, Gross-Umstadt, GER) durchgeführt. Der dazu verwendete Standard
besteht aus fünf Komponenten unterschiedlicher Molekülmaße, dargestellt in Tabelle 23.
Aufgrund eines Anlagenwechsels erfolgte die Übertragung der Methode auf eine Agilent 1100
Series Anlage (Vacuum Degasser, Binary Pump, UV/VIS-Detector, Manual Injector) (Agilent
Technologies Corporation, Santa Clara, USA).
Tabelle 23: Zusammensetzung des BioRad Gel-Filtration Standards # 151-1901 (BioRad Laboratories, Inc., n.d.)
Komponente Molare Maße (kDa)
(1) Thyroglobulin (Bovine) 670
(2) γ-Globulin (Bovine) 158
(3) Ovalbumin (Chicken) 44
(4) Myoglobin (Horse) 17
(5) Vitamin B12 1,35
Nachfolgend sind die Chromatogramme (jeweils links dargestellt, (a)) der SE-HPLC Analysen
des BioRad Gel-Filtration Standards dargestellt. Die Peak-Beschriftung (1 – 5) weist den
Peaks der Chromatogramme eine der in Tabelle 23 aufgeführten Komponenten zu. Zwischen
molarer Masse der Komponenten (1 – 5) und der korrelierenden Retentionszeit tr (x-Achse)
kann grafisch ein Zusammenhang dargestellt werden (jeweils rechts dargestellt, (b)). Je
linearer dieser Zusammenhang (bewertet anhand des Bestimmtheitsmaßes R2 der Trendlinie),
desto geeigneter ist die Kombination aus Säule und Laufmittel für die SE-HPLC-Analyse.
Neben der Linearität spielt die Trennschärfe eine wesentliche Rolle bei der Bewertung der
Chromatogramme. Wünschenswert ist hierbei eine möglichst vollständige Trennung der
einzelnen Komponenten (Peaks).

Ergebnisse
80
Aufgrund der schlecht lesbaren Achsenbeschriftung der Ausgangsgrafiken wurde diese zur
besseren Lesbarkeit nachgearbeitet. Jeder | auf der x-Achse entspricht einer Retentionszeit
∆tr von 5 min. Auf der y-Achse wird zur Wahrung der Übersicht nur der Maximalwert des
Detektorsignals angegeben (Einheit: µV).
4.1.1.1 Tosoh TSKgel G 3000 SWXL (7,8 mm x 30 mm, 5 µm)
Die TSKgel G 3000 SWXL von Tosoh reagiert sehr empfindlich auf Änderungen der
Pufferzusammensetzung. Besonders bei niedriger Molarität des Puffers, Abweichungen vom
idealen pH Wert 6,7, sowie dem Verzicht des Zusatzes von Na2SO4, kann es zu Unruhen
(Auftrennung der Peaks in mehrere „Unter-Peaks“) im Chromatogramm. Diese treten,
beispielhaft dargestellt in Abbildung 42 (a), besonders zwischen Thyroglobulin (1) und γ-
Globulin (2), aber auch rechts von Vitamin B12 (5) auf. Zudem laufen die Peaks (2) und (3),
sowie (4) und (5) ineinander, was für eine mangelnde Auftrennung der Komponenten spricht.
Das beispielhaft dargestellte Bestimmtheitsmaß von R2 = 0,934 des 0,05 mol/l
Natriumphosphatpuffer +0,05 % NaN3, pH 6,9 ist zwar zufriedenstellend, Abbildung 42 (b)
offenbart allerdings unerwünschte Abweichungen vom linearen Verlauf der Kalibriergeraden.
Legende: 1, Thyroglobulin (670 kDa). 2, γ-Globulin (158 kDa). 3, Ovalbumin (44 kDa). 4, Myoglobin (17 kDa).
5, Vitamin B12 (1,35 kDa)
Abbildung 42: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des BioRad Gel-Filtration Standards mittels Säule: Tosoh TSKgel G 3000 SWXL – Puffer: 0,05 mol/l Natriumphosphatpuffer +0,05 % NaN3, pH 6,7, quantitative Darstellung
Eine verlässliche Trennung der Komponenten des Standards wurde bei einer Molarität von 0,1
mol/l, pH 6,7 unter Zusatz von 0,1 mol/l Na2SO4 erreicht (Abbildung 43). Die Verwendung von
0,1 mol/l Na2SO4, sowie die erhöhte Molarität des Puffers resultiert im Vergleich zu Puffern
niedriger Molaritäten in einer differenzierteren Auftrennung der Komponenten, wenn auch γ-
Globulin (2) und Ovalbumin (3) nicht separiert dargestellt werden können. Das am Beispiel
des 0,1 mol/l Natriumphosphatpuffers mit 0,1 mol/l Na2SO4, pH 6,7 dargestellte
Bestimmtheitsmaß von R2 = 0,957 ist abermals zufriedenstellend, Abbildung 43 (b) offenbart
R² = 0,9340
2
4
6
8
10
12
14
16
1 10 100 1000
t r (m
in)
Mol. Maße (kDa)
0 5 10 15 20
1
3
4
5
2
a) b)
tr (min)
µV 1*105

Ergebnisse
81
aber ebenfalls geringfügige Abweichungen vom linearen Verlauf der Kalibriergeraden. Der
verwendete Laufpuffer entspricht den Empfehlungen des Herstellers (Tosoh Bioscience, 2010)
Legende: 1, Thyroglobulin (670 kDa). 2, γ-Globulin (158 kDa). 3, Ovalbumin (44 kDa). 4, Myoglobin (17 kDa).
5, Vitamin B12 (1,35 kDa)
Abbildung 43: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des BioRad Gel-Filtration Standards mittels Säule: Tosoh TSKgel G 3000 SWXL – Puffer: 0,1 mol/l Natriumphosphatpuffer + 0,1 mol/l Na2SO4 + 0,05 % NaN3, pH 6,7, quantitative Darstellung
Eine weitere Steigerung der Molarität des Puffers auf 0,2 mol/l führte sowohl mit als auch ohne
den Zusatz von 0,2 mol/l Na2SO4 zu breiteren, wenig differenzierten Peaks. Das
Bestimmtheitsmaß R2 ist für den 0,2 mol/l Puffer mit 0,2 mol/l Na2SO4, pH 7 mit 0,995 zwar
hoch (Abbildung 44 b), dies ist jedoch in einer mangelnde Schärfe der Trennung, sowie dem
pH-anhängigen Verlust (pH 6,7/pH 7,0) der Differenzierung von γ-Globulin (2) und Ovalbumin
(3) begründet (Abbildung 44 a) und somit nicht erwünscht.
Legende: 1, Thyroglobulin (670 kDa). 2, γ-Globulin (158 kDa). 3, Ovalbumin (44 kDa). 4, Myoglobin (17 kDa).
5, Vitamin B12 (1,35 kDa)
Abbildung 44: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des BioRad Gel-Filtration Standards mittels Säule: Tosoh TSKgel G 3000 SWXL – Puffer: 0,2 mol/l Natriumphosphatpuffer + 0,2 mol/l Na2SO4 + 0,05 % NaN3, pH 7, quantitative Darstellung
Die Analysedauer beträgt bei einem Anlagenfluss von 1,0 ml/min 20 min.
R² = 0,9570
2
4
6
8
10
12
14
16
1 10 100 1000
t r(m
in)
Mol. Maße (kDa)
R² = 0,9950
2
4
6
8
10
12
14
16
1 10 100 1000
t r(m
in)
Mol. Maße (kDa)
1
2
3
4 5
1 3
4
5
0 5 10 15
1,5*105 µV
tr (min)
a) b)
µV
0 5 10 15
1,5*105
a) b)
tr (min)

Ergebnisse
82
4.1.1.2 PSS Suprema 1000 Â (8 mm x 300 mm, 10 µm)
Die Säule Suprema 1000 Â von PSS reagiert weniger empfindlich auf Änderungen der
Pufferzusammensetzung. Der Zusatz von Na2SO4 (0,05 mol/l – 0,2 mol/l) oder die Variation
des pH-Werts (6,7 – 7,0) resultierten in keiner sichtbaren Veränderung der
chromatographischen Trennung. Die Molarität des Puffers (0,05 mol/l – 0,2 mol/l) wirkte sich
allerdings entscheidend auf die Trenneigenschaften der Säule aus und wird aus diesem Grund
nachfolgend betrachtet.
Eine differenzierte Auftrennung des Standards wurde, abweichend von den Angaben des
Herstellers, für eine Molarität von 0,05 mol/l bei einem pH-Wert von 6,9 erreicht (Abbildung
45 a) (PSS Polymer Standards Service GmbH, n.d.). Das Bestimmtheitsmaß R2 = 0,992 spricht
für die Eignung des Puffers zur chromatographischen Analyse des BioRad Gel-Filtration
Standards. Im Vergleich zu der vorangehend dargestellten Tosoh TSKgel G 3000 SWXL
erscheint die Auftrennung aufgrund enger zusammenliegender Peaks (1 – 3), sowie
mangelnder Wiederfindung von Myoglobin (4) jedoch weniger geeignet.
Legende: 1, Thyroglobulin (670 kDa). 2, γ-Globulin (158 kDa). 3, Ovalbumin (44 kDa). 4, Myoglobin (17 kDa).
5, Vitamin B12 (1,35 kDa)
Abbildung 45: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des BioRad Gel-Filtration Standards mittels Säule: PSS Suprema 1000 Â – Puffer: 0,05 mol/l Natriumphosphatpuffer +0,05 % NaN3, pH 6,9, quantitative Darstellung
Eine Steigerung der Molarität auf 0,1 mol/l führte darüber hinaus, beispielhaft dargestellt für
pH 7, zum Verlust des Myoglobin-Peaks (4), sowie zu geringerer Differenzierbarkeit von γ-
Globulin (2) und Ovalbumin (3) (Abbildung 46 a). Das Bestimmtheitsmaß R2 sinkt auf 0,984
ab.
R² = 0,9920
2
4
6
8
10
12
14
1 10 100 1000
t r(m
in)
Mol. Maße (kDa)1
2 & 3
4
5
0 5 10 15 20 tr (min)
µV
4,0*105
a) b)

Ergebnisse
83
Legende: 1, Thyroglobulin (670 kDa). 2, γ-Globulin (158 kDa). 3, Ovalbumin (44 kDa). 4, Myoglobin (17 kDa).
5, Vitamin B12 (1,35 kDa)
Abbildung 46: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des BioRad Gel-Filtration Standards mittels Säule: PSS Suprema 1000 Â – Puffer: 0,1 mol/l Natriumphosphatpuffer +0,05 % NaN3, pH 7,0, quantitative Darstellung
Eine Steigerung der Molarität des Puffers auf 0,2 mol/l resultierte im vollständigen Verlust der
Differenzierbarkeit von γ-Globulin (2) und Ovalbumin (3), sowie dem bereits vom 0,1 mol/l
Puffer bekannten Verlust des Myoglobin-Peaks (4) (Abbildung 47 a). Das Bestimmtheitsmaß
R2 sinkt auf 0,971. Die mangelnde Auflösung der Trennung spricht gegen eine Eignung des
Puffers für den Zweck der Trennung des BioRad Gel-Filtration Standards.
Legende: 1, Thyroglobulin (670 kDa). 2, γ-Globulin (158 kDa). 3, Ovalbumin (44 kDa). 4, Myoglobin (17 kDa).
5, Vitamin B12 (1,35 kDa)
Abbildung 47: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des BioRad Gel-Filtration Standards mittels Säule: PSS Suprema 1000 Â – Puffer: 0,2 mol/l Natriumphosphatpuffer +0,05 % NaN3, pH 7,0, quantitative Darstellung
Die Analysedauer beträgt bei einem Anlagenfluss von 1,0 ml/min 15 min.
4.1.1.3 Agiltent Zorbax GF 250 (4,6 mm x 250 mm, 4 µm)
Die Säule Zorbax GF 250 von Agilent liefert, weitestgehend unabhängig von pH-Wert, Na2SO4-
Zusatz und Molarität des Puffers, die performanteste Trennung der untersuchten Säulen.
Lediglich bei niedriger Molarität des Puffers (0,05 mol/l), pH 6,9 konnten Ovalbumin (3) und
R² = 0,9840
2
4
6
8
10
12
14
1 10 100 1000
t r(m
in)
Mol. Maße (kDa)
R² = 0,9710
2
4
6
8
10
12
14
1 10 100 1000
t r(m
in)
Mol. Maße (kDa)
1
2 & 3
5
1
3
5
0 5 10 15 tr (min)
µV
4,0*105
0 5 10 15 tr (min)
µV
5,0*105
a) b)
a) b)

Ergebnisse
84
Myoglobin (4) nicht differenziert werden (Abbildung 48 a). Das Bestimmtheitsmaß R2 = 0,971
ist jedoch als zufriedenstellend zu bewerten.
Legende: 1, Thyroglobulin (670 kDa). 2, γ-Globulin (158 kDa). 3, Ovalbumin (44 kDa). 4, Myoglobin (17 kDa).
5, Vitamin B12 (1,35 kDa)
Abbildung 48: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des BioRad Gel-Filtration Standards mittels Säule: Agilent Zorbax GF 250 – Puffer: 0,05 mol/l Natriumphosphatpuffer + 0,05 % NaN3, pH 6,9, quantitative Darstellung
Bei Molaritäten < 0,2 mol/l ist zudem ein unerwünschtes Tailing des Thyroglobulin-Peaks (1)
erkennbar (Abbildung 48 a, Abbildung 49 a). Ab einer Molarität des Puffers von 0,1 mol/l erfolgt
eine Differenzierung des Ovalbumin- (3) und Myoglobin-Peaks (4). Das Bestimmtheitsmaß R2
ist, beispielhaft dargestellt in Abbildung 49 b, mit 0,955 abermals als zufriedenstellend zu
bewerten.
Legende: 1, Thyroglobulin (670 kDa). 2, γ-Globulin (158 kDa). 3, Ovalbumin (44 kDa). 4, Myoglobin (17 kDa).
5, Vitamin B12 (1,35 kDa)
Abbildung 49: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des BioRad Gel-Filtration Standards mittels Säule: Agilent Zorbax GF 250 – Puffer: 0,1 mol/l Natriumphosphatpuffer + 0,05 % NaN3, pH 7,0, quantitative Darstellung
In Übereinstimmung mit den Empfehlungen des Herstellers erwies sich ein 0,2 mol/l
Natriumphosphatpuffer + 0,05 % NaN3, pH 7,0 als optimales Puffermedium (Abbildung 50 a)
R² = 0,9710
1
2
3
4
5
6
7
1 10 100 1000
t r(m
in)
Mol. Maße (kDa)
R² = 0,9550
1
2
3
4
5
6
7
1 10 100 1000
t r(m
in)
Mol. Maße (kDa)
1 2 4
5
1 2 3 & 4 5
a) b)
a) b)
0 5 10
0 5 10
µV
µV
8,0*105
8,0*105
tr (min)
tr (min)

Ergebnisse
85
(Agilent Technologies, n.d.). Das Bestimmtheitsmaß R2 genügt mit 0,967 den Anforderungen
an die Auftrennung des BioRad Gel-Filtration Standards.
Legende: 1, Thyroglobulin (670 kDa). 2, γ-Globulin (158 kDa). 3, Ovalbumin (44 kDa). 4, Myoglobin (17 kDa).
5, Vitamin B12 (1,35 kDa)
Abbildung 50: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des BioRad Gel-Filtration Standards mittels Säule: Agilent Zorbax GF 250 – Puffer: 0,2 mol/l Natriumphosphatpuffer + 0,05 % NaN3, pH 7,0, quantitative Darstellung
Die Analysedauer beträgt bei einem Anlagenfluss von 0,5 ml/min 10 min.
Basierend auf den Ergebnissen der Auftrennung des BioRad Gel-Filtration Standards wurde
die Kombination aus der Säule Agilent Zorbax GF 250, sowie einem 0,2 mol/l
Natriumphosphatpuffer + 0,05 % NaN3, pH 7,0 als SE-HPLC Methode für die Analysen im
Rahmen dieser Studie ausgewählt. Die gewählte Kombination gewährleistet eine differenzierte
Auftrennung des BioRad Gel-Filtration-Standard bei kurzer Analysezeit (10 min) und
zufriedenstellender Linearität (R2 = 0,967) der Kalibriergerade.
Die Tosoh TSKgel G 3000 SWXL Säule wurde trotz guten Trennergebnissen aufgrund der
erhöhten Analysezeit (20 min), sowie der Notwendigkeit des Zusatz von Na2SO4 nicht in
Betracht gezogen. Gleiches gilt für die PSS Suprema 1000 Â Säule, deren mangelnde
Auflösung gegen eine Verwendung im Versuchssetup spricht.
4.1.1.4 Übertragung der Methode auf eine Agilent 1100 Series HPLC
Aufgrund eines Anlagenwechsels erfolgte die Übertragung der Methode auf eine Agilent 1100
Series Anlage (Vaccum Degasser, Binary Pump, UV/VIS-Detector, Manual Injector) (Agilent
Technologies Corporation, Santa Clara, USA). Abbildung 51 a zeigt die Auftrennung des
Standards bei gleichen Untersuchungsparametern (Anlagenflux: 0,5 ml/min, Puffer: 0,2 mol/l
Natriumphosphatpuffer + 0,05 % NaN3, pH 7,0). Die Auftrennung des Standards erwies sich
in Zehnfachwiederholung als zuverlässig und wiederholbar (R2 = 0,996), wenn auch der
Myoglobin (4) Peak (ermittelt anhand des Vergleichs der Retentionszeit tR) unter Verwendung
der Agilent 1100 Series HPLC nicht ausreichend differenziert werden konnte. Die
R² = 0,9670
1
2
3
4
5
6
7
1 10 100 1000
t r(m
in)
Mol. Maße (kDa)
1 2 3 & 4 5
a) b)
0 5
µV 6,0*105
tr (min)

Ergebnisse
86
Standardabweichung der Retentionszeiten, dargestellt in Tabelle 24, liegt zwischen 0,51 %
(Thyroglobulin (1)) und 1,74 % (Vitamin B12 (5)).
Legende: 1, Thyroglobulin (670 kDa). 2, γ-Globulin (158 kDa). 3, Ovalbumin (44 kDa). 4, Myoglobin (17 kDa).
5, Vitamin B12 (1,35 kDa)
Abbildung 51: Chromatogramm (a) und Kalibriergerade (b) der SE-HPLC-Analyse des BioRad Gel-Filtration Standards mittels Säule: Agilent Zorbax GF 250 – Puffer: 0,2 mol/l Natriumphosphatpuffer + 0,05 % NaN3, pH 7,0, quantitative Darstellung
Tabelle 24: Retentionszeiten tr und prozentuale Standardabweichung (SD) der Komponenten des BioRad Gel-Filtration Standards
Komponente Molare Maße (kDa) tr MW (min) SD (%)
(1) Thyroglobulin (Bovine) 670 3,712 ± 0,019 0,51
(2) γ-Globulin (Bovine) 158 4,312 ± 0,071 1,65
(3) Ovalbumin (Chicken) 44 4,856 ± 0,062 1,28
(4) Myoglobin (Horse) 17 - -
(5) Vitamin B12 1,35 6,138 ± 0,107 1,74
R² = 0,9960
1
2
3
4
5
6
7
1 10 100 1000
t R (m
in)
Molare Maße (kDa)
1
2
3
5 (4)
0 5 10
a) b)
mA
U
350
tr (min)

Ergebnisse
87
4.1.2 Spektralphotometrische Proteingehaltbestimmung
Gemäß des Lambert-Beer-Gesetzes besteht bei konstanter Schichtdicke des durchstrahlten
Mediums ein proportionaler Zusammenhang zwischen der Extinktion eines
monochromatischen Lichtstrahls und der molaren Konzentration einer gelösten Substanz, die
eine Bestimmung der molaren Konzentration bei Vorhandensein einer Kalibrationsfunktion
erlaubt (Gressner & Arndt, 2007). Die im Rahmen dieser Studie ermittelten
Kalibrationsfunktionen sind nachfolgend dargestellt.
4.1.2.1 Molkenprotein-Isolat
Abbildung 52: Kalibrationsfunktion für die photometrische Bestimmung des Proteingehalts von hydrolysiertem Molkenprotein-Isolat bei 280 nm
Abbildung 52 zeigt die in Dreifachbestimmung ermittelte Kalibrationsfunktion für das
verwendete Molkenprotein-Isolat. Die Konzentration der Stammlösung wurde unter
Berücksichtigung des Proteingehalts des verwendeten Rohstoffs (Thermax 690, Glanbia
Nutritionals Ltd., IRL) eingestellt und ist somit als absolut zu betrachten. Durch Auflösung der
ermittelten Geradengleichung nach Cs (x) ist bei bekannter Extinktion eine Aussage über den
Proteingehalt der untersuchten Probe möglich. Das Bestimmtheitsmaß R2 der
Kalibrationsfunktion liegt bei 0,999. Dies spricht für die Eignung der spektralphotometrischen
Analyse zur Bestimmung des Proteingehalts von hydrolysierten Molkenprotein-Isolat-
Lösungen.
y = 0,7552xR² = 0,999
0,0
0,5
1,0
1,5
2,0
2,5
0,0 0,5 1,0 1,5 2,0 2,5 3,0
Exti
nkt
ion
bei
28
0 n
m
Konzentration CF (g/l)

Ergebnisse
88
𝐶𝑆 = 𝑦𝐸𝑥𝑡𝑖𝑛𝑘𝑡𝑖𝑜𝑛
0,7552
Formel 16: Kalibrationsfunktion zur photometrischen Bestimmung des Proteingehalts von hydrolysiertem Molkenprotein-Isolat
CS = spektralphotometrisch bestimmter Proteingehalt
yExtinktion = spektralphotometrisch ermittelte Extinktion
4.1.2.2 Alge (Chlorella vulgaris)
Abbildung 53: Kalibrationsfunktion zur photometrischen Bestimmung des Proteingehalts von hydrolysierter Alge (Chlorella vulgaris) bei 280 nm
Abbildung 53 zeigt die in Dreifachbestimmung ermittelte Kalibrationsfunktion zur Bestimmung
des Proteingehalts der hydrolysierten Alge (Chlorella vulgaris). Der Proteingehalt der
Stammlösung wurde extern per Kjeldahl-Stickstoffbestimmung ermittelt (von: KWALIS
Qualitätsforschung Fulda GmbH, Fulda, GER). Das Bestimmtheitsmaß R2 der
Kalibrationsfunktion liegt bei 0,999. Dies spricht für die Eignung der spektralphotometrischen
Analyse zur Bestimmung des Proteingehalts von hydrolysierter Alge (Chlorella vulgaris).
𝐶𝑆 = 𝑦𝐸𝑥𝑡𝑖𝑛𝑘𝑡𝑖𝑜𝑛
1,692
Formel 17: Kalibrationsfunktion zur photometrischen Bestimmung des Proteingehalts von hydrolysierter Alge (Chlorella vulgaris)
CS = spektralphotometrisch bestimmter Proteingehalt
yExtinktion = spektralphotometrisch ermittelte Extinktion
y = 1,692xR² = 0,999
0,0
0,5
1,0
1,5
2,0
2,5
3,0
0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4 1,6
Exti
nkt
ion
bei
28
0 n
m
Konzentration CF (g/l)

Ergebnisse
89
4.2 Hydrolyse
Nachfolgend sind die Ergebnisse der Versuchsreihe zum Einfluss von Enzym und Art der
Vorbehandlung (enzymatisch, unter Verwendung einer Pektinase; „sauer“, unter Verwendung
von HCl, pH 3) auf Hydrolysegrad und Molekülgrößenverteilung von Algenhydrolysaten
gewonnen aus der Alge Chlorella vulgaris dargestellt. Die Beurteilung der Eignung des
Hydrolysevorgangs zur Erzeugung eines geeigneten Feeds für den Fraktionierungsprozess
basiert nicht alleinig auf dem erreichten Hydrolysegrad. Vielmehr spielt die
Molekülgrößenverteilung des Feeds eine entscheidende Rolle. Ziel der Versuche war die
Erzeugung eines Hydrolysats mit möglichst breiter Molekülgrößenverteilung. Der Hintergrund
dieser Zielsetzung ist im Studienziel begründet. Zur Erzeugung unterschiedlicher
Peptidfraktionen eignet sich eine breite Größenverteilung, da potentiell mehr funktionelle
Fraktionen gewonnen werden können.
4.2.1 Alge (Chlorella vulgaris)
Mikroalgen, wie die im Rahmen dieser Studie verwendete Chlorella vulgaris, verfügen über
stabile, vorwiegend Cellulose-, Hemicellulose- und Pektin-basierte Zellwände (Bastos, 2018;
Domozych, 2016). Die Stabilität der Zellwand kann eine Digestion der Algenproteine
erschweren (Renneberg & Berkling, 2012; Morris et al., 2008; Tchorbanov et al., 1986).
Die im Rahmen dieser Studie durchgeführten Pilotversuche (Versuchsschema: siehe
Abbildung 39, Seite 62) ohne enzymatische oder saure Vorbehandlung der Algenzellwände
resultierten, unabhängig von den verwendeten Enzymen (Pancreatin, Pronase, Corolase) und
deren Konzentration (Pancreatin 0,125 % (10 USP), 0,25 % (20 USP), 0,5 % (40 USP);
Pronase 0,125 % (56,25 PU), 0,25 % (112,5 PU), 0,5 % (225 PU); Corolase 0,1 % (2,75 UHb),
1,5 % (41,25 UHb), 3,0 % (82,5 UHb)), sowie der Prozessdauer (45 min, 135 min), in einem
Hydrolysat das von Verbindungen im Größenbereich 1,35 kDa dominiert wurde. Dies geht aus
den Chromatogrammen der Hydrolysate, exemplarisch dargestellt in Abbildung 54 und
Abbildung 55, hervor.
Das Vorhandensein derart kleiner, niedermolekularer Verbindungen deutet, für sich
genommen, auf eine zu weit fortgeschrittene Hydrolyse hin. Bezieht man jedoch die Menge
des bei der Zentrifugation gewonnenen Sediments mit in die Betrachtung ein (> 50 % der
Einwaage), wird klar, dass vermutlich nur ein Bruchteil des Algenproteins für das jeweilige zur
Hydrolyse verwendete Enzym zur Verfügung stand, während ein Großteil der Algenzellen nicht
aufgeschlossen werden konnte. Der verbleibende, der Hydrolyse zugängliche Proteinanteil
wurde vermutlich, bedingt durch die Veränderung des Verhältnis Protein-zu-Enzym zu
niedermolekularen Verbindungen hydrolysiert.

Ergebnisse
90
Abbildung 54: Molekülgrößenverteilung – Hydrolysat der Alge Chlorella vulgaris mit Pronase (0,25 % (112,5 PU); 45 min), qualitative Darstellung
Abbildung 55: Molekülgrößenverteilung – Hydrolysat der Alge Chlorella vulgaris mit Pronase (0,50 % (225 PU); 45 min), qualitative Darstellung
Pronase 0,25 %
(112,5 PU), 45 min
670 kDa
158 kDa
44 kDa
1,35 kDa
Pronase 0,50 %
(225 PU), 45 min
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ

Ergebnisse
91
Diese Annahme wurde durch zwei je 10 minütige Hydrolysen mit Pancreatin (0,25 % (20 USP))
und Pronase (0,25 % (112,5 PU)) verifiziert.
In beiden Fällen resultierte die Hydrolyse in einem von niedermolekularen Strukturen (< 1,35
kDa) dominierten Hydrolysat, sowie hohem Sediment-Anteil (> 75 % der Einwaage),
dargestellt in Abbildung 56 und Abbildung 57. Der geringfügig erhöhte (in den Abbildungen
markierte) Anteil an Verbindungen > 1,35 kDa ist vermutlich auf einen unwesentlich geringeren
Hydrolysegrad der Algenproteine, bedingt durch die sehr kurze Reaktionszeit, zurückzuführen.
Zur Verbesserung des Hydrolyse-Prozesses wurde die Alge einer enzymatischen
Vorbehandlung (Pektinase, Rohament® PL) bzw. einer sauren Vorbehandlung (HCl, pH 3)
unterzogen um die Zellwände aufzubrechen und das Protein für die enzymatische Hydrolyse
zugänglich zu machen. Für den anschließenden Hydrolyseprozess wurde jeweils eine
Enzymkonzentration von 0,25 % Pronase (112,5 PU) bzw. 0,25 % Pancreatin (20 USP)
verwendet.

Ergebnisse
92
Abbildung 56: Molekülgrößenverteilung – Hydrolysat der Alge Chlorella vulgaris mit Pancreatin (0,25 % (20 USP); 10 min), qualitative Darstellung
Abbildung 57: Molekülgrößenverteilung – Hydrolysat der Alge Chlorella vulgaris mit Pronase (0,25 % (112,5 PU); 10 min), qualitative Darstellung
Pancreatin 0,25 %
(20 USP), 10 min
670 kDa
158 kDa
44 kDa
1,35 kDa
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
Pronase 0,25 %
(112,5 PU), 10 min
670 kDa
158 kDa
44 kDa
1,35 kDa
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)

Ergebnisse
93
4.2.1.1 Behandlung mit Rohament® PL
Wie bereits zu Beginn des Kapitels beschrieben, verfügen Mikroalgen über stabile, vorwiegend
Cellulose-, Hemicellulose- und Pektin-basierte Zellwände (Bastos, 2018; Domozych, 2016).
Vergleichbar der nachfolgend dargestellten Behandlung der Algensuspension mit HCl, pH 3
(Kapitel 4.2.1.2, Seite 97) wurde überprüft, ob die Verwendung der Pektinase Rohamant® PL
zu einem Aufschluss der Algenzellwand führt und somit die Zugänglichkeit der Algenproteine
für die Enzyme Pancreatin und Pronase erhöht.
In Abbildung 58 und Tabelle 25 sind die Hydrolysegrade des Algenhydrolysats gewonnen aus
Chlorella vulgaris in Abhängigkeit des Enzyms, sowie der Dauer der Vorbehandlung tEV
dargestellt.
Abbildung 58: Hydrolysegrad des Algen-Hydrolysats in Abhängigkeit des Enzyms und der Dauer der Vorbehandlung mit Rohament® PL
Sowohl für Pancreatin als auch für Pronase unterschieden sich die einer enzymatischen
Vorbehandlung mit der Pektinase Rohament® PL unterzogenen Hydrolysate nicht signifikant
in Abhängigkeit der Dauer der Vorbehandlung (18 h, 48 h, 72 h) (p > 0,05) (Abbildung 58). Für
Pronase und Pancreatin wurden jeweils ähnliche Hydrolysegrade ermittelt (Pancreatin,
Rohament® PL, 18 h: 15,32 ± 1,19, Pronase, Rohament® PL, 18 h: 14,63 ± 2,73; Pancreatin,
Rohament® PL, 48 h: 20,14 ± 1,56, Pronase, Rohament® PL, 48 h: 21,39 ± 1,77; Pancreatin,
Rohament® PL, 72 h: 19,81 ± 0,10, Pronase, Rohament® PL, 72 h: 19,01 ± 2,99).
0,00
5,00
10,00
15,00
20,00
25,00
Hyd
roly
segr
ad D
H (
%)

Ergebnisse
94
Verglichen mit der 48-stündigen Behandlung mit Rohament® PL, sinkt der Hydrolysegrad der
72-stündigen Behandlung für beide Enzyme geringfügig ab (Tabelle 25).
Tabelle 25: Hydrolysegrad des Algen-Hydrolysats in Abhängigkeit des Enzyms und der Dauer der Vorbehandlung mit Rohament® PL
Enzym n
Enzym-
wirkdauer
tE (min)
Vorbehandlung
Dauer der
Vorbehandlung tEV
(h)
Hydrolysegrad
DH (%)
Pancreatin 3 45 Rohament® PL 18 15,32 ± 1,19 a
Pancreatin 3 45 Rohament® PL 48 20,14 ± 1,56 a
Pancreatin 2* 45 Rohament® PL 72 19,81 ± 0,10 a
Pronase 3 45 Rohament® PL 18 14,63 ± 2,73 a
Pronase 3 45 Rohament® PL 48 21,39 ± 1,77 a
Pronase 2* 45 Rohament® PL 72 19,01 ± 2,99 a
Werte in einer Spalte mit unterschiedlichen Buchstaben a, b unterschieden sich signifikant (p < 0,05). * Ausschluss eines Messwerts aufgrund fehlerhafter Temperaturführung während des Vorbehandlungsprozesses.
Die einer enzymatische Behandlung mit der Pektinase Rohamant® PL unterzogenen
Hydrolysate verfügen über eine relativ schmale Molekülgrößenverteilungen (Abbildung 59 –
Abbildung 61). Sowohl für Pancreatin als auch für Pronase resultiert die enzymatische
Vorbehandlung unabhängig von der Dauer (18 h, 48 h, 72 h) in einem verschwindend kleinen
Anteil von Molekülen im Bereich von 158 kDa – 10 kDa. Strukturen mit einer Molekülgröße <
1,35 kDa sind stark repräsentiert. Mit zunehmender Dauer der Pektinase-Behandlung (48 h,
72 h) sind in den Chromatogrammen zudem (in Abbildung 60 & Abbildung 61 markierte)
Moleküle erkennbar, die weitaus kleiner als < 1,35 kDa sind (tr = 9,5 min – 12 min) (Abbildung
60, Abbildung 61). Unter Berücksichtigung der Hydrolysegrade (Pancreatin, Rohament® PL,
48 h: 20,14 ± 1,56, Pronase, Rohament® PL, 48 h: 21,39 ± 1,77; Pancreatin, Rohament® PL,
72 h: 19,81 ± 0,10, Pronase, Rohament® PL, 72 h: 19,01 ± 2,99) kann davon ausgegangen
werden, dass es sich hierbei um eine Anreicherung, bzw. einen erhöhten Anteil kleinster
Peptide oder Peptidbruchstücke handelt, deren Entstehung durch einen weiter
fortgeschrittenen Hydrolyseprozess begünstigt wurde.

Ergebnisse
95
Abbildung 59: Molekülgrößenverteilung – Hydrolysate der Alge Chlorella vulgaris mit a) Pancreatin (0,25 % (20 USP); 45 min; Behandlung: Rohament® PL, 18 h) und b) Pronase (0,25 % (112,5 PU); 45 min; Behandlung: Rohament® PL, 18 h), qualitative Darstellung
Abbildung 60: Molekülgrößenverteilung – Hydrolysate der Alge Chlorella vulgaris mit a) Pancreatin (0,25 % (20 USP); 45 min; Behandlung: Rohament® PL, 48 h) und b) Pronase (0,25 % (112,5 PU); 45 min; Behandlung: Rohament® PL, 48 h), qualitative Darstellung
a) Pancreatin 0,25 %, (20 USP),45 min, Rohament® PL 18 h
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
a) Pancreatin 0,25 %, (20 USP),45 min, Rohament® PL 48 h
b) Pronase 0,25 %, (112,5 PU),45 min, Rohament® PL 18 h
b) Pronase 0,25 %, (112,5 PU),45 min, Rohament® PL 18 h
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
qu
alit
ativ
qu
alit
ativ
qu
alit
ativ

Ergebnisse
96
Abbildung 61: Molekülgrößenverteilung – Hydrolysate der Alge Chlorella vulgaris mit a) Pancreatin (0,25 % (20 USP); 45 min; Behandlung: Rohament® PL, 72 h) und b) Pronase (0,25 % (112,5 PU); 45 min; Behandlung: Rohament® PL, 72 h), qualitative Darstellung
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
a) Pancreatin 0,25 %, (20 USP),45 min, Rohament® PL 72 h
b) Pronase 0,25 %, (112,5 PU),45 min, Rohament® PL 72 h
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
qu
alit
ativ

Ergebnisse
97
4.2.1.2 Behandlung mit HCl, pH 3
Um die Zugänglichkeit der Algenproteine für die Enzyme Pronase und Pancreatin zu
verbessern, wurde eine Behandlung mit HCl, pH 3 für 18 h, sowie 48 h durchgeführt. Hiermit
sollte überprüft werden, ob die Behandlung, vergleichbar mit einer sauren, besonders beim
Umsatz von Biomasse zur Glucose-Gewinnung Anwendung findenden Hydrolyse, zu
effektivem Aufschluss der Algenzellwand führt.
In Abbildung 62 und Tabelle 26 sind die Hydrolysegrade des Algenhydrolysats in Abhängigkeit
des Enzyms, sowie der Dauer der Vorbehandlung tEV dargestellt.
Abbildung 62: Hydrolysegrad des Algen-Hydrolysats in Abhängigkeit des Enzyms und der Dauer der Vorbehandlung mit HCl, pH 3
Sowohl für Pancreatin als auch für Pronase zeigt sich bei einer Vorbehandlung mit HCl, pH 3
ein geringfügig erhöhter, aber nicht signifikant unterschiedlicher, Hydrolysegrad (p > 0,05) in
Abhängigkeit der Dauer (Pancreatin, HCl, pH 3, 18 h: 10,29 ± 0,97, Pancreatin, HCl, pH 3, 48
h: 12,76 ± 1,21; Pronase, HCl, pH 3, 18 h: 17,04 ± 2,83, Pronase, HCl, pH 3, 48 h: 18,16 ±
2,59). Die Verwendung von Pronase führt im Vergleich zu Pancreatin zu signifikant höheren
Hydrolysegraden (Pancreatin, HCl, pH 3, 18 h: 10,29 ± 0,97, Pronase, HCl, pH 3, 18 h: 17,04
± 2,83; Pancreatin, HCl, pH 3, 48 h: 12,76 ± 1,21, Pronase, HCl, pH 3, 48 h: 18,16 ± 2,59) (p
< 0,05). Bei der Hydrolyse der Algenproteine nach einer Behandlung mit HCl, pH 3 blieben
nach der Zentrifugation des Hydrolysats nur geringe Menge Sediment zurück (Massen nicht
gravimetrisch ermittelt).
0,00
5,00
10,00
15,00
20,00
25,00
Hyd
roly
segr
ad D
H (
%)

Ergebnisse
98
Tabelle 26: Hydrolysegrad des Algen-Hydrolysats in Abhängigkeit des Enzyms und der Dauer der Vorbehandlung mit HCl, pH 3
Enzym n
Enzym-
wirkdauer
tE (min)
Vorbehandlung
Dauer der
Vorbehandlung tSV
(h)
Hydrolysegrad
DH (%)
Pancreatin 3 45 HCl, pH 3 18 10,29 ± 0,97 a
Pancreatin 3 45 HCl, pH 3 48 12,76 ± 1,21 a, b
Pronase 3 45 HCl, pH 3 18 17,04 ± 2,83 b, c
Pronase 3 45 HCl, pH 3 48 18,16 ± 2,59 c
Werte in einer Spalte mit unterschiedlichen Buchstaben a, b, c unterschieden sich signifikant (p < 0,05).
Die Erhöhung des Hydrolysegrads bei einer Behandlung mit HCl, pH 3 ist für beiden Enzyme
(Pancreatin, Pronase) auch chromatographisch erkennbar. Während bei einer 18-stündigen
Vorbehandlung lediglich eine geringer Anteil von Molekülen im Bereich von 44 – 10 kDa
erzeugt werden können (Abbildung 63 a, b), lassen sich beim 48-stündigen Prozess Moleküle
im Bereich von 158 kDa – 10 kDa nachweisen (Abbildung 64 a, b). Besonders bei 48-stündiger
Vorbehandlung resultiert die Verwendung von Pronase in einem vergleichsweise hohen Anteil
an Molekülverbindungen > 44 kDa (Abbildung 64 b).
Eine Zunahme des Anteils von Verbindungen dieser Größenordnungen erscheint, unter
Berücksichtigung des steigenden Hydrolysegrads, ungewöhnlich. Da nicht-native, denaturierte
oder teilgefaltete Proteine gemäß Roberts (2007), unter anderem bedingt durch frei
zugängliche hydrophobe Gruppen, jedoch anfällig für die Ausbildung von irreversiblen Protein-
Aggregaten sind, wird von einer verstärkten Aggregatbildung bei steigender Dauer der
Vorbehandlung ausgegangen.
Unabhängig vom verwendeten Enzym und der Dauer der Behandlung mit HCl, pH 3 sind
Strukturen mit einer Molekülgröße < 1,35 kDa stark repräsentiert.

Ergebnisse
99
Abbildung 63: Molekülgrößenverteilung – Hydrolysate der Alge Chlorella vulgaris mit a) Pancreatin (0,25 % (20 USP); 45 min; Behandlung: HCl, pH 3, 18 h) und b) Pronase (0,25 % (112,5 PU); 45 min; Behandlung: HCl, pH 3, 18 h), qualitative Darstellung
Abbildung 64: Molekülgrößenverteilung - Hydrolysate der Alge Chlorella vulgaris mit a) Pancreatin (0,25 % (20 USP); 45 min; Behandlung: HCl, pH 3, 48 h) und b) Pronase (0,25 % (112,5 PU); 45 min; Behandlung: HCl, pH 3, 48 h), qualitative Darstellung
a) Pancreatin 0,25 %, (20 USP), 45 min, HCl, pH 3, 18 h
670 kDa
158 kDa
44 kDa
1,35 kDa
b) Pronase 0,25 %, (112,5 PU), 45 min, HCl, pH 3, 18 h
670 kDa
158 kDa
44 kDa
1,35 kDa
a) Pancreatin 0,25 %, (20 USP), 45 min, HCl, pH 3, 48 h
b) Pronase 0,25 %, (112,5 PU), 45 min, HCl, pH 3, 48 h
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
qu
alit
ativ
qu
alit
ativ
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)

Ergebnisse
100
4.2.1.3 Vergleich – Behandlung mit HCl, pH 3 vs. Behandlung mit Rohament® PL
In Tabelle 27 sind die Hydrolysegrade des Algen-Hydrolysats in Abhängigkeit des Enzyms,
sowie der Dauer tEV bzw. tSV und Art (HCl, pH 3; Rohament® PL) der Vorbehandlung
dargestellt.
Die Behandlung der Algen (Chlorella vulgaris) für 18 h und 48 h mit der Pektinase Rohament®
PL führte, verglichen mit einer Behandlung durch HCl, pH 3 (18 h, 48 h), zu nicht signifikant
erhöhten Hydrolysegraden (p > 0,05). Die Hydrolysegrade der sauren Vorbehandlung (HCl,
pH 3) lagen dabei durchweg unter den Hydrolysegraden der mit der Pektinase Rohament® PL
behandelten Alge (Chlorella vulgaris) (ausgenommen: Pronase, HCl, pH 3, 18 h vs. Pronase,
Rohament® PL, 18 h).
Tabelle 27: Hydrolysegrad des Algen-Hydrolysats in Abhängigkeit des Enzyms und der Dauer der Vorbehandlung mit HCl, pH 3 und Rohament® PL
Enzym n
Enzym-
wirkdauer
tE (min)
Vorbehandlung
Dauer der
Vorbehandlung
tEV/tSV (h)
Hydrolysegrad
DH (%)
Pancreatin 3 45 HCl, pH 3 18 10,29 ± 0,97 a
Pancreatin 3 45 HCl, pH 3 48 12,76 ± 1,21 a, d
Pancreatin 3 45 Rohament® PL 18 15,32 ± 1,19 a, c
Pancreatin 3 45 Rohament® PL 48 20,14 ± 1,56 b, c
Pancreatin 2* 45 Rohament® PL 72 19,81 ± 0,10 b, c
Pronase 3 45 HCl, pH 3 18 17,04 ± 2,83 b, c, d
Pronase 3 45 HCl, pH 3 48 18,16 ± 2,59 b, c
Pronase 3 45 Rohament® PL 18 14,63 ± 2,73 a, c
Pronase 3 45 Rohament® PL 48 21,39 ± 1,77 b, c
Pronase 2* 45 Rohament® PL 72 19,01 ± 2,99 b, c
Werte in einer Spalte mit unterschiedlichen Buchstaben a, b, c, d unterschieden sich signifikant (p < 0,05). * Ausschluss eines Messwerts aufgrund fehlerhafter Temperaturführung während des Vorbehandlungsprozesses.
Die vorab dargestellte, verstärke Ausbildung von Peptiden < 1,35 kDa, sowie der geringe Anteil
an Peptiden im Bereich zwischen 158 – 10 kDa sprechen gegen die Anwendung der
Behandlung mit Rohament® PL, sodass im Rahmen dieser Studie, unter Berücksichtigung der
Zielsetzung „Erzeugung eines Hydrolysats mit moglichst breiter Molekülgroßenverteilung“,
eine Behandlung mit HCl, pH 3 für 48 h in Kombination mit einer anschließend durchgeführten
Hydrolyse durch Pronase 0,25 % (112,5 PU), 45 min Anwendung fand.

Ergebnisse
101
4.3 Crossflow-Diafiltration & Fraktionierung
Nachfolgend sind die Ergebnisse der Membrancharakterisierung, sowie die Versuchsreihe
zum Einfluss des pH-Werts und der Ionenstärke auf den Crossflow-Parameter Permeatflux
JF,t dargestellt. Darüber hinaus werden die Auswirkungen der Crossflow-Fraktionierung
anhand der SE-HPLC Daten, sowie der absoluten Proteingehalte von Retentat mRet- und
Permeat-Fraktion mPer beschrieben. In weiteren Schritten wird dargelegt, wie sich die
Verwendung von Ultraschall, sowie alternativer Membranmaterialien auf die Fraktionierung
auswirkt. Abschließend wird das Verfahren der Dialyse und dessen Einfluss auf die
Molekülgrößenverteilung des vorfiltrierten Feeds beleuchtet.
4.3.1 Membran- & Anlagen-Charakterisierung
Entscheidend für eine Crossflow Filtration ist das Operieren unterhalb des
deckschichtkontrollierten Bereichs. Im Rahmen dieser Studie wurde dazu der kritische Druck
pCP der Anlagenkonzeption für hydrolysiertes Molkenprotein-Isolat und hydrolysierte Alge
(Chlorella vulgaris) ermittelt. Hierfür wurde der Permeatflux JF,t in Abhängigkeit des
Transmembrandrucks pTMP für die Filtration mit Wasser und Feed bestimmt. Die Ergebnisse
der Versuche sind in Kapitel 4.3.1.1 und 4.3.1.2 (Abbildung 68 – Abbildung 69) dargestellt.
Den Charakterisierungs-Versuchen vorausgehen, erfolgte die Festlegung des MWCO der
verwendeten Membran, basierend auf den Ergebnissen der Pilot-Versuche, sowie der
Molekülgrößenverteilung der Hydrolysate von Molkenprotein-Isolat und der Alge Chlorella
vulgaris.
Die im Rahmen der Pilot-Versuche anhand des hydrolysierten Molkenprotein-Isolats „Thermax
690“ (Glanbia Nutritionals Ltd., IRL) bei einer Feed-Konzentration von 3 % (bezogen auf den
Proteingehalt), pH 8 auf derselben Anlagenkonzeption unter identischen Bedingungen mittels
einer Membran mit 50 kDa MWCO (Microdyn Nadir GmbH, Wiesbaden, GER) gewonnenen
Ergebnisse offenbarten, gelelektrophoretisch ermittelt (Abbildung 65), massive
Verschiebungen der Membrantrenngrenze, sowie einen umfassenden Rückhalt großer (> 220
kDa) aber auch kleiner (< 14 kDa) Moleküle über der Membran (im Retentat). In der Fraktion
< 50 kDa – 30 kDa konnten, basierend auf einer Bewertung der Farbintensität, nur geringe
Molekülmengen im Bereich > 14 kDa festgestellt werden. Der Anteil an Strukturen < 14 kDa
war gelelektrophoretisch nicht darstellbar. Zum Zeitpunkt der Versuchsdurchführung wurde
diese Feststellung auf eine mangelnde Färbung der Peptide im betroffenen Größenbereich
durch „Coomassie Brilliant Blau G 250“ zurückgeführt, da spektralphotometrisch ca. 6 – 7 %
des Gesamtproteingehalts aller Fraktionen in der betroffenen Fraktion festgestellt werden
konnten.

Ergebnisse
102
Legende: 1) Größenmarker, 2) „Thermax 690“, 3) > 50 kDa, 4) 50 kDa – 30 kDa, 5) 30 kDa – 20 kDa
6) 20 kDa – 10 kDa, 7) 10 kDa – 5 kDa, 8) 5 kDa – 4 kDa, 9) 4 kDa – 1 kDa, 10) Größenmarker
Abbildung 65: Gelelektrophorese der Fraktionen von hydrolysiertem Molkenprotein-Isolat
In einer nachgestellten SE-HPLC-Analyse der Fraktionen konnten in der Fraktion < 50 kDa –
30 kDa jedoch lediglich wenige Moleküle im Bereich < 17 kDa und kleiner nachgewiesen
werden. Der Großteil der Moleküle lag im Bereich < 1,35 kDa und kleiner (Abbildung 66 b).
Betrachtet man die Molekülgrößenverteilung der Fraktion > 50 kDa, liegt der Schluss nahe,
dass Moleküle im Bereich < 50 kDa - > 1,35 kDa die Membran nicht passieren konnten und
somit eine Verschiebung der Trenngrenze hin zu einem geringeren MWCO stattgefunden hat
(Abbildung 66 a).
Um die Problematik zu umgehen, wurde die Feed-Konzentration auf 1,5 % (bezogen auf den
Proteingehalt) herabgesetzt. Weiterhin wurde der MWCO der Membran auf 100 kDa
angehoben um einer Permeatfraktion mit zu schmaler Molekülgrößenverteilung vorzubeugen,
da in diesem Falle eine weitere Fraktionierung des Permeats aufgrund der beschriebenen
Problematik der Trenngrenzenverschiebung als nicht sinnvoll anzusehen ist.
Die unter den genannten Bedingungen durchgeführte Fraktionierung offenbarte ebenfalls eine
starke Verschiebung der Trenngrenze hin zu einem geringeren MWCO (Abbildung 67 a, b).
Trotzdem konnten durch die Reduktion der Feed-Konzentration ein geringer (in der Abbildung
markierte) Anteil von Molekülen im Bereich zwischen 17 kDa – 44 kDa festgestellt werden
(Abbildung 67 b).
Erst die, basierend auf umfangreichen Literaturrecherchen, zusammengefasst dargestellt in
Kapitel 2.1.4, vorgenommene Absenkung der Feed-Konzentration des hydrolysierten
Molkenprotein-Isolat-Feeds auf 0,15 % (bezogen auf den Proteingehalt) in Verbindung mit der
220 kDa (Myosin)
116 kDa (ß-Galactosidase)
66 kDa (Albumin)
55,6 kDa (Glutamat-Dehydrogenase)
29 kDa (α-Carboanhydrasen)
14 kDa (Lysozym)
1 2 3 4 5 6 7 8 9 10

Ergebnisse
103
Verwendung einer Membran mit einer Trenngrenze von 100 kDa führte zu einer breiteren
Molekülgrößenverteilung im Permeat (siehe Kapitel 4.3.2, Abbildung 78 - Abbildung 81).
Aufgrund einer vergleichbaren Molekülgrößenverteilung der hydrolysierten Alge (Chlorella
vulgaris) (siehe Abbildung 64 b) wurden Feed-Konzentration und MWCO der Membran für die
Fraktionierung des hydrolysierten Algen-Feeds (Chlorella vulgaris) übernommen.

Ergebnisse
104
Anmerkung: Die Zuweisung der Molekülgrößen erfolgte unter Verwendung der entsprechenden Retentionszeiten tr des BioRad Gel-Filtration-Standards. Aufgrund der schlecht
lesbaren Achsenbeschriftung der Ausgangsgrafiken wurde diese zur besseren Lesbarkeit nachgearbeitet. Jeder | auf der x-Achse entspricht einer Retentionszeit ∆tr von 5 min. Auf
der y-Achse wird zur Wahrung der Übersicht nur der Maximalwert des Detektorsignals angegeben (Einheit: µV).
Legende: 1, Thyroglobulin (670 kDa). 2, γ-Globulin (158 kDa). 3, Ovalbumin (44 kDa). 4, Myoglobin (17 kDa). 5, Vitamin B12 (1,35 kDa).
Abbildung 66: Molekülgrößenverteilung - SE-HPLC-Chromatogramme von fraktioniertem, hydrolysiertem Molkenprotein-Isolat (Feed-Konzentration
3 % (bezogen auf den Proteingehalt) a) Fraktion > 50 kDa b) Fraktion < 50 kDa – 30 kDa, quantitative Darstellung
0 5 10 0 5 10 tr (min) tr (min)
µV µV 2*104
3*105
1 2 3 4 5 1 2 3 4 5
a) b)

Ergebnisse
105
Abbildung 67: Molekülgrößenverteilung - SE-HPLC-Chromatogramme von fraktioniertem, hydrolysiertem Molkenprotein-Isolat (Feed-Konzentration 1,5 % (bezogen auf den Proteingehalt) a) Fraktion > 100 kDa b) Fraktion < 100 kDa, qualitative Darstellung
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
rot: BioRad Gel-Filtration Standard
blau: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
rot: BioRad Gel-Filtration Standard
blau: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
a) b)

Ergebnisse
106
4.3.1.1 Molkenprotein-Isolat
Zur Fraktionierung des hydrolysierten Molkenprotein-Isolats wurde, basierend auf der
Molekülgrößenverteilung des Feeds, sowie der vorausgehen dargestellten Erkenntnisse
(Kapitel 4.3.1), eine Membran mit einer Trenngrenze von 100 kDa (Microdyn Nadir GmbH,
Wiesbaden, GER) gewählt.
Abbildung 68: Membrancharakterisierung 100 kDa – hydrolysiertes Molkenprotein-Isolat
Zur Charakterisierung der Membran wurde der Permeatflux JF,t des Feeds bei steigendem
Transmembrandruck pTMP dokumentiert. Hierzu wurde pTMP, in Intervallen von ∆pTMP = 0,2 bar,
schrittweise von 0 bar bis max. 2 bar angehoben. Ab einem Transmembrandruck pTMP von 0,6
bar weicht der Flux JF,t des Feeds vom linearen Verlauf des Wasserflux JWvor ab. Der kritische
Druck pCP der Filtration von Molkenprotein-Isolat wurde auf 0,5 bar festgelegt.
4.3.1.2 Alge (Chlorella vulgaris)
Zur Fraktionierung der hydrolysierten Alge (Chlorella vulgaris) wurde, basierend auf der
Molekülgrößenverteilung des Feeds, sowie der vorausgehend dargestellten Erkenntnisse
(Kapitel 4.3.1), eine Membran mit einer Trenngrenze von 100 kDa (Microdyn Nadir GmbH,
Wiesbaden, GER) gewählt.
R² = 0,955
0
20
40
60
80
100
120
140
160
180
200
220
240
0 0,2 0,4 0,6 0,8 1 1,2 1,4 1,6 1,8 2
Flu
x J F
,t(m
l/m
in)
Druck pTMP (bar)
Flux Wasser MW (ml/min) Flux Medium MW (ml/min)

Ergebnisse
107
Abbildung 69: Membrancharakterisierung 100 kDa – hydrolysierte Alge (Chlorella vulgaris)
Zur Charakterisierung der Membran wurde der Permeatflux JF,t des Feeds bei steigendem
Transmembrandruck pTMP, vergleichbar der vorab dargestellten Charakterisierung der
Membran für hydrolysiertes Molkenprotein-Isolat, dokumentiert. Hierzu wurde pTMP, in
Intervallen von ∆pTMP = 0,2 bar, schrittweise von 0 bar bis max. 2 bar angehoben. Vergleichbar
mit der Filtration von hydrolysiertem Molkenprotein-Isolat weicht der Flux des Feeds JF,t ab
einem Transmembrandruck pTMP von 0,6 bar von linearen Verlauf des Wasserflux JWvor ab. Der
kritische Druck pCP der Filtration von hydrolysierter Alge (Chlorella vulgaris) wurde
dementsprechend auf 0,5 bar eingestellt.
4.3.2 Einfluss des pH-Werts & der Ionenstärke
Der Einfluss des pH-Wertes auf den Fouling-Prozess ist seit langem intensiver Gegenstand
der Forschung. Kernaussage vieler Studien ist, dass starkes Fouling in Verbindung mit
niedrigem Permeatflux bei nicht hydrolysierten Feeds grundsätzlich in der Nähe des IEP auftritt
und, dass sowohl über- als auch unterhalb des IEP eine vergleichsweise reduzierte
Auswirkung von Fouling auf dem Permeatflux JF zu beobachten ist (Wang & Tang, 2011; She
et al., 2009; Huisman et al., 2000; McDonogh et al., 1990; Palecek et al., 1993; Fane et al.,
1983).
In Verbindung mit dem pH-Wert spielt die Ionenstärke des Feeds eine wichtige Rolle bei der
Entstehung und Strukturbildung der Deckschicht. Eine allgemeingültige Aussage bezüglich
des Effekts der Ionenstärke auf den Fouling-Prozess ist, hinsichtlich widersprüchlicher
Forschungsergebnisse, unter anderem von She et al. (2009), Salgın (2007), Teng et al. (2006),
Tarabara et al. (2004), Chan & Chen (2001), Kwon et al. (2000), Palecek & Zydney (1994),
Heinemann et al. (1988) & Fane et al. (1983), aber nicht uneingeschränkt möglich.
R² = 0,997
0
20
40
60
80
100
120
140
160
180
200
0 0,2 0,4 0,6 0,8 1 1,2 1,4 1,6 1,8 2
Flu
x J F
,t (m
l/m
in)
Druck pTMP (bar)
Flux Wasser MW (ml/min) Flux Medium MW (ml/min)
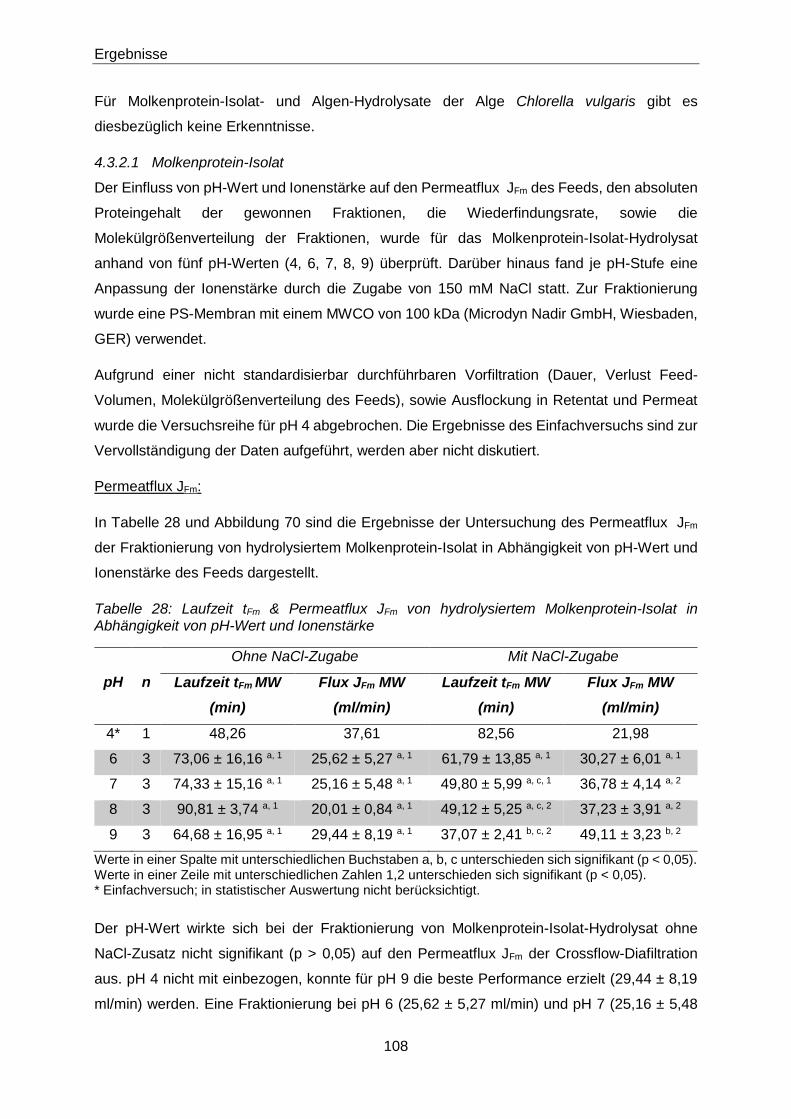
Ergebnisse
108
Für Molkenprotein-Isolat- und Algen-Hydrolysate der Alge Chlorella vulgaris gibt es
diesbezüglich keine Erkenntnisse.
4.3.2.1 Molkenprotein-Isolat
Der Einfluss von pH-Wert und Ionenstärke auf den Permeatflux JFm des Feeds, den absoluten
Proteingehalt der gewonnen Fraktionen, die Wiederfindungsrate, sowie die
Molekülgrößenverteilung der Fraktionen, wurde für das Molkenprotein-Isolat-Hydrolysat
anhand von fünf pH-Werten (4, 6, 7, 8, 9) überprüft. Darüber hinaus fand je pH-Stufe eine
Anpassung der Ionenstärke durch die Zugabe von 150 mM NaCl statt. Zur Fraktionierung
wurde eine PS-Membran mit einem MWCO von 100 kDa (Microdyn Nadir GmbH, Wiesbaden,
GER) verwendet.
Aufgrund einer nicht standardisierbar durchführbaren Vorfiltration (Dauer, Verlust Feed-
Volumen, Molekülgrößenverteilung des Feeds), sowie Ausflockung in Retentat und Permeat
wurde die Versuchsreihe für pH 4 abgebrochen. Die Ergebnisse des Einfachversuchs sind zur
Vervollständigung der Daten aufgeführt, werden aber nicht diskutiert.
Permeatflux JFm:
In Tabelle 28 und Abbildung 70 sind die Ergebnisse der Untersuchung des Permeatflux JFm
der Fraktionierung von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit von pH-Wert und
Ionenstärke des Feeds dargestellt.
Tabelle 28: Laufzeit tFm & Permeatflux JFm von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit von pH-Wert und Ionenstärke
pH n
Ohne NaCl-Zugabe Mit NaCl-Zugabe
Laufzeit tFm MW
(min)
Flux JFm MW
(ml/min)
Laufzeit tFm MW
(min)
Flux JFm MW
(ml/min)
4* 1 48,26 37,61 82,56 21,98
6 3 73,06 ± 16,16 a, 1 25,62 ± 5,27 a, 1 61,79 ± 13,85 a, 1 30,27 ± 6,01 a, 1
7 3 74,33 ± 15,16 a, 1 25,16 ± 5,48 a, 1 49,80 ± 5,99 a, c, 1 36,78 ± 4,14 a, 2
8 3 90,81 ± 3,74 a, 1 20,01 ± 0,84 a, 1 49,12 ± 5,25 a, c, 2 37,23 ± 3,91 a, 2
9 3 64,68 ± 16,95 a, 1 29,44 ± 8,19 a, 1 37,07 ± 2,41 b, c, 2 49,11 ± 3,23 b, 2
Werte in einer Spalte mit unterschiedlichen Buchstaben a, b, c unterschieden sich signifikant (p < 0,05). Werte in einer Zeile mit unterschiedlichen Zahlen 1,2 unterschieden sich signifikant (p < 0,05). * Einfachversuch; in statistischer Auswertung nicht berücksichtigt.
Der pH-Wert wirkte sich bei der Fraktionierung von Molkenprotein-Isolat-Hydrolysat ohne
NaCl-Zusatz nicht signifikant (p > 0,05) auf den Permeatflux JFm der Crossflow-Diafiltration
aus. pH 4 nicht mit einbezogen, konnte für pH 9 die beste Performance erzielt (29,44 ± 8,19
ml/min) werden. Eine Fraktionierung bei pH 6 (25,62 ± 5,27 ml/min) und pH 7 (25,16 ± 5,48

Ergebnisse
109
ml/min) liefert weitestgehend identische Ergebnisse. Eine Fraktionierung bei pH 8 führt zum
geringsten Permeatflux JFm (20,01 ± 0,84 ml/min) bei gleichzeitig sehr guter Wiederholbarkeit,
beurteilt anhand der geringen Standardabweichung.
Abbildung 70: Permeatflux JFm von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit von pH-Wert und Ionenstärke
Bei einer Fraktionierung unter Zusatz von 150 mM NaCl konnte für pH 9 abermals die beste
Performance JFm erreicht werden (49,11 ± 3,23 ml/min), die sich signifikant (p < 0,05) von den
weiteren pH-Stufen (6, 7, 8) unterschied. Des Weiteren ist anhand der Daten ein Ansteigen
des Permeatflux JFm mit zunehmendem pH-Wert erkennbar.
Für die pH-Stufen 7 (36,78 ± 4,14 ml/min), 8 (37,23 ± 3,91 ml/min) und 9 (49,11 ± 3,23 ml/min)
konnten durch das Erhöhen der Ionenstärke signifikant (p < 0,05) erhöhte Permeatflüsse JFm
bei gleichzeitig geringeren Standardabweichungen im Vergleich zu einer Fraktionierung ohne
150 mM NaCl erreicht werden (pH 7: 25,16 ± 5,48 ml/min; pH 8: 20,01 ± 0,84 ml/min; pH 9:
29,44 ± 8,19 ml/min). Für pH 6 führte der Zusatz von 150 mM NaCl nicht zu signifikant
erhöhtem Permeatflux JFm (pH 6: 25,62 ± 5,27 vs. pH 6:, NaCl: 30,27 ± 6,01).
Proteingehalt von Retentat mRet & Permeat mPer:
In Tabelle 29, sowie Abbildung 71 und Abbildung 72 sind die absoluten Proteingehalt der
Retentat (mRet)- und Permeatfraktionen (mPer) in Abhängigkeit von pH-Wert und Ionenstärke
dargestellt.
37,61
25,62
25,16
20,01
29,48
21,98
30,27
36,78
37,23
49,11
0 5 10 15 20 25 30 35 40 45 50 55
4
6
7
8
9
Permeatflux JFm (ml/min)
pH
-Wer
t
Flux (ml/min) - mit NaCl Flux (ml/min) - ohne NaCl

Ergebnisse
110
Tabelle 29: Absolute Proteingehalte von hydrolysiertem Molkenprotein-Isolat in Retentat und Permeat in Abhängigkeit von pH-Wert und Ionenstärke
pH n Ohne NaCl-Zugabe Mit NaCl-Zugabe
mRet (g) mPer (g) mRet (g) mPer (g)
4* 1 0,348 0,395 0,271 0,461
6 3 0,300 ± 0,056 a, c, 1 0,373 ± 0,050 a, 1 0,209 ± 0,031 a, 1 0,354 ± 0,014 a, 1
7 3 0,209 ± 0,060 a, 1 0,367 ± 0,047 a, 1 0,136 ± 0,037 a, 1 0,426 ± 0,039 a, 1
8 3 0,354 ± 0,067 b, c, 1 0,358 ± 0,026 a, 1 0,194 ± 0,019 a, 2 0,419 ± 0,037 a, 1
9 3 0,286 ± 0,012 a, c, 1 0,398 ± 0,024 a, 1 0,163 ± 0,049 a, 2 0,419 ± 0,045 a, 1
Werte in einer Spalte mit unterschiedlichen Buchstaben a, b, c unterschieden sich signifikant (p < 0,05). Werte in einer Zeile mit unterschiedlichen Zahlen 1,2 unterschieden sich signifikant (p < 0,05). * Einfachversuch; in statistischer Auswertung nicht berücksichtigt.
Der pH-Wert wirkte sich bei der Fraktionierung von Molkenprotein-Isolat-Hydrolysat ohne
NaCl-Zusatz in nur einem Fall (vergleiche: pH 7 vs. pH 8) signifikant (p < 0,05) auf den
Proteingehalt mRet der Retentat-Fraktionen aus. Der höchste Proteingehalt mRet wurde hierbei
für pH 8 (0,354 ± 0,067 g), der niedrigste für pH 7 (0,209 ± 0,060 g) festgestellt. Der
Proteingehalt der pH-Stufe 6 und 9 lag zwischen 0,286 ± 0,012 g (pH 9) und 0,300 ± 0,056 g
(pH 6).
Die Permeat-Fraktionen unterschieden sich hinsichtlich ihres Proteingehalts mPer nicht
signifikant (p > 0,05) und lagen im Bereich zwischen 0,373 ± 0,050 g (pH 6) und 0,398 ± 0,024
g (pH 9).
Abbildung 71: Absolute Proteinmenge der Retentat-Fraktionen von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit von pH-Wert und Ionenstärke
0,348
0,300
0,209
0,354
0,286
0,271
0,209
0,136
0,194
0,163
0,000 0,050 0,100 0,150 0,200 0,250 0,300 0,350 0,400 0,450
4
6
7
8
9
Proteingehalt Retentat (g)
pH
-Wer
t
Proteinmenge (g) Retentat - mit NaCl Proteinmenge (g) Retentat - ohne NaCl

Ergebnisse
111
Eine Fraktionierung unter Zusatz von 150 mM NaCl resultierte weder für Retentat- noch für
Permeatfraktion in signifikant unterschiedlichen Proteingehalten mRet/mPer (p > 0,05). Der
höchste Retentat-Proteingehalt wurde bei pH 6 (0,209 ± 0,031 g), der niedrigste bei pH 7
(0,136 ± 0,037 g) festgestellt. Generell lagen die Proteingehalte des Retentats bei einer
Fraktionierung unter Zusatz von 150 mM NaCl unter denen der Fraktionierung ohne Zusatz
von 150 mM NaCl. Für pH 8 (0,194 ± 0,019 g) und pH 9 (0,163 ± 0,049 g) konnten durch die
Erhöhung der Ionenstärke im Vergleich zu einer Fraktionierung ohne 150 mM NaCl (pH 8:
0,354 ± 0,067 g; pH 9: 0,286 ± 0,012 g) signifikant geringere Proteingehalte mRet (p < 0,05) im
Retentat festgestellt werden.
Der Proteingehalt mPer der zugehörigen Permeatfraktionen (pH 8: 0,419 ± 0,037 g; pH 9: 0,419
± 0,045 g) lag über dem der ohne NaCl-Zusatz erzeugten Fraktionen (pH 8: 0,358 ± 0,026 g;
pH 9: 0,398 ± 0,024 g), unterschied sich aber nicht signifikant (p > 0,05). Mit Ausnahme von
pH 6 wurden im Permeat des unter 150 mM NaCl-Zusatz fraktionierten Feed höhere
Proteingehalte festgestellt als in den Permeatfraktionen des ohne NaCl aufgetrennten Feeds.
Abbildung 72: Absolute Proteinmenge der Permeat-Fraktionen von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit von pH-Wert und Ionenstärke
Wiederfindungsrate:
Die Wiederfindungsrate, dargestellt in Tabelle 30, lag bei der Fraktionierung von
Molkenprotein-Isolat-Hydrolysat ohne NaCl-Zusatz zwischen 76, 91 ± 5,53 % (pH 7) und 94,85
± 4,63 % (pH 8) (pH 4 nicht miteinbezogen). Der pH-Wert beeinflusste die WFR nicht
signifikant (p> 0,05). Ein Großteil der Molekülverbindungen (47,67 ± 3,43 % (pH 8) - 53,02 ±
3,22 % (pH 9) passierte die Membran. Bei pH 7 ist der Anteil an Protein in der Retentatfraktion
am geringsten (27,92 ± 8,03 %). Dies resultierte jedoch nicht in einem erhöhten Proteinanteil
im Permeat. Darüber hinaus wurde bei pH 7 (76,91 ± 5,53 %) die geringste WFR festgestellt.
0,395
0,373
0,367
0,358
0,398
0,461
0,355
0,426
0,419
0,420
0,000 0,050 0,100 0,150 0,200 0,250 0,300 0,350 0,400 0,450 0,500
4
6
7
8
9
Proteingehalt Permeat (g)
pH
-Wer
t
Proteinmenge (g) Permeat - mit NaCl Proteinmenge (g) Permeat - ohne NaCl

Ergebnisse
112
Die höchste WFR, sowie der höchste Proteinanteil im Retentat wurde bei pH 8 (WFR: 94,85 ±
4,63 %; WFRRET: 47,18 ± 8,94 %), der höchste Proteinanteil im Permeat bei pH 9 (53,02 ±
3,22 %) festgestellt.
Tabelle 30: Wiederfindungsrate und Anteile der Retentat- und Permeat-Fraktion bei der Fraktionierung von hydrolysiertem Molkenprotein-Isolat
pH n Ohne NaCl-Zugabe Mit NaCl-Zugabe
WFR (%) WFRRet (%) WFRPer (%) WFR (%) WFRRet (%) WFRPer (%)
4* 1 99,10 46,41 52,69 97,70 36,17 61,53
6 3 89,64 ±
11,46 a, 1
39,96 ±
7,52 a, 1
49,68 ±
6,67 a, 1
75,14 ±
4,90 a, 1
27,87 ±
4,22 a, 1
47,27 ±
1,79 a, 1
7 3 76,91 ±
5,53 a, 1
27,92 ±
8,03 a, 1
48,99 ±
6,31 a, 1
74,88 ±
3,39 a, 1
18,10 ±
4,92 a, 1
56,78 ±
5,23 a, 1
8 3 94,85 ±
4,63 a, 1
47,18 ±
8,94 a, 1
47,67 ±
3,43 a, 1
81,74 ±
4,36 a, 2
25,91 ±
2,62 a, 2
55,83 ±
4,97 a, 1
9 3 91,10 ±
4,63 a, 1
38,07 ±
1,56 a, 1
53,02 ±
3,22 a, 1
77,67 ±
4,82 a, 2
21,72 ±
6,60 a, 2
55,95 ±
5,97 a, 1
Werte in einer Spalte mit unterschiedlichen Buchstaben a, b unterschieden sich signifikant (p < 0,05). Werte in einer Zeile mit unterschiedlichen Zahlen 1,2 unterschieden sich signifikant (p < 0,05). * Einfachversuch; in statistischer Auswertung nicht berücksichtigt.
Die Wiederfindungsrate bei der Fraktionierung von Molkenprotein-Isolat-Hydrolysat unter
NaCl-Zusatz lag, pH-abhängig, zwischen 74,88 ± 3,39 % (pH 7) und 81,74 ± 4,36 % (pH 8).
Der Großteil der Proteine findet sich auch in diesem Fall in der Permeatfraktion (47,27 ± 1,79
% (pH 6) - 56,78 ± 5,23 % (pH7)). Die höchste WFR wurde bei pH 8 (81,74 ± 4,36 %;
WFRRET: 47,18 ± 8,94 %), der höchste Proteinanteil im Retentat bei pH 6 (27,87 ± 4,22 %)
und der höchste Proteinanteil im Permeat bei pH 7 (56,78 ± 5,23 %) festgestellt.
Die Erhöhung der Ionenstärke resultierte in einer geringeren WFR im Vergleich zur
Fraktionierung ohne NaCl-Zusatz. Die pH-Stufen 8 (WFR (ohne NaCl): 94,85 ± 4,63 %, WFR
(mit NaCl): 81,74 ± 4,36 %) und 9 (WFR (ohne NaCl): 91,10 ± 4,63 %, WFR (mit NaCl): 77,67
± 4,82 %) unterschieden sich signifikant (p < 0,05). Die geringere WFR lässt sich auf signifikant
geringere Proteinanteile (p < 0,05) in der Retentat-Fraktion zurückführen.
Molekülgrößenverteilung der Fraktionen:
Zur Beurteilung der Güte der Fraktionierung wurden SE-HPLC Analysen der Retentat- und
Permeatproben durchgeführt. Die Ergebnisse der Analysen sind nachfolgend qualitativ
dargestellt.
Abbildung 73 - Abbildung 77 zeigen die Chromatogramme einer nicht-fraktionierten
Molkenprotein-Isolat-Hydrolysat-Probe (Nullprobe) nach der Vorfiltration. Die

Ergebnisse
113
Molekülgrößenverteilung der Nullproben wiesen keine deutlichen erkennbaren pH- und
ionenstärke-abhängigen Unterschiede auf und verfügten über eine breite
Molekülgrößenverteilung.
Lediglich bei pH 4, ohne NaCl wurde ein Großteil der Moleküle > 17 kDa bereits im Rahmen
der Vorfiltration abgetrennt (Abbildung 73 a). Wie bereits vorab dargestellt, wurde die
Fraktionierung bei pH 4 aufgrund der nicht standardisierbar durchführbaren Vorfiltration
(Dauer, Verlust Feed-Volumen, Molekülgrößenverteilung des Feeds), sowie Ausflockungen in
Retentat und Permeat abgebrochen.
Die Molekülgrößenverteilung der bei unterschiedlichen pH-Stufen erzeugten Feeds zeigt,
unabhängig von pH-Wert und Ionenstärke, dass Strukturen ≤ 1,35 kDa einen bestimmenden
Anteil des Feeds einnehmen (Abbildung 74 - Abbildung 77). Molekülstrukturen ≥ 20 kDa sind
jedoch in ausreichendem Maße vorhanden, um analytisch (SE-HPLC) nach der Fraktionierung
in den Retentatfraktionen abgebildet werden zu können.

Ergebnisse
114
Abbildung 73: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die Fraktionierung von hydrolysiertem Molkenprotein-Isolat, pH 4, a) ohne NaCl b) mit NaCl, qualitative Darstellung
Abbildung 74: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die Fraktionierung von hydrolysiertem Molkenprotein-Isolat, pH 6, a) ohne NaCl b) mit NaCl, qualitative Darstellung
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Molkenprotein-Isolat, pH 4,Nullprobe, ohne NaCl
Molkenprotein-Isolat, pH 4,Nullprobe, mit NaCl
Molkenprotein-Isolat, pH 6,Nullprobe, ohne NaCl
Molkenprotein-Isolat, pH 6,Nullprobe, mit NaCl
a) b)
a) b)

Ergebnisse
115
Abbildung 75: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die Fraktionierung von hydrolysiertem Molkenprotein-Isolat, pH 7, a) ohne NaCl b) mit NaCl, qualitative Darstellung
Abbildung 76: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die Fraktionierung von hydrolysiertem Molkenprotein-Isolat, pH 8, a) ohne NaCl b) mit NaCl, qualitative Darstellung
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Molkenprotein-Isolat, pH 7,Nullprobe, ohne NaCl
Molkenprotein-Isolat, pH 7,Nullprobe, mit NaCl
Molkenprotein-Isolat, pH 8,Nullprobe, ohne NaCl
Molkenprotein-Isolat, pH 8,Nullprobe, mit NaCl
a) b)
a) b)

Ergebnisse
116
Abbildung 77: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die Fraktionierung von hydrolysiertem Molkenprotein-Isolat, pH 9, a) ohne NaCl b) mit NaCl, qualitative Darstellung
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Molkenprotein-Isolat, pH 9,Nullprobe, ohne NaCl
Molkenprotein-Isolat, pH 9,Nullprobe, mit NaCl
a) b)

Ergebnisse
117
In Abbildung 78 - Abbildung 81 sind die Chromatogramme von Retentat- und Permeat-
Fraktionen des Molkenprotein-Isolat-Hydrolysats in Abhängigkeit von pH-Wert und
Ionenstärke des Feeds dargestellt.
Bei der Betrachtung der Chromatogramme der Retentat-Fraktion wird, weitestgehend
unabhängig vom pH-Wert, deutlich, dass neben Molekülverbindungen im Zielbereich auch
eine große Menge an Strukturen ≤ 1,35 kDa vorhanden sind (Abbildung 78 a, c - Abbildung 81
a, c). Diese konnten folglich, trotz der Verwendung des Diafiltrationsverfahrens, nicht aus den
Retentat-Fraktionen ausgewaschen werden. Eine Fraktionierung bei erhöhter Ionenstärke
(Zusatz von 150 mM NaCl) führte darüber hinaus, deutlich bei pH 7 und pH 8 (Abbildung 79 c,
Abbildung 80 c), zu einem geringeren Anteil an Strukturen ≥ 20 kDa.
Anhand der Permeat-Fraktionen ist eine deutliche Verschiebung der Membrantrenngrenze hin
zu einem niedrigeren MWCO erkennbar (Abbildung 78 b, d - Abbildung 81 b, d). Nur ein
geringer Anteil an Verbindungen ≥ 20 kDa ist in den Permeaten feststellbar. Nähert man sich
der nominalen Trenngrenze der Membran (100 kDa) an, nimmt der Anteil weiter ab.
Molekülstrukturen ≤ 1,35 kDa sind stark repräsentiert.
Die Molekülgrößenverteilung der Permeatfraktion ist pH- und ionenstärke-abhängig. Für pH 9
(Abbildung 81 b) konnte bei einer Fraktionierung ohne NaCl-Zusatz die anteilsmäßig größte
Menge an Strukturen ≥ 20 kDa die Membran passieren, wohingegen bei pH 6, 7, 8
anteilsmäßig nur sehr geringe Mengen festgestellt werde konnten (Abbildung 78 b - Abbildung
80 b).
Der Zusatz von 150 mM NaCl führte im Fraktionierungsprozess, weitestgehend unabhängig
vom pH-Wert, zu einem erhöhten Anteil an Strukturen ≥ 20 kDa im Permeat, der besonders
für pH 7 (Abbildung 79 d), pH 8 (Abbildung 80 d) und pH 9 (Abbildung 81 d) über dem Niveau
des ohne NaCl fraktionierten Feeds lag. Molekülstrukturen ≤ 1,35 kDa sind jedoch weiterhin
stark repräsentiert.

