molecular docking and_virtual_screening
TRANSCRIPT

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Molecular Docking
Virtual Screening

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Overview
1. Introduction2. Basic concepts3. Preparation steps of molecular docking
3.1. Basic knowledge3.2. Target structure
3.2.1. Source3.2.2. Resolution3.2.3. Treatment
3.3. Interacting site3.4. Ligand structure3.5. Flexibility
3.5.1. Ligand3.5.2. Macromolecule

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Overview
4. Manual docking5. Automatic docking
5.1. Rules5.2. Algorithms and methods
5.2.1. Grid method5.2.2. Sphere method5.2.3. Incremental method5.2.4. Genetic algorithm
5.3. Scoring5.3.1. Force-field5.3.2. Empirical potential5.3.3. Knowledge based
6. Applications6.1. Direct conception

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Overview
6.2. Virtual screening6.2.1. Rules6.2.2. Databases
6.2.2.1. 1D storage6.2.2.2. 3D storage
6.2.3. Filtering6.2.3.1. Redundancy6.2.3.2. Reactivity & toxicity6.2.3.3. Drug-like6.2.3.4. ADMET
6.2.4. Scoring6.2.5. Assessing quality
6.3. De novo design7. General conclusions8. References

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
1. Introduction

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
1. Introduction
molecular docking: prediction of the association between two molecules
Experimentally, the interaction process between two compounds is never easyand provides, no to few informations about the structure.
We use computational approaches to: Observe how a compound is structurally placed with (or inside) its partner Understand the recognition process and establish structure activity/property
relationships Predict on a database of chemical compounds which ones are the most able
to interact with the target
Molecular docking is mainly applied in the field of medicinal chemistry. However,we can apply this technique to study the biological interactions between twomacromolecules (protein/protein or DNA/protein) or any other interactions.

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
2. Basic concepts

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
2. Basic concepts
A drug always acts on a bio-macromolecule (protein, DNA or RNA) as a key (ligand)in a lock (target).
Most of the time we wish to directly compete with the substrate.
enzyme
+
Substrate
+
drug
Competitive inhibition:concentration and affinity are key elements for inhibiting the enzyme.
It's the most widespread case.

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
2. Basic concepts
A + B AB DG = DH -TDS
KD =A B
AB=1
KA
Even the most complex biomacromolecules obey to thermodynamic.
If DG is negative the reaction will be driven toward the AB formation.
If DG is decreased by 2.7 kcal/mol then the dissociation constant (KD) changefrom 100 to 1 and the association population evolve from 50% to 99% (Boltzmannstatistic's).
This logarithmic dependency shows the problem of accuracy in molecularmodeling
∆Gbind = RTlogKD

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking
3.1. Basic knowledge
We know the 3D structure of the target and we wish to simulate the interactionof a database of compounds (around 1 million!)
One naive approach is to perform molecular dynamics in explicit solvent
Protein is embedded in a box Ligand is randomly placed in this box MD predicts the interaction
This should work but this requires trajectory in thescale of ms to s whereas we generally perform nsto µs.See David Shaw
We need other methods, more direct, since theinteraction prediction of two molecules is highlycomplex and requires tremendous explorations

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking
3.2. Target structure3.2.1. Sources
A target 3D structure is required!The PDB (protein databank)
➔ Xray diffraction● No size limit●More accurate●Unique structure (of the crystal)●Crystallization problems●Hydrogen are missed
➔ NMR● Lowest accuracy●Solution structure●Size limit around 150 residues (for aprotein)●Average structure
➔ Homology modelling● Free and quick●No experimental●Low precision of sidechains●Sequence similarity or identity?

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking
Accuracy is an important parameter: RX
3.2. Target structure
3.2.2. Resolution
Here precision, accuracy is very good.

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
A protein alpha-helix with different resolution
3.2. Target structure
3.2.2. Resolution
3. Preparation steps of molecular docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking
In NMR the resolution is hard to determine numerically:Generally we look at the RMSD or the number of restraints by residue.
3.2. Target structure
3.2.2. Resolution

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking
3.2. Target structure
3.2.2. Resolution
For homology modelling (comparative modelling) the resolution has no realmeaning.
In all cases, it is essential to have a feeling of the target structure resolution at theitneracting site location. For enzyme, generally, this area is the best defined.
Beware: for Xray structures some protein parts or atoms may be missed. In thiscase, we choose to add or not these parts depending of their location or influencefor the chemical association.
To sum-up, it is always required to gather as much as you can information aboutthe target.

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking
3.2. Target structure
3.2.3. Treatment
Experimental structures are far frombeing perfect!
You can find in them:
o Ionso Watero Soapo Glycosylo Antibodyo Chaperon proteinso Missing atoms…
You must clean the pdb file

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking
Where is the interacting site on the protein?
Three major methods:
Experimental complex Safer method We need an identical mechanism for ligands
Analysis of structural properties Cavity detection is complex More an art than a definite method
Molecular docking of the whole protein Time consuming and boring Needs a lot of docking poses (~ 1000) to do statistics Generally we have “surprising” results
3.3. Interacting site:

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking
3.3. Interacting site
The cavity detection method "knob & hole".
Principe:
We consider a sphere of a given volume V. The center of this sphere is placed onthe molecular surface (Connoly). We roll this sphere around the molecular surfaceand we compute the common volume, Vcom, which belongs also to the protein.
0 < Vcom ≤V
3
V
3< Vcom <
2V
3
2V
3≤ Vcom < V
if
Knob
Plane
Hole

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking
3.3. Interacting site
"Knob & Hole" cavity detection technique
*
Knob
Hole

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking
3.4. Ligand structure
Ligands are generally molecular organic compounds. We use GUI software(Graphical User Interface), working with the molecular mechanic theory, such asMaestro, Sybyl, Accelrys, Moe, ICM...

