morphine withdrawal inhibits il-12 induction in a ... · pdf filemorphine withdrawal inhibits...
TRANSCRIPT

of May 12, 2018.This information is current as
a Mechanism That Involves cAMPthroughInduction in a Macrophage Cell Line
Morphine Withdrawal Inhibits IL-12
and Sabita RoyAnitha Krishnan, Richard Charboneau, Roderick A. Barke Jennifer Kelschenbach, Jana Ninkovic, Jinghua Wang,
http://www.jimmunol.org/content/180/6/3670doi: 10.4049/jimmunol.180.6.3670
2008; 180:3670-3679; ;J Immunol
Referenceshttp://www.jimmunol.org/content/180/6/3670.full#ref-list-1
, 15 of which you can access for free at: cites 32 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2008 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Morphine Withdrawal Inhibits IL-12 Induction in aMacrophage Cell Line through a Mechanism ThatInvolves cAMP1
Jennifer Kelschenbach,† Jana Ninkovic,† Jinghua Wang,* Anitha Krishnan,*Richard Charboneau,‡ Roderick A. Barke,*‡ and Sabita Roy2*†‡
There are very few studies that examine the effects that morphine withdrawal has on immune functioning, and of these even fewerdescribe the mechanisms by which withdrawal brings about these changes. Our previous work demonstrated that morphinewithdrawal contributed to Th cell differentiation by biasing cells toward the Th2 lineage. A major finding from these studies wasthat IL-12 was decreased following withdrawal, and it was concluded that this decrease may be a mechanism by which morphinewithdrawal is mediating Th2 polarization. Therefore, it was the aim of the current studies to develop an in vitro model to examinethe process of morphine withdrawal and to understand the signaling mechanisms that withdrawal may use to effect IL-12 pro-duction through the use of this model. It was demonstrated and concluded that morphine withdrawal may be effecting IL-12production by increasing cAMP levels, which activates protein kinase A. Protein kinase A activation then prevents the phosphor-ylation and subsequent degradation of I�B, which in turn prevents translocation of the NF-�B p65 subunit to the nucleus totransactivate the IL-12 p40 gene, ultimately resulting in decreased IL-12 production following LPS stimulation. The Journal ofImmunology, 2008, 180: 3670–3679.
A lthough chronic morphine use and abuse have been doc-umented to result in severe immune consequence (1–3)with increased prevalence of opportunistic infections
(4–6), very few studies have been dedicated to examining theeffects of opiate withdrawal on immune functioning. Initial studiesby Tomei and Renaud (7) demonstrated that opioid withdrawal inan in vitro model significantly decreases Fc�R-mediated macro-phage phagocytosis and the decrease correlated with altered intra-cellular cAMP levels (8). More recent studies from Eisenstein andcolleagues (9, 10) show that both abrupt and precipitated morphinewithdrawal results in significant immunosuppression and the ob-served dysfunction was a result of impaired macrophage func-tion, as evidenced by the findings that macrophages obtainedfrom withdrawn spleens displayed reduced expression of thecostimulatory molecule B7.2 and had depressed cytokine pro-duction. Additional studies from this group (11, 12) furtherdemonstrated that morphine-withdrawn mice when adminis-tered a sublethal dose of LPS exhibited 100% lethality that wasaccompanied by a decrease in IL-12 production. Similarly, wehave also recently shown that morphine withdrawal results in asignificant decrease in LPS-induced IL-12 synthesis in an invivo morphine-withdrawal model (13).
IL-12 is a pivotal cytokine involved in cell-mediated immunityand impairment in IL-12 synthesis results in increased suscepti-bility to intracellular bacterial pathogens (14–18). Thus, the ob-servation that morphine withdrawal results in reduction in IL-2synthesis is clinically significant and may account for the increasedsusceptibility to opportunistic infection in the drug abusepopulation.
IL-12 is a heterodimeric cytokine (referred to as IL-12p70) andis comprised of two disulfide-linked subunits known as p35 andp40, which corresponds to their approximate molecular mass (19,20). Although p35 is produced by most hematopoetic cells (21,22), IL-12p40 is tightly regulated and the active form of IL-12 isproduced only by APCs (19, 20). Most studies indicate that levelsof IL-12p40 are a better indicator of IL-12 production, and as aresult, the IL-12p40 gene has been studied for transcriptional reg-ulation of IL-12 (21).
The production of IL-12p40 in both macrophages and den-dritic cells is transcriptionally regulated by key transcriptionfactors which include AP-1, PU.1, C/EBP, and NF-�B (22–27).Recent studies (21) show that NF-�B activation is essential forLPS-induced IL-12 production and mutation of the IL-12 pro-moter at the NF-�B site significantly reduced IL-12 transcrip-tion (21).
The goal of the current studies was to initially establish an invitro model of withdrawal and using this model examine themechanism by which morphine withdrawal modulates IL-12production.
Materials and MethodsReagents
LPS (from Escherichia coli O127:B7) was obtained from Sigma-Aldrichand used as an immune stimulus. Protein kinase inhibitors (H-89,SB203580, Rp cAMP, and PD98059) were obtained from Calbiochem/EMD Biosciences and were used to assess cell signaling components in-volved in morphine-withdrawal mediated IL-12 production.
*Department of Surgery and †Department of Pharmacology, University of Min-nesota, Minneapolis, MN 55455; and ‡Veterans Affairs Medical Center, Minne-apolis, MN 55417
Received for publication January 31, 2007. Accepted for publication January 4, 2008.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by National Institutes of Health Grants R01DA12104,R01 DA022935, K02 DA015349, P50DA11806 (to S.R.), and T32 DA07097 (toJ.K. and J.N.).2 Address correspondence and reprint requests to Dr. Sabita Roy, Departments ofSurgery and Pharmacology, University of Minnesota, MMC 195, 420 DelawareStreet, Minneapolis, MN 55455. E-mail address: [email protected]
The Journal of Immunology
www.jimmunol.org
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Animals
Six- to 8-wk-old B6129SF2 male mice were used in the experiments de-scribed within. Animals were housed four animals per cage under con-trolled conditions of temperature and lighting (12-h light/dark) and givenfree access to standard food and tap water. All animals were allowed toacclimate to their environment for at least 7 days before any experimentalmanipulations. Sacrifice was performed by carbon dioxide asphyxiationand spleen tissue was harvested aseptically. Discomfort, distress, and in-jury to the animals were minimized. The Institutional Animal Care and UseCommittee at the University of Minnesota have approved all protocols inuse, and all procedures are in agreement with the guidelines set forth by theNational Institute of Health Guide for the Care and Use of LaboratoryAnimals.
Purification of macrophages from splenocytes
Spleens were removed aseptically and suspensions were prepared by forc-ing the tissue through a tissue sieve with a sterile syringe plunger. Spleno-cytes were allowed to adhere in serum-free medium for 1 h at 37°C. Non-adherent cells were washed and adherent cells were resuspended in RPMI1640 (Invitrogen Life Technologies) supplemented with 10% newborn calfserum and 1% penicillin-streptomycin (Sigma-Aldrich). The compositionof the adherent populations was evaluated using FACS analysis. Flow cy-tometric analysis revealed that adherent cells were primarily macrophages,with almost a 5-fold enhancement of F4/80 staining compared with thetotal splenocyte population. CD3� T lymphocytes were not detected in theadherent population. The adherent cells were plated at a concentration of1–2 � 106 cells/ml in triplicate onto 6-well culture plates. Cells weresubjected to the in vitro withdrawal paradigm described below and stim-ulated with LPS (Sigma-Aldrich) at 5 �g/ml (a concentration determined tohave significant stimulatory effect when measuring subsequent protein lev-els) and incubated overnight at 37°C in 5% CO2.
Cell culture
The mouse alveolar macrophage cell line CRL-2019 (American Type Cul-ture Collection) was also used to examine cell signaling properties andIL-12p40 promoter activities following morphine withdrawal. Cells weremaintained in RPMI 1640 (Invitrogen Life Technologies) supplementedwith 10% FBS and 1% penicillin-streptomycin (Sigma-Aldrich). Cellswere plated at a concentration of 1–2 � 106 cells/ml in triplicate onto6-well culture plates. Cells were subjected to the in vitro withdrawal par-adigm described below and stimulated with LPS (Sigma-Aldrich) at 5�g/ml (a concentration determined to have significant stimulatory effectwhen measuring subsequent protein levels) and incubated overnight at37°C in 5% CO2.
