modulationoftlr2proteinexpressionbymir-105inhuman ... · lation in human gingival keratinocytes....
TRANSCRIPT

Modulation of TLR2 Protein Expression by miR-105 in HumanOral Keratinocytes*
Received for publication, January 28, 2009, and in revised form, May 22, 2009 Published, JBC Papers in Press, June 9, 2009, DOI 10.1074/jbc.M109.013862
Manjunatha R. Benakanakere‡, Qiyan Li‡, Mehmet A. Eskan‡, Amar V. Singh§¶, Jiawei Zhao‡, Johnah C. Galicia‡,Panagiota Stathopoulou‡, Thomas B. Knudsen§�, and Denis F. Kinane‡1
From the ‡Center for Oral Health and Systemic Disease and the §Department of Molecular, Cellular, and Craniofacial Biology,University of Louisville School of Dentistry, Louisville, Kentucky 40202, the �National Center for Computational Toxicology,Environmental Protection Agency, Research Triangle Park, North Carolina 27711, and ¶Lockheed Martin,Research Triangle Park, North Carolina 27711
Mammalian biological processes such as inflammation,involve regulation of hundreds of genes controlling onset andtermination. MicroRNAs (miRNAs) can translationally represstarget mRNAs and regulate innate immune responses. Ourmodel system comprised primary human keratinocytes, whichexhibited robust differences in inflammatory cytokine produc-tion (interleukin-6 and tumor necrosis factor-�) following spe-cific Toll-like receptor 2 and 4 (TLR-2/TLR-4) agonist chal-lenge. We challenged these primary cells with Porphyromonasgingivalis (a Gram-negative bacterium that triggers TLR-2 andTLR-4) and performedmiRNA expression profiling.We identi-fied miRNA (miR)-105 as a modulator of TLR-2 protein trans-lation in human gingival keratinocytes. There was a stronginverse correlation between cells that had high cytokineresponses following TLR-2 agonist challenge and miR-105 lev-els. Knock-in and knock-down of miR-105 confirmed thisinverse relationship. In silico analysis predicted that miR-105had complementarity for TLR-2 mRNA, and the luciferasereporter assay verified this. Further understanding of the role ofmiRNA in host responses may elucidate disease susceptibilityand suggest new anti-inflammatory therapeutics.
The innate immune response is a crucial first line ofdefense against pathogens. Host detection of microbesoccurs through pattern recognition receptors, includingToll-like receptors (TLRs)2 that are expressed on many cells,including macrophages, monocytes (1), and keratinocytes(2). To date, 11 TLRs have been identified in humans, recog-nizing a range of distinct and conserved microbial molecules(3). TLRs responding to particular pathogens may activatecomplex networks of pathways and interactions, positiveand negative feedback loops, and multifunctional transcrip-
tional responses (4). Among the key downstream targets ofthese networks are NF-�B, mitogen-activated proteinkinases, and members of the IRF family (5). Proper regula-tion of the gene products comprising these networks by tran-scriptional and post-transcriptional processing is not onlyimportant for selective pathogen elimination but also for pre-venting excessive accumulation of cytokines such as interfer-on-�, interferon-�, IL-6, and TNF-� that initiate the hostdefense against microbial attack (6). Deregulated expression ofthese cytokines has been implicated in cancer, autoimmunity,and hyper-inflammatory states (7–9).MicroRNAs (miRNAs) have been implicated in pathway-
level regulation of complex biological processes (10). The roleof miRNA-based regulation of the innate immune responses isa current topic of investigation (11). MammalianmiRNAs are aclass of conserved, small noncoding RNA oligonucleotides thatfunction as negative regulators of translation formultiple targettranscripts (12). Asmany as 5000 distinctmiRNAsmay be tran-scribed and processed in mammalian cells (13–17). MaturemiRNAs bind to specific cognate sequences in the 3�-UTRs oftarget transcripts, resulting in either mRNA degradation orinhibition of translation (12).In mammalian cells, the miRNAs provide a key level of bio-
logical regulation in developmental and differentiation path-ways (18). Deregulation of specificmiRNA abundance has beenassociated with malignancies in the colon, breast, and lung (19,20). Recently, miRNAs have been shown to modulate theNF-�B pathway (miR-146a) (21) and negatively regulateTRAF6, IRAK1 (miR-155) (22), or SOCS3 (miR-203) (23). It ispresently unclear how miRNAs regulate cellular pathways ininnate and inflammatory processes, where precise control ofcomplex networks is needed to engage an appropriate responseto microbes that avoids a cytokine storm.Periodontitis is a common chronic inflammatory condition
affecting 50% of humans that results in loss of bone and teeth(24). This disease is initiated by dental plaque, a microbial bio-film composed mainly of Gram-negative anaerobic bacilli (25,26), including the pathogen Porphyromonas gingivalis. Individ-ual human variability in susceptibility to periodontitis is recog-nized (27) and may involve individual variation in the immuneresponse (25, 26). We identified innate immune variationswithin a bank of over 30 primary human gingival cell cultures(25) based on variations in cytokine response following TLRagonist challenge.
* This work was supported, in whole or in part, by National Institutes of HealthGrant DE017384 (to D. F. K.) from the United States Public Health Service,NIDCR.
1 To whom correspondence should be addressed: University of LouisvilleSchool of Dentistry, 501 South Preston St., Rm. 204, Louisville, KY 40202.Tel.: 502-852-3175; Fax: 502-852-5572; E-mail: [email protected].
2 The abbreviations used are: TLR, Toll-like receptor; HGEC, human gingivalepithelial cell; IL-6, interleukin 6; TNF-�, tumor necrosis factor �; miRNA,microRNA; hsa-miR, Homo sapiens microRNA; pre-miR, precursormicroRNA; UTR, untranslated region; SOCS3, suppressor of cytokine signal-ing 3; FSL-1, Pam2CGDPKHPKSF (a synthetic diacylated lipoprotein and aspecific ligand for TLR-2).
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 284, NO. 34, pp. 23107–23115, August 21, 2009Printed in the U.S.A.
AUGUST 21, 2009 • VOLUME 284 • NUMBER 34 JOURNAL OF BIOLOGICAL CHEMISTRY 23107
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

The present study tested the hypothesis that differentialexpression of miRNAs may account for some of the variabilityin innate immunity. To test this hypothesis, we selected three“normal” and three “diminished” cytokine-response pheno-types.We subjected the corresponding primary human gingivalcell cultures to TLR agonist challenge and profiled the expres-sion of 600 miRNAs. We found strong up-regulation of hsa-miR-105 specifically in the diminished cytokine-response phe-notype and furthermore showed that TLR-2 protein levels weredepressed. This implies a concordant logic circuit in whichmiR-105 inversely regulates TLR-2 function. A computational(in silico) search of the miRNA database revealed that TLR-2transcript is a potential target for miR-105 regulation at the3�-UTR. This binding was confirmed using a linked luciferasereporter gene, and through small interference RNA and inhib-itor (antagomir) studies a functional association with cell sur-face TLR-2 expression. We also confirmed this complementa-rity. We conclude that cell surface TLR-2 expression isinversely regulated by miR-105 expression in human gingivalepithelial cells. This mechanism may reduce inflammatorycytokine production and provide a novel target for therapeuticintervention.
