molecular characterization of the von hippel-lindau … · molecular characterization of the von...
TRANSCRIPT

Molecular Characterization of the von Hippel-Lindau Tumour Suppressor Protein
By
Ryan Charles Russell
A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy
Laboratory Medicine and Pathobiology University of Toronto
© Copyright by Ryan Charles Russell 2009

ii
Molecular Characterization of the von Hippel-Lindau
Tumour Suppressor Protein
Ryan Charles Russell
Doctor of Philosophy
Laboratory Medicine and Pathobiology University of Toronto
2009
Abstract
Inheritance of one mutant von Hippel-Lindau (VHL) allele gives rise to the development of the
autosomal dominant VHL disease, which affects approximately 1 in 36 000 individuals. The
VHL tumour suppressor protein plays a critical role in the E3 ubiquitin ligase-mediated
destruction of hypoxia-inducible factor (HIF) and the promotion of fibronectin extracellular
matrix assembly. A failure in either process is associated with oncogenic progression. Work
included in this thesis provides evidence that these tumour suppressor functions are mutually
exclusive. Additionally, post-translational modification of VHL by NEDD8 is shown to act as a
‘molecular switch’, altering VHL protein associations and providing a mechanism of pathway
segregation. As a result of HIF stabilization, the expression of a homophilic adhesion molecule
E-cadherin is significantly down-regulated in primary renal clear-cell carcinoma (RCC) upon
VHL loss. E-cadherin down-regulation is shown to increase the invasive potential and is of
prognostic value in RCC. Finally, VHL and SOCS1 are shown to dimerize and negatively
regulate the JAK2-STAT5 signalling cascade. Defects in this dimerization are shown to underlie
Chuvash polycythemia and provide a molecular understanding of the phenotypic observations
associated with VHL-related polycythemias.

iii
Acknowledgments
First, I would like to thank my supervisor, Dr. Michael Ohh, for his mentoring throughout my
PhD. His unquenchable enthusiasm for science and constant encouragement enabled me to push
my boundaries and become a better scientist. I would also like to thank my supervisory
committee, Dr. Eldad Zacksenhaus and Dr. Meredith Irwin, who have provided important
insight, advice and support that has been essential to my success. The members of the Ohh lab
have continually provided fruitful discussions that have impacted my studies. In particular I
would like to thank Dr. Olga Roche for her excellent collaboration in our joint study of E-
cadherin. I would also like to thank Roxana Sufan for her essential contributions to our joint
study of Chuvash Polycythemia. In addition, I would like to acknowledge the excellent
cooperation of Julie Metcalf in our joint study of many intriguing aspects of tumour biology, and
Stephanie Sybingco for her administrative prowess. I would also note our outstanding
collaborations with Drs. Andrew Evans, Kyle Furge and Bin Teh, which have given our research
a breadth and clinical significance that would be otherwise lacking.
I would like to thank my parents Tom and Laurie Russell for their constant support and
encouragement. Finally, I would like to thank my wife Kiely for her amazing patience,
understanding and support.

iv
Table of Contents
Abstract ........................................................................................................................................................................ii
Acknowledgments.......................................................................................................................................................iii
Table of Contents........................................................................................................................................................iv
List of Abbreviations .................................................................................................................................................vii
List of Tables...............................................................................................................................................................xi
List of Figures ............................................................................................................................................................xii
Chapter 1 Introduction to the von Hippel-Lindau tumour
suppressor ....................................................................................................................................................................1
1.1 VHL disease ....................................................................................................................................................1
1.1.1 History ...................................................................................................................................................1
1.1.2 Haemangioblastoma in VHL disease.....................................................................................................1
1.1.3 Phaeochromocytoma in VHL disease....................................................................................................2
1.1.4 Renal clear cell carcinoma in VHL disease ...........................................................................................2
1.1.5 Classification of VHL disease ...............................................................................................................3
1.2 Molecular function of VHL .............................................................................................................................5
1.2.1 The VHL gene and tumour suppressor protein......................................................................................5
1.2.2 VHL containing E3 ubiquitin ligase ......................................................................................................6
1.2.2.1 Intracellular oxygen levels dictate HIFα stability ........................................................................9
1.2.3 Fibronectin/collagen IV matrix deposition ..........................................................................................12
1.2.4 Neddylation of VHL ............................................................................................................................12
1.2.5 Microtubule stability and ciliogenesis .................................................................................................13
1.2.6 Regulation of PHD3 in phaeochromocytoma ......................................................................................14
1.2.7 Regulation of early endosome fusion ..................................................................................................14
1.2.8 Maintenance of renal intracellular junctions........................................................................................15
1.2.9 E-cadherin in epithelial cancer ............................................................................................................16
1.3 Polycythemia in VHL disease........................................................................................................................16
1.3.1 Primary and secondary polycythemia..................................................................................................16
1.3.2 Chuvash polycythemia (CP) ................................................................................................................19

v
Chapter 2 VHL Promotes E2 Box-dependent E-cadherin
Transcription by HIF-mediated Regulation of SIP1 and Snail .............................................................................20
2.1 Rationale .......................................................................................................................................................21
2.2 MATERIALS AND METHODS .....................................................................................................................21
2.2.1 Cell Culture..........................................................................................................................................21
2.2.2 Antibodies............................................................................................................................................22
2.2.3 Plasmids...............................................................................................................................................22
2.2.4 Immunoprecipitation and immunoblotting ..........................................................................................22
2.2.5 Hypoxia treatment of cells ...................................................................................................................23
2.2.6 Immunohistochemical staining ............................................................................................................23
2.2.7 Subcellular fractionation......................................................................................................................24
2.2.8 Dual-luciferase assay ...........................................................................................................................24
2.2.9 Microarray analysis .............................................................................................................................25
2.2.10 siRNA-mediated VHL knockdown .................................................................................................25
2.2.11 Quantitative real-time PCR.............................................................................................................26
2.2.12 Chromatin Immunoprecipitation (ChIP) .........................................................................................27
2.3 RESULTS AND DISCUSSION ......................................................................................................................28
2.3.1 Expression of E-cadherin is down-regulated in RCC and correlates with VHL status. .......................28
2.3.2 ‘Knockdown’ of endogenous VHL results in dramatic attenuation of E-cadherin expression. ...........32
2.3.3 shRNA-mediated down-regulation of E-cadherin increases the invasive potential of RCC................35
2.3.4 VHL regulates E-cadherin expression via HIF-dependent mechanism. ..............................................38
2.3.5 VHL down-regulates E-cadherin-specific transcriptional repressors Snail and SIP1..........................43
2.3.6 Wild-type, but not RCC-causing mutant VHL, induces transcriptional activation of E-cadherin. ......48
2.3.7 E-cadherin expression is cell density-dependent. ................................................................................50
2.3.8 Discussion............................................................................................................................................52
Chapter3 NEDD8 defines tumour suppressor function of VHL
.....................................................................................................................................................................................56
3.1 Rationale .......................................................................................................................................................57
3.2 Materials and Methods..................................................................................................................................57
3.2.1 Cells .....................................................................................................................................................57
3.2.2 Antibodies and reagents.......................................................................................................................57
3.2.3 Plasmids...............................................................................................................................................58
3.2.4 Immunoprecipitation and immunoblotting ..........................................................................................58
3.2.5 Affinity Purification.............................................................................................................................59
3.2.6 Metabolic labeling ...............................................................................................................................59
3.2.7 Subcellular fractionation......................................................................................................................59

vi
3.2.8 Confocal microscopy ...........................................................................................................................60
3.2.9 siRNA ..................................................................................................................................................60
3.3 RESULTS AND DISCUSSION ......................................................................................................................60
3.3.1 ECV- and FN-associated functions of VHL are mutually exclusive ...................................................60
3.3.2 Disruption of NEDD8 pathway abrogates FN binding to VHL, but not ECV formation ....................65
3.3.3 Neddylation of VHL prevents ECV complex formation via steric hindrance .....................................69
3.3.4 Cul2 is excluded from the VHL/FN complex......................................................................................72
3.3.5 Discussion............................................................................................................................................75
Chapter 4 VHL/SOCS1 Heterocomplex Degrades JAK2........77
4.1 Rationale .......................................................................................................................................................78
4.2 Materials and Methods..................................................................................................................................79
4.2.1 Cells. ....................................................................................................................................................79
4.2.2 Antibodies............................................................................................................................................79
4.2.3 Plasmids...............................................................................................................................................80
4.2.4 Immunoprecipitation and immunoblotting. .........................................................................................80
4.2.5 Metabolic labeling. ..............................................................................................................................80
4.2.6 In vitro ubiquitylation assay. ...............................................................................................................81
4.2.7 Generation of phenylhydrazine-primed splenic erythroblasts. ............................................................81
4.2.8 Cytokine deprivation and stimulation of murine splenic erythroblasts................................................81
4.3 RESULTS ......................................................................................................................................................82
4.3.1 CP-VHL mutants have reduced capacity to form ECV .......................................................................82
4.3.2 VHL binds JAK2 in a proteasome-sensitive manner...........................................................................85
4.3.3 VHL promotes ubiquitin-mediated degradation of pJAK2..................................................................85
4.3.4 VHL binds and requires SOCS1 to promote pJAK2 degradation........................................................90
4.3.5 CP-VHL/SOCS1 association inhibits pJAK2 binding and degradation ..............................................95
4.3.6 pJAK2 and pSTAT5 are elevated in CP-mice .....................................................................................98
4.3.7 Discussion............................................................................................................................................98
Chapter 5 Conclusions and future directions .........................102
5.1 E-cadherin loss in RCC...............................................................................................................................102
5.2 Uncovering the mechanism of VHL mediated FN assembly .......................................................................105
5.3 Characterization of VHL mutation in additional haematopoietic malignancies.........................................106
References ................................................................................................................................................................108

vii
List of Abbreviations
AffPD affinity pull-down
ALPHA-MEM alpha modification Eagle's medium
aPKC atypical protein kinase C
APP-BP1 APP binding protein 1
AR autoradiography
BAC bacterial artificial chromosomes
Bcl-xl B-cell lymphoma-extra large
BFU-E burst forming units-erythroid
BNIP3L Bcl2/adenovirus E1B interacting protein 3L
CA9 carbonic anhydrase 9
CBP CREB binding protein
CDC53 coil domain containing 53
cDNA complementary DNA
CFU-E colony forming units-erythroid
ChIP chomatin immunoprecipitation
cHL classical Hodgkin lymphoma
CHO Chinese hamster ovary
Chr chromosome
CITED2 Cbp/p300-interacting transactivator, with Glu/Asp-rich carboxy-terminal domain, 2
CLL chronic lymphocytic leukemia
CMV cytomegalovirus
CNS central nervous system
CO2 carbon dioxide
CoCl2 Cobalt Chloride
COLIV collagen IV
CP Chuvash polycythemia
cp ferroxidase
CR chromophobe RCC
cRNA complementary RNA
C-SRC cellular-sarcoma
Ct cycle threshold
C-TAD carboxy-terminal transactivation domain
Cul cullin
CXCR4 chemokine (CXC motif) receptor 4
DAB diaminobenzidine
delta-ef1 eukaryotic translation elongation factor 1, delta
DFO deferoxamine
DMEM Dulbecco's modification Eagle's medium
DNA deoxyribonucleic acid
dNTP deoxynucleotide triphosphate
DTT dichlorodiphenyltrichloroethane
E-CAD epithelial-cadherin
ECM extracellular matrix
ECS elongins BC/Cul2 or 5/SOCS1
ECV elongins BC/Cul2/VHL
EDTA ethylenediaminetetraacetic acid
EGLN egg laying nine
EMT epithelial-mesenchymal transition

viii
ENO1 enolase 1
EPO erythropoietin
EPOR erythropoietin receptor
ER endoplasmic reticulum
ET essential thrombocythemia
FBS fetal bovine serum
FER FPS/FES related tyrosine kinase
FIH factor inhibiting HIF
FISH fluorescent in situ hybridization
FN fibronectin
FYN fibroblast src/yes novel gene
GFP green fluorescent protein
GLUT glucose transporter 1/3
GSK3 glycogen synthase kinase
H &E hematoxylin and eosin
HA hemagglutinin
HDAC histone deacetylase
HER2 human epidermal growth factor receptor 2
HGF hepatocyte growth factor
HIF hypoxia-inducible factor
HMOX1 heme oxygenase (decycling) 1
HRE hypoxia-responsive elements
IB immunoblot
IGFBP1 insulin-like growth factor binding proteins 1
IGFBP2 insulin-like growth factor binding proteins 2
IgGL immunoglobulin G, light chain
IHC immunohistochemistry
IP immunoprecipitation
JAK2 Janus kinase 2
KIF1B kinesin family member 1B
LEF leukocyte enhancer factor
LGL large granular lymphocyte
Log logarithm
Luc luciferase
MDM mouse double minute
MMM myelosclerosis with myeloid metaplasia
MMP matrix metalloproteinase
mRNA messenger RNA
mTOR mammalian target of rapamycin
NAE NEDD8 activating enzyme
NCE NEDD8 conjugating enzyme
NEDD8 neural precursor cell expressed developmentally downregulated protein 8
NEDP1 NEDD8 protease 1
NEM N-ethyl maleimide
NGF nerve growth factor
NLE NEDD8 ligating enzyme
O2 oxygen
ODD oxygen-dependent degradation domain
ON oncocytoma
Opti-MEM Optimal modification Eagle's medium
p27 protein of 27 kilodaltons

ix
p300 protein of 300 kilodaltons
p53 protein of 53 kilodaltons
p73 protein of 73 kilodaltons
PAGE polyacrylamide gel electrophoresis
PBS phosphate buffered saline
PCR polymerase chain reaction
PDF portable document format
PDGF platelet-derived growth factor
PhD doctor of philosophy
PHD prolyl hydroxylase domain
PHZ phenylhydrazine
PI3K phosphatidylinositol-3-kinase
pJAK2 phosphorylated JAK2
PMBL primary mediastinal B-cell lymphoma
PML promyelocytic leukemia
POLII polymerase II
pSTAT5 phosphorylated STAT5
PTEN phosphatase and tensin homolog
PV polycythemia vera
PVDF polyvinylidene difluoride
qPCR quantatative PCR
RAB5 Ras associated protein 5
RB retinoblastoma suspectibility protein
RBC red blood cell
RBX1 RING box 1
RCC renal clear-cell carcinoma
RLU relative luminescence units
RNA ribonucleic acid
RPMI Roswell Park Memorial Institute growth medium
RTK receptor tyrosine kinase
SCF Skp1/Cdc53/F-box protein complex
SCID severe combined immunodeficiency
SDF-1 stromal cell-derived factor 1
SDH succinate dehydrogenase
SDS sodium dodecyl sulfate
shRNA short hairpin RNA
SIP1 Smad-interacting protein 1
siRNA small interfering RNA
SKP-1 S phase kinase-associated protein 1
SNP single nucleotide polymorphism
SOCS suppressor of cytokine signalling
STAT5 signal transducer and activator of transcription 5
TCF T-cell factor
TGF transforming-growth factor
TIMP tissue inhibitor of metalloproteinase
TMA tissue microarray
TSC2 tuberous sclerosis complex 2
Ub ubiquitin
UBCH5A ubiquitin conjugating enzyme homolog 5a
VBC VHL/elongins B/C
VEGF vascular endothelial growth factor
VHL von Hippel-Lindau

x
WCE whole cell extract
WCP whole chromosome paint
WNT wingless type
WT wildtype
ZEB-2 zinc finger homeo box 1B
ZFHX1A Zinc finger homeodomain enhancer binding

xi
List of Tables
Chapter 1 Page
Table 1.1 Classification of VHL disease 4

xii
List of Figures
Chapter 1 Pages
Figure 1.1 Mutations across VHL open reading frame 4
Figure 1.2 Similarities between ECV and SCF ligases 8
Figure 1.3 Intracellular oxygen levels dictate HIFα stability 11
Figure 1.4 JAK2-STAT5 signalling 18
Chapter 2
Figure 2.1 Expression of E-cadherin is down-regulated in RCC and
correlates with VHL status
30
Figure 2.2 Loss of VHL results in down-regulation of E-cadherin 33
Figure 2.3 Down-regulation of E-cadherin increases the migration of
embryonic kidney cells and invasion of RCC cells
36
Figure 2.4 VHL regulation of E-cadherin is HIF-mediated 40-41
Figure 2.5 VHL-mediated transcription of E-cadherin is attenuated by
Snail and SIP1 via the conserved E2 boxes
45-46
Figure 2.6 VHL activity is required for E-cadherin transcription 49
Figure 2.7 Cell confluency influences E-cadherin expression 51
Figure 2.8 VHL gatekeeper’s pathway in renal epithelium 54

xiii
Chapter 3 Pages
Figure 3.1 ECV- and FN-associated functions of VHL are mutually
exclusive
62-63
Figure 3.2 Restriction of a dynamic NEDD8 pathway results in the
attenuation of VHL binding to FN
66-67
Figure 3.3 NEDD8 modification of VHL generates steric hindrance
blocking the formation of ECV
70
Figure 3.4 VHL/FN complex excludes ECV component Cul2 73
Chapter 4
Figure 4.1 CP-VHL exhibits altered binding to ECV components and
JAK2
83-84
Figure 4.2 VHL promotes ubiquitin-mediated destruction of pJAK2 87-88
Figure 4.3 VHL and SOCS1 cooperate to degrade pJAK2 in vivo 92-93
Figure 4.4 CP-VHL mutants are defective in pJAK2 degradation and
R200W/R200W CP mice exhibit elevated pJAK2 and
pSTAT5 levels
96-97
Figure 4.5 The ‘SOCS groove’ and the revised molecular model of CP 100
Chapter 5
Figure 5.1 Role of VHL in the regulation of E-cadherin and β-catenin 104

1
Chapter 1 Introduction to the von Hippel-Lindau tumour suppressor
1.1 VHL disease
1.1.1 History
In 1894, a British ophthalmologist E. Treacher Collins described bilateral retinal haemangiomas
in two siblings, representing the first report of von Hippel-Lindau (VHL) disease1. VHL disease
is a rare heritable disorder with an incidence of approximately 1/36000 live births2,3. The disease
derives its name from the German ophthalmologist Eugene von Hippel, who further described
kindred displaying retinal haemangiomas, and the Swedish neuropathologist Arvind Lindau who
recognized the common origin of the tumours in families who displayed cerebellar
haemangiomas and those with retinal haemangiomas4,5. The two neoplasms have subsequently
been shown to display similar histopathology and as a result are often collectively referred to as
haemangioblastomas6. In addition to haemangioblastoma, VHL patients are predisposed to
develop highly vascularized tumours in multiple organs including: renal clear cell carcinoma
(RCC), phaeochromocytoma, endolymphatic sac tumour, pancreatic islet tumour, epididymal
cystadenoma, and a variety of other malignant and benign tumours7-10. The cardinal
manifestations of VHL disease and the basis by which the disease is classified are
haemangioblastomas of the central nervous system (CNS) and retina, renal clear cell carcinoma,
and phaeochromocytoma.
1.1.2 Haemangioblastoma in VHL disease
Haemangioblastomas arise upon VHL inactivation in the retina, cerebellum, and spinal chord6.
There are also rare occurrences of VHL haemangioblastomas in the pituitary, hypothalamus,
optic nerve, corpus callosum, and other areas of the brain 11-13. These tumours are often cystic
and highly angiogenic due to the secretion of growth factors such as VEGF and PDGF, which
are required for the stimulation and stabilization of the blood vessels within the tumour14. The
cell of origin for haemangioblastoma is poorly understood. It is currently believed that the
tumour arises from a developmentally arrested angioblast that maintains expression of EPO
receptor15. Interestingly, the tumours themselves secrete EPO, which acts along with TGF-α in
an autocrine loop to stimulate tumour cell growth16-19. Collectively, these observations suggest

2
that the alteration in growth factor secretion plays a key role in the genesis of
haemangioblastomas.
1.1.3 Phaeochromocytoma in VHL disease
Phaeochromocytoma is a tumour of the adrenal gland. Tumours arise from the chromaffin cells
of the sympathetic nervous system20. These lesions are usually benign; however, they often
cause a drastic increase in circulating hormones including norepinephrine, epinephrine,
dopamine, and metanephrines21. Elevation of these hormones can cause a variety of symptoms
including hypertension, nausea, headaches, or heart failure22. Phaeochromocytoma was
classically considered at ‘10% tumour’, where 10% were considered to be hereditary, bilateral,
malignant, or extra-adrenal. These numbers are now considered inaccurate and the percentage of
familial phaeochromocytoma is estimated to be between 15 and 25%23. Other clinical
syndromes with predisposition to phaeochromocytoma include: multiple endocrine neoplasia
Types 2A and 2B and neurofibromatosis Type 1. Additionally, mutations in the mitochondrial
complex II, namely succinate dehydrogenase subunits B-D, have recently been described to give
rise to familial phaeochromocytoma23. Recent work has proposed that a common defect in
embryonic culling of sympathetic neurons may represent a generalized defect present in all
hereditary syndromes displaying phaeochromocytoma (for details see section 1.26)24.
1.1.4 Renal clear cell carcinoma in VHL disease
The primary cause of morbidity and mortality in VHL kindred is due to RCC, which is resistant
to conventional chemo and radio theraputics25,26. Similar to haemangioblastomas RCC secretes
EPO, VEGF, PDGF, and TGFα27-29. Prior to RCC formation, VHL kindred develop renal cysts
that display loss of the remaining wild type VHL allele by immunohistochemistry30,31. It is
unclear, however, if RCC must first develop from a renal cyst, and the origin of RCC is still a
matter of debate32-34. It is commonly thought that RCC arises from renal tubular epithelial cells
and tumour cells display markers of both proximal and distal tubules. One hypothesis by
Maxwell and colleagues suggests that loss of VHL in the distal tubule is responsible for the
acquisition of proximal tubule markers and the loss of some distal tubule markers32. In support

3
of the distal tubule as the origin of RCC, cell division is described to be greatly enhanced upon
VHL loss in the distal tubule when compared to the proximal tubule32. Inactivation of the
remaining wildtype allele in the renal tubule does not initiate tumourigenesis, IHC analysis
shows that VHL loss occurs in numerous pre-neoplastic lesions that have not yet gained the
requirements for tumourigenesis32,35. This has lead to the commonly held believe that additional
mutations are required to drive the formation of RCC. It has recently been shown that VHL loss
can induce cellular senescence in murine fibroblasts via the dephosphorylation of RB in a p27
dependent manner36. It has been postulated that for tumourigenesis to occur RB-dependent
senescence must be overcome, although these observations have not been observed in human
renal cells36.
1.1.5 Classification of VHL disease
VHL disease can be divided into two subcategories depending on the risk of developing
phaeochromocytoma (see table 1). Individuals with Type 1 VHL disease are not predisposed to
develop phaeochromocytoma, while Type 2 patients have an increased propensity to develop
phaeochromocytoma37. Type 2 VHL disease is further subdivided into Type 2A, 2B, and 2C:
Type 2B patients also develop renal clear cell carcinoma (RCC) and Type 2C patients
exclusively develop phaeochromocytoma38,39 In addition, VHL patients with Types 1, 2A, and
2B have an increased predisposition to develop the two principal features of the disease, retinal
and CNS haemangioblastomas. Interestingly, a heritable polycythemic disorder, Chuvash
polycythemia, has recently been described as a VHL-related disorder that exists without an
increased cancer predisposition40. Due to the distinct nature of Chuvash polycythemia it can be
considered Type 3 VHL disease.

4
Table 1.1: Classification of VHL disease
Figure 1.1: Mutations across VHL open reading frame. Adapted from compilation of
mutational data obtained from Universal VHL-Mutation Database. Height of bars represents
numbers of afflicted families (ranging from 1-52). See text for additional details.

