modern approaches to the analysis of disinfection by...
TRANSCRIPT

Phil. Trans. R. Soc. A (2009) 367, 4097–4118doi:10.1098/rsta.2009.0130
REVIEW
Modern approaches to the analysisof disinfection by-products in drinking water
BY HOWARD S. WEINBERG*
Department of Environmental Sciences and Engineering,Gillings School of Global Public Health, University of North Carolina,
Chapel Hill, NC 27599-7431, USA
The discovery and study of disinfection by-products (DBPs) of health and regulatoryconcern in drinking water have often been hampered by the lack of appropriate analyticalmethods, but, with the new tools and expertise now available to the drinking waterindustry, there is an opportunity to plug a major gap in our knowledge of the natureand identity of these chemicals. The challenge is that less than half of the halogenatedby-products resulting from the chlorination of drinking water have been identified, andeven less is known about those produced in waters treated with ozone, chloramines orchlorine dioxide. For the DBPs that have been identified, very little or no occurrencedata exist for the unregulated chemicals to document how often a particular DBP isformed and in what quantity. The elucidation of the nature and identity of these by-products is hindered by two complicating factors. The first is the inherent aqueoussolubility of many of these compounds, which renders their efficient extraction fromwater difficult to achieve. The second is the lack of established identity of specificpotential by-products, which complicates targeted analytical approaches. This paperreviews existing and new methodologies that attempt to overcome some of thesechallenges.
Keywords: drinking water; disinfection by-products; analytical methods; chlorination;ozonation
1. Introduction
Disinfectants by design are strong oxidizing agents, which can react with thenatural organic matter (NOM) as well as bromide, iodide and backgroundpollutants in the water prepared for drinking to produce disinfection by-products(DBPs). The analysis of these DBPs provides a serious challenge to researchersbecause NOM is inherently complex, variable and typically poorly characterized.
One contribution of 12 to a Theme Issue ‘Emerging chemical contaminants in water andwastewater’.
This journal is © 2009 The Royal Society4097
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

4098 H. S. Weinberg
Tab
le1.
Ana
lyti
cal
frac
tion
sof
maj
ordi
sinf
ecti
onby
-pro
duct
s(D
BP
s)in
drin
king
wat
er.
(TH
M4,
four
chlo
rine
-an
dbr
omin
e-co
ntai
ning
trih
alom
etha
nes;
HA
Ns,
halo
acet
onit
rile
s;H
Ks,
halo
keto
nes;
CP,
chlo
ropi
crin
;C
H,
chlo
ral
hydr
ate;
I-T
HM
s,io
dine
-con
tain
ing
trih
alom
etha
nes;
Cl-H
AN
s,ch
lori
ne-c
onta
inin
gha
loac
eton
itri
les;
Cl-H
NM
s,ch
lori
ne-c
onta
inin
gha
loni
trom
etha
nes;
Br-
HA
Ns,
brom
ine-
cont
aini
ngha
loac
eton
itri
les;
Br-
HN
Ms,
brom
ine-
cont
aini
ngha
loni
trom
etha
nes;
HA
A9,
nine
chlo
rine
-an
dbr
omin
e-co
ntai
ning
halo
acet
icac
ids;
ND
MA
,N
-nit
roso
dim
ethy
lam
ine;
MtB
E,
met
hyl
tert
iary
buty
let
her;
KH
P,po
tass
ium
hydr
ogen
phth
alat
e;P
FB
HA
,pe
ntafl
uoro
benz
ylhy
drox
ylam
ine;
LLE
,liq
uid–
liqui
dex
trac
tion
;SP
E,s
olid
-pha
seex
trac
tion
;PC
R,p
ost-
colu
mn
reac
tion
;GC
/EC
D,g
asch
rom
atog
raph
yw
ith
elec
tron
capt
ure
dete
ctio
n;G
C/M
S,ga
sch
rom
atog
raph
yw
ith
mas
ssp
ectr
omet
ric
dete
ctio
n;C
I,ch
emic
alio
niza
tion
;IC
,io
nch
rom
atog
raph
y;N
/A,no
tap
plic
able
.)
pres
erva
tive
(s)
orex
trac
tion
min
.re
port
ing
anal
ytic
alfr
acti
onap
plic
able
DB
Ps
quen
chin
gag
ent
solv
ent,
reag
ents
leve
ls(μ
gl−
1 )m
etho
dci
tati
on
neut
ral-
extr
acta
ble
vola
tile
san
dse
mi-vo
lati
les
TH
M4,
HA
Ns,
HK
s,C
P,C
HN
H4C
l,bu
ffer
topH
5.2,
Na 2
SO3
LLE
,M
tBE
,N
a 2SO
4
1.0
for
TH
Ms,
othe
rwis
e0.
5by
GC
/EC
D
US
EPA
(199
5a)
acid
ic-e
xtra
ctab
levo
lati
les
and
sem
i-vo
lati
les
TH
M4
+I-
TH
Ms,
Cl-H
AN
s,H
Ks,
Cl-H
NM
s,C
H,
Br-
HA
Ns,
Br-
HN
Ms
asco
rbic
acid
+H
2SO
4to
pH3–
4,N
H4C
l/H
2SO
4
MtB
E,N
a 2SO
41.
0by
GC
/EC
D,
1.0–
2.5
byG
C/M
S
Kra
sner
etal
.(2
001)
,C
hinn
etal
.(2
007)
acid
-ext
ract
able
non-
vola
tile
sH
AA
9N
H4C
lM
tBE
,N
a 2SO
4,H
2SO
4(p
H<
0.5)
,H
2SO
4/C
H3O
H
1.0
for
allex
cept
ClA
Aan
dB
r 3A
A(2
.0)
byG
C/E
CD
US
EPA
(199
5b)
cyan
ogen
halid
esC
NC
l,C
NB
ras
corb
icac
id,
H2S
O4
(pH
3–4)
MtB
E,N
a 2SO
41.
0(G
C/E
CD
)Sc
limen
tiet
al.(1
995)
(Con
tinu
ed.)
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

Review. Modern analysis of by-products 4099
Tab
le1.
(Con
tinu
ed.)
pres
erva
tive
(s)
orex
trac
tion
min
.re
port
ing
anal
ytic
alfr
acti
onap
plic
able
DB
Ps
quen
chin
gag
ent
solv
ent,
reag
ents
leve
ls(μ
gl−
1 )m
etho
dci
tati
on
nitr
osam
ines
ND
MA
plus
six
tose
ven
othe
rsas
corb
icac
id(t
wo
wee
ks)
SPE
(Am
bers
orb)
0.00
2by
GC
/MS/
MS
(CI)
Am
eric
anP
ublic
Hea
lth
Ass
ocia
tion
(200
6),C
heng
etal
.(2
004)
,M
unch
&B
asse
tt(2
004)
oxyh
alid
esbr
omat
e,ch
lori
te,
chlo
rate
diet
hyla
min
eP
CR
0.1,
2.0
and
1.7
byIC
Wei
nber
g&
Yam
ada
(199
8),
Wag
ner
etal
.(2
002)
carb
onyl
sal
dehy
des,
halo
alde
hyde
sN
H4C
l,K
I,so
dium
azid
ehe
xane
,K
HP
/NaO
Hbu
ffer
(pH
6),
PFB
HA
1.0
byG
C/E
CD
Am
eric
anP
ublic
Hea
lth
Ass
ocia
tion
(200
6),
Wei
nber
get
al.(2
002)
poly
func
tion
alD
BP
sal
doke
toac
ids
NH
4Cl,
KI,
sodi
umaz
ide
MtB
E,P
FB
HA
,di
azom
etha
ne2.
0–3.
0by
GC
/EC
DW
einb
erg
&G
laze
(199
7),
Xie
&R
eckh
ow(1
992)
orga
nic
acid
sca
rbox
ylic
acid
sK
I,be
nzal
koni
umch
lori
deN
/A10
–20
byIC
Kuo
(199
8)
halo
acet
amid
esC
l-,B
r-an
dI-
cont
aini
ng(N
H4)
2SO
4LLE
,M
tBE
1.0–
5.0
byG
C/E
CD
Wei
nber
get
al.(2
002)
halo
fura
none
sM
X,B
MX
(NH
4)2S
O4
BF
3/M
eOH
,LLE
-hex
ane
0.02
byG
C/E
CD
Ons
tad
etal
.(2
008)
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

