molekularbiologie - casa-di-lago.purespace.de · 1 mm (0,74 g) na2-edta...
TRANSCRIPT

Molekularbiologie
Methoden
Dr. Thomas Seehaus

Molekularbiologische Methoden Seite 2
Aufgabe
Isolation des hypothetischen Gens KRYP1 aus Zucchini

Molekularbiologische Methoden Seite 3
DNA-Extraktion aus Geweben
Jede Zelle enthält im Zellkern DNA (Desoxyribonukleinsäure). Dazu kommen Proteine, Fette, Kohlenhydrate, Ionen.
Um die DNA freisetzen zu können, müssen zunächst die Zellwand, Zellmembran und die Kernmembran zerstört werden.
Zellwände werden mechanisch und/oder enzymatisch geöffnet. - Glasbeads, Mörsern, kalt, flüssiger Stickstoff oder Trockeneis- Enzyme
Da die Membranen aus amphiphilen Molekülen aufgebaut sind, können sie mit Detergentien (in herkömmlichen Spülmitteln) aufgelöst werden. Fette werden ebenfalls im Detergenz gelöst.
Die in den Spülmitteln enthaltenen Enzyme (Proteasen) zerstören außerdem begleitende Eiweiße (Proteine, u. a. DNasen!).
Aufreinigung der DNA durch Ethanol-Fällung (Dehydratation der DNA => fällt aus, Kohlenhydrate und Ionen bleiben gelöst)
Vorüberlegungen für einen Versuch mit Haushaltsmitteln

Molekularbiologische Methoden Seite 4
Besonderheiten bei RNA-Extraktion
RNA ist sehr viel schwieriger zu isolieren als DNA
- Cytoplasma enthält sehr viele RNasen (RNA abbauende Enzyme, u. a. zur Regulation der Proteinsynthese!)
- RNasen müssen inhibiert oder deaktiviert werden (Proteasen, RNase-Inhibitoren, Phenol)
- auf der Haut sind viele RNasen vorhanden!
- Handschuhe tragen, Gerätschaften und Lösungen sterilisieren!
RNA-Moleküle sind kleiner als DNA-Moleküle
- Ethanol-Fällung ist daher weniger effizient
- Ethanol muss sehr kalt sein (-20°C)
- zur Unterstützung höherer Salzgehalt

Molekularbiologische Methoden Seite 5
DNA-Isolierung aus Zucchini
Material/Geräte:
- Zucchini, (Kartoffel)reibe, Teesieb, Thermometer, 100 ml Messzylinder, zwei 100 ml Bechergläser, ein 250 ml Becherglas (niedrige Form), 60°C warmes Wasser, Spülmittel (das Proteasen enthält!), Kochsalz, Brennspiritus, Eiswürfel, evtl. kleine Styroporbox
Vorbereitungen:
- 20 ml Brennspiritus (oder 96 % Ethanol) eine Stunde vor dem Versuch ins Kühlfach (ca. -20°C) legen. Zum Transport auf Eis (in einer kleinen Styroporbox) legen.
- Wasser (60°C) bereitstellen (z. B. in einem Babyflaschenwärmer)
- Spülmittel-/Salz-Lösung zur Extraktion: 50 ml Wasser mit 5 ml Spülmittel (mit Proteasen) und 1 g Kochsalz gut mischen.

Molekularbiologische Methoden Seite 6
DNA-Isolierung aus Zucchini - Durchführung
Man reibt ein Zucchini-Stück und füllt einen Esslöffel in ein 100 ml Becherglas
- Durch das Reiben wird das Pflanzengewebe zerkleinert und die Zellwände teilweise aufgebrochen
und gibt 25 ml der Spülmittel-/Salz-Lösung hinzu
- Das Spülmittel enthält Detergentien, die aufgrund ihrer chemischen Struktur Lipide in Lösung bringen. Salze verstärken den Effekt

Molekularbiologische Methoden Seite 7
DNA-Isolierung aus Zucchini - Durchführung
Das Becherglas für 15 Minuten in ein 60°C warmes Wasserbad stellen. Sehr gut geeignet sind dafür (gebrauchte) Babyflaschenwärmer. (Kann man bei ebay ersteigern!)
- Die höhere Temperatur beschleunigt die Freisetzung der DNA und zerstört DNAsen (Enzyme, die DNA zersetzen.)
In Eiswasser abkühlen.
- Abkühlen verhindert DNA-Zerfall
Den Zucchini-Brei durch ein Teesieb passieren. Dazu das Teesieb auf ein 250 ml Becherglas (niedrige Form) legen und mit einem Teelöffel durch das Netz streichen.

Molekularbiologische Methoden Seite 8
DNA-Isolierung aus Zucchini - Durchführung
Einen Teil (ca. 1 Teelöffel) des Breis in ein kleines Becherglas o. ä. füllen und ganz vorsichtig mit tiefgekühlten (möglichst aus der Kühltruhe) Alkohol überschichten. Dazu den Alkohol langsam seitlich hinein fließen lassen. Besser geht es mit einer Pipette.
- DNA (und RNA) sind in Ethanol bei niedriger Temperatur unlöslich und fallen daher leicht aus.
Die DNA wird oberhalb des Breis als weißgelbe Flocken/Klümpchen sichtbar.
Durch Abgießen oder Herausfischen kann man die DNA in ein Röhrchen mit Alkohol überführen.

Molekularbiologische Methoden Seite 9
DNA-Isolierung: Quellen
Trolldenier, G.: Isolierung der Erbsubstanz aus Zucchini - die DNA-Küche (Industrieverband Agrar)
www.chempage.de/versuche/vdna.htm
www.chemicon.com/techsupp/genomic.asp
www.dialog-gentechnik.at/binaries/108927.pdf
Empfohlen wird auch die DNA-Extraktion aus anderen pflanzlichen Geweben, z. B. Tomaten, Kiwi, Zwiebeln usw.
Interessant bleibt auch die früher häufig vorgeschlagene Extraktion aus tierischem Material z. B. Kalbsbries.