5
1.2 Molecular function of VHL
1.2.1 The VHL gene and tumour suppressor protein
In 1988, Seizinger and colleagues mapped the locus of the putative VHL tumour suppressor gene
to a narrow region of chromosome 3p. In accord, deletions in this region have been observed in
RCC41. In 1993, Latif and colleagues identified and cloned the gene defective in VHL patients42.
Homozygous deletion of VHL in murine embryonic stem cells results in an embryonic lethal
phenotype43. Defects in extra embryonic vasculogenesis result in death in utero between days
10.5 to 12.5 of gestation43. VHL kindred inherit one mutant copy of the VHL gene and tumours
in this setting arise from the mutational inactivation, gene silencing, or loss of the remaining
wild-type VHL allele, in keeping with Knudson’s ‘Two-hit’ model of tumourigenesis.
The VHL gene contains three exons that produce a 4.5 kb mRNA (see figure 1.1). Two
translation products are observed from the human VHL gene: A full-length VHL of 213 amino
acids with a molecular weight of 30KDa (VHL30), and an internally translated shorter 160 amino
acid VHL of 19 kDa (VHL19), which results from an alternative start site at codon 5444,45. While
both VHL30 and VHL19 are functional tumour suppressors, there are subtle differences between
the two isoforms. For example, VHL19 is equally distributed in the nucleus and cytoplasm, while
VHL30 is found primarily in the cytoplasm with minor fractions localized in the nuclear and
membrane compartments44. VHL30 has the ability to shuttle between the nucleus and the
cytoplasm46. Recently it has been shown that under acidic conditions VHL30 and VHL19 can be
sequestered to nuclear loci, and upon reinstatement of neutral pH, VHL30 and VHL19 return to the
cytoplasm47. Unless otherwise specified, VHL will henceforth refer to both VHL19 and VHL30.
The crystal structure of VHL was determined in 1999 and showed that VHL contains two
functional domains48. The α domain (so named for the α helices that form this domain) was
predicted to function as a binding site for the adaptor elongin C. The β domain (so named for the
β-pleated sheets that form this domain) was predicted to function as a protein-protein interaction
interface. Tumour-derived mutations frequently occur on the surface residues within the α and β
domains, suggesting a significance for these regions in the tumour suppressor function of VHL48.
While the alpha and beta domains of VHL are considered mutational ‘hotspots’, mutations that

6
give rise to VHL disease have been found over the entire open reading frame of the VHL gene
(see figure 1.1). Despite the heterogeneity of these mutations, a phenotypic pattern that
corresponds to specific mutations has arisen. Type 1 VHL disease is often associated with
mutations resulting in gross truncations or even a complete loss of VHL. Mutations that cause
Type 2 VHL disease are frequently missense mutations. These differences in susceptibility to
develop phaeochromocytoma suggest a gain-of-function mutation, or that complete loss of VHL
function is not permissible for the development of phaeochromocytoma. Mutations that give rise
to Type 2B have been described to cause a more profound defect in VHL function compared to
Type 2A, providing an explanation for the difference in susceptibility to RCC49. Unlike Types 1
and 2, Type 3 disease has been described to be caused by inheritance of two point mutations and
therefore disease presents much earlier than Type 1 and 250,51.
1.2.2 VHL containing E3 ubiquitin ligase
As the initial discovery of the VHL gene sequence did not contain any known domains or give
any clues of possible functions, efforts to find VHL-associated proteins were made with the
supposition that these interacting proteins will have known functions or contain motifs with
predicted functions. It is now known that VHL forms a multiprotein complex with elongin C,
elongin B, Rbx1, and Cul252. The VHL complex (ECV) has high structural similarity with a
yeast multiprotein complex called SCF (Skp1/Cdc53/F-box protein) (see figure 1.2). Cul2 and
Cdc53 are members of the cullin family. Elongin C is an orthologue of yeast Skp1, and both
ECV and SCF contain a ring-finger protein Rbx1. SCF is a known E3 ubiquitin ligase complex
that targets substrates recruited via the F-box protein for ubiquitylation (see figure 1.2).
Ubiquitylation represents a common scheme for targeting proteins for rapid degradation by the
26S proteasome. Ubiquitylation of proteins is accomplished by the concerted action of a
common ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2) and ubiquitin-
ligating enzyme (E3 often referred to as E3 ligase)53. The ECV acts as an E3 ligase targeting the
HIFα family of transcription factors for polyubiquitylation (see section 1.2.2.1 for a detailed
description of HIFα mediated transcription and oxygen dependent post translational
modification). VHL acts as the substrate recognition component of the ECV binding directly to
HIFα via the β-domain. Interactions with elongins B and C act to dock VHL to Cul2, a

7
scaffolding component that brings also recruits the E2-ubiquitin conjugating enzyme UbcH5a54.
Thus, disruption of either ECV nucleation (α domain mutation) or HIFα binding (β domain
mutation) results in the stabilization of the HIFα transcription factors. Tumours devoid of VHL
show an upregulation of many hypoxia-responsive genes55.
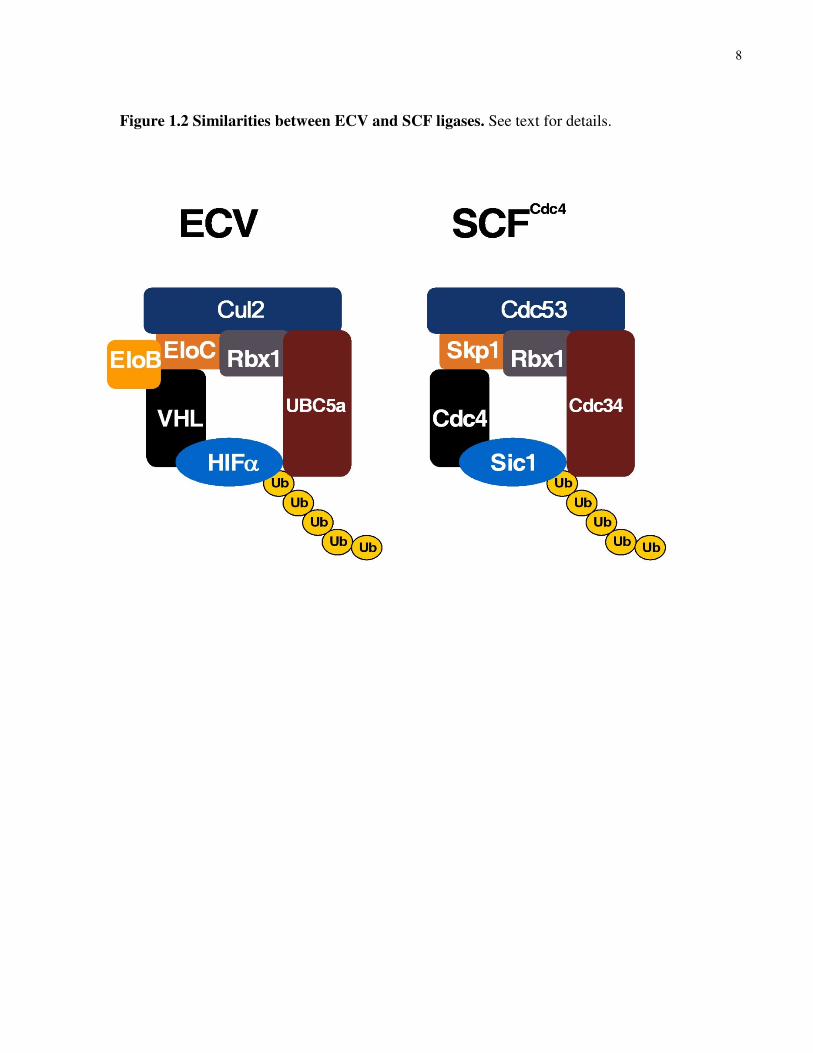
8
Figure 1.2 Similarities between ECV and SCF ligases. See text for details.

9
1.2.2.1 Intracellular oxygen levels dictate HIFα stability
Hypoxia-inducible factor (HIF) is a major regulator mediating the adaptive response to changing
oxygen tension55. HIF is a heterodimeric transcriptional activator, composed of constitutively
stable HIF-β subunit and labile HIF-α subunit, which is stabilized under hypoxia. Thus, the
activity of HIF is conferred by the oxygen-dependent stability of HIF-α. There are three
members of the HIF-α family: HIF-1α, -2α, and -3α55. Under hypoxia, HIF dimers bind to the
hypoxia-responsive elements (HRE) contained in the promoter/enhancer regions of many
hypoxia-responsive genes including VEGF, GLUT1, and EPO56.
As predicted from the VHL crystal structure, the β domain of VHL is necessary and sufficient to
bind the α subunit of HIF. However, this interaction is strictly dependent on oxygen tension57.
That is, under normal oxygen levels VHL recognizes HIF-α, while under reduced oxygen level
VHL fails to recognize HIF-α, explaining why HIF-α is no longer degraded under hypoxia58.
The selectivity of VHL binding of HIF-α was determined to be dependent on the hydroxylation
of conserved proline residues (402 and 564 based on HIF-1α sequence) within the LAPYIXMD
motif found within and near the oxygen-dependent degradation (ODD) domain of HIF-α59-61.
Prolyl-hydroxylation is carried out by a newly identified class of enzymes called prolyl
hydroxylases (PHD) 1, 2, and 3 in the presence of oxygen62. Interestingly, it was recently shown
that PHDs are upregulated during hypoxia63. The current explanation this upregulation of PHDs
is that upon recovering oxygen homeostasis there will be an abundance of PHDs ready to rapidly
hydroxylate HIF-α for subsequent ubiquitin-mediated destruction, thus curtailing the hypoxic
response.
HIF-α is also regulated in a VHL-independent manner through the C-terminal transactivation
domain (C-TAD), which is present in HIF-1α and HIF-2α, but not HIF-3α64. C-TAD interacts
with co-activators p300/CBP to effectively induce the transcription of hypoxia-inducible genes
via HRE55. Recently, it was shown that C-TAD was subjected to hydroxylation at an asparagine
residue at position 803 (based on HIF-1α sequence) under normoxia by Factor Inhibiting HIF-1
(FIH)65,66. Importantly, asparaginyl-hydroxylation of C-TAD prevented the recruitment of

10
p300/CBP via steric hindrance. This represents an added preventive mechanism to suppress the
transcriptional activity of HIF under normoxia, preventing triggering of hypoxia response during
normal oxygen levels (see Fig. 1.3).
Upon VHL inactivation in RCC, HIFα is constitutively stable and inappropriately activates the
hypoxic program under normoxic conditions. The constitutive overexpression of hypoxia
responsive genes such as VEGF and PDGF likely explains the angiogenic phenotype of VHL-
associated tumours, but also supports the notion that constitutive stabilization of HIF-α is
causally linked to tumourigenesis. In support, Kaelin and colleagues have shown that forced
stable expression of HIF-2α in RCC cells ectopically expressing wild-type VHL overrides the
tumour suppressor capacity of VHL and restores the tumourigenic potential of RCC cells in an
animal xenograft system 67. Conversely, shRNA-mediated knockdown of HIF-2α is sufficient to
suppress the tumourigenic capacity of RCC cells devoid of VHL 68. Notably, all RCC-causing
VHL mutants tested-to-date have shown a failure in either assembling of ECV complex or
binding to HIFα 69,70. However, the critical event(s) downstream of HIF that causes neoplastic
transformation of renal tubular epithelial cell is unclear.
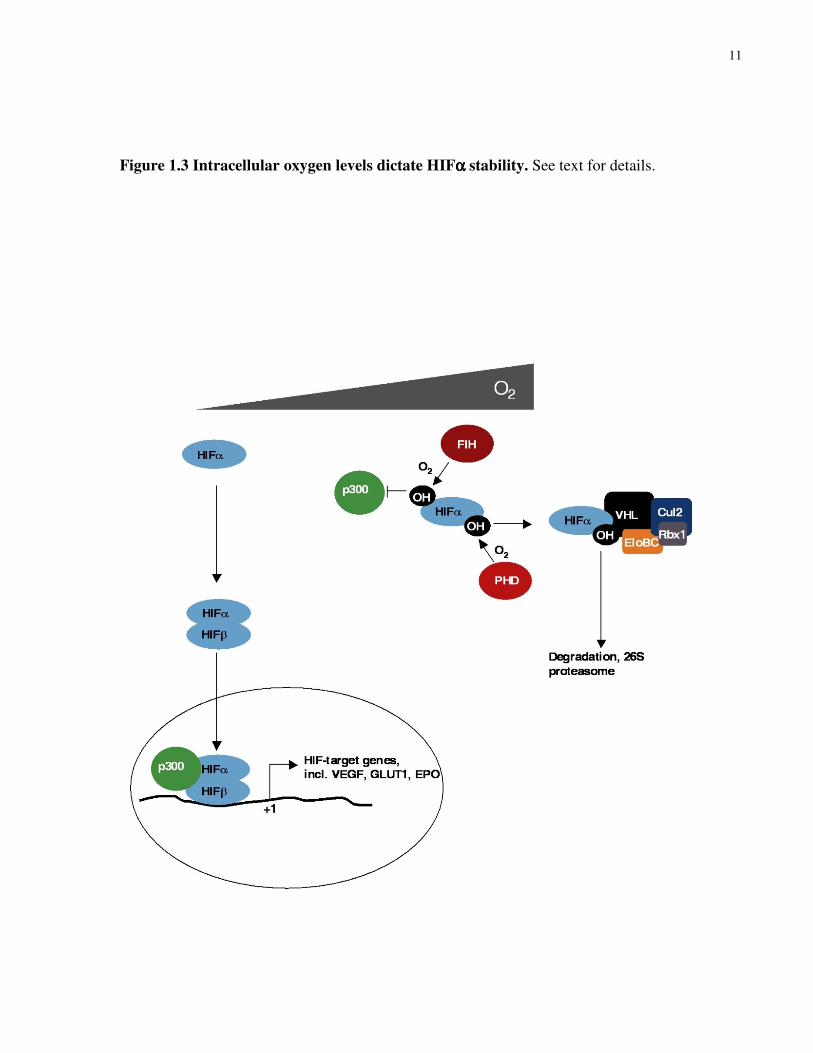
11
Figure 1.3 Intracellular oxygen levels dictate HIFαααα stability. See text for details.

12
1.2.3 Fibronectin/collagen IV matrix deposition
VHL binds fibronectin (FN) and this physical interaction is critical for the promotion of proper
extra cellular matrix (ECM) assembly71. All tumour-causing VHL mutants tested-to-date show a
striking failure in either binding to and/or assembly of FN69,70. It was recently shown that an
intact ECM attenuates angiogenesis of RCC lines by impeding the formation of new blood
vessels72. This inhibition of blood vessel formation was shown to be VHL-dependent, but HIF-
independent72. In addition, mice with a conditional knockout of VHL in endothelial cells
showed defects in vasculogenesis, which was correlated to a defect in ECM deposition. Addition
of exogenous FN can partially restore normal vascular phenotype of VHL-null cells73. These
two studies have helped show that the promotion of a normal ECM by VHL not only helps to
impede tumour formation but also represents a ubiquitous pathway necessary for embryonic
vascularization. The conservation of this pathway is highlighted by genomic clustering studies in
Caenorhabditis elegans that identified a discrete HIF-independent role of VHL in ECM
function74. Recently, VHL was shown to also interact with collagen IV (ColIV) to promote its
deposition in the extracellular space75,76. These findings expand the role of VHL-mediated ECM
assembly beyond FN. However, the mechanisms by which VHL promotes matrix deposition
remain poorly understood.
1.2.4 Neddylation of VHL
Analogous to the ubiquitin pathway, ubiquitin-like NEDD8 modification of proteins involves the
concerted actions of a common NEDD8-activating enzyme (E1 or NAE) a specific NEDD8-
conjugating enzyme (E2 or NCE) and a NEDD8-ligating enzyme or E3 ligase (E3 or NLE). The
classic targets of NEDD8 are the cullins, the scaffolding component of E3 ubiquitin ligases SCF
and ECV, where NEDD8 modification has been shown to affect E3 formation and activity 54,77.
In the case of Cul2, RBX1, a common component of cullin containing E3 ligases acts as the E3
for neddylation. Recently, evidence has begun to emerge for the role of NEDD8 in the
suppression of cancer through its recently identified targets VHL, p53, breast cancer-associated
protein 3, and p73. In each case NEDD8 modification has been shown to modulate the activity
of these genes, often impacting their viability as a tumour suppressor78-82. MDM2 acts as the E3

13
NEDD8 ligase for p53 and p73. Recently, we have shown that VHL is covalently modified by
NEDD8 on lysyl residues; however, the NEDD8 ligase for VHL has not yet been identified78.
NEDD8-conjugation, like many other ubiquitin like modifications occurs with a rapid turnover.
Endogenously neddylated VHL comprises less than 5% of total VHL at any given time within
the cell under physiologic conditions78,83. There are 3 lysyl residues on VHL (K159, K171, and
K196) of which K159 is the major acceptor site of NEDD8. Neddylation-defective VHL mutant
with lysine (K) to arginine (R) substitutions retains ‘wild-type’ level of ECV activity78.
However, neddylation-defective VHL mutant showed dramatic attenuation in binding FN. In
addition, RCC cells ectopically expressing neddylation-defective VHL consequently exhibited
reduced extracellular FN fibrillar array and more importantly, despite having ‘normal’ HIF
profile, grew as tumours in SCID mouse xenograft assay78, underscoring the importance of this
minor fraction of NEDD8-modified VHL in renal oncogenesis.
1.2.5 Microtubule stability and ciliogenesis
In addition to the development of RCC, VHL patients are predisposed to develop renal cysts84.
Development of renal cysts is often linked to defects in primary cilia, a sensory appendage with a
core of microtubules capable of measuring both biochemical and mechanical stimuli85-87.
Interestingly, RCC cell lines devoid of VHL do not display primary cilia. Stable re-constitution
of wild type VHL in this background is capable to rescuing native cilia formation85-87. Type 1
and Type 2A mutations of VHL display defects in the ability to promote ciliogenesis; however,
Type 2B mutations retain the ability to maintain cilia88. Interestingly, Type 2B mutations and
not Type 2A result in RCC, raising the possibility that defects in ciliogenesis may not be
necessary for RCC development and that RCC may develop independently of renal cysts. While
the exact mechanism by which VHL promotes ciliogenesis remains unclear, the ability to
maintain cilia appears to correlate with the ability to interact and stabilize microtubules at the
cell periphery87. The ability of VHL to stabilize microtubules is inhibited by phosphorylation on
two serines in the N-terminus. First, phosphorylation of VHL by casein kinase I ‘primes’ VHL
for phosphorylation on serine 69 by glycogen synthase kinase 3 (GSK3). Interestingly, these
phosphorylation events disrupt microtubule stability without disrupting the interaction of VHL
with tubules88. The exact mechanism by which GSK3-mediated phosphorylation inhibits VHL

14
function is unclear, but may be partially explained by the observation that N-terminally
phosphorylated VHL has a reduced capacity to bind HIFα.
1.2.6 Regulation of PHD3 in phaeochromocytoma
Sporadic mutations of VHL are common in RCC, haemangioblastomas and in other tumours
afflicting VHL kindred89. Phaeochromocytoma is a notable exception, where VHL mutations are
not a common cause of the sporadic form of this neoplasm90. This paradox has been attributed to
a loss or gain of function of VHL that must exert itself during embryogenesis in the neuronal
population of cells that give rise to phaeochromocytoma, setting the stage for disease. During
development an excess of sympathetic neurons are produced24,91. Following this expansion of
neurons a developmental cell death program is initiated as the availability of nerve growth factor
(NGF) becomes limiting91.
Molecularly, inadequate NGF levels initiate a JUN-dependent apoptotic program which depends
on PHD3 and downstream KIF1B-beta for apoptosis92. Type 2 VHL mutation results in an
increase in atypical protein kinase C (aPKC), which in turn elevates the levels of JUNB.
Accumulation of JUNB antagonizes c-JUN and inhibits the apoptotic signalling initiated by NGF
withdrawal24. Mutations or deletions that give rise to Type 1 VHL disease are also defective for
aPKC activity, but do not cause phaeochromocytoma. This may be due to a more drastic
stabilization of HIFα, which accompanies these mutations. PHD3 levels are increased by HIFα
and Type 1 mutations seem to recover enough PHD3 activity to allow apoptosis upon NGF
withdrawal93. Thus, the determining factor of whether a particular VHL mutation will give rise
to phaeochromocytoma is the extent to which the negative (through aPKC) and positive (through
HIFα) impact of VHL loss add up to affect PHD3 activity.
Mutation of VHL and other genes that give rise to familial phaeochromocytoma, such as NF1,
confer a resistance to NGF withdrawal94. It is currently believed that a common defect in
embryonic culling of sympathetic neurons is responsible for the survival of a unique subset of
cells that give rise to phaeochromocytoma in these disorders.
1.2.7 Regulation of early endosome fusion
VHL loss in renal cells has been described to increase the levels or activity of multiple receptor
tyrosine kinases (RTK)95,96. Receptor tyrosine kinases are cell surface receptors that bind a

15
variety of hormones, growth factors and cytokines to initiate receptor dimerization and
intracellular signalling. The negative regulation of RTK signalling can take place by
dephosphorylation, ubiquitin mediated degradation or endocytosis followed by lysosomal
destruction97,98. It has recently been shown that VHL loss reduces the rate of RTK turnover
through a general repression of the endocytic pathway99.
The rab family of proteins are responsible for the progression of the endocytic cycle100.
Perturbations of rab proteins severely alter the rate and progression of endosomes by regulation
of endosome fusion and cycling back to the plasma membrane101. The decreased rate of
endocytosis in the case of VHL loss was shown to be a result of transcriptional repression of
rabaptin-5, a Rab5 activator, critical for early endosome fusion99. Rabaptin-5 has an HRE
element in its promoter and the stabilization of HIFα that occurs upon VHL inactivation results
in a repression of rabaptin-5 expression in a HIFα dependent manner. Moreover, it was also
shown that rabaptin-5 is transcriptionally down-regulated in other solid tumours including breast
cancer and oncocytoma99. Thus, HIFα stabilization resulting from genetic alteration, as is the
case in tumours of VHL disease, or by limiting intracellular oxygen results in a generalized
increase in RTK-mediated signalling via the inhibition of early endosome fusion.
1.2.8 Maintenance of renal intracellular junctions
Loss of VHL in RCC cells leads to a loss of cell polarity and hallmarks of differentiation102,103.
These observations have been linked to the loss of both tight and adheren junctions upon VHL
loss104-106. Adheren junctions are known to participate in the signalling from extracellular cues.
One of the most important and well characterized mediators of these signalling cascades is
β-catenin107,108. β-catenin links cadherins to the actin cytosketelon and is important for the
integrity of the junction. When released from the cadherins β-catenin can activate the
transcription of genes by association with the transcription factor TCF (see discussion for
details)107. Disruption of intracellular junctions is associated with loss of epithelial
characteristics and an increase in cell migration and invasion109. The loss of cell-cell junctions
upon VHL inactivation has been hypothesized to occur through both HIF-dependent and
independent mechanisms104-106. It is currently proposed that perturbations in ECM, aPKC,
β -catenin ubiquitylation and or transcriptional regulation of junction members may all play a
role in the loss of adheren or tight junctions upon VHL loss104-106.

16
1.2.9 E-cadherin in epithelial cancer
The transmembrane protein E-cadherin is a major constituent of adherent cell-cell junctions,
which forms homophilic associations via its extracellular cadherin repeats110. The cytoplasmic
tail of E-cadherin associates with β-catenin, which links to α-catenin and actin forming a
dynamic junction108. Loss of E-cadherin is a hallmark of epithelial-mesenchymal transition
(EMT)111. EMT is a process essential in development for various morphogenic events allowing
programmed migration and invasion of cells during normal embryogenesis112. Paradoxically, a
similar program is seen in many cancers of the epithelial origin in which E-cadherin expression
is frequently lost113,114. However, the loss of expression is rarely due to germline or sporadic
mutations in E-cadherin gene, but rather by epigenetic alterations (e.g., CpG island methylation)
or upregulation of E-cadherin-specific transcriptional repressors114,115. The latter has been shown
to play a role in EMT observed several cancer types, including ovarian, breast, prostate, and
gastric cancers111,116-118.
1.3 Polycythemia in VHL disease
In recent years a unique subset of VHL kindred have been identified who do not develop the
classic tumour types associated with Types 1 and 2 VHL disease. Rather, these patients develop
a unique polycythemic disorder that has characteristics of both primary and secondary
polycythemia, caused by the inheritance of two VHL point mutations.
1.3.1 Primary and secondary polycythemia
Polycythemia is a condition characterized by a net increase in the total number of blood cells,
primarily red blood cells (RBCs) resulting in elevated haematocrit, and is generally categorized
as primary or secondary119. Primary polycythemia, the most common form of which is
polycythemia vera (PV), is defined by excessive erythrocytosis arising from an intrinsic defect in
erythroid progenitors rendering them hypersensitive to or independent of EPO stimulation 119.
Secondary polycythemia is defined as excessive erythrocytosis arising from increased production
of EPO 119. For example, perturbation of the oxygen-sensing pathway due to mutations in PHD2
and HIF2α has been identified in individuals with congenital secondary polycythemia 120-122.

17
Polycythemia can also develop secondary to increased EPO production by some renal tumours or
in mice with constitutive expression of HIF2α50,123. Recently, JAK2 mutations, predominated by
V617F, have been identified in the vast majority of PV patients that encode constitutively active
JAK2 124-128. JAK2 binds most prominently STAT5 transcription factors, which, upon
phosphorylation by JAK2, dimerize and translocate to the nucleus to regulate expression of
genes that control proliferation, differentiation and survival of haematopoietic cells (see Fig. 1.4) 129. STAT5 also triggers a negative feedback mechanism by transactivating the expression of
SOCS family members, which bind and inhibit activated JAKs 130. Notably, SOCS1 directly
binds and targets phosphorylated JAK2 for ubiquitin-mediated degradation via E3 ubiquitin
ligase ECS (Elongins BC/Cul2 or 5/SOCS1) 131,132. In addition, colony-forming units-erythroid
(CFU-E) cells from the fetal livers of SOCS1-/- mice were shown to be hyper-responsive to EPO 133. Moreover, JAK2(V617F) mutation induces PV phenotype in mouse bone marrow
transplantation assays, and the introduction of JAK2(V617F) into cytokine-dependent cell lines
promotes cytokine-independent signalling 134-137. JAK2(V617F) is constitutively phosphorylated
at Y1007, which is required for JAK2 activation 124-128,138. Regardless of JAK2(V617F) status,
high STAT5 phosphorylation is detected in bone marrow biopsies of PV patients 139. These lines
of evidence suggest that constitutive activation of JAK2-STAT5 signalling is a major causative
determinant of PV, and that increases in JAK2-STAT5 signalling represents a common
mechanism for the development of primary polycythemia.

18
Figure 1.4 JAK2-STAT5 signalling. See text for details.

19
1.3.2 Chuvash polycythemia (CP)
CP has features of both primary and secondary polycythemia40,140. Homozygous or compound
heterozygous germline mutations of VHL has recently been shown to cause of CP(REFs). The
best characterized of these mutations, R200W is carried with a particularly high frequency in the
Chuvash Autonomous Republic of the Russian Federation, causing an endemic polycythemia
disorder140. Additional mutations in the extreme C-terminus of VHL (i.e. H191D) have also
been described. Since its discovery the R200W mutation has been found in diverse ethnic
populations including anther endemic population in Italy141. The development of CP appears
distinct from the tumour Types 1 and 2 of VHL disease, and as such, CP-patients are not
afflicted with an increased risk of VHL-related tumours. However, Type 3 kindred have a
reduced lifespan due to polycythemia-related complications such as cerebral vascular events and
thrombosis. Current treatment is limited to Aspirin or phlebotomy. Retrospective analysis has
shown no increase in overall survival for either treatment. A mouse model of CP has been
developed by insertion of two alleles of R166W, the equivalent of the R200W mutation in
humans. To avoid confusion from this point the R166W mutation will be referred to as R200W,
in keeping with the human nomenclature of the original report142. CP-patients and
R200W/R200W mice that faithfully recapitulate the human CP condition have high EPO levels
and an intrinsic hypersensitivity to EPO displayed by burst forming units-erythroid (BFU-E)
cells, prominent features of secondary and primary polycythemia, respectively 40,142. The
secondary polycythemic feature was previously explained by Ang et al. who showed diminished
capacity of CP-VHL(R200W) to bind HIFα, resulting in mild HIFα stabilization and elevated
levels of EPO 40. However, HIF has not been associated with hypersensitivity of erythroid
progenitors to EPO and thus, the molecular mechanism underlying primary polycythemic
features of CP remains unknown and unexplained by the currently established functions of VHL,
which infer an additional yet-to-be-defined role(s) of VHL.