4100 H. S. Weinberg
Consequently, the initial starting materials of any chemical reaction withNOM are largely unknown. As a result, fewer than half of the halogenatedby-products by mass resulting from the chlorination of drinking waterhave been identified, and even less is known about the by-products inwaters treated with other disinfectants such as ozone, advanced oxidationprocesses, chloramines, chlorine dioxide, mixed oxidants or ultraviolet irradiation(Weinberg 1999).
In 1979, the United States Environmental Protection Agency (US EPA)began the first steps towards protecting consumers from exposures to DBPsin their drinking water by regulating the acceptable levels of some of thesecompounds based upon toxicological data and the availability of analyticaltechniques to measure them (US Environmental Protection Agency 1979). Asthe early targeted species were the thermally and chemically stable, volatile,neutral organic halogenated trihalomethanes (THMs), isolation from the aquaticmatrix (by purge-and-trap or liquid–liquid extraction (LLE)) and detection (usinga gas chromatograph equipped with electron capture detector, GC/ECD) wasrelatively straightforward. As drinking water treatment plants incorporated newdisinfection processes such as chlorine dioxide, ozonation and chloramination,driven, in part, to lower the levels of the regulated DBPs in their finishedproduct, alternative by-products became a concern. However, because DBPsvary in molecular weight, volatility and polarity, no single analytical methodcan be used for extracting and identifying them all. New methods, includingmore sophisticated analytical approaches, are necessary to isolate such by-products from the aquatic matrix and to convert them into a form thatpermits sensitive detection. Many of the products are unlikely to be suitedto gas chromatography–mass spectrometry (GC/MS) analysis, the standby foranalysis of unknowns, because some may be too polar, insufficiently volatileor of molecular weight beyond the range of detection (Khiari et al. 1997;Hua & Reckhow 2007). As long as the molecular weights are low, chemicalderivatization can be used to render the target chemicals more amenable tochromatographic separation and detection by GC, but at higher masses inexcess of 650 atomic mass units, liquid chromatography has to be examinedas an analytical option (Zhang & Minear 2002; Zhang et al. 2004). Hence, anapproach that uses a range of complementary techniques will be necessary tocharacterize the mixture of DBPs present in drinking water. If more DBPs ofhealth concern are identified through new analytical methods, occurrence datawill only be generated on a wide scale if the methods can be disseminatedaround a large user base. In other words, methods should be developed withthe user, such as drinking water utilities and their laboratories, in mind sothat they can be used quickly, cheaply and without extraordinarily high skilllevels. In the subsequent sections, the discussion of analytical methods used tomeasure DBPs generated from NOM is restricted to those for which analyticalreference standards are commercially available and which have been quantifiedin real drinking waters. Table 1 lists those DBPs discussed in this paper andsummarizes the major attributes of the methods used for their analysis. It isbeyond the scope of this paper to address methods developed only for bench-scale studies, for broad-screen analyses where confirmation of analyte identity isnot possible or for the potential by-products of reactions with micropollutants ofanthropogenic origin.
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

Review. Modern analysis of by-products 4101
2. Sample preservation
There are two aspects of sample collection that must not be overlooked ifanalyte concentration is to be preserved until the sample can be extracted.The prevention of continued formation of DBPs must be achieved by removingresidual disinfectant using a reagent that does not change the concentration ofany of the species targeted in that sample. Typical residual chlorine quenchingagents include salts of ammonia that form chloramines from free chlorine. Theiruse is appropriate if the presence of chloramines does not change the equilibriaestablished between DBPs and precursor material at the time of disinfectantquenching. An evaluation of the stability of the DBPs examined in each quenchedsample must be undertaken before using this reagent in the field. In somecases, a holding time study will determine the maximum length of time andconditions under which samples can be stored before extraction and similarly forthe extract. Published methods for the analysis of four THMs (US EnvironmentalProtection Agency 1995a) and nine haloacetic acids (HAAs) (US EnvironmentalProtection Agency 1995b) quench residual chlorine with salts of ammonia butdo not specify a reagent for removing chloramines. In fact, there is now someevidence that dihalogenated HAAs can form in chloraminated waters and thatammonium chloride can enhance the process (Hong et al. 2007). The qualityof the quenching agent must not be overlooked during selection. Some sources ofammonium chloride, for example, contain sufficiently high bromide contaminationthat residual chlorine can generate hypobromite as well as bromamines thatcan engage NOM in side reactions after ‘quenching’ (Cavanagh et al. 1992).Further stabilization of analytes can occur with pH adjustment as in the caseof analytes measured by EPA Method 551.1 (US Environmental ProtectionAgency 1995a). This prevents base-catalysed hydrolysis of the non-THM analytesamong those extracted in this method. Although sulphur-reducing agents wereused as disinfectant quenching agents in earlier versions of published methods,they were shown to destroy many of the DBPs these methods were targeting(Bieber & Trehy 1983; Croué & Reckhow 1989). For those DBPs more commonlyproduced during chloramination, such as cyanogen halides, iodinated DBPs andnitrosamines, ascorbic acid is usually the quenching reagent of choice, mostlikely releasing a small amount of ammonia into the water as the ascorbic acidbecomes oxidized. For some analytes, it is possible to use this same reagent undersomewhat controlled conditions for quenching both chlorine and chloramine as,for example, in the case of a method for the analysis of 35 halogenated volatileand semi-volatile DBPs (Chinn et al. 2007). This particular method underscoresthe need for a deep understanding of the complexities of DBP chemistry inaqueous matrices in order to ensure valid application of analytical methods foraccurate quantification of target analytes. In early method development, ascorbicand sulphuric acids were added together and placed in sample collection vialsshipped to sampling sites but it was found that the ascorbic acid could degradein the presence of sulphuric acid prior to the sample event and resulted innot fully dechlorinating samples at the time of sampling. Thus, it was decidedto add ascorbic acid to the sample vials that were shipped out and sulphuricacid was added in the field at the time of sampling. Additionally, only a slightexcess of the quenching agent should be present so some prior knowledge ofexpected disinfectant residual is required for the method performance to be
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