Ergebnisse
118
Abbildung 78: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und Permeatfraktion von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit der Ionenstärke, pH 6, a) Retentat ohne NaCl b) Permeat ohne NaCl c) Retentat mit NaCl d) Permeat mit NaCl, qualitative Darstellung
Molkenprotein-Isolat, pH 6 Retentat, ohne NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
Molkenprotein-Isolat, pH 6 Permeat, ohne NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
Molkenprotein-Isolat, pH 6 Retentat, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
Molkenprotein-Isolat, pH 6 Permeat, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
a) b)
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
c) d)

Ergebnisse
119
Abbildung 79: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und Permeatfraktion von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit der Ionenstärke, pH 7, a) Retentat ohne NaCl b) Permeat ohne NaCl c) Retentat mit NaCl d) Permeat mit NaCl, qualitative Darstellung
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
a) b)
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
c) d)
Molkenprotein-Isolat, pH 7 Retentat, ohne NaCl
Molkenprotein-Isolat, pH 7 Permeat, ohne NaCl
Molkenprotein-Isolat, pH 7 Retentat, mit NaCl
Molkenprotein-Isolat, pH 7 Permeat, mit NaCl

Ergebnisse
120
Abbildung 80: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und Permeatfraktion von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit der Ionenstärke, pH 8, a) Retentat ohne NaCl b) Permeat ohne NaCl c) Retentat mit NaCl d) Permeat mit NaCl, qualitative Darstellung
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
a) b)
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
c) d)
Molkenprotein-Isolat, pH 8 Retentat, ohne NaCl
Molkenprotein-Isolat, pH 8 Permeat, ohne NaCl
Molkenprotein-Isolat, pH 8 Retentat, mit NaCl
Molkenprotein-Isolat, pH 8 Permeat, mit NaCl

Ergebnisse
121
Abbildung 81: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und Permeatfraktion von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit der Ionenstärke, pH 9, a) Retentat ohne NaCl b) Permeat ohne NaCl c) Retentat mit NaCl d) Permeat mit NaCl, qualitative Darstellung
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
a) b)
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
c) d)
Molkenprotein-Isolat, pH 9 Retentat, ohne NaCl
Molkenprotein-Isolat, pH 9 Permeat, ohne NaCl
Molkenprotein-Isolat, pH 9 Retentat, mit NaCl
Molkenprotein-Isolat, pH 9 Permeat, mit NaCl

Ergebnisse
122
4.3.2.2 Alge (Chlorella vulgaris)
Der Einfluss von pH-Wert und Ionenstärke auf den Permeatflux JFm des Feeds, den absoluten
Proteingehalt der gewonnen Fraktionen, die Wiederfindungsrate, sowie die
Molekülgrößenverteilung der Fraktionen wurde für das Algen-Hydrolysat (Chlorella vulgaris)
anhand von fünf pH-Werten (3, 6, 7, 8, 11) überprüft. Darüber hinaus fand je pH-Stufe eine
Anpassung der Ionenstärke durch die Zugabe von 150 mM NaCl statt. Zudem wurde die
anhand einer grafischen Auswertung des Filtrationsprozess ermittelte Charakteristik des
Filtrationsprozesses bewertet. Zur Fraktionierung wurde eine PS-Membran mit einem MWCO
von 100 kDa (Microdyn Nadir GmbH, Wiesbaden, GER) verwendet.
Permeatflux JFm:
In Tabelle 31 und Abbildung 82 sind die Ergebnisse der Fraktionierung von hydrolysierter Alge
(Chlorella vulgaris) in Abhängigkeit von pH-Wert und Ionenstärke des Feeds dargestellt.
Der pH-Wert wirkte sich bei der Fraktionierung von Algen-Hydrolysat ohne NaCl-Zusatz,
abgesehen von einer Ausnahme, nicht signifikant (p > 0,05) auf den Permeatflux JFm der
Crossflow-Diafiltration aus. Lediglich die Fraktionierung bei pH 6 (42,91 ± 2,22 ml/min) führen
im Vergleich zu pH 3 (29,15 ± 0,35 ml/min) zu einem signifikant erhöhten Permeatflux (p <
0,05). Der höchste Permeatflux JFm wurde bei pH 6 (42,91 ± 2,22 ml/min), der niedrigste bei
pH 3 (29,15 ± 0,35 ml/min) beobachtet. Die pH-Stufen 7 (38,27 ± 2,86 ml/min), 8 (39,65 ±
4,19 ml/min) und 11 (37,94 ± 6,83 ml/min) unterschieden sich nicht signifikant (p > 0,05).
Tabelle 31: Laufzeit tFm & Permeatflux JFm von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit von pH-Wert und Ionenstärke
pH n
Ohne NaCl-Zugabe Mit NaCl-Zugabe
Laufzeit tFm MW (min)
Flux JFm MW (ml/min)
Laufzeit tFm MW (min)
Flux JFm MW (ml/min)
3 3 66,55 ± 0,81 a, 1 29,15 ± 0,35 a, 1 69,79 ± 9,20 a, 1 28,14 ± 3,96 a, 1
6 3 45,29 ± 2,30 b, c, 1 42,91 ± 2,22 b, c, 1 55,10 ± 7,95 a, 1 35,71 ± 5,22 a, 1
7 3 50,89 ± 3,94 a, c, 1 38,27 ± 2,86 a, c, 1 62,34 ± 2,34 a, 2 31,15 ± 1,16 a, 2
8 3 49,30 ± 5,26 a, c, 1 39,65 ± 4,19 a, c, 1 59,49 ± 9,81 a, 1 33,17 ± 5,04 a, 1
11 3 52,38 ± 10,48 a, c, 1 37,94 ± 6,83 a, c, 1 75,34 ± 18,31 a, 2 26,67 ± 5,68 a, 2
Werte in einer Spalte mit unterschiedlichen Buchstaben a, b, c unterschieden sich signifikant (p < 0,05). Werte in einer Zeile mit unterschiedlichen Zahlen 1,2 unterschieden sich signifikant (p < 0,05).
Bei einer Fraktionierung unter Zusatz von 150 mM NaCl konnte für pH 6 (35,71 ± 5,22 ml/min)
abermals die beste Performance JFm erreicht werden, die sich jedoch nicht signifikant (p >
0,05) von den weiteren pH-Stufen unterschied. Der Permeatflux bei einer Fraktionierung unter
Zusatz von 150 mM NaCl lag zwischen 26,67 ± 5,68 ml/min (pH 11) und 35,71 ± 5,22 ml/min
(pH 6).
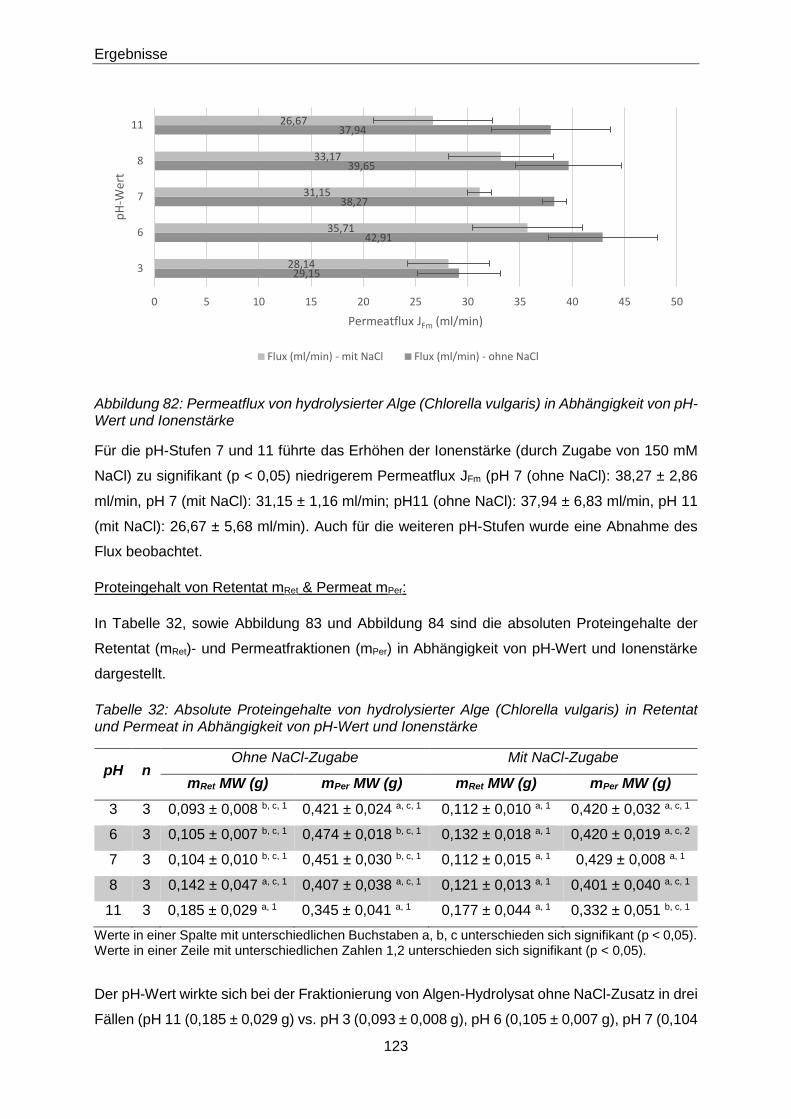
Ergebnisse
123
Abbildung 82: Permeatflux von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit von pH-Wert und Ionenstärke
Für die pH-Stufen 7 und 11 führte das Erhöhen der Ionenstärke (durch Zugabe von 150 mM
NaCl) zu signifikant (p < 0,05) niedrigerem Permeatflux JFm (pH 7 (ohne NaCl): 38,27 ± 2,86
ml/min, pH 7 (mit NaCl): 31,15 ± 1,16 ml/min; pH11 (ohne NaCl): 37,94 ± 6,83 ml/min, pH 11
(mit NaCl): 26,67 ± 5,68 ml/min). Auch für die weiteren pH-Stufen wurde eine Abnahme des
Flux beobachtet.
Proteingehalt von Retentat mRet & Permeat mPer:
In Tabelle 32, sowie Abbildung 83 und Abbildung 84 sind die absoluten Proteingehalte der
Retentat (mRet)- und Permeatfraktionen (mPer) in Abhängigkeit von pH-Wert und Ionenstärke
dargestellt.
Tabelle 32: Absolute Proteingehalte von hydrolysierter Alge (Chlorella vulgaris) in Retentat und Permeat in Abhängigkeit von pH-Wert und Ionenstärke
pH n Ohne NaCl-Zugabe Mit NaCl-Zugabe
mRet MW (g) mPer MW (g) mRet MW (g) mPer MW (g)
3 3 0,093 ± 0,008 b, c, 1 0,421 ± 0,024 a, c, 1 0,112 ± 0,010 a, 1 0,420 ± 0,032 a, c, 1
6 3 0,105 ± 0,007 b, c, 1 0,474 ± 0,018 b, c, 1 0,132 ± 0,018 a, 1 0,420 ± 0,019 a, c, 2
7 3 0,104 ± 0,010 b, c, 1 0,451 ± 0,030 b, c, 1 0,112 ± 0,015 a, 1 0,429 ± 0,008 a, 1
8 3 0,142 ± 0,047 a, c, 1 0,407 ± 0,038 a, c, 1 0,121 ± 0,013 a, 1 0,401 ± 0,040 a, c, 1
11 3 0,185 ± 0,029 a, 1 0,345 ± 0,041 a, 1 0,177 ± 0,044 a, 1 0,332 ± 0,051 b, c, 1
Werte in einer Spalte mit unterschiedlichen Buchstaben a, b, c unterschieden sich signifikant (p < 0,05). Werte in einer Zeile mit unterschiedlichen Zahlen 1,2 unterschieden sich signifikant (p < 0,05).
Der pH-Wert wirkte sich bei der Fraktionierung von Algen-Hydrolysat ohne NaCl-Zusatz in drei
Fällen (pH 11 (0,185 ± 0,029 g) vs. pH 3 (0,093 ± 0,008 g), pH 6 (0,105 ± 0,007 g), pH 7 (0,104
29,15
42,91
38,27
39,65
37,94
28,14
35,71
31,15
33,17
26,67
0 5 10 15 20 25 30 35 40 45 50
3
6
7
8
11
Permeatflux JFm (ml/min)
pH
-Wer
t
Flux (ml/min) - mit NaCl Flux (ml/min) - ohne NaCl

Ergebnisse
124
± 0,010 g)) signifikant (p < 0,05) auf den Proteingehalt der Retentat-Fraktionen mRet aus
(Abbildung 83).
Der höchste Retentat-Proteingehalt wurde für pH 11 (0,185 ± 0,029 g) festgestellt. Dieser
Proteingehalt liegt signifikant (p < 0,05) über den Proteingehalten der pH-Stufen 3 (0,093 ±
0,008 g), 6 (0,105 ± 0,007 g) und 7 (0,104 ± 0,010 g). Der bei pH 3 (0,093 ± 0,008 g) ermittelte
Proteingehalt des Retentats stellt den niedrigsten der Versuchsreihe dar.
Abbildung 83: Absolute Proteinmenge der Retentat-Fraktionen von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit von pH-Wert und Ionenstärke
Ein ähnliches Bild zeigt sich für die Permeat-Fraktionen, dargestellt in Abbildung 84.
Abbildung 84: Absolute Proteinmenge der Permeat-Fraktionen von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit von pH-Wert und Ionenstärke
Die Fraktionierung bei pH 11 (0,345 ± 0,041 g) resultierte hier, verglichen mit den pH-Stufen 3
(0,421 ± 0,024 g), 6 (0,474 ± 0,018 g) und 7 (0,451 ± 0,030 g), in einem signifikant niedrigeren
0,093
0,105
0,104
0,142
0,185
0,112
0,132
0,112
0,121
0,177
0,000 0,050 0,100 0,150 0,200 0,250
3
6
7
8
11
Proteingehalt Retentat (g)
pH
-Wer
t
Proteinmenge (g) Retentat - mit NaCl Proteinmenge (g) Retentat - ohne NaCl
0,421
0,474
0,451
0,407
0,345
0,420
0,420
0,429
0,401
0,332
0,000 0,100 0,200 0,300 0,400 0,500
3
6
7
8
11
Proteingehalt Permeat (g)
pH
-Wer
t
Proteinmenge (g) Permeat - mit NaCl Proteinmenge (g) Permeat - ohne NaCl

Ergebnisse
125
Permeat-Proteingehalt mPer (p < 0,05). Der höchste Permeat-Proteingehalt wurde bei pH 6
(0,474 ± 0,018 g) festgestellt.
Eine Fraktionierung unter Zusatz von NaCl resultierte für die Retentat-Fraktion nicht in
signifikant unterschiedlichen Proteingehalten mRet (p > 0,05). Der höchste Proteingehalt mRet
wurde hierbei für pH 11 (0,177 ± 0,044 g), der niedrigste für pH 3 (0,112 ± 0,010 g) und 7
(0,112 ± 0,015 g) festgestellt
Die Proteingehalte der Permeatfraktion mPer lagen im Bereich zwischen 0,332 ± 0,051 g (pH
11) und 0,429 ± 0,008 g (pH 7). Für diese pH-Stufen konnten signifikante Unterschiede
ermittelt werden (p < 0,05). Die Proteingehalte der weiteren unter Zusatz von 150 mM NaCl
gewonnenen Permeatfraktionen unterschieden sich nicht signifikant (p > 0,05). Im Vergleich
zu einer Fraktionierung ohne den Zusatz von 150 mM NaCl wurden im Permeat durchweg
geringere Proteingehalte festgestellt.
Die Erhöhung der Ionenstärke (durch Zusatz von 150 mM NaCl) führte jedoch lediglich für pH
6 zu signifikant geringerem Proteingehalt im Permeat (p < 0,05) (pH 6 (ohne NaCl): 0,474 ±
0,018 g, pH 6 (mit NaCl): 0,420 ± 0,019 g). Die weiteren pH-Stufen unterschieden sich nicht
signifikant (p > 0,05).
Wiederfindungsrate:
Die Wiederfindungsrate, dargestellt in Tabelle 33, lag bei der Fraktionierung von Algen-
Hydrolysat ohne NaCl-Zusatz zwischen 68, 64 ± 4,18 % (pH 3) und 77,11 ± 1,58 % (pH 6).
Der pH-Wert beeinflusste die WFR nicht signifikant (p> 0,05). Ein Großteil der
Molekülverbindungen (46,06 ± 5,43 % (pH 11) – 63,14 ± 2,42 % (pH 6)) passierte die
Membran. Die Transmission der Molekülverbindungen durch die Membran unterschied sich
für pH 11 signifikant von den pH-Stufen 6 und 7 (p < 0,05). Eine Fraktionierung bei pH 11
führte zu einer Verringerung des Proteinanteils im Permeat (pH 11: 46,06 ± 5,43 %, pH 6:
63,14 ± 2,42 %, pH 7: 60,14 ± 3,99 %) bei gleichzeitigem Anstieg des Gehalts im Retentat (pH
11: 24,61 ± 3,92 %, pH 3: 12,47 ± 1,04 %, pH 6: 13,96 ± 0,8 %, pH 7: 13,85 ± 1,30 %).
Für den Fraktionierungsprozess unter Zusatz von 150 mM NaCl wurde Vergleichbares
beobachtet. Die Wiederfindungsrate lag zwischen 67,83 ± 12,66 % (pH 11) und 73,51 ± 1,44
% (pH 6). Bis zu 32,17 % (pH 11) der Protein-Einwaage gingen folglich im
Fraktionierungsprozess verloren. 44,23 ± 6,77 % (pH 11) - 57,15 ± 1,12 % (pH 7) des im Feed
vorliegenden Proteins passierten die Membran. Abermals unterschieden sich lediglich die
Permeat-Proteinanteile der pH-Stufen pH 7 (57,15 ± 1,12 %) und pH 11 (44,23 ± 6,77 %).
Die Erhöhung der Ionenstärke resultierte nur für pH 6 (WFR (ohne NaCl): 77,11 ± 1,58 %,
WFR (mit NaCl): 73,51 ± 1,44 %) in einer signifikant niedrigeren WFR (p < 0,05). Die WFR

Ergebnisse
126
liegt bei einer Fraktionierung unter NaCl-Zusatz generell geringfügig unter der WFR bei einer
Fraktionierung ohne NaCl-Zusatz (ausgenommen pH 3).
Tabelle 33: Wiederfindungsrate und Anteile der Retentat- und Permeat-Fraktion bei der Fraktionierung von hydrolysierter Alge (Chlorella vulgaris)
pH n
Ohne NaCl-Zugabe Mit NaCl-Zugabe
WFR
MW (%)
WFRRet MW
(%)
WFRPer MW
(%)
WFR MW
(%)
WFRRet MW
(%)
WFRPer MW
(%)
3 3 68,64 ±
4,18 a, 1
12,47 ±
1,04 b, c, 1
56,18 ±
3,20 a, c, 1
70,96 ±
5,26 a, 1
14,99 ±
1,32 a, 1
55,98 ±
4,25 a, c, 1
6 3 77,11 ±
1,58 a, 1
13,96 ±
0,89 b, c, 1
63,14 ±
2,42 b, c, 1
73,51 ±
1,44 a, 2
17,54 ±
2,41 a, 1
55,96 ±
2,58 a, c, 2
7 3 73,99 ±
5,19 a, 1
13,85 ±
1,30 b, c, 1
60,14 ±
3,99 b, c, 1
72,03 ±
1,15 a, 1
14,89 ±
1,98 a, 1
57,15 ±
1,12 b, c, 1
8 3 73,16 ±
4,00 a, 1
18,95 ±
6,22 a, c, 1
54,20 ±
5,08 a, c, 1
69,54 ±
6,55 a, 1
16,07 ±
1,78 a, 1
53,47 ±
5,36 a, c, 1
11 3 70,67 ±
9,12 a, 1
24,61 ±
3,92 a, 1
46,06 ±
5,43 a, 1
67,83 ±
12,66 a, 1
23,61 ±
5,91 a, 1
44,23 ±
6,77 a, 1
Werte in einer Spalte mit unterschiedlichen Buchstaben a, b, c unterschieden sich signifikant (p < 0,05). Werte in einer Zeile mit unterschiedlichen Zahlen 1,2 unterschieden sich signifikant (p < 0,05).
Weitere Daten zur Charakterisierung des Filtrationsprozesses:
Zur Charakterisierung des Filtrationsprozesses wurde die Permeatmenge VPer zum Zeitpunkt
t über die Prozesszeit tF aufgetragen und die Regressionsfunktion der Permeatzunahme,
sowie deren Bestimmtheitsmaß R2 ermittelt. Darüber hinaus wurde der mittlere pH-Wert des
Retentats ermittelt, um etwaige pH-Wert-abhängige Effekte beurteilen zu können. Die
Ergebnisse sind in Tabelle 34 dargestellt.

Ergebnisse
127
Tabelle 34: Bestimmtheitsmaß R2 und pH-Mittelwert, -Minima und -Maxima der Fraktionierung von hydrolysierter Alge (Chlorella vulgaris)
pH n
Ohne NaCl-Zugabe Mit NaCl-Zugabe
R2 pH
R2 pH
MW Min Max MW Min Max
3 3 0,984 ±
0,002
3,11 ±
0,03
3,01 ±
0,01
3,12 ±
0,04
0,983 ±
0,003
3,21 ±
0,09
3,00 ±
0,01
3,25 ±
0,11
6 3 0,991 ±
0,004
6,23 ±
0,06
6,02 ±
0,03
6,41 ±
0,08
0,986 ±
0,008
6,27 ±
0,09
6,02 ±
0,01
6,45 ±
0,13
7 3 0,998 ±
0,003
7,13 ±
0,08
7,02 ±
0,01
7,29 ±
0,11
0,990 ±
0,003
7,11 ±
0,06
7,01 ±
0,00
7,18 ±
0,12
8 3 0,986 ±
0,002
7,84 ±
0,26
7,60 ±
0,34
8,01 ±
0,04
0,984 ±
0,005
7,73 ±
0,14
7,51 ±
0,24
8,02 ±
0,01
11 3 0,980 ±
0,008
11,00 ±
0,04
10,96
± 0,04
11,05
± 0,02
0,964 ±
0,012
10,96
± 0,01
10,92 ±
0,02
11,01 ±
0,01
Legende: Bestimmtheitsmaß, R2. Arithmetischer Mittelwert, MW. Minimum, Min. Maximum, Max.
Die Zunahme der Permeatmenge VPer entsprach, unabhängig vom pH-Wert, sowohl bei einer
Fraktionierung unter Zugabe von NaCl als auch ohne NaCl-Zusatz den Kriterien einer linearen
Funktion, gemessen am Bestimmtheitsmaß R2 der Regressionsgeraden (Min (ohne NaCl):
0,980 ± 0,008 (pH 11), Max (ohne NaCl): 0,998 ± 0,003 (pH 7); Min (mit NaCl): 0,964 ± 0,012
(pH 11), Max (mit NaCl): 0,990 ± 0,003 (pH 7)).
Der pH-Wert des Retentats blieb, unabhängig von der untersuchten pH-Stufe, über die Dauer
der Filtration für die Fraktionierung mit und ohne NaCl-Zusatz weitestgehend konstant. Die
größte Abweichung zwischen pH-Minima und pH-Maxima wurde bei pH 8 festgestellt (∆pH
(ohne NaCl): 0,41, ∆pH (mit NaCl): 0,51).
Molekülgrößenverteilung der Fraktionen:
Zur Beurteilung der Güte der Fraktionierung wurden SE-HPLC Analysen der Retentat- und
Permeatproben durchgeführt. Die Ergebnisse der Analysen sind nachfolgend dargestellt.
Abbildung 85 - Abbildung 89 zeigen die Chromatogramme nicht-fraktionierter Algen-
Hydrolysat-Proben (Nullproben) nach der Vorfiltration. Die Nullproben wiesen pH- und
ionenstärke-abhängige Unterschiede auf.
Bei extremen pH-Werten (pH 3, pH 11) ohne Zusatz von NaCl resultierte die Vorfiltration in
einer Abtrennung großer Moleküle, sodass für pH 3 und pH 11 nur ein geringer Anteil an
Molekülen ≥ 10 kDa bzw. ≥ 20 kDa im Feed verblieben (Abbildung 85 a, Abbildung 89 a). Auch
bei pH 8 wurden Moleküle ≥ 20 kDa durch die Vorfiltration reduziert, jedoch weitaus wenig

Ergebnisse
128
drastisch als für die untersuchten Extrem-pH-Werte (Abbildung 88 a). Die Nullproben der pH-
Stufen 6 und 7 verfügen über eine breite Verteilung unterschiedlicher Molekülstrukturen
(Abbildung 86 a, Abbildung 87 a).
Durch Zusatz von NaCl konnte der Anteil an Molekülen ≥ 20 kDa für pH 3 (Abbildung 85 b)
und pH 8 (Abbildung 88 b) deutlich erhöht werden. Lediglich bei pH 9 (Abbildung 89 (b) wurde
weiterhin ein Großteil der Strukturen ≥ 10 kDa bzw. ≥ 20 kDa aus dem Feed abgetrennt.

Ergebnisse
129
Abbildung 85: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die Fraktionierung von hydrolysierter Alge (Chlorella vulgaris), pH 3, a) ohne NaCl b) mit NaCl, qualitative Darstellung
Abbildung 86: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die Fraktionierung von hydrolysierter Alge (Chlorella vulgaris), pH 6, a) ohne NaCl b) mit NaCl, qualitative Darstellung
a) Alge (Chlorella vulgaris), pH 3, Nullprobe, ohne NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
b) Alge (Chlorella vulgaris), pH 3, Nullprobe, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa 670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
a) Alge (Chlorella vulgaris), pH 6, Nullprobe, ohne NaCl
b) Alge (Chlorella vulgaris), pH 6, Nullprobe, mit NaCl

Ergebnisse
130
Abbildung 87: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die Fraktionierung von hydrolysierter Alge (Chlorella vulgaris), pH 7, a) ohne NaCl b) mit NaCl, qualitative Darstellung
Abbildung 88: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die Fraktionierung von hydrolysierter Alge (Chlorella vulgaris), pH 8, a) ohne NaCl b) mit NaCl, qualitative Darstellung
a) Alge (Chlorella vulgaris), pH 8, Nullprobe, ohne NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
b) Alge (Chlorella vulgaris), pH 8, Nullprobe, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
a) Alge (Chlorella vulgaris), pH 7, Nullprobe, ohne NaCl
b) Alge (Chlorella vulgaris), pH 7, Nullprobe, mit NaCl
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
qu
alit
ativ
670 kDa
158 kDa
44 kDa
1,35 kDa
670 kDa
158 kDa
44 kDa
1,35 kDa

Ergebnisse
131
Abbildung 89: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Nullprobe für die Fraktionierung von hydrolysierter Alge (Chlorella vulgaris), pH 11, a) ohne NaCl b) mit NaCl, qualitative Darstellung
a) Alge (Chlorella vulgaris), pH 11, Nullprobe, ohne NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
b) Alge (Chlorella vulgaris), pH 11, Nullprobe, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
qu
alit
ativ

Ergebnisse
132
In Abbildung 90 - Abbildung 94 sind die Chromatogramme von Retentat- und Permeat-
Fraktionen des Algen-Hydrolysats dargestellt.
Bei der Betrachtung der Chromatogramme der Retentat-Fraktion wird, weitestgehend
unabhängig vom pH-Wert, deutlich, dass neben Molekülverbindungen im Zielbereich auch
eine große Menge an Strukturen ≤ 1,35 kDa vorhanden sind (Abbildung 90 a, c - Abbildung 94
a, c). Diese konnten trotz der Verwendung des Diafiltrationsverfahrens auch im Falle der
Fraktionierung hydrolysierter Alge nicht aus der Retentat-Fraktion ausgewaschen werden.
Des Weiteren sind Molekülstrukturen ≥ 20 kDa bei Fraktionierungen ohne NaCl-Zusatz stark
unterrepräsentiert, obwohl in den Nullproben, mit Ausnahme von pH 3 und pH 11, Strukturen
≥ 158 kDa nachgewiesen werden konnten (Abbildung 90 a - Abbildung 94 a).
In den unter NaCl-Zusatz bei erhöhter Ionenstärke gewonnenen Retentat-Fraktionen konnten
insbesondere bei pH 3 (Abbildung 90 c) und pH 6 (Abbildung 91 c), sowie in geringem Umfang
auch bei pH 8 (Abbildung 93 c) Molekülverbindungen ≥ 20 kDa konzentriert werden.
Anhand der Permeat-Fraktionen ist sowohl für die Fraktionierung unter Zusatz von 150 mM
NaCl, als auch für die Fraktionierung ohne NaCl-Zusatz eine deutliche Verschiebung der
Membrantrenngrenze hin zu einem niedrigeren MWCO erkennbar (Abbildung 90 b, d -
Abbildung 94 b, d). Molekülverbindungen ≥ 5 – 10 kDa sind unabhängig von pH-Wert und
Ionenstärke nicht, oder nur in verschwindend kleinen Anteilen nachweisbar (pH 7: Abbildung
92 d, pH 8: Abbildung 93 d). Molekülstrukturen ≤ 1,35 kDa sind stark repräsentiert.

Ergebnisse
133
Abbildung 90: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und Permeatfraktion von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit der Ionenstärke, pH 3, a) Retentat ohne NaCl b) Permeat ohne NaCl c) Retentat mit NaCl d) Permeat mit NaCl, qualitative Darstellung
a) Alge (Chlorella vulgaris), pH 3, Retentat, ohne NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
b) Alge (Chlorella vulgaris), pH 3, Permeat, ohne NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
c) Alge (Chlorella vulgaris), pH 3, Retentat, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
d) Alge (Chlorella vulgaris), pH 3, Permeat, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ

Ergebnisse
134
Abbildung 91: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und Permeatfraktion von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit der Ionenstärke, pH 6, a) Retentat ohne NaCl b) Permeat ohne NaCl c) Retentat mit NaCl d) Permeat mit NaCl, qualitative Darstellung
a) Alge (Chlorella vulgaris), pH 6, Retentat, ohne NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
b) Alge (Chlorella vulgaris), pH 6, Permeat, ohne NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
c) Alge (Chlorella vulgaris), pH 6, Retentat, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
d) Alge (Chlorella vulgaris), pH 6, Permeat, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ

Ergebnisse
135
Abbildung 92: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und Permeatfraktion von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit der Ionenstärke, pH 7, a) Retentat ohne NaCl b) Permeat ohne NaCl c) Retentat mit NaCl d) Permeat mit NaCl, qualitative Darstellung
670 kDa
158 kDa
44 kDa
1,35 kDa
b) Alge (Chlorella vulgaris), pH 7, Permeat, ohne NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
c) Alge (Chlorella vulgaris), pH 7, Retentat, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
d) Alge (Chlorella vulgaris), pH 7, Permeat, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
a) Alge (Chlorella vulgaris), pH 7, Retentat, ohne NaCl
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ

Ergebnisse
136
Abbildung 93: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und Permeatfraktion von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit der Ionenstärke, pH 8, a) Retentat ohne NaCl b) Permeat ohne NaCl c) Retentat mit NaCl d) Permeat mit NaCl, qualitative Darstellung
670 kDa
158 kDa
44 kDa
1,35 kDa
b) Alge (Chlorella vulgaris), pH 8, Permeat, ohne NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
c) Alge (Chlorella vulgaris), pH 8, Retentat, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
d) Alge (Chlorella vulgaris), pH 8, Permeat, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
a) Alge (Chlorella vulgaris), pH 8, Retentat, ohne NaCl
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
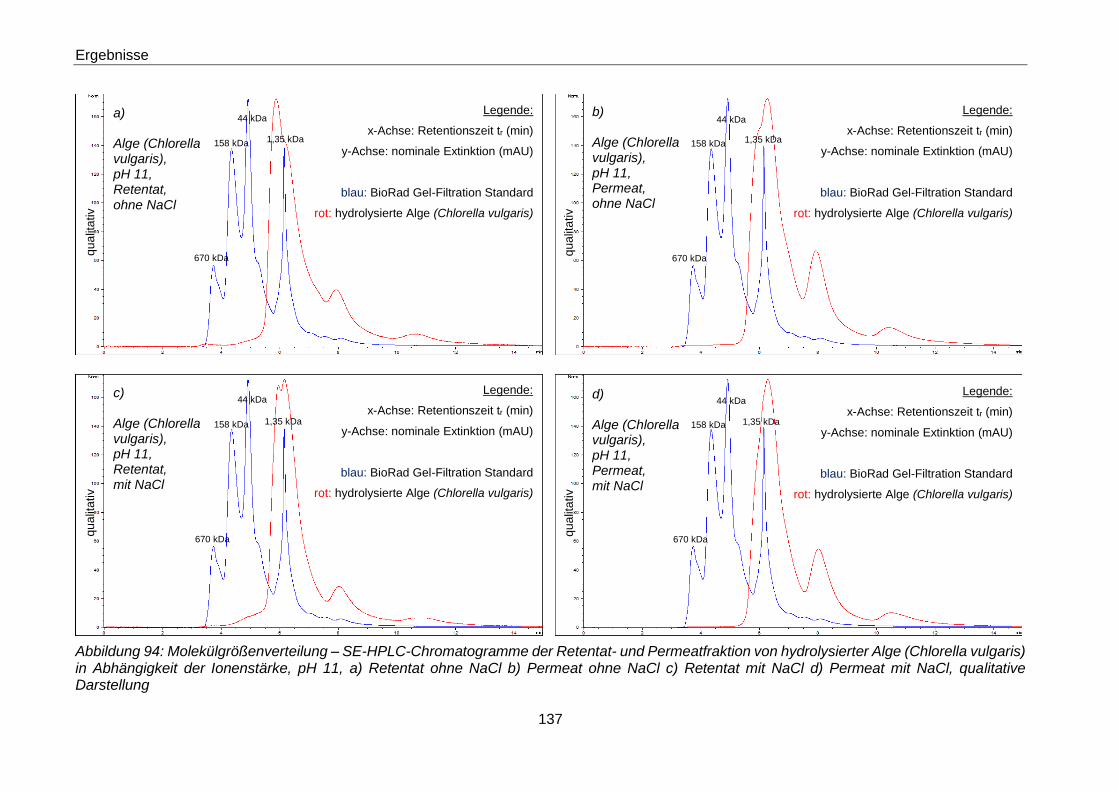
Ergebnisse
137
Abbildung 94: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und Permeatfraktion von hydrolysierter Alge (Chlorella vulgaris) in Abhängigkeit der Ionenstärke, pH 11, a) Retentat ohne NaCl b) Permeat ohne NaCl c) Retentat mit NaCl d) Permeat mit NaCl, qualitative Darstellung
670 kDa
158 kDa
44 kDa
1,35 kDa
b) Alge (Chlorella vulgaris), pH 11, Permeat, ohne NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
c) Alge (Chlorella vulgaris), pH 11, Retentat, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
d) Alge (Chlorella vulgaris), pH 11, Permeat, mit NaCl
670 kDa
158 kDa
44 kDa
1,35 kDa
a) Alge (Chlorella vulgaris), pH 11, Retentat, ohne NaCl
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysierte Alge (Chlorella vulgaris)
qu
alit
ativ

Ergebnisse
138
4.3.3 Einfluss der Ultraschall-Anwendung
Die Verwendung von Ultraschall hat sich bei der Optimierung von Filtrationsprozessen intakter
Proteine, besonders bei Crossflow-Systemen, als vielversprechend erwiesen. Die
fluxsteigernde Wirkung des Ultraschalls wird dabei auf eine Reduktion von
Konzentrationspolarisation und eine Abschwächung von Fouling durch Aufbrechen der
Deckschicht zurückgeführt (Kobayashi et al., 1999; Lamminen, Walker, & Weavers, 2004;
Muthukumaran et al., 2004).
In dieser Studie wurde der Einfluss von Ultraschall (35 kHz) auf den Prozessparameter
Permeatflux JFm, sowie auf die Trennschärfe der Fraktionierung für hydrolysiertes
Molkenprotein-Isolat untersucht. Abweichend zu den Hauptversuchen wurden für diese
Versuchsreihe zwei mit Polyethersulfon (PES) Membranen ausgestattete Crossflow-
Filtrationskammer (Vivaflow 50, Sartorius AG, Göttingen, GER) mit einer Trenngrenze von 100
kDa verwendet. Die Konzentration des vorfiltrierten Feeds (pH 7) aus hydrolysiertem
Molkenprotein-Isolat (Thermax 690, Glanbia Nutritionals Ltd., IRL) lag bei 0,30 % (w/v).
Versuchsaufbau und Durchführung sind in Kapitel 3.2.2.2 (Seite 56) ausführlich beschrieben.
Die Ergebnisse der Versuche sind in Tabelle 35 und Abbildung 95 – Abbildung 97 dargestellt.
Permeatflux JFm:
Tabelle 35: Laufzeit tFm, Permeatflux JFm und absolute Proteingehalte der Retentat- und Permeat-Fraktion von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit der Anwendung von Ultraschall
Probe n Laufzeit tFm MW (min)
Flux JFm MW
(ml/min) mRet MW (mg) mPer MW (mg)
Fraktionierung ohne Ultraschall
6 67,02 ± 15,55 a
6,38 ± 1,61 a
94,63 ± 9,14 a 183,78 ± 17,13 a
Fraktionierung mit Ultraschall
6 77,71 ± 27,40 a
5,81 ± 2,36 a
89,50 ± 10,03 a 187,24 ± 19,96 a
Werte in einer Spalte mit unterschiedlichen Buchstaben a, b unterschieden sich signifikant (p < 0,05).
Die Anwendung von Ultraschall resultierte in einem geringeren Permeatflux JFm bei gleichzeitig
hoher Standardabweichung (JFmUS: 5,81 ± 2,36 ml/min; JFmOUS: 6,38 ± 1,61 ml/min) (Abbildung
95). Eine signifikanter Unterschied hinsichtlich des Permeatflux konnte zwischen den
Fraktionierungsprozessen mit und ohne Ultraschall-Anwendung nicht festgestellt werden (p >
0,05).

Ergebnisse
139
Abbildung 95: Permeatflux JFm von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit einer Ultraschall-Anwendung
Proteingehalt von Retentat mRet & Permeat mPer:
Retentat- und Permeat-Fraktion unterschieden sich hinsichtlich der Proteingehalte der
Fraktionen mRet/mPer nicht signifikant (p > 0,05) (mRetUS: 89,50 ± 10,03 mg; mRetOUS: 94,63 ±
9,14 mg; mPerUS: 187,24 ± 19,96 mg; mPerOUS: 183,78 ± 17,13 mg). Im Falle der Ultraschall-
Anwendung wurden geringfügig niedrigere Proteingehalte im Retentat und geringfügig höhere
Proteingehalte im Permeat festgestellt (Abbildung 96, Abbildung 97).
Abbildung 96: Absolute Proteinmenge der Retentat-Fraktionen von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit einer Ultraschall-Anwendung
Abbildung 97: Absolute Proteinmenge der Permeat-Fraktionen von hydrolysiertem Molkenprotein-Isolat n in Abhängigkeit einer Ultraschall-Anwendung
Wiederfindungsrate:
Die Wiederfindungsrate, dargestellt in Tabelle 36, lag bei der Fraktionierung von
Molkenprotein-Isolat-Hydrolysat, in Abhängigkeit einer Ultraschall-Anwendung, zwischen
6,385,81
0 1 2 3 4 5 6 7 8 9
Flux (ml/min)
Flux (ml/min) - mit US Flux (ml/min) - ohne US
94,6389,50
0 10 20 30 40 50 60 70 80 90 100 110
Proteingehalt (mg)
Protein Retentat (mg) - mit US Protein Retentat (mg) - ohne US
183,78187,24
0 20 40 60 80 100 120 140 160 180 200 220
Proteingehalt (mg)
Protein Permeat (mg) - mit US Protein Permeat (mg) - ohne US

Ergebnisse
140
92,24 ± 7,65 % (mit US) und 92,80 ± 8,39 % (ohne US). 29,83 ± 3,34 % (mit US) - 31,54 ±
3,05 (ohne US) des Proteinanteils lagen im Retentat, 61,26 ± 5,71 % (ohne US) - 62,41 ± 6,65
% (mit US) im Permeat vor.
Tabelle 36: Wiederfindungsrate und Anteile der Retentat- und Permeat-Fraktion bei der Fraktionierung von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit der Anwendung von Ultraschall
Probe n WFR MW (%) WFRRet MW (%) WFRPer MW (%)
Fraktionierung mit Ultraschall 6 92,24 ± 7,65 a 29,83 ± 3,34 a 62,41 ± 6,65 a
Fraktionierung ohne Ultraschall 6 92,80 ± 8,39 a 31,54 ± 3,05 a 61,26 ± 5,71 a
Werte in einer Spalte mit unterschiedlichen Buchstaben a, b unterschieden sich signifikant (p < 0,05).
Molekülgrößenverteilung der Fraktionen:
Zur Beurteilung der Güte der Fraktionierung wurden SE-HPLC Analysen der Retentat- und
Permeatproben durchgeführt. Die Ergebnisse der Analysen sind in Abbildung 98 dargestellt.
Die Chromatogramme der Retentat- und Permeatfraktionen wiesen in Abhängigkeit der
Ultraschall-Anwendung keine deutlichen Unterschiede auf. In den Retentat-Fraktionen (rot)
wurden Molekülverbindungen ≥ 20 kDa in beiden Fällen aufkonzentriert. In den Permeat-
Fraktionen (grün) konnte eine Trenngrenzverschiebung des MWCO der Membran hin zu
einem deutlich niedrigeren MWCO beobachtet werden, sodass nur eine geringe
Molekülmenge ≥ 10 kDa in den Permeat-Fraktionen verblieb.
Strukturen ≤ 1,35 kDa sind in beiden Fraktionen stark repräsentiert und stellen, besonders in
den Permeatfraktionen, eine Großteil der Fraktion dar.
Die Performance der Vivaflow-Filtrationskammern veränderte sich im Versuchsprozess nicht
(beispielhaft dargestellt anhand der Chromatogramme von Versuchswiederholung 1 und
Versuchswiederholung 5; Abbildung 98).

Ergebnisse
141
Abbildung 98: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und Permeatfraktion von hydrolysiertem Molkenprotein-Isolat in Abhängigkeit der Anwendung von Ultraschall, pH 7, a) Retentat & Permeat ohne US, Wdh. 1 b) Retentat & Permeat mit US, Wdh. 1 c) Retentat & Permeat ohne US, Wdh. 5 d) Retentat & Permeat mit US, Wdh. 5, qualitative Darstellung
670 kDa
44 kDa
1,35 kDa
b) Molkenprotein-Isolat, pH 7, ohne US, Whd. 5
670 kDa
158 kDa
44 kDa
1,35 kDa
c) Molkenprotein-Isolat, pH 7, mit US, Wdh. 1
670 kDa
158 kDa
44 kDa
1,35 kDa
d) Molkenprotein, pH 7, mit US, Wdh. 5
670 kDa
158 kDa
44 kDa
1,35 kDa
a) Molkenprotein-Isolat, pH 7, ohne US, Whd. 1
158 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Retentat)
grün: hydrolysiertes Molkenprotein
(Permeat)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Retentat)
grün: hydrolysiertes Molkenprotein
(Permeat)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Retentat)
grün: hydrolysiertes Molkenprotein
(Permeat)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Retentat)
grün: hydrolysiertes Molkenprotein
(Permeat)
qu
alit
ativ

Ergebnisse
142
4.3.4 Eignung von Membranen aus regenerierter Cellulose
Neben synthetischen Polyethersulfon Membranen finden auch Filtermedien aus regenerierter
Cellulose Anwendung in der Proteinaufreinigung (Walter et al., 2012). In einem Pilotversuch
wurde untersucht, ob die Nutzung von RC-Membranen in einer Verbesserung der
Trennleistung resultiert.
Abweichend zu den Hauptversuchen wurde für diese Versuchsreihe eine mit Membranen aus
regenerierter Cellulose (RC) ausgestattete Crossflow-Filtrationskammer (Vivaflow 50,
Sartorius AG, Göttingen, GER) mit einer Trenngrenze von 100 kDa genutzt. Die Konzentration
des vorfiltrierten Feeds (pH 9) aus hydrolysiertem Molkenprotein-Isolat (Thermax 690, Glanbia
Nutritionals Ltd., IRL) lag bei 0,15 % (w/v). Versuchsaufbau und Durchführung sind in Kapitel
3.2.2.3 (Seite 58) beschrieben.
Die Ergebnisse der Versuche sind in Tabelle 37 und Abbildung 99 dargestellt.
Abbildung 99: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentat- und Permeatfraktion von hydrolysiertem Molkenprotein-Isolat unter Verwendung einer RC-Membran, pH 9, a) Retentat, b) Permeat, qualitative Darstellung
670 kDa
44 kDa
1,35 kDa
b) Molkenprotein-Isolat, pH 9, Permeat
670 kDa
158 kDa
44 kDa
1,35 kDa
a) Molkenprotein-Isolat, pH 9, Retentat
158 kDa
Molkenprotein-Isolat, pH 9, Nullprobe
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
qu
alit
ativ

Ergebnisse
143
Bei der Betrachtung der Chromatogramme der Retentat-Fraktion, dargestellt in Abbildung 99
a, wird abermals deutlich, dass neben Molekülverbindungen im Zielbereich auch eine große
Menge an Strukturen ≤ 1,35 kDa vorhanden sind. Ähnlich den Fraktionierungsprozessen unter
Verwendung von PS/PES-Membranen konnten in der Retentat-Fraktion Molekülverbindungen
≥ 20 kDa aufkonzentriert werden.
Anhand des Chromatogramms der Permeat-Fraktionen ist ebenfalls eine deutliche
Verschiebung der Membrantrenngrenze hin zu einem niedrigeren MWCO erkennbar
(Abbildung 99 b). Nur ein geringer Anteil an Verbindungen ≥ 20 kDa passierte die Membran.
Nähert man sich der nominalen Trenngrenze der Membran an, nimmt der Anteil weiter ab.
Molekülstrukturen ≤ 1,35 kDa sind in beiden Fraktionen stark repräsentiert.
Die Wiederfindungsrate lag bei 109,96 ± 17,00 %. 38,22 ± 7,28 % des Proteins lagen im
Retentat und 71,74 ± 10,27 % im Permeat vor. Die erhöhte WFR ist, unter Berücksichtigung
der Stammdaten, auf einen Ausreißer zurückzuführen, der den hier dargestellten Mittelwert
beeinflusst. Zudem können geringfügige Einwaagefehler aufgrund der geringen
Einwaagemenge von wenigen mg an dieser Stelle in einem erhöhten Endproteingehalt des
Feeds und somit höheren Proteingehalten in Retentat und Permeat resultieren.
Tabelle 37: Laufzeit tFm, Permeatflux JFm und absolute Proteingehalte der Retentat- und Permeat-Fraktion von hydrolysiertem Molkenprotein-Isolat
Probe n Laufzeit tFm
MW (min)
Flux JFm MW
(ml/min) mRet MW (mg) mPer MW (mg)
Fraktionierung –
RC Membran 3 48,85 ± 10,07 8,46 ± 1,60 57,33 ± 10,92 107,61 ± 15,41

Ergebnisse
144
4.3.5 Dialyse zur Abtrennung kleiner Moleküle (< 2 kDa) aus dem Feed
Um die Möglichkeit zu untersuchen, ob Moleküle < 2 kDa aus dem vorfiltrierten Feed
abgetrennt werden können, wurden Dialyseversuche unter Variation des Initial-pH-Werts,
sowie der Dialysedauer durchgeführt. Unter Initial-pH wird der pH-Wert der Dialyseprobe
verstanden, der zu Beginn des Dialyseprozess, sowie bei Wechsel der Dialyselösung
eingestellt wurde. Darüber hinaus sollte durch Überprüfung verschiedener Feed-
Konzentrationen festgestellt werden, ob die Dialyse zur Abtrennung kleiner Moleküle aus dem
vorfiltrierten Feed (Nullprobe) geeignet ist, da durch Adsorption von Molekülen in den Poren
der Membran die nominale Trenngrenze einer Membran herabgesetzt werden kann, sodass
zunehmend auch niedermolekulare Partikel zurückgehalten werden können (Wang &
Tarabara, 2008; Marshall et al., 1993). Die Dialyselosung „destilliertes Wasser“ wurde mit dem
Hintergrund gewählt, das Feed frei von den zur Herstellung von Dialysepuffern üblichen
Salzen, Aminen und ähnlichen Zusätzen mit Pufferwirkung zu halten, die eine Aufreinigung
des Retentats vor einem nachfolgenden Fraktionierungsprozess notwendig machen würden.
Tabelle 38: Absoluter Proteingehalt der Dialyseproben (Retentat) von hydrolysiertem Molkenprotein-Isolat vor (mvD) und nach (mnD) der Dialyse in Abhängigkeit von pH-Wert, Konzentration und Dialysedauer
pH n Dauer tDL
(h)
Konzentration CF
(%) mvD MW (mg) mnD MW (mg)
3 3 24 1 24,57 ± 0,29 c, 1 23,46 ± 0,80 a, 1
5 3 24 1 19,85 ± 0,38 a, 1 9,92 ± 0,27 c, 2
6 3 24 1 26,98 ± 0,73 b, 1 16,77 ± 0,38 b, 2
7,5 3 24 1 26,02 ± 0,49 b, c, 1 16,39 ± 0,74 b, 2
9 3 24 1 26,61 ± 0,50 b, 1 15,62 ± 0,57 b, 2
11 3 24 1 28,32 ± 1,06 b, 1 15,52 ± 0,36 b, 2
9 3 24 2 51,88 ± 0,39 a, α, 1 21,54 ± 1,71 a, α, 2
11 3 24 2 53,02 ± 0,13 b, α, 1 27,37 ± 1,04 b, α, 2
9 3 48 2 47,32 ± 1,27 a, β, 1 13,29 ± 0,68 a, β, 2
11 3 48 2 48,02 ± 1,48 a, β, 1 18,79 ± 1,41 b, β, 2
9 3 24 4 92,53 ± 0,24 a, 1 53,14 ± 2,97 a, 2
11 3 24 4 99,42 ± 0,72 b, 1 47,85 ± 1,22 b, 2
Werte einer Konzentration (%) & Dauer (h) in einer Spalte mit unterschiedlichen Buchstaben a, b, c unterschieden sich signifikant (p < 0,05). Werte einer Konzentration (%) und eines pH mit unterschiedlicher Dauer (h) mit unterschiedlichen Symbolen α, β unterschieden sich signifikant (p < 0,05). Werte in einer Zeile mit unterschiedlichen Zahlen 1,2 unterschieden sich signifikant (p < 0,05).
Die Ergebnisse der Studie sind in Tabelle 38, sowie Abbildung 101 – Abbildung 110 dargestellt.
Aufgrund von Peptid-Ausflockung bei Konzentrationen ≥ 2 % im Bereich pH 3 – pH 7,5 wurden

Ergebnisse
145
die Untersuchungen für Konzentrationen ≥ 2 % nur für die Initial-pH-Stufen 9 und 11
durchgeführt.
pH-Wert & Dialysezeit tDL:
Initial-pH-bedingt konnten bei einer Konzentration von 1 % (w/v) und einer Prozessdauer tDL
von 24 h signifikant unterschiedliche Proteingehalte der Nullprobe mvD (p < 0,05) für pH 3
(24,57 ± 0,29 mg) und pH 5 (19,85 ± 0,38 mg) im Vergleich zu den weiteren Initial-pH-Stufen
(für Vergleich pH 3: ausgenommen pH 7,5) festgestellt werden, wobei bei pH 5 (19,85 ± 0,38
mg) die geringste und bei pH 11 (28,32 ± 1,06 mg) die höchste Proteinmenge der Nullprobe
festgestellt wurde.
Für die Konzentrationen 2 % (w/v) und 4 % (w/v), 24 h wurden ebenfalls signifikant
unterschiedliche Proteingehalte des Feed „vor Dialyse“ (p < 0,05) in Abhängigkeit des Initial-
pH-Werts ermittelt (2% (w/v): pH 9: 51,88 ± 0,39 mg; pH 11: 53,02 ± 0,13 mg; 4 % (w/v): pH
9: 92,53 ± 0,24 mg; pH 11: 99,42 ± 0,72 mg).
In den Fraktionen „nach Dialyse“ wurden vergleichbare Effekte beobachtet. Der Proteingehalt
mnD unterschied sich für die pH-Werte pH 3 (23,46 ± 0,80 mg) und pH 5 (9,92 ± 0,27 mg)
signifikant von den weiteren Initial-pH-Stufen (p < 0,05). Hierbei wird deutlich, dass bei pH 3
beinahe die gesamte Proteinmenge im Retentat verblieb. Die geringste Proteinmenge „nach
Dialyse“ wurde bei pH 5 (9,92 ± 0,27 mg), die höchste bei pH 3 (23,46 ± 0,80 mg) festgestellt.
Eine Verdopplung der Dialysezeit auf 48 h führte für die Initial-pH Stufen 9 und 11, im Vergleich
zu 24 h, zu signifikant geringeren Proteingehalten mnD der Proben „nach Dialyse“ (p < 0,05)
(pH 9, 48 h: 13,29 ± 0,68 mg vs. pH 9, 24 h: 21,54 ± 1,71 mg; pH 11, 48 h: 18,79 ± 1,41 mg
vs. pH 11, 24 h: 27,37 ± 1,04 mg). Allerdings konnten hier bereits beim Vergleich der
Nullproben (mvD 24 h vs. mvD 48 h) signifikante Unterschiede (p < 0,05) festgestellt werden (pH
9, 48 h: 47,32 ± 1,27 mg; pH 9, 24 h: 51,88 ± 0,39 mg; pH 11, 48 h: 48,02 ± 1,48 mg; pH 11,
24 h: 53,02 ± 0,13 mg).
Für alle Initial-pH-Stufen und Konzentrationen (ausgenommen pH 3, 1 % (w/v)) führte die
Dialyse der Proben bei einer Prozessdauer von 24 h und 48 h zu einem signifikant niedrigerem
Proteingehalt mnD der Proben „nach Dialyse“ im Vergleich zu den Nullproben (p < 0,05).
Molekülgrößenverteilung der Fraktionen:
Die Chromatogramme ermöglichen eine nähere Beurteilung der Dialysevorgänge. Die
Nullprobe „vor Dialyse“ ist rot, das Retentat „nach Dialyse“ grün dargestellt. In Abbildung 101
- Abbildung 105 sind die Daten qualitativ, in Abbildung 106 - Abbildung 110 quantitativ
abgebildet.

Ergebnisse
146
Unabhängig vom Initial-pH-Wert führte der Dialyseprozess zu einer Abtrennung von Molekülen
≤ 1,35 kDa bei gleichzeitigem Rückhalt von Molekülen ≥ 20 kDa im Retentat. Dies wird
insbesondere aus den quantitativen Daten ersichtlich (Abbildung 106 - Abbildung 110). Im
Falle des Versuchs 1% (w/v), pH 3, 24 h wurde im Retentat „nach Dialyse“ ein über den
Ausgangsgehalt der Nullprobe erhöhter Anteil an Molekülstrukturen im Bereich > 20 kDa
festgestellt (Abbildung 106 a). Molekülverbindungen im Bereich ≤ 1,35 kDa sind pH-
unabhängig trotz einem MWCO von 2 kDa weiterhin in den dialysierten Probe vorhanden,
konnten jedoch deutlich reduziert werden (Abbildung 106 - Abbildung 110).
Die Abtrennung bzw. Reduzierung des Gehalts an Molekülen ≤ 2 kDa ist bis zu einer Feed-
Konzentration von 4 % möglich (vergleiche: Abbildung 107, Abbildung 108 & Abbildung 110).
Bei höherer Feed-Konzentration scheint die Abtrennung kleiner Moleküle (< 2 kDa) zudem
deutlicher ausgeprägt (vergleiche: Abbildung 107, Abbildung 108 & Abbildung 110).
Eine Steigerung der Dialysezeit von 24 h auf 48 h führte nicht zu einer weiteren Reduktion von
kleinen Molekülen (< 2 kDa) im Retentat „nach Dialyse“, sondern zu einer anteiligen
Verringerung von Molekülstrukturen > 2 kDa (vergleiche: Abbildung 109 & Abbildung 110).
Dies ist vermutlich auf eine Deckschichtbildung an der Membraninnenseite bei Dialysezeiten
> 24 h zurückzuführen die zur Ausflockung von Peptid-Verbindungen führte (Abbildung 100).
Die deutlich niedrigeren Proteingehalte mnD „nach Dialyse“ im Vergleich zum 24-stündigen
Dialyseprozess bestärken die Annahme (pH 9, 48 h: 13,29 ± 0,68 mg vs. pH 9, 24 h: 21,54 ±
1,71 mg; pH 11, 48 h: 18,79 ± 1,41 mg vs. pH 11, 24 h: 27,37 ± 1,04 mg).
Abbildung 100: Ausflockung einer Dialyseprobe (pH 9, 48 h)

Ergebnisse
147
Abbildung 101: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach Dialyse in Abhängigkeit des pH-Werts, a) pH 3, 1 %, 24 h, b) pH 5, 1 %, 24 h, c) pH 6, 1 %, 24 h, d) pH 7,5, 1 %, 24 h, qualitative Darstellung
670 kDa
44 kDa
1,35 kDa
a) Molkenprotein- Isolat 1 %, pH 3, 24 h
158 kDa
670 kDa
44 kDa
1,35 kDa
b) Molkenprotein- Isolat 1 %, pH 5, 24 h
158 kDa
670 kDa
44 kDa
1,35 kDa
c) Molkenprotein- Isolat 1 %, pH 6, 24 h
158 kDa
670 kDa
44 kDa
1,35 kDa
d) Molkenprotein- Isolat 1 %, pH 7,5, 24 h
158 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
alit
ativ

Ergebnisse
148
Abbildung 102: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach Dialyse in Abhängigkeit des pH-Werts, a) pH 9, 1 %, 24 h, b) pH 11, 1 %, 24 h, qualitative Darstellung
Abbildung 103: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach Dialyse in Abhängigkeit des pH-Werts, a) pH 9, 2 %, 24 h, b) pH 11, 2 %, 24 h, qualitative Darstellung
670 kDa
44 kDa
1,35 kDa
a) Molkenprotein- Isolat 1 %, pH 9, 24 h
158 kDa
670 kDa
44 kDa
1,35 kDa
b) Molkenprotein- Isolat 1 %, pH 11, 24 h
158 kDa
670 kDa
44 kDa
1,35 kDa
a) Molkenprotein- Isolat 2 %, pH 9, 24 h
158 kDa
670 kDa
44 kDa
1,35 kDa
b) Molkenprotein- Isolat 2 %, pH 11, 24 h
158 kDa
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
alit
ativ

Ergebnisse
149
Abbildung 104: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach Dialyse in Abhängigkeit des pH-Werts a) pH 9, 2 %, 48 h, b) pH 11, 2 %, 48 h, qualitative Darstellung
Abbildung 105: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach Dialyse in Abhängigkeit des pH-Werts, a) pH 9, 4 %, 24 h, b) pH 11, 4 %, 24 h, qualitative Darstellung
44 kDa
1,35 kDa
a) Molkenprotein- Isolat 2 %, pH 9, 48 h
158 kDa
44 kDa
1,35 kDa
b) Molkenprotein- Isolat 2 %, pH 11, 48 h 158 kDa
670 kDa 670 kDa
44 kDa
1,35 kDa
158 kDa
44 kDa
1,35 kDa
b) Molkenprotein- Isolat 4 %, pH 11, 24 h
158 kDa
670 kDa 670 kDa
a) Molkenprotein- Isolat 4 %, pH 9, 24 h
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
alit
ativ
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
alit
ativ

Ergebnisse
150
Abbildung 106: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach Dialyse in Abhängigkeit des pH-Werts, a) pH 3, 1 %, 24 h, b) pH 5, 1 %, 24 h, c) pH 6, 1 %, 24 h, d) pH 7,5, 1 %, 24 h, quantitative Darstellung
a) Molkenprotein- Isolat 1 %, pH 3, 24 h
b) Molkenprotein- Isolat 1 %, pH 5, 24 h
c) Molkenprotein- Isolat 1 %, pH 6, 24 h
d) Molkenprotein- Isolat 1 %, pH 7,5, 24 h
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
an
tita
tiv
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
an
tita
tiv
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
an
tita
tiv
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
an
tita
tiv
44 kDa
1,35 kDa
158 kDa
670 kDa
44 kDa
1,35 kDa
158 kDa
670 kDa
44 kDa
1,35 kDa
158 kDa
670 kDa
44 kDa
1,35 kDa
158 kDa
670 kDa
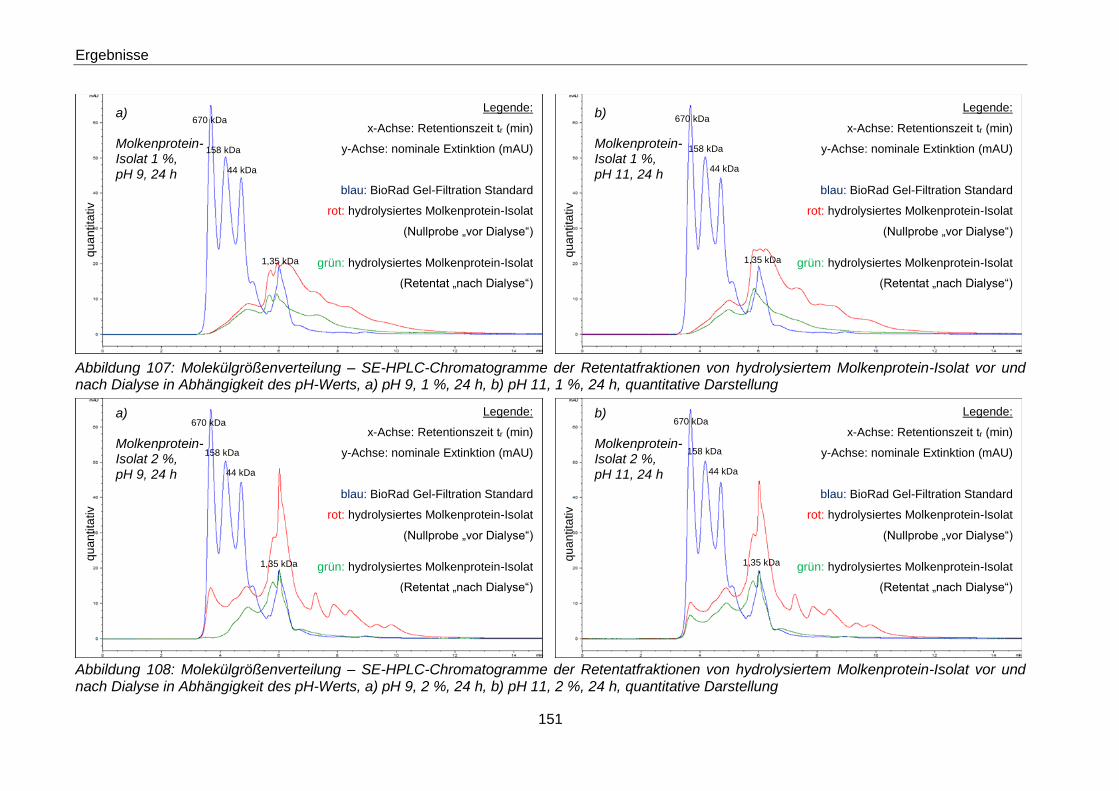
Ergebnisse
151
Abbildung 107: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach Dialyse in Abhängigkeit des pH-Werts, a) pH 9, 1 %, 24 h, b) pH 11, 1 %, 24 h, quantitative Darstellung
Abbildung 108: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach Dialyse in Abhängigkeit des pH-Werts, a) pH 9, 2 %, 24 h, b) pH 11, 2 %, 24 h, quantitative Darstellung
a) Molkenprotein- Isolat 1 %, pH 9, 24 h
b) Molkenprotein- Isolat 1 %, pH 11, 24 h
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
an
tita
tiv
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
an
tita
tiv
a) Molkenprotein- Isolat 2 %, pH 9, 24 h
b) Molkenprotein- Isolat 2 %, pH 11, 24 h
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
an
tita
tiv
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
an
tita
tiv
44 kDa
1,35 kDa
158 kDa
670 kDa
44 kDa
1,35 kDa
158 kDa
670 kDa
44 kDa
1,35 kDa
158 kDa
670 kDa
44 kDa
1,35 kDa
158 kDa
670 kDa

Ergebnisse
152
Abbildung 109: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach Dialyse in Abhängigkeit des pH-Werts a) pH 9, 2 %, 48 h, b) pH 11, 2 %, 48 h, quantitative Darstellung
Abbildung 110: Molekülgrößenverteilung – SE-HPLC-Chromatogramme der Retentatfraktionen von hydrolysiertem Molkenprotein-Isolat vor und nach Dialyse in Abhängigkeit des pH-Werts, a) pH 9, 4 %, 24 h, b) pH 11, 4 %, 24 h, quantitative Darstellung
a) Molkenprotein- Isolat 2 %, pH 9, 48 h
b) Molkenprotein- Isolat 2 %, pH 11, 48 h
b) Molkenprotein- Isolat 4 %, pH 11, 24 h
a) Molkenprotein- Isolat 4 %, pH 9, 24 h
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
an
tita
tiv
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
an
tita
tiv
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
an
tita
tiv
Legende:
x-Achse: Retentionszeit tr (min)
y-Achse: nominale Extinktion (mAU)
blau: BioRad Gel-Filtration Standard
rot: hydrolysiertes Molkenprotein-Isolat
(Nullprobe „vor Dialyse“)
grün: hydrolysiertes Molkenprotein-Isolat
(Retentat „nach Dialyse“)
qu
an
tita
tiv

Ergebnisse
153
4.4 Sensorik
Im Rahmen der Studie sollten die durch Crossflow-Diafiltration gewonnenen Fraktionen auf
vorhandene Geschmacksqualitäten („süß“, „sauer“, „salzig“, „bitter“, „umami“) überprüft
werden. Besonderes Augenmerk lag hierbei zudem auf einer den „bitter“-Geschmack
hemmenden Eigenschaft bestimmter Peptidfraktionen.
Nachfolgend sind die Ergebnisse der Sensorik Schulung, die daraus hervorgehende Auswahl
der Prüfer, sowie die Erkenntnisse der sensorische Bewertung der Peptidfraktionen
dargestellt. Die Auswahl der Prüfer basiert, gestützt durch statistische Verfahren, auf den
Ergebnisse der Skalenschulung. Ziel war die Zusammenstellung eines homogenen Panels für
jede Grundgeschmacksart. Aufgrund eines Abbruchs der Teilnahme an der Schulung erfolgt
die Auswertung für 23 Prüfpersonen.
4.4.1 Schulung des Sensorik-Panels
4.4.1.1 Prüfung auf Geschmacksblindheit (gemäß DIN EN ISO 8586:2014)
Die Prüfung auf Geschmacksblindheit diente zur Gewinnung einer Übersicht hinsichtlich des
Ist-Zustands des Panels und wurde nicht als Ausschlusskriterium gewertet. Gemäß DIN EN
ISO 8586:2014 müssen mindestens 80 % der Proben richtig erkannt werden, um als Prüfer
zugelassen zu werden (DIN Deutsches Institut für Normung e.V., 2014b). Die Ergebnisse der
Prüfung sind in Tabelle 39 dargestellt.
Das Prüfziel von 80 % richtigen Zuordnungen wurde von 22 der 23 Prüfer erreicht. In einem
Fall (Prüfer 7) wurden nur 70 % der Proben richtig erkannt. Zu fehlerhaften Zuordnungen kam
es überwiegend durch nicht-erkennen der „bitter“-Proben.
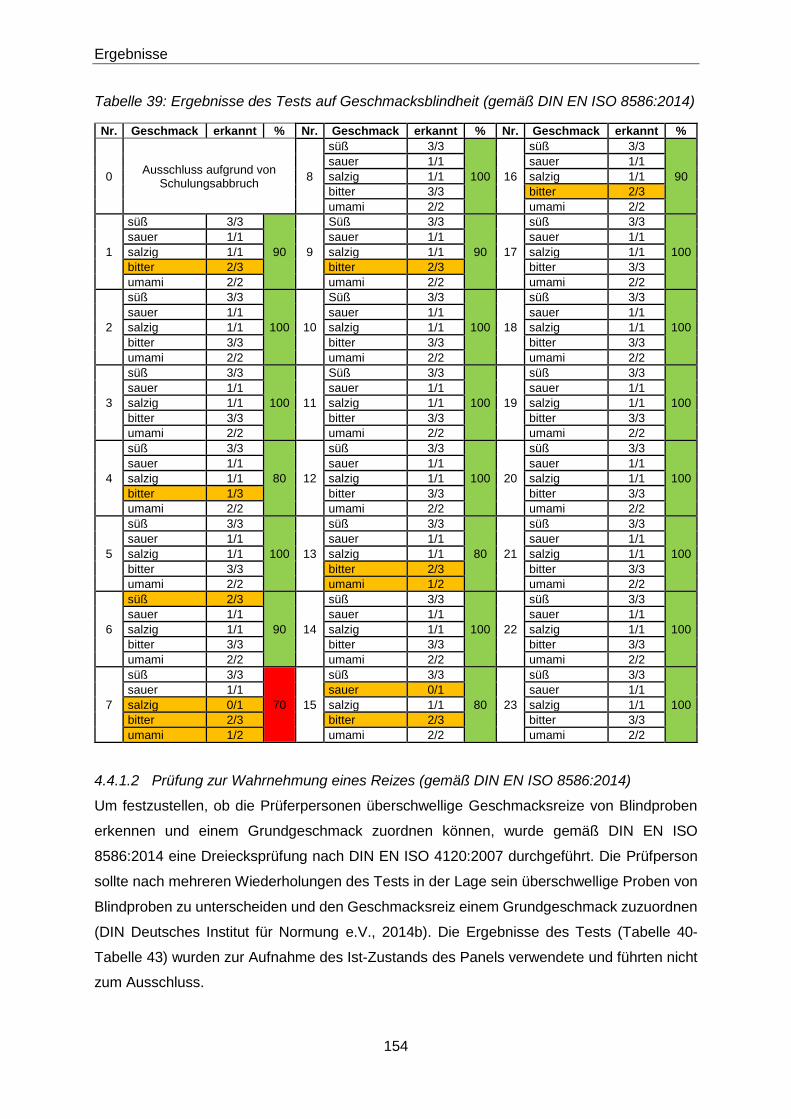
Ergebnisse
154
Tabelle 39: Ergebnisse des Tests auf Geschmacksblindheit (gemäß DIN EN ISO 8586:2014)
Nr. Geschmack erkannt % Nr. Geschmack erkannt % Nr. Geschmack erkannt %
0 Ausschluss aufgrund von
Schulungsabbruch 8
süß 3/3
100 16
süß 3/3
90
sauer 1/1 sauer 1/1
salzig 1/1 salzig 1/1
bitter 3/3 bitter 2/3
umami 2/2 umami 2/2
1
süß 3/3
90 9
Süß 3/3
90 17
süß 3/3
100
sauer 1/1 sauer 1/1 sauer 1/1
salzig 1/1 salzig 1/1 salzig 1/1
bitter 2/3 bitter 2/3 bitter 3/3
umami 2/2 umami 2/2 umami 2/2
2
süß 3/3
100 10
Süß 3/3
100 18
süß 3/3
100
sauer 1/1 sauer 1/1 sauer 1/1
salzig 1/1 salzig 1/1 salzig 1/1
bitter 3/3 bitter 3/3 bitter 3/3
umami 2/2 umami 2/2 umami 2/2
3
süß 3/3
100 11
Süß 3/3
100 19
süß 3/3
100
sauer 1/1 sauer 1/1 sauer 1/1
salzig 1/1 salzig 1/1 salzig 1/1
bitter 3/3 bitter 3/3 bitter 3/3
umami 2/2 umami 2/2 umami 2/2
4
süß 3/3
80 12
süß 3/3
100 20
süß 3/3
100
sauer 1/1 sauer 1/1 sauer 1/1
salzig 1/1 salzig 1/1 salzig 1/1
bitter 1/3 bitter 3/3 bitter 3/3
umami 2/2 umami 2/2 umami 2/2
5
süß 3/3
100 13
süß 3/3
80 21
süß 3/3
100
sauer 1/1 sauer 1/1 sauer 1/1
salzig 1/1 salzig 1/1 salzig 1/1
bitter 3/3 bitter 2/3 bitter 3/3
umami 2/2 umami 1/2 umami 2/2
6
süß 2/3
90 14
süß 3/3
100 22
süß 3/3
100
sauer 1/1 sauer 1/1 sauer 1/1
salzig 1/1 salzig 1/1 salzig 1/1
bitter 3/3 bitter 3/3 bitter 3/3
umami 2/2 umami 2/2 umami 2/2
7
süß 3/3
70 15
süß 3/3
80 23
süß 3/3
100
sauer 1/1 sauer 0/1 sauer 1/1
salzig 0/1 salzig 1/1 salzig 1/1
bitter 2/3 bitter 2/3 bitter 3/3
umami 1/2 umami 2/2 umami 2/2
4.4.1.2 Prüfung zur Wahrnehmung eines Reizes (gemäß DIN EN ISO 8586:2014)
Um festzustellen, ob die Prüferpersonen überschwellige Geschmacksreize von Blindproben
erkennen und einem Grundgeschmack zuordnen können, wurde gemäß DIN EN ISO
8586:2014 eine Dreiecksprüfung nach DIN EN ISO 4120:2007 durchgeführt. Die Prüfperson
sollte nach mehreren Wiederholungen des Tests in der Lage sein überschwellige Proben von
Blindproben zu unterscheiden und den Geschmacksreiz einem Grundgeschmack zuzuordnen
(DIN Deutsches Institut für Normung e.V., 2014b). Die Ergebnisse des Tests (Tabelle 40-
Tabelle 43) wurden zur Aufnahme des Ist-Zustands des Panels verwendete und führten nicht
zum Ausschluss.

Ergebnisse
155
Teilnehmer Geschmack Termin 1 Termin 2 Termin 3 % erkannt MW (%)
1
süß 1 1 1 100,00
60,00
sauer 0 1 1 66,67
salzig 1 1 1 100,00
bitter 0 0 0 0,00
umami 0 1 0 33,33
2
süß 1 1 1 100,00
80,00
sauer 0 0 1 33,33
salzig 1 1 1 100,00
bitter 1 1 1 100,00
umami 0 1 1 66,67
3
süß 1 1 1 100,00
100,00
sauer 1 1 1 100,00
salzig 1 1 1 100,00
bitter 1 1 1 100,00
umami 1 1 1 100,00
4
süß 1 1 1 100,00
66,67
sauer 0 0 1 33,33
salzig 1 1 1 100,00
bitter 0 0 0 0,00
umami 1 1 1 100,00
5
süß 1 1 0 66,67
80,00
sauer 1 1 1 100,00
salzig 1 1 1 100,00
bitter 1 1 1 100,00
umami 1 0 0 33,33
6
süß 1 0 1 66,67
66,67
sauer 1 1 1 100,00
salzig 1 1 1 100,00
bitter 0 0 1 33,33
umami 0 1 0 33,33
Tabelle 40: Ergebnisse des Test zur Prüfung der Wahrnehmung eines Reizes (gemäß DIN EN ISO 8586:2014)
Legende: 1, Ja (erkannt). 0, Nein (nicht erkannt). MW, arithmetischer Mittelwert.
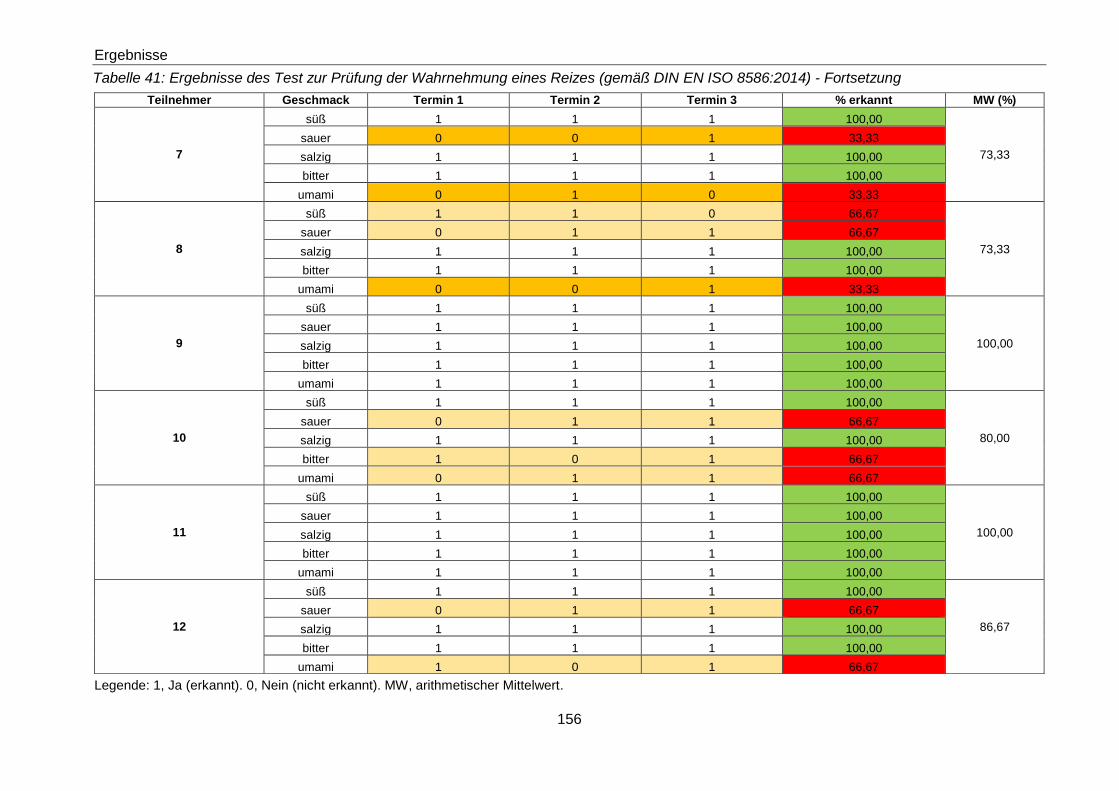
Ergebnisse
156
Teilnehmer Geschmack Termin 1 Termin 2 Termin 3 % erkannt MW (%)
7
süß 1 1 1 100,00
73,33
sauer 0 0 1 33,33
salzig 1 1 1 100,00
bitter 1 1 1 100,00
umami 0 1 0 33,33
8
süß 1 1 0 66,67
73,33
sauer 0 1 1 66,67
salzig 1 1 1 100,00
bitter 1 1 1 100,00
umami 0 0 1 33,33
9
süß 1 1 1 100,00
100,00
sauer 1 1 1 100,00
salzig 1 1 1 100,00
bitter 1 1 1 100,00
umami 1 1 1 100,00
10
süß 1 1 1 100,00
80,00
sauer 0 1 1 66,67
salzig 1 1 1 100,00
bitter 1 0 1 66,67
umami 0 1 1 66,67
11
süß 1 1 1 100,00
100,00
sauer 1 1 1 100,00
salzig 1 1 1 100,00
bitter 1 1 1 100,00
umami 1 1 1 100,00
12
süß 1 1 1 100,00
86,67
sauer 0 1 1 66,67
salzig 1 1 1 100,00
bitter 1 1 1 100,00
umami 1 0 1 66,67
Tabelle 41: Ergebnisse des Test zur Prüfung der Wahrnehmung eines Reizes (gemäß DIN EN ISO 8586:2014) - Fortsetzung
Legende: 1, Ja (erkannt). 0, Nein (nicht erkannt). MW, arithmetischer Mittelwert.

Ergebnisse
157
Teilnehmer Geschmack Termin 1 Termin 2 Termin 3 % erkannt MW (%)
13
süß 1 1 1 100,00
86,67
sauer 1 1 1 100,00
salzig 1 1 1 100,00
bitter 1 1 1 100,00
umami 0 0 1 33,33
14
süß 1 1 1 100,00
93,33
sauer 1 1 1 100,00
salzig 1 1 1 100,00
bitter 1 1 1 100,00
umami 1 0 1 66,67
15
süß 1 0 1 66,67
66,67
sauer 1 0 1 66,67
salzig 1 1 1 100,00
bitter 0 0 1 33,33
umami 1 0 1 66,67
16
süß 1 1 1 100,00
93,33
sauer 1 1 1 100,00
salzig 1 1 1 100,00
bitter 0 1 1 66,67
umami 1 1 1 100,00
17
süß 1 1 0 66,67
60,00
sauer 0 0 1 33,33
salzig 1 1 1 100,00
bitter 1 1 0 66,67
umami 0 0 1 33,33
18
süß 1 1 1 100,00
60,00
sauer 0 0 1 33,33
salzig 1 1 1 100,00
bitter 1 0 0 33,33
umami 1 0 0 33,33
Legende: 1, Ja (erkannt). 0, Nein (nicht erkannt). MW, arithmetischer Mittelwert.
Tabelle 42: Ergebnisse des Test zur Prüfung der Wahrnehmung eines Reizes (gemäß DIN EN ISO 8586:2014) - Fortsetzung

Ergebnisse
158
Teilnehmer Geschmack Termin 1 Termin 2 Termin 3 % erkannt MW (%)
19
süß 1 0 0 33,33
73,33
sauer 0 1 1 66,67
salzig 1 1 1 100,00
bitter 0 1 1 66,67
umami 1 1 1 100,00
20
süß 1 1 0 66,67
93,33
sauer 1 1 1 100,00
salzig 1 1 1 100,00
bitter 1 1 1 100,00
umami 1 1 1 100,00
21
süß 1 1 1 100,00
80,00
sauer 0 0 1 33,33
salzig 1 1 1 100,00
bitter 1 1 0 66,67
umami 1 1 1 100,00
22
süß 1 1 1 100,00
93,33
sauer 1 1 1 100,00
salzig 1 1 1 100,00
bitter 1 1 1 100,00
umami 1 0 1 66,67
23
süß 1 1 1 100,00
86,67
sauer 1 1 1 100,00
salzig 1 1 1 100,00
bitter 1 0 0 33,33
umami 1 1 1 100,00
Tabelle 43: Ergebnisse des Test zur Prüfung der Wahrnehmung eines Reizes (gemäß DIN EN ISO 8586:2014) - Fortsetzung
Legende: 1, Ja (erkannt). 0, Nein (nicht erkannt). MW, arithmetischer Mittelwert.

Ergebnisse
159
Zu mehrfach fehlerhaften Zuordnungen (33,33 % erkannt) kam es vor allem für die
Grundgeschmacksarten „umami“ (8 Fälle), „bitter“ (6 Fälle) und „sauer“ (6 Fälle). In einem Fall
wurde eine „süß“ Probe mehrfach nicht korrekt erkannt.
In Einzelfällen (66,67% erkannt) kam es für die Grundgeschmacksarten „umami“ (6 Fälle),
„süß“ (6 Fälle), „bitter“ (5 Fälle) und „sauer“ (5 Fälle) zu fehlerhaften Zuordnungen.
Tabelle 44: Gruppenauswertung des Test zur Prüfung der Wahrnehmung eines Reizes (gemäß DIN EN ISO 8586:2014)
Geschmack Mittelwert N (Prüfer)
„erkannt“
Mittelwert N (Prüfer)
„nicht erkannt“
Mittelwert
„% erkannt“
süß 19,33 ± 3,21 3,67 ± 3,21 84,06 ± 13,98
sauer 17,00 ± 5,57 6,00 ± 5,57 73,91 ± 24,21
salzig 23,00 ± 0,00 0,00 ± 0,00 100,00 ± 0,00
bitter 16,33 ± 1,15 6,67 ± 1,15 71,01 ± 5,02
umami 15,67 ± 2,08 7,33 ± 2,08 68,12 ± 9,05
Legende: N, Anzahl Prüfer.
Insgesamt erreichten 14 Prüfer ein Prüferergebnis ≥ 80 %, 3 Prüfer ≥ 73,33 % < 80 % und 6
Prüfer ≥ 60,00 % < 73,33 %. 3 Prüfer (3, 9, 11) konnten 100 % der Proben richtig zuordnen.
Die Grundgeschmacksarten „umami“, „bitter“ und „sauer“ bereiteten dem Panel die großten
Schwierigkeiten. Im Mittel wurden hier, dargestellt in Tabelle 44, 68,12 ± 9,05 % (umami) -
73,91 ± 24,21 % (sauer) der Prüflösungen korrekt wahrgenommen. Basierend auf
Rückmeldungen der Teilnehmer können die fehlerhaften Zuordnungen des Grundgeschmacks
„umami“ durch die Nähe zum Grundgeschmack „salzig“ herbeigeführt worden sein.
Die Wahrnehmung von „bitter“ verbesserte sich im Verlauf der drei Schulungstage nicht
wesentlich, wohingegen „umami“ und besonders „sauer“ am letzten Schulungstag zuverlässig
unterschieden werden konnten. Die Ergebnisse sind in Tabelle 45 dargestellt.
Tabelle 45: Anteil der Prüferpersonen die eine Geschmacksausprägung erkennen – Verlauf der Schulung - Tag 1 – Tag 3
Geschmack
Tag 1
„% erkannt“
/ N (Prüfer)
Tag 2
„% erkannt“
/ N (Prüfer)
Tag 3
„% erkannt“
/ N (Prüfer)
sauer 52,17 / 12 69,57 / 16 100,00 / 23
bitter 73,91 / 17 65,22 / 15 73,91 / 17
umami 65,22 / 15 60,87 / 14 78,26 / 18
Legende: N, Anzahl Prüfer.

Ergebnisse
160
Das Gesamt-Panel wies zu jedem Zeitpunkt der Schulung eine signifikante
Unterscheidungsfähigkeit (p < 0,05) hinsichtlich aller Grundgeschmacksarten auf. Hierzu sind
bei einem Panel mit 23 Prüfern mindestens 12 korrekte Antworten nötig.
4.4.1.3 Ermittlung der Reiz- und Erkennungsschwelle (gemäß ISO 3972:2011)
Um Kenntnisse bezüglich der Erkennungsschwellen (ES) der Prüfer zu gewinnen, sowie die
Unterscheidungsfähigkeit des gesamten Panels abzubilden, wurden Reiz- und
Erkennungsschwelle der Prüfer gemäß ISO 3972:2011 unter Verwendung einer einfachen
beschreibende Prüfung nach DIN 10964:2014 ermittelt. Die Erkenntnisse sind in Abbildung
111 – Abbildung 115 und Tabelle 46 –Tabelle 50 dargestellt.
Ein abweichender Geschmacksreiz wurde von den Prüfern, weitestgehend unabhängig von
der Grundgeschmacksart, bereits bei geringen Probenkonzentrationen festgestellt. Die
Reizschwelle lag, ausgenommen „süß“ und „umami“, zwischen Probe 1 und Probe 2
(Abbildung 111 b - Abbildung 115 b; Tabelle 46 - Tabelle 50). Prüferspezifische Unterschiede
resultierten jedoch bei allen Grundgeschmacksarten in deutlichen Standardabweichungen
(Abbildung 111 a, b - Abbildung 115 a, b). Eine oder mehrere Prüferpersonen waren folglich
nicht in der Lage auch bei erhöhten Konzentrationen einen Geschmacksreiz wahrzunehmen.
Dies wurde auch bei der Betrachtung der jeweiligen Erkennungsschwellen deutlich. Während
„sauer“ und „bitter“ relativ früh benannt werden konnten (zwischen Probe 2 und Probe 3),
hatten die Prüfer großere Schwierigkeiten „salzig“, „umami“ und besonders „süß“ zu
identifizieren.
Die Unterscheidungsfähigkeit verschiedener Konzentrationen ist ebenfalls stark
prüferabhängig. Während einige Prüfer bereits geringe Änderungen der Probenkonzentration
zuverlässig unterscheiden konnten, wurde bei anderen eine mangelnde
Diskriminierungsfähigkeit festgestellt (Tabelle A17 – Tabelle A19, Anhang, Seite 322 – 324).
Dies führte besonders bei „salzig“ und „umami“ zu deutlichen Standardabweichungen. „süß“
wurde panelübergreifend erst bei relativ hohen Konzentrationen identifiziert.

Ergebnisse
161
„süß“:
Abbildung 111: Grafische Darstellung der a) Prüfersensibilität, b) Reiz- und Erkennungsschwelle „süß“
Tabelle 46: Reiz- und Erkennungsschwelle "süß"
Reizschwelle
(Probe) Reizschwelle MW (g/l)
Bereich der RS (g/l) Erkennungsschwelle
(Probe)
Erkennungsschwelle (g/l) Bereich der ES (g/l)
Min Max Mittelwert Median Min Max
2,43 ± 1,77 0,70 ± 0,44 0,28 1,85 4,87 ± 1,66 2,41 ± 1,89 2,19 0,79 8,54
Legende: RS, Reizschwelle. ES, Erkennungsschwelle. MW, Mittelwert.
0
1
2
3
4
5
6
7
8
1 2 3 4 5 6 7 8
Bew
ertu
ng
Probe
0,0
2,0
4,0
6,0
8,0
10,0
12,0
14,0
1 2 3 4 5 6 7 8
Ko
nze
ntr
atio
n (
g/l)
Probe
Konzentrationsverlauf Reizschwelle Erkennungsschwelle
a) b)

Ergebnisse
162
„sauer“:
Abbildung 112: Grafische Darstellung der a) Prüfersensibilität, b) Reiz- und Erkennungsschwelle „sauer“
Tabelle 47: Reiz- und Erkennungsschwelle "sauer"
Reizschwelle
(Probe) Reizschwelle (g/l)
Bereich der RS (g/l) Erkennungsschwelle
(Probe)
Erkennungsschwelle (g/l) Bereich der ES (g/l)
Min Max Mittelwert Median Min Max
1,28 ± 0,98 0,14 ± 0,02 0,10 0,17 2,04 ± 1,17 0,16 ± 0,02 0,16 0,12 0,20
Legende: RS, Reizschwelle. ES, Erkennungsschwelle. MW, Mittelwert.
0
1
2
3
4
5
6
7
8
1 2 3 4 5 6 7 8
Bew
ertu
ng
Probe
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
1 2 3 4 5 6 7 8
Ko
nze
ntr
atio
n (
g/l)
Probe
Konzentrationsverlauf Reizschwelle Erkennungsschwelle
a) b)

Ergebnisse
163
„salzig“:
Abbildung 113: Grafische Darstellung der a)Prüfersensibilität, b) Reiz- und Erkennungsschwelle „salzig“
Tabelle 48: Reiz- und Erkennungsschwelle "salzig"
Reizschwelle
(Probe) Reizschwelle (g/l)
Bereich der RS (g/l) Erkennungsschwelle
(Probe)
Erkennungsschwelle (g/l) Bereich der ES (g/l)
Min Max Mittelwert Median Min Max
1,58 ± 1,21 0,20 ± 0,08 0,14 0,48 3,22 ± 1,56 0,36 ± 0,20 0,34 0,16 0,98
Legende: RS, Reizschwelle. ES, Erkennungsschwelle. MW, Mittelwert.
0
1
2
3
4
5
6
7
8
1 2 3 4 5 6 7 8
Bew
ertu
ng
Probe
0,0
0,5
1,0
1,5
2,0
2,5
1 2 3 4 5 6 7 8
Ko
nze
ntr
atio
n (
g/l)
Probe
Konzentrationsverlauf Reizschwelle Erkennungsschwelle
a) b)
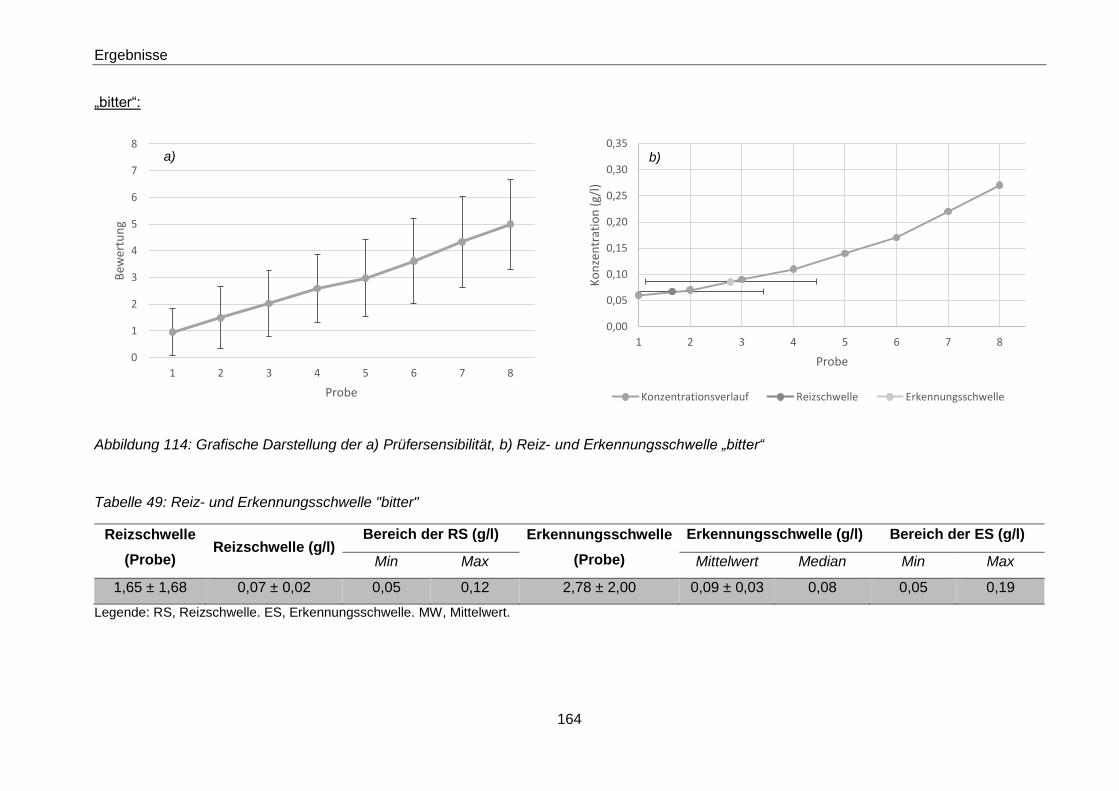
Ergebnisse
164
„bitter“:
Abbildung 114: Grafische Darstellung der a) Prüfersensibilität, b) Reiz- und Erkennungsschwelle „bitter“
Tabelle 49: Reiz- und Erkennungsschwelle "bitter"
Reizschwelle
(Probe) Reizschwelle (g/l)
Bereich der RS (g/l) Erkennungsschwelle
(Probe)
Erkennungsschwelle (g/l) Bereich der ES (g/l)
Min Max Mittelwert Median Min Max
1,65 ± 1,68 0,07 ± 0,02 0,05 0,12 2,78 ± 2,00 0,09 ± 0,03 0,08 0,05 0,19
Legende: RS, Reizschwelle. ES, Erkennungsschwelle. MW, Mittelwert.
0
1
2
3
4
5
6
7
8
1 2 3 4 5 6 7 8
Bew
ertu
ng
Probe
0,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
1 2 3 4 5 6 7 8
Ko
nze
ntr
atio
n (
g/l)
Probe
Konzentrationsverlauf Reizschwelle Erkennungsschwelle
a) b)

Ergebnisse
165
„umami“:
Abbildung 115: Grafische Darstellung der a) Prüfersensibilität, b) Reiz- und Erkennungsschwelle „umami“
Tabelle 50: Reiz- und Erkennungsschwelle "umami"
Reizschwelle
(Probe) Reizschwelle MW (g/l)
Bereich der RS (g/l) Erkennungsschwelle
(Probe)
Erkennungsschwelle (g/l) Bereich der ES (g/l)
Min Max Mittelwert Median Min Max
2,13 ± 1,62 0,12 ± 0,08 0,07 0,43 3,51 ± 2,00 0,20 ± 0,18 0,21 0,07 0,79
Legende: RS, Reizschwelle. ES, Erkennungsschwelle. MW, Mittelwert.
0
1
2
3
4
5
6
7
8
1 2 3 4 5 6 7 8
Bew
ertu
ng
Probe
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1 2 3 4 5 6 7 8
Ko
nze
ntr
atio
n (
g/l)
Probe
Konzentrationsverlauf Reizschwelle Erkennungsschwelle
a) b)

Ergebnisse
166
4.4.1.4 Prüfung zur Unterscheidung von Intensitätsstufen eines Reizes (gemäß DIN EN ISO
8586:2014)
Die Diskriminierungsfähigkeit zwischen Proben unterschiedlicher Konzentration wurde gemäß
DIN EN ISO 8586:2014 unter Anwendung eines Rangordnungstests nach DIN ISO 8587:2010
durchgeführt. Die Diskriminierungsfähigkeit der Prüfer wurde je Grundgeschmacksart mittels
Friedman-Test ermittelt (Tabelle 51). Im Falle signifikanter Ergebnisse wurden Wilcoxon-
Vorzeichen-Rangtests zur Ermittlung signifikanter Paardifferenzen durchgeführt (Tabelle 52)
(Quadt et al., 2009; Lawless & Heymann, 1999).
Tabelle 51: Diskriminierungsfähigkeit der Prüfer - Friedman-Test
Geschmack Chi-Quadrat p signifikant?
süß 156,830 0,000 Ja
sauer 160,874 0,000 Ja
salzig 172,254 0,000 Ja
bitter 65,566 0,000 Ja
umami 149,529 0,000 Ja

Ergebnisse
167
Tabelle 52: Diskriminierungsfähigkeit der Prüfer - Wilcoxon-Test
Geschmack Probenpaare Zb p signifikant?
süß
AB -1,923 0,054 Nein
AC -5,656 0,000 Ja
AD -7,320 0,000 Ja
BC -6,147 0,000 Ja
BD -7,625 0,000 Ja
CD -7,205 0,000 Ja
sauer
AB -4,275 0,000 Ja
AC -6,821 0,000 Ja
AD -7,494 0,000 Ja
BC -5,716 0,000 Ja
BD -7,415 0,000 Ja
CD -6,861 0,000 Ja
salzig
AB -5,498 0,000 Ja
AC -7,237 0,000 Ja
AD -7,481 0,000 Ja
BC -6,634 0,000 Ja
BD -7,495 0,000 Ja
CD -7,170 0,000 Ja
bitter
AB -1,848 0,065 Nein
AC -4,811 0,000 Ja
AD -5,804 0,000 Ja
BC -3,845 0,000 Ja
BD -4,990 0,000 Ja
CD -2,240 0,025 Ja
umami
AB -4,081 0,000 Ja
AC -7,043 0,000 Ja
AD -7,327 0,000 Ja
BC -5,281 0,000 Ja
BD -6,990 0,000 Ja
CD -5,832 0,000 Ja
Legende: b, basiert auf negativen Rängen
Friedman- und Wilcoxon-Test weißen auf eine gute Diskriminierungsfähigkeit des Panels
hinsichtlich aller Grundgeschmacksarten hin. Lediglich niedrige Konzentrationen der
Geschmacksausprägungen „süß“ und „bitter“ konnten nicht eindeutig voneinander
unterschieden werden. Bezieht man die mittleren Ränge der einzelnen Konzentrationen,
dargestellt in Tabelle 53, in die Bewertung mit ein wird klar, dass die Proben nicht nur
signifikant unterschiedlich wahrgenommen wurden, sondern auch betreffend ihrer
Konzentration korrekt gereiht werden konnten.