Molekularbiologische Methoden Seite 10
Pufferlösung zur Aufbewahrung von DNA
Tris-Puffer (EDTA-Tris-Acetat, pH 7.75)
10 mM (12,12 g) Tris-(hydroxy-methyl)-aminomethan
1 mM (0,74 g) Na2-EDTA (Dinatrium-Ethylendiamin-tetraacetat)
mit destilliertem Wasser auf 900 ml aufgefüllt
pH-Wert mit 2 M Essigsäure auf 7,75 einstellen
mit destilliertem Wasser auf 1000 ml Endvolumen auffüllen
Aufbewahrung von DNA-Proben
- in TE-Puffer bei -20°C
- alternativ gefällt unter Ethanol
- lyophilisiert
EDTA: KomplexbildnerTRIS: pH-Puffer

Molekularbiologische Methoden Seite 11
Nachweis von DNA
Feulgen Nuklealreaktion (Robert J. W. F., 1884-1955, physiol. Chemiker, Gießen)
mittels Photometrie quantitativ faßbare Färbung der DNA, der Zellkerne, Chromosomen, Plastiden, Bakterien u. Viren
nach (mild-)saurer hydrolytischer Abspaltung der Purinbasen erfolgt die Umsetzung der zurückbleibenden Desoxyribose (mit freier Aldehydgruppe an C1) mit Schiff-Reagens (fuchsinschweflige Säure; 1 Std.) zu rotviolettem Farbstoff

Molekularbiologische Methoden Seite 12
Konzentrationsbestimmung von DNA-Lösungen
UV-Spektrophotometrie
Nukleinsäuren besitzen ein Absorptionsmaximum bei 260 nm
optische Dichte (OD260) von 1 entspricht einer Konzentration von 50 μg dsDNA/ml, 35 μg RNA/ml oder 33 μg ssDNA/ml.
Es wird in der Regel eine 200 fache Verdünnung der Lösung in einem Endvolumen von 200 μl hergestellt und photometrisch in einer Quarzküvette gegen Wasser vermessen.
Die Reinheit der DNA-Lösung kann durch das Erstellen eines Absorptionsspektrums beurteilt werden, das bei 260 nm ein Absorptionsmaximum aufweisen sollte. Verunreinigung mit Protein lassen sich durch hohe Absorption bei 280 nm identifizieren.
Charakteristisch für eine saubere DNA-Lösung ist ein Wert des Quotienten 260nm/280nm nahe 2.

Molekularbiologische Methoden Seite 13
Übersicht Enzyme für die Molekularbiologie
Nukleasen
- Unspezifische Nukleasen (Endo- und Exonukleasen, Dnase, RNase)
- Spezifische Endonukleasen (Restriktions-Endonukleasen)
Ligasen
Polymerasen
- DNA-abhängige DNA-Polymerasen (Replikation)
- RNA-abhängige DNA-Polymerasen (Reverse Transkription)
- DNA-abhängige RNA-Polymerasen (Transkription)
Kinasen
Phosphatasen

Molekularbiologische Methoden Seite 14
Isolierte DNA „handlich“ machen
Unsere isolierte DNA besteht idealerweise aus Zucchini-Chromatiden (Länge mehrere cm/dm pro Molekül!)
- DNA-Moleküle können brechen/zerreißen
- gesuchtes Gen irgendwo auf den Chromatiden versteckt
- übliche Trennmethoden mit so langen DNA-Molekülen überfordert
Aufgabe:
- reproduzierbare Zerlegung der DNA in handliche Fragmente
Welche Methoden stehen zur Verfügung?

Molekularbiologische Methoden Seite 15
Unspezifische Nukleasen
Exonuclease III
- 3’-5’ Exonuklease
- dsDNA mit einem Einzelstrangbruch
- mit blunt ends
- 3’ recessed ends
Nuklease Bal 31 (Alteromonas expejiana)
- dsDNA => 3‘- und 5‘-Exonuklease
- ssDNA => Endonuklease
- Ca-abhängige Reaktionen, Stop durch EDTA
- Gezielte Verkürzung von DNA
Nuklease S1 (Aspergillus oryzae)
- ssDNA => Endonuclease (pH<4,5 / T<20°C !)
- Abbau ungepaarter Bereiche in DNA-RNAHybriden (cDNA-Synthese)

Molekularbiologische Methoden Seite 16
Unspezifische Nukleasen
DNase I (Rinder-Pankreas)
- dsDNA und ssDNA, zufälliges Schneiden beider Stränge
- Entfernen von DNA, Einführen statistischer Schnittstellen in einzelne Stränge
- Enzym besitzt 3 verschiedene Aktivitäten, die in gentechnischen Experimenten unterschiedlich genutzt werden können
- 5‘-3‘ Polymerase-Aktivität : freie 3‘ OH-Enden, freie Nukleotide, Mg2+ als Kofaktor, Einzelstrangmatritze
- 5‘-3‘ Exonuklease-Aktivität: Abbau von DNA im Doppelstrang (Reparatur)
- 3‘-5‘Exonuklease-Aktivität: Korrekturlesen (in Gegenwart von dNTP‘s gehemmt)
- Polymerase-und Exonuklease-Aktivitäten liegen im intakten Enzym im Gleichgewicht vor
- Polymerasefunktion kann durch Salzkonzentration und die Konzentration freier Nukleotide erhöht werden

Molekularbiologische Methoden Seite 17
λ-Phagen infizieren E. coli unterschiedlich effizient!
Paar von Enzymen schützt Bakterien
Methylase markiert eigene DNA an spezifischen Stellen
Endonuklease schneidet DNA an den spezifischen Stellen, wenn diese nicht methyliert sind
=> fremde DNA wird abgebaut!

Molekularbiologische Methoden Seite 18
Spezifische Nukleasen (Restriktionsenzyme)
Quelle: Bakterien
- Restriktion: Immunität gegen Phagen, Schutz vor fremder DNA
- Schutz der eigenen DNA durch spezifische Methylierungsmuster
Als Werkzeuge interessant:
- Typ II Endonukleasen (sequenzspezifische Bindung und Hydrolyse)
Erkennungssequenzen:
- Palindrome / Nicht-Palindrome
- 4-8 Basenpaare (4-, 5-, 6-, 8- cutter)
Isochizomere: Gleiche ES, gleiche Schnittstelle
Neochizomere: Gleiche ES, andere Schnittstelle
Schnitt-Frequenzen (statistisch!):
- 4-cutter = 1/256 bp, 6-cutter = 1/4096 bp, 8-cutter = 1/65536 bp
Entstehende Fragmente: 5‘-Phosphat, 3‘-OH