4102 H. S. Weinberg
assured. Moreover, some of the DBPs obtained during extraction in this method,most notably brominated analogues of trichloroacetonitrile and chloropicrin, areunstable in the presence of even a slight excess of ascorbic acid. Consequently,for their analysis, there is a second analytical fraction using ammonium chlorideand sulphuric acid (Krasner et al. 2001; Chinn et al. 2007).
The previous examples show how the complexities of sample preparationand handling can impact the validity of DBP field occurrence data, which arethen highly dependent on having trained sample collectors and strict chain ofcustody documentation with good channels of communication. Sample collectorsmust be given a detailed set of instructions that they must read ahead ofthe sampling event and confirm their understanding. This includes, of course,not rinsing out bottles that contain chemicals for sample preservation. As anaid for collectors, vials and bottles requiring pH adjustment and measurementcan have different colour caps. As part of quality assurance, sample pHshould be rechecked and adjusted if necessary when returned to the analyticallaboratory and all such actions noted on chain of custody documentation. Inthe absence of defined quality assurance in the field and the laboratory, thedata generated from the analysis of micropollutants and especially those DBPsthat are subject to stability and preservation issues cannot be guaranteed to bequantitative.
The second control for analyte preservation requires that no degradation ofthe target DBP occurs during sample holding. In addition to the concerns ofinduced degradation caused by inappropriate use of quenching agents, some DBPsare biodegradable, most notably dihalogenated acetic acids, aldehydes and acids,and, in the absence of a disinfectant residual after quenching, require a biocideto prevent bacterial regrowth (Brophy et al. 1999; US Environmental ProtectionAgency 1998).
3. Chlorination by-products
Chlorine reacts with NOM through a variety of mechanisms includingelectrophilic aromatic substitution, electrophilic addition, oxidation and freeradical reactions (Christman et al. 1983). It will react with both phenolic andnon-phenolic (including both aromatic and non-aromatic) structures in NOM,providing a source for chlorinated by-products produced during disinfection, thefirst of which were THMs identified in the mid-1970s using headspace (Rook 1974)or purge-and-trap extraction (Bellar et al. 1974) both with GC/MS analysis.LLE and GC/ECD were later used and eventually became the routine methodsfor THM analysis once these chemicals had been identified and confirmed tobe among the most prominent of the DBPs formed during the disinfectionof drinking water. Ultimately, another seven DBPs in this neutral extract ofwater were stabilized using ammonium chloride to quench residual chlorine andidentified using LLE–GC/ECD. However, this approach is limited to volatile orsemi-volatile chemicals with low water solubility and assumes that each speciescan be resolved on a GC column and that their retention times match thoseof verifiable standards. A dual-column GC method comparing two stationaryphases was developed by Krasner et al. (2001) because some species coelutedon one column (e.g. trichloro- (TCA) and bromochloroacetaldehydes (BCA) on a
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from
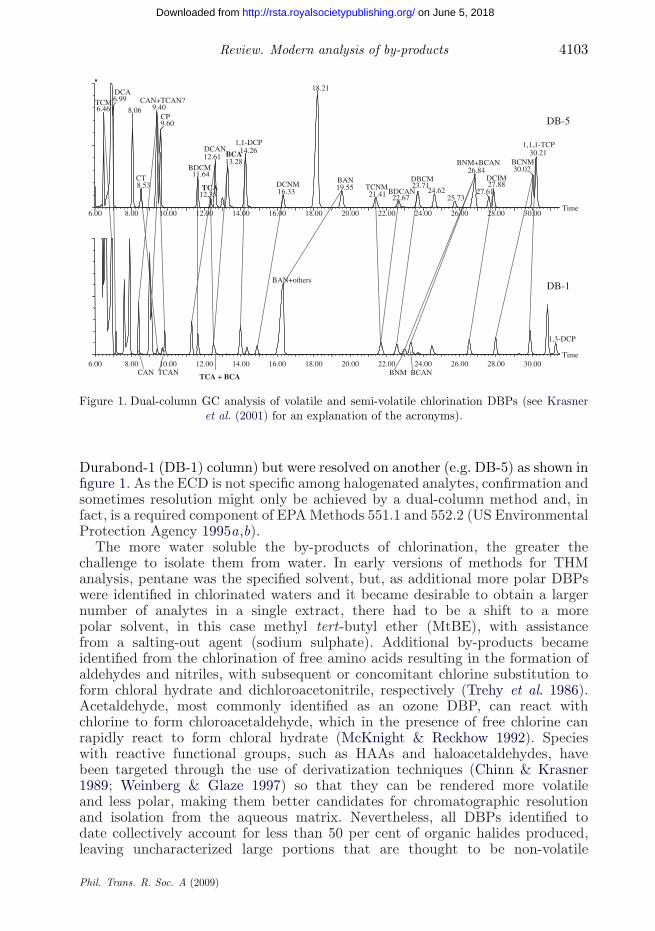
Review. Modern analysis of by-products 4103
DB-5
TCMDCA
CAN+TCAN?
CT
CP
BDCM
TCA
DCAN BCA
1,1-DCP
DCNM BAN TCNM
BDCAN
DBCM
BNM+BCAN
DCIM
BCNM
1,1,1-TCP
6.00 8.00 10.00 12.00 14.00 16.00 18.00 20.00 22.00 24.00 26.00 28.00 30.00Time
6.46
18.21
6.999.408.06
8.53
9.60
14.2612.61
11.64
12.35
13.28
16.33
30.21
26.84
19.55 23.7121.41 22.67
24.6225.73
30.02
27.8827.61
BAN+others
1,3-DCP
CAN TCAN BNM BCAN
DB-1
TCA + BCA
6.00 8.00 10.00 12.00 14.00 16.00 18.00 20.00 22.00 24.00 26.00 28.00 30.00Time
Figure 1. Dual-column GC analysis of volatile and semi-volatile chlorination DBPs (see Krasneret al. (2001) for an explanation of the acronyms).
Durabond-1 (DB-1) column) but were resolved on another (e.g. DB-5) as shown infigure 1. As the ECD is not specific among halogenated analytes, confirmation andsometimes resolution might only be achieved by a dual-column method and, infact, is a required component of EPA Methods 551.1 and 552.2 (US EnvironmentalProtection Agency 1995a,b).
The more water soluble the by-products of chlorination, the greater thechallenge to isolate them from water. In early versions of methods for THManalysis, pentane was the specified solvent, but, as additional more polar DBPswere identified in chlorinated waters and it became desirable to obtain a largernumber of analytes in a single extract, there had to be a shift to a morepolar solvent, in this case methyl tert-butyl ether (MtBE), with assistancefrom a salting-out agent (sodium sulphate). Additional by-products becameidentified from the chlorination of free amino acids resulting in the formation ofaldehydes and nitriles, with subsequent or concomitant chlorine substitution toform chloral hydrate and dichloroacetonitrile, respectively (Trehy et al. 1986).Acetaldehyde, most commonly identified as an ozone DBP, can react withchlorine to form chloroacetaldehyde, which in the presence of free chlorine canrapidly react to form chloral hydrate (McKnight & Reckhow 1992). Specieswith reactive functional groups, such as HAAs and haloacetaldehydes, havebeen targeted through the use of derivatization techniques (Chinn & Krasner1989; Weinberg & Glaze 1997) so that they can be rendered more volatileand less polar, making them better candidates for chromatographic resolutionand isolation from the aqueous matrix. Nevertheless, all DBPs identified todate collectively account for less than 50 per cent of organic halides produced,leaving uncharacterized large portions that are thought to be non-volatile
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

4104 H. S. Weinberg
elute resin with 1.5 ml of MtBEinto 2 ml autosampler vial
rinse 3 ml solid-phase cartridge with8 ml methanol
transfer the final extract to 100 µlvial insert inside autosampler vial
stored at –19°C for 1 h or more
36 l water sample
analysis byGC/MS
SPE extraction at 6 ml min–1
Figure 2. Summary of the SPE GC/MS method used for analysing 35+ chlorination DBPs indrinking water using Bond Elute PPL solid-phase resin (Chinn et al. 2007).
and/or polar and not readily amenable to these techniques of extraction andGC analysis. Some alternative approaches to address these challenges arenow presented.
(a) Solid-phase extraction
In order to attempt identification of levels of DBPs too low to be seen usingdirect extraction and analysis, disinfected water samples can be concentrated byextraction on a solid phase, as demonstrated in figure 2 (Chinn et al. 2007) for theisolation of more than 35 DBPs. The challenge with offline solid-phase extraction(SPE) is to minimize the loss of the target compounds during the severalhours when the aqueous sample is exposed to the laboratory environment ordirect vacuum.
The method of Chinn and colleagues achieved a reasonably high analyterecovery by protecting the extracts from exposure to heat and keeping asheadspace free as possible. This was achieved by using 100 μl conical autosamplervials (that hold approx. 300 μl when filled to the top) for storage rather thanthe typical 2 ml autosampler vials. In order to keep the extracts cool, most
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

Review. Modern analysis of by-products 4105
autosampler trays have the option for circulating a coolant and, as long asthe temperature is kept below 21.0◦C, analyte degradation or volatilization isminimized. An alternative approach is to minimize the number of vials kepton the autosampler tray at any one time. Finally, in this method, it appearsthat individual analyte concentrations up to 500 μg l−1 do not compromise theirsorbent recovery from a typical dechlorinated drinking water.
An alternative methodology for targeting unknown DBPs using a broad-screenapproach adsorbs a large volume of aqueous sample after adjustment to pH 2on XAD-8 over XAD-2, polymeric resins of differing polarity (Richardson et al.1999). A maximum ratio of 770 : 1 (v/v) of water to resin was used to maximizethe adsorption of organic compounds and to minimize the breakthrough. Thecolumns were eluted with ethyl acetate and residual water removed from the ethylacetate eluents by addition of sodium sulphate. Concentration of the extractswas by rotary evaporation followed by evaporation with a gentle stream ofnitrogen (to a final volume of 1 ml) or freeze drying. This approach is mostsuited to semi- or non-volatile chemicals, as off-gassing can occur during sampleprocessing.
With the variety of stationary phases now available, including those withhydrophilic functional groups that are unaffected by water, SPE can increasethe concentration factor for target analytes by up to 1000, achieving nanogramper litre detection of hitherto undetected DBPs. Furthermore, by using massspectrometry for halogenated analyte detection, identification is more specificcompared with the more commonly used ECD. An application to the analysisof HAAs in water that was adjusted between pH 1.5 and 2.0 used SPEwith LiChrolute EN stationary phase, which generated extracts whose analyteswere sensitively determined using ion-pair reverse-phase high-performance liquidchromatography (HPLC) and negative ionization electrospray mass spectrometry(Loos & Barcelo 2001). Quantitation limits were similar to those using LLEGC/ECD but without the need for derivatization.
Another approach to SPE uses a membrane, in the form of a fused silicarod mounted on a syringe device, which is coated with a small volume ofsorbent phase onto which organic components of water can partition whenthe rod is protruded into either the sample or the headspace above it. Theprocess, known as solid-phase microextraction (SPME), has only recentlybeen explored for the analysis of chlorination DBPs; owing to the design ofsorbent phases with varying polarities, SPME offers a solvent-free alternative toexisting extraction techniques, with the added potential of online monitoring.In the earliest reported application, Sclimenti et al. (2002) employed acarboxen/divinylbenzene/polydimethylsiloxane fibre to sample the headspace ofa 5 ml aqueous sample to which sodium chloride (25% by weight) had been added.The targeted DBPs were THMs (including iodinated species), haloacetonitriles,haloketones, halonitromethanes and some miscellaneous compounds, which,with a 30 min adsorption time and a 5 min desorption in a 200◦C injectorof a GC/ECD, produced detection limits below 1.0 μg l−1. In a more recentapplication, Niri et al. (2008) used the same fibre in the headspace of 20 mlsamples containing a 1 per cent salt solution at 35◦C and 2 min contact timefor the isolation of THMs and other volatile halogenated hydrocarbons fromchlorinated waters. The desorbed analytes were analysed on a GC time-of-flight MS instrument whose high sensitivity, fast acquisition rates and powerful
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