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking
3.4. Ligand structure
Not an easy step:
No or scarce experimental 3D structures (CSD) No absolute force-field parameters Sometimes stereochemistry is not an issue for organic chemist’s ; but not
for you. Ionization states? Physiological pH? Atomic type hybridization Tautomeric forms Partial atomic charges Resonance structures

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking
3.5. Flexibility
During the interaction, the ligand flexibility is highly engaged whereas theprotein (larger molecule) hardly moves.
Rigid docking
Flexible docking
Induced fit docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking
3.5. Flexibility3.5.1. Ligand flexibility
It is impossible to manage all ligand cartesian coordinates. Thus, only rotatabledihedral angles (torsion) move. Rings are maintained fixed so that they must becorrectly minimized.
Some questions remain:resonance angle, peptide bond, guanidinium... how to manage them, fixed orrotatable?

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
3. Preparation steps of molecular docking
Other anecdotic method: make a rigid docking with several ligand conformations.
Captopril
3.5. Flexibility3.5.1. Ligand flexibility

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Direct methods are still in development. In Autodock 4.2, the user can define, forfew protein residues inside the active site, sidechain torsion angles.
3.5. Flexibility3.5.2. Target flexibility
Advantage: You choose the amino-acids you
want to involve
Drawbacks: Difficult to choose which amino-
acids Only sidechain movements are
considered Possible explusion of the ligand
by collapse of the rotatableresidues
3. Preparation steps of molecular docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Indirect methods: A molecular docking is performed. Then a molecular dynamicsimulation of the obtained complexe is realized...
3.5. Flexibility3.5.2. Target flexibility
Advantages: With methods such as MMPBSA you
can determine (evaluate) bindingfree energy
You can explore the physicalchemistry of the recognition process
You have access to statistical view ofinteraction (hydrogen bond lifetime)
Drawback: If the starting structure is not
correct...
3. Preparation steps of molecular docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Indirect methods: A MD simulation is made with the apo protein. Representativestructures are then extracted and molecular docking is performed with thesetargets.
3.5. Flexibility3.5.2. Target flexibility
Advantage: Real consideration of the apo
protein
Drawback: How to extract "representative"
target conformations? What about the molecular docking
precision?
3. Preparation steps of molecular docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
4. Manual docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
4. Manual docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
4. Manual docking
Looks like a joke... The ligand is placed in the interacting site and the associationenergy is calculated at each steps.
The user manually moves, rotates or translates the compound inside the proteincavity. A new association energy is recorded... etc
Advantages: Quick (and dirty?) Can be very efficient if the user knows well the interacting site
Drawbacks: Users dependant You can really obtain stupid results
This rudimentary method surprisingly provided interesting results in the past. Itis still applicable if only small ligand modifications are explored.

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
5. Automatic docking
5.1. Rules
Principles:Ligand is automatically placed onto the macromolecule. More exhaustive andsafer this technique requires long CPU time.

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
5.1. Rules
Dreaming about a perfect molecular docking technique: Reasonable computation time The global minimum of the ligand/target interaction energy is reached The calculated free energies reproduce the experimental ones Experimental interaction patterns observed in XRay complexes are identical
Generally the molecular docking simulation can be shared in two steps.
DOCKING
Searching algorithm:- Conf ormational exploration- Several possible docking poses
Scoring function:- Energy quantification- Ranking of docking poses- Clustering
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
A box is drawn on the proteinmacromolecule. Therefore, theinteraction will be explored only on thisbox. This drastically limits thecomputational time.
Beware:o If the box is too small, docking will be
falseo If the box is too large, exploration
must be more intensive and couldprovides strange "false positive"ligand conformations
5.2. Algorithms and methods5.2.1. Grid method
5. Automatic docking
o Take care of the amino-acids you want to embedded in the box (especiallythe charged residues)

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
For all points (nodes) of the grida probe atom is positioned.
There are as many probes asligand atom types. Asupplemental probe of a +echarge is also considered for theelectrostatic computation.
The software places iterativelythe probe atom in each nodepoints and then compute theenergy. These values (tables) arerecorded in map files.
5.2. Algorithms and methods5.2.1. Grid method
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
C
H
H
O
1
2
34
The evaluation of the interaction energy is instantaneous:
𝐸𝑖𝑛𝑡𝑒𝑟𝑔𝑟𝑖𝑑= 𝐸𝑂4 + 𝐸𝐶
3 + 𝐸𝐻1 + 𝐸𝐻
2
Computationally, the energy calculation is made by tables summations. However, molecule is considered as a list of points without bonds.
5.2. Algorithms and methods5.2.1. Grid method
5. Automatic docking
Formol example:

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Now, we have to explore box in order to find the global optimum.
It's a "classical" molecular modelling problem... without absolute solution.
In docking several exploration methods are used: Molecular dynamics (global search) Simulated annealing (global search) Genetic algorithm (global search) Conjugated gradient (local serach)
Actually, the best method seems to be a genetic algorithm (Lamarckian)followed by some steps of conjugated gradient.
5.2. Algorithms and methods5.2.1. Grid method
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Dihedral angles are translated in genes (binary)
101001010101001011001100111010101010
A random initial population is easily generated
001011010111000101001010011101010101101010010111101110001010010100111010110101101011100010100101001110101010010111110110101110001010010100111010......