20
Chapter 2 VHL Promotes E2 Box-dependent E-cadherin Transcription by
HIF-mediated Regulation of SIP1 and Snail
This work is now published:
Andrew J. Evans*, Ryan C. Russell*, Olga Roche Losada*, T. Nadine Burry, Jason E. Fish, William Y. Kim, Mindy A. Maynard, Michelle L. Gervais, Roxana I. Sufan, Andrew M. Roberts, Leigh A. Wilson, Mark Betten, Cindy Vandewalle, Geert Berx, Philip A. Marsden, Meredith S. Irwin, Bin T. Teh, Michael A.S. Jewett, and Michael Ohh. 2007. VHL Promotes E2 Box-dependent E-cadherin Transcription by HIF-mediated Regulation of SIP1 and Snail. Mol Cell Biol 27(1): 157-169.
* These authors contributed equally to this work

21
2.1 Rationale
Proper regulation of cell-cell adhesion is vital during cell growth, differentiation, and tissue
development. Loss of cell-cell adhesion is frequently associated with tumour progression,
metastasis, and poor prognosis 143. Major constituents of the cell junctions in polarized epithelial
cells are E-cadherins, homophilic adhesion molecules, and their associated catenins 143.
Increased expression of E-cadherin is associated with the differentiation of mesenchymal cells
into tubular epithelial cells of the adult nephron. Conversely, the loss of E-cadherin is associated
with the progression of numerous carcinoma types 143. In addition, forced expression of E-
cadherin suppresses tumour development and invasion in various in vitro and in vivo tumour
model systems, establishing E-cadherin as a critical tumour suppressor of the epithelium 143.
Here, we show that the expression of E-cadherin is significantly down-regulated in human
primary RCC. siRNA-mediated knockdown of endogenous VHL or functional hypoxia resulted
in dramatic attenuation of E-cadherin expression. Importantly, re-introduction of wild-type
VHL, but not RCC-causing VHL mutant incapable of promoting HIF-α degradation, in RCC
(VHL-/-) cells fully restored E-cadherin transcription, in part, via HIF-dependent regulation of
transcriptional repressors Snail and SIP1 (Smad-interacting protein-1; also known as ZEB-2) and
the engagement of RNA Polymerase II on endogenous E-cadherin promoter/gene. These
findings reveal a potentially critical molecular pathway governing the development and
aggressive nature of RCC upon the loss of VHL function.
2.2 MATERIALS AND METHODS
2.2.1 Cell Culture
HEK293A embryonic kidney cells, U2OS osteosarcoma, and 786-O (VHL-/-) renal clear cell
carcinoma cell lines were obtained from the American Type Culture Collection (Rockville, MD)
and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-
inactivated fetal bovine serum (Sigma) at 37°C in a humidified 5% CO2 atmosphere. 786-O
subclones ectopically expressing wild-type HA-VHL (786-VHL) or mutant HA-VHL(C162F) or

22
(L188V) were previously described 70,144. RCC4 (VHL-/-) renal clear cell carcinoma subclones
stably expressing HA-VHL (RCC4-VHL) or empty plasmid (RCC4-MOCK) were previously
described 57. 786-VHL stably expressing HIF-2α(P531A) (786-VHL+HIF-2α) or empty control
(786-VHL+MOCK) via retrovirus were previously described 67 and generously provided by Dr.
William G. Kaelin. 786-O subclones stably expressing pRetroSUPER-empty or pRetroSUPER-
HIF2α shRNA were previously described 68.
2.2.2 Antibodies
Monoclonal anti-hemagglutinin (HA) antibody (12CA5) was obtained from Roche Molecular
Biochemicals. Monoclonal anti-VHL antibody (IG32) was as previously described 145. Anti-β-
catenin, anti-Lamin A/C and anti-α-tubulin antibodies were obtained from Santa Cruz (Santa
Cruz, CA), Abcam (Cambridge, MA), and Sigma-Aldrich (Oakville, Ontario, Canada),
respectively. Anti-E-cadherin antibody was obtained from BD Transduction Labs (Mississauga,
Canada). Anti-HIF-2α antibody was obtained from Novus Biologicals Inc. (Littleton, CO).
2.2.3 Plasmids
Mammalian expression plasmid pRc-CMV-HA-VHL(WT) was described previously 144. E-
cadherin core promoter (–308/+21)-luciferase reporter plasmids (WT and mut E2, which
contains inactivating mutations in both E2 boxes) and expression plasmid encoding SIP1 were
previously described 146. Expression plasmid encoding Snail was generously provided by Dr.
Paul Hamel.
2.2.4 Immunoprecipitation and immunoblotting
Immunoprecipitation and Western blotting were performed as described previously 71. In brief,
cells were lysed in EBC buffer (50 mM Tris [pH 8.0], 120 mM NaCl, 0.5% NP-40)
supplemented with a cocktail of protease and phosphatase inhibitors (Roche, Laval, Canada).

23
Immunoprecipitates immobilized on protein A-Sepharose beads (Amersham Biosciences,
Piscataway, NJ) were washed five times with NETN buffer (20 mM Tris [pH 8.0], 120 mM
NaCl, 1 mM ETDA, 0.5% NP-40), eluted by boiling in sodium dodecyl sulfate (SDS)-containing
sample buffer, and size-fractionated by SDS-polyacrylamide gel electrophoresis (PAGE).
Resolved proteins were then electro-transferred onto PVDF membrane (Bio-Rad Laboratories,
Hercules, CA), immunoblotted with the various antibodies, and visualized by
chemiluminescence (Amersham Biosciences, Piscataway, NJ).
2.2.5 Hypoxia treatment of cells
Cells were maintained at 1% O2 for indicated times in a ThermoForma (Marietta, OH) hypoxia
chamber (5% CO2, 10% H2, 85% N2). Cell lysates were prepared in the chamber in hypoxic
environment prior to further experimentation.
2.2.6 Immunohistochemical staining
Formalin-fixed paraffin-embedded sections from 13 nephrectomy specimens with renal cell
carcinoma of clear cell type (RCC) were obtained from the files of The Department of Pathology
and Laboratory Medicine at The University Health Network (Toronto, Canada). These tissue
blocks were used and processed in accordance with a University Health Network Research
Ethics Board-approved protocol concerning gene expression in renal cell carcinoma. Tissues
were fixed in 10% neutral buffered formalin for 24-36 h. Representative sections of tumour with
adjacent non-tumour renal parenchyma, 3-4 mm in thickness, were embedded in paraffin and 5-
micron sections were cut and placed on coated slides for light microscopy. Tumour morphology
and classification were assessed using standard hematoxylin and eosin (H&E) staining. The
tumours were classified as RCC according to criteria described in the World Health Organization
classification of renal tumours147. Immunohistochemical staining for E-cadherin and VHL was
performed manually using a standard avidin-biotin-peroxidase complex method. Sections were
incubated overnight in a humidified chamber with either unlabeled mouse anti-human E-
cadherin or mouse anti-human VHL antibodies, each at a 1:2000 dilution, following microwave
pretreatment for antigen retrieval. The sections were then incubated with a biotinylated
secondary antibody (horse anti-mouse IgG, 1:200 dilution) and the avidin-peroxidase complex.

24
The color reaction was visualized using diaminobenzidine (DAB) as the chromagen. The tissue
was then lightly counterstained with hematoxylin.
2.2.7 Subcellular fractionation
Cells were resuspended in Buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.34
M sucrose, 10% glycerol) supplemented with protease and phosphatase inhibitors (Roche, Laval,
Canada) and 1mM DTT and subsequently lysed with 0.1% Triton X-100. Samples were
incubated 7 min on ice and centrifuged. While the supernatant was recovered (cytoplasmic
fraction), the pellet was washed with Buffer A and resuspended in Buffer B (0.2 mM EGTA [pH
8], 3 mM EDTA [pH 8]) supplemented with protease and phosphatase inhibitors (Roche, Laval,
Canada) and 1 mM DTT. After 30 min incubation, samples were centrifuged and the resulting
supernatant was isolated (nuclear fraction).
2.2.8 Dual-luciferase assay
U2OS osteosarcoma cells grown on 6-well plates were transfected with a total of 2.5 µg of
expression plasmids using Fugene 6 (Roche). E-cad prom-luc WT or mutE2 (0.9 µg per
transfection) was used to measure E-cadherin core promoter-mediated transcription and 0.1 µg of
the renilla luciferase plasmid, pRL-SV40 (Promega), was used as a transfection control. An
empty pcDNA3.1 plasmid (Invitrogen) was used to maintain a constant final amount of
transfected DNA. Cells were lysed 48 h after transfection and luciferase assays performed using
the Dual-Luciferase Reporter Assay system (Promega) and the relative light units (RLUs)
measured using the lumat LB9507 luminometer (Berthold Technologies). Firefly luciferase
RLUs were normalized against Renilla luciferase RLUs and standardized to the result of the E-
cad prom-luc WT only transfection, which was arbitrarily set to 1.0. Experiments and
transfections were performed in triplicate with one representative experiment presented. Error
bars represent standard deviations.

25
2.2.9 Microarray analysis
We have established a large gene expression profiling database of renal tumours, some of which
have previously been published 148,149. For this study, we selected a total of 105 renal tumours of
clear-cell type and 12 normal kidney tissue samples. The Affymetrix HGU133 Plus 2.0
GeneChip oligonucleotide arrays were used for all 117 cases. The HGU133 Plus 2.0 arrays
contain 54,675 probe sets, representing approximately 47,000 transcripts and variants. The
manufacturer’s recommended protocol (GeneChip Expression Analysis Technical Manual,
Affymetrix, April 2003) was followed for expression profiling. Briefly, for oligonucleotide
expression profiling, 5-20 µg of total RNA was used to prepare antisense biotinylated RNA. A
subset of cases was spiked with external poly-A RNA positive controls (Affymetrix, CA).
Synthesis of complementary DNA was performed with the use of T7-oligo (dT) primer. In vitro
transcription was performed using Enzo Bioarray Transcript Labelling Kit (Enzo, NY). The
biotinylated cRNA was subsequently fragmented, and 15 ug was hybridized to each array at
45°C for 16 h. Scanning was performed in a GeneChip 3000 scanner. Quality assessment was
performed in GeneChip Operating System (GCOS) 1.4 (Affymetrix) using global scaling to a
target signal of 500. Quality assessment was also performed using denaturing gel
electrophoresis. Median background was 73, median scaling factor was 3.06 and median
GADPH 3’/5’ratio was 1.03, indicative of a high overall array and RNA quality.
Statistical analyses were performed in the statistical environment R 2.2, utilizing packages from
the Bioconductor project. The MAS 5 algorithm was used to perform pre-processing of the CEL
files, including background adjustment, quartile normalization and summarization. The means
and the standard errors for E-cadherin gene expressions were calculated for each of the group of
samples. A two-tailed Student’s t test was used to determine statistically significant differences
between various groups.
2.2.10 siRNA-mediated VHL knockdown
siGENOME SMARTpool targeted to VHL was used (Dharmacon, Austin, TX). A non-targeting
scrambled siRNA duplex was used as a negative control (5’-CCAUUCCGAUCCUGAUCCG-
3’). HEK293A (VHL+/+) cells grown on 6 well tissue culture plates were transfected with

26
scrambled and VHL siRNA at a final concentration of 200 nM. Briefly, 8 µL of Oligofectamine
(Invitrogen) was incubated with 48 µL of Opti-MEM I (Gibco/Invitrogen) for 8 min. The
oligofectamine mixture was added to the siRNA diluted in 175 µL of Opti-MEM I and incubated
for 20 min before adding to 800 µL of Opti-MEM I into the wells. After 3 h, 300 µL of DMEM
containing 30% heat-inactivated FBS (Sigma) was added to the plates. RNA was extracted 48 h
after transfection using the RNeasy kit (Qiagen, Mississauga, ON) treated with RNA-free DNase
(Ambion, TX, USA) and first-strand cDNA synthesis was performed.
2.2.11 Quantitative real-time PCR
First-strand cDNA synthesis: 1 µL of oligo(dT)23 primer (Sigma) was incubated with 5 µg of
RNA and dH2O (total reaction volume of 20 µL) for 10 min at 70°C in a thermal cycler (MJ
Research, Boston, MA). The mixture was cooled to 4°C at which time 4 µL of 5x 1st strand
reaction buffer, 2 µL of 0.1 M DTT, 1 µL of 10 mM dNTPs, and 1 µL Superscript II reverse
transcriptase (Invitrogen) were added. cDNA synthesis was performed for 1.5 h at 42°C,
followed by 15 min at 70°C in the thermal cycler. Human genomic DNA standards (human
genomic DNA was obtained from Roche, Mannheim, Germany) or cDNA equivalent to 20 ng of
total RNA were added to the qPCR reaction in a final volume of 10 µL containing 1x PCR buffer
(without MgCl2), 3 mM MgCl2, 0.25 units of Platinum Taq DNA polymerase, 0.2 mM dNTPs,
0.3 µL SYBR Green I, 0.2 µL ROX reference dye, and 0.5 µM each primer (Invitrogen).
Amplification conditions were performed as follows: 95°C (3 min), 40 cycles of 95°C (10 s),
65°C (15 s), 72°C (20 s), 95°C (15 s). qPCR was performed using the ABI Prism 7900HT
Sequence Detection System (Applied Biosystems, Foster City, CA). Gene-specific
oligonucleotide primers designed using Primer Express (Applied Biosystems) were as follows:
Snail primer set (5’-TTCAACTGCAAATACTGCAACAAG-3’ and 5’-
CGTGTGGCTTCGGATGTG-3’), SIP1 primer set (5’-CCACACTTCGCGGCTTCTT-3’ and
5’-CGATCTGCGAAGTCTTGTTTGT-3’), E-cadherin primer set (5’-
GTCATCCAACGGGAATGCA-3’ and 5’-TGATCGGTTACCGTG ATCAAAA-3’), GLUT-1
primer set (5'-CACCACCTCACTCCTGT-TACTT-3' and 5'-
CAAGCATTTCAAAACCATGTTTCTA-3'), VEGF primer set (5'-
CTCTCTCCCTCATCGGTGACA-3' and 5'-GGAGGGCAGAGCTGAGTGTTAG-3'), and

27
U1AsnRNP1 primer set (5’-CAACGACAGCCGAGACATGTA-3’ and 5’-
AGCCTCCATCAAATACCCATTC-3’). SYBR Green I fluoresces during each cycle of the
qPCR by an amount proportional to the quantity of amplified cDNA (the amplicon) present at
that time. The point at which the fluorescent signal is statistically significant above background
is defined as the cycle threshold (Ct). Expression levels of the various transcripts were
determined by taking the average Ct value for each cDNA sample performed in triplicate and
measured against a standard plot of Ct values from amplification of serially diluted human
genomic DNA standards. Since the Ct value is inversely proportional to the log of the initial
copy number, the copy number of an experimental mRNA can be obtained from linear regression
of the standard curve. A measure of the fold difference in copy number was determined for each
mRNA. Values were normalized to expression of U1AsnRNP1 mRNA and expressed relative to
scrambled siRNA samples (arbitrarily set to 1.0) and represented as the mean value of three
independent experiments performed in triplicate ± standard deviations.
2.2.12 Chromatin Immunoprecipitation (ChIP)
ChIP was performed as published previously using the Upstate ChIP assay kit 150. 5 µg of anti-
RNA Polymerase II (N-20) antibody (Santa Cruz) was added to sheared, formaldehyde cross-
linked chromatin preparations from 1 x 106 cells, and immunoprecipitation was performed
overnight at 4oC. A control immunoprecipitation without the addition of antibody was also
performed in parallel. An 18 µL aliquot (of 1800 µL total) of chromatin was removed prior to
immunoprecipitation to serve as an input control. The cross-links were reversed by addition of 2
µL of 5M NaCl, and the sample was diluted 1 in 10 before real-time PCR was performed.
Immune complexes were collected with protein A-agarose beads and, after extensive washing,
immune complexes were released, formaldehyde cross-links were reversed and DNA was
purified by phenol-chloroform extraction. Following ethanol precipitation, DNA was
resuspended in 30 µL of water. Real-time was performed on 2 µL of anti-Pol II
immunoprecipitated DNA, 2 µL of no antibody control and 2 µL of the diluted input sample.
Real-time PCR was performed in triplicate using SYBR green chemistry. Copies of the target
gene were determined using genomic DNA as a standard curve (where 1 ng of genomic DNA =
300 copies of a single copy gene). Immunoprecipitated DNA (IP DNA) was determined by

28
subtracting the number of copies from the no antibody control from the anti-Pol II
immunoprecipitated DNA and dividing by the number of copies in the diluted input sample.
Primers were designed to amplify the human E-cadherin promoter; forward: 5’-CCACGC
ACCCCCTCTCAGT-3’ and reverse: 5’-GAGCGGGCTGGAGTCTGAAC-3’, human E-
cadherin exon 10; forward: 5’-CCGTGGATGTGCTGGATGTGA-3’ and reverse: 5’-
TGGGCAGTGTAGGATGTGATTTC-3’ and the human Cyclophilin A promoter; forward: 5’-
CCTCATGTGTCGTCCCCATCA-3’ and reverse: 5’-CGCCCGTTTTATACCACGTTCG-3’.
2.3 RESULTS AND DISCUSSION
2.3.1 Expression of E-cadherin is down-regulated in RCC and correlates
with VHL status.
We have established a large gene expression profiling database of renal tumours, some of which
have previously been published 148,149. For this study, we selected a total of 105 human renal
tumours of clear-cell type and 12 normal kidney tissue samples. Using the Affymetrix HGU133
Plus 2.0 GeneChip oligonucleotide arrays for all 117 cases, we found that the expression of E-
cadherin transcripts was significantly down-regulated in RCC (Fig. 2.1a). This is consistent with
immunohistochemical studies that showed reduced E-cadherin staining in the vast majority of
RCC tumour samples and cell lines tested 151,152. However, the molecular mechanism that
accounts for the frequent loss of E-cadherin in RCC is unknown.
To date, VHL is the most frequently mutated gene in RCC and biallelic inactivation of the VHL
locus is associated with the development of greater than 80% of sporadic RCC. Thus, we asked
whether the expression of E-cadherin is associated with the VHL status. Hematoxylin and eosin
staining of representative sections from nephrectomy specimens from 13 patients confirmed the
characteristic morphologic features of RCC including nests of cells with abundant, optically
clear cytoplasm and delicate cell membranes surrounded by a network of small, thin-walled
blood vessels (data not shown). Each section studied by immunohistochemistry contained
normal renal parenchyma including core convoluted tubules within the renal cortex (Fig. 2.1b,
left lower half of the micrograph) adjacent to RCC (right upper half of the micrograph).
Membranous anti-E-Cadherin staining (upper panel) and cytoplasmic/membranous anti-VHL
staining (lower panel) shown by core convoluted tubules was used as an internal positive control

29
on each slide. Cells in this representative tumour sample showed correlative staining for E-
cadherin and VHL, where negative staining for VHL observed in RCC corresponded with
markedly reduced staining of E-cadherin (Fig. 2.1b, compare upper and lower panels).
To further validate the positive correlation between VHL and E-cadherin expression, TMAs
consisting of 56 RCC cores in quadruplicate were generated. Thirty-nine of the RCC samples
met the quality standard criteria (see Materials and Methods) and were analyzed for E-cadherin
and VHL protein expression patterns. While only 33% (5/15) of the tumours that stained
negative for VHL (15/39) stained positive for E-cadherin, the majority (67% or 16/24) of
tumours that stained positive for VHL (24/39) also stained positive for E-cadherin (Fig 2.1C).
However, a positive stain for VHL does not formally indicate the presence of a wild-type VHL,
as, for example, a subtle point mutation will likely produce a positive staining signal. Thus,
additional mutational analysis will be required to generate a more precise E-cadherin:VHL
correlation index.

30
Figure 2.1. Expression of E-cadherin is down-regulated in RCC and correlates with VHL
status. (A) 105 RCC tumour samples and 12 normal kidney tissue samples were analyzed using
Affymetrix HGU133 Plus 2.0 GeneChip oligonucleotide arrays. Mean E-cadherin expression

31
and standard error were calculated and two-tailed Student’s t test was used to determine
statistical significance between the two groups. (B) Immunohistochemical staining of a
representative RCC with anti-E-cadherin (top panel) and anti-VHL (bottom panel) antibodies.
Note the negative staining of the tumour cells (upper right in each image) with each marker, in
contrast to the positive staining shown by core tubule epithelium in the adjacent non-tumour
renal cortex (lower left in each image) (50x original magnification).

32
2.3.2 ‘Knockdown’ of endogenous VHL results in dramatic attenuation
of E-cadherin expression.
Reconstitution of 786-O (VHL-/-; HIF-1α-/-) or RCC4 (VHL-/-) renal carcinoma cells with HA-
VHL dramatically restored the expression of E-cadherin protein and mRNA, as measured by
Western blotting and quantitative real-time PCR, respectively (Fig. 2.2a and b). In addition,
siRNA-mediated knockdown of endogenous VHL in HEK293A embryonic kidney epithelial
cells resulted in marked down-regulation of E-cadherin expression (Fig. 2.2c). Microarray (Fig.
2.1a) and real-time PCR data strongly suggest that E-cadherin regulation by VHL is at the pre-
translational level. The cytoplasmic domain of E-cadherin is in a complex with β-catenin,
implicating a potential ‘outside-in’ signalling where a loss of E-cadherin would release β-catenin
to associate with the leukocyte enhancer factor (LEF)/T cell factor (TCF) to regulate the
transcription of cell cycle- (e.g., Cyclin D1) or invasion-related genes (e.g., metalloproteinase
matrilysin and FN) 143. Interestingly, increased level of Cyclin D1 has been observed in RCC
cells devoid of VHL at high cell density 153 and cells expressing tumour-causing VHL mutants
fail to assemble proper extracellular FN matrices 71,78. However, both the overall expression and
subcellular localization of β-catenin remained unaffected by VHL (Fig. 2.2a and d), suggesting
that β-catenin-mediated transcription is likely not involved in potential ‘outside-in’ signalling via
the loss of E-cadherin in the context of RCC.

33
Figure 2.2. Loss of VHL results in down-regulation of E-cadherin. (A) VHL-/- 786-O and
RCC4 cells stably expressing wild-type VHL or empty plasmid (MOCK) were lysed, equal
amounts of total cellular lysates separated on SDS-PAGE, and immunoblotted with the indicated
antibodies. Anti-α-tubulin immunoblot was performed as an internal loading control. (B)
Expression of E-cadherin was measured by quantitative real-time PCR in 786-MOCK and 786-
VHL cells and normalized to U1AsnRNP1 mRNA level. E-cadherin level in 786-VHL cells was
arbitrarily set to 1.0. Error bars represent standard deviations of the fold-changes between the

34
indicated cell types over three independent experiments. (C) Endogenous VHL in HEK293A
cells was knocked-down using VHL-specific siRNA or scrambled non-targeting control siRNA.
RNA was then extracted for cDNA synthesis and endogenous transcript levels of VHL, E-
cadherin, and U1AsnRNP1 measured. Error bars represent standard deviations of the fold-
changes between the expression of the indicated mRNA relative to its expression using control
siRNA (arbitrarily set to 1.0) over three independent experiments. (D) 786-MOCK and 786-
VHL cells were biochemically fractionated (see Materials and Methods) into cytoplasmic (C)
and nuclear (N) fractions. 100µg of each fraction were resolved on SDS-PAGE and
immunoblotted with anti-β-catenin (upper panel), anti-Lamin A/C (nuclear protein control;
middle panel) and anti-α-tubulin (cytoplasmic protein control; lower panel) antibodies. IB:
immunoblot.

35
2.3.3 shRNA-mediated down-regulation of E-cadherin increases the
invasive potential of RCC
The role of E-cadherin in modulating the migration and invasion properties of epithelial cells
is well established. However, it is not known whether E-cadherin has similar biological
effects in the context of kidney epithelial cells or RCC. Although E-cadherin expression can
be predictably determined by manipulating the status of VHL, altering the expression level of
VHL has other consequences that can influence the motility and invasion properties of RCC
(see discussion below). Thus, we used an shRNA approach to specifically down-regulate the
endogenous expression level of E-cadherin in HEK293A embryonic kidney epithelial cells,
which resulted in a significant enhancement of migration as measured by percent wound
closure (61.2% ± 4.8%) compared to cells expressing scrambled shRNA (43.0% ± 3.0%)
(Fig 2.3A and B). The change in motility was noticeable from the early time points,
suggesting that the effect of modulating the expression of E-cadherin is not only potent but
also immediate (Fig 2.3C). shRNA-mediated down-regulation of E-cadherin consistently
increased the motility of 786-VHL cells in a similar wound assay but was not statistically
significant (data not shown). However, the invasion potential was increased (2.4 ± 0.2%)-
fold in comparison to 786-VHL cells expressing the scrambled shRNA, as measured on the
standard matrigel invasion chambers (Fig. 2.3D and E). It should be noted that the changes
in motility and invasion are likely underestimated due to the incomplete knockdown of E-
cadherin (Fig. 2.3A and D). Nevertheless, these results suggest that the diminution of E-
cadherin expression would promote the invasive property of RCC.

36
Figure 2.3. Down-regulation of E-cadherin increases the migration of embryonic kidney
cells and invasion of RCC cells. (A) HEK293A cells were transiently transfected with a
plasmid encoding the scrambled shRNA or a cocktail of four E-cadherin-specific shRNAs.
Equal amounts of the whole-cell lysates were immunoprecipitated with an anti-E-cadherin

37
antibody, resolved by SDS-PAGE and immunoblotted with an anti-γ-tubulin antibody. E-
cadherin signal intensities were quantified using a Kodak Image Station 2000R densitometer
and normalized against the corresponding γ-tubulin signals; values are indicated in the
parentheses. (B) Wounds were created 48h post-transfection with the indicated plasmids.
Percent wound closure was determined by measuring the migration of cells from the wound
edge 25h post-wound scrape. Each wound measurement was taken in triplicate, and the
experiment was repeated three times. (C) Line graph representing early migration profile, as
indicated by percent wound closure, as measured in the experiment shown in panel B, of
HEK293A cells transfected with the indicated shRNA plasmids. (D) 786-VHL cells were
transiently transfected with a plasmid encoding the scrambled shRNA or a cocktail of four E-
cadherin-specific shRNAs. Equal amounts of the whole-cell lysates were
immunoprecipitated with an anti-E-cadherin antibody, resolved by SDS-PAGE and
immunoblotted with an anti-hnRNP antibody. E-cadherin signal intensities were quantified
using a Kodak Image Station 2000R densitometer and normalized against the corresponding
hnRNP signals; results are given in parentheses. (E) 786-VHL cells were transiently
transfected with the indicated plasmids as shown in panel D. Cells were counted 72h
postransfection, and 2.5 x 104 cells were seeded into BD Matrigel Invasion Chambers and
incubated for 22h. The invading cells were stained with 0.1% crystal violet, and images were
captured under an inverted light microscope. Cells were counted from photographs of the
membrane, and each experiment was repeated twice. The relative change in invasion was
determined by counting the number of invading cells transfected with E-cadherin-specific
shRNA and normalizing the value against the number of invading cells transfected with the
scrambled shRNA (arbitrarily set at 1.0). Anti-E-cad, anti-E-cadherin; IP,
immunoprecipitaion; IB, immunoblot; Anti-Tub, anti- γ-tubulin; shE-cad, E-cadherin-
specific shRNA; shScram, scrambled shRNA; T, time.