Molekularbiologische Methoden Seite 19
Restriktions-Enzyme
Typ I: Schneidet die DNA an einer zufälligen Stelle weit von der Erkennungssequenz entfernt. Benötigt ATP und transferiert eine Methylgruppe von S-Adenosyl-Methionin.
Typ II: Schneidet die DNA innerhalb der Erkennungssequenz. Benötigt kein ATP und hat keine Methyltransferase-Aktivität.
Typ III: Schneidet die DNA etwa 20 bis 25 Basenpaare von der Erkennungssequenz entfernt. Benötigt ATP und transferiert eine Methylgruppe von S-Adenosyl-Methionin.
Beispiele für Typ II:

Molekularbiologische Methoden Seite 20
Restriktion genomischer DNA
vollständige Restriktion mit einer Restriktionsendonuklease
vollständige Restriktion mit zwei ver-schiedenen Restriktionsendonukleasen
partielle Restriktion mit einer Restriktionsendonuklease

Molekularbiologische Methoden Seite 21
Auftrennen von DNA-Fragmenten
Agarose-Gel zur Trennung verschieden langer DNA-Fragmente
Trennende Kraft: elektrische Spannung
negativ geladene DNA wandert von der Anode zur Kathode
poröse Gelstruktur setzt der Wanderung der Fragmente Widerstand entgegen
je kleiner die Fragmente sind, desto schneller wandern sie
Molekularbiologische Methoden Seite 22
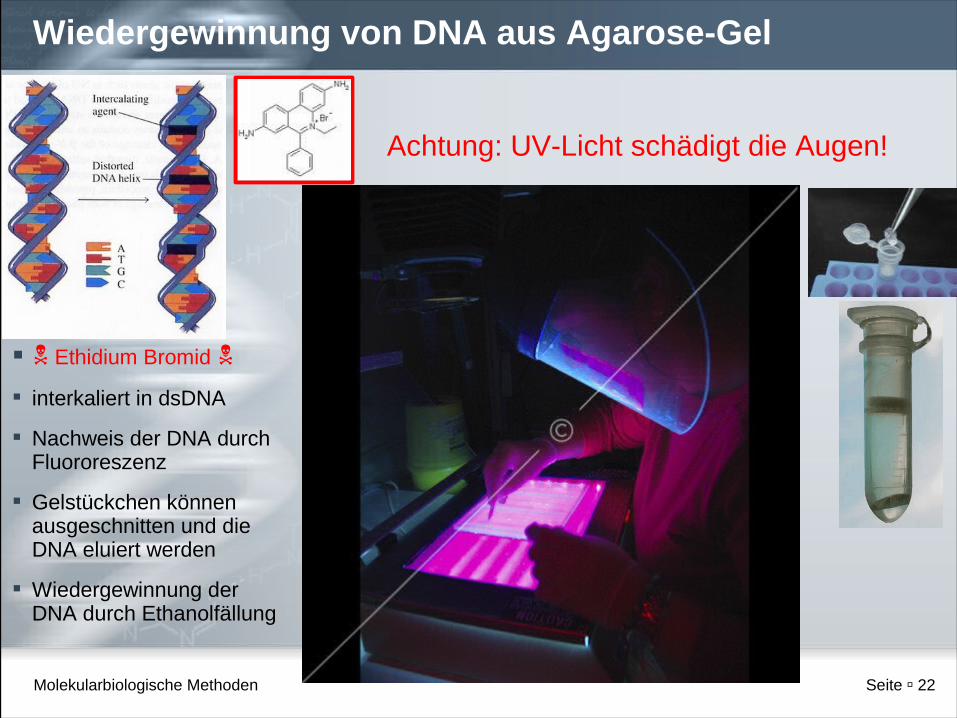
Wiedergewinnung von DNA aus Agarose-Gel
Ethidium Bromid
interkaliert in dsDNA
Nachweis der DNA durch Fluororeszenz
Gelstückchen können ausgeschnitten und die DNA eluiert werden
Wiedergewinnung der DNA durch Ethanolfällung
Achtung: UV-Licht schädigt die Augen!

Molekularbiologische Methoden Seite 23
Verbinden von DNA-Fragmenten
kompatible Enden von Restriktions-Fragmente hybridisieren miteinander
dsDNA wird gebildet
DNA-Ligase schließt Lücken im Zucker-Phosphat-Rückgrad der DNA
Doppelhelix aus zwei verschiedenen DNAs hergestellt

Molekularbiologische Methoden Seite 24
DNA-Ligasen
Verknüpfung des 3‘-OH eines DNA-Fragments mit dem 5‘-Phosphat eines anderen
benötigt ATP
Häufigste: T4-Ligase
Komplementäre „sticky ends“
- Raumtemperatur
- Reaktionszeit einige Stunden
Blunt ends
- 10°C oder Kühlschrank
- Reaktionszeit über Nacht
- Effizienz gering, da keine stabilisierenden Wasserstoffbrücken zwischen den zu verbindenden DNA-Fragmenten

Molekularbiologische Methoden Seite 25
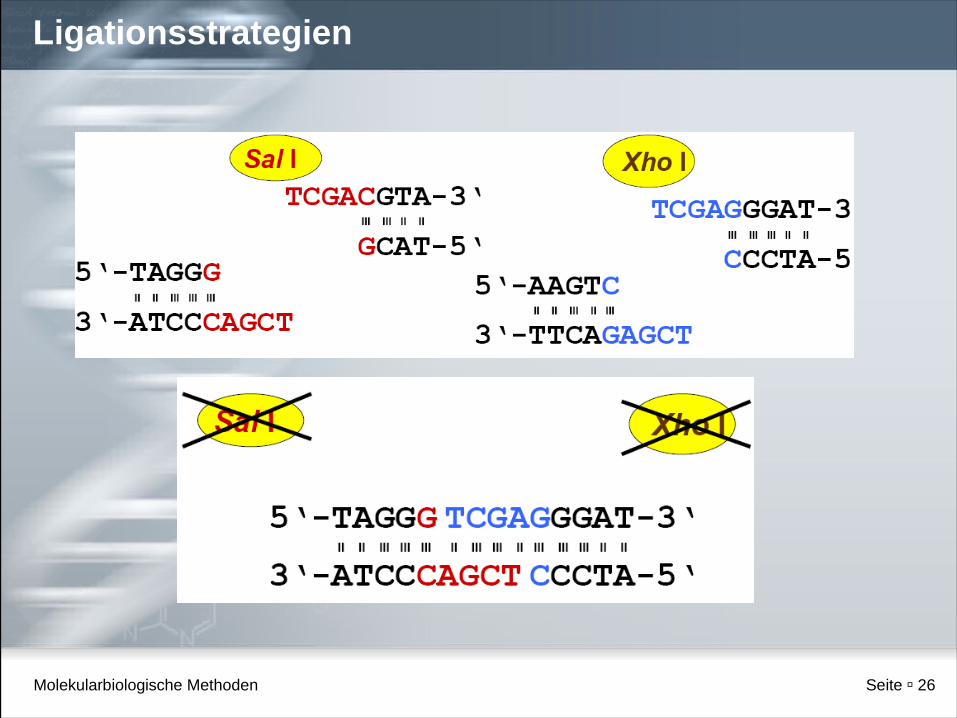
Ligation verschiedener Restriktionsfragmente
Molekularbiologische Methoden Seite 26
Ligationsstrategien