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Starting population
genotypes phenotypes
Parents selectionfitness fonction
Children
This process is stopped after several defined steps
translation
Crossingmutation

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Phenotype
Genotype

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Starting population
genotypes phenotypes
Parents selectionfitness fonction
Children
This process is stopped after several defined steps
translation
Crossingmutation Parents optimisation

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Phenotype
Genotype

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
5.2. Algorithms and methods5.2.2. Sphere method
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
5.2. Algorithms and methods5.2.2. Sphere method
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
**
*
** *
5.2. Algorithms and methods5.2.2. Sphere method
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
*
assumption: distances between center of spheres correspond to inter-atomsdistances (heavy atoms)
**
*
***
* **
*
***
5.2. Algorithms and methods5.2.2. Sphere method
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
5.2. Algorithms and methods5.2.2. Sphere method
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
The DOCK software used this method.
This technique acts more on the shape of molecules than on interactions complementarity.
Some issues: Sphere dimensions? Matching of sphere centers? Ligand flexibility?
5.2. Algorithms and methods5.2.2. Sphere method
5. Automatic docking
This old method has proven itsefficiency and is still employed.

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
5.2. Algorithms and methods5.2.3. Incremental method
5. Automatic docking
NH
N
O
OH
NH2 OH
O
O
OH
NH
NH2
N
OH
O
Fragments

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Definition of interactionsas "umbrellas"
5.2. Algorithms and methods5.2.3. Incremental method
5. Automatic docking
OH N

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
5.2. Algorithms and methods5.2.3. Incremental method
5. Automatic docking
NH
NH2
NH
NH2
The base fragment isplaced by triangulation

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
5.2. Algorithms and methods5.2.3. Incremental method
5. Automatic docking
NH
NH2
The second fragment islinked to the first.
Torsion exploration is madeto find the best pose for thisnew fragment
O
NH
NH2
OH
O
OH
NH
NH2

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
5.2. Algorithms and methods5.2.3. Incremental method
5. Automatic docking
O
NH
NH2
N
OO
-
OHThe ligand is then incrementallybuild In the protein

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
The target can only be a protein
Umbrella interactions: Hbond electrostatic hydrophobic contact
This method tends tooverestimate the importance ofHbonds regarding othersinteractions.
5.2. Algorithms and methods5.2.3. Incremental method
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Aims to describe and quantify the association.
Purpose:
Quick computation
Able to compare results with experimental data
Able to distinguish true inhibitors to false positive ligands
Able to rank the ligands
5.3. Scoring
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
A force-field (FF) is used to describe the interaction.
Based on classical FF such as AMBER or CHARMM.
Advantages:• Quick• Good parameterization based on empirical parameters
Drawbacks:• Electrostatic is generally overestimated• Entropy??
Example : Dock
5.3. Scoring5.3.1. Force-field
𝐸 =
𝑖
𝑁𝐵𝑂𝑁𝐷
𝑗=𝑖+1
𝑁𝐵𝑂𝑁𝐷𝑞𝑖𝑞𝑗
𝜀𝑖𝑗𝑟𝑖𝑗+𝐴𝑖𝑗
𝑟𝑖𝑗12 −𝐵𝑖𝑗
𝑟𝑖𝑗6
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
A function is designed to evaluate free energy of binding instead of interactionenergy.
5.3. Scoring5.3.2. Empirical potential
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
These functions are calibrated with experimental data.
Advantages: Safer evaluation of energy More physical effects are incorporated in the equation More accurate results
Drawbacks: The function is calibrated with a training set of data. Beware if your
system is not "classical". Sometimes the electrostatic effect is overestimated Estimation of entropy is far from being correct.
Example : FlexX, Autodock, Gold...
5.3. Scoring5.3.2. Empirical potential
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Used only for scoring (after the docking pose)
How it works:A statistical analysis is made on a dataset of complex structures form the PDB.ligand/protein atomic distances are recorded. According to the clouds found, ascore is given for the atomic distances found in the docking calculation.
5.3. Scoring5.3.2. Knowledge based
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
This technique works well but has no chemical meaning. This type of score ranksmore on drug-likeness than on interactions.
This technique is sensitive to the studied protein family type. For example,different scoring values are found depending the protein location in cell and itsfunction. This can be an advantage or a drawback.
This type of docking scoring (drugscore, ligscore,...) is usually used in consensusscoring
Compounds which have a goodrank with several scoring functionsmay be the best ones.
No physical interpretation.
5.3. Scoring5.3.2. Knowledge based
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Applications: Able to localize a ligand inside a biological macromolecule. Analysis of the interacting binding mode. Able to draw structure activity relationships.
Limitations: Target flexibility is never taken into account, or scarcely. Scoring functions are far from being perfect. Energetical interpretations
are thus questionable. Beware of searching parameters. Generally, several binding modes are proposed... which one should be
picked?Software:
Grid method: Autodock, Gold, ICM, Glide Sphere method: Dock Incremental construction: FlexX, Ludi
5.4 Conclusions
5. Automatic docking