38
2.3.4 VHL regulates E-cadherin expression via HIF-dependent
mechanism.
We next asked whether the regulation of E-cadherin expression by VHL is mediated through
the activity of HIF. RCC4-VHL and 786-VHL cells were maintained under normoxic (21%)
or hypoxic (1%) conditions for 16 h and analyzed by Western blotting (Fig. 2.4a and b). The
expression of E-cadherin was dramatically reduced under hypoxia while preserving the
expression status of VHL (Fig. 2.4a). The effect of hypoxic treatment was confirmed by the
increase in HIF-2α expression (Fig. 2.4a, upper panel). Although this result suggests that
hypoxia-induced stabilization of HIF results in repression of E-cadherin expression, it is
formally possible that VHL, independent of HIF, regulates the expression of E-cadherin in an
oxygen-dependent manner. Therefore, we examined various VHL mutants that have retained
or lost the ability to regulate HIF. Certain non-RCC-associated VHL mutants have been
shown to retain the ability to regulate HIF activity 69,70,78. For example, The L188V mutation
allows proper oxygen-dependent degradation of HIF-α and is associated with a sub-class of
VHL disease (Type 2C), which is clinically characterized by the exclusive development of
phaeochromocytoma 70. In contrast, invariably all RCC-associated VHL mutations test-to-
date, such as C162F, result in a complete loss of ability to mediate the destruction of HIF-α
via the ubiquitin-proteasome pathway 69,70. 786-O cells ectopically expressing VHL(C162F)
showed negligible expression of E-cadherin, while those expressing VHL(L188V) showed
higher detectable levels of E-cadherin, albeit at a lower level than observed in cells
expressing VHL(WT) (Fig. 2.4c). In addition, 786-O (VHL-/-; HIF-1α-/-) cells stably
expressing wild-type VHL (786-VHL) infected with retroviruses that express functional and
stable HIF-2α(P531A; escapes VHL recognition) demonstrated reduced level of E-cadherin
relative to 786-VHL cells infected with ‘empty’ retrovirus (Fig. 2.4d, compare lanes 1 and 2).
Notably, the level of E-cadherin was inversely proportional to the level of HIF-2α (Fig. 4d).
Conversely, 786-O subclones infected with retroviruses that express HIF-2α-specific shRNA
demonstrated markedly increased level of E-cadherin relative to 786-O cells infected with
‘empty’ retrovirus (Fig. 2.4e). In addition, the activity of the exogenous E-cadherin
promoter-driven luciferase reporter was much higher in 786-O cells reconstituted with wild-
type VHL (786-WT; low HIF activity) than in 786-MOCK (high HIF activity) cells (Fig.

39
2.4f). Taken together, these results strongly suggest that HIF negatively regulates E-cadherin
expression, at a minimum, at the level of transcription.

40

41
Figure 2.4. VHL regulation of E-cadherin is HIF-mediated. (A) RCC4-VHL cells were
maintained under normoxia (N; 21% O2) or hypoxia (H; 1% O2) for 16h, lysed, resolved on
SDS-PAGE, and immunoblotted with anti-HIF-2α (top panel), anti-E-cadherin (middle panel),
and anti-HA (bottom panel) antibodies. Asterisk denotes non-specific bands and illustrates equal
loading of total cellular extracts between lanes. (B) 786-MOCK and 786-VHL cells were
maintained under normoxia (N; 21% O2) or hypoxia (H; 1% O2) for 16h and lysed. Cell lysates
were equilibrated following Bradford protein assay and immunoprecipitated with anti-E-cadherin
antibody and resolved on SDS-PAGE. 100µg of cell lysates were also resolved on SDS-PAGE.
Proteins separated on SDS-PAGE were transferred onto PVDF membrane and immunoblotted
with anti-E-cadherin (top panel) and anti-HA (bottom panel) antibodies. IP:
immunoprecipitation; IB: immunoblot. (C) 786-O cells stably expressing HA-VHL(WT), HA-
VHL(C162F), or HA-VHL(L188V) were lysed, resolved on SDS-PAGE, and immunoblotted
with the indicated antibodies, where α-tubulin served as an internal loading control. (D) 786-
MOCK and 786-VHL cells infected with ‘empty’ retrovirus (786-VHL+EMPTY) or retrovirus
expressing constitutively stable and functional HIF-2α(P531A) were lysed, resolved on SDS-
PAGE, and immunoblotted with the indicated antibodies, where α-tubulin served as an internal
loading control. (E) 786-O (VHL -/-) subclones stably expressing pRetroSUPER-empty or

42
pRetroSUPER-HIF2α shRNA were lysed, comparable amounts of whole cell extracts
immunoprecipitated and immunoblotted with an anti-E-cadherin antibody (top panel). Equal
amounts of the whole cell extracts were also resolved on SDS-PAGE and immunoblotted with
anti-HIF-2α (middle panel) and anti-actin (bottom panel) antibodies. (F) Dual-luciferase assays
were performed in 786-MOCK and 786-VHL cells transfected with the firefly luciferase
construct (E-cad prom-luc) driven by the human E-cadherin promoter sequence. CMV-driven
renilla luciferase was used as an internal transfection control and the firefly luciferase relative
light units (RLUs) were normalized against Renilla luciferase RLUs. Experiments and
transfections were performed in triplicate with one representative experiment presented. Error
bars represent standard deviations.

43
2.3.5 VHL down-regulates E-cadherin-specific transcriptional repressors
Snail and SIP1.
Mutational analyses have shown that biallelic somatic inactivating mutations of E-cadherin are
rare 154,155, and emerging evidences suggest that the loss or reduction in E-cadherin expression in
cancer cells primarily occurs at the level of transcription 113,156-158. The two major regulators of
E-cadherin transcription are the zinc finger transcriptional repressors Snail and SIP1 (Smad-
interacting protein-1; also known as ZEB-2), which bind evolutionarily conserved E2 boxes
located within the E-cadherin core promoter resulting in the inhibition of E-cadherin
transcription 111,146,159. Moreover, hypoxic treatment of ovarian carcinoma cells was shown to
attenuate the expression of E-cadherin via the upregulation of Snail 116.
Quantitative real-time PCR analysis showed a significant attenuation of both Snail and SIP1 in
RCC 786-O cells restored with VHL (Fig. 2.5a). As expected, restoration of VHL reduced the
expression of HIF-target genes, VEGF and GLUT-1, and increased E-cadherin expression (Fig.
2.5a and as shown in Fig. 2.2). These results suggest the possibility that VHL may increase the
expression of E-cadherin by down-regulating the transcriptional repressors Snail and SIP1. In a
complementary experiment, we tested the ability of VHL in the transactivation of E-cadherin
promoter-driven luciferase reporter (Fig. 2.5b). As expected, E-cadherin promoter containing
both E2 boxes had lower basal transcriptional activity relative to E-cadherin promoter with
mutations in the E2 boxes that abrogate Snail/SIP1 binding (Fig. 2.5b). Importantly, the addition
of VHL markedly increased the wild-type E-cadherin promoter-driven luciferase transcription,
but had insignificant effect on the E2 mutant E-cadherin promoter (Fig. 2.5b). Moreover, the
increase in VHL-mediated transactivity of wild-type E-cadherin promoter-luciferase was
dampened by the addition of SIP1 or/and Snail in a dosage-dependent manner (Fig. 2.5c).
Coexpression analysis indicated that Snail or SIP1 had negligible effect on the steady-state level
of VHL (Fig. 2.5d). These results demonstrate that the E2 boxes are functionally important in
upregulating E-cadherin transcription by VHL, in part, via the down-regulation of SIP1 and/or
Snail. However, neither SIP1 nor Snail individually or in combination achieved a complete
inhibition of E-cadherin promoter-driven reporter activity. This suggests the existence of other
yet-to-be-defined VHL-HIF-mediated E-cadherin-specific transcriptional repressors or that full
repression requires the concerted actions of multiple repressors and involves, in addition to the

44
E2 boxes, other elements within the E-cadherin promoter. These results, however, do
demonstrate that the E2 boxes are functionally important in upregulating E-cadherin
transcription by VHL, in part, via the down-regulation of SIP1 and/or Snail.

45

46
Figure 2.5. VHL-mediated transcription of E-cadherin is attenuated by Snail and SIP1 via
the conserved E2 boxes. (A) Expression of E-cadherin, Snail, SIP1, VEGF, GLUT-1 were
measured by quantitative real-time PCR in 786-MOCK and 786-VHL cells and normalized to
U1AsnRNP1 mRNA expression. Solid bars represent expression of the indicated mRNA in 786-

47
MOCK cells relative to its expression in 786-VHL cells, which was arbitrarily set to 1.0. (B)
Dual-luciferase assays were performed in U2OS cells transfected with the indicated expression
plasmids. The firefly luciferase construct (E-cad prom-luc) was driven by the human E-cadherin
promoter sequence (WT) or the promoter with deletion of both E2 boxes (mut E2). CMV-driven
renilla luciferase was used as an internal transfection control and the firefly luciferase relative
light units (RLUs) were normalized against Renilla luciferase RLUs. Experiments and
transfections were performed in triplicate with one representative experiment presented. Error
bars represent standard deviations. (C) Performed as in B with increasing concentrations of
Snail/SIP-1 mixed into HA-VHL transfection reactions at a ratio of 1:2, 2:2, and 4:2 (Snail/SIP-
1:HA-VHL). Fold induction was standardized to E-cad prom-luc activity in the absence of
exogenous VHL. (D) U2OS cells were transfected with the expression plasmid encoding HA-
VHL alone or in combination with a plasmid encoding Snail in increasing amounts (represented
as a triangle). Equal amounts of the whole cell extracts were immunoprecipitated with an anti-
Snail antibody, resolved on SDS-PAGE and immunoblotted with an anti-Snail antibody (top
panel). Equal amounts of the remaining whole cell extracts were resolved on SDS-PAGE and
immunoblotted with an anti-VHL (middle panel) or anti-α-tubulin (bottom panel) antibody.

48
2.3.6 Wild-type, but not RCC-causing mutant VHL, induces
transcriptional activation of E-cadherin.
Principal mechanism by which transcriptional repressors attenuate the rate of transcription is by
blocking the engagement of RNA Polymerase II (Pol II) and associated factors to the promoter.
Notably, Snail has been shown to repress E-cadherin expression through the binding of
Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex 160, which is thought to impair
recruitment of Pol II and transcriptional initiation via repressive changes in chromatin structure.
Repressors can also act at a post-initiation step of transcription including inhibition of
phosphorylation of the Pol II holoenzyme, which represents a key step in promoter escape and
cessation in elongation 161. Therefore, we asked whether the engagement of Pol II on the
endogenous E-cadherin promoter/gene is influenced by VHL. Chromatin immunoprecipitation
(ChIP) assays at the E-cadherin locus was performed using antibodies recognizing the N-
terminus of Pol II in 786-O cells expressing wild-type or HIF regulation-defective mutant
VHL(C162F) (Fig. 2.6a). The total amount of Pol II at the E-Cadherin promoter and exon 10
was dramatically decreased in the absence of VHL functional activity (Fig. 2.6a). This was in
contrast to the promoter of the housekeeping gene, Cyclophilin A, where binding of Pol II was
similar in wild-type and mutant VHL cells (Fig. 2.6a). This suggests that VHL functional
activity to negatively regulate HIF is necessary for transcriptional activation of E-cadherin. To
further establish a functional role of VHL/HIF in E-cadherin gene transcription, 786-O cells
stably expressing wild-type VHL were exposed to hypoxia. Assessment of Pol II binding to
genomic regions corresponding to coding regions of E-cadherin (exon 10) revealed that hypoxia
decreased E-cadherin transcription (Fig. 2.6b). A similar decrease in Pol II binding was
demonstrated at the promoter of E-cadherin (data not shown). As expected, hypoxia elicited a
time-dependent increase in VEGF mRNA expression (Fig. 2.6b). Taken together, these results
suggest that VHL activity, specifically E3 ligase function to negatively regulate HIF, is required
for the transcription of the E-cadherin gene. Conversely, cellular hypoxia or loss of HIF-
associated function of VHL results in the activation of HIF and disengagement of Pol II from the
E-cadherin promoter, resulting in the down-regulation of E-cadherin transcription. However, it
is not formally known whether HIF-mediated engagement of Pol II on E-cadherin promoter is
SIP1/Snail-dependent.

49
Figure 2.6. VHL activity is required for E-cadherin transcription. (A) Chromatin
immunoprecipitation (ChIP) using anti-RNA polymerase II antibody was performed on
sheared chromatin from 786-O cell lines (VHL-/-) that had been stably transfected with wild-
type VHL (open bar) or mutant VHL(C162F) (solid bar). IP DNA was determined for the
promoter and exon 10 of E-cadherin and the promoter of Cyclophilin A using real-time PCR,
and the value in VHL(WT) cells was arbitrarily set to 1.0. (B) RNA polymerase II ChIPs
were performed in 786-VHL(WT) cells exposed to 4 or 20h of hypoxia (1% oxygen). IP
DNA for exon 10 of E-cadherin was normalized to the IP DNA for the Cyclophilin A
promoter (left graph). Normoxia was arbitrarily set to 1.0. Expression of VEGF was
assessed by real-time PCR as internal control for hypoxia treatment (right graph).

50
2.3.7 E-cadherin expression is cell density-dependent.
E-cadherin expression in RCC cells is also cell density-dependent as measured by Western
blotting and quantitative real-time PCR (Fig 2.7a and b). Interestingly, the expression of VHL is
strictly regulated by cell density where the steady-state amount of VHL in human renal proximal
tubule epithelial cells was shown to increase more than 100-fold in dense cultures relative to
sparse cultures 162. In addition, other components of the VHL E3 ligase complex showed a
similar cell density-dependent regulation 163. Importantly, HIF-2α level was elevated in sparsely
growing cells with low levels of VHL and significantly reduced or undetectable in confluent
cells containing abundant VHL 163. Moreover, the ability of VHL to shuttle between the nucleus
and the cytoplasm is also regulated by cell density 46, which in turn may influence the ability of
VHL to regulate HIF activity 164. Thus, cell density-dependent expression of E-cadherin may be
due to a corresponding cell density-dependent regulation of VHL stability/function.

51
Figure 2.7. Cell confluency influences E-cadherin expression. (A) 786-MOCK, 786-VHL,
RCC4-MOCK, and RCC4-VHL were grown to varying levels of confluency, lysed, equal
amounts (150µg) of total cell lysates separated on SDS-PAGE, and immunoblotted for E-
cadherin protein expression. (B) Representative experiment showing mRNA expression of E-
cadherin assayed using quantitative real-time PCR after RNA isolation from 786-VHL and 786-
MOCK cells that were grown to the indicated confluencies. Days grown past 100% confluency
are noted in parentheses. E-cadherin expression was normalized to U1AsnRNP1 mRNA
expression.

52
2.3.8 Discussion
VHL is a direct oxygen-dependent negative regulator of HIF-α via the ubiquitin pathway 89.
Loss of VHL or VHL mutations associated with the development of RCC invariably results in
the accumulation/hyper-activation of HIF due to a failure in VHL’s ability to either bind or
ubiquitylate HIF-α. Here, we propose that HIF - stabilized by hypoxia in the presence of wild-
type VHL - or upon mutation/loss of VHL activates the transcriptional repressors SIP1 and Snail
(likely via HIF-engagement to the HRE element (5’-GCGTG-3’) found in the Snail promoter at
position –86 to –82; SIP1 promoter/enhancer has not been defined), preventing PolII engagement
on E-cadherin promoter and resulting in the down-regulation of E-cadherin expression (see Fig.
2.8). There is, however, an alternate pathway to consider (described below).
Increased transforming-growth factor (TGF)-β signalling and expression of Snail and SIP-1, as
well as the loss of E-cadherin expression, have all been correlated with epithelial to
mesenchymal transition (EMT) process that occurs during normal development and acquisition
of invasive phenotype in epithelial cancers 111,146,159,165. Smad-mediated signalling by TGF-β has
been shown to induce the expression of the repressors Snail and SIP-1 146,166,167. HIF-1 has been
shown to upregulate the expression of the members of TGF-β family in a transcription-
dependent manner under hypoxic conditions. HIF-1 and Smad proteins cooperate in regulating
the expression of several hypoxia and TGF-β-regulated genes, including the expression of TGF-
β2 in human umbilical vein endothelial cells 168. In addition, HIF-1 has been shown to directly
bind to the TGF-β3 promoter and upregulate its expression under hypoxia during placental and
epithelial development processes 169,170. Therefore, increased transcriptional activity of HIF by
the functional loss of VHL in RCC may result in an upregulation of TGF-β signalling, resulting
in a Smad-mediated induction of SIP1 and Snail and subsequent loss of E-cadherin (Fig. 2.8).
Notably, VHL has also been shown to repress the expression of TGF-β1 via regulating its mRNA
stability (Fig. 2.8, dashed line) 171. Whether this process is HIF-mediated is currently unknown.
In the current work, we demonstrated that the loss of VHL leads to the dramatic down-regulation
of E-cadherin in RCC. VHL-dependent transactivation of E-cadherin was dependent on the
conserved E2 boxes known to recruit transcriptional repressors Snail and SIP1 to the promoter of
E-cadherin. Re-introduction of VHL in RCC cells devoid of VHL showed a reduction in the

53
expression of both Snail and SIP1 and thereby explaining, at least in part, the resulting
restoration of E-cadherin expression. Transcriptional repressors principally block transcription
by inhibiting the engagement of PolII to the promoter. In support, Snail has been shown to
repress E-cadherin expression through the binding of histone deacetylase, promoting repressive
changes in chromatin structure and thereby impairing the recruitment of PolII and transcription 160. Consistent with this view, wild-type VHL enhanced the recruitment of PolII to the E-
cadherin promoter/gene. However, hypoxia or tumour-causing VHL mutant with a failure in
targeting HIF-α for ubiquitin-mediated destruction dramatically decreased the association of
PolII with the E-cadherin gene. Thus, VHL directly affects PolII engagement on E-cadherin
DNA via HIF-dependent regulation of E-cadherin-specific transcriptional repressors, revealing a
previously unrecognized regulation of a major epithelial tumour suppressor E-cadherin.
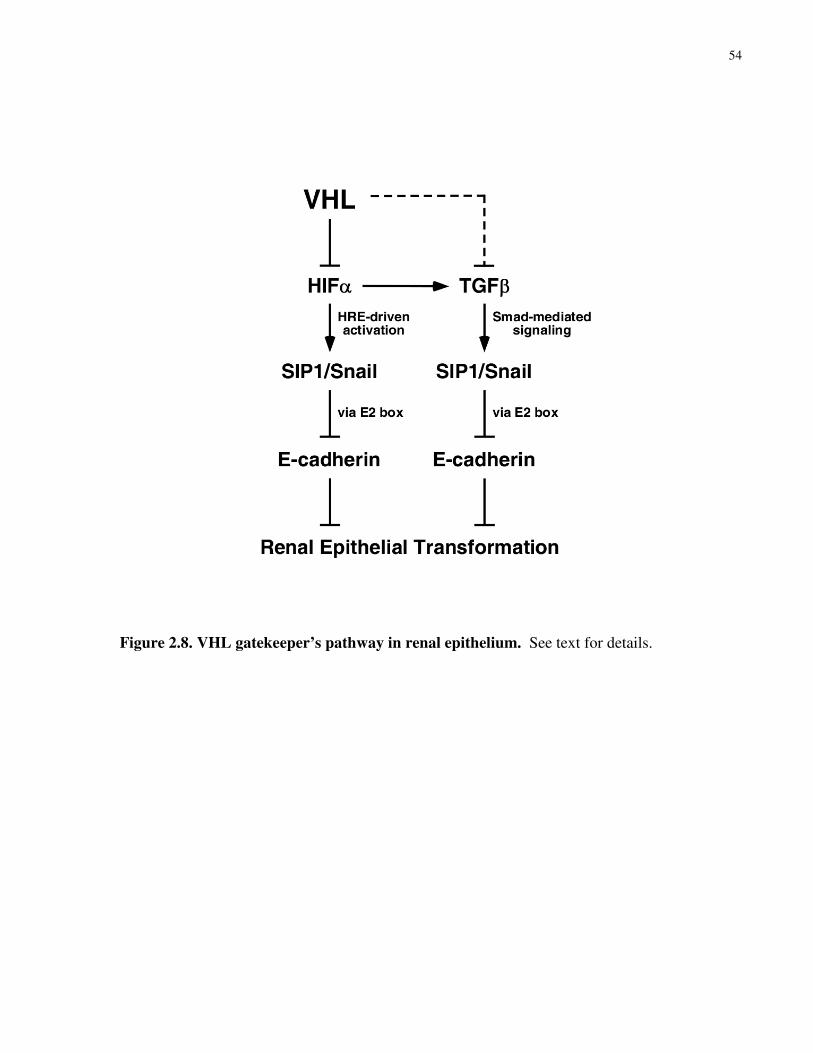
54
Figure 2.8. VHL gatekeeper’s pathway in renal epithelium. See text for details.

55
Although the loss of VHL-HIF-mediated regulation of E-cadherin likely provides an important
biological basis for the malignant nature of RCC, as well as the epithelial-to-mesenchymal
transition, there is one notable VHL-dependent event bearing on RCC progression to consider.
CXCR4 is a chemokine receptor that aids in the metastasis of tumour cells to organs abundant in
CXCR4-specific ligand, stromal cell-derived factor-1α (SDF-1α). Staller and colleagues
showed that the expression of chemokine receptor CXCR4 increases upon the loss of VHL,
suggesting a potential mechanism of RCC metastasis 172. In addition, Zagzag and colleagues
recently demonstrated that RCC and haemangioblastoma cells devoid of VHL overexpress not
only CXCR4, but also its ligand SDF-1α 173. These findings suggest that loss-of-function of
VHL can establish an autocrine signalling pathway providing selective survival advantage and
increased tendency for metastasis. The impact of the individual events (i.e., VHL-mediated E-
cadherin versus CXCR4/SDF-1α regulation) in RCC development/progression is not yet
established, but nevertheless remains an important question to address.
There are other examples of cancer-causing mutations (aside from inactivating mutations on
VHL) that often increase the expression of HIF-1α and provide mechanistic explanation for the
highly vascular tumours including RCC that develop in the absence of VHL mutations.
Mutations in TSC2 tumour suppressor gene increase the level of HIF-1α via the mammalian
target of Rapamycin (mTOR)-dependent and -independent mechanisms that may involve
chromatin remodelling 174. Loss of PTEN, which has been observed in the brain tumour
glioblastoma multiforme, results in increased HIF-1α levels via the activation of the Akt/protein
kinase B signalling cascade 175. The increased expression of HER2 receptor tyrosine kinase in
breast cancer and the loss of p53 in various tumours enhance HIF-1-dependent transcription,
often correlating with tumour aggressiveness 176,177. Although these examples support the notion
that there are multiple important regulators of HIF to ultimately promote oncogenic
transformation, whether non-VHL-associated HIF activation likewise results in the down-
regulation of E-cadherin via the activation of SIP1/Snail family of transcriptional repressors is an
important question that remains to be resolved.