Ergebnisse
168
Tabelle 53: Diskriminierungsfähigkeit der Prüfer - Mittlerer Rang
Probe Mittlerer Rang
süß sauer salzig bitter umami
Probe A 1,56 1,31 1,25 1,75 1,32
Probe B 1,77 1,92 1,92 2,08 1,96
Probe C 2,72 2,86 2,89 2,83 2,91
Probe D 3,97 3,91 3,94 3,34 3,80
Legende: Konzentrationen, D > C > B > A.
Die Ergebnisse erlauben keine Aussage hinsichtlich der Homogenität der Bewertungen.
Betrachtet man die Minima und Maxima der jeweiligen Proben in der deskriptiven Teststatistik,
dargestellt in Tabelle 54, wird klar, dass ein oder mehrere Prüfer Probleme hatten, die korrekte
Rangordnung der Proben herzustellen.
Tabelle 54: Diskriminierungsfähigkeit der Prüfer – Deskriptive Teststatistik der Rangordnungstests
Geschmack Proben n Mittelwert Minimum Maximum
süß
A 68 1,71 ± 0,69 1 3
B 68 1,92 ± 0,42 1 3
C 68 2,72 ± 0,54 1 4
D 68 3,97 ± 0,17 3 4
sauer
A 68 1,29 ± 0,55 1 3
B 68 1,90 ± 0,58 1 4
C 68 2,85 ± 0,63 1 4
D 68 3,94 ± 0,24 3 4
salzig
A 68 1,32 ± 0,50 1 3
B 68 1,97 ± 0,42 1 3
C 68 2,90 ± 0,43 1 4
D 68 3,94 ± 0,38 1 4
bitter
A 68 1,75 ± 0,87 1 4
B 68 2,06 ± 0,88 1 4
C 68 2,82 ± 0,96 1 4
D 68 3,31 ± 0,97 1 4
umami
A 68 1,38 ± 0,52 1 3
B 68 1,99 ± 0,74 1 4
C 68 2,93 ± 0,61 1 4
D 68 3,84 ± 0,48 2 4
Legende: Konzentrationen, D > C > B > A. n = 68, Bewertung eines Prüfers anhand von zwei Wiederholungen.
Besonders niedrige Konzentrationen konnten von einem oder mehreren Prüfern nicht
unterschieden werden.
Die Ergebnisse der Test zeigen eine vorhandene Diskriminierungsfähigkeit der Prüfer auf,
verdeutlichen aber auch die Notwendigkeit einer gezielten Prüferauswahl.
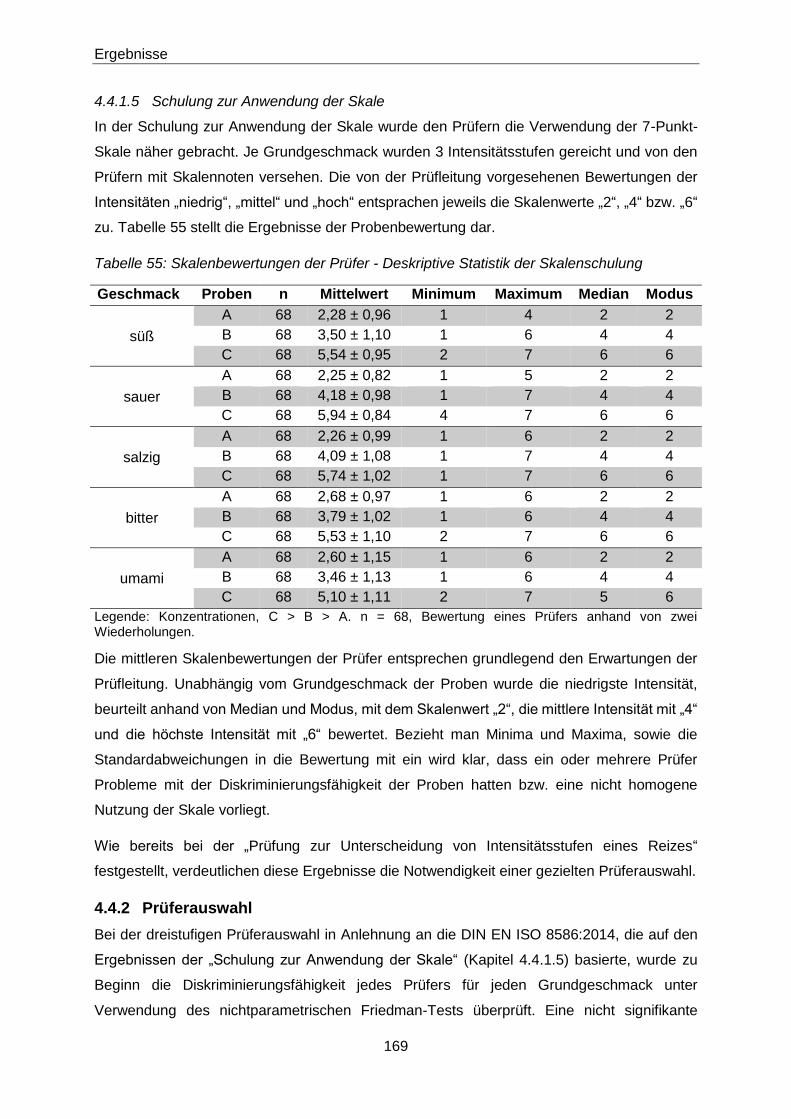
Ergebnisse
169
4.4.1.5 Schulung zur Anwendung der Skale
In der Schulung zur Anwendung der Skale wurde den Prüfern die Verwendung der 7-Punkt-
Skale näher gebracht. Je Grundgeschmack wurden 3 Intensitätsstufen gereicht und von den
Prüfern mit Skalennoten versehen. Die von der Prüfleitung vorgesehenen Bewertungen der
Intensitäten „niedrig“, „mittel“ und „hoch“ entsprachen jeweils die Skalenwerte „2“, „4“ bzw. „6“
zu. Tabelle 55 stellt die Ergebnisse der Probenbewertung dar.
Tabelle 55: Skalenbewertungen der Prüfer - Deskriptive Statistik der Skalenschulung
Geschmack Proben n Mittelwert Minimum Maximum Median Modus
süß
A 68 2,28 ± 0,96 1 4 2 2
B 68 3,50 ± 1,10 1 6 4 4
C 68 5,54 ± 0,95 2 7 6 6
sauer
A 68 2,25 ± 0,82 1 5 2 2
B 68 4,18 ± 0,98 1 7 4 4
C 68 5,94 ± 0,84 4 7 6 6
salzig
A 68 2,26 ± 0,99 1 6 2 2
B 68 4,09 ± 1,08 1 7 4 4
C 68 5,74 ± 1,02 1 7 6 6
bitter
A 68 2,68 ± 0,97 1 6 2 2
B 68 3,79 ± 1,02 1 6 4 4
C 68 5,53 ± 1,10 2 7 6 6
umami
A 68 2,60 ± 1,15 1 6 2 2
B 68 3,46 ± 1,13 1 6 4 4
C 68 5,10 ± 1,11 2 7 5 6
Legende: Konzentrationen, C > B > A. n = 68, Bewertung eines Prüfers anhand von zwei Wiederholungen.
Die mittleren Skalenbewertungen der Prüfer entsprechen grundlegend den Erwartungen der
Prüfleitung. Unabhängig vom Grundgeschmack der Proben wurde die niedrigste Intensität,
beurteilt anhand von Median und Modus, mit dem Skalenwert „2“, die mittlere Intensität mit „4“
und die hochste Intensität mit „6“ bewertet. Bezieht man Minima und Maxima, sowie die
Standardabweichungen in die Bewertung mit ein wird klar, dass ein oder mehrere Prüfer
Probleme mit der Diskriminierungsfähigkeit der Proben hatten bzw. eine nicht homogene
Nutzung der Skale vorliegt.
Wie bereits bei der „Prüfung zur Unterscheidung von Intensitätsstufen eines Reizes“
festgestellt, verdeutlichen diese Ergebnisse die Notwendigkeit einer gezielten Prüferauswahl.
4.4.2 Prüferauswahl
Bei der dreistufigen Prüferauswahl in Anlehnung an die DIN EN ISO 8586:2014, die auf den
Ergebnissen der „Schulung zur Anwendung der Skale“ (Kapitel 4.4.1.5) basierte, wurde zu
Beginn die Diskriminierungsfähigkeit jedes Prüfers für jeden Grundgeschmack unter
Verwendung des nichtparametrischen Friedman-Tests überprüft. Eine nicht signifikante

Ergebnisse
170
Diskriminierungsfähigkeit führte zum Ausschluss des Prüfers für den betroffenen
Grundgeschmack. Die Ergebnisse des Friedman-Tests sind in Tabelle 56 dargestellt.
Im Anschluss erfolgte ein Vergleich der Punktevergabe jeder Prüfperson und Intensitätsstufe
in Bezug auf den Gruppenmittelwert mit dem reduzierten Panel. Eine Abweichung > 0,6
Punkten je Konzentrationsstufe oder mehr als eine Abweichung > 0,5 Punkten führten zum
Ausschluss der betroffenen Prüferperson (Tabelle 57 – Tabelle 61).

Ergebnisse
171
Tabelle 56: Prüferauswahl - Friedman-Test zur Überprüfung der Diskriminierungsfähigkeit
Prüfer süß
Prüfer sauer
Prüfer salzig
Prüfer bitter
Prüfer umami
p signifikant? p signifikant? p signifikant? p signifikant? p signifikant?
1 0,3879 Nein 1 0,0970 Nein 1 0,0970 Nein 1 0,7165 Nein 1 0,7165 Nein
2 0,0498 Ja 2 0,0970 Nein 2 0,0970 Nein 2 0,0970 Nein 2 0,0970 Nein
3 0,0498 Ja 3 0,0498 Ja 3 0,0970 Nein 3 0,0970 Nein 3 0,0498 Ja
4 0,0970 Nein 4 0,0498 Ja 4 0,0498 Ja 4 0,2635 Nein 4 0,0498 Ja
5 0,0970 Nein 5 0,0970 Nein 5 0,0498 Ja 5 0,0498 Ja 5 0,0970 Nein
6 0,0970 Nein 6 0,0498 Ja 6 0,0498 Ja 6 0,7165 Nein 6 0,0970 Nein
7 0,0970 Nein 7 0,0498 Ja 7 0,0970 Nein 7 0,7165 Nein 7 0,2635 Nein
8 0,0970 Nein 8 0,0498 Ja 8 0,0970 Nein 8 0,0970 Nein 8 0,2635 Nein
9 0,0498 Ja 9 0,0498 Ja 9 0,7170 Nein 9 0,0498 Ja 9 1,0000 Nein
10 0,0970 Nein 10 0,0498 Ja 10 0,0970 Nein 10 0,0498 Ja 10 0,3678 Nein
11 0,0970 Nein 11 0,0970 Nein 11 0,0970 Nein 11 0,0498 Ja 11 0,0498 Ja
12 0,0498 Ja 12 0,0970 Nein 12 0,0970 Nein 12 0,0970 Nein 12 0,0970 Nein
13 0,0498 Ja 13 0,0498 Ja 13 0,0970 Nein 13 0,0970 Nein 13 0,3678 Nein
14 0,0970 Nein 14 0,0498 Ja 14 0,0498 Ja 14 0,0498 Ja 14 0,0970 Nein
15 0,0970 Nein 15 0,0970 Nein 15 0,0970 Nein 15 0,0970 Nein 15 0,0498 Ja
16 0,2331 Nein 16 0,1350 Nein 16 0,1350 Nein 16 0,2231 Nein 16 0,2231 Nein
17 0,0498 Ja 17 0,0498 Ja 17 0,0970 Nein 17 0,0970 Nein 17 0,0498 Ja
18 0,0970 Nein 18 0,0498 Ja 18 0,0498 Ja 18 0,0498 Ja 18 0,0970 Nein
19 0,0970 Nein 19 0,0498 Ja 19 0,2635 Nein 19 0,0498 Ja 19 0,0498 Ja
20 0,0498 Ja 20 0,0498 Ja 20 0,0498 Ja 20 0,0498 Ja 20 0,0498 Ja
21 0,0498 Ja 21 0,0498 Ja 21 0,0498 Ja 21 0,0970 Nein 21 0,0498 Ja
22 0,0498 Ja 22 0,0498 Ja 22 0,0970 Nein 22 0,0498 Ja 22 0,0970 Nein
23 0,0970 Nein 23 0,0498 Ja 23 0,0498 Ja 23 0,2635 Nein 23 0,3678 Nein
Summe p < 0,05 9 Summe p < 0,05 16 Summe p < 0,05 8 Summe p < 0,05 9 Summe p < 0,05 8

Ergebnisse
172
Tabelle 57: Prüferauswahl - Differenz der Prüfpersonen zum Gruppenmittelwert – „süß“
Anmerkung: Prüfer 2, 3, 9, 13, 22 ausgeschlossen
Tabelle 58: Prüferauswahl - Differenz der Prüfpersonen zum Gruppenmittelwert – „salzig“
Anmerkung: Prüfer 4, 18, 20 ausgeschlossen
Prüfer n
Probe A („niedrig“) Probe B („mittel“) Probe C („hoch“)
Mittelwert Differenz zum Mittelwert Mittelwert Differenz zum Mittelwert Mittelwert Differenz zum Mittelwert
2 3 2,00 ± 1,00 -0,07 4,00 ± 0,00 -0,44 6,33 ± 0,58 -0,85
3 3 3,00 ± 0,00 -1,07 4,00 ± 0,00 -0,44 5,33 ± 0,58 0,15
9 3 2,67 ± 0,58 -0,74 4,33 ± 0,58 -0,78 5,67 ± 0,58 -0,19
12 3 1,67 ± 0,58 0,26 3,33 ± 1,53 0,22 5,67 ± 0,58 -0,19
13 3 1,00 ± 0,00 0,93 2,67 ± 1,16 0,89 5,00 ± 01,73 0,48
17 3 1,67 ± 0,58 0,26 3,33 ± 0,58 0,22 5,67 ± 0,58 -0,19
20 3 1,33 ± 0,58 0,59 3,33 ± 1,16 0,22 5,67 ± 0,58 -0,19
21 3 1,67 ± 0,58 0,26 3,67 ± 0,58 -0,11 5,33 ± 1,53 0,15
22 3 2,33 ± 0,58 -0,41 3,33 ± 0,58 0,22 4,67 ± 0,58 0,81
Insgesamt 27 1,93 ± 0,78 3,56 ± 0,85 5,48 ± 0,89
Prüfer n
Probe A („niedrig“) Probe B („mittel“) Probe C („hoch“)
Mittelwert Differenz zum Mittelwert Mittelwert Differenz zum Mittelwert Mittelwert Differenz zum Mittelwert
4 3 1,67 ± 0,58 0,21 3,33 ± 0,58 0,71 6,00 ± 0,00 0,04
5 3 1,33 ± 0,58 0,54 3,67 ± 0,58 0,38 6,00 ± 0,00 0,04
6 3 2,00 ± 0,00 -0,13 4,00 ± 0,00 0,04 6,00 ± 0,00 0,04
14 3 2,33 ± 1,16 -0,46 4,33 ± 1,16 -0,29 6,00 ± 1,00 0,04
18 3 1,67 ± 1,16 0,21 5,00 ± 1,00 -0,96 7,00 ± 0,00 -0,96
20 3 1,67 ± 1,16 0,21 4,00 ± 0,00 0,04 5,00 ± 0,00 1,04
21 3 2,33 ± 0,58 -0,46 4,00 ± 0,00 0,04 6,33 ± 0,58 -0,29
23 3 2,00 ± 1,00 -0,13 4,00 ± 0,00 0,04 6,00 ± 0,00 0,04
Insgesamt 24 1,88 ± 0,80 4,04 ± 0,69 6,04 ± 0,62

Ergebnisse
173
Tabelle 59: Prüferauswahl - Differenz der Prüfpersonen zum Gruppenmittelwert – „sauer“
Anmerkung: Prüfer 3, 4, 8, 9, 13, 17, 18, 20, 22 ausgeschlossen
Prüfer n
Probe A („niedrig“) Probe B („mittel“) Probe C („hoch“)
Mittelwert Differenz zum Mittelwert Mittelwert Differenz zum Mittelwert Mittelwert Differenz zum Mittelwert
3 3 3,00 ± 0,00 -0,81 4,00 ± 0,00 0,06 5,33 ± 0,58 0,71
4 3 3,00 ± 0,00 -0,81 4,00 ± 0,00 0,06 5,67 ± 0,58 0,38
6 3 2,67 ± 0,58 -0,48 4,33 ± 0,58 -0,27 6,33 ± 0,58 -0,29
7 3 2,00 ± 0,00 0,19 4,00 ± 0,00 0,06 6,33 ± 0,58 -0,29
8 3 2,33 ± 0,58 -0,15 5,33 ± 0,58 -1,27 6,67 ± 0,58 -0,63
9 3 2,33 ± 0,58 -0,15 3,67 ± 0,58 0,40 5,33 ± 1,16 0,71
10 3 2,33± 0,58 -0,15 4,00 ± 0,00 0,06 5,67 ± 0,58 0,38
13 3 1,67 ± 1,16 0,52 4,67 ± 1,16 -0,60 6,33 ± 0,58 -0,29
14 3 2,00 ± 1,00 0,19 4,33 ± 0,58 -0,27 6,33 ± 0,58 -0,29
17 3 1,67 ± 0,58 0,52 4,00 ± 1,00 0,06 7,00 ± 0,00 -0,96
18 3 1,33 ± 0,58 0,85 3,00 ± 0,00 1,06 6,33 ± 0,58 -0,29
19 3 2,33 ± 0,58 -0,15 4,00 ± 1,00 0,06 6,00 ± 0,00 0,04
20 3 1,33 ± 0,58 0,85 3,33 ± 1,16 0,73 6,00 ± 0,00 0,04
21 3 1,67 ± 1,16 0,52 3,67 ± 0,58 0,40 5,67± 0,58 0,38
22 3 3,33 ± 1,53 -1,15 4,67 ± 1,16 -0,60 6,00 ± 1,00 0,04
23 3 2,00 ± 1,00 0,19 4,00 ± 0,00 0,06 5,67 ± 0,58 0,38
Insgesamt 48 2,19 ± 0,87 4,06 ± 0,78 6,04 ± 0,68

Ergebnisse
174
Tabelle 60: Prüferauswahl - Differenz der Prüfpersonen zum Gruppenmittelwert – „bitter“
Anmerkung: Prüfer 5, 11 ausgeschlossen
Tabelle 61: Prüferauswahl - Differenz der Prüfpersonen zum Gruppenmittelwert – „umami“
Anmerkung: Prüfer 3 ausgeschlossen (nach Überprüfung der Panelhomogenität)
Prüfer n
Probe A („niedrig“) Probe B („mittel“) Probe C („hoch“)
Mittelwert Differenz zum Mittelwert Mittelwert Differenz zum Mittelwert Mittelwert Differenz zum Mittelwert
5 3 1,67 ± 0,58 0,67 4,00 ± 0,00 -0,19 6,33 ± 1,16 -0,56
9 3 2,67 ± 0,58 -0,33 4,33 ± 0,58 -0,52 6,00 ± 1,00 -0,22
10 3 2,33 ± 0,58 0,00 3,67 ± 0,58 0,15 5,33 ± 1,53 0,44
11 3 2,67 ± 0,58 -0,33 3,67 ± 0,58 0,15 5,00 ± 0,00 0,78
14 3 2,67 ± 0,58 -0,33 4,00 ± 1,00 -0,19 6,33 ± 0,58 -0,56
18 3 2,33 ± 0,58 0,00 3,33 ± 0,58 0,48 6,00 ± 0,00 -0,22
19 3 2,00 ± 0,00 0,33 3,67 ± 0,58 0,15 6,00 ± 0,00 -0,22
20 3 2,67 ± 0,58 -0,33 4,00 ± 0,00 -0,19 5,33 ± 0,58 0,44
22 3 2,00 ± 0,00 0,33 3,67 ± 0,58 0,15 5,67 ± 0,58 0,11
Insgesamt 27 2,33 ± 0,56 3,81 ± 0,58 5,78 ± 0,80
Prüfer n
Probe A („niedrig“) Probe B („mittel“) Probe C („hoch“)
Mittelwert Differenz zum Mittelwert Mittelwert Differenz zum Mittelwert Mittelwert Differenz zum Mittelwert
3 3 2,67 ± 0,58 -0,46 4,00 ± 0,00 -0,17 5,00 ± 0,00 0,58
4 3 2,00 ± 0,00 0,21 3,33 ± 0,58 0,50 5,33 ± 0,58 0,25
11 3 2,67 ± 0,58 -0,46 4,00 ± 0,00 -0,17 5,33 ± 0,58 0,25
15 3 2,00 ± 0,00 0,21 3,33 ± 0,58 0,50 5,33 ± 0,58 0,25
17 3 2,00 ± 0,00 0,21 4,00 ± 0,00 -0,17 6,00 ± 0,00 -0,42
19 3 2,00 ± 1,00 0,21 4,33 ± 0,58 -0,50 6,00 ± 0,00 -0,42
20 3 1,67 ± 0,58 0,54 3,67 ± 0,58 0,17 5,67 ± 0,58 -0,08
21 3 2,67 ± 0,58 -0,46 4,00 ± 0,00 -0,17 6,00 ± 0,00 -0,42
Insgesamt 24 2,21 ± 0,59 3,83 ± 0,48 5,58 ± 0,50

Ergebnisse
175
Aus den Ergebnissen des Friedman-Tests ergab sich eine vorläufige
Panelzusammensetzung. Die Panelgröße variierte abhängig vom Grundgeschmack zwischen
8 – 16 Prüfern. Aufgrund von großen oder wiederholt auftretenden Abweichungen vom
Gruppenmittel erfolgte je Grundgeschmack eine Reduktion der Panelgröße auf 4 – 8 Prüfer.
Die Homogenität des jeweils resultierenden Panels wurde je Grundgeschmack mittels Kruskal-
Wallis H Tests überprüft. Bei unzureichender Homogenität der Merkmalsausprägung
(niedrige, mittlere, hohe Intensität) eines Grundgeschmacks erfolgte eine Überprüfung der
Panelzusammenstellung unter Verwendung von Mann-Whitney U Tests. Signifikante
Unterschiede zwischen den Prüfpersonen resultierten im Ausschluss der Prüfperson mit vom
Rangmittel abweichender Rangsumme.
Aufgrund signifikant vom Panel abweichender Bewertungen kam es für den Grundgeschmack
„umami“ zum Ausschluss eines weiteren Prüfers. Die Bewertungen der jeweiligen finalen
Panels sind in Tabelle 62 – Tabelle 66 dargestellt.
Tabelle 62: Prüferauswahl - Panelhomogenität "süß"
Probe N
(Prüfer) Mittelwert Minimum Maximum
Chi-
Quadrat Signifikanz
A („niedrig“) 4 1,58 ± 0,52 1 2 0,943 0,815
B („mittel“) 4 3,42 ± 0,90 2 5 0,388 0,943
C („hoch“) 4 5,58 ± 0,79 4 7 0,365 0,947
Tabelle 63: Prüferauswahl - Panelhomogenität "sauer"
Probe N
(Prüfer) Mittelwert Minimum Maximum
Chi-
Quadrat Signifikanz
A („niedrig“) 7 2,14 ± 0,73 1 3 3,326 0,767
B („mittel“) 7 4,05 ± 0,50 3 5 3,836 0,699
C („hoch“) 7 6,00 ± 0,55 5 7 6,667 0,353
Tabelle 64: Prüferauswahl - Panelhomogenität "salzig"
Probe N
(Prüfer) Mittelwert Minimum Maximum
Chi-
Quadrat Signifikanz
A („niedrig“) 5 2,00 ± 0,76 1 3 3,500 0,478
B („mittel“) 5 4,00 ± 0,54 3 5 2,333 0,675
C („hoch“) 5 6,07 ± 0,46 5 7 1,299 0,862

Ergebnisse
176
Tabelle 65: Prüferauswahl - Panelhomogenität "bitter"
Probe N
(Prüfer) Mittelwert Minimum Maximum
Chi-
Quadrat Signifikanz
A („niedrig“) 7 2,38 ± 0,50 2 3 6,538 0,366
B („mittel“) 7 3,81 ± 0,60 3 5 5,146 0,525
C („hoch“) 7 5,81 ± 0,75 4 7 4,694 0,584
Tabelle 66: Prüferauswahl - Panelhomogenität "umami"
Probe N
(Prüfer) Mittelwert Minimum Maximum
Chi-
Quadrat Signifikanz
A („niedrig“) 7 2,14 ± 0,57 1 3 8,102 0,231
B („mittel“) 7 3,81 ± 0,51 3 5 9,889 0,129
C („hoch“) 7 5,67 ± 0,48 5 6 8,571 0,199
Die gezielte Auswahl der Panelteilnehmer resultierte für alle Grundgeschmacksarten in einer
deutlichen Reduktion der Standardabweichungen bei guter bis sehr guter Panelhomogenität.
In Einzelfällen kommt es jedoch besonders bei niedrigen Intensitäten zu voneinander
abweichenden Bewertungen.
4.4.3 Sensorik der Peptidfraktionen
Im Rahmen der sensorischen Beurteilung der Fraktionen wurden je Proteinquelle vier Proben
zur Bewertung herangezogen. Neben der jeweiligen Permeat- (CPer: 1 g/l) und
Retentatfraktion (CRet: 1 g/l) wurde ein Bitterstandard (CBitter: 0,22 g/l) und eine Mischung aus
Bitterstandard und Permeat (CBitter/Per: 0,22 g/l/1 g/l) gereicht um zu überprüfen, ob die
Geschmacksausprägung „bitter“ durch Zusatz einer Permeatfraktion reduziert werden kann.
Wie in Kapitel 2.5.4 (Seite 47) dargelegt, verfügen besonders niedermolekulare
Peptidverbindungen über Geschmacksqualitäten. Den „bitter“-Geschmack hemmende
Eigenschaften, dargestellt in Kapitel 2.5.3.2 (Seite 44), werden ebenfalls mit
niedermolekularen Strukturen in Verbindung gebracht. Die Ergebnisse der sensorischen
Bewertung sind in Tabelle 67 – Tabelle 68 sowie Abbildung 116 - Abbildung 117 dargestellt.
Für die „Retentat“-Fraktion (> 100 kDa, pH 9) des hydrolysierten Molkenprotein-Isolats wurde
von den Prüfern eine sehr schwache „süß“ Wahrnehmung beschrieben (Tabelle 67, Abbildung
116). Weitere Grundgeschmackarten konnten nicht oder nur in geringem Umfang ermittelt
werden.
In der „Permeat“-Fraktion (< 100 kDa, pH 9) wurde ein schwacher „salzig“ Geschmack
festgestellt, der ebenfalls in der „Bitter-Standard & Permeat“ diagnostiziert wurde (Tabelle 67,

Ergebnisse
177
Abbildung 116). Die Zugabe von „Permeat“ zum „Bitter-Standard“ führte zu einer nicht
signifikanten Reduktion der „bitter“ Wahrnehmung (p > 0,05).
Für die „Retentat“-Fraktion (> 100 kDa, pH 6) der hydrolysierten Alge (Chlorella vulgaris) (>
100 kDa, pH 6) wurde von den Prüfern eine schwache „salzig“-, sowie eine sehr schwache
„umami“ Wahrnehmung beschrieben (Tabelle 68, Abbildung 117).
In der „Permeat“-Fraktion (< 100 kDa, pH 6) wurde eine schwache „salzig“-, sowie sehr
schwache „sauer“ und „umami“ Geschmacksausprägungen festgestellt (Tabelle 68, Abbildung
117). Der „salzig“ Geschmack blieb im „Bitter-Standard & Permeat“ erhalten und wurde dort
schwach bis mittel wahrgenommen. Die Zugabe von „Permeat“ zum „Bitter-Standard“ führte
zu einer signifikanten Reduktion der durch Koffein ausgelösten „bitter“ Wahrnehmung (p <
0,05).
Tabelle 67: Sensorische Bewertung – Fraktionen hydrolysierten Molkenprotein-Isolats (Retentat > 100 kDa, pH 9; Permeat < 100 kDa, pH 9)
Grundgeschmack N
(Prüfer) n
Retentat Permeat Bitter-
Standard
Bitter-Standard &
Permeat
Mittelwert Mittelwert Mittelwert Mittelwert
süß 4 3 1,08 ± 1,17
0,75 ± 1,22
0,34 ± 0,65 0,34 ± 0,89
sauer 7 3 0,38 ± 0,92
0,81 ± 1,29
0,57 ± 1,03 0,95 ± 1,20
salzig 5 3 0,67 ± 1,05
2,40 ± 1,60
0,27 ± 0,80 2,47 ± 2,13
bitter 7 3 0,67 ± 0,91
0,29 ± 0,72
2,81 ± 1,40 1 2,19 ± 1,81 1
umami 7 3 0,76 ± 1,00
0,81 ± 0,98
0,62 ± 0,87 1,19 ± 1,08
Werte in einer Zeile mit unterschiedlichen Zahlen 1,2 unterschieden sich signifikant (p < 0,05).
Tabelle 68: Sensorische Bewertung - Fraktionen hydrolysierter Alge (Chlorella vulgaris) (Retentat > 100 kDa, pH 6; Permeat < 100 kDa, pH 6)
Grundgeschmack N
(Prüfer) n
Retentat Permeat Bitter-
Standard
Bitter-Standard &
Permeat
Mittelwert Mittelwert Mittelwert Mittelwert
süß 4 3 0,42 ± 0,52
0,83 ± 0,94
0,00 ± 0,00 0,25 ± 0,45
sauer 7 3 0,57 ± 0,98
1,05 ± 1,60
0,33 ± 0,58 0,86 ± 1,49
salzig 5 3 1,40 ± 1,40
2,33 ± 2,38
0,40 ± 0,91 3,00 ± 1,93
bitter 7 3 0,29 ± 0,56
0,24 ± 0,44
2,48 ± 1,57 1 1,48 ± 1,50 2
umami 7 3 1,10 ± 1,18
1,62 ± 1,50
0,38 ± 0,67 1,57 ± 1,17
Werte in einer Zeile mit unterschiedlichen Zahlen 1,2 unterschieden sich signifikant (p < 0,05).

Ergebnisse
178
Abbildung 116: Sensorische Bewertung - Fraktionen hydrolysierten Molkenprotein-Isolats (Retentat > 100 kDa, pH 9; Permeat < 100 kDa, pH 9)
Abbildung 117: Sensorische Bewertung - Fraktionen hydrolysierter Alge (Chlorella vulgaris) (Retentat > 100 kDa, pH 6; Permeat < 100 kDa, pH 6)
0
1
2
3
4Süß
Sauer
SalzigBitter
Umami
Molkenprotein-Isolat
Retentat Permeat Bitter-Standard Bitter-Standard & Permeat
0
1
2
3
4Süß
Sauer
SalzigBitter
Umami
Alge (Chlorella vulgaris)
Retentat Permeat Bitter-Standard Bitter-Standard & Permeat

Diskussion
179
5. Diskussion
5.1 Analytik
Die Bewertung der in Kapitel 3.4.2 (Seite 64)/Kapitel 4.1.2 (Seite 87) beschriebenen
„Spektralphotometrischen Proteingehaltbestimmung“, sowie der in Kapitel 3.4.4 (Seite
67)/Kapitel 4.1.1 (Seite 79) dargelegten „Analytik der Peptidfraktionen mittels SE-HPLC“ findet
im Rahmen der abschließenden Diskussion der „Methodischen Aspekte“ (Kapitel 5.5.2, Seite
237) statt.
5.2 Hydrolyse
5.2.1 Alge (Chlorella vulgaris)
Mikroalgen der Gattung Chlorella, in die auch die in dieser Studie verwendete Chlorella
vulgaris fällt, sind, trotz großen Potentials im Lebensmittelbereich, nur wenig erforscht (Ursu
et al., 2014). Grund dafür ist, unter anderem, die starke Zellwandstruktur, die zur Nutzung
funktioneller Komponenten aufgebrochen werden muss (Morris et al., 2008).
In Pilotversuchen bestätigte sich die Problematik der Unzugänglichkeit des Algenproteins für
den enzymatischen Hydrolyseprozess, die auch durch erhöhte Enzymkonzentration von 0,5
% (Pronase (225 PU), Pancreatin (40 USP)), bzw. 3 % (Corolase (82,5 UHB)) nicht gelöst
werden konnte. In einer lichtmikroskopischen Analyse der Hydrolysate konnten lediglich
intakte Zellstrukturen festgestellt werden. Da Mikroalgen der Gattung Chlorella vorwiegend
über Cellulose-, Hemicelluslase- und Pektin-basierte Zellwände verfügen, wurde die
Wirksamkeit einer Pektinase-Behandlung (18 h, 48 h, 72 h) der Zellwand durch Rohament®
PL, sowie einer sauren Vorbehandlung unter Verwendung von HCl, pH 3 (18 h, 48 h) überprüft.
Zur Hydrolyse der Algenproteine wurden die Endopeptidasen Pancreatin (0,25 %, 20 USP)
und Pronase (0,25 %, 112,5 PU) verwendet.
Behandlung mit Rohament® PL:
Die enzymatische Vorbehandlung durch die Pektinase Rohament® PL mit anschließender 45-
minütiger Hydrolyse resultierte für Pronase und Pancreatin, abhängig von der Prozessdauer
tEV, in Hydrolysegraden zwischen 15,32 ± 1,19 % (Pancreatin, 18 h), bzw. 14,63 ± 2,73 %
(Pronase, 18 h) und 20,14 ± 1,56 % (Pancreatin, 48 h), bzw. 21,39 ± 1,77 % (Pronase, 48 h).
Eine weitere Steigerung der Prozesslaufzeit auf 72 h führte nicht zu einer weiteren Erhöhung
des Hydrolysegrads, sondern resultierte im Gegensatz dazu in einer geringfügigen Abnahme.
Die maximalen Hydrolysegrade entsprechen den Ergebnissen von Morris et al. (2008) und
Tchorbanov & Bozhkova (1988) die bei der Hydrolyse von mit organischen Lösungsmitteln
extrahierten Algenproteinen unter Verwendung von Pancreatin und Subtilisin Hydrolysegrade
von 20 – 22 % erreichten. Eine Steigerung der Prozesszeit führte nicht zu einer weiteren
Erhöhung des Hydrolysegrads und äußerte sich lediglich in einer gesteigerten Ausbeute

Diskussion
180
hydrolysierten Proteins (Tchorbanov & Bozhkova, 1988). Tchorbanov & Bozhkova (1988)
führten dies auf einen maximalen Hydrolysegrad zurück, der mit Subtilisin erreicht werden
kann. Diese Erkenntnisse stimmen mit den Ergebnissen anderer Autoren überein, die bei
langen Hydrolyseprozessen von Weizengluten eine Annäherung an einen enzym- und
substratspezifischen DH-Grenzwert feststellten (Koo et al., 2011; Kong, Zhou, & Qian, 2007a,
2007b). Die geringfügige Abnahme der Hydrolysegrade bei einer Steigerung der Dauer der
enzymatischen Vorbehandlung von 48 h auf 72 h bei den mit Pronase und Pancreatin
durchgeführten Hydrolyse ist vermutlich auf prozessbedingte Schwankungen, bei
gleichzeitiger Erreichung des vorab beschriebenen maximalen Hydrolysegrads der Enzyme
zurückzuführen. Darüber hinaus besteht die Möglichkeit, dass durch die Steigerung der Dauer
der enzymatischen Vorbehandlung mehr Protein bereitgestellt werden konnte, welches in der
anschließenden Hydrolyse aufgrund der Prozessbedingungen (Enzymkonzentration, Dauer
der Hydrolyse) nicht in vergleichbarer Weise umgesetzt werden konnte.
Die Chromatogramme der Hydrolysate weisen einen großen Anteil an Molekülen ≥ 1,35 kDa
auf. Mit zunehmender Dauer der Vorbehandlung ist zudem eine Zunahme noch kleinerer
Strukturen erkennbar (Abbildung 59, Abbildung 60, Abbildung 61; Seite 95 - 96). Die
Ergebnisse deuten in Verbindung mit den Hydrolysegraden darauf hin, dass der
Hydrolyseprozess weit fortgeschritten ist und Molekülstrukturen ≥ 1,35 kDa enzymatisch
abgebaut oder aufgrund mangelnder Löslichkeit durch den Zentrifugationsprozess abgetrennt
worden sind. Nach Ursu et al. (2014) liegt der IEP intakter Chlorella vulgaris Proteine zwischen
4,0 und 5,5. Vollständige Löslichkeit wurde bei pH 7,0 beobachtet. Der End-pH der
Hydrolysate fiel zu keinem Zeitpunkt unter pH 7 und bewegte sich zwischen 8,0 und 7,0. Eine
Abtrennung aufgrund des Zentrifugationsprozess erscheint somit nicht in mangelnder
Löslichkeit begründet.
Behandlung mit HCl, pH 3:
Die saure Vorbehandlung mit anschließender 45-minütiger Hydrolyse resultierte für Pronase
und Pancreatin in Hydrolysegraden zwischen 10,29 ± 0,97 (Pancreatin, 18 h) bzw. 17,04 ±
2,83 (Pronase, 18 h) und 12,76 ± 1,21 (Pancreatin, 48 h) bzw. 18,16 ± 2,59 (Pronase, 48 h).
Anhand des Hydrolysegrads beurteilt, resultiert eine Vorbehandlung für 18 h in
Hydrolysegraden im angestrebten Zielbereich zwischen 8 – 12 %. Die saure Hydrolyse von
Cellulose, die besonders beim Umsatz von Biomasse zur Glucose-Gewinnung Anwendung
findet, kann sowohl unter hohen (100 – 240 °C), als auch unter niedrigen (18 – 50 °C)
Temperaturen zum effektiven Aufschluss von Cellulose führen (Lai, 2000). Park et al. (2016)
konnten bei der Hydrolyse von Chlorella vulgaris zur Gewinnung fermentierbarer Zucker mit
HCl (2 %, 121 °C, 20 min) 92,5 % der Kohlenhydrate in reduzierende Zucker umsetzen.

Diskussion
181
Ähnlich hohe Umsätze (83 %) wurden von Zhou et al. (2011) bei einer Hydrolyse mit HCl (2
%, 180 °C, 10 min) im Beisein von 2.5% Magnesiumchlorid (MgCl2) erreicht.
Da der Fokus dieser Studie nicht auf dem Umsatz von Cellulose in reduzierende Zucker lag,
wurde zur sauren Vorbehandlung eine Temperatur von 45 °C verwendet. Die
Chromatogramme zeigen für die 18-stündige Vorbehandlung Molekülstrukturen im Bereich ≥
20 kDa – 158 kDa. Eine Steigerung der Dauer der Vorbehandlung mit HCl, pH 3 auf 48 h
führte, besonders für Pronase, zu einem deutlich höheren Anteil an Strukturen ≥ 44 kDa. Die
Ergebnisse deuten, unter Berücksichtigung der Erkenntnisse von Zhou et al. (2011) und Park
et al. (2016), darauf hin, dass die Zellwand der Algenzellen erfolgreich lysiert werden konnte.
Dies resultierte vermutlich in erhöhter Zugänglichkeit des Algenproteins für die zur Hydrolyse
verwendeten Endopeptidasen. Betrachtet man hierzu jedoch die von Morris et al. (2008)
ermittelte Molekülgrößenverteilung der Algenproteine (12 – 120 kDa) wird klar, dass,
zumindest im Hydrolysat (HCl, pH 3, 48 h, Pronase 0,25 % (112,5 PU)), neben Peptiden auch
Proteinaggregate vorliegen müssen (vergleiche: Abbildung 64 b, Seite 99). Nicht-native,
denaturierte oder teilgefaltete Proteine sind gemäß Roberts (2007), unter anderem bedingt
durch frei zugängliche hydrophobe Gruppen, anfällig für die Ausbildung von irreversiblen
Protein-Aggregaten. Unter Berücksichtigung dieser Erkenntnis kann nicht alleinig von einer
Steigerung der Zugänglichkeit der Proteine für das Enzym ausgegangen werden. Vielmehr
wird ein simultanes Auftreten beider Phänomene (erhöhte Zugänglichkeit der Proteine durch
saure Vorbehandlung mit HCl, pH 3, Aggregatbildung durch frei zugängliche hydrophobe
Gruppen) vermutet.
5.2.1.1 Zusammenfassung der Diskussionspunkte
Eine Hydrolyse von Algenprotein ist nicht ohne Aufbrechen der Algenzellwände
möglich.
Der IEP der Algenproteine führt nicht zur Interferenzen mit den pH-Optima gängiger
Enzyme.
Die Zellwand der Alge Chlorella vulgaris lässt sich durch saure Hydrolyse lysieren.
Die Bildung von Protein-Aggregaten ist während/nach dem Hydrolyseprozess nicht
auszuschließen.
5.2.1.2 Zusammenfassung & Darstellung der Ergebnisse
Eine Lyse der Zellwand durch Pektinase (Rohament® PL, 0,02 %, 14000 PGU)
erscheint effektiv, erwies sich aber unter den gegebenen Versuchsbedingungen als
ungeeignet.
Eine Lyse mit HCl (18 h, 48 h) mit anschließender Hydrolyse durch Pronase (0,25 %,
112,5 PU) oder Pancreatin (0,25 %, 20 USP) eignet sich zur Erzeugung von
Algenhydrolysaten mit Hydrolysegraden zwischen 10 – 20 %.

Diskussion
182
Eine Steigerung der Dauer der Vorbehandlung auf 48 h führt zu einer Zunahme an
Molekülstrukturen ≥ 44 kDa.
Eine 48-stündige Vorbehandlung resultiert vermutlich in der Ausbildung von Protein-
Aggregaten.
5.3 Crossflow-Diafiltration & Fraktionierung
5.3.1 Membran- & Anlagencharakterisierung
Für Reinstmedien steigt der Permeatflux JFm, proportional zum Transmembrandruck ∆pTMP.
Bei beladenen Medien zeigt sich, abhängig von Feed-Konzentration und
Strömungsgeschwindigkeit, eine Abweichung vom linearen Zusammenhang des Permeatflux
JF,t bei steigendem Transmembrandruck ∆pTMP. (Kofod Nielsen, 2004; Irmler, 2001; Lojkine et
al., 1992). Die Abnahme des Permeatflux JFm ist hierbei auf die Ausbildung einer
Konzentrationspolarisation an der Membranoberfläche und im weiteren Verlauf durch die
Ausbildung einer Foulingschicht auf der Membran zurückzuführen (Taddei et al., 1989). Die
Ausbildung von Fouling kann, durch eine Prozessführung unterhalb eines Drucks pCP reduziert
werden. Unterhalb eines kritischen Drucks pCP entspricht der kritische Flux JCF weitestgehend
dem limitierend Flux JLF. Unter limitierendem Flux JLF versteht man den Flux, für den eine
Steigerung des Transmembrandrucks ∆pTMP nicht länger in einem Anstieg des Permeatflux
resultiert (Vyas et al., 2000; Defrance & Jaffrin, 1999). Um die Ausbildung und den Einfluss
von Fouling auf den Fraktionierungsprozess zu reduzieren wurde die verwendete Membran
unter Verwendung des jeweiligen Feeds (0,15 % (w/v)) vor Beginn der Versuchsreihe
charakterisiert.
5.3.1.1 Molkenprotein-Isolat & Alge (Chlorella vulgaris)
Die in Abbildung 68 (Seite 106)/Abbildung 69 (Seite 107) dargestellten Flux-Charakteristika
von destilliertem Wasser, sowie hydrolysiertem Molkenprotein-Isolat, bzw. hydrolysierter Alge
(Chlorella vulgaris) entspricht den in der Literatur, unter anderem von Bacchin et al. (2006)
und Irmler (2001) dargestellten, Flux-Verläufen von Reinst- und beladenen Feeds. Der
Permeatflux von destilliertem Wasser verzeichnet in beiden Fällen eine lineare Zunahme bei
steigendem Druck pTMP. Eine Erhöhung des Drucks pTMP von 0,4 bar auf 0,6 bar resultierte
sowohl bei der Filtration von hydrolysiertem Molkenprotein-Isolat (Abbildung 68), als auch bei
der Filtration von hydrolysierter Alge (Chlorella vulgaris) (Abbildung 69) in einer Abweichung
des Permeatflux-Anstiegs vom linearen Fluxverlauf. Der kritische Druck pCP wurde infolge für
beide Feeds auf 0,5 bar festgelegt.
Wie bereits vorab beschrieben, ist die Abnahme des Permeatflux bei der Filtration beladener
Feeds, unter anderem unter Berufung auf Bacchin et al. (2006), Field et al. (1995), Howell
(1995) und Taddei et al. (1989) auf die Ausbildung von Konzentrationspolarisation und Fouling
zurückzuführen. In Anbetracht der Flux-Charakteristika ist in Anlehnung an Bacchin et al.

Diskussion
183
(2006) davon auszugehen, dass es sich um eine „harte/starke“ Form der Flux-Druck-
Abhängigkeit handelt, bei welcher der Feed-Flux analog des Reinstwasserflux bis zum
Erreichen des kritischen Flux JCF, bzw. kritischen Druck pCP ansteigt und nachfolgend deutlich
abnimmt (siehe auch: Abbildung 9, Seite 17). Bei beiden Membrancharakterisierungen wird
deutlich, dass der Abfall des Permeatflux nicht durch den Rückhalt nativer Proteine, sowie den
daraus durch Hydrolyse entstehenden Peptiden über der Membran, bzw. der Ausbildung einer
Deckschicht von aufgrund ihrer Molekülgröße zurückgehaltenen Proteinen/Peptiden
begründet werden kann, da die Molekülgrößen der Feed-Proteine unter der nominalen
Trenngrenze der Membran liegen. In der nachfolgenden Diskussion zum Einfluss des pH-
Werts und der Ionenstärke (Kapitel 5.3.2) werden die für den Flux-Abfall verantwortlichen
Effekte ausführlich diskutiert und in Kapitel 5.3.2.3 (Seite 201), sowie Kapitel 5.3.2.4 (Seite
201) abschließend zusammengefasst. Um der Diskussion nicht vorzugreifen, sowie
Wiederholungen zu vermeiden, wird an dieser Stelle auf die genannten Kapitel verwiesen.
5.3.2 Einfluss des pH-Werts & der Ionenstärke
In den folgenden Abschnitten wird der Einfluss von pH-Wert und Ionenstärke auf die
Parameter Permeatflux JFm, Proteingehalt in Retentat mRet und Permeat mPer (Transmission),
sowie die Wiederfindungsrate diskutiert. Effekte, die auf weitere, im Rahmen dieser Studie
nicht variierte, hydrodynamische und physikalisch-chemische Eigenschaften des
Filtrationsprozesses zurückgehen, werden im Kapitel 5.3.6 näher beleuchtet.
5.3.2.1 Molkenprotein-Isolat
Die Verwendung der Crossflow-Filtration zur Fraktionierung und Aufkonzentration von
Milchkomponenten oder Molkenproteinen hat sich in den vergangenen Jahren etabliert. Bei
der Fraktionierung intakter Molkenproteine (geklärte Molke) stellen Almécija et al. (2007) fest,
dass sich sowohl der Permeatflux, als auch die Proteinausbeute in der Retentat- und Permeat-
Fraktion pH-abhängig unterscheiden. Eine Filtration im Bereich des IEP resultierte in Fouling
und Aggregation von Proteinen, während der Permeatflux mit zunehmender Entfernung vom
IEP zunahm (pH 3, pH 9, pH 10). Die Transmission der Proteine war darüber hinaus ebenfalls
pH-abhängig unterschiedlich. Pouliot, Wijers, Gauthier, & Nadeau (1999) untersuchten den
Einfluss von Ionenstärke und pH auf die Fraktionierung eines Molkenproteinhydrolysat mit
Molekülstrukturen ≤ 10 kDa. Eine Erhöhung des pH von 5 auf 9 resultierte bei der
Fraktionierung mit einer Cellulose-Acetat-Membran (MWCO 1 – 5 kDa) in Permeatflux- und
Transmissionssteigerung, sowie einer Reduktion der Fouling-Tendenz. Eine Erhöhung der
Ionenstärke resultierte darüber hinaus in einer verbesserten Transmission, führte aber auch
zu verstärktem Fouling. Diese Erkenntnisse sind weitestgehend deckungsgleich mit den in
dieser Studie ermittelten, nachfolgend näher beschriebenen Beobachtungen.

Diskussion
184
Gemessen am technologischen Potential der Crossflow-Filtrationstechnik zur Fraktionierung
von Molkenproteinen und Proteinhydrolysaten mit dem Zweck der Gewinnung von bioaktiven
Substanzen und funktionellen Lebensmittelbestandteilen, ist die Anzahl der
Veröffentlichungen, insbesondere bei Hydrolysaten, überschaubar. Ein Großteil der
Veröffentlichungen fokussiert sich auf die Aufkonzentration intakter Proteine und/oder arbeitet
mit Modellsystemen.
Permeatflux JFm:
In der vorliegenden Arbeit wurde der Permeatflux JFm durch die Anpassung des pH-Werts nicht
signifikant beeinflusst und lag zwischen 20,01 ± 0,84 ml/min (pH 8) und 29,44 ± 8,19 ml/min
(pH 9). Eine Steigerung des Permeatflux für die pH-Stufe 9 im Vergleich zu darunter liegenden
pH-Werten wurde von Almécija et al. (2007) bei der Fraktionierung geklärter Molke festgestellt.
Der niedrigste Permeatflux wurde von den Autoren im Bereich des IEP der Molkenproteine
(pH 4 – pH 5) festgestellt. Die pH-abhängigen Unterschiede fielen für intakte Molkenproteine
mit einem minimalen Permeatflux JF,t von 25 - 51 l/(m2h) (pH 4, pH 5) und einem maximalen
Permeatflux JF,t von 89 – 125 l/(m2h) (pH 10) sehr deutlich aus. Die Autoren begründen dies
durch ladungsbedingtes Fouling im Bereich des IEP (pH 4, pH 5), bzw. ladungsbedingte
Abstoßungseffekte bei Extrem-pH-Werten (pH 3, pH 10). Pouliot et al. (1999) führten einen
Anstieg des Permeatflux bei der Erhöhung des pH von pH 5 auf pH 9 bei der Fraktionierung
von Molkenproteinhydrolysat mit Molekülstrukturen ≤ 10 kDa auf ladungsbedingte
Abstoßungseffekte zwischen Peptiden und Membran zurück. Ein pH-Wert von pH 9 resultierte
bei der Verwendung negativ geladener Cellulose-Acetat-Membranen in ebenfalls negativ
geladenen oder neutralen Peptiden. Bei pH 5 gingen die Autoren von größtenteils positiv
geladenen Peptiden aus, die durch Adsorption auf der Membran zur Reduktion des Flux durch
Fouling führten.
Bei der Filtration intakter Proteine beeinflusst der pH-Wert die Struktur der Foulingschicht. Bei
der Filtration von BSA-Molekülen unter Verwendung von Polysulfon-Membran (MWCO 300
kDa) stellten Huisman et al. (2000) mit Hilfe von rasterkraftmikroskopischen Aufnahmen pH-
abhängige Unterschiede der Struktur der Foulingschicht fest. Im Bereich des IEP (pH 5) bildete
sich eine dichte Foulingschicht aus, die zu einem minimalen Permeatflux führte. pH 3 führte
zu einer lockeren Foulingschicht mit hohen Fluxwerten. Die Trenngrenze der Membran hatte
nur geringfügigen Einfluss auf die Struktur der Foulingschicht.
Die Reduktion des Flux durch Fouling ist dabei abhängig von der Partikelgrößenverteilung.
Tarleton & Wakeman (1993) stellten bei der Filtration von Calcite-Suspensionen mit
verschiedenen Partikelgrößenverteilungen fest, dass besonders kleine Moleküle zur
Entstehung der Foulingschicht beitragen. Darüber hinaus spielt die Form der Partikel eine
entscheidende Rolle. Je geringer die Kugelhaftigkeit von Partikeln ist, desto höher ist die

Diskussion
185
Durchlässigkeit der Foulingschicht, sofern es sich nicht um plättchenförmige Partikel handelt
(Foust et al., 2008; Dullien, 1992). Proteinhydrolysate bestehen, abhängig vom Hydrolysegrad
und der Art des Enzyms (Exopeptidase, Endopeptidase), aus einer Vielzahl unterschiedlicher
Moleküle (Clemente, 2000). Diese können durch pH-abhängige Ladungsveränderung durch
ionische und hydrophobe Wechselwirkungen, sowie die Ausbildung von
Wasserstoffbrückenbindungen verschiedene Raumstrukturen annehmen (Schmid, 2016).
Bezieht man die Erkenntnisse der Autoren auf die in der vorliegenden Studie durchgeführte
Fraktionierung ohne NaCl-Zusatz (keine Erhöhung der Ionenstärke) legen diese die
Vermutung nahe, dass Unterschiede im Permeatflux JFm bei der Fraktionierung von
Molkenproteinhydrolysaten durch Fouling hervorgerufen wurden, sich verschiedene pH-Stufen
aber nicht signifikant auf die Struktur der Foulingschicht und der damit verbundenen pH-
abhängigen Reduktion des Permeatflux auswirken.
Die Ausbildung der Foulingschicht wird hauptsächlich durch kleine Moleküle induziert. Diese
resultieren durch ladungsbasierte Adsorption in der Verblockung einzelner Membranporen
(internem Fouling), gemäß des Modells von Wang & Tarabara (2008) (siehe Abbildung 5; Seite
9). In einem weiteren Schritt kommt es zur Ausbildung einer Deckschicht (externes Fouling),
deren Struktur durch die Partikelgrößenverteilung der Hydrolysatmoleküle abhängig ist. Die
Partikelgrößenverteilung ist wiederum pH-abhängig. Aufgrund einer Vielzahl an
verschiedenen Molekülstrukturen resultierte eine Änderung des pHs nicht in deutlichen
Veränderungen der Struktur der Foulingschicht. Die in dieser Arbeit festgestellten
geringfügigen Unterschiede des Permeatflux lassen sich auf ladungsbedingte Abstoßungs-
bzw. Adsorptionseffekte zwischen Molekül und Membran zurückführen, die vermutlich
besonders in der Startphase des Filtrationsprozesses eine Rolle spielen.
Die Erhöhung der Ionenstärke durch Zusatz von 150 mM NaCl resultierte in der vorliegenden
Studie in einem Permeatflux JFm zwischen 30,27 ± 6,01 ml/min (pH 6) und 49,11 ± 3,23 ml/min
(pH 9) und lagen für die pH-Stufen 7, 8, 9 signifikant über dem Permeatflux einer
Fraktionierung ohne NaCl-Zusatz. pH-Stufe 9 unterschied sich darüber hinaus signifikant von
den übrigen Stufen (pH 6, pH 7, pH 8). Vergleichbare Erkenntnisse stammen von She et al.
(2009), die bei der Filtration von BSA eine Steigerung des Permeatflux für die pH-Stufen 5,8
und 7 bei der Zugabe von NaCl beobachteten. Während die Konzentration bei pH 7 keinen
Einfluss besaß, resultierte eine Erhöhung der NaCl-Konzentration von 1 mM auf 10 mM und
weiter auf 100 mM in jeweils deutlichen Steigerungen des Permeatflux. Die Autoren führen
dies auf ein geringere Molekülgröße (hydrodynamisches Volumen) des BSA Moleküls zurück,
das in Folge nicht als Foulant auf der Membran zurückbleibt, sondern diese passiert. pH-
induzierte Abstoßungseffekte können dabei ebenfalls zu geringerer Foulingneigung und
höherem Permeatflux führen (Salgın, 2007). Es ist folglich davon auszugehen, dass beide

Diskussion
186
Effekte auch in der vorliegenden Arbeit eine Rolle spielen. Der Einfluss der Ionenstärke auf
den Permeatflux lässt sich nicht verallgemeinern. Bei der Fraktionierung von
Molkenproteinhydrolysat mit Molekülstrukturen ≤ 10 kDa stellten Pouliot et al. (1999) einen
signifikant niedrigeren Permeatflux für die pH-Stufe 9 fest, der darüber hinaus in einer
Zunahme des Widerstands durch Fouling führte. Die Autoren führen dies auf eine gesteigerte
Ionisation bzw. Abschirmung der ansonsten negativ geladenen Peptide zurück, die folglich,
aufgrund verringerter Interaktion zwischen Peptid und Membran, verstärkt zur Adsorption auf
der Membranoberfläche neigen. Teng et al. (2006) gehen darüber hinaus davon aus, dass
eine Erhöhung der Ionenstärke durch Abnahme des hydrodynamischen Volumens eines
Moleküls (hier: BSA) bei druckgetriebenen Filtrationsprozessen zur Ausbildung einer dichteren
Deckschicht führen, die zur Reduktion des Permeatflux führt.
Bezieht man die Erkenntnisse der Autoren auf die in der vorliegenden Studie durchgeführte
Fraktionierung mit NaCl-Zusatz (Erhöhung der Ionenstärke), legen diese die Vermutung nahe,
dass die Ionenstärke bei der Fraktionierung von Molkenproteinhydrolysaten einen
signifikanten Einfluss auf den Permeatflux JFm hat, der auf strukturellen Veränderungen der
Moleküle und ein verändertes Ladungsprofil zurückzuführen ist. Anders als bei intakten
Proteinen resultierte die Erhöhung der Ionenstärke nicht in einer Reduktion des Permeatflux,
sondern in einer Fluxsteigerung. Diese ist im Falle der im Rahmen dieser Arbeit
dokumentierten Ergebnisse pH-abhängig und nimmt mit steigenden pH-Wert zu. Die
Ausbildung einer dichteren Foulingschicht im Vergleich zu einer Filtration ohne NaCl-Zugabe
erscheint nicht wahrscheinlich. Vielmehr ist davon auszugehen, dass es durch die Erhöhung
der Ionenstärke zu einer Veränderung des Ladungsprofils des Hydrolysats kommt. Durch eine
ionenstärke-induzierte Reduktion des hydrodynamischen Radius kann es im Vorfeld zu der
Entstehung von Molekülaggregaten kommen, welche die Struktur der ausgebildeten
Foulingschicht durch einen erhöhten Anteil größerer Strukturen positiv beeinflussen. Darüber
hinaus führt eine Reduktion des hydrodynamischen Radius zu verbesserter Transmission.
Proteingehalt von Retentat mRet & Permeat mPer, Wiederfindungsrate:
Die Wiederfindungsrate wurde durch die Anpassung des pH-Werts nicht signifikant beeinflusst
und lag bei der Fraktionierung ohne NaCl-Zusatz zwischen 76,91 ± 5,53 % (pH 7) und 94,85
± 4,63 % (pH 8). 27,92 ± 8,03 % (pH 7) bis 47,18 ± 8,94 % (pH 8) der verbleibenden
detektierbaren Verbindungen lagen im Retentat, 47,67 ± 3,43 % (pH 8) bis 53,02 ± 3,22 % (pH
9) im Permeat vor. Der Proteingehalt im Retentat lag damit zwischen 0,209 ± 0,060 g (pH 7)
und 0,354 ± 0,067 g (pH 8), sowie zwischen 0,358 ± 0,026 g (pH 8) und 0,398 ± 0,024 g (pH
9) im Permeat. Der Proteingehalt des Retentats bei einer Fraktionierung bei pH 7 lag signifikant
unter den Gehalten der verbleibenden pH-Stufen.

Diskussion
187
Zahlreiche Studien bestätigen die Abhängigkeit der Transmission vom pH-Wert des Feeds.
Bei der Filtration von BSA am IEP erreichten de la Casa et al. (2007), Persson et al. (2003)
und Huisman et al. (2000) eine BSA-Transmission von 55 – 100 %. Laut Huisman et al. (2000)
lässt sich dies auf pH-induzierte Veränderungen der Molekülgröße von Proteinen und
Proteinaggregaten, der Protein-Membran-Interaktion und der Eigenschaften der externen
Foulingschicht zurückführen. Die Molekülgröße eines Proteins (hydrodynamischer Radius) ist
am IEP am kleinesten (Pincet, Perez, & Belfort, 1995; Pincet, Perez, & Belfort, 1994). Persson
et al. (2003) gehen darüber hinaus davon aus, dass die Verbesserung der Transmission im
Ausbleiben von Ladungseffekten zwischen Protein und Membran, sowie Protein und
Foulingschicht begründet ist. Die Transmission von BSA am IEP kann jedoch nicht losgelöst
von weiteren Faktoren beurteilt werden. Membrantrenngrenze und Dauer des Prozesses
spielen ebenfalls eine entscheidende Rolle. Huisman et al. (2000) stellten bei der Filtration von
BSA am IEP unter Verwendung einer Polysulfon-Membran mit einem MWCO von 300 kDa
nach 10 h die vergleichsweise niedrigste Transmission fest, obwohl zu Filtrationsbeginn die
höchste Transmission verzeichnet wurde. Die Autoren führen dies auf die Ausbildung einer
besonders dichten Foulingschicht zurück. Die Verwendung einer Trenngrenze von 100 kDa
resultierte hingegen in durchweg höherer Transmission. Unter den richtigen Bedingungen
(Membran, pH, Transmembrandruck) lässt sich auch ein Feed aus zwei Modellproteinen im
Bereich des IEP fraktionieren. Das Zielprotein wird dabei, bedingt durch eine Reduktion des
hydrodynamischen Radius, sowie einem neutralen Ladungszustand ins Permeat transportiert,
während die weiteren Proteine durch ladungsbedingte Abstoßung und größeren
hydrodynamischen Radius im Retentat zurückgehalten wird (Nyström et al., 1994). Eine
selektive Fraktionierung ist sogar gegen den Größengradienten möglich, wie Nyström et al.
(1994) für die Fraktionierung von Ovalbumin (45 kDa) und Myoglobin (17,4 kDa) bei einem pH
von 4,8 unter Kaliumchlorid (KCl) Zugabe zeigten. Saksena & Zydney (1994) stellten
vergleichbare Effekte bei der Fraktionierung von BSA (69 kDa) und IgG (155 kDa) unter
Verwendung einer Polyethersulfon Membran (MWCO 300 kDa), pH 7,4, 1,5 mM NaCl fest.
Abseits von Modellsystemen gestaltet sich eine selektive Fraktionierung am IEP schwierig. Bei
der Fraktionierung von geklärter Molke unter Verwendung einer Keramikmembran (MWCO
300 kDa) stellten Almécija et al. (2007) für die pH-Stufen 4 und 5 eine Retention von annährend
100 % für α-Lactalbumin, β-Lactoglobulin, BSA und IgG fest. Eine hohe Transmission für α-
Lactalbumin und β-Lactoglobulin wurde bei den pH-Stufen 7, 8 und 9 festgestellt. BSA wurde,
trotz einer Molekülgröße von ca. 69 kDa, in allen pH-Stufen (pH 3 – pH 10) zu 94 – 100 % im
Retentat zurückgehalten. Die Autoren führen dies für α-Lactalbumin und β-Lactoglobulin, unter
Berufung auf Taulier & Chalikian (2001), sowie Boye, Alli, & Ismail (1997), auf die Ausbildung
von Proteinaggregaten aus bis zu acht Molekülen zurück. Die Ausbildung weiterer Aggregate
erscheint ebenfalls möglich. Die Ergebnisse von Almécija et al. (2007) deuten darauf hin, dass

Diskussion
188
die in Modellsystemen ermittelten optimalen Filtrationsbedingungen nicht ohne Weiteres auf
komplexere Medien übertragbar sind. Vielmehr ist eine experimentelle Ermittlung der
optimalen Bedingungen in Abhängigkeit des Feeds erforderlich. Darüber hinaus wird anhand
BSA (69 KDa) deutlich, dass auch Moleküle deren Größe weit unter der Trenngrenze (MWCO
300 kDa) der Membran liegen nicht alleinig anhand ihrer Größe getrennt werden können.
Die Erhöhung der Ionenstärke durch Zusatz von 150 mM NaCl resultierte in der vorliegenden
Arbeit in einer Wiederfindungsrate zwischen 74,88 ± 3,39 % (pH 7) und 81,74 ± 4,36 % (pH
8). Für die pH-Stufen 8 und 9 führte die Erhöhung der Ionenstärke zu einer signifikant
niedrigeren WFR. 18,10 ± 4,92 % (pH 7) bis 27,87 ± 4,22 (pH 6) lagen im Retentat, 47,27 ±
1,79 % (pH 6) bis 56,78 ± 5,23 % (pH 7) im Permeat vor. Der Proteingehalt im Retentat lag
zwischen 0,136 ± 0,037 g (pH 7) und 0,209 ± 0,031 g (pH 6), sowie zwischen 0,354 ± 0,014 g
(pH 6) und 0,426 ± 0,039 g (pH 7) im Permeat. Die Erhöhung der Ionenstärke resultierte in
einem gesteigerten Proteinanteil im Permeat, bei gleichzeitig geringerem Proteingehalt im
Retentat. Für die pH-Stufen 6 und 9 wird deutlich, dass der geringere Proteinanteil im Retentat
nicht nur auf einen gesteigerten Proteingehalt im Permeat, sondern vielmehr auf den Verlust
von Protein zurückzuführen ist.
Der Einfluss der Ionenstärke auf die Transmission ist unter anderem abhängig von pH-Wert,
Salzkonzentration und der Art des Feeds. Bei der Filtration von BSA konnten Persson et al.
(2003) die Transmission für pH 3 und 7 unter Zugabe von 0,15 M NaCl auf über 80 % steigern.
Die Zugabe von NaCl führte jedoch, besonders bei pH 7, zu einer deutlichen Minderung des
Permeatflux. Die Autoren führen dies auf eine Abschirmung der geladenen Moleküle durch
NaCl-Ionen zurück, die dazu führt, dass die Moleküle zur Ausbildung von Aggregaten und
einem daraus resultierenden dichteren Filterkuchen neigen. Die erhöhte Transmission
resultiert aus verminderten Abstoßungseffekten zwischen Membran und Protein aufgrund der
Ladungsneutralität des Proteins. Die durch das Fouling hohe Proteinkonzentration an der
Membranoberfläche begünstigt dabei die Transmission. Der selbe Effekt scheint auch bei
Hydrolysaten zu greifen. Pouliot et al. (1999) stellten bei der Fraktionierung von
Molkenproteinhydrolysat mit Molekülstrukturen ≤ 10 kDa unter Zugabe von 0,5 M NaCl bei
einem pH von 9 ebenfalls eine Steigerung der Transmission fest. Die Neutralisierung von
Ladungen zur Steigerung der Transmission entspricht dem zuvor beschriebenen Prinzip der
gezielten Filtration am IEP des Zielproteins und resultiert ebenfalls in verstärktem Fouling und
daraus folgendem Absinken des Permeatflux. Aber auch am IEP des Zielproteins lässt sich
die Transmission über eine gewisse Filtrationsdauer durch Zugabe geringer Mengen NaCl
verbessern. Eine Zugabe von 5 mM NaCl führte für de la Casa et al. (2007) bei einer Filtration
von BSA (pH 4,9) nicht nur zu einer Steigerung der Transmission zu Filtrationsbeginn (71 %),
sondern ermöglichte eine hohe Transmission über den gesamten Filtrationsprozess (57 – 62

Diskussion
189
%). Die Autoren führen dies ebenfalls auf eine Abschirmung verbleibender Partialladungen
von Proteinen und Membran durch NaCl-Ionen zurück. Die Erkenntnisse von Martin-Orue et
al. (1998) gehen noch einen Schritt weiter. Bei der Nanofiltration von vier kurzkettigen
Modellpeptiden arbeiten die Autoren heraus, dass nicht Nettoladung, sondern die Anzahl und
Art (positiv, negativ) der Partialladung entscheidend für den Transmissionsprozess eines
Peptids ist. Dem pH-Wert des Feeds wird dabei der größte Einfluss zugeschrieben, da dieser
die Ionisation des Feeds und die Oberflächenladung der Membran beeinflusst. Die Anzahl
negativer Partialladungen scheint entscheidend für die verminderte Transmission eines
Peptids zu sein. Durch eine Anpassung der Ionenstärke konnten ladungsbedingte
Rückweisungseffekte reduziert werden. Aus den Erkenntnissen wird deutlich, dass mit
steigender Komplexität des Feeds auch ladungsbedingte Effekte komplexer ausfallen. So sind
bei komplexeren Medien auch Verluste von intakten Proteinen durch Adsorption auf der
Membran, Denaturierung durch Scherkräfte oder den Zusammenschluss mit anderen
Proteinen möglich (Almécija et al., 2007). Diese Verluste resultieren in einer Reduktion der
Wiederfindungsrate während des jeweiligen Prozesses.
Der Einfluss von pH-Wert und Ionenstärke auf den Filtrationsprozess im Rahmen dieser Studie
ist folglich auf mehrere Effekte zurückzuführen. Bei der Fraktionierung ohne NaCl-Zusatz
resultierte die Anpassung des pH-Werts in Veränderungen des Ladungszustands und des
hydrodynamischen Radius der Moleküle. Abhängig vom pH-Wert kommt es darüber hinaus
zur Beeinflussung der Zusammensetzung und Dichte der Foulingschicht. Da in einem
Hydrolysat verschiedenste, nicht näher kategorisierte Molekülstrukturen vorliegen, können die
Effekte ohne eine detaillierte Analyse der Hydrolysatzusammensetzung nicht auf bestimmte
Strukturen zurückgeführt werden. Die verhältnismäßig niedrige WFR in Verbindung mit dem
geringeren Proteingehalt des Retentats bei pH 7 ist diesbezüglich auf eine irreversible
Adsorption bestimmter Moleküle auf der Membran zurückzuführen. Die Erhöhung der
Ionenstärke führte zu geringfügig gesteigerter Transmission, aber gleichzeitig auch zu
durchweg geringerer WFR. Dies ist vermutlich auf eine Reduktion der zwischenmolekularen
Abstoßungseffekte, sowie der Peptid-Membraninteraktion zurückzuführen, die aus NaCl-
basierenden Abschirmungseffekten resultiert. Die geringere WFR lässt sich abermals durch
irreversible Adsorption bestimmter Moleküle auf der Membran erklären. Interessant ist die
Tatsache, dass die erhöhte Transmission nicht mit einer Reduktion des Permeatflux, sondern
vielmehr mit gesteigertem Flux einherging, obwohl anhand der WFR auf eine ausgeprägte
Foulingschicht geschlossen werden kann. Im Vergleich zu einer Filtration ohne NaCl-Zusatz
handelte es sich hierbei vermutlich um eine schwerpunktmäßig extern auftretende
Foulingschicht, die durch die Ausbildung von größeren Aggregaten eine gesteigerte
Permeation der Foulingschicht und der Membran zulässt. Im Fall der Filtration ohne NaCl-

Diskussion
190
Zusatz ist die Ausbildung von internem Fouling denkbar, die zu einer Reduktion des
Porendurchmessers der Membran und daraus folgend zu geringerem Permeatflux führte.
Molekülgrößenverteilung der Fraktionen:
Bei der Betrachtung der Chromatogramme der Retentat-Fraktion wurde, weitestgehend
unabhängig vom pH-Wert, eine große Menge an Strukturen ≤ 1,35 kDa festgestellt (Abbildung
78 a, c - Abbildung 81 a, c). Moleküle ≥ 20 kDa wurden in allen Versuchen, unabhängig von
pH-Wert und Ionenstärke, im Retentat zurückgehalten (Abbildung 78 a, c - Abbildung 81 a, c).
Für pH 9 (Abbildung 81 b) konnte bei einer Fraktionierung ohne NaCl-Zusatz die anteilsmäßig
größte Menge an Strukturen ≥ 20 kDa im Permeat festgestellt werden. Eine Fraktionierung bei
erhöhter Ionenstärke (Zusatz von 150 mM NaCl) führte, deutlich bei pH 7 und pH 8 (Abbildung
79 c, Abbildung 80 c) erkennbar, zu einem geringeren Anteil an Strukturen ≥ 20 kDa im
Retentat. Bei Betrachtung der Permeat-Fraktionen wurde eine Verschiebung der
Membrantrenngrenze hin zu einem niedrigeren MWCO festgestellt (Abbildung 78 b, d -
Abbildung 81 b, d). Der Zusatz von 150 mM NaCl führte im Fraktionierungsprozess,
insbesondere für pH 7 (Abbildung 79 d), pH 8 (Abbildung 80 d) und pH 9 (Abbildung 81 d), zu
erhöhten Anteilen von Strukturen ≥ 20 kDa. Die Molekülgrößenverteilung der Nullproben war,
mit Ausnahme des nicht zur Bewertung herangezogenen pH 4 (Abbildung 73 a, b),
weitestgehend identisch (Abbildung 74 a, b - Abbildung 77 a, b).
Der pH-Wert besaß im Rahmen dieser Arbeit durch seinen Einfluss auf den Ladungszustand
der Moleküle einen entscheidenden Einfluss auf die Zusammensetzung der Retentat- und
Permeat-Fraktionen, wie besonders am Vergleich der Fraktionierung ohne Zusatz von NaCl
bei pH 7 und pH 9 erkennbar ist. Die Retentat-Fraktion von pH 7 (Abbildung 79 a) zeichnet
sich durch einen besonders hohen Anteil an Molekülen ≥ 20 kDa aus, während der Anteil an
Molekülen ≥ 20 kDa im Permeat (Abbildung 79 b) gering ist. Bei pH 9 ist ein wesentlich höherer
Anteil an Molekülen ≥ 20 kDa im Permeat (Abbildung 81 b) vorzufinden, der in ähnlichem
Umfang nur bei einer Fraktionierung unter NaCl-Zusatz (pH 7 (mit NaCl): Abbildung 79 d, pH
8 (mit NaCl): Abbildung 80 d, pH 9 (mit NaCl): Abbildung 81 d) erreicht werden konnte. Das
lässt darauf schließen, dass die von Almécija et al. (2007) und Pouliot et al. (1999)
beschriebenen ladungsbedingten Abstoßungs- bzw. Interaktionseffekte zwischen
verschiedenen Molekülen, sowie zwischen Molekülen und Membran den
Fraktionierungsprozess maßgeblich beeinflusst haben. Die Transmission von Feed-Molekülen
ist unter Berufung auf die zitierten Autoren, abhängig von den Partialladungen der Moleküle,
sowie deren daraus resultierenden Interaktion mit der Membran, der Foulingschicht oder
anderen Molekülen und führt im Falle von pH 7 (Abbildung 79 a, b) zu einem vergleichsweise
hohen Rückhalt von Strukturen ≥ 20 kDa im Retentat, wohingegen im Falle der Fraktionierung
bei pH 9 (Abbildung 81 a, b) das Ladungsprofil bestimmter Moleküle eine Transmission durch

Diskussion
191
die Membran ermöglichte. In Anlehnung an die von Huisman et al. (2000) für die Fraktionierung
von BSA erarbeiteten Ergebnisse wird darüber hinaus davon ausgegangen, dass
Eigenschaften wie Dicke und Dichte der Foulingschicht den Fraktionierungsprozess in
Abhängigkeit des pHs maßgeblich beeinflussen. Diese Faktoren führen in Kombination mit
dem vorab beschriebenen Einfluss des Ladungsprofils der Moleküle auf deren Transmission
zu einer komplexen Kaskade an Abläufen während des Fraktionierungsprozesses, die pH-
abhängig in Rückhalt oder Transmission bestimmter Moleküle resultiert.
Die anhand der WFR beurteilte, geringfügig höhere Transmission (Tabelle 30, Seite 112) bei
der Filtration unter NaCl-Zugabe resultierte in einem höheren Anteil an Molekülstrukturen ≥ 20
kDa im Permeat, insbesondere anhand der Chromatogramme von pH 7 (Abbildung 79 d), pH
8 (Abbildung 80 d) und pH 9 (Abbildung 81 d) erkennbar, der wahrscheinlich auf die von de la
Casa et al. (2007) und Persson et al. (2003) beschriebenen NaCl-induzierte
Abschirmungseffekte von Partialladungen zurückzuführen ist. Dabei spielte vermutlich die
Zusammensetzung und Dichte der entstehenden Foulingschicht eine Rolle. Abhängig von
Ladungszustand und dem Vorhandensein von NaCl-Ionen ist hierbei, in Anlehnung an
Persson et al. (2003), die Ausbildung von Aggregaten denkbar, die durch Ablagerung auf der
Membran eine für ungeladene Moleküle durchlässige Foulingschicht bildeten. Es wird folglich
davon ausgegangen, dass die Partialladungen bestimmter Moleküle ≥ 20 kDa durch NaCl-
Ionen abgeschirmt wurden, sodass diese aufgrund geringerer Abstoßungseffekte zwischen
Membran und Molekül, bzw. Molekül und Foulingschicht, die Membran passieren konnten.
Vergleicht man die in Retentat und Permeat festgestellten Anteile an Molekülen ≥ 20 kDa der
Fraktionierung ohne Zusatz von NaCl mit den Anteilen an Molekülen ≥ 20 kDa der
Fraktionierung unter Zusatz von NaCl und bezieht die erhöhte WFR des Permeat bei
gleichzeitig reduzierter WFR im Retentat der Fraktionierung unter Zusatz von NaCl mit ein,
lässt dies darauf schließen, dass der erhöhte Anteil an Molekülen ≥ 20 kDa im Permeat auf
eine Transmission selbiger durch die Membran zurückzuführen ist. Die im Falle der
Fraktionierung mit NaCl ermittelte, deutliche niedrigere Gesamt-WFR (pH 8 (ohne NaCl):
94,85 ± 4,63 %, pH 8 (mit NaCl): 81,74 ± 4,36 %; pH 9 (ohne NaCl): 91,10 ± 4,63 %, pH 9 (mit
NaCl): 77,67 ± 4,82 %) ist vermutlich in einer irreversiblen Adsorption von Molekülen auf der
Membran, bzw. der Foulingschicht, vergleichbar den von Pouliot et al. (1999) für
Molkenprotein-Hydrolysat beschriebenen ladungsbedingten Effekte zurückzuführen, da
insbesondere die Molekül-Membran-Interaktion bei Filtrationsprozessen zu einer irreversiblen
Adsorption von Molekülen auf der Membran führen kann (Huisman et al., 2000)
Das Zurückbleiben kleiner Moleküle im Retentat (Abbildung 78 a, c - Abbildung 81 a, c) lässt
sich, in Anlehnung an Nyström et al. (1994) und Saksena & Zydney (1994), durch
Ladungseffekte erklären, die unter bestimmten Feed-Bedingungen, insbesondere dem pH-

Diskussion
192
Wert zu einer Auftrennung gegen den Größengradienten der Moleküle führen können. In Falle
dieser Studie ist davon auszugehen, dass Strukturen ≤ 1,35 kDa durch Abstoßungseffekte
zwischen Molekül-Membran oder Molekül-Foulingschicht im Retentat zurückgehalten wurden.
5.3.2.2 Alge (Chlorella vulgaris)
Für Molkenprotein-Isolat wurde der Einfluss von pH-Wert und Ionenstärke auf die Parameter
Permeatflux JFm, Proteingehalt in Retentat mRet und Permeat mPer (Transmission), sowie die
Wiederfindungsrate diskutiert. Im Falle der Alge Chlorella vulgaris wurden darüber hinaus
weitere Daten zur Charakterisierung des Filtrationsprozesses erfasst. Die Permeatmenge VPer
zum Zeitpunkt t wurde über die Prozesszeit tF aufgetragen und die Regressionsfunktion der
Permeatzunahme, sowie deren Bestimmtheitsmaß R2 ermittelt. Zudem wurde der mittlere pH-
Wert des Retentats bestimmt, um etwaige pH-Wert abhängige Effekte beurteilen zu können.
Auf membrangestützten Filtrationsverfahren basierende Prozesse werden im Falle von
Mikroalgen insbesondere bei der Erzeugung (z.B. Behandlung des Wachstumsmedium für die
Rezirkulation) und Gewinnung (z.B. Separation der Biomasse aus dem flüssigen
Wachstumsmedium) der Algen eingesetzt, wie Drexler & Yeh (2014) in einem aktuellen
Review bezüglich der Anwendung membranbasierter Prozesse im Zusammenhang mit Algen
darlegen. Zur Gewinnung bestimmter Peptidverbindungen aus Algen-Hydrolysaten sind
säulenbasierte Gel-Filtrationsverfahren in Kombination mit weiteren
säulenchromatographischen Verfahren beschrieben (Sheih, Fang, & Wu, 2009a; Sheih, Wu,
& Fang, 2009b). Bei der Gewinnung intakter Algenzellen aus dem flüssigen
Wachstumsmedium per „Ausflockung“ spielt der pH-Wert eine entscheidende Rolle, wie
Studien von Pérez, Salgueiro, Maceiras, Cancela, & Sánchez (2017), Liu et al. (2013) & Wu
et al. (2012) belegen. Wu et al. (2012) erreichten für Chlorella vulgaris, Scenedesmus sp. und
Chlorococcum sp. eine Ausflockungs-Effizienz von > 90 % für pH-Werte > 10,6. Der Beginn
der Agglomeration wurde von den Autoren bereits ab einem pH > 8,6 beobachtet. Die
Ausflockungs-Effizienz, sowie das Auftreten von Fouling bei membranbasierten Prozessen
können dabei nicht losgelöst vom Vorhandensein extrazellulärer polymerer Substanzen
(EPS)/extrazellulärer organischer Stoffe (EOM) betrachtet werden (Drexler & Yeh, 2014; Wu
et al., 2012). Wu et al. (2012) untersuchten den Einfluss von freien Polysacchariden (RPS) auf
die Ausflockungs-Effizienz von Chlorella vulgaris. Konzentrationen von 25 mg/l bzw. 70 mg/l
RPS resultierten in einer Reduktion der Ausflockungs-Effizienz bei pH 10,6 von > 90 % auf <
40 % bzw. < 10 % (Wu et al., 2012).
Für die vorliegende Studie muss folglich der Einfluss von im Feed vorliegenden EPS/RPS,
insbesondere unter Berücksichtigung des Kohlenhydratanteils (12 %) im Rohstoff „Chlorella
vulgaris Powder“ (Lot. 201410215) (Allma, Lissabon, PRT) miteinbezogen werden.