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications
An iterative work with experimental chemists is made. The purpose is to proposeoriginal ideas for getting more active compounds.
Requirements:
A collaboration with people from experimental fields (chemist/biologist). All people must understand each other! Not so obvious because each field of
research has its own logic. Structural analyses must be performed for "all ligands“
The pros and cons:
Provides more original compounds than screening. Safer interpretation of results when we compare to virtual screening (see
later). Real scientific interactions but needs human and computational time.
6.1. Direct conception

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Example 1: exploring a protein cavity with several moities.
6. Applications
6.1. Direct conception

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Example 2: Extend a ligand to pick up a new favourable interaction.
6. Applications
6.1. Direct conception

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Example 2: Extend a ligand to pick up a new favourable interaction.
6. Applications
6.1. Direct conception

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications
Ligand n° DG
123 -12.3 kcal/mol22 -11.7 kcal/mol13 -10.1 kcal/mol49 -9.3 kcal/mol76 -6.5 kcal/mol
6.2. Virtual screening6.2.1 Rules

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Instead of making molecular docking for a small set of defined ligands thiscomputation is extended to a large database.
The compounds which will have the best ranks will be purchased and biologicallytested.
Virtual screening is named by its analogy to all experimental screening methods.
Three major steps:1. Ligand database. If you remove the good ones... You will have nothing
at the end.2. Molecular docking. Even if your database is full of good compounds if
you are not able to correctly dock each one... You will have nothing atthe end.
3. Ranking. Even if the two previous steps were correctly made, if you arenot able to meaningfully rank the ligands... You will have nothing at theend.
6. Applications6.2. Virtual screening
6.2.1 Rules

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Chemical universe:10100 à 10400 compounds.
Organic molecules:1024 à 1040 compounds.
Synthesized molecules:106 compounds.
Acitve molecules:10? molécules.
6. Applications6.2. Virtual screening
6.2.1 Databases
We are looking of a needle in a haystack... if this needle exists.

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Numerous chemical databases exist. Some of them are commercial.
6. Applications6.2. Virtual screening
6.2.1 Databases
Name Type Number
Pubchem Public 30 million
ChEMBL Public 1 million
NCI set Public 140 000
ChemSpider Public 26 million
CoCoCo Public 7 million
TCM Public 32 000
ZINC Public 13 million
ChemBridge Commercial 700 000
Specs Commercial 240 000
Name Type Number
IUPHAR Public 3 180
Asinex Commercial 550 000
Enamine Commercial 1.7 million
Maybridge Commercial 56 000
WOMBAT Commercial 263 000
ChemDiv Commercial 1.5 million
Chemnavigator Commercial 55.3 million
ACD Commercial 3 870 000
MDDR Commercial 150 000

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6.Applications
There are problems of storingchemical data as 3D files: difficulty to compare
chemical composition it needs high hard-drive
access modification of databases
is hard to make
can we simplify?
Benzene example
6.2. Virtual screening6.2.2. Databases
6.2.2.1. 1D storage

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
The SMILES code gives a benzene with only one line.
SMILES: Simplified Molecular Input Line Entry System
Others coding system exist (SLN, WLN, STRAPS...), however, they share a similarphilosophy and the knowledge of their differences are not for the uninitiatedpeople.
6.Applications6.2. Virtual screening
6.2.2. Databases6.2.2.1. 1D storage
c1ccccc1 Cc1ccccc1
OH
Oc1ccccc1

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Example of SMILES code for a molecule:
This system has numerous advantages: Simple storage (1 line!) Easy to manage Generation of virtual library is very easy
6.Applications6.2. Virtual screening
6.2.2. Databases6.2.2.1. 1D storage

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
A chemical database is SMILES
6.Applications6.2. Virtual screening
6.2.2. Databases6.2.2.1. 1D storage

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Unfortunately, there are drawbacks of using SMILES coding: Hydrogens are added at the end for filling the chemical valences Software are required to transform 1D in 3D. These are generally commercial
and have their own drawbacks (CORINA, Omega, ROTATE, CAESAR...) Smile code is not (yet) unique!!! A molecule might be present twice (or more)
6.Applications6.2. Virtual screening
6.2.2. Databases6.2.2.1. 1D storage

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
The 3D storage partially solves the 1D problems... but
Storage problems: if a 1D database of 1.5 Go is transformed in 3D, the size isaround 132 Go.
Really more difficult to create virtual chemical databases comparing toSMILES code.
Still problem for tautomeric forms and charge
6.Applications6.2. Virtual screening
6.2.2. Databases6.2.2.1. 1D storage

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
The main problem of chemical databases is that they contain mainlyuninteresting compounds.We must filter them to:
Eliminate as much as possible uninteresting compounds Spend more computational time for molecular docking calculations.
First obvious filter is the redundancy: Sometimes, chemical databases containthe same compounds (even the commercial databases). Why?
1D databases → SMILES code is not unique3D database → Comparison of compounds is hard to perform
6.2.3.1. Redundancy
6.Applications6.2. Virtual screening
6.2.3. Filtering