In vitro withdrawal paradigm
To replicate conditions tested in vivo (13), either primary cells or the cellline was plated as described above. Following plating, cells were treatedwith 100 nM morphine sulfate (National Institute on Drug Abuse, ResearchTriangle Park, NC) once per day for 3 consecutive days. Parallel cultureswere treated with vehicle and designated vehicle control groups. Followingthe third day of morphine treatment, cells were vigorously washed three tofive times with PBS (Invitrogen Life Technologies) to simulate withdrawal.Parallel cultures were washed in the presence of morphine and incubatedwith morphine and was designated as the chronic morphine group. Cellswere then stimulated with 5 �g/ml LPS (Sigma-Aldrich; incubated eitherovernight or for various time points depending on the type of experimentat 37°C in 5% CO2.
ELISA
ELISAs were performed as described previously by our laboratory (13).Briefly, quantikine ELISA kits were obtained (R&D Systems) and assaysrun according to the manufacturer’s directions. Sample supernatant wasassayed in triplicate per experimental condition. Following the incubationperiod, plates were washed and 100 �l of detection Ab conjugated to HRPwas added to the wells. Finally, 100 �l of substrate solution was added andabsorbance was read at 450 nm using a standard plate reader (PackardSpectraCount Microplate Photometer). OD measurements for the standardswere used to generate a standard curve, and the concentration of the par-ticular cytokine in each of the samples was extrapolated from this standardcurve. Concentrations are presented as pg/ml.
RT-PCR
RT-PCR experiments were used to assess mRNA levels of cytokines, andwere performed as described previously by our laboratory (13). Briefly,
total RNA was extracted from splenocytes using an RNeasy Mini kit (Qia-gen), a system which uses spin columns to extract and purify RNA fromcells. Before RT-PCR, RNA samples were treated with deoxyribonucleaseI (DNase I; Invitrogen Life Technologies) according to the manufacturer’sinstructions. To ensure quality of RNA samples, 260/280 UV absorbancereadings were performed and the 260:280 ratios were in range for pureRNA. Total RNA (1 �g) was then reverse transcribed to synthesize first-strand cDNA (45°C, 45 min) using random hexamers (2.5 �M), Moloneymurine leukemia virus reverse transcriptase (2.5 U), and 1 mM each ofdATP, dCTP, dGTP, and dTTP for a final reaction volume of 40 �l (Ap-plied Biosystems). Following first-strand synthesis, the reaction mixturewas heated at 95°C for 5 min to inactivate reverse transcriptase. Amplifi-cation steps were performed using upstream and downstream primers spe-cific for mouse IL-12p40 (Clontech Laboratories) and �2-microglobulin(Clontech Laboratories). The primer sequences are as follows: IL-12p40,sense 5�-ACT CAC ATC TGC TGC TCC AC-3�, antisense 5�-CTG GTTTGA TGA TGT CCT G-3�; and �2-microglobulin, sense 5�-ATG GCTCGC TCG GTG ACC CTA-3�, antisense 5�-TCA TGA TGC TTG ATCCAT GTC TCG-3�. PCR buffer containing 2 mM MgCl2, 0.1 �M primer,and 2.5 U of AmpliTaq DNA polymerase was prepared and �5 �l of thefirst-strand cDNA reaction mixture was added for a final volume of 50 �l.PCR conditions consisted of 35 cycles of 94°C for 45 s (denaturation),60°C for 45 s (annealing), and 72°C for 45 s (extension), followed by afinal extension at 72°C for 15 min. Internal controls included both a neg-ative control (containing only PCR mix) and a control lacking reversetranscriptase used at the point of cDNA synthesis. PCR products wereseparated using 1.5% agarose gels and visualized by ethidium bromidestaining.
Real-time RT-PCR
Real-time RT-PCR experiments were used to assess mRNA levels of Il-12p40. Total cellular RNA was isolated using the RNeasy Minikit per themanufacturer’s instructions (Qiagen). Total RNA was DNase I treated perthe manufacturer’s instructions (Invitrogen Life Technologies) and quan-titated using the A260:A280 ratio. Reverse transcription and real-time PCRwas performed on Applied Biosystems Prism 7500 using SYBR GreenPCR Master Mix (Applied Biosystems). The established primers for IL-12and 18 S were used at a final concentration of 300 nM and 18 S at a finalconcentration of 100 nM. All samples were performed in triplicates orquadruplicates and normalized to ribosomal 18 S mRNA in the samecDNA set. Data are expressed as fold change over control, untreated cellsby the cycle threshold method. Three independent experiments wereperformed.
EMSA
Transcription factor interactions with DNA response elements were as-sessed using EMSAs as described previously by our laboratory (12).Briefly, nuclear extracts were prepared as described below. NF-�B con-sensus oligonucleotides were purchased (Santa Cruz Biotechnology) andend-labeled with 32P according to the manufacturer’s instructions (Pro-mega). Approximately 10 �g of nuclear extracts was incubated with 0.5 ngof labeled probe in binding buffer. DNA-protein complexes were resolvedusing nondenaturing acrylamide gels. Gels were then dried and visualizedby either autoradiography or phosphorimaging techniques.
cAMP immunoassay
Cells were exposed to withdrawal as described above. At 5, 10, and 30 minfollowing withdrawal, cells were washed in PBS and resuspended at adensity of 1 � 10 7 cells/ml and lysed in cell lysis buffer. The supernatantfrom lysed cells were analyzed using cAMP assay kits (R&D Systems)according to the manufacturer’s instructions.
Western blotting
Protein levels of various NF-�B subunits and I�B� were determined usingWestern blot techniques. First, nuclear protein extracts were prepared usingthe CellLytic Nuclear Extraction Kit (Sigma-Aldrich). Briefly, cells wereresuspended in lysis buffer containing DTT and protease inhibitors. Cellswere then homogenized and centrifuged to separate cytoplasmic and nu-clear proteins. Proteins were then separated by SDS-PAGE using 10% gelsand the gels were then transferred to nitrocellulose membranes using asemidry transfer procedure. Membranes were blocked using SuperblockStarting Buffer (Pierce) and probed with a 1/1000 dilution of a mouse �rabbit monoclonal primary Ab (Cell Signaling Technology). Membraneswere washed and probed with a 1/1500 dilution of a goat � rabbit sec-ondary Ab conjugated to HRP (Sigma-Aldrich). Membrane blots were then
3671The Journal of Immunology
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
exposed to ECL detection reagents (SuperSignal West Pico Chemilumi-nescent Substrate; Pierce) and visualized using x-ray films.
Transient transfection
The human IL-12p40 promoter-luciferase reporter plasmid was providedby Dr. A. Kumar (University of Ottawa, Ottawa, Ontario, Canada) and theconstruction of this plasmid has been previously described (21). CRL-2019cells were transfected with plasmids using the Effectene reagent (Qiagen)according to the manufacturer’s instructions. Briefly, 10 �g of IL-12p40promoter-firefly luciferase reporter plasmid and 0.5 �g of pRL-TK-Renillareniformis luciferase reporter internal control plasmid (Promega) were in-cubated for 10 min with Effectene reagent in standard RPMI 1640 mediumto allow formation of liposome complexes. Complexes were added directlyto each well of a 6-well plate and cells were maintained at 37°C in 5% CO2
culture conditions. Transfections were performed on day 3 of these 5-dayexperiments. Following the treatment paradigm, cells were lysed and lu-ciferase activity was measured using a Dual-Luciferase Reporter Assaysystem (Promega) and a Turner Biosystems TD 20/20 luminometer ac-cording to the manufacturers’ instructions. Data are presented as standard-ized luciferase activity and was determined by the ratio between fireflyluciferase and Renilla reniformis luciferase.
Statistics
Each cytokine supernatant protein concentration was expressed as mean �SEM, and comparisons between group means were assessed using an un-paired Student’s t test. Standardized luciferase activity � SEM was plottedaccording to treatment group and differences were assessed using an un-paired Student’s t test. Significance was set at p � 0.05.