56
Chapter3 NEDD8 defines tumour suppressor function of VHL
This work is now published: Ryan C. Russell and Michael Ohh. 2008. NEDD8 acts as a
‘molecular switch’ defining the functional selectivity of VHL. EMBO Reports 9(5):486-91

57
3.1 Rationale
Although the significance of the both the HIF and fibronectin functions of VHL in tumour
suppression has been well established, the mechanisms that determine the specific tumour
suppressive effects of VHL remain a mystery. It was recently shown that covalent modification
of VHL by NEDD8 is required for physical interaction with FN. Further, it was shown that
neddylation of VHL is not required for HIF function, providing the first hints at a molecular
determinant for the demarcation of these pathways. Here, we show that NEDD8 modification of
VHL acts as a ‘molecular switch’ where its covalent conjugation to VHL precludes ECV
function and concomitantly allows interaction with fibronectin, and thus providing the first
mechanistic step in the definition of functional selectivity of VHL.
3.2 Materials and Methods
3.2.1 Cells
786-O RCC, U2OS osteosarcoma and HEK293A embryonic kidney cell lines were obtained
from the American Type Culture Collection (Rockville, MD) and maintained in Dulbecco’s
modified Eagle’s medium supplemented with 10% heat-inactivated fetal bovine serum (Sigma,
Milwaukee, WI) at 37°C in a humidified 5% CO2 atmosphere. 786-O subclones ectopically
expressing wild-type VHL (786-WT) or empty plasmid (786-MOCK), RCC4 cells ectopically
expressing wild-type VHL (RCC4-WT) or empty plasmid (RCC4-MOCK), and ts41 Chinese
hamster ovary cells were as previously described69,71,178.
3.2.2 Antibodies and reagents
Monoclonal anti-haemagglutinin (HA) (12CA5) and anti-HIF1α antibodies were obtained from

58
Boehringer Ingelheim (Laval, QC) and Novus Biological (Littleton, CO), respectively.
Monoclonal anti-T7 antibody was obtained from Novagen (Madison, WI). Monoclonal anti-
vinculin, tubulin, and hnRNP were obtained from Abcam (Cambridge, MA). Polyclonal anti-
GLUT1 and anti-Cul2 antibodies were obtained from Alpha Diagnostics (San Antonio, TX) and
Zymed (San Francisco, CA), respectively. Monoclonal anti-VHL antibody (IG32) was as
previously described71. Polyclonal anti-Col IV, anti-luminal Calnexin, and anti-cyto Calnexin
were obtained from Abcam (Cambridge, MA). MG132 and NEDP1 were obtained from Boston
Biochem (Boston, MA). N-ethyl maleimide (NEM), cobalt chloride (CoCl2) and desferroxamine
(DFO) were obtained from Sigma (Oakville, ON). Trypsin was obtained from Invitrogen
(Burlington, ON).
3.2.3 Plasmids
Mammalian expression plasmids pRc-CMV-HA-VHL(WT), pRc-CMV-HA-VHL(C162F), pRc-
CMV-HA-VHL(RRR), pRc-CMV-HA-Cul2, and pRc-CMV-T7-VHL were described
previously71,78,144,179,180. pcDNA3-NEDD8 was generated by PCR from a human fetal brain
library using primers 5-ATGGATCCATGCTAATTAAAGTGAAGACGCTGAC-3 and 5-
TGAATTCGCTGCCTAAGACCACCTCCT-3. The PCR product was then ligated into the
BamHI and EcoRI sites in pcDNA3(-). All plasmids were confirmed by direct DNA sequencing.
3.2.4 Immunoprecipitation and immunoblotting
Immunoprecipitation and Western blotting were performed as described previously71. In brief,
cells were lysed in EBC buffer (50 mM Tris [pH 8.0], 120 mM NaCl, 0.5% NP-40)
supplemented with a cocktail of protease and phosphatase inhibitors (Roche, Laval, Canada).
Immunoprecipitates immobilized on protein A-Sepharose beads (Amersham Biosciences,
Piscataway, NJ) were washed five times with NETN buffer (20 mM Tris [pH 8.0], 120 mM
NaCl, 1 mM ETDA, 0.5% NP-40), eluted by boiling in sodium dodecyl sulfate (SDS)-containing
sample buffer, and size-fractionated by SDS-polyacrylamide gel electrophoresis (PAGE).
Resolved proteins were then electro-transferred onto PVDF membrane (Bio-Rad Laboratories,

59
Hercules, CA), immunoblotted with the various antibodies, and visualized by
chemiluminescence (Amersham Biosciences, Piscataway, NJ).
3.2.5 Affinity Purification
Gelatin-Sepherose beads (Amersham Pharmaceuticals, Piscataway, NJ) were used to affinity
purify FN from whole cell extracts by rocking at 4°C for 3 hours. FN complexes were eluted in
250mM Arginine in PBS, rocking for 10 min at 22°C, as previously described181.
3.2.6 Metabolic labeling
Metabolic labelling was performed as described previously. In brief, 786-O cells were
maintained in methionine-free Dulbecco's modified Eagle's medium for 45 min then
supplemented with 35S-methionine (100 µCi/ml of medium; Amersham Biosciences,
Buckinghamshire, United Kingdom) and 2% dialyzed fetal bovine serum for 3 h at 37 °C in a
humidified 5% CO2 atmosphere.
3.2.7 Subcellular fractionation
Cells were resuspended in Buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.34
M sucrose, 10% glycerol) supplemented with protease and phosphatase inhibitors (Roche, Laval,
Canada) and 1mM DTT and subsequently lysed with 0.1% Triton X-100. Samples were
incubated 7 min on ice and centrifuged. The supernatant was recovered (cytoplasmic fraction),
and the pellet was washed with Buffer A and resuspended in Buffer B (0.2 mM EGTA [pH 8], 3
mM EDTA [pH 8]) supplemented with protease and phosphatase inhibitors (Roche, Laval,
Canada) and 1 mM DTT. After a 30 min incubation period, samples were centrifuged and the
resulting supernatant isolated (nuclear fraction).

60
3.2.8 Confocal microscopy
Confocal images were acquired on a Zeiss LSM 510 Meta Laser Scanning confocal system (Carl
Zeiss, Thornwood, NJ, USA) with a 100× Plan-Apochromat 1.4 NA oil-immersion objective.
3.2.9 siRNA
SMARTpool Cul2-specific siRNA was obtained from Dharmacon (Chicago, IL) using
Oligofectamine (Invitrogen, Burlington, ON) or EXTREMEGENE (Roche, Laval, QU).
3.3 RESULTS AND DISCUSSION
3.3.1 ECV- and FN-associated functions of VHL are mutually exclusive
The ability of VHL to regulate HIF is dependent on its ability to form a functional ECV.
Therefore, we asked whether the ability of VHL to bind FN was likewise dependent of ECV.
VHL-associated FN was immunoprecipitated following siRNA-mediated knockdown of Cul2, a
scaffold component on ECV, in 35S-radiolabelled VHL-null RCC4 renal carcinoma cells
ectopically expressing HA-VHL(WT) or empty plasmid (MOCK). The level of FN co-
precipitating with VHL did not diminish despite marked reduction in Cul2 expression (Fig. 3.1
and Fig. 3.1d). As expected, siRNA-mediated knockdown of Cul2 attenuated VHL-dependent
ubiquitylation of HIF1αODD (data not shown). Furthermore, biotinylated HIF1αODD-OH
peptides co-precipitated ECV components without the presence of FN as compared to FN co-
precipitated from 35S-radiolabelled 786-VHL RCC cells using anti-HA antibody directed against
HA-VHL (Fig. 3.1c, compare lanes 2 and 3). Although formally possible that HIF1αODD-OH
peptides could have displaced FN via competition for VHL, hypoxia or hypoxia mimetic
(desferroxamine or CoCl2) treatment of 786-VHL cells did not increase VHL/FN interaction
(Fig. 1e). These results argue against the notion that VHL binding to FN is influenced by
competition with HIFα, and that ECV complex is not necessarily required for VHL to bind FN.
The identity of the high molecular weight protein co-precipitating with VHL was confirmed as
FN via immunoblotting and subsequent autoradiography of the same PVDF membrane (Fig. 3f).
FN is known to bind ColIV. However, affinity purified FN complex containing VHL generated

61
from 786-WT cells failed to show the presence of ColIV (data not shown), suggesting that
VHL/FN interaction is independent of ColIV.

62
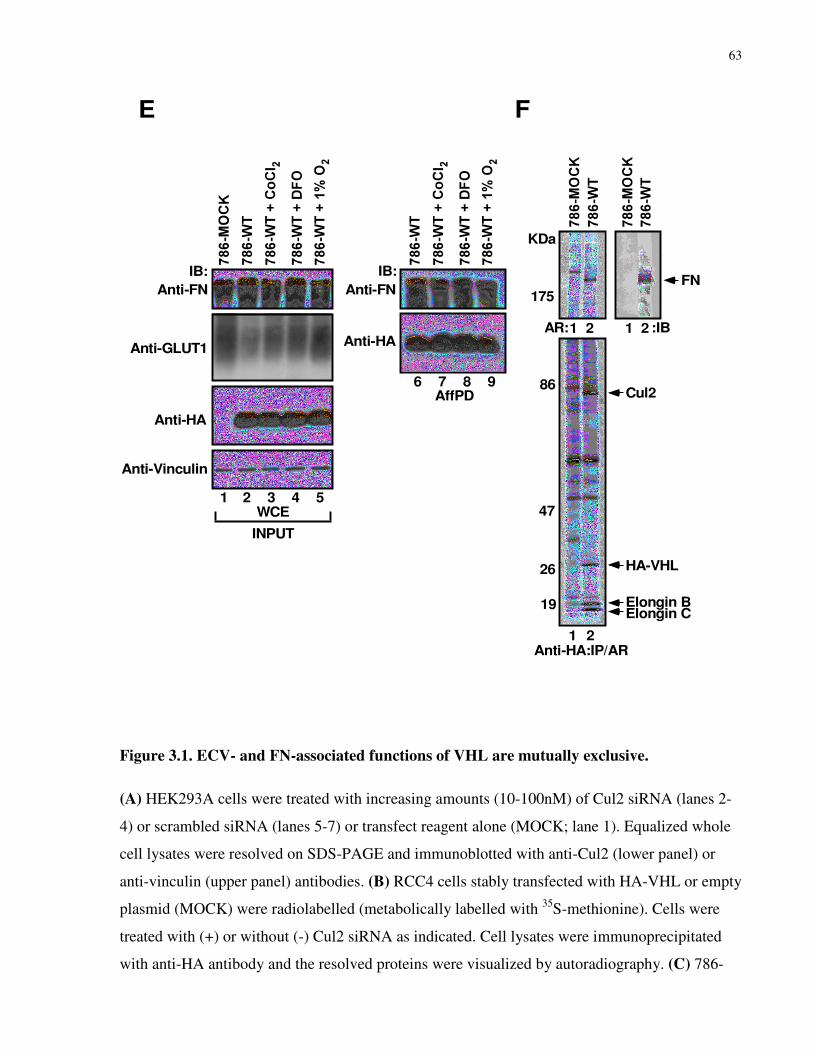
63
FN
Cul2
HA-VHL
Elongin BElongin C
KDa
175
86
47
26
19
Anti-HA:IP/AR
AR: :IB
1 2
1 2 1 2
E
Anti-HA
Anti-FN
Anti-Vinculin
Anti-GLUT1
WCE1 2 3 4 5
IB:
INPUT
AffPD6 7 8 9
Anti-HA
Anti-FN
IB:
F
Figure 3.1. ECV- and FN-associated functions of VHL are mutually exclusive.
(A) HEK293A cells were treated with increasing amounts (10-100nM) of Cul2 siRNA (lanes 2-
4) or scrambled siRNA (lanes 5-7) or transfect reagent alone (MOCK; lane 1). Equalized whole
cell lysates were resolved on SDS-PAGE and immunoblotted with anti-Cul2 (lower panel) or
anti-vinculin (upper panel) antibodies. (B) RCC4 cells stably transfected with HA-VHL or empty
plasmid (MOCK) were radiolabelled (metabolically labelled with 35S-methionine). Cells were
treated with (+) or without (-) Cul2 siRNA as indicated. Cell lysates were immunoprecipitated
with anti-HA antibody and the resolved proteins were visualized by autoradiography. (C) 786-

64
WT and 786-MOCK cells were metabolically labelled with 35S-methionine, lysed and
immunoprecipitated with an anti-HA antibody (lanes 1 and 2) or pulled-down with synthetic
HIF1α[ODD]-OH peptides (lanes 3 and 4). Bound proteins were resolved and visualized by
autoradiography. (D) Extracts prepared from RCC4-VHL or RCC4-MOCK cells treated with
Cul2-specific or scrambled siRNA in the presence of MG132 were immunoprecipitated with
anti-HA antibody and the resolved proteins were visualized by immunoblotting with the
indicated antibodies. (E) 786-MOCK and 786-WT cells were radiolabelled with 35S-Methionine
and lysates were immunoprecipitated with anti-HA antibody. Bound proteins were resolved by
SDS-PAGE, transferred to PVDF membrane and visualized by autoradiography (left panels).
The upper portion of the membrane (above 130KDa) was also immunoblotted with an anti-FN
antibody (right panel). (F) Equal amounts of whole cell extracts generated from 786-WT cells
treated with hypoxia mimetics DFO and CoCl2 or hypoxic conditions (1% Oxygen) were
resolved on SDS-PAGE and immunoblotted with the indicated antibodies (left panel).
Fibronectin complexes were affinity purified from the indicated whole cell extracts, bound
proteins resolved and immunoblotted with anti-FN (right, top panel) or anti-HA (right, bottom
panel) antibody. AffPd: FN affinity pull-down; IB: immunoblot; IP: immunoprecipitation; AR:
autoradiography; WCE: whole cell extract * denotes non-specific protein bands.

65
3.3.2 Disruption of NEDD8 pathway abrogates FN binding to VHL, but
not ECV formation
Recently, we have shown that mutations in VHL that disrupt NEDD8 conjugation lead to a
failure in binding FN78. To further address whether VHL binding to FN is dependent on the
NEDD8 pathway independent of ECV complex formation, Chinese hamster ovary (CHO) ts41
cells with a temperature-sensitive APP-BP1 (a component of NEDD8-activating enzyme;
NAE)178 were transiently transfected with plasmids encoding HA-VHL and GFP-FN. VHL
binding to FN was dramatically decreased in cells maintained under restrictive temperature as
compared to cells under permissive temperature (Fig. 3.2a), suggesting that an intact NEDD8
pathway is critical for promoting VHL binding to FN. As expected, neddylation of VHL and
Cul2 was curtailed under non-permissive temperature (Fig. 3.2b and c). Importantly, an intact
ECV capable of binding HIF1α was observed under both temperatures conditions (Fig. 3.2c),
suggesting that the ability of VHL to form an ECV is not sufficient for binding FN. Furthermore,
non-neddylatable VHL(RRR)78, while showing similar subcellular distribution pattern as
VHL(WT) has compromised ability to bind FN78 (data not shown). These results suggest that
neddylation of VHL does not promote FN binding by altering the subcellular localization of
VHL, which binds to the cytosol-exposed region of FN in ER/Golgi (data not shown).
Neddylated substrates are often de-conjugated post-lysis making detection of the neddylated
species technically challenging. Therefore, we performed our purification of the VHL/FN
complex in the presence of a deneddylase inhibitor NEM and show that VHL in complex with
FN is exclusively unmodified VHL (Fig. 3.2e). As a control, neddylated Cul2 was preserved in
the presence of NEM, even in the presence of a well-established purified deneddylase NEDP1
(Fig. 3.2d). The exclusive presence of unneddylated VHL in complex with FN suggests that the
neddylation of VHL is an intermediary step, which is proceeded by dennedylation of VHL
allowing unhindered association with FN.

66

67
Figure 3.2. Restriction of a dynamic NEDD8 pathway results in the attenuation of VHL
binding to FN. (A) Ts41 cells were transfected with plasmids encoding HA-VHL(WT) and
GFP-FN. Cells were grown at permissive or non-permissive temperature for 15 h and
radiolabelled. Cells lysates were immunoprecipitated with an anti-HA antibody and resolved
proteins were visualized by autoradiography. (B) Ts41 CHO cells were transfected with plasmids
encoding HA-VHL(WT) and NEDD8. Cells were grown at permissive (P; 33°C) or non-
permissive (NP; 39°C) temperature for 15 h, lysed, immunoprecipitated and immunoblotted with
an anti-HA antibody. (C) ts41 cells were transfected with plasmids encoding HA-VHL and
HIF1α. Cells were grown at permissive or non-permissive temperature for 15 h, then lysed, and
immunoprecipitated with anti-HIF1α antibody. Resolved proteins were visualized by
immunoblotting with anti-HIF1α (upper panel), anti-Cul2 (middle panel) or anti-HA (lower
panel) antibodies. (D) 786-WT cellular extracts were incubated with or without a purified

68
deneddylase NEDP1 in the presence or absence of a deneddylase inhibitor NEM for 30min at
37°C. Reaction mixtures were then resolved on SDS-PAGE and visualized by immunoblotting
with the indicated antibodies. (E) FN complexes were affinity purified from the indicated whole
cell extracts in the presence of excess NEM. Bound proteins were competitively eluted and
immunoprecipitated with anti-HA antibody and immunoblotted with anti-FN or anti-HA
antibody (lanes 1-3, top and bottom panel, respectively). Equal amounts of whole cell extracts
were also immunoblotted with anti-FN or anti-vinculin antibody (lanes 4-6, top and bottom
panel, respectively).* denotes uncharacterized protein band; IP: immunoprecipitation; IB:
immunoblot; AR: autoradiography; AffPd: FN affinity pull-down; Open arrow denotes predicted
molecular weight of NEDD8-conjugated HA-VHL.

69
3.3.3 Neddylation of VHL prevents ECV complex formation via steric
hindrance
VHL contains two major functional domains; α and β48. The β domain is required for binding
substrates48,179 and the α domain is required for binding Elongin C48, which serves as a bridge
connecting VHL to the rest of the ECV components. Residues spanning 158-172 (Elongin B/C-
box) within the α domain has been shown to be necessary and sufficient for binding Elongin C182
and K159 has been shown to be the major acceptor site of NEDD878. Structurally and
functionally, ECV is analogous to SCF. Although ECV has not been crystallized, SCF183 and the
VHL/Elongins B/C (VBC) complex48,184 have been solved. To determine possible effects of
NEDD8 conjugation to VHL, we superimposed the VBC complex against SCF. In particular,
Skp1 and its orthologue Elongin C polypeptide backbones were aligned within 1.3Å, giving
confidence that Cul1 would be positioned similarly to Cul2 in the context of ECV. Based on the
composite VBC-Cul1 structure, NEDD8 conjugation of VHL at K159 would create significant
steric hindrance that would prohibit the incorporation of Cul2 or possibly Elongin C to VHL
(Fig. 3.3a and b). Based on this prediction, a VHL mutant incapable of binding Elongin C due to
a mutation within the Elongin B/C-box (excluding K159 and K171) would be more accessible
for NEDD8 modification. HEK293A cells were transfected with plasmids encoding T7-NEDD8
in combination with plasmids encoding HA-VHL(WT), non-neddylatable HA-VHL(RRR), and
HA-VHL(C162F), a well-established α domain mutant incapable of binding Elongin C179,182. As
expected, HA-VHL(WT) generated a slower migrating T7-NEDD8-conjugated HA-VHL, while
HA-VHL(RRR) failed to generate a NEDD8-modified isoform (Fig. 3.3c). Consistent with the
steric hindrance model, HA-VHL(C162F) was neddylated to a greater extend in comparison to
VHL(WT) (Fig. 3.3c). Moreover, while the neddylated VHL comprises a minor fraction of total
VHL, significantly less neddylated VHL was found in complex with Cul2 (Fig. 3.3d, compare
lanes 1 and 2), suggesting an exclusion of neddylated VHL in ECV complex. These results
strongly suggest that neddylation of VHL generates a steric clash preventing its association into
the ECV complex.

70
Figure 3.3. NEDD8 modification of VHL generates steric hindrance blocking the formation
of ECV. (A) VBC (VHL/Elongins B/C) crystallized with HIF1αODD peptide was visualized
using DeepView/Swiss-PdbViewer v3.7. Complex was viewed with side chains and showing van
der Waals forces. Lysine 159, the primary site of neddylation, has been highlighted. (B) The
backbone of Elongin C in the VBC (1LM8.pdb) was overlaid with the backbone of Elongin C-
orthologue Skp1 in the SCF complex (1LDK.pdb). Using the iterative Magic Fit function of

71
DeepView/Swiss-PdbViewer, a fit was generated with an overlap consisting of 99 residues
between Elongin C and Skp1 with a RMS of 1.26 Å. (C) U2OS cells were transfected with
plasmids encoding HA-VHL(WT), HA-VHL(RRR), HA-VHL(C162F), T7-NEDD8, or empty
vector (MOCK). Cells were then lysed, immunoprecipitated and immunoblotted with anti-HA
antibody. (D) HEK293A cells were transfected with the indicated combination of plasmids
encoding HA-Cul2, T7-VHL, and NEDD8. Immunoprecipitation with anti-HA (lanes 2 and 3) or
anti-T7 (lanes 1 and 4) antibodies were performed on pooled lysates. Resolved proteins were
immunoblotted with an anti-Cul2 (top panel), anti-T7 (middle panel), or anti-VHL (bottom
panel) antibody. A long exposure of anti-T7 immunoblot was taken to better visualize neddylated
VHL. * denotes uncharacterized protein band; IP: immunoprecipitation; IB: immunoblot.

72
3.3.4 Cul2 is excluded from the VHL/FN complex
The NEDD8-induced steric hindrance model would predict an exclusion of one or more ECV
components, which may be necessary for the promotion of FN-mediated function. To directly
determine whether ECV components are excluded from the VHL-FN complex, we performed
affinity purification of intracellular FN from 786-MOCK, 786-WT and 786-C162F cells. FN-
containing complexes were then competitively eluted from the sepharose beads and
immunoprecipitated with an anti-HA antibody selecting for the FN complexes associated with
HA-VHL. As expected, HA-VHL(WT) was present in the affinity purified FN complex and co-
precipitated FN, while a diseasing-causing HA-VHL(C162F) mutant, which has an intrinsic
defect in FN binding78, was absent in the affinity purified FN complex (Fig. 3.4a, lanes 4 and 5).
Equal amounts of whole cell extracts were separated on SDS-PAGE and immunoblotted for total
FN and HA-VHL, which indicated the presence of FN in all of the indicated cell types (Fig. 3.4a,
lanes 1-3). Notably, FN is known to bind ColIV, which has been shown recently to interact with
VHL. However, the affinity purified FN co-precipitated via HA-VHL did not contain ColIV
(data not shown), suggesting that VHL binds FN independently of ColIV. Next, anti-HA
immunoprecipitations were performed on the whole cell extracts or affinity purified intracellular
FN complexes generated from 786-MOCK, 786-WT and 786-C162F cells (Fig. 3.4b). While,
HA-VHL(WT) co-precipitated Cul2 from the whole cell extracts as expected, HA-VHL(WT) in
the FN complex did not co-precipitate Cul2 (Fig. 3.4b, compare lanes 2 and 5). In parallel, an
anti-HA immunoprecipitation from the affinity purified FN complex from 35S-radiolabelled 786-
VHL cells showed an absence of Cul2, but a clear presence of Elongins B and C, in the HA-
VHL/FN complex (Fig. 3.4c). These results demonstrate that Cul2 is excluded from the VHL/FN
complex. NEDD8 conjugation to VHL precludes VHL from entering the ECV; we therefore
reasoned that additional means of removing Cul2 might rescue the FN binding of non-
neddylatable VHL. Immunoprecipitated HA-VHL was washed under high-salt/detergent buffer
condition and then mixed with whole cell extracts prepared from 786-MOCK cells radiolabelled
with 35S-Methionine. Post-lysis complexes were then washed, resolved by SDS-PAGE and
visualised by audioradiography (Fig. 3.4d, right panel vs. left panel). Notably, the exclusive
presence of unneddylated VHL in complex with FN (Fig. 3.4, Fig. 3.2e) also suggests that the
neddylation of VHL represents an intermediary step that prohibits Cul2 engagement, which is
proceeded by deneddylation of VHL allowing unhindered association with FN.

73
FN
HA-VHL
A
1 2 3 4 5 6
INPUT
AffPD
Anti-FN
IB:
Anti-HA
Anti-HA:IP
WCE
B
AffPD WCE
HA-VHLAnti-HA
Anti-Cul2 Cul2
1 2 3 4 5 6
IB:
Anti-HA:IP
IgG light
Cul2
HA-VHL
Elongin B
AffPD WCE1 2 3 4 5 6
Elongin C
*
Anti-HA:IP/AR
C
FN
5 642 31WCE-Anti-HA:IB
WASH-Anti-HA:IP/AR
HA-VHL
*
INPUT
FN*
HA-VHL
Anti-HA:IP/AR
Anti-HA:IP/AR
D
Figure 3.4. VHL/FN complex excludes ECV component Cul2. (A) Equal amounts of whole
cell extracts generated from 786-MOCK, 786-WT, and 786-C162F cells were resolved and
immunoblotted with anti-FN (top panel) or anti-HA (bottom panel) antibodies (lanes 1-3). FN
complexes were affinity purified from the indicated whole cell extracts, bound proteins
competitively eluted and immunoprecipitated with anti-HA antibody (lanes 4-6). Resolved
proteins were immunoblotted with anti-FN (top panel), or anti-HA (bottom panel) antibody. (B)
HA-VHL was immunoprecipitated with anti-HA antibody from either purified FN complexes
(lanes 1-3) or whole cell extract (lanes 4-6) generated from pooled cell lysate from the indicated

74
cell lines. Bound proteins were resolved and immunoblotted with anti-Cul2 (top panel) or anti-
HA (bottom panel) antibody. (C) Cells were radiolabelled and prepared as in B. Resolved
proteins were visualized by autoradiography. (D) Left: 786-O cells stably expressing the
indicated VHL mutants were metabolically labelled with 35S-Methionine, lysed and
immunoprecipitated with anti-HA antibody. Bound proteins were resolved on SDS-PAGE and
visualized by autoradiography. Right: HA-VHL was immunoprecipitated from the indicated cell
lysates and washed under high-salt/detergent buffer condition and equilibrated with PBS. HA-
VHL-bound beads were then mixed with whole cell extracts prepared from 786-MOCK cells
radiolabelled with 35S-Methionine. Post-lysis complexes were then washed, resolved by SDS-
PAGE and visualized by autoradiography (top panel). Whole cell extracts of the indicated cells
were also resolved on SDS-PAGE and immunoblotted using anti-HA antibody (bottom panel).
IP: immunoprecipitation; IB: immunoblot; AR: autoradiography.

75
3.3.5 Discussion
In keeping with the prediction based on the composite VBC-Cul1 structure (see Fig. 3), NEDD8
modification of VHL prevents Cul2 engagement and thus excluded from the ECV complex. This
‘freed’, albeit, minor pool of VHL binds FN, representing a requisite step in the eventual
assembly of the extracellular FN matrix. VHL in complex with FN is unmodified, which
suggests that VHL is transiently modified by a dynamic neddylation and deneddylation process.
In support, the inhibition of NEDD8 pathway or the ablation of the NEDD8-conjugation sites on
VHL markedly attenuated the ability of VHL to interact with FN while preserving ECV
integrity. The requirement of this dynamic process also explains why the non-neddylatable
VHL(RRR) mutant is defective in FN binding and assembly.
The preclusion of Cul2 in the VHL/FN complex also suggests that the physical presence of Cul2
may be inhibitory in the engagement of FN to VHL. In support of this notion, a near-complete
knockdown of Cul2 increased the amount of FN-bound to VHL (Fig 1d). In a complementary
experiment, HA-VHL from 786-WT and 786-RRR cells was immunoprecipitated and washed
under higher stringency salt and detergent conditions to strip away VHL-associated proteins. The
‘stripped’ VHL was then mixed with radiolabelled VHL-null 786-O cell lysates and re-
immunoprecipitated. Under such condition, VHL(RRR)’s ability to bind de novo FN was
restored to a level comparable to VHL(WT). VHL(C162F) was still incapable of binding FN
(Fig 4d), consistent with the notion that Elongins B/C are required for VHL/FN interaction.
Moreover, a direct interaction between FN and VHL was shown to not require Cul2 as VBC
complex lacking Cul2 was sufficient to bind FN70. These results suggest that the deficiency of
non-neddylatable VHL lies in the inability to disengage Cul2 or Cul2-associated inhibitory
factor(s) in the absence of dynamic NEDD8 processing. Elongins B/C are likely providing
stability to the unstable tertiary structure of VHL48,185 within the VHL/FN complex. The
structural requirement provided by Elongins B/C perhaps explains why α domain VHL mutants
including C162F fail to bind FN. In this regard, analogous to HIFα, FN binding by VHL requires
both direct physical interaction and the association of Elongins.
VHL plays a critical role in ECV-mediated destruction of HIFα and the assembly of FN ECM.
Neddylation of VHL prohibits the engagement of Cul2 and concomitantly activates the
association with FN. Thus, NEDD8 acts as a molecular switch that defines the functional

76
selectivity of VHL and provides the first mechanistic demarcation of the HIF-dependent and
HIF-independent pathways.