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Comparison of 3D information is hard to perform
6.Applications6.2. Virtual screening
6.2.3. Filtering
6.2.3.1. Redundancy

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Other types of redundancy
These three compounds may appear as different in a database!!!!!
6.Applications6.2. Virtual screening
6.2.3. Filtering
6.2.3.1. Redundancy

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6.Applications6.2. Virtual screening
6.2.3. Filtering
6.2.3.2. Reactivity and toxicity
Some chemical moieties areknown to be highly reactiveand/or toxic.
The compounds which carrythese moieties can thus bemoved apart.

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
The artemisinine counterexample (anti-paludic drug).
6.Applications6.2. Virtual screening
6.2.3. Filtering
6.2.3.2. Reactivity and toxicity
OO
O O
OH
H

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
The global assumption of this filtering step is that a biologically molecule lookslike... any other biologically active compounds.
From this idea (maybe false) several filters can be set: The 32 types of cycles The 34 types of moieties The Lipinski rule
From these filters, a score is determined. According to your defined thresholdsyou will get a database with more or less compounds.
6.Applications6.2. Virtual screening
6.2.3. Filtering
6.2.3.3. Drug-like

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6.Applications6.2. Virtual screening
6.2.3. Filtering
6.2.3.3. Drug-like
When a ligand interacts with its target it loses some degrees of freedom. Thisprocess decreases the association entropy variation and thus increase the freeenergy of binding.
To avoid this fact, there is no other way than to eliminate, as much as possible,ligand degrees of freedom by... making rings. But, keep in mind that: You must maintain a similar interaction scaffold (the bioactive
conformation) Generally, a ligand without flexibility has difficulties to pass through
membrane (distribution)
Making rings is thus a smart idea when you are designing biologically activecompounds. Some researchers made an inventory of the 32 classical ringsclassically encountered in drugs.

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6.Applications6.2. Virtual screening
6.2.3. Filtering
6.2.3.3. Drug-like
Compounds with one and two rings (5 or 6 membered).
( )n
n=1,2,3,4,5,6
( )n
n=1,2
Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6.Applications6.2. Virtual screening
6.2.3. Filtering
6.2.3.3. Drug-like
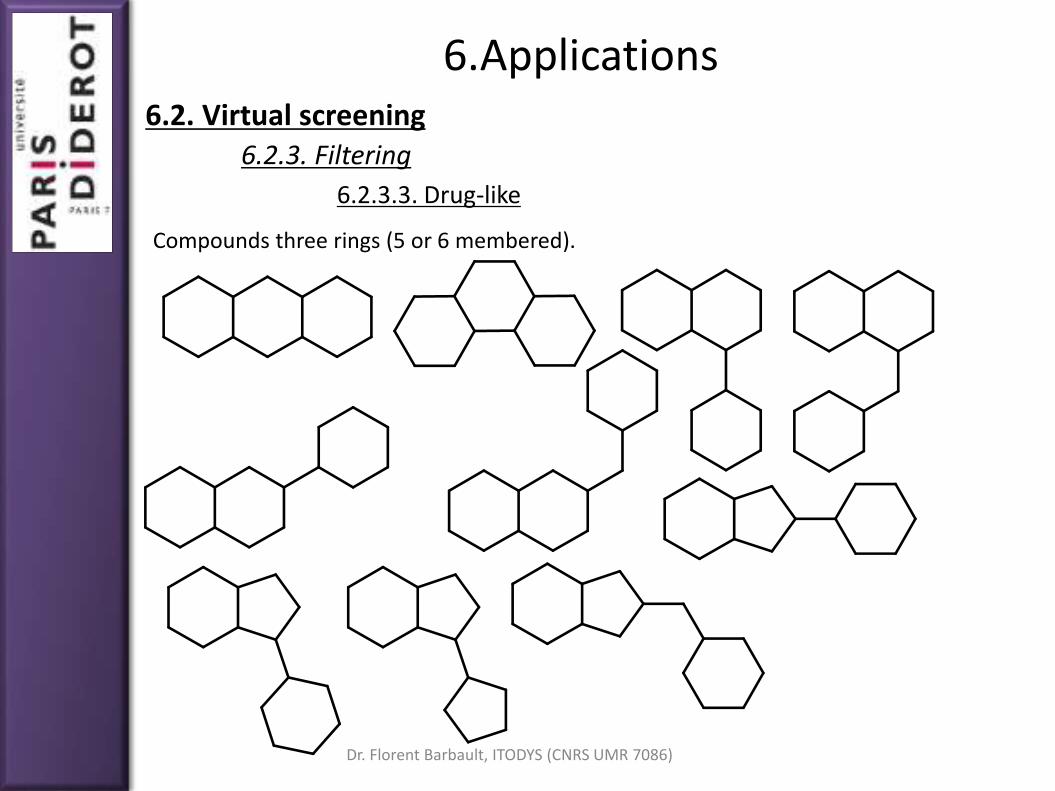
Compounds three rings (5 or 6 membered).