Diskussion
193
Permeatflux JFm:
Die Anpassung des pH-Werts resultierte bei der im Rahmen dieser Studie durchgeführten
Fraktionierung von Algen-Hydrolysat ohne NaCl-Zusatz lediglich im Falle von pH 6 (42,91 ±
2,22 ml/min) (höchster Permeatflux der Versuchsreihe) im Vergleich zu pH 3 (29,15 ± 0,35
ml/min) (niedrigster Permeatflux der Versuchsreihe) in signifikant erhöhten Permeatflux (p <
0,05). Die verbleibenden pH-Stufen unterschieden sich nicht signifikant (p > 0,05).
Im Falle der vorausgehend diskutierten Fraktionierung von Molkenprotein-Isolat-Hydrolysat
wurde der Einfluss des pH-Wert auf den Permeatflux durch das Auftreten von Fouling
begründet (Kapitel 5.3.2.1, Seite 183). Die durch Fouling hervorgerufene Verminderung des
Flux wurde unter Berufung auf Arbeiten von, unter anderem, Huisman et al. (2000), Pouliot et
al. (1999) und Tarleton & Wakeman (1993) auf den Einfluss des pH-Werts auf die
Partikelgrößenverteilung, ladungsbedingten Interaktions- und Abstoßungseffekten, sowie die
damit in Verbindung stehende Struktur der Foulingschicht zurückgeführt. Die für
Molkenprotein-Isolat erarbeitete Argumentationskette stellt auch für den Einfluss des pH-
Werts auf die Fraktionierung der Alge Chlorella vulgaris ein Erklärungsszenario dar, bedarf
zusätzlich aber weiterer Betrachtung.
Eine Erhöhung des pH-Werts führt für intakte Mikroalgenzellen in Abhängigkeit des
Vorhandenseins extrazellulärer Substanzen (z.B. freie Polysaccharide), wie vorab dargestellt,
zur Ausflockung von bis zu 90 % der Zellen (Wu et al., 2012). Setzt man eine durch die
Ausflockung bedingte Ausbildung einer Foulingschicht voraus, entsprechen die Ergebnisse
der Autoren den Beobachtungen der vorliegenden Studie, in der ein Abfall des Permeatflux
bei steigendem pH (von pH 6 auf pH 11) beobachtet wurde, dessen Ausprägung, unter
Berufung auf das für Molkenprotein-Isolat dargestellt Szenario, durch eine pH-abhängig
unterschiedlich strukturierte Foulingschicht und somit einen pH-abhängig Verminderung des
Permeatflux zurückgeführt werden kann. Die Erkenntnisse weiterer Autoren lassen darüber
hinaus darauf schließen, dass auch eine Absenkung des pHs in den sauren Bereich in einer
Ausflockung intakter Algenproteine resultiert (Pérez et al., 2017; Liu et al., 2013). Liu et al.
(2013) beobachteten für die Mikroalgen Chlorococcum ellipsoideum, Chlorococcum nivale und
Scenedesmus sp. eine beginnenden Ausflockung bei der Absenkung des pHs von pH 6,7 auf
pH 5. Eine weitere Absenkung des pHs resultierte in einer Ausflockung > 85 % - > 95 %
zwischen pH 4,5 und pH 1,5, in Abhängigkeit der Alge. In der vorliegenden Studie wurde ein
signifikanter Abfall des Permeatflux für pH 3 im Vergleich zu pH 6 festgestellt, der unter
Voraussetzung der Annahme der Ausbildung einer Foulingschicht durch Ausflockung, mit den
Erkenntnissen der Autoren übereinstimmt.
Die durch Fouling hervorgerufene Reduktion des Permeatflux kann nicht alleinig durch eine
pH-Wert-abhängige Ausbildung von Fouling begründet werden. Das Vorhandensein von

Diskussion
194
extrazellulären polymeren Substanzen trägt ebenso zum Foulingprozess bei (Drexler & Yeh,
2014). Diese EPS bestehen zum Großteil aus Polysacchariden und Proteinen (Wicaksana,
Fane, Pongpairoj, & Field, 2012; Her, Amy, Park, & Song, 2004). Wie bereits vorab dargestellt,
resultiert das Vorhandensein von freien Polysacchariden in Abhängigkeit der Konzentration
(25 mg/l bzw. 70 mg/l) in einer deutlichen Reduktion der Ausflockung um bis zu > 80 % bei
durch Erhöhung des pH-Werts ausgelöster Ausflockung (Wu et al., 2012). In Anbetracht der
Ergebnisse dieser Studie, in denen die pH-abhängige Veränderung des Permeatflux lediglich
in einem Fall (vergleiche: pH 3 vs. pH 6) signifikant ausfiel, sowie der Berücksichtigung der
Rohstoffzusammensetzung (Auszug: Proteingehalt: 52,0 %; Fett: 9,6 %; Kohlenhydrate: 12,0
%; Ballaststoffe: 13,6 %; Asche: 6,8 %), muss davon ausgegangen werden, dass das
Vorhandensein von EPS den Filtrationsprozess beeinflusst hat. Hierfür kommen insbesondere
während des Vorbehandlungs-, Filtrations- und Hydrolyseprozess freigesetzte freie
Polysaccharide infrage. Algenzellen (Microcystis aeruginosa, Chlorella vulgaris) tragen über
einen breiten pH-Bereich negative Ladungen auf der Oberfläche (Hadjoudja et al., 2010).
Diese verhindern eine Aggregation der Zellen. Eine Verminderung der Oberflächenladung
über einen bestimmten Punkt hinaus führt zur Ausflockung (Liu et al., 2013). Auch das
Vorhandensein von positiv geladenen Ionen führt zu einer Reduktion der Abstoßung zwischen
den Zellen und förderte deren Ausflockung. Bei der Ausflockung von intakten Mikroalgen-
Zellen „konkurrieren“ RPS mit den (negativ geladenen) Algenzellen um (positiv geladene)
multivalente Ionen und führend hierbei zu einer Reduktion der Ausflockung (Chen, Li, Liu, &
Jiao, 2009).
Das im Rahmen dieser Studie verwendete Feed aus hydrolysierter Alge (Chlorella vulgaris)
verfügt über eine komplexe Zusammensetzung unterschiedlicher Molekülstrukturen, deren
Ladungsprofil pH-abhängig ist. Die Erkenntnisse der vorab zitierten Autoren beruhen auf
Untersuchungen zu intakten Algenzellen und können somit nicht ohne Weiteres auf Algen-
Hydrolysate übertragen werden. Die aufgezeigten Übereinstimmungen hinsichtlich der pH-
Abhängigkeit des Permeatflux deuten jedoch darauf hin, dass die für intakte Algenzellen
bekannten Einflussfaktoren auch als Hinweis für das pH-abhängige Verhalten hydrolysierter
Algen, wie die im Fall dieser Studie verwendete Chlorella vulgaris, betrachtet werden können.
Das Vorhandensein von EPS/RPS vermindert im Rahmen dieser Studie vermutlich den Effekt
der pH-Anpassung. Es wird darüber hinaus angenommen, dass die Gegenwart von EPS/RPS
zu einer Stabilisierung des Filtrationsprozesses durch geringere Ausflockung von Feed-
Molekülen führt.
Die Erhöhung der Ionenstärke durch Zusatz von 150 mM NaCl resultierte in dieser Studie in
einem Permeatflux JFm zwischen 26,67 ± 5,68 ml/min (pH 11) und 35,71 ± 5,22 ml/min (pH 6)
und lagen für die pH-Stufen 7, 11 signifikant unter dem Permeatflux einer Fraktionierung ohne

Diskussion
195
NaCl-Zusatz JFm (pH 7 (ohne NaCl): 38,27 ± 2,86 ml/min, pH 7 (mit NaCl): 31,15 ± 1,16 ml/min;
pH 11 (ohne NaCl): 37,94 ± 6,83 ml/min, pH 11 (mit NaCl): 26,67 ± 5,68 ml/min). Auch für die
weiteren pH-Stufen wurde eine Abnahme des Flux beobachtet.
Im Falle der vorausgehend diskutierten Fraktionierung von Molkenprotein-Isolat-Hydrolysat
wurde der Einfluss der Ionenstärke auf den Permeatflux, unter Berufung auf, unter anderem,
She et al. (2009) und Salgın (2007) in einer ionenstärke-abhängigen Veränderung des
hydrodynamischen Radius und des Ladungsprofils der Moleküle im Hydrolysat zurückgeführt
die sich auf die Ausbildung und Struktur der Foulingschicht auswirken (Kapitel 5.3.2.1, Seite
183).
Chatsungnoen & Chisti (2016) stellten bei Ausflockungsversuchen für Chlorella vulgaris unter
Zuhilfenahme verschiedener Fällungsmittel (Aluminium-Sulfat, Eisenchlorid) einen Einfluss
der Ionenstärke des Mediums auf die Menge an benötigtem Fällungsmittel fest. Mit steigender
Ionenstärke wurde weniger Fällungsmittel benötigt. Bilanovic, Shelef, & Sukenik (1988)
untersuchten den Einfluss der Polymere Chitosan, Zetag 63 und Zetag 92 auf die Ausflockung
von Mikroalgen. Dabei stellten die Autoren fest, dass eine Erhöhung der Ionenstärke in einer
deutlichen Reduktion der Ausflockungs-Effizienz resultierte. Molina Grima, Belarbi, Acién
Fernández, Robles Medina, & Chisti (2003) führen eine Verminderung der Wirksamkeit
kationischer Polymere auf eine strukturelle Veränderung (Zusammenfalten) selbiger zurück.
Die Annahme vorausgesetzt, dass im Feed vorhandene RPS, sowie weitere EPS über
Ladungspotentiale verfügen und somit beispielsweise ähnlich kationischer Polymere, bzw. als
deren Konkurrenten um eine freie Bindungsstelle wirken, kann auch im Falle des in dieser
Studie verwendeten Feeds aus hydrolysierter Alge (Chlorella vulgaris) davon ausgegangen
werden, dass sich eine Veränderung der Ionenstärke auf das Ladungsprofil dieser Moleküle
auswirkt und somit zu einer Molekül-Molekül Interaktion führt, die in der Ausbildung einer pH-
und ionenstärke-abhängigen Foulingschicht resultiert. Wie bereits im Diskussionsteil für die
Versuche mit Molkenprotein-Isolat hergeleitet, ist darüber hinaus davon auszugehen, dass die
Erhöhung der Ionenstärke sich im Fall dieser Studie auf den hydrodynamischen Radius und
das Ladungsprofil der Feed-Moleküle auswirkt. Der Einfluss der Ionenstärke auf den
Permeatflux von hydrolysierter Alge (Chlorella vulgaris) ist somit vermutlich auf mehrere
Effekte zurückzuführen und resultiert in der Ausbildung einer pH-abhängig unterschiedlich
strukturierten Foulingschicht, die den Permeatflux maßgeblich beeinflusst. Diese Effekte
wirken sich im Vergleich zu einer Filtration ohne den Zusatz von 150 mM NaCl negativ auf den
Permeatflux aus, sodass dieser unter den korrespondierenden Flux-Werten einer
Fraktionierung ohne NaCl-Zusatz liegen.

Diskussion
196
Proteingehalt von Retentat mRet & Permeat mPer, Wiederfindungsrate:
Die Wiederfindungsrate wurde durch die Anpassung des pH-Werts nicht signifikant beeinflusst
und lag bei der Fraktionierung ohne NaCl-Zusatz zwischen 68,64 ± 4,18 % (pH 3) und 77,11
± 1,58 % (pH 6). 12,47 ± 1,04 % (pH 3) bis 24,61 ± 3,92 % (pH 11) der verbleibenden
detektierbaren Verbindungen lagen im Retentat, 46,06 ± 5,43 % (pH 11) bis 63,14 ± 2,42 %
(pH 6) im Permeat vor. Der Proteingehalt im Retentat lag damit zwischen 0,093 ± 0,008 g (pH
3) und 0,185 ± 0,029 g (pH 11), sowie zwischen 0,345 ± 0,041 g (pH11) und 0,474 ± 0,018 g
(pH 6) im Permeat. Der Proteingehalt des Retentats bei einer Fraktionierung bei pH 11 lag
signifikant über den Gehalten der pH-Stufen pH 3, pH 6, pH 7, der Proteingehalt des Permeats
für pH 6 und pH 7 signifikant darunter.
Eine (Crossflow-)Filtration intakter Algenzellen findet vornehmlich zur Gewinnung von Zellen
aus dem Wachstumsmedium statt. Eine Vielzahl von Studien, unter anderem von Zhao et al.
(2017), Wicaksana et al. (2012) und Castaing et al. (2010) beleuchtet beispielsweise den
Einfluss des MWCO und Typ der Membranen, der Strömungsgeschwindigkeit und der Feed-
Konzentration auf den Filtrationsprozess verschiedener Mikroalgen. Hierbei werden die
Algenzellen stets im Retentat zurückgehalten. Der bei Separierungsprozessen zur Bewertung
herangezogene Faktor der Transmission wird selten betrachtet und behandelt, wie im Falle
der Studie von Wicaksana et al. (2012), die Transmission von EPS ermittelt über den Gehalt
des „Gesamten organischen Kohlenstoffs“ (TOC). Die Diskussion des Einflusses des pH-
Werts auf die Transmission im Falle der in dieser Studie durchgeführte Fraktionierung von
hydrolysierter Alge (Chlorella vulgaris) basiert aufgrund der Quellenlage auf
Veröffentlichungen anderer pflanzlicher Proteine.
Im Falle der vorausgehend diskutierten Fraktionierung von Molkenprotein-Isolat-Hydrolysat
wurde der Einfluss des pH-Wert auf Transmission und WFR, unter Berufung auf, unter
anderem, Huisman et al. (2000), auf der pH-abhängigen Veränderungen der Molekülgröße
von Proteine und Proteinaggregaten, der Protein-Membran-Interaktion und der Eigenschaften
der externen Foulingschicht zurückgeführt (Kapitel 5.3.2.1, Seite 183). In diesem
Zusammenhang spielt der Ladungszustand der Moleküle eine entscheidende Rolle. Studien
von Nyström et al. (1994) und Saksena & Zydney (1994) belegen anhand von
Modellversuchen, dass in Abhängigkeit des Ladungszustands der Feed-Moleküle sogar eine
selektive Fraktionierung gegen den Größengradienten möglich ist. Unter den richtigen
Bedingungen (Membran, pH, Transmembrandruck) lässt sich ein Feed aus zwei
Modellproteinen im Bereich des IEP fraktionieren. Hierbei wird das Zielprotein bedingt durch
eine Reduktion des hydrodynamischen Radius, sowie neutralem Ladungszustand ins Permeat
transportiert. Weitere Proteine werden durch ladungsbedingte Abstoßung und größeren
hydrodynamischen Radius im Retentat zurückgehalten (Nyström et al., 1994). Für komplexere

Diskussion
197
Feeds wie Molke ist eine selektive Fraktionierung nicht ohne Weiteres möglich, wie Ergebnisse
von Almécija et al. (2007) belegen.
Nach Ursu et al. (2014) liegt der IEP eines Großteils der Proteine der Alge Chlorella vulgaris
bei 4,0 – 5,5, sowie für einen kleineren Teil der Proteine bei 6,0 – 8,0. Im Rahmen dieser
Studie wurden im pH-Bereich 6 – 8 bei einer Fraktionierung ohne Zusatz von 150 mM NaCl
die höchste WFR, sowie eine hohe Transmission festgestellt, während eine Fraktionierung bei
pH 11 in verminderter WFR und Transmission resultierte. Anders als bei den von Nyström et
al. (1994) und Saksena & Zydney (1994) anhand von Modelllösungen aus Ovalbumin und
Myoglobin, sowie BSA und IgG durchgeführten Versuchen handelt es sich in dieser Studie um
ein Feed aus hydrolysierter Alge (Chlorella vulgaris), dessen molekulare Zusammensetzung
nicht näher charakterisiert ist. Es wird jedoch vermutet, dass der Einfluss des pHs auf
Transmission und WFR, unter Berufung auf die zitierten Autoren, sowie die bereits für die
Fraktionierung von Molkenprotein-Isolat entwickelte Hypothese in einer pH-induzierten
Änderung des hydrodynamischen Radius, sowie Abstoßungs- und Adhäsions-Effekten
resultiert, welche die Transmission, sowie die Foulingneigung und Struktur der Foulingschicht
beeinflussen. Die Gegenwart von RPS/EPS besitzt darüber hinaus vermutlich ebenfalls, wie
vorab dargestellt, einen Einfluss auf die pH-Stabilität des Feeds und beeinflusst somit auch
die Neigung zur Deckschichtbildung und Deckschichtstruktur.
Die Erhöhung der Ionenstärke durch Zusatz von NaCl resultierte in einer Wiederfindungsrate
zwischen 67,83 ± 12,66 % (pH 11) und 73,51 ± 1,44 % (pH 6). 14,89 ± 1,98 % (pH 7) bis 23,61
± 5,91 (pH 11) lagen im Retentat, 44,23 ± 6,77 % (pH 11) bis 57,15 ± 1,12 % (pH 7) im Permeat
vor. Der Proteingehalt im Retentat lag zwischen 0,112 ± 0,010 g (pH 3)/0,112 ± 0,015 g (pH
7) und 0,177 ± 0,044 g (pH 11), sowie zwischen 0,332 ± 0,051 g (pH 11) und 0,429 ± 0,008 g
(pH 7) im Permeat. Die Erhöhung der Ionenstärke resultierte in einem geringeren Proteinanteil
im Permeat.
Im Falle der vorausgehend diskutierten Fraktionierung von Molkenprotein-Isolat-Hydrolysat
wurde der Einfluss der Ionenstärke auf Transmission und WFR, unter Berufung auf, unter
anderem, de la Casa et al. (2007), Persson et al. (2003) und Pouliot et al. (1999) auf eine
Abschirmung geladener Moleküle durch NaCl-Ionen zurückgeführt. Die Abschirmung der
Partialladung resultierte in einer veränderten Struktur der Foulingschicht, beeinflusste die
Foulingneigung, sowie die Molekül-Molekül und Molekül-Membran-Interaktion und sorgte
somit für veränderte Transmissionseigenschaften. Im Falle des in dieser Studie verwendeten
Feeds aus hydrolysierter Alge (Chlorella vulgaris) ist zusätzlich davon auszugehen, dass der
vorab dargestellt Einfluss von EPS/RPS sich auch bei einer Fraktionierung unter Zusatz von
150 mM NaCl auf das Ladungsprofil der Feed-Moleküle auswirkt und somit den
Foulingprozess und daraus resultierende Einflüsse auf die Transmission und WFR

Diskussion
198
mitbestimmt. Die im Vergleich mit der bei einer Fraktionierung ohne Zusatz von 150 mM NaCl
niedrigere WFR, sowie die geringeren Proteinanteile im Permeat bei gleichzeitig ähnlichen
Proteinanteilen im Retentat sind damit vermutlich in einer geringeren Transmission begründet
die durch die Ausbildung einer weniger durchlässigen Foulingschicht aufgrund von Molekül-
Molekül-, sowie Molekül-Membran-Interaktion zustande kommt, welche teilweise zu einer
irreversible Adsorption bestimmter Moleküle auf der Membran führen. Im Falle einer Filtration
intakter Mikroalgen scheint der durch Fouling verursachte Einfluss auf den
Filtrationsprozesses hauptsächlich in der Ausbildung eines Filterkuchens begründet (Ahmad,
Mat Yasin, Derek, & Lim, 2012; Castaing et al., 2010). Aufgrund der komplexeren
Zusammensetzung des Feeds in dieser Studie, sind den Filtrationsprozess beeinflussende
Effekte, die durch Adsorption auf der Membran, bzw. in den Poren der Membran gemäß des
Modells von Wang & Tarabara (2008) auftreten, jedoch nicht auszuschließen.
Molekülgrößenverteilung der Fraktionen:
Die Beurteilung der „Güte der Fraktionierung“ findet im Rahmen dieser Studie unter
Zuhilfenahme der für die Fraktionierung hydrolysierter Alge (Chlorella vulgaris) zusätzlich
ermittelten und im Ergebnis-Abschnitt „Weitere Daten zur Charakterisierung des
Filtrationsprozesses“ dargestellten Daten statt.
Bei der Betrachtung der Chromatogramme der Retentat-Fraktion wurde vergleichbar der
anhand von Molkenprotein-Isolat gewonnenen Ergebnisse, weitestgehend unabhängig vom
pH-Wert eine große Menge an Strukturen ≤ 1,35 kDa festgestellt (Abbildung 90 a, c - Abbildung
94 a, c). Moleküle ≥ 20 kDa wurden im Falle einer Fraktionierung ohne 150 mM NaCl-Zusatz
nicht (pH 3: Abbildung 90 a, pH 8: Abbildung 83 a, pH 11: Abbildung 94 a) oder nur in sehr
geringen Anteilen (pH 6: Abbildung 91 a, pH 7: Abbildung 92 a) im Retentat zurückgehalten
bzw. festgestellt. Die Beobachtungen decken sich mit den Auswertungen zum
Bestimmtheitsmaß R2, das eine Aussage über die Linearität der Permeatzunahme ermöglicht
und somit eine Aussage über den Einfluss von Fouling auf den Filtrationsprozess ermöglicht.
Hier wurde für pH 6 (0,991 ± 0,004) und pH 7 (0,998 ± 0,003) die höchste Linearität der
Permeatzunahme festgestellt. Eine Fraktionierung bei erhöhter Ionenstärke (Zusatz von 150
mM NaCl) führte für pH 3 (Abbildung 90 c), pH 6 (Abbildung 91 c) und in geringerem Umfang
pH 8 (Abbildung 93 c) zu einem erhohten Anteil an Strukturen ≥ 20 kDa im Retentat. Über das
höchste Bestimmtheitsmaß R2 verfügte die bei pH 7 (0,990 ± 0,003) durchgeführte
Fraktionierung. Anhand der Permeat-Fraktionen ist, sowohl für die Fraktionierung unter Zusatz
von 150 mM NaCl als auch für die Fraktionierung ohne NaCl-Zusatz, eine deutliche
Verschiebung der Membrantrenngrenze hin zu einem niedrigeren MWCO erkennbar
(Abbildung 90 b, d - Abbildung 94 b, d). Molekülverbindungen ≥ 5 – 10 kDa konnten
unabhängig von pH-Wert und Ionenstärke nicht, oder nur in verschwindend kleinen Anteilen

Diskussion
199
nachgewiesen werden (pH 7: Abbildung 92 d, pH 8: Abbildung 93 d). Molekülstrukturen ≤ 1,35
kDa sind stark repräsentiert.
Der mangelnde Rückhalt, bzw. das Fehlen von Strukturen ≥ 20 kDa im Retentat ist im Falle
der Fraktionierung für pH 3, pH 8 und pH 11 ohne Zusatz von NaCl auf den anteilsmäßig nicht
nachweisbaren (pH 3, pH 11)/geringen (pH 8) Gehalt an Strukturen ≥ 20 kDa in der Nullprobe
zurückzuführen (pH 3: Abbildung 85 a, pH 8: Abbildung 88 a, pH 11: Abbildung 89 a). Die
Abtrennung von Molekülen ≥ 20 kDa ist unter Berufung auf Liu et al. (2013) bzw. Wu et al.
(2012) möglicherweise auf Ausflockung von Feed-Molekülen im Rahmen der Vorfiltration
zurückzuführen. Die Autoren beobachteten bei intakten Algenzellen eine Ausflockungs-
Effizienz von > 85 - 95 % bei Absenken des pHs unter pH 4,5, bzw. Anheben des pHs über
10,6. Für pH 6 (Abbildung 86 a) und pH 7 (Abbildung 87 a) verfügten die Nullproben über eine
breite Molekülgrößenverteilung. In Anbetracht der im Vergleich zur Filtration von
hydrolysiertem Molkenprotein-Isolat deutlich reduzierten WFR (vergleiche: WFR
(Molkenprotein-Isolat, pH 6: 89,64 ± 11,46 %, WFR (Alge (Chlorella vulgaris), pH 6: 77,11 ±
1,58 %) wird vermutet, dass Strukturen ≥ 20 kDa durch Molekül-Membran-, ähnlich der von
Pouliot et al. (1999) für Molkenprotein-Hydrolysat beschriebenen ladungsbedingten Effekte,
Molekül-Foulingschicht-Interaktion oder eine hohe Dichte der Foulingschicht als Teil einer
irreversiblen Deckschicht auf der Membran zurückgehalten wurden. Insbesondere die
Molekül-Membran-Interaktion kann bei Filtrationsprozessen zu einer irreversiblen Adsorption
von Molekülen auf der Membran führen (Huisman et al., 2000). Der bei erhöhter Ionenstärke
(Zusatz von 150 mM NaCl) für pH 3 (Abbildung 90 c), pH 6 (Abbildung 91 c) und in geringerem
Umfang pH 8 (Abbildung 93 c) nachgewiesene erhohte Anteil an Strukturen ≥ 20 kDa im
Retentat kann, vergleichbar der Fraktionierung von Molkenprotein-Isolat, auf die von de la
Casa et al. (2007) und Persson et al. (2003) beschriebenen, NaCl-induzierte
Abschirmungseffekte von Partialladungen zurückgeführt werden. So wird im Rahmen dieser
Studie davon ausgegangen, dass eine Abschirmung der Partialladung bestimmter Moleküle
zu einer Reduktion der Molekül-Membran-Interaktion führe. Strukturen ≥ 20 kDa wurden in
Folge aufgrund einer Reduktion des MWCO der Membran durch Ausbildung einer
Foulingschicht im Retentat zurückgehalten ohne eine irreversible Interaktion mit Membran
oder Foulingschicht einzugehen.
Die Chromatogramme der Permeat-Fraktionen werden unabhängig von pH und Ionenstärke
von Molekülen ≤ 1,35 kDa dominiert. Für die pH-Stufen pH 3, pH 8 und pH 11 ohne Zusatz
von NaCl ist der Mangel an Molekülen ≥ 1,35 kDa auf die anteilsmäßig nicht nachweisbaren
(pH 3, pH 11)/geringen (pH 8) Gehalte an Strukturen ≥ 20 kDa in der Nullprobe zurückzuführen
(pH 3: Abbildung 85 a, pH 8: Abbildung 88 a, pH 11: Abbildung 89 a). Diese gilt jedoch nicht
für die Nullproben von pH 3 (Abbildung 85 b), pH 8 (Abbildung 88 b) und pH 11 (Abbildung 89

Diskussion
200
b), die unter Zusatz von 150 mM NaCl gewonnen wurden. Unter Berufung auf die vorab
genannten Autoren wird davon ausgegangen, dass die geringen/nicht feststellbaren Anteil an
Strukturen ≥ 20 kDa im Permeat auf ein komplexes Zusammenspiel der beschriebenen Effekte
beruht. Kernpunkt der Argumentation ist dabei der pH- und ionenstärke-abhängige Einfluss
der Partialladungen, bzw. der NaCl-induzierten Abschirmungen von geladenen Molekülen.
Diese resultieren in einer pH- und ionenstärke-abhängigen Ausbildung einer Foulingschicht
durch Molekül-Molekül-, Molekül-Membran- und im weiteren Prozessverlauf Molekül-
Foulingschicht-Interaktion. Die Transmission von Molekülen ≥ 20 kDa wird in Folge durch die
pH- und ionenstärke-abhängige Molekülladung, sowie ladungsbedingte Interaktions- und
Abstoßungseffekte zwischen Molekülen, bzw. zwischen Molekülen und Membran bestimmt.
Wie bereits vorab dargestellt, wird dabei davon ausgegangen, dass bestimmte Moleküle
aufgrund irreversibler Adsorption auf der Membran bzw. durch Rückhalt aufgrund eines durch
Fouling reduzierten MWCO über der Membran zurückgehalten werden. Das Vorhandensein
von EPS/RPS im Feed, sowie die komplexe Feed-Zusammensetzung erschwert eine
dezidierte Zuweisung der Effekte zu bestimmten Verbindungen.
Zusätzlich zu der dargestellten Hypothese muss ein weiterer Faktor bei der Bewertung der
Zusammensetzung der Retentat- und Permeat-Fraktionen miteinbezogen werden. In Kapitel
5.2.1 (Seite 179) wurde unter Berufung auf Morris et al. (2008) und Roberts (2007)
herausgearbeitet, dass die Molekülgrößenverteilung der hydrolysierten Alge (Chlorella
vulgaris) für den Fall des im Rahmen dieser Studie verwendeten Hydrolyseverfahrens
(Vorbehandlung: HCl, pH 3, 48 h; Hydrolyse: Pronase 0,25 % (112,5 PU), 45 min) auf das
Vorhandensein von Aggregaten schließen lässt. Hinsichtlich des geringen Anteils an
Molekülen ≥ 20 kDa in Permeat und Retentat der Fraktionen hydrolysierter Alge (Chlorella
vulgaris) besteht die Möglichkeit, dass sich Aggregate, sowie auch langkettige Peptide durch
die Hydrodynamik des Prozesses in Verbindung mit den physikalisch-chemischen
Eigenschaften von Feed und Membran strukturell verändert haben. Meireles, Aimar, &
Sanchez (1991) stellten bei der Ultrafiltration von Bovin-Serum-Albumin (BSA) eine
Denaturierung des Proteins in Abhängigkeit der Strömungsgeschwindigkeit fest. Die
Aggregation von Proteinen, beispielweise ausgelöst durch elektrostatische oder hydrophobe
Wechselwirkungen, gilt zudem in Abhängigkeit von pH-Wert, Salzgehalt, der Größe des
Aggregats, sowie der Phase der Aggregation als reversibel (Wang, Nema, & Teagarden,
2010). Eine Veränderung der Molekülgrößenverteilung durch hydrodynamische, sowie
chemisch-physikalische Einflussfaktoren während der Fraktionierung kann, insbesondere in
Anbetracht der hohen WFR im Permeat (Fraktionierung ohne 150 mM NaCl: 46,06 ± 5,43 %
(pH 11) - 63,14 ± 2,42 % (pH 6); Fraktionierung mit 150 mM NaCl: 44,23 ± 6,77 % (pH 11) -
57,15 ± 1,12 % (pH 7)), nicht ausgeschlossen werden. Es wird vermutet, dass dieser Vorgang
parallel zu der vorab dargestellten pH- und ionenstärke-abhängigen Beeinflussung des

Diskussion
201
Fraktionierungsprozess durch ladungs- und abschirmungsbedingte Molekül-Molekül, Molekül-
Membran und Molekül-Foulingschicht-Interaktionen stattfindet.
5.3.2.3 Zusammenfassung der Diskussionspunkte
Permeatflux und Proteinausbeute in Retentat- und Permeat-Fraktion unterschieden
sich pH- und ionenstärke-abhängig.
Fouling spielt im Hinblick auf Permeatflux und Transmission eine entscheidende Rolle.
Die initiale Ausbildung einer Foulingschicht wird insbesondere durch kleine Moleküle
initiiert.
pH-Wert und Ionenstärke wirken auf die molekulare (Raum-)Struktur, beeinflussen die
Ausbildung und Struktur der Foulingschicht und somit auch die Transmission.
Ladungsbedingte Abstoßungs- bzw. Adsorptionseffekte bestimmten maßgeblich die
Filtrationscharakteristika und die Transmission von Proteinen.
Die Komplexität der während des Fraktionierungsprozess auftretenden Effekte steigt
mit der Komplexität der Feed-Zusammensetzung.
5.3.2.4 Zusammenfassung & Darstellung der Ergebnisse
Molkenprotein-Isolat:
o Der pH-Wert beeinflusst den Permeatflux von hydrolysiertem Molkenprotein-
Isolat nicht signifikant. Der höchste Permeatflux wurde für pH 9 (29,44 ± 8,19
ml/min), der niedrigste für pH 8 (20,01 ± 0,84 ml/min) festgestellt.
o Der pH-Wert beeinflusst die Transmission, sowie die Wiederfindungsrate in
Retentat und Permeat nicht signifikant.
o Der pH-Wert beeinflusst die Molekülgrößenverteilung der Fraktionen
geringfügig. Bei der Fraktionierung kommt es zu einer
Trenngrenzenverschiebung der Membran hin zu einem deutlich kleineren
MWCO. Molekülstrukturen ≤ 1,35 kDa sind in Retentat- und Permeatfraktion
stark repräsentiert.
o Die Ionenstärke beeinflusst den Permeatflux in Abhängigkeit des pHs
signifikant. Unter Zusatz von 150 mM wurde für pH 9 (49,11 ± 3,23 ml/min) der
höchste und für pH 6 (30,27 ± 6,01 ml/min) der niedrigste Permeatflux
festgestellt. Der Zusatz von 150 mM NaCl führt in Abhängigkeit des pH-Werts
zu signifikant erhöhtem Permeatflux im Vergleich zu einer Fraktionierung ohne
NaCl-Zusatz.
o Die Ionenstärke beeinflusst die Transmission, sowie die Wiederfindungsrate in
Retentat und Permeat in Abhängigkeit des pHs nicht signifikant. Der Zusatz von
150 mM NaCl führt in Abhängigkeit des pH-Werts zu signifikant niedrigerer
Wiederfindungsrate (pH 8, pH 9) im Vergleich zu einer Fraktionierung ohne

Diskussion
202
NaCl-Zusatz, ausgelöst durch geringere Proteingehalte im Retentat bei
gleichzeitig nur geringfügig erhöhten Proteingehalten im Permeat.
o Die Ionenstärke beeinflusst die Molekülgrößenverteilung der Fraktionen in
Abhängigkeit des pHs geringfügig. Der Zusatz von 150 mM NaCl führt in
Abhängigkeit des pH-Wert zu einem erhöhten Anteil an Strukturen ≥ 20 kDa im
Permeat im Vergleich zu einer Fraktionierung ohne NaCl-Zusatz.
Molekülstrukturen ≤ 1,35 kDa sind in Retentat- und Permeatfraktion stark
repräsentiert.
Alge (Chlorella vulgaris):
o Der pH-Wert beeinflusst den Permeatflux von hydrolysierter Alge (Chlorella
vulgaris) mit Ausnahme des Vergleichs der pH Stufen pH 6 (42,91 ± 2,22
ml/min) (höchster Permeatflux der Versuchsreihe) und pH 3 (29,15 ± 0,35
ml/min) (niedrigster Permeatflux der Versuchsreihe) nicht signifikant.
o Der pH-Wert beeinflusst die Wiederfindungsrate nicht signifikant. Die
Transmission wird durch den pH-Wert signifikant beeinflusst und resultiert in
einem höheren Proteingehalt im Retentat bei gleichzeitig niedrigerem
Proteingehalt im Permeat für pH 11 im Vergleich zu pH 3, pH 6, pH 7.
o Der pH-Wert beeinflusst die Molekülgrößenverteilung der Fraktionen
geringfügig. Bei der Fraktionierung kommt es zu einer
Trenngrenzenverschiebung der Membran hin zu einem deutlich kleineren
MWCO. Molekülstrukturen ≤ 1,35 kDa sind in Retentat- und Permeatfraktion
stark repräsentiert. Der pH-Wert besitzt jedoch maßgeblichen Einfluss auf die
Molekülgrößenverteilung der vorfiltrierten Nullprobe und führt für die pH Stufen
3, 8 und 11 zu einem nicht nachweisbaren (pH 3, pH 11)/geringen (pH 8) Anteil
an Strukturen ≥ 20 kDa.
o Die Ionenstärke beeinflusst den Permeatflux in Abhängigkeit des pHs nicht
signifikant. Der Zusatz von 150 mM NaCl führt in Abhängigkeit des pH-Werts
für die pH Stufen 7 und 11 zu signifikant niedrigerem Permeatflux im Vergleich
zu einer Fraktionierung ohne NaCl-Zusatz.
o Die Ionenstärke beeinflusst die Transmission, sowie die Wiederfindungsrate in
Retentat und Permeat in Abhängigkeit des pHs lediglich in einem Fall signifikant
(Permeat-Proteingehalt: pH 11 (0,332 ± 0,051) vs. pH 7 (0,429 ± 0,008 g)). Der
Zusatz von 150 mM NaCl führt in Abhängigkeit des pH-Werts zu signifikant
niedrigerer Wiederfindungsrate (pH 6) im Vergleich zu einer Fraktionierung
ohne NaCl-Zusatz.
o Die Ionenstärke beeinflusst die Molekülgrößenverteilung der Fraktionen in
Abhängigkeit des pHs geringfügig. Lediglich bei pH 3, pH 6 und, in geringem

Diskussion
203
Umfang, pH 8 wurden erhöhte Anteile an Strukturen ≥ 20 kDa im Retentat
festgestellt. Der Zusatz von 150 mM NaCl führt in Abhängigkeit des pH-Werts
nicht zu einem erhöhten Anteil an Strukturen ≥ 20 kDa im Permeat im Vergleich
zu einer Fraktionierung ohne NaCl-Zusatz. Molekülstrukturen ≤ 1,35 kDa sind
in Retentat- und Permeatfraktion stark repräsentiert.
Hypothese zum Einfluss von pH- und Ionenstärke auf den Fraktionierungsprozess:
o pH-Wert und Ionenstärke beeinflussen ladungsbedingte Abstoßungs- und
Adsorptionseffekte während der Fraktionierung und nehmen somit Einfluss auf
die Ausbildung und Struktur der Foulingschicht.
o Die Ladungsprofile der Moleküle, Membran und Foulingschicht, sowie deren
Interaktionseffekte bestimmten den Fraktionierungs- und
Transmissionsprozess.
o Die komplexe Zusammensetzung und Molekülgrößenverteilung von
Hydrolysaten erschwert die Identifikation fouling-induzierender Verbindungen
in Abhängigkeit der physikalisch-chemischen Feed-Charakteristika.
o Im Falle der Fraktionierung hydrolysierter Alge (Chlorella vulgaris) ist der
Einfluss von extrazellulären polymeren Substanzen (EPS)/freien
Polysacchariden (RPS) auf den Fraktionierungsprozess nicht auszuschließen.
o „Ideale“ Fraktionierungsbedingungen für Hydrolysate konnen unter
Berücksichtigung aktueller Primärliteratur nur durch „trial and error“ identifiziert
und durch auf den Erkenntnissen basierenden Folgestudien reduziert werden.
5.3.3 Einfluss der Ultraschall-Anwendung
Die Verwendung von Ultraschall kann bei Filtrationsprozessen in einer Erhöhung des
Permeatflux resultieren, wie Muthukumaran, Kentish, Ashokkumar, et al. (2005), Kobayashi et
al. (1999) und Chai et al. (1998) bei der Filtration von Molke bzw. Dextran-Lösungen
feststellten. Die Wirksamkeit der Ultraschallanwendung ist dabei unter anderem abhängig von
der US-Frequenz, der Feed-Konzentration und dem Transmembrandruck (Kyllönen et al.,
2005; Muthukumaran, Kentish, Ashokkumar, et al., 2005). Neben der Anwendung von
Ultraschall während des Filtrationsprozesses erwies sich auch die US-Vorbehandlung von
erhitzter Molke als gangbarer Weg zur Reduktion von Porenverblockung und Wachstum des
Filterkuchens, führte aber zu geringerem Permeatflux (Koh et al., 2014). Erkenntnisse zur
Filtration von Molkenprotein-Hydrolysaten unter Ultraschall existieren bisher nicht.
Permeatflux JFm:
Der Permeatflux JFm wurde in der vorliegenden Arbeit durch die Anwendung von Ultraschall
nicht signifikant beeinflusst und lag zwischen 6,38 ± 1,61 ml/min (ohne US) und 5,81 ± 2,36
ml/min (mit US). Die Ergebnisse stehen im Widerspruch zu den Erkenntnissen von

Diskussion
204
Muthukumaran, Kentish, Ashokkumar, et al. (2005), die bei der Filtration von Molke (intakte
Proteine) eine Fluxsteigerung um Faktor 1,4 – 1,7 feststellten. Die Erkenntnisse weiterer
Autoren beziehen sich ebenfalls auf die Filtration intakter Proteine (z. B. BSA) oder verwenden
Modellsysteme (z. B. Sulfat-Polystyren-Latex-Partikel) (Lamminen et al., 2004; Matsumoto et
al., 1996). Die Wirksamkeit der Ultraschall-Anwendung ist abhängig von einer Vielzahl an
Faktoren, die von Kyllönen et al. (2005) in einem umfangreichen Review-Artikel
zusammengefasst wurden. Entscheidend für ultraschall-basierende Effekte sind unter
anderem die US-Frequenz, die Leistungsstärke der US-Einheit, Feed- und Membran-
Eigenschaften, sowie die Prozesshydrodynamik. Muthukumaran, Kentish, Ashokkumar, et al.
(2005) führen die Wirkweise des Ultraschall auf den Einfluss von Kavitation, akustischer
Gleichströmung, Agglomerations- und Suspensionseffekt zurück. Diese Effekte treten mitunter
gleichzeitig auf und wirken synergetisch (Kyllönen et al., 2005). Gemäß Lamminen et al. (2004)
tragen Kavitations-Mechanismen zur Ablösung von Partikeln von der Membranoberfläche bei,
die darauf folgend, bedingt durch akustische Gleichströmung, von der Membranoberfläche
abgetragen werden. Kavitation tritt bei US-Frequenzen zwischen 20 kHz und 1000 kHz auf
(Kyllönen et al., 2005). Der Kavitation-Effekt ist druckabhängig und nimmt mit steigendem
externen Druck ab (Leighton, 1997). Muthukumaran, Kentish, Ashokkumar, et al. (2005)
gewichten den Einfluss der akustischen Gleichströmung höher, da durch Ultraschall auch bei
erhöhtem Transmembrandruck und damit verringerter Kavitation einen gleichbleibend hoher
Permeatflux erzielt werden konnte. Der Einfluss von US auf die Proteinstruktur und die damit
verbundenen Agglomerations- bzw. Suspensionseigenschaften wird kontrovers diskutiert.
Chandrapala, Zisu, Palmer, Kentish, & Ashokkumar (2011) stellten bei der US-Behandlung
(20 kHz) von Molkenproteinkonzentrat nur geringfügige Änderungen der Sekundärstruktur der
Proteine fest. Eine lange Prozessdauer führte jedoch zur Ausbildung von Proteinaggregaten.
Die Bildung von Agglomeraten kann generell durch eine durch hydrodynamische Turbulenzen
verstärke Kollision strukturell veränderter Proteine ausgelöst werden (Jambrak, Mason, Lelas,
Paniwnyk, & Herceg, 2014). Dem entgegen stehen Jambrak et al. (2014) die bei der US-
Behandlung (20 kHz und 40 kHz) von Molkenproteinkonzentrat eine Abnahme des
Molekülgewichts und der Partikelgrößen, sowie eine Verengung der Partikelgrößenverteilung
feststellten. Die Autoren führten die Abnahme der Partikelgröße auf das Lösen
intermolekularer, hydrophober Wechselwirkungen zurück. Die Abnahme der
Molekülgrößenverteilung erfolgt durch Aufspaltung der Peptidbindungen. Hiervon waren
besonders große Moleküle (14 – 70 kDa) betroffen (Jambrak et al., 2014).
Die US-Behandlung hat im Rahmen dieser Studie nur einen vernachlässigbaren Einfluss auf
den Permeatflux JFm. Es ist davon auszugehen, dass der Filtrationsprozess von
Molkenproteinhydrolysaten, vergleichbar mit der zuvor diskutierten Filtration im größeren
Maßstab, durch ladungsbasierte Abstoßungs- und Interaktionseffekte zwischen Membran und

Diskussion
205
Molekül, bzw. zwischen verschiedenen Molekülen, wie von Almécija et al. (2007) und Pouliot
et al. (1999) beschrieben, zurückzuführen ist, die den Prozess aufgrund der komplexen
Zusammensetzung des Feeds dominieren. Der in der vorliegenden Studie festgestellte
geringere Permeatflux unter US Anwendung mit verhältnismäßig großer Standardabweichung
ist durch Foulingeffekte gemäß des Modells von Wang & Tarabara (2008) erklärbar. Bei der
Filtration mit US wurde eine deutliche Abnahme des Permeatflux zu Versuchsende, bei
gesteigertem Flux zu Versuchsbeginn festgestellt. Hierbei scheint vor allem internes Fouling
unter Einfluss akustischer Gleichströmung eine Rolle zu spielen. Ein mögliches Szenario ist
nachfolgend dargestellt.
Zu Versuchsbeginn erfolgt unter dem Einfluss akustischer Gleichströmung ein erhöhter
Transport von Molekülen zur Membran (Lamminen et al., 2004). Kleine Partikel penetrieren
die Membran und adsorbieren in den Membranporen. Während der Einfluss externen Foulings
durch US-Behandlung verringert werden kann, lässt sich der Einfluss internen Foulings durch
US nicht mindern (Muthukumaran et al., 2007; Muthukumaran, Kentish, Ashokkumar, et al.,
2005; Muthukumaran et al., 2004). Mit zunehmender Prozesslaufzeit wächst der Fouling-
Widerstand durch zusätzlich auftretendes, vergleichsweise vermindertes externes Fouling und
resultiert in Verbindung mit vergleichsweise stärkerem internem Fouling in einem deutlichen
Abfall des Permeatflux. Durch Ausbleiben US-induzierter Effekte, passiert die Ausbildung von
externem und internem Fouling bei der Filtration ohne US gleichmäßig über den
Prozesszeitraum. Durch den für die Versuche gewählten pH-Wert (pH 7) ist
prozessübergreifend, unter Berufung auf die zuvor dargestellten Ergebnisse, davon
auszugehen, dass externes Fouling maßgeblichen Einfluss auf den Filtrationsprozess hat.
Proteingehalt von Retentat mRet & Permeat mPer, Wiederfindungsrate:
Die Wiederfindungsrate wurde durch die Anwendung von Ultraschall nicht signifikant
beeinflusst und lag zwischen 92,24 ± 7,65 % (mit US) und 92,80 ± 8,39 % (ohne US). 29,83 ±
3,34 % (mit US) - 31,54 ± 3,05 (ohne US) des Proteinanteils lagen im Retentat, 61,26 ± 5,71
% (ohne US) - 62,41 ± 6,65 % (mit US) im Permeat vor. Der Proteingehalt im Retentat lag
damit zwischen 89,50 ± 10,03 mg (mit US) und 94,63 ± 9,14 mg (ohne US), sowie zwischen
183,78 ± 17,13 mg (ohne US) und 187,24 ± 19,96 mg (mit US) im Permeat.
Während der Einfluss des pHs und der Ionenstärke auf die Transmission von Molkenproteinen
und Proteinmodellsystemen, wie zuvor dargestellt, umfangreich behandelt wurde, gibt es keine
Erkenntnisse über den Einfluss einer Ultraschall-Anwendung auf die Transmission von
Molkenproteinen und Molkenprotein-Hydrolysaten. Kobayashi et al. (1999) konnten aus den
Ergebnissen der Filtration einer Dextran-Lösung unter US lediglich eine verminderte
Konzentration gelöster Stoffe an der Membranoberfläche herleiten. Dies würde, ausgehend
von der Annahme des überwiegenden Vorhandenseins ladungsbedingt zurückgewiesener

Diskussion
206
Moleküle, in einer Steigerung der Transmission resultieren (Ghosh, 2003). Im Gegenzug
begünstigt eine erhöhte Konzentration gelöster Moleküle an der Membranoberfläche die
Transmission, wenn keine ladungsbedingten Rückweisungseffekte auftreten (Persson et al.,
2003). In dieser Studie wurde kein US-Einfluss auf die Transmission von Molkenprotein-
Hydrolysaten durch eine PES-Membran (MWCO 100 kDa) festgestellt. Dies ist, unter Berufung
auf die im vorhergehenden Abschnitt hergeleiteten Effekte, vermutlich auf den dominanten
Einfluss ladungsbedingter Molekül-Membran- und Molekül-Molekül-Interaktionen
zurückzuführen, die Geschwindigkeit und Art des Membranfoulings beeinflussen. Der US ist
somit nicht in der Lage eine Steigerung der Transmission bei komplexen Feed-Medien
herbeizuführen, da ladungsbedingte Interaktions- und damit verbundene Fouling-Effekte
prozessbestimmend sind.
Molekülgrößenverteilung der Fraktionen:
Die Chromatogramme weisen, vergleichbar mit der WFR und den Proteingehalten der
Fraktionen, keine US-induzierten Unterschiede auf. Ähnlich der Filtration von Molkenprotein-
Isolat-Hydrolysat in größerem Maßstab, wurden im Retentat Moleküle ≥ 44 kDa, sowie eine
große Menge an Verbindungen ≤ 1,35 kDa zurückgehalten. Im Permeat ist lediglich eine
geringe Molekülmenge ≥ 10 kDa vertreten. Die Fraktion wird von Molekülen ≤ 1,35 kDa
dominiert. Auf einen die Molekülgrößenverteilung oder den Transmissionsprozess
beeinflussenden Effekt des Ultraschalls, durch beispielweise kavitationsbedingte Ablösung
von Partikeln von der Membranoberfläche oder Agglomeration von Proteinen und damit in
einer veränderten Struktur der Foulingschicht resultierenden Vorgang, kann anhand der
Chromatogramme nicht geschlossen werden, sodass die Beobachtungen auf die in den
vorhergehenden Abschnitten, sowie in Kapitel 5.3.2.1 bereits ausführlich beleuchten Effekte
zurückgeführt werden.
5.3.3.1 Zusammenfassung der Diskussionspunkte
Die fluxsteigernde Eigenschaft des US lässt sich auf Kavitation, akustischer
Gleichströmung, Agglomerations- und Suspensionseffekt zurückführen.
Der Einfluss der akustischen Gleichströmung wird höher gewichtet als
Kavitationseffekte.
US kann zur Agglomeration von Proteinen, sowie zu strukturellen Veränderung und
einer Abnahme des Molekülgewichts führen.
Ladungs- und ionenstärkeabhängige Interaktionseffekte sind auch durch die
Einwirkung von US nicht zu verhindern.

Diskussion
207
5.3.3.2 Zusammenfassung & Darstellung der Ergebnisse
Permeatflux, WFR, Proteingehalte in Retentat und Permeat, sowie die
Molekülgrößenverteilung der Fraktionen sind durch die Anwendung von US bei der
Fraktionierung von Molkenprotein-Isolat-Hydrolysaten nicht signifikant beeinflussbar.
Die Anwendung von US resultierte in vergleichsweise geringerem Permeatflux bei
höheren Standardabweichungen.
Bei der Fraktionierung von hydrolysiertem Molkenprotein-Isolat sind ladungs- und
ionenstärke-induzierte Effekte stärker ausgeprägt als US-Effekte.
5.3.4 Eignung von Membranen aus regenerierter Cellulose
Hydrophile PS/PES- (Polysulfon/Polyethersulfon) und RC- (regenerierte Cellulose)
Membranen werden in der Filtrationstechnik häufig eingesetzt. Neben Cellulose Acetat (CA)
ist regenerierte Cellulose das hydrophilste Membranmaterial (Walter et al., 2012). CA-
Membranen sind jedoch aufgrund ihrer geringeren pH-Stabilität nicht für alle
Filtrationsprozesse geeignet (Pearce, 2007).
Im Rahmen dieser Studie wurden hydrophilierte PS bzw. PES Membranen verwendet. PES
Membranen verfügen über eine höhere nichts-spezifische Bindungsaffinität als RC
Membranen, neigen somit also eher zu Protein-Membran-Interaktionen (Pitt, 1987). Durch
eine in der Herstellungspraxis gängige gezielte Oberflächenbehandlung lassen sich die nicht-
spezifische Bindungsaffinität der PES Membran jedoch deutlich mindern (Walter et al., 2012).
Die Eignung von Membranen aus regenerierter Cellulose für den Fraktionierungsprozess von
Molkenprotein-Isolat-Hydrolysaten wurde exemplarisch für die pH-Stufe 9 unter Verwendung
von kleinvolumigen Membrantrennkammern überprüft.
Permeatflux JFm:
Der Permeatflux JFm bei der Fraktionierung von Molkenproteinhydrolysat lag in der
vorliegenden Studie bei 8,46 ± 1,60 ml/min. Aufgrund der Durchführung des Versuchs im
Kleinmaßstab ist kein direkter Vergleich zum Permeatflux der Anlagenkonzeption der
Hauptversuche möglich. Der Flux liegt jedoch über dem Wert (6,38 ± 1,61 ml/min) der im
Rahmen der US-Versuche unter Verwendung von PES-Membranen ohne US-Anwendung
ermittelt wurde. Hier wurde allerdings eine höhere Feed-Konzentration verwendet. Die
Performance von PES-Membranen kann sich herstellerbedingt unterscheiden (Jönsson &
Jönsson, 1995). Dizge, Soydemir, Karagunduz, & Keskinler (2011) führen Unterschiede des
Initial- und Steady-State-Flux bei der Filtration von Belebtschlamm auf
Oberflächeneigenschaften wie Rauigkeit, Porengeometrie und Regelmäßigkeit zurück.
Unregelmäßige, raue Membranoberfläche führen dabei zu verstärktem Fouling und daraus
resultierendem Fluxabfall. Der Einfluss des hydrophilen Charakters einer Membran scheint nur

Diskussion
208
zu Beginn eines Prozess entscheidend für adsorptionsbedingtes Fouling zu sein. Bei der
Filtration von BSA stellten Nakamura & Matsumoto (2006) fest, dass die Adsorption von
Protein nur in der Startphase durch den hydrophoben/hydrophilen Charakter der Membran
beeinflusst wird, im weiteren Verlauf aber pH- und ionenstärke-abhängig ist. Der Permeatflux
von hydrophilen Membran ist jedoch vergleichsweise höher (Jönsson & Jönsson, 1995).
Proteingehalt von Retentat mRet & Permeat mPer, Wiederfindungsrate:
Die Wiederfindungsrate lag bei 109,96 ± 17,00 %. 38,22 ± 7,28 % des Proteins lagen im
Retentat und 71,74 ± 10,27 % im Permeat vor. Dies entsprach 57,33 ± 10,92 mg Protein im
Retentat und 107,61 ± 15,41 mg im Permeat. Die erhöhte WFR ist, unter Berücksichtigung der
Stammdaten, auf einen Ausreißer zurückzuführen, der den hier dargestellten Mittelwert
beeinflusst. Zudem können geringfügige Einwaagefehler aufgrund der geringen
Einwaagemenge von wenigen mg an dieser Stelle in einem erhöhten Endproteingehalt des
Feeds und somit höheren Proteingehalten in Retentat und Permeat resultieren. Das Verhältnis
zwischen Transmission und Rückhalt der Molekülverbindungen entspricht in etwa den
Ergebnissen der Hauptversuche für pH 9 (WFR: 91,10 ± 4,63 %, Retentat: 38,07 ± 1,56 %,
Permeat: 53,02 ± 3,22 %). Die die Transmission der Proteine beeinflussenden Prozesse
lassen sich auf die in Kapitel 5.3.2 beschriebenen Effekte zurückführen.
Molekülgrößenverteilung der Fraktionen:
Die Chromatogramme der Retentat- und Permeatfraktion unter Verwendung von RC
Membranen entsprechen den Chromatogramme der Fraktionen, die unter Verwendung von
hydrophilierten PS-Membranen ermittelt wurden. In der Retentatfraktion wurden
Molekülstrukturen ≥ 44 kDa zurückgehalten, Strukturen ≤ 1,35 kDa sind weiterhin stark
repräsentiert. In der Permeatfraktion wurde nur eine geringfügige Menge an Strukturen ≥ 20
kDa detektiert, während Verbindungen ≤ 1,35 kDa überrepräsentiert sind. Die Ergebnisse der
Versuche sind aufgrund der anlagenbedingten Unterschiede der Versuchssysteme als
Hinweis zu werten. Unter Berücksichtigung der Gegebenheiten offenbarte die Verwendung
von RC-Membranen keinen Vorteil gegenüber der Verwendung von hydrophilierten PS-
Membranen.
5.3.4.1 Zusammenfassung der Diskussionspunkte
Hydrophile Membranen werden in Filtrationsprozessen aufgrund geringer
Foulingneigung und hohem Permeatflux bevorzugt.
Unterschiedliche Membranmaterialien beeinflussen Permeatflux und Fouling.
Die Adsorption von Protein auf Membranen ist nur in der Startphase der Filtration
abhängig von hydrophoben/hydrophilen Eigenschaften der Membran.

Diskussion
209
5.3.4.2 Zusammenfassung & Darstellung der Ergebnisse
RC-Membranen verfügen im Vergleich zu PS-Membranen, unter Berücksichtigung der
Versuchsbedingungen, nicht über veränderte Trenncharakteristika.
5.3.5 Dialyse zur Abtrennung kleiner Moleküle (< 2 kDa) aus dem Feed
Die Dialyse ist ein auf dem Diffusionsprinzip basierendes Trennverfahren zur Abtrennung
niedermolekularer Substanzen (Klein, Ward & Lacey, 1987). Die Trennung im Dialyse-Prozess
erfolgt durch einen Konzentrationsgradienten zwischen zwei membrangetrennten Phasen
(Baynes & Dominiczak, 2014). Die Effektivität der Dialyse nimmt mit steigender Molekülgröße
(> 1000 Da) ab (Nath, 2008).
Im Rahmen dieser Studie wurde überprüft, ob niedermolekulare Bestandteile (< 2 kDa) aus
einem zur Fraktionierung vorgesehenen vorfiltrierten Feed unter Variation des Initial-pH-Werts
per Dialyse abgetrennt werden können. Darüber hinaus wurde der Einfluss einer Variation der
Dialysedauer und der Konzentration des Feeds betrachtet. Hintergrund des Versuchs war die
Überprüfung der Eignung der Dialyse zur Abtrennung kleiner Moleküle (< 2 kDa) aus der zur
Fraktionierung vorgesehenen Nullprobe.
pH-Wert, Dialysezeit tDL & Molekülgrößenverteilung der Fraktionen:
Die pH-Versuche fanden unter Verwendung eines „Initial-pHs“ statt. Unter Initial-pH wird der
pH-Wert der Dialyseprobe verstanden der zu Beginn des Dialyseprozess, sowie bei Wechsel
der Dialyselösung eingestellt wurde. Der pH-Wert der Dialyseprobe ist somit über die Dauer
des Prozesses als nicht konstant zu betrachten, wurde jedoch zu definierten Zeitpunkten
(Wechsel der Dialyselosung) neu justiert. Die Dialyselosung „destilliertes Wasser“ wurde mit
dem Hintergrund gewählt, das Feed frei von den zur Herstellung von Dialysepuffern üblichen
Salzen, Aminen und ähnlichen Zusätzen mit Pufferwirkung zu halten, die eine Aufreinigung
des Retentats vor einem nachfolgenden Fraktionierungsprozess notwendig machen würden.
Die für die untersuchten pH-Stufen identifizierten Effekte werden nachfolgend dargestellt.
In Abhängigkeit des Initial-pH-Werts wurde für die pH-Stufen 3 und 5 (1 % (w/v)
Molkenprotein-Isolat, 24 h) eine Veränderung der Molekülgrößenverteilung der Nullprobe
festgestellt. Diese ist vermutlich auf eine Abtrennung von Molekülen ≥ 20 kDa im Rahmen der
Vorfiltration der Proben zurückzuführen. Der IEP der meisten intakten Molkenproteine liegt
zwischen pH 4 und pH 5 (Töpel, 2004). Ein Ausfall intakter Moleküle erscheint möglich. Die
Löslichkeit von Molkenproteinhydrolysaten nimmt jedoch mit steigendem Hydrolysegrad,
weitestgehend unabhängig vom pH-Wert zu und liegt bei einem DH von 8 – 12 % zwischen
70 % und 85 % (Perea, Ugalde, Rodriguez, & Serra, 1993). Hinsichtlich dieser Erkenntnis
erscheinen ladungsbedingte Interaktions- und Abstoßungseffekte zwischen Molekül und
Membran, sowie Molekül und Molekül, wie von Pouliot et al. (1999) und Pincet et al. (1995)

Diskussion
210
beschrieben, wahrscheinlicher. Eine daraus resultierende Deckschichtbildung kann darüber
zu einer Reduktion der nominalen Trenngrenze der Filtrationsmembran führen (Wang &
Tarabara, 2008; Marshall et al., 1993). Dies ermöglicht den Rückhalt von Molekülverbindungen
≥ 20 kDa während der Vorfiltration.
Die Nullproben der Initial-pH-Stufen 9 und 11 (2 % (w/v) Molkenprotein-Isolat, 48 h) wiesen
ebenfalls eine veränderte Molekülgrößenverteilung auf. Bei den Nullproben derselben Initial-
pH-Stufen wurden für den 24-stündigen Prozess, sowie bei einer Konzentration von 4 % (w/v)
keine Abweichungen von der erwarteten Molekülgrößenverteilung der Nullprobe festgestellt.
Eine Abtrennung im Rahmen der Vorfiltration durch ungenügend in Lösung gebrachtes
Probenmaterial, könnte bei der Vorbereitung des 48-stündigen Prozess in einer Abtrennung
von Molekülverbindungen ≥ 20 kDa resultiert haben. Dies erscheint in Anbetracht der
Durchführung separater Dreifachversuche jedoch unwahrscheinlich.
Feed-Konzentrationen ≥ 2 % (w/v) führten für die Initial-pH-Stufen < 9 zum Ausfall von gelösten
Stoffen im Verlauf des Dialyseprozesses. Während des Dialyseprozesses wurden vom
Ausgangswert abhängige Veränderungen (pH-Abfall) des pH-Werts, basierend auf dem pH-
Unterschied zwischen Probe und Dialyselösung, beobachtet. Vergleichbares wurde von Craig,
King, & Stracher (1957) bei der Verwendung der gleichen Dialyselösung beobachtet. Abhängig
vom pH-Wert kann es zur Aggregation einzelner Peptide kommen (Fung, Hong, Keyes-Baig,
& Chen, 2005). Die pH-bedingte Ausbildung einer Peptid-Peptid-Interaktion ist abhängig von
der Art der Peptidverbindung (Sette et al., 1992). Ein Aggregatbildung kann, vergleichbar zum
Löslichkeitsverhalten intakter Proteine, zu einem Ausfall des Peptid-Aggregats führen
(Samanen, 1990). Das Auftreten von Ausfällungserscheinungen wird folglich auf eine
Verringerung der Löslichkeit gelöster Stoffe durch eine pH-induzierte Ausbildung von Peptid-
Peptid-Aggregaten zurückgeführt.
Der Dialyseprozess führte für alle Initial-pH-Stufen (ausgenommen pH 3, 1 % (w/v)
Molkenprotein-Isolat) zu einer signifikanten Reduktion des Proteingehalts mnD im Retentat
„nach Dialyse“. Diese Reduktion kann, unter Berufung auf die chromatographisch ermittelte
Probenzusammensetzung, auf ein Auswaschen kleiner Moleküle durch den Dialyseprozess
zurückgeführt werden. Der Auswaschungsprozess erscheint, übereinstimmend mit Nath
(2008), besonders für Strukturen ≤ 1 kDa effektiv zu sein. Der Grad der Auswaschung
unterscheidet sich bei einer Feed-Konzentration von 1 % (w/v) nicht pH-abhängig. Bei einer
Steigerung der Feed-Konzentration wurden signifikanten Unterschiede in Abhängigkeit des
Initial-pHs und der Dialysedauer tDL festgestellt. Eine höhere Dialysezeit resultierte Initial-pH-
abhängig in signifikant niedrigeren Konzentrationen im Retentat „nach Dialyse“. Der Einfluss
des Initial-pH kann für eine Feed-Konzentration von 2 % (w/v) nicht alleinig auf den pH-Wert
zurückgeführt werden, da sich die Proteingehalte der Nullproben bereits pH-bedingt

Diskussion
211
unterscheiden. Selbiges gilt für den Einfluss der Dialysedauer. Bei einer Feed-Konzentration
von 4 % (w/v) erscheint der Einfluss des Initial-pHs jedoch gesichert. Trotz höherem
Anfangsproteingehalt resultierte die Dialyse bei pH 11 im Vergleich zu pH 9 in einem
geringeren Proteingehalt mnD „nach Dialyse“. Darüber hinaus konnte bei erhöhtem
Anfangsproteingehalt (2 % (w/v) Molkenprotein-Isolat, 4 % (w/v) Molkenprotein-Isolat) eine
schärfere Abtrennung von Molekülen ≤ 1 kDa festgestellt werden. Bei einer Dialyse von
Hämoglobin stellten Guidotti & Craig (1963), in Übereinstimmung mit dieser Studie, eine
vergleichsweise höhere Transmission bei pH 11 im Vergleich zu pH 9 fest. Die treibende Kraft
des Dialyseprozesses ist der transmembrane Konzentrationsgradient. Eine Erhöhung der
Feed-Konzentration resultierte, bis zum Erreichen eines Grenzwertes, in einem effektiveren
Dialyseprozess (Klein, Ward & Lacey, 1987). Dies wurde auch im Rahmen dieser Studie
beobachtet. Darüber hinaus spielen bei der Dialyse, vergleichbar zu druckgetriebenen
Filtrationsprozessen, ladungsbedingte Abstoßungs- und Interaktionseffekte eine Rolle.
Dialyse-Membranen sind meist negativ geladen. Dies führt zu einer Rückweisung von
Molekülen negativer Ladung, sowie zu einer Abnahme der effektiven Porengröße für das
betroffenen Molekül (Mota & Simaes Goncalves, 1996). Die Initial-pH-abhängigen,
signifikanten Unterschiede zwischen pH 9 und pH 11 (4 % (w/v) Molkenprotein-Isolat)
hinsichtlich des Proteingehalts lassen sich folglich, wie für druckgetriebenen
Filtrationsprozessen bereits ausführlich dargestellt wurde (Kapitel 5.3.2), auf unterschiedliche
Ladungsprofile der Feed-Medien zurückführen. Chromatographisch waren keine pH-
abhängigen Unterschiede erkennbar. Bereits Craig & King (1955) führten unterschiedliche
Transmissionsraten (hier: „escape rate“) in Dialyseprozessen von Strukturen ähnlichen
Molekülgewichts auf Membraninteraktion und ladungsbedingte Effekte zurück. Darüber hinaus
führten die Autoren, sowie auch Craig, King, & Stracher (1957) strukturelle Veränderungen als
Grund für unterschiedliche Transmissionsraten (hier: „escape rate“) ins Feld. Diese resultieren
in einer Auftrennung bei der nicht direkt auf das Molekülgewicht der die Membran
passierenden Moleküle geschlossen werden kann.
Geringere Protein-Gehalte im Retentat „nach Dialyse“ können beim 48-stündigen
Dialyseprozess auf eine verstärkte, ladungsinduzierte Peptid-Membran-Interaktionen
zurückgeführt werden, wie sie auch bei druckgetriebenen Membrantrennverfahren auftreten.
Hierbei kommt es durch elektrostatische Interaktion zwischen Membran und Molekül zur
Anlagerung des Moleküls (Jones & O’Melia, 2000). Darüber hinaus wurde beim 48-stündigen
Prozess ein Ausfall gelöster Stoffe zum Versuchsende beobachtet. Bei 65-stündigen
Diffusionsversuchen von humanem Albumin (Fraktion V) wurde von Hoch & Turner (1960)
ebenfalls Trübung und „Unloslichkeit“ der Feed-Moleküle zu Prozessende festgestellt. Im
Rahmen dieser Studie ist die Beobachtung vermutlich auf eine Aggregatbildung verschiedener
Peptide zurückzuführen, die das Löslichkeitsverhalten beeinflusst (Samanen, 1990). Denkbar

Diskussion
212
wäre auch eine Kombination beider Phänomene, bei der es zu Beginn zu einer Anlagerung
von Molekülen auf der Membranoberfläche kommt. Daraus resultiert eine Deckschichtbildung
die den gradientengetriebenen Transportprozess von Molekülen zur Membran in einer
erhöhten Kontaktrate zwischen Peptiden und somit verstärkt zur Ausbildung von Peptid-Peptid
Aggregaten führt.
In Anbetracht der Ergebnisse ist die Anwendung der Dialyse hinsichtlich eines Einsatzes bei
der Abtrennung von niedermolekularen Strukturen (< 2 kDa) aus dem Feed (Nullproben)
geeignet. Hierfür können höher konzentrierte, kleine Flüssigkeitsmengen dialysiert und darauf
folgend auf die gewünschte Feed-Konzentration verdünnt werden. Im Falle einer Abtrennung
niedermolekularer Strukturen aus Retentat- oder Permeatfraktionen sind diese bevorzugt in
eingeengtem Zustand zur dialysieren, da der Dialyseprozess bei erhöhter Konzentration und
damit höherem Konzentrationsgradienten effektiver arbeitet.
5.3.5.1 Zusammenfassung der Diskussionspunkte
Die Abtrennung von Molekülen ≤ 1,35 kDa erfolgt in Dialyseprozessen besonders
effektiv.
Abhängig vom Initial-pH-Wert kommt es während des Dialyseprozesses zum Ausfall
gelöster Stoffe, der auf Peptid-Peptid-Interaktion und daraus resultierender
Aggregatbildung zurückzuführen ist.
Initial-pH-abhängige Performance-Unterschiede sind, vergleichbar druckgetriebenen
Trennverfahren, auf ladungsbedingten Interaktions- und Abstoßungseffekten zwischen
Molekülen, bzw. Molekül und Membran zurückzuführen.
5.3.5.2 Zusammenfassung & Darstellung der Ergebnisse
Durch Dialyse lassen sich Moleküle ≤ 1,35 kDa aus Molkenprotein-Isolat-Hydrolysaten
auswaschen.
Eine Dialysezeit tDL von 24 h ist dafür ausreichend. Erhöhte Dialysezeit führt zum
Ausfall von gelösten Stoffen durch Aggregatbildung.
Hohe Konzentrationen (4 % (w/v)) resultierten im Vergleich mit niedrigeren
Konzentrationen (1 % (w/v)) in verbesserter Auswaschung.
Initial-pH-bedingte Unterschiede sind chromatographisch nicht oder nur schwer
erkennbar und spiegeln sich nur in signifikant unterschiedlichem Proteingehalt der
Retentatfraktion „nach Dialyse“ wider. In Abhängigkeit des Initial-pH wurde jedoch ein
Ausfall gelöster Stoffe beobachtet.
5.3.6 Einfluss weiterer Faktoren
Nachfolgend wird der Einfluss weiterer filtrationsrelevanter Faktoren diskutiert, die im Rahmen
dieser Studie, basierend auf Literaturempfehlungen, ausgewählt wurden.

Diskussion
213
Strömungsgeschwindigkeit ʋ:
Die Strömungsgeschwindigkeit ʋ korreliert mit dem Permeatflux. Eine Steigerung der
Strömungsgeschwindigkeit ʋ resultiert in einem gesteigerten Permeatflux, sowie einem
höheren kritischen Flux JCF (Choi et al., 2005; Madaeni, Fane, & Wiley, 1999; Li, Fane, Coster,
& Vigneswaran, 1998). Höherer Permeatflux, bzw. JCF in Verbindung mit einer Erhöhung der
Strömungsgeschwindigkeit, werden auf ein Abtragen von Partikeln von der
Membranoberfläche durch gesteigerte Scherkräfte zurückgeführt (Melin & Rautenbach, 2007;
Wang & Song, 1999).
Im Rahmen dieser Studie wurde eine Strömungsgeschwindigkeit ʋ von 0,94 m/s verwendet.
Madaeni et al. (1999) und Choi et al. (2005) stellten bei der Filtration von Belebtschlamm einen
Anstieg des JCF für Strömungsgeschwindigkeiten ʋ zwischen 0,5 m/s und 1,5 m/s bzw. 0,1
m/s und 4,5 m/s fest. Der Anstieg des Permeatflux fiel bei Choi et al. (2005) für
Mikrofiltrationsmembranen deutlicher aus als für Ultrafiltrationsmembranen. Auch in
Modellsystemen unter Verwendung von Latexpartikeln (3 µm, 6,4 µm, 11,9 µm) wurde von Li
et al. (1998) ein Anstieg des Permeatflux bei steigenden Strömungsgeschwindigkeiten ʋ bis
0,82 m/s festgestellt. Darüber hinaus stieg der Permeatflux in Abhängigkeit der
Strömungsgeschwindigkeit ʋ mit zunehmender Partikelgröße unter Verwendung einer
Membran mit einem MWCO von 0,02 µm.
Die Strömungsgeschwindigkeit ʋ kann nicht losgelöst von weiteren Kenngrößen beurteilt
werden. Ein Anheben der Strömungsgeschwindigkeit resultierte für Madaeni et al. (1999) in
einem Anstieg des Transmembrandrucks ΔpTMP. Dieser Anstieg hat lässt sich auf
verschiedene Ursachen zurückführen. Insbesondere beim Rücktransport größerer Partikel
kann es bei gesteigerter Strömungsgeschwindigkeit zu einer Erhöhung des
Deckschichtwiderstands durch Verminderung der Deckschichtporosität kommen (Fane et al.,
2000; Lojkine et al., 1992). Trotz gesteigertem Permeatflux kann laut Thomassen, Faraday,
Underwood, & Cleaver (2005) eine daraus resultierende Erhöhung des Transmembrandrucks
ΔpTMP zu verminderter Transmission führen. Darüber hinaus ist bei hohen
Strömungsgeschwindigkeiten eine Denaturierung gelöster Moleküle möglich, wie Meireles et
al. (1991) bei der Ultrafiltration von BSA feststellten.
Feed-Konzentration CF:
Hohe Feed Konzentrationen CF resultieren in Abhängigkeit von Strömungsgeschwindigkeit
und den Charakteristika des Feeds in vergleichsweise niedrigem Permeatflux (Harker et al.,
2013; Wakeman & Tarleton, 2005; Tarleton & Wakeman, 1994).
Die während der Vorarbeit für diese Studie gewählte Feed-Konzentration CF von 3 % (w/v)
stellte sich als zu hoch für die Fraktionierung von Molkenprotein-Isolat-Hydrolysaten heraus.

Diskussion
214
Die SE-HPLC Analyse offenbarte eine starke Verschiebung der Trenngrenze der Membran,
sodass ein Großteil der Moleküle > 10 kDa in der Retentatfraktion verblieb.
Zur Optimierung der Fraktionierung wurde im Rahmen dieser Studie eine Feed-Konzentration
von 0,15 % (w/v) verwendet. Bei der Fraktionierung von Protein-Modelllösungen oder
Hydrolysaten werden häufig Feed-Konzentrationen < 1 % verwendet. Pouliot et al. (1999)
nutzten bei der Ultra- und Nanofiltration von Molkenproteinhydrolysat zur Bestimmung der
individuellen Filtrationsperformance verschiedener Membranen unter Variation der
Ionenstärke und des pH-Werts eine Feed-Konzentration von 1 % (w/v). Wijers et al. (1998)
arbeiteten bei der Entsalzung von Molkenproteinhydrolysaten mittels Nanofiltration mit einer
Feed-Konzentration von 0,5 % (w/v). Huisman et al. (2000) verwendeten zur Identifizierung
von Protein-Protein- und Protein-Membran-Interaktionseffekte bei der Mikrofiltration von BSA
eine Feed-Konzentration von 0,05 % (w/v). Selbst bei Feed-Konzentrationen < 0,001 % (w/v)
sinkt der Permeatflux im Laufe eines Filtrationsprozesses deutlich ab und nähert sich einem
Steady-State Flux an, wie She et al. (2009) bei der Ultrafiltration von BSA feststellten.
Die Wahl der optimalen Feed-Konzentration für die Fraktionierung ist des Weiteren von
verschiedenen Feed-Charakteristika abhängig. Tarleton & Wakeman (1993) beobachteten bei
Filtration von Kalzit-Partikeln eine Erhöhung des Permeatflux mit Zunahme der Partikelgröße.
Außerdem stellten die Autoren fest, dass bei unterschiedlicher Partikelgrößenverteilung die
Ausbildung von Fouling hauptsächlich von kleinen Partikeln bestimmt wird. Darüber hinaus
beeinflussen Strömungsgeschwindigkeit und Transmembrandruck Deckschichtcharakteristika
und resultieren damit bei gleicher Feed-Konzentration in unterschiedlichen
Filterkuchenwiderständen und Transmissionseigenschaften (Park et al., 2008; Lawler &
Kweon, 2004; Vyas et al., 2000; Tarleton & Wakeman, 1993).
Die Wahl der Feed-Konzentration kann unter den dargestellten Gesichtspunkten folglich nicht
alleinig anhand des Einflusses der Feed-Konzentration auf den Permeatflux beurteilt werden.
Die Partikelgrößenverteilung, sowie die Wahl der Strömungsgeschwindigkeit und des
Transmembrandrucks, wirken sich auf Transmission und Eigenschaften der Deckschicht aus
und beeinflussen somit den Trennprozess. Zur Ermittlung der Performance einer neuen
Anlagenkonzeption erscheinen niedrige Feed-Konzentrationen daher sinnvoll. Bei Erreichen
des gewünschten Trennziels kann eine schrittweise Erhöhung der Feed-Konzentration die
Wirtschaftlichkeit der Methode erhöhen.
Temperatur T:
Eine gezielte Temperatursteuerung des Prozesses kann, durch Viskosität-Abnahme, zu einem
erhöhtem Permeatflux führen. Dies wird unter anderem auf eine Verringerung der
Deckschichtdicke bei geringerer Viskosität zurückgeführt (Vetier et al., 1988). Wie bereits in

Diskussion
215
Kapitel 2.1.4.2 dargestellt, kann der Einfluss der Temperatur dabei nicht losgelöst von pH-Wert
und Ionenstärke betrachtet werden, da strukturelle Veränderungen von Molekülen zu einer
Verminderung elektrostatischer Abstoßung zwischen Molekül und Membran, bzw. zwischen
Molekülen, führen können (Mo et al., 2008).
Im Rahmen dieser Studie wurde die Feed-Lösung auf 45 °C temperiert. Butylina et al. (2006)
wählten bei der Fraktionierung von Molkenpeptiden eine Prozesstemperatur von 50 °C um
bakterielles Wachstum einzuschränken und industriellen Anforderungen zu genügen. Atra et
al. (2005) stellten bei der Ultrafiltration von Milch und Molke eine Steigerung des Permeatflux
bei einer Prozesstemperatur von 50 °C im Vergleich zu 30 °C und 40 °C fest. Die Autoren
führen dies auf eine Abnahme der Viskosität sowie verbesserte Diffusionseigenschaften und
gesteigerten Rücktransport gelöster Stoffe zurück.
Pelegrine & Gasparetto (2005) untersuchten das Löslichkeitsverhalten von Molkenproteinen
im Bereich zwischen 40 °C – 60 °C für verschiedene pH-Werte im Bereich zwischen 3,5 – 7,8.
Bei 43 °C lag die Proteinlöslichkeit pH-abhängig zwischen 78,78 ± 0,41 % (pH 4,5) und 88,94
± 0,35 % (pH 7,8). Erst ab Temperaturen > 50 °C kam es pH-abhängig zu einem deutlichen
Abfall der Löslichkeit für die pH-Stufen 4,5 und 6,8. Die Autoren führen dies auf gesteigerte
Protein-Protein Interaktion aufgrund geringer elektrostatischer Abstoßungskräfte für die
betroffenen pH-Stufen zurück. Hydrolysate sind weniger temperaturempfindlich als intakte
Proteine. Die Hydrolyse von Molkenprotein erhöht die Temperaturstabilität und Löslichkeit
(Adler-Nissen, Eggum, & Munck, 2014; Hidalgo & Gamper, 1977). Im Vergleich zu
unhydrolysiertem Molkenprotein verfügt mit Trypsin hydrolysiertes Molkenprotein über hohe
Löslichkeit bei 80 °C in einem pH Bereich von 2 – 12 (Adler-Nissen et al., 2014; Hidalgo &
Gamper, 1977). Auch bei Weizengluten führt bereits eine Teilhydrolyse (DH 2,8 %) zu
deutlicher verbesserter Wasserlöslichkeit zwischen 40 % (pH 6) und 80 % (pH 10).
Temperaturen > 50 °C führen jedoch zu einer Abnahme der Löslichkeit (Wang et al., 2006).
Unter den Gesichtspunkten der Löslichkeitseigenschaften, der Temperaturstabilität der
Hydrolysate und mikrobiologischen Gesichtspunkten erscheint die Wahl einer
Prozesstemperatur von 45 °C geeignet für die Filtration von Proteinhydrolysaten. Ein deutlich
erhöhter Permeatflux durch Reduktion der Viskosität des Feeds erscheint bei der gewählten
Feed-Konzentration CF von 0,15 % unwahrscheinlich.
Diafiltration:
Die Trennschärfe von hoch- und niedermolekularen Substanzen kann bei Crossflow-
Prozessen durch das Verfahren der Diafiltration verbessert werden (Irmler, 2001). Abhängig
von der Häufigkeit der Zugabe eines Diavolumens lässt sich, unter Verwendung des
Diafiltrationsverfahren, eine erhöhte Reinheit der Retentatfraktion erreichen (Baldasso,

Diskussion
216
Barros, & Tessaro, 2011; Venkiteshwaran, Heider, Teysseyre, & Belfort, 2008; Wakeman &
Tarleton, 2005; van Eijndhoven, Saksena, & Zydney, 1995).
Im Rahmen dieser Studie wurde auf eine diskontinuierliche Diafiltration unter Verwendung von
vier Diavolumina VDia (Nd = 4) zurückgegriffen. Goodnight Jr., Hartman Jr., & Marquardt (1976)
nutzen eine Kombination aus Ultra- und Diafiltration zur Abreicherung von Kohlenhydraten in
einer wässrigen Soja-Protein-Suspension. Bereits ab einer Zugabe von eineinhalb VDia (Nd =
1,5) stieg die Reinheit des Retentats von 77 % auf 95 %. Durch die Verwendung von drei VDia
(Nd = 3) wurde eine Reinheit von 97 % erreicht. Baldasso et al. (2011) verwendeten ebenfalls
eine Kombination aus Ultra- und Diafiltration zur Abreicherung von Lactose bei der
Aufreinigung von Molkenproteinen. Die Autoren steigerten die Proteinkonzentration im
Retentat von 15 % auf bis zu 71 % unter Verwendung von vier Diafiltrationsschritten. Hierbei
wurde in den ersten beiden Schritten je ein VDia und in den darauf folgenden je ein halbes VDia
zugegeben (∑ Nd = 3). Darüber hinaus erwies sich die Diafiltration kleinerer Volumina (5 l
verglichen mit 6 l) als geeignet für die Aufkonzentration von Molkenproteinen. Venkiteshwaran
et al. (2008) nutzten das Diafiltrationsverfahren bei der Mikrofiltration zur Trennung von
ausgefälltem IgG und BSA bei der Filtration von Rinderserum. Durch die Zugabe von fünf VDia
(Nd = 5) gelang es 97 % des BSA aus dem Retentat zu entfernen und eine Reinheit des IgG
von 93 % zu erreichen. Die Autoren führten den Verbleib von geringen BSA Menge im Retentat
unter Berufung auf Saksena & Zydney (1994) auf die Ausbildung von BSA-Aggregaten, bzw.
BSA-IgG-Interaktion, zurück.
Das Verfahren der Diafiltration eignet sich folglich zur Auswaschung niedermolekularer
Komponenten aus komplexen Feed-Medien. Ein komplettes Entfernen niedermolekularer
Bestandteile ist dennoch nicht möglich, deren Anteil kann aber deutlich reduziert werden
(Walter et al., 2012). Bereits durch die Zugabe von 3 – 5 VDia (Nd = 3 – 5) lassen sich, in
Abhängigkeit des Feeds, Retentat-Reinheiten von bis zu 97 % erreichen (Baldasso et al.,
2011; Venkiteshwaran et al., 2008; Goodnight Jr. et al., 1976).