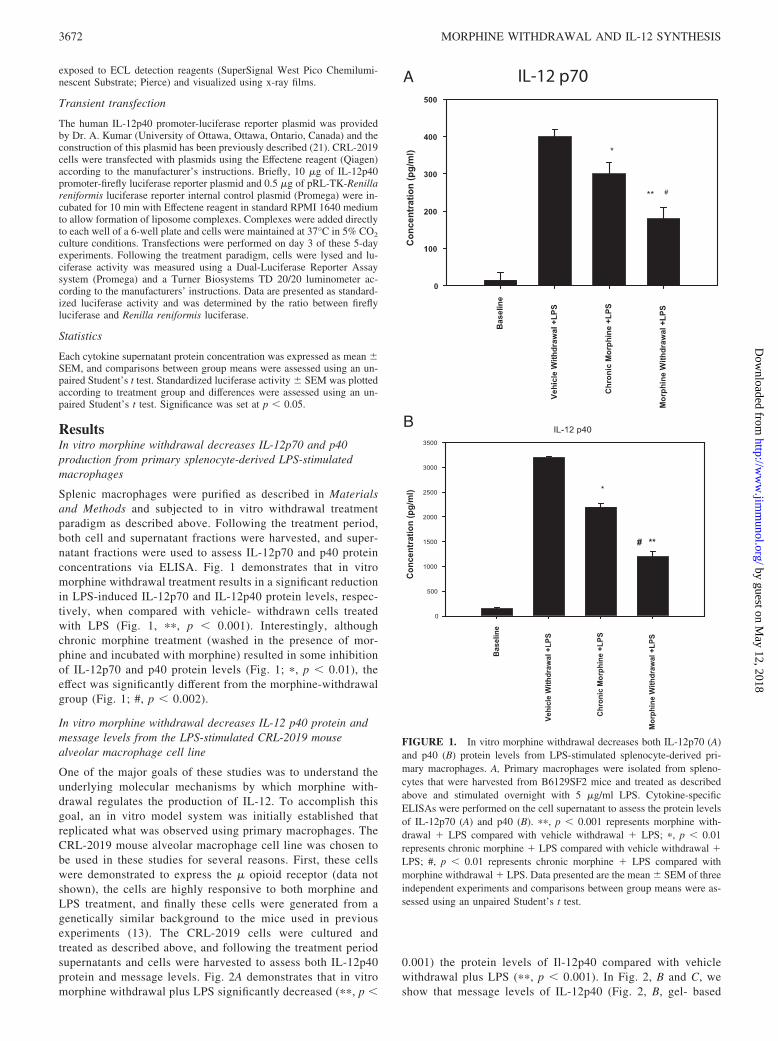
ResultsIn vitro morphine withdrawal decreases IL-12p70 and p40production from primary splenocyte-derived LPS-stimulatedmacrophages
Splenic macrophages were purified as described in Materialsand Methods and subjected to in vitro withdrawal treatmentparadigm as described above. Following the treatment period,both cell and supernatant fractions were harvested, and super-natant fractions were used to assess IL-12p70 and p40 proteinconcentrations via ELISA. Fig. 1 demonstrates that in vitromorphine withdrawal treatment results in a significant reductionin LPS-induced IL-12p70 and IL-12p40 protein levels, respec-tively, when compared with vehicle- withdrawn cells treatedwith LPS (Fig. 1, ��, p � 0.001). Interestingly, althoughchronic morphine treatment (washed in the presence of mor-phine and incubated with morphine) resulted in some inhibitionof IL-12p70 and p40 protein levels (Fig. 1; �, p � 0.01), theeffect was significantly different from the morphine-withdrawalgroup (Fig. 1; #, p � 0.002).
In vitro morphine withdrawal decreases IL-12 p40 protein andmessage levels from the LPS-stimulated CRL-2019 mousealveolar macrophage cell line
One of the major goals of these studies was to understand theunderlying molecular mechanisms by which morphine with-drawal regulates the production of IL-12. To accomplish thisgoal, an in vitro model system was initially established thatreplicated what was observed using primary macrophages. TheCRL-2019 mouse alveolar macrophage cell line was chosen tobe used in these studies for several reasons. First, these cellswere demonstrated to express the � opioid receptor (data notshown), the cells are highly responsive to both morphine andLPS treatment, and finally these cells were generated from agenetically similar background to the mice used in previousexperiments (13). The CRL-2019 cells were cultured andtreated as described above, and following the treatment periodsupernatants and cells were harvested to assess both IL-12p40protein and message levels. Fig. 2A demonstrates that in vitromorphine withdrawal plus LPS significantly decreased (��, p �
0.001) the protein levels of Il-12p40 compared with vehiclewithdrawal plus LPS (��, p � 0.001). In Fig. 2, B and C, weshow that message levels of IL-12p40 (Fig. 2, B, gel- based
IL-12 p70A
B
FIGURE 1. In vitro morphine withdrawal decreases both IL-12p70 (A)and p40 (B) protein levels from LPS-stimulated splenocyte-derived pri-mary macrophages. A, Primary macrophages were isolated from spleno-cytes that were harvested from B6129SF2 mice and treated as describedabove and stimulated overnight with 5 �g/ml LPS. Cytokine-specificELISAs were performed on the cell supernatant to assess the protein levelsof IL-12p70 (A) and p40 (B). ��, p � 0.001 represents morphine with-drawal � LPS compared with vehicle withdrawal � LPS; �, p � 0.01represents chronic morphine � LPS compared with vehicle withdrawal �LPS; #, p � 0.01 represents chronic morphine � LPS compared withmorphine withdrawal � LPS. Data presented are the mean � SEM of threeindependent experiments and comparisons between group means were as-sessed using an unpaired Student’s t test.
3672 MORPHINE WITHDRAWAL AND IL-12 SYNTHESIS
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
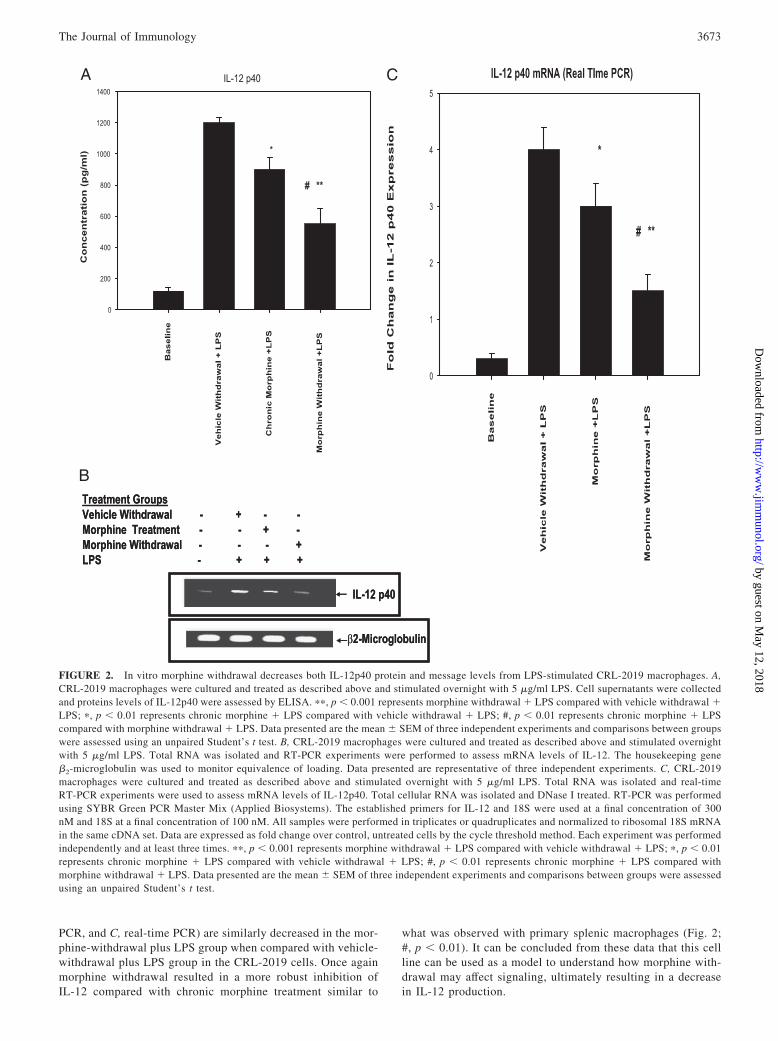
PCR, and C, real-time PCR) are similarly decreased in the mor-phine-withdrawal plus LPS group when compared with vehicle-withdrawal plus LPS group in the CRL-2019 cells. Once againmorphine withdrawal resulted in a more robust inhibition ofIL-12 compared with chronic morphine treatment similar to
what was observed with primary splenic macrophages (Fig. 2;#, p � 0.01). It can be concluded from these data that this cellline can be used as a model to understand how morphine with-drawal may affect signaling, ultimately resulting in a decreasein IL-12 production.