EXPERIMENTAL PROCEDURES
Cell Culture and Challenge Assays—A total of 13 human gin-gival epithelial cells (keratinocytes), with University of Louis-ville IRB approval, were obtained from healthy patients afterthird molar extraction. They were grown as previouslydescribed (28) to sub-confluence, sub-cultured, and challengedas described (2, 29, 30). At confluence, they were challengedwith heat-inactivated P. gingivalis (strain 33277) or 1 �g/mlFSL-1 (Pam2CGDPKHPKSF, a synthetic diacylated lipoproteinand a specific ligand for TLR-2) (InvivoGen, CA). Cells werechallenged for 24 h, and culture supernatants were subjected toIL-6 and TNF-� cytokine levels were measured by enzyme-linked immunosorbent assay (BD Biosciences). The transcrip-tion factor NF-�B assay was performed using a modified elec-trophoretic mobility shift assay technique with TransAMTM
NF-�B enzyme-linked immunosorbent assay kit from Active-Motif (Carlsbad, CA) according to the manufacturer’s instruc-tions. HEK-293 (ATCCnumber: CRL-1573) cells were culturedfollowing ATCC protocol. Briefly, the cell monolayer waswashed and incubatedwith 2–3ml of trypsin-EDTA solution tothe flask and neutralized with trypsin inhibitor after 5min. Thecells were centrifuged and suspended in ATCC-formulatedEagle’s minimum essential medium (catalogue no. 30-2003)with 10% fetal bovine serum (complete medium). The cellswere propagated in complete medium until they were ready fortransfection.miRNA Array Profiling/Analysis—Total RNA was collected
by the TRIzol method and purified with a Qiagen purificationkit (Qiagen), and total RNA quality was analyzed using a Bio-analyzer 2100 (Agilent). Equal amounts of each sample wereused to generate a reference pool. For each array to be hybrid-ized, 2 �g of total RNA from each sample, and the referencepool were labeled with Hy3TM and Hy5TM fluorescent label,respectively, using the miRCURYTM LNA Array labeling kit(Exiqon, Denmark) following the manufacturer’s instructions.
The Hy3TM-labeled sample and the Hy5TM-labeled referencepool RNAweremixed and hybridized to themiRCURYTMLNAarray version 8.1 (Exiqon). The hybridization was performedaccording to the miRCURYTM LNA array manual using aTecan HS4800 hybridization station (Tecan, Austria). ThemiRCURYTM LNA array microarray slides were scanned by aScanArray 4000 XL scanner (Packard Biochip Technologies),and the image analysis was carried out using the ImaGene 7.0software (BioDiscovery, Inc.). Expression ratios were deter-mined for microarray data by computing the background-cor-rected fluorescent signal from the query sample (Q)/referencesample (R). Ratiometric data were transformed to log 2 to pro-duce a continuous spectrum of up- and down-regulated values.Data were normalized by plotting the difference, log 2(Q/R),against the average, (1/2) log 2(Q*R) followed by the applicationof locally weighted regression (lowness) to smooth intensity-dependent ratios. The log 2(Hy3/Hy5) intensity data wereuploaded intoGeneSpring v7.3 for two-way analysis of variance(factors � cell-line and second treatment), using a parametrictest with variances assumed equal, � cutoff (p � 0.05) to gen-erate a heat-map through bi-directional hierarchical clustering(31, 32).TLR-2 mRNA and miR-105 Real-time PCR—Total RNA was
extracted fromcultured cells usingTRIzol reagent (Invitrogen).The isolated total RNA samples were used for first strandcDNA synthesis with specific miR-105 hairpin loop primers(Applied Biosystems, Foster City, CA). Real-time PCRwas per-formed by using 1 ng of cDNAwith anmiR-105-specific primerand probe on an ABI 7500 system (Applied Biosystems) in thepresence of TaqManDNApolymerase. The data were analyzedby normalizing miRNA level to miRNA RNU48 (small nucleo-lar RNA used as internal control, which has least variabilityacross the cell types and challenges). For TLR-2 mRNA quan-tification, the total RNA was converted to single-strandedcDNA using a cDNA archive kit (Applied Biosystems) and 100ng of cDNA to quantify TLR-2 mRNA using the TaqManmethod (Applied Biosystems). Glyceraldehyde-3-phosphatedehydrogenase was the internal control, and –fold increase wascalculated as described (33).Transfection of miRNA—Epithelial cells were transfected
with 100 pmol of miR-105mimic (UCAAAUGCUCAGACUC-CUGUGGU) and miR-105 inhibitor (AGTTTACGAGTCT-GAGGACACCA) (Dharmacon, CA) and co-transfected with100 pmol of small interferenceRNAcontrol, labeledwith 6-car-boxyfluorescein to monitor transfection efficiency. The trans-fection reaction was performed using FuGENE 6 reagent(Roche Applied Science). Cells were challenged with P. gingiva-lis/FSL-1 for 24 h following transfection.Immunohistochemistry—The cells were seeded onto collag-
en-coated chamber glass slides (Lab-TekTM II Chamber Slide�,Nalgene Nunc International, Rochester, NY). At 50–60% con-fluence, the cells were transfected either with miR-105 mimicormiR-105 inhibitor orwith scrambled small interferenceRNAusing FuGENE 6 transfection reagent as described above. Thetransfection reactionwas performed for up to 24 h and replacedwith fresh medium. The challenge assay was performed after48 h of transfection with FSL-1 (0.5 �g/ml, InvitroGen), theywere fixed in 4% paraformaldehyde, permeabilized, and stained
miR-105 in Human Oral Keratinocytes
23108 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 34 • AUGUST 21, 2009
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

with anti-human TLR-2 antibody overnight at 4 °C followed byAlexa Fluor� 488 anti-mouse IgG in 3% bovine serum albumin(1/500, InvitroGen) for 1 h at room temperature and SYTO�83-orange for 15 min. The stained cultures were photographedusing a Confocal Laser Scanning Microscope (FV500, Olym-pus, Melville, NY).Western Blotting—Total protein was extracted from cells
using radioimmune precipitation assay buffer after 24 h of chal-lenge. The Western blot was performed by loading 25 �g oftotal proteins on to each lane. Blottedmembraneswere blockedusing 5% nonfat milk and incubated at 4 °C overnight in anti-TLR-2 antibody (Cell Signaling Technology, Danvers, MA).Themembranes were washed and incubated in anti-mouse IgGconjugated with horseradish peroxidase (Cell Signaling Tech-nology) secondary antibody and signal was developed usingECL plusTM Western blotting detection reagent (AmershamBiosciences). The ratiometric analysis of band intensitywas cal-culated using FluorChem HD software (Alpha Innotech).Luciferase Reporter Assay—The putative miRNA-105 target
site of 52 bp within the 3�-UTR region of human TLR-2mRNA(Ensembl transcript ID: ENST 00000260010) were synthesized
with flanking SpeI andHindIII restriction enzyme sites. In addi-tion, the primers with their putative binding site mutated, werealso synthesized from Integrated DNA Technologies Inc.(sense primers: 5�-gctgactagtCATAGATGATCAAGTCCCT-TATAAGAGTGGCATAGTATTTGCATATAACaagcttggac-3�; antisense primer: 5�-gtccaagcttGTTATATGCAAATACT-ATGCCACTCTTATAAGGGACTTGATCATCTATGactag-tcagc-3�; mutated sense primer: 5�-gctgactagtCATAGATGA-TCAAGTCCCTTATAAGAGTGGCATAGTCATATAACaa-gcttggac-3�; and mutated antisense primer: 5�-gtccaagcttGTT-ATATGACTATGCCACTCTTATAAGGGACTTGATCAT-CTATGactagtcagc-3�). The sense and antisense strands of theoligonucleotides were annealed (34). The annealed oligonu-cleotides were digested with SpeI and HindIII and ligated intothe multiple cloning site of the pMIRREPORT Luciferase vec-tor (Ambion, Inc.). The post-transcriptional regulation ofpMIRREPORT luciferase vector was potentially regulated bymiRNA interactions with the TLR-2 3�-UTR. We then trans-fected cultured HEK293 cells with each of these reporter con-structs (pMIR-TLR2 or pMIR-mutTLR-2), as well as co-trans-fecting them with pMIF-cGFP-Zeo-miR-105 (pMIF-miR-105)
FIGURE 1. Normal and diminished cytokine response cells were challenged with heat-inactivated P. gingivalis (strain 33277) at 100 multiplicity ofinfection for 24 h, and RNA was miRNA microarray profiled using the miRCURY LNA Array (Exiqon). The heat map shows two-way hierarchical clusteringof genes and samples (rows � miRNA, columns � sample). The color scale indicates relative expression: yellow, above mean; blue, below mean; and black, belowbackground. Global microarray expression revealed distinct profiles for 109 miRNAs expressed among these cells (p � 0.05) out of which, 26 well annotatedmiRNAs revealed distinct patterns (p � 0.0037), and miR-105 is represented with a rectangular dotted box (A). This signal is up-regulated in the diminishedcytokine response phenotype after challenging with heat-inactivated P. gingivalis and down-regulated in normal response cells after challenge (p � 0.0017) (B).The stem loop of miR-105 with complimentarity to TLR-2 mRNA and sequence of antagomir used for the present study are shown (C).
