msa & rooted/unrooted tree
TRANSCRIPT


"Phylogenetics" is the study or estimation of the evolutionary history that underlies that biological diversity.
The results of phylogenetic analysis are usually presented as a collection of nodes and branches. That is, a tree
In such tree, taxa that are closely related in an evolutionary sense appear close to each other, and taxa that are distantly related are in different (far) branches of the trees
Phylogenetic trees are also important for multiple sequence alignment
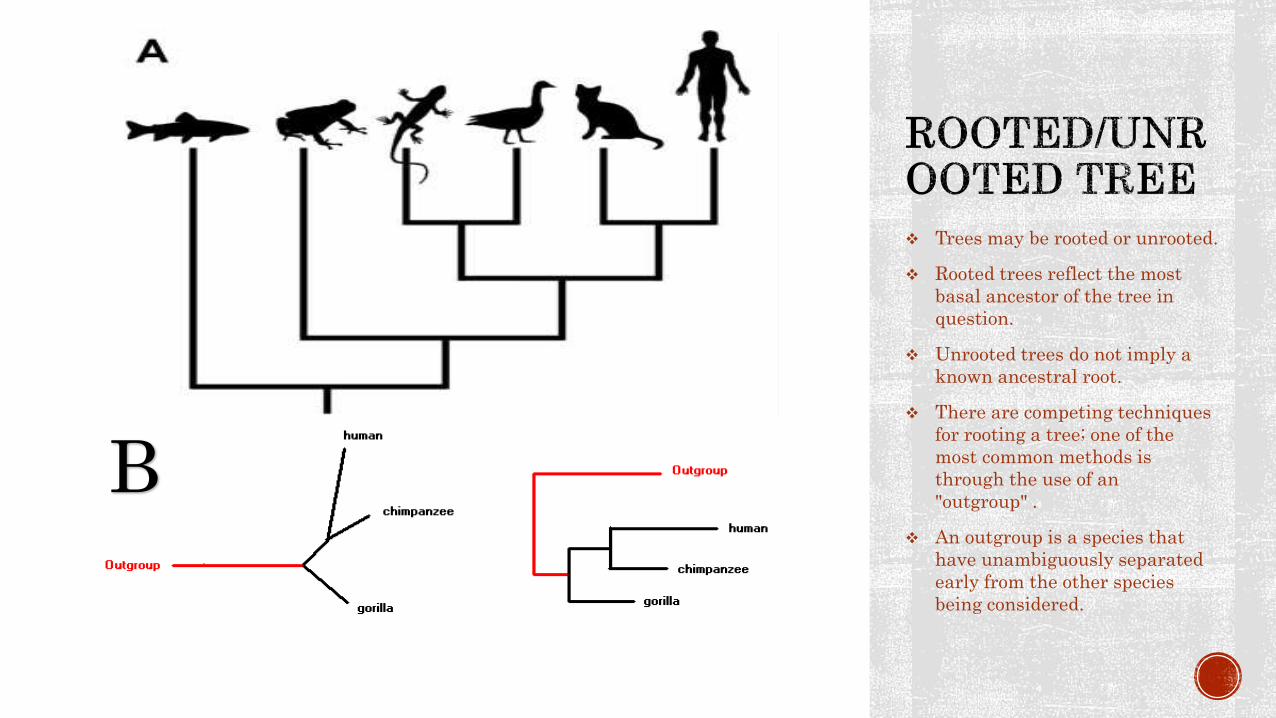
Trees may be rooted or unrooted.
Rooted trees reflect the most
basal ancestor of the tree in
question.
Unrooted trees do not imply a
known ancestral root.
There are competing techniques
for rooting a tree; one of the
most common methods is
through the use of an
"outgroup" .
An outgroup is a species that
have unambiguously separated
early from the other species
being considered.
B

Multiple sequence alignment can be viewed as an extension of pairwise sequence alignment, but the complexity of the computation grows exponentially with the number of sequences.
MSA applies both to nucleotide and amino acid sequences
One of the most essential tools in molecular biology that is used since 1987.
MSA can help us to reveal biological facts about proteins, like analysis of the secondary/tertiary structure.
MSA helps us to do a phylogenetic analysis of the sequences so as to construct evolutionary trees.