Diskussion
217
5.4 Sensorik
5.4.1 Schulung des Sensorik-Panels
Prüfung auf Geschmacksblindheit (gemäß DIN EN ISO 8586:2014):
Zur Aufnahme des Ist-Zustand des Panels wurde eine Prüfung auf Geschmacksblindheit
gemäß DIN EN ISO 8586:2014 unter Verwendung überschwelliger Konzentrationen wässriger
Standards durchgeführt.
Die daraus hervorgehenden Ergebnisse bestätigen grundlegend die Eignung aller
Prüfpersonen für die Teilnahme an einer sensorischen Studie. Lediglich in einem Fall wurde
der Grenzwert von 80 % korrekter Zuordnungen unterschritten (DIN Deutsches Institut für
Normung e.V., 2014b). Das Nichterkennen einer Geschmacksausprägung wurde überwiegend
für „bitter“ beobachtet. Erkenntnisse von Hall, Bartoshuk, Cain, & Stevens (1975) deuten auf
eine bimodale Verteilung des Schwellenwerts der Bitterstoffe Phenylthiocarbamid (PTC) und
Koffein hin. Dies resultiert, besonders bei niedrigen Konzentrationen der Bitterstoffe, in einem
Erkennen bzw. Nichterkennen des Grundgeschmacks „bitter“ in Abhängigkeit des individuellen
Schwellenwerts (Hall et al., 1975). Die Ergebnisse von Tanimura & Mattes (1993) bestätigten
diese These. Die Autoren untersuchten die „bitter“-Sensibilität von Koffein- und Nicht-Koffein-
Konsumenten und ermittelten einen signifikant niedrigeren „bitter“-Schwellenwert für die
Population der Nicht-Koffein-Konsumenten. Für PTC sind mehrere Arten von Nicht-
Schmeckern bekannt. Frank & Korchmar (1985) unterteilten die Gruppe der „PTC-Nicht-
Schmecker“ in zwei Untergruppen, basierend auf einer Reaktionszeit-Analyse. Ähnliche
Unterschiede hinsichtlich der Sensibilität gegenüber Koffein sind denkbar. Reed, Tanaka, &
McDaniel (2006) gehen davon aus, dass eine unterschiedliche „bitter“-Wahrnehmung in
Abhängigkeit individueller genetischer Unterschiede besteht.
Die Prüfung auf Geschmacksblindheit wurde nicht als Ausschlusskriterium für die Teilnahme
am Panel gewertet, da von einer Verbesserung der Panel-Performance hinsichtlich
individueller Schwellenwerte und Vorhersagegenauigkeit im Rahmen der Prüferschulung
ausgegangen wurde (Labbe, Rytz, & Hugi, 2004; Pangborn, 1959).
Prüfung auf Wahrnehmung eines Reizes (gemäß DIN EN ISO 8586:2014):
Zur Schulung des Panels wurde eine Prüfung auf Wahrnehmung eines Reizes gemäß DIN EN
ISO 8586:2014 unter Anwendung einer Dreiecksprüfung gemäß DIN EN ISO 4120:2007
durchgeführt. Die dabei verwendeten überschwelligen Probenkonzentrationen orientierten
sich an den Vorgaben der Norm.
Die Ergebnisse belegen grundsätzlich die Eignung des gesamten Panels zur signifikanten
Unterscheidung (p < 0,05) von Prüflösungen. Hierfür sind bei einer Panelgröße von 23 Prüfern

Diskussion
218
mindestens 12 korrekte Antworten (je Prüfkategorie) notwendig (Quadt et al., 2009). Mehrfach
auftretende fehlerhafte Zuordnungen einiger Prüferpersonen verdeutlichen dennoch die
Sinnhaftigkeit einer gezielten Prüferauswahl im Anschluss an den Schulungsprozess.
Für die Grundgeschmacksarten „umami“, „sauer“ und „bitter“ kam es am häufigsten zu
fehlerhaften Zuordnungen. Die bei einigen Teilnehmern ersichtlichen Probleme bei der „bitter“-
Wahrnehmung können, wie bereits dargestellt, auf das Vorhandensein unterschiedlicher
Schwellenwerte für Koffein, sowie individuelle genetische Unterschiede zurückgeführt werden
(Reed et al., 2006; Hall et al., 1975). Fehlerhafte Zuordnungen von „umami“ resultierten nach
Angaben der Prüfer auf die Ähnlichkeit der Geschmacksausprägung zu „salzig“. Durch die
Wiederholung des Tests verbesserte sich der Anteil an korrekten „umami“-Zuordnungen von
65,22 % (Tag 1) auf 78,26 % (Tag 3). Ergebnisse von Kobayashi & Kennedy (2002) bestätigen
den positiven Effekt von sensorischer Erfahrung auf die Wahrnehmung von „umami“-Reizen
in Lebensmitteln. Darüber hinaus ist eine geringe Sensibilität für „umami“, ähnlich der von Hall
et al. (1975) beschriebenen „bitter“-Blindheit in der Literatur belegt. Sinesio, Comendador,
Peparaio, & Moneta (2009) schulten 12 Prüfpersonen auf die qualitative und quantitative
Wahrnehmung von „umami“. Im Rahmen ihrer Schulung stellten die Autoren fest, dass 14 %
der nicht geschulten Teilnehmer eine MSG-Konzentration von 1,87 g/l nicht von einer
Wasserprobe unterscheiden konnten. Lugaz, Pillias, & Faurion (2002) wiesen nach, dass 27
% eines 109-kopfigen Panels nicht spezifisch zwischen „salzig“ (NaCl) und „umami“ (MSG)
diskriminieren können und identifizierten „umami-Nicht-Schmecker“. Die Performance des
Gesamt-Panels für „sauer“ konnte von 52,17 % (Tag 1) auf 100,00 % (Tag 3) verbessert
werden. Der Schwellenwert für saure Komponenten variiert von Person zu Person. Ergebnisse
von Tempere et al. (2011) belegen, dass selbst erfahrene Experten stark unterschiedliche
Schwellenwerte für saure Komponenten wie Weinsäure aufweisen. Im Falle der „sauer“-
Wahrnehmung kann, gemessen an den Ergebnissen, ebenfalls von einem Trainingseffekt im
Rahmen der Prüferschulung ausgegangen werden. Die gereichte überschwellige
Konzentration wurde am Ende des Schulungsabschnitts zuverlässig erkannt.
Ermittlung der Reiz- und Erkennungsschwelle (gemäß ISO 3972:2011):
Um Kenntnisse bezüglich der Erkennungsschwellen der Prüfer zu erlangen, wurde eine
Ermittlung der Reiz- und Erkennungsschelle gemäß ISO 3972:2011 unter Verwendung einer
einfachen beschreibenden Prüfung gemäß DIN 10964:2014 durchgeführt.
Ein vom Neutralgeschmack abweichender Reiz (Reizschwelle) wurde von den Prüfern bereits
bei den niedrigsten Konzentrationsstufen festgestellt. Der von „süß“ und „umami“ ausgehende
Reiz wurde im Vergleich zu den übrigen Geschmacksausprägungen später, sprich bei einer
höheren Konzentrationsstufe, wahrgenommen. Die prüferspezifischen
Wahrnehmungsunterschiede resultierten stellenweise in deutlichen Standardabweichungen

Diskussion
219
und individuellen Erkennungsschwellen. Von Schiffman, Crumbliss, Warwick, & Graham
(1990), Paulus & Reisch (1980), Pfaffmann, Bartoshuk, & McBurney (1971) dokumentierte
Konzentrationen der Erkennungsschwelle verschiedener Geschmacksstoffe sind beispielhaft
in Tabelle 69 dargestellt.
Tabelle 69: Konzentrationen der Erkennungsschwelle verschiedener Geschmacksstoffe (Schiffman et al., 19901; Paulus & Reisch, 19802; Pfaffmann et al., 19713)
Geschmack Substanz Mittelwert (g/l) Median (g/l) Bereich (g/l)
Min Max
süß3 Saccharose - 5,82 4,11 12,66
salzig3 Natriumchlorid - 1,76 0,18 4,97
sauer2 Zitronensäuremonohydrat 0,31 ± 0,16 - 0,07 0,50
bitter3 Koffein - 0,14 0,06 0,19
umami1 L-Mononatriumglutamat 0,08 ± 0,03a 0,61 ± 0,13b
- - -
Legende: a, erfahrene, junge Panelisten. b, unerfahrene, junge Panelisten.
Median (0,08 g/l) und Mittelwert (0,09 ± 0,03 g/l) von „bitter“ (Koffein) lagen in dieser Studie
geringfügig unter den Referenzwerten von Pfaffmann et al. (1971), Paulus & Haas (1980)
(MW: 0,16 ± 0,12 g/l), sowie Paulus & Reisch (1980) (MW: 0,17 ± 0,12 g/l; Min: 0,05 g/l; Max:
0,30 g/l), während die maximale (0,19 g/l) und minimale Erkennungsschwelle (0,05 g/l)
(annährend) deckungsgleich war. Die im Vergleich zu Paulus & Haas (1980) und Paulus &
Reisch (1980) geringe Standardabweichung bestätigt die Leistungsfähigkeit des Panels.
Unterschiede in der „bitter“-Wahrnehmung lassen sich, wie in den vorausgehenden
Abschnitten dargelegt, auf individuell verschiedene Schwellenwerte, sowie genetische
Unterschiede zurückführen.
Der Mittelwert (0,20 ± 0,18 g/l) für „umami“ lag im von Schiffman et al. (1990) für erfahrene
und unerfahrene Panelisten ermittelten Wertebereich, unterschied sich jedoch von Hong et al.
(2005) (MW: 1,47 ± 2,06 g/l). Die im Vergleich zu Schiffman et al. (1990) erhöhte
Standardabweichung deutet auf prüferspezifische Unterschiede in der „umami“-
Wahrnehmung hin. Diese Annahme findet in den Minima- und Maximawerten der
Erkennungsschwelle (Min: 0,07 g/l; Max: 0,79 g/l) Bestätigung. Eine unterschiedliche
Erkennungsschwelle für „umami“ kann, wie bereits dargelegt, nach Lugaz et al. (2002) auf
individuell unterschiedliche Erkennungsschwellen, sowie Probleme bei der Unterscheidung
von „salzig“ und „umami“ zurückgeführt werden. Die Erkenntnisse bestätigen die
Notwendigkeit einer gezielten Auswahl der Prüfpersonen zur Bewertung von „umami“-
Eindrücken.
Der Mittelwert (0,16 ± 0,02 g/l), sowie die maximale Erkennungsschwelle (0,20 g/l) für „sauer“
lag in dieser Studie deutlich unter den Referenzwerten von Paulus & Reisch (1980), während
die minimale Erkennungsschwelle (0,12 g/l) höher ausfiel. Wise & Breslin (2013) wiesen in

Diskussion
220
einer aktuellen Veröffentlichung noch niedrigere Werte aus (MW: 0,04 g/l; Min: 0,03 g/l; Max:
0,05 g/l). Die Autoren griffen jedoch auf ein erfahrenes Panel zurück. Die Erkennungsschwelle
für saure Geschmacksstoffe ist abhängig von der Erfahrung der Prüfer (Tempere et al., 2011).
Die niedrige Standardabweichung, sowie die geringe Differenz zwischen Minimum und
Maximum der Erkennungsschwelle, deuten auf eine gute Leistungsfähigkeit des Panels für die
Bewertung von „sauer“-Eindrücken hin.
Für die Geschmacksausprägung „süß“ lagen die Werte für Median (2,19 g/l), Minimum (0,79
g/l) und Maximum (8,54 g/l) der Erkennungsschwelle deutlich unter den Referenzwerten von
Pfaffmann et al. (1971). Eine größere Übereinstimmung wird im Vergleich zu Paulus & Reisch
(1980) besonders hinsichtlich von Minimum (1,2 g/l) und Maximum (8,8 g/l) der
Erkennungsschwelle deutlich. Die von Paulus & Haas (1980), sowie Paulus & Reisch (1980)
ermittelten Mittelwerte (5,17 ± 4,03 g/l bzw. 4,60 ± 3,12 g/l) liegen dennoch über dem Mittelwert
des Panels (2,41 ± 1,89 g/l). Die Wahrnehmung von durch Saccharose verursachten „süß“-
Reizen kann sich in Abhängigkeit genetischer Faktoren unterscheiden (Fushan, Simons,
Slack, & Drayna, 2010). Angaben zu Erkennungsschwellenwerten sind zudem
studienabhängig unterschiedlich, wie die Ergebnisse von Hong et al. (2005), Paulus & Haas
(1980), Paulus & Reisch (1980), Pfaffmann et al. (1971) und Henkin, Gill, & Bartter (1963)
belegen. Gemessen an der Erkennungsschwelle ist das Panel ausreichend sensibel für „süß“-
Reize. Eine deutliche Standardabweichung bestätigt jedoch die Notwendigkeit einer gezielten
Auswahl der Prüfpersonen.
Median (0,34 g/l) und die maximale Erkennungsschwelle (0,98 g/l) von „salzig“ lagen unter den
Referenzwerten von Pfaffmann et al. (1971), während die minimale Erkennungsschwelle (0,16
g/l) vergleichbar ausfiel. Vergleichbar zu „süß“ existieren in der Literatur auch bei „salzig“
teilweise deutlich voneinander abweichende Angaben für die Erkennungsschwelle. Wise &
Breslin (2013) ermittelten bei jungen Prüfern eine mittlere Erkennungsschwelle von 0,83 g/l
(Min: 0,54 g/l; Max: 1,27 g/l), während Hong et al. (2005) von 1,33 ± 1,05 g/l ausgehen.
Schiffman et al. (1990) nennen, in Abhängigkeit der Erfahrung der Prüfpersonen mittlere
Erkennungsschwellen zwischen 0,21 ± 0,04 g/l (erfahrene, junge Prüfer) und 0,74 ± 0,19 g/l
(unerfahrene, junge Prüfer). Diese Erkenntnisse decken sich mit den Ergebnissen der
vorliegenden Studie (MW: 0,36 ± 0,20 g/l). Die Erkenntnisse bestätigen im Hinblick auf die
Standardabweichung, ähnlich „umami“, die Notwendigkeit einer gezielten Auswahl der
Prüfpersonen zur Bewertung von „salzig“-Eindrücken.
Prüfung zur Unterscheidung von Intensitätsstufen eines Reizes (gemäß DIN EN ISO
8586:2014):
Die Kenntnis der Erkennungsschwelle ermöglicht lediglich eine Aussage bezüglich der
Sensibilität einer Prüfperson für eine bestimmte Geschmacksausprägung. Anhand der

Diskussion
221
Prüfung zur Unterscheidung von Intensitätsstufen eines Reizes kann, unter Verwendung einer
Rangordnungsprüfung gemäß DIN ISO 8587:2010, die tatsächliche Diskriminierungsfähigkeit
der Prüfperson ermittelt werden (DIN Deutsches Institut für Normung e.V., 2014b).
Um die Diskriminierungsfähigkeit des Panels zu bewerten ist ein mehrstufiger
Auswertungsprozess notwendig. Die grundlegende Diskriminierungsfähigkeit wurde mittels
Friedman-Test ermittelt. Der Test bestätigte, dass die Prüfpersonen die Proben aller
Geschmacksausprägungen signifikant (p < 0,05) voneinander unterscheiden konnten (Tabelle
51, Seite 166). Auf Basis des signifikanten Friedman-Tests erfolgte je
Geschmacksausprägung eine Betrachtung der einzelnen Probenpaare unter Verwendung des
Wilcoxon-Vorzeichen-Rangtests. Lediglich bei den Grundgeschmacksarten „süß“ und „bitter“
konnten die niedrigsten Konzentrationsstufen (Probenkombination: AB) nicht signifikant
voneinander differenziert werden (Tabelle 52, Seite 167). Die Ergebnisse deuten auf eine gute
Diskriminierungsfähigkeit des Panels hin. Diese Aussage wird durch die Betrachtung der
„mittleren Ränge“ (Tabelle 53, Seite 168), bei der jeweils eine Reihung der Konzentrationen D
> C > B > A vorgenommen wurde, bestätigt. Betrachtet man Minima und Maxima der Ränge
wird jedoch klar, dass ein oder mehrere Prüfer nicht in der Lage waren eine korrekte Reihung
der Proben vorzunehmen (Tabelle 54, Seite 168). Hiervon sind insbesondere niedrige
Probenkonzentrationen (A, B) betroffen. Unter Berücksichtigung der ermittelten mittleren
Erkennungsschwellen der Geschmacksausprägungen (Kapitel 4.4.1.3) wird deutlich, dass die
verwendete Probenkonzentration A (süß: 0,55 g/l; sauer: 0,16 g/l; salzig: 0,24 g/l; bitter: 0,07
g/l; umami: 0,12 g/l) stellenweise unter der Erkennungsschwelle einiger Prüfpersonen lag (süß
(MW): 2,41 ± 1,89 g/l; sauer (MW): 0,16 ± 0,02 g/l; salzig (MW): 0,36 ± 0,20 g/l; bitter (MW):
0,09 ± 0,03 g/l; umami (MW): 0,20 ± 0,18 g/l), sodass Verwechslungen der niedrigen
Probenkonzentrationen A und B nicht in mangelnder Differenzierung einer
Geschmacksausprägung sondern in einem Nicht-Erkennen des Geschmacks begründet sein
können. Gründe für prüferspezifische Unterschiede der Erkennungsschwellen wurden in den
vorausgehenden Abschnitten bereits näher beleuchtet. Prüferspezifische Unterschiede in der
Diskriminierungsfähigkeit von Geschmacksausprägungen lassen sich grundlegend auf
ähnliche Effekte zurückführen. Genetische Faktoren verfügen über einen signifikanten Einfluss
auf die Wahrnehmung und Intensität von Geschmackseindrücken (Bartoshuk, 2000). Miller Jr.
& Reedy Jr. (1990) untersuchten den Einfluss der prüferspezifisch vorhandenen Anzahl an
Geschmacksknospen auf die Intensitätswahrnehmung der Grundgeschmacksarten „süß“,
„salzig“, „sauer“ und „bitter“. Die Autoren verwiesen auf signifikant unterschiedliche Intensitäts-
Ratings in Abhängigkeit der Dichte an Geschmacksknospen bei der Beurteilung von
Saccharose-, NaCl- und Propylthiouracil-Lösungen durch zwei Gruppen von Prüferpersonen
(Gruppe 1 – hohe Dichte an Geschmacksknospen: 374 ± 134 Geschmacksknospen/cm2;
Gruppe 2 – niedrige Dichte an Geschmacksknospen: 135 ± 43 Geschmacksknospen/cm2).

Diskussion
222
Schulung zur Anwendung der Skale:
Um die Geschmackswahrnehmungen der Prüfpersonen quantifizieren zu können, wurden die
Prüfer auf die Nutzung einer ordinalen 7-Punkt Skale zur Intensitätsmessung trainiert. Die
Schulung erfolgte in Anlehnung an Rummel (2002) „Ausbildung eines deskriptiven Panels –
Schulungsphase: Training von Intensitätsmessungen“.
Die Prüfer erhielten je Grundgeschmacksart drei Proben unterschiedlicher Konzentration und
sollten diese zuerst hinsichtlich ihrer Intensität den Kategorien „schwach“, „mittel“ und „stark“
zuzuordnen. Im nächsten Schritt wurden die Prüfer gebeten auf einer 7-Punkt Skale
anzugeben, wie ausgeprägt sie den jeweiligen Geschmackseindruck empfunden haben. Die
mittleren Skalenbewertungen entsprachen den vorgesehenen Skalenwerten 2 „niedrig“, 4
„mittel“ und 6 „hoch“ (Tabelle 55, Seite 169). Einzelne Prüfpersonen hatten, beurteilt anhand
von Minima und Maxima der vergebenen Skalenwerte, jedoch Probleme bei der korrekten
Verwendung der Skale. Zu einer deutlichen Unterschätzung der Konzentrationen „mittel“ und
„hoch“ kam es jedoch nur in Einzelfällen, wie sich anhand Median und Modus der jeweiligen
Probenbewertungen erkennen lässt.
Voneinander abweichende Skalenbewertungen lassen sich einerseits auf prüferspezifisch
unterschiedlich wahrgenommene Intensitäten der jeweiligen Geschmacksreize zurückführen.
Gründe hierfür wurden bereits in den vorangegangenen Abschnitten dargestellt. Ebenso spielt
die individuell unterschiedliche Nutzung der Skale eine Rolle (Romano, Brockhoff, Hersleth,
Tomic, & Næs, 2008). Von Bedeutung sind hierbei, unter anderem, Level-Effekt (Wahl des
Skalenmittelpunkts), Scaling-Effekt (Ausnutzung der Skalenspanne), sowie eine individuell
unterschiedliche Empfindung der Differenz der Geschmacksausprägung zwischen Proben
(Næs, 1990). Eine Vermeidung der Vergabe extremer Skalenbewertungen kann ebenfalls zu
individuellen Unterschieden der Skalenbewertungen führen (Lawless & Heymann, 1999)
Darüber hinaus spielt die Reproduzierbarkeit der Bewertung bei identischen
Geschmacksreizen eine Rolle (Næs & Solheim, 1991). Skaleneffekte lassen sich durch
zielgerichtetes Training reduzieren, können aber niemals vollkommen ausgeschlossen werden
(Romano et al., 2008). Um Skaleneffekte im Rahmen dieser Studie zu minimieren, wird eine
gezielte Auswahl der Prüferpersonen vorgenommen.
5.4.1.1 Zusammenfassung der Diskussionspunkte
Die Performance von Prüfpersonen unterscheidet sich individuell. Gründe hierfür sind,
unter anderen, unterschiedliche Sensibilitäten für bestimmte
Geschmacksausprägungen, sensorische Vorkenntnisse, Art und Umfang
sensorischen Trainings, sowie genetische Unterschiede zwischen den Prüfpersonen.

Diskussion
223
Erkenntnisse bezügliche den Erkennungsschwellen der fünf
Geschmacksausprägungen unterscheiden sich in Abhängigkeit der gewählten Quelle
teilweise deutlich.
Skaleneffekte (Unterschiedliche Verwendung der Prüfskale) beeinflussen individuell
die Nutzung der Prüfskale und können auch durch Training nicht vollständig vermieden
werden.
5.4.1.2 Zusammenfassung & Darstellung der Ergebnisse
Die Panelteilnehmer sind in der Lage die fünf Grundgeschmacksarten zu erkennen und
unterschiedliche Geschmacksausprägungen zu unterscheiden.
Einige Prüfer hatten zur Beginn der Schulung Schwierigkeiten bei der zuverlässigen
Bestimmung von „umami“-, „sauer“- und „bitter“-Reizen.
Die mittleren Erkennungsschwellen des Panels decken sich mit Literaturerkenntnissen
und bestätigen grundsätzlich die Eignung des Panels.
Das Panel ist in der Lage unterschiedliche Intensitäten einer Geschmacksausprägung
voneinander zu unterscheiden und bezüglich der Intensität zu reihen.
Das Panel ist in der Lage unterschiedlichen Intensitäten einer Geschmacksausprägung
einen einheitlichen Skalenwert zuzuweisen.
Die Angaben einiger Prüfer weichen vom Mittel des Panels ab, sodass eine gezielte
Auswahl der am Panel teilnehmenden Prüfer notwendig ist.
5.4.2 Prüferauswahl
Die Ergebnisse der Prüferschulung bestätigten die Notwendigkeit einer gezielten Auswahl der
Prüfpersonen für die finalen Panels. Als Grundlage für die Auswahl wurden die Ergebnisse der
Schulung zur Anwendung einer Skale (Kapitel 4.4.1.5) genutzt. Die Bewertung und Auswahl
der Prüfpersonen erfolgte in Anlehnung an die DIN EN ISO 8586:2014. Je
Geschmackausprägung wurde aus dem Prüferpool ein Panel zusammengestellt. Aufgrund
des ordinalen Skalenniveaus und der Verletzung der Annahme der Normalverteilung der
Daten wurden verteilungsunabhängige, nichtparametrische Testverfahren zur Auswertung
herangezogen.
Die Vorauswahl basierte auf dem nichtparametrischen Friedman-Test und diente dem
Ausschluss von Prüfpersonen mit geringer Diskriminierungsfähigkeit. Hieraus resultierten je
Geschmacksausprägung vorläufige Panels aus 8 – 16 Prüfern. Durch einen Vergleich der
individuellen Bewertungen mit dem Gruppenmittel je Geschmacksausprägung und
Merkmalsausprägung (niedrige, mittlere, hohe Intensität) wurden stark vom Gruppenmittel
abweichende Prüfer aus dem betroffenen Panel ausgeschlossen. Hieraus resultierten aus 4 –
8 Prüfern bestehende Panels. Die Homogenität der Panelbewertung wurde mittels Kruskal-
Wallis H Test überprüft. Im Falle von unzureichender Homogenität erfolgte ein Vergleich der

Diskussion
224
Bewertungen der einzelnen Prüfpersonen mittels Mann-Whitney U Test. Signifikante
Unterschiede zwischen den Prüfpersonen resultierten im Ausschluss der Prüfperson mit vom
Rangmittel abweichender Rangsumme.
Die für die Auswertung dieser Studie angewendete DIN EN ISO 8586:2014 gilt als
allgemeingültige Richtlinie, bleibt aber, besonders im Falle von ordinalen Skalenniveaus und
Verletzungen der Annahme der Normalverteilung, relativ vage. Als grundlegende
Anforderungen bleiben
eine Analyse der Varianz jeder Prüfperson zur Beurteilung der Verlässlichkeit der
Bewertungen,
eine statistische Auswertung der Unterschiede zwischen Prüfpersonen,
eine statistische Auswertung der Unterschiede zwischen den Proben, sowie
eine Auswertung bezüglich der Eignung einer Prüfperson in Bezug auf das
Gruppenergebnis.
Hierfür dürfen parametrische und nicht-parametrische Testverfahren verwendet werden.
Grundlegend ist gemäß DIN EN ISO 8586:2014 sicherzustellen, dass die Prüfpersonen
verlässliche, wiederholbare Bewertungen abgeben und in Form eines Panels als homogene
Einheit agieren. Die Norm empfiehlt für ein finales Panel eine Prüferanzahl von mindestens 10
Prüfern, sofern dies im Rahmen des Studiendesigns realisierbar ist (DIN Deutsches Institut für
Normung e.V., 2014b).
Die Auswertung der Diskriminierungsfähigkeit der Prüfpersonen verdeutlichte die individuell
unterschiedliche Leistungsfähigkeit der Prüfer in Abhängigkeit der Geschmacksausprägung.
Während bei „sauer“ ein Großteil des Panels (16 Prüfer) zur Differenzierung der
unterschiedlichen Merkmalsausprägungen imstande war, konnten bei den übrigen
Geschmacksausprägungen jeweils nur 8 – 9 Prüfer zwischen den verschiedenen
Konzentrationsstufen unterscheiden. Nur eine Prüfperson (Prüfer 20) verfügte dabei über eine
statistisch nachgewiesene Diskriminierungsfähigkeit für alle Geschmacksausprägungen. Auch
die Homogenität der Bewertungen unterschied sich in Abhängigkeit der
Geschmacksausprägung. Für „bitter“ und „umami“ wurden nur wenige Prüfpersonen aufgrund
von abweichenden Bewertungen vom Gruppenmittel aus den Panels ausgeschlossen (1 – 2
Prüfer). Im Falle von „süß“ und „sauer“ resultierten abweichende Bewertungen in einer
Halbierung der Anzahl der Prüfpersonen. Die anhand der Vorgaben der Norm erarbeiteten,
finalen Panels genügten, abgesehen von der Mindestanzahl von zehn Prüfern je Panel, den
Anforderungen der Norm und verfügten besonders hinsichtlich der Geschmacksausprägungen
„süß“, „sauer“ und „salzig“ über eine sehr homogene Bewertungsvergabe (p = 0,353 – 0,947).
Diese konnte lediglich für die Geschmacksausprägung „umami“ nicht in vergleichbarer Weise

Diskussion
225
erreicht werden (p = 0,129 – 0,231). Abweichende Punktevergaben, die in einer Reduzierung
der Panelhomogenität resultierten, wurden im Falle von „sauer“ bei „hoher Intensität“ (p =
0,353) und in Falle von „bitter“ bei „niedriger Intensität“ (p = 0,366) festgestellt.
Hinsichtlich der Auswahl von Prüfpersonen und Bewertung der Schulungsergebnisse sind
Problemstellungen bekannt. Traditionell werden beispielsweise Rangordnungstests
verwendetet, um die Differenzierungsfähigkeiten der Prüfer zu ermitteln und die
Leistungsfähigkeit eines einzelnen Prüfers in Bezug auf das Gruppenmittel des Panels zu
bewerten (Stone & Sidel, 2004). Hierbei wird das Gruppenmittel als Referenz für den Vergleich
genutzt. Die Verwendung des Gruppenmittels resultiert, gemäß Mangan (1992), jedoch in zwei
grundlegenden Auffälligkeiten:
Die Bewertung jedes Prüfers ist im Mittel enthalten, wodurch bei Vergleichen dieser Art
keine statistisch unabhängigen Variablen verglichen werden.
Die Bewertung basiert auf der Annahme, dass das Mittel ein akkurater
Vergleichsstandard für die Bewertung einer individuellen Prüfperson darstellt.
Problematisch bei sensorischen Bewertungen ist grundlegend die nicht vorhandene Kenntnis
bezüglich der „wahren“ Ausprägung eines Merkmals. Die Annahme, dass das Gruppenmittel
dem wahren Wert eines Merkmals entspricht, birgt die Gefahr des Ausschlusses einzelner
Prüfpersonen mit geringer Varianz und folglich hoher Vorhersagegenauigkeit die jedoch nicht
dem Mittel entsprechen (Mangan, 1992). Die beschriebenen Problemstellungen führten zu
intensiven Forschungen hinsichtlich alternativer Auswertungsmöglichkeiten. Latreille et al.
(2006), Brockhoff (2003), Schlich (1994) und Mangan (1992) erarbeiteten, unter anderen,
komplexe statistische Methoden um die genannten Problemstellungen einzudämmen und die
Aussagekraft sensorischer Bewertungen durch Panels zu verbessern. Die Komplexität dieser
Methoden erschwert jedoch deren Anwendung in sensorischen Kleinstudien. Die Ergebnisse
statistischer Test können darüber hinaus, in Abhängigkeit der gewählten Methode, zu
unterschiedlichen Aussagen führen, sodass die finale Bewertung über die Eignung eines
Prüfers letztendlich der Erfahrung des Prüfleiters überlassen bleibt. Die grundlegenden
Anforderungen an die Prüfer, wie Differenzierungsfähigkeit und Wiederholungspräzision,
sowie die Homogenität des Panels bleiben, unabhängig von der Art der Auswertung, erhalten
(Bárcenas, Elortondo, & Albisu, 2000).
Die in Kapitel 5.4.1 und Kapitel 5.4.2 dargestellte Bewertung der Ergebnisse der
Panelschulung und Prüferauswahl belegen die Konformität der finalen Panels mit den
Anforderungen der DIN EN ISO 8586:2014. Die Güte der Ergebnisse für die jeweiligen
Geschmacks- und Merkmalsausprägungen muss bei der Bewertung der sensorischen
Ergebnisse beachtet werden, um die Belastbarkeit der getroffenen Aussagen zu
gewährleisten.

Diskussion
226
5.4.2.1 Zusammenfassung der Diskussionspunkte
Die Auswahl und Bewertung der Performance sensorischer Prüfer und Panels
beinhaltet, je nach Anforderung, komplexe statistische Methoden.
Maßgeblich für die Bewertung sind, unabhängig von der Auswertungsmethode,
Differenzierungsfähigkeit und Wiederholungspräzision der Prüfer, sowie die
Homogenität des Panels.
5.4.2.2 Zusammenfassung & Darstellung der Ergebnisse
Zur Sicherstellung der Homogenität der Panelaussagen war der Ausschluss von
Prüfern aus den Panels notwendig.
Je Geschmacksausprägung wurden individuelle Panels aus 4 – 7 Prüfern
zusammengestellt.
Besonders hinsichtlich der Geschmacksausprägungen „süß“, „sauer“ und „salzig“
wurden sehr homogene Bewertungsvergabe (p = 0,353 – 0,947) erreicht.
Bei der Bewertung der sensorischen Ergebnisse muss die Güte der Bewertungen
einzelner Geschmacks- und Merkmalsausprägungen beachtet werden.
5.4.3 Sensorik der Peptidfraktionen
Retentat- und Permeatfraktion der fraktionierten Proteinhydrolysate wurden sensorisch
hinsichtlich der Geschmacksausprägungen „süß“, „sauer“, „salzig“, „bitter“ und „umami“
untersucht. Zusätzlich wurde ein Bitterstandard, sowie ein Gemisch aus Bitterstandard und
Permeat gereicht um zu überprüfen, ob die Permeatfraktion in der Lage ist, die „bitter“-
Wahrnehmung zu reduzieren.
Molkenprotein-Isolat:
Die Retentat- und Permeatfraktion des hydrolysierten Molkenprotein-Isolats wurde sensorisch
als überwiegend geschmacksneutral bewertet. Im Retentat wurde eine sehr schwache
„süß“-, im Permeat eine schwache „salzig“-Komponente wahrgenommen. Die „bitter“-
Ausprägung des Standards konnte durch die Zugabe von Permeat nicht signifikant verringert
werden, wurde jedoch als weniger intensiv empfunden.
Die sensorischen Eigenschaften von Molkenprodukten, Molkenproteinkonzentraten (WPC)
und Molkenproteinisolaten (WPI) sind in der Literatur bereits umfassend dokumentiert. Drake,
Karagul-Yuceer, Cadwallader, Civille, & Tong (2003) erarbeiteten eine standardisierte
Prüfsprache für getrocknete Milchpulver und getrocknete Milcherzeugnisse. Die
Geschmacksauprägungen „süß“, „salzig“, „gekocht“, „adstringierend“ und „kartonartig“ wurden
in den untersuchten 63 Produkten am häufigsten festgestellt. Untersuchungen von Russell,
Drake, & Gerard (2006) bestätigten diese Erkenntnisse und ergänzten die
Geschmacksausprägungen um „metallisch“ und „seifig“.

Diskussion
227
Mortenson, Vickers, & Reineccius (2008) verarbeiteten Molkenproteinisolate und
–Konzentrate zu Cheddar und Mozzarella um den Einfluss des Verarbeitungsprozesses auf
die sensorischen Eigenschaften der Endprodukte zu untersuchen. Bei den Endprodukten
wurden vor allem „Milch“- und „Koch“-Aromen, sowie ein „süß“-Geschmack festgestellt. Die
weiteren Grundgeschmacksausprägungen wurden nur in geringem Maße wahrgenommen.
Off-Flavours bei WPIs wie „kartonartig“ können, unter anderem auf die Entstehung von
Maillard-Reaktionsprodukten, Oxidation von Inhaltsstoffen und weitere prozessbedingte
Veränderungen des Rohstoffs zurückgeführt werden (Morr & Ha, 1991).
Molkenproteinhydrolysate werden aufgrund ihres Gehalts an kurzkettigen, hydrophoben
Peptiden häufig als „bitter“ empfunden (Matoba & Hata, 1972). Leksrisompong, Miracle, &
Drake (2010) arbeiteten bei der umfangreichen Charakterisierung von 22
Molkenproteinhydrolysaten darüber hinaus die Geschmacksausprägungen „sauer“,
„adstringierend“, „malzig“, „käsig“, „kartoffelig“ und „verbrannt“ heraus. Eine „bitter“
Geschmacksausprägung wurde im Rahmen dieser Studie für Molkenproteine weder im
Retentat noch im Permeat festgestellt, obwohl, bezugnehmend auf die Ergebnisse der SE-
HPLC Analytik, vom Vorhandensein kurzkettiger Di- und Tripeptidverbindungen ausgegangen
werden muss. Der verhältnismäßig hohe Anteil an Leucin (ca. 9,6 %) im Ausgangsprodukt
scheint ebenfalls nicht in einer erhohten „bitter“-Ausprägung zu resultieren (Glanbia
Nutritionals Ltd., 2012). L-Leucin ist dafür bekannt eine bitteren Geschmack zu verursachen
(Warendorf, Belitz, Gasser, & Grosch, 1992). Die sehr schwach empfundene „süß“-
Komponente der Retentatfraktion ist möglicherweise durch das Vorhandensein geringer
Mengen Lactose (ca. 1,5 %) im Rohstoff erklärbar. Lactose verfügt über eine leicht süßen
Geschmack (Shallenberger, 1993). Dagegen spricht, bezugnehmend auf die Trenngrenze der
Membran (100 kDa), die molare Masse von ca. 342 Da (Butylina et al., 2006). Es muss davon
ausgegangen werden, dass die im Feed vorhandene Lactose die Membran nahezu vollständig
passiert hat und sich im Permeat anreichert. Dafür sprechen auch Ergebnisse von Chollangi
& Hossain (2007), die bei der Ultrafiltration von Abwässern der Milchproduktherstellung bereits
bei einem MWCO von 10 kDa eine Lactoserückgewinnung im Permeat von 100 % erreichten.
Das Vorhandensein von Lactose im Retentat kann, betrachtet man die SE-HPLC
Chromatogramme, jedoch nicht ausgeschlossen werden, da in der Retentatfraktion
Verbindungen im Bereich von ca. 300 – 400 Da nachgewiesen wurden. Die schwache „salzig“
Komponente kann, unter anderem, auf im Rohstoff befindliche Salze zurückzuführen werden,
die sich im Permeat anreichen. Hellemann (1992) stelle fest, dass der wahrgenommene
„salzig“-Eindruck von NaCl durch den Zusatz geringer Mengen Säure (Milchsäure, Essigsäure)
verstärkt wird. Smith, Metzger, & Drake (2016) arbeiteten heraus, dass Molke- und
Milchpermeate die „salzig“ Wahrnehmung von Suppen steigern und führten dies, unter
anderem auf den Milchsäure-Gehalt der Permeate zurück. Eine vergleichbare, säureinduzierte

Diskussion
228
Erhohung des „salzig“-Eindrucks erscheint auch im Falle des verkosteten Permeats möglich.
Andererseits besteht die Moglichkeit, dass der „salzig“ Eindruck durch niedermolekulare
Peptidverbindungen zustande kommt. Tamura et al. (1989) identifizierten „salzig“/“umami“
schmeckende Dipeptide aus Glutamin- (Glu) und Asparaginsäure (Asp). Nakata et al. (1995)
stellten bei der Untersuchung des „schmackhaften Peptides“ welchem ein „umami“- und
„salzig“-Geschmack zugeschrieben wird fest, dass die Position der Aminosäuren
Glutaminsäure und Asparaginsäure eine wesentlichen Einfluss auf den „umami“/“salzig“-
Eindruck des Peptids haben. Hohe Konzentrationen, der durch den Hydrolyseprozess frei
zugänglichen Aminosäuren Glutaminsäure (16,7 %) und Asparaginsäure (10,7 %) im Rohstoff
legen die Vermutung einer ladungsbedingten Ausbildung von Asp-Glu-Dipeptiden oder, in
Verbindung mit NaCl, die Ausbildung von Dipeptidsalzen nahe.
Alge (Chlorella vulgaris):
Die Retentatfraktion der hydrolysierten Alge (Chlorella vulgaris) wurde sensorisch als schwach
„salzig“, sowie sehr schwach „umami“ bewertet. Das Permeat wurde als schwach „salzig“-,
sowie sehr schwach „sauer“ und „umami“ empfunden. Die „bitter“-Ausprägung des Standards
konnte durch die Zugabe von Permeat signifikant verringert werden, während „salzig“ als
schwach bis mittel empfunden wurde.
Der Geschmack von Algen bzw. Algenprotein ist, bedingt durch das ernährungsphysiologische
Potential des Rohstoffs, besonders in Sachbüchern charakterisiert. Die Beschreibungen
reichen von „charakteristisch“ bis „salzig“ und „fischig“ (Donhauser, 2017; Kreischer,
Schuttelaar, & Farm, 2016). Darüber hinaus wird eine „umami“-Geschmacksauprägung
beschrieben (Kreischer et al., 2016). In der wissenschaftlichen Primärliteratur sind
Algenproteine und –Hydrolysate bis dato nicht umfassend charakterisiert. Becker (2007) zieht
den fischigen Geruch, die pulverförmige Konsistenz und die dunkelgrüne Farbe als Grund für
den geringen Einsatz von getrocknetem Algenprotein in Lebensmitteln heran. Die Einarbeitung
von Algenprotein in Lebensmitteln ist, trotz der genannten Nachteile, intensiver Gegenstand
der Forschung. Morimura & Tamiya (1954) arbeiteten Chlorella Pulver in Brot, Nudeln und
Kekse ein. Fradique et al. (2010) bewerteten die sensorischen Eigenschaften von mit Chlorella
vulgaris und Spirulina maxima versetzten Nudelprodukten als positiv hinsichtlich der
Produktakzeptanz. Zumeist wirkt sich der Zusatz von Algenprotein jedoch nachteilig auf die
sensorischen Attribute des Produkts aus (Beheshtipour, Mortazavian, Mohammadi,
Sohrabvandi & Khosravi-Darani, 2013).
Weder Retentat noch Permeat des Algenproteinhydrolysats wurden als „bitter“ empfunden.
Die für einen bitteren Geschmack bekannten Aminosäuren L-Try, L-Phe, L-Tyr und L-Leu sind
in Algenprotein, mit Ausnahme von Leucin mit 7,5 – 9,5 %, nur schwach repräsentiert (Safi et
al., 2014). Glutaminsäure und Asparaginsäure stellt im Algenprotein mit 9,1 – 13,7 % bzw. 9,3

Diskussion
229
– 10,9 % den größten Anteil dar (Safi et al., 2014). Die in Retentat und Permeat identifizierte
„salzig“/“umami“-Ausprägung kann von Dipeptiden aus Glu und Asp hervorgerufen werden.
Da der Na-Gehalt des Rohstoffes nicht aus der Spezifikation hervorgeht, kann das
Zustandekommen des „salzig“-Geschmacks durch Na-Verbindungen jedoch nicht
ausgeschlossen werden (Allma, 2014). Der im Permeat festgestellte „sauer“-Eindruck ist
möglichweise durch Glu- und Asp-haltige Oligopeptide zu begründen (Tamura et al., 1989).
Auch frei vorliegende Glu, Asp und Asn oder eine Verbindung aus His und HCl, sowie weitere
Dipeptide auf Glu-, Asp-, sowie Ser- und Ala-Basis kommen dafür in Frage (Kirimura et al.,
1969).
Die „bitter“-Geschmacksauprägung von Koffein wurde durch Zugabe der Permeatfraktion
signifikant reduziert. Eine Reduzierung des „bitter“-Geschmacks ist, wie in Kapitel 2.5.3
dargestellt, durch verschiedene Mechanismen möglich. Hiervon erscheinen besonders
Modulationen auf molekularer Ebene als wahrscheinlich. Darunter fallen das Einwirken von
Molekülen die eine Komplexbildung bewirken, Bitterstoffe zersetzen oder deren Transport zum
Rezeptor einschränken, als Gegenspieler der T2R-Rezeptor Bindungsstellen wirken, sowie
Moleküle, welche die T2R-Rezeptorbindungsstellen verändern, Moleküle die weitere Proteine
der Transduktionskette modulieren, Moleküle die die Funktion des TRPM5-Ionenkanals oder
die Ausschüttung, Bindung und Wiederaufnahme von Neurotransmittern beeinflussen (Ley,
2008). Natrium-Salze sind dafür bekannt die „bitter“-Wahrnehmung zu reduzieren (Keast et
al., 2001; Breslin & Beauchamp, 1995). Keast et al. (2001) setzten, unter anderem, NaCl,
Natrium-Acetat und Natrium-Glucanat dazu ein um die „bitter“-Ausprägung von KCl,
Harnsäure und Amilorid zu reduzieren, während der „bitter“-Eindruck von Koffein und Chinin-
HCl nur geringfügig vermindert werden konnte. Breslin & Beauchamp (1995) gelang es
darüber hinaus die durch Koffein erzeugte „bitter“-Wahrnehmung durch die Zugabe von 0,3 M
– 0,5 M NaCl um 55 ± 6 % zu reduzieren. Der Effekt scheint insbesondere von den Na+-Ionen
auszugehen (Breslin & Beauchamp, 1995). Keast et al. (2001) postulierten verschiedene
Wirkmechanismen, darunter die Ausbildung eines ladungsbedingten Abschirmungseffekts am
Rezeptor, sowie die Modulation der am Transduktionsprozess beteiligten Ionen-Kanäle.
Aufgrund der niedrigen Na-Konzentration von ca. 0,1 % im Rohstoff wird nicht von einer auf
der Wirkung von Natrium-Salzen basierende Reduktion der „bitter“-Geschmacksausprägung
ausgegangen. Eine Reduktion des „bitter“-Eindrucks durch strukturelle Änderungen des
Bitterstoffs erscheint aufgrund der chemischen Stabilität von Koffein unwahrscheinlich. Koffein
liegt bis 141 °C in der β-Form vor und wandelt sich bei Überschreitung der Grenztemperatur
in die α-Form (Epple, Cammenga, Sarge, Diedrich, & Balek, 1995). Der Siedepunkt liegt
zwischen 234 – 236 °C (Bothe & Cammenga, 1979; Anselmino, Biberfeld, & Gilg, 1911).
Binello, Cravotto, Nano, & Spagliardi (2004) gelang eine Abschwächung des „bitter“-Eindrucks
von Koffein um ca. 90 % durch Zugabe von 0,4 % β-Cyclodextrin oder 0,4 % eines Chitosan-

Diskussion
230
β-Cyclodextrin Polymer zu einer 0,05 %-Koffeinlösung. Basierend auf Bibby, Davies, & Tucker
(2000) ist die Ausbildung nicht-kovalenter Komplexe denkbar, die in einer Reduktion des
„bitter“-Eindrucks resultiert. Eine Verminderung des „bitter“-Geschmacks durch
Komplexbildung erscheint unter Berücksichtigung der bekannten Komplexbildner und der
chemischen Zusammensetzung des Rohstoffes unwahrscheinlich. Mattes (2007) erreichte
eine Abschwächung der „bitter“-Wahrnehmung von Koffeinmodelllösungen, sowie eine
Erhohung der „bitter“-Erkennungsschwelle durch die Zugabe von 1% Linolsäure. Der Zusatz
von Linolsäure erhoht darüber hinaus auch die Erkennungsschwellen für „sauer“ und „salzig“.
Zur Herstellung homogener Probenlösungen wurde ein Emulgator verwendet. Linolsäure, eine
zweifach ungesättigte Fettsäure, ist nur schlecht wasserlöslich (Belitz et al., 2008). Der
Mechanismus der Reduktion der „bitter“-Wahrnehmung durch Fettsäuren ist nicht geklärt.
Mattes (2007) führt, unter Berufung auf Koriyama, Wongso, Watanabe, & Abe (2002), Metcalf
& Vickers (2002), Lynch, Liu, Mela, & MacFie (1993), sowie Forss (1973), Möglichkeiten zur
Modulation der Geschmackswahrnehmung durch Fettsäuren auf. Diese beinhalten
eine Reduktion der Konzentration des für den Reiz verantwortlichen Stoffes durch eine
fettsäureninduzierte Anregung des Speichelflusses,
die Ausbildung einer Barriere, die den Zugang von reizauslösenden Molekülen zu den
Rezeptoren hemmt, oder
die Modulation der Raumstruktur einer Geschmackskomponente, sowie der effektiven
Konzentration.
Der zur Hydrolyse verwendete Rohstoff verfügt über einen Fettgehalt von ca. 9,6 % (Allma,
2014). Aufgrund der mangelnden Wasserlöslichkeit von Fetten wird vermutet, dass der
Fettsäureanteil im Hydrolysat durch den Aufbereitungsprozess (Zentrifugation, Filtration)
abgetrennt wurde. Der Effekt von Fettsäuren ist, in Abhängigkeit der Fettsäure, zudem nicht
besonders stark und an erhöhte Konzentrationen gebunden (Ley, 2008). Es wird folglich nicht
davon ausgegangen, dass die notwendigen Fettsäure-Konzentrationen erreicht werden, um
eine signifikante Reduktion der „bitter“-Wahrnehmung auszulösen. Am wahrscheinlichsten ist
eine Reduktion der „bitter“-Wahrnehmung durch im Permeat vorliegende Peptidverbindungen.
Für die Bitterwahrnehmung sind Rezeptoren der T2R-Familie verantwortlich (Mueller et al.,
2005). Eine Reduktion des „bitter“-Geschmacks auf molekularer Ebene kann durch die
Hemmung der Bindung von Bitterstoffen an die relevanten Rezeptoren erreicht werden
(Nadathur & Carolan, 2016). Als „bitter“-Blocker sind mittlerweile sieben Substanzen bekannt,
deren Wirkung auf eine Interaktion mit bestimmten „bitter“-Rezeptoren zurückzuführen ist.
Darunter fallen
GIV3727 (4-(2,2,3-tri-Methylcyclopentyl)-Buttersäure) als Gegenspieler des
hTAS2R31 (Slack et al., 2010),

Diskussion
231
Probenecid (4-(di-Propylsulfamoyl)benzoesäure), welches, vermutlich über einen
allosterischem Mechanismus mit hTAS2R16 interagiert (Greene et al., 2011),
4‘-fluoro-6-Methoxyflavanon, welches die „bitter“-Wahrnehmung an hTAS2R39
beeinflusst (Roland et al., 2014),
die Sesquiterpenlactone (STL) 3β-Hydroxydihydrocostunolid (3HDC) und 3β-
hydroxypelenolid (3HD) die an hTAS2R46 wirken (Brockhoff et al., 2011), sowie
die Aminosäurenderivate α-Aminobuttersäure (GABA) und Nα,Nα-bis(Carboxymethyl)-
L-Lysin (BCML) an T2R4 (Pydi et al., 2014)
Zudem berichteten Belikov & Gololobov (1986) von einer Eliminierung der „bitter“-
Geschmacksausprägung von Koffein, Brucin und Proteinhydrolysaten durch das Dipeptid L-
Glu-L-Glu. Die Aussage der Autoren ist jedoch nicht durch quantitative Daten belegt.
Die genannten Substanzen reduzieren die „bitter“-Wahrnehmung nicht nur an den genannten
Rezeptor-Typen. GIV3727, 3HD und 3HDC wirken beispielsweise übergreifend an hTAS2R31,
hTAS2R40 und hTAS2R43 (Brockhoff et al., 2011; Slack et al., 2010). Die Wirkweise der
verschiedenen Verbindungen ist nicht abschließend geklärt. Brockhoff et al. (2011) vermuten
beispielsweise ein antagonistisches Prinzip, bei dem 3HD oder 3HDC an die Bindungsstelle
der Rezeptoren andocken, dort aber nicht zu einer Aktivierung führen. Dies führen die Autoren
auf eine strukturelle Ähnlichkeit der Antagonisten zu den eine „bitter“-Wahrnehmung
auslösenden Agonisten zurück. Die Annahmen fußen auf einer Hypothese von Brockhoff,
Behrens, Niv, & Meyerhof (2010) die von dem Vorhandensein nur einer Bindungsstelle an
hTAS2Rs ausgehen. Von der Existenz eines universalen „bitter“-Blockers wird nicht
ausgegangen, da die bekannten Substanzen nicht für alle 25 T2Rs Wirkung zeigten, sodass
eine komplette Blockierung des „bitter“-Geschmacks, im besten Fall, nur durch eine
Kombination verschiedener Substanzen erreicht werden kann (Pydi et al., 2014).
Aufgrund der Zusammensetzung des Rohstoffs, sowie der daraus vermuteten
Zusammensetzung des Hydrolysats wird abschließend davon ausgegangen, dass die Wirkung
als „bitter“-Blocker von einer oder mehreren Peptidverbindungen ausgelöst wurde. In Frage
kommen hierfür, unter Berücksichtigung des hohen Glu-Anteils in Algen der Gattung Chlorella
vulgaris und Berufung auf Belikov & Gololobov (1986), das Dipeptid L-Glu-L-Glu. Um diese
Hypothese bestätigen zu können, ist eine Sequenzierung der in der Permeatfraktion
vorhandenen Verbindungen, sowie weitere sensorische Versuche mit den identifizierten und
isolierten Reinsubstanzen notwendig. Es wird nicht davon ausgegangen, dass eine
Verminderung des „bitter“-Geschmacks auf den von Mattes (2007) identifizierten Effekte der
„bitter“-Reduktion durch das Vorhandensein von Fettsäuren, insbesondere Linolsäure
zurückzuführen ist. Aufgrund der mangelnden Wasserlöslichkeit von Fetten wird auch an
dieser Stelle vermutet, dass der Fettsäureanteil im Hydrolysat durch den

Diskussion
232
Aufbereitungsprozess (Zentrifugation, Filtration) abgetrennt wurde. Darüber hinaus führt der
Zusatz von Linolsäure zu einer Erhohung der Erkennungsschwellen für „sauer“ und „salzig“
(Mattes, 2007). Im Falle einer Reduktion der „bitter“-Ausprägung durch Linolsäure wären
folglich auch die Geschmacksausprägungen „sauer“ und „salzig“ in der Probe „Bitter-Standard
& Permeat“ niedriger bewertet worden. Denkbar ist ebenfalls eine Reduktion der „bitter“-
Wahrnehmung durch „salzig“ schmeckende Peptide, ähnlich der Erkenntnisse von Keast et
al. (2001) und Breslin & Beauchamp (1995) für Natrium-Verbindungen. Eine Reduktion des
bitteren Geschmacks durch Na-Salze, kann aufgrund der fehlenden Kenntnis hinsichtlich des
Na-Gehalt des Rohstoffs nicht vollständig ausgeschlossen werden.
Zusammenfassende Theorie zur Hemmung der „bitter“-Ausprägung durch Fraktionen von
hydrolysierter Alge (Chlorella vulgaris):
Die Wirkmechanismen „bitter“-blockender Verbindungen sind, trotz intensiver
Forschungsbemühungen, nicht abschließend geklärt. Aktuelle Veröffentlichungen zum Thema
gehen zumeist von einer Rezeptorinteraktion der „bitter“-blockenden Substanzen aus. Neben
der Möglichkeit einer allosterischen Hemmung der Enzymfunktion stellt insbesondere das
„Gegenspieler-Prinzip“ eine nachvollziehbare Theorie dar (Brockhoff et al., 2011; Slack et al.,
2010). Hierbei blockiert der „bitter“-Blocker, bedingt durch strukturelle Ähnlichkeit, die
Bindungsstelle des Rezeptors und sorgt somit für eine Abschwächung der „bitter“-
Wahrnehmung (Brockhoff et al., 2011). Die Annahmen fußen auf einer Hypothese von
Brockhoff, Behrens, Niv, & Meyerhof (2010), die von dem Vorhandensein nur einer
Bindungsstelle am Rezeptor hTAS2Rs ausgehen. Im Hinblick auf die in humansensorischen
Studien festgestellte Reduktion aber nicht vollständige Maskierung einer „bitter“-Ausprägung
erscheint ein derartiger Mechanismus stimmig, da dieser aufgrund der Vielzahl an Rezeptoren
der T2R-Familie nicht für alle Rezeptortypen greift. Eine von Ley (2008) angeführte Modulation
von „second-messenger“-Botenstoffen, sowie der Ausschüttung und Wiederaufnahme von
Neurotransmittern gilt ebenfalls als schlüssig, ist aufgrund er Komplexität der Vorgänge des
Transduktionsprozesses vermutlich nur schwer nachweisbar.
Im Rahmen dieser Studie wird, wie vorab dargestellt, davon ausgegangen, dass
Peptidverbindungen für die Reduktion der „bitter“-Wahrnehmung verantwortlich sind. In
Anbetracht der angeführten Moglichkeit zur Hemmung einer „bitter“-Ausprägung erscheint
besonders das von Brockhoff et al. (2011) bereits vorab beschriebene antagonistische Prinzip
einen denkbaren Lösungsansatz zu liefern. Hierbei ist das Vorhandensein eines oder mehrerer
Antagonisten denkbar. Unter Berücksichtigung der SE-HPLC-Analysen der Permeatfraktionen
wird davon ausgegangen, dass die verantwortliche(n) Struktur(en) < 1,35 kDa sind oder aus
Fragmenten < 1,35 kDa bestehen, die sich beispielweise durch ladungsbedingte ionische
Bindungen oder Wasserstoffbrückenbindungen zu komplexeren Strukturen

Diskussion
233
zusammengeschlossen haben, welche in der Lage sind die Bindungsstelle eines oder
mehrerer Rezeptortypen zu besetzen.
Um diese Aussagen zu verifizieren, sind neben der Sequenzierung der verwendeten
Fraktionen weitere, umfangreiche Untersuchungen hinsichtlich der Interaktion der
identifizierten Strukturen mit den Bindungsstellen verschiedener Rezeptortypen, sowie deren
Interaktion bei bestimmten Milieuverhältnissen notwendig.
5.4.3.1 Zusammenfassung der Diskussionspunkte
„bitter“-Geschmack kann durch niedermolekulare Peptidverbindungen ausgelöst
werden.
Die Peptide L-Tryptophan (Try), L-Phenylalanin (Phe), L-Tyrosin (Tyr), L-Leucin (Leu)
werden mit einem „bitter“-Geschmackseindruck in Verbindung gebracht.
Glu und Asp-haltige Di- und Oligopeptide verfügen über „salzig“, „umami“ und „sauer“
Geschmacksausprägungen.
5.4.3.2 Zusammenfassung & Darstellung der Ergebnisse
Aus hydrolysiertem Molkenprotein-Isolat gewonnene Fraktionen verfügen nicht über
nennenswerte Geschmacks- oder geschmacksmodulatorische Eigenschaften. Im
Retentat wurde eine sehr schwache „süß“-Ausprägung im Permeat ein schwacher
„salzig“-Geschmack identifiziert.
Aus hydrolysierter Alge (Chlorella vulgaris) gewonnene Fraktionen weisen „salzig“,
„umami“ und „sauer“ Ausprägungen auf. Diese werden vermutlich durch Glu und Asp-
haltige Di- und Oligopeptide ausgelöst.
Permeatfraktionen (< 100 kDa) aus hydrolysierter Alge (Chlorella vulgaris) führten zu
einer Reduktion der „bitter“-Wahrnehmung.
Der Wirkmechanismus der „bitter“-Reduktion durch niedermolekulare
Peptidverbindungen ist bis dato nicht abschließend geklärt.
Das Agieren der Peptidverbindungen als Antagonisten zu bestimmten Bitterstoffen,
vergleichbar des für 3β-Hydroxydihydrocostunolid und 3β-hydroxypelenolid
postulierten Mechanismus, erscheint möglich.

Diskussion
234
5.5 Evaluation der Ergebnisse
In den folgenden Abschnitten werden die Ergebnisse in Bezug auf die formulierten Hypothesen
bewertet und abschließend zusammengefasst.
5.5.1 Evaluation der Ergebnisse in Bezug auf die formulierten Hypothesen
Nachfolgend werden die Ergebnisse in Bezug auf die zu Beginn formulierten Hypothesen
betrachtet.
Hypothese 1: Proteinhydrolysate lassen sich durch Filtration in Fraktionen auftrennen.
Status: teilweise verifiziert
Grundlegend eignet sich die erarbeitete Methode zur Auftrennung von Proteinhydrolysaten.
Die im Rahmen dieser Studie untersuchten Hydrolysate konnten per Crossflow-Filtration in
zwei Fraktionen getrennt werden. Hierzu wurde ein MWCO von 100 kDa verwendet. Zur
Optimierung des Fraktionierungsprozesses wurde der Einfluss von pH-Wert und Ionenstärke
auf die Filtrationsparameter Permeatflux JFm, Transmission, sowie die
Molekülgrößenverteilung der Fraktionen, überprüft.
Entgegen den Erwartungen konnten jedoch nicht mehr als zwei Fraktionen pro Rohstoff
erzeugt werden. Zudem wurden für Permeatflux JFm und Transmission rohstoffabhängig
teilweise signifikante Einflüsse von pH-Wert und Ionenstärke festgestellt, die, vermutlich
aufgrund des dominanten Einflusses ladungsbedingter Molekül-Membran- und Molekül-
Molekül-Interaktionen, jedoch keinen maßgeblichen Einfluss auf die per SE-HPLC ermittelte
Molekülgrößenverteilung der Fraktionen haben. Entscheidend für die Güte der Fraktionierung
ist die Beschaffenheit der Foulingschicht. Unter Berufung auf die Erkenntnisse von She et al.
(2009), Salgın (2007), Huisman et al. (2000) und Pouliot et al. (1999) wird davon ausgegangen,
dass pH-Wert, sowie Nähe zum IEP der gelösten Stoffe, Ionenstärke und pH-induzierte
Abstoßungseffekte die Foulingneigung und Struktur der Foulingschicht beeinflussen. Während
sich deren Einfluss in zumeist unter Verwendung von BSA ermittelten Modellsystem
reduzieren lässt, deuten unter anderem Erkenntnisse von Almécija et al. (2007) darauf hin,
dass sich die in Modellsystemen ermittelten optimalen Filtrationsbedingungen nicht ohne
Weiteres auf komplexere Medien übertragen lassen. Bei komplexen Medien ist diesbezüglich
eine Minimierung der Foulingeinfüsse, jedoch keine vollständige Vermeidung selbiger zu
erwarten. Dies resultiert in teilweise deutlichen Verschiebungen des MWCO und konnte auch
durch die Anwendung von Ultraschall während der Filtration nicht umgangen werden. Die
Erzeugung von mehreren, hinsichtlich der Molekülgrößenverteilung enger definierter
Fraktionen, gestaltet sich somit schwierig. Einen Ansatzpunkt zur Verbesserung der
Molekülgrößenverteilung der Fraktionen stellt die Dialyse des Feeds vor dem eigentlichen
Filtrationsprozess dar. Hierbei lassen sich bereits durch 24-stündige Dialyse unter

Diskussion
235
Verwendung eines MWCO von 2 kDa Moleküle ≤ 1,35 kDa zuverlässig auswaschen. Die
Ausbildung von internem Fouling kann so potentiell reduziert werden. Eine Verifizierung dieser
These steht jedoch noch aus. Unabhängig davon kann die zu Beginn aufgestellte Hypothese
unter Berücksichtigung des aktuellen Kenntnisstands somit teilweise verifiziert werden.
Hypothese 2: Durch Hydrolyse kann aus der Alge (Chlorella vulgaris) ein für die
Fraktionierung nutzbares Feed gewonnen werden.
Status: verifiziert
Durch Hydrolyse der Alge (Chlorella vulgaris) konnte ein für Fraktionierungszwecke nutzbares
Feed unter Verwendung Pronase erzeugt werden.
Eine Hydrolyse der Alge Chlorella vulgaris resultiert bedingt durch die geringe Zugänglichkeit
des Proteins in einer komplexen Abfolge mehrerer Verfahrensschritte. Eine der Hydrolyse
vorausgehende Vorbehandlung mit HCl, pH 3 ist notwendig um die Zellwandstrukturen der
Alge zu lysieren. Die daraus resultierende Molekülgrößenverteilung verfügt über ein breites
Molekülgrößenspektrum mit Strukturen von bis zu 670 kDa. In Anbetracht der von Morris et al.
(2008) durchgeführten Charakterisierung von Chlorella vulgaris Proteinen muss davon
ausgegangen werden, dass die Ausbildung von Proteinaggregaten nicht auszuschließen ist.
Unter Berücksichtigung der Zielsetzung der Fraktionierung kann die aufgestellt Hypothese
verifiziert werden.
Hypothese 3: Die anhand von hydrolysiertem Molkenprotein-Isolat gewonnenen
Kenntnisse hinsichtlich der Fraktionierung lassen sich auf weitere Proteinquellen
übertragen.
Status: teilweise verifiziert
Die grundlegenden Betriebsparameter Pumpenfördermenge Q/Strömungsgeschwindigkeit ʋ,
Transmembrandruck pTMP, Feed-Temperatur TCF, sowie die Anzahl der verwendeten
Diavolumina VDia wurden bei allen Versuchen beibehalten.
pH-Wert und Ionenstärke des Feeds besitzen, wie bereits unter Hypothese 1 dargestellt,
jedoch einen maßgeblichen Einfluss auf Permeatflux JFm, Transmission, sowie die
Molekülgrößenverteilung der Fraktionen. Eine vollständige Verifizierung der Hypothese ist
somit nicht möglich.

Diskussion
236
Hypothese 4: Einzelne Peptid-Fraktionen verfügen über unterschiedliche
Geschmacksqualitäten.
Status: verifiziert
Die sensorisch untersuchten Retentat- und Permeatfraktionen wurden als „süß“ (Retentat
hydrolysiertes Molkenprotein-Isolat), „salzig“ (Permeat hydrolysiertes Molkenprotein-Isolat,
Retentat/Permeat hydrolysierte Alge (Chlorella vulgaris)), „sauer“ (Permeat hydrolysierte Alge
(Chlorella vulgaris)) und „umami“ (Retentat/Permeat hydrolysierte Alge (Chlorella vulgaris))
charakterisiert.
Während die „süß“-Komponente von hydrolysiertem Molkenprotein-Isolat vermutlich auf im
Retentat befindliche Lactose-Rückstände zurückzuführen ist, wird davon ausgegangen, dass
die weiteren Geschmackseindrücke überwiegend auf Glu und Asp-haltige Di- und
Oligopeptidverbindungen zurückzuführen ist. Um diese Aussage zu verifizieren, ist eine
Sequenzierung der Strukturen mit anschließender sensorischer Bemusterung notwendig. Die
Hypothese kann diesbezüglich unter Vorbehalt verifiziert werden.
Hypothese 5: Eine Peptidfraktion besitzt die Eigenschaft eines Bittergeschmack-
Blockers.
Status: verifiziert
Die aus hydrolysiertem Molkenprotein-Isolat, sowie hydrolysierter Alge (Chlorella vulgaris)
gewonnenen Permeatfraktionen wurden hinsichtlich ihrer „bitter“-blockenden Eigenschaften
auf den durch Koffein hervorgerufenen Bittergeschmack untersucht. Hierzu wurde ein flüssiger
„bitter“-Standard (0,22 g/l Koffein) mit „bitter“-Standard/Permeat-Lösungen (0,22 g/l
Koffein/1,0 g/l fraktioniertes Proteinhydrolysat) bezüglich der „bitter“-Ausprägung vergleichend
bewertet.
Durch Zusatz von fraktioniertem Proteinhydrolysat aus Alge (Chlorella vulgaris) konnte der
„bitter“-Eindruck von Koffein signifikant um 40,3 % verringert werden. Dies ist vermutlich auf
das Wirken einer oder mehrerer Peptidverbindung als Bitterstoff-Antagonist zurückzuführen.
Diese blockieren, wie von Brockhoff et al. (2011) beschrieben, die Bindungsstelle bestimmter
Bitterrezeptoren und beeinflussen somit den Transduktionsprozess. Um diese Aussagen zu
verifizieren, sind neben der Sequenzierung der Fraktionen weitere, umfangreiche
Untersuchungen hinsichtlich der Interaktion der identifizierten Strukturen mit den
Bindungsstellen verschiedener Rezeptortypen notwendig. Die Hypothese kann diesbezüglich
unter Vorbehalt verifiziert werden.

Diskussion
237
Hypothese 6: Die Peptidfraktionen lassen sich mit der erarbeiteten Methode in
produktionstauglichem Maßstab herstellen.
Status: falsifiziert
Im Rahmen dieser Studie wurde, aufgrund des bereits unter Hypothese 1 dargestellten
Einfluss von Fouling auf die Molekülgrößenverteilung der Fraktionen, auf eine Feed-
Konzentration von 0,15 % zurückgegriffen.
Die Widerfindungsrate bei der Fraktionierung der als „bitter“-Blocker wirksamen Algen-
Fraktionen (Chlorella vulgaris) lag bei 77,11 ± 1,58 %. Hiervon entfallen 63,14 ± 2,42 % auf
die jeweiligen Permeatfraktionen. Zur Herstellung von ca. 0,1 %igen Lösungen der
Permeatfraktion waren bei der verwendeten Anlagenkonzeption 45,29 ± 2,30 min notwendig.
Nachfolgend sind weitere Aufarbeitungsschritte, beispielsweise eine Aufkonzentration des
durch Diafiltration verdünnten Permeats, notwendig. Eine Umsetzung der Methodik unter
Verwendung höherer Feed-Konzentrationen erscheint aufgrund der erreichten Güte der
Fraktionierung unwahrscheinlich. Die Hypothese muss diesbezüglich unter dem jetzigen
Kenntnisstand falsifiziert werden.
5.5.2 Methodische Aspekte
Im nachfolgenden Abschnitt werden methodische Aspekte, die potentiell einen wesentlichen
Einfluss auf die Ergebnisse haben könnten, bewertet.
5.5.2.1 Rohstoffe
Molkenprotein-Isolat:
Für die Etablierung des Crossflow-Filtrations-Prozesses wurden aufgrund der
standardisierten, von produzierenden Unternehmen analytisch kontrollierten
Zusammensetzung, der kommerziellen Erhältlichkeit, sowie dem breiten Angebot und der
preiswerten Anschaffung hydrolysierte Molkenprotein-Isolate verwendet. Im Rahmen der
Vorarbeiten für diese Studie wurden verschiedene hydrolysierte Molkenprotein-Isolate auf ihre
Eignung überprüft. Die Auswahl erfolgte bedingt durch den im Vergleich zu
Konkurrenzprodukten geringen „bitter“-Geschmack, sowie das gute Lösungsverhalten des
Rohstoffs in wässrigen Medien. „Thermax 690“ (Lot. 02923210) (Glanbia Nutritionals Ltd., IRL)
verfügt über einen hohen Protein-Anteil (> 90 %) bei gleichzeitig niedrigem Gehalt an Fett (<
1 %) und weiteren niedermolekularen Bestandteilen (Tabelle 2, Seite 49). Die
Amionsäurenzusammensetzung (Tabelle 3, Seite 50), sowie der Anteil an Mineralstoffen ist
zudem bekannt (Glanbia Nutritionals Ltd. 2012).
Die Wahl von „Thermax 690“ (Lot. 02923210) (Glanbia Nutritionals Ltd., IRL) kann im Hinblick
auf die Eignung des Rohstoffs, sowie die umfangreiche Kenntnis der Zusammensetzung als

Diskussion
238
zufriedenstellend betrachtet werden. Der hohe Anteil an Protein bei gleichzeitig niedrigem
Anteil weiterer Komponenten vereinfacht die Herleitung der während des
Fraktionierungsprozess auftretenden Effekte, da diese mit sehr hoher Wahrscheinlichkeit auf
die variierten physikalisch-chemischen Eigenschaften des Feeds (pH-Wert, Ionenstärke),
sowie die Prozesshydrodynamik zurückgeführt werden können. Der Einfluss des „Nicht-
Protein-Anteils“ des Rohstoffs, insbesondere des Gehalts an Salzen, auf die Sensorik der
Retentat- & Permeatfraktionen, kann im Rahmen dieser Studie nicht vollständig
ausgeschlossen werden, erscheint im Hinblick auf die in der Spezifikation festgehaltenen
geringen Mengen jedoch als zu vernachlässigen.
Alge (Chlorella vulgaris):
Neben Molkenprotein-Isolat wurde in dieser Studie das Hydrolysat der Alge Chlorella vulgaris
fraktioniert und die gewonnenen Fraktionen sensorisch bewertet. Der Bezug
versuchsdienlicher Mengen an Alge bzw. Algenprotein stellte sich zum Studienzeitpunkt als
aufwendig heraus. Unter Berücksichtigung der Rohstoffzusammensetzung wurde „Chlorella
vulgaris Powder“ (Lot. 201410215) (Allma, Lissabon, PRT) für die beschriebenen Versuche
verwendet.
„Chlorella vulgaris Powder“ (Allma, Lissabon, PRT) verfügt über eine ähnliche
Zusammensetzung wie Konkurrenzprodukte (z.B. „Protein Rich Whole Algae“ (AlgaVia®, San
Francisco, USA)) und eignete sich grundlegend für die Versuchszwecke. Der vergleichsweise
hohe Gehalt an Kohlenhydraten und Ballaststoffen (Tabelle 4, Seite 50) erschwerten allerdings
die präzise Zuweisung der beobachteten Effekte, sowie der sensorischen Profile der
Fraktionen, da deren Einfluss auf den Fraktionierungsprozess, bzw. die
Geschmacksausprägungen der Fraktionen, nicht vollständig ausgeschlossen werden konnten.
Nichtsdestotrotz ließen sich die im Rahmen dieser Studie dokumentierten Beobachtungen
unter Berufung auf aktuelle Primärliteratur einordnen und diskutieren.
5.5.2.2 Crossflow-Diafiltration
Membran- & Anlagen-Charakterisierung:
Die im Rahmen dieser Studie durchgeführte Membrancharakterisierung lieferte jeweils in
Dreifachwiederholung reproduzierbare Ergebnisse mit sehr geringen Standardabweichungen
und erwies sich im Rahmen dieser Studie als geeignet. Alternativ zu der aufgrund der
einfacheren Durchführung angewandten Charakterisierung der Membran über den kritischen
Druck pCP besteht die Möglichkeit der Membrancharakterisierung mit dem von Field et al.
(1995) und Howell (1995) beschriebenen Verfahren der Charakterisierung des
Filtrationsvorgangs anhand des kritischen Flux JCF.

Diskussion
239
Einfluss von pH-Wert & Ionenstärke:
Der Einfluss von pH-Wert und Ionenstärke auf den Filtrationsprozess wurde anhand von je 5
pH-Stufen (Molkenprotein-Isolat: 4, 6, 7, 8, 9; Alge (Chlorella vulgaris): 3, 6, 7, 8, 11), sowie
einer NaCl-Konzentration (150 mM) überprüft. Die Auswahl der Parameter basierte auf
Literaturrecherchen unter Berücksichtigung des Einflusses der physikalisch-chemischen
Eigenschaften des Feeds auf die Struktur und Eigenschaften der im Feed enthaltenen
Moleküle. Die Auswahl der pH-Stufen und Ionenstärke erwies sich als geeignet um den
Einfluss der Faktoren auf den Fraktionierungsprozess abzubilden. Im Falle der Fraktionierung
von hydrolysiertem Molkenprotein-Isolat konnten die im Rahmen dieser Studie beobachteten
Erkenntnisse mit den anhand von intakten Proteinen oder Modellsystemen ermittelten
Ergebnissen aktueller Primärliteratur abgeglichen werden. Im Falle der Fraktionierung von
hydrolysierter Alge (Chlorella vulgaris) wurden die beobachteten Effekte unter Zuhilfenahme
verwandter Proteinquellen, sowie grundlegender Erkenntnisse zu Mikroalgen hergeleitet.
Zur Optimierung des Prozesses im Hinblick auf Permeatflux, Transmission und Ausbeute sind
kleinstufigere pH-Studien, sowie eine breiter aufgestellte Versuchsreihe zum Einfluss der
Ionenstärke denkbar.
Einfluss der Ultraschall-Anwendung:
Der Einfluss der Ultraschall-Anwendung auf die Fraktionierung von hydrolysiertem
Molkenprotein-Isolat wurde anhand eines Modellversuchs in kleinem Maßstab (Methodik:
Kapitel 3.2.2.2, Seite 56) untersucht. Die ermittelten Ergebnisse stehen im Wiederspruch zu
den Erkenntnissen vieler Autoren, darunter Muthukumaran, Kentish, Ashokkumar, et al.
(2005), Lamminen et al. (2004) und Matsumoto et al. (1996). Diese arbeiteten jedoch mit
intakten Proteinen, weniger komplexen Feeds oder verwendeten Modellsysteme. Die
Fraktionierung hydrolysierter Proteinquellen unter Einsatz von Ultraschall ist in der
Primärliteratur bis dato nicht dokumentiert. Die in der vorliegenden Studie ermittelten
Ergebnisse besitzen, aufgrund des geringen Umfangs der Untersuchungen, Hinweischarakter.
Um die Aussagekraft zu erhöhen, sind weitere Versuchsreihen notwendig, bei denen neben
dem Einfluss des Ultraschalls auch Faktoren wie die US-Frequenz, die Leistungsstärke der
US-Einheit, weitere Feed- und Membran-Eigenschaften, sowie die Prozesshydrodynamik
näher betrachtet werden müssen. Nichtsdestotrotz liefern die Ergebnisse wichtige
Erkenntnisse bezüglich des Einflusses des Ultraschalls auf die Fraktionierung von
Proteinhydrolysaten.
Eignung von Membranen aus regenerierter Cellulose:
Die Foulingneigung einer Membran wird neben den Trenneigenschaften auch durch deren
Morphologie und hydrophobe/hydrophile Eigenschaften beeinflusst (Melin & Rautenbach,

Diskussion
240
2007). Regenerierte Cellulose ist neben Cellulose Acetat das hydrophilste Membranmaterial
(Walter et al., 2012). Neben den im Rahmen dieser Studie verwendeten PS- bzw. PES-
Membranen, wurde anhand eines Modellversuchs in kleinem Maßstab (Methodik: Kapitel
3.2.2.3, Seite 58) eine RC-Membran auf ihre Eignung zur Filtration von hydrolysiertem
Molkenprotein-Isolat überprüft. Vergleichbar der Versuchsreihe zum Einfluss von Ultraschall
besitzen die Ergebnisse, aufgrund des geringen Untersuchungsumfangs, Hinweischarakter.
Zur Erhöhung der Aussagekraft sind weitere Versuche unter Variation der physikalisch-
chemischen Eigenschaften des Feeds, der Prozesshydrodynamik, sowie der Verwendung
verschiedener Membranen notwendig.
Dialyse zur Abtrennung kleiner Moleküle (< 2 kDa) aus dem Feed:
Um zu überprüfen, ob kleine Moleküle (< 2 kDa) aus dem Feed durch Dialyse abgetrennt
werden können, wurde der Einfluss von pH-Wert und Dialysedauer auf die
Molekülgrößenverteilung der Retentatfraktion überprüft. Die pH-Versuche fanden unter
Verwendung eines „Initial-pHs“ statt. Unter Initial-pH wird der pH-Wert der Dialyseprobe
verstanden, der zu Beginn des Dialyseprozess, sowie bei Wechsel der Dialyselösung
eingestellt wurde. Der pH-Wert der Dialyseprobe ist somit über die Dauer des Prozesses als
nicht konstant zu betrachten, wurde jedoch zu definierten Zeitpunkten (Wechsel der
Dialyselösung) neu justiert. Die in Abhängigkeit des pH-Werts ermittelten Ergebnisse basieren
somit nicht rein auf dem Einfluss des pH-Werts, sondern auch auf der im Laufe des
Dialyseprozess stattfindenden pH-Änderung. Nichtsdestotrotz können die in Abhängigkeit des
pH-Werts ermittelten Ergebnisse auf einen definierten Einflussfaktor zurückgeführt werden. In
Anbetracht des geringen Versuchsumfangs besitzen die Erkenntnisse Hinweischarakter. Um
die Aussagen zu verifizieren, sowie den Prozess zu optimieren, sind weitere Versuchsreihen
unter Verwendung von pH-stabilen Pufferlösungen denkbar.
Einfluss weiterer Faktoren:
Der Einfluss der konstant gehaltenen Filtrationsparameter Strömungsgeschwindigkeit ʋ, Feed-
Konzentration CF, Temperatur des Feeds TCF, sowie die Anzahl der verwendeten Diavolumina
VDia wurde bereits in Kapitel 5.3.6 näher betrachtet. Die Auswahl der Parameter wurde anhand
des aktuellen Kenntnisstands einschlägiger Fachliteratur (unter anderem: Baldasso, Barros, &
Tessaro, 2011; Butylina, Luque, & Nyström, 2006; Choi, Zhang, Dionysiou, Oerther, & Sorial,
2005; Pelegrine & Gasparetto, 2005; Huisman, Prádanos, & Hernández, 2000; Madaeni, Fane,
& Wiley, 1999; She, Tang, Wang, & Zhang, 2009) unter Berücksichtigung der
Anlagenkonzeption und der Eigenschaften der verwendeten Rohstoffe getroffen. Die im
Rahmen dieser Studie erarbeitet Erkenntnisse verifizieren und erweitern den literaturseitig
dokumentierten Kenntnisstand.

Diskussion
241
5.5.2.3 Hydrolyse der Alge Chlorella vulgaris
Mikroalgen verfügen über stabile, vorwiegend cellulosebasierte Zellwände, die eine Digestion
der Algenproteine erschweren können (Renneberg & Berkling, 2012; Morris et al., 2008;
Tchorbanov et al., 1986). Um die Proteine/Peptide der Alge Chlorella vulgaris als Feed nutzen
zu können, war die Entwicklung eines Hydrolyseverfahrens notwendig. Für Chlorella vulgaris
erwies sich eine saure Vorbehandlung (HCl, pH 3, 48 h) mit anschließender Hydrolyse durch
Pronase (0,25 %, 112,5 PU, 45 min) als geeignet.
Das erarbeitete Hydrolyseverfahren lieferten wiederholbare Ergebnisse hinsichtlich
Hydrolysegraden, Standardabweichungen und Molekülgrößenverteilungen, die mit aktueller
Primärliteratur wie beispielsweise Morris et al. (2008) vergleichbar sind. Um den
Vorbehandlungs- und Hydrolyseprozess zu optimieren, ist die Überprüfung weiterer
leistungsfähiger Enzyme, wie beispielsweise Alcalase oder Subtilisin, denkbar.
5.5.2.4 Instrumentelle Analytik & Probenaufbereitung
Spektralphotometrische Proteingehaltbestimmung:
Die Proteingehalte von Feed, Retentat und Permeat wurden spektralphotometrisch bei einer
Wellenlänge von 280 nm ermittelt. Die zur Bestimmung der Proteingehalte verwendete
spektralphotometrische Methode verfügt über eine solide Kalibrierfunktionen, deren Eignung
durch Bestimmtheitsmaße R2 zwischen 0,997 (Alge (Chlorella vulgaris)) und 0,999
(Molkenprotein-Isolat), bei gleichzeitig sehr geringen Standardabweichungen, belegt werden.
Bei der quantitativen Bestimmung der Proteingehalte einer Probe ist die Analyse bei 280 nm
am weitesten verbreitet (Simonian, 2002). Im Bereich der Wellenlänge 280 nm sind
schwerpunktmäßig die Aminosäuren Tryptophan, Tyrosin, sowie Disulfid-Brücken für die
Extinktion verantwortlich (Aitken & Learmonth, 2009; Simonian, 2002). Eine Alternative hierzu
stellt die Bestimmung des Proteingehalts im Fern-UV-Bereich, z.B. bei 205 nm dar (Aitken &
Learmonth, 2009; Ahmed, 2004). Für die Extinktion sind bei dieser Wellenlänge
schwerpunktmäßig (in absteigender Reihenfolge) die Aminosäuren Tryptophan, Phenylalanin,
Tyrosin, Histidin, Cystein, Methionin und Arginin verantwortlich (Aitken & Learmonth, 2009).
Die Messung bei 205 nm gilt als sehr sensibel und ist insbesondere für Proben mit niedrigen
Proteingehalten geeignet, wird jedoch durch in der Probe enthaltene Reagenzien (z.B.
Puffersubstanzen) beeinträchtigt (Ahmed, 2004; Simonian, 2002). Whitaker & Granum (1980)
etablierten eine Methode, bei welcher die Bestimmung des Proteingehalts einer Probe ohne
Referenzproteine oder die Erstellung einer Standardkurve vorgenommen werden kann. Hierzu
wird die Extinktion der Probe bei 280 nm und 235 nm gemessen und der Proteingehalt
rechnerisch ermittelt. Vorteil der Methode ist unter anderem der Ausschluss von durch
Nukleinsäuren ausgelöste Interferenzen, sowie der Ausschluss des Einfluss verschiedener
Aminosäurenzusammensetzungen auf die Genauigkeit des Messergebnis (Whitaker &

Diskussion
242
Granum, 1980). Für nachfolgenden Versuche ist eine Verifizierung der Messergebnisses
durch Verwendung einer der vorab beschriebenen Methoden denkbar.
Bestimmung des Hydrolysegrads der Proteinhydrolysate:
Zur Schnell-Bestimmung des Hydrolysegrads wurde die von Nielsen et al. (2001)
beschriebene OPA-Methode verwendet. Die OPA-Methode gilt als einfach, genau, wenig
fehleranfällig und liefert vergleichbare Ergebnisse zu anderen Bestimmungsmethoden
(Nielsen et al., 2001). Bei Cystein-reichen Proteinen wird der durch die OPA-Methode
ermittelte Hydrolysegrad im Vergleich zur TNBS- und pH-stat-Methode geringfügig
unterschätzt (Spellman et al., 2003). Spellman et al. (2003) führen dies auf die instabile
Bindung von Cystein und OPA zurück. Im Rahmen dieser Studie ist ein Einfluss des Cystein-
Gehalts auf den per OPA-Methode ermitteln Hydrolysegrad, in Anbetracht des
Aminosäurenprofils des Rohstoffs Molkenprotein-Isolat, bzw. der bekannten
Aminosäurenzusammensetzung der Alge Chlorella vulgaris, als gering zu bewerten.
Analytik der Peptidfraktionen mittels SE-HPLC:
Die SE-HPLC gilt als leistungsfähiges Verfahren zur Größencharakterisierung einer
Probensubstanz (Hong, Koza, & Bouvier, 2012). Die Auftrennung einer Probe erfolgt dabei, im
Idealfall, lediglich anhand des hydrodynamischen Radius der Moleküle (Tieke, 2014; Yau &
Kirkland, 1981). In der Praxis lassen sich Wechselwirkungen zwischen Probe und stationärer
Phase jedoch nicht vollkommen ausschließen. Um dadurch entstehende nachteilige Effekte
auf die Vorhersage zu minimieren, ist diese beispielsweise durch experimentelle Anpassung
von Ionenstärke und pH-Wert des Puffers zu reduzieren. In Kapitel 4.1.1 (Seite 79) ist die im
Rahmen dieser Studie durchgeführte SE-HPLC-Methodenerstellung dargestellt. Die Eignung
und Zuverlässigkeit der SE-HPLC-Methode wurde durch sehr geringe Standardabweichungen
der Retentionszeiten tR (SD: 0,51 - 1,74 %) bei gleichzeitig höchst linearem Verlauf der
Kalibriergeraden (R2 = 0,996) im Zehnfachversuch belegt. Die geringfügigen,
anlagespezifischen Performance-Unterschiede (Auftrennung des Myoglobin-Peaks) zwischen
den beiden Test-Systemen sind auf die gerätespezifisch unterschiedliche Bauform
zurückzuführen.
5.5.2.5 Sensorik
Schulung des Sensorik-Panels:
Die im Rahmen dieser Studie durchgeführte sensorische Schulung der Prüfpersonen basierte
auf der DIN EN ISO 8586:2014. Zusätzlich zu den in der vorab erwähnten Norm beschriebenen
und im Kapitel 3.5.1 (Seite 68) dargestellten Prüfungen wurde das Schulungskonzept um die
Prüfung „Ermittlung der Reiz- und Erkennungsschwelle“ gemäß ISO 3972:2011 erweitert, um
die Leistung der Prüfpersonen, sowie des Panels besser charakterisieren zu können. Die

Diskussion
243
Skalen-Schulung wurde in Anlehnung an das Kapitel „Ausbildung eines deskriptiven Panels -
Schulungsphase: Training von Intensitätsmessungen“ von Rummel (2002) durchgeführt.
Geschult wurden 24 Teilnehmer (8 männlich, 16 weiblich) im Alter von 19 – 29 (MW: 22,75 ±
2,44) Jahren. Die Teilnehmer wurden in zwei Gruppe à 12 Teilnehmern aufgeteilt. Die
Schulung der Gruppen erfolgte an aufeinanderfolgenden Tagen.
Das gewählte Schulungsprocedere eignete sich, gemessen an den Ergebnissen der Schulung,
für den vorgesehenen Schulungszweck. Bei der „Prüfung der Wahrnehmung eines Reizes“
konnte der Anteil korrekter Zuordnungen insbesondere bei „sauer“ und „umami“ deutlich
verbessert werden. Die Grundgeschmacksarten „salzig“ und „süß“ wurden bereits zu Beginn
des Schulungsprozesses von einem Großteil der Panelteilnehmer korrekt bestimmt. Die im
Anschluss durchgeführte Prüfung „Ermittlung der Reiz- und Erkennungsschwelle“ (gemäß ISO
3972:2011) offenbarte prüferspezifische Unterschiede hinsichtlich der Reiz- und
Erkennungsschwelle, insbesondere für „salzig“, „umami“ und „süß“, welche die Notwendigkeit
einer gezielten Prüferauswahl verdeutlichten. Die Diskriminierungsfähigkeit des Panels
konnte, beurteilt anhand der „Prüfung zur Unterscheidung von Intensitätsstufen eines Reizes“,
jedoch mit Ausnahme niedriger Konzentrationen von „bitter“ und „süß“ gewährleistet werden.
Gemessen an den Ergebnissen der Skalenschulung (Mittelwert, Modus, Median; Tabelle 55,
Seite 169) konnten den Prüfpersonen der Zusammenhang zwischen Geschmackseindruck
und korrespondierendem Skalenwert verdeutlich werden.
Prüferauswahl:
Aufgrund des ordinalen Skalenniveaus und der Verletzung der Annahme der Normalverteilung
der Daten wurden verteilungsunabhängige, nichtparametrische Testverfahren zur Auswertung
herangezogen. Die Prüferauswahl erfolgte mehrstufig in Anlehnung an die Vorgaben der DIN
EN ISO 8586:2014. Der zur Überprüfung der Diskriminierungsfähigkeit der Prüfpersonen
zwischen Proben mit den Intensitätsstufen „niedrig“, „mittel“, „hoch“ durchgeführte Friedman-
Test gilt als nicht-parametrische Alternative der ANOVA bei der Analyse verbundener
Stichproben (Kühlmeyer, 2001). Der zur Bestimmung der Panelhomogenität verwendete
Kruskal-Wallis H Test stellt eine nicht-parametrische Alternative der ANOVA bei der Analyse
unverbundener Stichproben dar (MacFarland & Yates, 2016; Corder & Foreman, 2009). Da
der Kruskal-Wallis H Test eine Aussage hinsichtlich der gesamten Stichprobe (hier: Panel)
trifft, wurde in Anlehnung an Corder & Foreman (2009) im Bedarfsfall eine paarweise
Überprüfung der Bewertungsunterschiede zwischen Prüfteilnehmern durchgeführt, um deren
Eignung für das jeweilige Panel zu bestimmen. Die Teststärke nicht-parametrischer Tests gilt
generell als geringer als die des zugehörigen parametrischen Tests (Cunningham & Aldrich,
2013). Gemäß Kraemer & Thiemann (1987) sind die geringen Verlust der Teststärke in
Anbetracht des Erhalts der Validität der Daten jedoch vernachlässigbar. Unter

Diskussion
244
Berücksichtigung der Datenlage sind nicht-parametrische Test parametrischen Verfahren
vorzuziehen, wenn die Anforderung der Normalverteilung verletzt wird (Ryan, 2013; Cohen &
Holliday, 1996). Die Auswertung der Daten mit nicht-parametrischen Test gestaltet sich
komplexer und aufwändiger als eine vergleichbare Analyse unter Verwendung parametrischer
Verfahren. Die Anforderungen der DIN EN ISO 8586:2014 hinsichtlich der Auswertung
sensorischer Daten, sowie der Zusammenstellung eines Panels konnten jedoch eingehalten
werden.
Die Homogenität der Panel-Aussage hinsichtlich der Intensitäts-Bewertung der
Geschmacksausprägungen „niedrig“, „mittel“ und „hoch“ der fünf Grundgeschmacksarten ist
insbesondere für „süß“, „sauer“ und „salzig“ gegeben. Lediglich im Falle von „umami“ wurde
eine deutlich geringere Aussage-Homogenität festgestellt. Dies ist vermutlich auf eine von den
Normvorgaben abweichende Prüferanzahl je Panel zurückzuführen. Die in der DIN EN ISO
8586:2014 geforderte Prüferanzahl von mindestens 10 Prüfern pro Panel konnte im Rahmen
dieser Studie, insbesondere Aufgrund der begrenzten Verfügbarkeit der Panelisten nicht
gewährleistet werden. Für nachfolgende Studien unter vergleichbaren Umständen empfiehlt
sich eine frühzeitige Rekrutierung und Schulung eines größeren Prüferpools, z.B. durch
Rekrutierung von Studierenden des 1. und 2. Semesters zu Beginn einer Studie. Diese
könnten über einen längeren Zeitraum intensiver geschult werden, wodurch potentiell die
Sicherheit der Prüfaussage, sowie die Homogenität des Prüfergebnisses verbessert werden
könnten. Schwierig dabei ist jedoch vermutlich der Erhalt der Motivation der Panelteilnehmer
über eine längere Zeitspanne.
Sensorik der Peptidfraktionen:
Im Rahmen dieser Studie wurden je Proteinquelle (Molkenprotein-Isolat, Alge „Chlorella
vulgaris“) Retentat- und Permeatfraktion der fraktionierten Proteinhydrolysate sensorisch
hinsichtlich der Geschmacksausprägungen „süß“, „sauer“, „salzig“, „bitter“ und „umami“
untersucht. Zusätzlich wurde ein Bitterstandard, sowie ein Gemisch aus Bitterstandard und
Permeat verkostet, um zu überprüfen, ob die Permeatfraktion in der Lage ist, die
Bitterwahrnehmung zu reduzieren. Retentat- und Permeatfraktion wurden in einer
Konzentration von 1g/l gereicht. Der Bitterstandard entsprach der Konzentrationsstufe D2
(0,22 g/l Koffein) der Prüfung „Ermittlung der Reiz- und Erkennungsschwelle“ und somit einer
hohen Geschmacksintensität. Das Gemisch aus Bitterstandard und Permeat bestand aus 1 g/l
fraktioniertem Proteinhydrolysat und 0,22 g/l Koffein. Die Wahl der Konzentration basiert auf
einer rechnerischen ermittelten, maximalen Probenkonzentration unter Berücksichtigung der
verfügbaren Probenmengen. In Anbetracht der zumeist mit niedrigen Skalennoten bewerteten
Geschmacksausprägungen wäre eine sensorische Beurteilung höher konzentrierter Proben
der verkosteten Fraktionen interessant. Hierdurch könnten die im Rahmen dieser Studie

Diskussion
245
erlangten Ergebnisse verifiziert werden, da die Aussagegenauigkeit der Prüfteilnehmer mit
steigender Intensität der Geschmacksausprägung an Sicherheit gewinnt. Diese Annahme
setzt voraus, dass eine höhere Konzentration an hydrolysiertem, fraktioniertem Protein über
eine höhere Geschmacksintensität verfügt. Aufgrund des aufwendigen und zeitintensiven
Gewinnungsprozesses war dies im Zuge dieser Studie nicht möglich. Die Untersuchung der
Permeatfraktion auf „bitter“-blockende Eigenschaften basierte auf Literaturerkenntnissen
(unter anderem dargestellt in Kapitel 2.5.3.2, Seite 44) die insbesondere niedermolekulare
Substanzen mit einer „bitter“-blockenden Eigenschaft in Verbindung bringen.
5.5.3 Ausblick
Die Ergebnisse der vorliegenden Studie liefern Ansatzpunkte für weitere Forschungsarbeiten
zur Optimierung des Fraktionierungsprozesses von Proteinhydrolysaten.
Nachfolgende Studien sollten die Eignung eines vorfiltrierten, dialysierten Feeds für den
Crossflow-Fraktionierungsprozess näher betrachten. In der vorliegenden Studie konnte
anhand der Dialyse verschieden konzentrierter Feeds hydrolysierten Molkenprotein-Isolats
gezeigt werden, dass sich der Anteil an Strukturen ≤ 1,35 kDa durch eine 24 h Dialyse bis zu
einer Feed-Konzentration von 4 % (w/v) reduzieren lässt. Unter der Annahme, dass
insbesondere kleine Moleküle für die Ausbildung von Fouling und die damit in Verbindung
stehende Reduktion des MWCO verantwortlich sind, besteht die Möglichkeit einer Optimierung
des Fraktionierungsprozesses, insbesondere im Hinblick auf die Molekülgrößenverteilung der
gewonnenen Fraktionen.
Der Einsatz von Ultraschall (35 kHz) zur Verminderung des Foulingeinflusses und Steigerung
des Permeatflux erwies sich im Rahmen dieser Studie als nicht zielführend. Unter
Berücksichtigung der zuvor beschriebenen Verwendung vorfiltrierter, dialysierter Feeds bedarf
diese Beobachtung allerdings einer erneuten Bewertung. Selbiges gilt für den Einsatz
alternativer Membranmaterialien, wie beispielsweise die im Rahmen dieser Studie untersuchte
Membran aus regenerierter Cellulose.
Um die im Zuge dieser Untersuchung festgestellten Geschmackseigenschaften der
Fraktionen, sowie den „bitter“-blockenden Effekt der Permeatfraktion hydrolysierter Alge
Chlorella vulgaris bestimmten Molekülen zuzuordnen ist eine Sequenzierung der Fraktionen,
sowie eine umfangreiche Untersuchungen hinsichtlich der Interaktion der identifizierten
Strukturen mit den Bindungsstellen verschiedener Rezeptortypen notwendig. Weitere
sensorische Studien zur Verifizierung des beschriebenen Effekts unter Verwendung eines
Expertenpanels werden empfohlen.