miR-105 in Human Oral Keratinocytes
AUGUST 21, 2009 • VOLUME 284 • NUMBER 34 JOURNAL OF BIOLOGICAL CHEMISTRY 23109
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from
plasmid (System Biosciences) following transfection withFuGENE 6 as noted above. Luciferase expression was assessedby confocal microscopy 24 h after transfection by use of anti-Luciferase antibody (Abcam). The protein extract from thetransfected cells was collected using radioimmune precipita-tion assay buffer and equal amounts of protein were tested by aLuciferase activity assay kit following the manufacturer’sinstructions (Stratagene).Statistical Analysis—The microarray statistical analysis is
detailed under “miRNA Array Profiling/Analysis” above. ThemRNA -fold increase data were calculated according to the��CTmethod (33). Cytokine data were evaluated by analysis ofvariance using the InStat program (GraphPad, San Diego, CA)with Bonferroni corrections applied. Statistical differenceswere declared significant at p � 0.05 level. Statistically signifi-
cant data are indicated by asterisks (p � 0.05 (*), p � 0.01 (**),and p � 0.001 (***)).
RESULTS
MiR-105 Is Up-regulated in Human Gingival Keratinocyteswith a Diminished Inflammatory Response—We hypothesizedthat miRNAs may play a role in innate immune response vari-ation (25) and could differentiate periodontitis disease-suscep-tible and disease-resistant subjects. From a bank of over 30 pri-mary cell cultures of human gingival epithelial cells (HGECs)(2) we selected cultures having a diminished cytokine responsetype and “normal” response type as reported previously. Briefly,the rule specification for the latter selected cells that up-regu-lated IL-6 and TNF-� production by at least 2-fold after 4-hchallenge with TLR agonists, and the rule for the former
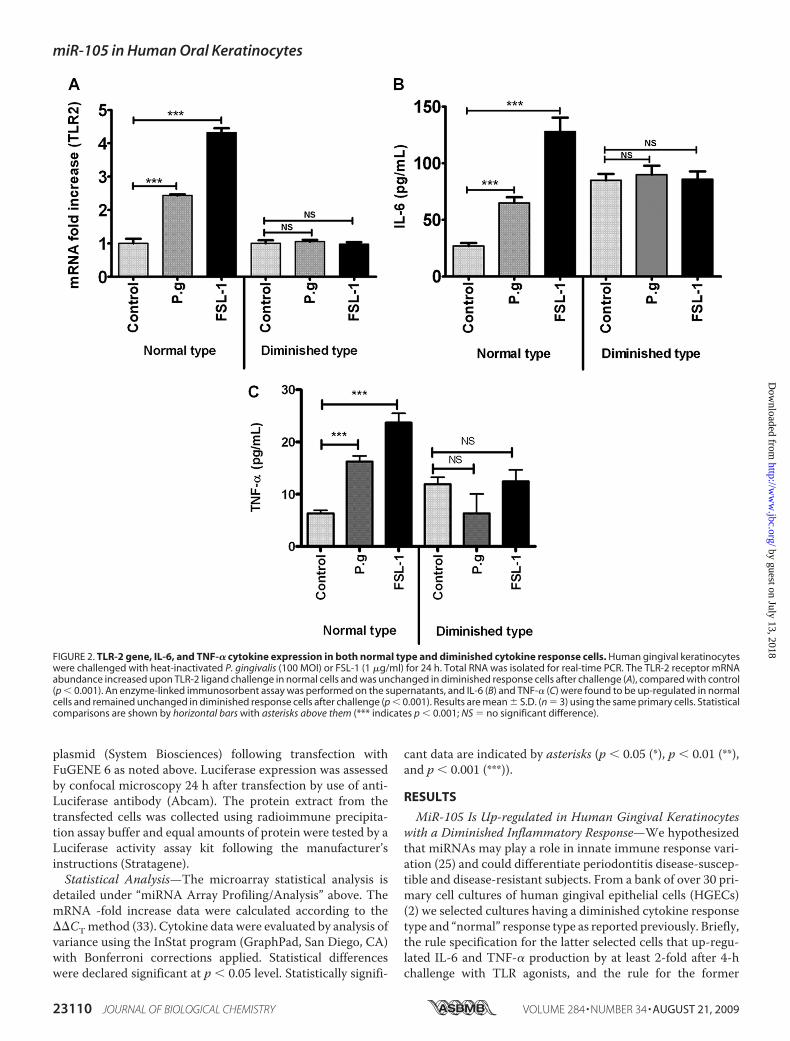
FIGURE 2. TLR-2 gene, IL-6, and TNF-� cytokine expression in both normal type and diminished cytokine response cells. Human gingival keratinocyteswere challenged with heat-inactivated P. gingivalis (100 MOI) or FSL-1 (1 �g/ml) for 24 h. Total RNA was isolated for real-time PCR. The TLR-2 receptor mRNAabundance increased upon TLR-2 ligand challenge in normal cells and was unchanged in diminished response cells after challenge (A), compared with control(p � 0.001). An enzyme-linked immunosorbent assay was performed on the supernatants, and IL-6 (B) and TNF-� (C) were found to be up-regulated in normalcells and remained unchanged in diminished response cells after challenge (p � 0.001). Results are mean � S.D. (n � 3) using the same primary cells. Statisticalcomparisons are shown by horizontal bars with asterisks above them (*** indicates p � 0.001; NS � no significant difference).