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6.Applications6.2. Virtual screening
6.2.3. Filtering
6.2.3.3. Drug-like
Other scaffolds

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6.Applications6.2. Virtual screening
6.2.3. Filtering
6.2.3.3. Drug-like

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
From a statistical study of 2548 commercially avaible orally active substances,Lipinski defined a rule: the "Lipinski rule's of five".
If you want to design an orally available active substance it must follow at least4 of these 5 points:
A molecular weight lower than 500 g/mol
A logP lower than 5
A number of hydrogen bond donors atoms lower than 5
A number of hydrogen bond acceptors atoms lower than 10
A polar surface lower than 150 Ų
6.Applications6.2. Virtual screening
6.2.3. Filtering
6.2.3.3. Drug-like

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
ADMET:
Adsorption Desorption Metabolism Excretion Toxicity
Usually, drugs failed to be marketed during the clinical tests. It is thus essential toremove compounds that have bad AMDET properties.
QSAR 2D equations are used to defined the several ADMET properties.
With all of these properties a chemical space can be defined. Some software arededicated to predict pharmacokinetic properties (Volsurf) or toxicity (CORAL)
This space is useful to visualize the chemical space and get diverse or similarcompounds.
6.Applications6.2. Virtual screening
6.2.3. Filtering
6.2.3.4. ADMET

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
Chemical descriptors labelthe axis and colors of thechemical space
Statistical tools are useful toanalyze this chemical space
6.Applications6.2. Virtual screening
6.2.3. Filtering
6.2.3.4. ADMET

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications
The molecular docking calculation is a long step.
You can decrease the computational time by two ways: A low generation of docking poses... In this case, you have to be lucky to get
a right-first-time molecular docking calculation. A highly filtered databases... In this case, you have "few" compounds but,
you have to be lucky that the good molecules are not discarded.
To sum-up, you have to be lucky (or gifted).
The scoring part is the Achille's heel of the structure-based virtual screening.
There are 3 main methods of scoring (see previous slides). A consensus scoringis certainly the best way to avoid the major drawbacks of each techniques.
6.2. Virtual screening6.2.4. Scoring

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.2. Virtual screening
6.2.5. Assessing quality
𝑅𝑀𝑆𝐷 =
𝑖=1
𝑁𝑟𝑖 − 𝑟𝑖𝑜
2
𝑁
Like "classical" molecular docking calculations, if experimental structures of acomplex are known, it's interesting to add these compounds in your database.
These compounds, normally, mustn't be discarded during the filteringprocesses.
We can compare the predicted docked position and the experimentalstructure. A root mean square deviation (RMSD) can thus be determined:
The user should define its threshold value, generally between 0.3 to 2 Å. Despiteits simplicity, this metric is far from being perfect (size, interaction, symetry...).

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.2. Virtual screening
6.2.5. Assessing quality
Number of compounds
Interaction energy or scoreThreshold

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.2. Virtual screening
6.2.5. Assessing quality
False positive compounds

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.2. Virtual screening
6.2.5. Assessing quality
False negative compounds

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.2. Virtual screening
6.2.5. Assessing quality
How to evaluate a good virtual screening procedure? Several groups havedeveloped the used of decoys in the VS strategy.
The decoys have been designed to display similar physico-chemical properties ofknown ligands.
For example, the DUD-E (Directory of Useful Decoy Enhanced) contains:
around 102 protein systems (classical drug-target)
for each system, several known ligands are put in a database (average 13)
for each ligand, around 50 decoys (with similar properties) are added
The databases contain, on average, 650 compounds

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.2. Virtual screening
6.2.5. Assessing quality
We can compute the Enrichment Factor for x% of selected compounds:
𝐸𝐹𝑥% =𝑅𝑎𝑡𝑖𝑜 𝑜𝑓 𝑎𝑐𝑡𝑖𝑣𝑒 𝑐𝑜𝑚𝑝𝑜𝑢𝑛𝑑𝑠 𝑖𝑛 𝑡ℎ𝑒 𝑥% 𝑜𝑓 𝑠𝑒𝑙𝑒𝑐𝑡𝑒𝑑
𝑅𝑎𝑡𝑖𝑜 𝑜𝑓 𝑎𝑐𝑡𝑖𝑣𝑒 𝑐𝑜𝑚𝑝𝑜𝑢𝑛𝑑𝑠 𝑖𝑛 𝑡ℎ𝑒 𝑤ℎ𝑜𝑙𝑒 𝑑𝑎𝑡𝑎𝑏𝑎𝑠𝑒
EF must be up to 1, because if EF=1 then your molecular docking is equivalent toa random choice!
EF is easy to calculate but: Requires active compounds Not easy to compare virtual screening methods with distinct databases All active compounds are considered similarly. However, some active
molecules are not so much active...