77
Chapter 4 VHL/SOCS1 Heterocomplex Degrades JAK2
Ryan C. Russell*, Roxana I. Sufan*, Olga Roche, Terri D. Richmond, Dwayne L. Barber,
Meredith S. Irwin, and Michael Ohh. VHL and SOCS1 cooperate to degrade JAK2:
implications for polycythemia. In preparation.
*Authors contributed equally to this work.

78
4.1 Rationale
Mutations of VHL leading to CP cause a disease with features of both primary and secondary
polycythemia. CP patients and the CP mouse have high EPO levels and an intrinsic
hypersensitivity to EPO 40,142. Secondary polycythemic features have been explained by a
diminished capacity of CP-VHL(R200W) to bind HIFα resulting in mild HIFα stabilization and
elevation of EPO levels 40. However, HIFα stabilization has not been associated with
hypersensitivity of erythroid progenitors to EPO. Therefore, we biochemically characterized
mutants that give rise to autosomal recessive polycythemia in order to address the molecular
mechanism underlying primary polycythemic features of CP. Here, we reveal that the wild-type
VHL and tumor-causing VHL mutants form a complex with SOCS1 to target phosphorylated
JAK2 for ubiquitin-mediated destruction. We further show that a select cluster of VHL mutants
including CP-VHL(R200W and H191D) mutants form a defective heterodimer with SOCS1,
severely compromising JAK2 degradation and consequently enhancing JAK2-STAT5 signalling
pathway. These findings provide the mechanism underlying primary polycythemic features of
CP and introduce VHL as a novel regulator of JAK2.

79
4.2 Materials and Methods
4.2.1 Cells.
786-O RCC and HEK293A cells were obtained from the American Type Culture Collection
(Rockville, MD, USA) and maintained in Dulbecco’s modified Eagle’s medium supplemented
with 10% heat-inactivated fetal bovine serum (FBS) (Sigma, Milwaukee, WI, USA) at 37°C in a
humidified 5% CO2 atmosphere. 786-O subclones ectopically expressing HA-VHL(WT), HA-
VHL(C162F) or empty plasmid were previously described 38. 786-O subclones ectopically
expressing HA-VHL(R200W) and HA-VHL(H191D) were generated as previously described 38.
Ba/F3 pro B cells were obtained from the American Type Culture Collection (Rockville, MD,
USA) and maintained in RPMI 1640 supplemented with 10% FBS and 0.5 U/ml recombinant
human EPO (Janssen Ortho, Toronto, ON, Canada).
4.2.2 Antibodies.
Rabbit antibodies against JAK2, VHL, pJAK2, and pSTAT5 were obtained from Cell Signalling
Technologies (Danvers, MA, USA). Polyclonal antibodies against ubiquitin, Elongin B and
HIF2α antibodies were obtained from DAKO Canada (Mississauga, ON, Canada), Santa Cruz
Biotechnology (Santa Cruz, CA, USA) and Novus Biologicals (Littleton, CO, USA),
respectively. Monoclonal antibodies against HA (12CA5), T7 and VHL(IG32) were obtained
from Boehringer Ingelheim (Laval, QC, Canada), Novagen (Madison, WI, USA) and BD
Biosciences (Mississauga, ON, Canada), respectively. Monoclonal anti-α-tubulin antibody was
obtained from Abcam (Cambridge, MA, USA). Polyclonal anti-Cul2 and anti-SOCS1 antibodies
were obtained from Invitrogen (Burlington, ON, Canada) and Novus Biologicals (Littleton, CO,
USA), respectively. MG132 proteasome inhibitor was obtained from Boston Biochem
(Cambridge, MA, USA).

80
4.2.3 Plasmids.
Plasmid encoding HA-SOCS1 was generously provided by Dr. Robert Rottapel (Ontario Cancer
Institute, Toronto, ON, Canada). T7-VHL and HA-VHL(WT, R64P, V74G, Y98H, S111H,
Y112H, Y112N, F119S, L128F, L158S, K159E, C162F, L188V) were previously described 38,78,186,187. HA-VHL(R200W, H191D) were generated using QuikChange site-directed
mutagenesis kit (Stratagene, La Jolla, CA, USA) and mutations verified by direct DNA
sequencing.
4.2.4 Immunoprecipitation and immunoblotting.
Immunoprecipitation and Western blotting were performed as described previously 188. In brief,
cells were lysed in EBC buffer (50 mM Tris, pH 8.0; 120 mM NaCl; and 0.5% NP-40)
supplemented with protease and phosphatase inhibitors (Roche, Laval, Canada). Cell lysates
were immunoprecipitated with indicated antibodies in the presence of Protein-A agarose beads
(Waltham, MA, USA). Bound proteins were washed five times with NETN buffer (20 mM Tris,
pH 8.0; 120 mM NaCl; 1 mM EDTA; and 0.5% NP-40), eluted by boiling in sodium dodecyl
sulfate (SDS)-containing sample buffer, and resolved by SDS polyacrylamide gel electrophoresis
(PAGE).
4.2.5 Metabolic labeling.
Metabolic labeling was performed as described previously 188. In brief, 786-O cells were
maintained in methionine-free Dulbecco's modified Eagle's medium for 45 min then
supplemented with 35S-methionine (100 µCi/ml of medium; Amersham Biosciences,
Buckinghamshire, United Kingdom) and 2% dialyzed fetal bovine serum for 3 h at 37 °C in a
humidified 5% CO2 atmosphere.

81
4.2.6 In vitro ubiquitylation assay.
T7-JAK2 and T7-pJAK2 were purified on Protein-A agarose beads (Waltham, MA, USA) with
anti-T7 antibody from HEK293 cells transfected with T7-JAK2 and EPOR stimulated with or
without EPO. Ubiquitylation reaction was then performed as described previously 189 on JAK2 or
pJAK2 bound on beads.
4.2.7 Generation of phenylhydrazine-primed splenic erythroblasts.
Mice were injected intraperitoneally with 50 mg/kg phenylhydrazine hydrochloride (Sigma-
Aldrich, Oakville, ON, Canada) in PBS on days 1 and 2, as previously described 142. Mice were
sacrificed and spleens removed on day 4 under sterile conditions. Single-cell suspensions were
generated using a 70-µm cell strainer for further analysis.
4.2.8 Cytokine deprivation and stimulation of murine splenic erythroblasts.
Cells were washed twice in PBS, starved in α-MEM supplemented with 2% FCS for 4 hrs at
37°C, and then stimulated with various concentrations of EPO for 15 min at 37°C. Cells were
pelleted at 6000 rpm for 1 min and lysed in 1% Triton X-100 lysis buffer supplemented with
phosphatase inhibitors (Laval, Canada) supplemented with 20mM Na3P2O2, 10mM NaF and
1mM Na3VO4.

82
4.3 RESULTS
4.3.1 CP-VHL mutants have reduced capacity to form ECV
VHL(R200W and H191D) mutants showed diminished association with Elongins B/C and Cul2,
the core components of ECV, when expressed in human embryonic kidney epithelial cells
HEK293 or RCC 786-O(VHL-/-) cells (Fig. 4.1a, b and c). Tumor-associated VHL(C162F)
mutant, which is known to be defective in forming an ECV complex 190, served as control. Thus,
in addition to the previously reported defect in HIFα binding, CP-VHL mutants are
compromised in ECV assembly, which is also likely to contribute to HIFα stabilization.
Furthermore, all tumour-associated VHL mutants tested-to-date have invariably shown a failure
in binding to FN and formation of FN fibrillar array in the extracellular space 188. In contrast,
CP-VHL mutants, but as expected not VHL(C162F), showed intact interaction with FN and
robust extracellular FN matrix deposition (Fig. 4.1d). Thus, VHL(R200W) and VHL(H191D)
are the first naturally occurring VHL mutants exhibiting proper FN matrix deposition, which is
consistent with the absence of cancer predisposition in individuals with CP.

83

84
Figure 4.1. CP-VHL exhibits altered binding to ECV components and JAK2.
(A) HEK293 cells transfected with the indicated plasmids were lysed, immunoprecipitated with
anti-HA antibody and immunoblotted with indicated antibodies. (B,C) 35S-radiolabelled 786-O
subclones stably expressing indicated HA-VHL were immunoprecipitated with anti-HA
antibody, resolved by SDS-PAGE and visualized by autoradiography. (D) 786-O subclones
stably expressing the indicated HA-VHL were grown on glass coverslips and immunostained for
FN (red) and visualized by fluorescent microscopy. DAPI (blue) staining indicates nuclei.
(E) HEK293 cells transfected with the indicated combination of plasmids were treated with (+)
or without (-) MG132. Equal amounts of cell lysates were immunoprecipitated with anti-VHL
antibody and immunoblotted with the indicated antibodies. (F) HEK293 cells transfected with
the indicated plasmids were lysed in the absence of MG132, immunoprecipitated with anti-HA
antibody and immunoblotted with the indicated antibodies. WCE: whole cell extract; IP:
immunoprecipitation; IB: immunoblot; AR: autoradiography. Asterisk denotes non-specific
protein bands.

85
4.3.2 VHL binds JAK2 in a proteasome-sensitive manner
In addition to reduced Cul2 binding, 35S-metabolic labelling of 786-O cells stably expressing
VHL(R200W or H191D) revealed an associated protein of 120kDa in the absence of proteasome
inhibitor (Fig. 4.1c). JAK2 is approximately 120KDa and aberrant JAK2-STAT5 signalling has
been reported to cause hypersensitivity in BFU-E cells to EPO in PV patients 191. Similarly,
BFU-E cells of CP patients are also hypersensitive to EPO 40 and thus, we asked whether VHL
interacts with JAK2. HEK293 cells transfected with plasmids encoding HA-VHL(WT) and T7-
JAK2 were treated with or without proteasome inhibitor MG132 and immunoprecipitated with
anti-VHL antibody. HA-VHL(WT) co-precipitated JAK2 preferentially in the presence of
MG132 (Fig. 4.1e). HA-VHL(R200W) and HA-VHL(H191D) showed increased association
with JAK2 in comparison to VHL(WT) in the absence of MG132 (Fig. 4.1f). These results
identify JAK2 as a novel substrate of VHL and suggest that CP-VHL mutants have a diminished
capacity to promote proteasome-dependent degradation of JAK2.
4.3.3 VHL promotes ubiquitin-mediated degradation of pJAK2
The level of total JAK2 remained unaffected by ectopic expression of VHL (Fig. 4.1e and f,
bottom panels) suggesting that VHL promotes diminution of a select population of JAK2 upon
engagement. We asked whether VHL promoted degradation of activated JAK2, which is defined
by Y1007/1008 phosphorylation 192. Introduction of HA-VHL(WT) in HEK293 cells resulted in
a dramatic loss of phosphorylated JAK2 (pJAK2) (Fig. 4.2a). We next asked whether the loss of
pJAK2 was due to VHL-mediated ubiquitylation of pJAK2. HEK293 cells were co-transfected
with plasmids encoding T7-JAK2 and EPOR and stimulated with EPO (20U/ml) for 15 min to
generate robust levels of pJAK2 (Fig. 4.2b, left panel), which was subsequently isolated via anti-
T7 immunoprecipitation. The enriched T7-pJAK2 was then subjected to an in vitro
ubiquitylation reaction using S100 extracts devoid of or reconstituted with VHL(WT) (Fig. 4.2b,
right panel). While the total JAK2 levels were unaffected, the level of pJAK2 decreased
dramatically in the presence of VHL(WT), which was accompanied by the appearance of pJAK2
polyubiquitylation (Fig. 4.2b, lane 4). Notably, the low level of VHL-dependent ubiquitylation
observed in the absence of EPO is likely due to limited spontaneous JAK2 autophosphorylation,

86
commonly observed upon ectopic JAK2 expression (Fig. 4.2b, right panel, lane 3). These results
demonstrate that VHL promotes pJAK2 ubiquitylation.
We next investigated the effect of CP-VHL mutants on pJAK2 stability and observed that while
VHL(WT) co-precipitated negligible levels of pJAK2 in the absence of MG132, both CP-VHL
mutants R200W and H191D co-precipitated higher levels of pJAK2, supporting the notion that
CP-VHL mutants have a diminished capacity to promote pJAK2 degradation (Fig. 4.2c).
VHL patients rarely develop polycythemia despite harboring VHL mutations that abolish HIFα
degradation 142. Thus, tumor-causing VHL mutants incapable of binding or ubiquitylating HIFα
are predicted to retain the ability to promote ubiquitin-mediated destruction of pJAK2. A panel
of VHL substitution mutants spanning the open reading frame were tested for their ability to
degrade pJAK2. Consistent with our prediction, expression of VHL mutants, with the exception
of F119S and L128F (discussed below), resulted in negligible levels of pJAK2 in the absence of
MG132 (Fig. 4.2d). In addition, a panel of tumor-causing VHL mutants retained binding to
JAK2 in the presence of MG132 (Fig. 4.2e). Notably, well established α domain VHL mutants
C162F and L158S, which cannot form an ECV 186,190, decreased pJAK2 levels comparable to
that of VHL(WT). These results infer that a novel, ECV-independent, mechanism is responsible
for VHL-mediated pJAK2 degradation.

87

88
Figure 4.2. VHL promotes ubiquitin-mediated destruction of pJAK2.
(A) HEK293 cells transfected with the indicated plasmids were lysed and immunoblotted with
the indicated antibodies. (B) HEK293 cells transfected with the indicated plasmids were treated
with (+) or without (-) EPO and pJAK2 was isolated via anti-T7 immunoprecipitation (left
panel), which was then added to an in vitro ubiquitylation reaction containing proteasome-
depleted S100 fractions containing (+) or not containing (-) VHL (right panels). Reaction
mixtures were then re-immunoprecipitated with anti-T7 antibody, resolved by SDS-PAGE and
immunoblotted with the indicated antibodies. (C) HEK293 cells transfected with the indicated
plasmids were lysed in the absence of MG132, immunoprecipitated with anti-HA antibody and
immunoblotted with the indicated antibodies. (D) HEK293 cells transfected with the indicated
plasmids encoding various tumor-causing HA-VHL mutants were lysed, equal amount of whole
cell extracts resolved by SDS-PAGE and immunoblotted with the indicated antibodies. WCE:
whole cell extract; IP: immunoprecipitation; IB: immunoblot; AR: autoradiography. Asterisk
denotes non-specific protein bands. (E) HEK293 cells transfected with the indicated plasmids
encoding various tumor-causing HA-VHL mutants in combination with T7-JAK2 were treated
with MG132. Equal amounts of cell lysates were immunoprecipitated with anti-HA antibody

89
and immunoblotted with the indicated antibodies. IP: immunoprecipitation; IB: immunoblot;
WCE: whole cell extract.

90
4.3.4 VHL binds and requires SOCS1 to promote pJAK2 degradation
The F-box protein SOCS1 is the principal negative regulator of pJAK2 via ubiquitin-mediated
degradation. VHL, as well as other F-box proteins that confer substrate specificity, have been
shown to homodimerize 193-197. Moreover, homodimerization of entire E3 enzymes such as the
SCF (Skp1/Cdc53 or Cul1/F-box protein) has been shown to increase the efficiency of
ubiquitylation by improving spatial orientation of substrate to active site 197. We asked whether
SOCS1 interacts with VHL to promote ECV-independent degradation of pJAK2. T7-VHL co-
precipitated HA-SOCS1 when ectopically expressed in HEK293 cells (Fig. 4.3a, left panel), and
similar results were obtained by reciprocal immunoprecipitation (Fig. 4.3a, right panel). We
then asked whether VHL/SOCS1 interaction occurred under physiologic conditions. BaF3 cells
that stably express EPOR were treated with or without MG132. Cell lysates were
immunoprecipitated with anti-VHL or isotype-matched control antibody and bound proteins
were visualized by Western blot analysis, which showed endogenous VHL co-precipitating
SOCS1 in the presence of MG132 (Fig. 4.3b). Notably, VHL/SOCS1 interaction was
significantly reduced in the absence of MG132, suggesting perhaps that the complex is sensitive
to proteasomal degradation. The endogenous binding of VHL and SOCS1 will be repeated using
affinity purification for hydroxylated HIF ODD-OH and phosphor tyrosine JAK2 peptides to
improve clarity of band visualization by removing immunoglobulin light chain.
VHL(F119S) and VHL(L128F) mutants are capable of forming an intact ECV and targeting
HIFα for degradation (Fig. 4.3c and 3d), but fail to promote pJAK2 degradation (Fig. 4.3e and
see Fig. 4.2d) and thus, supporting again the notion that the defect in pJAK2 regulation is
independent of ECV. One possibility is that the failure in pJAK2 degradation is due to a defect
in F119S and L128F to engage SOCS1. As predicted, unlike VHL(WT), both F119S and L128F
mutants were severely compromised in binding SOCS1 (Fig. 4.3f), underscoring the potential
requirement of SOCS1, rather than ECV complex formation, in the degradation of pJAK2.
Notably, F119S and L128F mutants retained the ability to bind JAK2 (data not shown).
We asked whether the ability of SOCS1 to recruit the E3 ubiquitin ligase components was
required for VHL-dependent pJAK2 degradation. Analogous to the α domain of VHL, the
SOCS-box of SOCS1 facilitates the recruitment of the various ECS components including Cul5

91
or Cul2, Elongins BC and Rbx1 132,198,199. While both VHL(WT) and VHL(C162F; α domain
mutant that cannot form an ECV) mutant promoted pJAK2 degradation when co-expressed with
wild-type SOCS1, co-expression of SOCS1∆SOCS-box mutant abrogated pJAK2 degradation
(Fig. 4.3g). These results suggest that SOCS-box is required for enzymatic activity of the
VHL/SOCS1 heterodimer for the degradation of pJAK2.

92

93
Figure 4.3. VHL and SOCS1 cooperate to degrade pJAK2 in vivo.

94
(A) HEK293 cells transfected with the indicated plasmids were lysed, immunoprecipitated with
either anti-T7 (left panels) or anti-HA (right panels) antibody, resolved by SDS-PAGE, and
immunoblotted with the indicated antibodies. (B) BaF3 cells stably expressing EPOR were
stimulated with EPO in the presence (+) or absence (-) of MG132. Cells were lysed and
immunoprecipitated with anti-VHL or isotype-matched control antibody and immunoblotted
with the indicated antibodies. (C) 786-O subclones stably expressing the indicated HA-VHL
were lysed, immunoprecipitated with anti-HA antibody and immunoblotted with indicated
antibodies. (D) 786-O subclones stably expressing the indicated HA-VHL were lysed and
immunoblotted with indicated antibodies. (E) Equal amounts of whole cell extracts prepared
from HEK293 cells transfected with the indicated plasmids were resolved by SDS-PAGE and
immunoblotted with the indicated antibodies. (F) HEK293 cells transfected with the indicated
plasmids in combination with a plasmid encoding HA-SOCS1 were lysed, immunoprecipitated
with anti-VHL antibody and immunoblotted with anti-HA antibody. (G) Equal amounts of
whole cell extracts prepared from HEK293 cells transfected with the indicated plasmids and
stimulated with EPO were resolved by SDS-PAGE and immunoblotted with the indicated
antibodies. IP: immunoprecipitation; IB: immunoblot; WCE: whole cell extract. Asterisk
denotes non-specific protein bands.

95
4.3.5 CP-VHL/SOCS1 association inhibits pJAK2 binding and
degradation
We asked whether the observed defect in pJAK2 degradation via CP-VHL was due to a failure in
binding SOCS1. Unexpectedly, both VHL(R200W) and VHL(H191D) mutants showed a
dramatic increase in SOCS1 binding in comparison to their wild-type VHL counterpart (Fig.
4.4a), which suggests that CP-causing mutations confer significantly higher affinity for SOCS1.
We next asked whether this altered affinity of CP-VHL for SOCS1 affected pJAK2 recruitment.
HEK293 cells transfected with plasmids encoding EPOR, T7-JAK2 and HA-SOCS1 in
combination with plasmids encoding HA-VHL(WT or R200W or H191D) were stimulated with
EPO in the presence of MG132 to minimize the degradation of pJAK2. pJAK2 co-precipitated
significantly lower levels of CP-VHL mutants in comparison to VHL(WT), suggesting that the
abnormal association between CP-VHL and SOCS1 hinders pJAK2 substrate binding (Fig. 4.4b).
We next directly compared the efficiency of VHL(WT)/SOCS1 against CP-VHL/SOCS1 in
promoting pJAK2 degradation. T7-pJAK2 was first generated by ectopic expression of EPOR
and T7-JAK2 in HEK293 cells followed by EPO stimulation. Cells were lysed and
immunoprecipitated with an anti-T7 antibody. T7-pJAK2 enriched on beads were washed and
equally distributed into 4 reaction tubes, as confirmed by comparable levels of IgGL (Fig. 4.4c,
bottom panel), and mixed with HEK293 cell lysates expressing empty plasmid (MOCK), HA-
VHL(WT), HA-VHL(R200W) or HA-VHL(H191D) in combination with HA-SOCS1.
VHL(WT)/SOCS1 containing lysate markedly reduced the level of pJAK2 in comparison to CP-
VHL/SOCS1 or SOCS1 only containing lysates (Fig. 4.4c). These results collectively suggest
that the CP-VHL/SOCS1 heterocomplex is defective in promoting pJAK2 degradation.
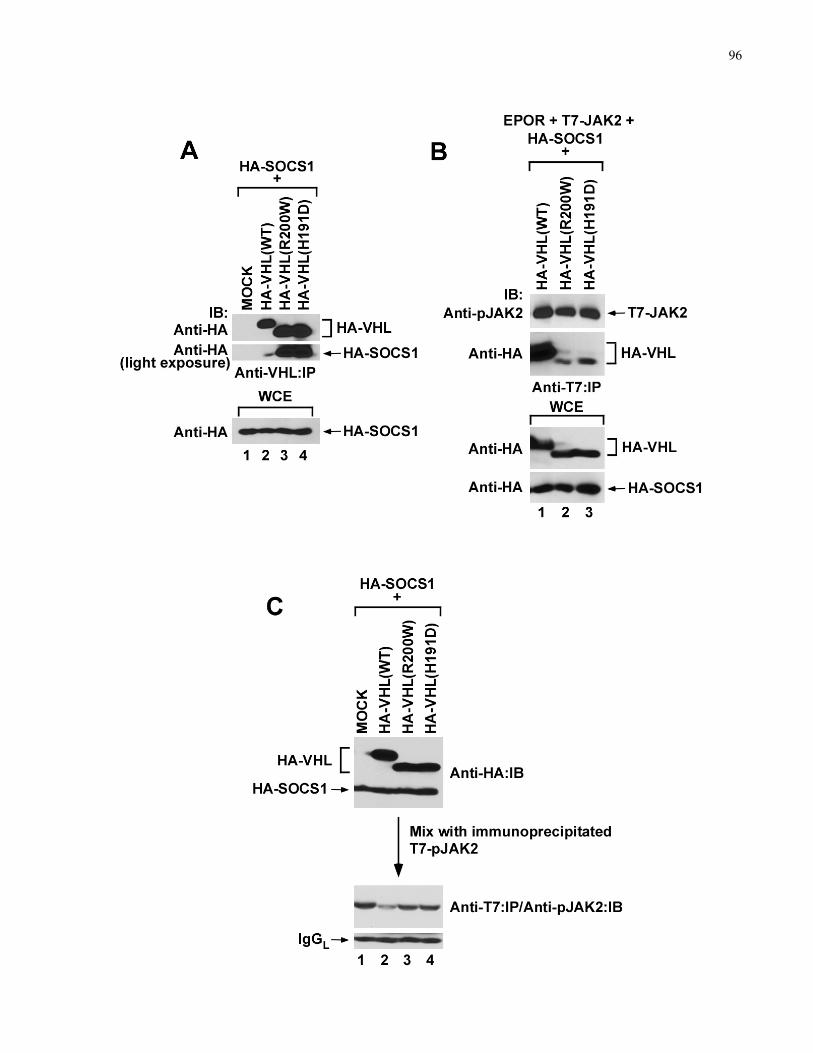
96

97
Figure 4.4. CP-VHL mutants are defective in pJAK2 degradation and R200W/R200W CP
mice exhibit elevated pJAK2 and pSTAT5 levels.
(A) HEK293 cells transfected with the indicated plasmids in combination with HA-SOCS1 were
lysed, immunoprecipitated with anti-VHL antibody and blotted with anti-HA antibody. (B)
HEK293 cells transfected with the indicated plasmids were treated with EPO and MG132, lysed,
immunoprecipitated with anti-T7 antibody, and immunoblotted with the indicated antibodies.
(C) T7-pJAK2 was first generated by ectopic expression of EPOR and T7-JAK2 in HEK293
cells followed by EPO stimulation. Cells were lysed and immunoprecipitated with an anti-T7
antibody. T7-pJAK2 enriched on beads were washed and equally distributed into 4 reaction
tubes, as confirmed by comparable levels of IgGL (bottom panel), and mixed with HEK293 cell
lysates expressing empty plasmid (MOCK), HA-VHL(WT), HA-VHL(R200W) or HA-
VHL(H191D) in combination with HA-SOCS1. (D) Single cell suspensions enriched with
erythroid progenitors generated from spleens of phenylhydrazine-treated R200W/R200W or WT
mice were washed in cytokine-free media to remove any residual cytokines. Cells were cytokine
starved for additional 4 h to purge any pre-existing stimulation of JAK2-STAT5 pathway and
subsequently treated with increasing concentrations of exogenous EPO for 15 min. Equal
amounts of cell lysates were resolved on SDS-PAGE and immunoblotted with the indicated
antibodies. IP: immunoprecipitation; IB: immunoblot; WCE: whole cell extract. Asterisk
denotes non-specific protein bands.