miR-105 in Human Oral Keratinocytes
23110 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 34 • AUGUST 21, 2009
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

(diminished) phenotype was no significant increase in pro-in-flammatory cytokine production after challenge. We thustested in depth the 3 diminished cytokine response primarycultures against 3 representatives of the normal HGEC typechosen from themedian of the range of the 13 available cultures(2).We performed chip-based miRNA profiling from normal
and diminished cytokine response cells challenged with heat-inactivated P. gingivalis, a TLR-2 and TLR-4 agonist. The datawere deposited in the Gene Expression Omnibus data baseunder the platform GPL7423 and series GSE13042(www.ncbi.nlm.nih.gov/geo). In the preliminary statisticalanalysis, the normalized miRNA data were analyzed by two-way analysis of variance (phenotype, treatment, and interac-tion) for all miRNA chips. Of the 600 miRNA species tested onthe platform, 95 miRNAs were significantly different betweenphenotypes and 45 between treatments. Among the 109miRNAs that were differentially expressed only 26 were wellannotated. These 26 were significantly altered by challengewith heat-inactivated P. gingivalis when compared withunstimulated cells (p � 0.0038). ThemiR-105 signal was mark-edly down-regulated in normal versus diminished cells afterTLR agonist challenge (Fig. 1A) (p � 0.0017).In silico analyses revealed 1060 hits for miR-105 targets
and predicted complimentarity to TLR-2 (Fig. 1C). Because
we noted miR-105 up-regulationin the diminished cytokine re-sponse cells, we constructed acomputational matrix for miR-105targets, which revealed 37 poten-tial mRNA targets across species.In matrix, hsa-miR-577 and hsa-miR-19b were also differentiallyexpressed between normal anddiminished cells but to a lesserdegree thanmiR-105 (seedotted boxin Fig. 1A). Numerical expressionvalues were plotted to show the dif-ference between normal and dimin-ished cell types (Fig. 1B). For thisreason, we focused on miR-105.TLR-2 mRNAUp-regulation Cor-
relates with Pro-inflammatoryCytokines inGingivalKeratinocytes—Because P. gingivalis signalsthrough both TLR-2 and TLR-4 (2,35) we evaluated TLR-2 mRNA byquantitative real-time PCR afterchallenging both cell types withFSL-1, a specific agonist to TLR-2.Analysis of TLR-2 mRNA abun-dance between phenotypes revealeda 2.5-fold up-regulation with P. gin-givalis challenge and a 4.5-fold up-regulation with FSL-1 challenge rel-ative to the unstimulated control(p � 0.001) (Fig. 2A). In strikingcontrast, diminished cytokine
response cells did not show this up-regulation (Fig. 2A). TLR-2induction upon P. gingivalis or FSL-1 stimulation is consistentwith TLR-2 recognition of P. gingivalis (36, 37). Furthermore,IL-6 cytokine protein levels corresponded to correspondingmRNA levels. Normal response cells up-regulated their IL-6(Fig. 2B) andTNF-� (Fig. 2C) production following P. gingivalisor FSL-1 challenge, and again this response was not evident indiminished response cells (Fig. 2, B and C). We also exploredIL-12p40 secretion in the gingival epithelial cells after P. gingi-valis and FSL-1 challenge. The primary gingival epithelial cellsdo not secret IL-12p40.3 This has to be further verified andhence not included in the present study.Modulation of TLR-2 Protein Expression by miR-105—Con-
firmation of the differential expression of miR-105 was soughtby a non-array method. The real-time PCR data indicated an8-fold up-regulation of miR-105 following P. gingivalis chal-lenge, and an 11-fold increase following FSL-1 challenge, indiminished-response cells relative to an internal benchmark,miRNA RNU48 (Fig. 3, A and B). Consistent with the microar-ray data, the normal response cells did not show significantup-regulation of miR-105 (Fig. 3).
3 M. R. Benakanakere, Q. Li, M. A. Eskan, A. V. Singh, J. Zhao, J. C. Galicia, P.Stathopoulou, T. B. Knudsen, and D. F. Kinane, unpublished data.
FIGURE 3. miR-105 and TLR2 expression in normal and diminished cytokine response cells. The cells weresubjected to P. gingivalis and FSL-1 treatment for 24 h and quantitated the miR-105 expression and Westernblot for TLR-2. Total RNA was amplified with specific miR-105 hairpin loop primers and subjected to real-timePCR. miR-105 up-regulated in diminished cytokine response cells after challenge. The miR-105 expressionshowed minimal or no change in normal cells (A) (p � 0.05). Three normal and three diminished cell types werecompared after P. gingivalis challenge for miR-105 gene expression (B) and TLR-2 protein expression, usingWestern blot (C) with ratio metric analysis (�-Actin/TLR-2) (D). Statistical comparisons are shown by bars withasterisks above them (* indicates p � 0.05; NS � no significant difference).
miR-105 in Human Oral Keratinocytes
AUGUST 21, 2009 • VOLUME 284 • NUMBER 34 JOURNAL OF BIOLOGICAL CHEMISTRY 23111
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from
Search of the open miRNA data base revealed over 1060potential target transcripts for miR-105. These potential targetgenes were loaded onto Ingenuity pathway analysis (IngenuitySystems Inc.) software to discover the most significant path-ways associated with the global miR-105 target genes. Thisanalysis revealed a preponderance of immune diseases (datanot shown) and identified TLR-2 as one of the important tar-gets. Because microarray profiling revealed miR-105 as themost significant miRNA discriminating between normal anddiminished HGEP phenotypes, we tested for evidence thatmiR-105 expressionwould down-regulateTLR-2 aswell as pro-inflammatory cytokines in the 3 selected cell types, as well asacross the broader panel of 13 HGECs available. This analysisrevealed a generalized, strong inverse correlation betweenTLR2 protein and miR-105 gene expression (Fig. 3, B and C).