4106 H. S. Weinberg
deconvolution software were instrumental in achieving the limits of detection formany of the target analytes at an order of magnitude lower than those obtainedby LLE.
(b) Derivatizations
In the analysis of the regulated five HAAs, the prescribed US EPA Method552 indicates the preferred use of acidic methanol for esterification of theextracted acids in preparation for GC separation (US Environmental ProtectionAgency 1995b). Although this approach has been extended in EPA Method552.3 (US Environmental Protection Agency 2003) for the determination of thenine chlorine- and bromine-containing HAAs using MtBE to allow for higherderivatization temperatures, high conversion of the bromine-containing speciesto their ester counterparts had been previously demonstrated with diazomethane(Brophy et al. 1999). This latter method can be extended for the analysis of3,3-dichloropropenoic acid (Weinberg et al. 2002) and some of the iodine-containing acids (Weissbach & Weinberg 2007). Methylation with eitherdiazomethane or boron trifluoride in methanol has been used for carboxylic acidsand 12 halogenated furanones (Kanniganti et al. 1992; Onstad et al. 2008).
Pentafluorobenzylhydroxylamine (PFBHA) has been demonstrated to be avery effective fingerprinting derivatizing agent for polar aldehydes and ketones(Cancilla & Que Hee 1992; Weinberg & Glaze 1997) and was expanded for theanalysis of haloacetaldehydes (Weinberg et al. 2002). More details are providedon this technique in §5 describing ozonation.
(c) Gas chromatography–mass spectrometry analysis
Although GC has been the traditional separation technique of choice forchlorination-generated DBPs, it is limited by each of the three major hardwarecomponents. The target analytes that are usually concentrated either in asolvent or on a resin need to be volatilized so that they enter the carriergas in their vapour state. Although derivatization can be used to generate amore volatile product than the parent DBP, the injected analytes must stillhave vapour pressures that are compatible with the temperature limitations ofthe injection port, analytical column and detector. This means that DBPs ortheir derivatives with boiling points in excess of 200◦C are the least amenableto GC analysis. Another limitation of the technique is that multiple analytesare collected in a single extract and some of the components in that extractmay be thermally labile, causing them to degrade before they enter thechromatographic column. Trihalonitromethanes fall into this category (Chenet al. 2002) and should be introduced into the injection port of a GC at atemperature no higher than 140◦C. While this might limit the method to theanalysis of those analytes that are more stable, there is also the possibilitythat the degraded compound could generate one of the other analytes in thefraction, leading to a false-positive bias. An example would be the generation oftribromomethyl radicals from bromopicrin in the injector port, which togetherwith an abstracted hydrogen atom from the injected solvent would generatebromoform. Nevertheless, many low-molecular-weight volatile and semi-volatileDBPs have been identified using GC/MS and high-resolution GC/electronionization (EI) MS operating at an accelerating voltage of 8 kV and 10 000
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

Review. Modern analysis of by-products 4107
resolution. This is an extremely powerful tool for broad-screen analysis of DBPsgenerated from the treatment of different drinking waters prior to tentativeidentification (Weinberg et al. 2002).
The primary analytical technique for quantitation of regulated DBP analysis isGC/ECD since the use of GC/MS is sometimes limited. For example, errors canoccur when using single ion monitoring (SIM), the most sensitive technique oncetarget ions associated with the DBP are identified, due to the scan speed. Duringthe acquisition of ions, the dwell time for each charge-to-mass (m/z) measurementmust balance sampling the ion population with collecting as many data pointsper eluting peak as possible. Richardson (2002) determined that, by setting aresidence time of 50 ms per m/z measured and allowing time for the magnetto switch to the next mass, the scan time of 2.17 s/scan only permitted five toeight data points per chromatographic peak, too few for an accurate measure ofintegrated peak area. This is a challenge when attempting to measure a largenumber of analytes in a single chromatographic run, especially if resolution forsome of them is not complete. One approach to offset this is to target differentquantitation masses for the coeluting or closely resolved peaks, assuming that thetargeted ion for one of the analytes is either not present or negligible in the spectraof the chromatographically coeluting analytes. For example, chloropicrin can betargeted at m/z = 117 while bromodichloroacetonitrile, a coeluting species, canbe targeted at m/z = 108.
A slightly different approach from using GC/MS for volatile DBP analysis isthe use of a solvent-free extraction technique such as purge-and-trap or SPME.The latter technique was discussed briefly earlier and has not been widely used forDBP analysis. On the other hand, purge-and-trap techniques have been publishedand applied to the extraction of non-polar volatile DBPs such as THMs using aprocess similar to that depicted in figure 3.
An inert gas is bubbled through the aqueous sample and vaporizes the volatileconstituents, which are then trapped on an adsorbent such as Tenax or activatedcharcoal and condensed. The retained components are then rapidly desorbedinto the injection port of a GC for analysis. The technique, which can be fullyautomated and interfaced with a mass spectrometer, is also appropriate for theanalysis of the iodinated THMs and others listed in table 1. A variation of thistechnique is that of closed-loop stripping (CLS) in which a gas is continuouslycirculated (up to 90 min) through the aqueous sample (typically heated to 60◦C)and through a trap containing a very small amount of activated charcoal. Thevolatiles, trapped on the charcoal, are extracted with 50–100 μl solvent by forcingit backwards and forwards through the charcoal. Although CLS has been usedextensively for taste and odour analysis, it is much more time intensive thanpurge-and-trap analysis, and, since the development of SPME, has not beenwidely used for DBP analysis.
(d) Liquid chromatography/mass spectrometric analysis
Among the known chlorination by-products, some, such as HAAs, aresufficiently polar that they have to be derivatized before being analysed byGC. LC is more amenable to the analysis of polar hydrophilic DBPs in waterowing to the ability to differentiate species based on their polarity indicesusing chromatographic systems with wider choices of stationary and mobile
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

4108 H. S. Weinberg
25 ml sample aliquot
analysis by GC/MS
sparge with helium for11 min onto
VOCARB 4000 trap
desorb trap at 240°C for 4 min
transfer to GC injector
1 µl‘fortification solution’
Figure 3. Flow chart demonstrating the purge-and-trap GC/MS method for analysing volatileDBPs in drinking water.
phases. Liquid chromatography/mass spectrometry (LC/MS) is also not limitedby analyte molecular weight, thermal lability or volatility, yet few DBPs aremeasured using this technique. Part of the challenge is to isolate trace levelsof DBPs in a drinking water matrix where the salt content is several orders ofmagnitude higher. Nine HAAs were successfully isolated from drinking waterby SPE and, using ion-pair HPLC with negative ionization electrospray MS,were detected without the need to derivatize the target analytes (Loos & Barcelo2001). More recently, a chlorinated drinking water containing background levelsof bromide of less than 100 μg l−1 was evaluated for bromine-containing haloacidsafter extracting 1.7 l at a pH below 0.5 by LLE using MtBE. The extract wasroto-evaporated with acetonitrile down to a final volume of 1 ml after mixingwith deionized water and then initially analysed at charge-to-mass ratios of 79and 81 (representing the formation of bromide ion) in the ion scan from anelectrospray ionization (negative potential) triple quadrupole MS. By adjustingthe cone voltages, it was found that lower collision energies were required forgenerating the bromide ion than for chloride (Zhang et al. 2008).
4. Chloramination
Although chloramination is widely practised by allowing a short contact timewith free chlorine before the addition of ammonia, there have been somedemonstrated by-products associated with this process that are distinct from
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