Treatment GroupsVehicle Withdrawal - + - -Morphine Treatment - - + -Morphine Withdrawal - - - +LPS - + + +
IL-12 p40
β2-Microglobulin
Treatment GroupsVehicle Withdrawal - + - -Morphine Treatment - - + -Morphine Withdrawal - - - +LPS - + + +
IL-12 p40
β2-Microglobulin
A
B
C
FIGURE 2. In vitro morphine withdrawal decreases both IL-12p40 protein and message levels from LPS-stimulated CRL-2019 macrophages. A,CRL-2019 macrophages were cultured and treated as described above and stimulated overnight with 5 �g/ml LPS. Cell supernatants were collectedand proteins levels of IL-12p40 were assessed by ELISA. ��, p � 0.001 represents morphine withdrawal � LPS compared with vehicle withdrawal �LPS; �, p � 0.01 represents chronic morphine � LPS compared with vehicle withdrawal � LPS; #, p � 0.01 represents chronic morphine � LPScompared with morphine withdrawal � LPS. Data presented are the mean � SEM of three independent experiments and comparisons between groupswere assessed using an unpaired Student’s t test. B, CRL-2019 macrophages were cultured and treated as described above and stimulated overnightwith 5 �g/ml LPS. Total RNA was isolated and RT-PCR experiments were performed to assess mRNA levels of IL-12. The housekeeping gene�2-microglobulin was used to monitor equivalence of loading. Data presented are representative of three independent experiments. C, CRL-2019macrophages were cultured and treated as described above and stimulated overnight with 5 �g/ml LPS. Total RNA was isolated and real-timeRT-PCR experiments were used to assess mRNA levels of IL-12p40. Total cellular RNA was isolated and DNase I treated. RT-PCR was performedusing SYBR Green PCR Master Mix (Applied Biosystems). The established primers for IL-12 and 18S were used at a final concentration of 300nM and 18S at a final concentration of 100 nM. All samples were performed in triplicates or quadruplicates and normalized to ribosomal 18S mRNAin the same cDNA set. Data are expressed as fold change over control, untreated cells by the cycle threshold method. Each experiment was performedindependently and at least three times. ��, p � 0.001 represents morphine withdrawal � LPS compared with vehicle withdrawal � LPS; �, p � 0.01represents chronic morphine � LPS compared with vehicle withdrawal � LPS; #, p � 0.01 represents chronic morphine � LPS compared withmorphine withdrawal � LPS. Data presented are the mean � SEM of three independent experiments and comparisons between groups were assessedusing an unpaired Student’s t test.
3673The Journal of Immunology
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Morphine withdrawal decreases NF-�B binding to responseelements in LPS-treated CRL-2019 cells
As mentioned, morphine withdrawal decreased the message levelsof the transcriptionally regulated IL-12 subunit p40. Therefore, theability of morphine withdrawal to mediate transcription factorbinding to response elements was assessed. Several transcriptionfactors are involved in the transactivation of IL-12p40 includingAP-1, C/EBP, PU.1, and NF-�B. All of these transcription factorinteractions were measured following withdrawal periods of 4 and8 h. Earlier time periods were used in an attempt to reveal whatwas occurring at the time of transcription of IL-12. Fig. 3A dis-plays that morphine withdrawal greatly reduced NF-�B binding toresponse elements at both the 4- and 8-h withdrawal time points.Fig. 3B reveals that PU.1 binding is decreased following morphinewithdrawal of both 4 and 8 h. In contrast, morphine withdrawal didnot significantly alter binding of either AP-1 or C/EBP to theirconsensus oligonucleotide (data not shown). The results obtainedfor PU.1 binding were interesting given the findings that PU.1 actsas a repressor at the � opioid receptor (MOR)3 gene promoterthrough a PU.1 binding site (17). Under the current circumstances,morphine withdrawal by decreasing binding of PU.1 should re-move the repression exerted by PU.1 on the MOR gene and pos-sibly lead to an increase in MOR expression. This possibility hasnot been explored, but this mechanism may serve as a cellularadaptation to morphine withdrawal in immune cells. In can beconcluded from these findings that morphine withdrawal decreasesthe ability of the key IL-12 transcription factors NF-�B and PU.1to bind to DNA response elements.
Morphine withdrawal inhibits translocation of the NF-�Bsubunit p65 into the nuclear compartment of LPS-stimulatedCRL-2019 macrophages
Given the findings that morphine withdrawal decreased binding ofNF-�B to DNA response elements, the effect of withdrawal ontranslocation of NF-�B subunits into the nucleus was assessed.CRL-2019 cells were treated as described above and, after 24 h ofin vitro withdrawal, cells were stimulated with LPS for 10, 30, and
60 min. Following the stimulation, the cells were lysed, nuclearprotein was harvested and separated on SDS-PAGE gels, and fi-nally probed with anti-p65 Abs as described. Fig. 4A demonstratesthe effects of in vitro morphine withdrawal on nuclear transloca-tion of the p65 NF-�B protein subunit. LPS treatment alone re-sulted in an increased translocation of the NF-�B p65 subunit intothe nucleus. This increase was observed at all LPS treatment timepoints examined (10, 30, and 60 min) when compared with base-line activity. In contrast, in the morphine-withdrawal plus LPSgroups, the expression of p65 was markedly reduced when com-pared with the matched vehicle-withdrawn LPS-treated cells.These results provide support for the idea that morphine with-drawal may be inhibiting the production of IL-12p40 by effectingthe transcriptional regulation of this cytokine through a mechanismthat involves NF-�B.
Morphine withdrawal modulates NF-�B inhibitory proteinI�B� levels in the cytosolic compartment of LPS- stimulatedCRL-2019 macrophages
Translocation of NF-�B to the nucleus is regulated by the inhib-itory protein I�B and the associated I�B kinases (IKKs). Whencells are in a stimulus-free environment, NF-�B is sequestered tothe cytoplasm by I�B. Following stimulation, IKKs become acti-vated and phosphorylate I�B, which triggers the ubiquitination anddegradation of I�B and ultimately allows for the translocation ofNF-�B to the nucleus where it transactivates NF-�B-responsivegenes. Given the observations that the p65 subunit translocationinto the nucleus is blunted following withdrawal, the levels of I�Bin the cytoplasm were examined. It can be expected that if theNF-�B subunit expression is blunted in the nucleus there would beincreased detection of I�B levels in the cytoplasm facilitating thisblunted response. CRL-2019 cells were treated as described aboveand, after 24 h of in vitro withdrawal, cells were stimulated withLPS for 5, 10, and 20 min. Following the stimulation, the cellswere lysed, whole cell lysates were prepared and separated on
3 Abbreviations used in this paper: MOR, � opioid receptor; IKK, I�B kinase; PKA,protein kinase A.
Treatment Groups
Vehicle Withdrawal + + Morphine Withdrawal + +LPS - + + + +
Treatment Groups
Vehicle Withdrawal + + Morphine Withdrawal + +LPS - + + + +
Treatment Groups
Vehicle Withdrawal + + Morphine Withdrawal + +LPS - + + + +
Treatment Groups
Vehicle Withdrawal + + Morphine Withdrawal + +LPS - + + + +
A
B
FIGURE 3. In vitro morphine withdrawal results in decreased consen-sus sequence binding of NF-�B (A) and PU.1 (B). CRL-2019 macrophageswere cultured and treated as described above and stimulated overnight with5 �g/ml LPS. Nuclear extracts were prepared and incubated with 32P-labeled consensus sequences for NF-�B (A) and PU.1 (B). DNA-proteincomplexes were run on nondenaturing acrylamide gels and visualized byautoradiography.
FIGURE 4. In vitro morphine withdrawal decreases expression of theNF-�B subunit p65 in the nuclear compartment (A) and decreases degra-dation of I�B in the cytoplasm (B). CRL-2019 macrophages were culturedand treated as described above. Nuclear extracts (A) or whole cell lysates(B) were prepared and separated by SDS-PAGE. Blots were then probedwith a rabbit anti-mouse primary mAb, followed by incubation with a goatanti-rabbit secondary Ab, and visualized using ECL detection reagents.Blots were then stripped and probed with a �-actin primary Ab to ensurefor equivalence of protein loading.
3674 MORPHINE WITHDRAWAL AND IL-12 SYNTHESIS
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

SDS-PAGE gels, and finally probed with anti-I�B� Abs as de-scribed. Fig. 4B demonstrates the levels of I�B� from both cellsthat underwent morphine withdrawal and were stimulated withLPS, and those cells that were saline treated and washed and stim-ulated with LPS for the indicated times. As can be seen, vehiclewithdrawn plus LPS treatment resulted in decreased levels of I�B�in the cytoplasm. At 5 and 10 min after LPS treatment, barelydetectable levels of I�B� were observed, indicating stimulation-induced degradation of I�B�. Twenty minutes after LPS treat-ment, an increased detectable level of I�B� was observed as com-pared with the 5- and 10-min groups, suggesting that mechanismsthat return this signaling pathway to baseline have begun to betriggered. In contrast, the morphine- withdrawal plus LPS groupsdisplay increased protein levels of I�B� in the cytoplasm whencompared with the LPS alone at all treated times. These data in-dicated that morphine withdrawal by preventing the degradation ofthe NF-�B inhibitory protein I�B in the cytosolic compartmentserves as a mechanism by which withdrawal hinders the transcrip-tional regulation of NF-�B, ultimately resulting in decreased pro-duction of IL-12p40 following LPS treatment.