98
4.3.6 pJAK2 and pSTAT5 are elevated in CP-mice
Erythroid progenitors from PV patients are hypersensitive to EPO due to JAK2 activating
mutations associated with increased levels of phosphorylated JAK2 and STAT5 139. Erythroid
progenitors from CP patients or R200W/R200W mice have likewise been shown to be
hypersensitive to EPO 40,142. Single cell suspensions enriched with erythroid progenitors were
generated from spleens of phenylhydrazine (PHZ)-treated R200W/R200W or WT mice and
residual cytokines were removed by washes in cytokine-free media. Cells were cytokine starved
for additional 4 h to purge any pre-existing stimulation of the JAK2-STAT5 pathway and
subsequently treated with increasing concentrations of exogenous EPO for 15 min. Expression
levels of pJAK2 and pSTAT5 were noticeably higher in R200W/R200W compared to the WT
erythroid progenitor-enriched cell lysates (Fig. 4.4d, compare lanes 3 and 4 against 7 and 8).
Densitometry performed on unsaturated exposures of the immunoblots validated the observed
trend (data not shown). These results demonstrate that homozygous inheritance of CP-causing
R200W mutation increases JAK2-STAT5 signalling pathway in vivo. Further experiments will
be conducted in order to determine JAK2-STAT5 sensitivity using both cellular methods (colony
forming assays) and additional biochemical stimulation assays.
4.3.7 Discussion
Mapping of VHL disease-causing mutations on VHL/Elongin B/Elongin C (VBC) crystal
structure engaged with HIF1α peptide has revealed two major domains α and β required for
Elongin C and HIF1α binding, respectively 48,184,200. VHL mutations that disrupt (F119S and
L128F) or enhance (R200W and H191D) SOCS1 binding interestingly clustered to a unique
region of VHL, revealing a likely interface or ‘SOCS groove’ required for the engagement of
SOCS1 (Fig. 4.5a). Notably, the SOCS groove does not overlap with Elongin C or HIF1α
binding interface. This is consistent with the observed autonomy of HIF- and JAK2-associated
functions of VHL clearly revealed by specific mutants F119S and L128F, which retain the ability
to degrade HIFα but fail to degrade pJAK2 despite their ability to form ECV. Conversely,
C162F retains the ability to degrade pJAK2 despite its inability to form ECV or degrade HIFα.

99
Thus, mutations within the groove may alter VHL’s affinity for SOCS1 positively or negatively
via steric conformational change.
We propose the following model of CP. In normal individuals, VHL forms a proper ECV
complex and negatively regulate HIFα via the ubiquitin pathway. In contrast, CP-associated
mutations (e.g., R200W) attenuate HIFα binding and ECV complex formation, causing the
reported mild stabilization of HIFα, which leads to the overproduction of HIF-target EPO in the
kidney and secondary polycythemia (Fig. 4.5b). In normal individuals, VHL also binds SOCS1
through its SOCS groove and together recognize pJAK2 for ubiquitin-mediated degradation, and
thus negatively regulate the JAK2-STAT pathway. The R200W mutation in CP patients causes
conformational change within the SOCS groove, leading to an inordinately tight CP-
VHL/SOCS1 association and thereby blocking pJAK2 recruitment and degradation. Resulting
pJAK2 stabilization promotes hyperactivation of the JAK2-STAT pathway in erythroid
progenitors, causing hypersensitivity to EPO and primary polycythemia.

100
Figure 4.5. The ‘SOCS groove’ and the revised molecular model of CP.
(A) Mutations (red) that influence SOCS1 binding are indicated on the VHL/Elongin B/Elongin
C (VBC) crystal structure bound to HIF1α peptide and cluster within the ‘SOCS-groove’.
Analyzed using DeepView/Swiss-PdbViewer v4.0. (B) Molecular model of CP. See text for
details.

101
The present findings also provide molecular explanations to several mysteries and paradoxes in
VHL and CP fields. For example, it has been unclear why polycythemia is rarely observed in
VHL patients despite the fact that mutations that promote HIFα stabilization were common
among VHL patients 38,69,142. We show here that most tumour-causing VHL mutants, including
those that have lost the ability to degrade HIFα, retain the ability to negatively regulate pJAK2
downstream of EPO signalling, which likely explains the rarity of polycythemia among VHL
patients (see Fig. 4.5b). Furthermore, R200W/WT heterozygous mice, which do not show
detectable HIFα accumulation or EPO overproduction, have BFU-E cells that are modestly
hypersensitive to EPO ex vivo 142. Consistent with this observation is a report describing a
woman with Y175C/WT VHL genotype who has polycythemia without an elevated level of
serum EPO 201. These findings support the notion that HIF is unlikely involved in
hypersensitivity of erythroid progenitors to EPO, a hallmark feature of primary polycythemia.
PV-associated JAK2(V617F) mutation causes uncontrolled expansion of RBCs, but also gives
rise to pleomorphic and clustered megakaryocytes hypersensitive to thrombopoietin, which,
similar to EPO, signals through JAK2 134. Abnormal megakaryocyte function is thought to be
critical in thrombotic complications frequently observed in PV patients 192. Strikingly,
R200W/R200W mice exhibit increased number of megakaryocytes that cluster and CP patients,
like PV patients, often present with thrombotic complications 40,142. In contrast, secondary
polycythemia associated with elevated EPO does not give rise to megakaryocytic defects; an
observation supported in mice with constitutive overexpression of EPO that do not develop
thrombotic complications despite inordinately high RBC count 202. These observations suggest
that the hyperactive JAK2-STAT signalling, but not the increased EPO production due to a mild
defect in HIF regulation, is the principal mechanism underlying thrombotic complications
observed in CP patients.
The discovery of JAK2 mutations in PV patients has certainly expedited the clinical trials of
JAK2 inhibitors in the management of PV. However, despite clinical features shared between
PV and CP, including hypersensitivity to erythropoietin and megakaryocytic defects associated
with thrombotic complications, JAK2 inhibitors have not been considered for CP. Thus, the
present findings linking VHL to JAK2-STAT5 pathway provide a biochemical rationale for
JAK2-targeted therapies in CP.

102
Chapter 5 Conclusions and future directions
5.1 E-cadherin loss in RCC
In addition to our publication of the HIFα-mediated repression of E-cadherin, two independent
reports were published showing a very similar mechanism responsible for E-cadherin loss in
RCC35,203,204. In all three publications E-cadherin expression was recovered by reconstitution of
RCC cell lines with wild type-VHL. However, some differences were noted. Krishnamachary et
al. argue that the regulation of E-cadherin is strictly mediated through HIF1α since restoration of
VHL in HIF1α-deficient 786-O RCC cell line did not rescue E-cadherin expression203. In
contrast, Estaban et al. demonstrated both HIF1α and HIF2α were capable of repressing E-
cadherin expression35. Intriguingly, we were able to observe restoration of E-cadherin
expression upon reintroduction of wild-type VHL in 786-O cells, which do not express HIF1α,
suggesting that downregulation of HIF2α is sufficient for promotion of E-cadherin
expression204,205. It is possible that after extended passaging some immortalized cell lines may
lose the ability to recover E-cadherin expression, explaining the differences in these
observations. Similar to our findings that HIF2α drives the expression of E2 box-specific
transcriptional repressors, Krishnamachary et al. found elevation of SIP1 as well as modest
induction of two additional repressors TCF-3 and δEF1 (ZFHX1A). Interestingly, in our
luciferase assay we saw an incomplete repression of the E-cadherin promoter with SIP1 alone,
indicating that additional repressors may play a role in HIF-mediated repression of E-cadherin.
Taken together, these reports highlight a previously unknown link between the major cause of
RCC development (i.e., VHL inactivation) and the loss of a critical invasion suppressor, E-
cadherin. Early loss of both VHL and E-cadherin observed in the pre-malignant foci of VHL
patients undoubtedly sets the stage for disease progression.
The VHL-mediated regulation of E-cadherin is dependent on HIF, which also governs the
transcription of more than 60 genes, including VEGF, PDGFβ, GLUT1, transferrin and its
receptor, EPO, TGFα/β3, and CXCR4 and its ligand SDF1α55,173. In addition, VHL also
regulates the expression of genes independent of HIF, such as MMPs, TIMPs and FN55. Many

103
of these genes, both HIF-dependent and independent, have been shown to affect the metastatic
potential of RCC; however, the biological contribution of an individual gene product upon VHL
loss during RCC progression is at present unclear and difficult to discern. Interestingly, shRNA-
targeted reduction of E-cadherin in RCC cells with a restored VHL-HIF pathway markedly
increased the invasive potential, suggesting that the loss of E-cadherin plays a critical role in
promoting the malignant behavior of RCC. In keeping with this hypothesis, it was recently
shown that a lack of E-cadherin and VHL staining by IHC correlated with high grade tumours
and poor prognosis206.
Loss of E-cadherin, in addition to promoting invasiveness and EMT, may also influence
the Wnt signalling pathway (see Fig. 5.1)143. For example, phosphorylation of β-catenin via
receptor tyrosine kinases (e.g., c-Met, fyn, fer) or c-src can disassociate β-catenin from the
cytoplasmic tail of E-cadherin and translocate to the nucleus where it binds TCF to drive the
transcription of genes responsible for proliferation and differentiation108,143. Peruzzi et al.
showed that the loss of VHL in RCC triggers HGF-driven β-catenin signalling that induced
branched morphogenesis. Reintroduction of VHL repressed the accumulation of the active
cytoplasmic β-catenin and the disruption of adherens junction106. Nakaigawa et al. showed that
c-Met is phosphorylated upon VHL loss in the absence of HGF ligand stimulation. The
inhibition of c-Met using a c-Met inhibitor attenuated the growth of VHL-null RCC tumors in
nude mice, implicating the importance of c-Met signaling in renal epithelial oncogenesis207. It is
tempting to speculate that a loss of VHL may not only decrease the adhesive potential (and
therefore, increase invasive potential), but also simultaneously increase the availability of active
β-catenin (and β-catenin-mediated gene transcription, such as Cyclin D1) by either constitutive
c-Met phosphorylation and/or inhibition of β-catenin degradation. Thus, the inactivation of a
single gene VHL may cause a pronounced shift from a normal epithelial homeostasis towards an
invasive de-differentiated cellular state with enhanced proliferative capacity. These recent
studies have begun to unravel the molecular pathways regulating the development of aggressive
RCC upon VHL inactivation, involving both HIF-dependent and -independent mechanisms.
Determining the pathophysiologic relevance, as well as the relative contribution, of these distinct
pathways will undoubtedly shed important insight into the understanding the molecular basis of
EMT in kidney cancer.
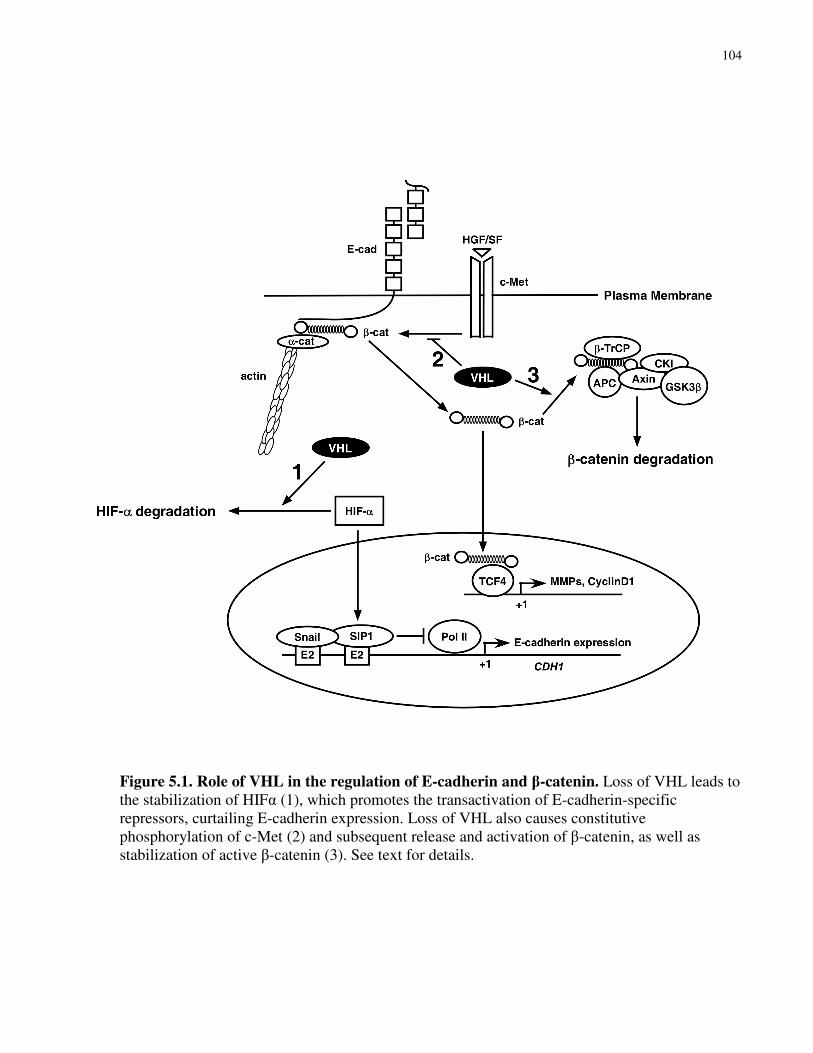
104
Figure 5.1. Role of VHL in the regulation of E-cadherin and β-catenin. Loss of VHL leads to the stabilization of HIFα (1), which promotes the transactivation of E-cadherin-specific repressors, curtailing E-cadherin expression. Loss of VHL also causes constitutive phosphorylation of c-Met (2) and subsequent release and activation of β-catenin, as well as stabilization of active β-catenin (3). See text for details.

105
5.2 Uncovering the mechanism of VHL mediated FN assembly
The dual role of VHL in the promotion of ECM and the negative regulation of the hypoxic
response is well established. While the mechanisms that underlie HIFα destruction are well
understood, the mechanism by which VHL is able to promote an ECM is significantly less clear.
We have demonstrated that the neddylation of VHL promotes direct association with FN by
displacing Cul2; however, the processes that regulate NEDD8 conjugation to VHL remain
unclear. The specificity and regulation of NEDD8 conjugation remains at the level of the E3-
ligase. Therefore, discovery and characterization of the E3 for VHL neddylation will likely yield
clues as to what physiologic conditions promote VHL-FN interaction and subsequent
extracellular fibril formation.
VHL binds FN via its beta-domain, while VHL itself has no known enzymatic function, it likely
acts as an adaptor bringing in additional proteins through its association with elongins B and C.
As the discovery of VHL associated proteins yielded significant clues to the function of VHL in
the ECV, it stands to reason that identification of additional proteins in the VHL-FN complex
will shed light on the function of the VHL-FN complex. Intriguingly, the mechanisms that have
been proposed for FN and ColIV interaction with VHL differ greatly. VHL has been proposed
to bind hydroxylated collagen and the competition for VHL by HIF and ColIV determines the
ability of VHL to bind ColIV. We have not seen a difference in VHL-FN association under
hypoxia, which is in keeping with the observation that overexpression of HIFα does not affect
FN deposition. The differences in proposed mechanisms are in contrast to the striking similarity
that is seen in the defects and production of FN and ColIV matrices. While it is certainly
possible that the rules that govern VHL interaction with the two matrix proteins are entirely
divergent, there is also the possibility that a greater understanding of VHL matrix function will
produce a more unified theory of VHL matrix function. It is interesting to note that VHL-ColIV
interaction was not tested under limiting oxygen tension in vivo, instead a potent iron chelator,
DFO was used as a mimetic for hypoxia. Treatment of DFO resulted in a shift of approximately
30KDa in the ColIV protein. This shift is likely due to a inhibition of glycosylation, which
requires hydroxylated prolines as a substrate for the addition of glycosyl groups in the ER208. It

106
is interesting to note that a Far Western of DFO-treated lysates revealed that VHL only
associates with the upper-glycosylated ColIV band and this association does not appear to be
lessened by DFO treatment, which would be expected if DFO inhibited the hydroxylation of
ColIV. An alternate explanation is that DFO reduces glycosylation and that VHL specifically
recognizes the glycosylated form of ColIV. Interestingly, we have seen VHL colocalize with FN
specifically in the Golgi. Given the role of the Golgi in the refinement of glycosylation and the
fact that both ColIV and FN are heavily glycosylated proteins, it is tempting to hypothesize that
future work may uncover a role of VHL in promoting matrix deposition via refinement of
glycosylation of target proteins in the Golgi.
5.3 Characterization of VHL mutation in additional haematopoietic malignancies
Mutations in JAK2 have been identified in a majority of patients with PV, essential
thrombocythemia (ET) and myelofibrosis with myeloid metaplasia (MMM)127. Increases in
signalling downstream of JAK2 have been observed in mediastinal B-cell lymphoma (PMBL),
classical Hodgkin lymphoma (cHL), chronic lymphocytic leukemia (CLL), and large granular
lymphocyte (LGL) leukemia209. Constitutive signalling downstream of JAK2 in leukemia has
been shown to prevent apoptosis of leukemic cells to stimulus, such as Fas ligand, via the
upregulation of anti-apoptotic proteins including Bcl-xL210,211.
Interestingly, increased signalling of tyrosine kinases including JAK2 has recently been
described in Hodgkin’s lymphoma in the absence of activating mutations of JAK2212. Although
JAK2 is often amplified due to inherent genomic instability, the activation status of JAK2 does
not correlate with copy number213. This paradox was understood upon the identification of
SOCS1 mutation in nearly 50% of cHL samples214. However, JAK2 expression is elevated in
over 85% of cHL allowing for the possibility that additional mutations contribute to the increase
of tyrosine kinase activity in this lymphoma. Given the importance of proper VHL-SOCS1
function in polycythemia, it stands to reason that VHL mutations could affect a similar
hyperactivation of JAK2 in the background of cHL.
Our understanding of the role of JAK2 deregulation in haematopoietic malignancies has rapidly
expanded in the past four years. The negative regulators of JAK2, VHL and SOCS1 represent an

107
important impediment for haematopoietic disease development. Our identification of the role of
VHL in a myeloproliferative disorder brings CP in line with our understanding of MPDs.

108
References
1. Collins, E.T. Intra-ocular growths (two cases, brother and sister, with peculiar vascular new growth, probably retinal, affecting both eyes). Trans. Ophthal. Soc. U.K. 14, 141-149 (1894).
2. Neumann, H.P.H. & Wiestler, O.D. Clustering of features of von Hippel-Lindau disease: evidence of a complex genetic locus. Lancet 337, 1052-1054 (1991).
3. Maddock, I., et al. A genetic register for von Hippel-Lindau disease. J Med Genet 33 33, 120-127 (1996).
4. von Hippel, E. Ueber eine sehr seltene Erkrankung der Nethaut. Graefe Arch Ophthal 59, 83-106 (1904).
5. Lindau, A. Zur Frage der Angiomatosis Retinae und ihrer Hirnkomplikation. Acta Opthal 4, 193-226 (1927).
6. Richard, S., Campello, C., Taillandier, L., Parker, F. & Resche, F. Haemangioblastoma of the central nervous system in von Hippel-Lindau disease. French VHL Study Group. J
Intern Med 243, 547-553 (1998).
7. Frew, I.J., et al. Combined VHLH and PTEN mutation causes genital tract cystadenoma and squamous metaplasia. Mol Cell Biol 28, 4536-4548 (2008).
8. Hough, D.M., Stephens, D.H., Johnson, C.D. & Binkovitz, L.A. Pancreatic lesions in von Hippel-Lindau disease: prevalence, clinical significance, and CT findings. AJR Am J
Roentgenol 162, 1091-1094 (1994).
9. Neumann, H.P.H., et al. Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Landau Disease. N. Engl. J. Med. 329, 1531-1538 (1993).
10. Manski, T., et al. Endolymphatic sac tumors - A source of morbid hearing loss in von Hippel-Lindau disease. JAMA 277, 1461-1466 (1997).
11. Sung, D.I., Chang, C.H. & Harisiadis, L. Cerebellar hemangioblastomas. Cancer 49, 553-555 (1982).
12. Dan, N.G. & Smith, D.E. Pituitary hemangioblastoma in a patient with von Hippel-Lindau disease. Case report. J Neurosurg 42, 232-235 (1975).
13. Ginzburg, B.M., et al. Diagnosis of von Hippel-Lindau disease in a patient with blindness resulting from bilateral optic nerve hemangioblastomas. AJR Am J Roentgenol 159, 403-405 (1992).
14. Benjamin, L.E. & Keshet, E. Conditional switching of vascular endothelial growth factor (VEGF) expression in tumors: induction of endothelial cell shedding and regression of

109
hemangioblastoma-like vessels by VEGF withdrawal. Proc Natl Acad Sci (USA) 94, 8761-8766 (1997).
15. Vortmeyer, A.O., et al. Developmental arrest of angioblastic lineage initiates tumorigenesis in von Hippel-Lindau disease. Cancer Res 63, 7051-7055 (2003).
16. Reifenberger, G., Reifenberger, J., Bilzer, T., Wechsler, W. & Collins, V. Coexpression of transforming growth factor-alpha and epidermal growth factor receptor in capillary hemangioblastomas of the central nervous system. Am J Pathol 147, 245 (1995).
17. Wizigmann-Voos, S., Breier, G., Risau, W. & Plate, K. Up-regulation of vascular endothelial growth factor and its receptors in von Hippel-Lindau disease-associated and sporadic hemangioblastomas. Cancer Res 55, 1358-1364 (1995).
18. Bohling, T., et al. Expression of growth factors and growth factor receptors in capillary hemangioblastoma. J Neuropathol Exp Neurol 55, 522-527 (1996).
19. Krieg, M., Marti, H. & KH, P. Coexpression of erythropoietin and vascular endothelial growth factor in nervous system tumors associated with von hippel-lindau tumor suppressor gene loss of function. Blood 92, 3388-3393 (1998).
20. Glenn, F. Pheochromocytoma, a chromaffin tumor of the adrenal medulla. Harlem Hosp
Bull 10, 1-20 (1957).
21. Reisch, N., Peczkowska, M., Januszewicz, A. & Neumann, H.P. Pheochromocytoma: presentation, diagnosis and treatment. J Hypertens 24, 2331-2339 (2006).
22. Zelinka, T., Eisenhofer, G. & Pacak, K. Pheochromocytoma as a catecholamine producing tumor: implications for clinical practice. Stress 10, 195-203 (2007).
23. Gimm, O. Pheochromocytoma-associated syndromes: genes, proteins and functions of RET, VHL and SDHx. Fam Cancer 4, 17-23 (2005).
24. Lee, S., et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell 8, 155-167 (2005).
25. Cohen, H.T. & McGovern, F.J. Renal-cell carcinoma. N Engl J Med 353, 2477-2490 (2005).
26. Brugarolas, J. Renal-cell carcinoma--molecular pathways and therapies. N Engl J Med 356, 185-187 (2007).
27. Gunaratnam, L., et al. Hypoxia inducible factor activates the transforming growth factor-alpha/epidermal growth factor receptor growth stimulatory pathway in VHL(-/-) renal cell carcinoma cells. J Biol Chem 278, 44966-44974 (2003).

110
28. Wiesener, M.S., et al. Paraneoplastic erythrocytosis associated with an inactivating point mutation of the von Hippel-Lindau gene in a renal cell carcinoma. Blood 99, 3562-3565 (2002).
29. Lager, D.J., Slagel, D.D. & Palechek, P.L. The expression of epidermal growth factor receptor and transforming growth factor alpha in renal cell carcinoma. Mod Pathol 7, 544-548 (1994).
30. Choyke, P.L., et al. The natural history of renal lesions in von Hippel-Lindau disease: a serial CT study in 28 patients. AJR Am J Roentgenol 159, 1229-1234 (1992).
31. Paraf, F., et al. Renal lesions in von Hippel-Lindau disease: immunohistochemical expression of nephron differentiation molecules, adhesion molecules and apoptosis proteins. Histopathology 36, 457-465 (2000).
32. Mandriota, S.J., et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell 1, 459-468 (2002).
33. Lee, Y.S., et al. Coexpression of erythropoietin and erythropoietin receptor in von Hippel-Lindau disease-associated renal cysts and renal cell carcinoma. Clin Cancer Res 11, 1059-1064 (2005).
34. Nogueira, E., Klimek, F., Weber, E. & Bannasch, P. Collecting duct origin of rat renal clear cell tumors. Virchows Arch B Cell Pathol Incl Mol Pathol 57, 275-283 (1989).
35. Esteban, M.A., et al. Regulation of E-cadherin expression by VHL and hypoxia-inducible factor. Cancer Res 66, 3567-3575 (2006).
36. Young, A.P., et al. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat Cell Biol 10, 361-369 (2008).
37. Neumann, H.P. & Wiestler, O.D. Clustering of features of von Hippel-Lindau syndrome: evidence for a complex genetic locus. Lancet 337, 1052-1054 (1991).
38. Hoffman, M.A., et al. von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet 10, 1019-1027 (2001).
39. Brauch, H., et al. von Hippel-Lindau disease with pheochromocytoma in the Black Forest region in Germany: evidence for a founder effect. Hum Genet 95, 551-556 (1995).
40. Ang, S.O., et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet 32, 614-621 (2002).
41. Seizinger, B.R., et al. Von-hippel lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature 332, 268-269 (1988).
42. Latif, F., et al. Identification of the von Hippel-Lindau Disease Tumor Suppressor Gene. Science 260, 1317-1320 (1993).

111
43. Gnarra, J., et al. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc Natl Acad Sci U S A 94, 9102-9107 (1997).
44. Iliopoulos, O., Ohh, M. & Kaelin, W. pVHL19 is a biologically active product of the von Hippel-Lindau gene arising from internal translation initiation. Proc Natl Acad Sci U S A 95, 11661-11666 (1998).
45. Schoenfeld, A., Davidowitz, E.J. & Burk, R.D. A second major native von Hippel-Lindau gene product, initiated from an internal translation start site, functions as a tumor suppressor. Proc Natl Acad Sci U S A 95, 8817-8822. (1998).
46. Lee, S., et al. Nuclear/cytoplasmic localization of the von Hippel-Lindau tumor suppressor gene product is determined by cell density. Proc Natl Acad Sci 93, 1770-1775 (1996).
47. Mekhail, K., Gunaratnam, L., Bonicalzi, M.E. & Lee, S. HIF activation by pH-dependent nucleolar sequestration of VHL. Nat Cell Biol 6, 642-647 (2004).
48. Stebbins, C.E., Kaelin, W.G. & Pavletich, N.P. Structure of the VHL-ElonginC-elonginB complex: implications for VHL tumor suppressor function. Science 284, 455-461 (1999).
49. Li, L., et al. Hypoxia-inducible factor linked to differential kidney cancer risk seen with type 2A and type 2B VHL mutations. Mol Cell Biol 27, 5381-5392 (2007).
50. Pastore, Y.D., et al. Mutations in the VHL gene in sporadic apparently congenital polycythemia. Blood 101, 1591-1595 (2003).
51. Pastore, Y., et al. Mutations of von Hippel-Lindau tumor-suppressor gene and congenital polycythemia. Am J Hum Genet 73, 412-419 (2003).
52. Ohh, M. & Kaelin, W.G., Jr. VHL and kidney cancer. Methods Mol Biol 222, 167-183 (2003).
53. Jariel-Encontre, I., Bossis, G. & Piechaczyk, M. Ubiquitin-independent degradation of proteins by the proteasome. Biochim Biophys Acta 1786, 153-177 (2008).
54. Sufan, R.I. & Ohh, M. Role of the NEDD8 modification of Cul2 in the sequential activation of ECV complex. Neoplasia 8, 956-963 (2006).
55. Maynard, M.A. & Ohh, M. von Hippel-Lindau tumor suppressor protein and hypoxia-inducible factor in kidney cancer. Am J Nephrol 24, 1-13 (2004).
56. Maynard, M.A. & Ohh, M. The role of hypoxia-inducible factors in cancer. Cell Mol Life
Sci 64, 2170-2180 (2007).
57. Maxwell, P., et al. The von Hippel-Lindau gene product is necessary for oxgyen-dependent proteolysis of hypoxia-inducible factor α subunits. Nature 399, 271-275 (1999).