Review. Modern analysis of by-products 4109
those found when using chlorine alone, most notably N -nitrosodimethylamine(NDMA) (Mitch et al. 2003). Moreover, if ozone is used as a pre-oxidant,in the USA it is more likely to be followed by chloramination than by theuse of a free chlorine disinfectant. This combination of processes can changethe dynamics of DBP formation as demonstrated by the following examples.Ozone can produce formaldehyde and acetaldehyde, which, provided there is nosubsequent biofiltration, can react with chloramines to form cyanogen chlorideand dichloroacetaldehyde, respectively, as secondary by-products (Pedersen et al.1999; Krasner et al. 2006). Because chloramine is a much weaker oxidantthan chlorine, the rates of its reaction with precursors are different fromthose of chlorine, which in turn affects the pathways of DBP formation. Forexample, whereas chlorine will rapidly oxidize iodide to iodate, which is ‘inert’to further reaction, chloramine can engage iodide in a series of reactions withNOM to produce iodine-containing DBPs (Bichsel & von Gunten 2000). Theconcentrations of these DBPs can be very much reduced if chloramine is precededby ozonation, since the latter will rapidly oxidize iodide to iodate. DihalogenatedHAAs (Hong et al. 2007) and halonitromethanes (Krasner et al. 2006) are amongthe other DBPs associated with chloramination.
(a) Mass spectrometry
Given that many of the DBPs associated with chloramine have elevatedlevels of geno- and cytotoxicity compared with chlorination by-products (Plewaet al. 2008), there will most likely be a push to reconsider the list of DBPsfor which maximum contaminant levels (MCLs) are established. Most of theidentified chloramination by-products are routinely measured by GC/ECDanalysis and, as highlighted earlier, unless dual-column methods are applied,the identity of detected species can be wrongly attributed. Even with a dual-column method, there may be unknown DBPs or other chemical artefacts thatcould coelute with a target species creating false-positive quantitations. The useof MS detection provides an additional dimension for analyte identification, and,although not full proof, should be considered as a tool for analyte confirmation,especially during this age of more accessible instrumentation. In most cases,MS is less sensitive than ECD, which necessitates a higher degree of analyteconcentration, as illustrated in the analysis of bromoiodoacetamide, observedin some chloraminated waters (Weinberg et al. 2002), in which GC/ECD wascompared with GC/EI-MS (SIM). A particular focus for GC/MS methods hasbeen placed on identifying nitrosamines at low to sub-nanogram per litre levels.One of the earliest publications in this area was for the development of a high-resolution GC/MS method (Taguchi et al. 1994) and later one employing chemicalionization (Yoo et al. 2000). A more sensitive method developed by Cheng et al.(2004) used Ambersorb to achieve minimum reporting levels (MRLs) of 2 ng l−1
for multiple species including NDMA and N -nitrosopiperidine.Zhao et al. (2006) used SPE with LC/MS/MS and identified a new nitrosamine
(N -nitrosodiphenylamine) in chloraminated drinking water, which had beenmissed in GC analysis owing to its thermal instability. Some of the MRLs usingthis approach are not yet as low as those achieved by GC/MS, but with theavailability of higher-capacity solid phases and more sensitive instrumentation,LC/MS is becoming a viable option for analysing some of the more polar DBPs.
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

4110 H. S. Weinberg
Membrane introduction MS uses online pre-concentration through a siliconetube separating the aqueous phase from the ion-trap MS operated in EI modefor cyanogen halides, haloacetonitriles and chloropicrin (Yang et al. 2007).Chloramines are pre-concentrated on the membrane material before thermaldesorption (Riter et al. 2001). This technique has also been used to analyse forvolatile DBPs formed in swimming pools, where the chlorine disinfectant hasreacted with organic nitrogen in the pool to form inorganic chloramines andorganic by-products (Li & Blatchley 2007).
5. Ozonation
Reactions of NOM in drinking water with ozone introduce additional challengesto DBP analysis owing to a multitude of possible reaction pathways. In water,molecular ozone, a highly specific oxidant, can decompose to form hydroxylradicals, which react very quickly with many dissolved species (von Gunten2003a,b). The relative importance of molecular ozone and hydroxyl radicals isdependent on the precise aqueous chemistry. Therefore, poor characterizationof starting materials is further complicated by the typically poor predictabilityof the significance of precise reaction pathways. The combination of thesefactors renders the prediction of the chemical products of drinking waterozonation a challenging task, and in the case of ozone less than 40 percent of its by-products in drinking water, as measured by assimilable organiccarbon, have been identified (Weinberg & Glaze 1997). The elucidation ofthe nature and identity of these ozonation by-products is hindered by twocomplicating factors. The first is the inherent aqueous solubility of manyof these compounds, which renders their efficient extraction from waterdifficult to achieve. The second is the lack of established identity of specificpotential by-products, which complicates targeted analytical approaches. Thesechallenges can be overcome, to some extent, by using methods involving aqueousfunctional group-specific derivatization reactions. These procedures enhancethe extractability of the analytes and provide an analytical ‘flag’ for thespecific compound classes. Derivatized compounds are extracted by LLE orSPE and, in some cases, after further derivatization, analysed by GC/ECDor GC/MS.
Aldehydes and ketones are two classes of organic chemicals known to be formedas by-products of drinking water ozonation (Weinberg et al. 2002). However,current methods of analysis are highly limited in their ability to distinguishbetween specific compounds. The method currently supported by the US EPAincorporates an aqueous-phase derivatization of the carbonyl groups of aldehydesand ketones, which is achieved by the nucleophilic addition of PFBHA to forman oxime, as depicted in figure 4, which is selectively extracted and analysed byGC/ECD (Sclimenti et al. 1990; US Environmental Protection Agency 1998).
By adding a large hydrophobic moiety, PFBHA selectively renders the targetDBPs much more extractable from water. Furthermore, it provides a highlydetectable and identifiable moiety that is sensitive and characteristic to bothelectron capture and MS detectors. In the EPA version of this method (USEnvironmental Protection Agency 1998), specific species’ identification relies onthe comparison of GC peak retention time with authentic analytical standards
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

Review. Modern analysis of by-products 4111
NH2
O
F
F
F
F
F
..
pentafluorobenzylhydroxylamine
pKb = 3.2
C O+HR1
R2
carbonyl attacked fromeither side of plane
pH 4
NH2+
O
F
F
F
F
R1 R2
OH
NHO
F
F
F
F
R1 R2
O+H2
..NH
O
F
F
F
F
F C
F C
F C
R1 R2
–H2O+
(E- and Z-pentafluorobenzyloxime)
NO
F
F
F
F
F C
R1 R2
..
–H+
Figure 4. PFBHA derivatization of carbonyl compounds.
taken through the same method, which has the inherent issue of analytemisidentification. In fact, the issue is exacerbated in this method because ofthe formation of stereoisomers, some of which resolve from each other andsome of which do not. As quantitation is performed through a calibrationproduced external to the sample matrix, it is possible that the relative proportionsof stereoisomers could be different for the same aldehyde concentration ineach matrix. Although the method uses a surrogate standard to identify anypotential matrix impacts on method performance, it may not capture allthese subtleties.
An adaptation of this method using chemical ionization ion-trap MShas been responsible for identifying new ozonation by-products because amolecular ion specific to the oxime product of derivatization is generated. Inthis category, dimethyl glyoxal, trans-2-hexenal and haloacetaldehydes weredetermined (Weinberg et al. 2002). For multifunctional by-products, PFBHA canbe used in conjunction with additional moiety-targeted reactions. In this class,aldo- and ketoacids are first extracted as their oximes from water and the MtBEextract reacted with diazomethane to generate the methyl ester (Weinberg &Glaze 1997). Silylating agents have been employed for targeting hydroxyl-containing molecules (Weinberg & Glaze 1997) and epoxides (Khan et al. 2006),the latter suspected of being transition by-products associated with ozonation.
2,4-Dinitrophenylhydrazine (2,4-DNPH) has been used to target multi-functional carbonyl-containing compounds, which can then be analysed byLC/MS (Zweiner et al. 2002). In this way, using collisionally induced dissociationmass spectrometry, aldehydes and ketones of the same molecular mass could bedistinguished (Richardson 2002).
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