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.2. Virtual screening
6.2.5. Assessing quality
ROC (Receiver Operating Characteristic) curves:
% of false positive: 𝐹𝑃 = 1 −𝑁𝑖𝑛𝑎𝑐𝑡𝑖𝑣𝑒 𝑢𝑛𝑠𝑒𝑙𝑒𝑐𝑡𝑒𝑐𝑡
𝑁𝑡𝑜𝑡𝑎𝑙 𝑜𝑓 𝑖𝑛𝑎𝑐𝑡𝑖𝑣𝑒
% of active compounds
Random
Ideal ROC
Classical good ROC

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications
Moon and Howe: ‘‘Given detailed structural knowledge of the target receptor, itshould be possible to construct a model of a potential ligand, by algorithmicconnection of small molecular fragments, that will exhibit the desired structuraland electrostatic complementarity with the receptor.’’
De novo design purpose and challenge: Build an ideal compound inside theprotein. If synthesized, this one should be a perfect inhibitor.
There are several methods to do this task. All of them have advantages anddrawbacks. To date, nothing is perfect!
Examples of software: LUDI, CAVEAT, SPROUT, MCSS... Here, we will see only theMCSS strategy.
MCSS: Multiple Copy Simultaneaous Search
6.3. De novo design

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.3. De novo design
We start from an empty protein binding site
NH3
+
Thr
Lys
Phe
Trp
Leu Ile
Val
Ser
OH
OH

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.3. De novo design
The binding site is filled with a lot of identical fragments
NH3
+
Thr
Lys
Phe
Trp
Leu Ile
Val
Ser
OH
OH
O O-

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.3. De novo design
The binding site is filled with a lot of identical fragments
NH3
+
Thr
Lys
Phe
Trp
Leu Ile
Val
Ser
OH
OH
O O-
O O-
O O-
O O-
O O-
O O-
O
O-O
O-
O
O-
O
O-
O
O-
O
O-
O
O-
O
O-
O
O-

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.3. De novo design
Only the best position of the fragment is kept
NH3
+
Thr
Lys
Phe
Trp
Leu Ile
Val
Ser
OH
OH
O O-
O O-
O O-
O O-
O O-
O O-
O
O-O
O-
O
O-
O
O-
O
O-
O
O-
O
O-
O
O-
O
O-

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.3. De novo design
Step by step, the protein binding site is completely filled with fragments at optimalpositions.
NH3
+
Thr
Lys
Phe
Trp
Leu Ile
Val
Ser
OH
OH
O O-
N
H
H

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.3. De novo design
Last step: linkage of all elements
O O-
N
H
H
NH3
+
Thr
Lys
Phe
Trp
Leu Ile
Val
Ser
OH
OH

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.3. De novo design
First example: the design of novel inhibitor of hepatitis C virus helicase (HCV).
Ligbuilder 1st proposition
N
OH
NH
O
NH
OH
NH2
OH
SHH
H
OH

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.3. De novo design
First example: the design of novel inhibitor of hepatitis C virus helicase (HCV).
Ligbuilder 2nd proposition
O
OH
O
OH
O
O
NH
O
NH
O
Human enhancement
IC50 = 260 nM

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications6.3. De novo design
Second example: the design of inverse agonist of cannabinoid receptor 1 (CB1).
TOPAS proposition Human enhancement
IC50 = 4 nMIC50 = 1500 nM
N
OO
O
F N
O
O
F
Cl
Cl

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
6. Applications
Advantages:
Quick Provides new ideas of chemical scaffolds Compounds are here original. It is not the case of virtual screening
Drawbacks:
How to synthetize them? New softwares attempt to follow some chemicalrules of synthesis...
Sometimes the molecules are generated only for filling the protein cavity andnot for inhibit the enzyme.
A final human design is always required
6.3. De novo design

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
7. General conclusions

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
7. General conclusions
Molecular docking is an efficient method topredict the structural interaction of anorganic molecule inside a biomacromoleculebinding site.
However, molecular docking has a weaknessfor the determation of the interaction energy(scoring function).
Generally, molecular docking calculations andtheir applications don't give an uniquesolution but rather several solutions. Humanhas the last word.
Molecular docking is mainly applied for thedrug-design and get many success.

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
7. General conclusions
Some successful drugs through molecular docking between 1995 and 2009.

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
7. General conclusions
Some successful drugs through molecular docking between 1995 and 2009.

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
7. General conclusions
Some successful drugs through molecular docking between 1995 and 2009.
1) LMC : Leucémie Myéloïde Chronique2) EGFR : récepteur au facteur de croissance endothélial3) Cancer pulmonaire non à petites cellules4) VEGFR : récepteur au facteur de croissance endothélial vasculaire5) Cancer gastro-intestinal résistant à l’imatinib6) Lymphome cutané à cellules T7) INNTI : inhibiteur non nucléosidique de la transcriptase inverse.