Morphine withdrawal decreases IL-12p40 promoter activity inLPS-stimulated CRL-2019 macrophages
Given the results observed following in vitro morphine withdrawalwith regard to NF-�B DNA response element binding and expres-sion of subunits in the nuclear compartment, we next investigatedthe effects of withdrawal on IL-12p40 promoter activity. CRL-2019 macrophages were transfected as described above and un-derwent morphine withdrawal for 24 h followed by 4 h of LPSstimulation. Following treatment, cells were lysed and lucif-
erase activity was measured as described previously. Standard-ized luciferase activity measurements were calculated and areindicative of IL-12p40 promoter activity. Fig. 5 demonstratesthat in the vehicle-withdrawal plus LPS treatment group a sig-nificant increase in promoter activity was observed when com-pared with baseline activity (�, p � 0.001), whereas morphinewithdrawal plus LPS treatment resulted in a significant decreasein LPS-induced IL-12p40 promoter activity when comparedwith vehicle treatment plus LPS group (��, p � 0.001). Theseresults provide evidence that morphine withdrawal decreasesthe promoter activity of IL-12p40 following LPS stimulationand adds further support to the idea that withdrawal is acting tosuppress IL-12p40 production by inhibiting transcriptional reg-ulation presumably provided by NF-�B.
Overexpression of the p65 subunit of NF-�B partially rescuesmorphine withdrawal-induced IL-12 p40 promoter activity
Given the results described with regard to the contribution ofNF-�B to this system, the effects of the overexpression of the p65subunit of NF-�B on IL-12p40 promoter activity were examined.CRL-2019 cells were treated and transfected as described aboveand Fig. 6 displays the results. As was seen in the previous figure,vehicle withdrawn plus LPS treatment alone significantly in-creased (#, p � 0.001) IL-12p40 promoter activity when comparedwith baseline activity. In contrast, morphine withdrawal plus LPSstimulation significantly reduced this activity when compared withcells treated with vehicle withdrawn plus LPS (�, p � 0.001). Over-expression of p65 enhanced IL-12p40 promoter activity as would beexpected given that there are increased levels of this transcriptionfactor which can also serve to transactivate the IL-12p40 promoter-luciferase reporter (��, p � 0.01). Vehicle withdrawn plus LPS
FIGURE 5. Morphine withdrawal decreases IL-12p40 promoter activityin LPS-stimulated CRL-2019 macrophages. CRL-2019 macrophages werecultured, transfected, and treated as described above and stimulated for 4 hwith 5 �g/ml LPS. Data are presented as standardized luciferase activity,which are values that were calculated by the ratio between firefly andRenilla luciferase and represents IL-12p40 promoter activation. Eachtreatment group was tested in triplicate and results are representative ofat least three independent experiments. �, p � 0.005 represents vehiclewithdrawal � LPS compared with baseline; ��, p � 0.01 representsvehicle withdrawal � LPS compared with morphine withdrawal � LPS.
Vehicle Withdrawal - + - + + - + -
Morphine Withdrawal - - + - - + - +
LPS - + + - + + + +
p65 overexpression - - - + + + + +
p50 overexpression - - - - - - + +
0
0.2
0.4
0.6
0.8
1
1.2
A b c c1 d e f g
Sta
nd
ard
ized
Lu
cife
rase
Act
ivit
y
*
**
#***
Vehicle Withdrawal - + - + + - + -
Morphine Withdrawal - - + - - + - +
LPS - + + - + + + +
p65 overexpression - - - + + + + +
p50 overexpression - - - - - - + +
0
0.2
0.4
0.6
0.8
1
1.2
A b c c1 d e f g
*
**
#***
FIGURE 6. NF-�B p65 overexpression partially reverses morphine-withdrawal-mediated decreases in IL-12p40 promoter activity. CRL-2019macrophages were cultured, transfected, and treated as described aboveand stimulated for 4 h with 1 �g/ml LPS. Data are presented as standard-ized luciferase activity, which are values that were calculated by the ratiobetween firefly and Renilla luciferase and represents IL-12p40 promoteractivation. Each treatment group was tested in triplicate and results are themean of three independent experiments. �, p � 0.001 represents vehiclewithdrawal � LPS compared with morphine withdrawal � LPS; ��, p �0.01 vehicle withdrawal � LPS � p65 overexpression compared with mor-phine withdrawal � LPS � p65 overexpression.
3675The Journal of Immunology
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

stimulation of cells that overexpressed p65 only marginally in-creased promoter activity over NF-�B p65 expression alone, indi-cating that a ceiling in promoter activity may have been reached.
Overexpression of p65-NF-�B in cells that were morphine with-drawn plus LPS- treated partially but significantly rescued mor-phine withdrawal-induced inhibition of IL-12p40 promoter activ-ity (���, p � 0.01). These results indicate that a more robust effectmay not have been observed given the idea that morphine with-drawal is preventing the degradation of I�B (Fig. 4B) and thereforesequestering NF-�B p50, a necessary binding partner for p65 thatis required for appropriate promoter activation. Overexpression ofboth p65 and p50 significantly and completely rescued morphinewithdrawal-induced inhibition of Il-12p40 promoter activity (Fig.6). These results provide further support for the idea that morphinewithdrawal impairs IL-12p40 production by impeding the tran-scriptional regulation of the IL-12p40 promoter by NF-�B.
Morphine withdrawal increases intracellular cAMP levels
cAMP has been shown to be elevated following periods of chronicmorphine treatment or morphine withdrawal, a phenomenonknown as adenylyl cyclase superactivation (28). The contributionof this phenomenon to morphine-withdrawal-mediated reductionsin IL-12p40 seemed to be a logical path to examine given thecombined observations that withdrawal increases cAMP, whichcan act to decrease IL-12 40 (8). To initially determine whethermorphine withdrawal results in an increase in intracellular cAMP,CRL-2019 cells were treated with morphine and withdrawn as de-scribed above. Our results show a time-dependent increase incAMP following morphine withdrawal (Fig. 7), with almost a3-fold increase in cAMP levels seen at 30 min following with-drawal. We next compared intracellular cAMP levels followingchronic morphine with that observed following morphine with-drawal. Interestingly, the increase in cAMP following withdrawalwas almost 3-fold greater when compared with the vehicle treat-ment plus withdrawal group and 2-fold greater than the chronicmorphine treatment group (Fig. 7B).
To expand on these findings and to provide further evidence thatit is the ability of morphine withdrawal to elevate cAMP, which isthe ultimate causative factor in impairing IL-12p40 production, theeffect of the cAMP inhibitor Rp cAMP was investigated on mor-phine withdrawal-induced decrease in IL-12 synthesis. CRL-2019cells were pretreated with varying doses of Rp cAMP at the timeof withdrawal for 1 h and then stimulated with LPS for 24 h.IL-12p40 was measured as described above. Data shows thatthe cAMP antagonist rescued morphine-induced inhibition ofIL-12 synthesis in a concentration-dependent manner, provid-ing strong support for our hypothesis that morphine withdrawalmodulates IL-12 synthesis through a mechanism that involvescAMP (Fig. 7C).