4112 H. S. Weinberg
Highly fluorinated chloroformates were shown to effectively convert a varietyof polar low-molecular-weight compounds in water into more easily extractablederivatives that contained fingerprint moieties that made them easier to identifyin negative chemical ionization MS. Carboxylic acids, alcohols (including phenols)and amines were converted into the corresponding esters, carbonates andcarbamates, respectively, and in bench-scale ozonation of humic acids, new by-products were identified using this technique. These were malic acid, tricarballylicacid and 1,2,3-benzenetricarboxylic acid (Vincenti et al. 2005).
(a) Solid-phase extraction
Aldehydes have been successfully isolated as oximes (after derivatization withPFBHA) in the headspace of aqueous samples by use of a divinylbenzene-polydimethylsilane SPME fibre inserted directly into the injection port of a GCwith ECD. Detection limits can be an order of magnitude lower than thoseobtained by LLE, although fewer analytes have to date been evaluated usingthis technique (Cancho et al. 2001) and the MRLs are very much influenced bythe background aldehyde levels in the blanks.
(b) Mass spectrometry
LC/MS tools have been used to attempt characterization of the highly polarDBP fraction believed to be a major component of the unidentified assimilableorganic carbon generated by ozone. Richardson et al. (1999) described thedevelopment of a 2,4-DNPH derivatization using C18 SPE followed by LC/ESI-MS for identifying highly polar carbonyl DBPs in drinking water. Highly polaruncharged carbonyl compounds were isolated from salts that would otherwiseinterfere with the electrospray process, and low concentrations of analytes couldbe concentrated and measured. Using a gradient LC programme, aldehydeswere easily distinguished from ketones both by their different chromatographicbehaviour and by the presence of a key ion in the collision-induced dissocia-tion mass spectra. One new DBP, 1,3-dihydroxyacetone, along with otherhighly polar DBPs (including pyruvic acid, glyoxylic acid, ketomalonic acid,5-ketohexanal, 6-hydroxy-2-hexanone) were identified in drinking waters treatedwith ozone.
(c) Ion chromatography
Bromate is most often identified as a DBP resulting from the reaction of ozonewith bromide (von Gunten 2003b). Early methods used ion chromatography (IC)with conductivity detection and selective pre-treatment to remove chloride forachieving limits of detection in the 5–10 μg l−1 range (Kuo et al. 1991). With theneed to reach one to two orders of magnitude more sensitivity, procedures weredeveloped that used multiple injections and pre-concentration (Hautman 1992),electrospray IC (Charles et al. 1996) or offline derivatization GC/MS (Nymanet al. 1996), which converts the target compound to another easily detectablespecies. Unfortunately, these methods use several reaction steps involving multiplechemicals, some of which are toxic. Chlorpromazine is one such reagent thatwas used in an IC post-column reaction (PCR) method achieving bromatedetection down to 5.0 μg l−1 (Walters et al. 1997). In a highly sensitive approach,
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

Review. Modern analysis of by-products 4113
a high-capacity anion exchange column was used, allowing larger injectionvolumes of samples to be loaded (Weinberg & Yamada 1998). The resolvedoxyhalide species first pass through a conductivity detector and then undergoPCR with sodium bromide in the presence of nitrous acid. The acidic conditionsare generated from sodium nitrite via a chemical suppressor unit. A stable andhighly chromophoric tribromide species is formed and detected by UV at 267 nm.The commonly occurring anions in typical drinking water samples (i.e. chloride,sulphate, phosphate and nitrate) are invisible to the UV detector and so it is notnecessary to employ chloride removal techniques or pre-concentration to achievesensitive measurements. This approach was used to determine the presence oflow levels of bromate in hypochlorite used for chlorination and suggested thatlowering the MCL for this DBP to meet prescribed cancer risks could not beachieved with the current level of contamination in the commercially availabledisinfectant (Weinberg et al. 2003). EPA Methods 317.0 and 326 were built onthis approach, the former using o-dianisidine, a potential human carcinogen (USEnvironmental Protection Agency 2001), and the latter a catalysed PCR withpotassium iodide (Wagner et al. 2002). IC has also been used in conjunction withinductively coupled plasma–mass spectrometry (ICP/MS) by using large injectionvolumes to achieve sub-microgram per litre detection of bromate (Heitkemperet al. 1994). However, this type of instrumentation is expensive to acquire aswell as to maintain. When IC is coupled with negative ion ESI-MS, limits ofdetection for oxyhalides are in the range 0.05–1 μg l−1 after pre-treatment toremove salts present at one to two orders of magnitude higher concentration(Roehl et al. 2002).
IC is also used for the analysis of low-molecular-weight carboxylic acids withsuppressed conductivity detection. Using a high-capacity ion exchange column,sensitivity of detection can reach 10–20 μg l−1 for formic, propionic, acetic andoxalic acids (Kuo 1998). Many of the acids elute close to the water ‘dip’ atthe front end of the chromatogram, and so the development of higher-resolutioncolumns has permitted better separation of these acids as well as the inclusionof glycolic acid. Their separation from the typical anions present in water at twoto three orders of magnitude higher concentrations and even some of the aldo-and ketoacids was achieved by this technique but sensitivity is not as low asfor those methods already described that use derivatization with LLE GC/ECD.The interface of IC with ESI-MS may expand its evolution as an approach forstudying additional acidic or basic DBPs in drinking water.
6. Conclusions
Approaches to DBP analysis have traditionally focused on those species amenableto solvent extraction and analysis by GC, with perhaps some manipulationof sample pH and/or conversion of polar functional groups into moieties thatenhance isolation from the aquatic matrix and enable detection by a non-specificbut low-priced detector. The literature is awash with descriptions of exotictechnologies to lower detection limits or simplify sample handling, but, otherthan the use of MS detection after the handling of large volumes of samplesto meet sensitivity limitations, there is no widespread use of new techniques tohelp elucidate the identity of the missing DBPs. Instead, broad-screen assays
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

4114 H. S. Weinberg
using large sample volumes that identify potential ‘new’ DBPs at nanogram perlitre concentrations with high cyto- or genotoxic properties have pushed the useof existing analytical approaches to grow ever more sensitive and precise. Therecognition that insufficiently specific detectors such as ECD cannot adequatelyconfirm the presence of a DBP continues to drive the need for MS detection andhigher quality control of the whole sample and data collection process. Drinkingwater is a highly challenging matrix to analyse confidently owing to the presenceof chemicals that can affect analyte stability and the sometimes questionableeffectiveness of extraction and analysis. The more a sample can be analysedwithout human contact, the less prone to contamination and imprecision theapproach will be, and many of the newer techniques described in this paper offerpromising approaches in this direction.
This review benefits from the excellent scientists behind much of the DBP analytical research overthe last 30 years, many of whose papers are cited here, and also others who have toiled silentlyfor years at the laboratory bench. I would like especially to acknowledge those close collaborators(Stuart Krasner at Metropolitan Water District of Southern California based in LaVerne, CA,and Susan Richardson of the US Environmental Protection Agency’s National Exposure ResearchLaboratory in Athens, GA) and the loyal core of graduate students and postdocs who havedeveloped some of these analytical methods in my laboratory. These include (in alphabetical order)Nathalie Bodin, Katherine Brophy, Carrie Delcomyn, Alice Harris, Stuart Khan, Gretchen Onstad,Vasanthi Unnam, Lauren Weinrich and Katja Weissbach.
References
American Public Health Association (APHA), American Water Works Association (AWWA) &Water Environment Federation (WEF). 2006 Standard methods for the examination of waterand wastewater. Washington, DC: APHA.
Bellar, T. A., Lichtenberg, J. J. & Kroner, R. C. 1974 The occurrence of organohalides inchlorinated drinking water. US Environmental Protection Agency Research Report, EPA-670/4-74-008.
Bichsel, Y. & von Gunten, U. 2000 Formation of iodo-trihalomethanes during disinfection andoxidation of iodide-containing waters. Environ. Sci. Technol. 34, 2784–2791. (doi:10.1021/es9914590)
Bieber, T. I. & Trehy, M. L. 1983 Dihaloacetonitriles in chlorinated natural waters. In Waterchlorination: environmental impact and health effects, vol. 4 (eds R. L. Jolley et al.). Ann Arbor,MI: Ann Arbor Science.
Brophy, K. S., Weinberg, H. S. & Singer, P. C. 1999 Quantification of nine haloacetic acids usinggas chromatography with electron capture detection. In Natural organic matter and disinfectionby-products, ACS Symposium Series 761, pp. 343–355. Washington, DC: American ChemicalSociety.
Cancho, B., Ventura, F. & Galceran, M. T. 2001 Determination of aldehydes in drinkingwater using pentafluorobenzylhydroxylamine derivatization and solid-phase microextraction.J. Chromatogr. A 943, 1–13. (doi:10.1016/S0021-9673(01)01437-6)
Cancilla, D. A. & Que Hee, S. S. 1992 O-(2,3,4,5,6-pentafluorophenyl)methylhydroxylaminehydrochloride: a versatile reagent for the determination of carbonyl-containing compounds. J.Chromatogr. A 627, 1–16. (doi:10.1016/0021-9673(92)87181-7)
Cavanagh, J. E., Weinberg, H. S., Gold, A., Sangaiah, R., Marbury, D. & Glaze, W. H. 1992Ozonation by-products: identification of bromohydrins from the ozonation of natural waterswith enhanced bromide levels. Environ. Sci. Technol. 26, 1658–1682. (doi:10.1021/es00032a027)
Charles, L., Pepin, D. & Casetta, B. 1996 Electrospray ion chromatography–tandem massspectrometry of bromate at sub-ppb levels in water. Analyt. Chem. 68, 2554–2558. (doi:10.1021/ac960175k)
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