Molekularbiologische Methoden Seite 27
Plasmide
ringförmige, extrachromosomal lokalisierte DNA-Moleküle, die in den meisten gram-positiven und gram-negativen Bakterien, in einigen Hefen, aber nicht in höheren Eukaryoten vorkommen
wurden Anfang der 50-er Jahre durch das Auftreten und die schnelle Ausbreitung von Antibiotika-Resistenzen entdeckt
verschaffen den Wirtszellen in entsprechender Umgebung (Anwesenheit des Antibiotikums) einen Wachstumsvorteil
Unterscheidung von konjugativen (F-Plasmid) und nicht-konjugativen Plasmiden
konjugative Plasmide können durch Transfer von einer Donorzelle in eine Rezipientenzelle übertragen werden (Konjugation)
Verwendung von nicht-konjugativen Plasmiden in der Gentechnik (biologische Sicherheit)
Molekularbiologische Methoden Seite 28
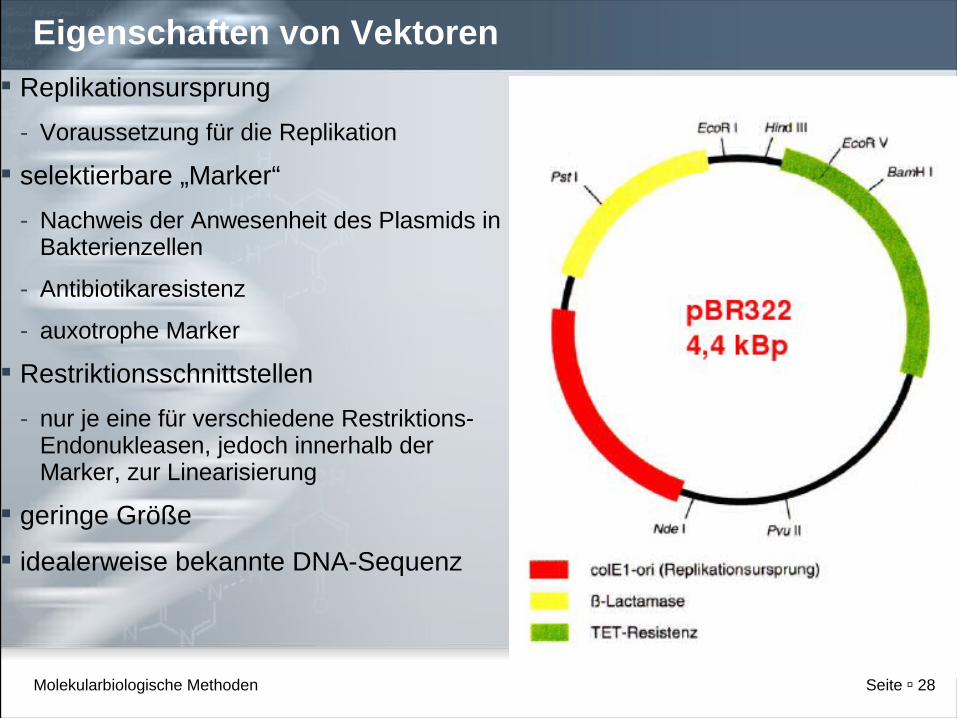
Eigenschaften von Vektoren
Replikationsursprung
- Voraussetzung für die Replikation
selektierbare „Marker“
- Nachweis der Anwesenheit des Plasmids in Bakterienzellen
- Antibiotikaresistenz
- auxotrophe Marker
Restriktionsschnittstellen
- nur je eine für verschiedene Restriktions-Endonukleasen, jedoch innerhalb der Marker, zur Linearisierung
geringe Größe
idealerweise bekannte DNA-Sequenz

Molekularbiologische Methoden Seite 29
Verknüpfung von DNA-Fragmenten
T4 Ligase als meist genutztes Enzym für die Verknüpfungsreaktion
Abhängigkeit der Verknüpfungsreaktion von der Konzentration der DNA-Fragmente
Bevorzugung von inter- oder intramolekularer Reaktion ist von der DNA-Konzentration und der Fragmentgröße abhängig
konstante DNA-Konzentration
Größe des DNA-Fragmentes Neigung zur intramolekularen Reaktion
konstante DNA-Fragmentlänge
DNA-Konzentration Neigung zur intramolekularen Reaktion
hohe DNA-Konzentrationen – Bevorzugung intermolekularer Reaktion
bei Klonierungsexperimenten ist bevorzugt eine bimolekulare Reaktion erwünscht

Molekularbiologische Methoden Seite 30
optimale Konzentrationen für bimolekulare Reaktion

Molekularbiologische Methoden Seite 31
Prinzip der DNA-Klonierung
Linearisierung des Vektors und der Fremd-DNA mit gleicher oder kompatibler Restriktions-endonuklease
Mischen der beiden DNAs in optimaler Konzentration
Ligierung mit T4-DNA-Ligase
Transformation in kompatible Wirtszellen (meist E.coli)

Molekularbiologische Methoden Seite 32
Transformation
einige Bakterien besitzen die Eigenschaft DNA aus der Umgebung aufnehmen zu können
Labormethoden zur Erhöhung der Aufnahmeeffizenz:
Hitzeschock
- Bakterienzellen werden mit Calciumchlorid oder effektiver mit Rubidiumchlorid behandelt, so dass zwischen der negativ geladenen DNA und der negativ geladenen Zellmembran weniger abstoßende Kräfte bestehen. Bei einem kurzen Hitzeschock (41–43 °C für 45–90 Sekunden) entstehen Poren in der Membranen, so dass die DNA in die Zellen gelangen kann.
Elektroporation
- Hierbei werden die Bakterien mit einem elektrischen Schock behandelt (2000-2500 V für einige Millisekunden), um die Membran zu öffnen. Diese Methode ist effektiver als die chemische Methode
Anschließend Kultur unter Selektionsbedingungen