The protein kinase A (PKA) inhibitor H-89 reverses morphinewithdrawal-mediated decreases in IL-12p40 promoter activity
To further examine the signaling mechanisms involved in the ob-served reductions in IL-12p40 as a result of morphine withdrawal,the effects of several protein kinase inhibitors on promoter activitywere tested. Specifically, both H-89 (PKA inhibitor, 1 �M) andSB203580 (p38 MAPK inhibitor, 1 �M) were examined by treat-ing the CRL-2019 cells with the particular inhibitor at the statedconcentration for 1 h immediately after withdrawal treatment. Fig.8A demonstrates the results, which once again displays that LPSsignificantly increases promoter activity, whereas morphine with-drawal significantly reduces the activity as compared with LPStreatment alone. Pretreatment with the PKA inhibitor H-89 wasable to reverse morphine withdrawal-induced reduction in IL-12promoter activity, demonstrated by the nonsignificant differencebetween LPS alone and morphine-withdrawal plus LPS plusH-89 treatment groups. Finally, the p38 MAPK inhibitor
05
101520253035
Baseli
ne
Vehicl
e
Chronic
Mor
phine
Vehicl
e with
drawn
Morphin
e With
drawn
cAM
P (p
mol
/ml)
*
*
0
500
1000
1500
2000
2500
3000
A B C D E F G H
IL-1
2 p4
0 (p
g/m
l)
*
*
*
*
Vehicle Withdrawal - + + - - - - -
Morphine Withdrawal - - - + + + + +
LPS - + + + + + + +
Rp cAMP (uM) - - 100 - 1 10 50 100
0
500
1000
1500
2000
2500
3000
A B C D E F G H
IL-1
2 p4
0 (p
g/m
l)
*
*
*
*
Vehicle Withdrawal - + + - - - - -
Morphine Withdrawal - - - + + + + +
LPS - + + + + + + +
Rp cAMP (uM) - - 100 - 1 10 50 100
A
B
C
FIGURE 7. Role of cAMP in morphine-withdrawal-mediated effect onIL-12 40 promoter activity. A, Effect of morphine withdrawal on intracellularcAMP in CRL-2019 macrophages. B, Comparison of cAMP levels in mor-phine-withdrawn cells to cells chronically treated with morphine. C, Effect ofcAMP antagonist Rp cAMP on morphine-withdrawal-mediated decreases inIL-12p40 protein synthesis. A, CRL-2019 macrophages were subjected to mor-phine withdrawal as described above. At 5, 10, and 30 min following with-drawal, cells were washed in PBS and spun and resuspended at a final densityof 1 � 107 cells/ml and lysed in cell lysis buffer. The supernatant of lysed cellswas analyzed using cAMP assay kits (��, p � 0.01). B, CRL-2019 macro-phages were subjected to either morphine withdrawal or chronically treatedwith morphine for 24 h. At 30 min following morphine withdrawal or 24 hfollowing morphine treatment, cells were washed in PBS and spun and resus-pended at a final density of 1 � 107 cells/ml and lysed in cell lysis buffer. Thesupernatant of lysed cells was analyzed using cAMP assay kits (�, p � 0.01).C, CRL-2019 macrophages were subjected to either vehicle withdrawal ormorphine withdrawal and immediately treated with varying concentrations ofRp cAMP (10–100 �M) for 1 h. Cells were then stimulated with LPS (5�g/ml) for 24 h. Culture supernatants were collected and IL-12p40 proteinlevels were determined using ELISA. Each treatment group was tested intriplicate and results are the mean of three independent experiments. Compar-isons between groups were assessed using an unpaired Student’s t test.
3676 MORPHINE WITHDRAWAL AND IL-12 SYNTHESIS
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
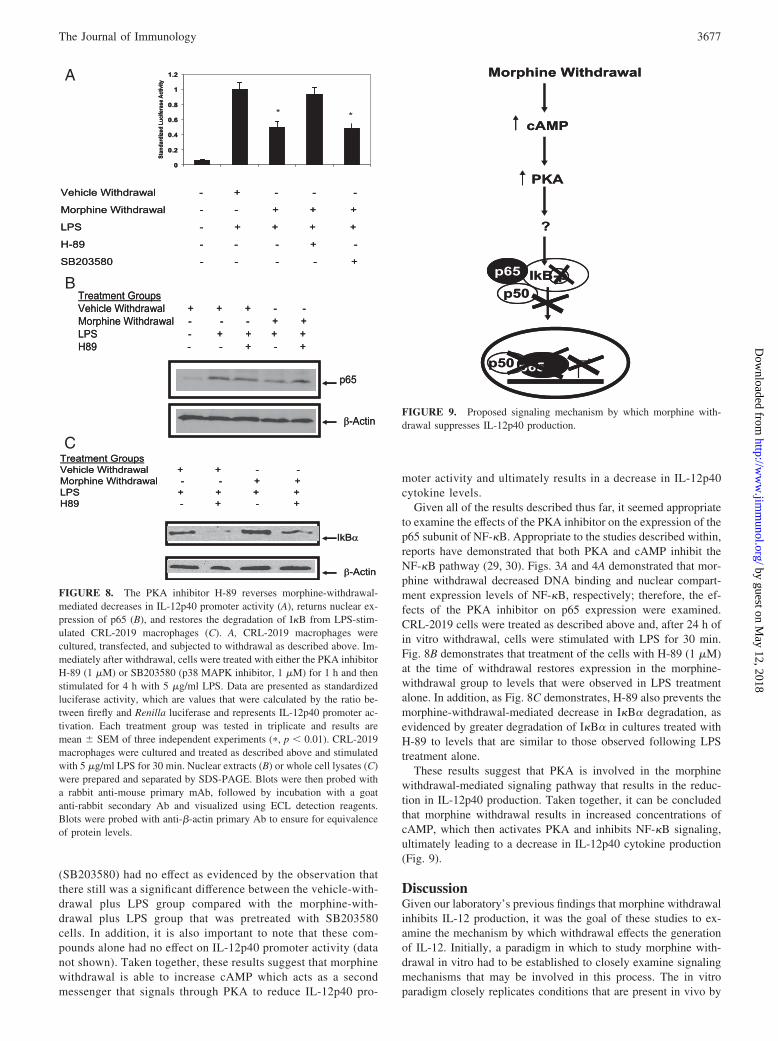
(SB203580) had no effect as evidenced by the observation thatthere still was a significant difference between the vehicle-with-drawal plus LPS group compared with the morphine-with-drawal plus LPS group that was pretreated with SB203580cells. In addition, it is also important to note that these com-pounds alone had no effect on IL-12p40 promoter activity (datanot shown). Taken together, these results suggest that morphinewithdrawal is able to increase cAMP which acts as a secondmessenger that signals through PKA to reduce IL-12p40 pro-
moter activity and ultimately results in a decrease in IL-12p40cytokine levels.
Given all of the results described thus far, it seemed appropriateto examine the effects of the PKA inhibitor on the expression of thep65 subunit of NF-�B. Appropriate to the studies described within,reports have demonstrated that both PKA and cAMP inhibit theNF-�B pathway (29, 30). Figs. 3A and 4A demonstrated that mor-phine withdrawal decreased DNA binding and nuclear compart-ment expression levels of NF-�B, respectively; therefore, the ef-fects of the PKA inhibitor on p65 expression were examined.CRL-2019 cells were treated as described above and, after 24 h ofin vitro withdrawal, cells were stimulated with LPS for 30 min.Fig. 8B demonstrates that treatment of the cells with H-89 (1 �M)at the time of withdrawal restores expression in the morphine-withdrawal group to levels that were observed in LPS treatmentalone. In addition, as Fig. 8C demonstrates, H-89 also prevents themorphine-withdrawal-mediated decrease in I�B� degradation, asevidenced by greater degradation of I�B� in cultures treated withH-89 to levels that are similar to those observed following LPStreatment alone.
These results suggest that PKA is involved in the morphinewithdrawal-mediated signaling pathway that results in the reduc-tion in IL-12p40 production. Taken together, it can be concludedthat morphine withdrawal results in increased concentrations ofcAMP, which then activates PKA and inhibits NF-�B signaling,ultimately leading to a decrease in IL-12p40 cytokine production(Fig. 9).