Exhaustive search: extension of DP to multiple dimensions.
Progressive alignment: compute tree of sequences, based on hierarchical clustering, and then merge closest first, greedily. E.g. ClustalW
Block-based global alignment find highly conserved regions and then grow alignment around these regions. E.g. BLAST
Iterative search: based on genetic algorithm search.
• Local alignments Profile analysis
Block analysis
Patterns searching and/or Statistical methods

VTISCTGSSSNIGAG-NHVKWYQQLPG
VTISCTGTSSNIGS--ITVNWYQQLPG
LRLSCSSSGFIFSS--YAMYWVRQAPG
LSLTCTVSGTSFDD--YYSTWVRQPPG
PEVTCVVVDVSHEDPQVKFNWYVDG--
ATLVCLISDFYPGA--VTVAWKADS--
AALGCLVKDYFPEP--VTVSWNSG---
VSLTCLVKGFYPSD--IAVEWWSNG--

Alignment of 2 sequences is represented as a
2-row matrix
In a similar way, we represent alignment of 3 sequences as a 3-row matrix
A T _ G C G _
A _ C G T _ A
A T C A C _ A
Score: more conserved columns, better alignment
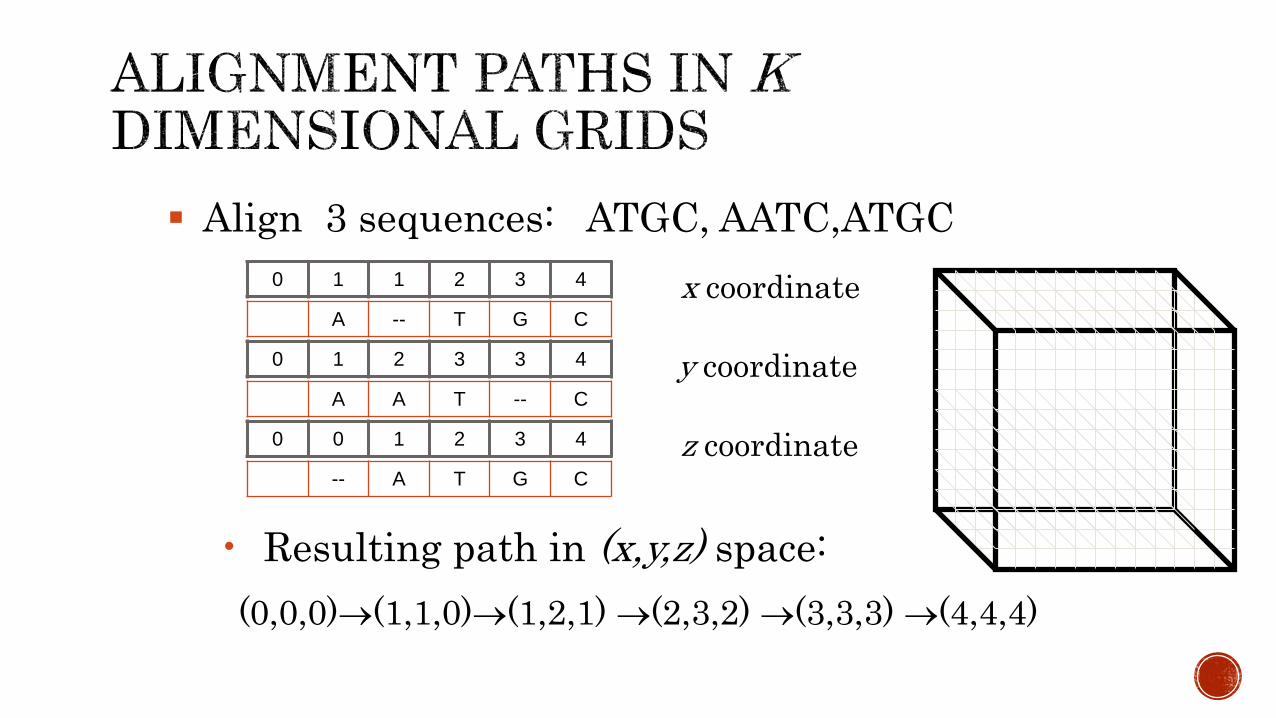
Align 3 sequences: ATGC, AATC,ATGC
0 1 1 2 3 4
0 1 2 3 3 4
A A T -- C
A -- T G C
0 0 1 2 3 4
-- A T G C
• Resulting path in (x,y,z) space:
(0,0,0)(1,1,0)(1,2,1) (2,3,2) (3,3,3) (4,4,4)
x coordinate
y coordinate
z coordinate
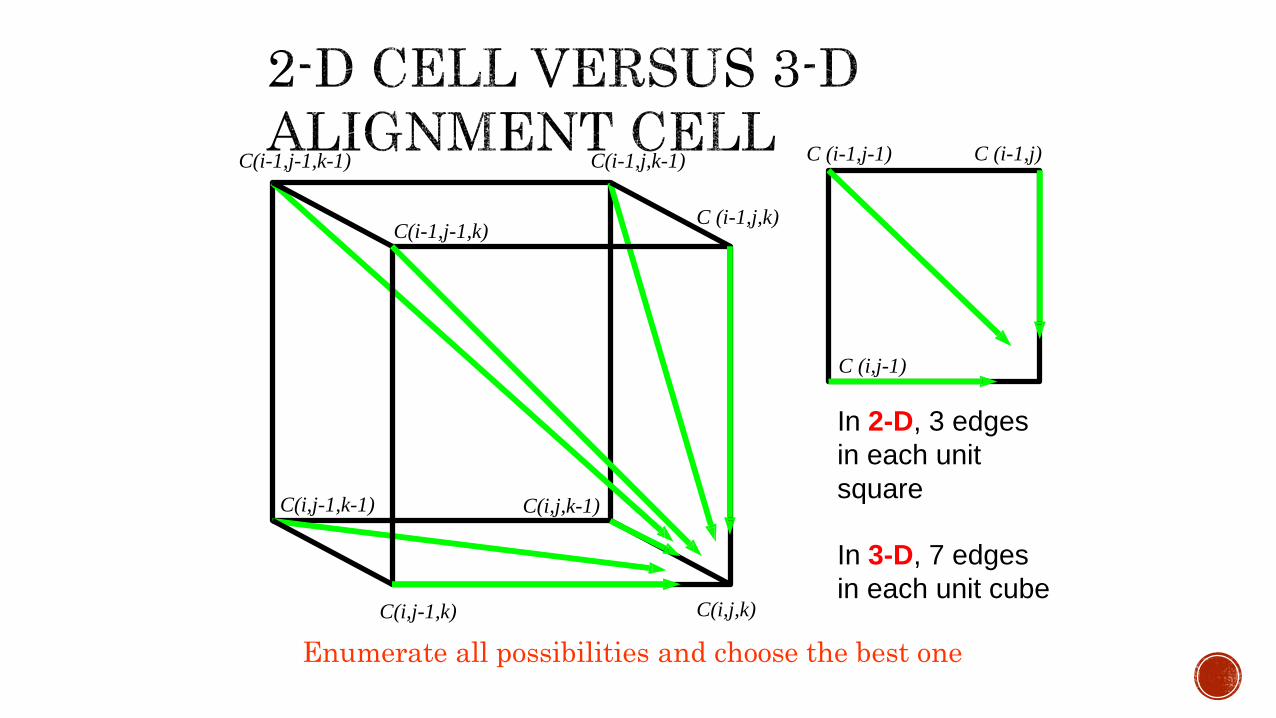
In 3-D, 7 edges
in each unit cube
In 2-D, 3 edges
in each unit
square
C(i-1,j-1,k-1) C(i-1,j,k-1)
C(i,j-1,k)
C(i-1,j-1,k)C (i-1,j,k)
C(i,j,k)
C(i,j,k-1)C(i,j-1,k-1)
Enumerate all possibilities and choose the best one
C (i-1,j-1) C (i-1,j)
C (i,j-1)