Molekularbiologische Methoden Seite 33
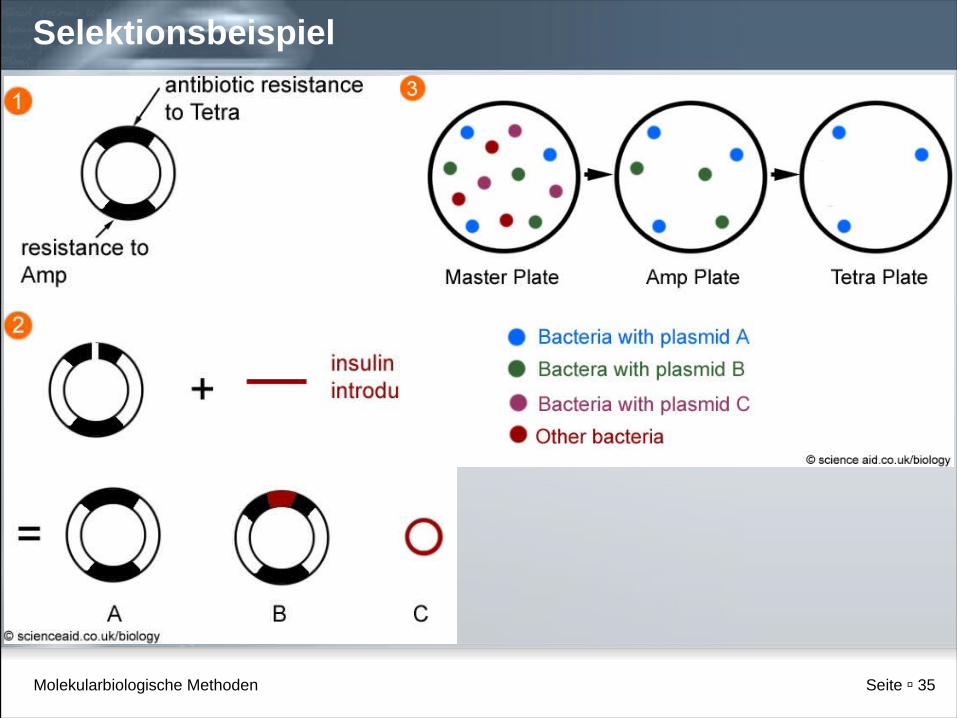
Selektion der gesuchten Plasmide
Mögliche Ligationsergebnisse:
1.Vektor religiert
2.Fremd-DNA religiert
3.Vektor + Fremd-DNA ligiert
Gesucht: 3.
2. keine Replikation in Bakterien (ORI fehlt)
Bei Klonierung in BamH1-Stelle:
- 1. Klone Ampicillin- und Tetrazyklin resistent
- 3. Klone nur Ampicillin resistent
Identifikation der Klone durch Stempeln auf Ampicillin- und Tetrazyklin-Platten

Molekularbiologische Methoden Seite 34
Replikaplattierung
Molekularbiologische Methoden Seite 35
Selektionsbeispiel

Molekularbiologische Methoden Seite 36
Kinasen / Phosphatasen
Kinasen
- Anhängen eines Phosphats an 5‘-OH einer Nukleinsäure
- unter Verbrauch von ATP
- Häufigste: T4-Polynucleotidkinase
- Anwendung:
- Markierung mit radioaktivem 32P
- Auffüllen dephosphorylierter 5'-Enden
Phosphatasen
- Abspalten eines Phosphats vom 5‘-Ende einer Nukleinsäure
- BAP (Bacterial Alkaline Phosphatase)
- CIP (Calf Intestine Phosphatase)
- Anwendung: Verhinderung unerwünschter Ligation

Molekularbiologische Methoden Seite 37
Verhinderung der Vektor-Religierung
Alkalische Phosphatase entfernt 5'-Phosphat
linearisierter Vektor kann nicht mehr rezyklisiert werden
Bei Hybriden mit Fremd-DNA kann zumindest je Strang eine Lücke geschlossen werden

Molekularbiologische Methoden Seite 38
Nachweis bestimmter DNA-Sequenzen
Nach der Klonierung unseres Gemisches von DNA-Fragmenten aus Zucchini erhalten wir eine Fülle von Klonen, von denen einige wenige das oder die gesuchten Fragmente enthalten sollten
Wie können wir diese identifizieren?
einige Möglichkeiten:
- Komplementation auxotropher Mutanten (Mangelmutanten)
- Fragment muss intaktes Gen enthalten, das auxotrophe Mutation komplementieren kann
- Zielorganismus (E.coli) muss Gen des Spenders (Zucchini) „verstehen“
- Vektor muss zur Genexpression (Transkriptions-Signale) fähig sein
- Direkte Identifikation durch DNA-DNA-Hybridisierung mit „Sonden-DNA“
- Sequenz muss zumindest teilweise bekannt sein, z. B. Ableitung aus Proteinsequenz, bekannte Sequenz eines nahe verwandten Organismus
- Sonde muss markiert sein

Molekularbiologische Methoden Seite 39
DNA-Polymerasen
DNA-Polymerase I aus E. coli (Kornberg-Polymerase)
- DNA-Synthese 5‘=>3‘ an DNA-Matrize und Primer
- 5‘-3‘-Exonuklease
- 3‘-5‘-Exonuklease
- Nick-Translation
Klenow-Polymerase
- Proteolyse der DNA-Polymerase I
- Klenow-Fragment => stark verminderte Exonuklease
- Auffüllen von Einzelsträngen, benötigt Primer
Thermostabile DNA-Polymerasen
- PCR-Enzyme !