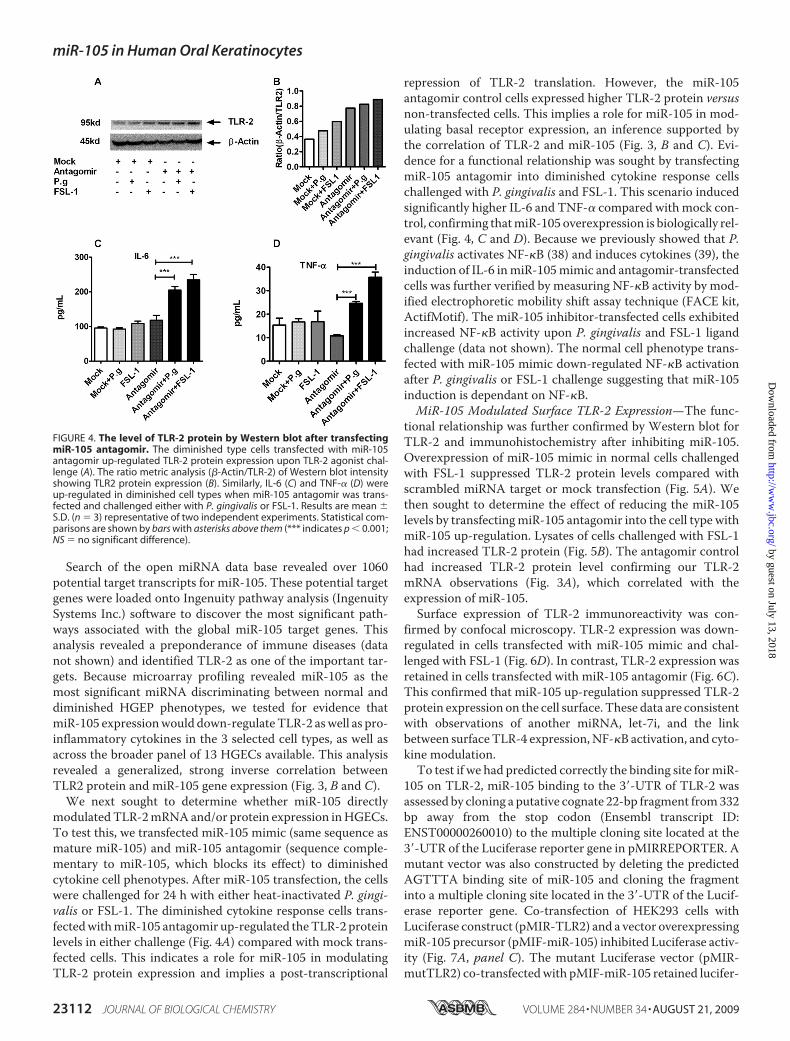
We next sought to determine whether miR-105 directlymodulatedTLR-2mRNAand/or protein expression inHGECs.To test this, we transfected miR-105 mimic (same sequence asmature miR-105) and miR-105 antagomir (sequence comple-mentary to miR-105, which blocks its effect) to diminishedcytokine cell phenotypes. After miR-105 transfection, the cellswere challenged for 24 h with either heat-inactivated P. gingi-valis or FSL-1. The diminished cytokine response cells trans-fectedwithmiR-105 antagomir up-regulated theTLR-2 proteinlevels in either challenge (Fig. 4A) compared with mock trans-fected cells. This indicates a role for miR-105 in modulatingTLR-2 protein expression and implies a post-transcriptional
repression of TLR-2 translation. However, the miR-105antagomir control cells expressed higher TLR-2 protein versusnon-transfected cells. This implies a role for miR-105 in mod-ulating basal receptor expression, an inference supported bythe correlation of TLR-2 and miR-105 (Fig. 3, B and C). Evi-dence for a functional relationship was sought by transfectingmiR-105 antagomir into diminished cytokine response cellschallenged with P. gingivalis and FSL-1. This scenario inducedsignificantly higher IL-6 and TNF-� compared with mock con-trol, confirming thatmiR-105 overexpression is biologically rel-evant (Fig. 4, C and D). Because we previously showed that P.gingivalis activates NF-�B (38) and induces cytokines (39), theinduction of IL-6 inmiR-105mimic and antagomir-transfectedcells was further verified by measuring NF-�B activity by mod-ified electrophoretic mobility shift assay technique (FACE kit,ActifMotif). The miR-105 inhibitor-transfected cells exhibitedincreased NF-�B activity upon P. gingivalis and FSL-1 ligandchallenge (data not shown). The normal cell phenotype trans-fected with miR-105 mimic down-regulated NF-�B activationafter P. gingivalis or FSL-1 challenge suggesting that miR-105induction is dependant on NF-�B.MiR-105 Modulated Surface TLR-2 Expression—The func-
tional relationship was further confirmed by Western blot forTLR-2 and immunohistochemistry after inhibiting miR-105.Overexpression of miR-105 mimic in normal cells challengedwith FSL-1 suppressed TLR-2 protein levels compared withscrambled miRNA target or mock transfection (Fig. 5A). Wethen sought to determine the effect of reducing the miR-105levels by transfectingmiR-105 antagomir into the cell type withmiR-105 up-regulation. Lysates of cells challenged with FSL-1had increased TLR-2 protein (Fig. 5B). The antagomir controlhad increased TLR-2 protein level confirming our TLR-2mRNA observations (Fig. 3A), which correlated with theexpression of miR-105.Surface expression of TLR-2 immunoreactivity was con-
firmed by confocal microscopy. TLR-2 expression was down-regulated in cells transfected with miR-105 mimic and chal-lenged with FSL-1 (Fig. 6D). In contrast, TLR-2 expression wasretained in cells transfected with miR-105 antagomir (Fig. 6C).This confirmed that miR-105 up-regulation suppressed TLR-2protein expression on the cell surface. These data are consistentwith observations of another miRNA, let-7i, and the linkbetween surfaceTLR-4 expression,NF-�B activation, and cyto-kine modulation.To test if we had predicted correctly the binding site formiR-
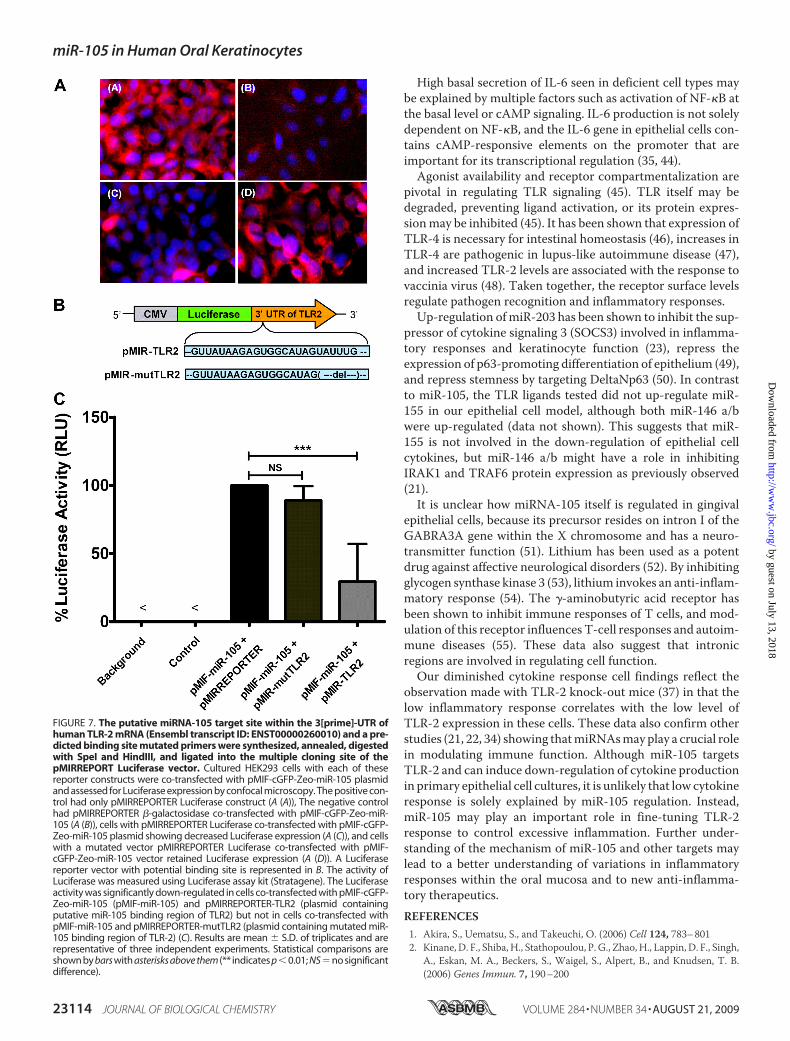
105 on TLR-2, miR-105 binding to the 3�-UTR of TLR-2 wasassessed by cloning a putative cognate 22-bp fragment from332bp away from the stop codon (Ensembl transcript ID:ENST00000260010) to the multiple cloning site located at the3�-UTR of the Luciferase reporter gene in pMIRREPORTER. Amutant vector was also constructed by deleting the predictedAGTTTA binding site of miR-105 and cloning the fragmentinto a multiple cloning site located in the 3�-UTR of the Lucif-erase reporter gene. Co-transfection of HEK293 cells withLuciferase construct (pMIR-TLR2) and a vector overexpressingmiR-105 precursor (pMIF-miR-105) inhibited Luciferase activ-ity (Fig. 7A, panel C). The mutant Luciferase vector (pMIR-mutTLR2) co-transfectedwith pMIF-miR-105 retained lucifer-
FIGURE 4. The level of TLR-2 protein by Western blot after transfectingmiR-105 antagomir. The diminished type cells transfected with miR-105antagomir up-regulated TLR-2 protein expression upon TLR-2 agonist chal-lenge (A). The ratio metric analysis (�-Actin/TLR-2) of Western blot intensityshowing TLR2 protein expression (B). Similarly, IL-6 (C) and TNF-� (D) wereup-regulated in diminished cell types when miR-105 antagomir was trans-fected and challenged either with P. gingivalis or FSL-1. Results are mean �S.D. (n � 3) representative of two independent experiments. Statistical com-parisons are shown by bars with asterisks above them (*** indicates p � 0.001;NS � no significant difference).