Review. Modern analysis of by-products 4115
Chen, P. H., Richardson, S. D., Krasner, S. W., Majetich, G. & Glish, G. L. 2002 Hydrogenabstraction and decomposition of bromopicrin and other trihalogenated disinfection byproductsby GC/MS. Environ. Sci. Technol. 36, 3362–3371. (doi:10.1021/es0205582)
Cheng, R. C., Andrews-Tate, C., Hwang, C. J., Guo, Y. C., Grebel, J. E. & Suffet, I. H. M. 2004Development of low cost method(s) for NDMA analysis: round-robin testing. In Proc. 2004AWWA Water Quality Technology Conf. Denver, CO: AWWA.
Chinn R. & Krasner S. W. 1989 A simplified technique for the measurement of halogenated organicacids in drinking water by electron capture gas chromatography. Presented at 28th Pacific Conf.on Chemistry and Spectroscopy, Pasadena, CA.
Chinn, R., Lee, T., Krasner, S. W., Dale, M., Richardson, S., Pressman, J., Speth, T., Miltner,R. & Simmons, J. E. 2007 Solid-phase extraction of 35 DBPs with analysis by GC/ECD andGC/MS. In Proc. 2007 AWWA Water Quality Technology Conf. Denver, CO: AWWA.
Christman, R. F., Norwood, D. L., Millington, D. S., Johnson, J. D. & Stevens, A. A. 1983 Identityand yields of major halogenated products of aquatic fulvic acid chlorination. Environ. Sci.Technol. 17, 625–628. (doi:10.1021/es00116a012)
Croué, J.-P. & Reckhow, D. A. 1989 Destruction of chlorination byproducts with sulfite. Environ.Sci. Technol. 23, 1412. (doi:10.1021/es00069a014)
Hautman, D. 1992 Analysis of trace bromate in drinking water using selective anion concentrationand ion chromatography. In Proc. 1992 AWWA Water Quality Technology Conf., Toronto,Canada. Denver, CO: AWWA.
Heitkemper, D. T., Kaine, L. A., Jackson, D. S. & Wolnik, K. A. 1994 Practical applicationsof element-specific detection by inductively coupled plasma atomic emission spectroscopy andinductively coupled plasma mass spectrometry to ion chromatography of foods. J. Chromatogr.A 671, 101–108. (doi:10.1016/0021-9673(94)80227-0)
Hong, Y., Liu, S., Song, H. & Karanfil, T. 2007 HAA formation during chloramination—significanceof monochloramines’s direct reaction with DOM. J. AWWA 99, 57.
Hua, G. & Reckhow, D. A. 2007 Characterization of disinfection byproduct precursorsbased on hydrophobicity and molecular size. Environ. Sci. Technol. 41, 3309–3315.(doi:10.1021/es062178c)
Kanniganti, R., Johnson, J. D., Ball, L. M. & Charles, M. J. 1992 Identification of compoundsin mutagenic extracts of aqueous monochloraminated fulvic acid. Environ. Sci. Technol. 26,1998–2004. (doi:10.1021/es00034a018)
Khan, S., Weinberg, H. S. & Bedford, E. C. 2006 Aqueous-phase aminolysis: approach for theanalysis of epoxides in water. Analyt. Chem. 78, 2608–2616. (doi:10.1021/ac0516990)
Khiari, D., Krasner, S. W., Hwang, C. J., Chinn, R. & Barrett, S. E. 1997 Effects of chlorinationand chloramination on the molecular weight distribution of natural organic matter and theproduction of high-molecular-weight disinfection by-products. In Proc. 1996 AWWA WaterQuality Technology Conf. Denver, CO: AWWA.
Krasner, S. W., Pastor, S., Chinn, R., Sclimenti, M. J., Weinberg, H. S., Richardson, S. D. &Thruston Jr, A. D. 2001 The occurrence of a new generation of DBPs (beyond the ICR). InProc. 2001 AWWA Water Quality Technology Conf. Denver, CO: AWWA.
Krasner, S. W., Weinberg, H. S., Richardson, S. D., Pastor, S. J., Chinn, R., Sclimenti, M. J.,Onstad, G. D. & Thruston Jr, A. D. 2006 Occurrence of a new generation of disinfectionbyproducts. Environ. Sci. Technol. 40, 7175–7185. (doi:10.1021/es060353j)
Kuo, C.-Y. 1998 Improved application of ion chromatographic determination of carboxylic acids inozonated drinking water. J. Chromatogr. A 804, 265–272. (doi:10.1016/S0021-9673(98)00089-2)
Kuo, C.-Y., Krasner, S. W., Stalker, G. A. & Weinberg, H. S. 1991 Analysis of inorganic disinfectionby-products in ozonated drinking water by ion chromatography. Proc. 1991 AWWA WaterQuality Technology Conf., San Diego, CA. Denver, CO: AWWA.
Li, J. & Blatchley, III E. 2007 Volatile disinfection byproduct formation resulting from chlorinationof organic-nitrogen precursors in swimming pools. Environ. Sci. Technol. 41, 6732–6739.(doi:10.1021/es070871+)
Loos, R. & Barcelo, D. J. 2001 Determination of haloacetic acids in aqueous environments bysolid-phase extraction followed by ion-pair liquid chromatography–electrospray ionization massspectrometric detection. J. Chromatogr. A 938, 45–55. (doi:10.1016/S0021-9673(01)01092-5)
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

4116 H. S. Weinberg
McKnight, A. & Reckhow, D. A. 1992 Reactions of ozonation by-products with chlorine andchloramines. In Proc. 1992 AWWA Annual Conf. (Water Quality), pp. 399-409. Denver, CO:AWWA.
Mitch, W. A., Sharp, J. O., Trussell, R. R., Valentine, R. L., Alvarez-Cohen, L. & Sedlak, D. L.2003 N -Nitrosodimethylamine as a drinking water contaminant: a review. Environ. Eng. Sci.20, 389–404. (doi:10.1089/109287503768335896)
Munch, J. W. & Bassett, M. V. 2004 Method 521: Determination of nitrosamines in drinkingwater by solid phase extraction and capillary column gas chromatography with large volumeinjection and chemical ionization tandem mass spectrometry (MS/MS). Report EPA/600/R-05/054. USEPA Office of Research and Development, National Exposure Research Laboratory,Cincinnati, OH.
Niri, V. H., Bragg, L. & Pawliszyn, J. 2008 Fast analysis of volatile organic compoundsand disinfection by-products in drinking water using solid phase microextraction–gas chromatography/time-of-flight mass spectrometry. J. Chromatogr. A 1201, 222–227.(doi:10.1016/j.chroma.2008.03.062)
Nyman P. J., Canas, B. J., Joe Jr, F. L. & Diachenko, G. W. 1996 Screening method for thegas chromatographic/mass spectrometric determination of microgram/litre levels of bromate inbottled water. Food Addit. Contam. 13, 23–31.
Onstad, G. D., Weinberg, H. S. & Krasner, S. W. 2008 Occurrence of halogenated furanones inU.S. drinking waters. Environ. Sci. Technol. 42, 3341–3348. (doi:10.1021/es071374w)
Pedersen, E. J., Urbansky, E. T., Marinas, B. J. & Margerum, D. W. 1999 Formation of cyanogenchloride from the reaction of monochloramine with formaldehyde. Environ. Sci. Technol. 33,4239–4249. (doi:10.1021/es990153q)
Plewa, M. J., Muellner, M. G., Richardson, S. D., Fasano, F., Buettner, K. M., Woo, Y.-T.,McKague, A. B. & Wagner, E. D. 2008 Occurrence, synthesis, and mammalian cell cytotoxicityand genotoxicity of haloacetamides: an emerging class of nitrogenous drinking water disinfectionbyproducts. Environ. Sci. Technol. 42, 955–961. (doi:10.1021/es071754h)
Richardson, S. D. 2002 The role of GC-MS and LC-MS in the discovery of drinking waterdisinfection by-products. J. Environ. Monit. 4, 1–9.
Richardson, S. D. et al. 1999 Identification of new ozone disinfection byproducts in drinking water.Environ. Sci. Technol. 33, 3368–3377. (doi:10.1021/es981218c)
Riter, L. S., Charles, L., Turowski, M. & Cooks, R. G. 2001 External interface for trap-and-releasemembrane introduction mass spectrometry applied to the detection of inorganic chloramines andchlorobenzenes in water. Rapid Commun. Mass Spectrom. 15, 2290–2295. (doi:10.1002/rcm.489)
Roehl, R., Slingsby, R., Avdalovic, N. & Jackson, P. E. 2002 Applications of ion chromatographywith electrospray mass spectrometric detection to the determination of environmentalcontaminants in water. J. Chromatogr. A 956, 245–254. (doi:10.1016/S0021-9673(02)00041-9)
Rook, J. J. 1974 Formation of haloforms during chlorination of natural water. Water Treat. Exam.23, 234–243.
Sclimenti, M. J., Krasner, S. W., Glaze, W. H. & Weinberg, H. S. 1990 Ozone disinfection by-products: optimization of the PFBHA derivatization method for the analysis of aldehydes. InProc. 1990 AWWA Water Quality Technology Conf., pp. 477–501. Denver, CO: AWWA.
Sclimenti, M. J., Hwang, C. J., Speitel, G. E. & Diehl, A. C. 1995 The simultaneousdetermination of cyanogen chloride and cyanogen bromide in chloraminated waters by asimplified microextraction GC/ECD technique. In Proc. 1994 AWWA Water Quality TechnologyConf. Denver, CO: AWWA.
Sclimenti, M. J., Krasner, S. W. & Richardson, S. D. 2002 The determination of DBPs using asolid-phase microextraction (SPME)–GC/ECD technique. In Proc. 2002 AWWA Water QualityTechnology Conf. Denver, CO: AWWA.
Taguchi, V. Y., Jenkins, S. W. D., Wang, D. T., Palmentier, B. & Reiner, E. J. 1994 Determinationof N -nitrosodimethylamine by isotope dilution, high-resolution mass spectrometry. Can. J.Appl. Spectrosc. 39, 87.
Trehy, M. L., Yost, R. A. & Miles, C. J. 1986 Chlorination byproducts of amino acids in naturalwaters. Environ. Sci. Technol. 20, 1117–1122. (doi:10.1021/es00153a006)
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