Molekularbiologische Methoden Seite 40
nick translation
A) DNase I erzeugt Einzelstrang-Brüche (nicks) indem sie die Phosphodiester-Bindungen (p) spaltet. Dabei entsteht eine 5'-Phosphat-Gruppe und ein 3'-Hydroxyl-Ende. Zugabe von E. coli DNA Polymerase I liefert zwei Enzymaktivitäten: i. 5'3' Exonuclease entfernt vom freien 5'-Ende
sequentiell Nukleotide in 5'3' Richtung
ii. DNA Polymerase baut von der freien 3'-Hydroxyl Gruppe in 5'3' Richtung neue Nukleotide ein und füllt die, durch Exonuklease erzeugte, Lücke wieder auf (translation)
B) Random primed labeling. Klenow-Polymerase synthetisiert radioaktiv markierte DNA-Stränge anhand hitze-denaturierter Einzelstrang-Matritzen mit Hexanukleotid Primern mit zufälligen Sequenzen

Molekularbiologische Methoden Seite 41
End-Markierung von DNA
A) Markierung von Oligonukleotiden durch Kinase.
• Das 5'-terminale Phosphat wird in einer Austauschreaktion durch 32P-markiertes -Phosphate aus [-32P]-ATP ersetzt. Auf gleiche Weise können beide 5'-Enden doppel-strängiger DNA markiert werden.
B) Auffüllen überhängender Enden mit Klenow-Polymerase.
• DNA wird mit einer passenden Restriktions Endonuclease gepalten um 5'-Überhänge zu erzeugen. Diese dienen als Primer für Klenow_Polymerase um markierte Nukleotide ein zu bauen. Fragmente, die nur an einem Ende markiert sind, können durch internes Schneiden in einer passenden Restriktionsstelle erzeugt werden, um zwei unterschiedlich große Fragments zu erhalten, die leicht nach der Größe fraktioniert werden können.

Molekularbiologische Methoden Seite 42
radioaktiv:
- 32P ([-32P]ATP)
- 35S (ersetzt O- in [-P]ATP)
- Nachweis durch Autoradiographie (Schwärzung eine Films durch radioaktive Substanzen)
nicht radioaktiv
- Fluoreszierende Gruppe
- anitgene Gruppe (Digoxigenin, Nachweis mit anti-Digoxigenin-Antikörper, der an Reportermolekül gekoppelt ist, das wiederum Farbreaktion auslöst)
- ...
Markierungsmöglichkeiten für DNA

Molekularbiologische Methoden Seite 43
Hybridisierung
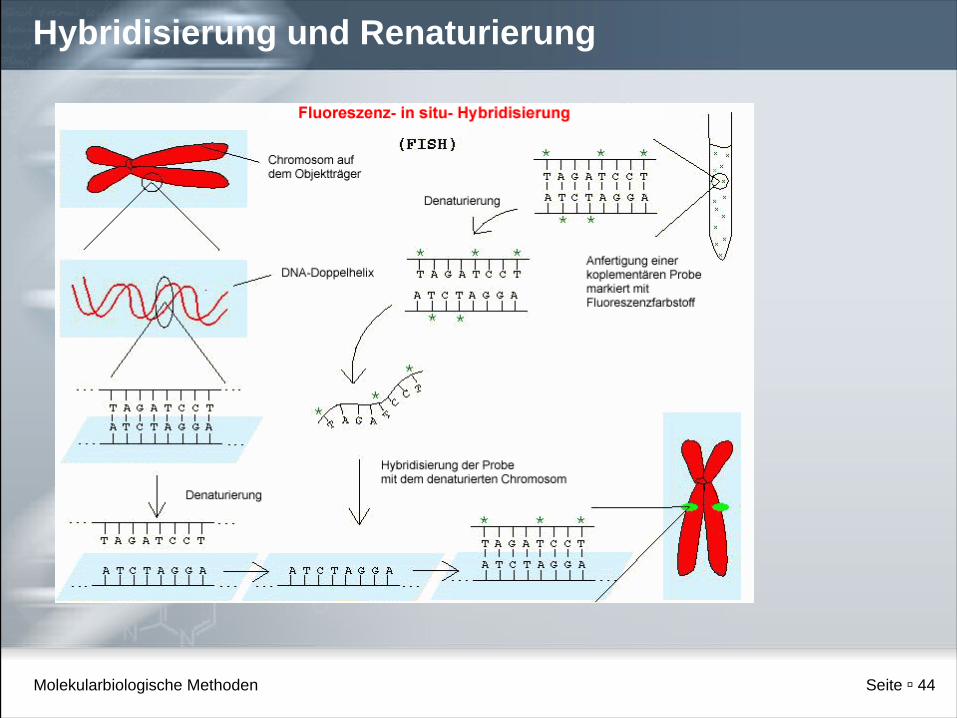
Molekularbiologische Methoden Seite 44
Hybridisierung und Renaturierung

Molekularbiologische Methoden Seite 45
Southern Transfer

Molekularbiologische Methoden Seite 46
Hybridiserung und Autoradiografie

Molekularbiologische Methoden Seite 47
Southern - Ergebnisse

Molekularbiologische Methoden Seite 48
RFLPs: Restriction Fragments Length Polymorphisms
verschiedene Allele eines Gens können verschiedene Restriktionsmuster produzieren
diese Muster sind vererbbar
Verwendung:
- Genomkartierung- Analyse von Varianten- Verwandschaftsanalyse- DNA-Fingerprinting
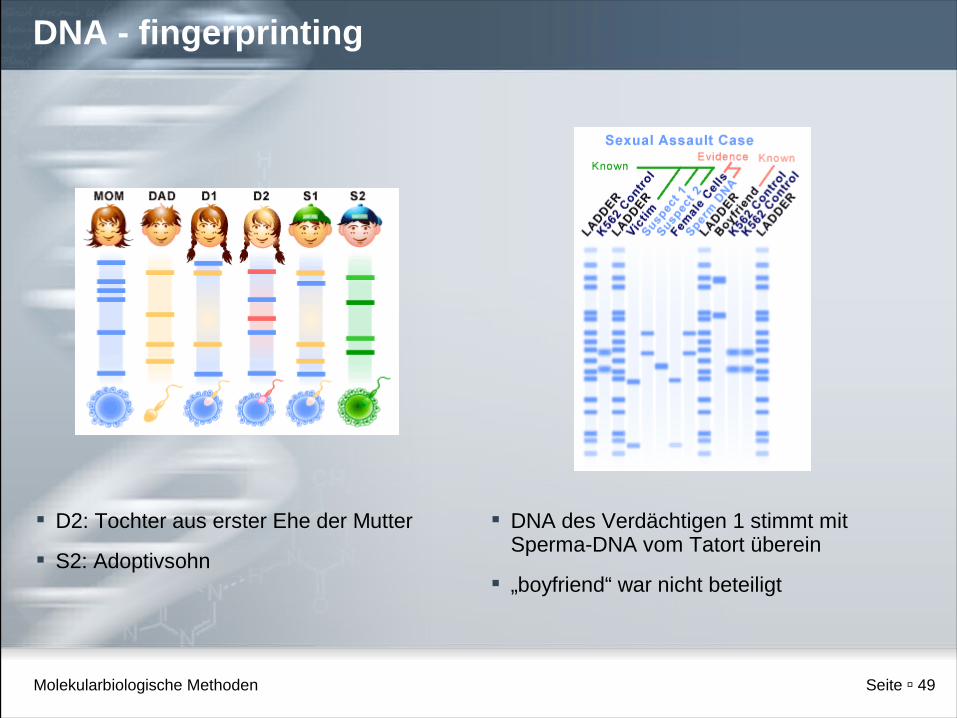
Molekularbiologische Methoden Seite 49
DNA - fingerprinting
D2: Tochter aus erster Ehe der Mutter
S2: Adoptivsohn
DNA des Verdächtigen 1 stimmt mit Sperma-DNA vom Tatort überein
„boyfriend“ war nicht beteiligt