Zusammenfassung
246
6. Zusammenfassung
Bestimmte Peptidverbindungen, wie beispielweise das Dipeptid Aspartam („süß“), verfügen
über Geschmacksqualitäten welche sie für einen Einsatz in Lebensmitteln qualifizieren. Neben
„süß“ und „bitter“ schmeckenden Verbindungen wurden bereits Stoffe identifiziert, die einen
„umami“ und „sauer“ Eindruck auslosen. Neben der chemischen Synthese konnen
Peptidverbindungen durch eine Hydrolyse intakter Proteine gewonnen werden. Durch
chromatographische Verfahren wie beispielsweise die Gel-Permeation Chromatography
können aus hydrolysierten Feeds einzelne Peptidverbindungen isoliert werden. Im Hinblick auf
eine großtechnische Gewinnung geschmacksaktiver Peptide stellen Membrantrennverfahren
eine vielversprechende, skalierbare Alternative dar.
Im Rahmen der vorliegenden Studie wurde deshalb ein Crossflow-Membrantrennverfahren zur
Fraktionierung von hydrolysierten Protein-Feeds unter Verwendung von hydrophilierten
Polysulfon-Membranen (MWCO: 100 kDa) entwickelt. Als Proteinquellen dienten
hydrolysiertes Molkenprotein-Isolat, sowie die Alge Chlorella vulgaris, für deren Hydrolyse ein
eigenes Verfahren erarbeitet wurde. Zur Optimierung des Fraktionierungsprozesses wurde der
Einfluss von pH-Wert und Ionenstärke (Anpassung durch Zugabe von 150 mM NaCl) auf
Permeatflux, Wiederfindungsrate, Transmission und die molekulare Zusammensetzung der
Fraktionen beider Proteinquellen ermittelt. Die Molekülgrößenverteilung der Fraktionen wurde
mittels SE-HPLC analysiert. Ausgewählte Retentat- und Permeatfraktionen wurden durch ein
im Rahmen der Studie geschultes Sensorik-Panel im Hinblick auf das Vorhandensein der fünf
Grundgeschmacksausprägungen „süß“, „salzig“, „sauer“, „bitter“ und „umami“ untersucht.
Darüber hinaus wurde geprüft, ob die Permeatfraktionen über eine den „bitter“-Geschmack
blockende Eigenschaft verfügen. In kleinem Maßstab wurde für hydrolysiertes Molkenprotein-
Isolat untersucht ob sich der Fraktionierungsprozess hinsichtlich Permeatflux und molekularer
Zusammensetzung der Fraktionen durch den Einsatz von Ultraschall (35 kHz) und alternativer
Membranmaterialien (regenerierte Cellulose) verbessern lässt. Zudem wurde die Eignung
einer Dialyse zur Abtrennung kleiner Moleküle (< 2 kDa) aus dem vorfiltrierten Feed unter
Variation von Initial-pH-Wert, Feed-Konzentration und Dialysedauer (24 h, 48 h) für
hydrolysiertes Molkenprotein-Isolat bewertet.
Aufgrund von während der Pilot-Versuche identifizierten, massiven Verschiebungen der
Trenngrenze der Membran (50 kDa) hin zu einem deutlich kleineren MWCO (< 17 kDa) bei
einer Feed-Konzentration von 3 % (w/v) (bezogen auf den Proteingehalt) und 1,5 % (w/v)
(bezogen auf den Proteingehalt), wurde die Feed-Konzentration auf 0,15 % (w/v) (bezogen
auf den Proteingehalt) gesenkt.
Der Einfluss des pH-Werts auf Permeatflux, Wiederfindungsrate und molekulare
Zusammensetzung der Fraktionen wurde je Proteinquelle (Molkenprotein-Isolat, Alge

Zusammenfassung
247
Chlorella vulgaris) anhand von fünf pH-Stufen untersucht (Molkenprotein-Isolat: pH 4, pH 6,
pH 7, pH 8, pH 9; Alge Chlorella vulgaris: pH 3, pH 6, pH 7, pH 8, pH 11).
Bei der Fraktionierung von hydrolysiertem Molkenprotein-Isolat beeinflusste der pH-Wert
Permeatflux, Transmission und Wiederfindungsrate nicht signifikant. Die chromatographisch
ermittelte Molekülgrößenverteilung der Fraktionen offenbarte einen weitestgehend pH-
unabhängigen Rückhalt von Strukturen ≥ 20 kDa im Retentat bei gleichzeitig hohem Rückhalt
von Strukturen ≤ 1,35 kDa. Im Permeat wurden mit Ausnahme von pH 9 lediglich geringe
Anteile an Strukturen ≥ 20 kDa festgestellt. Strukturen im Bereich ≤ 1,35 kDa dominierten die
Fraktion.
Eine Erhöhung der Ionenstärke durch Zugabe von 150 mM NaCl führte zu einer signifikanten
Erhöhung des Permeatflux im Vergleich zu einer Fraktionierung ohne NaCl-Zusatz für die pH-
Stufen pH 7, pH 8, pH 9. Darüber hinaus wurde unter Zusatz von 150 mM NaCl ein Anstieg
des Permeatflux mit steigendem pH-Wert festgestellt. Im Vergleich zu einer Fraktionierung
ohne NaCl-Zusatz führte die Erhöhung der Ionenstärke jedoch zu einer niedrigeren
Wiederfindungsrate, die sich für die pH-Stufen pH 8, pH 9 signifikant unterschied. Hierbei
wurden geringere Proteingehalte im Retentat bei gleichzeitig nur geringfügig erhöhten
Proteingehalten im Permeat festgestellt. Die chromatographisch ermittelte
Molekülgrößenverteilung zeigte, vergleichbar zu der Fraktionierung ohne NaCl-Zusatz einen
Rückhalt von Strukturen ≥ 20 kDa im Retentat bei gleichzeitig hohem Rückhalt von Strukturen
≤ 1,35 kDa. Im Permeat wurden jedoch durchweg höhere Anteile an Strukturen ≥ 20 kDa
festgestellt.
Bei der Fraktionierung von hydrolysierter Alge Chlorella vulgaris beeinflusste der pH-Wert den
Permeatflux nur in einem Fall signifikant. Für pH 6 wurden im Vergleich zu pH 3 signifikant
höhere Fluxwerte erzielt. Die Wiederfindungsrate unterschied sich in Abhängigkeit des pH-
Werts nicht signifikant, es wurden jedoch ein signifikant erhöhter Proteingehalt im Retentat bei
gleichzeitig niedrigerem Proteingehalt im Permeat für die pH-Stufe 11, im Vergleich zu pH 3,
pH 6 und pH 7 festgestellt. Der pH-Wert beeinflusste die Molekülgrößenverteilung der
Fraktionen nur geringfügig. Unabhängig von der pH-Stufe wurden in Retentat und Permeat
hauptsächlich Strukturen ≤ 1,35 kDa festgestellt. Dies ist nicht alleinig auf den Einfluss des
pH-Werts während der Fraktionierung, bzw. den Fraktionierungsprozess zurückzuführen.
Bereits nach der Vorfiltration des Feeds konnten in der Nullprobe für die pH-Stufen pH 3, pH
11 chromatographisch keine und für die pH-Stufe pH 8 nur geringe Anteile an Strukturen ≥ 20
kDa ermittelt werden.
Eine Erhöhung der Ionenstärke durch Zugabe von 150 mM NaCl führte zu einem signifikant
niedrigeren Permeatflux im Vergleich zu einer Fraktionierung ohne NaCl-Zusatz für die pH-
Stufen pH 7 und pH 11. Auch für die weiteren pH-Stufen wurde eine Reduktion des

Zusammenfassung
248
Permeatflux festgestellt. Wiederfindungsrate und Transmission wurden durch die Erhöhung
der Ionenstärke im Vergleich zu einer Fraktionierung ohne NaCl-Zusatz lediglich im Falle einer
Prozessführung bei pH 6 signifikant beeinflusst. Die chromatographisch ermittelte
Molekülgrößenverteilung unterschied sich in Abhängigkeit der Ionenstärke nur geringfügig.
Lediglich für pH 3, pH 6 und, in geringem Umfang, pH 8 wurden erhöhte Anteile an Strukturen
≥ 20 kDa im Retentat festgestellt. Die Permeatfraktionen wurden von Strukturen ≤ 1,35 kDa
dominiert.
Für die Fraktionierung von hydrolysiertem Molkenprotein-Isolat und hydrolysierter Alge
Chlorella vulgaris wird von einem dominanten Einfluss ladungsbedingter Abstoßungs- und
Adsorptionseffekte ausgegangen, welche den Fraktionierungsprozess durch Ausbildung einer
Foulingstruktur, sowie Interaktionseffekten zwischen Molekül und Membran bzw. Molekül und
Molekül maßgeblich beeinflussen. Die komplexe Zusammensetzung des Feeds erschwert
dabei die Identifikation fouling-induzierender Verbindungen. Im Falle der Fraktionierung
hydrolysierter Alge Chlorella vulgaris ist der Einfluss extrazellulärer polymerer Substanzen
(EPS)/freien Polysacchariden (RPS) auf den Fraktionierungsprozess nicht auszuschließen.
Bei der sensorischen Bewertung der Fraktionen aus hydrolysiertem Molkenprotein-Isolat
wurde im Retentat ein sehr schwacher „süß“-, im Permeat ein schwacher „salzig“-Geschmack
identifiziert. Die „bitter“-Ausprägung des Koffein-Standards (0,22 g/l) wurde durch Zugabe von
1 g/l Permeat nicht signifikant reduziert. Die Retentatfraktion hydrolysierter Alge Chlorella
vulgaris wurden als schwache „salzig“-, sowie sehr schwache „umami“ schmeckend, die
Permeatfraktion als schwach „salzig“-, sowie sehr schwach „sauer“ und „umami“ beschrieben.
Die „bitter“-Ausprägung des Koffein-Standard (0,22 g/l) konnte durch Zugabe von Permeat
(1g/l) signifikant reduziert werden.
Der Wirkmechanismus der „bitter“-Reduktion durch niedermolekulare Peptidverbindungen ist
bis dato nicht abschließend geklärt. Im Rahmen dieser Studie erscheint das von Brockhoff et
al. (2011) beschriebene „Gegenspieler-Prinzip“, sprich die Blockierung der (einzigen)
Bindungsstelle bestimmter Rezeptoren und die damit verbundene Maskierung des
Geschmacksreizes ein schlüssiges Erklärungskonzept zu liefern. Es wird vermutet, dass die
im Rahmen dieser Studie beschriebenen Effekte durch Moleküle < 1,35 kDa ausgelöst werden
oder aus Verbindungen < 1,35 kDa bestehen, die sich zu komplexeren Strukturen
zusammengeschlossen haben, welche in der Lage sind, die Bindungsstelle eines oder
mehrerer Rezeptortypen zu besetzen. Um diese Aussage zu verifizieren, sind weitere
Untersuchungen notwendig.
Aus den zuvor beschriebenen Erkenntnissen dieser Arbeit geht hervor, dass die
Fraktionierung von hydrolysierten Protein-Feeds unter Verwendung eines Crossflow-
Filtrationsverfahrens grundsätzlich möglich ist. Aufgrund der komplexen Zusammensetzung

Zusammenfassung
249
des Feeds können die optimalen Prozessbedingungen allerdings nur per „trial-and-error“ unter
Zuhilfenahme aktueller Primärliteratur ermittelt werden. Eine Übertragung der Erkenntnisse
auf weitere Proteinquellen ist in Ansätzen möglich, benötigt aber weitere Feinabstimmung zur
Optimierung des Prozesses. Die im Rahmen dieser Studie sensorisch bewerteten Fraktionen
verfügten über unterschiedliche Geschmacksqualitäten, sowie „bitter“-blockende
Eigenschaften (Permeat (< 100 kDa): hydrolysierte Alge Chlorella vulgaris).

Literaturverzeichnis
250
Literaturverzeichnis
Adler-Nissen, J. (1979). Determination of the degree of hydrolysis of food protein
hydrolysates by trinitrobenzenesulfonic acid. Journal of Agricultural and Food
Chemistry, 27(6), 1256–1262. https://doi.org/10.1021/jf60226a042
Adler-Nissen, J. (1986). Enzymic hydrolysis of food proteins. London; New York: Elsevier.
Adler-Nissen, J., Eggum, B. O., & Munck, L. (2014). Biochemical Aspects of New Protein Food:
Federation of European Biochemical Societies, 11th Meeting, Copenhagen 1977,
Volume 44, Symposium A3. New York: Elsevier.
Agilent Technologies. (n.d.). Agilent ZORBAX Bio Series GF-250 - Datasheet.
Ahmad, A. L., Mat Yasin, N. H., Derek, C. J. C., & Lim, J. K. (2012). Crossflow microfiltration
of microalgae biomass for biofuel production. Desalination, 302, 65–70.
https://doi.org/10.1016/j.desal.2012.06.026
Ahmed, H. (2004). Principles and Reactions of Protein Extraction, Purification, and
Characterization. Boca Raton: CRC Press.
Aidley, D. J. (1998). The Physiology of Excitable Cells. Fourth Edition. Cambridge: Cambridge
University Press.
Aimar, P., Meireles, M., Bacchin, P., & Sanchez, V. (1994). Fouling and Concentration
Polarisation in Ultrafiltration and Microfiltration. In J.G. Crespo, K.W. Böddeker (Eds.),
Membrane Processes in Separation and Purification (pp. 25-57). Dordrecht: Springer
Science & Business Media.
Aitken, A., & Learmonth, M. (2009). Protein Determination by UV Absorption. In J. M. Walker
(Ed.), The Protein Protocols Handbook. Third Edition (pp. 3-6). New York: Humana
Press.
Allma. (2014, July 30). Spezifikation - Chlorella vulgaris.
Almécija, M. C., Ibáñez, R., Guadix, A., & Guadix, E. M. (2007). Effect of pH on the fractionation
of whey proteins with a ceramic ultrafiltration membrane. Journal of Membrane
Science, 288(1–2), 28–35. https://doi.org/10.1016/j.memsci.2006.10.021

Literaturverzeichnis
251
Amersham Pharmacia Biotech. (1999). Reversed Phase Chromatography. Uppsala:
Amersham Pharmacia Biotech AB.
Anema, S. G., & Klostermeyer, H. (1997). Heat-Induced, pH-Dependent Dissociation of Casein
Micelles on Heating Reconstituted Skim Milk at Temperatures below 100 °C. Journal
of Agricultural and Food Chemistry, 45(4), 1108–1115.
https://doi.org/10.1021/jf960507m
Anselmino, O., Biberfeld, J., & Gilg, E. (1911). Kommentar zum Deutschen Arzneibuch: Auf
Grundlage der Hager-Fischer-Hartwichschen Kommentare der früheren Arzneibücher.
5. Auflage. Berlin; Heidelberg: Springer-Verlag.
Arakawa, T., Ejima, D., Li, T., & Philo, J. S. (2010). The critical role of mobile phase
composition in size exclusion chromatography of protein pharmaceuticals. Journal of
Pharmaceutical Sciences, 99(4), 1674–1692. https://doi.org/10.1002/jps.21974
Assadi-Porter, F. M., Aceti, D. J., Cheng, H., & Markley, J. L. (2000). Efficient Production of
Recombinant Brazzein, a Small, Heat-Stable, Sweet-Tasting Protein of Plant Origin.
Archives of Biochemistry and Biophysics, 376(2), 252–258.
https://doi.org/10.1006/abbi.2000.1725
Atra, R., Vatai, G., Bekassy-Molnar, E., & Balint, A. (2005). Investigation of ultra- and
nanofiltration for utilization of whey protein and lactose. Journal of Food Engineering,
67(3), 325–332. https://doi.org/10.1016/j.jfoodeng.2004.04.035
Bacchin, P., Aimar, P., & Field, R. W. (2006). Critical and sustainable fluxes: Theory,
experiments and applications. Journal of Membrane Science, 281(1–2), 42–69.
https://doi.org/10.1016/j.memsci.2006.04.014
Balakrishnan, M., & Agarwal, G. P. (1996). Protein fractionation in a vortex flow filter. I: Effect
of system hydrodynamics and solution environment on single protein transmission.
Journal of Membrane Science, 112(1), 47–74. https://doi.org/10.1016/0376-
7388(95)00266-9

Literaturverzeichnis
252
Baldasso, C., Barros, T. C., & Tessaro, I. C. (2011). Concentration and purification of whey
proteins by ultrafiltration. Desalination, 278(1–3), 381–386.
https://doi.org/10.1016/j.desal.2011.05.055
Ballew, H. W., Martinez, F. J., Markee, C., & Eddleman, R. T. (2002). The ABCs of Filtration &
Bioprocessing for the third Millenium. Rancho Dominguez: Spectrum Laboratories, Inc.
Barashkov, V. A., Trubachev, I. N., & Gitel’zon, I. I. (1979). Characteristics of the proteins of
unicellular organisms as potential components of ecological life-support systems.
Kosmicheskaia biologiia i aviakosmicheskaia meditsina, 13(3), 75–80.
Bárcenas, P., Elortondo, F. j. P., & Albisu, M. (2000). Selection and Screening of a Descriptive
Panel for Ewes Milk Cheese Sensory Profiling. Journal of Sensory Studies, 15(1), 79–
99. https://doi.org/10.1111/j.1745-459X.2000.tb00411.x
Bartoshuk, L. M. (2000). Comparing Sensory Experiences Across Individuals: Recent
Psychophysical Advances Illuminate Genetic Variation in Taste Perception. Chemical
Senses, 25(4), 447–460. https://doi.org/10.1093/chemse/25.4.447
Bastos, R. G. (2018). Biofuels from Microalgae: Biodiesel. In E. Jacob-Lopes, L. Queiroz
Zepka, & M. I. Queiroz (Eds.), Energy form microalgae (pp. 229-246). Basel: Springer
International Publishing
Baynes, J., & Dominiczak, M. H. (2014). Medical Biochemistry. Fourth Edition. Edinburgh;
London; New York, Oxford; Philadelphia; St. Louis; Sydney; Toronto: Elsevier Health
Sciences.
Becker, E. W. (2007). Micro-algae as a source of protein. Biotechnology Advances, 25(2),
207–210. https://doi.org/10.1016/j.biotechadv.2006.11.002
Becker-Carus, C., & Wendt, M. (2017). Allgemeine Psychologie: Eine Einführung. 2. Auflage.
Berlin; Heidelberg: Springer-Verlag.
Beheshtipour, H., Mortazavian, A. M., Mohammadi, R., Sohrabvandi, S., & Khosravi-Darani,
K. (2013). Supplementation of Spirulina platensis and Chlorella vulgaris Algae into
Probiotic Fermented Milks. Comprehensive Reviews in Food Science and Food Safety,
12(2), 144–154. https://doi.org/10.1111/1541-4337.12004

Literaturverzeichnis
253
Belikov, V. M., & Gololobov, M. Y. (1986). Plasteins. Their preparation, properties, and use in
nutrition. Food / Nahrung, 30(3–4), 281–287.
https://doi.org/10.1002/food.19860300320
Belitz, H.-D., Grosch, W., & Schieberle, P. (2008). Lehrbuch der Lebensmittelchemie. 6.
Auflage. Berlin; Heidelberg: Springer-Verlag.
Bewicke, D., & Potter, B. (1984). Chlorella: The Emerald Food. Berkeley: Ronin Publishing.
Bhave, R. M. (1997). Crossflow Filtration. In H. C. Vogel, & C. M. Todaro (Eds.), Fermentation
and Biochemical Engineering Handbook, Second Edition: Principles, Process Design
and Equipment (pp. 271-348). Westwood: Noyes Publications.
Bibby, D. C., Davies, N. M., & Tucker, I. G. (2000). Mechanisms by which cyclodextrins modify
drug release from polymeric drug delivery systems. International Journal of
Pharmaceutics, 197(1), 1–11. https://doi.org/10.1016/S0378-5173(00)00335-5
Bilanovic, D., Shelef, G., & Sukenik, A. (1988). Flocculation of microalgae with cationic
polymers — Effects of medium salinity. Biomass, 17(1), 65–76.
https://doi.org/10.1016/0144-4565(88)90071-6
Binello, A., Cravotto, G., Nano, G. M., & Spagliardi, P. (2004). Synthesis of chitosan–
cyclodextrin adducts and evaluation of their bitter-masking properties. Flavour and
Fragrance Journal, 19(5), 394–400. https://doi.org/10.1002/ffj.1434
BioRad Laboratories, Inc. (n.d.). Gel Filtration Standard. Instruction Manual. Catalog # 151-
1901. Retrieved from http://www.bio-rad.com/webroot/web/pdf/lsr/literature/MSLIT-
102E.pdf (26.06.2018)
Birbaumer, N., & Schmidt, R. F. (1991). Biologische Psychologie. 2. Auflage. Berlin;
Heidelberg: Springer-Verlag.
Bodanszky, M. (1984). Principles of Peptide Synthesis. Berlin; Heidelberg; New York, Tokyo:
Springer-Verlag.
Boron, W. F., & Boulpaep, E. L. (2016). Medical Physiology. Third Edition. Philadelphia:
Elsevier Health Sciences.

Literaturverzeichnis
254
Bothe, H., & Cammenga, H. K. (1979). Phase transitions and thermodynamic properties of
anhydrous caffeine. Journal of Thermal Analysis and Calorimetry, 16(2), 267–275.
https://doi.org/10.1007/BF01910688
Bowen, W. R., Calvo, J. I., & Hernández, A. (1995). Steps of membrane blocking in flux decline
during protein microfiltration. Journal of Membrane Science, 101(1–2), 153–165.
https://doi.org/10.1016/0376-7388(94)00295-A
Boye, J. I., Alli, I., & Ismail, A. A. (1997). Use of Differential Scanning Calorimetry and Infrared
Spectroscopy in the Study of Thermal and Structural Stability of α-Lactalbumin. Journal
of Agricultural and Food Chemistry, 45(4), 1116–1125.
https://doi.org/10.1021/jf960360z
Bradley, P., Desai, M. A. (2000). Application of HPLC in the Purification of Biomolecules. In M.
A. Desai (Ed.), Downstream Processing of Proteins: Methods and Protocols (pp. 141-
160). Totowa: Humana Press.
Brans, G., Schroën, C. G. P. H., van der Sman, R. G. M., & Boom, R. M. (2004). Membrane
fractionation of milk: state of the art and challenges. Journal of Membrane Science,
243(1–2), 263–272. https://doi.org/10.1016/j.memsci.2004.06.029
Breslin, P. a. S., & Beauchamp, G. K. (1995). Suppression of Bitterness by Sodium: Variation
Among Bitter Taste Stimuli. Chemical Senses, 20(6), 609–623.
https://doi.org/10.1093/chemse/20.6.609
Britten, M., Giroux, H. J., & Gaudin, V. (1994). Effect of pH During Heat Processing of Partially
Hydrolyzed Whey Protein. Journal of Dairy Science, 77(3), 676–684.
https://doi.org/10.3168/jds.S0022-0302(94)76999-X
Brockhoff, A., Behrens, M., Niv, M. Y., & Meyerhof, W. (2010). Structural requirements of bitter
taste receptor activation. Proceedings of the National Academy of Sciences, 107(24),
11110–11115. https://doi.org/10.1073/pnas.0913862107
Brockhoff, A., Behrens, M., Roudnitzky, N., Appendino, G., Avonto, C., & Meyerhof, W. (2011).
Receptor Agonism and Antagonism of Dietary Bitter Compounds. Journal of

Literaturverzeichnis
255
Neuroscience, 31(41), 14775–14782. https://doi.org/10.1523/JNEUROSCI.2923-
11.2011
Brockhoff, P. B. (2003). Statistical testing of individual differences in sensory profiling. Food
Quality and Preference, 14(5–6), 425–434. https://doi.org/10.1016/S0950-
3293(03)00007-7
Bryant, R. W., Palmer, R. K., Cerne, R., Atwal, K. S., Lee, S. P., & Atwal, A. B. (2007). US
2007/0207093 A1. Retrieved from http://www.google.com/patents/US20070207093
(26.06.2018)
Buddrus, J. (2011). Grundlagen der organischen Chemie. 4. überarbeitete und aktualisierte
Auflage. Berlin ; New York: De Gruyter.
Bujas, Z., Szabo, S., Ajdukovć, D., & Mayer, D. (1989). Individual gustatory reaction times to
various groups of chemicals that provoke basic taste qualities. Perception &
Psychophysics, 45(5), 385–390. https://doi.org/10.3758/BF03210710
Büsing, C. M. & Heinzeller, T. (2001). Histologie, Histopathologie und Zytologie für den
Einstieg. Stuttgart: Georg Thieme Verlag.
Butylina, S., Luque, S., & Nyström, M. (2006). Fractionation of whey-derived peptides using a
combination of ultrafiltration and nanofiltration. Journal of Membrane Science, 280(1–
2), 418–426. https://doi.org/10.1016/j.memsci.2006.01.046
Caicedo, A., Kim, K.-N., & Roper, S. D. (2002). Individual mouse taste cells respond to multiple
chemical stimuli. The Journal of Physiology, 544(2), 501–509.
https://doi.org/10.1113/jphysiol.2002.027862
Castaing, J.-B., Massé, A., Pontié, M., Séchet, V., Haure, J., & Jaouen, P. (2010). Investigating
submerged ultrafiltration (UF) and microfiltration (MF) membranes for seawater pre-
treatment dedicated to total removal of undesirable micro-algae. Desalination, 253(1),
71–77. https://doi.org/10.1016/j.desal.2009.11.031
Chai, X., Kobayashi, T., & Fujii, N. (1998). Ultrasound effect on cross-flow filtration of
polyacrylonitrile ultrafiltration membranes. Journal of Membrane Science, 148(1), 129–
135. https://doi.org/10.1016/S0376-7388(98)00145-8

Literaturverzeichnis
256
Chan, R., & Chen, V. (2001). The effects of electrolyte concentration and pH on protein
aggregation and deposition: critical flux and constant flux membrane filtration. Journal
of Membrane Science, 185(2), 177–192. https://doi.org/10.1016/S0376-
7388(00)00645-1
Chandrapala, J., Zisu, B., Palmer, M., Kentish, S., & Ashokkumar, M. (2011). Effects of
ultrasound on the thermal and structural characteristics of proteins in reconstituted
whey protein concentrate. Ultrasonics Sonochemistry, 18(5), 951–957.
https://doi.org/10.1016/j.ultsonch.2010.12.016
Chatsungnoen, T., & Chisti, Y. (2016). Harvesting microalgae by flocculation–sedimentation.
Algal Research, 13, 271–283. https://doi.org/10.1016/j.algal.2015.12.009
Chen, H. (2014). Biotechnology of Lignocellulose. Dodrecht: Springer Science & Business
Media.
Chen, K. L., Mylon, S. E., & Elimelech, M. (2006). Aggregation Kinetics of Alginate-Coated
Hematite Nanoparticles in Monovalent and Divalent Electrolytes. Environmental
Science & Technology, 40(5), 1516–1523. https://doi.org/10.1021/es0518068
Chen, L., Li, P., Liu, Z., & Jiao, Q. (2009). The released polysaccharide of the cyanobacterium
‘Aphanothece halophytica’ inhibits flocculation of the alga with ferric chloride. Journal
of Applied Phycology, 21(3), 327–331. https://doi.org/10.1007/s10811-008-9371-z
Chen, V., Fane, A. G., Madaeni, S., & Wenten, I. G. (1997). Particle deposition during
membrane filtration of colloids: transition between concentration polarization and cake
formation. Journal of Membrane Science, 125(1), 109–122.
https://doi.org/10.1016/S0376-7388(96)00187-1
Cheremisinoff, N. P. (1998). Liquid Filtration. Second Edition. Oxford: Butterworth-Heinemann.
Cheryan, M. (1990). Handbuch Ultrafiltration. Hamburg: Behr's Verlag.
Cheryan, M. (1998). Ultrafiltration and Microfiltration Handbook. Second Edition. Boca Raton:
CRC Press.

Literaturverzeichnis
257
Cho, M. J., Unklesbay, N., Hsieh, F., & Clarke, A. D. (2004). Hydrophobicity of Bitter Peptides
from Soy Protein Hydrolysates. Journal of Agricultural and Food Chemistry, 52(19),
5895–5901. https://doi.org/10.1021/jf0495035
Choi, H., Zhang, K., Dionysiou, D. D., Oerther, D. B., & Sorial, G. A. (2005). Influence of cross-
flow velocity on membrane performance during filtration of biological suspension.
Journal of Membrane Science, 248(1–2), 189–199.
https://doi.org/10.1016/j.memsci.2004.08.027
Chollangi, A., & Hossain, M. M. (2007). Separation of proteins and lactose from dairy
wastewater. Chemical Engineering and Processing: Process Intensification, 46(5),
398–404. https://doi.org/10.1016/j.cep.2006.05.022
Choo, K.-H., & Lee, C.-H. (1996). Membrane fouling mechanisms in the membrane-coupled
anaerobic bioreactor. Water Research, 30(8), 1771–1780.
https://doi.org/10.1016/0043-1354(96)00053-X
Choo, K.-H., & Lee, C.-H. (2000). Understanding Membrane Fouling in Terms of Surface Free
Energy Changes. Journal of Colloid and Interface Science, 226(2), 367–370.
https://doi.org/10.1006/jcis.2000.6845
Chudacek, M. W., & Fane, A. G. (1984). The dynamics of polarisation in unstirred and stirred
ultrafiltration. Journal of Membrane Science, 21(2), 145–160.
https://doi.org/10.1016/S0376-7388(00)81551-3
Church, F. C., Porter, D. H., Catignani, G. L., & Swaisgood, H. E. (1985). An o-phthalaldehyde
spectrophotometric assay for proteinases. Analytical Biochemistry, 146(2), 343–348.
https://doi.org/10.1016/0003-2697(85)90549-4
Clemente, A. (2000). Enzymatic protein hydrolysates in human nutrition. Trends in Food
Science & Technology, 11(7), 254–262. https://doi.org/10.1016/S0924-
2244(01)00007-3
Cohen, L., & Holliday, M. (1996). Practical Statistics for Students: An Introductory Text.
Thousand Oaks: SAGE.

Literaturverzeichnis
258
Corder, G. W., & Foreman, D. I. (2009). Nonparametric Statistics for Non-Statisticians: A Step-
by-Step Approach. Hoboken: John Wiley & Sons.
Craig, L. C., & King, T. P. (1955). Some Dialysis Experiments with Polypeptides. Journal of the
American Chemical Society, 77(24), 6620–6624. https://doi.org/10.1021/ja01629a062
Craig, L. C., King, T. P., & Stracher, A. (1957). Dialysis Studies. II. Some Experiments Dealing
with the Problem of Selectivity. Journal of the American Chemical Society, 79(14),
3729–3737. https://doi.org/10.1021/ja01571a033
Cunningham, J. B., & Aldrich, J. O. (2013). Using SPSS: An Interactive Hands-On Approach.
Thousand Oaks: SAGE.
de Castro, M. D. L., Capote, F. P., & Ávila, N. S. (2008). Is dialysis alive as a membrane-based
separation technique? TrAC Trends in Analytical Chemistry, 27(4), 315–326.
https://doi.org/10.1016/j.trac.2008.01.015
de la Casa, E. J., Guadix, A., Ibáñez, R., & Guadix, E. M. (2007). Influence of pH and salt
concentration on the cross-flow microfiltration of BSA through a ceramic membrane.
Biochemical Engineering Journal, 33(2), 110–115.
https://doi.org/10.1016/j.bej.2006.09.009
Defrance, L., & Jaffrin, M. Y. (1999). Comparison between filtrations at fixed transmembrane
pressure and fixed permeate flux: application to a membrane bioreactor used for
wastewater treatment. Journal of Membrane Science, 152(2), 203–210.
https://doi.org/10.1016/S0376-7388(98)00220-8
DeMan, J. M. (1999). Principles of food chemistry. Third Edition. Gaithersburg: Aspen
Publishers.
Demirbas, A., & Demirbas, M. F. (2010). Algae Energy: Algae as a New Source of Biodiesel.
London: Springer-Verlag.
Dennison, C. (2003). A Guide to Protein Isolation. Dordrecht: Springer Science & Business
Media.
DIN Deutsches Institut für Normung e.V. (2007). DIN EN ISO 4120:2007 - Sensorische
Analyse - Prüfverfahren - Dreiecksprüfung. Berlin: Beuth Verlag.

Literaturverzeichnis
259
DIN Deutsches Institut für Normung e.V. (2010). DIN ISO 8587:2010-08 - Sensorische Analyse
- Prüfverfahren - Rangordnungsprüfung. Berlin: Beuth Verlag.
DIN Deutsches Institut für Normung e.V. (2014a). DIN 10964:2014 - Sensorische
Prüfverfahren - Einfache beschreibende Prüfung. Berlin: Beuth Verlag.
DIN Deutsches Institut für Normung e.V. (2014b). DIN EN ISO 8586:2014 - Sensorische
Analyse - Leitfaden für die Auswahl, Schulung und Überprüfung ausgewählter Prüfer
und Sensoriker. Berlin: Beuth Verlag.
Dizge, N., Soydemir, G., Karagunduz, A., & Keskinler, B. (2011). Influence of type and pore
size of membranes on cross flow microfiltration of biological suspension. Journal of
Membrane Science, 366(1–2), 278–285. https://doi.org/10.1016/j.memsci.2010.10.010
Domozych, D. S. (2016). Biosynthesis of the Cell Walls of the Algae. In M. A. Borowitzka, J.
Beardall, & J. A. Raven (Eds.), The Physiology of Microalgae (pp. 47-63). Basel:
Springer International Publishing
Donhauser, R. M. (2017). Superfoods: Alles über die exotischen & heimischen Kraftpakete -
Mit über 80 Rezepten. München: Südwest Verlag.
Dosmar, M., & Pinto, S. (2007). Crossflow Filtration. In M. J. Jornitz, & T. H. Meltzer (Eds.),
Filtration and Purification in the Biopharmaceutical Industry, Second Edition (pp. 495-
543). Boca Raton: CRC Press.
Drake, M. A., Karagul-Yuceer, Y., Cadwallader, K. R., Civille, G. V., & Tong, P. S. (2003).
Determination of the Sensory Attributes of Dried Milk Powders and Dairy Ingredients.
Journal of Sensory Studies, 18(3), 199–216. https://doi.org/10.1111/j.1745-
459X.2003.tb00385.x
Drexler, I. L. C., & Yeh, D. H. (2014). Membrane applications for microalgae cultivation and
harvesting: a review. Reviews in Environmental Science and Bio/Technology, 13(4),
487–504. https://doi.org/10.1007/s11157-014-9350-6
Dullien, F. A. L. (1992). Porous Media: Fluid Transport and Pore Structure. Second Edition.
San Diego: Academic Press.

Literaturverzeichnis
260
Dürrschmid, K. (2009). Gustatorische Wahrnehmung gezielt abwandeln. Hamburg: Behr’s
Verlag.
Eckstein, P. P. (2016). Angewandte Statistik mit SPSS: Praktische Einführung für
Wirtschaftswissenschaftler. 8. Auflage. Wiesbaden: Springer-Verlag.
Epple, M., Cammenga, H. K., Sarge, S. M., Diedrich, R., & Balek, V. (1995). The phase
transformation of caffeine: Investigation by dynamic X-ray diffraction and emanation
thermal analysis. Thermochimica Acta, 250(1), 29–39. https://doi.org/10.1016/0040-
6031(94)01958-J
Faheed, F. A. (Sohag U. (Egypt) B. D. ), & Fattah, Z. A. (Sohag U. (Egypt) B. D. ). (2008).
Effect of Chlorella vulgaris as bio-fertilizer on growth parameters and metabolic aspects
of lettuce plant. Journal of Agriculture and Social Sciences (Pakistan). Retrieved from
http://agris.fao.org/agris-search/search.do?recordID=PK2009000203 (26.06.2018)
Fane, A. G., Beatson, P., & Li, H. (2000). Membrane fouling and its control in environmental
applications. Water Science and Technology, 41(10–11), 303–308.
Fane, A. G., Fell, C. J. D., & Suki, A. (1983). The effect of ph and ionic environment on the
ultrafiltration of protein solutions with retentive membranes. Journal of Membrane
Science, 16, 195–210. https://doi.org/10.1016/S0376-7388(00)81310-1
Feher, J. J. (2017). Quantitative Human Physiology: An Introduction. Waltham; San Diego;
London: Academic Press.
Field, R. W., Wu, D., Howell, J. A., & Gupta, B. B. (1995). Critical flux concept for microfiltration
fouling. Journal of Membrane Science, 100(3), 259–272. https://doi.org/10.1016/0376-
7388(94)00265-Z
Fields, R. (1971). The measurement of amino groups in proteins and peptides. Biochemical
Journal, 124(3), 581–590. https://doi.org/10.1042/bj1240581
Foegeding, E. A., Davis, J. P., Doucet, D., & McGuffey, M. K. (2002). Advances in modifying
and understanding whey protein functionality. Trends in Food Science & Technology,
13(5), 151–159. https://doi.org/10.1016/S0924-2244(02)00111-5

Literaturverzeichnis
261
Foley, G. (2013). Membrane Filtration: A Problem Solving Approach with MATLAB.
Cambridge: Cambridge University Press.
Forss, D. A. (1973). Odor and flavor compounds from lipids. Progress in the Chemistry of Fats
and Other Lipids, 13, 177–258. https://doi.org/10.1016/0079-6832(73)90007-4
Foust, A. S., Wenzel, L. A., Clump, C. W., Maus, L., & Andersen, L. B. (2008). Principles Of
Unit Operations. Second Edition. New Delhi: Wiley India Pvt. Limited.
Fradique, M., Batista, A. P., Nunes, M. C., Gouveia, L., Bandarra, N. M., & Raymundo, A.
(2010). Incorporation of Chlorella vulgaris and Spirulina maxima biomass in pasta
products. Part 1: Preparation and evaluation. Journal of the Science of Food and
Agriculture, 90(10), 1656–1664. https://doi.org/10.1002/jsfa.3999
Frank, R. A., & Korchmar, D. L. (1985). Gustatory processing differences in PTC tasters and
non-tasters: A reaction time analysis. Physiology & Behavior, 35(2), 239–242.
https://doi.org/10.1016/0031-9384(85)90343-9
Frings, S., & Müller, F. (2013). Biologie der Sinne: Vom Molekül zur Wahrnehmung. Berlin,
Heidelberg: Springer-Verlag.
Fujimaki, M., Arai, S., Yamashita, M., Kato, H., & Noguchi, M. (1973). Taste Peptide
Fractionation from a Fish Protein Hydrolysate. Agricultural and Biological Chemistry,
37(12), 2891–2898. https://doi.org/10.1080/00021369.1973.10861085
Fung, S. Y., Hong, Y., Keyes-Baig, C., Chen, P. (2005). Self-assembly of peptides and ist
potential application. In P. Chen (Ed.), Molecular Interfacial Phenomena of Polymers
and Biopolymers (pp. 421-462). Sawston: Woodhead Publishing Limited; Boca Raton:
CRC Press.
Fushan, A. A., Simons, C. T., Slack, J. P., & Drayna, D. (2010). Association between Common
Variation in Genes Encoding Sweet Taste Signaling Components and Human Sucrose
Perception. Chemical Senses, 35(7), 579–592. https://doi.org/10.1093/chemse/bjq063
Galensa, R., Bahadir, M., Engelhardt, U., & Böhm, H. (1995). Lebensmittel- und
Umweltanalytik mit der HPLC: Tips, Tricks und Beispiele für die Praxis. Weinheim:
Wiley VCH Verlag.

Literaturverzeichnis
262
Garson, M. J. (1989). Biosynthetic studies on marine natural products. Natural Product
Reports, 6(2), 143–170. https://doi.org/10.1039/NP9890600143
Gekle, M., Wischmeyer, M., Gründer, S., Petersen, M., Schwab, A., Markwardt, F., Klöckner,
N., Baumann, R., Marti, H. (2010). Taschenlehrbuch Physiologie. Stuttgart: Georg
Thieme Verlag.
Ghosh, R. (2003). Protein Bioseparation Using Ultrafiltration: Theory, Applications and New
Developments. Singapur: World Scientific.
Gilbertson, T. A., Boughter, J. D., Zhang, H., & Smith, D. V. (2001). Distribution of Gustatory
Sensitivities in Rat Taste Cells: Whole-Cell Responses to Apical Chemical Stimulation.
Journal of Neuroscience, 21(13), 4931–4941.
Glanbia Nutritionals Ltd. (2012). Spezifikation - Thermax 690. Glanbia Nutritionals Ltd.
Golovchenko, N. P., Kataeva, I. A., & Akimenko, V. K. (1992). Analysis of pH-dependent
protein interactions with gel filtration medium. Journal of Chromatography A, 591(1),
121–128. https://doi.org/10.1016/0021-9673(92)80229-N
Goodnight Jr., K. C., Hartman Jr., G. H., & Marquardt, R. F. (1976). US3995071 A. Retrieved
from https://www.google.com/patents/US3995071 (26.06.2018)
Graham, E. R. B., Malcolm, G. N., & McKenzie, H. A. (1984). On the isolation and conformation
of bovine β-casein A1. International Journal of Biological Macromolecules, 6(3), 155–
161. https://doi.org/10.1016/0141-8130(84)90058-8
Greene, T. A., Alarcon, S., Thomas, A., Berdougo, E., Doranz, B. J., Breslin, P. A. S., & Rucker,
J. B. (2011). Probenecid Inhibits the Human Bitter Taste Receptor TAS2R16 and
Suppresses Bitter Perception of Salicin. PLOS ONE, 6(5), e20123.
https://doi.org/10.1371/journal.pone.0020123
Gressner, A. M., & Arndt, T. (Eds.) (2007). Lexikon der Medizinischen
Laboratoriumsdiagnostik. Band 1: Klinische Chemie. Heidelberg: Springer-Verlag.
Gritter, R. J., Bobbitt, J. M., & Schwarting, A. E. (1987). Einführung in die Chromatographie.
Berlin; Heidelberg: Springer-Verlag.

Literaturverzeichnis
263
Guadagni, D. G., Horowitz, R. M., Gentili, B., & Maier, V. P. (1979). Method of reducing
bitterness and off-after-taste. Retrieved from
http://www.google.com/patents/US4154862 (26.06.2018)
Güell, C., & Davis, R. H. (1996). Membrane fouling during microfiltration of protein mixtures.
Journal of Membrane Science, 119(2), 269–284. https://doi.org/10.1016/0376-
7388(96)80001-J
Guidotti, G., & Craig, L. C. (1963). Dialysis Studies. VIII. The Behavior Of Solutes Which
Associate. Proc Natl Acad Sci, 50(1), 46–54. Retrieved from
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC300652/pdf/pnas00174-0057.pdf
(26.06.2018)
Guigoz, Y., & Solms, J. (1976). Bitter Peptides, Occurence And Structure. Chemical Senses,
2(1), 71–84. https://doi.org/10.1093/chemse/2.1.71
Hadjoudja, S., Deluchat, V., & Baudu, M. (2010). Cell surface characterisation of Microcystis
aeruginosa and Chlorella vulgaris. Journal of Colloid and Interface Science, 342(2),
293–299. https://doi.org/10.1016/j.jcis.2009.10.078
Hall, M. J., Bartoshuk, L. M., Cain, W. S., & Stevens, J. C. (1975). PTC taste blindness and
the taste of caffeine. Nature, 253(5491), 442–443. https://doi.org/10.1038/253442a0
Harker, J. H., Backhurst, J. R., & Richardson, J. F. (2013). Chemical Engineering, Volume 2.
Fifth Edition. Oxford: Butterworth-Heinemann.
Harlin, M. M., & Darley, W. M. (1998). The algae: An overview. In C. A. Lembi, & J. R. Waaland,
J. R. (Eds.), Algae and Human Affairs (5-27). Cambridge: Cambridge University Press.
Harnedy, P. A., & FitzGerald, R. J. (2011). Bioactive Proteins, Peptides, and Amino Acids from
Macroalgae. Journal of Phycology, 47(2), 218–232. https://doi.org/10.1111/j.1529-
8817.2011.00969.x
Hartmann, R., & Meisel, H. (2007). Food-derived peptides with biological activity: from
research to food applications. Current Opinion in Biotechnology, 18(2), 163–169.
https://doi.org/10.1016/j.copbio.2007.01.013

Literaturverzeichnis
264
Hatt, H (2006). Geschmack. In R. F. Schmidt, & H. G. Schaible (Eds.), Neuro- und
Sinnesphysiologie. 5. Auflage (pp.328-340). Berlin; Heidelberg: Springer-Verlag.
Hatt, H. (2011). Geschmack & Geruch. In R. F. Schmidt, F. Lang, & M. Heckmann (Eds.),
Physiologie des Menschen: Mit Pathophysiologie. 31. Auflage (pp. 386-400). Berlin,
Heidelberg: Springer-Verlag.
Heinemann, P., Howell, J. A., & Bryan, R. A. (1988). Microfiltration of protein solutions: effect
of fouling on rejection. Desalination, 68(2), 243–250. https://doi.org/10.1016/0011-
9164(88)80058-4
Hellemann, U. (1992). Perceived taste of NaCl and acid mixtures in water and bread.
International Journal of Food Science & Technology, 27(2), 201–211.
https://doi.org/10.1111/j.1365-2621.1992.tb01196.x
Henkin, R. I., Gill, J. R., & Bartter, F. C. (1963). Studies On Taste Thresholds In Normal Man
and In Patients With Adrenal Cortical Insufficience: The Role Of Adrenal Cortical
Steroids and Of Serum Sodium Concentration. Journal of Clinical Investigation, 42(5),
727–735.
Her, N., Amy, G., Park, H.-R., & Song, M. (2004). Characterizing algogenic organic matter
(AOM) and evaluating associated NF membrane fouling. Water Research, 38(6),
1427–1438. https://doi.org/10.1016/j.watres.2003.12.008
Herrero, C., Prádanos, P., Calvo, J. I., Tejerina, F., & Hernández, A. (1997). Flux Decline in
Protein Microfiltration: Influence of Operative Parameters. Journal of Colloid and
Interface Science, 187(2), 344–351. https://doi.org/10.1006/jcis.1996.4662
Herskovits, T. T. (1966). On the Conformation of Caseins. Optical Rotatory Properties*.
Biochemistry, 5(3), 1018–1026. https://doi.org/10.1021/bi00867a030
Hidalgo, J., & Gamper, E. (1977). Solubility and Heat Stability of Whey Protein Concentrates.
Journal of Dairy Science, 60(10), 1515–1518. https://doi.org/10.3168/jds.S0022-
0302(77)84061-7
Hiersig, H. M. (Ed.) (1995). Lexikon Produktionstechnik Verfahrenstechnik. Berlin; Heidelberg:
Springer-Verlag.

Literaturverzeichnis
265
Ho, C.-C., & Zydney, A. L. (1999). Effect of membrane morphology on the initial rate of protein
fouling during microfiltration. Journal of Membrane Science, 155(2), 261–275.
https://doi.org/10.1016/S0376-7388(98)00324-X
Hoch, H., & Turner, M. E. (1960). Studies of rates of escape of proteins through membranes.
Biochimica et Biophysica Acta, 38, 410–419. https://doi.org/10.1016/0006-
3002(60)91276-2
Hoek, C., Mann, D., & Jahns, H. M. (1995). Algae: An Introduction to Phycology. Cambridge:
Cambridge University Press.
Hofmann, T., Ottinger, H., Frank, O., Soldo, T., Blank, I., Villard, R., & Robert, F. (2007).
Pyridinium-Betain compounds for use as taste modulators. Retrieved from
http://www.google.com/patents/US7175872 (26.06.2018)
Holt, C., de Kruif, C. G., Tuinier, R., & Timmins, P. A. (2003). Substructure of bovine casein
micelles by small-angle X-ray and neutron scattering. Colloids and Surfaces A:
Physicochemical and Engineering Aspects, 213(2–3), 275–284.
https://doi.org/10.1016/S0927-7757(02)00520-4
Hong, J.-H., Chung, J.-W., Kim, Y.-K., Chung, S.-C., Lee, S.-W., & Kho, H.-S. (2005). The
relationship between PTC taster status and taste thresholds in young adults. Oral
Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontology, 99(6),
711–715. https://doi.org/10.1016/j.tripleo.2004.08.004
Hong, P., Koza, S., & Bouvier, E. S. P. (2012). Size-Exclusion Chromatography for the Analysis
of Protein Biotherapeutics and their Aggregates. Journal of Liquid Chromatography &
Related Technologies, 35(20), 2923–2950.
https://doi.org/10.1080/10826076.2012.743724
Horne, D. S. (1998). Casein Interactions: Casting Light on the Black Boxes, the Structure in
Dairy Products. International Dairy Journal, 8(3), 171–177.
https://doi.org/10.1016/S0958-6946(98)00040-5
Howell, J. A. (1995). Sub-critical flux operation of microfiltration. Journal of Membrane Science,
107(1–2), 165–171. https://doi.org/10.1016/0376-7388(95)00114-R

Literaturverzeichnis
266
Huisman, I. H., Prádanos, P., & Hernández, A. (2000). The effect of protein–protein and
protein–membrane interactions on membrane fouling in ultrafiltration. Journal of
Membrane Science, 179(1–2), 79–90. https://doi.org/10.1016/S0376-7388(00)00501-
9
Hwang, S.-J., Chang, D.-J., & Chen, C.-H. (1996). Steady State permeat flux for particle cross-
flow filtration. The Chemical Engineering Journal and the Biochemical Engineering
Journal, 61(3), 171–178. https://doi.org/10.1016/0923-0467(95)03047-6
International Organization for Standardization. (2005). Sensory analysis - Methodology -
General guidance - ISO 6658:2005. Berlin: Beuth Verlag.
International Organization for Standardization. (2011). Sensory analysis - Methodology -
Method of investigating sensitivity of taste - ISO 3972:2011. Berlin: Beuth Verlag.
Irmler, H. W. (2001). Dynamische Filtration mit keramischen Membranen. Essen: Vulkan
Verlag.
Jacobsen, C. F., Léonis, J., Linderstrøm-Lang, K., & Ottesen, M. (1957). The pH-Stat and Its
Use in Biochemistry. In D. Glick (Ed.), Methods of Biochemical Analysis (pp. 171–210).
Hoboken: John Wiley & Sons.
Jambrak, A. R., Mason, T. J., Lelas, V., Paniwnyk, L., & Herceg, Z. (2014). Effect of ultrasound
treatment on particle size and molecular weight of whey proteins. Journal of Food
Engineering, 121, 15–23. https://doi.org/10.1016/j.jfoodeng.2013.08.012
Janssen, J., & Laatz, W. (2007). Statistische Datenanalyse mit SPSS für Windows. 6. Auflage.
Berlin; Heidelberg: Springer-Verlag.
Jones, K. L., & O’Melia, C. R. (2000). Protein and humic acid adsorption onto hydrophilic
membrane surfaces: effects of pH and ionic strength. Journal of Membrane Science,
165(1), 31–46. https://doi.org/10.1016/S0376-7388(99)00218-5
Jönsson, C., & Jönsson, A.-S. (1995). Influence of the membrane material on the adsorptive
fouling of ultrafiltration membranes. Journal of Membrane Science, 108(1), 79–87.
https://doi.org/10.1016/0376-7388(95)00144-X

Literaturverzeichnis
267
Jung, G., & Beck-Sickinger, A. G. (1992). Multiple Peptide Synthesis Methods and Their
Applications. New Synthetic Methods (87). Angewandte Chemie International Edition
in English, 31(4), 367–383. https://doi.org/10.1002/anie.199203673
Keast, R. S. J., Breslin, P. A. S., & Beauchamp, G. K. (2001). Suppression of Bitterness Using
Sodium Salts. CHIMIA International Journal for Chemistry, 55(5), 441–447.
Kelly, S. T., & Zydney, A. L. (1995). Mechanisms for BSA fouling during microfiltration. Journal
of Membrane Science, 107(1), 115–127. https://doi.org/10.1016/0376-7388(95)00108-
O
Kienle, D. U. (2010). Ein Naturstoff macht Karriere. Journal für Verbraucherschutz und
Lebensmittelsicherheit, 5(2), 199–203. https://doi.org/10.1007/s00003-010-0550-x
Kirimura, J., Shimizu, A., Kimizuka, A., Ninomiya, T., & Katsuya, N. (1969). Contribution of
peptides and amino acids to the taste of foods. J. Agric. Food Chem., 17(4), 689–695.
https://doi.org/10.1021/jf60164a031
Kjeldahl, J. (1883). A new method for the determination of nitrogen in organic matter, Zeitschrift
für Analytische Chemie, 22(1), 366–382.
Klein, E., Ward, R. A., & Lacey, R. E. (1987). Membrane processes - Dialysis and
Electrodialysis. In R. W. Rousseau (Ed.), Handbook of Separation Process Technology
(pp. 954-982). Hoboken: John Wiley & Sons.
Kobayashi, C., & Kennedy, L. M. (2002). Experience-induced changes in taste identification of
monosodium glutamate. Physiology & Behavior, 75(1–2), 57–63.
https://doi.org/10.1016/S0031-9384(01)00634-5
Kobayashi, T., Chai, X., & Fujii, N. (1999). Ultrasound enhanced cross-flow membrane
filtration. Separation and Purification Technology, 17(1), 31–40.
https://doi.org/10.1016/S1383-5866(99)00023-4
Kobayashi, Takaomi, Kobayashi, T., Hosaka, Y., & Fujii, N. (2003). Ultrasound-enhanced
membrane-cleaning processes applied water treatments: influence of sonic frequency
on filtration treatments. Ultrasonics, 41(3), 185–190. https://doi.org/10.1016/S0041-
624X(02)00462-6

Literaturverzeichnis
268
Kofod Nielsen, W. (2004). Membranfiltration und verwandte molekulare Separationsverfahren.
Silkeborg: Invensys APV Systems.
Koh, L. L. A., Nguyen, H. T. H., Chandrapala, J., Zisu, B., Ashokkumar, M., & Kentish, S. E.
(2014). The use of ultrasonic feed pre-treatment to reduce membrane fouling in whey
ultrafiltration. Journal of Membrane Science, 453, 230–239.
https://doi.org/10.1016/j.memsci.2013.11.006
Kong, X., Zhou, H., & Qian, H. (2007a). Enzymatic hydrolysis of wheat gluten by proteases
and properties of the resulting hydrolysates. Food Chemistry, 102(3), 759–763.
https://doi.org/10.1016/j.foodchem.2006.06.062
Kong, X., Zhou, H., & Qian, H. (2007b). Enzymatic preparation and functional properties of
wheat gluten hydrolysates. Food Chemistry, 101(2), 615–620.
https://doi.org/10.1016/j.foodchem.2006.01.057
Koo, S. H., Bae, I. Y., Lee, S., Lee, D.-H., Hur, B.-S., & Lee, H. G. (2011). Evaluation of wheat
gluten hydrolysates as taste-active compounds with antioxidant activity. Journal of
Food Science and Technology, 51(3), 535–542. https://doi.org/10.1007/s13197-011-
0515-9
Koolman, J., & Röhm, K.-H. (2003). Taschenatlas der Biochemie. 3. Auflage. Stuttgart: Georg
Thieme Verlag.
Kopaciewicz, W., & Regnier, F. E. (1982). Nonideal size-exclusion chromatography of proteins:
Effects of pH at low ionic strength. Analytical Biochemistry, 126(1), 8–16.
https://doi.org/10.1016/0003-2697(82)90102-6
Koriyama, T., Wongso, S., Watanabe, K., & Abe, H. (2002). Fatty Acid Compositions of Oil
Species Affect the 5 Basic Taste Perceptions. Journal of Food Science, 67(2), 868–
873. https://doi.org/10.1111/j.1365-2621.2002.tb10691.x
Kostanski, L. K., Keller, D. M., & Hamielec, A. E. (2004). Size-exclusion chromatography - A
review of calibration methodologies. Journal of Biochemical and Biophysical Methods,
58(2), 159–186. https://doi.org/10.1016/j.jbbm.2003.10.001

Literaturverzeichnis
269
Kraemer, H. C., & Thiemann, S. (1987). How Many Subjects?: Statistical Power Analysis in
Research. Newbury Park: SAGE.
Kreischer, L., Schuttelaar, M., & Farm, N. S. (2016). Ocean Greens: Explore the World of
Edible Seaweed and Sea Vegetables: A Way of Eating for Your Health and the
Planet’s. New York: Workman Publishing.
Kremer, B. P., & Bannwarth, H. (2014). Einführung in die Laborpraxis: Basiskompetenzen für
Laborneulinge. 3. Auflage. Berlin; Heidelberg: Springer-Verlag.
Kromidas, S. (2008). More Practical Problem Solving in HPLC. Weinheim: Wiley VCH Verlag.
Kromidas, S. (2012). Handbuch Validierung in der Analytik. Weinheim: Wiley VCH Verlag.
Kromidas, S. (2006). HPLC richtig optimiert: Ein Handbuch für Praktiker. Weinheim: WileyVCH
Verlag.
Kromidas, S., & Kuss, H.-J. (2008). Chromatogramme richtig integrieren und bewerten: Ein
Praxishandbuch für die HPLC und GC. Weinheim: Wiley VCH Verlag.
Kühlmeyer, M. (2001). Statistische Auswertungsmethoden für Ingenieure: mit
Praxisbeispielen. Berlin; Heidelberg: Springer-Verlag.
Kwon, D. Y., Vigneswaran, S., Fane, A. G., & Aim, R. B. (2000). Experimental determination
of critical flux in cross-flow microfiltration. Separation and Purification Technology,
19(3), 169–181. https://doi.org/10.1016/S1383-5866(99)00088-X
Kyllönen, H. M., Pirkonen, P., & Nyström, M. (2005). Membrane filtration enhanced by
ultrasound: a review. Desalination, 181(1–3), 319–335.
https://doi.org/10.1016/j.desal.2005.06.003
Labbe, D., Rytz, A., & Hugi, A. (2004). Training is a critical step to obtain reliable product
profiles in a real food industry context. Food Quality and Preference, 15(4), 341–348.
https://doi.org/10.1016/S0950-3293(03)00081-8
Lahl, W. J., & Braun, S. D. (1994). Enzymatic Production of Protein Hydrolysates for Food Use,
Food Technol., 48(10), 68–71.

Literaturverzeichnis
270
Lai, Y. Z. (2000). Chemical Degradation. In D. N.-S. Hon, & N. Shiraishi (Eds.), Wood and
Cellulosic Chemistry, Second Edition, Revised, and Expanded (pp. 443-513). Boca
Raton: CRC Press.
Lamminen, M. O., Walker, H. W., & Weavers, L. K. (2004). Mechanisms and factors influencing
the ultrasonic cleaning of particle-fouled ceramic membranes. Journal of Membrane
Science, 237(1–2), 213–223. https://doi.org/10.1016/j.memsci.2004.02.031
Latreille, J., Mauger, E., Ambroisine, L., Tenenhaus, M., Vincent, M., Navarro, S., & Guinot, C.
(2006). Measurement of the reliability of sensory panel performances. Food Quality
and Preference, 17(5), 369–375. https://doi.org/10.1016/j.foodqual.2005.04.010
Lawler, D., & Kweon, J. (2004). Integrated Water Treatment: Softening and Ultrafiltration.
London: IWA Publishing.
Lawless, H. T., & Heymann, H. (1999). Sensory Evaluation of Food: Principles and Practices.
New York: Springer Science+Business Media LLC.
Le Ray, C., Maubois, J.-L., Gaucheron, F., Brulé, G., Pronnier, P., & Garnier, F. (1998). Heat
stability of reconstituted casein micelle dispersions: changes induced by salt addition.
Le Lait, 78(4), 375–390. https://doi.org/10.1051/lait:1998437
Leighton, T. (1997). The Acoustic Bubble. San Diego, London: Academic Press.
Leksrisompong, P. P., Miracle, R. E., & Drake, M. (2010). Characterization of Flavor of Whey
Protein Hydrolysates. Journal of Agricultural and Food Chemistry, 58(10), 6318–6327.
https://doi.org/10.1021/jf100009u
Ley, J. P. (2008). Masking Bitter Taste by Molecules. Chemosensory Perception, 1(1), 58–77.
https://doi.org/10.1007/s12078-008-9008-2
Li, H., Fane, A. G., Coster, H. G. L., & Vigneswaran, S. (1998). Direct observation of particle
deposition on the membrane surface during crossflow microfiltration. Journal of
Membrane Science, 149(1), 83–97. https://doi.org/10.1016/S0376-7388(98)00181-1
Linne von Berg, K.-H., Hoef-Emden, K., & Melkonian, M. (2012). Der Kosmos-Algenführer:
Süßwasseralgen unter dem Mikroskop ; ein Bestimmungsbuch. Stuttgart: Kosmos.

Literaturverzeichnis
271
Liu, J., Zhu, Y., Tao, Y., Zhang, Y., Li, A., Li, T., … Zhang, C. (2013). Freshwater microalgae
harvested via flocculation induced by pH decrease. Biotechnology for Biofuels, 6, 98.
https://doi.org/10.1186/1754-6834-6-98
Lojkine, M. H., Field, R. W., & Howell, J. A. (1992). Crossflow microfiltration of cell
suspensions: a review of models with emphasis on particle size effects, 70, 149–164.
Lugaz, O., Pillias, A.-M., & Faurion, A. (2002). A New Specific AgeusiaSome Humans Cannot
Taste L-Glutamate. Chemical Senses, 27(2), 105–115.
https://doi.org/10.1093/chemse/27.2.105
Luo, J., Wu, C., Xu, T., & Wu, Y. (2011). Diffusion dialysis-concept, principle and applications.
Journal of Membrane Science, 366(1–2), 1–16.
https://doi.org/10.1016/j.memsci.2010.10.028
Lynch, J., Liu, Y.-H., Mela, D. J., & MacFie, H. J. H. (1993). A time—intensity study of the effect
of oil mouthcoatings on taste perception. Chemical Senses, 18(2), 121–129.
https://doi.org/10.1093/chemse/18.2.121
MacFarland, T. W., & Yates, J. M. (2016). Introduction to Nonparametric Statistics for the
Biological Sciences Using R. Basel: Springer International Publishing.
Madaeni, S. S., Fane, A. G., & Wiley, D. E. (1999). Factors influencing critical flux in membrane
filtration of activated sludge. Journal of Chemical Technology & Biotechnology, 74(6),
539–543. https://doi.org/10.1002/(SICI)1097-4660(199906)74:6<539::AID-
JCTB70>3.0.CO;2-X
Mangan, P. a. P. (1992). Performance Assessment of Sensory Panelists. Journal of Sensory
Studies, 7(3), 229–252. https://doi.org/10.1111/j.1745-459X.1992.tb00534.x
Marshall, A. D., Munro, P. A., & Trägårdh, G. (1993). The effect of protein fouling in
microfiltration and ultrafiltration on permeate flux, protein retention and selectivity: A
literature review. Desalination, 91(1), 65–108. https://doi.org/10.1016/0011-
9164(93)80047-Q

Literaturverzeichnis
272
Martin-Orue, C., Bouhallab, S., & Garem, A. (1998). Nanofiltration of amino acid and peptide
solutions: mechanisms of separation. Journal of Membrane Science, 142(2), 225–233.
https://doi.org/10.1016/S0376-7388(97)00325-6
Mason, T. J., & Lorimer, J. P. (1988). Sonochemistry: Theory, Applications and Uses of
Ultrasound in Chemistry. Chichester: Ellis Horwood.
Mata, T. M., Martins, A. A., & Caetano, N. S. (2010). Microalgae for biodiesel production and
other applications: A review. Renewable and Sustainable Energy Reviews, 14(1), 217–
232. https://doi.org/10.1016/j.rser.2009.07.020
Matoba, T., & Hata, T. (1972). Relationship between Bitterness of Peptides and their Chemical
Structures. Agricultural and Biological Chemistry, 36(8), 1423–1431.
https://doi.org/10.1080/00021369.1972.10860410
Matsumoto, Y., Miwa, T., Nakao, S., & Kimura, S. (1996). Improvement of Membrane
Permeation Performance by Ultrasonic Microfiltration. Journal of Chemical Engineering
of Japan, 29(4), 561–567. https://doi.org/10.1252/jcej.29.561
Mattes, R. D. (2007). Effects of linoleic acid on sweet, sour, salty, and bitter taste thresholds
and intensity ratings of adults. American Journal of Physiology - Gastrointestinal and
Liver Physiology, 292(5), G1243–G1248. https://doi.org/10.1152/ajpgi.00510.2006
Matthiasson, E. (1983). The role of macromolecular adsorption in fouling of ultrafiltration
membranes. Journal of Membrane Science, 16, 23–36. https://doi.org/10.1016/S0376-
7388(00)81297-1
Matthiasson, E., & Sivik, B. (1980). Concentration polarization and fouling. Desalination, 35,
59–103. https://doi.org/10.1016/S0011-9164(00)88604-X
Maximous, N., Nakhla, G., & Wan, W. (2009). Comparative assessment of hydrophobic and
hydrophilic membrane fouling in wastewater applications. Journal of Membrane
Science, 339(1–2), 93–99. https://doi.org/10.1016/j.memsci.2009.04.034
Mazur, R., Schlatter, J., & Goldkamp, A. (1969). Structure-taste relationships of some
dipeptides. J. Am. Chem. Soc., 91(10), 2684–2691. https://doi.org/
10.1021/ja01038a046

Literaturverzeichnis
273
McDonogh, R. M., Bauser, H., Stroh, N., & Chmiel, H. (1990). Concentration polarisation and
adsorption effects in cross-flow ultrafiltration of proteins. Desalination, 79(2), 217–231.
https://doi.org/10.1016/0011-9164(90)85007-W
Mehrens, H. A. (2004). Komponenten aus Magermilch und Molke. In R. Heiss (Ed.),
Lebensmitteltechnologie: biotechnologische, chemische, mechanische und thermische
Verfahren der Lebensmittelverarbeitung. 6. Auflage (pp. 37-50). Berlin; New York:
Springer-Verlag.
Meireles, M., Aimar, P., & Sanchez, V. (1991). Albumin denaturation during ultrafiltration:
Effects of operating conditions and consequences on membrane fouling.
Biotechnology and Bioengineering, 38(5), 528–534.
https://doi.org/10.1002/bit.260380511
Meisel, H. (1997). Biochemical properties of regulatory peptides derived from milk proteins.
Peptide Science, 43(2), 119–128. https://doi.org/10.1002/(SICI)1097-
0282(1997)43:2<119::AID-BIP4>3.0.CO;2-Y
Melin, T., & Rautenbach, R. (2007). Membranverfahren: Grundlagen der Modul- und
Anlagenauslegung. 3., aktualisierte und erw. Auflage. Berlin ; New York: Springer
Verlag.
Metcalf, K. L., & Vickers, Z. M. (2002). Taste Intensities of Oil-in-Water Emulsions with Varying
Fat Content. Journal of Sensory Studies, 17(5), 379–390.
https://doi.org/10.1111/j.1745-459X.2002.tb00354.x
Meyer, V. R. (1988). Praxis der Hochleistungsflüssigkeitschromatographie. 5. Auflage.
Frankfurt am Main/Aarau: Verlag Moritz Diesterweg GmbH & Co. KG/Verlag
Sauerländer AG.
Miller Jr., I. J., & Reedy Jr., F. E. (1990). Variations in human taste bud density and taste
intensity perception. Physiology & Behavior, 47(6), 1213–1219.
https://doi.org/10.1016/0031-9384(90)90374-D

Literaturverzeichnis
274
Ming, D., & Hellekant, G. (1994). Brazzein, a new high-potency thermostable sweet protein
from Pentadiplandra brazzeana B. FEBS Letters, 355(1), 106–108.
https://doi.org/10.1016/0014-5793(94)01184-2
Mo, H., Tay, K. G., & Ng, H. Y. (2008). Fouling of reverse osmosis membrane by protein (BSA):
Effects of pH, calcium, magnesium, ionic strength and temperature. Journal of
Membrane Science, 315(1–2), 28–35. https://doi.org/10.1016/j.memsci.2008.02.002
Molina Grima, E., Belarbi, E.-H., Acién Fernández, F. G., Robles Medina, A., & Chisti, Y.
(2003). Recovery of microalgal biomass and metabolites: process options and
economics. Biotechnology Advances, 20(7), 491–515. https://doi.org/10.1016/S0734-
9750(02)00050-2
Morimura, Y., & Tamiya, N. (1954). Preliminary experiments in the use of Chlorella as human
food, Food Technol., 8, 179–182.
Morr, C. V., & Ha, E. Y. W. (1991). Off-flavors of whey protein concentrates: A literature review.
International Dairy Journal, 1(1), 1–11. https://doi.org/10.1016/0958-6946(91)90024-3
Morris, H. J., Almarales, A., Carrillo, O., & Bermúdez, R. C. (2008). Utilisation of Chlorella
vulgaris cell biomass for the production of enzymatic protein hydrolysates. Bioresource
Technology, 99(16), 7723–7729. https://doi.org/10.1016/j.biortech.2008.01.080
Mortenson, M. A., Vickers, Z. M., & Reineccius, G. A. (2008). Flavor of whey protein
concentrates and isolates. International Dairy Journal, 18(6), 649–657.
https://doi.org/10.1016/j.idairyj.2007.12.003
Mota, A.M., Simaes Goncalves, M. L. (1996). Direct methods of specification of heavy metals
in natural waters. In S. Caroli (Ed.), Element Speciation in Bioinorganic Chemistry (pp.
21-87). Hoboken: John Wiley & Sons.
Mueller, K. L., Hoon, M. A., Erlenbach, I., Chandrashekar, J., Zuker, C. S., & Ryba, N. J. P.
(2005). The receptors and coding logic for bitter taste. Nature, 434(7030), 225–229.
https://doi.org/10.1038/nature03352
Mulder, M. (1996). Basic Principles of Membrane Technology. Dordrecht: Kluwer Academic
Publishers.

Literaturverzeichnis
275
Muthukumaran, S., Kentish, S. E., Ashokkumar, M., & Stevens, G. W. (2005). Mechanisms for
the ultrasonic enhancement of dairy whey ultrafiltration. Journal of Membrane Science,
258(1–2), 106–114. https://doi.org/10.1016/j.memsci.2005.03.001
Muthukumaran, S., Kentish, S. E., Stevens, G. W., Ashokkumar, M., & Mawson, R. (2007).
The application of ultrasound to dairy ultrafiltration: The influence of operating
conditions. Journal of Food Engineering, 81(2), 364–373.
https://doi.org/10.1016/j.jfoodeng.2006.11.008
Muthukumaran, S., Kentish, S., Lalchandani, S., Ashokkumar, M., Mawson, R., Stevens, G.
W., & Grieser, F. (2005). The optimisation of ultrasonic cleaning procedures for dairy
fouled ultrafiltration membranes. Ultrasonics Sonochemistry, 12(1–2), 29–35.
https://doi.org/10.1016/j.ultsonch.2004.05.007
Muthukumaran, S., Yang, K., Seuren, A., Kentish, S., Ashokkumar, M., Stevens, G. W., &
Grieser, F. (2004). The use of ultrasonic cleaning for ultrafiltration membranes in the
dairy industry. Separation and Purification Technology, 39(1–2), 99–107.
https://doi.org/10.1016/j.seppur.2003.12.013
Nadathur, S., & Carolan, M. (2016). Flavors, taste preferences, and the consumer: Taste
modulation and influencing change in dietary patterns for a sustainable earth. In S.
Nadathur, J. P. D. Wanasundara, & L. Scanlin (Eds.), Sustainable Protein Sources (pp.
377-391). London, San Diego, Cambridge, Oxford: Academic Press.
Næs, T. (1990). Handling individual differences between assessors in sensory profiling. Food
Quality and Preference, 2(3), 187–199. https://doi.org/10.1016/0950-3293(90)90023-N
Næs, T., & Solheim, R. (1991). Detection and Interpretation of Variation Within and Between
Assessors in Sensory Profiling. Journal of Sensory Studies, 6(3), 159–177.
https://doi.org/10.1111/j.1745-459X.1991.tb00512.x
Naik, S. N., Goud, V. V., Rout, P. K., & Dalai, A. K. (2010). Production of first and second
generation biofuels: A comprehensive review. Renewable and Sustainable Energy
Reviews, 14(2), 578–597. https://doi.org/10.1016/j.rser.2009.10.003

Literaturverzeichnis
276
Nakamura, K., & Matsumoto, K. (2006). Properties of protein adsorption onto pore surface
during microfiltration: Effects of solution environment and membrane hydrophobicity.
Journal of Membrane Science, 280(1–2), 363–374.
https://doi.org/10.1016/j.memsci.2006.01.039
Nakata, T., Takahashi, M., Nakatani, M., Kuramitsu, R., Tamura, M., & Okai, H. (1995). Role
of Basic and Acidic Fragments in Delicious Peptides (Lys-Gly-Asp Glu-Glu-Ser-Leu-
Ala) and the Taste Behavior of Sodium and Potassium Salts in Acidic Oligopeptides.
Bioscience, Biotechnology, and Biochemistry, 59(4), 689–693.
https://doi.org/10.1271/bbb.59.689
Nakatsuka, S., & Michaels, A. S. (1992). Transport and separation of proteins by ultrafiltration
through sorptive and non-sorptive membranes. Journal of Membrane Science, 69(3),
189–211. https://doi.org/10.1016/0376-7388(92)80039-M
Nath, K. (2008). Membrane Seperation Processes. New Delhi: PHI Learning Pvt. Ltd.
Ney, K. H. (1971). Voraussage der Bitterkeit von Peptiden aus deren
Aminosäurezusammensetzung. Zeitschrift für Lebensmittel-Untersuchung und
Forschung, 147(2), 64–68. https://doi.org/10.1007/BF01879606
Nielsen, P. M. (1997). Functionality of protein hydrolysates. In S. Damodaran, A. Paraf (Eds.),
Food Proteins and Their Applications (pp. 443-472). Boca Raton: CRC Press.
Nielsen, P. M., Petersen, D., & Dambmann, C. (2001). Improved Method for Determining Food
Protein Degree of Hydrolysis. Journal of Food Science, 66(5), 642–646.
https://doi.org/10.1111/j.1365-2621.2001.tb04614.x
Nielsen, S. (2010). Food Analysis Laboratory Manual. New York: Springer Science & Business
Media.
Nigam, M. O., Bansal, B., & Chen, X. D. (2008). Fouling and cleaning of whey protein
concentrate fouled ultrafiltration membranes. Desalination, 218(1–3), 313–322.
https://doi.org/10.1016/j.desal.2007.02.027

Literaturverzeichnis
277
Nomura, T., Fujii, T., & Suzuki, M. (1997). Application of the ceramic membrane with
hydrophobic skin layer to separation of activated sludge. Water Science and
Technology, 35(8), 137–144.
Nyström, M., Pihlajamäki, A., & Ehsani, N. (1994). Characterization of ultrafiltration
membranes by simultaneous streaming potential and flux measurements. Journal of
Membrane Science, 87(3), 245–256. https://doi.org/10.1016/0376-7388(94)87031-4
O’Callaghan, D. M., Donnelly, W. J., Slattery, H. M., & Mulvihill, D. M. (1995). Non-Size
Exclusion Effects During Gel Permeation Chromatography of Milk Protein Hydrolysates
on an FPLC Superose 12 Column. Journal of Liquid Chromatography, 18(8), 1543–
1562. https://doi.org/10.1080/10826079508009294
Oehlenschläger, J., & Schneider-Häder, B. (2012). Grundlagenvokabular Sensorik. Frankfurt
am Main: DLG-Verlag.
Ohyama, S., Ishibashi, N., Tamura, M., Nishizaki, H., & Okai, H. (1988). Synthesis of Bitter
Peptides Composed of Aspartic Acid and Glutamic Acid. Agricultural and Biological
Chemistry, 52(3), 871–872. https://doi.org/10.1271/bbb1961.52.871
Ousman, M., & Bennasar, M. (1995). Determination of various hydraulic resistances during
cross-flow filtration of a starch grain suspension through inorganic membranes. Journal
of Membrane Science, 105(1–2), 1–21. https://doi.org/10.1016/0376-7388(94)00266-2
Palecek, S. P., Mochizuki, S., & Zydney, A. L. (1993). Effect of ionic environment on BSA
filtration and the properties of BSA deposits. Desalination, 90(1), 147–159.
https://doi.org/10.1016/0011-9164(93)80172-J
Palecek, S. P., & Zydney, A. L. (1994). Intermolecular Electrostatic Interactions and Their
Effect on Flux and Protein Deposition during Protein Filtration. Biotechnology Progress,
10(2), 207–213. https://doi.org/10.1021/bp00026a010
Pangborn, R. M. (1959). Influence of Hunger on Sweetness Preferences and Taste
Thresholds. The American Journal of Clinical Nutrition, 7(3), 280–287.

Literaturverzeichnis
278
Park, C., Lee, Y. H., Lee, S., & Hong, S. (2008). Effect of cake layer structure on colloidal
fouling in reverse osmosis membranes. Desalination, 220(1), 335–344.
https://doi.org/10.1016/j.desal.2007.01.038
Park, C., Lee, J. H., Yang, X., Yoo, H. Y., Lee, J. H., Lee, S. K., & Kim, S. W. (2016).
Enhancement of hydrolysis of Chlorella vulgaris by hydrochloric acid. Bioprocess and
Biosystems Engineering, 39(6), 1015–1021. https://doi.org/10.1007/s00449-016-1570-
4
Pasupuleti, V. K., Holmes, C., Demain, A. L. (2010). Applications of protein hydrolysates in
biotechnology. In V. K. Pasupuleti, & A. L. Demain (Eds.), Protein Hydrolysates in
Biotechnology (pp. 1-11). Dordrecht: Springer Science & Business Media.
Paulus, K., & Haas, E.-M. (1980). The influence of solvent viscosity on the threshold values of
primary tastes. Chemical Senses, 5(1), 23–32. https://doi.org/10.1093/chemse/5.1.23
Paulus, K., & Reisch, A. M. (1980). The influence of temperature on the threshold values of
primary tastes. Chemical Senses, 5(1), 11–21. https://doi.org/10.1093/chemse/5.1.11
Pearce, G. (2007). Introduction to membranes: Membrane selection. Filtration & Separation,
44(3), 35–37. https://doi.org/10.1016/S0015-1882(07)70083-6
Pelegrine, D. H. G., & Gasparetto, C. A. (2005). Whey proteins solubility as function of
temperature and pH. LWT - Food Science and Technology, 38(1), 77–80.
https://doi.org/10.1016/j.lwt.2004.03.013
Pelegrine, D. H. G., & Gomes, M. T. M. S. (2012). Analysis of Whey Proteins Solubility at High
Temperatures. International Journal of Food Engineering, 8(3).
https://doi.org/10.1515/1556-3758.1265
Perea, A., Ugalde, U., Rodriguez, I., & Serra, J. L. (1993). Preparation and characterization of
whey protein hydrolysates: Applications in industrial whey bioconversion processes.
Enzyme and Microbial Technology, 15(5), 418–423. https://doi.org/10.1016/0141-
0229(93)90129-P

Literaturverzeichnis
279
Pérez, L., Salgueiro, J. L., Maceiras, R., Cancela, Á., & Sánchez, Á. (2017). An effective
method for harvesting of marine microalgae: pH induced flocculation. Biomass and
Bioenergy, 97, 20–26. https://doi.org/10.1016/j.biombioe.2016.12.010
Persson, A., Jönsson, A.-S., & Zacchi, G. (2003). Transmission of BSA during cross-flow
microfiltration: influence of pH and salt concentration. Journal of Membrane Science,
223(1–2), 11–21. https://doi.org/10.1016/S0376-7388(03)00268-0
Pfaffmann, C., Bartoshuk, L. M., & McBurney, D. H. (1971). Taste Psychophysics. In L. M.
Beidler (Ed.), Taste (pp. 75–101). Berlin; Heidelberg: Springer-Verlag.
Pförtsch, W., & Müller, I. (2006). Die Marke in der Marke - Bedeutung und Macht des Ingredient
Branding. Berlin: Springer-Verlag.
Pharmacia LBK Biotechnology. (1991). Gel Filtration - Principles and Methods (5th ed.).
Uppsala: Pharmacia LKB Biotechnology.
Pilz, G. (2010). Biotechnologie - Elektronische Ressource Anwendung, Branchenentwicklung,
Investitionschancen. München: Oldenbourg.
Pincet, F., Perez, E., & Belfort, G. (1994). Do Denatured Proteins Behave Like Polymers?
Macromolecules, 27(12), 3424–3425. https://doi.org/10.1021/ma00090a042
Pincet, F., Perez, E., & Belfort, G. (1995). Molecular Interactions between Proteins and
Synthetic Membrane Polymer Films. Langmuir, 11(4), 1229–1235.
https://doi.org/10.1021/la00004a031
Pingoud, A., & Urbanke, C. (1997). Arbeitsmethoden der Biochemie. Berlin: Walter de Gruyter.
Pitt, A. M. (1987). The Nonspecific Protein Binding of Polymeric Microporous Membranes. PDA
Journal of Pharmaceutical Science and Technology, 41(3), 110–113.
Plaza, M., Cifuentes, A., & Ibáñez, E. (2008). In the search of new functional food ingredients
from algae. Trends in Food Science & Technology, 19(1), 31–39.
https://doi.org/10.1016/j.tifs.2007.07.012
Polinelli, S., Broxterman, Q. B., Schoemaker, H. E., Boesten, W. H. J., Crisma, M., Valle, G.,
… Kamphuis, J. (1992). New aspartame-like sweeteners containing L-(αMe)Phe.