112
58. Maxwell, P.H., et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271-275 (1999).
59. Masson, N., Willam, C., Maxwell, P.H., Pugh, C.W. & Ratcliffe, P.J. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 20, 5197-5206 (2001).
60. Ivan, M., et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464-468. (2001).
61. Jaakkola, P., et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468-472. (2001).
62. Epstein, A.C., et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. [see comments]. Cell 107, 43-54 (2001).
63. D'Angelo, G., Duplan, E., Boyer, N., Vigne, P. & Frelin, C. Hypoxia up-regulates prolyl hydroxylase activity: a feedback mechanism that limits HIF-1 responses during reoxygenation. J Biol Chem 278, 38183-38187 (2003).
64. Maynard, M.A., et al. Multiple splice variants of the human HIF-3alpha locus are targets of the VHL E3 ubiquitin ligase complex. J Bio Chem 278, 11032-11040 (2003).
65. Lando, D., Peet, D.J., Whelan, D.A., Gorman, J.J. & Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 295, 858-861 (2002).
66. Lando, D., et al. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 16, 1466-1471 (2002).
67. Kondo, K., Klco, J., Nakamura, E., Lechpammer, M. & Kaelin Jr, W.G. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 1, 237-246 (2002).
68. Kondo, K., Kim, W.Y., Lechpammer, M. & Kaelin, W.G., Jr. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol 1, E83 (2003).
69. Clifford, S.C., et al. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet 10, 1029-1038 (2001).
70. Hoffman, M.A., et al. von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet 10, 1019-1027. (2001).
71. Ohh, M., et al. The von Hippel-Lindau Tumor Suppressor Protein is Required for Proper Assembly of an Extracellular Fibronectin Matrix. Mol. Cell 1, 959-968 (1998).

113
72. Kurban, G., Hudon, V., Duplan, E., Ohh, M. & Pause, A. Characterization of a von Hippel Lindau pathway involved in extracellular matrix remodeling, cell invasion, and angiogenesis. Cancer Res 66, 1313-1319 (2006).
73. Lieubeau-Teillet, B., et al. von Hippel-Lindau gene-mediated growth suppression and induction of differentiation in renal cell carcinoma cells grown as multicellular tumor spheroids. Cancer Res 58, 4957-4962 (1998).
74. Bishop, T., et al. Genetic Analysis of Pathways Regulated by the von Hippel-Lindau Tumor Suppressor in Caenorhabditis elegans. PLoS Biol 2, E289 (2004).
75. Grosfeld, A., et al. Interaction of hydroxylated collagen IV with the von hippel-lindau tumor suppressor. J Biol Chem 282, 13264-13269 (2007).
76. Kurban, G., et al. Collagen matrix assembly is driven by the interaction of von Hippel-Lindau tumor suppressor protein with hydroxylated collagen IV alpha 2. Oncogene (2007).
77. Pan, Z.Q., Kentsis, A., Dias, D.C., Yamoah, K. & Wu, K. Nedd8 on cullin: building an expressway to protein destruction. Oncogene 23, 1985-1997 (2004).
78. Stickle, N.H., et al. pVHL modification by NEDD8 is required for fibronectin matrix assembly and suppression of tumor development. Mol Cell Biol 24, 3251-3261 (2004).
79. Xirodimas, D.P., Saville, M.K., Bourdon, J.C., Hay, R.T. & Lane, D.P. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell 118, 83-97 (2004).
80. Watson, I.R., Blanch, A., Lin, D.C., Ohh, M. & Irwin, M.S. Mdm2-mediated NEDD8 modification of TAp73 regulates its transactivation function. J Biol Chem 281, 34096-34103 (2006).
81. Oved, S., et al. Conjugation to Nedd8 instigates ubiquitylation and down-regulation of activated receptor tyrosine kinases. J Biol Chem 281, 21640-21651 (2006).
82. Gao, F., Cheng, J., Shi, T. & Yeh, E.T. Neddylation of a breast cancer-associated protein recruits a class III histone deacetylase that represses NFkappaB-dependent transcription. Nat Cell Biol 8, 1171-1177 (2006).
83. Russell, R.C. & Ohh, M. NEDD8 acts as a 'molecular switch' defining the functional selectivity of VHL. EMBO Rep 9, 486-491 (2008).
84. Neumann, H.P.H. & Zbar, B. Renal cysts, renal cancer and von Hippel-Lindau disease. Kidney Intl. 51, 16-26 (1997).
85. Esteban, M.A., Harten, S.K., Tran, M.G. & Maxwell, P.H. Formation of primary cilia in the renal epithelium is regulated by the von Hippel-Lindau tumor suppressor protein. J
Am Soc Nephrol 17, 1801-1806 (2006).

114
86. Lutz, M.S. & Burk, R.D. Primary cilium formation requires von hippel-lindau gene function in renal-derived cells. Cancer Res 66, 6903-6907 (2006).
87. Schermer, B., et al. The von Hippel-Lindau tumor suppressor protein controls ciliogenesis by orienting microtubule growth. J Cell Biol 175, 547-554 (2006).
88. Thoma, C.R., et al. pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat Cell Biol 9, 588-595 (2007).
89. Kaelin, W.G., Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat Rev
Cancer 2, 673-682 (2002).
90. Hofstra, R.M.W., et al. Extensive mutation scanning of RET in sporadic medullary thyroid carcinoma and of RET and VHL in sporadic pheochromocytoma reveals involvement of these genes in only a minority of cases. Journal of Clinical
Endocrinology and Metabolism 81, 2881-2884 (1996).
91. Estus, S., et al. Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol 127, 1717-1727 (1994).
92. Schlisio, S., et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev 22, 884-893 (2008).
93. Kaelin, W.G., Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer 8, 865-873 (2008).
94. Vogel, K.S., Brannan, C.I., Jenkins, N.A., Copeland, N.G. & Parada, L.F. Loss of neurofibromin results in neurotrophin-independent survival of embryonic sensory and sympathetic neurons. Cell 82, 733-742 (1995).
95. Smith, K., et al. Silencing of epidermal growth factor receptor suppresses hypoxia-inducible factor-2-driven VHL-/- renal cancer. Cancer Res 65, 5221-5230 (2005).
96. Oh, R.R., et al. Expression of HGF/SF and Met protein is associated with genetic alterations of VHL gene in primary renal cell carcinomas. APMIS 110, 229-238 (2002).
97. Mukhopadhyay, D. & Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 315, 201-205 (2007).
98. Wiley, H.S. & Burke, P.M. Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic 2, 12-18 (2001).
99. Wang, Y., et al. Regulation of endocytosis via the oxygen-sensing pathway. Nat Med 15, 319-324 (2009).
100. Bucci, C., et al. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 70, 715-728 (1992).

115
101. Ceresa, B.P. Regulation of EGFR endocytic trafficking by rab proteins. Histol
Histopathol 21, 987-993 (2006).
102. Davidowitz, E.J., Schoenfeld, A.R. & Burk, R.D. VHL induces renal cell differentiation and growth arrest through integration of cell-cell and cell-extracellular matrix signaling. Mol Cell Biol 21, 865-874. (2001).
103. Lieubeau-Teillet, B., et al. von Hippel-Lindau gene-mediated growth suppression and induction of differentiation in renal cell carcinoma cells grown as multicellular tumor spheroids. Cancer Res. 58, 4957-4962 (1998).
104. Harten, S.K., et al. Regulation of renal epithelial tight junctions by the von Hippel-Lindau tumor suppressor gene involves occludin and claudin 1 and is independent of E-cadherin. Mol Biol Cell 20, 1089-1101 (2009).
105. Calzada, M.J., et al. von Hippel-Lindau tumor suppressor protein regulates the assembly of intercellular junctions in renal cancer cells through hypoxia-inducible factor-independent mechanisms. Cancer Res 66, 1553-1560 (2006).
106. Peruzzi, B., Athauda, G. & Bottaro, D.P. The von Hippel-Lindau tumor suppressor gene product represses oncogenic beta-catenin signaling in renal carcinoma cells. Proc Natl
Acad Sci U S A 103, 14531-14536 (2006).
107. Jin, T., George Fantus, I. & Sun, J. Wnt and beyond Wnt: multiple mechanisms control the transcriptional property of beta-catenin. Cell Signal 20, 1697-1704 (2008).
108. Brembeck, F.H., Rosario, M. & Birchmeier, W. Balancing cell adhesion and Wnt signaling, the key role of beta-catenin. Curr Opin Genet Dev 16, 51-59 (2006).
109. Barasch, J. Genes and proteins involved in mesenchymal to epithelial transition. Curr
Opin Nephrol Hypertens 10, 429-436 (2001).
110. Nollet, F., Berx, G. & van Roy, F. The role of the E-cadherin/catenin adhesion complex in the development and progression of cancer. Mol Cell Biol Res Commun 2, 77-85 (1999).
111. Cano, A., et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2, 76-83 (2000).
112. Takeichi, M. Morphogenetic roles of classic cadherins. Curr Opin Cell Biol 7, 619-627 (1995).
113. Yoshiura, K., et al. Silencing of the E-cadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc Natl Acad Sci U S A 92, 7416-7419 (1995).
114. Strathdee, G. Epigenetic versus genetic alterations in the inactivation of E-cadherin. Semin Cancer Biol 12, 373-379 (2002).

116
115. Moreno-Bueno, G., et al. Genetic profiling of epithelial cells expressing e-cadherin repressors reveals a distinct role for snail, slug, and e47 factors in epithelial-mesenchymal transition. Cancer Res 66, 9543-9556 (2006).
116. Imai, T., et al. Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells. Am J Pathol 163, 1437-1447 (2003).
117. Come, C., et al. Snail and slug play distinct roles during breast carcinoma progression. Clin Cancer Res 12, 5395-5402 (2006).
118. Rosivatz, E., et al. Differential expression of the epithelial-mesenchymal transition regulators snail, SIP1, and twist in gastric cancer. Am J Pathol 161, 1881-1891 (2002).
119. Messinezy, M. & Pearson, T.C. The classification and diagnostic criteria of the erythrocytoses (polycythaemias). Clin Lab Haematol 21, 309-316 (1999).
120. Percy, M.J., et al. A novel erythrocytosis-associated PHD2 mutation suggests the location of a HIF binding groove. Blood 110, 2193-2196 (2007).
121. Percy, M.J., et al. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med 358, 162-168 (2008).
122. Percy, M.J., et al. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci U S A 103, 654-659 (2006).
123. Kim, W.Y., et al. Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation in vivo. EMBO J 25, 4650-4662 (2006).
124. Baxter, E.J., et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365, 1054-1061 (2005).
125. James, C., et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434, 1144-1148 (2005).
126. Kralovics, R., et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 352, 1779-1790 (2005).
127. Levine, R.L., et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer
Cell 7, 387-397 (2005).
128. Zhao, R., et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol
Chem 280, 22788-22792 (2005).
129. Watowich, S.S., et al. Cytokine receptor signal transduction and the control of hematopoietic cell development. Annu Rev Cell Dev Biol 12, 91-128 (1996).
130. Constantinescu, S.N., Girardot, M. & Pecquet, C. Mining for JAK-STAT mutations in cancer. Trends Biochem Sci 33, 122-131 (2008).

117
131. Ungureanu, D., Saharinen, P., Junttila, I., Hilton, D.J. & Silvennoinen, O. Regulation of Jak2 through the ubiquitin-proteasome pathway involves phosphorylation of Jak2 on Y1007 and interaction with SOCS-1. Mol Cell Biol 22, 3316-3326 (2002).
132. Kamizono, S., et al. The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. Journal of Biological Chemistry 276, 12530-12538 (2001).
133. Sarna, M.K., et al. Differential regulation of SOCS genes in normal and transformed erythroid cells. Oncogene 22, 3221-3230 (2003).
134. Wernig, G., et al. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood 107, 4274-4281 (2006).
135. Lacout, C., et al. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood 108, 1652-1660 (2006).
136. Bumm, T.G., et al. Characterization of murine JAK2V617F-positive myeloproliferative disease. Cancer Res 66, 11156-11165 (2006).
137. Tiedt, R., et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood 111, 3931-3940 (2008).
138. Feng, J., et al. Activation of Jak2 catalytic activity requires phosphorylation of Y1007 in the kinase activation loop. Mol Cell Biol 17, 2497-2501 (1997).
139. Teofili, L., et al. Different STAT-3 and STAT-5 phosphorylation discriminates among Ph-negative chronic myeloproliferative diseases and is independent of the V617F JAK-2 mutation. Blood 110, 354-359 (2007).
140. Sergeyeva, A., et al. Congenital polycythemia in Chuvashia. Blood 89, 2148-2154 (1997).
141. Perrotta, S., et al. Von Hippel-Lindau-dependent polycythemia is endemic on the island of Ischia: identification of a novel cluster. Blood 107, 514-519 (2006).
142. Hickey, M.M., Lam, J.C., Bezman, N.A., Rathmell, W.K. & Simon, M.C. von Hippel-Lindau mutation in mice recapitulates Chuvash polycythemia via hypoxia-inducible factor-2alpha signaling and splenic erythropoiesis. J Clin Invest 117, 3879-3889 (2007).
143. Nelson, W.J. & Nusse, R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303, 1483-1487 (2004).
144. Lonergan, K.M., et al. Regulation of Hypoxia-Inducible mRNAs by the von Hippel-LIndau Protein Requires Binding to Complexes containing Elongins B/C and Cul2. Mol.
Cell. Biol. 18, 732-741 (1998).
145. Kibel, A., Iliopoulos, O., DeCaprio, J.D. & Kaelin, W.G. Binding of the von Hippel-Lindau tumor suppressor protein to elongin B and C. Science 269, 1444-1446 (1995).

118
146. Comijn, J., et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 7, 1267-1278 (2001).
147. Eble, J.N., Sauter, G., Epstein, J.I. & Sesterhenn, I.A. World Health Organization Classification of Tumors: Pathology and Genetics of Tumors of the Urinary System and Male Genital Organs. IRAC press, Lyon (2004).
148. Furge, K.A., et al. Robust classification of renal cell carcinoma based on gene expression data and predicted cytogenetic profiles. Cancer Res 64, 4117-4121 (2004).
149. Takahashi, M., et al. Gene expression profiling of clear cell renal cell carcinoma: gene identification and prognostic classification. Proc Natl Acad Sci U S A 98, 9754-9759 (2001).
150. Fish, J.E., et al. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J Biol Chem 280, 24824-24838 (2005).
151. Shimazui, T., Giroldi, L.A., Bringuier, P.P., Oosterwijk, E. & Schalken, J.A. Complex cadherin expression in renal cell carcinoma. Cancer Res 56, 3234-3237 (1996).
152. Morell-Quadreny, L., et al. Disruption of basement membrane, extracellular matrix metalloproteinases and E-cadherin in renal-cell carcinoma. Anticancer Res 23, 5005-5010 (2003).
153. Baba, M., et al. Loss of von Hippel-Lindau protein causes cell density dependent deregulation of CyclinD1 expression through hypoxia-inducible factor. Oncogene 22, 2728-2738 (2003).
154. Berx, G., et al. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. Embo J 14, 6107-6115 (1995).
155. Berx, G., et al. E-cadherin is inactivated in a majority of invasive human lobular breast cancers by truncation mutations throughout its extracellular domain. Oncogene 13, 1919-1925 (1996).
156. Behrens, J., Lowrick, O., Klein-Hitpass, L. & Birchmeier, W. The E-cadherin promoter: functional analysis of a G.C-rich region and an epithelial cell-specific palindromic regulatory element. Proc Natl Acad Sci U S A 88, 11495-11499 (1991).
157. Graff, J.R., et al. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 55, 5195-5199 (1995).
158. Ji, X., Woodard, A.S., Rimm, D.L. & Fearon, E.R. Transcriptional defects underlie loss of E-cadherin expression in breast cancer. Cell Growth Differ 8, 773-778 (1997).
159. Batlle, E., et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2, 84-89 (2000).

119
160. Peinado, H., Ballestar, E., Esteller, M. & Cano, A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol
Cell Biol 24, 306-319 (2004).
161. Luecke, H.F. & Yamamoto, K.R. The glucocorticoid receptor blocks P-TEFb recruitment by NFkappaB to effect promoter-specific transcriptional repression. Genes Dev 19, 1116-1127 (2005).
162. Baba, M., et al. Tumor suppressor protein VHL is induced at high cell density and mediates contact inhibition of cell growth. Oncogene 20, 2727-2736 (2001).
163. Mohan, S. & Burk, R.D. von Hippel-Lindau protein complex is regulated by cell density. Oncogene 22, 5270-5280 (2003).
164. Groulx, I. & Lee, S. Oxygen-dependent ubiquitination and degradation of hypoxia-inducible factor requires nuclear-cytoplasmic trafficking of the von Hippel-Lindau tumor suppressor protein. Mol Cell Biol 22, 5319-5336 (2002).
165. Vandewalle, C., et al. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res 33, 6566-6578 (2005).
166. Spagnoli, F.M., Cicchini, C., Tripodi, M. & Weiss, M.C. Inhibition of MMH (Met murine hepatocyte) cell differentiation by TGF(beta) is abrogated by pre-treatment with the heritable differentiation effector FGF1. J Cell Sci 113 ( Pt 20), 3639-3647 (2000).
167. Jamora, C., et al. A signaling pathway involving TGF-beta2 and snail in hair follicle morphogenesis. PLoS Biol 3, e11 (2005).
168. Zhang, H., et al. Cellular response to hypoxia involves signaling via Smad proteins. Blood 101, 2253-2260 (2003).
169. Scheid, A., et al. Physiologically low oxygen concentrations in fetal skin regulate hypoxia-inducible factor 1 and transforming growth factor-beta3. Faseb J 16, 411-413 (2002).
170. Nishi, H., et al. Hypoxia-inducible factor-1 transactivates transforming growth factor-beta3 in trophoblast. Endocrinology 145, 4113-4118 (2004).
171. Ananth, S., et al. Transforming growth factor beta1 is a target for the von Hippel-Lindau tumor suppressor and a critical growth factor for clear cell renal carcinoma. Cancer Res 59, 2210-2216 (1999).
172. Staller, P., et al. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature 425, 307-311 (2003).
173. Zagzag, D., et al. Stromal cell-derived factor-1alpha and CXCR4 expression in hemangioblastoma and clear cell-renal cell carcinoma: von Hippel-Lindau loss-of-function induces expression of a ligand and its receptor. Cancer Res 65, 6178-6188 (2005).

120
174. Brugarolas, J.B., Vazquez, F., Reddy, A., Sellers, W.R. & Kaelin, W.G., Jr. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 4, 147-158 (2003).
175. Zundel, W., et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 14, 391-396 (2000).
176. Ravi, R., et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev 14, 34-44 (2000).
177. Laughner, E., Taghavi, P., Chiles, K., Mahon, P.C. & Semenza, G.L. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Molecular & Cellular Biology 21, 3995-4004 (2001).
178. Chen, Y., McPhie, D., Hirschberg, J. & Neve, R. The amyloid precursor protein-binding protein APP-BP1 drives the cell cycle through the S-M checkpoint and causes apoptosis in neurons. J Biol Chem 275, 8929-8935 (2000).
179. Ohh, M., et al. Ubiquitination of HIF requires direct binding to the von Hippel-Lindau protein beta domain. Nature Cell Biology 2, 423-427 (2000).
180. Ohh, M., et al. An intact NEDD8 pathway is required for Cullin-dependent ubiquitylation in mammalian cells. EMBO Reports 3, 177-182 (2002).
181. Vuento, M. & Vaheri, A. Dissociation of fibronectin from gelatin-agarose by amino compounds. Biochem J 175, 333-336 (1978).
182. Ohh, M., et al. Synthetic peptides define critical contacts between elongin C, elongin B, and the von hippel-lindau protein. J Clin Invest 104, 1583-1591 (1999).
183. Zheng, N., et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 416, 703-709 (2002).
184. Min, J.H., et al. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science 296, 1886-1889 (2002).
185. Feldman, D.E., Spiess, C., Howard, D.E. & Frydman, J. Tumorigenic mutations in VHL disrupt folding in vivo by interfering with chaperonin binding. Mol Cell 12, 1213-1224 (2003).
186. Lonergan, K.M., et al. Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol Cell Biol 18, 732-741 (1998).
187. Rathmell, W.K., et al. In vitro and in vivo models analyzing von Hippel-Lindau disease-specific mutations. Cancer Res 64, 8595-8603 (2004).

121
188. Ohh, M., et al. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell 1, 959-968 (1998).
189. Ohh, M., et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol 2, 423-427 (2000).
190. Ohh, M., et al. Synthetic peptides define critical contacts between elongin C, elongin B, and the von Hippel-Lindau protein. J Clin Invest 104, 1583-1591 (1999).
191. Prchal, J.F. & Axelrad, A.A. Letter: Bone-marrow responses in polycythemia vera. N
Engl J Med 290, 1382 (1974).
192. Bellucci, S. & Michiels, J.J. The role of JAK2 V617F mutation, spontaneous erythropoiesis and megakaryocytopoiesis, hypersensitive platelets, activated leukocytes, and endothelial cells in the etiology of thrombotic manifestations in polycythemia vera and essential thrombocythemia. Semin Thromb Hemost 32, 381-398 (2006).
193. Chung, J., Roberts, A.M., Chow, J., Coady-Osberg, N. & Ohh, M. Homotypic association between tumour-associated VHL proteins leads to the restoration of HIF pathway. Oncogene 25, 3079-3083 (2006).
194. Dixon, C., et al. Overproduction of polypeptides corresponding to the amino terminus of the F-box proteins Cdc4p and Met30p inhibits ubiquitin ligase activities of their SCF complexes. Eukaryot Cell 2, 123-133 (2003).
195. Kominami, K., Ochotorena, I. & Toda, T. Two F-box/WD-repeat proteins Pop1 and Pop2 form hetero- and homo-complexes together with cullin-1 in the fission yeast SCF (Skp1-Cullin-1-F-box) ubiquitin ligase. Genes Cells 3, 721-735 (1998).
196. Suzuki, H., et al. Homodimer of two F-box proteins betaTrCP1 or betaTrCP2 binds to IkappaBalpha for signal-dependent ubiquitination. J Biol Chem 275, 2877-2884 (2000).
197. Tang, X., et al. Suprafacial orientation of the SCFCdc4 dimer accommodates multiple geometries for substrate ubiquitination. Cell 129, 1165-1176 (2007).
198. Kamura, T., et al. Muf1, a novel Elongin BC-interacting leucine-rich repeat protein that can assemble with Cul5 and Rbx1 to reconstitute a ubiquitin ligase. J Biol Chem 276, 29748-29753 (2001).
199. Rui, L., Yuan, M., Frantz, D., Shoelson, S. & White, M.F. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem 277, 42394-42398 (2002).
200. Hon, W.C., et al. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 417, 975-978 (2002).
201. Bento, M.C., et al. Congenital polycythemia with homozygous and heterozygous mutations of von Hippel-Lindau gene: five new Caucasian patients. Haematologica 90, 128-129 (2005).

122
202. Shibata, J., et al. Hemostasis and coagulation at a hematocrit level of 0.85: functional consequences of erythrocytosis. Blood 101, 4416-4422 (2003).
203. Krishnamachary, B., et al. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res 66, 2725-2731 (2006).
204. Evans, A.J., et al. VHL promotes E2 box-dependent E-cadherin transcription by HIF-mediated regulation of SIP1 and snail. Mol Cell Biol 27, 157-169 (2007).
205. Evans, A.J., et al. VHL Promotes E2 Box-dependent E-cadherin Transcription by HIF-mediated Regulation of SIP1 and Snail. Mol Cell Biol (2006).
206. Gervais, M.L., et al. Nuclear E-cadherin and VHL immunoreactivity are prognostic indicators of clear-cell renal cell carcinoma. Lab Invest 87, 1252-1264 (2007).
207. Nakaigawa, N., et al. Inactivation of von Hippel-Lindau gene induces constitutive phosphorylation of MET protein in clear cell renal carcinoma. Cancer Res 66, 3699-3705 (2006).
208. Prockop, D.J. & Kivirikko, K.I. Collagens: molecular biology, diseases, and potentials for therapy. Annu Rev Biochem 64, 403-434 (1995).
209. Ferrajoli, A., Faderl, S., Ravandi, F. & Estrov, Z. The JAK-STAT pathway: a therapeutic target in hematological malignancies. Curr Cancer Drug Targets 6, 671-679 (2006).
210. Mullighan, C.G., et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 106, 9414-9418 (2009).
211. Epling-Burnette, P.K., et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest 107, 351-362 (2001).
212. Melzner, I., Weniger, M.A., Menz, C.K. & Moller, P. Absence of the JAK2 V617F activating mutation in classical Hodgkin lymphoma and primary mediastinal B-cell lymphoma. Leukemia 20, 157-158 (2006).
213. Renne, C., et al. High expression of several tyrosine kinases and activation of the PI3K/AKT pathway in mediastinal large B cell lymphoma reveals further similarities to Hodgkin lymphoma. Leukemia 21, 780-787 (2007).
214. Mottok, A., Renne, C., Willenbrock, K., Hansmann, M.L. & Brauninger, A. Somatic hypermutation of SOCS1 in lymphocyte-predominant Hodgkin lymphoma is accompanied by high JAK2 expression and activation of STAT6. Blood 110, 3387-3390 (2007).