Molekularbiologische Methoden Seite 50
Übersicht Blotting - Methoden
Prinzip
- Trennung von Makromolekülen durch Elektrophorese
- Transfer der Moleküle aus dem Gel auf Nitrozellulosemembran
- Nachweis spezifischer Moleküle
Southern
- Makromoleküle: DNA
- Nachweis: Hyridisierung mit markierten Sonden (DNA, Oligonukleotide)
Northern
- Makromoleküle: RNA
- Nachweis: Hyridisierung mit markierten Sonden (DNA, Oligonukleotide)
Western
- Makromoleküle: Proteine
- Nachweise: Antikörper

Molekularbiologische Methoden Seite 51
Verschiedene Vektortypen
Expressionsvektoren
- klonierte DNA soll in Wirtszellen exprimiert werden- Probleme:
- Wirtszelle muss DNA-Regulation des Ausgangsorganismus „verstehen“- gesamte Regulationseinheit muss kloniert sein
- Lösung: Klonierung des prozessierten Gens: mRNA
Vektor Einbaugröße (kb) Plasmid <10 kb
Bacteriophage 9-20 kb Cosmids 33-47 kb
BACs (künstlicheBakterienchromosomen) 75-125 kb
YACS (künstliche Hefechromosomen) 100-1000 kb
Cosmide sind Plasmid-Vektoren, die cos-Sequenzen (Phage λ)enthalten. Diese sind für die Integration in Phagen notwendig

Molekularbiologische Methoden Seite 52
Klonierung von prozessierten Genen: cDNA
Isolation von mRNA
- Regulation hat stattgefunden
- Splicing hat stattgefunden
- endgültige kodierende Sequenz
- Polyadenylierung durchgeführt
DNA-Synthese
- poly-dT als Primer!
- Reverse Transkriptase als DNA-Polymerase
zweite Syntheserunde zur Produktion von dsDNA
Klonierung der „fertigen Gene“ in Expressionsvektoren

Molekularbiologische Methoden Seite 53
RNA-abhängige DNA-Polymerasen
Reverse Transkriptasen
- Umschreibung retrovorale RNA in DNA
- Laboranwendung: cDNA-Synthese aus RNA
Enzme:
AMV-RT (Avian Myeloblastosis Virus)
- RNase H, (DNA-abh. DNA-Pol.)
- T = 42°C, niedrigere Prozessivität (1-2 kb)
MMLV-RT (Moloney Mouse Leukemia Virus)
- geringere RNase H
- T = 37°C, höhere Prozessivität (> 2 kb)
Modifizierte MMLV-RT (SuperScript etc.)
- keine RNase H !
- Produktlängen bis 20 kb !

Molekularbiologische Methoden Seite 54
Primer
Oligo-dT-Primer
- 15-20 dT
- Hybridisierung mit 3‘-PolyA-Schwanz der mRNA
- Möglichkeit der Full-length-cDNA
Random-Primer
- Statistische Hexamere
- Alle Bereiche von mRNA und Nicht-mRNA vertreten
Spezifische Primer
- Bei bekannter Sequenz
- Niedrige Ausbeuten
Anwendungen:
- cDNA-Banken
- RT-PCR

Molekularbiologische Methoden Seite 55
DNA-abhängige RNA-Polymerasen
Enzyme der regulären Transkription
Verwendung als Werkzeuge:
Polymerasen aus Phagen
- SP6-Polymerase
- T3-Polymerase
- T7-Polymerase
Erkennung spezifischer Promotor-Sequenzen
RNA-Synthese 3‘ von diesen Sequenzen

Molekularbiologische Methoden Seite 56
„DNA-Bibliotheken“
Quelle:- genomische DNA- cDNA
Ziel:- möglichst jede DNA-Sequenz eines Organismus in Plasmid kloniert (Genomsequenzierung)- möglichst jede mRNA eines Organismus als Klon (Physiologie, Zellzyklus, etc)

Molekularbiologische Methoden Seite 57
Expression humaner Antikörper in Bakterien
Klonierung des Vektors pSEX in E.coli führt zu Produktion veränderter M13-Phagen
Ein Teil der Bindeprotein pIII trägt eine humane erste Antikörperdomäne mit Antigenbindestelle

Molekularbiologische Methoden Seite 58
DNA-Bibiliothek in YACs
Mit Hilfe dieser Methode können extrem lange DNA-Fragmente bis zu 1 MBb (z. B. zur Erzeugung von Genombibliotheken) kloniert werden
Erzeugung langer Fragmente durch partiellen Restriktionsverdau
Es können Regulationsmechanismen größerer Geneinheiten untersucht werden

Molekularbiologische Methoden Seite 59
Expression in Pflanzen
● Agrobacterium tumefaciens (erzeugt Tumore in den Wirtspflanzen)
● Bei der Infektion einer Pflanze durch das Bakterium verlässt ein Stück DNA (= T-DNA) mit 30,000 Basenpaaren das Plasmid und integriert sich in die DNA der pflanzlichen Wirtszelle.
● Die Tumorgene der T- DNA müssen inaktiviert werden
● Fremdgene müssen an dieselbe Stelle des Ti-Plasmids integriert werden. Das rekombinante Plasmid mit dem integrierten Gen muß dann wieder in die A. tumefaciens Zelle transferiert werden. Die Bakterienzelle muß nun in das Protoplasma der Pflanzenzelle gebracht werden.
● Die Wirtspflanzen exprimieren die eingebarchten Gene