For three sequences of length n, the run time is proportional to the number of edges in the 3-D grid. i. e 7n .
For a k-way alignment, build a k-dimensional Manhattan graph with n nodes
Most nodes have 2 -1 incoming edges
Runtime: 0(2 n )
Consider 2 protein sequences of 100 amino acids in length.
If it takes 1002 (103) seconds to exhaustively align these sequences, then it will take 104 seconds to align 3 sequences, 105 to align 4 sequences, etc.
It will take ~1021 seconds to align 20 sequences. One year is ~3x107 seconds. The age of the visible universe is ~.4x1018 seconds.
k
k k
k

Greedy method follows the problem solving heuristic of
making the locally optimal choice at each stage of ksequences with the hope of finding a global optimum to
an alignment of of k-1 sequences/profiles.
u1= ACGTACGTACGT…
u2 = TTAATTAATTAA…
u3 = ACTACTACTACT…
…
uk = CCGGCCGGCCGG
u1= ACg/tTACg/tTACg/cT…
u2 = TTAATTAATTAA…
…
uk = CCGGCCGGCCGG…
k
k-1

• Consider these 4 sequences
s1 GATTCA
s2 GTCTGA
s3 GATATT
s4 GTCAGC

• There are = 6 possible alignments2
4
s2 GTCTGA
s4 GTCAGC (score = 2)
s1 GAT-TCA
s2 G-TCTGA (score = 1)
s1 GAT-TCA
s3 GATAT-T (scores3 GAT-ATT
= 1) s4 G-TCAGC
s1 GATTCA--
s4 G—T-CAGC(score = 0)
s2 G-TCTGA
s3 GATAT-T (score = -1)
(score = -1)
Match= +1
Mismatch/gap= -1

s2 and s4 are closest; combine:
s2 GTCTGA
s4 GTCAGCs GTCt/aGa/c2,4(profile)
s1 s3s2,4
GATTCA GATATTGTCt/aGa/c
new set of 3 sequences:

s1
s3
s2,4
GATTCA GATATTGTCt/aGa/c
s1 GATTC- - A
s2,4 G -T -CTGA(score = 0)
s3 GATATT -
s2,4 G -TCTGA(score = -1)
s1 and s2,4 are closest; combine:
s1 GATTC- - A
S2,4 G -T -CTGAS1,2,4 Ga/-Tt/-ct/-g/-A
s3
S1,2,4
GATATTGa/-Tt/-ct/-g/-A
s3 GATAT –T- -
S1,2,4 GAT-TCTGA(score = 1)
S1,2,3,4 GATa/-Tc/-Tg/-a/-
Final Alignment:

Computationally complex
If msa includes matches, mismatches and gaps and alsoaccounts the degree of variation then msa can be appliedto only a few sequences
Difficult to score
Multiple comparison necessary in each column of the msa for a cumulative score
Placement of gaps and scoring of substitution is more difficult
Difficulty increases with diversity
Relatively easy for a set of closely related sequences
Identifying the correct ancestry relationships for a setof distantly related sequences is more challenging
Even difficult if some members are more alike compared to others

EMBL-EBI http://www.ebi.ac.uk/clustalw/
BCM Search Launcher: Multiple Alignment
http://dot.imgen.bcm.tmc.edu:9331/multi-align/multi-align.html
Multiple Sequence Alignment for Proteins (Wash. U. St. Louis) http://www.ibc.wustl.edu/service/msa/
web.warwick.ac.uk/telri/Bioinfo/
http://science.marshall.edu/murraye/
http://www.cs.iastate.edu/~cs544/Lectures/