miR-105 in Human Oral Keratinocytes
23112 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 34 • AUGUST 21, 2009
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

ase activity (Fig. 7A, panelD). Cell lysates had significantly lessluciferase from samples co-transfected with pMIR-TLR2and pMIF-miR-105 (Fig. 7B). This confirms our in silico pre-
diction of the binding site. Wemay conclude that modulation ofTLR-2 expression by miR-105occurs through binding to the3�-UTR of TLR-2 mRNA, thusinhibiting TLR-2 translation.
DISCUSSION
This study identified miR-105 as amodulator of TLR-2 protein transla-tion in human gingival keratinocytes.There was a strong inverse correla-tion between cells that naturally hadhigh cytokine responses followingTLR-2 agonist challenge and miR-105 levels. Knock-in andknock-downof miR-105 confirmed this inverserelationship. In silico analysis pre-dicted thatmiR-105hadcomplemen-tarity forTLR-2mRNA, and the lucif-erase reporter assay verified this.Recently, miRNAs have been
shown to fine-tune innate immuneresponses (40). For example, miR-146a/b was up-regulated in an NF-�B-dependent manner (21). Inanother study, IL-6 induced let-7a-modulated apoptosis in cholan-giocytes (41) and up-regulatedmiR-19a and -19bwhile down-regulatingSOCS1 (suppressor of cytokine sig-naling 1), a gene important in nega-tive regulation of TLR signaling(42). The present study adds miR-105 to the panel of miRNA speciesknown to influence innate immunefunction.Human miR-105 is located on
the intronic region of GABRA3A(�-aminobutyric acid receptor 3�),which resides on the X chromosome.Certain typesof tumorcellshavebeenshown to transcribemiR-105 but lackprocessing machinery in the nucleusto formmature miRNA (43). It is stillunclear how miR-105 is processedand exported out of the nucleus ingingival epithelial cells. Perhaps it isanalogous to miR-155, which is pres-ent on the B-cell integration clustertranscript up-regulated with polyri-boinosinic:polyribocytidylic acidor thecytokine interferon-� challenge.The up-regulated B-cell integrationcluster transcript withmiR-155 pre-
cursor undergoes processing to export mature miR-155 out ofthe nucleus, which suppresses the macrophage inflammatoryresponse via c-Jun NH2-terminal kinase pathway (21).
FIGURE 5. The expression of TLR-2in epithelial cells following miR-105 mimic and antagomir transfection.The normal cells were transfected with miR-105 mimic, and diminished cells were transfected with miR-105antagomir and challenged with FSL-1 (1 �g/ml) for 24 h. Protein (20 �g) was loaded onto each well and detected byanti-TLR-2 antibody for chemiluminescence detection. The normal cells transfected with miR-105 down-regulatedTLR-2 protein expression (A), and diminished cell type transfected with miR-105 antagomir up-regulated TLR-2protein level (B), ratios of Western blot intensity data (�-Actin/TLR2) are represented in C and D, respectively.
FIGURE 6. Surface expression of TLR-2 in gingival keratinocytes. Normal cells transfected with either miR-105mimic or antagomir and stained with antiTLR-2 antibody-clone TL2.3 (eBiosciences) with proper Isotype control(Mouse IgG2a). TLR-2 was detected by immunohistochemistry and photographed by confocal microscopy. Control(A), FSL-1-treated cells increased TLR-2 expression (B) miR-105 inhibitor did not affect the surface expression ofTLR-2, but the expression was maximized after FSL-1 challenge (C), miR-105 mimic suppressed TLR-2 surface expres-sion even after challenging with FSL-1 as seen by confocal microscopy (D). Bright field overlay with merged SYTO�83-orange-stained nucleus and Alexa Fluor� 488-stained TLR2 (i), SYTO� 83-orange (ii), Alexa Fluor� 488 (iii), andmerged image (iv).