DiscussionGiven our laboratory’s previous findings that morphine withdrawalinhibits IL-12 production, it was the goal of these studies to ex-amine the mechanism by which withdrawal effects the generationof IL-12. Initially, a paradigm in which to study morphine with-drawal in vitro had to be established to closely examine signalingmechanisms that may be involved in this process. The in vitroparadigm closely replicates conditions that are present in vivo by
0
0.2
0.4
0.6
0.8
1
1.2
A B C D E
Stan
dard
ized
Luci
fera
se A
ctiv
ity
* *
Vehicle Withdrawal - + - - -
Morphine Withdrawal - - + + +
LPS - + + + +
H-89 - - - + -
SB203580 - - - - +
**
0
0.2
0.4
0.6
0.8
1
1.2
A B C D E
Stan
dard
ized
Luci
fera
se A
ctiv
ity
**
Vehicle Withdrawal - + - - -
Morphine Withdrawal - - + + +
LPS - + + + +
H-89 - - - + -
SB203580 - - - - +
**
p65
β-Actin
Treatment GroupsVehicle Withdrawal + + + - -Morphine Withdrawal - - - + +LPS - + + + +H89 - - + - +
p65
β-Actin
Treatment GroupsVehicle Withdrawal + + + - -Morphine Withdrawal - - - + +LPS - + + + +H89 - - + - +
IkBα
β-Actin
Treatment GroupsVehicle Withdrawal + + - -Morphine Withdrawal - - + + LPS + + + + H89 - + - +
IkBα
β-Actin
Treatment GroupsVehicle Withdrawal + + - -Morphine Withdrawal - - + + LPS + + + + H89 - + - +
A
B
C
FIGURE 8. The PKA inhibitor H-89 reverses morphine-withdrawal-mediated decreases in IL-12p40 promoter activity (A), returns nuclear ex-pression of p65 (B), and restores the degradation of I�B from LPS-stim-ulated CRL-2019 macrophages (C). A, CRL-2019 macrophages werecultured, transfected, and subjected to withdrawal as described above. Im-mediately after withdrawal, cells were treated with either the PKA inhibitorH-89 (1 �M) or SB203580 (p38 MAPK inhibitor, 1 �M) for 1 h and thenstimulated for 4 h with 5 �g/ml LPS. Data are presented as standardizedluciferase activity, which are values that were calculated by the ratio be-tween firefly and Renilla luciferase and represents IL-12p40 promoter ac-tivation. Each treatment group was tested in triplicate and results aremean � SEM of three independent experiments (�, p � 0.01). CRL-2019macrophages were cultured and treated as described above and stimulatedwith 5 �g/ml LPS for 30 min. Nuclear extracts (B) or whole cell lysates (C)were prepared and separated by SDS-PAGE. Blots were then probed witha rabbit anti-mouse primary mAb, followed by incubation with a goatanti-rabbit secondary Ab and visualized using ECL detection reagents.Blots were probed with anti-�-actin primary Ab to ensure for equivalenceof protein levels.
Morphine Withdrawal
cAMP
PKA
?
IkB-p
p65
p65
p50
p50
Morphine Withdrawal
cAMP
PKA
?
IkB-p
p65
p65
p50
p50
FIGURE 9. Proposed signaling mechanism by which morphine with-drawal suppresses IL-12p40 production.
3677The Journal of Immunology
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

maintaining the same treatment time course and using plasma con-centrations of morphine that were observed following in vivo ma-nipulations, i.e., surgical s.c. implantation of 75 mg of morphinepellets. This treatment paradigm referred to throughout as in vitromorphine withdrawal was a successful recapitulation of in vivoconditions, given the evidence that withdrawal decreased both IL-12p70 and p40 cytokine levels when compared with vehicle-with-drawal plus LPS treatment. This model was further extended to themouse alveolar macrophage cell line CRL-2019 due to the fact thattransfection experiments needed to be performed and this cell lineprovided an accessible and efficient transfection model. Onceagain, in vitro morphine withdrawal decreased both the protein andmessage levels of IL-12p40 as compared with vehicle-withdrawalplus LPS treatment. Taken together, these results indicate that thein vitro model developed was adequate and replicated what hadbeen observed in vivo.
Using this model, we show that transcription of IL-12p40 wassignificantly reduced following withdrawal and that this reductioncorrelated with decreased binding of both NF-�B and PU.1 to con-sensus oligonucleotide sequences. Furthermore, we also show de-creased translocation of the active form of NF-�B, a p65 subunitinto the nucleus of cells that are morphine withdrawn. Interest-ingly, this decrease in p65 translocation in the morphine-with-drawal treatment group was accompanied by a concurrent increasein cytosolic levels of I�B�, suggesting that withdrawal may beinhibiting the degradation of I�B� (Fig. 4B). Hindering degrada-tion of I�B� would therefore prevent the dissociation betweenI�B� and p65 and would therefore sequester p65 in the cytoplas-mic compartment. This hypothesis was validated since overexpres-sion of p65 only partially rescued morphine-withdrawal-inducedinhibition of IL-12 promoter activity; however, overexpression ofboth p50 and p65 completely reversed the observed inhibition bymorphine withdrawal. The idea that morphine withdrawal is hin-dering the degradation of I�B� and ultimately resulting in a de-crease in IL-12p40 production is a worthy explanation given theobservations that both vasoactive intestinal peptide and pituitaryadenylate cyclase-activating polypeptide act via a similar mecha-nism to reduce IL-12p40 production (31). These authors demon-strated that treatment of IFN-�-primed and LPS-stimulated RAW264.7 macrophages with these compounds resulted in decreasedI�B degradation accompanied by decreased expression levels ofp65 in the nuclear compartment. It was concluded that this may beone of the mechanisms by which these compounds act to decreaseIL-12p40 production.
Given the expression pattern results for NF-�B and I�B� fol-lowing morphine withdrawal, we sought to determine the mecha-nism by which morphine withdrawal is effecting these alterations.We show that, following both chronic morphine treatment andmorphine withdrawal, there is an increase in the second messengercAMP (32). This phenomenon known as adenylyl cyclase super-activation is well characterized in the CNS, and it is speculated thatit is a process that works to counteract the effects of acute mor-phine treatment and is also considered the cellular hallmark ofmorphine tolerance (28). Furthermore, this process has been dem-onstrated to occur in cells of the immune system following chronicmorphine treatment (32). We further show that Rp cAMP, a cAMPantagonist, significantly reversed morphine-induced inhibition in aconcentration-dependent manner. It is also well documented thatcAMP and cAMP- elevating agents are known to decrease theproduction of IL-12 (11, 19, 27). Taken together, it can be spec-ulated that it is the ability of morphine withdrawal to increase theintracellular concentrations of cAMP, which ultimately decreasesIL-12 production possibly via a NF-�B pathway. It can be con-cluded that the cAMP-modulating effects of morphine withdrawal
may be decreasing IL-12p40 promoter activity and ultimately re-sulting in decreased cytokine levels of IL-12p40.
Next, we sought to link the effects morphine withdrawal has oncAMP with what was observed with regard to the NF-�B pathway.Previous studies have demonstrated that cAMP is a potent inhib-itor of the NF-�B pathway by blocking the phosphorylation andsubsequent degradation of I�B and that PKA is a downstreameffector in this process (16). In addition, it has also been demon-strated that increases in cAMP activate PKA which decreases thetransactivation potential of the p65 subunit of NF-�B. Interest-ingly, however, these studies demonstrated that PKA did not affectI�B� phosphorylation, degradation, nor NF-�B/DNA binding(26). Given these previous findings, the role of PKA in mor-phine-withdrawal-mediated IL-12p40 suppression was investi-gated. It was demonstrated that the PKA inhibitor H-89 pre-treatment before withdrawal was able to restore both IL-12p40promoter activity, as well as p65 translocation into the nucleus.Furthermore, it was demonstrated that H-89 was able to preventthe decrease in I�B� degradation, as evidenced by a return ofexpression levels of I�B� similar to those observed following LPStreatment alone. The mechanism by which PKA is acting is yetunknown but it can be speculated that there are several possiblymechanisms. First, PKA may be activating a phosphatase whichprevents the phosphorylation and hence degradation of I�B. Sec-ond, PKA may be altering the activity of IKKs, the kinase whichphosphorylates I�B. Finally, PKA may interrupt the ubiquitinationof I�B, which is a crucial signal ultimately resulting in the deg-radation of the protein. Taken together, it can be concluded thatmorphine withdrawal acts to increase cAMP, which activatesPKA, thus activating a yet unknown protein or series of proteinswhich ultimately prevents the degradation of I�B. The increasedstabilization of the I�B/NF-�B-p65 complex prevents nucleartranslocation of p65 and thus decreases the transactivation of IL-12p40 and ultimately hinders the production of IL-12p40 follow-ing LPS stimulation (Fig. 9).
In conclusion, it was the goal of these studies to understand themolecular mechanisms by which morphine withdrawal decreasesIL-12 production. It was demonstrated using an in vitro model ofwithdrawal that the increases in cAMP generated following with-drawal may be acting through PKA to inhibit the NF-�B pathwayand thus IL-12p40 production. Understanding the mechanisms bywhich morphine withdrawal brings about immune system failure isa worthwhile endeavor given the wide use and abuse of morphine.
AcknowledgmentsWe thank Dr. A. Kumar for providing us with the IL-12p40 promoter-luciferase reporter plasmid and Richard Charboneau for technical support.
DisclosuresThe authors have no financial conflict of interest.
References1. Roy, S., J. Wang, J. Keschenbach, L. Koodie, and J. Martin. 2006. Modulation of
immune function by morphine: implications for susceptibility to infection.J. Neuroimmune Pharm. 1: 77–89.