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
8. References

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
8. References
Articles:
Shoichet, B.K., D.L. Bodian, and I.D. Kuntz, J. Comp. Chem., 1992. 13(3): p. 380-397. Meng, E.C., B.K. Shoichet, and I.D. Kuntz, J. Comp. Chem., 1992. 13: p. 505-524. Kuntz, I.D., J.M. Blaney, S.J. Oatley, R. Langridge, and T.E. Ferrin, J. Mol. Biol., 1982. 161: p.
269-288. Meng, E.C., D.A. Gschwend, J.M. Blaney, and I.D. Kuntz, Proteins, 1993. 17(3): p. 266-278. F. Barbault, C. Landon, M. Guenneugues, M. Legrain, et al, Biochemistry 2003. 42 14434-42 D. Eisenberg, E. Schwarz, M. Komaromy and R. Wall, J. Mol. Biol. 1984. 179 125-142. F. Barbault, B. Ren, J. Rebehmed, C. Teixeira, Y. Luo, et al, Eur. J. Med. Chem. 2008 .43 1648-
56. W. Humphrey, A. Dalke, K. Schulten, J. Mol. Graph. 1996 (14) 33-8 C. Teixeira, N. Serradji, F. Maurel, F. Barbault, Eur. J. Med. Chem. 2009 . 44 3524-32 Hu R., Barbault F., Delamar M., Zhang R. Bioorg. Med. Chem. 2009. 17 2400–9 Morris G.M., Huey R., Lindstrom W., Sanner M.F,et al, J Comput Chem 2009. 30 2785–91. Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K., Olson A.J., J. Comput.
Chem. 1998. 19 1639–62 T. Cheng, Q. Li, Z. Zhou, Y. Wang, SH. Bryan., AAPS Journal 2012. 14 133-41 Crivori P, Cruciani G, Carrupt P-A, Testa B., J Med Chem. 2000;43(11):2204–2216. Toropova AP, Toropov AA, Lombardo A, Roncaglioni A, Benfenati E, Gini G. , J Comput. Chem.
2012. doi:10.1002/jcc.22953. Sharman JL, Benson HE, Pawson AJ, et al. , Nucleic Acids Res. 2013;41(D1):D1083–D1088

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
8. References
Articles:
Nesrine Ben Nasr, "Optimisation de méthodes de criblage virtuel et synthèse de moléculesà visée thérapeutique pour le traitement des maladies auto-immunes", 2013, Thesis, CNAM
OMEGA135,213, ROTATE195 , CAESAR214 Boström J, Greenwood JR, Gottfries J., J. Mol. Graph. Model. 2003;21(5):449–462 CORINA - http://www.molecular-networks.com/products/corina Renner S, Schwab CH, Gasteiger J, Schneider G., J Chem Inf Model. 2006;46(6):2324–2332 Hawkins PCD, Skillman AG, Warren GL, et al, J Chem Inf Model. 2010;50(4):572–584 Li J, Ehlers T, Sutter J, Varma-O’brien S, Kirchmair J., J Chem Inf Model. 2007;47(5):1923–1932 Brooijmans N, Kuntz ID., Annu Rev Biophys Biomol Struct. 2003;32(1):335–373 Boström J., J Comput Aided Mol Des. 2001;15(12):1137–1152 Bissantz C, Folkers G, Rognan D. , J Med Chem. 2000;43(25):4759–4767 Pham TA, Jain AN., J Med Chem. 2006;49(20):5856–5868 Irwin JJ, Raushel FM, Shoichet BK., Biochemistry (Mosc). 2005;44(37):12316–12328 Huang N, Shoichet BK, Irwin JJ., J Med Chem. 2006;49(23):6789–6801 DUD - A Directory of Useful Decoys. http://dud.docking.org/ Spitzer R, Jain AN., J Comput Aided Mol Des. 2012;26(6):687–699 Neves MAC, Totrov M, Abagyan R., J Comput Aided Mol Des. 2012;26(6):675–686 Brozell SR, Mukherjee S, Balius TE, et al, J Comput Aided Mol Des. 2012;26(6):749–773

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)
8. References
Articles:
Fan H, Irwin JJ, Webb BM, Klebe G, Shoichet BK, Sali A., J Chem Inf Model. 2009;49(11):2512–2527.
DUD-E: A Database of Useful (Docking) Decoys — Enhanced. http://dude.docking.org/ Mysinger MM, Carchia M, Irwin JJ, Shoichet BK., J Med Chem. 2012;55(14):6582–6594 Triballeau N, Acher F, Brabet I, Pin J-P, Bertrand H-O. J Med Chem. 2005;48(7):2534–2547 Kirchmair J, Distinto S, Markt P, et al. J Chem Inf Model. 2009;49(3):678–692 Giganti D, Guillemain H, Spadoni J-L, et al., J Chem Inf Model. 2010;50(6):992–1004 Böhm HJ. J Comput Aided Mol Des. 1992;6(1):61–78 Böhm HJ. J Comput Aided Mol Des. 1992;6(6):593–606 Miranker, A.; Karplus, M., PROTEINS: Struct, Funct, and Gene 1991 11:29–34 Bohacek, R. S. & McMartin, C., J Am Chem Soc 1994 116:5560–71 Gisbert Schneider and Karl-Heinz Baringhaus, "De Novo Design: From Models to
Molecules"(book) Kandil, S., Biondaro, S., Vlachakis, D., et al, Bioorg. Med. Chem. Lett 2009 19:2935–7 Wang, R., Gao, Y., and Lai, L., J Mol Model 2000 6:498–516 Rogers-Evans, M., Alanine, A., Bleicher, et al, QSAR Comb Sci 2004 26:426–30 Schneider, G., Neidhart, W., Giller, T., et al, Angew. Chem Int Ed 1999 38:2894–6 Alig, L., Alsenz, J., Andjelkovic, M., Bendels, S., Benardeau, A., et al, J Med Chem 2008
51:2115–27 Alex AA, Millan DS. In: Drug Design Strategies.; 2011 (chapter book)

Dr. Florent Barbault, ITODYS (CNRS UMR 7086)