miR-105 in Human Oral Keratinocytes
AUGUST 21, 2009 • VOLUME 284 • NUMBER 34 JOURNAL OF BIOLOGICAL CHEMISTRY 23113
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from
High basal secretion of IL-6 seen in deficient cell types maybe explained by multiple factors such as activation of NF-�B atthe basal level or cAMP signaling. IL-6 production is not solelydependent on NF-�B, and the IL-6 gene in epithelial cells con-tains cAMP-responsive elements on the promoter that areimportant for its transcriptional regulation (35, 44).Agonist availability and receptor compartmentalization are
pivotal in regulating TLR signaling (45). TLR itself may bedegraded, preventing ligand activation, or its protein expres-sionmay be inhibited (45). It has been shown that expression ofTLR-4 is necessary for intestinal homeostasis (46), increases inTLR-4 are pathogenic in lupus-like autoimmune disease (47),and increased TLR-2 levels are associated with the response tovaccinia virus (48). Taken together, the receptor surface levelsregulate pathogen recognition and inflammatory responses.Up-regulation ofmiR-203 has been shown to inhibit the sup-
pressor of cytokine signaling 3 (SOCS3) involved in inflamma-tory responses and keratinocyte function (23), repress theexpression of p63-promoting differentiation of epithelium (49),and repress stemness by targeting DeltaNp63 (50). In contrastto miR-105, the TLR ligands tested did not up-regulate miR-155 in our epithelial cell model, although both miR-146 a/bwere up-regulated (data not shown). This suggests that miR-155 is not involved in the down-regulation of epithelial cellcytokines, but miR-146 a/b might have a role in inhibitingIRAK1 and TRAF6 protein expression as previously observed(21).It is unclear how miRNA-105 itself is regulated in gingival
epithelial cells, because its precursor resides on intron I of theGABRA3A gene within the X chromosome and has a neuro-transmitter function (51). Lithium has been used as a potentdrug against affective neurological disorders (52). By inhibitingglycogen synthase kinase 3 (53), lithium invokes an anti-inflam-matory response (54). The �-aminobutyric acid receptor hasbeen shown to inhibit immune responses of T cells, and mod-ulation of this receptor influences T-cell responses and autoim-mune diseases (55). These data also suggest that intronicregions are involved in regulating cell function.Our diminished cytokine response cell findings reflect the
observation made with TLR-2 knock-out mice (37) in that thelow inflammatory response correlates with the low level ofTLR-2 expression in these cells. These data also confirm otherstudies (21, 22, 34) showing thatmiRNAsmayplay a crucial rolein modulating immune function. Although miR-105 targetsTLR-2 and can induce down-regulation of cytokine productionin primary epithelial cell cultures, it is unlikely that low cytokineresponse is solely explained by miR-105 regulation. Instead,miR-105 may play an important role in fine-tuning TLR-2response to control excessive inflammation. Further under-standing of the mechanism of miR-105 and other targets maylead to a better understanding of variations in inflammatoryresponses within the oral mucosa and to new anti-inflamma-tory therapeutics.REFERENCES1. Akira, S., Uematsu, S., and Takeuchi, O. (2006) Cell 124, 783–8012. Kinane, D. F., Shiba, H., Stathopoulou, P. G., Zhao,H., Lappin, D. F., Singh,
A., Eskan, M. A., Beckers, S., Waigel, S., Alpert, B., and Knudsen, T. B.(2006) Genes Immun. 7, 190–200
FIGURE 7. The putative miRNA-105 target site within the 3[prime]-UTR ofhuman TLR-2 mRNA (Ensembl transcript ID: ENST00000260010) and a pre-dicted binding site mutated primers were synthesized, annealed, digestedwith SpeI and HindIII, and ligated into the multiple cloning site of thepMIRREPORT Luciferase vector. Cultured HEK293 cells with each of thesereporter constructs were co-transfected with pMIF-cGFP-Zeo-miR-105 plasmidand assessed for Luciferase expression by confocal microscopy. The positive con-trol had only pMIRREPORTER Luciferase construct (A (A)), The negative controlhad pMIRREPORTER �-galactosidase co-transfected with pMIF-cGFP-Zeo-miR-105 (A (B)), cells with pMIRREPORTER Luciferase co-transfected with pMIF-cGFP-Zeo-miR-105 plasmid showing decreased Luciferase expression (A (C)), and cellswith a mutated vector pMIRREPORTER Luciferase co-transfected with pMIF-cGFP-Zeo-miR-105 vector retained Luciferase expression (A (D)). A Luciferasereporter vector with potential binding site is represented in B. The activity ofLuciferase was measured using Luciferase assay kit (Stratagene). The Luciferaseactivity was significantly down-regulated in cells co-transfected with pMIF-cGFP-Zeo-miR-105 (pMIF-miR-105) and pMIRREPORTER-TLR2 (plasmid containingputative miR-105 binding region of TLR2) but not in cells co-transfected withpMIF-miR-105 and pMIRREPORTER-mutTLR2 (plasmid containing mutated miR-105 binding region of TLR-2) (C). Results are mean � S.D. of triplicates and arerepresentative of three independent experiments. Statistical comparisons areshown by bars with asterisks above them (** indicates p�0.01; NS�no significantdifference).
miR-105 in Human Oral Keratinocytes
23114 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 34 • AUGUST 21, 2009
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

3. Akira, S., and Takeda, K. (2004) Nat. Rev. Immunol. 4, 499–5114. Bartel, D. P. (2004) Cell 116, 281–2975. Liew, F. Y., Xu, D., Brint, E. K., andO’Neill, L. A. (2005)Nat. Rev. Immunol.
5, 446–4586. Lynn, D. J., Winsor, G. L., Chan, C., Richard, N., Laird, M. R., Barsky, A.,
Gardy, J. L., Roche, F.M., Chan, T. H., Shah, N., Lo, R., Naseer, M., Que, J.,Yau, M., Acab, M., Tulpan, D., Whiteside, M. D., Chikatamarla, A., Mah,B., Munzner, T., Hokamp, K., Hancock, R. E., and Brinkman, F. S. (2008)Mol. Syst. Biol. 4, 218
7. Farh, K. K., Grimson, A., Jan, C., Lewis, B. P., Johnston, W. K., Lim, L. P.,Burge, C. B., and Bartel, D. P. (2005) Science 310, 1817–1821
8. Gregory, R. I., Yan, K. P., Amuthan, G., Chendrimada, T., Doratotaj, B.,Cooch, N., and Shiekhattar, R. (2004) Nature 432, 235–240
9. Chendrimada, T. P., Gregory, R. I., Kumaraswamy, E., Norman, J., Cooch,N., Nishikura, K., and Shiekhattar, R. (2005) Nature 436, 740–744
10. Ambros, V. (2004) Nature 431, 350–35511. Fritz, J. H., Girardin, S. E., and Philpott, D. J. (2006) Sci STKE 2006, pe2712. Lee, Y., Kim, M., Han, J., Yeom, K. H., Lee, S., Baek, S. H., and Kim, V. N.
(2004) EMBO J. 23, 4051–406013. Fazi, F., Rosa, A., Fatica, A., Gelmetti, V., DeMarchis, M. L., Nervi, C., and
Bozzoni, I. (2005) Cell 123, 819–83114. O’Donnell, K. A., Wentzel, E. A., Zeller, K. I., Dang, C. V., and Mendell,