Literaturverzeichnis
280
Bioorganic & Medicinal Chemistry Letters, 2(5), 453–456.
https://doi.org/10.1016/S0960-894X(00)80168-7
Pouliot, Y., Wijers, M. C., Gauthier, S. F., & Nadeau, L. (1999). Fractionation of whey protein
hydrolysates using charged UF/NF membranes. Journal of Membrane Science, 158(1–
2), 105–114. https://doi.org/10.1016/S0376-7388(99)00006-X
PSS Polymer Standards Service GmbH. (n.d.). Benutzer-Manual PSS SUPREMA (Lux) GPC-
Säulen.
Punidadas, P., & Rizvi, S. S. H. (1998). Separation of milk proteins into fractions rich in casein
or whey proteins by cross flow filtration. Food Research International, 31(4), 265–272.
https://doi.org/10.1016/S0963-9969(98)00088-X
Pydi, S. P., Sobotkiewicz, T., Billakanti, R., Bhullar, R. P., Loewen, M. C., & Chelikani, P.
(2014). Amino Acid Derivatives as Bitter Taste Receptor (T2R) Blockers. Journal of
Biological Chemistry, 289(36), 25054–25066. https://doi.org/10.1074/jbc.M114.576975
Quadt, A., Schönberger, S., & Schwarz, M. (2009). Statistische Auswertungen in der Sensorik:
Leitfaden für die Praxis. Hamburg: Behr's Verlag.
Reed, D. R., Tanaka, T., & McDaniel, A. H. (2006). Diverse tastes: Genetics of sweet and bitter
perception. Physiology & Behavior, 88(3), 215–226.
https://doi.org/10.1016/j.physbeh.2006.05.033
Rehner, G., & Daniel, H. (2010). Biochemie der Ernährung. 3. Auflage. Wiesbaden: Springer-
Verlag.
Renneberg, R., & Berkling, V. (2012). Biotechnologie für Einsteiger. 4. Auflage. Heidelberg:
Spektrum Akademischer Verlag.
Ricq, L., Narçon, S., Reggiani, J.-C., & Pagetti, J. (1999). Streaming potential and protein
transmission ultrafiltration of single proteins and proteins in mixture: β-lactoglobulin and
lysozyme. Journal of Membrane Science, 156(1), 81–96.
https://doi.org/10.1016/S0376-7388(98)00341-X

Literaturverzeichnis
281
Riedl, K., Girard, B., & Lencki, R. W. (1998). Influence of membrane structure on fouling layer
morphology during apple juice clarification. Journal of Membrane Science, 139(2),
155–166. https://doi.org/10.1016/S0376-7388(97)00239-1
Roberts, C. J. (2007). Nonnative protein aggregation. In R. Murphy, & A. Tsai (Eds.),
Misbehaving Proteins: Protein (Mis)Folding, Aggregation, and Stability (pp. 17-46).
New York: Springer Science & Business Media.
Rogers, K. (Ed.) (2011). Fungi, Algae, and Protists. New York: The Rosen Publishing Group.
Roland, W. S. U., Gouka, R. J., Gruppen, H., Driesse, M., Buren, L. van, Smit, G., & Vincken,
J.-P. (2014). 6-Methoxyflavanones as Bitter Taste Receptor Blockers for hTAS2R39.
PLOS ONE, 9(4), e94451. https://doi.org/10.1371/journal.pone.0094451
Romano, R., Brockhoff, P. B., Hersleth, M., Tomic, O., & Næs, T. (2008). Correcting for
different use of the scale and the need for further analysis of individual differences in
sensory analysis. Food Quality and Preference, 19(2), 197–209.
https://doi.org/10.1016/j.foodqual.2007.06.008
Rudolf, M., & Kuhlisch, W. (2008). Biostatistik: Eine Einführung für Biowissenschaftler.
München: Pearson Studium.
Rummel, C. (2002). Profilprüfung - Ausbildung eines deskriptiven Panels. In M. Busch-
Stockfisch (Ed.), Praxishandbuch Sensorik in der Produktentwicklung und
Qualitätssicherung (pp. 1-31). Hamburg: Behr's Verlag.
Russell, T. A., Drake, M. A., & Gerard, P. D. (2006). Sensory Properties of Whey and Soy
Proteins. Journal of Food Science, 71(6), S447–S455. https://doi.org/10.1111/j.1750-
3841.2006.00055.x
Ryan, T. P. (2013). Sample Size Determination and Power. Hoboken: John Wiley & Sons.
Sablani, S., Goosen, M., Al-Belushi, R., & Wilf, M. (2001). Concentration polarization in
ultrafiltration and reverse osmosis: a critical review. Desalination, 141(3), 269–289.
https://doi.org/10.1016/S0011-9164(01)85005-0
Safi, C., Charton, M., Pignolet, O., Silvestre, F., Vaca-Garcia, C., & Pontalier, P.-Y. (2012).
Influence of microalgae cell wall characteristics on protein extractability and

Literaturverzeichnis
282
determination of nitrogen-to-protein conversion factors. Journal of Applied Phycology,
25(2), 523–529. https://doi.org/10.1007/s10811-012-9886-1
Safi, C., Zebib, B., Merah, O., Pontalier, P.-Y., & Vaca-Garcia, C. (2014). Morphology,
composition, production, processing and applications of Chlorella vulgaris: A review.
Renewable and Sustainable Energy Reviews, 35, 265–278.
https://doi.org/10.1016/j.rser.2014.04.007
Saksena, S., & Zydney, A. L. (1994). Effect of solution pH and ionic strength on the separation
of albumin from immunoglobulins (IgG) by selective filtration. Biotechnology and
Bioengineering, 43(10), 960–968. https://doi.org/10.1002/bit.260431009
Salgın, S. (2007). Effects of Ionic Environments on Bovine Serum Albumin Fouling in a Cross-
Flow Ultrafiltration System. Chemical Engineering & Technology, 30(2), 255–260.
https://doi.org/10.1002/ceat.200600342
Samanen, J.M. (2000). Physical biochemistry of peptide drugs: Structure, Properties, ans
stabilities of peptides compared with proteins. In V. Lee (Ed.), Peptide and Protein Drug
Delivery (pp. 137-167). Boca Raton: CRC Press.
Samarakoon, K., & Jeon, Y.-J. (2012). Bio-functionalities of proteins derived from marine algae
— A review. Food Research International, 48(2), 948–960.
https://doi.org/10.1016/j.foodres.2012.03.013
Samuelsson, G., Dejmek, P., Trägårdh, G., & Paulsson, M. (1997). Minimizing whey protein
retention in cross-flow microfiltration of skim milk. International Dairy Journal, 7(4),
237–242. https://doi.org/10.1016/S0958-6946(97)00009-5
Saxena, A., Tripathi, B. P., Kumar, M., & Shahi, V. K. (2009). Membrane-based techniques for
the separation and purification of proteins: An overview. Advances in Colloid and
Interface Science, 145(1–2), 1–22. https://doi.org/10.1016/j.cis.2008.07.004
Schiffman, S. S., Crumbliss, A. L., Warwick, Z. S., & Graham, B. G. (1990). Thresholds for
sodium salts in young and elderly human subjects: correlation with molar conductivity
of anion. Chemical Senses, 15(6), 671–678. https://doi.org/10.1093/chemse/15.6.671

Literaturverzeichnis
283
Schlich, P. (1994). Grapes: A Method and a Sas® Program for Graphical Representations of
Assessor Performances. Journal of Sensory Studies, 9(2), 157–169.
https://doi.org/10.1111/j.1745-459X.1994.tb00238.x
Schlichtherle-Cerny, H., & Amadò, R. (2002). Analysis of Taste-Active Compounds in an
Enzymatic Hydrolysate of Deamidated Wheat Gluten. Journal of Agricultural and Food
Chemistry, 50(6), 1515–1522. https://doi.org/10.1021/jf010989o
Schmid, R. D. (2016). Taschenatlas der Biotechnologie und Gentechnik. 3. Auflage.
Weinheim: Wiley VCH Verlag.
Schuchmann, H. P., & Schuchmann, H. (2005). Lebensmittelverfahrenstechnik: Rohstoffe,
Prozesse, Produkte. Weinheim: Wiley VCH Verlag.
Schulz, H., Hundeiker, M., & Kreusch, J. (2016). Kompendium der Dermatoskopie. Berlin;
Heidelberg: Springer-Verlag.
Schütte, L., & Paschke, A. (2006). Reducing allergens in milk and milk products. In C. Mills, H.
Wichers, K. Hoffmann-Sommergruber (Eds.), Managing allergens in food (pp. 159-
178). Cambridge: Woodhead Publishing.
Scott, K. (2004). The sweet and the bitter of mammalian taste. Current Opinion in
Neurobiology, 14(4), 423–427. https://doi.org/10.1016/j.conb.2004.06.003
Sette, A., Southwood, S., O’Sullivan, D., Gaeta, F. C., Sidney, J., & Grey, H. M. (1992). Effect
of pH on MHC class II-peptide interactions. The Journal of Immunology, 148(3), 844–
851.
Shaaban, M. M. (2001). Green microalgae water extract as foliar feeding to wheat plants,
Pakistan Journal of Biological Sciences, 4(6), 628–632.
Shallenberger, R. S. (1993). Taste Chemistry. London: Chapman & Hall.
She, Q., Tang, C. Y., Wang, Y.-N., & Zhang, Z. (2009). The role of hydrodynamic conditions
and solution chemistry on protein fouling during ultrafiltration. Desalination, 249(3),
1079–1087. https://doi.org/10.1016/j.desal.2009.05.015

Literaturverzeichnis
284
Sheih, I.-C., Fang, T. J., & Wu, T.-K. (2009a). Isolation and characterisation of a novel
angiotensin I-converting enzyme (ACE) inhibitory peptide from the algae protein waste.
Food Chemistry, 115(1), 279–284. https://doi.org/10.1016/j.foodchem.2008.12.019
Sheih, I.-C., Wu, T.-K., & Fang, T. J. (2009b). Antioxidant properties of a new antioxidative
peptide from algae protein waste hydrolysate in different oxidation systems.
Bioresource Technology, 100(13), 3419–3425.
https://doi.org/10.1016/j.biortech.2009.02.014
Shirazi, S., Lin, C.-J., & Chen, D. (2010). Inorganic fouling of pressure-driven membrane
processes — A critical review. Desalination, 250(1), 236–248.
https://doi.org/10.1016/j.desal.2009.02.056
Silverthorn, D. U. (2009). Physiologie. 4. Auflage. München: Pearson Studium.
Simonian, M. H. (2002). Spectrophotometric Determination of Protein Concentration. Current
Protocols in Food Analytical Chemistry, 15(1), A.3B.1-A.3B.7.
https://doi.org/10.1002/0471143030.cba03bs15
Sinesio, F., Comendador, F. J., Peparaio, M., & Moneta, E. (2009). Taste Perception of
Umami-Rich Dishes in Italian Culinary Tradition. Journal of Sensory Studies, 24(4),
554–580. https://doi.org/10.1111/j.1745-459X.2009.00226.x
Singh, N. K., Donovan, R., & MacRitchie, F. (1990). Use of sonication and size-exclusion high-
performance liquid chromatography in the study of wheat flour proteins. II. Relative
quantity of glutenin as a measure of breadmaking quality. Cereal Chemistry, 67(2),
161–170. Retrieved from
https://www.aaccnet.org/publications/cc/backissues/1990/Documents/67_161.pdf
(26.06.2018)
Siouffi, A. M. (2000). HPLC. In L. M. L. Nollet (Ed.), Food Analysis by HPLC - Second Edition
(pp. 1-55). Boca Raton: CRC Press.
Slack, J. P., Brockhoff, A., Batram, C., Menzel, S., Sonnabend, C., Born, S., … Meyerhof, W.
(2010). Modulation of Bitter Taste Perception by a Small Molecule hTAS2R Antagonist.
Current Biology, 20(12), 1104–1109. https://doi.org/10.1016/j.cub.2010.04.043

Literaturverzeichnis
285
Slattery, C. W. (1976). Review: Casein Micelle Structure; An Examination of Models. Journal
of Dairy Science, 59(9), 1547–1556. https://doi.org/10.3168/jds.S0022-
0302(76)84403-7
Smith, S. T., Metzger, L., & Drake, M. A. (2016). Evaluation of whey, milk, and delactosed
permeates as salt substitutes. Journal of Dairy Science, 99(11), 8687–8698.
https://doi.org/10.3168/jds.2016-10904
Smithers, G. W. (2008). Whey and whey proteins—From ‘gutter-to-gold’. International Dairy
Journal, 18(7), 695–704. https://doi.org/10.1016/j.idairyj.2008.03.008
Solms, J., Vuataz, L., & Egli, R. H. (1965). The taste of L- and D-amino acids. Experientia,
21(12), 692–694. https://doi.org/10.1007/BF02138474
Spellman, D., McEvoy, E., O’Cuinn, G., & FitzGerald, R. J. (2003). Proteinase and
exopeptidase hydrolysis of whey protein: Comparison of the TNBS, OPA and pH stat
methods for quantification of degree of hydrolysis. International Dairy Journal, 13(6),
447–453. https://doi.org/10.1016/S0958-6946(03)00053-0
Stone, H., & Sidel, J. L. (2004). Sensory Evaluation Practices: Food and Science Technology
Series. Third Edition. London, Waltham, San Diego: Academic Press.
Suzuki, N., Fujimura, A., Nagai, T., Mizumoto, I., Itami, T., Hatate, H., … Yoda, B. (2004).
Antioxidative activity of animal and vegetable dietary fibers. Biofactors, 21(1–4), 329–
333.
Taddei, C., Daufin, G., Aimar, P., & Sanchez, V. (1989). Role of some whey components on
mass transfer in ultrafiltration. Biotechnology and Bioengineering, 34(2), 171–179.
https://doi.org/10.1002/bit.260340205
Tamura, M., Nakatsuka, T., Tada, M., Kawasaki, Y., Kikuchi, E., & Okai, H. (1989). The
Relationship between Taste and Primary Structure of ‘Delicious Peptide’ (Lys-Gly-Asp-
Glu-Glu-Ser-Leu-Ala) from Beef Soup. Agricultural and Biological Chemistry, 53(2),
319–325. https://doi.org/10.1271/bbb1961.53.319

Literaturverzeichnis
286
Tanford, C. (1962). Contribution of Hydrophobic Interactions to the Stability of the Globular
Conformation of Proteins. Journal of the American Chemical Society, 84(22), 4240–
4247. https://doi.org/10.1021/ja00881a009
Tanimura, S., & Mattes, R. D. (1993). Relationships Between Bitter Taste Sensitivity and
Consumption of Bitter Substances. Journal of Sensory Studies, 8(1), 31–41.
https://doi.org/10.1111/j.1745-459X.1993.tb00200.x
Tarabara, V. V., Koyuncu, I., & Wiesner, M. R. (2004). Effect of hydrodynamics and solution
ionic strength on permeate flux in cross-flow filtration: direct experimental observation
of filter cake cross-sections. Journal of Membrane Science, 241(1), 65–78.
https://doi.org/10.1016/j.memsci.2004.04.030
Tarleton, E. S., & Wakeman, R. J. (1993). Understanding flux decline in crossflow
microfiltration. Part 1 - Effects of particle and pore size. Retrieved from
https://dspace.lboro.ac.uk/dspace-jspui/handle/2134/4843 (26.06.2018)
Tarleton, E. S., & Wakeman, R. J. (1994). Understanding flux decline in crossflow
microfiltration. Part 2 - Effects of process parameters. Retrieved from
https://dspace.lboro.ac.uk/dspace-jspui/handle/2134/4842 (26.06.2018)
Taulier, N., & Chalikian, T. V. (2001). Characterization of pH-induced transitions of β-
lactoglobulin: ultrasonic, densimetric, and spectroscopic studies1. Journal of Molecular
Biology, 314(4), 873–889. https://doi.org/10.1006/jmbi.2001.5188
Tavano, O. L. (2013). Protein hydrolysis using proteases: An important tool for food
biotechnology. Journal of Molecular Catalysis B: Enzymatic, 90, 1–11.
https://doi.org/10.1016/j.molcatb.2013.01.011
Tayyab, S., Qamar, S., & Islam, M. (1991). Size exclusion chromatography and size exclusion
HPLC of proteins. Biochemical Education, 19(3), 149–152.
https://doi.org/10.1016/0307-4412(91)90060-L
Tchorbanov, B., & Bozhkova, M. (1988). Enzymatic hydrolysis of cell proteins in green algae
Chlorella and Scenedesmus after extraction with organic solvents. Enzyme and
Microbial Technology, 10(4), 233–238. https://doi.org/10.1016/0141-0229(88)90072-5

Literaturverzeichnis
287
Tchorbanov, B., Bozhkova, M., Dilov, C., & Litchev, V. (1986). Protein hydrolyzates from green
algae chlorella and scenedesmus. Food / Nahrung, 30(3–4), 405–408.
https://doi.org/10.1002/food.19860300358
Tempere, S., Cuzange, E., Malak, J., Bougeant, J. C., Revel, G. de, & Sicard, G. (2011). The
Training Level of Experts Influences their Detection Thresholds for Key Wine
Compounds. Chemosensory Perception, 4(3), 99. https://doi.org/10.1007/s12078-011-
9090-8
Temussi, P. A. (2002). Why are sweet proteins sweet? Interaction of brazzein, monellin and
thaumatin with the T1R2-T1R3 receptor. FEBS Letters, 526(1–3), 1–4.
https://doi.org/10.1016/S0014-5793(02)03155-1
Temussi, P. A. (2009). Sweet, bitter and umami receptors: a complex relationship. Trends in
Biochemical Sciences, 34(6), 296–302. https://doi.org/10.1016/j.tibs.2009.02.005
Temussi, P. A. (2011). The good taste of peptides. Journal of Peptide Science, 18(2), 73–82.
https://doi.org/10.1002/psc.1428
Teng, M.-Y., Lin, S.-H., Wu, C.-Y., & Juang, R.-S. (2006). Factors affecting selective rejection
of proteins within a binary mixture during cross-flow ultrafiltration. Journal of Membrane
Science, 281(1–2), 103–110. https://doi.org/10.1016/j.memsci.2006.03.019
Ternes, W. (2008). Naturwissenschaftliche Grundlagen der Lebensmittelzubereitung. 3.
Auflage. Hamburg: Behr's Verlag.
Thomassen, J. K., Faraday, D. B. F., Underwood, B. O., & Cleaver, J. A. S. (2005). The effect
of varying transmembrane pressure and crossflow velocity on the microfiltration fouling
of a model beer. Separation and Purification Technology, 41(1), 91–100.
https://doi.org/10.1016/j.seppur.2004.05.002
Tieke, B. (2014). Makromolekulare Chemie: Eine Einführung. 3. Auflage. Weinheim: Wiley
VCH Verlag.
Toledo, R. T. (2004). Verfahrenstechnische Grundlagen der Lebensmittelproduktion.
Hamburg: Behr’s Verlag.

Literaturverzeichnis
288
Tomchik, S. M., Berg, S., Kim, J. W., Chaudhari, N., & Roper, S. D. (2007). Breadth of Tuning
and Taste Coding in Mammalian Taste Buds. Journal of Neuroscience, 27(40), 10840–
10848. https://doi.org/10.1523/JNEUROSCI.1863-07.2007
Toniolo, C., Formaggio, F., Crisma, M., Valle, G., Boesten, W. H. J., Schoemaker, H. E., …
Précigoux, G. (1993). Bioactive and model peptides characterized by the helicogenic
(αMe)Phe residue. Tetrahedron, 49(17), 3641–3653. https://doi.org/10.1016/S0040-
4020(01)90220-0
Töpel, A. (2004). Chemie und Physik der Milch: Naturstoff - Rohstoff - Lebensmittel. 3. Auflage.
Hamburg: Behr's Verlag.
Tosoh Bioscience. (2010, November). Operating Conditions and Specifications TSK-Gel
G3000SWXL. Retrieved from
http://www.separations.eu.tosohbioscience.com/NR/rdonlyres/B6D4A616-F002-
40A4-A160-20B63EDB93A1/0/DS1009G3000SWXL.pdf (26.06.2018)
Tracey, E. M., & Davis, R. H. (1994). Protein Fouling of Track-Etched Polycarbonate
Microfiltration Membranes. Journal of Colloid and Interface Science, 167(1), 104–116.
https://doi.org/10.1006/jcis.1994.1338
Ursu, A.-V., Marcati, A., Sayd, T., Sante-Lhoutellier, V., Djelveh, G., & Michaud, P. (2014).
Extraction, fractionation and functional properties of proteins from the microalgae
Chlorella vulgaris. Bioresource Technology, 157, 134–139.
https://doi.org/10.1016/j.biortech.2014.01.071
Uversky, V. N., & Ptitsyn, O. B. (1994). ‘Partly Folded’ State, a New Equilibrium State of Protein
Molecules: Four-State Guanidinium Chloride-Induced Unfolding of .beta.-Lactamase at
Low Temperature. Biochemistry, 33(10), 2782–2791.
https://doi.org/10.1021/bi00176a006
van Boxtel, A. J. B., Otten, Z. E. H., & van der Linden, H. J. L. J. (1991). Evaluation of process
models for fouling control of reverse osmosis of cheese whey. Journal of Membrane
Science, 58(1), 89–111. https://doi.org/10.1016/S0376-7388(00)80639-0

Literaturverzeichnis
289
van den Oord, A. H. A., & van Wassenaar, P. D. (1997). Umami peptides: assessment of their
alleged taste properties. Zeitschrift Für Lebensmitteluntersuchung Und -Forschung A,
205(2), 125–130. https://doi.org/10.1007/s002170050138
van Eijndhoven, H. C. M., Saksena, S., & Zydney, A. (1995). Protein fractionation using
membrane filtration: role of electrostatic interactions, Biotechnol. Bioeng., 48, 406-414.
https://doi.org/10.1002/bit.260480413
van Oort, M. (2009). Enzymes in food technology - Introduction. In R. J. Whitehurst, & M. van
Oort (Eds.), Enzymes in Food Technology (pp. 1-18). Hoboken: Wiley-Blackwell.
van Reis, R., & Zydney, A. (2001). Membrane separations in biotechnology. Current Opinion
in Biotechnology, 12(2), 208–211. https://doi.org/10.1016/S0958-1669(00)00201-9
Venkiteshwaran, A., Heider, P., Teysseyre, L., & Belfort, G. (2008). Selective precipitation-
assisted recovery of immunoglobulins from bovine serum using controlled-fouling
crossflow membrane microfiltration. Biotechnology and Bioengineering, 101(5), 957–
966. https://doi.org/10.1002/bit.21964
Verordnung (EU) Nr. 1131/2011. VO (EU) Nr. 1131/2011 der Kommission vom 11. November
2011 zur Änderung von Anhang II der Verordnung (EG) Nr. 1333/2008 des
Europäischen Parlaments und des Rates hinsichtlich Steviolglycosiden (2011).
Retrieved from http://eur-
lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2011:295:0205:0211:DE:PDF
(26.06.2018)
Vetier, C., Bennasar, M., & de la Fuente, B. T. (1988). Study of the fouling of a mineral
microfiltration membrane using scanning electron microscopy and physicochemical
analyses in the processing of milk. Journal of Dairy Research, 55(03), 381–400.
https://doi.org/10.1017/S0022029900028648
Vyas, H. K., Bennett, R. J., & Marshall, A. D. (2000). Influence of feed properties on membrane
fouling in crossflow microfiltration of particulate suspensions. International Dairy
Journal, 10(12), 855–861. https://doi.org/10.1016/S0958-6946(01)00030-9

Literaturverzeichnis
290
Wakeman, R. J., & Tarleton, E. S. (1991). An experimental study of electroacoustic crossflow
microfiltration, Chemical Engineering Research and Design, 69, 387–397. Retrieved
from https://dspace.lboro.ac.uk/2134/5246 (26.06.2018)
Wakeman, R. J., & Tarleton, E. S. (2005). Solid/Liquid Separation: Principles of Industrial
Filtration. Oxford; New York; Tokyo: Elsevier.
Walter, J. K., Jin, Z., Jornitz, M. W., Gottschalk, U. (2012). Membrane Separations. In J. C.
Janson (Ed.), Protein Purification: Principles, High Resolution Methods, and
Applications. Third Edition (pp. 281-319). Hoboken: John Wiley & Sons.
Wang, F., & Tarabara, V. V. (2008). Pore blocking mechanisms during early stages of
membrane fouling by colloids. Journal of Colloid and Interface Science, 328(2), 464–
469. https://doi.org/10.1016/j.jcis.2008.09.028
Wang, J., Zhao, M., Yang, X., & Jiang, Y. (2006). Improvement on functional properties of
wheat gluten by enzymatic hydrolysis and ultrafiltration. Journal of Cereal Science,
44(1), 93–100. https://doi.org/10.1016/j.jcs.2006.04.002
Wang, L., & Song, L. (1999). Flux decline in crossflow microfiltration and ultrafiltration:
experimental verification of fouling dynamics. Journal of Membrane Science, 160(1),
41–50. https://doi.org/10.1016/S0376-7388(99)00075-7
Wang, W., Nema, S., & Teagarden, D. (2010). Protein aggregation—Pathways and influencing
factors. International Journal of Pharmaceutics, 390(2), 89–99.
https://doi.org/10.1016/j.ijpharm.2010.02.025
Wang, Y.-N., & Tang, C. Y. (2011). Protein fouling of nanofiltration, reverse osmosis, and
ultrafiltration membranes—The role of hydrodynamic conditions, solution chemistry,
and membrane properties. Journal of Membrane Science, 376(1–2), 275–282.
https://doi.org/10.1016/j.memsci.2011.04.036
Warendorf, T., Belitz, H. D., Gasser, U., & Grosch, W. (1992). The taste of beef bouillon. 2.
Sensory analysis of the constituents and imitation of a broth, Zeitschrift für
Lebensmitteluntersuchung und -Forschung, 195, 215–223.

Literaturverzeichnis
291
Watson, J. D., Baker, T. A., Bell, S. P., Gann, A., Levine, M., Losick, R. (2010).
Molekularbiologie. 6. Auflage. München: Pearson Studium.
Webb, M. F., Naeem, H. A., & Schmidt, K. A. (2002). Food Protein Functionality in a Liquid
System: A Comparison of Deamidated Wheat Protein with Dairy and Soy Proteins.
Journal of Food Science, 67(8), 2896–2902. https://doi.org/10.1111/j.1365-
2621.2002.tb08835.x
Whitaker, J. R., & Granum, P. E. (1980). An absolute method for protein determination based
on difference in absorbance at 235 and 280 nm. Analytical Biochemistry, 109(1), 156–
159. https://doi.org/10.1016/0003-2697(80)90024-X
Wicaksana, F., Fane, A. G., Pongpairoj, P., & Field, R. (2012). Microfiltration of algae (Chlorella
sorokiniana): Critical flux, fouling and transmission. Journal of Membrane Science,
387–388, 83–92. https://doi.org/10.1016/j.memsci.2011.10.013
Wijers, M. C., Pouliot, Y., Gauthier, S. F., Pouliot, M., & Nadeau, L. (1998). Use of nanofiltration
membranes for the desalting of peptide fractions from whey protein enzymatic
hydrolysates. Le Lait, 78(6), 621–632. https://doi.org/10.1051/lait:1998655
Wise, P. M., & Breslin, P. A. S. (2013). Individual Differences in Sour and Salt Sensitivity:
Detection and Quality Recognition Thresholds for Citric Acid and Sodium Chloride.
Chemical Senses, 38(4), 333–342. https://doi.org/10.1093/chemse/bjt003
Wong, C. H., Whitesides, G. M. (1994). Enzymes in Synthetic Organic Chemistry, Volume 12.
Oxford, New York, Tokyo: Academic Press.
Wu, Z., Zhu, Y., Huang, W., Zhang, C., Li, T., Zhang, Y., & Li, A. (2012). Evaluation of
flocculation induced by pH increase for harvesting microalgae and reuse of flocculated
medium. Bioresource Technology, 110, 496–502.
https://doi.org/10.1016/j.biortech.2012.01.101
Yamaguchi, K. (1997). Recent advances in microalgal bioscience in Japan, with special
reference to utilization of biomass and metabolites: a review. Journal of Applied
Phycology, 8(6), 487–502. https://doi.org/10.1007/BF02186327

Literaturverzeichnis
292
Yamamoto, M., Kurihara, I., & Kawano, S. (2005). Late type of daughter cell wall synthesis in
one of the Chlorellaceae, Parachlorella kessleri (Chlorophyta, Trebouxiophyceae).
Planta, 221(6), 766–775. https://doi.org/10.1007/s00425-005-1486-8
Yau, W. W., & Kirkland, J. J. (1981). Comparison of sedimentation field flow fractionation with
chromatographic methods for particulate and high-molecular-weight macromolecular
characterizations. Journal of Chromatography A, 218, 217–238.
https://doi.org/10.1016/S0021-9673(00)82058-0
Zayas, J. F. (1997). Functionality of proteins in food. New York: Springer-Verlag.
Zhang, L., Xia, Y., & Peterson, D. G. (2014). Identification of Bitter Modulating Maillard-
Catechin Reaction Products. Journal of Agricultural and Food Chemistry, 62(33),
8470–8477. https://doi.org/10.1021/jf502040e
Zhang, Y., Hoon, M. A., Chandrashekar, J., Mueller, K. L., Cook, B., Wu, D., … Ryba, N. J. P.
(2003). Coding of Sweet, Bitter, and Umami Tastes. Cell, 112(3), 293–301.
https://doi.org/10.1016/S0092-8674(03)00071-0
Zhao, F., Chu, H., Yu, Z., Jiang, S., Zhao, X., Zhou, X., & Zhang, Y. (2017). The filtration and
fouling performance of membranes with different pore sizes in algae harvesting.
Science of The Total Environment, 587–588, 87–93.
https://doi.org/10.1016/j.scitotenv.2017.02.035
Zhao, G. Q., Zhang, Y., Hoon, M. A., Chandrashekar, J., Erlenbach, I., Ryba, N. J. P., & Zuker,
C. S. (2003). The Receptors for Mammalian Sweet and Umami Taste. Cell, 115(3),
255–266. https://doi.org/10.1016/S0092-8674(03)00844-4
Zhou, N., Zhang, Y., Wu, X., Gong, X., & Wang, Q. (2011). Hydrolysis of Chlorella biomass for
fermentable sugars in the presence of HCl and MgCl2. Bioresource Technology,
102(21), 10158–10161. https://doi.org/10.1016/j.biortech.2011.08.051
Zydney, A., & Colton, C. K. (1986). A Concentration Polarization Model for the Filtrate Flux in
Cross-Flow Microfiltration of Particulate Suspensions. Chemical Engineering
Communications, 47(1–3), 1–21. https://doi.org/10.1080/00986448608911751

Literaturverzeichnis
293
Zydney, A. L. (1998). Protein Separations Using Membrane Filtration: New Opportunities for
Whey Fractionation. International Dairy Journal, 8(3), 243–250.
https://doi.org/10.1016/S0958-6946(98)00045-4

Anhang
294
Anhang
Crossflow-Fraktionierung - Versuchsdaten
Tabelle A1: Versuchsdaten – Fraktionierung von hydrolysiertem Molkenprotein-Isolat
Probenname CF (%) WH MWCO pH tF (min) tF-korrigiert (min) tFm MW (min) JFm (ml/min) JFm MW (ml/min) VPer (ml) JWvor (ml/min)
WPI, pH 4, WH 1 0,15 1 100 4 47 48,26
48,26
37,61
37,61
1735 81
WPI, pH 4, WH 2 0,15 2 100 4 - - - - -
WPI, pH 4, WH 3 0,15 3 100 4 - - - - -
WPI, pH 6, WH 1 0,15 1 100 6 66 68,32
73,06 ± 16,16
26,57
25,62 ± 5,27
1640 85
WPI, pH 6, WH 2 0,15 2 100 6 88 91,06 19,93 1680 83
WPI, pH 6, WH 3 0,15 3 100 6 57 59,80 30,35 1495 92
WPI, pH 7, WH 1 0,15 1 100 7 58 58,12
74,33 ± 15,16
31,23
25,16 ± 5,48
1694 85
WPI, pH 7, WH 2 0,15 2 100 7 78 88,15 20,59 1680 76
WPI, pH 7, WH 3 0,15 3 100 7 71 76,73 23,65 1690 79
WPI, pH 8, WH 1 0,15 1 100 8 78 86,59
90,81 ± 3,74
20,96
20,01 ± 0,84
1688 77
WPI, pH 8, WH 2 0,15 2 100 8 78 93,72 19,37 1668 72
WPI, pH 8, WH 3 0,15 3 100 8 77 92,13 19,70 1675 72
WPI, pH 9, WH 1 0,15 1 100 9 45 47,22
64,68 ± 16,95
38,44
29,48 ± 8,19
1677 82
WPI, pH 9, WH 2 0,15 2 100 9 58 65,75 27,60 1720 74
WPI, pH 9, WH 3 0,15 3 100 9 64 81,08 22,39 1675 68
WPI, pH 4, WH 1, 0,15 M NaCl 0,15 1 100 4 81 82,56
82,56
21,98
21,98
1815 78
WPI, pH 4, WH 2, 0,15 M NaCl 0,15 2 100 4 - - - - -
WPI, pH 4, WH 3, 0,15 M NaCl 0,15 3 100 4 - - - - -
WPI, pH 6, WH 1, 0,15 M NaCl 0,15 1 100 6 48 53,28
61,79 ± 13,85
34,07
30,27 ± 6,01
1605 81
WPI, pH 6, WH 2, 0,15 M NaCl 0,15 2 100 6 65 77,77 23,34 1675 72
WPI, pH 6, WH 3, 0,15 M NaCl 0,15 3 100 6 46 54,33 33,41 1490 82
WPI, pH 7, WH 1, 0,15 M NaCl 0,15 1 100 7 60 56,70
49,80 ± 5,99
32,01
36,78 ± 4,14
1735 88
WPI, pH 7, WH 2, 0,15 M NaCl 0,15 2 100 7 48 46,57 38,97 1690 88
WPI, pH 7, WH 3, 0,15 M NaCl 0,15 3 100 7 42 46,11 39,36 1685 78
WPI, pH 8, WH 1, 0,15 M NaCl 0,15 1 100 8 46 48,37
49,12 ± 5,25
37,52
37,23 ± 3,91
1694 81
WPI, pH 8, WH 2, 0,15 M NaCl 0,15 2 100 8 43 44,28 40,99 1730 81
WPI, pH 8, WH 3, 0,15 M NaCl 0,15 3 100 8 41 54,70 33,18 1690 64
WPI, pH 9, WH 1, 0,15 M NaCl 0,15 1 100 9 34 39,35
37,07 ± 2,41
46,13
49,11 ± 3,23
1685 74
WPI, pH 9, WH 2, 0,15 M NaCl 0,15 2 100 9 35 34,54 52,54 1700 86
WPI, pH 9, WH 3, 0,15 M NaCl 0,15 3 100 9 38 37,31 48,65 1670 88
Legende: CF, Feed-Konzentration. WH, Wiederholung. MWCO, Molecular Weight Cut-Off. tF, Unkorrigierte Prozesslaufzeit. tF-korrigert, korrigierte Prozesslaufzeit. tFm, mittlere korrigerte Prozesslaufzeit. JFm, mittlerer Permeatflux. VPer, Permeat Volumen. JWvor, Permeatflux von dest. Wasser einer neuen Membran. MW, arithmetischer Mittelwert.

Anhang
295
Tabelle A2: Versuchsdaten – Fraktionierung von hydrolysierter Alge (Chlorella vulgaris)
Probenname CF (%) WH MWCO pH tF (min) tF-korrigiert (min) tFm MW (min) JFm (ml/min) JFm MW (ml/min) VPer (ml) JWvor (ml/min)
Alge, pH 3, WH 1 0,15 1 100 3 65 65,63
66,55 ± 0,81
29,56
29,15 ± 0,35
1820 48
Alge, pH 3, WH 2 0,15 2 100 3 65 66,93 28,99 1900 45
Alge, pH 3, WH 3 0,15 3 100 3 68 67,10 28,91 1940 46
Alge, pH 6, WH 1 0,15 1 100 6 48 45,86
45,29 ± 2,30
42,30
42,91 ± 2,22
1880 49
Alge, pH 6, WH 2 0,15 2 100 6 49 47,25 41,06 1820 50
Alge, pH 6, WH 3 0,15 3 100 6 47 42,76 45,37 1921 50
Alge, pH 7, WH 1 0,15 1 100 7 46 47,79
50,89 ± 3,94
40,59
38,27 ± 2,86
1744 48
Alge, pH 7, WH 2 0,15 2 100 7 48 55,32 35,07 1881 40
Alge, pH 7, WH 3 0,15 3 100 7 45 49,55 39,15 1840 43
Alge, pH 8, WH 1 0,15 1 100 8 44 48,80
49,30 ± 5,26
39,76
39,65 ± 4,19
1640 48
Alge, pH 8, WH 2 0,15 2 100 8 45 44,31 43,78 1880 47
Alge, pH 8, WH 3 0,15 3 100 8 50 54,79 35,41 1760 45
Alge, pH 11, WH 1 0,15 1 100 11 46 64,45
52,38 ± 10,48
30,10
37,94 ± 6,83
1440 43
Alge, pH 11, WH 2 0,15 2 100 11 37 45,61 42,54 1501 47
Alge, pH 11, WH 3 0,15 3 100 11 50 47,09 41,20 1841 50
Alge, pH 3, WH 1, 0,15 M NaCl 0,15 1 100 3 65 59,42
69,79 ± 9,20
32,65
28,14 ± 3,96
1920 49
Alge, pH 3, WH 2, 0,15 M NaCl 0,15 2 100 3 73 73,00 26,58 1940 44
Alge, pH 3, WH 3, 0,15 M NaCl 0,15 3 100 3 77 76,96 25,21 1941 44
Alge, pH 6, WH 1, 0,15 M NaCl 0,15 1 100 6 59 55,22
55,10 ± 7,95
35,13
35,71 ± 5,22
1900 48
Alge, pH 6, WH 2, 0,15 M NaCl 0,15 2 100 6 60 62,99 30,80 1920 42
Alge, pH 6, WH 3, 0,15 M NaCl 0,15 3 100 6 48 47,09 41,20 1883 46
Alge, pH 7, WH 1, 0,15 M NaCl 0,15 1 100 7 57 60,17
62,34 ± 2,34
32,24
31,15 ± 1,16
1740 46
Alge, pH 7, WH 2, 0,15 M NaCl 0,15 2 100 7 64 64,82 29,93 1841 45
Alge, pH 7, WH 3, 0,15 M NaCl 0,15 3 100 7 50 62,03 31,27 1681 40
Alge, pH 8, WH 1, 0,15 M NaCl 0,15 1 100 8 46 55,14
59,49 ± 9,81
35,18
33,17 ± 5,04
1600 44
Alge, pH 8, WH 2, 0,15 M NaCl 0,15 2 100 8 68 70,72 27,43 1840 44
Alge, pH 8, WH 3, 0,15 M NaCl 0,15 3 100 8 49 52,60 36,88 1861 42
Alge, pH 11, WH 1, 0,15 M NaCl 0,15 1 100 11 80 96,48
75,34 ± 18,31
20,11
26,67 ± 5,68
1756 40
Alge, pH 11, WH 2, 0,15 M NaCl 0,15 2 100 11 47 64,92 29,88 1420 45
Alge, pH 11, WH 3, 0,15 M NaCl 0,15 3 100 11 46 64,62 30,02 1381 46
Legende: CF, Feed-Konzentration. WH, Wiederholung. MWCO, Molecular Weight Cut-Off. tF, Unkorrigierte Prozesslaufzeit. tF-korrigert, korrigierte Prozesslaufzeit. tFm, mittlere korrigerte Prozesslaufzeit. JFm, mittlerer Permeatflux. VPer, Permeat Volumen. JWvor, Permeatflux von dest. Wasser einer neuen Membran. MW, arithmetischer Mittelwert.

Anhang
296
Tabelle A3: Versuchsdaten – Fraktionierung von hydrolysiertem Molkenprotein-Isolat mit/ohne Ultraschall
Probenname CF (%) WH MWCO pH tF (min)
tVF (min) tVF-korrigiert (min) tFm MW (min) JFm (ml/min) JFm MW (ml/min) VV-Per-korrigiert (ml) JV-W-Initial (ml/min) JV-W-W (ml/min)
WPI – OUS, WH 1 0,30 1 100 7 68 54 54,00
67,02 ± 15,55
7,64
6,38 ± 1,61
412,73 9,0 - WPI – OUS, WH 2 0,30 2 100 7 80 64 66,11 6,04 399,55 - 9,2
WPI – OUS, WH 3 0,30 3 100 7 65 46 46,31 8,85 409,98 - 8,0
WPI – OUS, WH 4 0,30 4 100 7 90 66 66,78 6,11 407,94 - 8,2
WPI – OUS, WH 5 0,30 5 100 7 106 82 83,81 4,82 403,81 - 8,8
WPI – OUS, WH 6 0,30 6 100 7 114 84 85,14 4,78 407,21 - 8,0
WPI – US, WH 1 0,30 1 100 7 50 50 51,04
77,71 ± 27,40
7,85
5,81 ± 2,36
400,55 11,0 -
WPI – US, WH 2 0,30 2 100 7 100 96 96,97 4,17 404,82 - 10,5
WPI – US, WH 3 0,30 3 100 7 47 44 44,00 9,29 408,91 - 10,2
WPI – US, WH 4 0,30 4 100 7 100 100 107,17 3,56 381,54 - 11,5
WPI – US, WH 5 0,30 5 100 7 62 62 64,46 6,10 393,32 - 11,0
WPI – US, WH 6 0,30 6 100 7 91 92 98,32 3,89 382,61 - 11,5
Legende: CF, Feed-Konzentration. WH, Wiederholung. MWCO, Molecular Weight Cut-Off. tF, Unkorrigierte Prozesszeit. tVF, Unkorrigierte Prozesslaufzeit nach Performance-Anpassung. tVF-korrigert, korrigierte Prozesslaufzeit. tFm, mittlere korrigerte Prozesslaufzeit. JFm, mittlerer Permeatflux. VV-Per-korrigiert, korrigiertes Permeat Volumen. JV-W-Initial, Initialflux von dest. Wasser einer Vivaflow-Einheit. JV-V-W, Permeatflux von dest. Wasser vor Start eines Wiederholungsversuchs. MW, arithmetischer Mittelwert.
Tabelle A4: Versuchsdaten – Fraktionierung von hydrolysiertem Molkenprotein-Isolat mit RC-Membran
Probenname CF (%) WH MWCO pH tF (min) tRCF (min) tRCF-korrigiert (min) tFm MW (min) JFm (ml/min) JFm MW (ml/min) VV-Per-korrigiert (ml) JV-W-Initial (ml/min) JV-W-W (ml/min)
WPI, pH 9, WH 1 0,15 1 100 9 40 40 40,42
48,85 ± 10,07
9,90
8,46 ± 1,60
400,18 12,0 - WPI, pH 9, WH 2 0,15 2 100 9 62 46 46,14 8,74 403,19 - 9,3
WPI, pH 9, WH 3 0,15 3 100 9 64 60 60,00 6,74 404,41 - 11,3 Legende: CF, Feed-Konzentration. WH, Wiederholung. MWCO, Molecular Weight Cut-Off. tF, Unkorrigierte Prozesszeit. tVF, Unkorrigierte Prozesslaufzeit nach Performance-
Anpassung. tVF-korrigert, korrigierte Prozesslaufzeit. tFm, mittlere korrigerte Prozesslaufzeit. JFm, mittlerer Permeatflux. VV-Per-korrigiert, korrigiertes Permeat Volumen. JV-W-Initial, Initialflux von dest. Wasser einer Vivaflow-Einheit. JV-V-W, Permeatflux von dest. Wasser vor Start eines Wiederholungsversuchs. MW, arithmetischer Mittelwert.

Anhang
297
Tabelle A5: Versuchsdaten – Proteingehalt der hydrolysiertem Molkenprotein-Isolat-Fraktionen
Probenname yEx-Ret yEx-Per CS-Ret (g/l) CS-Per (g/l) VRet (ml) VPer (ml) mRet (g) mRet MW (g) mPer (g) mPer MW (g)
WPI, pH 4, WH 1 0,381 0,172 0,505 0,228 690 1735 0,348
0,348
0,395
0,395 WPI, pH 4, WH 2 - - - - - - - -
WPI, pH 4, WH 3 - - - - - - - -
WPI, pH 6, WH 1 0,487 0,198 0,645 0,262 485 1640 0,313
0,300 ± 0,056
0,430
0,373 ± 0,050 WPI, pH 6, WH 2 0,554 0,157 0,734 0,208 475 1680 0,348 0,349
WPI, pH 6, WH 3 0,473 0,171 0,626 0,226 380 1495 0,238 0,339
WPI, pH 7, WH 1 0,294 0,185 0,389 0,245 485 1694 0,189
0,209 ± 0,060
0,415
0,367 ± 0,047 WPI, pH 7, WH 2 0,395 0,144 0,523 0,191 530 1680 0,277 0,320
WPI, pH 7, WH 3 0,272 0,164 0,360 0,217 450 1690 0,162 0,367
WPI, pH 8, WH 1 0,575 0,157 0,761 0,208 540 1688 0,411
0,354 ± 0,067
0,351
0,358 ± 0,026 WPI, pH 8, WH 2 0,47 0,152 0,622 0,201 595 1668 0,370 0,336
WPI, pH 8, WH 3 0,455 0,174 0,602 0,230 465 1675 0,280 0,386
WPI, pH 9, WH 1 0,397 0,168 0,526 0,222 530 1677 0,279
0,286 ± 0,012
0,373
0,398 ± 0,024 WPI, pH 9, WH 2 0,393 0,175 0,520 0,232 536 1720 0,279 0,399
WPI, pH 9, WH 3 0,354 0,19 0,469 0,252 638 1675 0,299 0,421
WPI, pH 4, WH 1, 0,15 M NaCl 0,471 0,192 0,624 0,254 435 1815 0,271
0,271
0,461
0,461 WPI, pH 4, WH 2, 0,15 M NaCl - - - - - - - -
WPI, pH 4, WH 3, 0,15 M NaCl - - - - - - - -
WPI, pH 6, WH 1, 0,15 M NaCl 0,436 0,169 0,577 0,224 425 1605 0,245
0,209 ± 0,032
0,359
0,355 ± 0,013 WPI, pH 6, WH 2, 0,15 M NaCl 0,392 0,153 0,519 0,203 374 1675 0,194 0,339
WPI, pH 6, WH 3, 0,15 M NaCl 0,388 0,185 0,514 0,245 365 1490 0,188 0,365
WPI, pH 7, WH 1, 0,15 M NaCl 0,181 0,204 0,240 0,270 472 1735 0,113
0,136 ± 0,037
0,469
0,426 ± 0,039 WPI, pH 7, WH 2, 0,15 M NaCl 0,258 0,175 0,342 0,232 522 1690 0,178 0,392
WPI, pH 7, WH 3, 0,15 M NaCl 0,201 0,187 0,266 0,248 435 1685 0,116 0,417
WPI, pH 8, WH 1, 0,15 M NaCl 0,246 0,197 0,326 0,261 527 1694 0,172
0,194 ± 0,020
0,442
0,419 ± 0,037 WPI, pH 8, WH 2, 0,15 M NaCl 0,289 0,164 0,383 0,217 534 1730 0,204 0,376
WPI, pH 8, WH 3, 0,15 M NaCl 0,372 0,196 0,493 0,260 420 1690 0,207 0,439
WPI, pH 9, WH 1, 0,15 M NaCl 0,227 0,173 0,301 0,229 528 1685 0,159
0,163 ± 0,049
0,386
0,420 ± 0,045 WPI, pH 9, WH 2, 0,15 M NaCl 0,182 0,209 0,241 0,277 480 1700 0,116 0,470
WPI, pH 9, WH 3, 0,15 M NaCl 0,259 0,182 0,343 0,241 625 1670 0,214 0,402
Legende: yEx-Ret, Extinktion Retentat. yEx-Per, Extinktion Permeat. CS-Ret, Spektralphotometrisch ermittelte Konzentration des Retentats. CS-Per, Spektralphotometrisch ermittelte Konzentration des Permeats. VRet, Retentat Volumen. VPer, Permeat Volumen. mRet, absolute Proteinmenge Retentat. mPer, absolute Proteinmenge Permeat. MW, arithmetischer Mittelwert.
Anhang
298
Tabelle A6: Versuchsdaten – Proteingehalt der Fraktionen hydrolysierter Alge (Chlorella vulgaris)
Probenname yEx-Ret yEx-Per CS-Ret (g/l) CS-Per (g/l) VRet (ml) VPer (ml) mRet (g) mRet MW (g) mPer (g) mPer MW (g)
Alge, pH 3, WH 1 0,413 0,366 0,244 0,216 350 1820 0,085
0,093 ± 0,008
0,394
0,421 ± 0,024 Alge, pH 3, WH 2 0,393 0,386 0,232 0,228 405 1900 0,094 0,433
Alge, pH 3, WH 3 0,402 0,381 0,238 0,225 425 1940 0,101 0,437
Alge, pH 6, WH 1 0,447 0,422 0,264 0,249 410 1880 0,108
0,105 ± 0,007
0,469
0,474 ± 0,018 Alge, pH 6, WH 2 0,449 0,426 0,265 0,252 410 1820 0,109 0,458
Alge, pH 6, WH 3 0,421 0,435 0,249 0,257 390 1920 0,097 0,494
Alge, pH 7, WH 1 0,442 0,409 0,261 0,242 355 1744 0,093
0,104 ± 0,010
0,422
0,451 ± 0,030 Alge, pH 7, WH 2 0,456 0,433 0,270 0,256 410 1881 0,110 0,481
Alge, pH 7, WH 3 0,453 0,414 0,268 0,245 405 1840 0,108 0,450
Alge, pH 8, WH 1 0,496 0,431 0,293 0,255 335 1640 0,098
0,142 ± 0,047
0,418
0,407 ± 0,038 Alge, pH 8, WH 2 0,573 0,394 0,339 0,233 405 1880 0,137 0,438
Alge, pH 8, WH 3 0,851 0,350 0,503 0,207 380 1760 0,191 0,364
Alge, pH 11, WH 1 0,975 0,369 0,576 0,218 266 1440 0,153
0,185 ± 0,029
0,314
0,345 ± 0,041 Alge, pH 11, WH 2 0,999 0,373 0,590 0,220 320 1501 0,189 0,331
Alge, pH 11, WH 3 0,873 0,360 0,516 0,213 410 1840 0,212 0,391
Alge, pH 3, WH 1, 0,15 M NaCl 0,428 0,343 0,253 0,203 400 1920 0,101
0,112 ± 0,010
0,389
0,420 ± 0,032 Alge, pH 3, WH 2, 0,15 M NaCl 0,467 0,395 0,276 0,233 420 1940 0,116 0,453
Alge, pH 3, WH 3, 0,15 M NaCl 0,478 0,364 0,283 0,215 425 1940 0,120 0,417
Alge, pH 6, WH 1, 0,15 M NaCl 0,543 0,357 0,321 0,211 475 1900 0,152
0,132 ± 0,018
0,401
0,420 ± 0,019 Alge, pH 6, WH 2, 0,15 M NaCl 0,476 0,369 0,281 0,218 430 1920 0,121 0,419
Alge, pH 6, WH 3, 0,15 M NaCl 0,472 0,395 0,279 0,233 435 1883 0,121 0,440
Alge, pH 7, WH 1, 0,15 M NaCl 0,527 0,422 0,311 0,249 355 1740 0,111
0,112 ± 0,015
0,434
0,429 ± 0,008 Alge, pH 7, WH 2, 0,15 M NaCl 0,551 0,385 0,326 0,228 390 1841 0,127 0,419
Alge, pH 7, WH 3, 0,15 M NaCl 0,492 0,436 0,291 0,258 335 1680 0,097 0,433
Alge, pH 8, WH 1, 0,15 M NaCl 0,472 0,375 0,279 0,222 400 1600 0,112
0,121 ± 0,013
0,355
0,401 ± 0,040 Alge, pH 8, WH 2, 0,15 M NaCl 0,582 0,390 0,344 0,230 395 1840 0,136 0,424
Alge, pH 8, WH 3, 0,15 M NaCl 0,502 0,386 0,297 0,228 385 1860 0,114 0,424
Alge, pH 11, WH 1, 0,15 M NaCl 0,975 0,374 0,576 0,221 395 1756 0,228
0,177 ± 0,044
0,388
0,332 ± 0,051 Alge, pH 11, WH 2, 0,15 M NaCl 0,894 0,378 0,528 0,223 300 1420 0,159 0,317
Alge, pH 11, WH 3, 0,15 M NaCl 0,846 0,355 0,500 0,210 290 1381 0,145 0,290
Legende: yEx-Ret, Extinktion Retentat. yEx-Per, Extinktion Permeat. CS-Ret, Spektralphotometrisch ermittelte Konzentration des Retentats. CS-Per, Spektralphotometrisch ermittelte Konzentration des Permeats. VRet, Retentat Volumen. VPer, Permeat Volumen. mRet, absolute Proteinmenge Retentat. mPer, absolute Proteinmenge Permeat. MW, arithmetischer Mittelwert.

Anhang
299
Tabelle A7: Versuchsdaten – Proteingehalt der hydrolysiertem Molkenprotein-Isolat-Fraktionen mit/ohne Ultraschall
Probenname yEx-Ret yEx-Per CS-Ret (mg/ml) CS-Per (mg/ml) VRet (ml) VPer (ml) mRet (mg) mRet MW (mg) mPer (mg) mPer MW (mg)
WPI – OUS, WH 1 0,830 0,374 1,099 0,495 93 400 102,21
94,63 ± 9,14
198,09
183,78 ± 17,13
WPI – OUS, WH 2 0,812 0,320 1,075 0,424 85 400 91,39 169,49
WPI – OUS, WH 3 0,692 0,333 0,916 0,441 89 400 81,55 176,38
WPI – OUS, WH 4 0,817 0,325 1,082 0,430 82 393 88,71 172,14
WPI – OUS, WH 5 0,848 0,340 1,123 0,450 87 396 97,69 180,08
WPI – OUS, WH 6 0,764 0,404 1,012 0,535 105 395 106,22 213,98
WPI – US, WH 1 0,770 0,427 1,020 0,565 87 400 88,70
89,50 ± 10,03
226,17
187,24 ± 19,96
WPI – US, WH 2 0,781 0,356 1,034 0,471 102 400 105,48 188,56
WPI – US, WH 3 0,689 0,328 0,912 0,434 94 400 85,76 173,73
WPI – US, WH 4 0,816 0,327 1,081 0,433 89 399 96,17 173,20
WPI – US, WH 5 0,773 0,352 1,024 0,466 82 393 83,93 186,44
WPI – US, WH 6 0,807 0,338 1,069 0,448 72 400 76,94 179,03
Legende: yEx-Ret, Extinktion Retentat. yEx-Per, Extinktion Permeat. CS-Ret, Spektralphotometrisch ermittelte Konzentration des Retentats. CS-Per, Spektralphotometrisch ermittelte Konzentration des Permeats. VRet, Retentat Volumen. VPer, Permeat Volumen. mRet, absolute Proteinmenge Retentat. mPer, absolute Proteinmenge Permeat. MW, arithmetischer Mittelwert.
Tabelle A8: Versuchsdaten – Proteingehalt der hydrolysierten Molkenprotein-Isolat-Fraktionen bei der Filtration mittels RC-Membran
Probenname yEx-Ret yEx-Per CS-Ret (mg/ml) CS-Per (mg/ml) VRet (ml) VPer (ml) mRet (mg) mRet MW (mg) mPer (mg) mPer MW (mg)
WPI, pH 9, WH 1 0,495 0,189 0,655 0,250 72 401 47,19
57,33 ± 10,92
100,36
107,61 ± 15,41 WPI, pH 9, WH 2 0,598 0,236 0,792 0,313 87 401 68,89 125,31
WPI, pH 9, WH 3 0,603 0,203 0,749 0,268 70 401 55,89 97,17
Legende: yEx-Ret, Extinktion Retentat. yEx-Per, Extinktion Permeat. CS-Ret, Spektralphotometrisch ermittelte Konzentration des Retentats. CS-Per, Spektralphotometrisch ermittelte Konzentration des Permeats. VRet, Retentat Volumen. VPer, Permeat Volumen. mRet, absolute Proteinmenge Retentat. mPer, absolute Proteinmenge Permeat. MW, arithmetischer Mittelwert.

Anhang
300
Tabelle A9: Versuchsdaten – Wiederfindungsrate bei der Filtration von hydrolysiertem Molkenprotein-Isolat
Probenname WFR (%) WFR MW (%) WFRRet (%) WFRRet MW (%) WFRPer (%) WFRPer MW (%)
WPI, pH 4, WH 1 99,10
99,10
46,41
46,41
52,69
52,69 WPI, pH 4, WH 2 - - -
WPI, pH 4, WH 3 - - -
WPI, pH 6, WH 1 99,03
89,64 ± 11,46
41,70
39,96 ± 7,52
57,33
49,68 ± 6,67 WPI, pH 6, WH 2 93,03 46,46 46,57
WPI, pH 6, WH 3 76,87 31,73 45,14
WPI, pH 7, WH 1 80,50
76,91 ± 5,53
25,17
27,92 ± 8,03
55,33
48,99 ± 6,31 WPI, pH 7, WH 2 79,67 36,96 42,71
WPI, pH 7, WH 3 70,54 21,61 48,93
WPI, pH 8, WH 1 101,61
94,85 ± 6,43
54,82
47,18 ± 8,94
46,79
47,67 ± 3,43 WPI, pH 8, WH 2 94,14 49,37 44,76
WPI, pH 8, WH 3 88,81 37,35 51,46
WPI, pH 9, WH 1 86,89
91,10 ± 4,63
37,15
38,07 ± 1,56
49,74
53,02 ± 3,22 WPI, pH 9, WH 2 90,33 37,19 53,14
WPI, pH 9, WH 3 96,06 39,88 56,19
WPI, pH 4, WH 1, 0,15 M NaCl 97,70
97,70
36,17
36,17
61,53
61,53 WPI, pH 4, WH 2, 0,15 M NaCl - - -
WPI, pH 4, WH 3, 0,15 M NaCl - - -
WPI, pH 6, WH 1, 0,15 M NaCl 80,60
75,14 ± 4,90
32,72
27,87 ± 4,22
47,89
47,27 ± 1,79 WPI, pH 6, WH 2, 0,15 M NaCl 71,13 25,88 45,25
WPI, pH 6, WH 3, 0,15 M NaCl 73,67 25,00 48,67
WPI, pH 7, WH 1, 0,15 M NaCl 77,57
74,88 ± 3,39
15,08
18,10 ± 4,92
62,49
56,78 ± 5,23 WPI, pH 7, WH 2, 0,15 M NaCl 75,99 23,78 52,22
WPI, pH 7, WH 3, 0,15 M NaCl 71,07 15,44 55,63
WPI, pH 8, WH 1, 0,15 M NaCl 81,81
81,74 ± 4,36
22,89
25,91 ± 2,62
58,92
55,83 ± 4,97 WPI, pH 8, WH 2, 0,15 M NaCl 77,34 27,25 50,09
WPI, pH 8, WH 3, 0,15 M NaCl 86,07 27,58 58,48
WPI, pH 9, WH 1, 0,15 M NaCl 72,63
77,67 ± 4,82
21,16
21,72 ± 6,60
51,47
55,95 ± 5,97 WPI, pH 9, WH 2, 0,15 M NaCl 78,15 15,42 62,73
WPI, pH 9, WH 3, 0,15 M NaCl 82,24 28,58 53,66
Legende: WFR, Wiederfindungsrate. WFRRet, Wiederfindungsrate im Retentat. WFRPer, Wiederfindungsrate im Permeat. MW, arithmetischer Mittelwert

Anhang
301
Tabelle A10: Versuchsdaten – Wiederfindungsrate bei der Filtration von hydrolysierter Alge (Chlorella vulgaris)
Probenname WFR (%) WFR MW (%) WFRRet (%) WFRRet MW (%) WFRPer (%) WFRPer MW (%)
Alge, pH 3, WH 1 63,88
68,64 ± 4,18
11,39
12,47 ± 1,04
52,49
56,18 ± 3,20 Alge, pH 3, WH 2 70,34 12,54 57,79
Alge, pH 3, WH 3 71,71 13,46 58,25
Alge, pH 6, WH 1 76,96
77,11 ± 1,58
14,44
13,96 ± 0,89
62,52
63,14 ± 2,42 Alge, pH 6, WH 2 75,60 14,51 61,10
Alge, pH 6, WH 3 78,75 12,94 65,82
Alge, pH 7, WH 1 68,57
73,99 ± 5,19
12,36
13,85 ± 1,30
56,21
60,14 ± 3,99 Alge, pH 7, WH 2 78,92 14,73 64,18
Alge, pH 7, WH 3 74,49 14,46 60,03
Alge, pH 8, WH 1 68,79
73,16 ± 4,00
13,09
18,95 ± 6,22
55,70
54,20 ± 5,08 Alge, pH 8, WH 2 76,66 18,29 58,37
Alge, pH 8, WH 3 74,03 25,48 48,54
Alge, pH 11, WH 1 62,31
70,67 ± 9,12
20,44
24,61 ± 3,92
41,87
46,06 ± 5,43 Alge, pH 11, WH 2 69,31 25,19 44,12
Alge, pH 11, WH 3 80,40 28,21 52,20
Alge, pH 3, WH 1, 0,15 M NaCl
65,39
70,96 ± 5,26
13,49
14,99 ± 1,32
51,90
55,98 ± 4,25 Alge, pH 3, WH 2, 0,15 M NaCl
75,84 15,46 60,39
Alge, pH 3, WH 3, 0,15 M NaCl
71,66 16,01 55,65
Alge, pH 6, WH 1, 0,15 M NaCl
73,78
73,51 ± 1,44
20,33
17,54 ± 2,41
53,45
55,96 ± 2,58 Alge, pH 6, WH 2, 0,15 M NaCl
71,96 16,13 55,83
Alge, pH 6, WH 3, 0,15 M NaCl
74,79 16,18 58,61
Alge, pH 7, WH 1, 0,15 M NaCl
72,61
72,03 ± 1,15
14,74
14,89 ± 1,98
57,86
57,15 ± 1,12 Alge, pH 7, WH 2, 0,15 M NaCl
72,79 16,93 55,85
Alge, pH 7, WH 3, 0,15 M NaCl
70,71 12,99 57,72
Alge, pH 8, WH 1, 0,15 M NaCl
62,16
69,54 ± 6,55
14,88
16,07 ± 1,78
47,28
53,47 ± 5,36 Alge, pH 8, WH 2, 0,15 M NaCl
74,66 18,12 56,55
Alge, pH 8, WH 3, 0,15 M NaCl
71,81 15,23 56,58
Alge, pH 11, WH 1, 0,15 M NaCl
82,10
67,83 ± 12,66
30,35
23,61 ± 5,91
51,75
44,23 ± 6,77 Alge, pH 11, WH 2, 0,15 M NaCl
63,43 21,13 42,30
Alge, pH 11, WH 3, 0,15 M NaCl
57,97 19,33 38,63
Legende: WFR, Wiederfindungsrate. WFRRet, Wiederfindungsrate im Retentat. WFRPer, Wiederfindungsrate im Permeat. MW, arithmetischer Mittelwert

Anhang
302
Tabelle A11: Versuchsdaten – Wiederfindungsrate bei der Filtration von hydrolysiertem Molkenprotein-Isolat mit/ohne Ultraschall
Probenname WFR (%) WFR MW (%) WFRRet (%) WFRRet MW (%) WFRPer (%) WFRPer MW (%)
WPI – OUS, WH 1 100,10
93,22 ± 8,50
34,07
31,54 ± 3,05
66,03
61,68 ± 5,80
WPI – OUS, WH 2 86,96
30,46
56,50 WPI – OUS, WH 3 85,98
27,18
58,79
WPI – OUS, WH 4 86,95
29,57
57,38 WPI – OUS, WH 5 92,59
32,56
60,03
WPI – OUS, WH 6 106,74
35,41
71,33 WPI – US, WH 1 104,96
92,45 ± 7,56
29,57
29,83 ± 3,34
75,39
62,62 ± 6,60
WPI – US, WH 2 98,01
35,16
62,85 WPI – US, WH 3 86,50
28,59
57,91
WPI – US, WH 4 89,79
32,06
57,73 WPI – US, WH 5 90,12
27,98
62,15
WPI – US, WH 6 85,32
25,65
59,68 Legende: WFR, Wiederfindungsrate. WFRRet, Wiederfindungsrate im Retentat. WFRPer, Wiederfindungsrate im Permeat. MW, arithmetischer Mittelwert
Tabelle A12: Versuchsdaten – Wiederfindungsrate bei der Filtration von hydrolysiertem Molkenproteinen-Isolat mittels RC-Membran
Probenname WFR (%) WFR MW (%) WFRRet (%) WFRRet MW (%) WFRPer (%) WFRPer MW (%)
WPI, pH 9, WH 1 98,37
109,96 ± 17,00
31,46
38,22 ± 7,28
66,90
71,74 ± 10,27 WPI, pH 9, WH 2 129,47 45,93 83,54
WPI, pH 9, WH 3 102,04 37,26 64,78
Legende: WFR, Wiederfindungsrate. WFRRet, Wiederfindungsrate im Retentat. WFRPer, Wiederfindungsrate im Permeat. MW, arithmetischer Mittelwert

Anhang
303
Tabelle & Abbildung A1: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 3, WH 1
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 532,21 70 0 3,00 55 3,11 1 59,01 20 663,86 75 5 3,11 60 3,10 2 111,61 25 793,89 80 10 3,12 65 3,11 3 154,92 30 918,11 85 15 3,12 70 4 193,48 35 1070,57 90 20 3,11 75 5 230,61 40 1207,87 95 25 3,11 80 6 265,84 45 1333,88 100 30 3,11 85 7 300,99 50 1466,68 105 35 3,12 90 8 333,74 55 1593,39 110 40 3,12 95 9 365,29 60 1710,43 115 45 3,11 100 10 395,41 65 1820,68 120 50 3,11 105
Tabelle & Abbildung A2: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 3, WH 2
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 568,17 70 0 3,01 55 3,16 1 61,05 20 717,40 75 5 3,12 60 3,16 2 112,74 25 861,14 80 10 3,13 65 3,16 3 172,13 30 997,52 85 15 3,13 70 4 200,17 35 1166,08 90 20 3,15 75 5 241,41 40 1283,29 95 25 3,15 80 6 281,02 45 1417,02 100 30 3,15 85 7 317,04 50 1549,81 105 35 3,16 90 8 355,95 55 1667,73 110 40 3,16 95 9 388,79 60 1788,79 115 45 3,16 100 10 425,59 65 1900,35 120 50 3,16 105
Tabelle & Abbildung A3: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 3, WH 3
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 562,92 68 1940,30 0 3,01 55 3,08 1 59,49 20 708,66 75 5 3,09 60 3,08 2 113,05 25 846,53 80 10 3,09 65 3,08 3 158,32 30 977,92 85 15 3,09 68 3,07 4 200,15 35 1114,03 90 20 3,09 75 5 238,87 40 1246,87 95 25 3,09 80 6 276,06 45 1377,03 100 30 3,09 85 7 313,79 50 1503,89 105 35 3,09 90 8 352,35 55 1629,98 110 40 3,09 95 9 381,13 60 1754,88 115 45 3,09 100 10 414,94 65 1871,03 120 50 3,09 105
R² = 0,986
0 10 20 30 40 50 60 70
3,0
3,2
3,4
0
500
1000
1500
2000
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,983
2,8
3,0
3,2
3,4
0
500
1000
1500
2000
0 10 20 30 40 50 60 70
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,984
3,0
3,2
0
500
1000
1500
2000
0 10 20 30 40 50 60 70
pH
Per
mea
tmen
ge [
g]
Zeit [min]

Anhang
304
Tabelle & Abbildung A4: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 6, WH 1
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 718,23 70 0 6,05 55 1 58,14 20 911,45 75 5 6,16 60 2 115,22 25 1093,18 80 10 6,20 65 3 168,48 30 1288,26 85 15 6,22 70 4 221,09 35 1463,96 90 20 6,27 75 5 267,34 40 1630,04 95 25 6,30 80 6 319,91 45 1787,07 100 30 6,33 85 7 367,82 48 1880,51 105 35 6,36 90 8 413,41 55 110 40 6,41 95 9 455,82 60 115 45 6,45 100
10 498,35 65 120 48 6,49 105
Tabelle & Abbildung A5: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 6, WH 2
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 675,91 70 0 6,01 55 1 58,45 20 851,99 75 5 6,12 60 2 113,82 25 1035,83 80 10 6,16 65 3 161,70 30 1211,45 85 15 6,18 70 4 206,14 35 1383,46 90 20 6,18 75 5 249,75 40 1539,16 95 25 6,23 80 6 293,68 45 1696,34 100 30 6,24 85 7 336,96 49 1820,96 105 35 6,26 90 8 382,49 55 110 40 6,31 95 9 432,57 60 115 45 6,35 100
10 465,00 65 120 49 6,40 105
Tabelle & Abbildung A6: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 6, WH 3
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 702,34 70 0 5,99 55 1 62,50 20 893,35 75 5 6,10 60 2 118,87 25 1067,34 80 10 6,13 65 3 167,56 30 1236,04 85 15 6,12 70 4 212,47 35 1458,78 90 20 6,14 75 5 256,05 40 1659,27 95 25 6,16 80 6 298,60 45 1842,13 100 30 6,22 85 7 339,13 47 1920,86 105 35 6,22 90 8 378,56 55 110 40 6,24 95 9 416,98 60 115 45 6,33 100 10 486,26 65 120 47 6,33 105
R² = 0,988
6,0
6,2
6,4
6,6
0
500
1000
1500
2000
0 10 20 30 40 50
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,990
0 10 20 30 40 50
5,8
6,0
6,2
6,4
6,6
0
500
1000
1500
2000
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,995
5,8
6,0
6,2
6,4
0
500
1000
1500
2000
0 10 20 30 40 50
pH
Per
mea
tmen
ge [
g]
Zeit [min]

Anhang
305
Tabelle & Abbildung A7: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 7, WH 1
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 676,13 70 0 7,03 55 1 57,47 20 858,64 75 5 7,06 60 2 111,45 25 1062,5 80 10 7,08 65 3 158,91 30 1249,01 85 15 7,11 70 4 204,24 35 1422,97 90 20 7,17 75 5 247,80 40 1584,01 95 25 7,21 80 6 294,01 45 1730,66 100 30 7,22 85 7 336,34 46 1740,32 105 35 7,27 90 8 388,23 55 110 40 7,31 95 9 434,52 60 115 45 7,40 100 10 478,34 65 120 46 7,40 105
Tabelle & Abbildung A8: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 7, WH 2
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 729,15 70 0 7,02 55 1 54,96 20 918,40 75 5 7,09 60 2 110,23 25 1095,38 80 10 7,12 65 3 162,54 30 1299,17 85 15 7,14 70 4 210,56 35 1477,03 90 20 7,16 75 5 257,89 40 1635,50 95 25 7,20 80 6 315,64 45 1784,53 100 30 7,20 85 7 365,99 48 1881,67 105 35 7,21 90 8 413,32 55 110 40 7,24 95 9 458,70 60 115 45 7,27 100 10 505,12 65 120 48 7,30 105
Tabelle & Abbildung A9: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 7, WH 3
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 757,03 70 0 7,02 55 1 56,55 20 945,55 75 5 7,11 60 2 113,72 25 1151,99 80 10 7,08 65 3 169,35 30 1340,01 85 15 7,08 70 4 222,12 35 1519,42 90 20 7,11 75 5 277,14 40 1690,81 95 25 7,08 80 6 330,72 45 1840,44 100 30 7,10 85 7 381,23 50 105 35 7,10 90 8 431,13 55 110 40 7,11 95 9 480,86 60 115 45 7,18 100 10 537,65 65 120 50 105
R² = 0,991
7,0
7,2
7,4
7,6
0
500
1000
1500
2000
0 10 20 30 40 50
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,988
7,0
7,2
7,4
0
500
1000
1500
2000
0 10 20 30 40 50
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,986
7,0
7,2
7,4
0
500
1000
1500
2000
0 10 20 30 40 50
pH
Per
mea
tmen
ge [
g]
Zeit [min]

Anhang
306
Tabelle & Abbildung A10: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 8, WH 1
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 681,53 70 0 8,03 55 1 57,32 20 858,11 75 5 8,00 60 2 111,24 25 1047,72 80 10 8,08 65 3 157,80 30 1221,93 85 15 8,05 70 4 202,07 35 1383,02 90 20 8,03 75 5 245,55 40 1530,54 95 25 8,01 80 6 291,41 44 1640,16 100 30 8,02 85 7 335,62 50 105 35 8,02 90 8 379,74 55 110 40 8,00 95 9 422,96 60 115 45 8,02 100 10 474,06 65 120 50 105
Tabelle & Abbildung A11: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 8, WH 2
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 778,70 70 0 8,01 55 1 59,37 20 979,00 75 5 7,88 60 2 119,21 25 1196,65 80 10 7,79 65 3 170,13 30 1394,87 85 15 7,73 70 4 221,06 35 1575,65 90 20 7,66 75 5 280,52 40 1737,22 95 25 7,58 80 6 338,95 45 1880,52 100 30 7,53 85 7 390,38 50 105 35 7,50 90 8 440,29 55 110 40 7,43 95 9 492,23 60 115 44 7,43 100
10 541,69 65 120 50 105
Tabelle & Abbildung A12: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 8, WH 3
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 653,55 70 0 8,01 55 1 59,65 20 826,75 75 5 7,95 60 2 117,02 25 987,96 80 10 7,82 65 3 167,42 30 1154,6 85 15 7,70 70 4 213,80 35 1310,22 90 20 7,60 75 5 258,47 40 1464,79 95 25 7,55 80 6 301,71 45 1638,67 100 30 7,46 85 7 344,81 50 1760,66 105 35 7,44 90 8 385,02 55 110 40 7,41 95 9 424,56 60 115 45 7,39 100 10 465,97 65 120 50 7,38 105
R² = 0,988
7,8
8,0
8,2
0
500
1000
1500
2000
0 10 20 30 40 50
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,986
7,4
7,6
7,8
8,0
8,2
0
500
1000
1500
2000
0 10 20 30 40 50
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,985
7,2
7,4
7,6
7,8
8,0
8,2
0
500
1000
1500
2000
0 10 20 30 40 50 60
pH
Per
mea
tmen
ge [
g]
Zeit [min]

Anhang
307
Tabelle & Abbildung A13: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 11, WH 1
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 603,82 70 0 11,01 55 1 57,42 20 754,83 75 5 11,04 60 2 108,54 25 934,89 80 10 11,03 65 3 153,06 30 1073,53 85 15 10,98 70 4 197,39 35 1202,7 90 20 10,97 75 5 239,29 40 1318,89 95 25 10,93 80 6 279,01 45 1425,21 100 30 10,93 85 7 314,62 46 1440,4 105 35 10,94 90 8 346,97 55 110 40 10,94 95 9 381,98 60 115 45 10,94 100 10 427,84 65 120 46 10,96 105
Tabelle & Abbildung A14: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 11, WH 2
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 710,88 70 0 11,00 55 1 60,67 20 869,32 75 5 11,03 60 2 120,33 25 1106,02 80 10 11,07 65 3 172,08 30 1283,92 85 15 11,06 70 4 222,31 35 1451,16 90 20 11,03 75 5 264,92 37 1501,04 95 25 11,05 80 6 307,04 45 100 30 11,03 85 7 346,10 50 105 35 11,05 90 8 388,03 55 110 37 11,03 95 9 453,41 60 115 45 100 10 505,08 65 120 50 105
Tabelle & Abbildung A15: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 11, WH 3
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 736,69 70 0 10,99 55 1 67,89 20 901,75 75 5 11,00 60 2 132,22 25 1048,66 80 10 11,05 65 3 188,53 30 1238,10 85 15 11,00 70 4 240,06 35 1391,04 90 20 10,99 75 5 286,98 40 1547,79 95 25 10,99 80 6 338,82 45 1683,54 100 30 10,97 85 7 387,35 50 1840,50 105 35 10,96 90 8 434,56 55 110 40 10,96 95 9 477,74 60 115 45 10,96 100 10 532,79 65 120 50 10,97 105
R² = 0,978
10,8
11,0
11,2
0
500
1000
1500
2000
0 10 20 30 40 50
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,989
10,8
11,0
11,2
0
500
1000
1500
2000
0 10 20 30 40
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,974
10,8
11,0
11,2
0
500
1000
1500
2000
0 20 40 60
pH
Per
mea
tmen
ge [
g]
Zeit [min]

Anhang
308
Tabelle & Abbildung A16: Versuchsdaten – Crossflow-Filtration, Algen (Chlorella vulgaris), pH 3, NaCl, WH 1
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 573,13 70 0 3,01 55 3,26 1 63,81 20 731,91 75 5 3,21 60 3,26 2 120,59 25 889,79 80 10 3,24 65 3,26 3 167,99 30 1037,14 85 15 3,25 70 4 211,58 35 1197,57 90 20 3,25 75 5 250,93 40 1336,84 95 25 3,25 80 6 290,91 45 1463,09 100 30 3,26 85 7 327,77 50 1589,5 105 35 3,26 90 8 363,07 55 1709,13 110 40 3,26 95 9 396,49 60 1820,01 115 45 3,26 100 10 428,56 65 1920,17 120 50 3,26 105
Tabelle & Abbildung A17: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 3, NaCl, WH 2
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 510,51 70 1876,14 0 3,00 55 3,34 1 54,96 20 659,79 73 1940,35 5 3,20 60 3,34 2 104,92 25 803,87 80 10 3,24 65 3,35 3 145,23 30 923,82 85 15 3,27 70 3,35 4 181,81 35 1073,12 90 20 3,29 73 3,35 5 215,68 40 1211,06 95 25 3,31 80 6 249,68 45 1322,77 100 30 3,31 85 7 283,11 50 1437,61 105 35 3,33 90 8 315,32 55 1553,21 110 40 3,33 95 9 346,12 60 1662,99 115 45 3,33 100 10 376,24 65 1773,29 120 50 3,34 105
Tabelle & Abbildung A18: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 3, NaCl, WH 3
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 514,60 70 1814,44 0 3,00 55 3,13 1 54,97 20 660,24 75 1907,32 5 3,10 60 3,13 2 104,11 25 786,01 77 1940,69 10 3,11 65 3,13 3 145,98 30 906,20 85 15 3,11 70 3,13 4 183,39 35 1020,96 90 20 3,12 75 3,13 5 218,16 40 1150,57 95 25 3,12 77 3,13 6 254,49 45 1277,22 100 30 3,12 85 7 288,25 50 1387,68 105 35 3,13 90 8 319,75 55 1499,34 110 40 3,13 95 9 350,00 60 1611,75 115 45 3,13 100 10 379,47 65 1714,88 120 50 3,13 105
R² = 0,981
2,8
3,0
3,2
3,4
0
500
1000
1500
2000
0 10 20 30 40 50 60 70
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,986
2,8
3,0
3,2
3,4
3,6
0
500
1000
1500
2000
0 10 20 30 40 50 60 70 80
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,983
3,0
3,2
3,4
0
500
1000
1500
2000
0 10 20 30 40 50 60 70 80
pH
Per
mea
tmen
ge [
g]
Zeit [min]

Anhang
309
Tabelle & Abbildung A19: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 6, NaCl, WH 1
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 655,79 70 0 6,02 55 6,54 1 56,94 20 828,11 75 5 6,19 59 6,57 2 111,42 25 972,99 80 10 6,27 65 3 156,74 30 1148,11 85 15 6,29 70 4 196,08 35 1282,29 90 20 6,33 75 5 247,54 40 1419,77 95 25 6,35 80 6 301,73 45 1554,37 100 30 6,38 85 7 348,29 50 1677,81 105 35 6,40 90 8 394,28 55 1801,25 110 40 6,42 95 9 436,29 59 1900,19 115 45 6,45 100 10 472,93 65 120 50 6,48 105
Tabelle & Abbildung A20: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 6, NaCl, WH 2
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 577,02 70 0 6,02 55 6,27 1 55,41 20 766,11 75 5 6,11 60 6,32 2 109,96 25 914,41 80 10 6,13 65 3 154,93 30 1048,02 85 15 6,14 70 4 196,50 35 1232,29 90 20 6,15 75 5 234,85 40 1381,62 95 25 6,16 80 6 270,85 45 1526,02 100 30 6,18 85 7 305,78 50 1655,6 105 35 6,18 90 8 337,59 55 1794,82 110 40 6,19 95 9 369,38 60 1920,84 115 45 6,21 100 10 399,92 65 120 50 6,24 105
Tabelle & Abbildung A21: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 6, NaCl, WH 3
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 733,84 70 0 6,03 55 1 60,98 20 884,23 75 5 6,16 60 2 111,38 25 1051,34 80 10 6,21 65 3 163,83 30 1259,62 85 15 6,24 70 4 207,07 35 1444,11 90 20 6,27 75 5 249,02 40 1603,62 95 25 6,30 80 6 299,59 45 1777,93 100 30 6,31 85 7 356,11 48 1883,35 105 35 6,33 90 8 408,60 55 110 40 6,41 95 9 455,92 60 115 45 6,42 100 10 493,88 65 120 48 6,46 105
R² = 0,977
5,8
6,0
6,2
6,4
6,6
6,8
0
500
1000
1500
2000
0 10 20 30 40 50 60
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,991
6,0
6,2
6,4
0
500
1000
1500
2000
0 10 20 30 40 50 60 70
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,990
6,0
6,2
6,4
6,6
0
500
1000
1500
2000
0 10 20 30 40 50
pH
Per
mea
tmen
ge [
g]
Zeit [min]

Anhang
310
Tabelle & Abbildung A22: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 7, NaCl, WH 1
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 558,04 70 0 7,01 55 7,30 1 58,98 20 725,80 75 5 7,10 57 7,32 2 109,62 25 864,13 80 10 7,12 65 3 155,21 30 1037,77 85 15 7,14 70 4 194,82 35 1185,80 90 20 7,16 75 5 233,14 40 1322,62 95 25 7,18 80 6 272,10 45 1449,40 100 30 7,19 85 7 312,45 50 1570,84 105 35 7,20 90 8 348,50 55 1687,90 110 40 7,22 95 9 383,23 57 1740,58 115 45 7,24 100 10 414,47 65 120 50 7,27 105
Tabelle & Abbildung A23: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 7, NaCl, WH 2
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 515,08 70 0 7,01 55 7,11 1 55,52 20 671,12 75 5 7,07 60 7,11 2 100,41 25 812,63 80 10 7,08 64 7,12 3 139,01 30 944,78 85 15 7,09 70 4 175,88 35 1098,39 90 20 7,08 75 5 210,86 40 1239,82 95 25 7,08 80 6 246,73 45 1378,64 100 30 7,09 85 7 280,23 50 1514,01 105 35 7,08 90 8 314,04 55 1637,74 110 40 7,08 95 9 345,52 60 1757,49 115 45 7,10 100 10 376,36 64 1841,50 120 50 7,10 105
Tabelle & Abbildung A24: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 7, NaCl, WH 3
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 607,31 70 0 7,01 55 1 48,98 20 770,55 75 5 7,07 60 2 96,20 25 918,50 80 10 7,07 65 3 139,76 30 1095,9 85 15 7,07 70 4 179,91 35 1254,55 90 20 7,07 75 5 221,64 40 1407,94 95 25 7,08 80 6 263,97 45 1552,43 100 30 7,05 85 7 306,27 50 1680,82 105 35 7,07 90 8 348,80 55 110 40 7,08 95 9 387,02 60 115 45 7,08 100 10 420,75 65 120 50 7,10 105
R² = 0,986
6,8
7,0
7,2
7,4
0
500
1000
1500
2000
0 10 20 30 40 50 60
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,992
7,0
7,2
0
500
1000
1500
2000
0 10 20 30 40 50 60 70
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,991
7,0
7,2
0
500
1000
1500
2000
0 20 40 60
pH
Per
mea
tmen
ge [
g]
Zeit [min]

Anhang
311
Tabelle & Abbildung A25: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 8, NaCl, WH 1
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 691,86 70 0 8,02 55 1 58,87 20 824,21 75 5 8,01 60 2 119,92 25 1001,26 80 10 7,96 65 3 166,17 30 1159,17 85 15 7,93 70 4 209,01 35 1302,24 90 20 7,90 75 5 253,08 40 1440,47 95 25 7,89 80 6 295,41 45 1569,00 100 30 7,86 85 7 333,69 50 1600,13 105 35 7,84 90 8 377,90 55 110 40 7,81 95 9 421,65 60 115 45 7,79 100
10 468,10 65 120 46 7,78 105
Tabelle & Abbildung A26: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 8, NaCl, WH 2
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 526,58 70 1840,04 0 8,02 55 7,41 1 53,10 20 666,64 75 5 7,98 60 7,38 2 102,93 25 794,05 80 10 7,90 65 7,35 3 145,36 30 920,04 85 15 7,83 68 7,33 4 191,53 35 1047,52 90 20 7,75 75 5 225,48 40 1173,67 95 25 7,70 80 6 263,07 45 1294,93 100 30 7,63 85 7 296,06 50 1413,34 105 35 7,57 90 8 328,11 55 1528,19 110 40 7,52 95 9 359,29 60 1637,13 115 45 7,48 100 10 389,10 65 1751,73 120 50 7,45 105
Tabelle & Abbildung A27: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 8, NaCl, WH 3
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 702,82 70 0 8,03 55 1 54,51 20 895,52 75 5 7,97 60 2 110,68 25 1069,23 80 10 7,86 65 3 155,67 30 1269,51 85 15 7,79 70 4 202,84 35 1437,04 90 20 7,72 75 5 246,98 40 1603,45 95 25 7,65 80 6 300,09 45 1746,48 100 30 7,58 85 7 345,01 49 1860,56 105 35 7,53 90 8 395,23 55 110 40 7,47 95 9 443,42 60 115 45 7,44 100 10 488,97 65 120 49 7,41 105
R² = 0,980
7,6
7,8
8,0
8,2
0
500
1000
1500
2000
0 10 20 30 40 50
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,984
7,2
7,4
7,6
7,8
8,0
8,2
0
500
1000
1500
2000
0 10 20 30 40 50 60 70
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,989
7,2
7,4
7,6
7,8
8,0
8,2
0
500
1000
1500
2000
0 10 20 30 40 50
pH
Per
mea
tmen
ge [
g]
Zeit [min]