2. Sharp, B. M., S. Roy, and J. M. Bidlack. 1998. Evidence for opioid receptors oncells involved in host defense and the immune system. J. Neuroimmunol. 183:45–56.
3. Friedman, H., C. Newton and T. W. Klein. 2003. Microbial infections, immu-nomodulation, and drugs of abuse. Clin. Microbiol. Rev. 16: 209–219.
4. Quaglio, G., F. Lugoboni, G. Talamini, A. Lechi, and P. Mezzelani. 2002. Prev-alence of tuberculosis infection and comparison of multiple-puncture liquid tu-berculin test and Mantoux test among drug users. Scand. J. Infect. Dis. 34:574–576.
5. Georges, H., O. Leroy, C. Vandenbussche, B. Guery, S. Alfandari, L. Tronchon,and G. Beaucaire. 1999. Epidemiological features and prognosis of severe com-munity-acquired pneumococcal pneumonia. Intensive Care Med. 25: 198–206.
3678 MORPHINE WITHDRAWAL AND IL-12 SYNTHESIS
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

6. Nath, A., K. F. Hauser, V. Wojna, R. M. Booze, W. Maragos, M. Prendergast,W. Cass and J. T. Turchan. 2002. J. Acquired Immune Defic. Syndr. 31(Suppl. 2):S62–S69.
7. Tomei, E. Z., and F. L. Renaud. 1997. Effect of morphine on Fc-mediated phago-cytosis by murine macrophages in vitro. J. Neuroimmunol. 74: 111–116.
8. Lugo-Chinchilla, A. M., D. Baez, M. Velez, C. Ildefonso, and F. L. Renaud.2006. Altered subcellular signaling in murine peritoneal macrophages uponchronic morphine exposure. J. Neuroimmunol. 176: 86–94.
9. Rahim, R. T., M. W. Adler, J. J Meissler, Jr., A. Cowan, T. J. Rogers,E. B. Geller, and T. K. Eisenstein. 2002. Abrupt or precipitated withdrawal formmorphine induces immunosuppression. J. Neuroimmunol. 127: 88–95.
10. Rahim, R. T., J. J. Meissler, Jr., L. Zhang, M. W. Adler, T. J. Rogers, andT. K. Eisenstein. 2003. Withdrawal from morphine in mice suppresses splenicmacrophage function, cytokine production, and costimulatory molecules. J. Neu-roimmunol. 144: 16–27.
11. Feng, P., J. J. Meissler, Jr., M. W. Adler, and T. K Eisenstein. 2005. Morphinewithdrawal sensitizes mice to lipopolysaccharide: elevated TNF-� and nitric ox-ide with decreased IL-12. J. Neuroimmunol. 164: 57–65.
12. Feng, P., A. L. Truant, J. J Meissler, J. P. Gaughan, M. W. Adler, andT. K. Eisenstein. 2006. Morphine withdrawal lowers host defense to enteric bac-teria: spontaneous sepsis and increased sensitivity to oral Salmonella entericaserovar Typhimurium infection. Infect. Immun. 74: 5221–5226.
13. Kelschenbach. J., R. A. Barke, and S. Roy. 2005. Morphine withdrawal contrib-utes to T helper cell differentiation by biasing cells toward the Th2 lineage.J. Immunol. 175: 2655–2665.
14. Gately, M. K., L. M. Renzetti, J. Magram, A. S. Stern, L. Adorni, U. Gubler, andD. H. Presky. 1998. The interleukin-12/interleukin-12 receptor system: role innormal and pathologic immune responses. Annu. Rev. Immunol. 16: 495–521.
15. Gillessen, S., D. Carvajal, P. Ling, F. J. Podlaski, D. L. Stremlo, P. C. Familletti,U. Gubler, A. S. Stern and M. K. Gately. 1995. Mouse interleukin 12 p40 ho-modimer: a potent IL-12 antagonist. Eur. J. Immunol. 25: 200–206.
16. Hasko, G., and C. Szabo. 1999. IL-12 as a therapeutic target for pharmacologicalmodulation in immune-mediated and inflammatory diseases: regulation of Thelper 1/T helper 2 responses. Br. J. Pharmacol. 127: 1295–1304.
17. Hwang, C. K., C. S Kim, H. S. Choi, S. R. McKercher, and H. H. Loh. 2004.Transcriptional regulation of mouse � opioid receptor gene by PU.1. J. Biol.Chem. 279: 19764–19774.
18. Kang, B. Y., E. Kim, and T. S. Kim. 2005. Regulatory mechanisms and theirtherapeutic implications of interleukin-12 production in immune cells. Cell. Sig-nal. 17: 665–673.
19. Gubler, U., A. O. Chua, D. S. Schoenhaut, C. M. Dwyer, W. McComas,R. Motyka, N. Nabavi, A. G. Wolitzky, P. M. Quinn, and P. C. Familletti. 1991.Coexpression of two distinct genes is required to generate secreted bioactivecytotoxic lymphocyte maturation factor. Proc. Natl. Acad. Sci. USA 88:4143–4147.
20. Aste-Amezaga, M., X. Ma, A. Sartori, and G. Trinchieri. 1998. Molecular mech-anisms of the induction of IL-12 and its inhibition by IL-10. J. Immunol. 160:5936–5944.
21. Ma, W., K. Gee, W. Lim, K. Chambers, J. B. Angel, M. Kozlowski, andA. Kumar. 2004. Dexamethasone inhibits IL-12 p40 production in lipopolysac-charide-stimulated human monocytic cells by down-regulating the activity ofc-Jun N-terminal kinase, the activation protein-1, and NF-�B transcription fac-tors. J. Immunol. 172: 318–330.
22. Bonizzi, G., and M. Karin. 2004. The two NF-�B activation pathways and theirrole in innate and adaptive immunity. Trends Immunol. 25: 280–288.
23. Mason, N., J. Aliberti, J. C. Caamano, H. Liou, and C. A. Hunter. 2002. Cuttingedge: identification of c-Rel dependent and independent pathways of IL-12 pro-duction during infectious and inflammatory stimuli. J. Immunol. 168:2590–2594.
24. Murphy, T. L., M. G. Cleveland, P. Kulesza, J. Magram, and K. M. Murphy.1995. Regulation of interleukin 12 p40 expression through an NF-�B half site.Mol. Cell. Biol. 15: 5258–5267.
25. Plevy, S. E., J. H. Gemberling, S. Hsu, A. J. Dorner, and S. T. Smale. 1997.Multiple control elements mediate activation of the murine and human interleukin12 p40 promoters: C/EBP and Rel proteins. Mol. Cell. Biol. 17: 4572–4588.
26. Sanjabi, S., A. Hoffmann, H. C. Liou, D. Baltimore, and S. T. Smale. 2000.Selective requirement for c-Rel during IL-12p40 gene induction in macrophages.Proc. Natl. Acad. Sci. USA 97: 12705–12710.
27. Zhu, C., K. Gagnidze, J. H. Gemberling, and S. E. Plevy. 2001. Characterizationof an activation protein-1 binding site in the murine interleukin-12 p40 promoter:demonstration of novel functional elements by a reductionist approach. J. Biol.Chem. 276: 18519–18528.
28. Avidor-Reiss, T., L. Nevo, R. Levy, T. Pfeuffer, and Z. Vogel. 1996. Chronicopioid treatment induces adenylyl cyclase V superactivation: involvement ofG��. J. Biol. Chem. 271: 21309–21315.
29. Minguet, S., M. Huber, L. Rosenkrans, W. W. A. Schamel, M. Reth, andT. Brummer. 2005. Adenosine and cAMP are potent inhibitors of the NF-�Bpathway downstream of immunoreceptors. Eur. J. Immunol. 35: 31–41.
30. Takashi, N., T. Tetsuka, H. Uranishi, and T. Okamoto. 2002. Inhibition of theNF-�B transcriptional activity by protein kinase A. Eur. J. Biochem. 269:4559–4565.
31. Delgado, M., and D. Ganea. 1999. Vasoactive intestinal peptide and pituitaryadenylate cyclase-activating polypeptide inhibit interleukin-12 transcription byregulating nuclear factor �B and Ets activation. J. Biol. Chem. 274:31930–31940.
32. Roy, S., J. Wang, R. Charboneau, H. H. Loh, and R. A. Barke. 2005. Morphineinduces CD4� T cell IL-4 expression through an adenylyl cyclase mechanismindependent of the protein kinase A pathway. J. Immunol. 175: 6361–6367.
3679The Journal of Immunology
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from