J. T. (2005) Nature 435, 839–84315. Pasquinelli, A. E., Hunter, S., and Bracht, J. (2005) Curr. Opin. Genet. Dev.
15, 200–20516. Chen, C. Z., Li, L., Lodish, H. F., and Bartel, D. P. (2004) Science 303,
83–8617. Monticelli, S., Ansel, K. M., Xiao, C., Socci, N. D., Krichevsky, A.M., Thai,
T. H., Rajewsky, N., Marks, D. S., Sander, C., Rajewsky, K., Rao, A., andKosik, K. S. (2005) Genome Biol. 6, R71
18. Esau, C., Kang, X., Peralta, E., Hanson, E.,Marcusson, E. G., Ravichandran,L. V., Sun, Y., Koo, S., Perera, R. J., Jain, R., Dean, N. M., Freier, S. M.,Bennett, C. F., Lollo, B., and Griffey, R. (2004) J. Biol. Chem. 279,52361–52365
19. Calin, G. A., Sevignani, C., Dumitru, C. D., Hyslop, T., Noch, E., Yen-damuri, S., Shimizu, M., Rattan, S., Bullrich, F., Negrini, M., and Croce,C. M. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 2999–3004
20. Lu, J., Getz, G., Miska, E. A., Alvarez-Saavedra, E., Lamb, J., Peck, D.,Sweet-Cordero, A., Ebert, B. L.,Mak, R.H., Ferrando, A. A., Downing, J. R.,Jacks, T., Horvitz, H. R., and Golub, T. R. (2005) Nature 435, 834–838
21. Taganov, K. D., Boldin, M. P., Chang, K. J., and Baltimore, D. (2006) Proc.Natl. Acad. Sci. U.S.A. 103, 12481–12486
22. O’Connell, R. M., Taganov, K. D., Boldin, M. P., Cheng, G., and Baltimore,D. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 1604–1609
23. Sonkoly, E.,Wei, T., Janson, P. C., Saaf, A., Lundeberg, L., Tengvall-Linder,M., Norstedt, G., Alenius, H., Homey, B., Scheynius, A., Ståhle, M., andPivarcsi, A. (2007) PLoS ONE 2, e610
24. Kinane, D. F. (2000) Ann. R. Australas Coll. Dent. Surg. 15, 42–5025. Kinane, D. F., Galicia, J. C., Gorr, S. U., Stathopoulou, P. G., and Benaka-
nakere, M. (2008) Front. Biosci. 13, 966–98426. Spratt, D. (2003) in Medical Biofilms: Detection, Prevention, and Control
(Jass, J., Surman, S., andWalker, J., eds) pp. 175–198,Wiley and Sons, Ltd.,London
27. Kinane,D. F., andHart, T. C. (2003)Crit. Rev.Oral Biol.Med.14, 430–44928. Shiba, H., Venkatesh, S. G., Gorr, S. U., Barbieri, G., Kurihara, H., and
Kinane, D. F. (2005) J. Periodontal Res. 40, 153–15729. Eskan, M. A., Rose, B. G., Benakanakere, M. R., Zeng, Q., Fujioka, D.,
Martin, M. H., Lee, M. J., and Kinane, D. F. (2008) Eur. J. Immunol. 38,1138–1147
30. Eskan,M. A., Benakanakere, M. R., Rose, B. G., Zhang, P., Zhao, J., Statho-poulou, P., Fujioka, D., and Kinane, D. F. (2008) Infect. Immun. 76,2080–2089
31. Eisen, M. B., Spellman, P. T., Brown, P. O., and Botstein, D. (1998) Proc.Natl. Acad. Sci. U.S.A. 95, 14863–14868
32. Singh, A. V., Green, M., States, J. C., and Knudsen, T. B. (2007) BirthDefects Res. Part A. Clin. Mol. Teratol. 79, 319–350
33. Livak, K. J., and Schmittgen, T. D. (2001)Methods 25, 402–40834. Chen, X. M., Splinter, P. L., O’Hara, S. P., and LaRusso, N. F. (2007) J. Biol.
Chem. 282, 28929–2893835. Eskan,M. A., Hajishengallis, G., and Kinane, D. F. (2007) Infect Immun 75,
892–89836. Darveau, R. P., Pham, T. T., Lemley, K., Reife, R. A., Bainbridge, B. W.,
Coats, S. R., Howald, W. N., Way, S. S., and Hajjar, A. M. (2004) Infect.Immun. 72, 5041–5051
37. Burns, E., Bachrach, G., Shapira, L., and Nussbaum, G. (2006) J. Immunol.177, 8296–8300
38. Brozovic, S., Sahoo, R., Barve, S., Shiba, H., Uriarte, S., Blumberg, R. S., andKinane, D. F. (2006)Microbiology 152, 797–806
39. Sandros, J., Karlsson, C., Lappin, D. F., Madianos, P. N., Kinane, D. F., andPapapanou, P. N. (2000) J. Dent. Res. 79, 1808–1814
40. Gantier, M. P., Sadler, A. J., andWilliams, B. R. (2007) Immunol. Cell Biol.85, 458–462
41. Meng, F., Henson, R., Wehbe-Janek, H., Smith, H., Ueno, Y., and Patel, T.(2007) J. Biol. Chem. 282, 8256–8264
42. Pichiorri, F., Suh, S. S., Ladetto, M., Kuehl, M., Palumbo, T., Drandi, D.,Taccioli, C., Zanesi, N., Alder, H., Hagan, J. P., Munker, R., Volinia, S.,Boccadoro, M., Garzon, R., Palumbo, A., Aqeilan, R. I., and Croce, C. M.(2008) Proc. Natl. Acad. Sci. U.S.A. 105, 12885–12890
43. Lee, E. J., Baek,M., Gusev, Y., Brackett, D. J., Nuovo, G. J., and Schmittgen,T. D. (2008) RNA 14, 35–42
44. Krueger, J., Ray, A., Tamm, I., and Sehgal, P. B. (1991) J. Cell. Biochem. 45,327–334
45. Miggin, S. M., and O’Neill, L. A. (2006) J. Leukoc. Biol. 80, 220–22646. Rakoff-Nahoum, S., Paglino, J., Eslami-Varzaneh, F., Edberg, S., and
Medzhitov, R. (2004) Cell 118, 229–24147. Liu, B., Yang, Y., Dai, J., Medzhitov, R., Freudenberg, M. A., Zhang, P. L.,
and Li, Z. (2006) J. Immunol. 177, 6880–688848. Zhu, J., Martinez, J., Huang, X., and Yang, Y. (2007) Blood 109, 619–62549. Yi, R., Poy, M. N., Stoffel, M., and Fuchs, E. (2008) Nature 452, 225–22950. Lena, A.M., Shalom-Feuerstein, R., di Val Cervo, P., Aberdam, D., Knight,
R. A., Melino, G., and Candi, E. (2008) Cell Death Differ. 15, 1187–119551. Bell,M. V., Bloomfield, J.,McKinley,M., Patterson,M.N., Darlison,M.G.,
Barnard, E. A., and Davies, K. E. (1989) Am. J. Hum. Genet. 45, 883–88852. Rowe, M. K., and Chuang, D. M. (2004) Expert Rev. Mol Med. 6, 1–1853. Phiel, C. J., and Klein, P. S. (2001) Annu. Rev. Pharmacol. Toxicol. 41,
789–81354. Martin, M., Rehani, K., Jope, R. S., andMichalek, S. M. (2005)Nat. Immu-
nol. 6, 777–78455. Tian, J., Chau, C., Hales, T. G., and Kaufman, D. L. (1999) J. Neuroimmu-
nol. 96, 21–28
miR-105 in Human Oral Keratinocytes
AUGUST 21, 2009 • VOLUME 284 • NUMBER 34 JOURNAL OF BIOLOGICAL CHEMISTRY 23115
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

KinaneZhao, Johnah C. Galicia, Panagiota Stathopoulou, Thomas B. Knudsen and Denis F. Manjunatha R. Benakanakere, Qiyan Li, Mehmet A. Eskan, Amar V. Singh, Jiawei
KeratinocytesModulation of TLR2 Protein Expression by miR-105 in Human Oral
doi: 10.1074/jbc.M109.013862 originally published online June 9, 20092009, 284:23107-23115.J. Biol. Chem.
10.1074/jbc.M109.013862Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/284/34/23107.full.html#ref-list-1
This article cites 53 references, 20 of which can be accessed free at
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from