Anhang
312
Tabelle & Abbildung A28: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 11, NaCl, WH 1
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 470,84 70 1575,38 0 10,99 55 10,92 1 51,40 20 587,19 75 1664,03 5 11,00 60 10,91 2 99,24 25 694,00 80 1756,35 10 10,99 65 10,90 3 139,28 30 796,68 85 15 10,97 70 10,90 4 175,18 35 895,33 90 20 10,98 75 10,90 5 208,54 40 997,46 95 25 10,97 80 10,89 6 243,87 45 1098,22 100 30 10,95 85 7 270,52 50 1197,5 105 35 10,95 90 8 299,26 55 1295,28 110 40 10,95 95 9 326,84 60 1391,64 115 45 10,94 100 10 352,99 65 1484,18 120 50 10,93 105
Tabelle & Abbildung A29: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 11, NaCl, WH 2
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 610,79 70 0 11,00 55 1 57,99 20 767,61 75 5 11,02 60 2 117,40 25 912,43 80 10 11,00 65 3 169,85 30 1045,11 85 15 10,98 70 4 216,66 35 1167,41 90 20 10,97 75 5 262,86 40 1273,32 95 25 10,95 80 6 304,48 45 1378,94 100 30 10,95 85 7 342,50 47 1420,54 105 35 10,94 90 8 379,83 55 110 40 10,93 95 9 419,43 60 115 45 10,93 100
10 456,57 65 120 47 10,92 105
Tabelle & Abbildung A30: Versuchsdaten – Crossflow-Filtration, Alge (Chlorella vulgaris), pH 1, NaCl, WH 3
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 15 616,61 70 0 11,00 55 1 59,07 20 763,27 75 5 11,02 60 2 120,38 25 909,25 80 10 11,01 65 3 172,36 30 1043,63 85 15 10,98 70 4 219,47 35 1156,71 90 20 10,99 75 5 264,60 40 1258,65 95 25 10,95 80 6 305,12 45 1329,75 100 30 10,95 85 7 342,36 46 1381,03 105 35 10,94 90 8 377,86 55 110 40 10,94 95 9 410,37 60 115 45 10,94 100 10 452,39 65 120 46 10,94 105
R² = 0,978
10,8
11,0
11,2
0
500
1000
1500
2000
0 10 20 30 40 50 60 70 80 90
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,960
10,8
11,0
11,2
0
500
1000
1500
2000
0 10 20 30 40 50
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,954
10,8
11,0
11,2
0
500
1000
1500
2000
0 10 20 30 40 50
pH
Per
mea
tmen
ge [
g]
Zeit [min]

Anhang
313
Tabelle & Abbildung A31: Versuchsdaten – Crossflow-Filtration, Molken-Isolat, pH 7, US, WH 1
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 22 171,66 44 355,45 66 88 0 6,98 55 2 17,50 24 189,24 46 - 68 90 5 - 60 4 40,13 26 206,44 48 382,51 70 92 10 - 65 6 46,95 28 224,54 50 394,30 72 94 15 7,10 70 8 63,20 30 242,13 52 400,55 74 96 20 7,19 75
10 76,72 32 259,82 54 76 98 25 7,24 80 12 90,84 34 277,20 56 78 100 30 7,36 85 14 104,65 36 298,90 58 80 102 35 7,44 90 16 119,59 38 311,29 60 82 104 40 7,48 95 18 136,32 40 326,06 62 84 106 45 7,50 100 20 153,88 42 341,79 64 86 108 50 7,55 105
Tabelle & Abbildung A32: Versuchsdaten – Crossflow-Filtration, Molkenprotein-Isolat, pH 7, US, WH 2
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 22 112,17 44 213,1 66 290,54 88 362,44 0 7,00 55 7,53 2 18,13 24 121,5 46 217,33 68 297,52 90 368,71 5 7,12 60 7,55 4 29,20 26 130,2 48 225,07 70 303,01 92 374,38 10 7,11 65 7,58 6 38,35 28 139,39 50 232,49 72 311,33 94 380,32 15 7,16 70 7,61 8 48,17 30 147,95 52 240,05 74 317,9 96 386,42 20 7,22 75 7,65
10 57,46 32 157,37 54 247,27 76 324,98 98 392,37 25 7,26 80 7,69 12 66,51 34 164,76 56 255,35 78 331,26 100 400,25 30 7,28 85 7,70 14 74,80 36 173,33 58 261,92 80 337,71 102 35 7,31 90 7,75 16 84,00 38 181,79 60 268,82 82 344,1 104 40 7,38 95 7,78 18 92,10 40 190,42 62 276,62 84 350,54 106 45 7,42 100 7,86 20 101,54 42 202,29 64 283,53 86 357,07 108 50 7,44 105
Tabelle & Abbildung A33: Versuchsdaten – Crossflow-Filtration, Molkenprotein-Isolat, pH 7, US, WH 3
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 22 209,41 44 379,17 66 88 0 7,02 55 2 21,20 24 225,18 46 394,18 68 90 5 7,08 60 4 41,68 26 244,57 47 400,08 70 92 10 7,11 65 6 70,85 28 258,81 50 72 94 15 7,18 70 8 80,53 30 275,86 52 74 96 20 7,25 75
10 97,02 32 292,48 54 76 98 25 7,27 80 12 115,48 34 305,28 56 78 100 30 7,30 85 14 138,10 36 322,76 58 80 102 35 7,37 90 16 159,33 38 335,46 60 82 104 40 7,42 95 18 174,45 40 351,64 62 84 106 45 7,46 100 20 190,90 42 364,93 64 86 108 47 7,54 105
R² = 0,998
6,8
7,0
7,2
7,4
7,6
0
100
200
300
400
500
0 20 40 60
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,981
6,87,07,27,47,67,88,0
0
100
200
300
400
500
0 20 40 60 80 100
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,992
6,8
7,0
7,2
7,4
7,6
0
100
200
300
400
500
0 20 40 60
pH
Per
mea
tmen
ge [
g]
Zeit [min]

Anhang
314
Tabelle & Abbildung A34: Versuchsdaten – Crossflow-Filtration, Molkenprotein-Isolat, pH 7, US, WH 4
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 22 157,91 44 245,59 66 306,37 88 367,40 0 7,01 55 7,55 2 23,81 24 167,94 46 251,68 68 311,92 90 372,79 5 7,09 60 7,60 4 39,39 26 176,34 48 256,78 70 320,48 92 378,12 10 7,10 65 7,65 6 56,19 28 188,38 50 262,46 72 322,74 94 383,10 15 7,22 70 7,70 8 70,05 30 198,32 52 268,13 74 328,11 96 389,04 20 7,25 75 7,71
10 82,75 32 208,13 54 273,85 76 335,31 98 393,81 25 7,29 80 7,76 12 96,48 34 213,70 56 280,04 78 340,23 100 398,88 30 7,36 85 7,79 14 109,99 36 221,76 58 284,86 80 346,26 102 35 7,43 90 7,84 16 124,20 38 227,57 60 290,88 82 351,14 104 40 7,49 95 7,86 18 134,45 40 233,58 62 295,08 84 356,04 106 45 7,50 100 7,86 20 146,98 42 240,33 64 300,43 86 361,80 108 50 7,53 105
Tabelle & Abbildung A35: Versuchsdaten – Crossflow-Filtration, Molkenprotein-Isolat, pH 7, US, WH 5
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 22 172,16 44 298,50 66 88 0 7,00 55 7,58 2 24,73 24 184,55 46 306,91 68 90 5 7,07 60 7,67 4 41,68 26 198,57 48 314,90 70 92 10 7,13 62 7,74 6 55,17 28 210,28 50 334,13 72 94 15 7,23 70 8 72,43 30 222,16 52 343,78 74 96 20 7,27 75
10 87,10 32 233,85 54 354,45 76 98 25 7,35 80 12 104,25 34 244,51 56 365,15 78 100 30 7,37 85 14 119,26 36 256,25 58 374,35 80 102 35 7,37 90 16 135,44 38 266,45 60 383,93 82 104 40 7,43 95 18 147,18 40 276,99 62 393,32 84 106 45 7,49 100 20 158,73 42 288,29 64 86 108 50 7,54 105
Tabelle & Abbildung A36: Versuchsdaten – Crossflow-Filtration, Molkenprotein-Isolat, pH 7, US, WH 6
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 22 114,67 44 210,78 66 299,06 88 386,72 0 7,02 55 7,55 2 15,25 24 124,15 46 219,06 68 307,23 90 395,16 5 7,11 60 7,58 4 27,73 26 133,68 48 227,50 70 315,03 91 400,00 10 7,15 65 7,65 6 40,80 28 142,65 50 236,97 72 322,13 94 15 7,21 70 7,67 8 49,44 30 150,64 52 243,15 74 329,79 96 20 7,28 75 7,70
10 58,18 32 160,04 54 259,17 76 337,41 98 25 7,33 80 7,73 12 68,24 34 168,67 56 251,35 78 344,89 100 30 7,37 85 7,76 14 78,23 36 177,61 58 266,43 80 352,40 102 35 7,41 90 7,80 16 87,14 38 185,57 60 274,91 82 360,02 104 40 7,45 91 7,82 18 95,72 40 193,81 62 283,81 84 366,20 106 45 7,48 100 20 104,69 42 202,24 64 291,08 86 375,58 108 50 7,53 105
R² = 0,863
6,8
7,0
7,2
7,4
7,6
7,8
8,0
0
100
200
300
400
500
0 20 40 60 80 100
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,979
6,8
7,0
7,2
7,4
7,6
7,8
0
100
200
300
400
500
0 10 20 30 40 50 60 70
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,990
6,87,07,27,47,67,88,0
0
100
200
300
400
500
0 20 40 60 80 100
pH
Per
mea
tmen
ge [
g]
Zeit [min]
Anhang
315
Tabelle & Abbildung A37: Versuchsdaten – Crossflow-Filtration, Molkenprotein-Isolat, pH 7, OUS, WH 1
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 22 137,05 44 265,80 66 391,18 88 0 6,97 55 7,54 2 15,40 24 149,31 46 - 68 400,48 90 5 - 60 7,58 4 35,35 26 162,61 48 289,30 70 92 10 - 65 7,62 6 39,78 28 173,46 50 300,60 72 94 15 7,09 68 7,64 8 53,13 30 185,51 52 312,47 74 96 20 7,22 75
10 65,97 32 197,10 54 324,12 76 98 25 7,27 80 12 78,49 34 209,37 56 337,69 78 100 30 7,35 85 14 90,50 36 219,72 58 348,00 80 102 35 7,40 90 16 102,03 38 229,22 60 359,05 82 104 40 7,45 95 18 113,19 40 241,33 62 370,24 84 106 45 7,45 100 20 125,41 42 253,92 64 380,87 86 108 50 7,50 105
Tabelle & Abbildung A38: Versuchsdaten – Crossflow-Filtration, Molkenprotein-Isolat, pH 7, OUS, WH 2
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 22 130,9 44 249,82 66 342,15 88 0 6,97 55 7,57 2 16,50 24 141,8 46 254,27 68 350,73 90 5 7,08 60 7,58 4 29,90 26 152,48 48 263,6 70 358,84 92 10 7,11 65 7,64 6 41,13 28 162,59 50 272,88 72 367,45 94 15 7,13 70 7,69 8 53,40 30 172,83 52 281,81 74 375,6 96 20 7,20 75 7,70
10 65,77 32 183,63 54 290,85 76 383,8 98 25 7,25 80 7,81 12 76,63 34 192,78 56 299,73 78 391,58 100 30 7,30 85 14 87,50 36 202,66 58 308,23 80 399,62 102 35 7,32 90 16 98,50 38 212,24 60 316,83 82 104 40 7,36 95 18 109,02 40 222,24 62 325,85 84 106 45 7,40 100 20 119,70 42 236,96 64 334,17 86 108 50 7,44 105
Tabelle & Abbildung A39: Versuchsdaten – Crossflow-Filtration, Molkenprotein-Isolat, pH 7, OUS, WH 3
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 22 165,53 44 286,66 65 400,38 88 0 7,02 55 7,58 2 19,10 24 177,23 46 298,17 68 90 5 7,09 60 7,63 4 35,95 26 190,37 48 309,45 70 92 10 7,13 65 7,69 6 60,03 28 200,13 50 320,06 72 94 15 7,18 70 8 67,73 30 212,18 52 331,4 74 96 20 7,21 75
10 80,84 32 224,18 54 342,05 76 98 25 7,28 80 12 95,52 34 233,64 56 353,78 78 100 30 7,35 85 14 111,58 36 246,43 58 362,88 80 102 35 7,38 90 16 124,90 38 255,57 60 372,99 82 104 40 7,41 95 18 137,48 40 266,29 62 383,25 84 106 45 7,45 100 20 148,92 42 276,28 64 392,41 86 108 50 7,53 105
R² = 0,999
6,8
7,0
7,2
7,4
7,6
7,8
0
100
200
300
400
500
0 10 20 30 40 50 60 70
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,989
6,8
7,0
7,2
7,4
7,6
7,8
8,0
0
100
200
300
400
500
0 20 40 60 80 100
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,983
6,8
7,0
7,2
7,4
7,6
7,8
0
100
200
300
400
500
0 10 20 30 40 50 60 70
pH
Per
mea
tmen
ge [
g]
Zeit [min]

Anhang
316
Tabelle & Abbildung A40: Versuchsdaten – Crossflow-Filtration, Molkenprotein-Isolat, pH 7, OUS, WH 4
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 22 117,81 44 213,14 66 304,1 88 386,25 0 7,01 55 7,54 2 16,46 24 126,85 46 223,35 68 312,28 90 393,47 5 7,05 60 7,59 4 27,63 26 135,27 48 230,41 70 317,21 92 10 7,09 65 7,64 6 39,37 28 144,89 50 238,46 72 327,8 94 15 7,18 70 7,69 8 49,54 30 153,79 52 246,91 74 335,69 96 20 7,24 75 7,72
10 58,93 32 163,6 54 255,09 76 346,13 98 25 7,27 80 7,76 12 68,22 34 171,28 56 264,82 78 353,22 100 30 7,31 85 7,79 14 77,62 36 181,95 58 271,92 80 361,75 102 35 7,38 90 7,84 16 87,95 38 189,64 60 280,76 82 368,95 104 40 7,44 95 18 98,39 40 197,54 62 286,87 84 376,25 106 45 7,46 100 20 108,05 42 206,52 64 295,41 86 384,58 108 50 7,49 105
Tabelle & Abbildung A41: Versuchsdaten – Crossflow-Filtration, Molkenprotein-Isolat, pH 7, OUS, WH 5
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 22 94,60 44 179,98 66 267,80 88 341,98 0 7,00 60 7,53 2 13,19 24 102,07 46 187,56 68 275,92 90 347,92 5 7,06 65 7,57 4 21,64 26 110,80 48 194,92 70 282,86 92 354,48 10 7,10 70 7,63 6 30,28 28 117,71 50 209,20 72 290,84 94 360,10 15 7,18 75 7,66 8 38,02 30 126,82 52 216,42 74 296,98 96 366,60 20 7,21 80 7,68
10 45,28 32 134,75 54 224,46 76 304,07 98 372,38 25 7,27 85 7,72 12 53,86 34 142,19 56 232,60 78 310,45 100 378,23 30 7,35 90 7,73 14 62,63 36 150,29 58 239,28 80 316,52 102 384,39 35 7,38 95 7,76 16 72,20 38 157,60 60 246,08 82 323,05 104 390,21 40 7,44 100 7,79 18 79,53 40 164,69 62 253,09 84 329,24 106 396,14 45 7,47 105 7,83 20 86,48 42 172,77 64 260,76 86 335,89 108 50 7,48 106 7,85 55 7,49 115
R² = 0,990
6,8
7,0
7,2
7,4
7,6
7,8
8,0
0
100
200
300
400
500
0 20 40 60 80 100
pH
Per
mea
tmen
ge [
g]
Zeit [min]
R² = 0,994
6,8
7,0
7,2
7,4
7,6
7,8
8,0
0
100
200
300
400
500
0 20 40 60 80 100120
pH
Per
mea
tmen
ge [
g]
Zeit [min]

Anhang
317
Tabelle & Abbildung A42: Versuchsdaten – Crossflow-Filtration, Molkenprotein-Isolat, pH 7, OUS, WH 6
tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) VPer (ml) tF (min) pH tF (min) pH
0 0,00 26 99,15 52 196,24 78 277,3 104 365,86 0 7,02 60 7,54 2 12,77 28 105,67 54 189,66 80 284,04 106 371,51 5 7,09 65 7,56 4 21,62 30 112,56 56 202,13 82 289,13 108 377,75 10 7,13 70 7,57 6 30,08 32 119,46 58 210,01 84 296,15 110 383,52 15 7,18 75 7,58 8 37,57 34 126,68 60 217,06 86 306,84 112 389,21 20 7,22 80 7,60
10 44,43 36 133,04 62 224,06 88 314,33 114 394,49 25 7,31 85 7,62 12 51,48 38 139,01 64 230,88 90 321,63 116 30 7,38 90 7,63 14 58,86 40 146,87 66 237,78 92 330,25 118 35 7,42 95 7,66 16 65,75 42 154,69 68 244,39 94 336,03 120 40 7,47 100 7,71 18 72,37 44 161,88 70 251,14 96 341,99 122 45 7,48 105 7,72 20 79,14 46 169,11 72 257,35 98 348,01 124 50 7,50 110 7,73 22 84,56 48 177,25 74 263,86 100 353,89 126 55 7,52 114 7,76
24 92,15 50 182,58 76 270,73 102 359,88 128
R² = 0,998
6,8
7,0
7,2
7,4
7,6
7,8
8,0
0
100
200
300
400
500
0 20 40 60 80 100120
pH
Per
mea
tmen
ge [
g]
Zeit [min]

Anhang
318
Dialyse - Versuchsdaten
Tabelle A13: Versuchsdaten – Dialyse von Molkenprotein-Isolat
Probenname WH pH CF (%) tDL (h) VD yEx-vD CS-vD (mg/ml) mvD (mg) mvD MW (mg) VnDL (ml) yEx-nD CS-nD (mg/ml) mnD (mg) mnD MW (mg)
WPI - pH 3 1 3 1 24 -
-
-
-
-
-
0,814 1,078 24,79
24,57 ± 0,29
20 0,917 1,214 24,28
23,46 ± 0,80 WPI - pH 3 2 3 1 24 - 0,796 1,054 24,24 20 0,857 1,135 22,70
WPI - pH 3 3 3 1 24 - 0,810 1,073 24,67 20 0,883 1,169 23,38
WPI - pH 5 1 5 1 24 - 0,666 0,882 20,28
19,85 ± 0,38
20 0,379 0,502 10,04
9,92 ± 0,27 WPI - pH 5 2 5 1 24 - 0,646 0,855 19,67 20 0,363 0,481 9,61
WPI - pH 5 3 5 1 24 - 0,643 0,851 19,58 20 0,382 0,506 10,12
WPI - pH 6 1 6 1 24 - 0,884 1,171 26,92
26,98 ± 0,73
20 0,617 0,817 16,34
16,77 ± 0,38 WPI - pH 6 2 6 1 24 - 0,911 1,206 27,74 20 0,638 0,845 16,90
WPI - pH 6 3 6 1 24 - 0,863 1,143 26,28 20,5 0,629 0,833 17,07
WPI - pH 7,5 1 7,5 1 24 - 0,865 1,145 26,34
26,02 ± 0,49
20 0,618 0,818 16,37
16,39 ± 0,74 WPI - pH 7,5 2 7,5 1 24 - 0,862 1,141 26,25 20 0,647 0,857 17,13
WPI - pH 7,5 3 7,5 1 24 - 0,836 1,107 25,46 20,5 0,577 0,764 15,66
WPI - pH 9 1 9 1 24 - 0,860 1,139 26,19
26,61 ± 0,50
20 0,59 0,781 15,63
15,62 ± 0,57 WPI - pH 9 2 9 1 24 - 0,892 1,181 27,17 20,5 0,596 0,789 16,18
WPI - pH 9 3 9 1 24 - 0,869 1,151 26,47 20 0,568 0,752 15,04
WPI - pH 11 1 11 1 24 - 0,919 1,217 27,99
28,32 ± 1,06
20,5 0,564 0,747 15,31
15,52 ± 0,36 WPI - pH 11 2 11 1 24 - 0,902 1,194 27,47 21 0,551 0,730 15,32
WPI - pH 11 3 11 1 24 - 0,969 1,283 29,51 20 0,602 0,797 15,94
WPI - pH 9 1 9 2 24 - 1,701 2,252 51,80
51,88 ± 0,39
22,5 0,779 1,032 23,21
21,54 ± 1,71 WPI - pH 9 2 9 2 24 - 1,717 2,274 52,29 22,5 0,726 0,961 21,63
WPI - pH 9 3 9 2 24 - 1,692 2,240 51,53 22,5 0,664 0,879 19,78
WPI - pH 11 1 11 2 24 - 1,744 2,309 53,11
53,02 ± 0,13
22,5 0,915 1,212 27,26
27,37 ± 1,04 WPI - pH 11 2 11 2 24 - 1,736 2,299 52,87 22,5 0,955 1,265 28,45
WPI - pH 11 3 11 2 24 - 1,743 2,308 53,08 21 0,949 1,257 26,39
WPI - pH 9 1 9 2 48 - 1,590 2,105 48,42
47,32 ± 1,27
20,5 0,461 0,610 12,51
13,29 ± 0,68 WPI - pH 9 2 9 2 48 - 1,563 2,070 47,60 20 0,512 0,678 13,56
WPI - pH 9 3 9 2 48 - 1,508 1,997 45,93 20 0,521 0,690 13,80
WPI - pH 11 1 11 2 48 - 1,627 2,154 49,55
48,02 ± 1,48
20,5 0,759 1,005 20,60
18,97 ± 1,41 WPI - pH 11 2 11 2 48 - 1,573 2,083 47,91 20,5 0,673 0,891 18,27
WPI - pH 11 3 11 2 48 - 1,530 2,026 46,60 20,5 0,665 0,881 18,05
WPI - pH 9 1 9 4 24 1:5 0,584 3,867 92,80
92,53 ± 0,24
22,5 1,898 2,513 56,55
53,14 ± 2,97 WPI - pH 9 2 9 4 24 1:5 0,582 3,853 92,48 23 1,678 2,222 51,10
WPI - pH 9 3 9 4 24 1:5 0,581 3,847 92,32 23 1,700 2,251 51,77
WPI - pH 11 1 11 4 24 1:5 0,63 4,171 100,11
99,42 ± 0,72
23 1,525 2,019 46,44
47,85 ± 1,22 WPI - pH 11 2 11 4 24 1:5 0,621 4,111 98,68 23,5 1,561 2,067 48,57
WPI - pH 11 3 11 4 24 1:5 0,626 4,145 99,47 24,5 1,496 1,981 48,53
Legende: WH, Wiederholung. CF, Feed-Konzentration. tDL, Dauer der Dialyse. VD, Verdünnungsfaktor. yEx-vD, Extinktion Feed vor Dialyse. CS-vD, Spektralphotometrisch ermittelte Konzentration des Feeds. mvD, absolute Proteinmenge Feed. MW, arithmetischer Mittelwert. VnDL, Retentat Volumen nach Dialyse.yEx-nD, Extinktion Retentat nach Dilayse.CS-nD, Spektralphotometrisch ermittelte Konzentration des Retentats nach Dialyse. mnD, absolute Proteinmenge Retentat nach Dilayse.

Anhang
319
Hydrolyse - Versuchsdaten
Tabelle A14: Versuchsdaten – Hydrolyse der Alge (Chlorella vulgaris)
Probenname WH pH tEVtSV (min) tD (h) TOH (°C) DH (%) DH MW (%)
HCl 18 h + Pancreatin 45 min 1 8 45 18 40 10,25
10,29 ± 0,97 HCl 18 h + Pancreatin 45 min 2 8 45 18 40 9,34
HCl 18 h + Pancreatin 45 min 3 8 45 18 40 11,28
HCl 48 h + Pancreatin 45 min 1 8 45 48 40 12,54
12,76 ± 1,12 HCl 48 h + Pancreatin 45 min 2 8 45 48 40 11,68
HCl 48 h + Pancreatin 45 min 3 8 45 48 40 14,07
Rohament 18 h + Pancreatin 45 min 1 8 45 18 40 16,49
15,32 ± 1,19 Rohament 18 h + Pancreatin 45 min 2 8 45 18 40 14,10
Rohament 18 h + Pancreatin 45 min 3 8 45 18 40 15,38
Rohament 48 h + Pancreatin 45 min 1 8 45 48 40 21,61
20,14 ± 1,56 Rohament 48 h + Pancreatin 45 min 2 8 45 48 40 20,29
Rohament 48 h + Pancreatin 45 min 3 8 45 48 40 18,51
Rohament 72 h + Pancreatin 45 min 1 8 45 72 40 19,74
19,81 ± 0,10 Rohament 72 h + Pancreatin 45 min 2 8 45 72 40 19,88
Rohament 72 h + Pancreatin 45 min 3 8 45 72 40 -3,93*
HCl 18 h + Pronase 45 min 1 8 45 18 40 14,77
17,04 ± 2,83 HCl 18 h + Pronase 45 min 2 8 45 18 40 16,13
HCl 18 h + Pronase 45 min 3 8 45 18 40 20,21
HCl 48 h + Pronase 45 min 1 8 45 48 40 15,32
18,16 ± 2,59 HCl 48 h + Pronase 45 min 2 8 45 48 40 20,41
HCl 48 h + Pronase 45 min 3 8 45 48 40 18,73
Rohament 18 h + Pronase 45 min 1 8 45 18 40 11,63
14,63 ± 2,73 Rohament 18 h + Pronase 45 min 2 8 45 18 40 16,96
Rohament 18 h + Pronase 45 min 3 8 45 18 40 15,30
Rohament 48 h + Pronase 45 min 1 8 45 48 40 20,21
21,39 ± 1,77 Rohament 48 h + Pronase 45 min 2 8 45 48 40 23,42
Rohament 48 h + Pronase 45 min 3 8 45 48 40 20,55
Rohament 72 h + Pronase 45 min 1 8 45 72 40 16,89
19,01 ± 2,99 Rohament 72 h + Pronase 45 min 2 8 45 72 40 21,12
Rohament 72 h + Pronase 45 min 3 8 45 72 40 0,10*
Legende: WH, Wiederholung. tE, Dauer der Enzymeinwirkung. TEV/tSV, Dauer der Vorbehandlung. TOH, Temperaturoptimum des Enzyms der Hydrolyse. DH, Hydrolysegrad. * Ausschluss eines Messwerts aufgrund fehlerhafter Temperaturführung während des Vorbehandlungsprozesses.

Anhang
320
Sensorik - Versuchsdaten
Tabelle A15: Versuchsdaten – Überprüfung auf Geschmacksblindheit
Teilnehmer GSA Anzahl Proben
erkannt nicht
erkannt Teilnehmer GSA
Anzahl Proben
erkannt nicht
erkannt
0
süß 3 2 1
12
süß 3 3 0
sauer 1 1 0 sauer 1 1 0
salzig 1 1 0 salzig 1 1 0
bitter 3 0 3 bitter 3 3 0
umami 2 0 2 umami 2 2 0
1
süß 3 3 0
13
süß 3 3 0
sauer 1 1 0 sauer 1 1 0
salzig 1 1 0 salzig 1 1 0
bitter 3 2 1 bitter 3 2 1
umami 2 2 0 umami 2 1 1
2
süß 3 3 0
14
süß 3 3 0
sauer 1 1 0 sauer 1 1 0
salzig 1 1 0 salzig 1 1 0
bitter 3 3 0 bitter 3 3 0
umami 2 2 0 umami 2 2 0
3
süß 3 3 0
15
süß 3 3 0
sauer 1 1 0 sauer 1 0 1
salzig 1 1 0 salzig 1 1 0
bitter 3 3 0 bitter 3 2 1
umami 2 2 0 umami 2 2 0
4
süß 3 3 0
16
süß 3 3 0
sauer 1 1 0 sauer 1 1 0
salzig 1 1 0 salzig 1 1 0
bitter 3 1 2 bitter 3 2 1
umami 2 2 0 umami 2 2 0
5
süß 3 3 0
17
süß 3 3 0
sauer 1 1 0 sauer 1 1 0
salzig 1 1 0 salzig 1 1 0
bitter 3 3 0 bitter 3 3 0
umami 2 2 0 umami 2 2 0
6
süß 3 2 1
18
süß 3 3 0
sauer 1 1 0 sauer 1 1 0
salzig 1 1 0 salzig 1 1 0
bitter 3 3 0 bitter 3 3 0
umami 2 2 0 umami 2 2 0
7
süß 3 3 0
19
süß 3 3 0
sauer 1 1 0 sauer 1 1 0
salzig 1 0 1 salzig 1 1 0
bitter 3 2 1 bitter 3 3 0
umami 2 1 1 umami 2 2 0
8
süß 3 3 0
20
süß 3 3 0
sauer 1 1 0 sauer 1 1 0
salzig 1 1 0 salzig 1 1 0
bitter 3 3 0 bitter 3 3 0
umami 2 2 0 umami 2 2 0
9
süß 3 3 0
21
süß 3 3 0
sauer 1 1 0 sauer 1 1 0
salzig 1 1 0 salzig 1 1 0
bitter 3 2 1 bitter 3 3 0
umami 2 2 0 umami 2 2 0
10
süß 3 3 0
22
süß 3 3 0
sauer 1 1 0 sauer 1 1 0
salzig 1 1 0 salzig 1 1 0
bitter 3 3 0 bitter 3 3 0
umami 2 2 0 umami 2 2 0
11
süß 3 3 0
23
süß 3 3 0
sauer 1 1 0 sauer 1 1 0
salzig 1 1 0 salzig 1 1 0
bitter 3 3 0 bitter 3 3 0
umami 2 2 0 umami 2 2 0
Legende: GSA, Geschmacksausprägung.
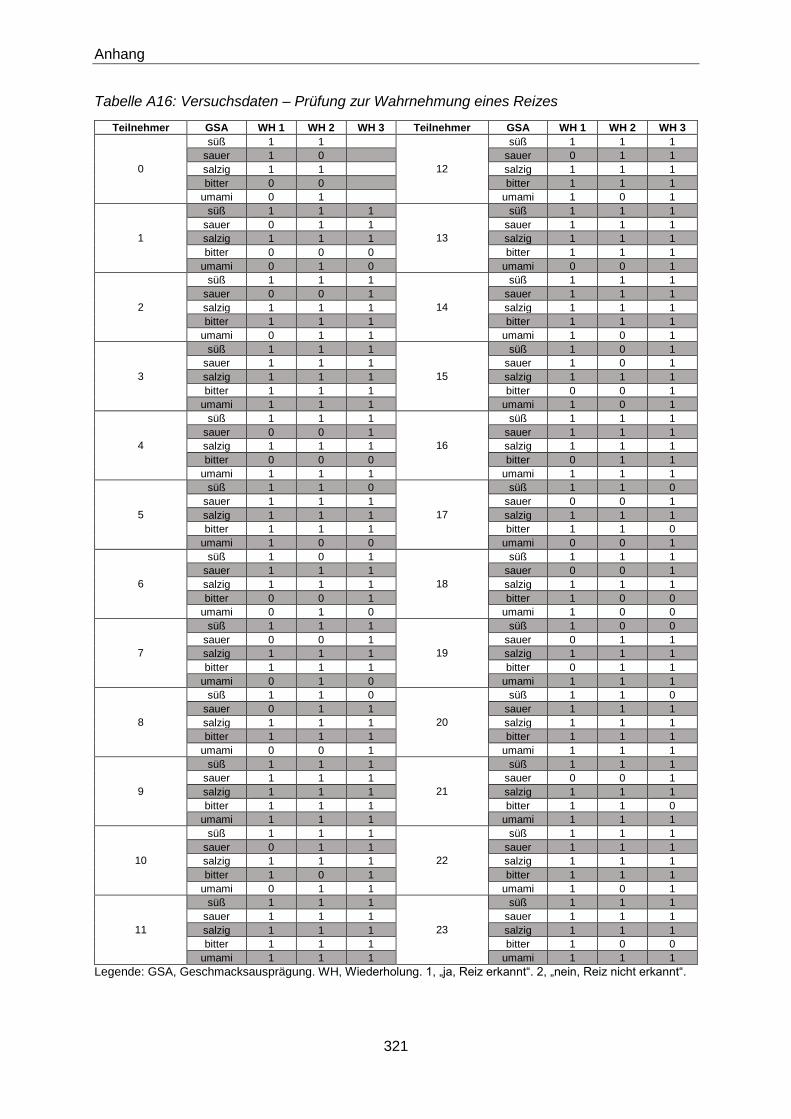
Anhang
321
Tabelle A16: Versuchsdaten – Prüfung zur Wahrnehmung eines Reizes
Teilnehmer GSA WH 1 WH 2 WH 3 Teilnehmer GSA WH 1 WH 2 WH 3
0
süß 1 1
12
süß 1 1 1
sauer 1 0 sauer 0 1 1
salzig 1 1 salzig 1 1 1
bitter 0 0 bitter 1 1 1
umami 0 1 umami 1 0 1
1
süß 1 1 1
13
süß 1 1 1
sauer 0 1 1 sauer 1 1 1
salzig 1 1 1 salzig 1 1 1
bitter 0 0 0 bitter 1 1 1
umami 0 1 0 umami 0 0 1
2
süß 1 1 1
14
süß 1 1 1
sauer 0 0 1 sauer 1 1 1
salzig 1 1 1 salzig 1 1 1
bitter 1 1 1 bitter 1 1 1
umami 0 1 1 umami 1 0 1
3
süß 1 1 1
15
süß 1 0 1
sauer 1 1 1 sauer 1 0 1
salzig 1 1 1 salzig 1 1 1
bitter 1 1 1 bitter 0 0 1
umami 1 1 1 umami 1 0 1
4
süß 1 1 1
16
süß 1 1 1
sauer 0 0 1 sauer 1 1 1
salzig 1 1 1 salzig 1 1 1
bitter 0 0 0 bitter 0 1 1
umami 1 1 1 umami 1 1 1
5
süß 1 1 0
17
süß 1 1 0
sauer 1 1 1 sauer 0 0 1
salzig 1 1 1 salzig 1 1 1
bitter 1 1 1 bitter 1 1 0
umami 1 0 0 umami 0 0 1
6
süß 1 0 1
18
süß 1 1 1
sauer 1 1 1 sauer 0 0 1
salzig 1 1 1 salzig 1 1 1
bitter 0 0 1 bitter 1 0 0
umami 0 1 0 umami 1 0 0
7
süß 1 1 1
19
süß 1 0 0
sauer 0 0 1 sauer 0 1 1
salzig 1 1 1 salzig 1 1 1
bitter 1 1 1 bitter 0 1 1
umami 0 1 0 umami 1 1 1
8
süß 1 1 0
20
süß 1 1 0
sauer 0 1 1 sauer 1 1 1
salzig 1 1 1 salzig 1 1 1
bitter 1 1 1 bitter 1 1 1
umami 0 0 1 umami 1 1 1
9
süß 1 1 1
21
süß 1 1 1
sauer 1 1 1 sauer 0 0 1
salzig 1 1 1 salzig 1 1 1
bitter 1 1 1 bitter 1 1 0
umami 1 1 1 umami 1 1 1
10
süß 1 1 1
22
süß 1 1 1
sauer 0 1 1 sauer 1 1 1
salzig 1 1 1 salzig 1 1 1
bitter 1 0 1 bitter 1 1 1
umami 0 1 1 umami 1 0 1
11
süß 1 1 1
23
süß 1 1 1
sauer 1 1 1 sauer 1 1 1
salzig 1 1 1 salzig 1 1 1
bitter 1 1 1 bitter 1 0 0
umami 1 1 1 umami 1 1 1
Legende: GSA, Geschmacksausprägung. WH, Wiederholung. 1, „ja, Reiz erkannt“. 2, „nein, Reiz nicht erkannt“.

Anhang
322
Tabelle A17: Versuchsdaten – Ermittlung der Reiz- & Erkennungsschwelle
WH 1 WH 2 WH 3 Teilnehmer GSA H2O D8 D7 D6 D5 D4 D3 D2 D1 H2O D8 D7 D6 D5 D4 D3 D2 D1 H2O D8 D7 D6 D5 D4 D3 D2 D1
0
süß 1 1 2 3 3 4 5 6 7 0 0 0 0 1 2 2 3 4
sauer 1 1 1 2 3 4 5 6 7 0 0 0 1 2 3 3 4 4
salzig 2 2 3 3 3 4 5 6 7 1 1 1 1 1 1 1 2 3
bitter 1 1 1 2 3 3 4 5 5 0 0 2 2 2 2 2 3 3
umami 0 0 0 0 0 0 0 0 0 0 0 0 1 1 2 3 3 3
1
süß 1 2 2 2 2 3 3 4 5 0 0 0 0 0 2 3 4 5 0 0 1 1 1 2 3 4 5
sauer 2 2 3 4 5 6 7 8 9 0 0 2 3 4 5 6 7 8 2 3 4 4 5 5 6 7 8
salzig 1 2 3 3 4 5 6 7 8 0 2 2 2 3 4 5 6 7 0 2 3 4 5 6 7 8 9
bitter 1 1 2 2 3 3 3 4 5 1 2 3 3 3 4 5 5 6 0 2 2 3 4 4 5 6 7
umami 1 1 1 1 2 3 4 5 6 0 0 0 1 2 3 4 5 6 0 0 0 0 2 3 4 5 6
2
süß 1 2 3 3 4 4 5 6 6 0 1 1 2 3 4 4 5 6 0 0 0 0 0 0 0 2 2
sauer 1 1 1 2 2 3 3 4 5 0 1 1 2 3 4 5 5 6 2 3 4 5 6 6 7 8 9
salzig 0 1 1 2 3 4 4 5 6 1 1 1 2 2 3 4 4 5 0 2 2 3 3 4 5 5 6
bitter 1 2 2 3 4 4 4 5 5 1 2 3 3 4 4 4 5 6 0 1 2 3 3 4 5 6 7
umami 0 2 3 4 4 5 6 7 9 1 2 3 4 4 5 6 6 7 0 1 2 3 4 5 5 6 7
3
süß 0 0 0 0 1 1 2 3 4 0 0 0 0 0 2 3 3 4 0 1 1 2 3 4 5 6 6
sauer 0 1 1 2 3 4 5 6 7 0 0 0 2 2 3 4 5 6 0 2 3 4 4 5 5 6 6
salzig 0 0 1 1 2 3 4 5 6 0 0 0 1 1 2 2 3 4 0 0 1 2 2 3 4 5 6
bitter 0 1 2 3 4 4 5 6 7 0 2 3 4 4 4 5 6 6 1 2 2 3 3 4 5 6 6
umami 0 1 2 3 3 4 4 5 5 0 2 3 3 4 4 5 5 6 0 2 3 4 4 5 5 5 6
4
süß 0 0 0 0 1 2 3 4 5 0 0 0 0 0 0 1 2 3 0 0 1 1 1 2 3 4 5
sauer 0 0 0 1 2 3 3 4 4 0 0 0 1 2 3 4 4 4 0 2 2 3 4 4 5 5 6
salzig 0 0 1 1 2 2 2 3 4 0 0 1 1 2 2 3 3 4 0 0 1 2 2 3 3 4 5
bitter 0 0 0 0 1 1 1 2 2 0 0 0 1 2 2 2 3 4 0 0 0 0 1 1 2 2 3
umami 0 0 0 1 2 2 3 4 5 0 0 0 1 2 3 3 4 5 0 0 1 2 2 3 4 4 5
5
süß 0 0 1 1 1 1 2 3 4 0 0 0 2 2 3 4 4 5 0 1 1 1 2 2 2 3 4
sauer 0 0 2 2 3 4 5 6 7 0 1 2 3 4 5 6 7 8 1 2 3 4 5 6 7 8 9
salzig 0 0 1 2 2 3 4 5 6 0 1 1 2 3 4 5 6 7 1 1 1 2 3 4 5 6 7
bitter 1 1 2 2 2 3 3 4 4 0 0 1 2 3 4 4 5 6 0 0 2 2 3 4 5 6 7
umami 1 2 3 4 5 6 6 7 8 0 2 2 3 4 5 6 7 8 0 2 3 4 5 6 7 8 9
6
süß 0 1 1 1 1 1 1 2 3 0 1 1 1 1 1 1 1 2 0 0 1 1 1 1 1 2 3
sauer 0 1 1 2 3 4 5 6 7 1 1 2 3 4 5 5 5 6 0 2 2 3 3 3 4 4 4
salzig 0 2 2 2 3 4 4 4 5 0 2 2 3 4 5 6 7 8 0 1 1 1 1 2 3 4 5
bitter 0 0 0 1 1 1 1 1 2 0 0 0 2 2 3 4 4 4 0 0 0 0 1 1 1 1 2
umami 0 0 0 1 1 1 1 1 2 0 0 0 0 1 1 2 2 3 0 0 1 1 1 1 1 1 2
7
süß 0 0 2 2 2 3 3 4 4 0 0 1 2 2 2 3 4 5 1 1 1 2 2 3 4 5 6
sauer 0 1 2 3 4 5 6 7 8 0 1 2 3 4 5 6 7 8 0 2 3 3 4 5 6 7 7
salzig 0 1 2 2 2 3 4 5 6 1 2 3 4 5 6 7 8 9 0 2 3 4 5 6 7 8 9
bitter 1 1 2 2 3 3 4 5 6 2 3 4 5 5 6 7 8 9 0 2 3 4 5 6 7 8 9
umami 1 2 3 4 4 5 6 7 8 0 0 2 3 4 5 6 7 8 0 0 2 3 4 5 6 7 8
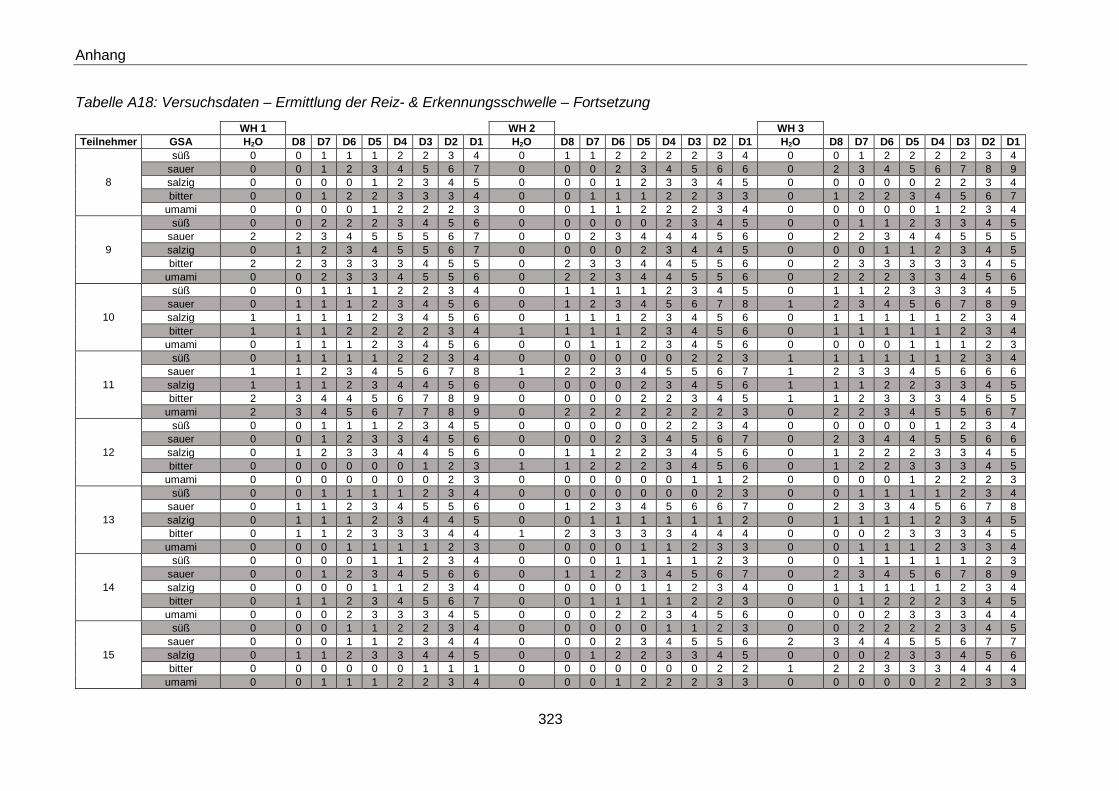
Anhang
323
Tabelle A18: Versuchsdaten – Ermittlung der Reiz- & Erkennungsschwelle – Fortsetzung
WH 1 WH 2 WH 3
Teilnehmer GSA H2O D8 D7 D6 D5 D4 D3 D2 D1 H2O D8 D7 D6 D5 D4 D3 D2 D1 H2O D8 D7 D6 D5 D4 D3 D2 D1
8
süß 0 0 1 1 1 2 2 3 4 0 1 1 2 2 2 2 3 4 0 0 1 2 2 2 2 3 4
sauer 0 0 1 2 3 4 5 6 7 0 0 0 2 3 4 5 6 6 0 2 3 4 5 6 7 8 9
salzig 0 0 0 0 1 2 3 4 5 0 0 0 1 2 3 3 4 5 0 0 0 0 0 2 2 3 4
bitter 0 0 1 2 2 3 3 3 4 0 0 1 1 1 2 2 3 3 0 1 2 2 3 4 5 6 7
umami 0 0 0 0 1 2 2 2 3 0 0 1 1 2 2 2 3 4 0 0 0 0 0 1 2 3 4
9
süß 0 0 2 2 2 3 4 5 6 0 0 0 0 0 2 3 4 5 0 0 1 1 2 3 3 4 5
sauer 2 2 3 4 5 5 5 6 7 0 0 2 3 4 4 4 5 6 0 2 2 3 4 4 5 5 5
salzig 0 1 2 3 4 5 5 6 7 0 0 0 0 2 3 4 4 5 0 0 0 1 1 2 3 4 5
bitter 2 2 3 3 3 3 4 5 5 0 2 3 3 4 4 5 5 6 0 2 3 3 3 3 3 4 5
umami 0 0 2 3 3 4 5 5 6 0 2 2 3 4 4 5 5 6 0 2 2 2 3 3 4 5 6
10
süß 0 0 1 1 1 2 2 3 4 0 1 1 1 1 2 3 4 5 0 1 1 2 3 3 3 4 5
sauer 0 1 1 1 2 3 4 5 6 0 1 2 3 4 5 6 7 8 1 2 3 4 5 6 7 8 9
salzig 1 1 1 1 2 3 4 5 6 0 1 1 1 2 3 4 5 6 0 1 1 1 1 1 2 3 4
bitter 1 1 1 2 2 2 2 3 4 1 1 1 1 2 3 4 5 6 0 1 1 1 1 1 2 3 4
umami 0 1 1 1 2 3 4 5 6 0 0 1 1 2 3 4 5 6 0 0 0 0 1 1 1 2 3
11
süß 0 1 1 1 1 2 2 3 4 0 0 0 0 0 0 2 2 3 1 1 1 1 1 1 2 3 4
sauer 1 1 2 3 4 5 6 7 8 1 2 2 3 4 5 5 6 7 1 2 3 3 4 5 6 6 6
salzig 1 1 1 2 3 4 4 5 6 0 0 0 0 2 3 4 5 6 1 1 1 2 2 3 3 4 5
bitter 2 3 4 4 5 6 7 8 9 0 0 0 0 2 2 3 4 5 1 1 2 3 3 3 4 5 5
umami 2 3 4 5 6 7 7 8 9 0 2 2 2 2 2 2 2 3 0 2 2 3 4 5 5 6 7
12
süß 0 0 1 1 1 2 3 4 5 0 0 0 0 0 2 2 3 4 0 0 0 0 0 1 2 3 4
sauer 0 0 1 2 3 3 4 5 6 0 0 0 2 3 4 5 6 7 0 2 3 4 4 5 5 6 6
salzig 0 1 2 3 3 4 4 5 6 0 1 1 2 2 3 4 5 6 0 1 2 2 2 3 3 4 5
bitter 0 0 0 0 0 0 1 2 3 1 1 2 2 2 3 4 5 6 0 1 2 2 3 3 3 4 5
umami 0 0 0 0 0 0 0 2 3 0 0 0 0 0 0 1 1 2 0 0 0 0 1 2 2 2 3
13
süß 0 0 1 1 1 1 2 3 4 0 0 0 0 0 0 0 2 3 0 0 1 1 1 1 2 3 4
sauer 0 1 1 2 3 4 5 5 6 0 1 2 3 4 5 6 6 7 0 2 3 3 4 5 6 7 8
salzig 0 1 1 1 2 3 4 4 5 0 0 1 1 1 1 1 1 2 0 1 1 1 1 2 3 4 5
bitter 0 1 1 2 3 3 3 4 4 1 2 3 3 3 3 4 4 4 0 0 0 2 3 3 3 4 5
umami 0 0 0 1 1 1 1 2 3 0 0 0 0 1 1 2 3 3 0 0 1 1 1 2 3 3 4
14
süß 0 0 0 0 1 1 2 3 4 0 0 0 1 1 1 1 2 3 0 0 1 1 1 1 1 2 3
sauer 0 0 1 2 3 4 5 6 6 0 1 1 2 3 4 5 6 7 0 2 3 4 5 6 7 8 9
salzig 0 0 0 0 1 1 2 3 4 0 0 0 0 1 1 2 3 4 0 1 1 1 1 1 2 3 4
bitter 0 1 1 2 3 4 5 6 7 0 0 1 1 1 1 2 2 3 0 0 1 2 2 2 3 4 5
umami 0 0 0 2 3 3 3 4 5 0 0 0 2 2 3 4 5 6 0 0 0 2 3 3 3 4 4
15
süß 0 0 0 1 1 2 2 3 4 0 0 0 0 0 1 1 2 3 0 0 2 2 2 2 3 4 5
sauer 0 0 0 1 1 2 3 4 4 0 0 0 2 3 4 5 5 6 2 3 4 4 5 5 6 7 7
salzig 0 1 1 2 3 3 4 4 5 0 0 1 2 2 3 3 4 5 0 0 0 2 3 3 4 5 6
bitter 0 0 0 0 0 0 1 1 1 0 0 0 0 0 0 0 2 2 1 2 2 3 3 3 4 4 4
umami 0 0 1 1 1 2 2 3 4 0 0 0 1 2 2 2 3 3 0 0 0 0 0 2 2 3 3

Anhang
324
Tabelle A19: Versuchsdaten – Ermittlung der Reiz- & Erkennungsschwelle – Fortsetzung
WH 1 WH 2 WH 3
Teilnehmer GSA H2O D8 D7 D6 D5 D4 D3 D2 D1 H2O D8 D7 D6 D5 D4 D3 D2 D1 H2O D8 D7 D6 D5 D4 D3 D2 D1
16
süß 0 0 0 0 1 2 2 3 4 0 0 0 0 2 2 2 3 4 0 0 0 1 1 1 2 3 4
sauer 0 0 2 2 3 3 4 4 5 0 0 2 2 3 4 5 6 7 0 2 2 3 4 5 5 5 6
salzig 0 2 2 2 3 3 3 4 5 0 0 0 1 1 1 1 1 1 0 0 0 1 1 2 2 3 4
bitter 0 0 0 0 0 0 2 2 2 0 0 0 2 2 3 3 3 3 0 2 3 3 4 4 4 4 5
umami 0 0 0 0 2 2 3 3 4 0 0 0 0 1 1 1 1 1 0 0 1 1 1 2 2 3 4
17
süß 0 0 0 0 2 2 3 3 4 0 0 0 0 2 3 3 4 5 0 0 0 2 2 2 3 3 4
sauer 0 0 2 3 4 4 5 6 6 0 0 0 2 3 4 5 6 7 2 3 3 4 5 5 6 7 8
salzig 0 0 0 2 3 3 4 5 6 0 0 0 0 2 3 4 5 6 1 2 2 2 3 3 4 5 6
bitter 0 2 2 2 2 3 3 3 4 0 0 0 0 2 2 3 3 4 0 2 2 3 4 5 6 7 8
umami 0 0 2 2 3 4 5 6 7 0 0 2 3 4 5 5 6 7 0 0 0 0 1 2 3 4 5
18
süß 1 1 1 1 1 1 2 3 4 0 0 1 1 1 1 1 2 3 1 1 1 1 1 1 2 3 4
sauer 0 0 1 1 1 1 1 1 1 0 1 1 2 3 3 4 4 5 0 2 2 3 3 3 4 4 6
salzig 0 2 2 3 3 4 5 5 6 0 1 1 2 2 3 3 4 5 0 0 1 2 3 3 4 4 5
bitter 0 1 1 1 2 2 3 3 4 0 0 0 1 1 1 1 2 2 0 1 2 3 3 3 4 5 5
umami 0 1 1 2 2 3 4 5 5 0 1 1 1 1 1 1 1 1 1 1 2 3 3 3 4 5 5
19
süß 0 1 1 2 2 2 2 3 4 0 0 1 1 2 3 3 4 5 0 1 1 1 1 1 1 2 3
sauer 0 0 1 1 2 3 4 4 4 0 0 1 2 3 4 5 6 7 1 2 2 3 4 4 4 5 5
salzig 0 1 2 2 3 3 3 4 4 0 2 2 3 4 4 5 5 6 0 2 2 2 3 3 4 4 4
bitter 0 1 1 2 3 3 3 4 5 1 1 2 3 4 4 5 5 6 0 2 2 3 4 4 4 5 5
umami 0 2 2 2 2 3 4 4 4 0 0 1 2 2 3 4 4 5 0 0 1 1 2 3 4 4 5
20
süß 0 0 0 1 1 2 3 3 4 0 1 1 2 2 2 2 3 4 0 1 1 1 2 2 2 3 3
sauer 0 0 1 2 2 3 3 4 4 0 2 2 3 3 3 4 4 4 0 2 3 3 4 4 4 5 5
salzig 1 1 1 1 1 2 2 3 4 0 1 2 2 3 3 4 5 6 0 1 1 1 2 3 4 4 5
bitter 0 2 3 3 3 3 4 5 5 0 1 2 2 2 3 3 4 5 0 0 1 2 2 3 4 5 5
umami 1 1 2 2 3 3 4 4 5 0 0 0 0 2 2 3 4 5 1 1 1 2 3 3 4 4 5
21
süß 0 1 1 1 1 1 2 3 4 0 1 1 1 2 2 3 3 4 0 0 1 1 1 2 2 3 3
sauer 0 0 0 2 3 4 4 5 5 0 0 1 2 3 3 4 4 5 0 2 3 3 4 4 4 5 5
salzig 0 0 1 1 2 3 3 3 4 0 1 2 2 2 2 2 3 3 0 1 1 1 2 3 3 4 4
bitter 0 1 1 2 3 3 4 5 5 0 1 2 2 3 3 4 4 4 0 1 2 3 3 4 4 5 6
umami 0 1 1 2 2 3 3 4 4 1 1 1 1 2 2 3 4 5 0 0 0 1 1 1 2 3 4
22
süß 0 0 0 0 2 3 3 4 5 0 1 1 1 1 1 1 1 2 0 0 0 1 1 1 2 3 4
sauer 1 1 1 2 3 4 5 5 6 0 0 1 2 3 4 5 6 7 0 2 3 4 5 6 7 8 9
salzig 0 1 1 1 1 2 3 4 5 0 1 1 1 2 3 4 5 6 0 0 1 2 2 3 4 5 6
bitter 2 3 4 5 6 7 8 9 10 0 0 0 1 2 3 4 5 6 0 1 2 3 4 5 6 7 8
umami 1 1 1 2 2 3 4 5 5 0 1 1 1 2 3 4 5 6 0 0 0 0 0 1 2 3 4
23
süß 1 1 1 1 1 1 1 2 3 0 1 1 2 2 2 3 4 5 0 0 0 1 1 2 3 4 5
sauer 0 0 0 1 2 3 4 5 6 1 1 2 3 4 5 6 7 7 1 2 3 4 4 5 5 6 7
salzig 0 0 1 2 3 4 5 6 7 0 0 1 2 2 3 4 5 6 1 1 2 3 3 4 5 5 6
bitter 0 0 0 0 2 3 4 5 6 0 0 0 0 1 1 1 2 3 0 0 1 2 2 2 3 3 4
umami 1 1 1 1 1 2 3 4 5 0 0 1 2 2 3 4 4 5 0 1 2 2 2 3 4 5 6

Anhang
325
Tabelle A20: Versuchsdaten – Prüfung zur Unterscheidung von Intensitätsstufen eines
Reizes
WH 1 WH 2 WH 3 Teilnehmer GSA D7 D5 D3 D1 D7 D5 D3 D1 D7 D5 D3 D1
0
süß
sauer
salzig
bitter
umami
1
süß 1,5 1,5 3,0 4,0 1,5 1,5 3,0 4,0 1,0 2,5 2,5 4,0
sauer 1,0 2,0 3,5 3,5 3,0 1,0 2,0 4,0 1,0 2,0 3,5 3,5
salzig 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 2,0 1,0 4,0 3,0 2,0 4,0 3,0 1,0 3,0 2,0 1,0 4,0
umami 2,0 1,0 3,0 4,0 2,0 1,0 3,0 4,0 1,0 2,0 3,0 4,0
2
süß 1,0 2,0 3,0 4,0 3,0 1,5 1,5 4,0 1,0 2,0 3,0 4,0
sauer 1,0 3,0 2,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
salzig 2,0 1,0 3,0 4,0 2,0 1,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 1,0 2,5 4,0 2,5 1,0 2,0 3,0 4,0 1,0 3,0 4,0 2,0
umami 1,0 2,0 3,5 3,5 2,0 1,0 3,0 4,0 2,0 1,0 3,0 4,0
3
süß 1,0 2,5 2,5 4,0 1,0 2,0 3,0 4,0 2,0 1,0 3,0 4,0
sauer 1,0 2,0 3,0 4,0 2,0 3,0 1,0 4,0 1,0 2,0 3,0 4,0
salzig 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 2,0 1,0 3,0 4,0
bitter 1,0 2,0 4,0 3,0 2,0 1,0 3,0 4,0 2,0 1,0 4,0 3,0
umami 1,0 2,0 3,0 4,0 1,0 3,5 3,5 4,0 1,0 2,0 3,0 4,0
4
süß 2,0 1,0 3,0 4,0 1,0 2,5 2,5 4,0 1,0 2,0 3,0 4,0
sauer 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
salzig 2,5 2,5 1,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 2,0 1,0 4,0 3,0 1,0 3,5 3,5 2,0 1,0 3,0 2,0 4,0
umami 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
5
süß 1,5 1,5 3,0 4,0 1,5 1,5 3,0 4,0 1,5 1,5 3,0 4,0
sauer 1,0 2,0 4,0 3,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
salzig 1,5 3,0 1,5 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 4,0 2,0 1,0 3,0 2,0 4,0 1,0 3,0 1,0 2,0 3,0 4,0
umami 1,0 3,0 2,0 4,0 1,5 1,5 3,0 4,0 3,0 1,0 2,0 4,0
6
süß 3,0 1,0 2,0 4,0 1,5 3,0 1,5 4,0 1,0 2,0 3,0 4,0
sauer 1,0 4,0 2,0 3,0 3,0 1,0 2,0 4,0 1,0 2,0 3,0 4,0
salzig 1,0 3,0 2,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 3,0 2,0 1,0 4,0 3,0 4,0 2,0 1,0 2,0 1,0 4,0 3,0
umami 1,0 2,0 3,0 4,0 1,0 3,0 2,0 4,0 2,0 1,0 3,0 4,0
7
süß 3,0 1,0 2,0 4,0 1,5 3,0 1,5 4,0 1,0 2,0 3,0 4,0
sauer 2,0 3,0 1,0 4,0 2,0 1,0 3,0 4,0 1,0 2,0 4,0 3,0
salzig 1,0 3,0 2,0 4,0 1,5 1,5 3,0 4,0 1,0 2,0 3,0 4,0
bitter 3,0 1,0 2,0 4,0 4,0 3,0 2,0 1,0 1,0 2,0 3,0 4,0
umami 1,0 2,0 3,0 4,0 1,0 4,0 2,0 3,0 1,0 3,0 2,0 4,0
8
süß 3,0 1,5 1,5 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
sauer 1,0 2,0 3,0 4,0 2,0 1,0 3,0 4,0 1,0 2,0 3,0 4,0
salzig 1,0 2,5 2,5 4,0 2,0 1,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 1,0 2,5 2,5 4,0 2,5 4,0 1,0 2,5 1,0 3,0 2,0 4,0
umami 1,5 4,0 1,5 3,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
9
süß 3,0 2,0 1,0 4,0 1,0 2,0 4,0 3,0 1,0 2,0 3,0 4,0
sauer 1,0 2,0 3,5 3,5 1,0 2,0 3,0 4,0 1,0 3,0 2,0 4,0
salzig 1,5 1,5 3,0 4,0 1,5 1,5 3,0 4,0 2,0 3,0 4,0 1,0
bitter 1,0 2,0 3,0 4,0 3,0 4,0 1,0 2,0 2,5 1,0 2,5 4,0
umami 1,0 2,0 3,5 3,5 2,0 1,0 3,0 4,0 1,0 3,0 2,0 4,0
10
süß 1,0 2,0 3,0 4,0 1,0 2,5 2,5 4,0 1,0 2,0 3,0 4,0
sauer 1,0 2,0 3,0 4,0 2,5 1,0 2,5 4,0 1,0 2,5 2,5 4,0
salzig 1,0 2,5 2,5 4,0 3,0 1,5 1,5 4,0 1,0 2,0 3,0 4,0
bitter 3,0 1,0 2,0 4,0 2,5 2,5 1,0 4,0 3,0 2,0 4,0 1,0
umami 1,0 2,5 4,0 2,5 1,0 2,5 2,5 4,0 2,0 1,0 3,0 4,0
11
süß 1,0 2,0 3,0 4,0 1,5 1,5 3,0 4,0 1,5 1,5 3,0 4,0
sauer 1,0 2,5 2,5 4,0 1,0 2,0 3,0 4,0 1,0 2,0 4,0 3,0
salzig 1,5 3,0 1,5 4,0 1,5 1,5 3,0 4,0 1,0 2,0 3,0 4,0
bitter 1,5 3,0 1,5 4,0 1,0 2,5 4,0 2,5 1,0 2,0 3,0 4,0
umami 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
Legende: GSA, Geschmacksausprägung. WH, Wiederholung.

Anhang
326
Tabelle A21: Versuchsdaten – Prüfung zur Unterscheidung von Intensitätsstufen eines Reizes – Fortsetzung
WH 1 WH 2 WH 3
Teilnehmer GSA D7 D5 D3 D1 D7 D5 D3 D1 D7 D5 D3 D1
12
süß 1,5 1,5 3,0 4,0 1,5 1,5 3,0 4,0 1,0 2,0 3,0 4,0
sauer 1,0 2,0 3,5 3,5 2,0 1,0 3,0 4,0 1,0 2,0 3,0 4,0
salzig 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 1,0 2,5 4,0 2,5 2,5 1,0 2,5 4,0 1,5 1,5 3,0 4,0
umami 1,5 1,5 3,0 4,0 1,5 1,5 4,0 3,0 2,0 1,0 3,0 4,0
13
süß 3,0 1,5 1,5 4,0 2,5 1,0 2,5 4,0 1,5 1,5 3,0 4,0
sauer 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
salzig 2,0 1,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 1,0 2,0 3,0 4,0 3,0 1,0 4,0 2,0 2,5 1,0 4,0 2,5
umami 1,0 2,5 2,5 4,0 1,5 4,0 3,0 1,5 1,0 2,0 3,0 4,0
14
süß 3,0 1,5 1,5 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
sauer 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
salzig 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 1,0 2,0 3,0 4,0 2,0 1,0 4,0 3,0 1,0 2,0 3,0 4,0
umami 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
15
süß 2,0 1,0 3,0 4,0 1,5 1,5 3,0 4,0 1,0 3,0 2,0 4,0
sauer 2,0 1,0 3,0 4,0 2,0 1,0 3,0 4,0 1,0 2,0 3,0 4,0
salzig 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 1,0 2,0 4,0 3,0 1,0 3,0 3,0 3,0 1,0 3,0 2,0 4,0
umami 2,0 1,0 3,0 4,0 2,0 3,0 1,0 4,0 1,0 2,0 3,0 4,0
16
süß 1,5 1,5 3,0 4,0 3,0 1,5 1,5 4,0
sauer 1,0 2,0 3,0 4,0 1,5 3,0 1,5 4,0
salzig 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 3,0 1,5 4,0 1,5 3,5 1,5 3,5 1,5
umami 1,0 2,5 2,5 4,0 1,0 2,5 2,5 4,0
17
süß 1,5 1,5 3,0 4,0 1,5 1,5 3,0 4,0 1,5 1,5 3,0 4,0
sauer 2,0 1,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
salzig 1,5 1,5 4,0 3,0 1,5 1,5 3,0 4,0 1,0 2,0 3,0 4,0
bitter 1,0 2,5 2,5 4,0 3,0 1,0 2,0 4,0 1,0 3,0 2,0 4,0
umami 1,0 2,0 3,0 4,0 1,5 1,5 3,0 4,0 2,0 1,0 3,0 4,0
18
süß 1,0 2,5 2,5 4,0 3,0 1,5 1,5 4,0 1,0 2,0 3,0 4,0
sauer 2,5 1,0 2,5 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
salzig 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 1,5 3,0 1,5 4,0 2,0 1,0 3,0 4,0 2,0 1,0 3,0 4,0
umami 1,5 1,5 3,5 3,5 1,0 2,0 3,0 4,0 2,0 1,0 3,0 4,0
19
süß 1,5 1,5 3,0 4,0 1,5 1,5 3,0 4,0 1,0 2,0 3,0 4,0
sauer 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
salzig 2,0 1,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 2,0 1,0 3,0 4,0
umami 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
20
süß 1,0 2,0 3,0 4,0 2,0 1,0 3,0 4,0 1,0 2,0 3,0 4,0
sauer 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
salzig 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 1,0 2,0 4,0 3,0 2,0 1,0 3,0 4,0 1,0 2,0 3,0 4,0
umami 1,0 2,0 3,0 4,0 2,0 1,0 3,0 4,0 2,0 1,0 3,0 4,0
21
süß 2,0 2,0 2,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
sauer 2,5 1,0 2,5 4,0 2,0 1,0 3,0 4,0 1,0 2,0 3,0 4,0
salzig 1,5 1,5 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 1,0 2,0 3,0 4,0 1,0 2,5 4,0 2,5 1,0 2,0 3,0 4,0
umami 1,0 2,0 4,0 3,0 1,5 1,5 3,5 3,5 1,0 2,0 3,0 4,0
22
süß 2,0 2,0 2,0 4,0 2,0 1,0 3,0 4,0 1,5 1,5 3,0 4,0
sauer 1,0 2,5 2,5 4,0 3,0 1,0 2,0 4,0 1,0 2,0 3,0 4,0
salzig 1,5 1,5 3,0 4,0 2,0 1,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 1,0 2,0 3,0 4,0 2,0 1,0 3,0 4,0 1,5 1,5 3,0 4,0
umami 2,0 1,0 4,0 3,0 1,0 3,0 4,0 2,0 1,0 2,0 3,0 4,0
23
süß 1,5 1,5 3,0 4,0 1,0 2,0 4,0 3,0 2,0 1,0 3,0 4,0
sauer 1,0 2,0 3,0 4,0 2,0 1,0 3,0 4,0 1,0 2,0 3,0 4,0
salzig 1,5 1,5 3,0 4,0 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0
bitter 1,0 2,0 3,0 4,0 1,0 2,5 2,5 4,0 1,0 3,0 2,0 4,0
umami 1,0 2,0 3,0 4,0 1,0 2,0 3,0 4,0 2,0 2,0 2,0 4,0
Legende: GSA, Geschmacksausprägung. WH, Wiederholung.

Anhang
327
Tabelle A22: Versuchsdaten – Schulung zur Anwendung der Skale
Intensität "schwach" "mittel "stark" Teilnehmer WH 1 WH 2 WH 3 WH 1 WH 2 WH 3 WH 1 WH 2 WH 3
0
1 2 4 3 3 6 4 7 2 7
2 1 3 2 4 4 4 7 6 6
3 3 3 3 4 4 4 6 5 5
4 1 4 3 3 3 5 7 5 6
5 3 4 2 4 3 4 6 7 6
6 4 2 2 2 4 4 5 6 5
7 4 2 2 2 4 4 5 6 6
8 3 3 2 6 2 4 7 5 6
9 3 2 3 4 4 5 5 6 6
10 1 3 4 3 2 3 6 5 5
11 2 3 3 3 5 4 7 4 5
12 2 1 2 3 2 5 6 5 6
13 1 1 1 2 2 4 3 6 6
14 1 3 2 3 2 3 5 6 6
15 3 2 1 2 4 4 5 6 6
16 1 2 3 1 5 4
17 1 2 2 3 3 4 5 6 6
18 1 3 2 3 1 5 6 5 6
19 2 2 3 3 4 6 6 6 4
20 1 1 2 2 4 4 6 6 5
21 1 2 2 3 4 4 4 5 7
22 3 2 2 4 3 3 5 5 4
23 1 4 4 4 2 5 5 6 6
Legende: WH, Wiederholung.

Anhang
328
Tabelle A23: Versuchsdaten – Sensorische Bewertung der Fraktionen – Molkenprotein-Isolat
Teilnehmer 1
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 0 0 0 2 0 1 2 3 5 0 0 0 4 5
Per. 2 1 0 1 0 0 6 0 3 4 3 0 6 0 6
"bitter" 0 0 0 1 1 1 3 2 0 5 7 6 0 0 0
Per. + "bitter" 2 0 0 2 0 1 7 3 2 6 0 0 7 6 6
Teilnehmer 2
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 1 0 0 2 0 0 2 1 1 0 2 2 0 1 0
Per. 2 1 1 0 1 1 1 0 1 0 0 0 2 0 0
"bitter" 0 0 0 0 0 0 0 0 0 3 2 2 3 0 0
Per. + "bitter" 0 1 0 0 0 1 0 0 0 3 2 2 2 0 2
Teilnehmer 3
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 1 2 1 0 0 0 2 1 1 1 0 0 4 3 1
Per. 0 2 1 0 0 0 1 2 0 0 1 0 4 1 2
"bitter" 0 0 0 0 0 0 2 1 1 5 4 1 0 0 0
Per. + "bitter" 1 2 0 0 1 0 3 1 1 3 1 1 1 0 0
Teilnehmer 4
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 1 1 1 0 0 0 0 1 0 2 1 2 0 0 1
Per. 0 0 0 0 0 0 1 1 1 1 2 1 0 2 0
"bitter" 0 0 0 0 0 1 0 0 0 3 2 3 0 0 1
Per. + "bitter" 0 0 0 0 0 0 0 0 1 2 1 2 1 1 2
Teilnehmer 5
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 4 1 3 0 0 0 2 1 2 3 0 0 5 2 2
Per. 1 1 0 0 1 1 4 4 2 0 0 0 5 2 1
"bitter" 0 1 0 1 0 1 1 0 0 3 3 3 3 0 0
Per. + "bitter" 0 3 4 0 0 0 4 1 0 4 1 2 2 2 1
Teilnehmer 6
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 0 1 0 0 1 0 0 0 1 2 0 0 3 3
Per. 0 0 2 0 0 0 3 3 0 1 1 1 0 2 3
"bitter" 0 0 0 0 0 2 0 0 0 3 5 3 1 0 0
Per. + "bitter" 0 0 2 0 0 2 3 2 0 4 5 2 0 3 4
Teilnehmer 7
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 1 3 0 3 1 1 5 4 5 4 2 0 3 5
Per. 3 6 3 2 1 2 5 3 5 4 2 2 6 4 6
"bitter" 0 1 1 1 3 0 2 4 2 5 6 6 1 2 2
Per. + "bitter" 4 5 4 4 2 1 6 3 2 5 2 4 6 3 3
Legende: GSA, Geschmacksausprägung. WH, Wiederholung. Ret., Retentat. Per., Permeat.

Anhang
329
Tabelle A24: Versuchsdaten – Sensorische Bewertung der Fraktionen – Molkenprotein-Isolat – Fortsetzung
Teilnehmer 8
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 0 0 0 0 0 0 2 3 0 0 1 2 5 6
Per. 0 2 0 0 2 0 2 1 2 0 0 0 2 4 7
"bitter" 0 0 0 0 0 0 0 0 0 4 6 3 0 0 1
Per. + "bitter" 0 0 0 0 0 0 2 3 4 0 2 2 4 4 7
Teilnehmer 9
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 3 2 0 2 2 2 0 0 3 0 0 0 0 4
Per. 0 3 1 0 0 0 2 2 1 3 0 0 0 0 3
"bitter" 0 1 0 5 1 3 0 0 0 2 4 4 0 0 0
Per. + "bitter" 1 2 1 0 0 0 0 2 1 1 0 4 1 0 4
Teilnehmer 10
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 2 3 2 0 0 0 0 0 0 1 0 0 0 0 0
Per. 2 0 3 0 1 0 0 3 0 1 1 0 0 1 0
"bitter" 0 0 0 1 0 3 0 0 0 4 4 2 2 1 0
Per. + "bitter" 2 2 3 1 1 1 2 0 0 0 2 0 0 0 0
Teilnehmer 11
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 1 0 0 0 0 0 0 0 2 1 1 1 1 0 2
Per. 1 2 2 0 3 1 0 4 1 1 1 0 1 0 0
"bitter" 2 2 2 2 2 2 1 1 1 4 1 3 2 0 2
Per. + "bitter" 1 1 1 0 0 1 0 2 2 0 3 3 1 2 3
Teilnehmer 12
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 4 0 1 3 0 0 4 2 1 1 3 1 1 4 5
Per. 2 0 1 1 1 2 3 1 1 1 3 1 4 6 5
"bitter" 2 0 0 1 0 1 3 1 0 5 6 5 3 1 3
Per. + "bitter" 1 0 0 3 0 0 3 3 2 5 7 4 5 6 6
Teilnehmer 13
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 0 0 0 0 0 1 0 2 0 1 0 1 1 4
Per. 0 1 0 0 0 0 1 2 0 1 0 0 2 1 2
"bitter" 0 0 0 1 1 1 0 0 0 4 3 3 0 0 0
Per. + "bitter" 0 0 0 0 0 0 1 1 0 0 0 3 4 3 2
Teilnehmer 14
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 1 0 0 0 0 0 0 0 0 0 1 3 0 2 2
Per. 0 0 0 0 1 0 0 2 1 0 0 0 3 4 2
"bitter" 0 0 0 0 0 0 0 0 0 2 4 3 0 1 0
Per. + "bitter" 0 0 0 0 1 0 0 0 0 1 5 3 5 3 3
Legende: GSA, Geschmacksausprägung. WH, Wiederholung. Ret., Retentat. Per., Permeat.

Anhang
330
Tabelle A25: Versuchsdaten – Sensorische Bewertung der Fraktionen – Molkenprotein-Isolat – Fortsetzung
Teilnehmer 15
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 1 0 0 0 0 0 0 0 2 0 0 0 0 0
Per. 0 0 0 1 0 1 0 3 1 0 0 0 0 0 0
"bitter" 0 0 0 0 1 0 0 0 0 2 3 5 0 0 2
Per. + "bitter" 0 0 0 0 0 2 2 3 0 1 3 1 0 0 0
Teilnehmer 16
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 1 0 1 0 0 2 0 1 1 2 2 0 0 3
Per. 0 0 0 0 2 0 1 0 1 2 0 0 0 2 2
"bitter" 0 0 0 0 0 1 0 0 0 2 0 1 0 0 0
Per. + "bitter" 0 0 0 0 0 0 2 0 0 1 2 2 0 2 3
Teilnehmer 17
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 2 0 0 2 0 0 3 1 0 0 0 0 0 0 1
Per. 0 0 0 0 0 0 2 3 1 3 0 0 0 3 1
"bitter" 0 0 0 0 0 0 0 0 0 4 3 3 2 1 0
Per. + "bitter" 0 0 0 0 0 0 0 1 0 2 3 1 3 1 2
Teilnehmer 18
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 4 4 0 0 1 1 1 1 1 1 1 1 0 0
Per. 0 4 4 1 0 1 3 2 2 0 0 1 2 0 0
"bitter" 0 0 0 0 3 1 1 5 0 4 4 3 5 2 4
Per. + "bitter" 1 0 0 3 3 2 1 5 2 4 5 5 1 6 4
Teilnehmer 19
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 1 0 0 0 0 1 0 0 1 0 0 2 0 0
Per. 0 2 0 0 0 0 2 2 1 0 0 0 2 1 1
"bitter" 1 0 0 0 0 0 0 0 0 4 4 3 0 0 0
Per. + "bitter" 1 2 0 0 0 0 3 1 1 3 4 3 1 0 1
Teilnehmer 20
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 1 1 0 0 0 0 1 1 0 1 1 2 3 2
Per. 0 1 0 0 1 0 1 2 1 0 0 0 2 2 2
"bitter" 0 1 1 1 0 0 0 0 1 3 3 2 1 2 0
Per. + "bitter" 0 0 0 0 1 0 0 2 1 2 1 2 2 2 3
Teilnehmer 21
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 1 1 2 0 0 0 0 0 0 0 3 0 2 0 0
Per. 0 4 1 0 0 0 1 5 2 2 0 2 0 0 0
"bitter" 0 0 0 2 0 0 0 0 0 1 5 5 0 0 0
Per. + "bitter" 0 3 0 0 0 0 3 5 5 3 5 6 0 0 0
Legende: GSA, Geschmacksausprägung. WH, Wiederholung. Ret., Retentat. Per., Permeat.

Anhang
331
Tabelle A26: Versuchsdaten – Sensorische Bewertung der Fraktionen – Molkenprotein-Isolat – Fortsetzung
Teilnehmer 22
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 0 0 0 0 0 0 1 1 0 0 0 0 1 0
Per. 0 0 0 0 0 0 1 2 1 0 0 0 0 2 0
"bitter" 0 0 0 0 0 0 0 2 0 0 0 0 0 2 0
Per. + "bitter" 0 0 0 0 0 0 0 2 0 0 1 0 0 2 0
Teilnehmer 23
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 2 2 0 3 0 3 3 0 3 0 1 5 5 6
Per. 1 3 2 2 4 4 2 5 2 0 1 0 4 5 4
"bitter" 0 0 0 0 0 0 0 3 0 2 6 5 5 2 0
Per. + "bitter" 0 1 0 1 3 3 4 5 5 3 1 1 4 5 5
Legende: GSA, Geschmacksausprägung. WH, Wiederholung. Ret., Retentat. Per., Permeat.

Anhang
332
Tabelle A27: Versuchsdaten – Sensorische Bewertung der Fraktionen – Alge (Chlorella vulgaris)
Teilnehmer 1
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 7 2 0 0 0 0 3 0 1 4 5 2 0 3 5
Per. 0 0 1 0 1 0 7 4 2 4 0 0 4 7 5
"bitter" 0 1 0 1 0 1 0 0 0 7 5 4 1 2 3
Per. + "bitter" 0 0 0 2 0 0 4 5 2 5 2 1 4 4 6
Teilnehmer 2
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 1 1 0 2 0 1 2 0 0 0 0 2 0 0 0
Per. 1 0 2 0 0 2 2 1 0 0 1 1 1 1 0
"bitter" 0 0 0 0 0 1 0 0 1 3 2 3 1 0 0
Per. + "bitter" 0 0 0 0 0 1 3 0 1 2 0 1 1 1 3
Teilnehmer 3
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 3 2 2 1 0 0 2 1 1 0 0 0 2 2 0
Per. 1 0 1 1 1 0 4 1 0 1 1 0 2 2 0
"bitter" 1 0 0 0 1 1 2 2 1 5 3 3 0 0 0
Per. + "bitter" 2 0 1 1 1 0 3 2 2 3 4 0 0 0 0
Teilnehmer 4
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 2 0 0 0 0 1 1 0 0 1 0 2 1 1 1
Per. 0 0 0 0 0 0 3 0 2 1 2 0 2 2 1
"bitter" 0 0 0 0 0 0 0 0 0 4 3 2 1 2 0
Per. + "bitter" 0 0 0 0 0 1 1 1 1 3 1 3 1 2 2
Teilnehmer 5
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 1 3 0 0 0 3 0 1 0 0 0 2 2 2
Per. 3 2 0 0 0 0 6 0 3 0 0 0 0 2 3
"bitter" 0 0 1 0 0 0 0 0 0 5 3 5 0 2 0
Per. + "bitter" 0 1 1 0 0 0 5 1 3 1 0 0 2 2 1
Teilnehmer 6
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 0 1 0 0 0 4 2 3 1 1 1 3 2 2
Per. 0 1 1 0 0 2 5 1 0 2 0 1 1 3 2
"bitter" 0 0 0 2 0 0 0 0 0 3 3 2 0 0 0
Per. + "bitter" 0 0 3 0 0 0 5 1 2 2 2 4 0 3 5
Teilnehmer 7
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 7 4 2 1 2 1 3 6 4 4 2 3 0 5 6
Per. 2 4 4 6 1 2 5 3 5 3 1 4 3 3 3
"bitter" 0 0 1 1 1 1 0 5 2 7 2 6 1 2 2
Per. + "bitter" 2 2 4 5 1 1 3 6 2 6 5 3 3 5 6
Legende: GSA, Geschmacksausprägung. WH, Wiederholung. Ret., Retentat. Per., Permeat.

Anhang
333
Tabelle A28: Versuchsdaten – Sensorische Bewertung der Fraktionen – Alge (Chlorella vulgaris) - Fortsetzung
Teilnehmer 8
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 1 0 0 0 0 1 1 3 0 0 0 3 4 5
Per. 0 0 0 0 0 0 0 3 4 0 0 0 6 6 6
"bitter" 0 0 0 0 0 2 1 0 1 5 4 3 1 0 3
Per. + "bitter" 0 0 0 0 0 0 3 2 4 1 2 4 6 5 7
Teilnehmer 9
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 2 1 0 0 0 0 3 1 1 0 0 0 2 3 3
Per. 0 3 2 0 0 0 3 0 0 0 0 0 2 3 4
"bitter" 0 1 0 2 1 2 0 0 0 2 0 1 0 0 0
Per. + "bitter" 2 0 2 1 2 0 1 2 2 0 0 0 2 4 4
Teilnehmer 10
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 3 2 1 0 1 0 3 1 1 0 0 0 0 0 3
Per. 2 3 2 1 0 3 5 1 0 1 0 0 1 0 1
"bitter" 0 1 0 0 0 0 1 0 0 5 3 4 0 1 1
Per. + "bitter" 0 2 2 4 0 2 3 0 0 1 0 1 0 2 1
Teilnehmer 11
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 2 2 0 2 0 2 4 1 1 0 1 1 3 2 0
Per. 1 3 2 3 2 1 5 0 0 0 1 0 4 1 1
"bitter" 0 1 1 0 1 0 1 1 1 5 2 3 2 1 0
Per. + "bitter" 0 2 2 3 0 2 6 0 3 3 3 2 2 3 1
Teilnehmer 12
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 1 0 0 3 0 0 4 2 0 4 1 1 6 4 3
Per. 0 1 1 1 0 1 3 3 2 3 1 1 4 5 6
"bitter" 0 0 0 1 0 0 3 2 0 4 5 3 2 3 1
Per. + "bitter" 0 1 0 0 0 0 3 1 2 4 6 5 6 3 7
Teilnehmer 13
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 0 0 0 0 0 1 1 1 1 0 0 2 2 1
Per. 0 0 0 0 0 0 1 0 1 0 0 0 1 2 2
"bitter" 0 0 0 2 1 1 0 0 0 1 2 4 0 0 0
Per. + "bitter" 0 0 0 0 0 0 2 1 3 2 1 0 2 1 4
Teilnehmer 14
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 0 0 0 0 1 1 0 0 0 0 0 4 4 4
Per. 0 1 0 0 0 0 0 0 0 0 0 0 4 4 3
"bitter" 1 0 0 0 0 0 0 0 0 4 1 1 0 0 4
Per. + "bitter" 0 0 0 0 0 0 0 0 2 4 2 2 4 3 5
Legende: GSA, Geschmacksausprägung. WH, Wiederholung. Ret., Retentat. Per., Permeat.

Anhang
334
Tabelle A29: Versuchsdaten – Sensorische Bewertung der Fraktionen – Alge (Chlorella vulgaris) – Fortsetzung
Teilnehmer 15
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 1 1 1 0 0 0 0 0 0 0 0 0 0 0 0
Per. 0 1 1 2 0 0 2 0 0 0 1 0 0 0 0
"bitter" 0 0 0 0 0 0 0 0 0 0 0 3 0 0 0
Per. + "bitter" 1 1 1 0 0 0 0 1 0 4 0 2 0 0 0
Teilnehmer 16
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 0 0 0 0 0 3 0 0 3 1 2 1 2 2
Per. 0 0 1 0 0 0 1 1 0 1 2 1 0 2 3
"bitter" 0 0 0 1 0 0 0 0 0 2 1 0 0 0 0
Per. + "bitter" 0 0 0 1 0 1 1 1 2 2 3 0 0 1 3
Teilnehmer 17
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 0 0 0 0 0 3 2 1 1 1 0 2 1 1
Per. 0 0 1 0 0 0 2 3 0 3 0 0 5 2 3
"bitter" 0 0 0 1 0 0 0 1 0 5 3 3 0 0 1
Per. + "bitter" 0 0 0 0 0 0 2 1 3 3 0 0 4 2 3
Teilnehmer 18
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 3 0 3 2 1 1 0 2 0 1 1 1 0 1 0
Per. 2 0 3 0 2 1 1 3 4 1 0 1 0 0 0
"bitter" 0 0 0 1 3 3 0 0 2 3 4 4 4 2 5
Per. + "bitter" 0 1 1 2 1 2 1 0 5 4 3 4 4 2 2
Teilnehmer 19
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 2 0 0 0 0 0 0 1 1 0 0 0 1 1 2
Per. 1 0 0 0 0 0 2 4 2 0 0 0 2 2 1
"bitter" 0 0 0 1 0 0 0 0 0 4 4 3 0 0 0
Per. + "bitter" 0 0 0 0 0 0 1 1 1 2 3 0 2 1 1
Teilnehmer 20
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 1 0 1 0 1 1 1 2 2 0 1 0 4 3
Per. 0 0 1 1 1 0 1 1 1 1 0 1 0 3 4
"bitter" 0 0 0 2 1 1 0 0 0 2 3 3 0 1 0
Per. + "bitter" 0 0 1 0 0 0 1 1 2 1 3 1 2 3 2
Teilnehmer 21
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 1 1 1 0 0 3 0 2 0 0 0 1 0 0 0
Per. 2 3 1 0 0 0 2 0 3 0 0 3 0 1 0
"bitter" 0 0 0 1 0 0 1 0 0 1 2 6 0 0 0
Per. + "bitter" 0 1 0 0 0 3 4 4 3 1 0 5 2 0 0
Legende: GSA, Geschmacksausprägung. WH, Wiederholung. Ret., Retentat. Per., Permeat.

Anhang
335
Tabelle A30: Versuchsdaten – Sensorische Bewertung der Fraktionen – Alge (Chlorella vulgaris) – Fortsetzung
Teilnehmer 22
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 0 0 0 0 0 0 0 1 1 0 0 0 1 0 1
Per. 0 0 0 0 0 0 0 0 0 0 0 0 1 1 0
"bitter" 0 1 0 1 0 0 0 0 0 0 1 0 0 0 0
Per. + "bitter" 0 0 0 0 0 0 1 2 1 0 0 0 1 2 1
Teilnehmer 23
GSA süß sauer salzig bitter umami
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
WH 1
WH 2
WH 3
Ret. 2 1 0 3 0 0 0 2 3 1 1 0 4 5 6
Per. 2 0 3 3 1 3 4 5 6 0 0 0 4 4 4
"bitter" 2 0 0 0 0 0 0 2 3 5 4 5 4 2 0
Per. + "bitter" 2 0 2 0 2 0 6 5 4 2 2 2 5 3 6

Publikationsliste
336
Publikationsliste
Teile dieser Arbeit wurde bereits veröffentlicht:
Vorträge :
Friedrich, Markus (2015): Erzeugung von Peptidfraktionen per Cross-Flow Diafiltration. „Tag
der Forschung“ des Fachbereich „Lebensmitteltechnologie“ der Hochschule Fulda, Fulda,
04.11.2015.