Molekularbiologische Methoden Seite 60
Transgene Pflanzen: einige Beispiele
Anti-Matsch-TomatePolygalacturonase (PG)
Bt-Mais („Genmais“)
Transgene SojapflanzenResistenz gegen Roundup,Liberty
Molekularbiologische Methoden Seite 61
Polymerase Kettenreaktion
PCR

Molekularbiologie
Methoden
Polymerase-Kettenreaktion (PCR)
Dr. Thomas Seehaus

Molekularbiologische Methoden Seite 63
Geschichte
1970 Kjell Kleppe: Vermehrung von DNA durch zwei flankierende Primer, wegen fehlender technischer Möglichkeiten geriet die Methode wieder in Vergessenheit
1983 Kary Banks Mullis: erneute Erfindung der Polymerase-Kettenreaktion, Idee: künstliche Vervielfältigung von DNA durch wiederholte Verdopplung in mehreren Zyklen mit Hilfe von DNA-Polymerase
Verbesserung: Verwendung von thermostabilen DNA-Polymerasen aus thermophilen Bakterien
Taq-Polymerase aus Thermus aquaticus, fehleranfällig -> Mutationen!
Pwu-Polymerase aus Pyrococcus woesei, Korrekturmechanismus!
Pfu-Polymerase aus Pyrococcus furiosus, ebenfalls Korrekturmechanismus

Molekularbiologische Methoden Seite 64
PCR in der Praxis
Vervielfältigung von kurzen (bis 3000 bp), genau definierten Teilen eines DNA-Stranges
Benötigte Komponenten:
- Original-DNA
- zwei Primer, die den zu vermehrenden Bereich berenzen
- thermostabile Polymerase
- Nukleotide
- Mg2+-Ionen (Kofaktor der Polymerasen)
- Pufferlösungen (geeignete chemische Umgebung für Polymerasen)
- Thermocycler

Molekularbiologische Methoden Seite 65
PCR - Beispiel
1,0 µl DNA-Vorlage (100 ng/µl)
2,0 µl pro Primer (10 µM)
1,0 µl Pfu-, Taq- oder andere thermostabile Polymerase (1-5 U/µl)
1,0 µl 10 mM Desoxy-Nukleotide (dATP, dGTP, dCTP, dTTP), „dNTP“
5,0 µl 10fach konzentrierte Polymerase-Pufferlösung
38,0 µl H2O
ad 50,0 µl Gesamtvolumen
Ein 200 µl-Reaktionsgefäß mit den 50 µl Gemisch wird in den Thermocycler gestellt.

Molekularbiologische Methoden Seite 66
Ablaufschema der PCR
(1) Schmelzen (Denaturierung) bei ca. 96 °C.
(2) Anlagerung (Primerhybridisierung) bei ca. 68 °C.
(3) Verlängerung (Elongation) bei ca. 72 °C (P=Polymerase).
(4) Der erste Zyklus ist beendet.

Molekularbiologische Methoden Seite 67
Schritte des PCR-Prozesses
Die folgenden Angaben sind als Richtwerte gedacht. Meist muss eine PCR auf die spezifische Reaktion hin optimiert werden. Für eine normale Präparation eines Amplifikats reicht folgendes Programm aber aus:
Initialisierung. Das Gemisch wird ca. 2 Minuten lang auf 96 °C erhitzt, um sicherzustellen, dass sich die DNA-Doppelstränge getrennt haben. Manche (sogenannte hot start-) Polymerasen müssen auch durch eine noch längere anfängliche Erhitzungs-Phase (bis zu 15 Minuten) erst aktiviert werden.
Denaturierung. Die Temperatur ca. 30 Sekunden lang auf 96 °C halten. Das genügt gewöhnlich, um die DNA während der Zyklen zu zerlegen.
Anlagerung (annealing). Die Temperatur ca. 30 Sekunden lang auf einer Temperatur halten, die eine spezifische Anlagerung der Primer an die DNA erlaubt. Die genaue Temperatur wird hierbei durch die Länge und die Sequenz der Primer bestimmt (bzw. der passenden Nukleotide im Primer, wenn durch diesen Mutationen eingeführt werden sollen = site-directed Mutagenesis). Wird die Temperatur zu niedrig gewählt, können sich die Primer u. U. auch an nicht 100%-komplementären Sequenzen anlagern und so zu unspezifischen Produkten („Geisterbanden“) führen. Wird die Temperatur zu hoch gewählt, ist die thermische Bewegung der Primer u. U. so gross, dass sie sich nicht richtig anheften können, so dass es zu gar keiner oder nur ineffizienter Produktbildung kommt. Meist liegt die Temperatur bei 55°C bis 65°C.

Molekularbiologische Methoden Seite 68
Schritte des PCR-Prozesses
Verlängerung (elongation). Die Temperatur für ca. 30 Sekunden je 500 Basenpaare auf 68-72 °C (je nach Polymerase) halten.
Die Schritte 2-4 werden 25-40 mal wiederholt.
Das Gemisch wird bei 4-8 °C aufbewahrt. Man kann die PCR am Abend vor dem Verlassen des Labors einleiten, so dass sie über Nacht läuft. Die DNA wird bei 4-8 °C nach nur einer Nacht nicht beschädigt.
Das PCR-Produkt kann durch Agarose-Gelelektrophorese anhand seiner Größe identifiziert werden. (Die Agarose-Gelelektrophorese ist ein Verfahren, bei der DNA in ein Agarose-Gel eingebracht wird und anschließend eine Spannung angelegt wird. Dann bewegen sich die kürzeren DNA-Stränge schneller als die längeren auf den Pluspol zu.) Die Menge des PCR-Produkts kann durch einen Vergleich mit einer DNA-Leiter, die DNA-Fragmente bekannter Größe enthält und parallel zur Probe im Gel mitläuft, bestimmt werden.
Das PCR-Produkt in niedriger (Bande 2) und hoher (Bande 3)Konzentration im Vergleich mit der DNA-Leiter (Bande 1) in Agarose-Gel.