Review. Modern analysis of by-products 4117
US Environmental Protection Agency. 1979 National interim primary drinking waterregulations. Control of trihalomethanes in drinking water. Final rule. Fed. Regist. 44,68 624–68 642.
US Environmental Protection Agency. 1995a Method 551.1: Determination of chlorinationdisinfection byproducts, chlorinated solvents, and halogenated pesticides/herbicides in drinkingwater by liquid–liquid extraction and gas chromatography with electron capture detection. InMethods for the Determination of Organic Compounds in Drinking Water, Supplement III.Report EPA-600/R-95/131. USEPA Office of Research and Development, National ExposureResearch Laboratory, Cincinnati, OH.
US Environmental Protection Agency. 1995b Method 552.2: Determination of haloacetic acids anddalapon in drinking water by liquid–liquid extraction, derivatization, and gas chromatographywith electron capture detection. USEPA Office of Research and Development, National ExposureResearch Laboratory, Cincinnati, OH.
US Environmental Protection Agency. 1998 Method 556: Determination of carbonyl compoundsin drinking water by pentafluorobenzylhydroxylamine derivatization and capillary gaschromatography with electron capture detection. USEPA Office of Research and Development,National Exposure Research Laboratory, Cincinnati, OH.
US Environmental Protection Agency. 2001 Method 317.0: Determination of inorganic oxyhalidedisinfection by-products in drinking water using ion chromatography with the addition of apostcolumn reagent for trace bromate analysis. USEPA Office of Ground Water and DrinkingWater, Cincinnati, OH.
US Environmental Protection Agency. 2003 Method 552.3: Determination of haloacetic acidsand dalapon in drinking water by liquid–liquid microextraction, derivatization, and gaschromatography with electron capture detection. USEPA Office of Ground Water and DrinkingWater, Cincinnati, OH.
Vincenti, M., Biazzi, S., Ghiglione, N., Valsania, M. C. & Richardson, S. D. 2005 Comparison ofhighly-fluorinated chloroformates as direct aqueous sample derivatizing agents for hydrophilicanalytes and drinking-water disinfection by-products. J. Am. Soc. Mass Spectrom. 16, 803–813.(doi:10.1016/j.jasms.2005.02.004)
von Gunten, U. 2003a Ozonation of drinking water: part I. Oxidation kinetics and productformation. Water Res. 37, 1443–1467. (doi:10.1016/S0043-1354(02)00457-8)
von Gunten, U. 2003b Ozonation of drinking water: part II. Disinfection and by-productformation in presence of bromide, iodide or chlorine. Water Res. 37, 1469–1487. (doi:10.1016/S0043-1354(02)00458-X)
Wagner, H. P., Pepich, B. V., Hautman, D. P., Munch, D. J., Salhi, E. & von Gunten, U. 2002Method 326.0: Determination of inorganic oxyhalide disinfection by-products in drinking waterusing ion chromatography incorporating the addition of a suppressor acidified post columnreagent for trace bromate analysis. USEPA Office of Ground Water and Drinking Water,Technical Support Center, Cincinnati, OH.
Walters, B. D., Gordon, G. & Bubnis, B. 1997 An ion chromatographic method formeasuring <5 micrograms/L bromate ion in drinking water. Analyt. Chem. 69, 4275–4277.(doi:10.1021/ac9703008)
Weinberg, H. 1999 Disinfection byproducts in drinking water: the analytical challenge. Analyt.Chem. 4, 801A–808A.
Weinberg, H. S. & Glaze, W. H. 1997 A unified approach to the analysis of polar organicbyproducts of oxidation in aqueous matrixes. Water Res. 31, 1555–1572. (doi:10.1016/S0043-1354(96)00262-X)
Weinberg, H. & Yamada, H. 1998 Post ion chromatography derivatization for the analysisof oxyhalides at sub ppb levels in drinking water. Analyt. Chem. 70, 1–6. (doi:10.1021/ac970651m)
Weinberg, H. S., Krasner, S. W., Richardson, S. D. & Thruston Jr, A. D. 2002 The occurrenceof disinfection by-products (DBPs) of health concern in drinking water: results of a nationwideDBP occurrence study. Report EPA/600/R-02/068. USEPA National Exposure ResearchLaboratory, Athens, GA.
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from

4118 H. S. Weinberg
Weinberg, H. S., Unnam, V. P. &. Delcomyn, C. D. 2003 Bromate in chlorinated drinkingwaters: occurrence and implications for future regulation. Environ. Sci. Technol. 37, 3104–3110.(doi:10.1021/es026400z)
Weissbach, K. & Weinberg, H. S. 2007 Iodoacids in our drinking water: the challenge to identifylow-level by-products of chronic health concern. In Proc. 2007 AWWA Water Quality TechnologyConf. Denver, CO: AWWA.
Xie, Y. & Reckhow, D. A. 1992 A new class of ozonation byproducts: the ketoacids. Proc. 1992AWWA Annual Conf. (Water Quality). Denver, CO: AWWA.
Yang, X., Shang, C. & Westerhoff, P. 2007 Factors affecting formation of haloacetonitriles,haloketones, chloropicrin and cyanogen halides during chloramination. Water Res. 41, 1193–1200. (doi:10.1016/j.watres.2006.12.004)
Yoo, L. J., Fitzsimmons, S. & Shen, Y. 2000 Determination of NDMA at part-per-trillion levelsusing positive chemical ionization from aqueous samples. In Proc. 2000 AWWA Water QualityTechnology Conf. Denver, CO: AWWA.
Zhang, X. & Minear, R. A. 2002 Characterization of high molecular weight DBPs resultingfrom chlorination of aquatic humic substances. Environ. Sci. Technol. 36, 4033–4038.(doi:10.1021/es025598k)
Zhang, X., Minear, R. A., Guo, Y., Hwang, C. J., Barrett, S. E., Ikeda, K., Shimizu, Y. &Matsui, S. 2004 An electrospray ionization–tandem mass spectrometry method foridentifying chlorinated drinking water disinfection byproducts. Water Res. 38, 3920–3930.(doi:10.1016/j.watres.2004.06.022)
Zhang, X., Talley, J. W., Boggess, B., Ding, G. & Birdsell, D. 2008 Fast selective detection of polarbrominated disinfection byproducts in drinking water using precursor ion scans. Environ. Sci.Technol. 42, 6598–6603. (doi:10.1021/es800855b)
Zhao, Y.-Y., Boyd, J., Hrudey, S. E. & Li, X.-F. 2006 Characterization of new nitrosamines indrinking water using liquid chromatography tandem mass spectrometry. Environ. Sci. Technol.40, 7636–7641. (doi:10.1021/es061332s)
Zweiner, C., Glauner, T. & Frimmel, F. H. 2002 Method optimization for the determination ofcarbonyl compounds in disinfected water by DNPH derivatization and LC-ESI-MS-MS. Analyt.Bioanalyt. Chem. 372, 615–621. (doi:10.1007/s00216-002-1233-y)
Phil. Trans. R. Soc. A (2009)
on June 5, 2018http://rsta.royalsocietypublishing.org/Downloaded from